Изобретение относится к группе новых соединений пиперазина и пиперидина, которые обладают интересными фармакологическими свойствами.
Установлено, что соединения формулы (а)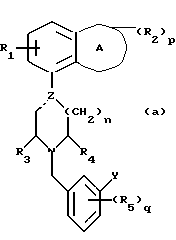
в которой A обозначает гетероциклическую группу с 5-7 атомами в кольце, содержащую 1-2 гетероатома из группы О, N и S;
R1 обозначает водород или фтор;
R2 обозначает C1-4-алкил или оксогруппу и p равен 0, 1 или 2;
Z обозначает углерод или азот, и пунктирная линия обозначает простую связь, когда Z является азотом, и простую или двойную связь, когда Z является углеродом;
R3 и R4 независимо обозначают водород или C1-4алкил;
n равен 1 или 2;
R5 обозначает галоген, гидрокси, C1-4 алкокси или C1-4алкил, и q равен 0, 1.
Y обозначает фенил, который может быть замещен 1-2 заместителями из группы гидрокси, галоген, C1-4алкокси, циано, аминокарбонил, ди-C1-4алкиламинокарбонил; или фурил, или тиенил и их соли обладают интересными фармакологическими свойствами.
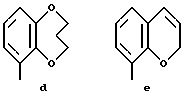
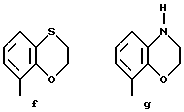
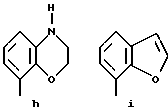
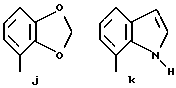
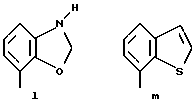
Предпочтительными соединениями по данному изобретению являются соединения формулы (а), в которой A вместе с фенильной группой обозначает группу формулы b-m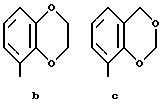
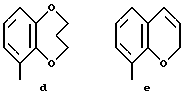
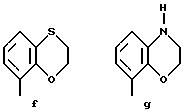
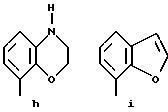
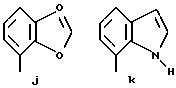
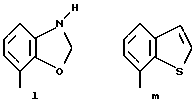
в которой R1, (R2)p, R3, R4, (R5)q, Y и Z имеют указанные выше значения и n равен 1, и их соли.
Особенно предпочтительными являются соединения формулы (а), в которой A вместе с фенильной группой является группой формулы (b) или группой формулы (I), замещенной в гетерокольце оксогруппой, Y обозначает фенил, который может быть замещен так, как указывалось выше, n равен 1, R3 и R4 обозначают водород, R5 обозначает гидрокси, метокси или галоген, q равен 0 или 1, Z обозначает азот, и их соли.
Еще более предпочтительными являются соединения формулы (а), в которой A вместе с фенильной группой является группой формулы (I), замещенной в гетерокольце оксогруппой, q равен 0, и Y обозначает фенил, и их соли.
Из европейского патента N 0650964 известно, что соединения формулы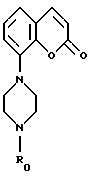
где R0 обозначает C1-4алкил, которые могут быть замещены в фенильной группе, и/или гетероциклической группе, и/или пиперазиновой группе, воздействуют на центральную нервную систему посредством связывания с рецепторами 5-НТ. Эти соединения, в частности, связываются с субтипами рецепторов 5-НТ, то есть с рецепторами 5-HT1A и 5-HT1D.
В настоящее время неожиданно установлено, что соединения по данному изобретению демонстрируют высокую степень сродства как к рецепторам допамина D2, так и к рецепторам серотонина 5-HT1A (в диапазоне pKi 7,0-9,5 для рецепторов обоих типов). Такое сочетание свойств полезно для лечения шизофрении и других психотических расстройств и может позволить более полное лечение всех симптомов болезни (например, позитивных симптомов, негативных симптомов и дефицита познавательной способности).
Эти соединения обнаруживают различные активности в качестве частичных агонистов или антагонистов при рецепторах допамина D2, D3 и D4. Некоторые соединения оказывают воздействие на рецепторы допамина, подобное агонистам, однако, они оказывают сильное антагонистическое действие на вызываемое апоморфином поведение лазанья у мышей (значения ED50 < 1 мг/кг при пероральном введении). Эти соединения по-разному действуют в качестве агонистов рецепторов 5-HT1A и вызывают признаки поведенческого синдрома, характерного для серотинина, с разной степенью интенсивности.
Указанные соединения являются активными на терапевтических моделях, чувствительных к применяемым в клинике антипсихотическим средствам (например, при лечении реакции обусловленного избегания; Van der Heyden & Bradford, Behav. Brain Res. , 1988, 31: 61-67), к антидепрессантам (например, при дифференцированном усилении замедленных реакций; Van Hest et al. , Psychopharmacology, 1992, 107: 474-479) и к анксиолитикам (например, подавление вызванной стрессом реакции повышения голоса; Van der Poel et al. , Psychopharmacology, 1989, 97: 147-148).
В отличие от применяемых в клинических условиях антагонистов рецепторов допамина D2 рассматриваемые соединения менее предрасположены вызывать каталепсию у грызунов, а следовательно, вероятно, вызывают меньшие экстрапирамидальные побочные эффекты, чем существующие антипсихотические средства.
Присущим этим соединениям агонизмом к рецепторам 5-НТ1A можно объяснить то, что они имеют пониженную тенденцию вызывать экстрапирамидальные и терапевтические эффекты, наблюдаемые у поведенческих моделей, чувствительных, или к антидепрессантам, или к анксиолитикам.
Эти соединения, вероятно, представляют ценность для лечения расстройств или болезней центральной нервной системы, вызываемых нарушениями в допаминергической или серотинергической системах, например болезни Паркинсона, агрессивности, страха, аутизма, головокружения, депрессии, нарушения познавательной способности или памяти, и особенно шизофрении и других психотических расстройств.
Подходящими кислотами, с которыми соединения могут образовывать фармацевтически приемлемые соли присоединения кислоты, являются, например, соляная кислота, серная кислота, фосфорная кислота, азотная кислота и органические кислоты, такие как лимонная кислота, фумаровая кислота, малеиновая кислота, винная кислота, уксусная кислота, бензойная кислота, паратолуолсульфокислота, метансульфокислота и нафталинсульфокислота. Из соединений по данному изобретению можно обычными способами изготовить лекарственные средства для введения с использованием вспомогательных веществ, таких как жидкие и твердые носители.
Соединения по данному изобретению можно получить с помощью ряда описанных ниже способов (A-E). Пиперазины, гомопиперазины и пиперидины, используемые при осуществлении этих способов, имеют обозначения от I-H до XIX-H, где римскими цифрами от I до XIX указаны следующие группы: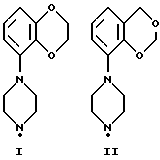
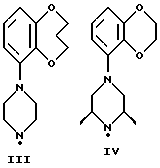
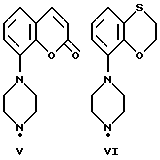
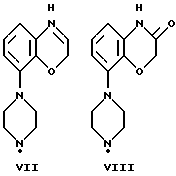
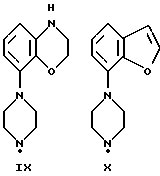
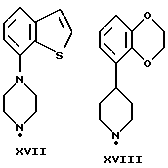
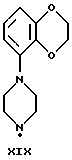
Пиперидины XVIII-H и XIX-H (фиг. A1), используемые при получении соединений по данному изобретению, синтезируются в соответствии с процедурой, аналогичной описанной в WO 94-GB 1507.
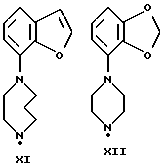
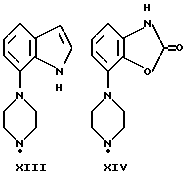
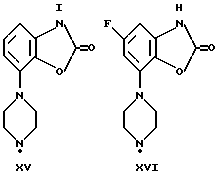
Синтез пиперазинов (фиг. A1), используемых при получении соединений по данному изобретению, описывается в ЕР0189612, за исключением соединений XI-H, XIII-H и XV-H (см. ниже).
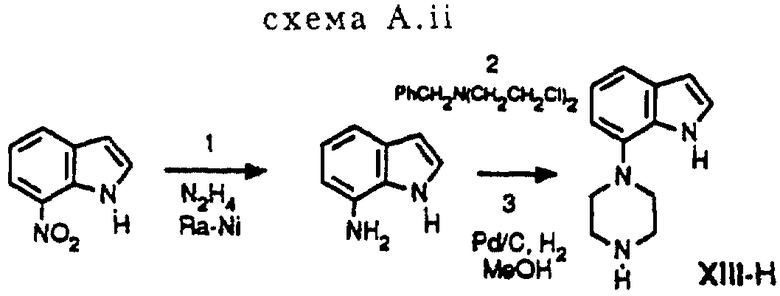
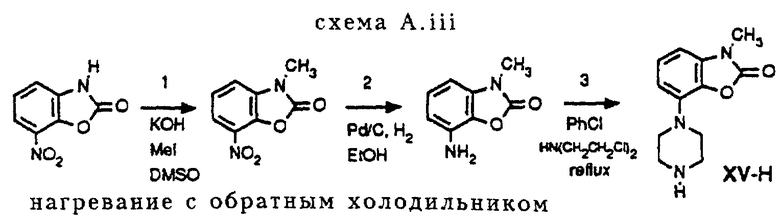
Гомопиперазин XI-H и пиперазины XIII-H и XV-H являются новыми соединениями, и способы их получения приведены ниже (схемы A. i- A. iii).
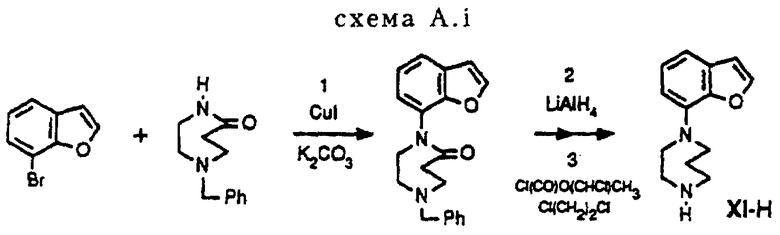
Способ получения соединения XI-H (см. схему A. i, представленную в конце описания).
Способ получения соединения XIII-H (см. схему A. ii, представленную в конце описания).
Стадии 1-3 (схема A. ii)
7-Нитроиндол описан С. М. Пармертером и др. (S. M. Parmerter et al. , J. Am. Chem. Soc. 80, (1958), 4621-2). Стадии 1, 2 и 3 выполняются аналогично синтезу, описанному в публикации европейского патента N 0650964.
Способ получения соединения XV-H (см. схему A. iii, представленную в конце описания).
Стадии 1-3 схемы A. i и стадия 1 схемы A. iii подробно описаны в примерах, и процедуры стадии 2 и 3 схемы A. iii аналогичны описанным в европейском патенте ЕРО 189612.
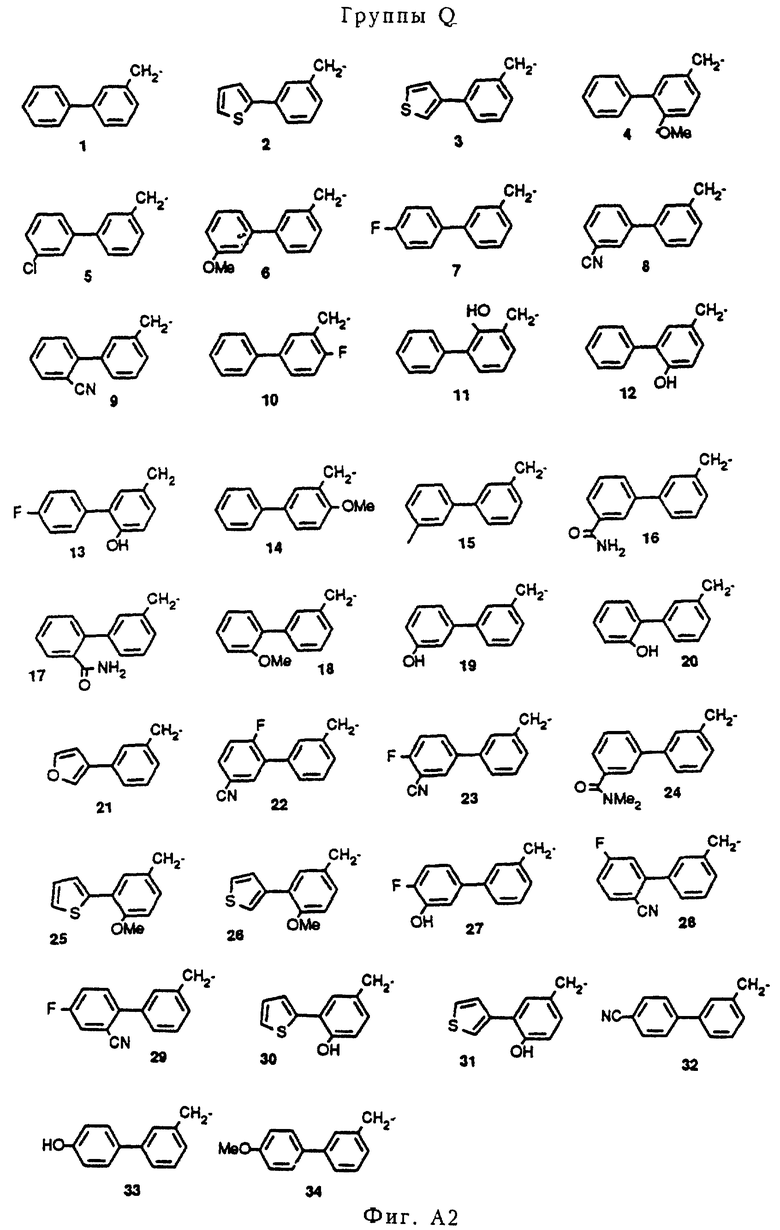
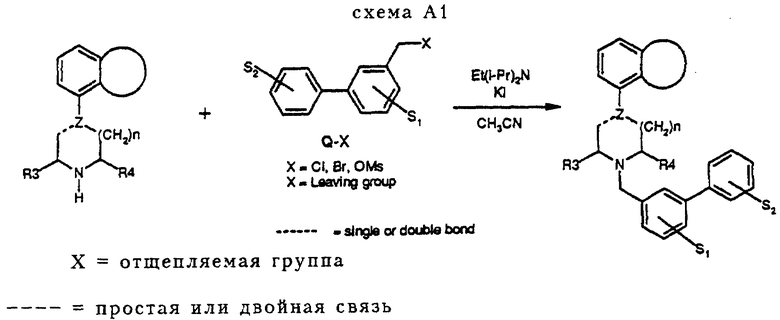
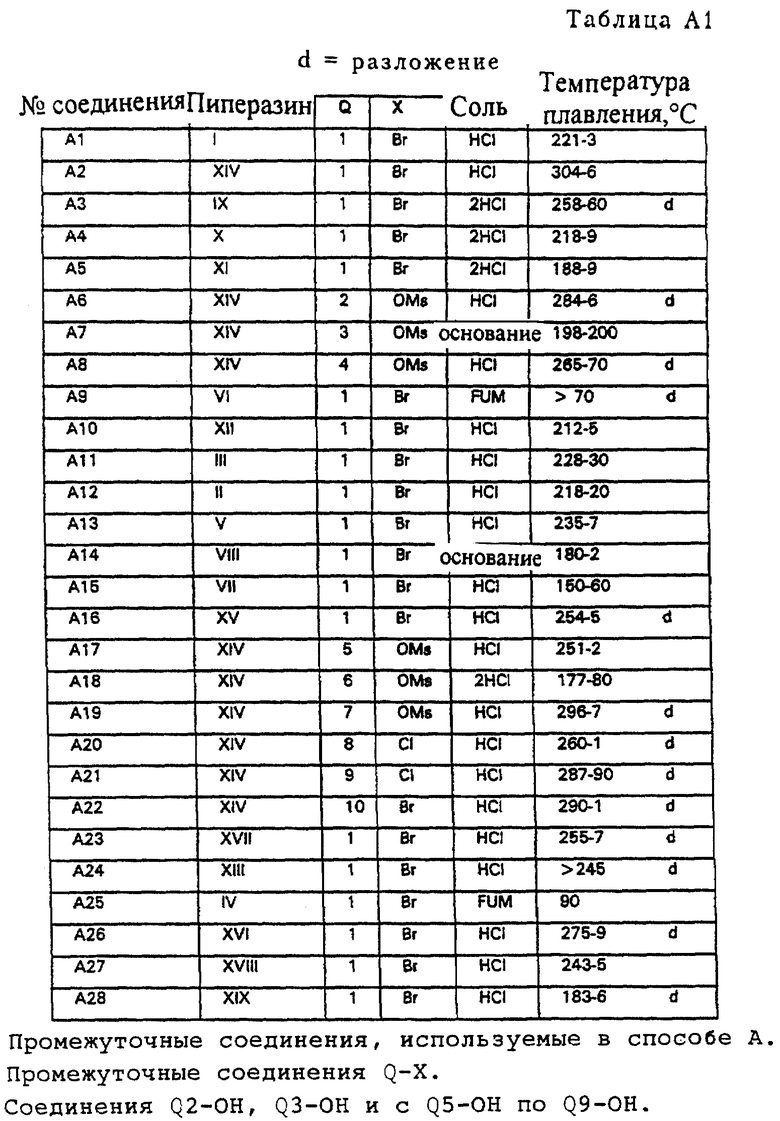
Атом водорода фрагмента N-H соединений с I-H по XIX-H можно заменить группой Q пятью разными химическими способами (A, B, C, D и Е, см. ниже), при осуществлении которых получают соединения изобретения. На фигуре A2 показаны значения групп с Q1 по Q34 (см. в конце описания)
Метод синтеза A
Соединения A1-A14 и A16-A28 получали в соответствии с методом синтеза, изображенным на схеме A1 (см. в конце описания). Пиперазин (соединения с I-H по VI-H и с VIII-H по XVII-H) подвергался взаимодействию с соединением Q-X (X= Cl, Br, OMs) в ацетонитриле с Et(i-Pr)2N, действующим в качестве основания, и в некоторых случаях добавлялся K1. Вместо Et(iPr)2N можно использовать Et3N.
Следующие методы синтеза В-Е не ограничиваются получением пиперазинов, их можно использовать также для получения пиперидинов.
Метод синтеза B
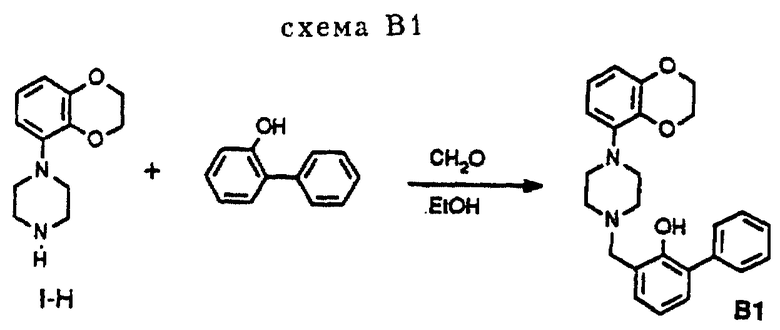
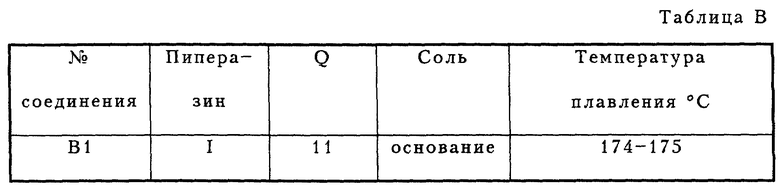
Эти соединения можно также получить в соответствии с методом синтеза, изображенным на схеме В1 (см. в конце описания). Пиперазин I-Н подвергался взаимодействию с 2-фенилфенолом и формальдегидом в EtOH.
Метод синтеза C
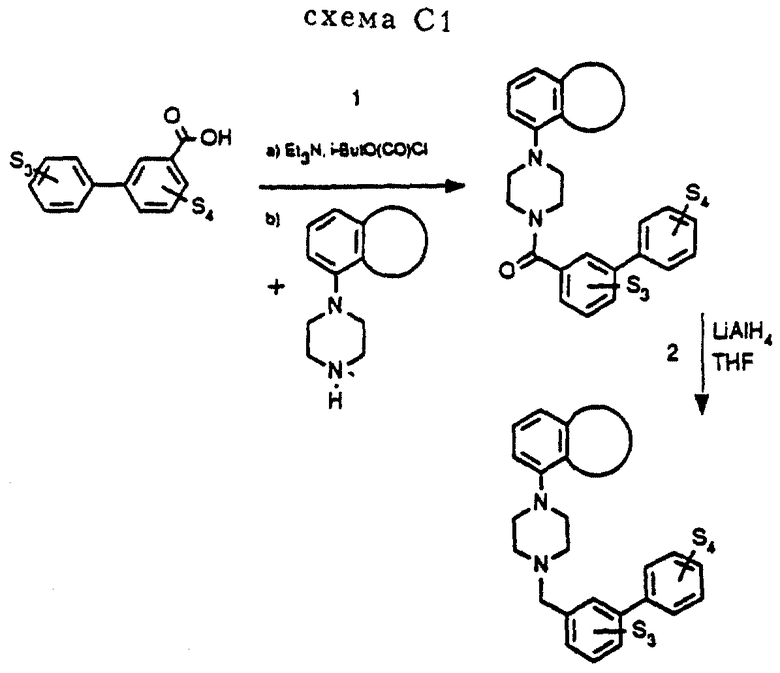
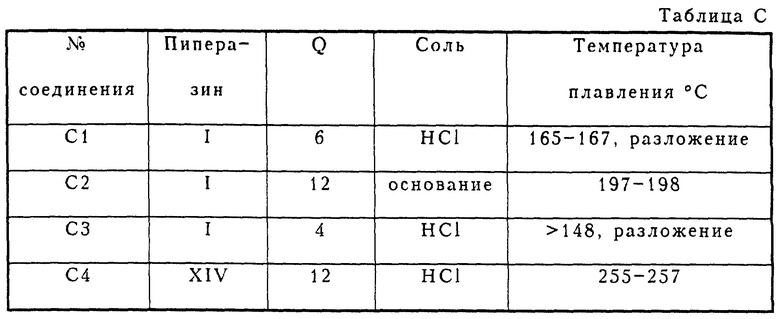
Соединения C1-C4 получались в соответствии с методом синтеза, изображенным на схеме C1 (см. в конце описания). Фенилпиперазины подвергались взаимодействию с несколькими хлорангидридами метазамещенной фенилбензойной кислоты, давая соответствующие амиды. Амиды затем восстанавливались в соединения C1-C4 при помощи LiAlH4.
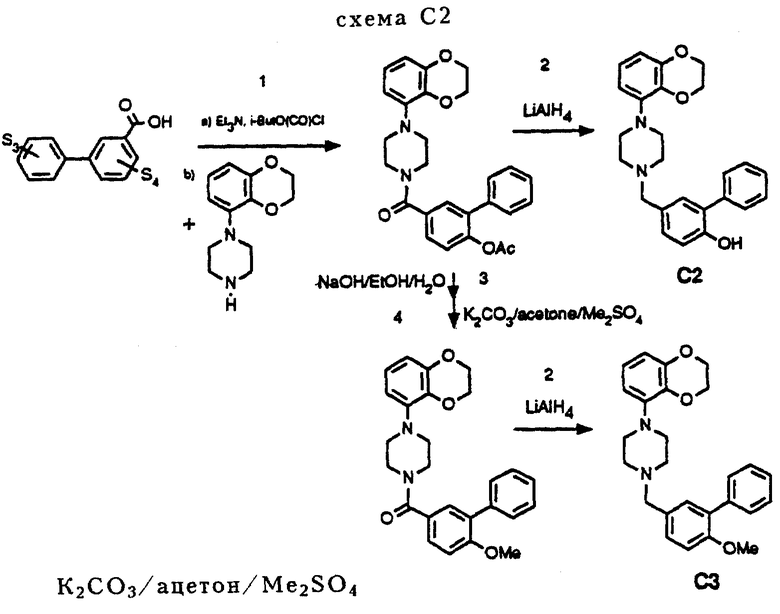
Соединения C2 и C3 получались так, как это показано на схеме C2 (см. в конце описания).
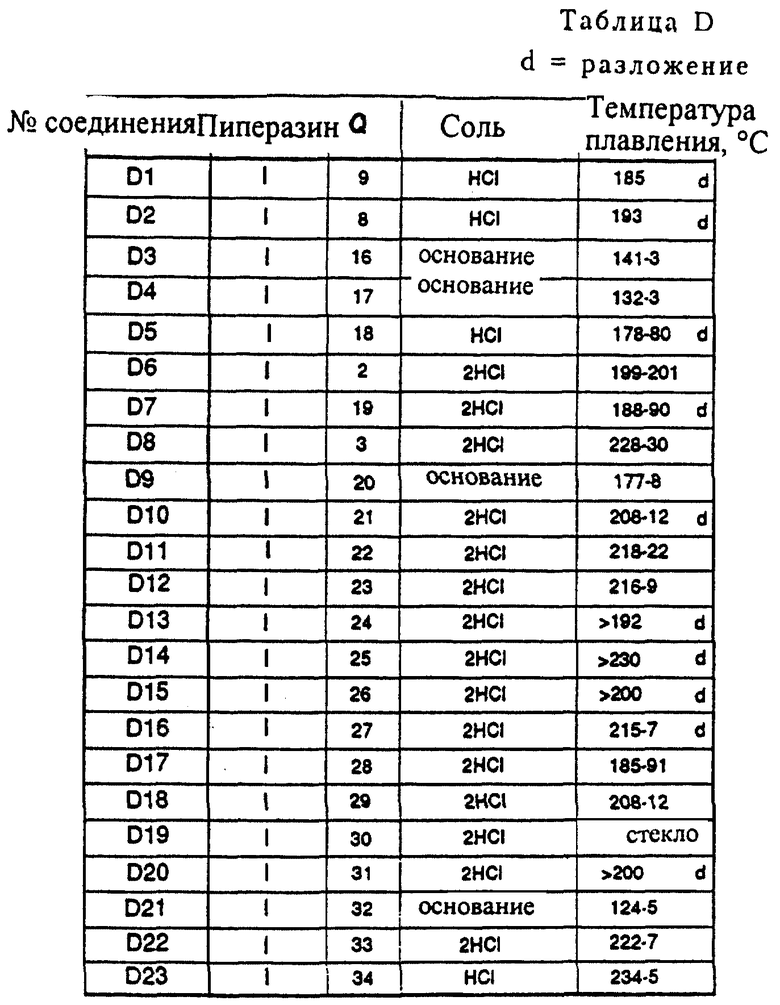
Метод синтеза D
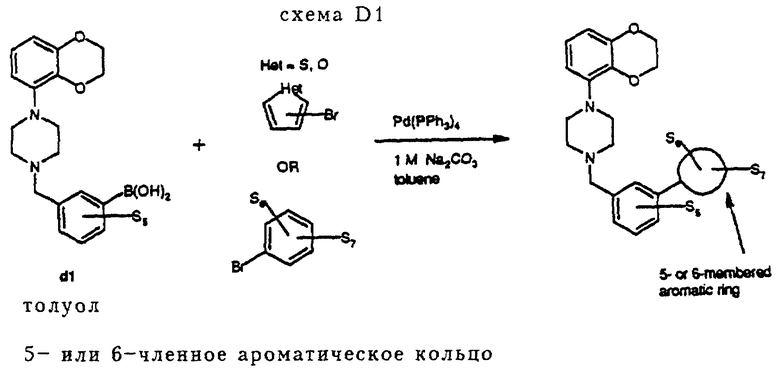
Соединения D1-D18 и D21-D23 получались в соответствии с методом синтеза, изображенным на схеме D1 (см. в конце описания). Арилбороновую кислоту подвергали взаимодействию с ароматическим бромидом в щелочных условиях в присутствии каталитического количества Pd(PPh3)4. В результате осуществления так называемой реакции "Сузуки" получали конечные продукты D с углерод-углеродной связью.
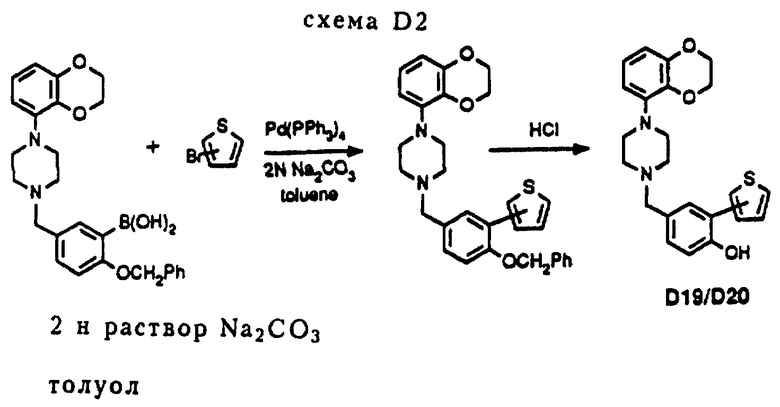
Соединения D19 и D20 получались в соответствии с модифицированным методом синтеза, который изображен на схеме D2 (см. в конце описания).
После осуществления вышеописанной реакции Сузуки дополнительный гидролиз удаляет защитную бензильную группу с помощью стандартных приемов (например, при помощи горячей концентрированной HCl), см. например, процедуру E2 (схема E2).
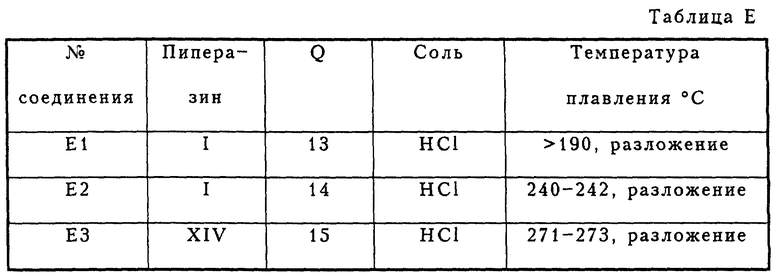
Метод синтеза E
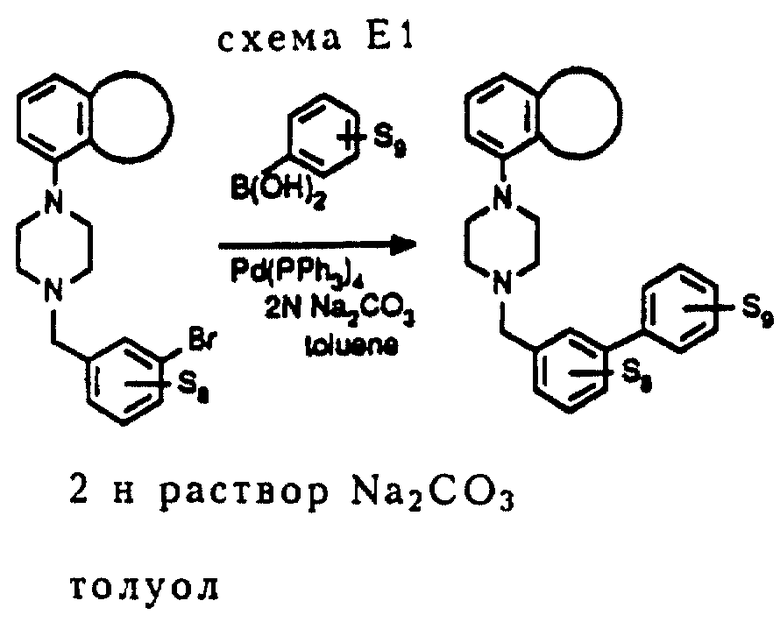
Соединения E2 и E3 получались в соответствии с методом синтеза, изображенным на схеме E1 (см в конце описания). В этом случае применяют ту же реакцию Сузуки, описанную в методе синтеза D, хотя промежуточные соединения являются иными.
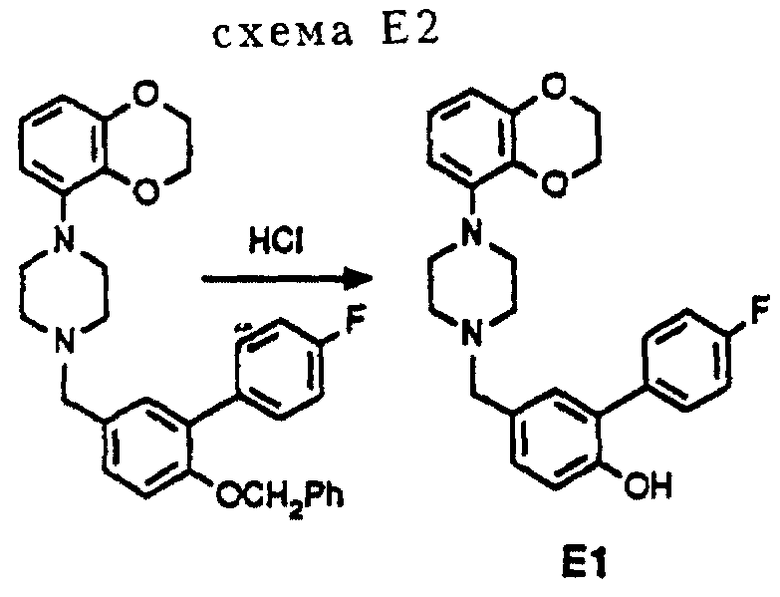
Соединение E1 получалось в соответствии с модифицированным методом синтеза, который изображен на схеме E2 (см. в конце описания). Дополнительной стадией к методу синтеза, показанному на схеме E1, был гидролиз защитной бензильной группы.
В приводимых ниже примерах подробно описываются способы получения соединений формулы (а) и ряда промежуточных соединений.
Пример 1
Процедура А1 (схема A1)
К 20 мл CH3CN добавляли 1,0 г (4,3 ммоль) пиперазина III-Н и 1,2 г (4,7 ммоль) соединения Q1-Br, после чего добавлялось 0,52 г (5,1 ммоль) Et3N и небольшое количество K1. Реакционная смесь перемешивалась и нагревалась с обратным холодильником в атмосфере азота в течение 16 часов. После охлаждения смеси растворитель удалялся в вакууме, оставляя остаток, который растворялся в CH2Cl2 и последовательно промывался 0,5 н. раствором NaOH и насыщенным раствором соли (2 раза). Органическую фракцию сушили на MgSO4. После удаления осушающего агента растворитель удалялся в вакууме, давая остаток. Полученный остаток давал после флэш-хроматографии на колонках (SiO2, элюент: смесь CH2Cl2 и MeOH, 99: 1) соединение A11 (свободное основание, см. таблицу A1)). Остаток растворялся в простом эфире, к которому добавлялся один эквивалент 1 норм. HCl EtOH. Выпавший осадок давал 0,98 г (2,3 ммоль, 52%) чистого соединения A11. HCl, т. п. 228-230oC.
1H-ЯМР (CDCl3, δ ): 2.18 (м. 2H), 3.09 (широкий, 2H), 3.3-3.7 (широкий кластер, 6H), 4.21 (м. 4H), 4.30 (с, 2H), 6.59 (дд, J= 1 и 8 Гц, 1H), 6.71 (дд, J= 1 и 8 Гц, 1H), 6.82 (т. J= 8 Гц, 1H), 7.37 (м, 1H), 7.47 (м, 2H), 7.53 (т. J= 8 Гц, 1H), 7.62-7.74 (кластер, 4H), 7.90 (т. J= 2 Гц, 1H), 12.9 (широкий, 1H).
В соответствии с описанным выше способом аналогично получали соединения A1-A14 и A16-A28, которые приведены в таблице A1. Соединение A15 получали из соединения A14 путем восстановления смесью LiAlH4 ТГФ аналогично процедуре A5 (см. ниже, восстановление A14 осуществлялось при температуре кипения с обратным холодильником вместо комнатной температуры).
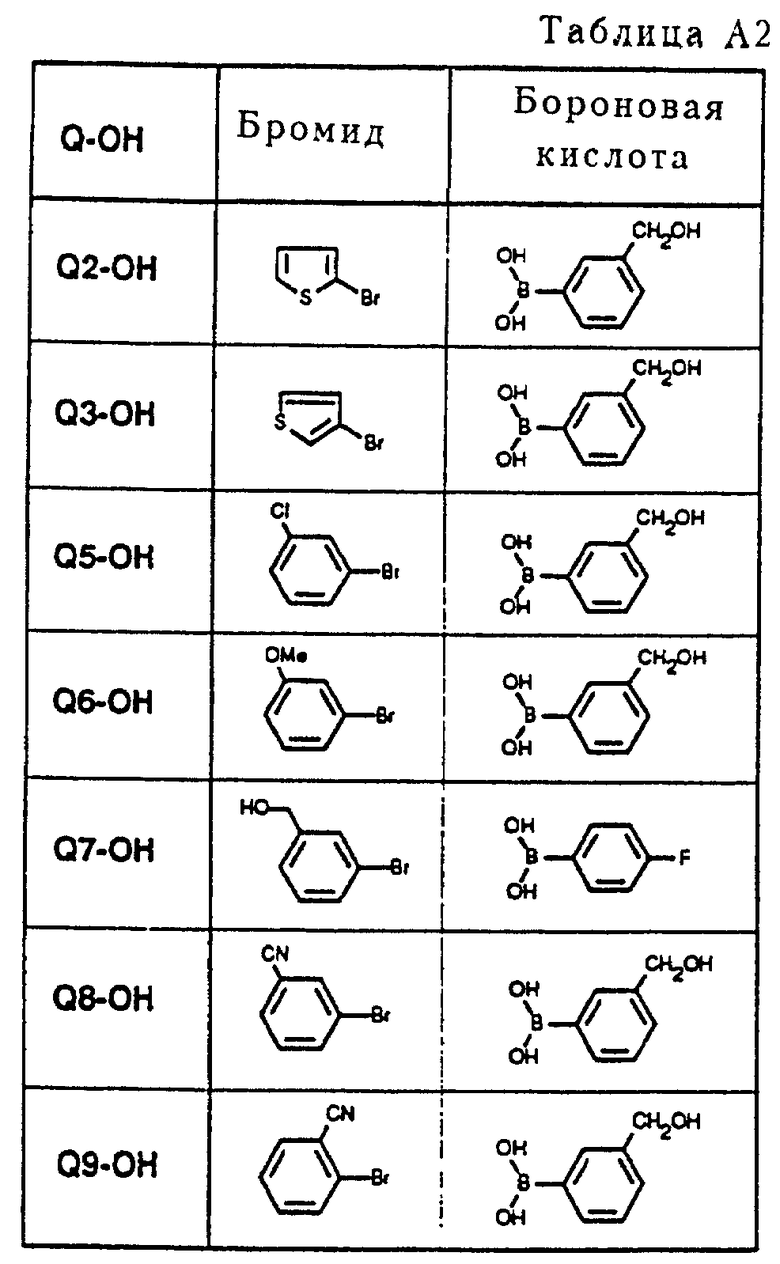
Пример получения соединения Q2-OH (см. схему A2 в конце описания).
Арилбороновую кислоту подвергали взаимодействию с ароматическим бромидом в щелочных условиях в присутствии каталитического количества Pd(PPh3)4. В результате осуществления так называемой реакции "Сузуки" получали промежуточные соединения Q-OH с углерод-углеродной связью. Используемые бороновые кислоты можно легко получить при помощи соответствующих бромидов. Общие способы получения описаны в статье D. Janietz et al. , Synthesis, (1993), 33 и в приведенных там ссылках.
Процедура A2 (схема A2).
10 мл диметоксиэтана нагревали с обратным холодильником в атмосфере азота, после чего растворитель оставляли охлаждаться. Добавляли 0,85 мл (1,43 г, 8,8 ммоль) 2-бромтиофена и в течение 10 минут через раствор барботировали азот. Затем к раствору добавляли 0,4 г (0,35 ммоль, 0,04 экв. ) Pd(PPh3)4. После перемешивания в течение 10 минут к реакционной смеси добавляли 8,5 мл 2 норм. Na2CO3/H2O и 1,25 г (8,2 ммоль) 3-(гидроксиметил)фенилбороновой кислоты, растворенной примерно в 2 мл EtOH. Реакционную смесь нагревали и оставляли нагреваться при температуре кипения с обратным холодильником в течение 4 часов, затем нагревание прекращалось, реакционная смесь перемешивалась еще 16 часов при комнатной температуре. Образовавшийся осадок фильтровался через целит, и фильтр промывался смесью EtOAc/H2O. Фильтрат экстрагировался EtOAc, объединенные органические фракции сушились на MgSO4. После удаления осушающего агента фильтрат давал после выпаривания растворителя 2,1 г масла. Флэш-хроматография (SiO2, элюент: смесь метил-трет-бутилового эфира и гексана, 1: 1) давала 0,85 г (4,5 ммоль, 51%) желаемого продукта Q2-OH.
Аналогичным образом получали следующие метазамещенные бензиловые спирты Q-ОН из сочетаний ароматических бромидов и бороновых кислот, указанных в таблице A2.
Все соединения Q-OH, указанные в таблице A2, успешно превращались в соответствующие мезилаты с помощью стандартных процедур способами (например, с использованием MsCl и Et3N в EtOAc). Однако в случае соединений Q8-OH и Q9-OH получали не мезилаты, а соответствующие хлориды Q8-Cl и Q9-Cl вследствие обработки 2 норм. HCl. Два последних соединения были также великолепными алкилирующими агентами при осуществлении реакции, изображенной на схеме A1.
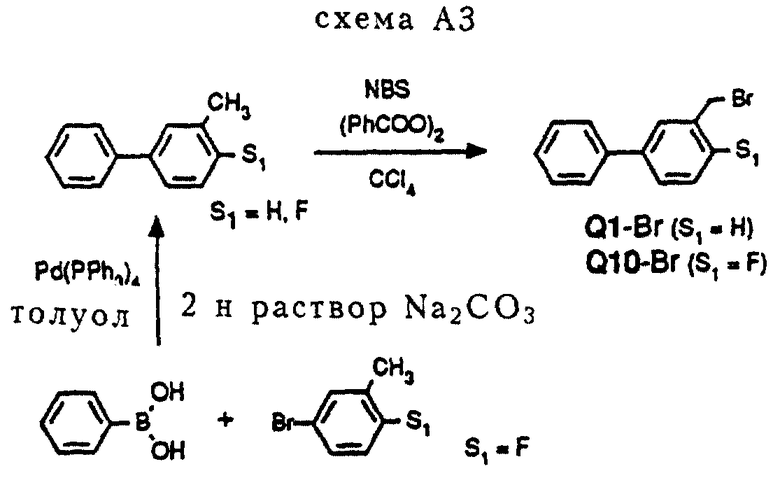
Промежуточное соединение Q1-Br (см. схему A3 в конце описания).
Метафенилтолуол (S1= H) бромировался с помощью действия N-бромсукцинимида (NBS) в присутствии каталитического количества дибензоилпероксида.
Процедура A3 (схема A3)
Соединение Q1-Br
3 г (29,8 ммоль)3-фенилтолуола и 5,3 г (29,8 ммоль) N-бромсукцинимида (NBS) растворялись в 30 мл CCl4. Добавлялось небольшое количество дибензилпероксида, и реакционную смесь нагревали с обратным холодильником в течение 10 часов. В течение данного периода дополнительно добавлялось еще немного дибензоилпероксида. После охлаждения реакционная смесь разбавлялась CCl4 и водой. Двухфазная система подщелачивалась 2 норм. NaOH, после чего она перемешивалась. Органический слой промывался 1 норм. NaOH и водой и впоследствии сушился над MgSO4. После удаления осушающего агента растворитель удалялся в вакууме, давая 8,0 г остатка. Последний очищался с помощью хроматографии на колонке (SiO2, элюент: смесь Et2O и петролейного эфира, 1: 9), давая 5,3 г (21,5 ммоль, 72%) чистого промежуточного соединения Q1-Br.
В случае соединения Q10-Br требуемый 2-фтор-5-фенилтолуол (S1= F) получался из фенилбороновой кислоты и 2-фтор-5-бромтолуола по реакции Сузуки аналогично процедуре A2. См. схему A3.
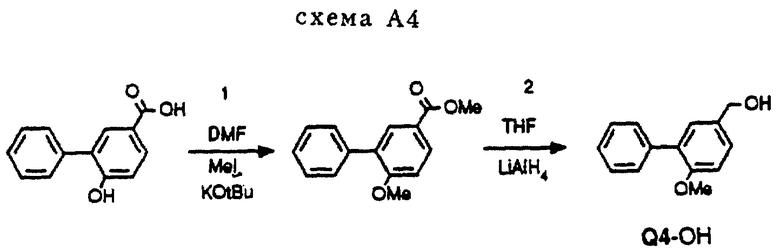
Соединение Q4
Пример получения (Q4-OH) (см. схему A4 в конце описания).
Диметилирование 3-фенил-4-гидроксибензойной кислоты (способ получения см. в патенте США N 4873367) под действием Mel/KOtBu давало соответствующий метиловый эфир метоксибензойной кислоты, который в свою очередь можно было восстановить (LiAlH4) в соединение Q4-OH.
Процедура A4 (схема A4)
Стадия 1
4,0 г (19 ммоль) 3-фенил-4-гидроксибензойной кислоты растворялось в 70 мл ДМФ, к которому добавлялось 4,6 г (41 ммоль) KOtBu, и смесь перемешивалась в течение 30 минут. После этого добавлялось 3,0 г (21 ммоль) Mel, и реакционная смесь перемешивалась в течение 14 часов при комнатной температуре, в течение этого периода добавлялся второй эквивалент Mel. Растворитель удалялся в вакууме, давая остаток, который растворялся в EtOAc. Полученный раствор встряхивался с 2 норм. NaOH. Органическую фракцию сушили над Na2SO4. После удаления осушающего агента и растворителя получалось 3,65 г (16,0 ммоль, 84%) довольно чистого метилового эфира 3-фенил-4-метоксибензойной кислоты. Данную порцию использовали без дальнейшей очистки для восстановления, описанного для процедуры A5.
Процедура A5 (схема A4)
Стадия 2
0,68 г (18 ммоль) LiAlH4 добавляли к 20 мл сухого ТГФ и перемешивали в атмосфере азота. Затем к смеси LiAlH4 и ТГФ по каплям добавляли 3,65 г (16,0 ммоль) метилового эфира З-фенил-4-метоксибензойной кислоты, растворенного в 60 мл сухого ТГФ. Перемешивание продолжалось в течение 1 часа при комнатной температуре. Реакционная смесь охлаждалась (смесь воды со льдом) и добавлялось 0,7 мл воды в смеси с ТГФ и 1,4 мл 2 норм. NaOH. Затем смесь нагревали с обратным холодильником в течение 10 минут, после чего ее фильтровали для удаления солей. Соли промывались горячим ТГФ, и промывочные воды объединялись с фильтратом. После удаления растворителя в вакууме получалось 3,1 г (14,5 ммоль, 90%) довольно чистого Q4-OH. Данную порцию использовали без дальнейшей очистки для получения мезилата Q4-OMs, который в свою очередь использовался в реакции, изображенной на схеме A1, дающей соединение A8.
Пример 2
Процедура В1 (схема В1)
3,74 г (17,0 ммоль) пиперазина I-H и 3,0 г (17,0 ммоль) 2-фенилфенола растворяли в 80 мл абсолютного этанола. При перемешивании раствора добавлялось 2,0 мл (24,0 ммоль) смеси 37% CH2O/H2O, продолжали перемешивать в течение 48 часов. После данного периода реакционную смесь концентрировали в вакууме с получением остатка, который подвергался флэш-хроматографии на колонке (SiO2, элюент: смесь CH2Cl2 и петролейного эфира, 1: 1). Сначала отделялась непрореагировавшая часть 2-фенилфенола при замене элюента, начиная с 100% CH2Cl2 до смеси CH2Cl2 и MeOH, 99: 1, давая 1,70 г (4,2 ммоль, 25%) соединения B1 в виде свободного основания, т. п. 174-175oC.
1H-ЯМР(CDCl3, δ): 2.65 (кластер, 8H), 3.83 (с, 2H), 4.27 (м, 4H), 6.48 (дд, J= 1.5 и J= 8 Гц, 1H), 6.59 (дд, J= 1,5 и J= 8 Гц, 1H), 6.76 (т. J= 8 Гц, 1H), 6.87 (т, J = 8 Гц, 1H), 7.05 (дд, J= 1.5 и J= 8 Гц, 1H), 7.28 (дд, J = 1,5 и J= 8 Гц, 1H), 7.32 (м, 1H) 7.42 (т. J= 8 Гц, 2H), 7.61 (м, 2H), 11.4 (широкий с, 1H).
Пример 3
Процедура C1 (схема C1).
Стадия 1.
В атмосфере азота 0,8 г (3,4 ммоль)3-(3-метоксифенил)бензойной кислоты растворялось в 15 мл сухого ТГФ вместе с 0,65 мл Et3N. Раствор охлаждался до 0oC и перемешивался при добавлении 0,42 мл изо-ButO(CO)Cl. Через 30 минут к полученному раствору добавлялось 0,71 г (3,2 ммоль) I-Н, растворенного в 5 мл сухого ТГФ. Реакционную смесь оставляли нагреваться до комнатной температуры, и перемешивание продолжалось в течение 16 часов. После этого реакционная смесь обрабатывалась 2 норм. NaOH, после чего двухфазную систему экстрагировали EtOAc. Органическую фракцию сушили над MgSO4. После удаления осушающего агента и удаления растворителя в вакууме оставался остаток, который подвергался хроматографии на колонке (SiO2, элюент: смесь EtOAc и петролейного эфира, 1: 1), давая 0,75 г (1,7 ммоль, 52%) соответствующего амида.
Стадия 2
0,9 г LiAlH4 суспендировалось в 20 мл сухого ТГФ, полученная суспензия доводилась до температуры кипения с обратным холодильником, после чего добавлялось 0,7 г (1,6 ммоль) амида (продукт стадии 1), растворенного в 15 мл сухого ТГФ. Реакционную смесь продолжали нагревать с обратным холодильником в течение 15 минут, после чего смесь охлаждалась (смесь воды со льдом) и очень осторожно по каплям добавлялось 0,9 г H2O. Затем добавлялось 1,8 мл 2 норм. NaOH и 0,9 г H2O, после чего смесь снова доводили до температуры кипения с обратным холодильником в течение 20 минут. Охлаждение до комнатной температуры и фильтрование давали остаток, который промывался EtOAc. Объединенный фильтрат и промывочные воды сушились над MgSO4. После удаления осушающего агента и удаления растворителя в вакууме оставался остаток, который подвергался хроматографии на колонке (SiO2, элюент: EtOAc), давая 0,57 г (1,4 ммоль, 85%) чистого свободного основания C1. Основание растворялось в EtOAc и превращалось в хлористо-водородную соль добавлением одного эквивалента 1 норм. HCl/метанол, давая 0,50 г чистого Cl•HCl, т. пл. 165-167oC (разложение).
1H-ЯМР (CDCl3, δ ): 3.24(широкий, 2H), 3.42-3.58 (кластер, 4H), 3.64-3,84 (широкий, 2H), 3.90 (с. 3H), 4.26 (м. 4H), 4.30 (с. 2H), 6.67 (широкий д. J= 8 Гц, 2H), 6.79 (t, J= 8 Гц, 1H), 6.93 (м. 1H), 7.23 (м. 2H), 7.38 (т. J= 8 Гц, 1H), 7.52 (т. J= 8 Гц, 1H), 7.65 (широкий д. J= 8 Гц, 1H), 7.69 (широкий д. J= 8 Гц, 1H), 7.92 (широкий с. 1H), 13.2 (широкий, 1H).
Процедура C2 (схема C2)
Стадии 1 и 2
Эти реакции аналогичны процедуре C1, стадии 1 и 2 (схема C1).
Стадия 3
1,1 г (2,4 ммоль) эфира уксусной кислоты суспендировалось в 150 мл этанола вместе с 15 мл H2O, после чего добавлялось 1,5 г (37,5 ммоль) NaOH. Реакционную смесь перемешивали в течение 16 часов, после чего этанол удалялся в вакууме. Оставшуюся фракцию обрабатывали насыщенным раствором NH4Cl и экстрагировали CH2Cl2. Объединенные органические фракции промывались насыщенным NaHCO3, сушились над MgSO4. После удаления осушающего агента и удаления растворителя в вакууме получалось 0,97 г (2,3 ммоль, 97%) остатка, содержащего соответствующее чистое производное фенола.
Стадия 4
0,98 г (2,3 ммоль) производного фенола (полученного на стадии 3) растворяли в 15 мл ацетона, к которому добавляли 1,5 г порошка K2CO3. При перемешивании добавляли 0,3 мл (CH3)2SO4, после чего реакционную смесь нагревали с обратным холодильником в течение 2 часов. После того как реакционная смесь достигала комнатной температуры, растворитель удаляли в вакууме. К оставшейся фракции добавляли 30 мл H2O и смесь впоследствии кипятилась в течение 45 минут. После охлаждения смеси осуществлялась экстракция CH2Cl2, органическая фракция сушилась над MgSO4. После удаления осушающего агента и удаления растворителя в вакууме оставался остаток 0,95 г (2,2 ммоль, 96%) соответствующего чистого О-метилированного производного фенола.
Соединение C4 синтезировалось в соответствии с процедурой, аналогичной описанной для соединения C2.
В таблице C приведены полученные соединения.
Промежуточные соединения, используемые в способе C
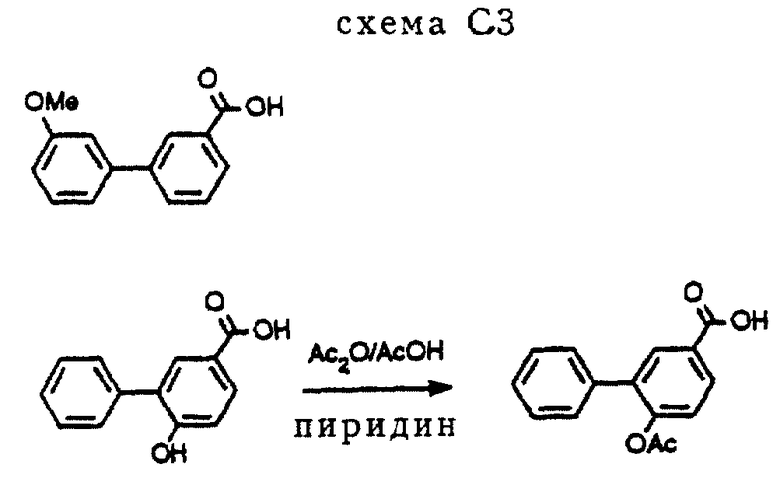
3-(3-Метоксифенил)бензойную кислоту (см. схему C3 в конце описания) получали в соответствии с процедурой, аналогичной описанной W. G. Dauben et al. , J. Am. Chem. Soc. , 75(1953), 4969-73. 3-Фенил-4-ацетоксибензойную кислоту (схема C3) получали с помощью стандартных процедур из 3-фенил-4-гидроксибензойной кислоты (см. схему C3). Синтез последнего соединения описан в патенте США N 4873367.
Пример 4
Процедура D1 (схема D1)
В атмосфере азота 0,4 г (2,8 ммоль) 4-бромфенола растворяли в 5 мл толуола. К полученному раствору добавлялось 97,5 мг (0,084 ммоль, 0,03 экв. ) Pd(PPh3)4, 2,8 мл 2 н. Na2CO3 и 1,0 г (2,8 ммоль) бороновой кислоты d1 (S5 = H), растворенной в 5 мл горячего этанола. Полученную смесь интенсивно перемешивали при 90oC в течение 4 часов. После того как реакционная смесь достигала комнатной температуры, она разбавлялась EtOAc и небольшим количеством воды. Затем осуществлялась экстракция EtOAc, объединенные органические фракции промывались насыщенным раствором соли и сушились над MgSO4. После удаления осушающего агента и удаления растворителя в вакууме оставалось 1,52 г остатка, который подвергался хроматографии на колонке (SiO2, элюент: смесь EtOAc и петролейного эфира, 1: 1), давая 0,53 г (1,3 ммоль, 47%) чистого свободного основания D22. Свободное основание превращалось в его дигидрохлоридную соль (кристаллизация из смеси EtOAc и простого эфира), давая соединение D22•2HCl, т. пл. 222-227oC.
1H-ЯМР (d16-ДМСО/CDCl3 4/1, δ): 3.14-3.30 (широкий кластер, 4H), 3.34-3.56 (широкий кластер, 4H), 4.23 (м. 4H), 4.42 (д. J= 4 Гц, 2H), 6.46-6.58 (кластер, 2H), 6.73 (т. J= 8 Гц, 1H), 6.89 (м. 2H), 7.47 (т. J= 7 Гц, 1H), 7.52-7.66 (кластер, 4H), 7.99 (т. J= 1 Гц, 1H), 9.40 (широкий,  ), 11.5 (широкий, 1H).
), 11.5 (широкий, 1H).
Согласно данному выше синтезу получались следующие соединения (таблица D).
Промежуточные соединения, используемые в способе D
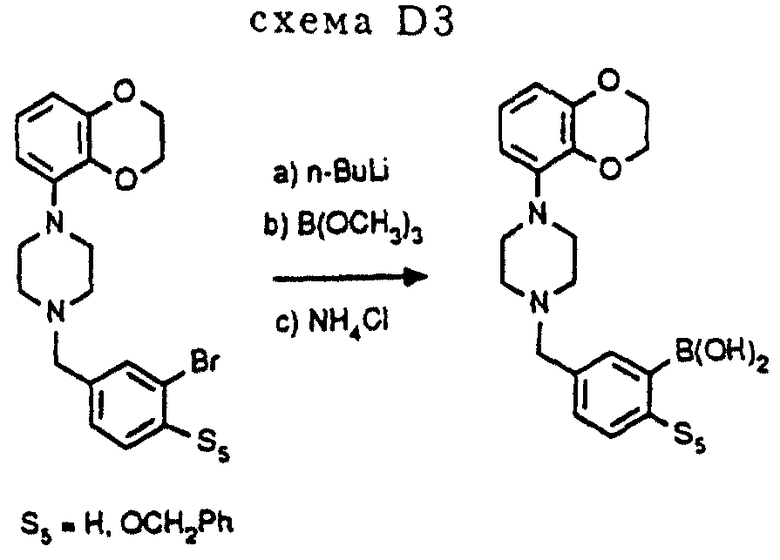
Бромиды, использованные в схеме D1 и D2, можно легко получить стандартными методами или приобрести коммерческим путем. Бороновые кислоты, использованные в схемах D1 и D2, можно легко получить через соответствующие бромиды (см. схему D3 в конце описания). Общие процедуры описаны в статье D. Janietz et al. , Synthesis, (1993), 33 и в приведенных там ссылках.
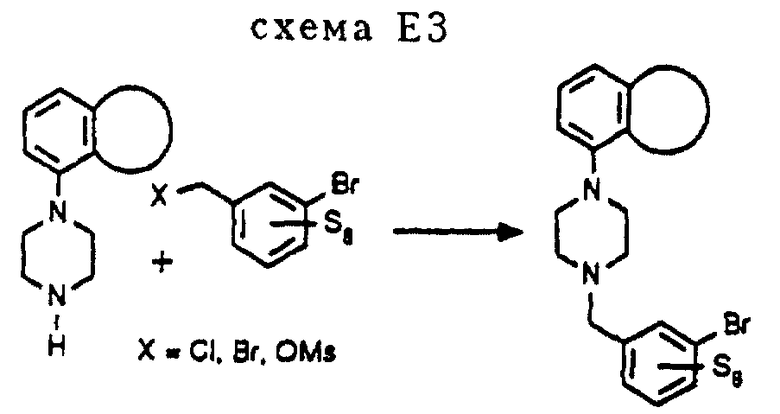
Использованные бромиды (S5= H, OCH2Ph, схема D3) синтезируются в соответствии с процедурой, аналогичной описанной в процедуре E3 (схема E3).
Пример 5
Процедура E1 (схема Е1)
5,1 г (12,0 ммоль) 1-[(2-метокси-5-бромфенил)метил] -4-(2,3-дигидро-1,4-бензодиоксин-5-ил)пиперазина растворяли в 20 мл толуола, к которому добавляли 12 мл 2 н. Na2CO3/H2O и 0,45 г (0,39 ммоль, 0,03 экв. ) Pd(PPh3)4. Затем к раствору добавляли 1,46 г (12,0 ммоль) фенилбороновой кислоты, растворенной в 3 мл теплого этанола. Реакционную смесь энергично перемешивали при 85oC. Через 4 часа двухфазную реакционную смесь оставляли достигать комнатной температуры, после чего отделяли органическую фракцию (толуол). Водный слой экстрагировали EtOAc. Объединенные толуольные и EtOAc фракции соответственно промывали водой и насыщенным раствором соли, после чего органическую фракцию сушили над Na2SO4. После удаления осушающего агента и последующего удаления растворителя в вакууме оставался остаток, который подвергался хроматографии на колонке (SiO2, элюент: смесь EtOAc и петролейного эфира, 1: 2). Выделенное чистое свободное основание соединения Е2 растворялось в смеси EtOAc и этанола (1: 1) и полученный раствор обрабатывался 1 эквивалентом смеси 1 н. HCl/этанол, давая 1,43 г (3,2 ммоль, 26%) соединения Е2•HCl, т. пл. 240-242oC (разложение).
1H-ЯМР (d6-DMCO/CDCl3 4/1, δ): 3.1-3.3 (кластер, 4H), 3.48 (кластер, 4H), 3.93 (с. 3H), 4.23 (м. 4H), 4.41 (д. J= 5 Гц, 2H), 6.48 (дд. J= 1 Гц, J= 8 Гц, 1H), 6.55 (дд, J= 1 Гц, J= 8 Гц, 1H), 6.73 (т. J= 8 Гц, 1H), 7.20 (д. J= 9 Гц, 1H), 7.32 (м, 1H), 7.40 (т. J= 8 Гц, 2H), 7.71 (м. 2H), 7.75 (дд, J= 2 Гц, J= 9 Гц, 1H), 8.04 (д. J= 2 Гц, 1H), 11.1 (широкий, 1H).
Процедура Е2 (схема Е2)
3,0 г (5,9 ммоль) О-бензил-защищенного соединения Е1 растворялось в 35 мл концентрированной HCl, после чего смесь перемешивалась и доводилась до температуры кипения с обратным холодильником. Через 45 минут дополнительно добавлялось 30 мл концентрированной HCl и нагревание с обратным холодильником продолжалось еще 45 минут. Затем реакционную смесь оставляли охлаждаться до комнатной температуры, и растворитель удалялся в вакууме. Остаток обрабатывают насыщенным раствором NaHCO3, и раствор экстрагировался EtOAc. Органическую фракцию промывали насыщенным раствором соли, сушили над MgSO4. После удаления осушающего агента и последующего удаления растворителя в вакууме оставался остаток, который подвергался флэш-хроматографии на колонке (SiO2 элюент: CH2Cl2/MeOH, 95: 5). Свободное основание Е1 отделялось и превращалось в HCl соль путем обработки смесью 1 н. HCl/EtOH. Перекристаллизация из смеси EtOAc и H2O давала 1,45 г (3,2 ммоль, 54%) чистого соединения Е1•HCl.
Указанные выше соединения приведены в таблице Е.
Промежуточные соединения, использованные в способе Е
Бромиды, использованные для осуществления реакции Сузуки, изображенной на схеме Е1, можно синтезировать из фенилпиперазинов и соответствующих замещенных 3-бромфенилметил-X промежуточных соединений, в которых X может быть Cl, Br или OMs (см. схему E3 в конце описания).
Процедура E3 (схема E3)
К 80 мл CH3CN добавляют 6,6 г (23,0 ммоль) (2-метокси-5-бромфенил)-метилбромида и 5,4 г (21 ммоль) I-Н•HCl, после чего вводят 5,2 г (51,0 ммоль) Et3N и небольшое количество KI. Реакционную смесь перемешивают и выдерживают при температуре кипения с обратным холодильником в течение 16 часов. Затем полученную смесь фильтруют и фильтрат концентрируют в вакууме. Остаток подвергают хроматографии на колонке (SiO2, элюент: EtOAc/петролейный эфир, 1: 2), что дает 5,1 г (12,2 ммоль, 58%) чистого 1-[(2-метокси-5-бромфенил)метил] -4- (2,3-дигидро-1,4-бензодиоксин-5-ил)пиперазина.
Используемые бороновые кислоты легко получить при помощи соответствующих бромидов, например, в соответствии с общими процедурами, описанными D. Janietz et. al. , Synthesis, (1993), 33, и приведенными там ссылками.
Получение промежуточного соединения XI-H в соответствии со схемой A. i
Стадия 1 (схема A. i)
5,1 г (25 ммоль) 1-(фенилметил)гексагидро-5Н-1,4-диазепин-5-она (для ознакомления со способом см. Dickerman et. al. , J. Org. Chem. , 19, (1954), 1855-61)) и 7,39 г (37,5 ммоль) 7-бромбензофурана вместе с 3,45 г (25 ммоль) высушенного K2CO3 и 0,48 г (2,5 ммоль) Cul помещают в колбу и полученную смесь, перемешивая, нагревают при температуре 120oC в течение 90 часов. После того как реакционная смесь доводилась до комнатной температуры, добавлялось 40 мл толуола. Полученную суспензию фильтруют через целит и остаток промывают теплым толуолом. Объединенные промывочные воды и фильтрат упаривают в вакууме, получая 12,4 г коричневого масла. Масло разбавляют CH2Cl2 и последовательно обрабатывают 2 н. NaOH, NaHCO3 (насыщенный) и водой. Органическую фракцию сушат над MgSO4. После удаления осушающего агента и выпаривания растворителя в вакууме получалось 11,7 г коричневого масла. Полученный остаток подвергался флэш-хроматографии на колонке (SiO2, элюент: CH2Cl2/MeOH, 98: 2), давая 5,7 г (83%) желаемого продукта.
Стадия 2 (схема. A. i)
5,9 г (18,6 ммоль) продукта стадии 1, растворенного в 40 мл сухого ТГФ, по каплям добавляют к смеси 2,14 г (55,8 ммоль) LiAlH4 в 100 мл Et2O и перемешивают в течение 3 часов. Затем реакционную смесь последовательно обрабатывают 2,1 мл H2O в ТГФ, 4,2 мл 2 н. NaOH и 2,4 мл H2O. Смесь продолжают перемешивать еще 2 часа, после чего ее фильтруют через целит, остаток последовательно промывают ТГФ и CH2Cl2. Объединенные промывочные воды и фильтрат упаривают в вакууме с получением 5,4 г коричневого масла. Полученный остаток подвергают флэш-хроматографии на колонке (SiO2, элюент: CH2Cl2/MeOH, 98: 1), что дает 4,83 г (85%) аналога диазепина.
Стадия 3 (схема A. i)
4,83 г (15,8 ммоль) продукта стадии 2 растворяют при перемешивании в 65 мл 1,2-дихлорэтана. В атмосфере азота при 2-4oC в течение 10 минут к полученному раствору добавляют 2,3 г (15,8 ммоль) Cl(CO)O(CHCl)CH3 ("ACE-хлорид"), растворенного в 25 мл 1,2-дихлорэтана. Затем реакционную смесь нагревают с обратным холодильником в течение 10 часов и концентрируют в вакууме с получением 5,1 г остатка. Остаток разбавляют метанолом и полученный раствор нагревают с обратным холодильником в течение 16 часов. После того как реакционную смесь доводят до комнатной температуры, растворитель удаляют в вакууме с получением 4,2 г остатка, который подвергают флэш-хроматографии на колонке (SiO2, элюент: CH2Cl2/MeOH/NH4OH, 92: 7,5: 0,5), что дает 2,8 г (82%) 1-(7-бензофуранил)гексагидро-1,4-диазепина.
Получение XV-H, см. схему A. iii
Стадия 1 (схема A. iii)
3,94 г (21,9 ммоль) 7-нитро-2-бензоксазолинона (для ознакомления со способом получения этого соединения см. Европейский патент ЕР0189612 и приведенные в нем ссылки) растворяют в 40 мл ДМСО и добавляют 1,72 г 85% порошка KOH (26,2 ммоль). К полученному раствору при перемешивании и охлаждении (водой) в течение 10 минут по каплям добавляют 3,72 г (26,2 ммоль) Mel, растворенного в 6 мл ДМСО. Реакционную смесь продолжают перемешивать при комнатной температуре в течение 16 часов, в течение последнего периода добавляют дополнительное количество Mel (0,5 г). После окончания реакции реакционную смесь разбавляют водой и экстрагируют CH2Cl2. Объединенные органические фракции последовательно промывают водой и насыщенным раствором соли, после чего органическую фракцию сушат над MgSO4. После удаления осушающего агента и выпаривания растворителя в вакууме получают 4,1 г твердого остатка. Флэш-хроматография последнего на колонке (SiO2, элюент: CH2Cl2) дает 3,6 г (85%) чистого 3-метил-7-нитро-2-бензоксазолинона.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА | 1998 |
|
RU2197488C2 |
| 2,3-ДИГИДРО-1,4-БЕНЗОДИОКСИ-5-ИЛ-ПИПЕРАЗИНОВЫЕ ПРОИЗВОДНЫЕ ИЛИ ИХ СОЛИ | 1994 |
|
RU2118322C1 |
| СПОСОБ СТЕРЕОСЕЛЕКТИВНОГО ПОЛУЧЕНИЯ ЭНАНТИОМЕРА ГЕТЕРОБИЦИКЛИЧЕСКОГО СПИРТА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2185379C2 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| АДДУКТЫ-АЛЬДЕГИДЫ В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ В СИНТЕЗЕ ПРОИЗВОДНЫХ ВИТАМИНА D | 1990 |
|
RU2036904C1 |
| АЛКИЛ-ПИПЕРАЗИНИЛ БЕНЗОКСАЗОЛОНОВЫЕ И АЛКИЛ-ПИПЕРИДИНИЛ БЕНЗОКСАЗОЛОНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СПОСОБ ЛЕЧЕНИЯ | 1999 |
|
RU2225406C2 |
| МАКРОЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГЕПАТИТА C | 2000 |
|
RU2247126C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |
| ПРОИЗВОДНЫЕ ГАЛИХОНДРИНА B, ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННЫХ ПИРАНОВ (ВАРИАНТЫ), ПРОИЗВОДНЫЕ ГИДРИРОВАННОГО ФУРАНА, ПРОИЗВОДНЫЕ ГИДРИРОВАННОГО ПИРАНА, ПРОИЗВОДНЫЕ ЕНОНА ГАЛИХОНДРИНА B, ПРОИЗВОДНЫЕ ЕНОНА НОРГАЛИХОНДРИНА | 1993 |
|
RU2112773C1 |
| МОДУЛЯТОРЫ CCR2 | 2016 |
|
RU2726206C2 |
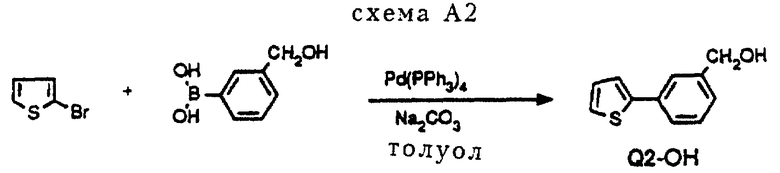
Изобретение относится к производным пиперазина и пиперидина общей формулы (а), в которой А обозначает гетероциклическую группу с 5-7 атомами в кольце, содержащую 1-2 гетероатома из группы О, N и S; R1 обозначает водород или фтор; R2 обозначает оксогруппу или С1-4алкил и р = 0 или 1; Z обозначает углерод или азот, и пунктирная линия обозначает простую связь, когда Z является азотом, и простую или двойную связь, когда Z является углеродом; R3 и R4 независимо друг от друга обозначают водород или С1-4алкил; n = 1 или 2; R5 обозначает С1-4алкокси, С1-4алкил, галоген или гидрокси и q = 0 или 1; Y обозначает фенил, замещенный 1-2 заместителями из группы гидрокси, галоген, С1-4алкокси, циано, аминокарбонил, ди-С1-4алкиламино-карбонил; фурил или тиенил и их соли. Также описывается способ получения этих соединений. Заявленные соединения обладают высокой степенью сродства как к рецепторам допамина D2, так и к рецепторам серотонина 5-НТ1А. 2 с. и 4 з. п. ф-лы, 6 табл.
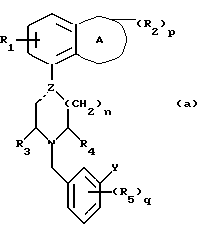
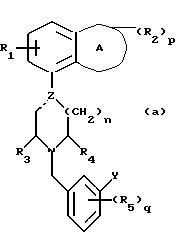
в которой А обозначает гетероциклическую группу с 5-7 атомами в кольце, содержащую 1-2 гетероатома из группы O, N и S;
R1 обозначает водород или фтор;
R2 обозначает оксогруппу или C1-4 алкил;
p = 0 или 1;
Z обозначает углерод или азот и пунктирная линия обозначает простую связь, когда Z является азотом, и простую или двойную связь, когда Z является углеродом;
R3 и R4 независимо друг от друга обозначают водород или C1-4алкил;
n = 1 или 2;
R5 обозначает C1-4 алкокси, C1-4 алкил, галоген или гидрокси;
q = 0 или 1;
Y обозначает фенил замещенный 1-2 заместителями из группы гидрокси, галоген, C1-4 алкокси, циано, аминокарбонил, ди-C1-4 алкиламинокарбанил; фурил или тиенил и их соли.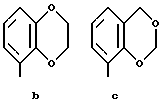
в которой R1 и (R2)р имеют значения, указанные в п. 1, n = 1, R3, R4, (R5)q, Y и Z имеют значения, указанные в п. 1, и их соли.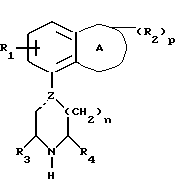
подвергают взаимодействию с соединением формулы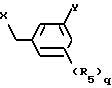
в которой Х является отщепляемой группой.
| RU 2056420 С1, 20.03.1996 | |||
| Фазный ротор машины переменного тока | 1971 |
|
SU466585A1 |
| WO 9304682 А1, 18.03.1993. | |||

