Изобретение относится к пептидным соединениям, в частности LHRH-антагонистам, их получению и применению в качестве лекарств, далее к аминокислотам с азотом в боковой цепи и их получению (LHRH = Luteinizing Hormone Releasing Hormone - гормон выделения лютеинизирующего гормона).
Гормон выделения лютеинизирующего гормона пиро
Glu1-His2-Trp3-Ser4-Tyr5- Gly6-Leu7-Arg8-Pro9-Gly10- NH2
производство у млекопитающих в гипеталамусе. Он стимулирует в гипофизе высвобождение лютеинизирующего гормона (LH) и фолликулостимулирующего гормона (FSH). Последние в свою очередь контролируют производство андрогенов и эстрогенов в половых органах.
Введением разовой дозы LHRH или синтетических агонистов достигается увеличение производства стероидных гормонов (например тестостерона). Постоянное введение, напротив, приводит к снижению производства гормонов. Этот эффект используется с некоторого времени для лечения гормонозависимых опухолей (простата CA).
Сопровождающим эффектом этой терапии является начальная стимуляция гормонов, которые подлежат подавлению. Этот эффект, приводящий к временному росту опухоли у гормонозависимых опухолей, можно избежать применением - антагонистов. Картен и др. (Endoczine Rev., 7, 44, 1986) и Лутта (Drugs of the Fufure, 13, 761, 1988) описали разработку LHRH - антагонистов.
Действенные LHRH антагонисты, имеющие основные аминокислоты в положениях 6 и 8, выделяют, однако, нежелательно большие количества гистамина. Предпринимались различные усилия по снижению выделения гистамина. В заявке на Европейский патент EP-OS097 03 описываются производные аргинина в положении 6. В EP-OS 0277829 описаны основные производные аминокислот в положениях 6 и 8. В заявке EP-OS 0299402 раскрывается комбинация цитруллина в положении 6 с аргинином в положении 6 с аргинином в положении 8, причем выделяется довольно мало гистамина.
Теперь неожиданно найдено, что путем обмена положений 6 и 8 с производными лизина, замещенными у омега-азота, может быть достигнуто повышение эффективности или снижение выделения гистамина.
Согласно изобретению предлагаются Производные декапептидов общей формулы 1
X - X1-X2-X3-Ser-Tyr-X6-Leu- X8-Pro-X10
где
X = ацил с 1-7 атомами углерода
X1 = D-(1) - Nal D-(2) - Nal
X2 = D=Cpa,
X3 = D-(3)-Pal
X6 = D-Cit, D-Hci, D-Orn, D-Lys, D-Neu,
X8 = D-Orn, Arg, Lys, L-Neu,
X10 = D-AlaNH2
при этом Neu является остатком формулы IX или IV:
при этом Z1 является группой
n = 1-6.
Описание содержит сокращения, значения которых излагаются ниже. При этом соблюдаются правила, установленные комиссией ИЮПАК-МЮБ для биологической номенклатуры (Biochemistry 11: 1726 (1972)) и (Biochem. Y. 219; 345 (1984)). Дополнительно применяют следующие сокращения и их комбинации:
Ape - 2-амино-пентановая кислота
Ahx - 2-амино-гексановая кислота
Ahp - 2-амино-гептановая кислота
Aoc - 2-амино-октановая кислота
Ano - 2-амино-нонановая кислота
Mor - морфолин-4-ил
Pip - пиперидин-1-ил-
Pyr - пирролидин-1-ил-
Tht - тетрагидро-1,4-тиазин-4-ил-
Mpz - 4-метил-пиперазин-1-ил-
Pon - 4-пиперидон-1-ил
Hpi - 4-гидрокси-пиперидин-1-ил-
Aps - 4-аза-пентаметиленсульфон-4-ил
(1)-Nal - 3-(нафт-1-ил)-аланин
(2)-Nal - 3-(нафт-2-ил)-аланин
(3)-Pal - 3-(3-пиридил)-аланин
(3)-Qal - 3-(хинол-3-ил)-аланин
Hei - гомоцитруллин
1 Im - имидазол-1-ил-
4Tr - 1,3,4-триазол-4-ил-
1Tr - 1,3,4-триазол-1-ил-
1Py - пиразол-1-ил-
CpZ - 4-4-карбамоил-пиперазин-1-ил-
Cpa - 4-хлор-фенилаланин
Так, например Aoc (Mor) обозначает 6-морфолин-4-ил-2-амино-октановую кислоту, Ape (Pip) = 5-пиперидин-4-ил-2-амино-пентановую кислоту и Ahx (1 Im) = 6-(имидазол-1-ил)-2-аминогексановую кислоту.
Пептиды представлены в сокращенной форме, причем приводятся аминоксилоты, измененные в сравнении с LHRH, и их положение. Так, например, пиро Glu1-His2-Trp3-Ser4- Tyr5-D-Nal6-Leu7-Arg8-Pro8-D- Ala10-NH2
становится [D-Nal6-D-Ala10]LHRH
Упоминаемые в тексте алкильные группы являются линейными или разветвленными и обозначают метил, этил, пропил, изопропил, н-бутил, втор.-бутил, изобутил, трет.-бутил, пентил, изопентил, неопентил, гексил, изогексил, гептил, изогептил.
Преимущества пептидных соединений согласно изобретению состоят в том, что фармакологическое действие пептидных соединений очень высоко и побочные действия в форме выделения гистамина удерживаются на низком уровне.
Оптимальны пептидные соединения, в которых Neu обозначает остаток формулы IX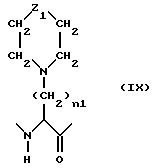
где
Z1 представляет собой группу
n1 представляет собой число от 3 до 6.
Предпочтительны пептидные соединения, у которых Neu обозначает группу формулы X
где
n1 представляет собой число от 3 до 6,
Более предпочтительны пептидные соединения, у которых Neu обозначает остаток.

Наиболее предпочтительны пептидные соединения, у которых D- или L-Neu обозначает X6 и X8.
Еще один предпочтительный вариант состоит в том, что Neu обозначает формулу XIII
Предпочтительно X6 и X8 означают Neu.
Наиболее предпочтительно пептидное соединение представляет собой
Ac-D-Nal-D-Cpa-D-Pal-Ser-Tyr-D-Cit-Leu-Axh (Mor)-Pro-D-Ala-NH2
Далее изобретение включает также производное аминокислот, встречающиеся в качестве структурных элементов в пептидных соединениях согласно изобретению.
Производные аминокислот относятся к группе общей формулы XVII
где
W представляет собой остаток
Z обозначает группу
n обозначает число от 1 до 6;
m обозначает число 2;
p обозначает число 2;
R3 обозначает H, защитную группу или карбонильную группу как часть пептидной связи пептидной цепи,
R4 обозначает O - R4 или аминогруппу как часть пептидной связи пептидной цепи,
W обозначает остаток
Защитные группы описаны в энциклопедии Губен-Вейля (1974) издательство Георг Тиме, 4-е издание, перечисление защитных групп в указателе литературы частично раскрывает изобретение.
Предпочтительны производные аминокислот с общей формулой XVIII.

где
Z1 представляет собой группу 
n обозначает число от 3 до 6;
R3 и R4 имеют вышеуказанные значения.
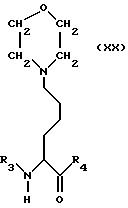
Более предпочтительны производные аминокислот с общей формулой XIX
где
n1 обозначает число от 3 до 6;
R3 и R4 имеют вышеуказанные значения.
Наиболее предпочтительны производные аминокислот формулы XX, причем остатки R3 и R4 имеют вышеуказанные значения.
Другие варианты предусматривают производные аминокислот согласно изобретению, имеющие общую формулу XXII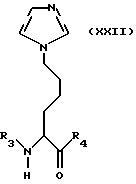
причем остатки R3 и R4 имеют вышеуказанные значения.
Применение пептидных соединений:
а) Изобретение включает фармацевтические композиции
Фармацевтические композиции обладают LHRH-антагонистической активностью и содержат пептидное соединение и целевые добавки. При этом композиции содержат производные декапептидов общей формулы I в эффективном количестве.
Производные декапептидных соединений согласно изобретению, их соли и смеси с фармацевтически приемлемыми вспомогательными веществами и носителями, имеют эффективное и длительное LHRH антагонистическое действие.
Пептидные соединения должны применяться при лечении доброкачественных гипертрофий простаты и карцином простаты. Поэтому проверяется тестостеронпонижающий потенциал. Для этого требуются в общем заметно более высокие дозировки одного и того же антагониста, чем для индукции торможения овуляции. В применяемом здесь способе испытания подкожно обрабатываются здоровые взрослые самцы крыс однократно исследуемым веществом. Воздействие на концентрацию сывороточного тестостерона через 24 часа определяются радиоиммунологически (инструментарий фирмы Бирманн).
Вещество примера 13 (пункт 6.1) индуцирует в дозировочных пределах от 0,5 до 5 мг/кг веса тела торможение концентрации сывороточного тестостерона на 80-97% в сравнении с контролем. Но и при дозе 0,25 мг/кг веса тела можно наблюдать еще торможение на 26%.
Наряду с понижением величины тестостерона имеет значение выделения гистамина. Так, ни при одной из испытанных дозировок (0,1 - 5 мг/кг веса тела подкожно) не было отечных изменений в лице и/или на конечностях, как это обычно имеет место при выделении гистамина. Этому тесту ин-виво придается существенно более высокое значение, чем многократно применяемому тесту на гранулярную соединительную ткань. Однако и этот последний тест на гранулярную соединительную ткань показывает, что при применении пептидного соединения примера 13 ЕД50 выделения гистамина при 0,01 мг/мл еще не достигнуто.
Другие способы испытания действенности LHPH-антагонистов включают: 1) ингибирование выделения FSH и LH в крысах, которое индуцируется гормоном LHPH (Vilchej-Martinej J.A. et al. (1975) Endocrinology. 96, 1130) и 2) ингибирование выделения LH и FSH распределенными ранними культурами клеток гипофиза, как это делается в радиоиммунном тесте (Vale et. al., Endocrinology, 91:562).
Действенность пептидных соединений согласно изобретению, изложенная выше, приводит к ряду вытекающих отсюда применений:
аа) терапия доброкачественной гипертрофии простаты;
бб) терапия болезней, вызываемых повышенным производством гормона репродуктивной железы в особей обоего пола, в частности карциномы простаты;
вв) лечение эндометриозы;
гг) контроль плодовитости у женщин;
дд) подавление овуляции или замедление овуляции;
ее) синхронизация овуляции;
жж) подавление эстроза;
зз) стимуляция роста самок животных;
ии) индукция менструации;
кк) ранний, в течение первых трех месяцев, аборт;
лл) лечение кист в груди;
мм) лечение поликистового синдрома яичника (Штейн-Левенталь);
нн) контроль плодовитости у мужчин;
оо) функциональная кастрация самцов животных в производстве мяса и
пп) подавление симптомов менопаузы.
Особенно предпочтительно лечение карциномы простаты и эндометриозы.
На практике человеку или животному, нуждающимся в таком лечении, вводятся эффективное количество пептидного соединения согласно формуле I или эффективное количество смеси, содержащей пептидное соединение формулы I и носитель и/или добавки. Пептидное соединение или смесь могут вводиться различными способами, т.е. орально, внутривенно, подкожно, внутримышечно, внутривагинально, ректально или назально. Соответствующий вид введения определяется формой лечения и дозой. Смотря по применению могут быть использованы форма депо, имплантат или лекарственная форма, медленно выделяющая активное вещество.
При лечении карциномы простаты у человека (лечение с высокими дозами) вводятся ежедневные дозы в пределах от 1 до 10, предпочтительно от 2 до 4 мг на человека.
Точная доза и форма введения зависят, в частности, от пептидного соединения согласно формуле I, от вида введения (путь в кровеносную систему) и от вида и тяжести заболевания.
б) Изобретение охватывает далее применение пептидного соединения согласно формуле I в соответствии с одним из пунктов аа) - пп). Изобретение охватывает также применение пептидного соединения согласно формуле I для изготовления лекарственного препарата для терапевтического применения по одному из пунктов аа) - пп). Изобретение также касается способа применения по одному из пунктов аа) - пп) у человека и млекопитающих животных, нуждающихся в таком применении, причем применение включает введение фармакологически надежного и эффективного количества пептидного соединения согласно формуле I человеку и млекопитающим.
в) Изобретение предпочтительно включает применение пептидного соединения согласно формуле I для лечения карциномы простаты. Изобретение также охватывает применение пептидного соединения согласно формуле I для применения лекарственного средства для лечения карциномы простаты. Также изобретение касается способа лечения карциномы простаты у человека и млекопитающих, нуждающихся в таком лечении, причем лечение включает введение фармакологически надежного и эффективного количества пептидного соединения согласно формуле I человеку и млекопитающим.
г) Изобретение предпочтительно охватывает применение пептидного соединения согласно формуле I для лечения эндометриозы. Изобретение касается также способа лечения эндометриозы у человека и млекопитающих, нуждающихся в таком лечении, причем лечение включает введение фармакологически надежного и эффективного количества пептидного соединения согласно формуле I человеку и млекопитающим.
д) Изобретение предпочтительно включает применение пептидного соединения согласно формуле I для контроля плодовитости. Изобретение также охватывает применение пептидного соединения согласно формуле 1 для изготовления лекарственного средства для контроля плодовитости у людей и млекопитающих, нуждающихся в таком лечении, причем лечение включает введение фармакологически надежного и эффективного количества пептидных соединений согласно формуле I человеку и млекопитающим.
Другим объектом изобретения является способ изготовления N-6-замещенных производных лизина общей формулы XXIV
где
R5 обозначает атом водорода или арилсульфонильный остаток общей формулы XXV
SO2-C6H5-R8, (XXV),
в которой
R8 имеет значение атома водорода или метильной группы и
где
R6 и R7 одинаковы или различны и обозначают максимально два атома водорода и/или максимально два содержащих до двенадцати углеродных атомов углеводородных остатка, возможно прерываемых максимально тремя атомами кислорода, азота или серы и/или замещенные максимально двумя гидрокси-, циано- и/или оксогруппами, отличающийся тем, что 3-амино-гексагидро-2-азепинон подвергают реакции в присутствии оснований с хлорангидридом арилсульфокислоты общей формулы XXVI
Cl - So2 - C6H5R8 (XXVI)
где
R8 имеет вышеуказанное значение,
образовавшийся 3-арилсульфонамидогексагидро-2-азепинон общей формулы XXVII
где
R8 имеет вышеуказанное значение,
расщепляют посредством минеральных кислот в N 6-назамещенное N2-арилсульфонилпроизводное общей формулы XXVIII
где
R8 имеет вышеуказанное значение, последнее по желанию N-алкилируют или N-ацилируют в N6- арилсульфониллизинпроизводной общей формулы XXIX
где
R9 и R10 имеют то же значение, что и R6 и R7 в той мере, что по меньшей мере один из заместителей R9 и R10 отличен от водорода и R8 имеет вышеуказанное значение, и это производное при необходимости переводят реакцией с натрием в аммиаке в N6-замещенное лизинпроизводное общей формулы XXX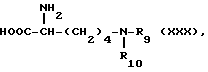
где
R9 и R10 имеют вышеуказанное значение.
Далее изобретение касается N6-замещенных N2-арилсульфонилпроизводных общей формулы XXIX, где R8, R9 и R10 имеют вышеуказанное значение, за исключением N6-бензилоксикарбонил-N2-тозил-лизина и N6, N6-диметил-N2-тозил-лизина.
Способ согласно изобретению позволяет синтезировать эти N6-замещенные лизин-производные общей формулы XXX простым путем с получением хороших выходов. Способ согласно изобретению может применяться универсально и годится для синтеза оптически активных лизин-производных общей формулы XXX высокой чистоты.
Исходными соединениями для способа согласно изобретению является 3-амино-гексагидро-2-азепинон (= α -амино- ε -капролактам), который представляет собой как рацемат, так и в форме его оптически активных антиподов готовый продукт. Этот лактам подвергают реакции в присутствии оснований с хлорангидридом арилсульфокислоты общей формулы XXVI (хлорангидрид бензолсульфокислоты или предпочтительно хлорангидрид п-толуолсульфокислоты) при хорошо известных специалисту условиях (Methoden der Organischen Chemic (Houben-Weyl); George Thieme Verlag, DE -Stuffgart; 4 Auflage, Band XV/1, 1974, seite 223).
Подходящим способом является, например, превращение лактама в водной фазе с избытком арилсульфонилхлорида в присутствии натровой щелочи.
Таким образом получают 3-арилсульфонамидо-гексагидро-2-азепинон общей формулы XXVII, который, как неожиданно оказалось, может быть расщеплен почти количественно в соответствующее N2-арилсульфониллизин-производное общей формулы XXVIII.
Подходящей минеральной кислотой является, например, соляная кислота с концентрацией до 12 вес.%. Целесообразно проводить реакции в кипящем растворе. Если применяют при этой реакции оптически активные 3-арилсульфонамидо-2-азепиноны, то получаются соответствующие оптически активные N2-арилсульфониллизин-производные.
N2-арилсульфониллизин-производне общей формулы XXVIII уже известны. Однако они применялись не для синтеза N6-замещенных лизин-производных общей формулы XXX, а служили для получения 3-арилсульфонамидо-гексагидро-2-азепинов общей формулы XXVII (J. Chem. Soc, 1957, 4830 - 4), или для получения оптически активной L-2-пиперидилкарбоновой кислоты (L-пипеколовая кислота; Bull. Chem. Soc. Japan 48, 1975, 1341-2).
N2-арилсульфониллизин-производные общей формулы XXIX могут быть преведены в соответствующие N2-арилсульфониллизин-производные общей формулы XXIX путем их N-алкилирования или N-ацилирования.
Для специалиста очевидно, что N6-замещенные N2-арилсульфониллизин-производные в качестве заместителей R6 и R7 могут содержать самые различные группы.
Так, например, заместитель R6 может быть атомом водорода. Заместитель R7 может быть углеводородным остатком с числом углеродных атомов до 12, возможно прерываемым максимально тремя атомами кислорода, азота или серы и/или замещенным максимально двумя гидроксигруппами и/или оксигруппами. Этот углеводородный остаток может быть насыщенным или ненасыщенным, а также алициклическим, циклическим или смешанно циклическим. Циклические или смешанно циклически-ациклические углеводороды могут содержать неароматические, ароматические и/или гетероциклические кольцевые системы.
Углеводороды, прерываемые кислородными атомами, - это например такие, которые присоединены в N6-аминогруппе лизина через карбонильную группу. С другой стороны, такими углеводородами могут быть также углеводороды, содержащие амидные группы или карбонилоксигруппы.
Углеводороды, прерываемые атомами азота, - это, например, углеводороды, содержащие диалкиламиногруппы, как например диметиламиногруппы, пирролиногруппы (Houben - Weyl, 4 Auflage, Band, XVII, 1974, стр. 293) или дибензиламиногруппы.
К остаткам этого рода относятся также ароматические N-гетероциклы или углеводороды, содержащие такого рода N-гетероциклы.
Углеводородные остатки, прерываемые атомами серы, - это например, тиоэфиры и такие углеводороды, которые содержат тиофеновые кольца.
С другой стороны, N6-замещенные N2-арилсульфониллизин-производные общей формулы XXIX в качестве заместителей R9 и R10 могут иметь также два из вышеуказанных органических остатка. Эти остатки могут быть одинаковыми или различными. Далее оба остатка R9 и R10 совместно могут обозначать углеводородный остаток, возможно прерванный атомами кислорода, азота или серы и/или замещенный гидроксигруппами и/или оксогруппами, и тем самым образовывать совместно с атомом азота предпочтительно пяти-семичленный гетероцикл, который в свою очередь снова может быть замещен вышеописанным образом.
N6-замещенные N2-арилсульфониллизин-производные общей формулы XXIX, - это, например, такие соединения общей формулы XXXI
где
R8 имеет вышеуказанное значение и оба остатка Ra обозначают соответственно алкильные группы с числом углеродных атомов до 4. Эти соединения, например, могут быть получены из N6-замещенных соединений общей формулы XXVIII реакцией с алкилбромидами или алкилиодидами при известных условиях (Houben - Weyl, 4 Auflage, Band XI/1, 1957, p. 24 ff).
Если хотят получить N, N-диметильные соединения общей формулы XXXI (Ra = CH3), это может быть сделано выгодно путем восстановительного алкилирования N6-незамещенного соединения формальдегидом и каталитически активированным водородом или формальдегидом в присутствии муравьиной кислоты (Houben-Weyl, 4 Auflage, Band XI/2, 1958, p. 330 и 331).
N6-замещенные N2-арилсульфониллизин-производные общей формулы XXIX - это далее соединения общей формулы XXXII
где R8 имеет вышеуказанное значение, R11 и R12 одинаковы или различны и обозначают атом водорода, цианогруппу, алкильную группу с максимально 6 углеродным атомами, возможно замещенную одной-тремя алкильными группами с максимально 4 углеродными атомами или одной-тремя алкоксигруппами с максимально 4 углеродными атомами фенольную группу или пиридильную группу и где (Z)n обозначает углерод-углеродную связь, метиленовую группу, кислородный атом или атом серы.
Такого рода соединения - это например соединения, которые замещены в 6-м положении пирролидиновой группой, 3-пиридинил-пирролидиновой группой, пиперидоновой группой, морфолиновой группой, 3-цианоморфолиновой группой, или 1,4-тетрагидротиазиновой группой.
Эти соединения могут быть получены восстановительным алкилированием из соответствующих дикарбонильных соединений общей формулы XXXIII
R11CO-CH2(Z)n-CH2COR12 (XXXIII),
где
R11, R12 и (Z)n имеют вышеуказанное значение, и N6-незамещенных N2-арилсульфониллизин-производных путем реакции этих компонентов с гидридами металлов, натрийборгидридом или, оптимально, натрийцианоборгидридом (Syuthesis, 1975, 135 - 146; J.Am. Chem. 29, 1986, 1225 - 1230, J. Org. Chem. 28, 1963, 3259 - 3261).
Так, например, это восстановительное алкилирование может быть проведено путем воздействия на компоненты в полярном - предпочтительно водосодержащем - инертном растворителе, как-то: триамид гексамметилфосфорной кислоты, ацетонитрил и т.д., или даже в воде при pH от 6 до 8 натрийцианборгидридом при комнатной температуре.
Достойные упоминания N6-замещенные N2-арилсульфониллизин-производные общей формулы XXIX - это также такие соединения общей формулы XXXIV
где
R8 имеет вышеуказанное значение и
где
R13 и R14 совместно с -CH-группой образуют 5- и/или 6-членную изоциклическую кольцевую систему или где
R13 обозначает алкильный остаток с числом углеродных атомов до 6 или возможно замещенный одной-тремя алкильными группами с числом углеродных атомов до 4 или одной-тремя алкоксигруппами с числом углеродных атомов до 4 фенильный остаток или пиридильный остаток и R14 имеет то же значение, что и R13 или представляет собой атом водорода.
Такого рода соединения являются, например, соединением общей формулы XXXIV, замещенным в 6-м положении алкиламиногруппой, как-то: метиламиногруппой, этиламиногруппой, пропиламиногруппой, изопропиламиногруппой, бутиламиногруппой, трет.-бутиламиногруппой, циклоалкиламиногруппой, как например, циелопентиламиногруппа, 2-адаматиламиногруппой или 1-фенилэтиламиногруппой.
Эти соединения могут быть получены восстановительным алкилированием из соответствующих карбонильных соединений общей формулы XXXV
R13-CO-R14 (XXXV),
где
R13 и R14 имеют вышеуказанное значение, и N6-незамещенных N2-арилсульфониллизин-производных путем реакции компонентов при уже упоминавшихся условиях, либо с металлгидридами, как-то: натрийборгидрид или натрийцианборгидрид, либо с каталитически активированным водородом.
Это может происходить, например, путем реакции компонентов в полярном растворителе, как-то: уксусная кислота, с натрийборгидридом при комнатной температуре.
В качестве N6-замещенных N-2-арилсульфониллизин-производных общей формулы XXXIV следует упомянуть еще N6-ацилированные соединения общей формулы XXXVI
где
R8 имеет вышеуказанное значение и R15 представляет собой остаток карбоновой кислоты R15COOH с максимально 12 углеродными атомами.
Такого рода остатки представляют собой, например, алкильные группы с числом углеродных атомов до 6, как-то: метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу или трет.-бутильную группу циклопентильную группу или циклогексильную группу, циклоалкильные группы, как-то: циклопентилметильную группу или 2-циклопропил-этильную группу или возможно замещенные гидроксигруппами одно-тремя алкильными группами с числом углеродных атомов до 4 или одной-термя алкоксигруппами с числом углеродных атомов до 4 фенильные остатки, 1- или 2-нафтильные остатки или пиридильные остатки.
Эти амиды синтезируются из соответствующих реакционноспособных производных карбоновых кислот, как например, хлорангидридов или ангидридов кислот, методами, хорошо известными специалисту (Houben-Weyl, Band XV/1, 1974).
Из N6-замещенных N2-арилсульфониллизин-производных общей формулы XXXIX можно отщепить сульфокислотные группы. Это осуществляется целесообразно путем реакции с натрием в кипящем аммиаке (Houben-Weyl, Band XV/1, 1974, p. 228 ff) и получают с хорошими выходами и хорошей оптической чистотой 6-заммещенные лизин-производные общей формулы XXX.
Для специалиста это неожиданно, так как общеизвестно, что "относительно сложное, осуществляемое лишь при жестких условиях и редко свободное от побочных реакций снятие N-тозильной маркировки все более и более приводит к тому, что отказываются от применения этой защитной группы - также и для блокировки, ω -амино- или соответственно гуанидо-функции - в пользу других остатков (Houben-Weyl, Band XV/1, 1974, p. 241).
Далее изобретение включает способ получения аминокислотного производного согласно изобретению, причем N-α- защищенная ω-амино-α- аминокислота подвергается взаимодействию с диальдегидом в присутствии восстановителя с последующим отщеплением защитной группы.
Изобретение также включает способ получения пептидного соединения согласно изобретению с применением известных аминокислотных производных и по меньшей мере одного аминокислотного производного согласно изобретению, который заключается в том, что аминокислотные производные конденсируют в однородной фазе или по твердофазному методу,
причем
а) карбоксильный конец сопрягаемого производного аминокислоты, аминогруппы и возможно функциональные группы боковой цепи которой несут защитную группу, реагирует со свободными аминным концом сопрягаемого аминокислотного производного или сопрягаемого пептидного фрагмента в присутствии реагента конденсации
и б) затем отщепляется α- амино-защитная группа сопряженного аминокислотного производного
и
возможно после вышеописанных обеих стадий к синтезируемой пептидной цепи присоединяют другие аминокислотные производные и после присоединения последней аминокислоты в случае твердофазного метода пептидное соединение отщепляется от твердой фазы.
Следующие примеры осуществления изобретения служат более подробному пояснению заявляемого способа.
Общий синтез пептидов.
Пептиды изобретения могут быть получены методами, которые уже известны специалистам в области синтеза пептидов. Сводку многих этих методов можно прочесть у Джей М.Стюарта и Джей Д.Янга, Сан-Франциско, 1969, и Джей, Мейенгофера Hormonal Proteins and Peptides, Vol. 2, p. 46 Academic Press (New Jork) 1973 по твердофазным методам и у Е.Шродера и К.Лубке Peptides, Vol 1, Academic Press (New-Jork, 1965, по жидкофазным методам, стадии синтеза описаны в EP-00097031.
Общие стадии процесса из европейской выложенной заявки могут быть по аналогии перенесены на синтезы описываемых здесь пептидных соединений согласно изобретению.
Более конкретно пептиды согласно изобретению получаются следующим образом.
Пептиды синтезируются на бензгидриламиновой смоле, содержащей около 0,5 миллиэкв NH2/г на ACT-синтезаторе, причем согласно описанному способу начинают с FMOC-D-Ala.
Сопряжения проводятся согласно схеме A следующим образом:
Схема A
Реагент
1. FMOC-аминокислота (2 - 3 ммоля/г смолы);
2. 4-эквивалента гидроксибензотриазол-гидрата, в пересчете на примененные аминокислоты;
3. 4 эквивалента реагента;
4. 4 эквивалента диизопропилэтиламина.
В качестве растворителя применяют N,N-диметилформамид. Время сопряжения составляет около 30 минут.
Деблокирование проводится согласно схеме B.
Схема B.
5. Промывание диметилформамидом (2 раза);
6. 20% пиперидин в диметилформамиде, 3 раза через 3 минуты;
7. Промывка диметилформамидов (2 раза).
Резюмируя, следует сказать, что FMOC применяется для защиты α- аминогрупп, tBu используется в качестве защитной группы для гидроксигруппы Ser и фенольной гидроксигруппы Tyr. Для защиты гуанидо-функции Arg применяется Mtr - группа.
Для расщепления и снятия защиты с защищенной пептидной смолы, ее обрабатывают трифторуксусной кислотой в течение по меньшей мере одного часа. Трифторуксусная кислота отделяется от нерастворенной смолы и упаривается в вакууме досуха. Из остатка изолируют известными способами жидкостной хроматографии высокого давления искомый пептид в чистой форме.
Примеры.
1.1. Получение этилового эфира 5-(1-аза-циклоалк-1-ил)-2- ацетамидо-2-этоксикарбонил-пентановой кислоты
(Общее предписание)
5,0 г (23 ммоля) ацетамидо-малонового эфира добавляют вначале 10 мл бензола и 1 ммоль натрий-метоксида. Затем прикапывают 23 ммоля акролеина в течение 30 минут, при этом благодаря наружному охлаждению ледяной водой температура реакции не превышает +35oC. Затем перемешивают в течение 60 минут (при этом температура поднимается до приблизительно +10oC) и устанавливают pH уксусной кислотой на 7 единиц (смоченная индикаторная бумажка). Реакционный раствор упаривают в вакууме и маслянистый остаток растворяют в 20 мл метанола (высушенном над молекулярными, 
Снова охлаждают ледяной водой и добавляют последовательно 23 ммоля аза-циклоалкана, 46 ммоля ацетата натрия, 2,5 г молекулярного сита  и, наконец, 46 ммоля натрийцианоборгидрида. По окончании выделения газа перемешивают 16 часов при +20oC. С помощью водного раствора карбоната натрия устанавливают pH 10, трижды экстрагируют каждый раз с помощью 100 мл этилового эфира ледяной уксусной кислоты, объединяют органические фазы, экстрагируют насыщенным раствором хлорида натрия, сушат над сульфатом натрия и упаривают фильтрат.
и, наконец, 46 ммоля натрийцианоборгидрида. По окончании выделения газа перемешивают 16 часов при +20oC. С помощью водного раствора карбоната натрия устанавливают pH 10, трижды экстрагируют каждый раз с помощью 100 мл этилового эфира ледяной уксусной кислоты, объединяют органические фазы, экстрагируют насыщенным раствором хлорида натрия, сушат над сульфатом натрия и упаривают фильтрат.
1.2. Получение трех специальных этиловых эфиров пентановой кислоты.
1.2.1. Пример 1: Получение этилового эфира 5-(морфолин-4-ил)- 2-ацетамидо-2-этоксикарбонил-пентановой кислоты.
Исходя из 21,7 г ацетамидомалонового эфира, получают 34,5 г сырого продукта, из которого получают хроматографически на кизельгеле (дихлорметан, метанол 95 : 5, об./об.) 112,8 г чистого продукта (масло). 1.2.2. Пример 2. Получение этилового эфира 5-(пирродилин-1-ил)-2- ацетамидо-2-этоксикарбонил-пентановой кислоты.
Исходя из 5,0 г ацетамидомалонового эфира получают 6,5 г сырого продукта, который затем подвергается реакции без дальнейшей очистки.
1.2.3. Пример 3. Получение этилового эфира 5-(тиоморфолин-4- ил)-2-ацетамидо-2-этоксикарбонил-пентановой кислоты.
Исходя из 5,0 г ацетамидомалонового эфира получают 7,5 г сырого продукта, из которого получают хроматографически на кизельгеле (дихлорметан, метанол 95 : 5 об./об.) чистый продукт.
2. Получение этилового эфира 6-бром-2-этоксикарбонил-гексановой кислоты (пример 4).
Смесь из 86,9 г ацетамидо-малонового эфира, 215,9 г 1,4-дибромбутана, 4 г триэтил-бензил-аммонийхлорида, 82,8 г карбоната калия и 400 мл ацетонитрила нагревают в течение 24 часов с обратным холодильником. Нерастворенные составные части отфильтровывают на целитах, фильтрат упаривают в вакууме и остаток трижды упаривают в вакууме каждый раз с 500 мл воды. Остаток переваривают с 500 мл диэтилового эфира и оставляют стоять на ночь при +5oC. Фильтрат от нерастворимых веществ упаривают в вакууме. Остаток очищают хроматографически на кизельегеле (трет.-бутилматиловый эфир, гексан; 7 : 3; об./об. ). Получают 64 г чистого продукта. Температура плавления 61 - 62oC.
3.1. Получение этилового эфира 6-(1-аза-циклоалк-1-ил)-2- ацетамидо-2-этоксикарбонил-гексановой кислоты
(Общее предписание)
352 мг этилового эфира 6-бром-2-ацетамидо-2-этоксикарбонил-гексановой кислоты (сравнительный пример 4) добавляют к раствору 2 мл аза-циклоалкана в 2 мл диэтилового эфира и перемешивают 12 часов при 20oC. Реакционный раствор упаривают в вакууме и остаток смешивают с 10 мл воды. После трехкратной экстракции с каждый раз 10 мл ледяным этилацетатом органические фазы объединяют и сушат над сульфатом натрия. Фильтрат упаривают в вакууме.
3.2. Получение нескольких специальных этиловых эфиров гаксановой кислоты.
3.2.1. Пример 5. Получение этилового эфира 6-(морфолин-4-ил)-2- ацетамидо-2-этоксикарбонил-гексановой кислоты.
Исходя из 70 г этилового эфира 6-бром-2-ацетамидо-2-ацетамидов- 2-этоксикарбонил-гексановой кислоты получают после реакции с морфолином и хроматографии на кизельгеле (дихлорметан, метанол/градиент: 0 на 10% метанола; об. /об.) 35 г чистого продукта. Температура плавления 58 - 59oC.
3.2.2. Пример 6. Получение этилового эфира 6-(пирролидин-1-ил)- 2-ацетамидо-2-этоксикарбонилгексановой кислоты.
Исходя из 352 мг этилового эфира 6-бром-2-ацетамидо-2- этоксикарбонил-гексановой кислоты получают после реакции с пирролидином и хроматографии на кизельгеле (дихлорметан, метанол 7 : 3; об./об.) 251 мг чистого продукта. Температура плавления 79 - 81oC.
3.2.3. Пример 7. Получение этилового эфира 6-(метил-пиперазин-4- ил)-2-ацетамидо-2-этоксикарбонил-гексановой кислоты.
Исходя из 352 мг этилового эфира 6-бром-2-ацетамидо-2- этоксикарбонилгексановой кислоты получают после реакции с 1-метил-пиперазином и хроматографии на кизельгеле (дихлорметан, метанол 7 : 3; об./об.) чистый продукт.
3.2.4. Пример 8. Получение этилового эфира 6-(пиперидин-1-ил)- 2-ацетамидо-2-этоксикарбонил-гексановой кислоты.
Исходя из 352 мг этилового эфира 6-бром-2-ацетамидо-2- этоксикарбонил-гексановой кислоты получают после реакции с пиперидином и хроматографии на кизельгеле (дихлорметан, метанол 7 : 3; об./об.) 148 мг чистого продукта. Температура плавления 73 - 75oC.
3.2.5. Пример 9. Получение этилового эфира 6-(имидазол)-2- ацетамидо-2-этоксикарбонил-гексановой кислоты.
Исходя из 352 мг этилового эфира 6-бром-2-ацетамидо-2- этоксикарбонил-гексановой кислоты получают после реакции с имидазолом и хроматографии на кизельгеле (дихлорметан, метанол/градиент: 0 на 10% метанол; об./об.) чистый продукт.
3.2.6. Пример 10. Получение этилового эфира 6-(пиразол)-2- ацетамидо-2-этоксикарбонил-гексановой кислоты.
Исходя из 352 мг этилового эфира 6-бром-2-ацетамидо-2- -этоксикарбонил-гексановой кислоты получают после реакции с пиразолом и хроматографии на кизельгеле (дихлорметан, метанол 7 : 3, об./об.) чистый продукт.
4. Получение (S)-6-(тиоморфолин- бензилоксикарбониламиногексановой кислоты).
4.1. Получение бензилового эфира (S)-6-амино-2- бензилоксикарбониламино-гексановой кислоты.
(Z - Lys - Ozl)
Соединение получают по известному способу, например: E. Wunsch, b "Methoden den Organischen Chemil", Band XV/1: "Synthese von Peptiden" (Georg Thime Verlag, 1974). B. Byas, Z. Servas, J. Amer. chem. Soc. 83, 719 (1961).
4.2. Бензиловый эфир (S)-6-(тиоморфолин-1,1-диоксид-4-ил)- 2-бензилоксикарбониламино-гексановой кислоты.
5,6 г Z - Lys - Obzl растворяют в смеси 750 мл метанола и 750 мл дихлорметана. После прибавления 1,8 г дивинилсульфона перемешивают 6 часов при +20oC. Растворитель отгоняют в вакууме и остаток очищают хроматографически на кизельгеле (градиент 0 - 10% этилацетат/трет.бутил-мктилэфир). Выход: 1,8 г (масло).
4.3. (S)-6-(тиоморфолин-1,1-диоксид-4-ил)-2-амино-гексановая кислота
Отщепление защитных групп производится по известному способу, например: E. Wunsch b: "Methoden den Organischen Chemil, Band XV/1: Synthese von Peptiden (Georg Thime Verlag, 1974).
5. Превращение этиловых эфиров (1-аза-циклоалк-1-ил)-2- ацетамидо-2-токсикарбонил-пентановой или -гексановой кислот в соответствующие незащищенные α- аминокислоты.
Названные предшественники, которые в соответствии со способом получения присутствуют в виде смесей энантиомеров, вначале частично омывают способами, хорошо известными химику, в этиловые эфиры (1-аза-циклоалк-1-ил)-2-ацетамидо-2-карбоксипентановой или -гексановой кислоты и затем декарбоксилируют в (1-аза-циклоалк-илил)-2-ацетамидо-пентановую или -гексановую кислоту. После энзиматического разделения рацемата получают полным гидролизом энантиомерно чистые аминокислоты.
Эти способы описаны, например, Си. Кей, Акоста и др. J. Chem. Research (M) 11, 914-934 (1991), Кей. Фолкерс и др. Int. J. Pept. Prot. Res. 24, 197-200 (1984).
6. Получение пептидов
Пептиды могут быть получены либо по твердофазной технологи, либо по классической технологии из раствора.
Метод твердой фазы описан, например, J.M. Steward u .D.Joung, Solid. Phase Peptide Synthesis, Perce Chem. Company, Rockford III 1984, метод раствора изложен, например, в Methoden den Organischen Chemie (Houben/Weyl), Bd 15/Nr 1 и 2, E.Wunsch (Arsg), Thieme Verlag Stuttgart, 1974).
Общим признаком всех этих систем является блокирование α- аминогруппы и возможно имеющихся реактивных групп боковых цепей таким образом, что α- аминогруппа может быть выделена избирательно. Эта стратегия делает возможной активирование и избирательную реакцию карбоксильной группы N-защищенных аминокислот со свободной α- аминогруппой другой аминокислоты. После состоявшегося сопряжения α- аминозащитная группа может быть отщеплена и выполнено следующее сопряжение. В случае твердофазного синтеза C-концевая карбоксильная группа связана со смолой-носителем, а в методике с раствором она может быть защищена подходящей группой. В обоих методах вместо отдельных аминокислот могут быть также связаны подходящие пептидные фрагменты. Обоими методами получают полипептиды с защищенными или частично защищенными побочно-ценными функциями. После отщепления защитных групп целевой продукт может быть получен в чистом виде жидкостной хроматографией высокого давления.
Примеры 12 - 15.
В качестве примера пептидного соединения приводятся три декапептида, получение которых описано выше. Свойства трех декапептидов приведены в таблице. Пояснения сокращений даются после примера 15.
6.1. Пример 13.
Ac-D-Nal-D-Cpa-D-Pal-Ser-Tyr-D-Cit-Leu-Ahx (Mor)-Pro-D-Ala- Nr2
D-Nal D-Cpa D-Pal Ser Tyr D-Cit Leu Ahx (Mor) Pro D-Ala
ASA расч. 1,00 1,00 1,00 1,00 1,00 1,00 1,00 1,00 1,00 1,00
ASAa) найд. 1,03 0,98 1,01 0,95 0,97 1,03 1,01 0,96 1,00 0,97
RACb) < 1, 1,2 2,5 < 1 < 1 c) < 1 c) 1,1 1,3
FAB-MS молекулярный пик m/e 1472,6 (+H). Рассчитанная молекулярная масса 1473,1.
Пептидное соединение примера 13 является предпочтительной формой выполнения изобретения.
6.2 Пример 14.
Ac-D-Nal-D-Cpa-D-Pal-Ser-Tyr-D-Ahx(Mor)-Leu-Arg-Pro-D-Ala- NH2
D-Nal D-Cpa Ser Tyr D-Ahx(Mor) Leu Arg Рro D-Ala
ASA расч. 1,00 1,00 1,00 1,00 1,00 1,00 1,00 1,00 1,00 1,00
ASAa) найд. 1,05 1,02 1,03 0,95 0,96 0,97 1,00 0,96 0,98 0,95
RACb) < 1 1,6 3,3 < 1 0,8 1,0 < 1 < 1 < 1 1,3
FAB-MS молекулярный пик m/e 1471,9 (+H). Рассчитанная молекулярная масса 1472,2.
6.3 Пример 15.
Ac-D-Nal-D-Cpa-D-Pal-Ser-Tyr-D-Ahx(Mor)-Leu-Ahx(Mor)-Pro-D-Ala- NH2
D-Nal D-Cpa D-Pal Ser Tyr D-Ahx (Mor)Leu Pro D-Alg
ASA расч. 1,00 1,00 1,00 1,00 1,00 2,00 1,00 1,00 1,00
ASAa) найд. 1,05 1,01 1,01 0,95 0,95 1,93 0,97 0,98 0,96
RACb) 1,6 1,5 2,6 < 1 0,8 d) < 1 < 1,1
FAB-MS молекулярный пик m/e 1513,8 (Х+H). Рассчитанная молекулярная масса 1514,2.
a) ASA - аминокислотный анализ гидролизата. Условия гидролиза: 6M раствор HCl, 110oC, 24 часа.
b) RAC = доля нежелательных энзиотимеров в процентах. Определение по H. Frank, G.J.Nichelson and E.Bayer J. Chromatogs. Science, 15, 174 (1977)
c) Определение невозможно, так как исследуемые производные Cit и Ahx (deor) показывают одинаковые хроматогроафические времена удержания.
d) Соотношение D-Ahx(Mor); L - Ahx (Mor) составляет 1 : 1.
7. Получение N6-замещенных лизин-производных
7.1. Пример 16.
Получение D-3-тозиламидогексагидро-2-азепинона.
18 г D-3-амино-гексагидро-2-азепинона растворяют в 180 мл воды и смешивают с 5,6 г тонкогранулированного гидроксида натрия и 29,5 г тозилхлорида. Суспензию энергично перемешивают. Как только pH уменьшиться, его устанавливают на величину pH 9. Смесь перемешивают всю ночь, отфильтровывают выпавший в осадок продукт, промывают его водой и кристаллизуют или соответственно переваривают из горячего метанола. Получают таким образом 25,5 г D-3-тозиламидо-гексагидро-2-азепинона с температурой плавления 213oC;
[α]
7.2. Пример 17. Получение N2-тозил-D-3-лизин-гидрохлорида.
Суспензию 14,8 г D-3-тозиламидогексагидро-2-азепинона в 1,2 л 12%-ной соляной кислоты нагревают от 1,5 до 2 часов с обратным холодильником, пока не образуется прозрачный раствор. Затем концентрируют досуха раствор в вакууме. Остаток переваривают с горячим гексаном/изопропанолом, белые кристаллы отсасывают, так что фильтрат несколько сгущается, и при -20oC подвергают дальнейшей кристаллизации. Получают таким образом 15,1 г N2-тозил-D-лизин-гидрохлорид с температурой плавления 189oC.
[α]
7.3. Пример 18. Получение 2,2'-оксибис(ацетальдегид)а
25 г 1,4-ангидромезо-эритита растворяют в 400 мл воды. Затем к раствору при охлаждении льдом прибавляют 41,1 г твердого периодата натрия, смесь перемешивают в течение ночи и устанавливают ее с помощью твердого бикарбоната натрия на pH 7,4. Смесь смешивают с 400 мл ацетонитрила, отфильтровывают неорганические соли и получают таким образом раствор 2,2-оксибис(ацетальдегид)а.
7.4. Пример 19. Получение N2-тозил-6-(морфолин-1-ил)-2- D-аминогексановой кислоты
37,8 г N2-тозил-D-лизина растворяют в 22 л тридистиллированной воды и раствор устанавливают на pH 7,4 с помощью бикарбоната натрия. Затем прибавляют 14,1 г натрийцианоборгидрида и свежеприготовленный раствор 2,2-оксибис(ацельдегид)а. Реакционную смесь сохранят 12 дней при комнатной температуре и затем по частям концентрируют в вакууме. Остаток сушат в глубоком вакууме, собирают в абсолютном метаноле/дихлорметане (1+1), неорганические соли отфильтровывают и полученный сырой продукт грубо хроматографируют через колонку и кизельгелем посредством абсолютного метанола/дихлорметана 1+1.
Получают таким образом 25,6 г N6-тозил-6-(морфолин-1-ил)-2-D-аминогкексановой кислоты в качестве сырого материала с температурой разложения от 120oC.
[α]
7.5. Пример 20. Получение 6-(морфолин-1-ил)-2-D-аминогексановой кислоты
16,8 г N2-тозил-6-(морфолин-1-ил)-2-D-аминогексановой кислоты растворяют при -70oC в 500 мл жидкого аммиака (высушенного над гидроксидом калия) и затем при -40oC до -33oC смешивают с небольшими кусочками натрия до тех пор, пока не появится интенсивное синее окрашивание, не исчезающее по меньшей мере в течение 3 минут. Затем раствор обесцвечивают, добавляя несколько капель ледяной уксусной кислоты, и дают испариться аммиаку в течение ночи. Полученный остаток освобождают от остатков аммиака под высоким вакуумом, собирают в тридистиллированной воде и устанавливают разбавленной соляной кислотой на pH 4. Аминокислоту поглощают сильнокислотным ионообменником, последний промывают в колонке с 3%-ной соляной кислотой и водой и соединение элюируют 3н водным аммиаком. Растворитель вытягивают в вакууме и затем в высоком вакууме и получают 87,7 г 6-(морфолин-1-ил)-2-D-аминогексановой кислоты, которую переваривают в небольшом количестве метанола/дихлорметана. Температура плавления выше 325oC.
[α]
Избыток энантиомеров 100%.
7.6. Пример 21. Получение N6-изопропил-N2-тозил- D-лизина.
2 г N-тозил-D-лизин-гидрохлорида смешивают в 5 мл ледяной уксусной кислотой, 1,5 г безводного ацетата натрия, 10 мл воды и 5 мл ацетона. Затем прибавляют при перемешивании столько ледяной уксусной кислоты, что раствор становится прозрачным. Смесь охлаждают до 0oC и добавляют при перемешивании 2 г натрийборгидрида маленькими долями. Затем добавляют еще 5 мл ацетона и еще маленькими порциями 2 г натрийборгидрида.
Полученную суспензию сгущают под вакуумом и затем под глубоким вакуумом, остаток растворяют в горячем метаноле. При охлаждении до 10oC выкристаллизовываются 1,2 г N6-изопропил-N2-тозил-D-лизина.
Белые иглы с температурой плавления 251oC.
[α]
Из маточных растворов могут быть получены кристаллизацией дальнейшие количества соединения.
7.7. Пример 22. Получение N6-изопропил-D-лизина.
Вусловияхпримера7,5реакцииподвергают342мгN6-изопропил-N2-тозил-D-лизина с температурой плавления 224oC.
[α]
Избыток энантиомеров 98,6%.
Изобретение относится к пептидным соединениям, а именно к декапептидам формулы I: Х-Х1-X2-Х3-Ser-Tyr-Х6 -Leu-X8-Pro-X10 (I), где Х- ацил; X1=D-Nal; Х2=D-Cpa; Х3=D-Рal; Х6=D-Cit; D-Hei, D-Orn, D-Lys, D-Neu; Х8= Orn, Arg, Lys, L-Neu ; X10= D-AlaNH2, Neu является остатком формулы IX или IV; Z - группа -O- или -S-, п = 1-6. Фармацевтическая композиция обладает LHRH - антагонистической активностью. Преимущество соединений 1 в том, что их фармакологическое действие очень высоко и побочные действия в форме выделения гистамина удерживаются на низком уровне. 2 с. и 4 з.п.ф-лы.
X - X1 - X2 - X3 - Ser - Tyr - X6 - Leu - X8 - Pro - X10
где Х - ацил с 1 - 7 атомами углерода;
X1 = D-(1)-Nal, D-(2)-Nal;
X2 = D-Cpa;
X3 = D-(3)-Pal;
X6 = D-Cit, D-Hci, D-Orn, D-Lys, D-Neu;
X8 = Orn, Arg, Lys, L-Neu;
X10 = D-AlaNH2 ,
при этом Neu является остатком формулы IX или IV:
Z1 является группой -О- или -S-; n = 1 - 6.
где n = 3 - 6.
4. Производные декапептидов общей формулы 1 по п.1, у которых X6 или X8 означают соответственно D- или L - Neu.
6. Фармацевтическая композиция, обладающая LHRH антагонистической активностью, содержащая пептидное соединение и целевые добавки, отличающаяся тем, что в качестве пептидного соединения она содержит производные декапептидов общей формулы 1 по п.1 в эффективном количестве.
| Способ получения нонапептидных или декапептидных производных гормона LH - RH или их фармацевтически приемлемых солей | 1980 |
|
SU1681733A3 |
| СПОСОБ ЗАГРУЗКИ И МОРСКОЙ ТРАНСПОРТИРОВКИ САХАРА-СЫРЦА В ТАНКЕРАХ | 0 |
|
SU299402A1 |
| Способ получения полимеров | 1976 |
|
SU664968A1 |
| DE 3700166 A, C 07 K 7/20, 1987 | |||
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| US 4721478 C 07 K 7/20, 1988. | |||

