Изобретение относится к способу стереоселективного получения энантиомера гетеробициклического спирта с помощью хиральных строительных блоков. Изобретение относится также к способу получения исходного соединения для реакции с хиральным строительным блоком.
Различные биологически активные вещества, которые могут использоваться, например, в фармацевтических или ветеринарных композициях, содержат в своей молекулярной структуре хиральный центр и соответственно порождают оптический изомеризм. Специалистам хорошо известно, что зачастую лишь один из энантиомеров обладает необходимой оптимальной биологической активностью. Присутствие другого оптического антипода в составе или средстве может порождать или усиливать побочные явления, нанося тем самым вред реципиенту, т.е. организму человека или животного. В целом все более четко появляется тенденция вводить биологически активное вещество в виде по существу чистого энантиомера, в наибольшей степени обладающего нужной биологической активностью. Поэтому получение по существу энантиометрически чистого соединения часто является важной стадией в процессе производства фармакологически активных веществ. В большинстве случаев энантиомеры получают разрешением рацемата на составляющие энантиомеры.
Известно несколько способов разрешения рацематов на составляющие энантиомеры. Первый из них - разрешение на основе различий в физиологических свойствах, например кристаллической структуре, - применяется лишь изредка.
Второй, гораздо более распространенный способ разрешения включает реакцию с промышленно выпускаемым оптически активным реактивом с получением диастереомеров, обладающих различными физическими свойствами. Полученные таким образом диастереомеры можно разделить, например, перекристаллизацией, после чего можно регенерировать соответствующие энантиомеры последующей химической обработкой. Очевидно, какой способ разрешения рацематов трудоемок и дорог, в первую очередь ввиду использования дорогостоящего оптически активного реактива.
В недавно предложенном более экономичном третьем способе разрешения один из энантиомеров в рацемате подвергают селективной химической модификации с помощью ферментов, а затем отделяют модифицированный энантиомер от немодифицированного. Например, Бьянки и др. (J.Org. Chem. 1988, 53, 5531-5534) сообщали об использовании ангидридов карбоновых кислот в качестве ацилирующих агентов при селективной эстерификации рацемических спиртов с липазным катализатором. Усовершенствование этого способа описано в европейской заявке 0605033.
Другой способ получения энантиомеров состоит в использовании хиральных строительных блоков. По сравнению с тремя вышеописанными этот способ обладает тем важным преимуществом, что в ходе реакции образуется исключительно или главным образом целевой энантиомер. Тем самым предотвращается образование значительных количеств (до 50%) нежелательного энантиомера, соединения, которое приходится рассматривать как химический отход или, если рацемизация возможна, регенерировать в одну или несколько трудоемких стадий.
Задачей настоящего изобретения является создание экономичного способа стереоселективного получения энантиомера гетероциклического спирта, служащего промежуточным соединением при синтезе фармакологически активных веществ, например флезиноксана.
Поставленная задача достигается использованием в способе хирального строительного блока для введения хирального центра. Способ в соответствии с настоящим изобретением отличается тем, что по существу чистый энантиомер общей формулы
где Х -O, S, NH или N-(C1-C4)алкил;
Y1 и Y2 каждый независимо друг от друга водород или заместители, выбранные из группы, включающей галоген, (C1-C4)алкил, (C1-C4)алкокси, (C1-C4)галоалкил-, формил-, нитро-, и цианогруппы;
атом С* имеет конфигурацию К либо S,
получают из соединения общей формулы
где Х, У1 и У2 имеют те же значения, что определены выше;
R1 - водород или приемлемая защитная группа;
R2 - водород;
или R1 и R2 вместе образуют по выбору моно- или ди-(C1-C3)алкилзамещенный метиленовый мостик, путем следующих поочередных стадий:
(1) реакции с соединением общей формулы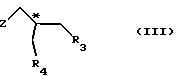
где Z - гидроксильная или приемлемая отщепляемая группа;
R3 - гидроксильная защитная группа;
R4 - атом галогена;
или где R3 и R4 вместе образуют валентную связь или бирадикал формулы - С(R11)2-О-, где R11 - прямая или разветвленная (C1-C4)алкильная группа;
атом С* имеет конфигурацию R либо S, в результате которой образуется соединение общей формулы
где Х, Y1, Y2, R3 и R4 определены выше;
R1 - водород или защитная группа;
(II) снятия защиты/реакции замыкания цикла полученного таким образом соединения;
(III) произвольное снятие защиты с гидроксильной группы продукта с замкнутым циклом.
Защита гидроксильной группы приемлемой защитной группой общеизвестна, например, описана в справочнике: Greene and Wuts "Protective Groups in Organic Sinthesis" (J. Willey & Sons, Inc., N.Y., 2nd ed., 1991). В качестве примеров защитных групп можно привести возможно замещенные бензил, ацетил, бутаноил, производно замещенный бензоил (например, 2,6-ди-хлорбензоил), метоксиизопропил (МИП), трет-бутил-диметилсилил и тетрагидропирадинил. Для защитной группы R1 предпочтительны направленно защитные бензил и бензоил, для защитной группы R3-МИП.
Реакцию (1) между соединениями II и III можно проводить в системе гомогенных растворителей, в полярных растворителях, например, N-метилпирролидона (N - МП), ДМСО и ДМФ, либо в гетерогенной системе неполярный растворитель/вода с помощью катализатора фазового перехода и под воздействием основания. Из неполярных растворителей предпочтителен толуол. Катализаторами фазового перехода могут служить соли тетрабутиламмония, предпочтительно бисульфат тетрабутиламмония. В качестве оснований можно использовать К2СО3, NaOH и NaH.
Если R2 - водород, то группой Z в соединении III должна быть приемлемая отщепляемая группа. Таковыми могут служить гало- и сульфанатные отщепляемые группы, например галогены, мезилокси-, тозилокси- и нозилоксигруппы. Предпочтительна в роли отщепляемой нозилоксигруппа. Когда R1 и R2 вместе образуют метиленовый мостик, Z может быть гидроксилом.
Когда образуется соединение IV, необходимо провести реакцию (II) снятия защиты/замыкания цикла, чтобы получить продукт с замкнутым циклом. Удаление всех защитных групп обязательно. Его можно осуществить известными способами. Защитные группы можно удалить одновременно или последовательно в зависимости от вида группы. Если это сложноэфирная защитная группа, можно установить основную среду, что ведет к снятию защиты с обеих групп и последовательному замыканию цикла до бициклической половины. Если защитной группой служит МИН или R3 и R4, образуют бирадикал формулы -С(R11)2-, где R11 определено выше, то защитную группу можно удалить в кислой среде, например в 45%-ном растворе МВr в уксусной кислоте. Реакция замыкания цикла осуществляется созданием основной среды.
В рекомендуемом варианте способа в соответствии с настоящим изобретением по существу чистый энантиомер общей формулы I, как указано выше, где Х-О получают вышеописанным способом, отличающимся тем, что по существу чистый энантиомер общей формулы I, как указано выше, где Х-О, получают реакцией производного катехина общей формулы
где Y1 и Y2 определены выше;
R5 - гидроксильная защитная группа, с соединением общей формулы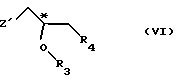
где R3 и R4 определены выше;
Z' - галоген или сульфатная отщепляемая группа, предпочтительно тозилокси-, нозилокси- или метилоксигруппа, после которой полученное промежуточное соединение общей формулы
подвергают последовательно реакциям II и III, как описано выше.
Приемлемые хиральные строительные блоки общей формулы IV для осуществления вышеописанной реакции можно описать следующими формулами: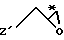
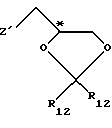

где С* имеет конфигурацию R либо S;
Z' определен выше (предпочтительно нозилокси- или тозилоксигруппа);
R12 -(C1-C4) алкил, предпочтительно метил или изопропил;
R13 - приемлемая гидроксильная защитная группа, предпочтительно метоксиизопропил (МИП).
Предпочтительные условия реакции соединения формулы V с вышеуказанными хиральными строительными блоками следующие: органический растворитель или смесь растворителей, например толуол, метилизобутилкетон (МИБК) или смесь толуол-ДМФ; температура реакции от комнатной до флегмы, предпочтительно температура флегмы; присутствие основания, например, NaOH, КОН, К2СО3 или NaH (в по меньшей мере эквимолярном количестве); при желании присутствие катализатора фазового перехода. Хорошими катализаторами фазового перехода могут служить четвертичные аммонийные соли, например бисульфат тетрабутиламмония и бромид тетрабутиламмония.
Приемлемые защитные группы описаны выше. Как и для R1, для защитной группы R5 предпочтительны возможно замещенные бензил и бензоил, лучше всего возможно замещенный бензил. В этом случае снятие защиты с R5 одновременно с эпоксидным или солькетальным расщеплением предпочтительно осуществляют в присутствии минеральной кислоты, например НСl или НВr, в полярном растворителе или смеси растворителей, например в уксусной кислоте или ее смеси с N-МП, при температуре от комнатной до флегмы. Последующую реакцию замыкания цикла можно проводить в том же растворителе, предпочтительно под воздействием основания, например КОН, NaOH и т.п.
В другом, не менее перспективном варианте осуществления настоящего изобретения по существу чистый энантиомер общей формулы I, как указано выше, где Х есть О, получают реакцией производного катехина общей формулы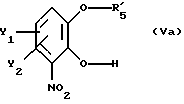
где Y1 и Y2 имеют те же самые значения, что определены в п.1;
R'5 - гидроксильная защитная группа, выбранная из группы, состоящей из (C1-C8-)алкилкарбонила и арилкарбонила, где арильная группа может быть замещена одним или несколькими заместителями из группы, включающей (C1-C4)алкоксигруппу и галоген;
с соединением общей формулы
где R3 и R4 имеют выше упомянутые значения; и
Z' - галоген или сульфонатная отщепляемая группа, предпочтительно выбранная из тозилокси-, ноизлокси- и мезилоксигруппы;
после чего полученное промежуточное соединение общей формулы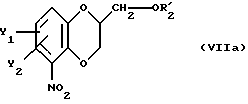
подвергают удалению защитной гидроксильной группы из продукта с замкнутым циклом.
Приемлемые хиральные строительные блоки общей формулы VI и предпочтительные условия реакции те же, что и для реакции соединений общей формулы V, описанной выше.
Предпочтительны в качестве R'5 - гидроксильных защитных, в этой реакции алкилкарбонильные группы. Удаление гидроксильных защитных групп R'5 осуществляется известным способом.
В еще одном допустимом варианте способа в соответствии с настоящим изобретением по существу чистый энантиомер общей формулы I, как описано выше, где Х это NH или N-(C1-C4) алкил, получают вышеописанным способом, отличающимся тем, что по существу чистый энантиомер общей формулы I, как указано выше, где Х это NH или N-(C1-C4)алкил, получают реакцией соединения общей формулы
где Y1 и Y2 имеют значения, приведенные в п.1; и
R'5 - гидроксильная защитная аминогруппа;
R6 - водород или (C1-C4) алкил;
с соединением общей формулы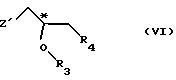
где R3 и R4 имеют выше упомянутые значения; и
Z' - галоген или сульфонатная отщепляемая группа, предпочтительно выбранная из тозилокси-, нозилокси- и мезилоксигруппы;
после чего полученное промежуточное соединение общей формулы
подвергают удалению защитной гидроксильной группы из продукта с замкнутым циклом.
Приемлемыми защитными аминогруппами могут быть ацильные, например арилкарбонильные, алкилкарбонильные (например, ацетил) и алкилсульфонильные группы. Предпочтительные алкилкарбонильные группы.
В следующем, в равной степени перспективном варианте способа в соответствии с настоящим изобретением по существу чистый энантиометр общей формулы I, как описано выше, где Х это О, можно получить реакцией бензодиоксольного соединения общей формулы
где Y1 и Y2 определены выше;
R7 и R8 - каждый независимо друг от друга водород или метил;
со следующими хиральным строительным блоком соединением формулы
или
где R11 - прямой или разветвленный (C1-C4)алкил.
Эту реакцию предпочтительно проводят в полярном органическом растворителе, например NaOH, КОН, NaH или К2СО3, при температуре от 0оС до флегмы. В случае использования соединения формулы ХIIb последующую солькетальную дециклизацию и реакцию замыкания цикла с соединением формулы I (Х=0) осуществляют, как описано выше, например, обработкой раствором минеральной кислоты в, например, уксусной кислоте и затем обработкой водным раствором основания, например NaOH.
Изобретение также относится к соединениям, используемым в вышеописанном способе в качестве промежуточных и имеющих общую формулу: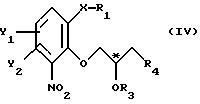
Эти новые соединения можно получить вышеописанным способом, т.е. способом получения вышеописанного соединения, отличающимся тем, что соединение общей формулы
в котором значения символов определены выше, вводят в реакцию с соединением общей формулы
в котором значения символов также определены выше.
Далее изобретение относится также к соединениям, используемым в качестве промежуточных в вышеописанном способом и имеющим общую формулу
в которых значения символов определены выше. Эти новые соединения можно получать, как описано выше, т.е. способом получения вышеприведенных соединений, отличающимся тем, что соединение общей формулы
в котором значения символов определены выше, вводят в реакцию с соединением общей формулы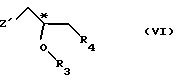
в котором значения символов также определены выше.
Для вышеописанной реакции V+VI--->VII в качестве исходного соединения требуется защищенное моногидроксильной группой производное катехина формулы V. В роли защитных пригодны алкилкарбонильные (например, ацетил), (возможно замещенные) бензильные и триалкилсилильные группы.
Получение моноацетатов гидрохинона и катехина изучалось, судя по предшествующим публикациям, с различными результатами. Олткотт (J.Am.Chem.Soc. 59, 1937. 392-393) установил, что моноацетилирование этих двух дигидробензолов можно осуществить путем осторожного ведения реакции с ацетилирующим веществом, например уксусным ангидридом, в водном растворе щелочи. Выход целевого моноацетата составил всего 20-30%, большей частью получался диацетат. Джонстону (Chem. Ind. 1982, 1000) удалось повысить выход моноацетата гидрохинона из гидрохинона и уксусного ангидрида, используя в качестве основания триэтиламин, а в роли катализатора 4-диметиламинпиридин (ДМАП) в полярном органическом растворителе, а именно в растворе этилацетата. В таких условиях Джонстон сумел добиться выхода моноацетата гидрохинона 58-65%.
Очевидно и такие результаты малопривлекательны с точки зрения экономики производства монозащищенных дигидрооксибензолов.
Установлено, что в дополнительном аспекте настоящего изобретения монозащищенные производные катехина, имеющие общую формулу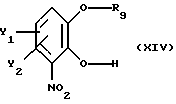
где Y1 и Y2 определены выше,
R9 - свободно замещенная бензоильная группа, (C1-C4)алкилкарбонильная или три(C1-C4)алкилсилильная группа; могут быть легко получены с высоким выходом путем реакции замещенного катехина общей формулы
где Y1 и Y2 определены выше,
р и q равны 0 или 1,
с соединением общей формулы
(R'9)2О или R'9Hal или (R14)3SiHal
где R'9 - произвольно замещенная бензоильная или (C1- C4)алкилкарбонильная группа;
R14 - (C1-C4)алкил;
Наl - галоген,
в присутствии органического основания, предпочтительно третамина или смеси органических оснований в количестве, достаточном для каталитического действия,
причем способ отличается тем, что реакцию проводят в неполярном органическом растворителе или без растворителя, после чего полученное соединение общей формулы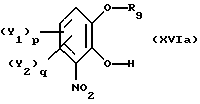
последовательно подвергают реакции ароматического замещения с целью введения при необходимости заместителей Y1 и Y2, в ароматическое ядро и нитрированию.
Если желательна ацильная защитная группа, реакцию моноацилирования предпочтительно проводят без растворителя, например, в расплаве, если реакционная смесь по условиям реакции жидкая. Температура реакции может колебаться от комнатной до около 150оС в зависимости от применяемых реагентов; в целом реакцию обычно ведут при слегка повышенной температуре.
Можно также проводить реакцию в неполярном органическом растворителе, например в углеводороде (толуол, ксилол) или в диалкилэфире-метил-трет-бутилэфире (МТБЭ).
Приемлемыми органическими основаниями при подобной монозащите катехина могут служить амины, например триэтиламин (ТЭА), диэтиламин, триизобутиламин (ТИБА), пиридин, 2,6-лутидин,диметиламин (ДМА), ДМПА и смеси ДМАП с ТЭА или ТИБА. Для целей моноацилирования амин должен присутствовать, в по меньшей мере, каталитическом количестве по отношению к исходному катехину; исходный ацилирующий агент предпочтительно содержится в небольшом избытке против эквимолярного количества. При моноацилировании катехина можно легко получить выход свыше 85% в расчете на исходный катехин.
Для получения моно (триалкилсилил) эфира катехина можно использовать различные галиды триалкилсилила, например галогенид трет-бутилметилсилила. Эту реакцию удобно вести в неполярном органическом растворителе типа диалкилэфира, например метил-трет-бутилэфире, в присутствии эквивалентного количества органического основания, как указано выше. При повышенной температуре, например при температуре флегмы, можно получить выход целевого моносилилэфира катехина свыше 90%.
Монозащиту можно получить также путем монобензилирования. Для получения катехина с монобензильной защитой используют (произвольно замещенный) бензилхлорид в присутствии основания. Растворителями служат спирты, предпочтительно метоксиэтинол. Либо можно использовать двухфазную систему углеводород/вода, предпочтительно толуол/вода, с применением катализатора фазового перехода, например соли тетрааллилкиламмония. Возможно также моноалкилирование без растворителя в присутствии катализатора фазового перехода. Основаниями служат гидроксиды и карбонаты, например NaOH, KHCO3. Добавление какого-либо йодида в каталитическом количестве помогает ускорить реакцию. Таким способом можно осуществить моноалкилирование катехина с выходом свыше 80%.
Последующее введение заместителей Y1 и Y2, если эти заместители не присутствовали в ходе монозащиты катехина, а также последующего нитрирования, можно осуществлять известными способами. Электрофильное ароматическое замещение с целью введения Y1 и Y2 хорошо известно специалистам; например, несложно провести хлорирование с помощью приемлемого хлорирующего агента, например сульфурилхлорида и т.п. Заключительное нитрирование также можно осуществить известными приемами, например, с помощью концентрированной азотной кислоты. Удобным растворителем при обоих ароматических замещениях служит уксусная кислота. Если монозащиту катехина осуществляют путем реакции моноацилирования в расплаве (без растворителя), то последующее введение Y1 и Y2, если это требуется, и NO2 выполняется в одной и той же среде без выделения промежуточных компонентов.
Далее настоящее изобретение относится к промежуточному соединению, используемому в вышеописанном способе, т.е. к промежуточным соединениям общей формулы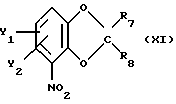
Это соединение формулы Х1 можно получить из катехина формулы ХУ в следующие стадии:
i) селективная защита одной из свободных гидроксильных групп;
ii) возможное введение одного или обоих заместителей Y1 и Y2;
iii) селективное нитрирование ортоположения незамещенной гидроксильной группы;
iv) снятие защитной гидроксильной группы;
v) образование бензофеоксольной части путем реакции с соединением общей формулы
где R7 и R8 определены выше;
R9 и R10 каждый независимо друг от друга - хлор, бром или (C1-C4)алкоксигруппа, либо они образуют совместно атом водорода.
Вышеперечисленные стадии (i), (ii), (iii) и (iv) описаны ранее. Стадию (v) реакции предпочтительно осуществляют путем реакции замещенного катехина, полученного на стадии (iv) и имеющего общую формулу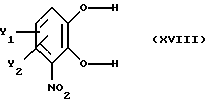
где Y1 и Y2 определены выше,
с образующим бензодиоксил соединением вышеприведенной формулы XYII, предпочтительно донором метилена, например СН2Сl2 или СН2Вr2, в присутствии неорганического основания, например NaOH, КОН, или К2СО3 в органическом растворителе. Приемлемые органические растворители данной реакции - ДМСО, ДМФ, N-МП и толуол при желании в присутствии агента фазового перехода. Оптимальные растворители - ДМВ или толуол в присутствии агента фазового перехода. Реакция протекает гладко при повышенной температуре, например при температуре флегмы.
Далее изобретение будет описано более подробно со ссылками на нижеследующие примеры.
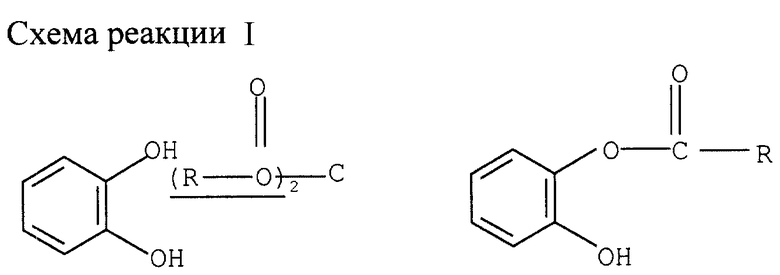
Пример I
Моноацилирование катехина (см. схему реакции I в конце описания).
Смесь 45 ммолей катехина и 53 ммолей ацилангидрида охлаждают до 2-8оС в водно-ледяной бане. Затем добавляют 1,5 ммоля органического основания в качестве катализатора. Реакционную смесь перемешивают 3 ч при комнатной температуре. После последовательного добавления 200 мл этилацетата и 150 мл воды разделяют фазы. Органическую фазу дважды промывают 50 мл 5%-ного водного раствора NaHCO3 и затем дважды 50 мл воды. Слитую водную фазу дважды экстрагируют 50 мл этилацетата. Органические фазы сливают и упаривают досуха при пониженном давлении, получая целевой моноацилированный катехин. Получают следующие результаты:
С уксусным ангидридом (без дополнительного растворителя)
Катализатор - Выход, %*
Триэтиламин (ТЭА) - 90
Диизопропилэтиламин (ДИЭА) - 92
Триизобутиламин (ТИБА) - 93
Пиридин - 67
2,6-лутидин - 91
Диметиламин (ДМА) - 90
4-диметиламинпиридин (ДМАП) - 84
ДМАП/ТЭА (1:1) - 88
ДМАП/ТИБА - 88
Имидазол - 75
* Выход измерялся газовой хроматографией (ГХ)
Если растворителем служит толуол, а катализатором - ТЭА, то моноацилированный катехин получают с выходом 86%.
Ту же реакцию ацилирования с ТЭА катализатором проводят с использованием различных ацилангидридов без растворителя, за исключением монобензилирования, где растворителем служит толуол. Получают следующие результаты:
Ацилангидрид - Выход, %*
Уксусный ангидрид - 90
Пропионовый ангидрид - 90
Масляный ангидрид - 90
Валериановый ангидрид - 93
Изомасляный ангидрид - 87
Бензойный ангидрид - 86
* Выход измерителя ГХ анализом
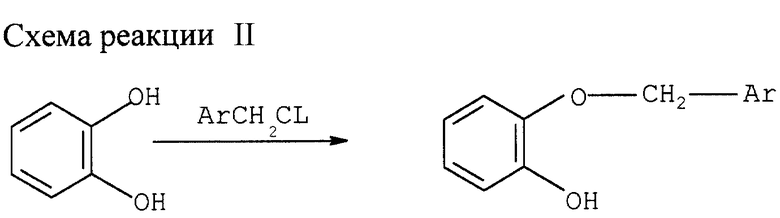
Пример II
Алкилирование катехина (см. схему реакции II в конце описания)
А. Моноалкилирование катехина в органическом растворителе
К смеси 45 ммолей катехина, 58,5 ммолей NaHCO3 и 50 мл растворителя добавляют 49,5 ммоля алкилирующего агента. Смесь нагревают до 85оС при перемешивании. После 20 ч реакции смеси дают остыть и добавляют последовательно 200 мл толуола и 150 мл воды. Разделяют фазы. Органическую фазу промывают 50 мл воды. Слитую водную фазу дважды экстрагируют 50 мл толуола. Слитые органические фазы упаривают досуха при 40оС и пониженном давлении, получая целевой моноэфир катехина.
Монобензилирование катехина бензилхлоридом проводят в различных растворителях со следующими результатами:
Растворитель - Выход, %*
Этанол - 78
Изопропанол - 72
Метоксиэтанол - 81
Этиленгликоль/изопропанол(1:4) - 72
* Выход измерялся ГХ анализом.
Моноалкилирование катехина проводят в растворе метоксиэтанола, начиная с различных замещенных бензилхлоридов:
(Замещенный) бензилхлорид - Выход, %*
незамещенный - 77
4-хлор - 75
4-метил - 74
3-хлор - 72
3-метокси - 76
2,4-дихлор- - 75
2-хлор- - 75
2-фтор - 73
* Выход измерялся ГХ анализом.
В. В толуоле в присутствии катализатора фазового перехода
Смесь 45 ммолей катехина, 4,1 ммоля бисульфата тетрабутиламмония (ВС ТБА) и 25 ммолей К2СО3 в 150 мл толуола нагревают 2 ч с обратным холодильником в условиях азеотропной дистилляции воды (устройство Дина-Старка). Затем добавляют 49,5 молей алкилирующего агента и нагревают смесь около 20 ч. С обратным холодильником при перемешивании. Смеси дают остыть и добавляют последовательно 100 мл толуола и 150 мл воды. Фазы разделяют, органический слой дважды промывают 50 мл 5%-ного раствора NaHCO3 и затем дважды 50 мл воды. Слитые водные фазы дважды экстрагируют 50 мл толуола. Слитые органические фазы упаривают досуха при 40оС и пониженном давлении, получая целевой моноэфир катехина.
Если алкилирующим агентом служит бензилхлорид, выход целевого монобензилэфира катехина составляет 85%.
С. Моноалкилирование катехина в присутствии катализатора фазового перехода без растворителя
Смесь 45 ммолей катехина, 59,5 ммолей КНСО3 и 4,5 ВС ТВА перемешивают 15 мин при 115оС. Затем добавляют 40,5 ммоля алкилирующего агента и перемешивают реакционную смесь 2 ч при 115оС. Смеси дают остыть и добавляют последовательно 150 мл воды. Разделяют фазы, органическую фазу промывают дважды 50 мл 5%-ного раствора NaHCO3 и дважды водой. Слитые водные фазы дважды экстрагируют 50 мл толуола. Слитые органические фазы упаривают досуха при 40оС и пониженном давлении, получая целевой моноалкилэфир катехина.
Вместо ВС ТБА в качестве катализатора фазового перехода можно использовать бромид или хлорид тетрабутиламмония.
Получены следующие результаты:
(Замещенный) бензилхлорид - Выход, %
незамещенный - 72
3-хлор- - 71
4-хлор- - 72
2,4-дихлор- - 76
4-нитро- - 70
* Выход измерялся ГХ анализом.
D. Алкилирование катехина в воде
К смеси 45 ммолей катехина, 58,5 ммолей NaHCO3 и 50 мл воды добавляют 49,5 ммоля алкилирующего агента. Смесь нагревают от 1,5 до 22 ч с обратным холодильником при перемешивании. После добавления последовательно 200 мл этилацетата и 100 мл воды фазы разделяют. Органическую фазу промывают дважды 50 мл 5%-ного раствора NaHCO3 и дважды водой. Слитые водные фазы дважды экстрагируют 50 мл толуола. Слитые органические фазы упаривают досуха при 40оС и пониженном давлении, получая целевой моноалкилэфир катехина.
Получены результаты, представленные в таблице в конце описания.
Пример III
Моносилирование катехина (см. схему реакции III в конце описания).
К смеси 45 ммолей катехина и 54 ммолей третбутилдиметилсилилхлорида в 50 мл метил-трет-бутилэфира (МТБЭ) добавляют в течение 10 мин 45 ммолей триэтиламина. Смесь нагревают 16 ч при перемешивании с обратным холодильником и затем дают остыть. После добавления последовательно 150 мл МТБЭ и 150 мл воды фазы разделяют. Органическую фазу промывают дважды 50 мл 5%-ного раствора NaHCO3 и дважды 50 мл воды. Слитые водные фазы дважды экстрагируют 50 мл МТБЭ. Слитые органические фазы упаривают досуха при 40оС и пониженном давлении, получая 10,6 г целевого моно(триалкилсилил)эфира катехина в виде желтого масла чистотой 87% (выход 91%). 1Н ЯМР (δ, СDCl3)=0,27 (S, 6H), 1,02 (S, 9H), 6,74(t, 1H, J=8 Гц), (d, 1H, J=8Гц), 6,93 (d, 1H, J=8 Гц)
Пример IV
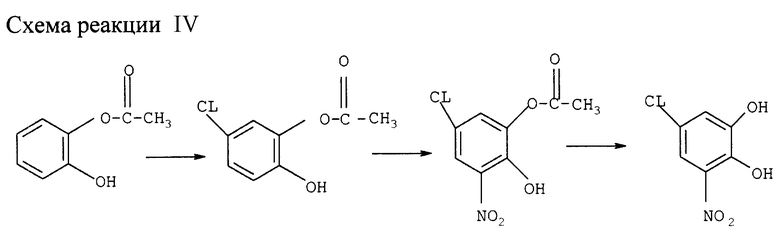
Получение 5-хлор-3-нитрокатехина (см. схему реакции IV в конце описания)
а) Получение 3-нитро-5-хлор-моноацетилкатехина
110 г катехина (1 моль) растворяют в 110 мл уксусной кислоты. Добавляют 4,4 мл (0,03) триэтиламина и 110 мл (1,19 моля) уксусного ангидрида и нагревают смесь за 60 мин до 80оС. После охлаждения до 15оС добавляют 330 мл уксусной кислоты и дозируют в течение 25 мин 82,5 мл (1,4 моля) сульфурилхлорида, поддерживая температуру 15oС, после чего перемешивают 40 мин при 15oС.
Готовят раствор 115,5 мл (1,8 моля) 80%-ной HNO3 в 165 мл уксусной кислоты, поддерживая температуру ниже 20оС. В эту смесь дозируют в течение 60 мин при 15-20оС полученную выше реакцией хлорирования смесь, а затем еще перемешивают 30 мин при 15-20оС. Добавляют 275 мл толуола и 550 мл воды и разделяют фазы. Водную фазу экстрагируют 140 мл толуола (трижды), а слитые органические фазы четырежды промывают 140 мл воды. Толуол отгоняют вакуумной дистилляцией, добавляют 200 мл этанола и также отгоняют под вакуумом. Добавляют 275 мл этанола, нагревают смесь до полного растворения материалов, охлаждая при перемешивании до 0-5оС и перемешивают еще час, образующиеся кристаллы отфильтровывают, промывают холодным этанолом и сушат, получая 106,5 г (выход 46%) 3-нитро-5-хлор-моноацетилкатехина.
1H-ЯМР: (δ, ДМСО/CDCl3 = 3:1) = 7,07 (d, 1H, J=2 Гц), 7,34 (d, 1H, J=2 Гц).
b) Получение 3-нитро-5-хлоркатехина
Полученную выше кристаллическую кашу суспендируют в 200 мл воды. Через 15 мин добавляют 82 мл 50%-ного NaOH при перемешивании, поддерживая температуру ниже 30оС, затем перемешивают еще 15 мин. Через 10 мин. Добавляют 155 мл концентрированной соляной кислоты, поддерживая температуру ниже 35оС, после чего охлаждают до 25оС. Добавляют 135 мл метил-трет-бутилэфира (МТБЭ) и перемешивают смесь 15 мин, затем разделяют фазы. Водную фазу дважды экстрагируют МТБЭ Слитые органические фазы концентрируют выпаркой, получая 85,6 г (45% от исходного катехина) твердого желтого продукта с чистотой 99%.
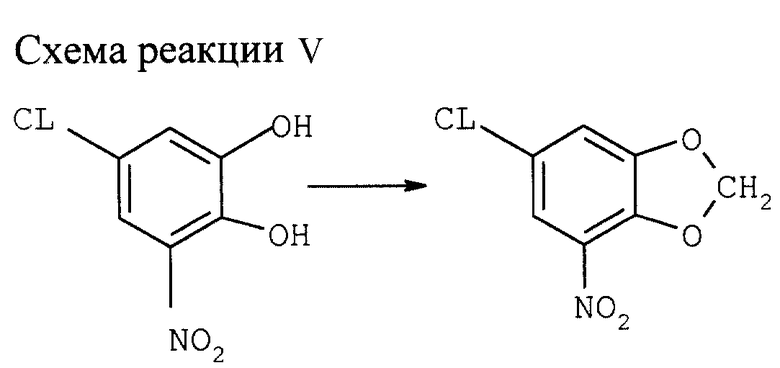
Пример V
Синтез 6-хлор-4-нитро-1,3-бензодиоксола (см. схему реакции V в конце описания)
5-хлор-3-нитрокатехин можно получать двумя способами:
1) Из салицилового альдегида хлорированием, натрированием и окислением по Дакину (см. M. Nikaido et al., J.Org.Chem. 1984, 49, 4740-4741), либо исходя из монозащищенного катехина, как описано в примере 1.
2) Синтез указанного соединения с использованием СН2Сl2:
В атмосфере N2 8,0 г порошка NaOH растворяют в 80 мл ДМСО либо в N-метилпирролидозе (N-МП) при 80оС. Через 15 мин. Добавляют раствор 20,0 г 5-хлор-3-нитрокатехина в 20 мл ДМСО (N-МП) и 100 мл СН2С12 при 80оС.После охлаждения до комнатной температуры добавляют последовательно 200 мл воды и 400 мл толуола. Перемешивают 5 мин, разделяют фазы, экстрагируют водную фазу 100 мл толуола, слитую органическую фазу промывают дважды 50 мл насыщенного водного раствора NaCl, 50 мл воды и дважды 100 мл насыщенного раствора NaCl. После упарки растворителя (100 мбар, 40оС) получают целевой продукт с выходом 70,3% и чистотой 84%.
1Н-ЯМР (δ, ДМСО/СDCl3 = 4:1) = 6,39 (s, 2H), 8,46 (d, 1H, J=2Гц).
b) Синтез указанного соединения с использованием СН2Br2.
В атмосфере N2 200 г 5-хлор-3-нитрокатехина растворяют в 1050 мл ДМФ. К раствору добавляют при перемешивании 204 г порошка безводного К2СО3 и 220 мл СН2Br2. Смесь нагревают 1 ч с обратным холодильником при 140оС и затем охлаждают до 80оС. После добавления 800 мл воды смесь охлаждают до комнатной температуры. Кристаллический продукт отфильтровывают и промывают последовательно дважды 600 мл воды, дважды 250 мл этанола и дважды 300 мл n-гексана. После сушки при 60оС и пониженном давлении получают целевой продукт с выходом 82% и чистотой 95%. Столь же успешные результаты получаются с толуолом вместо ДМФ в качестве растворителя в условиях фазового перехода.
Пример VI
Конверсия монозащищенного хлорнитрокатехина в соединении с формулой VII (см. схему реакции VI в конце описания)
а) Реакция с энантиометрически чистыми соединениями глицифила (Z-нозил; R3 и R4 cовместно образуют валентную связь; R5 - бензил).
8,94 ммоля 5-хлор -3-нитро-монобензилкатехина, 9,78 ммоля К2СО3, 23 молярных % БС ТБА и 100 мл толуола нагревают при перемешивании 30 мин с обратным холодильником, одновременно удаляя воду (устройство Дина-Старка). Смесь охлаждают до 80оС и добавляют 8,94 ммоля (R)-глицифилнозилата. Смесь перемешивают 30 мин при 100оС, охлаждают и разбавляют смесью этилацетата с рассолом. Водную фазу дважды экстрагируют этилацетатом. Слитые органические фазы дважды промывают рассолом, сушат над MgSO4 и фильтруют через 30 г кремнезем. Фильтр промывают этилацетатом, фильтрат упаривают. Осадок кристаллизуют из 20 мл 96%-ного этанола, получая 2,7 г (выход 90%) желтоватых игольчатых кристаллов с ее > 97% (определено ЯМР).
1Н ЯМР (δ, CDCl3) = 2,61 (dd, 1H, J=5 Гц, J=3 Гц), 2,80 (dd, 1H, J=5 Гц, J= 5 Гц, J=5 Гц) 3,3 (кластер, 2Н), 4,24 (m, 1H), 5,13 (s, 2H), 7,15 (d, 1H, J=2Гц), 7,36 (d, 1H, J=2 Гц), 7,37-7,48 (кластер, 5Н).
Ту же реакцию проводят в смеси 1:1 ДМФ/толуол без катализатора фазового перехода. Через 18 ч при 110оС получают целевой продукт с выходом 73%; ее= 98%.
Аналогичный опыт с исходным (R)-глицидилтозилатом, в толуоле и в присутствии ВСТВА дает через 46 ч 82%-ный выход целевого продукта; ее=98%.
Соответствующий (S)-глицидилэфир 5-хлор-3-нитро-монобензилкатехина получают аналогичной реакцией исходного монозащищенного катехина с (S)-глицидилтозилатом в толуоле в присутствии ВСТВА. Выход через 15 ч, 77%; ее=98%. В смеси 1:1 ДМФ/толуол без ВСТВА тот же продукт (ее=99%) получают через 46 ч с выходом 73%.
Ь) Дериватизации 5-хлор-2-гидрокси-3-нитрофенилбензилэфира солькательным соединением (Z-нозил; R3 и R4 совместно образуют бирадикал формулы С(СН3)2-О-R5 -бензил).
Таким же образом, как описано в (а),получают солькетальэфир 5-хлор-3-нитро-монобензилкатехина; 1,1 - 1,2 г-экв К2СO3, 10 мол. % БС ТБА, толуол, нагрев с обратным холодильником. При использовании (S)-солькетальнозелата получают целевой (S)-солькетальэфир 5-хлор-3-нитро-монобензинкатехина с выходом 83%; ее=98%.
Соответствующий (R)-солькетальэфир получают из (R)-солькетальтозилата с выходом 68% после 38 ч реакции; ее=98%. Выход можно увеличить до 80%, проводят реакцию 49 ч обратным холодильником.
С) Дериватизация 5-хлор-2-гидрокси-3-нитрофенолбензилэфира моноксиизопропиловым эфиром 1-хлор-3-тозилоксипропан-2-ина (ТКА-МИП эфиром) (Z-тозил; R3 - C(CH3)2OCH3; R4 - хлор; R5 - бензил).
Таким же образом, как описано в (а), получают вышеуказанное соединение из 5-хлор-3-нитро-монобензилкатехина и хирального ТКА-МИП эфира; К2СО3, БС ТБА, толуол, нагрев 24 ч с обратным
холодильником. Выход целевого энантиометрически чистого продукта 53%.
Пример VII
Конверсия монозащищенного хлорнитрокатехина в соединение с формулой VIIа (см. схему реакции VII в конце описания)
Реакция с энантиометрически чистыми соединениями глицидила (Z-нозил; R3 и R4 совместно образуют валентную связь, R5-бензоил).
А) Получение (S-7-хлор-2,3-дигидро-5-нитробензодиоксин-2-метанолбензойного эфира
2,0 г 5-хлор-3-нитро-нонобензоилкатехина (6,3 ммоля), 0,9 г К2СО3 (6,3 ммоля) и 0,2 г БС ТБА (0,63 ммоля) суспендируют в 75 мл толуола и нагревают с обратным холодильником и одновременным удалением воды (устройство Дина-Старка). После удаления 25 мл толуола оранжевую смесь охлаждают до 60оС и добавляют 1,7 г S(+) нозил-глицидилэфира. После нагрева 23 ч с обратным холодильником реакционную смесь разбавляют 10 мл насыщенного рассола и 10 мл воды и экстрагируют 50 мл этилацетата. Органическую фазу промывают дважды 10 мл насыщенного рассола и дважды 15 мл воды, подкисленной 2,5 мл 2NНC1. Органическую фазу упаривают, получая 2,52 г коричнево-оранжевого сиропа. Этот сироп очищают испарительной хроматографией в колонке 4,0 х 30 см из силикогеля со смесью 1:1 петройного эфира и диэтилэфира. Получают 0.75 г (выход 68%) твердого желтого продукта - (S)-7-хлор-2,3-дигидро-5-нитробензодиоксин-2-метанолбензойного эфира; точка плавления = 83-88oC; ее=96%; [α]D 25= +110,3 (EtoOAc, c=10 г/л, d=20 см).
1H ЯМР (δ, CDCl3) = 8,02 (dd), 7,60 (t), 4,62 (m), 4,27 (dd).
Пример VIII
Конверсия аминозащищенного хлорнитроаминофенола в соединение с формулой Х (см. схему реакции VIII в конце описания)
Суспензию 34,6 ммолей N-ацетил-2-гидрокси-3-нитро-5-хлоранилина 40,6 молей R2CO3, 80 мл NMP и 80 мл толуола нагревают 1 ч с обратным холодильником и удалением воды (устройство Дина-Старка). Толуол отгоняют дистилляцией, реакционную смесь охлаждают до 110оС. Добавляют 40,8 ммоля (S)-глидицидилтозилата. Реакционную смесь перемешивают 4,5 ч при 120оС, затем охлаждают, разбавляют водой и этилацетатом и корректируют рН до 4-6 разбавленной соляной кислотой. Водную фазу дважды экстрагируют этилацетатом. Смешанные органические фазы трижды промывают солевым раствором и сушат над MgSO4. После фильтрования и упарки растворителя при пониженном давлении получают 10,8 г темно-коричневого масла. После хроматографии с SiO2 (смесь 1:3 этилацетата и петролейного эфира получают 3-ацетоксиметил-6-хлор-8-нитро-2,3-дигидро-1,4-бензоксазин с выходом 42%, ее =86, [α]D 20 =-11,6 (с = 0,86, 96%-ный этанол), точка плавления 76-84оС. 1Н ЯМР (δ, CDCl3) = 2,12 (s, H), 3,79 (m, 1H), 4,07-4,35 (кластер, 4Н), 4,58 (широкая s, 1H, NH), 6,78 (d, 1H, J=2Hz), δ 7,21 (d, 1H, J=2Hz).
Пример IX
Дериватизация от исходного замещенного 1,3 бензодиоксола
а) Реакция с производными солькеталя (см. схему реакции IX в конце описания)
6-хлор-4-нитро-1,3-бензодиоксол подвергают дериватизации путем реакции хиральным солькеталем, т. е. хиральным 4-гидроксиметил-2.2-диметил-1,3-диоксоланом. Реакцию ведут в приемлемом органическом растворителе, например ДМФ, N-МП или толуоле, в присутствии основания, например, К2СО3, Li2CO3, NaH и т.п. Если толуол содержит растворителем, предпочтительно использовать катализатор фазового перехода, например БТА или БС ТБА.
Так, 4,6 г (3 г-экв).
Порошка К2СО3 суспендируют в 20 мл ДМФ в атмосфере азота при комнатной температуре. К суспензии добавляют 1,46 мл (1г-экв) S-солькеталя, а затем раствор 2,2 г 6-хлор-4-нитро-1,3-бензодиоксола в 20 мл ДМФ. Смесь нагревают 70 ч при 90оС. После охлаждения до комнатной температуры добавляют 50 мл толуола и 50 мл насыщенного раствора NaCl и перемешиват 5 мин. рН корректируют до 5-6 15 мл 2 н. раствора HCl. Разделяют фазы и дважды экстрагируют водную фазу 25 мл толуола. Концентрированные органические фазы трижды промывают 25 мл насыщенного раствора NaCl. Органическую фазу упаривают и получают 3,27 г (выход 92%) (S)-(4-хлор-2-гидрокси-6-нитро)-феноксиметил-2,2-диметил-1,3-диоксолана.
1Н ЯМР (δ, CDCl3): 1,48 (s, 3H), 1,54 (s, 3H), 3,91 (t, 1H, J=8Hz), 4,08 (dd, 1H, J=6Hz, J=11), 4,18 (t, 1H, J=8Hz), 4,37 (dd, 1H, J=11Hz), J=3Hz), 4,52 (m, 1H), 7,18 (d, 1H, J=2Hz), 7,36 (d, 1H, J=Hz), 8,74 (s, 1H).
После 5 ч реакции с N-МП в качестве растворителя выход такого же продукта составляет 79%.
Соответствующий (R)-солькетальэфир получают аналогичным образом из исходного (R)-солькеталя.
Подобным же образом можно получить энантиометрически чистые соединения 4-хлор-2-гидрокси-6-нитрофеноксиметил-2,2-диизопропил-1,3-диоксолана из 6-хлор-4-нитро-1,3-бенздиоксола и хирального 4-гидроксиметил-2,2-диизопропил-1,3-диоксолана.
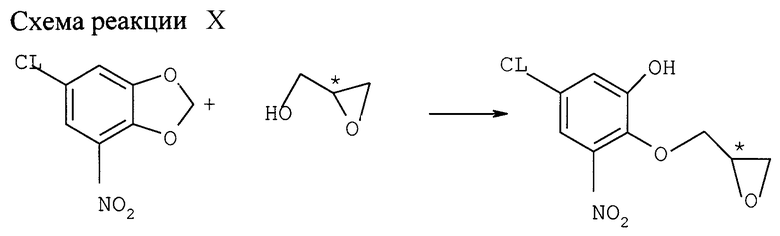
b) Реакция с глицилом (см. схему реакции X в конце описания)
Раствор 6-хлор-4-нитро-1,3-бензодиоксола (2,48 ммоля) в 10 мл ДМФ охлаждают на ледяной бане. Добавляют 5,5 ммоля 60%-ной дисперсии NaН в минеральном масле и сразу после этого раствор (S)-глицидола (3,13 ммоля, ее=84%) в 5 мл ДМФ. Через 1,5 ч перемешивания при 0оС добавляют лед с солевым раствором и 2 н. НСl, пока рН не станет ниже 5. Водную фазу трижды экстрагируют этилацетатом. Слитые органические фазы трижды промывают солевым раствором, сушат над MgSO4 и фильтруют. После фильтрации над Al2О3 и упарки растворителя при пониженном давлении получают 0,4 темно-коричневого масла. Выход (S)-хлор-2,3-дигидро-5-нитробензодиоксин-2-метанола составляет 35% после хроматографии через SiO2, ее=74%.
Пример Х
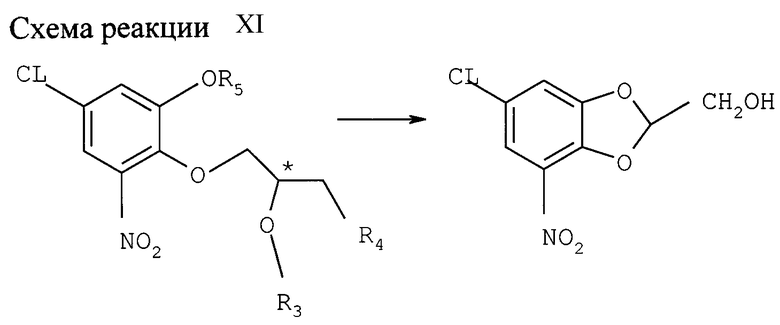
Снятие защиты и замыкание цикла; образование замещенного бензодиоксин-2-метанола и связанных с ним соединений (см. схему реакции XI в конце описания)
(а) С продукта, полученного в примере VI (а), снимают защиту и одновременно замыкают цикл эпоксидом с помощью минеральной кислоты. Хлор-нитро-монобензил-оксифенил-(R )-глицидиловый эфир легко конвертируется в соответствующий монохлоргидрид катехина нагревом 48 ч с обратным холодильником в смеси 1,1 пропанола с водой под воздействием 30 г-экв НСl; выход 87%, ее= 98%; после нагрева с обратным холодильником в течение 3 ч защита уже снимается на 83%. Соответствующий (S)-энантиомер получают после 9 ч нагрева с обратным холодильником при выходе 83% (ее=86%). Растворителем может также служить уксусная кислота. После этого снятие защиты и замыкание цикла эпоксидом протекает гладко уже при 35оС в течение 1 ч.
Вышеописанный хлоргидрин замыкает цикл в основной среде, давая значительный выход целевого хирального 7-хлор-2,3-дигидро-5-нитробензодиоксин-2-метанола. Замыкание цикла легко протекает при комнатной температуре в смеси 1: 1 этанола с водой под воздействием около 2 г-экв NaOH или КОН; время реакции около 18 ч.
(b) Снятие защиты и замыкание цикла солькетальэфира, полученного по примеру VI (b).
Снятие защиты осуществляется в соответствующем растворителе или смеси растворителей, например, в уксусной кислоте, под воздействием НBr или NaCl, предпочтительно при слегка повышенной температуре. Так, (R)-солькетальэфир монобензил-хлор-нитрокатехина дебензилируют с одновременным отщеплением изопропиловой группы из селькетальной части до соответствующего бромгидринацетата обработкой 8 г -экв 45% -ной НBr в уксусной кислоте при 35оС; выход составляет 88% через 0,25 ч. С соответствующего (S) - солькетальэфира защита снимается за 1,5 ч (выход 78%).
Последующая реакция замыкания цикла полученного таким образом соединения проводится в щелочной среде. Так, целевой (R) - 7-хлор-2,3-дигидро-5-нитробензодиоксин -2-метанол получают замыканием цикла в этаноле под воздействием 17 г-экв 2 н. NaOH в течение 1 ч при комнатной температуре, ее= 96%. Выход, включая снятие защиты, 60%.
Соответствующий (S)-стереоизомер получают с выходом 90% (ее=92%) реакцией с 7 г-экв 2 н. NaOH в этаноле при 23оС в течение 2 ч.
Солькетальэфиры, полученные в примере IХ, последовательно подвергают отщеплению солькеталя до соответствующего галоацетата, предпочтительно обработкой НСI или НBr в уксусной кислоте, и реакции замыкания цикла, как описано выше. Общий выход до 80%. Селективность: ее=89%.
Таким же образом замыкают цикл у диалкилсолькетальэфиров из примера IХ, получая целевой 7-хлор-2,3-дигидро-5-нитробензодиоксин-2-метанол-энантиомер с общим выходом около 55%, ее=98%.
c) ТКА-МИП эфир, полученный в примере VI (с),подвергают тем же реакциям снятия защиты и замыкания цикла: 45%-ная НBr в АсОН и 2 н. NaOH в этаноле последовательно. Получают хиральный замещенный бензодиоксан-метанол с общим выходом 90%, ее > 85%.






Изобретение относится к способу стереоселективного получения энантиомера гетеробициклического спирта. Более конкретно изобретение касается способа получения по существу чистого энантиомера общей формулы I, где Х - О, S, NH или N-(С1-С4)алкил; Y1 и Y2 каждый независимо друг от друга водород или заместители, выбранные из группы, включающей галоген, (С1-С4)алкил, (С1-С4)алкокси- (С1-С4)галоалкил-, формил-, нитро- и цианогруппы; атом С* имеет конфигурацию R либо S, реакцией соединения общей формулы II, где X, Y1 и Y2 имеют те же значения, что определены выше; R1 - водород или приемлемая защитная группа; R2 - водород; или R1 и R2 вместе образуют по выбору моно- или ди-(С1-С3)алкилзамещенный метиленовый мостик, с соединением общей формулы III, где Z - гидроксильная или приемлемая отщепляемая группа; R3 - гидроксилзащитная группа; R4 - атом галогена; или где R3 и R4 вместе образуют валентную связь или би-радикал формулы -C(R11)2-O-, где R11 - прямая или разветвленная (С1-С4)алкильная группа; атом С* имеет конфигурацию R либо S, в результате которой образуется соединение общей формулы IV, где X, Y1, Y2, R3 и R4 определены выше; R1 - водород или защитная группа; с последующим снятием защиты/реакции замыкания цикла полученного таким образом соединения; и необязательным снятием защиты с гидроксильной группы продукта с замкнутым циклом. Изобретение также относится к энантиометрически чистым промежуточным соединениям, способам их получения, получению исходного соединения. Технический результат - создание экономичного способа получения энантиомерного спирта, являющегося промежуточным соединением в синтезе биологически активных соединений, например флезиноксана. 7 с. и 5 з.п.ф-лы, 1 табл.





где Х - О, S, NH или N-(С1-С4)алкил;
Y1 и Y2 каждый независимо друг от друга представляет водород или заместители, выбранные из группы, включающей галоген, (С1-С4)алкил, (С1-С4)алкокси, (С1-С4)-галоалкил, формил, нитро и цианогруппы;
атом С* имеет конфигурацию R либо S,
отличающийся тем, что соединение общей формулы II
где X, Y1 и Y2 имеют те же значения, что определены выше;
R1 - водород или приемлемая защитная группа;
R2 - водород;
или R1 и R2 вместе образуют необязательно моно- или ди-(С1-С3)алкилзамещенный метиленовый мостик,
подвергают последовательно следующим стадиям:
(I) реакции с соединением общей формулы III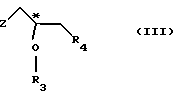
где Z - гидроксильная или приемлемая отщепляемая группа;
R3 - гидроксилзащитная группа,
R4 - атом галогена;
или R3 и R4 вместе образуют валентную связь или бирадикал формулы -С(R11)2-O-, где R11 - прямая или разветвленная (С1-С4)алкильная группа;
атом С* имеет конфигурацию R либо S, в результате которой образуется соединение общей формулы IV
где X, Y1, Y2, R3 и R4 определены выше;
R1 - водород или защитная группа;
(II) снятию защиты и реакции замыкания цикла полученного таким образом соединения;
(III) необязательному снятию защиты с гидроксильной группы продукта с замкнутым циклом.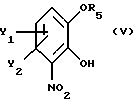
где Y1 и Y2 имеют те же самые значения, что определены в п. 1;
R5 - подходящая гидроксилзащитная группа;
с соединением общей формулы VI
где R3 и R4 имеют те же самые значения, которые определены в п. 1;
Z' - галоген или сульфонатная отщепляемая группа, предпочтительно тозилокси-, нозилокси- или мезилоксигруппа,
после которой полученное промежуточное соединение общей формулы VII
где Y1, Y2, R3, R4 и R5 имеют вышеуказанные значения,
подвергают последовательно реакционным стадиям II и III, как описано в п. 1.
где Y1 и Y2 имеют те же самые значения, что определены в п. 1;
R5' - гидроксилзащитная группа, выбранная из группы, состоящей из (С1-С8)алкилкарбонила и арилкарбонила, где арильная группа необязательно замещена одним или несколькими заместителями из группы, включающей (С1-С4)алкоксигруппу и галоген,
с соединением общей формулы VI
где R3 и R4 имеют те же самые значения, которые определены в п. 1;
Z' - галоген или сульфонатная отщепляемая группа, предпочтительно выбранная из тозилокси-, нозилокси- и мезилоксигруппы;
после чего полученное промежуточное соединение общей формулы VIIa
где Y1, Y2 и R5' имеют вышеуказанные значения,
подвергают удалению защищающей гидроксил группы из продукта с замкнутым циклом.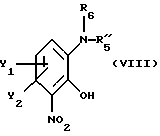
где Y1 и Y2 имеют значения, приведенные в п. 1;
R5'' - подходящая амино-защитная группа;
R6 - водород или (С1-С4)алкил;
с соединением общей формулы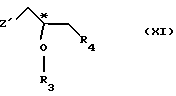
где R3 и R4 имеют те же самые значения, что указаны в п. 1;
Z' - галоген или сульфонатная отщепляемая группа, предпочтительно выбранная из тозилокси-, нозилокси- и мезилоксигруппы,
после чего полученное промежуточное соединение общей формулы Х
где Y1, Y2, R5'' и R6 имеют вышеуказанные значения,
подвергают удалению защищающей гидроксил группы из продукта с замкнутым циклом.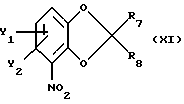
где Y1 и Y2 имеют те же самые значения, что определены в п. 1;
R7 и R8 каждый независимо друг от друга - водород или метил,
с хиральным строительным блоком формулы XIIa или XIIb
или
где R11 - прямая или разветвленная (С1-С4)алкильная группа;
атом С* имеет R или S-конфигурацию,
с получением промежуточного соединения общей формулы XIIIa или XIIIb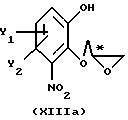
или
где R11, Y1 и Y2 имеют вышеуказанные значения,
которое затем подвергают реакционной стадии II, как определено в п. 1.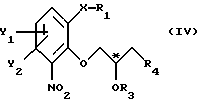
где Y1 - галоген;
Y2 - водород;
R3 - гидроксилзащитная группа;
R4 - галоген;
или R3 и R4 образуют валентную связь;
R1 имеет значения, определенные в п. 1,
Х - О или NH;
атом С* имеет R или S-конфигурацию.
где X, Y1, Y2, R1, R3 и R4 имеют значения, указанные в п. 1,
отличающийся тем, что соединение общей формулы II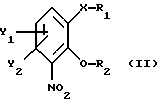
где X, Y1, Y2, R1 и R2 имеют значения, указанные в п. 1,
подвергают реакции с соединением общей формулы III
где символы R3, R4 и Z также имеют значения, указанные в п. 1.
где символы Y1, Y2 и R5' имеют значения, указанные в п. 3,
подвергают реакции с соединением общей формулы VI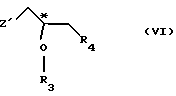
где символы Z', R3 и R4 имеют значения, указанные в п. 2.
где Y1 и Y2 имеют значения, определенные в п. 1;
R9 - необязательно замещенная бензоильная группа, (C1-C4)алкилкарбонильная группа или три(С1-С4)алкилсилильная группа,
путем реакции замещенного катехина общей формулы XV
где Y1 и Y2 имеют значения, определенные в п. 1;
р и q = 0 или 1,
с соединением общей формулы
(R9')2O или
R9'Hal или
(R14)3SiHal
где R9' - необязательно замещенная бензоильная группа или (C1-C6)алкилкарбонильная группа;
R14 - (С1-С4)алкил;
Hal - галоген,
в присутствии органического основания, предпочтительно третичного амина, или смеси органических оснований в каталитически достаточном количестве, отличающийся тем, что реакцию проводят в неполярном органическом растворителе или без растворителя с получением соединения общей формулы XVIa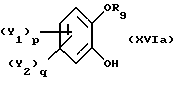
где Y1, Y2 и R9 имеют вышеуказанные значения,
которое последовательно подвергают реакции ароматического замещения с целью ввести заместители Y1 и Y2 в ароматическое ядро, при необходимости, и нитрованию.
где Y1 - галоген;
Y2 - водород;
R7 и R8 каждый независимо друг от друга водород или метил.
где Y1, Y2 имеют значения, указанные в п. 1;
R7 и R8 каждый независимо друг от друга водород или метил,
отличающийся тем, что соединение катехина общей формулы ХV по п. 10 подвергают следующим реакциям: (I) селективной защите одной из свободных гидроксильных групп; (II) необязательному введению одного или обоих заместителей Y1 и Y2; (III) селективному нитрованию орто-положения по отношению к незамещенной гидроксильной группе; (IV) снятию защиты с защищенной гидроксильной группы; (V) образованию бензодиоксольной части путем реакции с соединением общей формулы XVII
где R7 и R8 имеют значения, определенные в п. 11;
R9 и R10 каждый по отдельности - хлор, бром или (C1-С4)алкоксигруппа или они совместно образуют атом кислорода.
| СИДЕНЬЯ ДЛЯ УЧЕБНЫХ МАСТЕРСКИХ И ДРУГИХ ПОМЕЩЕНИЙ | 0 |
|
SU182302A1 |
| СПОСОБ ТЕРМИЧЕСКОЙ ОБРАБОТКИ ЖАРОПРОЧНЫХ ДИСПЕРСИОННОТВЕРДЕЮЩИХ СПЛАВОВ | 0 |
|
SU221725A1 |
| Дозатор жидкости | 1974 |
|
SU498770A1 |
| 2-/2-Цианоэтил/-4,5-бензо-1,3-диоксолан, обладающий акарицидной активностью против иксодовых клещей рода Шаломма | 1989 |
|
SU1703648A1 |
| КОНТАКТНЫЙ АППАРАТ ВИХРЕВОГО ТИПА | 0 |
|
SU203622A1 |

