Область техники, к которой относится изобретение.
Изобретение относится к антагонистам LH-RH с улучшенной растворимостью, способам получения этих соединений, лекарственным средствам, в которых содержатся указанные соединения, а также к применению лекарственного средства при лечении гормонзависимых опухолей и незлокачественных гормонзависимых заболеваний, таких как доброкачественная гиперплазия простаты (ВРН) и эндометриоз.
Уровень техники.
Номенклатура, используемая при описании пептидов, совпадает с номенклатурой, которая вошла в состав Биохимической Номенклатуры, утвержденной комиссией ИЮПАК-ИЮБ (European J. Biochem. (1984), т.138, стр. 9-37), в соответствии с которой является общепринятым, что N-концевая аминогруппа располагается слева, а С-концевая карбоксильная группа располагается справа. Антагонисты LH-RH, так же как и пептиды по настоящему изобретению, включают в себя как природные, так и синтетические аминокислоты, причем первые включают Ala, Val, Leu, Ile, Ser, Thr, Lys, Arg, Asp, Asn, Glu, Gln, Cys, Met, Phe, Tyr, Pro, Trp и His. Сокращенные названия отдельных аминокислотных остатков основаны на тривиальных названиях аминокислот, например Ala=аланин, Arg=аргинин, Gly=глицин, Leu=лейцин, Lys=лизин, Pal(3)=3-(3-пиридил)аланин, Nal(2)=3-(2-нафтил)аланин), Phe=фенилаланин, Сра=4-хлорфенилаланин, Pro=пролин, Ser=серин, Тhr=треонин, Тrp=триптофан, Тyr=тирозин и Sar=саркозин. Если не указано иное, все описанные в заявке аминокислоты относятся к L-ряду. Например, сокращенное название D-Nal(2) означает 3-(2-нафтил)-D-аланин, а сокращенное название Ser означает L-серин. Заместители по ε-аминогруппе боковой цепи лизина приведены после термина "Lys" в скобках, по выбору в сокращенной форме.
Прочие сокращения, использованные в тексте заявки:
Ас ацетил
В 4-(4-амидинофенил)амино-1,4-диоксобутил
Воc трет-бутилоксикарбонил
Вор гексафторфосфат бензотриазол-1-окситрис(диметиламино)фосфония
DCC дициклогексилкарбодиимид
ДХМ дихлорметан
Ddz диметоксифенилдиметилметиленоксикарбонил(диметоксидиметил-Z)
DIC диизопропилкарбодиимид
DIPEA N,N-диизопропилэтиламин
ДМФ диметилформамид
Fmoc флуоренилметилоксикарбонил
HF фтористоводородная кислота
HOBt 1-гидроксибензотриазол
ВЭЖХ высокоэффективная жидкостная хроматография
Me метил
ТФУ трифторуксусная кислота
Z бензилоксикарбонил
Пептиды по настоящему изобретению представляют собой аналоги релизинг-фактора лютеинизирующего гормона (LH-RH), как показано на следующей структуре:
p-Glu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2, (LH-RH, гонадорелин).
Селективные потенциальные антагонисты декапептида LH-RH исследуют уже более 20 лет (статья М.Karten, J.E.Rivier, Endocrine Reviews (1986), т.7, стр.44-66). Большой интерес к таким антагонистам обусловлен необходимостью их использования в области эндокринологии, гинекологии, предупреждения беременности и терапии онкологических заболеваний. В качестве потенциальных антагонистов LH-RH получено множество соединений. Наиболее интересными соединениями, которые найдены к настоящему времени, являются такие соединения, структура которых является модификацией структуры LH-RH.
Первую серию потенциальных антагонистов получали введением ароматических аминокислот в положения 1, 2, 3, и 6 или 2, 3 и 6 полипептидной цепи. Принятый способ описания таких соединений заключается в следующем: прежде всего указывают аминокислоты, на которые заменены первоначальные аминокислоты в пептидной цепи LH-RH, причем положения, в которых произошли замены, обозначают цифрами, расположенными в верхнем индексе. Затем указывают термин "LH-RH", обозначающий тот факт, что речь идет об аналоге LH-RH, содержащем указанные замены.
Известными антагонистами являются следующие соединения:
[Ac-D-Cpa1,2, D-Trp3,6]LH-RH (статья D.H.Coy и соавт., "Peptides; Proceedings of the 6th American Peptid Symposium" (Пептиды, Материалы 6-го американского симпозиума по пептидам), стр.775-779, ред. Gross E., Meienhofer J., Pierce Chem. Co., Rockville III (1979);
[Ac-Pro1, D-Cpa2, D-Nal(2)3,6]LH-RH (патент США №4419347) и [Ac-Pro1, D-Cpa2, D-Trp3,6] LH-RH (статья J.L.Pineda и соавт., J.Clin. Endocrinol. Metab. (1983), т.56, стр.420).
Для усиления действия антагонистов позднее в положение 6 стали вводить основные аминокислоты, например D-Arg. Например, получены: [Ac-D-Cpa1,2, D-Trp3, D-Arg6, D-Ala10]LH-RH (ORG-30276) (статья D.H.Coy и соавт., Endocrinology (1982), т.100, стр.1445), и [Ac-D-Nal(2)1, D-Phe(4-F)2, D-Trp3, D-Arg6] LH-RH (ORF 18260) (статья J.E.Rivier и соавт., в книге: "LHRH and its Analogs" (LHRH и его аналоги), стр.11-12, ред. Vickery B.H., Nestor Jr. J.J., Hafez E.S.E., MTS Press, Lancaster, UK, (1984)).
Прочие потенциальные антагонисты LH-RH описаны в заявках WO 92/19651, WO 94/19370, WO 92/17025, WO 94/14841, WO 94/13313, US-A 5300492, US-A 5140009, ЕР 0413209 А1 и DE 19544212 А1.
Позднее описаны соединения с модифицированными остатками орнитина и лизина в положении 6, представленные следующей формулой: Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-Tyr5-D-Xxx6-Leu7-Arg8-Pro9-D-Ala10-NH2, гдe D-Xxx является аминокислотным остатком общей формулы (VI)
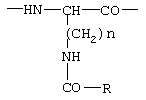
Другими известными антагонистами LH-RH являются антареликс, ганиреликс и цетрореликс.
Антареликс® (Международное непатентованное наименование (INN): Тевереликс):
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-Tyr5-D-Hci6-Leu7-Lys(iPr)8-Pro9-D-Ala10-NH2,
Ганиреликс:
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)з-Ser4-Tyr5-D-hArg(Et)2 6-Leu7-hArg(Et)2 8-Pro9-D-Ala10-NH2,
Цетрореликс:
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-Tyr5-D-Cit6-Leu7-Arg8-Pro9-D-Ala10-NH2.
Сущность изобретения
Целью изобретения является разработка новых антагонистов LH-RH, обладающих повышенной устойчивостью к действию ферментов и значительно более высокой растворимостью в воде.
Цель изобретения достигается получением соединений следующей общей формулы (I),
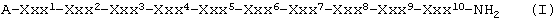
где А означает ацетильную группу,
Ххх1 означает D-Nal(2),
Ххх2 означает D-Cpa,
Ххх3 означает D-Pal(3),
Ххх4 означает Ser,
Ххх5 означает N-Ме-Туr,
Ххх6 означает D-Cit, D-Hci или D-[ε-N'-4-(4-амидинофенил)амино-1,4-диоксобутил]-Lys (сокращенно: D-Lys(B)),
Ххх7 означает Leu или NIe,
Ххх8 означает Arg или Lys(iPr),
Ххх9 означает Pro и
Ххх10 означает D-Ala или Sar,
при условии, что если Ххх6 означает D-Lys(B), то Ххх7 означает NIe, если Ххх6 означает D-Cit, то Ххх7 означает NIe и Ххх10 означает D-Ala, или если Ххх6 означает D-Hci, то Ххх7 означает Leu и Ххх10 означает D-Ala,
и их соли с фармацевтически приемлемыми кислотами, прежде всего ацетаты, эмбонаты и трифторацетаты.
Согласно другому аспекту настоящего изобретения предпочтительными соединениями и их солями с фармацевтически приемлемыми кислотами являются следующие соединения:
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)з-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Arg8-Pro9-Sar10-NH2,
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Arg8-Pro9-D-Ala10-NH2,
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Lys(iPr)8-Pro9-Sar10-NH2,
Ac-D-Nal(2)1-D-Phe(4-CI)2-D-Pal(3)з-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Lys(iPr)8-Pro9-D-Ala10-NH2,
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Cit6-Nle7-Arg8-Pro9-D-Ala10-NH2,
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Hci6-Leu7-Arg8-Pro9-D-Ala10-NH2,
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Cit6-Nle7-Lys(iPr)8-Pro9-D-Ala10-NH2,
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Hci6-Leu7-Lys(iPr)8-Pro9-D-Ala10-NH2.
Согласно другому аспекту изобретения соединения по настоящему изобретению существуют в виде ацетата, трифторацетата или эмбоната.
Согласно другому аспекту изобретения соединения по настоящему изобретению могут быть использованы в качестве лекарственных средств или фармацевтических композиций.
Согласно другому аспекту изобретения представлены фармацевтические композиции, содержащие по крайней мере одно соединение по настоящему изобретению и общепринятые носители и вспомогательные вещества.
Согласно другому аспекту изобретения представлен способ получения соединений по настоящему изобретению общей формулы I, который заключается в том, что фрагмент, состоящий из остатков Хххm с соответствующими защитными группами, где m означает целое число от 1 до 10 и Ххх1 ацетилирован, синтезируют на твердом носителе или в растворе по общепринятым методам, а затем фрагменты конденсируют на твердом носителе и после получения соединений общей формулы I их отщепляют от твердого носителя по общепринятому методу путем амидирования по остатку Ххх10.
Согласно другому аспекту изобретения представлено применение соединений по настоящему изобретению для получения лекарственных средств для лечения гормонально зависимых опухолей, прежде всего карциномы предстательной железы или рака молочной железы, а также при незлокачественных показаниях, для лечения которых требуется подавление гормона LH-RH.
Согласно другому аспекту изобретения представлен способ получения фармацевтических композиций, в которых по крайней мере одно соединение по пунктам 1-10 смешивают с общепринятыми носителем и вспомогательными веществами и обрабатывают для получения лекарственного средства.
Согласно другому аспекту изобретения представлен способ лечения гормонально зависимых опухолей, прежде всего, карциномы предстательной железы, рака молочной железы или миомы матки, а также незлокачественных показаний, при лечении которых требуется подавление гормона LH-RH, таких как эндометриоз, доброкачественная гиперплазия предстательной железы (ГПЖ), а также при лечении бесплодия у женщин или мужчин, для лечения млекопитающих, прежде всего человека, причем способ лечения заключается во введении действующей дозы по крайней мере одного соединения по настоящему изобретению.
Соединения по настоящему изобретению могут быть использованы для лечения гормонально зависимых опухолей, прежде всего карциномы предстательной железы, рака молочной железы или миомы матки, а также незлокачественных показаний, при лечении которых требуется подавление гормона LH-RH, таких как эндометриоз или доброкачественная гиперплазия предстательной железы (ГПЖ). Кроме того, такие соединения могут быть использованы для лечения бесплодия у женщин или мужчин, например, для контролируемой овариальной суперстимуляции при искуственном оплодотворении (оплодотворение in vitro). При этом такие соединения могут быть смешаны известными способами с подходящими носителем или вспомогательными веществами и обработаны для получения лекарственного средства.
Сведения, подтверждающие возможность осуществления изобретения
Синтез соединений формулы (I) можно осуществлять как с помощью классической конденсации фрагментов, так и твердофазным синтезом по Меррифильду с последовательным наращиванием цепи с использованием D-лизина, ацилированного по боковой цепи карбоновой кислотой общей формулы R1-COOH, а также посредством модификации декапептидного фрагмента соответствующими карбоновыми кислотами за счет образования амидной связи с боковой цепью D-лизина6. Благодаря этому введение R1-СО-группы можно осуществлять на трех различных стадиях синтеза: перед конденсацией отдельных фрагментов с образованием пептида, после введения в цепь лизина или орнитина, но перед конденсацией последующих фрагментов или после конденсации всех фрагментов.
Соединения формулы (I) синтезируют известными методами, например исключительно твердофазным методом с частичным использованием твердофазного метода (так называемой конденсацией фрагментов) или с помощью классического синтеза в растворе [см. в книге М.Bodanszky, "Principles of Peptide Synthesis" (Принципы пептидного синтеза), Springer Verlag, (1984)].
Например, методы твердофазного синтеза описаны в руководстве J.M.Stewart, J.D.Young "Solid Phase Peptide Synthesis" ("Твердофазный синтез пептидов"), Pierce Chem. Company, Rockford, III, (1984) и в книге G.Barany, R.B.Merrifield, "The peptides" ("Пептиды"), гл.1, стр.1-285 (1979), Academic Press Inc. Классический синтез в растворе подробно описан в руководстве "Methoden der Organischen Chemie (Houben-Weyl), Synthese von Peptiden" ("Методы органической химии (Губен-Вейль), синтез пептидов"), ред. Е.Wunsch, (1974), Georg Thieme Verlag, Stuttgart, BRD.
Ступенчатый синтез проводят, например, следующим образом, прежде всего С-концевую аминокислоту с защищенной α-аминогруппой ковалентно присоединяют к одному из обычных нерастворимых носителей, затем от этой аминокислоты отщепляют α-аминозащитную группу, и к образовавшейся свободной аминогруппе присоединяют по карбоксильной группе следующую аминокислоту с защищенной аминогруппой, и таким образом, наращивая пептид, стадия за стадией присоединяют в правильной последовательности остальные аминокислоты, а после присоединения всех аминокислот готовый пептид отщепляют от носителя и по выбору отщепляют остальные присутствующие защитные группы боковых цепей аминокислот. Ступенчатую конденсацию проводят обычным способом из соответствующих должным образом защищенных аминокислот.
Конденсацию отдельных аминокислот друг с другом проводят обычными методами, которые прежде всего включают:
- метод с использованием симметричного ангидрида в присутствии дициклогексилкарбодиимида или диизопропилкарбодиимида (DCC, DIC),
- общий карбодиимидный метод,
- карбодиимид-гидроксибензотризольный метод (см. в книге "The Peptides" (Пептиды), т.2, ред. Е.Gross, J.Meienhofer).
При конденсации фрагментов предпочтительно используют не сопровождающийся рацемизацией азидный метод или метод с использованием DCC-1-гидроксибензотриазола или соответственно DCC-3-гидрокси-4-оксо-3,4-дигидро-1,2,3-бензотриазина. Кроме того, можно использовать активированный эфир фрагмента.
Для ступенчатой конденсации аминокислот наиболее всего подходят активированные эфиры N-защищенных аминокислот, например N-гидроксисукцинимидный эфир или 2,4,5-трихлорфениловый эфир. Для эффективного катализа аминолиза используют N-гидроксисоединения, обладающие кислотностью, приблизительно равной кислотности уксусной кислоты, например 1-гидроксибензотриазол.
В качестве промежуточных аминозащитных групп предложены группы, удаляемые гидрированием, например бензилоксикарбонильный остаток (=Z-остаток) или группы, отщепляемые в слабой кислоте. В качестве защитных групп для α-аминогруппы используют, например, следующие группы: трет-бутилоксикарбонил, флуоренилметилоксикарбонил, карбобензокси, соответственно карбобензтио (по выбору с п-бром- или п-нитробензильным остатком), трифторацетил, фталильный остаток, о-нитрофеноксиацетил, тритил, п-толуолсульфонил, бензил, бензил, имеющий заместители в бензольном кольце (п-бром- или п-нитробензильный остаток) и α-фенилэтил. Кроме того, методы защиты аминогрупп описаны в следующих книгах: Jesse Р.Greenstein, Milton Winitz, "Chemistry of Amino Acids" (Химия аминокислот), New York, (1961), John Wiley and Sons, Inc., т.2, например, стр.883 и др., "Principles of Peptide Synthesis" (Принципы пептидного синтеза), Springer Verlag (1984), "Solid Phase Peptide Synthesis" (Твердофазный синтез пептидов), J.M.Stewart, J.D.Young, Pierce Chem. Company, Rockford, III, (1984), G. Barany, R.B. Merrifield, "The Peptides" (Пептиды), гл. 1, стр.1-285, (1979), Academic Press Inc., а также The Peptides (Пептиды), т.2, ред. Е.Gross, J.M.Meienhofer, Academic Press, New York. В основном эти же защитные группы используют и для защиты других функциональных боковых групп (ОН-групп, NH2-групп) соответствующих аминокислот.
Имеющиеся гидрокси группы (серина и треонина) предпочтительно защищают с помощью бензильных групп и аналогичных групп. Прочие не α-аминогруппы (например, аминогруппы в ω-положении, гуанидиногруппы аргинина) предпочтительно защищают ортогонально.
Отдельные производные аминокислот за исключением лизина, модифицированного R1-СО-группами, имеются в продаже.
Возможная схема процесса получения лизина, модифицированного R1-СО-группами, является следующей:
1. α-Карбоксильную группу блокируют подходящим методом, например, с помощью этерификации.
2. ε-Аминогруппу блокируют, например, с использованием Z-группы.
3. α-Аминогруппу блокируют таким образом (например, с использованием Вос-группы), чтобы обеспечить селективность относительно последующего отщепления аминозащитной группы.
4. От ε-аминогруппы отщепляют Z-группу.
5. К ε-аминогруппе присоединяют требуемую группу R1-CO-.
6. От α-аминогруппы отщепляют защитную группу.
7. α-Аминогруппу при необходимости обратимо модифицируют, например, с использованием Z-группы.
Для введения R1-СО-группы с целью модификации аминогруппы лизина соответствующей карбоновой кислотой или производным карбоновой кислоты в основном используют способы, описанные для конденсации аминокислот. Однако наиболее предпочтительна конденсация с использованием карбодиимида, например 1-этил-3-(3-диметиламинопропил)карбодиимида, и 1-гидроксибензотриазола.
Реакцию конденсации аминокислот проводят в обычных нейтральных растворителях и суспендирующих средствах (например, в дихлорметане), причем по выбору для повышения растворимости может быть добавлен диметилформамид.
В качестве синтетических материалов-носителей используют нерастворимые полимеры, например набухающую в органическом растворителе полистирольную смолу в форме гранул (например, сополимеризат полистирола и 1% дивинилбензола). Синтез защищенного декапептидамида, имеющего после отщепления от носителя в присутствии HF требуемую С-концевую амидную группу, на метилбензгидриламидной смоле (МВНА-смола, т.е. полистирольная смола, содержащая метилбензгидриламидные группы) может быть осуществлен по следующей постадийной схеме:
Протокол пептидного синтеза с использованием Вос-защищенных аминокислот
Nα-Boc-защищенные аминокислоты обычно конденсируют в присутствии трехкратного молярного избытка диизопропилкарбодиимида (DIC) и 1-гидроксибензотриазола (HOBt) в СН2Сl2/ДМФ в течение 90 мин, а Вос-защитную группу отщепляют под действием 50%-ной трифторуксусной кислоты (ТФУ) в CH2Cl2. Для контроля полноты прохождения реакции используют хлоранильный тест по Кристенсену и нингидриновый тест по Кайзеру. Остаток свободной аминогруппы блокируют ацетилированием в присутствии пятикратного избытка ацетилимидазола в CH2Cl2. Последовательность реакционных стадий наращивания пептидной цепи на смоле приведена на постадийной схеме. Для отщепления связанного со смолой пептида конечный продукт твердофазного синтеза сушат в вакууме над Р2О5 и обрабатывают 500-кратным избытком смеси HF/анизол, 10:1/об.:об. в течение 60 мин при 0°С.
После упаривания HF и анизола в вакууме пептидамид выпадает в осадок при перемешивании с безводным этиловым эфиром в виде твердого вещества белого цвета; отделение от осадка полимерного носителя осуществляют промыванием 50%-ной водной уксусной кислотой. После осторожного концентрирования раствора в уксусной кислоте в вакууме пептид может быть получен в виде очень вязкого масла, которое при добавлении абсолютного эфира на холоду превращается в твердое вещество белого цвета.
Дальнейшую очистку проводят известным методом высокоэффективной жидкостной хроматографии (ВЭЖХ).
Перевод пептида в его кислотно-аддитивную соль может быть осуществлен взаимодействием с кислотами по известному способу. И наоборот, свободный пептид может быть получен взаимодействием его кислотно-аддитивной соли с основанием. Эмбонат пептида может быть получен взаимодействием соли пептида и трифторуксусной кислоты (соль с ТФУ) с эмбоновой кислотой (памовой кислотой) или соответствующей динатриевой солью эмбоновой кислоты. Для этого соль пептида с ТФУ в водном растворе обрабатывают раствором динатриевой соли эмбоновой кислоты в полярном апротонном растворителе, предпочтительно диметилацетамиде, и выделяют образующийся осадок светло-желтого цвета.
Следующие примеры приведены для пояснения изобретения без ограничения объема притязаний изобретения.
Пример 1 (D-68968)
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Arg8-Pro9-Sar10-NH2
Синтез декапептида осуществляют на полимерном носителе, причем нагрузка составляет 0,55 ммоль/г (смола, содержащая аминометильные группы, защитные Fmoc-группы, тип D-1675, производство фирмы Bachem). Лизин присоединяют в виде Fmoc-D-Lys(Boc)-OH, защитные Fmoc-группы отщепляют в смеси 20% пиперидин/ДМФ. После одновременного отщепления всех защитных групп боковых цепей и отделения от полимерного носителя выделенный неочищенный пептид очищают с использованием препаративной ВЭЖХ. После лиофильного высушивания получают декапептид с чистотой 98,5%.
Замещение D-лизина по атому ε-азота с использованием 4-(4-аминофенил)амино-1,4-диоксомасляной кислоты проводят в присутствии РуВор в ДМФ и в смеси с DIPEA. Очистку выделенного неочищенного пептида осуществляют с использованием препаративной ВЭЖХ. После лиофильного высушивания получают продукт в виде трифторацетата, чистота составляет приблизительно 99%, брутто-формула С82Н106СIN19О15, скорректированный MC-FAB 1633 (М+Н) (рассчит. 1631,78096).
Пример 2 (D-68969)
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Arg8-Pro9-D-Ala10-NН2
Синтез декапептида осуществляют на полимерном носителе, причем нагрузка составляет 0,55 ммоль/г (смола, содержащая аминометильные группы, защитные Fmoc-группы, тип D-1675, производство фирмы Bachem). Лизин присоединяют в виде Fmoc-D-Lys(Boc)-OH, защитные Fmoc-группы отщепляют в смеси 20% пиперидин/ДМФ. После одновременного отщепления всех защитных групп боковых цепей и отделения от полимерного носителя выделенный неочищенный пептид, содержание которого составляет 71% (ВЭЖХ), в дальнейшем используют без очистки.
Замещение боковых цепей D-лизина с использованием 4-(4-аминофенил)амино-1,4-диоксомасляной кислоты проводят в присутствии РуВор в ДМФ и в смеси с DIPEA. Очистку выделенного неочищенного пептида осуществляют с использованием препаративной ВЭЖХ. После лиофильного высушивания получают продукт в виде трифторацетата, чистота которого составляет приблизительно 98,8%, брутто-формула C82H106CIN19O15, скорректированный MC-FAB 1633 (М+Н) (рассчит. 1631,78096).
Пример 3 (D-68971)
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Lys(iPr)8-Pro9-Sar10-NH2
Синтез декапептида осуществляют на полимерном носителе, причем нагрузка составляет 0,55 ммоль/г (смола, содержащая аминометильные группы, защитные Fmoc-группы, тип D-1675, производство фирмы Bachem). Лизин присоединяют в виде Fmoc-D-Lys(Boc)-OH, защитные Fmoc-групп отщепляют в смеси 20% пиперидин/ДМФ. После одновременного отщепления всех защитных групп боковых цепей и отделения от полимерного носителя выделенный неочищенный декапептид, содержание которого составляет приблизительно 59% (ВЭЖХ), очищают методом препаративной ВЭЖХ. После лиофильного высушивания получают декапептид с чистотой 95%.
Замещение боковых цепей D-лизина с использованием 4-(4-аминофенил)амино-1,4-диоксомасляной кислоты проводят в присутствии РуВор в ДМФ и в смеси с DIPEA. Очистку выделенного неочищенного пептида осуществляют с использованием препаративной ВЭЖХ. После лиофильного высушивания получают продукт в виде трифторацетата, чистота составляет приблизительно 96,6%, брутто-формула C85H112CIN17O15, скорректированный MC-FAB 1648 (М+Н) (рассчит. 1645,8218).
Пример 4 (D-68987)
Ac-D-Nal(2)1-D-Phe(4-CI)2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Lys(iPr)8-Pro9-D-Ala10-NH2
Синтез D-Lys-6-незамещенных декапептидов осуществляют с использованием 9,09 г полимерного носителя, причем нагрузка составляет 0,55 ммоль/г, лизин6 присоединяют в виде Fmoc-D-Lys(Boc)-OH. После отщепления смолы выделяют 8,15 г неочищенного пептида. Очистку неочищенного пептида осуществляют с использованием препаративной ВЭЖХ.
Замещение боковых цепей D-лизина с использованием 4-(4-аминофенил)амино-1,4-диоксомасляной кислоты проводят в присутствии РуВор в ДМФ и в смеси с DIPEA. Очистку выделенного неочищенного пептида осуществляют с использованием препаративной ВЭЖХ. После лиофильного высушивания получают продукт в виде трифторацетата, чистота составляет 94,6%, брутто-формула С85H112CIN17О15, соответственный MC-FAB 1646,8 (М+Н) (рассчит. 1645,82).
Пример 5
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Cit6-Nle7-Arg8-Pro9-Ala10-NH2
Пример 6
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Hci6-Leu7-Arg8-Pro9-D-Ala10-NH2
Пример 7
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)з-Ser4-N-Me-Tyr5-D-Cit6-Nle7-Lys(iPr)8-Pro9-D-Ala10-NH2
Пример 8
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Hci6-Leu7-Lys(iPr)8-Pro9-D-Ala10-NH2.
Общие рабочие методики получения пептида согласно Примерам 5-8.
Декапептиды могут быть получены как методом твердофазного синтеза Меррифилда, так и классическим методом конденсации фрагментов в растворе. С экономической точки зрения предпочтительным является синтез пептидной цепи на полимерном носителе, и в основном для присоединения D-аланина по С-концевой группе может быть по выбору использована 1) стратегия с защитными Вос-группами или 2) стратегия с защитными Fmoc-группами; соответственным образом может быть использована смола с метилбензгидриламиногруппами (для стратегии 1) или смола с Fmoc-2,4-диметокси-4'-(карбоксиметилокси)бензгидриламиногруппами (для статегии 2).
Твердофазный синтез Меррифилда
Декапептид синтезируют в стандартных условиях (постадийная схема, Таблица 1) твердофазного синтеза с использованием стратегии с защитными Fmoc-группами и с использованием 5 г полимерного носителя, а именно смолы с Fmос-2,4-диметокси-4'-(карбоксиметилокси)бензгидриламино-группами типа D1675 производства фирмы Bachem, нагрузка составляет 0,55 ммоль/г, размер гранул смолы 200-400 меш.
Ступенчатый синтез пептидной цепи на смоле осуществляют с использованием аминокислот, содержащих Na-Fmoc-защитные группы, согласно следующей постадийной схеме:
Таблица 1
Ниже приведены типичные (повторяемые) параметры условий реакции твердофазного синтеза декапептида по вышеуказанной схеме:
- Отщепление защитных Fmoc-групп в 20% пиперидине в ДМФ, 2×5 мин при комнатной температуре (стадия 2).
- Конденсация в использованием трехкратного молярного избытка Fmoc-аминокислоты в присутствии диизопропилкарбодиимида (DIC) и гидроксибензотриазола (HOBt) (стадия 8).
- Отщепление по С-концевому остатку от полимерного носителя, включая удаление защитных групп боковых цепей аминокислот в присутствии трифторуксусной кислоты (ТФУ).
После отделения полимерного носителя при использовании 5 г смолы получают приблизительно 5-6 г неочищенной пептидной смеси, в которой содержание требуемого компонента составляет приблизительно 70-80%, который выделяют с использованием препаративной ВЭЖХ.
Очистка декапептида методом препаративной ВЭЖХ, условия хроматографии
Препаративная ВЭЖХ, колонка Dynamax RP18, производства фирмы Shimadzu, 12 мкм, 300  длина 250 мм, внутренний диаметр 41,4 мм.
длина 250 мм, внутренний диаметр 41,4 мм.
Система градиента с временной программой, 40% В→90% В, 50 мин.
Смесь элюента А: 970 мл H2O+30 мл CH3CN+1 мл СF3СООН.
Смесь элюента В: 300 мл Н2О+700 мл CH3CN+1 мл СF3СООН.
УФ-детектирование, λ=220 нм, скорость потока 60 мл/мин.
Полученные фракции концентрируют в вакууме и высушивают лиофильно.
Декапептид получают в виде легкого бесцветного материала.
В заключение превращение продукта в требуемую для фармакологического применения ацетатную соль проводят с помощью ионнообменной хроматографии.
Исследование биологического действия
Соединения по настоящему изобретению формулы 1 исследуют по их связыванию с рецептором. Использованные способы относятся к известным методам, описанным в статье Beckers и соавт., Eur. J.Biochem., (1995) т.231, стр.535-543. Цетрореликс, синтезированный, как описано выше, иодируют [125I] (производства фирмы Amersham, удельная активность 80,5 Бк/фмоль) с использованием реагента IоdоGen (производствафирмы Pierce). Реакционную смесь очищают методом высокоэффективной жидкостной хроматографии с обращенной фазой, при этом получают моноиодированный цетрореликс без примеси немеченного пептида. При необходимости приблизительно 80% [125I]-цетрореликса и немеченного соединения по изобретению являются пригодными для специфического связывания с рецептором.
Соединения по настоящему изобретению могут быть исследованы по их действию in vitro с использованием следующих методов 1 и 2, причем аффинность связывания определяют методом анализа связывания с [125I]-цетрореликсом (метод 1), а функциональную активность определяют с использованием трипторелина в качестве агонистического стимулятора (метод 2).
Метод 1 (Определение КD на примере цетрореликса):
Анализ связывания с рецептором по статье Beckers Т., Marheineke К., Reilander H., Hilgard P. "Selection and characterization of mammalian cell lines with stable overexpression of human pituitary receptors for gonadoliberin (GnRH)" (Отбор и характеристика линий клеток млекопитающих, обладающих устойчивой сверхэкспрессией рецепторов гонадолиберина (GnRH) гипофиза человека) Eur. J.Biochem., (1995), т. 231, стр.535-543.
Для исследования связывания с рецептором цетрореликс иодируют [125I] (производства фирмы Amersham, удельная активность 80,5 Бк/фмоль) с использованием реагента lodoGen (производства фирмы Pierce). Реакционную смесь очищают методом высокоэффективной жидкостной хроматографии с замещенными фазами, причем получают моноиодированный цетрореликс без примеси немеченого пептида. Приблизительно 80% [125I]-цетрореликса являются пригодными для специфического связывания с рецептором.
Анализ связывания с рецептором проводят с использованием интактных клеток в физиологических условиях по описанной методике (Beckers и соавт., 1995). Субконфлюентные культуры стабильных трансфертных клеток LTK, которые экспрессируют на поверхности рецептор LH-RH человека, отделяют инкубированием в присутствии NaCl/Pi (137 мМ NaCl, 2,7 мМ KCl, 8,1 мМ Na2HPO4, 11,47 мМ KH2PO4)/1 мМ ЭДТУ и собирают центрифугированием. Осадок клеток ресуспендируют в буфере для связывания (DM ЕМ без Н2СО3, содержащая 4,5 г/л глюкозы, 10 мМ Hepes pH 7,5, 0,5 мас./об.% БСА, 1 г/л бацитрацина, 0,1 г/л SBTI, 0,1 мас./об.% NаN3. Для анализа по механизму замещения суспензию 0,25×106 клеток/100 мкл, содержащую приблизительно 225 пМ [125I]-цетрореликса (удельная активность (5-10)×105 импульс/мин/пмоль), инкубируют в присутствии различных концентраций немеченого соединения по настоящему изобретению в качестве конкурирующего соединения. Суспензию клеток в 100 мкл среды для связывания наслаивают в пробирки для исследования объемом 400 мкл на 200 мкл смеси 84 об.% силиконового масла (Merck Тур 550)/16 об.% парафинового масла. После инкубирования в течение 1 ч при 37°С при непрерывном медленном встряхивании клетки отделяют от инкубационной среды путем центрифугирования в течение 2 мин при 9000 об/мин (центрифуга Rototyp HTA13,8, производства Heraeus Sepatec, Osterode, Германия). Верхние части пробирок, содержащие осадок клеток, отрезают. Затем в осадке клеток и надосадочной жидкости определяют число импульсов γ-излучения. Затем определяют количество неспецифически связанных соединений по включению немеченного цетрореликса при конечной концентрации 1 мкМ, которое в типичном случае составляет ≤10% от общего количества связанных соединений. Для анализа данных по связыванию используют аналитическую программу EBDA/Ligand (Biosoft V3,0).
Величина КD для цетрореликса составляет 170 пмоль/л (пМ) (число проведенных независимых экспериментов составляет 21).
Метод 2 (Функциональный анализ для определения антагонистического действия (величина IC50)):
Анализ проводят с некоторыми модификациями, как описано в статье Beckers Т., Reilander H., Hilgard P. "Characterization of gonadotropin-releasing hormone analogs based on a sensitive cellular luciferase reporter gene assay" (Характеристика аналогов релизинг-гормона гонадотропина, основанная на чувствительном анализе репортерного гена клеточной люциферазы) (1997), Analyt. Biochem., т. 251, стр.17-23 (Beckers и соавт. 1997). Клетки (по 10000 клеток в лунке), которые содержат на поверхности LH-RH-рецептор человека и репортерный ген люциферазы человека, культивируют в микропланшете в течение 24 ч с использованием среды DMEM с добавками и 1 об./об.% эмбриональной сыворотки коров (FCS). Затем клетки стимулируют в течение 6 ч 1 нМ раствором [D-Trp6]LH-RH. Перед стимулированием добавляют антагонистические соединения по настоящему изобретению, и наконец, клетки лизируют для количественного определения активности клеточной люциферазы. Расчет величины IC50 проводят с использованием кривой дозозависимого действия и нелинейного регрессионного анализа Hill-Modells (программа EDX 2,0, С.Grunwald, Arzneimittelwerk, Dresden).
Количественное определение активности люциферазы в основном определяют по известному методу (Promega Technical Bulletins, No 101/161) с использованием повторных экспериментов и современных систем для анализа люциферазы (Promega E4030). При добавлении коэнзима А (СоА) происходит окисление люциферил-СоА с благоприятной кинетикой. После удаления культуральной среды из микропланшета клетки лизируют путем добавления 100 мкл буфера для лизиса (25 мМ трис-фосфат, рН 7,8, 2 мМ дитиотреитол, 2 мМ 1,2-диаминоциклогексан-N,N,N',N'-тетрауксусная кислота (CDTA), 10 об./об.% глицерин, 1 об./об.% тритон Х-100). После инкубирования в течение 15 мин при комнатной температуре 10 мкл клеточного лизата переносят в микропланшет белого цвета, пригодный для люминометрического определения (Dynatech). Ферментативную реакцию инициируют путем добавления 50 мкл буфера для анализа (20 мМ трицин, рН 7,8, 1,07 мМ (МgСО3)4Мg(ОН)2, 2,67 мМ МgSO4, 0,1 мМ этилендиаминтетрауксусная кислота (ЭДТУ), 33,3 мМ дитиотреитол, 270 мкМ коэнзим А, 470 мкМ люциферин из светляков Photinus pyralis, 530 мкМ rATPNa2). Через 1 мин определяют люминисценцию в течение общего времени 1 сек, причем длительность сигнала на половине высоты амплитуды составляет 5 мин, при использовании MicroLumat LB 96 Р, фирма EG&G Berthold.
В Таблице 2 представлены физико-химические характеристики и данные действия соединений по настоящему изобретению in vitro. IC50 означает функциональную активность, а термин "пМ" означает пикомоль в литре. Растворимость в воде определяют методом, описанным в примечании 2).
1) В скобках представлено число независимых друг от друга экспериментов.
2) Растворимость в воде определяют по описанному ниже методу.
Растворимость в растворе Рингера, определенная по стандартному методу.
Для определения растворимости по стандартному методу исследуемое соединение в избытке смешивают с инертным носителем, например песком, и смесью заполняют стеклянную колонку (объемом приблизительно 10 мл). Перед этой операцией на дно колонки помещают слой из ваты и стекловолоконный фильтр. Смесь исследуемого соединения и песка пропитывают 1,0 мл растворителя, растворимость в котором следует определить, в течение 1 ч. Затем в колонку наносят 10 мл растворителя и раствор прокачивают в циклическом режиме с помощью перистальтического насоса. Данный метод определения проводят при комнатной температуре (приблизительно 20°С). Растворимость определяют, когда массовая концентрация в последовательно полученных фракциях составляет постоянную величину. Массовую концентрацию определяют с помощью описанного ниже метода ВЭЖХ.
Метод ВЭЖХ
Оборудование
Система ВЭЖХ Hewlett Packard 1100, детектор Hewlett Packard DAD, серия 1100.

55% Мобильная Фаза А: смешивают 970 мл H2O Milli-Q, 30 мл ацетонитрила и 1 мл трифторуксусной кислоты. Величина рН составляет приблизительно 1,9.
45% Мобильная Фаза В: смешивают 300 мл Н2О Milli-Q, 700 мл ацетонитрила и 1 мл трифторуксусной кислоты. Величина рН составляет приблизительно 1,8.
Введение соединений по настоящему изобретению можно осуществлять в различной форме, подходящей для активного пептида. Подходящие способы введения известны специалистам в данной области техники. Например, введение можно осуществлять с помощью инъекции. Введение можно осуществлять также парентеральным способом. При этом предпочтительными являются подкожный (п.к.), внутримышечный (в.м.), внутривенный (в.в.), буккальный (например, подъязычный) или ректальный способы. Наиболее предпочтительными являются п.к. и в.м.
Соединения по настоящему изобретению подходят для изготовления различных форм применения, например лиофильно высушенных препаратов, растворов или суспензий. Подходящие формы применения и способы их получения известны специалистам в данной области техники. В качестве вспомогательных веществ и носителей пригодными являются, например, гекситы, такие как маннит, прежде всего D-маннит, L-маннит или D,L-маннит, сорбит, такой как D-сорбит, D- или L-альтрит, идит, глюцит и дульцит.
Изготовление осуществляют по известным методам, например с помощью смешивания, суспендирования или лиофилизации.
Пример А: лиофилизированный препарат для получения раствора для п.к. инъекции.
1 мг соединения согласно Примеру 1 (0,26-0,27 мг ацетата соответственно); 0-16,9 мас.ч. D-маннита, предпочтительно 0,1-7 мас.ч., при необходимости соотнесенные к Примеру 1, и вода для инъекций (для приготовления инъекционных растворов из лиофилизированных препаратов). Получение: 1,62 г соединения согласно Примеру 1 растворяют в 30%-ном растворе уксусной кислоты (приблизительно 1,5л воды для инъекций и 91,17 г уксусной кислоты). Раствор разбавляют 1,5 л воды. Добавляют 82,2 г маннита, стерилизуют и разливают по 2 мл в стерильные флаконы для инъекций в асептических условиях, высушивают лиофильно. В каждом флаконе содержится 1 мг лиофилизированного препарата соединения согласно Примеру 1.
Соединения по настоящему изобретению, например, подходят для лечения злокачественных или доброкачественных гормонально зависимых заболеваний, например для лечения карциномы молочной железы, карциномы предстательной железы, эндометриоза, миомы матки, доброкачественной гиперплазии предстательной железы (ГПЖ), а также для лечения бесплодия у женщин или мужчин, например для профилактики преждевременного отторжения яйцеклетки у пациенток, которые происходят при контролируемой овариальной стимуляции, при которой у пациенток извлекают яйцеклетки и используют вспомогательные методы воспроизводства. Упомянутые методы лечения могут быть использованы для лечения млекопитающих, прежде всего человека.
Изобретение относится к антагонистам LHRH - декапептидам общей формулы I: AXxx1-Xxx2-Xxx3-Xxx4-Xxx5-Xxx6-Xxx7-Xxx8-Xxx9-Xxx10-NH2, где А - ацетильная группа, Xxx1=D-Nal(2), Xxx2=D-Cpa, Xxx3=D-Pal(3), Xxx4=Ser, Xxx5=N-Me-Tyr, Xxx6 означает D-Cit, D-Hci, D-[ε-N1-4-(4-амидинофенил)амино-1,4-диоксобутил]-Lys [D-Lys(B)], Xxx7=Leu, Nle; Xxx8=Arg, Lys (iPr); Xxx9=Pro, Xxx10=D-Ala или Sar, при условии, что если Xxx6=D-Lys(B), то Xxx7 означает Nle, если Xxx6=D-Cit, то Xxx7=Nle и Xxx10=D-Ala, или если Xxx6 означает D-Hci, то Xxx7 означает Leu и Xxx10 означает D-Ala, а также их солям с фармацевтически приемлемыми кислотами; фармацевтической композиции, способу получения фармацевтической композиции, способу лечения гормонально зависимых опухолей у млекопитающих. Соединения по изобретению обладают повышенной растворимостью в воде. 5 с. и 11 з.п. ф-лы, 2 табл.
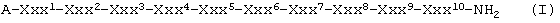
где
А означает ацетильную группу,
Ххх1 означает D-Nal(2),
Ххх2 означает D-Cpa,
Ххх3 означает D-Pal(3),
Ххх4 означает Ser,
Ххх5 означает N-Me-Tyr,
Ххх6 означает D-Cit, D-Hci или D-[ε-N'-4-(4-амидинофенил)амино-1,4-диоксобутил]-Lys (сокращенно: D-Lys(B)),
Ххх7 означает Leu или Nle,
Ххх8 означает Arg или Lys(iPr),
Ххх9 означает Pro и
Ххх10 означает D-Ala или Sar,
при условии, что если Ххх6 означает D-Lys(B), то Ххх7 означает Nle, если Ххх6 означает D-Cit, то Ххх7 означает Nle и Ххх10 означает D-Ala, или если Ххх6 означает D-Hci, то Ххх7 означает Leu и Ххх10 означает D-Ala, a также их соли с фармацевтически приемлемыми кислотами.
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Arg8-Pro9-Sar10-NH2
или его соль с фармацевтически приемлемыми кислотами.
Ac-D-Nal(2)1-D-Cpa2-D-Pal(3)3-Ser4-N-Me-Tyr5-D-Lys(B)6-Nle7-Arg8-Pro9-D-Ala10-NH2
или его соль с фармацевтически приемлемыми кислотами.
| СПОСОБ ТВЕРДОФАЗНОГО СИНТЕЗА ПЕПТИДОВ | 1991 |
|
RU2043362C1 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ, СПИДа И/ИЛИ СПИД-АССОЦИИРОВАННОГО КОМПЛЕКСА (ВАРИАНТЫ), ПЕПТИД ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИМЕНИМАЯ СОЛЬ (ВАРИАНТЫ) И НОНА- И ДЕКАПЕПТИДЫ - ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ, СПИДа И/ИЛИ СПИД-АССОЦИИРОВАННОГО КОМПЛЕКСА | 1994 |
|
RU2154492C2 |
| Способ протравливания зерна | 1935 |
|
SU49628A1 |
| 1971 |
|
SU413209A1 | |
| Экономайзер | 0 |
|
SU94A1 |
| Экономайзер | 0 |
|
SU94A1 |
| US 5300492, 05.04.1994 | |||
| Способ измерения разности фаз | 1985 |
|
SU1298683A1 |

