Изобретение касается ферментативного способа стереоизбирательного получения гетеробициклического спиртового энантиомера. Изобретение, далее, касается существенно чистого спиртового энантиомера и использования этого энантиомера для получения фармакологически активного пиперазинового производного.
Различные биологически активные вещества, которые могут быть использованы, например, в фармацевтических составах, предназначенных для человеческого и ветеринарного применения, содержат хиральный центр в своей молекулярной структуре и, следовательно, характеризуются появлением оптического изомеризма. В этой области техники в общем известно, что часто только один из энантиомеров обладает желаемой оптимальной биологической активностью. Присутствие другого оптического антипода в композиции или в веществе может вести к появлению или провоцировать появление определенных побочных эффектов и отягощать реципиента, т.е. человека или животного. В общем все сильнее и сильнее ощущается необходимость введения биологически активного вещества в виде в существенной мере чистого энантоиомера, который специфически проявляет требуемую биологическую активность. Следовательно, операция разделения рацемата на его энантиомеры часто является важным этапом процесса получения фармакологически активных веществ.
В основном известны три метода, пригодные для разделения рацематов на их соответствующие энантиомеры. Первый из этих методов, а именно метод разделения, основанный на различии физических свойств, например на различии кристаллического строения, применяется лишь иногда.
Второй и пока в общем наиболее используемый метод разделения включает в себя взаимодействие с промышленно выпускаемым оптически активным реактивом, при котором получают диастереоизомеры, которые различаются по своим физическим свойствам. Тем самым, диастереоизомеры, полученные этим способом, могут быть разделены, например, перекристаллизацией, после чего надлежащей химической обработкой могут быть вновь получены соответствующие энантиомеры. Можно видеть, что такой метод разделения рацематов является и трудоемким, и дорогим, что, в общем, обусловлено использованием и извлечением дорогого оптически активного реактива.
Недавно в более экономичном методе разделения стали применять ферменты, под воздействием которых избирательно осуществляется химическое модифицирование одного энантиомера рацемата, после чего проводится отделение модифицированного энантиомера от немодифицированного. В качестве примера можно привести работу Бианчи (Bianchi) и др. (J. Org. Chem., 1988, 53, 5531-5534), которые сообщают об использовании ангидридов карбоновой кислоты в качестве ацилирующих веществ при проведении катализируемой липазой избирательной этерификации рацемических спиртов. Им удалось получить ряд первичных и вторичных спиртов с высокой степенью оптической чистоты, а именно, с энантиомерной чистотой более 95%. В самом деле, в случае большинства фармацевтических применений требуется энантиомерная чистота, равная по крайней мере 95%. Хотя несколько результатов, полученных Бианчи с сотр., и являются обещающими, в случае некоторых спиртов не наблюдается или наблюдается недостаточное стереоизбирательное превращение. В двух недавних публикациях Энниса (Ennis) и др. (Tetrahedron Lett., 1992, 33, 6283-6286 и 6287-6290) подход с ферментативным разделением был применен к 2-гидроксиметил-1,4-бензодиоксанам, используемым в качестве подложек. Авторы установили, что использованием этого метода разделения не могут быть достигнуты требуемые стандарты оптической чистоты, в силу чего необходимо провести повторение процесса ферментативного разделения.
Для содействия процессу отделения сложного эфира, получаемого из оставшегося спирта Терао (Terao) и др. (Chem. Pharm. Bull., 1989, 37, 1653-1655) использовали янтарный ангидрид для получения янтарного моносложноэфирного энантиомера, который легко мог быть отделен от другого непровзаимодействовавшего спиртового энантиомера посредством промывки щелочным раствором. Этим способом желательный активный энантиомер может быть отделен от нежелательного неактивного энантиомера с меньшими усилиями, хотя в общем результаты, касающиеся оптической чистоты, т.е. энантиомерной чистоты, и являются неудовлетворительными. Лишь при использовании одной подложки, а именно (1-гидроксиэтил) бензола, являющегося вторичным спиртом, способ ферментативного разделения рацемата посредством энантиоизбирательной этерификации с янтарным ангидридом оказался удовлетворительным.
Вдобавок к часто непредсказуемым результатам ферментативного разделения спиртового рацемата, как это может быть заключено из приведенных выше публикаций, еще одна проблема является присущей процессу отделения требуемого спиртового энантиомера от его соответствующего рацемата. В самом деле, при каждом рацематическом разделении помимо требуемого энантиомера получается нежелательный оптический антипод, который в общем случае является ненужным. Сказанное означает, что по крайней мере 50% в общем случае дорогого субстрата следует считать химическим отходом или, иначе говоря, выход у процесса рацематического разделения, если говорить о выделении активного материала, составляет самое большее 50%. Сказанное четко иллюстрируется таблицами, приведенными в указанных выше публикациях Энниса и др., из которых следует, что исходный рацемат может дать 50% требуемого энантиомера (самое большее), получаемого либо в виде непревращенного спирта, либо после превращения в виде сложного эфира.
Цель настоящего изобретения сводится к разработке экономически оправданного способа стереоизбирательного получения гетеробициклического спиртового энантиомера.
Эта цель может быть достигнута проведением ферментативного способа, как он был определен выше, где способ в соответствии с настоящим изобретением характеризуется тем, что в существенной мере чистый энантиомер с общей формулой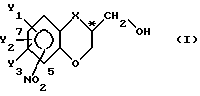
в которой
X - O, S, NH, N-(C1-C4)-алкильная группа или группа CH2;
Y1, Y2 и Y3 независимо представляют собой в каждом случае водород или являются заместителями, выбранными из галогена, (C1-C4)-алкильной группы, (C1-C4)-алкоксигруппы, (C1-C4)-галоалкильной группы, нитрогруппы и цианогруппы; NO2-заместитель присоединяется к бициклической кольцевой системе в 5- или 7-положении и C*-атом находится либо в R-, либо в S-конфигурации, готовят из его соответствующего спиртового рацемата, проводя последовательно следующие стадии взаимодействия:
I) ацилирование упомянутого рацемата ацилирующим веществом при воздействии фермента, обладающего стереоизбирательной этерификационной активностью;
II) отделение не претерпевшего этерификацию соединения от образовавшегося сложного эфира и выделение требуемого в существенной мере чистого спиртового энантиомера с формулой I или его сложного эфира;
III) гидролизование образовавшегося сложного эфира с превращением тем самым упомянутого сложного эфира в соответствующий спиртовой энантиомер, и
IV) превращение нежелательного спиртового энантиомера в исходный спиртовой рацемат при основных условиях, чтобы оказалось возможным его повторное использование.
Вопреки всем ожиданиям указанная выше основная обработка (стадия IV) привела к рацемизации нежелательного спиртового энантиомера. Это явление не может быть объяснено, поскольку протон, присоединенный к хиральному центру (C*) вообще не является кислотным. Спиртовой рацемат, полученный таким способом, может быть вновь использован в качестве исходного материала в последующей реакции разделения. Очевидно, что это изобретение делает процесс катализируемого ферментом стереоизбирательного получения спиртового энантиомера осуществимым процессом как с экономической, так и с экологической точки зрения.
Подходящими ацилирующими веществами для проведения указанной выше реакции ацилирования являются ангидриды карбоновой кислоты, как это будет проиллюстрировано ниже, и виниловые сложные эфиры, такие как винилацетат, винилпропионат, винилбутират, винилизобутират и им подобные вещества.
Реакцию ацилирования желательно вести в системах с органическим растворителем, содержащих небольшие количества воды или водного буферного раствора.
Фермент наиболее часто используют в виде сырого твердого препарата, который поставляет промышленность, чем облегчается его извлечение. Упомянутый фермент, однако, может быть также применен в иммобилизированном состоянии, например в ковалентно связанном состоянии или в адсорбированном состоянии с нанесением на надлежащий носитель. Соединение, не претерпевшее этерификацию, может быть отделено от полученного сложного эфира использованием различных способов, которые, как это известно, применяются для разделения родственных соединений, таких как экстракция, перекристаллизация, препаративная колонная хроматография и т.д.
Существенно чистый спиртовой энантиомер, как упоминали выше, относится, как это понимают, к спиртовым соединениям с энантиомерной чистотой, превышающей примерно 95%. Если в ферментативном способе, отвечающем настоящему изобретению, такая энантиомерная чистота не достигается, то тогда энантиомерная чистота может быть в общем случае доведена до требуемого уровня простым проведением перекристаллизации. Следовательно, процесс выделения требуемого существенно чистого спиртового энантиомера, как это было описано выше, может также включать в себя стадию перекристаллизации, что ведет к повышению энантиомерной чистоты и к удалению второстепенных примесей.
Критическая стадия взаимодействия, а именно рацемизация нежелательного спиртового энантиомера при основных условиях, может быть легко осуществлена как в апротонных, так и протонных условиях. К подходящим основаниям относятся гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид аммония и им подобные вещества; их используют растворенными в воде или в водных смесях растворителя, состоящих из смешиваемых с водой органических растворителей, таких как спирты. Примерами оснований, которые могут быть использованы в апротонных системах, являются: а) гидриды, например гидрид натрия, находящийся в апротонных растворителях, таких как диметилсульфоксид, b) алкоксиды калия, такие как трет-бутоксид калия и метил-2-бутоксид калия, находящиеся в апротонных растворителях, таких как простые эфиры (например, тетрагидрофуран), и с) алкиллитиевые соединения и алкиламиды лития, такие как метиллитий, различные бутиллитиевые соединения и диизопропиламид лития, также находящиеся в апротонных растворителях, например в тетрагидрофуране. Спиртовой рацемат может быть извлечен с хорошими выходами после нейтрализации кислотой, например экстракцией подходящим органическим растворителем из водной фазы и затем при необходимости с проведением испарения растворителя, пригодного для повторного использования.
Гидролиз полученного сложного эфира, как это упоминали выше на стадии III, может быть проведен обычным образом в кислых условиях или при слабо основных условиях, чтобы исключалась рацемизация получаемого спиртового энантиомера.
В качестве специального варианта осуществления настоящего изобретения, однако, указанный выше гидролиз сложного эфира и рацемизация спиртового энантиомера могут быть объединены. В этом способе указанные выше стадии взаимодействия III и IV могут быть объединены, чем может быть достигнуто понижение числа стадий на одну. Для одновременного осуществления обоих операций, а именно гидролиза и рацемизации, необходимо создать достаточно сильные основные условия, как это было указано выше при рассмотрении реакции рацемизации.
Способ, отвечающий настоящему изобретению, преимущественно предназначен для стереоизбирательного получения существенно чистого спиртового энантиомера со структурой бензодиоксана в виде соединения с общей формулой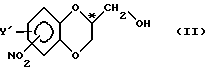
в которой
Y' - водород или заместитель, выбранный из хлорсодержащей, фторсодержащей и метильной группы;
NO2-заместитель присоединяется к бензодиоксановому кольцу в 5- или 7-положении, и
C*-атом находится либо в R-, либо в S-конфигурации,
посредством проведения последовательных стадий взаимодействия, как они были определены ранее.
Желательно, чтобы фермент представлял собой твердое вещество, поскольку в этом случае он может быть легко извлечен с целью повторного использования. Извлечение фермента удобно проводить после осуществления указанной выше стадии взаимодействия I, т.е. после завершения стадии ацилирования, что достигается использованием надлежащей методики, пригодной для этой цели, такой как простое фильтрование. Если используют фермент, который связан с надлежащим носителем, таким как целит (см. указанную выше публикацию Бианчи и др.) или стеклянные шарики, то тогда фермент может быть также извлечен простым фильтрованием, что, при желании, может сопровождаться промывкой фильтрата, свободного от примесей.
Предпочтение следует отдавать использованию ангидридов карбоновых кислот в сравнении с применением виниловых сложных эфиров в качестве ацилирующих веществ, поскольку ангидриды карбоновых кислот, такие как уксусный ангидрид, пропионовый ангидрид, масляный ангидрид, изомасляный ангидрид или гексановый ангидрид, обычно лучше действуют в присутствии надлежащего фермента. Для содействия отделению соединения, не претерпевшего этерификацию, от образовавшегося сложного эфира предпочтение следует отдавать ангидридам циклической карбоновой кислоты, в частности янтарному ангидриду или глутаровому ангидриду. Образовавшийся сложный моноэфир, полученный этим способом, может быть легко отделен от соединения, не претерпевшего этерификацию, экстракцией слабым щелочным раствором при таких условиях, при которых сложный эфир остается в сохранности.
Подходящими ферментами для проведения стереоизбирательной этерификации являются гидролазы, такие как встречающиеся в природе и синтезированные липазы и эстеразы. Примерами подходящих липаз являются Aspergillus niger, Candida cylindracea (например, мейто® MY 30 или амано® AY (торговые марки материалов)), Candida lipolytica, Chromobacterum viscosum, Geotrichum candidum, Humicola lanuginosa, Mucor miehei, Mucor javanicus (например, амано® M), свиная панкреатическая липаза, Penicillium cyclopium, Penicillium roqueforti, Pseudomonas cepacia (амано® PS), Pseudomonas fluorescence (например, амано® P), Rhizopus niveus (например, амано® N), Rhizopus javanicus (например, амано® F), Rhizopus arrhizus и Rhizopus delemar. В противоположность тому, что предлагается в публикации Бианчи и др., теперь установлено, что некоторые липазы, в частности липаза Candida cylindracea, обладает способностью давать S-энантиомер. Такие липазы обладают, следовательно, способностью стереоизбирательно осуществлять этерификацию S-энантиомера, в результате чего с высоким выходом и высокой стереохимической чистотой может быть получен в остатке R-энантиомер. Другие липазы, например Pseudomonas fluorescence и многие другие липазы, предпочитают осуществлять превращение R-энантиомера и, следовательно, являются вполне пригодными для выделения S-энантиомера, в равной мере с высоким выходом и высокой стереохимической чистотой.
Настоящее изобретение касается также существенно чистого спиртового энантиомера с общей формулой I, представленной ранее, в которой группа X и заместители Y имеют смысл, оговоренный выше, заместитель NO2 присоединяется к бициклической кольцевой системе в 5- или 7-положении и C*-атом находится в R-конфигурации. Этот энантиомер может быть легко получен по ферментативному способу, отвечающему настоящему изобретению. Этот энантиомер может быть использован в качестве ключевого промежуточного соединения в способе получения некоторых фармакологически активных пиперазиновых производных, о чем будет говориться ниже.
В литературе (Drugs of the Future, 1988, 13, 31-33) описан синтез солянокислого флезиноксана, который является сильнодействующим 5-HTIA-агонистом, проявляющим активность при оральном применении. Рацемический бензодиоксан, отвечающий соединению с приведенной выше формулой II, где Y' - 7-хлорсодержащий заместитель, сначала превращают, воздействуя хлористым бензоилом, что делается с целью защиты его спиртовой группы. Затем проведением каталитической гидрогенизации с последующим взаимодействием с бис(хлорэтил)амином получают рацемическое пиперазиновое соединение. На этом этапе проводят разделение пиперазинового рацемата, что делают с использованием (+)-камфорсульфоновой кислоты. После проведения нескольких перекристаллизаций получают оптически чистый R-(+)-энантиомер. Взаимодействием этого энантиомера с N-(4-фторбензоил)азиридином, разблокированием гидроксигруппы посредством омыления бензоатного сложного эфира и, наконец, обработкой хлористо-водородной кислотой получают требуемый существенно чистый (+)-энантиомер, а именно флезиноксан•HCl. В упомянутой выше недавно сделанной публикации Энниса (Ennis) и др. (Tetrahedron Lett., 1992, 33, 6287-6290) описано ферментативное разделение флезиноксана и его оптического антипода, что является конечной стадией разделения. После трудоемкого двухкратного проведения ферментативного процесса требуемый флезиноксан может быть получен с удовлетворительной энантиомерной чистотой.
Из сказанного выше становится очевидным, что описанный способ получения флезиноксана является трудоемким и дорогим, что, в частности, связано с проведением трудоемкого разделения рацемата в предложенной там стадии многоступенчатого процесса синтеза. Становится очевидным, что неизбежные потери активного материала при разделении составляют наиболее отрицательную сторону предложенной стадии процесса синтеза.
Теперь установлено, что существенно чистый спиртовой энантиомер с общей формулой I может быть легко использован в качестве ключевого промежуточного соединения в синтезе фармакологически активных пиперазиновых производных, поскольку устраняется трудоемкое разделение рацемата в предложенной стадии многоступенчатого синтеза.
Следовательно, настоящее изобретение касается также использования существенно чистого спиртового энантиомера с общей формулой I, представленной ранее, в которой группа X и заместители Y имеют смысл, оговоренный выше, заместитель NO2 присоединяется к бициклической кольцевой системе в 5-положении, и C*-атом находится в R-конфигурации, для получения фармакологически активного пиперазинового производного посредством проведения с использованием упомянутого энантиомера последовательности следующих взаимодействий:
I) защита свободной гидроксигруппы надлежащей гидроксизащищающей группой при сохранении фактической конфигурации C*-атома с целью получения соединения с общей формулой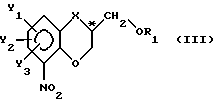
где
R1 - гидроксизащищающая группа;
II) восстановление нитрозаместителя с целью превращения упомянутого существенно чистого энантиомера с формулой III в аминосоединение с общей формулой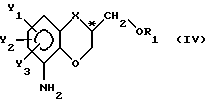
при сохранении фактической конфигурации у C*-атома;
III) превращение упомянутого выше аминосоединения с формулой IV в пиперазиновое соединение с общей формулой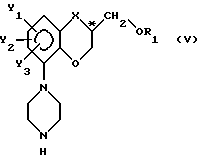
при сохранении фактической конфигурации у C*-атома;
IV) образование производного у упомянутого пиперазинового соединения с формулой Y при сохранении фактической конфигурации у C*-атома с получением пиперазинового производного с общей формулой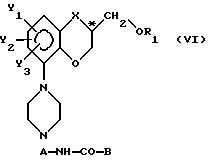
в которой
A - (C2-C4)-алкиленовая группа с прямой или разветвленной цепью;
B - фенильная группа или гетероциклическая группа, выбранная из тиенильной, пиранильной, фурильной, пирролильной, пиридильной и пиразинильной группы, которая может быть замещенной одним или несколькими заместителями, выбранными из галогена, (C1-C3)-алкильной группы, (C1-C3)-галоалкильной группы, цианогруппы, нитрогруппы, гидроксигруппы, подвергнутой этерификации гидроксигруппы и (C1-C3)-алкоксигруппы, посредством взаимодействия упомянутого пиперазинового соединения с формулой V либо
a) с соединением с общей формулой
L-A-NH-CO-B, (VII)
в которой
L - отщепляемая группа, желательно выбранная из хлорсодержащей, мезилатной и тозилатной группы, либо
b) с соединением с общей формулой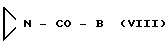
с образованием пиперазинового производного с общей формулой VI, в которой
A - этиленовая группа, и, наконец,
V) разблокирование упомянутого соединения с формулой VI с образованием свободного спиртового энантиомера с общей формулой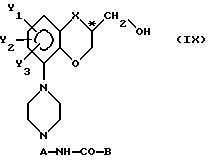
в которой
C*-атом находится в R-конфигурации.
Из сказанного выше становится очевидным, что приведенная выше последовательность стадий взаимодействия может быть легко осуществлена с сохранением фактически существующей конфигурации у C*-атома, чем устраняется опасность ухудшения энантиомерной чистоты у конечного пиперазинового производного.
Свободная гидроксигруппа может быть защищена (стадия взаимодействия I) использованием надлежащей эфирной группы сложного или простого эфира. Примерами приемлемых гидроксизащищающих групп являются (тригидрокарбил)силильная, (дигидрокарбил) (гидрокарбилокси)силильная, третичная (C4-C12)-алкильная, (произвольно замещенная)фенокси ((C2-C8)-диалкил)-этильная, (C1-C4)-алкокси ((C2-C8)-диалкил)метильная, (тио)-ацетилсодержащая группы, такие как ди- и тетрагидропиран-2-ил, и ди- и тетрагидрофур-2-ил и эфирсодержащие группы от сложных эфиров, являющиеся производными от моно-, ди- или тризамещенной уксусной кислоты, в случае чего заместителей преимущественно выбирают из (C1-C12)-алкильной группы и произвольно замещенных одним или несколькими заместителями замещенной фенильной группы, произвольно замещенных одной или несколькими метильными группами циклогексанкарбоновой кислоты или адамантанкарбоновой кислоты. Приведенный выше термин "гидрокарбил" распространяется на (C1-C8)-алкильную, (C2-C8)-алкенильную, (C2-C8)-алкинильную, фенильную группу и фенильную группу, замещенную одним или несколькими заместителями. Подходящими заместителями для названной выше фенильной группы и феноксигруппы, являются гидроксигруппа, алкоксигруппа, алкилкарбонилоксигруппа, аминогруппа, алкиламиногруппа, диалкиламиногруппа, алкилкарбониламиногруппа, алкилсульфониламиногруппа, нитрогруппа, алкилсульфонильная группа, алкилкарбонильная группа, галоген, цианогруппа, алкильная группа, у которых алкильные заместители содержат от одного до пяти атомов углерода, и (C5-C12)-циклоалкильная группа.
Восстановление нитрогруппы до аминогруппы (стадия II) может быть легко осуществлено воздействием водородом в присутствии подходящего металлического катализатора, например палладия, нанесенного на углерод, в надлежащем полярном органическом растворителе, например в спирте.
Превращение аминосоединения в пиперазиновое соединение (стадии III) может быть легко осуществлено с использованием, например, бис(2-хлорэтил)амина, находящегося в подходящем органическом растворителе, например в ароматическом углеводороде, таким как толуол, хлорбензол и им подобные вещества.
Стадию взаимодействия, указанную выше под номером IV, желательно проводить так, как это описано в Европейской патентной заявке 138280, а именно в инертном органическом растворителе или без использования растворителя и при условиях взаимодействия, описанных в упомянутой патентной заявке.
Конечное разблокирование гидрокси группы может быть осуществлено использованием веществ, пригодных для расщепления сложных или простых эфиров. Сложные эфиры могут быть легко подвергнуты гидролизу, что может быть осуществлено в слабощелочных или слабокислых условиях при сохранении фактически существующей конфигурации у C*-атома. Расщепление простого эфира желательно проводить воздействием сильных кислот, находящихся в органических растворителях.
Настоящее изобретение касается, далее, новых промежуточных соединений, участвующих в указанной выше последовательности взаимодействий, а именно существенно чистого энантиомера с общими формулами III и IV, представленными ранее, в которых
X и заместители Y имеют смысл, оговоренный выше, и конфигурация C*-атома совпадает с R-конфигурацией C*-атома у соединения с упомянутой выше формулой I.
Изобретение, наконец, касается способа получения существенно чистого пиперазинового производного энантиомера с общей формулой IX, представленной выше, путем сначала получения существенно чистого спиртового энантиомера с общей формулой I, представленной выше, в которой C*-атом находится в R-конфигурации, из его соответствующего спиртового рацемата путем последовательного проведения стадий взаимодействия, как они были определены выше, с последующим превращением упомянутого соединения с формулой I в требуемое пиперазиновое производное через последовательность взаимодействий, как это было определено выше.
Теперь изобретение будет более подробно описано со ссылкой на соответствующие специфические примеры.
Пример I.
Раствор, содержащий 125 mМ (±)-2,3-дигидро-5-нитро-7- хлор-1,4-бензодиоксан-2-метанола, 250 mМ пропионового ангидрида и 0,2% (по весу на объем) липазы Pseudomonas fluorescens (вещество с торговым названием амано® P) в смеси, состоящей из простого трет-бутилметилового эфира, гексана и воды (взятыми в объемном отношении 50:50:0,1), инкубировали при 37oC, проводя перемешивание. После превращения на 80% (происходила этерификация спирта) реакцию останавливали, отфильтровывая фермент. Полученный сложный эфир и оставшийся спирт разделяли на колонке с зорбаксом® C-8 (торговое название материала). Энантиомерную чистоту оставшегося спирта устанавливали, используя хиральную α-гликопротеидную колонку. Энантиомерную чистоту оставшегося спирта и полученного сложного эфира определяли также методом ядерного магнитного резонанса на ядрах водорода-1 (1-H-ЯМР), не проводя разделение, для чего в качестве хирального разделяющего вещества использовали (+)- или (-)-трифторметил-9-антраценметанол. Оставшийся спирт содержит S-(-)-спирт с энантиомерной чистотой 97,5%.
Пример II.
Поступая как и в случае примера 1, проводили этерификацию, используя 0,2% (по весу в расчете на объем) липазы Candida cylindracea (с торговым названием мейто® MY). После превращения спирта на 69% реакцию останавливали. Оставшийся спирт содержит R-(+)-спирт с энантиомерной чистотой 97,5%. Полученный R-(+)-спирт характеризуется таким же 1H-ЯМР-спектром, что и описанный в случае примера I. Удельное вращение плоскости поляризации света у R-(+)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола в ацетонитриле найдено следующим [α]
По тому же способу, который использовали в случае получения R-(+)-2,3-дигидро-5-нитро-7-метил-1,4-бензодиоксан-2-метанола, получили R-(+)-2,3-дигидро-5-нитро-1,4-бензодиоксан-2-метанол с одинаково высокой энантиомерной чистотой.
Пример III.
Раствор, содержащий 250 mМ (±)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола, 500 mМ ангидрида масляной кислоты и 0,5% (по весу в расчете на объем) липазы Candida cylindracea (с торговым названием мейто® MY) в смеси гексана, этилацетата и воды (взятыми в объемном отношении 50:50:0,2), инкубировали при 25oC в условиях перемешивания. После превращения спирта на 65% реакцию останавливали. Оставшийся спирт содержит R-(+)-спирт с энантиомерной чистотой 97,5%.
Пример IV.
По способу, описанному в примере III, 250 mМ (±)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола инкубировали совместно с 500 mМ изомасляного, соответственно гексанового, ангидрида. После превращения спирта на 63%, соответственно на 60%, реакцию останавливали. Оставшийся спирт содержит R-(+)-спирт, который в обоих случаях характеризуется энантиомерной чистотой в 97,5%.
Пример V.
Раствор, содержащий 350 mМ (±)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола, 600 mМ янтарного ангидрида и 2,4% (по весу в расчете на объем) липазы Candida cylindracea (с торговым названием мейто® MY) в смеси, состоящей из простого трет-бутилметилового эфира, ацетонитрила и воды (находящимися в объемном соотношении 90:10:0,6), инкубировали при комнатной температуре в условиях перемешивания. После превращения спирта на 70% реакцию останавливали, проводя фильтрование. Оставшийся спирт содержит R-(+)-энантиомер с энантиомерной чистотой, составляющей 98%.
Пример VI.
Энантиоизбирательная этерификация
Уравнение взаимодействия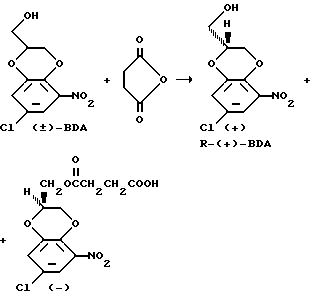
где
BDA - 2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанол.
Раствор, содержащий 15,2 кг (±)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола ((±)-BDA), 7,6 кг янтарного ангидрида и 3,7 кг липазы Candida cylidracea (с торговым названием мейто® МУ) в смеси, состоящей из 200 л простого трет-метилэтилового эфира, 17,5 л ацетонитрила и 925 мл воды, инкубировали в среде азота при комнатной температуре в реакционном сосуде. После достижения степени превращения в 60 - 63% (жидкостная хроматография высокого давления, примерно 20 ч) реакцию останавливали, отфильтровывая фермент. Фермент дважды промывали 10 л простого трет-бутилметилового эфира, и органический слой последовательно промывали 90 л и 30 л водного раствора карбоната натрия (150 г Na2CO3 в 1 л воды). Раствор карбоната натрия дважды экстрагировали 10 л простого трет-бутилметилового эфира. Затем объединенные органические слои последовательно промывали 30 л воды, содержащей разбавленную хлористо-водородную кислоту, полученную растворением 40 мл 36%-ной HCl в 15 л воды, и 10 л воды. Простой трет-бутилметиловый эфир отгоняли в вакууме при 60oC. Кристаллический остаток (4,6 кг) растворяли в 15 л 96%-ного этанола при 60oC; к этому раствору добавляли 10 л н-гексана, что делали при перемешивании. Смесь охлаждали примерно до 10oC, и после перемешивания в течение промежутка времени от 2 до 10 ч кристаллическое вещество подвергали отсосу, последовательно промывали 10 л смеси этанола с гексаном (при объемном соотношении 15: 35) и 5 л н-гексана и сушили. Кристаллическое вещество представляет собой чистый (с энантиомерной чистотой 98%) (+)-энантиомер, а именно R-(+)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанол (R-(+)-BDA); выход примерно составляет 4 кг.
Температура плавления составляет 116,0oC; [α]
Пример VII.
Омыление полученного сложного S-(-)-2,3-дигидро-5-нитро-7-хлор- 1,4-бензидиоксан-2-метанольного эфира
Уравнение взаимодействия
К объединенным водным слоям, взятым из эксперимента примера VI, добавляли 15 л 50%-ного NaOH, что делали примерно при 23oC. Реакционную смесь перемешивали в течение примерно 15 ч при 23oC и затем охлаждали до 5oC. После прививки смесь перемешивали в течение 3 ч при 5oC. Кристаллическое вещество подвергали отсосу, промывали 60 л воды и сушили. Результирующий спирт, содержащий избыток S-(-)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2- метанольного энантиомера (S-(-)-BDA-энантиомера), получали с выходом порядка 10 кг.
Пример VIII.
Рацемизация S-(-)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан- 2-метанольного энантиомера
Уравнение взаимодействия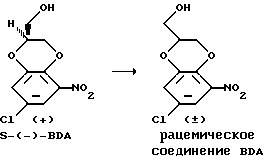
Энантиомер S(-)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан- 2-метанол (S-(-)-BDA), полученный по способу примера VII, в количестве 1 кг растворяли в 6 л н-пропанола в среде азота в условиях нагревания в сосуде с обратным холодильником. К этому раствору добавляли 235 мл 2н. водного раствора гидроксида натрия, что делали примерно за 15 минут. Раствор затем грели в сосуде с обратным холодильником в течение 1,5 ч. После охлаждения примерно до 40oC добавляли 47 мл раствора концентрированной HCl (до величины pH, равной 3). Пропанол отгоняли в вакууме при температуре порядка 60oC. К остатку добавляли 4 л н-гексана, и раствор подвергали прививке, охлаждая при этом до 20oC и медленно перемешивая. После перемешивания в течение 2 ч при 20oC и в течение ночи при 0oC кристаллическое вещество подвергали отсосу и дважды промывали 0,5 л н-гексана. Кристаллическое вещество затем перемешивали с 7,5 л воды, что делали примерно при 70oC в течение одного часа. После охлаждения до 20oC добавляли 350 мл н-гексана, и после перемешивания еще в течение одного часа кристаллическое вещество отфильтровывали и дважды промывали 0,5 л н-гексана. После сушки получали требуемый рацемический (±)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанол (рацемический BDA) с выходом 850 г; содержание составляет 95%; энантиомерная чистота равна 0. Температура плавления составляет 108,2oC.
Рацемизация в равной мере идет успешно при использовании диизопропиламида лития в качестве основания и использования тетрагидрофурана в качестве растворителя; реакция проводится при 40oC; полная рацемизация достигается через 5,5 ч.
Пример IX.
Омыление и одновременная рацемизация сложного S-(-)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанольного эфира
К аликвоте, которую получали смешиванием 43,5 г (126 ммоль) S-(-)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанольного сложного эфира, 620 мл водного раствора карбоната натрия (150 г Na2CO3 в 1 л воды), 150 мл воды и 59 мл ацетонитрила, взятого из основного водного слоя, полученного так, как это делали в примере VI, добавляли раствор, содержащий 250 мл этанола и 50 мл 50%-ного (по весу в расчете на объем) водного раствора гидроксида натрия. Реакционную смесь перемешивали при нагревании в сосуде с обратным холодильником в течение 16 ч. После охлаждения до 40oC осторожно добавляли 160 мл 12н. водного раствора хлористо-водородной кислоты (величина pH примерно составляла 5). Реакционную смесь охлаждали до комнатной температуры, после чего твердое вещество подвергали отсосу, промывали водой и сушили. Получали23,8гсветло-коричневогорацемического(±)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола с энантиомерной чистотой, равной нулю.
Пример X.
Получение флезиноксана из R-(+)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола
Уравнения взаимодействия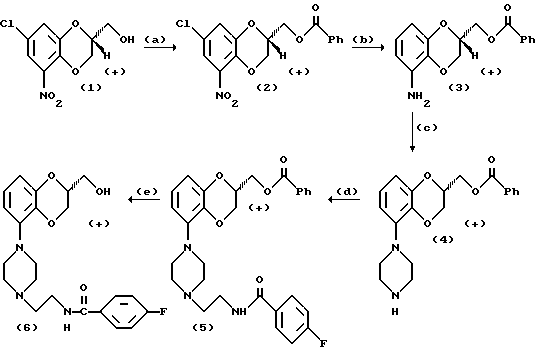
A. Бензоилирование R-(+)-2,3-дигидро-5-нитро-7-хлор-1,4- бензодиоксан-2-метанола (1) хлористым бензоилом в хлористом метилене, используемом в качестве растворителя, с целью получения соединения (2).
К раствору, содержащему 20 г (0,081 моля) соединения (1) в 250 мл дихлорметана и 12 мл триэтиламина, по каплям добавляли 10,1 мл (0,086 моля) бензоилхлорида, что делали при температуре 25oC. После добавления 10 мл воды и перемешивания в течение 10 минут слои разделяли. Органический слой промывали 50 мл воды, и объединенные водные слои экстрагировали 25 мл дихлорметана. Органические слои соединяли и упаривали при давлении 100 мбар (10 кН/м2) и температуре 30oC. После добавления 100 мл толуола продукт упаривали до сухого состояния (10 мбар (1 кН/м2), 50oC).
Требуемое соединение (2) получали с выходом 97,3%; чистота составляла 97,5. При проведении тонкослойной хроматографии элюирование проводили используя смесь, состоящую из дихлорметана, метанола и гидроксида аммония, взятых в соотношении 94:5:1; Rf = 0,71.
B. Восстановление нитросоединения (2) до соответствующего аминосоединения (3) воздействием водорода в присутствии каталитически эффективного количества палладия, нанесенного на углерод, и при использовании этанола в качестве растворителя.
К раствору, содержащему 6,0 г (16,7 ммоль) соединения (2) в 120 мл этанола и 40 мл этилацетата, добавляли 1,50 г препарата палладия, нанесенного на углерод (39,1% палладия, 10% углерода и 60,9% воды). После перемешивания в течение 5 мин добавляли 10,8 г (10 экв.) формиата аммония, и смесь перемешивали по крайней мере один час при температуре окружающей среды, а затем в течение двух часов при 40oC. Реакционную смесь охлаждали до 20 - 25oC, и палладиевый катализатор, нанесенный на углерод, отфильтровывали и промывали 50 мл этанола. Этанол выпаривали при давлении 100 мбар (10 кН/м2) и температуре 50oC. Остаток растворяли в 75 мл этилацетата и 15 мл 2н. водного раствора гидроксида натрия. После разделения слоев водный слой дважды экстрагировали 10 мл этилацетата. Объединенные органические слои дважды промывали 25 мл воды и доводили до сухого состояния при давлении 100 мбар (10 кН/м2) и температуре 50oC. После сушки в вакууме при 50oC получали требуемый продукт (3) с чистотой 96,0% и выходом 97,0%.
Проведением тонкослойной хроматографии (см. выше) получали: Rf = 0,67. Температура плавления солянокислой соли найдена равной 218 - 223oC. Величина [α]
C. Превращение аминосоединения (3) в соответствующее пиперазиновое соединение (4) при использовании солянокислого (бис(2-хлорэтил)амина с взаимодействием в ксилоле, выполняющем роль растворителя.
К раствору, содержащему 4,40 г (14,8 ммоль) соединения (3) в 50 мл ксилола, добавляли 2,8 г (14,8 ммоль) бис(2-хлорэтил)амин•HCl. Реакционную смесь грели в сосуде с обратным холодильном в течение 48 ч в среде азота. После охлаждения реакционной смеси до 35oC добавляли 1,36 мл 50%-ного водного раствора гидроксида натрия, находящегося в 25 мл 5%-ного водного раствора бикарбоната натрия. Реакционную смесь перемешивали при 35oC в течение трех часов, после чего добавляли 10 мл 2н. водного раствора гидроксида натрия и 20 мл воды. После перемешивания при 35oC в течение 10 мин реакционную смесь охлаждали до 20 - 25oC, и слои разделяли. Ксилоловый слой промывали трижды 25 мл воды. Органический слой доводили до сухого состояния (использовали 100%-ный этанол в качестве азеотропообразователя) при давлении 10 мбар (1 кН/м2) и температуре 50oC. Требуемый продукт (4) с чистотой 85,5% получали с выходом 82,3%.
Проведением тонкослойной хроматографии (см. выше) получали: Rf = 0,07. Температура плавления солянокислой соли найдена равной 183 - 186oC. Величина [α]
D. Взаимодействие пиперазинового соединения (4) с 4-фторбензоилазиридином с образованием соединения (5).
К 100,7 г (284 ммоль) соединения (4) добавляли п-фторбензоилазиридин (53,8 г, 325 ммоль) и 200 мл толуола. Реакционную смесь держали при пониженном давлении с температурой 80oC (использовали роторный испаритель); испаряли 150 мл вещества. После добавления 100 мл толуола реакционную смесь обрабатывали так, как это описано выше, еще в течение двух часов. После упаривания досуха к остатку добавляли метанол, и продукту предоставляли возможность закристаллизоваться при 5oC. Продукт подвергали отсосу, промывали метанолом (200 мл) и гексаном (400 мл), делая это последовательно, и сушили. Требуемое соединение (5) с чистотой 82% получали с выходом 105 г (71%). Переработкой маточной жидкости получали дополнительное количество требуемого продукта.
Проведением тонкослойной хроматографии (см. выше) получали: Rf = 0,59. Температура плавления составляла 126 - 127oC. Величина [α]
E. Омыление сложного эфира (5) с использованием этанольного раствора гидроксида калия с последующим подкислением этанольным раствором HCl с целью получения флезиноксана (6).
К суспензии, состоящей из 104 г (0,2 моль) соединения (5) в 1500 мл 96%-ного этанола, добавляли раствор, состоящий из 14 г (0,25 моль) KOH в 10 мл воды. После перемешивания при 20 - 25oC в течение 3,5 ч этанол выпаривали при 100 мбар (10 кН/м2) и температуре 50oC. К остатку добавляли воду (500 мл) и дихлорметан (200 мл) и реакционную смесь перемешивали в течение 5 минут. После разделения слоев водный слой экстрагировали 250 мл дихлорметана. Объединенные органические слои дважды промывали 100 мл воды. После сушки органический раствор упаривали до достижения остаточного объема величиной порядка 200 мл. К этому остатку добавляли 300 мл этилацетата и 100 мл жидкости выпаривали. После добавления 100 мл н-гексана продукту предоставляли возможность кристаллизоваться в течение ночи при 5oC. Кристаллический продукт отфильтровывали, последовательно промывали 30 мл холодного этилацетата и 200 мл н-гексана и сушили при 30oC. Флезиноксан (с чистотой 78%) получали с выходом 73 г.
Проведением тонкослойной хроматографии (см. выше) получали: Rf = 0,67. Температура плавления составляла 183-185oC. Величина [α]
Пример XI.
Раствор, содержащий 0,2 М 5-хлор-2,3-дигидро-7-нитро- 1,4-бензодиоксан-2-метанола, 0,34 М янтарного ангидрида и 2% липазы Candifa cylindracea (торговое название мейто® MY) в смеси, состоящей из простого трет-бутилметилового эфира, ацетонитрила и воды (находящихся в объемном соотношении 90:10: 0,3), инкубировали при комнатной температуре в условиях перемешивания. После превращения спирта на 41% (что устанавливали использованием колонки с зорбаксом C-8) реакцию останавливали фильтрованием. Оставшийся спирт содержит (+)-энантиомер с энантиомерной чистотой 38% (найденной в условиях использования колонки с чирацелом®-OD (торговое название материала)).
Пример XII.
Получение 6-хлор-2,3-дигидро-8-нитро-1,4-бензоксазин-3- метанола
Схема взаимодействия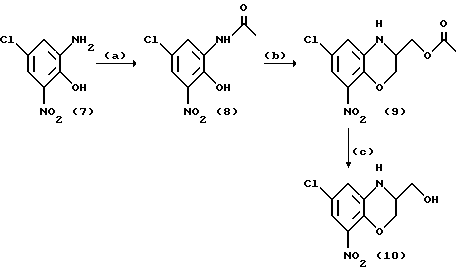
A. К суспензии, состоящий из 18,5 г (91 ммоль) соединения (7), находящегося в 50 мл толуола, добавляли 30 мл (314 ммоля) уксусного ангидрида. После прогрева в течение 4 ч при 100oC добавляли еще 10 мл уксусного ангидрида. Нагревание продолжали еще в течение двух часов. После удаления греющей ванны осторожно добавляли примерно 25 мл этанола. После охлаждения до комнатной температуры реакционную смесь обрабатывали этилацетатом и водой. Органический слой промывали дважды водой и сушили над сульфатом магния. После фильтрования растворитель отгоняли под вакуумом. К 18,23 г светло-коричневого твердого вещества добавляли 75 мл этанола и 80 мл 2н. водного раствора гидроксида натрия. Темно-красную суспензию перемешивали в течение ночи при комнатной температуре. После охлаждения до 0oC добавляли 90 мл 2н. раствора водной хлористо-водородной кислоты. Твердое вещество подвергали отсасыванию и дважды промывали водой. После сушки при комнатной температуре и нормальном давлении получали 16,5 г оранжевого порошка соединения (8).
Проведением тонкослойной хроматографии (элюирование проводили при 40 - 65oC смесью, состоящей из этилацетата и петролейного эфира, взятых в соотношении 50: 50) получали: Rf = 0,3. Температура плавления составляла 156-160oC.
B. К раствору, состоящему из 8 г (34,5 ммоль) соединения (8), находящегося в смеси, образованной из 80 мл толуола и 80 мл 1-метил-2-пирролидона, добавляли 5,6 г (40 ммоль) порошкообразного карбоната калия. Реакционную смесь перемешивали при температуре возврата флегмы в сосуде с обратным холодильником, и воду удаляли, используя аппарат Дина-Старка (Dean-Stark). Толуол отгоняли при атмосферном давлении. После охлаждения до 100oC добавляли 9,3 г (41 ммоль) глицидилтозилата. После перемешивания при 120oC в течение 4,5 ч суспензию охлаждали до комнатной температуры. Реакционную смесь разбавляли водой и этилацетатом и доводили величину pH до 5, добавляя 2н. водный раствор хлористо-водородной кислоты. Водный слой дважды экстрагировали этилацетатом. Соединенные органические слои промывали солевым раствором и сушили над сульфатом магния. После отфильтровывания сульфата магния и выпаривания растворителя под вакуумом получали 10,86 г темно-коричневой маслянистой жидкости. Очисткой методом жидкостной хроматографии высокого давления (элюировали при 40 - 65oC смесью этилацетата и петролейного эфира, взятыми в соотношении 25:75) получали 4,18 г соединения (9) в виде красных пластинчатых кристаллов.
Температура плавления составляла 76 - 84oC.
Проведением тонкослойной хроматографии (см. выше) получали: Rf = 0,15.
C. К суспензии, состоящей из 3 г (10 ммоль) соединения (9), находящегося в смеси, образованной из 100 мл метанола и 30 мл воды, добавляли 1,44 г порошкообразного карбоната калия. После перемешивания при комнатной температуре в течение 1,5 ч реакционную смесь обрабатывали, разбавляя водой и дважды подвергая экстракции этилацетатом. Объединенные органические слои трижды промывали разбавленным солевым раствором и сушили над сульфатом магния. После отфильтровывания сульфата магния и выпаривания растворителя под вакуумом получали 2,53 г соединения (10) (по данным метода ЯМР) в виде оранжевого твердого вещества.
Проведением тонкослойной хроматографии (элюировали при 40 - 65oC смесью этилацетата с петролейным эфиром, взятыми в соотношении 75:25) получали: Rf = 0,3.
1H-ЯМР: δ (миллионные доли) составляет 6,99 (дублет, 1H, в кольце); 6,90 (синглет, 1H, NH); 6,88 (дублет, 1H, в кольце); 5,02 (триплет, 1H, CH2OH); 4,19 (дублет из дублета, 1H, OCH2OH); 4,11 (дублет из дублета, 1H, OCH2CH); 3,40/3,50 (кластер, 3H, CHCH2OH).
Пример XIII.
Раствор, состоящий из 0,35 М 6-хлор-2,3-дигидро-8-нитро-1,4-бензоксазин-3-метанола, 0,6 М янтарного ангидрида и 3,3% (по весу в расчете на объем) липазы Candida cylindracea (с торговым названием мейто® MY) в смеси, образованной простым трет-бутилметиловым эфиром, ацетонитрилом и водой (взятыми в объемном отношении 90:10:0,6), инкубировали при комнатной температуре в условиях перемешивания. После превращения спирта на 47% (что устанавливали, используя колонку с зорбаксом C-8) реакцию останавливали фильтрованием. Оставшийся спирт содержит (+)-энантиомер с энантиомерной частотой 39% (устанавливали, используя колонку с материалом под торговым названием чирацел®-OD).
Пример XIV.
Раствор, состоящий из 0,13 М 2,3-дигидро-7-нитро-1,4-бензодиоксан-2-метанола, 0,24 М ангидрида масляной кислоты и 25% липазы в смеси, образованной простым диизопропиловым эфиром, ацетонитрилом и водой (взятыми в соотношении 50:50:0,5), инкубировали при комнатной температуре в условиях перемешивания. После превращения спирта на 64% реакцию останавливали фильтрованием. Оставшийся спирт содержит (+)-энантиометр с энантиомерной чистотой 42,4%.
Пример XV.
Рацемизация (+)-2,3-дигидро-7-нитро-1,4-бензодиоксин-2-метанола
К раствору, состоящему из 0,1 г (47 ммоль) (+)-2,3-дигидро-7-нитро-1,4-бензодиоксин-2-метанола ([α]
При использовании н-пропанола в качестве растворителя продолжительность взаимодействия составляет 30 часов.
Пример XVI.
Рацемизация (+)-5-хлор-2,3-дигидро-7-нитро-1,4-бензодиоксан-2- метанола
К раствору, содержащему 0,85 г (3,46 ммоль) (+)-5-хлор-2,3-дигидро-7-нитро-1,4-бензодиоксан-2-метанола ([α]
Пример XVII.
Рацемизация (+)-6-хлор-2,3-дигидро-8-нитро-1,4-бензоксазин-3- метанола
К раствору, содержащему 2 г (8,18 ммоль) (+)-6-хлор-2,3-дигидро-8-нитро-1,4-безноксазин-3-метанола ([α]
Объединенные органические слои дважды промывали разбавленным солевым раствором и сушили над сульфатом магния. После фильтрования с целью удаления сульфата магния и выпаривания растворителя в вакууме получали 1,83 г оранжево-коричневого твердого вещества. Удельное вращение плоскости поляризации света является равным нулю; и проведением хирального анализа на колонке с веществом чирацел-OD было установлено, что энантиомерная чистота вещества равна нулю.
Пример XVIII.
Рацемизация посредством использования гидрида натрия
К смеси, состоящей из 0,2 г (0,8 ммоль) R-(+)-2,3-дигидро-5-нитро-7-хлор-1,4-бензодиоксан-2-метанола и 0,01 г (0,5 эквивалента) 60%-ной суспензии гидрида натрия в минеральном масле, добавляли 5 мл диметилформамида. После завершения выделения газа оранжевый раствор перемешивали при комнатной температуре. Рацемизация завершалась за 0,75 ч, что было установлено проведением анализа на колонке с материалом чирацел-OD.
При использовании в качестве растворителя тетрагирофурана реакция протекает одинаково успешно.
Изобретение касается ферментативного способа стереоизбирательного получения гетеробициклического спиртового энантиомера, отличающегося тем, что существенно чистый энантиомер с общей формулой I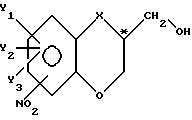
в которой X - кислород, группа NH, N-(C1-C4)-алкильная группа; Y1, Y2 и Y3 в каждом случае независимо представляют собой водород или галоген или (C1-C4)-алкил; заместитель NO2 присоединяется к бициклической кольцевой системе в 5- или 7-положении, C*-атом находится либо в R-, либо в S-конфигурации, получают из его соответствующего спиртового рацемата, проводя следующие последовательные реакционные стадии: 1) стереоизбирательную этерификацию, 2) отделение спирта от полученного сложного эфира, 3) гидролиз упомянутого сложного эфира с целью получения соответствующего спиртового энантиомера и 4) превращение упомянутого спиртового энантиомера в исходный рацемат в основных условиях для обеспечения возможности его нового использования. Изобретение также касается существенно чистого спиртового энантиомера с формулой I, используемого для получения из упомянутого энантиомера фармакологически активного пиперазинового производного и существенно чистых энантиомерных промежуточных соединений. Поскольку существенно чистый спиртовой энантиомер может быть легко использован в качестве ключевого промежуточного соединения в синтезе фармакологически активных пиперазиновых производных, устраняется трудоемкое разделение рацемата в предложенной стадии многоступенчатого синтеза. 3 с. и 6 з.п. ф-лы.
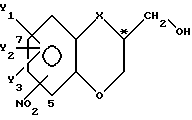
в которой X - атом кислорода, группа - NH или -N-(C1-C4) алкильная группа;
Y1, Y2 и Y3 в каждом случае независимо представляет собой атом водорода, галоген или (C1-C4) алкильную группу;
заместитель NO2 присоединен к бициклической кольцевой системе в 5- или 7-положении;
C* - атом находится либо в R-, либо в S-конфигурации,
отличающийся тем, что соответствующий спиртовой рацемат подвергают последовательно следующим реакционным стадиям: I) ацилируют упомянутый рацемат ацилирующим агентом под влиянием фермента, обладающего стереоизбирательной этерификационной активностью; II) отделяют соединение, не претерпевшее этерификации, от полученного сложного эфира и выделяют желаемый в существенной мере чистый спиртовой энантиомер формулы I или его сложный эфир; III) проводят гидролиз полученного сложного эфира с превращением упомянутого сложного эфира в соответствующий спиртовый энантиомер и IV) превращают нежелательный спиртовый энантиомер в исходный спиртовый рацемат в основных условиях для его повторного использования.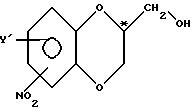
в которой Y1 - атом водорода или хлора, или фтора, или метильная группа;
заместитель NO2 присоединяется к бициклической кольцевой системе в 5- или 7-положении;
C* - атом находится либо в R-, либо S-конфигурации.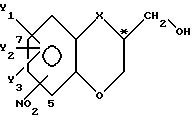
где X, Y1, Y2 и Y3 имеют значения, указанные в п.1;
заместитель NO2 присоединен к бициклической кольцевой системе в 5- или 7-положении;
C* - атом находится в R-конфигурации.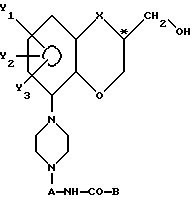
где X, Y1, Y2 и Y3 имеют значения, указанные в п.1;
A - (C2-C4) адкиленовая группа с прямой или разветвленной цепью;
B - фенил, который может быть замещен атомами галогена;
C* - атом обладает R-конфигурацией,
отличающийся тем, что существенно чистый спиртовый энантиомер с общей формулой I по п.1, в котором C* - атом обладает R-конфигурацией, получают из его соответствующего спиртового рацемата таким проведением последовательных реакционных стадий, как они определены в п.1, после чего свободную гидроксигруппу у спиртового энантиомера, полученного таким способом, защищают гидроксизащищающей группой при сохранении фактически существующей конфигурации у C* - атома, что обеспечивает получение соединения с общей формулой III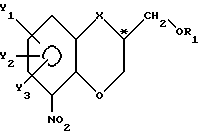
где R1 - гидроксизащитная группа;
X, Y1, Y2 и Y3 имеют значения, указанные в п.1,
после чего существенно чистый энантиомер, полученный таким образом, подвергают восстановлению по нитрозаместителю и получают аминосоединение формулы IV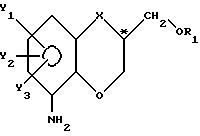
где X1 , Y1, Y2 и Y3 имеют указанные значения,
при сохранении фактически существующей конфигурации у C* - атома, с последующим превращением его в пиперазиновое соединение с общей формулы V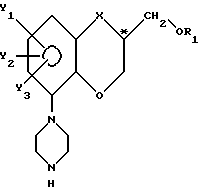
где R1, X, Y1, Y2 и Y3 имеют указанные значения,
при сохранении фактически существующей конфигурации у C* - атома, после чего из упомянутого пиперазинового соединения получают производное при сохранении фактически существующей конфигурации, для чего проводят взаимодействие с соединением с общей формулой VII
L-A-NH-CO-B VII,
где L - отщепляемая группа, желательно выбранная из атома хлора, мезилатной и тозилатной группы;
А и В имеют указанные значения,
либо с соединением общей формулы VIII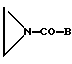
где В имеет указанные значения,
с получением пиперазинового производного с общей формулой VI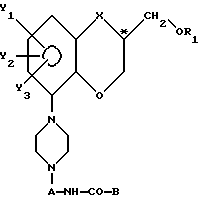
где А, В, R1, X, Y1, Y2 и Y3 имеют указанные значения,
с последующим деблокированием полученного соединения формулы VI и получением свободного спиртового энантиомера общей формулы IX, в которой X, Y1, Y2, Y3, А и В имеют указанные значения, при сохранении R-конфигурации у C* - атома.
| EP, 138280 A2, 1985 | |||
| EP, 221725 A2, 1987 | |||
| Michael D | |||
| Ennis et al, "The Synthesis of (+)- and (-) - Flesinoxan | |||
| Application Methodology", Tetrahedron Letters, 1992 | |||
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |

