Настоящее изобретение описывает производные антибиотика A 40926 формулы (I):
где
R1 представляет собой водород или защитную группу аминогруппы;
R2 представляет собой (C9-C12) алкил;
M представляет собой водород, α-D- маннопиранозил или 6-O-ацетил -α-D- маннопиранозил;
Y - представляет собой карбокси, (C1-C4) алкоксикарбонил, аминокарбонил, (C1-C4) алкиламинокарбонил, ди(C1-C4) алкиламинокарбонил, где алкильный фрагмент может иметь заместитель, выбираемый из гидрокси, амино, (C1-C4) алкиламино и ди (C1-C4) алкиламино или гидроксиметила;
X - представляет собой гидрокси или аминный остаток формулы
-NR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W,
где
R3 представляет собой водород или (C1-C4) алкил;
alk1, alk2 и alk3 каждый независимо представляет собой линейный или разветвленный алкилен с 2-10 атомами углерода;
p и q являются целыми числами, которые независимо друг от друга могут иметь значения 0 или 1;
R4 и R5 независимо представляют собой водород, (C1-C4) алкил, или R3 и R4 вместе представляют собой (C2-C4) алкиленовый фрагмент, соединяющий два атома азота, при условии, что p равно 1; или R4 и R5 вместе представляют собой (C2-C4) алкиленовый фрагмент, соединяющий два атома азота, при условии, что и p и q равны 1; W представляет собой водород, (C1-C4) алкил, амино, (C1-C4) - алкиламино, ди (C1-C4) алкиламино, аминогруппу, замещенным одним или двумя амино-(C2-C4) алкильными заместителями или одним или двумя (C1-C4) алкиламино-(C2-C4) алкильными заместителями, или же одним или двумя ди (C1-C4) алкиламино-(C2-C4) алкильными заместителями, или, если и p, и q имеют значение 0, то вместе с группой -NR3-alk1 - этот радикал может также представлять собой пиперазино или 4-метилпиперазино,
при условии, что, когда X представляет собой гидрокси, Y представляет собой гидроксиметил,
Z представляет собой водород или группу формулы
где
A⊖ представляет собой анион минеральной или органической кислоты, или, когда присутствует карбоксикислотная группа в оставшейся части антибиотика, этот радикал может также представлять собой внутренний анион, получаемый из этой карбоксикислотной группы;
а также фармацевтически-приемлемые соли этих производных, являющиеся продуктами присоединения.
Цифры в скобках в вышеприведенной формуле (I) и в последующих формулах означают обычную нумерацию соответствующих атомов углерода в молекулярной структуре антибиотика A-40926 и его производных.
Антибиотик A 40926 представляет собой гликопептидный антибиотический комплекс, выделенный из культуры Actinomadura под названием Actinomadura sp. АТСС 39727 в культуральной среде, содержащей ассимилируемые источники углерода, азота и неорганических солей (см. EP-177882). Согласно процедуре, описанной в вышеуказанном патенте, в процессе выделения антибиотического комплекса, основные факторы которого получили названия фактор A, фактор B, фактор B0, фактор B1, фактор PA и фактор PB, культуральные жидкости, после фильтрации или после предварительной очистки подвергали афинной хроматографии на неподвижном D-аланил-D-аланине.
Идентифицированные к настоящему времени факторы антибиотика A 40926 можно описать формулой (II) ниже, где R'1 представляет собой водород, X' - гидрокси, Y' - карбокси, R'2 представляет собой (C9-C12) алкильную группу, а M' представляет собой α-D-маннопиранозил или 6-O-ацетил -α-D-/ маннопиранозильную группу.

Конкретно, фактор A антибиотика A 40926 является соединением вышеуказанной формулы (II), где R'1 - водород. X' - гидрокси, Y' - карбокси, R'2 представляет собой n-децил, а M' представляет собой α-D- маннопиранозил. Согласно самым последним исследованиям вещество, идентифицированное как фактор Bo антибиотика A 40926 в вышеуказанном патенте EP-177882 в действительности состоит из двух близких компонентов. Фактор Bo антибиотика A 40926 в действительности является основным компонентом фактора B и соответствует соединению вышеуказанной формулы (II), где R'1 - водород, X' - гидрокси, Y' - карбокси, R'2 представляет собой 9-метилдецил, а M' представляет собой α-D-маннопиранозил.
Второстепенный компонент фактора B называется фактором B1 и отличается от фактора B0 только тем, что R'2 представляет собой n-ундецил (см. Е.Рива и др., "Chromatographia", том 24, с. 295, 1987).
Фактор PA и фактор PB антибиотика A 40926 отличаются от соответствующих фактора A и B тем, что маннозная группа у них замещена на 6-O-ацетил-α-D-маннопиранозную группу.
Факторы PA и PB антибиотика A 40926 по меньшей мере при определенных условиях ферментации являются основными антибиотическими продуктами микроорганизма, продуцирующего A 40926.
Факторы A и B антибиотика A 40926 в основном являются продуктами трансформации факторов PA и PB антибиотика A 40926 соответственно и часто уже присутствуют в культуральных средах.
Все сахарные фрагменты соединены с ядром антибиотика A 40926 посредством O-гликозидных связей.
Обнаружено, что фактор PA антибиотика A 40926 можно трансформировать в фактор A антибиотика A 40926, а фактор PB антибиотика A 40926 можно трансформировать в фактор B антибиотика A 40926 в щелочной среде, где происходит удаление ацетильной группы маннозного фрагмента без смещения ацильной группы на аминоглюкуронильном фрагменте.
В результате, если культуральную среду или антибиотик A 40926, содержащий ее экстракт или концентрат, оставить на некоторое время при щелочных условиях (напр., в водном растворе нуклеофильного основания при pH > 9 в течение ночи), то можно получить антибиотик A 40926, насыщенный фактором A и фактором B антибиотика A 40926.
Фактор A антибиотика A 40926 можно получить из комплекса A 40926 путем хроматографического разделения с помощью способа, описанного в EP-177882. Чистый фактор B0, который в вышеописанных условиях (согласно патенту EP-177882) составляет около 90% фактора A, можно получить путем дальнейшей очистки фактора B, например, несколько раз проведя хроматографию с обращенной фазой.
Новейшие исследования (Л. Зерилли и др., Rapid Communications in Mass Spectrometry, том 6, с. 109, 1992) показали, что в антибиотическом комплексе A 40926 присутствуют также некоторые второстепенные факторы, которые идентифицируют с акронимами A1, RS-1, RS-2 и RS-3 соответственно. Эти второстепенные факторы разделили с помощью ВЭЖХ, а структуры этих факторов определили с помощью газовой хроматографии / масс-спектрометрического анализа метанолизатов комплекса A-40926. Все вышеуказанные второстепенные факторы имели структуры, соответствующие структурам основных факторов, A, B0 и B1, за исключением того, что остатки жирных кислот были связаны с аминоглюкуронильным фрагментом. В частности, со ссылкой на формулу (II) радикалы R'1, X' и Y' имеют вышеуказанные значения, а R'2 представляет собой 8-метилнонил в факторе A1, 7-метилоктил в факторе RS-1, n-нонил в факторе RS-2 и n-додецил в факторе RS-3.
Несмотря на то, что в препаратах антибиотического комплекса A 40926, полученных с использованием условий, описанных в EP 177882, сильно преобладают факторы, где R'2 представляет собой (C10-C11) алкил, можно изменить условия ферментации с тем, чтобы повысить содержание второстепенных компонентов, в которых R'2 представляет собой C9 или C12 алкил.
В процессе обычной очистки антибиотического комплекса A 40926 факторы PA и PB обычно преобразуются в факторы A и B.
Кроме того, было обнаружено, что можно трансформировать антибиотический комплекс A 40926, отдельные его факторы или смесь этих факторов в любой пропорции в соответствующий N-ациламиноглюкуронилагликоновый комплекс AB, N-ациламиноглюкуронилагликоновый фактор A, N-ациламиноглюкуронилагликоновый фактор B и маннозилагликон антибиотика A 40926 путем контролируемого кислотного гидролиза одного из сахарных фрагментов исходного материала (см. EP-A-240609 и EP-A-228015).
Предпочитаемые условия гидролиза для получения N-ациламиноглюкуронилагликонов предполагают использование смеси диметилсульфоксид/концентрированная соляная кислота в соответствии от 8:2 до 9,5:0,5 при температуре от 40 до 80oC.
N-ациламиноглюкуронилагликоны антибиотика A 40926 выражаются вышеуказанной формулой (II), где R'1 и M' представляют собой атомы водорода, X' - гидрокси, Y' - карбокси, а R'2 представляет собой (C9-C12) алкил.
Полное отщепление всех сахарных фрагментов антибиотика A 40926 дает агликон. Этот процесс гидролиза описан в EP-A-240609.
Антибиотический комплекс A 40926, его факторы, соответствующие N-ациламиноглюкуронилагликоны, маннозилагликоны, агликон и их смеси в любой пропорции проявляют основную активность против грамположительных бактерий и Neisseriae.
В заявке на международный патент N PCT/EP92/00374 заявлен приоритет, согласно EP сер. N 91104857, на эфирные производные антибиотика A 40926 (эстерифицированные в положении 6B, т.е. карбоксильная группа присутствует на N-ацетиламиноглюкуронильном фрагменте) и описан N-ациламиноглюкуронилагликон этого антибиотика, напр. соединения формулы (II), где X' является ОН, Y' является (C1-C4) алкоксикарбонил, а R'1, R'2 и M' имеют те же значения, что и радикалы R1, R2 и M, описанные выше.
Эти эфирные производные получают путем взаимодействия аминного субстрата A 40926, имеющего защитную группу в N15 (в настоящем описании термин "N15" относится к атому азота в аминогруппе, присоединенному к атому углерода молекулы A 40926, который обычно обозначается номером 15) или не имеющего такой группы в N15, или деманнозильного производного этого вещества (т.е. N-ациламиноглюкуронилагликона) с алканолом в кислой среде, или производного A 40926 с защитной группой в N15, или его деманнозильного аналога, с алкилгалогенидом (предпочтительно, бромидом, хлоридом или иодидом), возможно, в присутствии акцептора галогеноводородной кислоты, в частности, при избытке выбранного алканола в присутствии концентрированной минеральной кислоты при температуре от 0oC до комнатной.
Эти эфирные производные антибиотика A 40926, полученные вышеупомянутым способом, используются в качестве исходных материалов для получения производных формулы (1) антибиотика A 40926.
Контролируемая эстерификация, используемая для получения эфирных производных антибиотика A 40926 и эфирных производных деманнозильного антибиотика A 40926, которые являются исходными материалами для получения соединений по изобретению, включает реакции эстерификации, в ходе которых субстрат A 40926 соединяют с избытком выбранного алканола в присутствии концентрированной минеральной кислоты при температуре от 0oC до комнатной в течение периода времени, длительность которого зависит от стерической сложности группы, которую предстоит ввести.
В некоторых случаях удобно ввести защитную группу в аминогруппу в положении 15 предшественника A 40926 с тем, чтобы по возможности избежать нежелательных побочных реакций. Это можно осуществить известными способами, например способами, описанными в таких справочниках, как Т.У. Грин "Protective Groups in Organic Synthesis", изд-во "Джо Уили энд Санз", Нью-Йорк, 1981 и М. Макоми "Protectig Groups in Organic Chemistry", изд-во "Пленум Пресс", Нью-Йорк, 1973. Эти защитные группы должны проявлять стабильность в условиях реакционных процессов, они не должны мешать протеканию основной реакции и должны обладать такими свойствами, которые позволят легко удалить эти группы в конце основной реакции.
В числе подходящих аминозащитных групп такие группы, как трет-бутоксикарбонил (±BOC), карбобензилокси (CBz) и арилалкил. Бензилирование с возможно замещенными бензилгалогенидами в присутствии основания протекает гладко с количественным выходом и приводит исключительно к образованию соответствующего N15-бензильного производного без сопутствующего образования бензильного эфира карбоксильных групп.
Селективную защиту аминогруппы в положении 15 можно осуществить посредством взаимодействия с бензилбромидом в присутствии акцептора галоидоводорода (т.е. третичного амина) без сопутствующей эстерификации двух карбоксильных групп.
Удаление защитных групп в N15 производят одним из известных способов удаления аминозащитных групп, но предварительно необходимо оценить химическую активность других групп, присутствующих в молекуле.
Исходное эфирное соединение формулы (II), где M' представляет собой α-D-маннопиранозил или 6-O-ацетил -α-D- маннопиранозил, а Y' представляет собой (C1-C4) алкоксикарбонил, можно преобразовать в соответствующее соединение, в котором M' - водород, посредством селективного кислотного гидролиза. Как описано в EP-A-240609, для проведения гидролиза для получения деманнозильных производных антибиотика A 40926 (напр., N-ациламиноглюкуронилагликона) предпочтительно использовать смесь диметилсульфоксида с концентрированной соляной кислотой в соотношении от 8:2 (о/о) до 9,5:0,5 (о/о) при температуре от 40 до 80oC.
Соответственно деманнозильные производные эфиров A 40926 можно получить в смеси с соответствующим агликоном и разделить посредством препаративной ВЭЖХ.
Условия проведения гидролиза можно изменять с тем, чтобы получать различные соотношения продуктов реакции. Например, если в качестве исходного материала использовать A 40926, эстерифицированный в положении 6B, увеличить соотношение растворитель/соляная кислота до 78:1, поддерживать температуру реакции ниже 60oC и увеличить время протекания реакции до около 7 дней, то отношение искомых деманнозильных производных A 40926, эстерифицированного в положении 6B к нежелательному агликону A 40926 будет приблизительно составлять 1,4:1,0.
Ход реакции контролируют с применением ВЭЖХ с помощью одного из известных способов. На основе результатов этих анализов специалист может дать оценку ходу реакции и решить, когда следует остановить реакцию и приступить к обработке реакционной смеси известными способами, включающими, например, экстрагирование растворителями, осаждение нерастворившихся компонентов, последующее разделение и очистку с применением хроматографии.
Эфирные производные, используемые в качестве исходных материалов для получения соединений формулы (I), могут представлять собой индивидуальные соединения, соответствующие каждому из нескольких факторов предшественника антибиотического комплекса A 40926, или смеси из двух или более компонентов в любой пропорции, соответствующих различным факторам предшественника A 40926. Указанные смеси эфирных производных можно получить при использовании комплекса A 40926, или смеси факторов предшественника комплекса A 40926 в процессе получения эфира в 6B, или с помощью особых условий выделения/очистки полученного эфирного продукта (изменение этих условий может изменять начальные пропорции факторов, характерные для предшественника комплекса A 40926), или же посредством смешивания в нужных пропорциях чистых эфирных продуктов, выделенных хроматографией с обращенной фазой или полученных при использовании чистых факторов A 40926 в качестве предшественников.
В настоящем описании и формуле изобретения термин "алкил", взятый отдельно или в комбинации с другими заместителями, включает углеводородные группы с прямой и разветвленной цепью; более конкретно, термин "(C1-C4) алкил" обозначает алифатический углеводород с прямой или разветвленной цепью, содержащей от 1 до 4 атомов углерода, такой как метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 1,1-диметилэтил, и 2-метилпропил.
В настоящем тексте термины "alk1", "alk2", "alk3" обозначают алкилен с прямой или разветвленной цепью, содержащий 2-10 атомов углерода, такой как, например:
-CH2-CH2-,
-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-,
-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-,



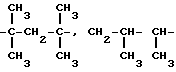
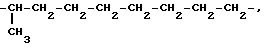
Термины "(C2-C4) алкильные фрагменты" и "(C2-C4 алкиленовые фрагменты" в настоящем тексте обозначают алифатические фрагменты с прямой или разветвленной цепью, содержащей 2-4 атома углерода. Примеры таких фрагментов содержатся в вышеприведенном списке.
Выражение "(C1-C4) алкоксикарбонил" включает алкоксикарбонильные группы с прямой и разветвленной цепью, такие как, например, метоксикарбонил, этоксикарбонил, пропилоксикарбонил, изопропилоксикарбонил, бутоксикарбонил, изобутоксикарбонил и трет-бутоксикарбонил.
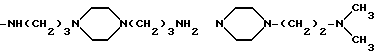
Ниже представлены некоторые примеры аминных остатков
-NR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W
согласно вышеприведенному определению:
-NH-(CH2)2-NH2
-NH-(CH2)3-NH2
-NH-(CH2)4-NH2
-NH-(CH2)5-NH2
-NH-(CH2)2-N(CH3)2
-NH-(CH2)3-N(CH3)2
-NH-(CH2)2-N(C2H5)2
-NH-(CH2)7-N(CH3)2
-NH-(CH2)2-N(C4H9)2
-NH-(CH2)3-N(C2H5)2
-NH-(CH2)3-N(C4H9)2
-N(CH3)-(CH2)2-NH2
-N(CH3)-(CH2)3-NH2
-N(CH3)-(CH2)2-N(CH3)2
-N(CH3)-(CH2)3-N(CH3)2
-NH-(CH2)n-CH3
n = 0, 1, 2, 3, 4 or 5
n = 0, 1, 2, 3, 4 or 5
m = 0, 1, 2, or 3
-NH-(CH2)2-NH-(CH2)2-NH2
-NH-(CH2)2-NH-(CH2)3-NH2
-NH-(CH2)2-NH-(CH2)4-NH2
-NH-(CH2)4-NH-(CH2)2-NH2
-NH-(CH2)3-NH-(CH2)4-NH2
-NH-(CH2)2-NH-(CH2)3-NH- (CH2)2-NH2
-NH-(CH2)2-NH-(CH2)4-NH- (CH2)2-NH2
-NH-(CH2)3-NH-(CH2)3-NH- (CH2)3-NH2
-NH-(CH2)3-NH-(CH2)4-NH- (CH2)3-NH2
-NH-(CH2)2-NH-(CH2)3-NH- (CH2)4-NH2
-NH-(CH2)4-NH-(CH2)3-NH- (CH2)4-NH2
-NH-(CH2)3-NH-(CH2)9-NH- (CH2)3-NH2
-NH-(CH2)3-NH-(CH2)10-NH- (CH2)3-NH2
-NH-(CH2)2-[NH(CH2)2]2-NH2
-NH-(CH2)3-[NH(CH2)3]3-NH2
-NH-(CH2)2-N[(CH2)2NH2]2
-NH-(CH2)3-N[(CH2)2NH2]2
-NH-(CH2)2-N[(CH2)3NH2]2
-NH-(CH2)2-N[(CH2)4NH2]2
-NH-(CH2)3-N[(CH2)3NH2]2
-NH-(CH2)4-N[(CH2)2NH2]2
-NH-(CH2)4-N[(CH2)3NH2]2
-NH-(CH2)2- N[(CH2)2N(CH3)2]2
-NH-(CH2)2- N[(CH2)3N(CH3)2]2
-NH-(CH2)3- N[(CH2)2N(CH3)2]2
-NH-(CH2)3- N[(CH2)3N(CH3)2]2
-NH-(CH2)2- N[(CH2)2N(C2H5)2]2
-N(CH3)(CH2)2- N[(CH2)2NH2]2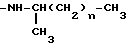
n = 0, 1, 2, or 3
-NH-(CH2)n-NHCH3
n = 2, 3 or 4
-NH-(CH2)n-NHiC3H7
n = 2, 3 or 4
n = 1, 2 or 3
n = 1, 2 or 3
-NH(CH3)-(CH2)n-NHCH3
n = 2, 3 or 4
-N(CH2)n-NHC2H5
n = 2, 3, or 4
and the like.
Когда R3 и R4 (или R4 и R5) вместе представляют собой (C2-C4) алкиленовый фрагмент, соединяющий два тома азота, то насыщенный гетероциклический фрагмент, образованный в комбинации с частями alk1 (или alk2) и двумя соседними атомами азота, предпочтительно, представляет собой пиперазиновое кольцо.
Например, когда R3 и R4 (или R4 и R5) вместе представляют собой (C2-C4) алкиленовый фрагмент, соединяющий два атома азота, или когда оба p и q равны O, W вместе с фрагментом -NR3-alk1- представляет собой пиперазино или 4-метилпиперазино, то аминный остаток формулы
-NR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W
идентифицирует следующие группы:
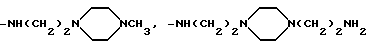





Настоящее изобретение охватывает отдельные соединения формулы (I), получаемые из отдельных факторов, предшественника антибиотического комплекса A 40926, а также смеси соединений формулы (I), полученных из самого комплекса A 40926 или из смеси двух или более факторов в любой пропорции. Соответственно варианты пропорциональных соотношений компонентов смесей соединений формулы (I), соответствующих факторов комплекса A 40926, получают в результате использования различных условий ферментации, восстановления, выделения и очистки предшественника антибиотического комплекса A 40926, или в результате смешивания отдельных выделенных факторов исходных эфиров формулы (II) в нужных пропорциях до их преобразования в соединения формулы (I), или же в результате смешивания чистых индивидуальных факторов соединений по изобретению формулы (I) в нужных пропорциях.
Предпочитаемыми соединениями по изобретению являются соединения формулы (I), где
R1 представляет собой водород или защитную группу аминного фрагмента;
R2 представляет собой (C9-C12) алкил;
M представляет собой водород, α-D-маннопиранозил или 6-O-ацетил α-D-маннопиранозил;
Y представляет собой карбокси, (C1-C4) алкоксикарбонил, аминокарбонил, (C1-C4) алкиламинокарбонил, ди (C1-C4) алкиламинокарбонил, где алкильный фрагмент может иметь заместитель, выбираемый из гидрокси, амино (C1-C4) алкиламино и ди (C1-C4) алкиламино или гидроксиметила;
X представляет собой гидрокси или аминный остаток формулы
-NR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W
где
R3, R4 и R5 представляет собой водород;
alk1, alk2 и alk3 каждый независимо представляет собой линейный или разветвленный алкилен с 2-4 атомами углерода;
p и q являются целыми числами, которые независимо друг от друга равны нулю или 1;
W представляет собой водород, (C1-C4)алкил, амино, (C1-C4)алкиламино, ди(C1-C4) алкиламино, аминогруппу, замещенную одним или двумя амино-(C2-C4)алкильными фрагментами или одним или двумя (C1-C4)алкиламино-(C2-C4)алкильными фрагментами, или, когда оба p и q равны 0, то вместе с фрагментом -NR3-alk1- этот радикал может представлять собой пиперазино или 4-метил-пиперазино,
при условии, что, когда X представляет собой гидрокси, Y представляет собой гидроксиметил;
Z представляет собой водород или группу формулы:
где
A⊖ представляет собой анион минеральной или органической кислоты или, когда присутствует карбоксикислотная группа в оставшейся части антибиотика, этот радикал может представлять собой внутренний анион, получаемый из указанной карбоксикислотной группы;
и фармацевтически приемлемые соли этих соединений.
Другая предпочитаемая группа соединений по изобретению включает те производные формулы (I), где R2 представляет собой (C10-C11)алкил, M представляет собой α-D- маннопиранозил, а R1, X, Y, и Z имеют вышеприведенные значения, и фармацевтически приемлемые соли этих соединений.
Следующая предпочитаемая группа соединений по изобретению охватывает те соединения формулы (1), где:
R1 представляет собой водород или защитную группу аминогруппы, предпочтительно водород;
R2 представляет собой 7-метилоктил, n-нонил, 8-метилнонил, n-децил, 9-метилдецил, n-ундецил, или n-додецил, предпочтительно, n-децил, 9-метилдецил, или n-ундецил, лучше всего 9-метилдецил; M представляет собой водород или α-D- маннопиранозил, предпочтительно α-D- маннопиранозил;
Y представляет собой карбокси, (C1-C4) алкоксикарбонил, аминокарбонил, (C1-C4) алкиламинокарбонил, ди (C1-C4) алкиламинокарбонил, где алкильный фрагмент может иметь заместитель, выбираемый из гидрокси, амино (C1-C4) алкиламино или ди (C1-C4) алкиламино или гидроксиметила, предпочтительно, карбокси, метоксикарбонил, аминокарбонил, метиламинокарбонил, диметиламинокарбонил, (диметиламино) этиламинокарбонил или гидроксиметил;
X является аминным остатком -NR3-alk1- (NH-alk2)p-(NH-alk3)q-W, где R3 является водородом;
alk1, alk2 и alk3 независимо представляет собой линейный алкилен с 2-4 атомами углерода;
p и q каждый независимо равны 0 или 1;
W представляет собой амино, (C1-C4) алкиламино, ди-(C1-C4)-алкиламино, аминогруппу, замещенную одним или двумя амино-(C2-C4)-алкильными фрагментами или, когда p и q равны 0, то вместе с фрагментом -NR3-alk1- этот радикал может представлять собой пиперазино или 4-метилпиперазино; лучше всего, чтобы X представлял собой аминный остаток, выбираемый из:
-NH-(CH2)3-N(CH3)2,
-NH-(CH2)3-[NH(CH2)3]2-NH2,
-NH-(CH2)3-N[(CH2)3NH2]2,
Z представляет собой водород;
и фармацевтически приемлемые соли этих соединений.
Соединения формулы (1), где Y представляет собой (C1-C4)-алкоксикарбонил, R1, R2, M и Z имеют вышеуказанные значения, а X представляет собой аминный остаток формулы:
-NR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W
где
R3, R4, R5, alk1, alk2, alk3, p, q и W имеют значения, приведенные в начале описания, получают посредством амидирования соответствующих производных формулы (II) выше, где R'1, R'2 и M' имеют те же значения, что и R1, R2 и M, X' представляет собой гидрокси, а Y' является (C1-C4) алкоксикарбонилом.
Эти исходные материалы формулы (II) получают вышеуказанным способом, а некоторые примеры этих соединений описаны в упомянутой заявке на международный патент N PCT/EP92/00374.
Процесс амидирования включает конденсацию указанных исходных материалов формулы (II) соответствующим амином формулы (III):
NHR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W (III),
где
R3, R4, R5, alk1, alk2, alk3, p и q и W имеют вышеуказанные значения, в присутствии конденсирующего агента, или через образование "активированного эфира" указанной исходной C63 карбоновой кислоты формулы (II) в инертном органическом растворителе.
Для реакции амидирования используют те инертные органические апротонные растворители, которые не оказывают неблагоприятного действия на ход реакции и по меньшей мере частично растворят исходные материалы.
Примеры таких инертных органических растворителей включают органические амиды, эфиры гликолей и полиолов, фосфорамиды и сульфоксиды. Предпочитаемыми органическим растворителями являются например, диметилформамид, диметоксиэтан, гексанметилфосфорамид, диметилсульфоксид и их смеси.
Конденсирующим агентом в процессе по изобретению является агент, подходящий для образования амидных связей в органических соединениях, и в частности, в синтезе пептидов.
В числе примеров конденсирующих агентов диизопропилкарбодиимид (DIC), дициклогексилкарбодиимид (DCC) в присутствии гидроксибензотриазола (HBT), бензотриазолилокси-трис-(диметиламино) фосфонийгексафторфосфат, бензотриазолилокси-трис-(пирролидино)фосфоний- гексафторфосфат и (C1-C4) алкил, фенил или гетероциклические фосфоразидаты, такие как дифенилфосфоразидат, диэтилфосфоразидат, ди-(4-нитрофенил)фосфоразидат, диморфолил-фосфоразидат и дифенилфосфорохлоридат. Предпочитаемыми конденсирующими агентами являются дифенилфосфоразидат, т. е. дифенилэфиразид фосфорной кислоты (DPPA), бензотриазолилокси-трис-(диметиламино)фосфоний-гексафторфосфат (BOP) и бензотриазолалокси-трис-(пирролидино)фосфоний-гексафторфосфат (Py BOP).
Из двух последних конденсирующих агентов Py BOP является наиболее предпочтительным, поскольку получаемый побочный продукт пирролидин обладает меньшей токсичностью, чем диметиламин.
В процессе амидирования по изобретению, аминный реактив обычно используется в мольном избытке, хотя в некоторых случаях реакцию можно проводить, получая хороший выход, и при использовании аминного реактива в эквимолярном соотношении или при слабом молярном избытке, в частности, если в качестве конденсирующих агентов используются ВОР или Py BOP.
В общем, если аминный реактив достаточно недорогой или легкодоступен, используют 2-х - 10-кратный избыток амина (III), хотя предпочтительным является 3-х - 4-х кратный избыток.
Для амидирования вышеуказанного исходного материала формулы (II) амином (III) в присутствии конденсирующего агента, необходимо, чтобы аминный реактив был способен образовывать соль с карбоксильной группой (X'-гидрокси) исходного материала. В том случае, когда аминный реактив недостаточно сильный для образования такой соли в реакционной среде, необходимо добавить солеобразующее основание (напр. , третичный алифатический или гетероциклический амин, такой как триэтиламин, N-метилпирролидин или N-метилпиперазин, который не может образовывать амидную связь с карбоксильной группой), солеобразующее основание добавляют в по меньшей мере эквимолярном количестве по отношению к исходному материалу.
Использование аминного реактива в низкомолярном избыточном количестве с добавлением солеобразующего основания наиболее целесообразно в тех случаях, когда аминный реактив очень дорого стоит или его трудно достать.
В числе примеров вышеуказанных солеобразующих оснований третичные органические алифатические или гетероциклические амины, такие как триметиламин, триэтиламин, N-метилпирролидин или пиколин и т.п.
Конденсирующий агент обычно используется в эквимолярном количестве или при слабом молярном избытке, например, от 1,1 до 1,7 раз, предпочтительно в 1,2 - 1,5 раз по отношению к исходному соединению A 40926. В частности, отмечено, что при использовании исходных материалов формулы (II), где Y' представляет собой (C1-C4) алкоксикарбонил, и Py BOP в качестве конденсирующего агента в большом избытке (напр., в 3-х кратном молярном избытке) и при большом избытке аминного реактива (напр., 6-ти - 10-ти кратном молярном избытке), конечные амидные продукты формулы (I), где Z представляет собой:
где
A⊖ имеет вышеуказанное значение,
получают почти количественные выходы.
Аминный реактив можно ввести в реакционную среду и в виде соответствующей кислой соли, например гидрохлорида. В этом случае аминный реактив добавляют по меньшей мере в двойной молярной пропорции, а сильное основание, способное высвобождать амин из соли, добавляют при 2-х - 4-х кратном молярном избытке. В этом же случае подходящим основанием является обычно третичный алифатический или гетероциклический амин, который не способен образовывать амидную связь с карбоксильной группой в отличии от других вышеперечисленных оснований. Фактически использование солей амина с последующим высвобождением амина на месте при помощи указанного основания является предпочтительным способом, особенно в тех случаях, когда соль является более стабильной, чем соответствующий свободный амин.
Температура реакции может быть различной в зависимости от типа исходных материалов и условий протекания реакции. В целом предпочтительно проводить реакцию при температурах от 0 до 30oC.
Время протекания реакции тоже может быть различным и зависит от типа конденсирующего агента и других параметров реакции. Обычно реакция конденсации завершается в течение периода времени, составляющего от одного часа до 24-48 часов.
В любом случае ход реакции контролируют с помощью тонкослойной хроматографии или, предпочтительно, ВЭЖХ с использованием любого известного способа.
На основе результатов хроматографических анализов специалист может оценить ход реакции и решить, когда следует остановить реакцию и приступить в обработке реакционной смеси по любому известному способу, например, такой способ может включать экстрагирование растворителями, осаждение путем добавки осадителей и т.п. с последующим разделением и очисткой, напр., с применением колоночной хроматографии.
Обычно при использовании конденсирующих агентов типа тех, которые перечислены выше, отпадает необходимость в защите аминогруппы в N15 исходного эфира формулы (II). Однако иногда желательно использовать исходные эфиры, имеющие соответствующую защитную группу, в частности в тех случаях, когда эти эфиры получены непосредственно на предыдущем этапе реакций, в ходе которого эфиры получают из предшественника антибиотика A 40926. Более того, могут быть и другие случаи, когда условия реакции амидирования требуют или по меньшей мере предполагают защиту аминогруппы в N15 исходного эфира формулы (II).
В указанных случаях введение защитной группы в положение N15 аминогруппы производят одним из известных способов, которые описаны в справочниках и упомянуты в настоящем описании в связи с защитой предшественника A 40926 для получения эфиров формулы (II), где Y' представляет собой (C1-C4) алкоксикарбонил.
N-защитные группы должны проявлять стабильность в условиях реакции, они не должны оказывать неблагоприятного действия на протекание реакции амидирования, они должны обладать такими свойствами, которые позволят легко удалить эти группы из реакционной среды в конце реакции, не вызывая изменений в новообразованной амидной связи и в общей структуре соединений, напр. сахарных фрагментах.
Примерами N-защитных групп, которые можно с успехом использовать в процессе по изобретению для защиты первичных аминогрупп в положении N15 исходных эфиров и при необходимости для защиты любых других аминогрупп, которые не должны вступать в реакцию амидирования, являются карбамат-образующие реактивы, характеризующиеся следующими оксикарбонильными группами: 1,1-диметилпропинилоксикарбонил, трет-бутилоксикарбонил, винилоксикарбонил, циннамилоксикарбонил, бензилоксикарбонил, p-нитробензилоксикарбонил, 3,4-диметокси-6-нитробензилоксикарбонил, 2,4-дихлорбензилоксикарбонил, 5-бензилоксазолилметилоксикарбонил, 9-антранилметилоксикарбонил, дифенилметилоксикарбонил, изоникотиноилоксикарбонил, дифенилметилоксикарбонил, S-бензилоксикарбонил и т.п.
Обычно эти защитные группы можно удалить после завершения реакции амидирования посредством обработки сильными органическими кислотами, такими как трифторуксусная кислота (ТГА), или разбавленными минеральными кислотами. Чтобы избежать опасности гидролиза сахарных фрагментов, присоединенных к ядру молекулы антибиотика, можно также производить удаление некоторых защитных групп в различных условиях, таких как каталитическое гидрирование, используя, например, палладий на углероде в качестве катализатора. Иначе можно удалить аминозащитные группы из числа вышеуказанных групп путем контролируемого подкисления, например, при низких температурах и/или сокращенном времени взаимодействия.
Когда реакцию амидирования осуществляют через промежуточное образование "активированного эфира" исходного соединения формулы (II), такой "активированный эфир" обычно образуется на месте или, в альтернативном варианте, такой эфир сначала выделяют, а затем взаимодействуют с амином формулы (III). При этом в аминогруппу в положении N15 исходного материала формулы (II) сначала вводят защитную группу, чтобы избежать какого-либо взаимодействия между реактивом, активирующим образование эфира, и аминогруппой в N15. Защиту аминогруппы можно обеспечить любым из известных способов, которые описаны выше.
Образование "активированных эфиров" карбоновых кислот в общих чертах описано в работе Физер и Физер, "Reagent for organic syntjesis" изд. "Джон Уили энд Санз", стр. 129-130 (1967).
Примеры указанных реактивов, образующих активированные эфиры, которые могут быть использованы в процессе по изобретению, приведены в работе Р. Швизер и др. "Helv. Chim. Acta", 1955, 38, 69-70 и включают те эфирные производные формулы (II), где X' представляет собой CH2CN, CH2COOC2H5, CH2(COOC2H5)2, CH2COCH3, СН2CH2N(C2H5)2
СН2CH2N(C2H5)2
которые могут быть получены из исходных материалов формулы (II), где R'1 представляет собой соответствующую защитную группу, а X' представляет собой гидрокси посредством взаимодействия с ClCH2CN, BrCH2COOC2H5, BrCH(COOC2H5)2,
ClCH2COCH3, ClCH2CH2N(C2H5)2
ClCH2CH2N(C2H5)2
соответственно, в присутствии акцептора кислоты в растворителе.
Предпочитаемым реактивом этого типа является хлорацетонитрил.
В этом случае сам хлорацетонитрил, диметилформамид (DMF) или димителсульфоксид (DMSO) используют в качестве предпочитаемых растворителей.
Обычно для образования "активированных эфиров" используют инертные органические апротонные растворители, которые не оказывают нежелательного действия на ход реакции и способны по меньшей мере частично растворять карбоновые кислоты, являющиеся исходным материалом.
Примеры таких инертных органических растворителей включают органические амиды, эфиры гликолей и полиолов, фосфорамиды, сульфоксиды и ароматические соединения. Предпочитаемыми органическими растворителями являются диметилформамид, диметоксиэтан, гексаметилфосфорамид, диметилсульфоксид, бензол, толуол и их смеси.
Более предпочтительными растворителями являются ацетонитрил, диметилсульфоксид, диметилформамид. Получение активированного эфира обычно осуществляют в присутствии основания, которое не вступает в реакцию, такого как триалкиламин, напр., триэтиламин, карбонат или бикарбонат натрия или калия. Обычно основание применяют в 2-х - 6-ти кратной молярной пропорции к исходному материалу и предпочтительно оно используется приблизительно в трехкратном молярном избытке. Предпочитаемым основанием является триэтиламин.
Реактив, образующий "активированный эфир", используется в большом избытке по отношению к исходному материалу формулы (II), содержащему карбоновую кислоту в C63. Обычно этот реактив используется в 5-35 молярной пропорции, предпочтительно в 20-30 кратном молярном избытке. Реакцию проводят при температуре от 10 до 60oC, предпочтительно при 15-30oC. Как обычно, время протекания реакции зависит от других параметров реакции и может варьироваться от 3 до 48 часов.
Ход протекания реакции контролируют с помощью ВЭЖХ или тонкослойной хроматографии, чтобы определить момент завершения реакции и приступить к восстановлению искомого промежуточного соединения. Промежуточный "активированный эфир" можно непосредственно использовать в той же реакционной среде, где он был получен, однако обычно его выделяют осаждением с применением осадителей или путем экстрагирования растворителями, после чего полученный "активированный эфир" используют на следующих этапах без последующей очистки. Однако при желании эфир можно подвергнуть очистке с помощью колоночной флэш-хроматографии или колоночной хроматографии с обращенной фазой.
Затем полученный промежуточный "активированный эфир" взаимодействуют с аминным производным формулы (III) (в молярном избытке) в присутствии органического полярного растворителя при температуре от 5 до 60oC, предпочтительно от 10 до 30oC.
В этом случае органический полярный растворитель может быть полярным протонным или апротонным растворителем.
Предпочитаемыми органическими полярными протонными растворителями являются низшие (C2-C4) алканолы, такие как этанол, n-пропанол, изопропанол, n-бутанол и т.п., или их смеси, предпочтительно используемые в сухом виде.
Предпочитаемыми органическими полярными апротонными растворителями являются N,N-диметилформамид (ДМФ), гексаметилфосфорамид (HMPA) или их смеси, 1,3-диметил-3,4,5,6-тетрагидро- 2-(1H)-пиримидин (DMPU), диметилсульфоксид (DMSO) или диметоксиэтан (DME).
Реакцию "активированного эфира" с выбранным амином формулы (III) можно проводить при температуре от 5 до 60oC, но предпочтительно при температуре от 10 до 30oC, лучше всего при 20-25oC, а предпочтительным молярным соотношением между промежуточным "активированным эфиром" и амином (III) является соотношение от 1:5 до 1:30, более предпочтительно от 1:10 до 1:20. Ход реакции, как обычно, контролируют с применением тонкослойной хроматографии или ВЭЖХ.
В случае, если аминным реактивом является полиамин формулы (III), то одна или более из аминных групп этого соединения, которые не участвуют в образовании амидной связи, могут быть защищены. В этих случаях подходящие защитные группы следует выбирать из групп, перечисленных для положения N15.
Соответственно, полученные амидные производные, содержащие защитные группы в N63, затем подвергаются обработке с целью удалить эти защитные группы, удаление защитных групп производят в тех же условиях, которые описаны выше в отношении удаления защитных групп в положении 15.
Соединения формулы (I), где Y представляет собой гидроксиметил, R1, R2, M, X и Z имеют значения, приведенные в начале описания, можно получить путем восстановления соответствующих производных формулы (I), где R2, M, X и Z имеют вышеуказанные значения, Y представляет собой (C1-C4) алкоксикарбонил, а R1 представляет собой подходящую защитную группу для аминогруппы в N15, с помощью боргидрида щелочного металла, предпочтительно, вбираемого из группы: боргидрид натрия, боргидрид калия и цианоборгидрид натрия, при температуре от 0 до 40oC в водной или водно-спиртовой среде. Удаление защитной группы аминогруппы N15 можно осуществить в вышеописанных условиях.
Использование этого способа необходимо при получении соединений формулы (I), где Y представляет собой гидроксиметил, X - гидрокси, R1, R2 и M имеют значения, приведенные в начале описания, а Z является водородом. В этом случае исходным материалом, который подвергают восстановлению в вышеописанных условиях, является соединение формулы (II), где Y' представляет собой (C1-C4) алкоксикарбонил, X' - гидрокси, R'2 и M' имеют те же значения, что R2 и M соответственно, а R'1 представляет собой подходящую защитную группу для аминогруппы в N15. Получение указанного исходного соединения описано в Заявке на международный патент N PCT/EP92/00374 и проводится в соответствии с общим способом получения исходного эфира формулы (II), как описано выше.
Обычно водоспиртовая среда, используемая в восстановительных реакциях, упомянутых выше, представляет собой смесь воды и водорастворимого или частично смешиваемого с водой низшего алканола, причем отношение вода/низший алканол варьируется от 40/60 до 90/10 (о/о), предпочтительно от 60/40 до 68/32 (о/о), лучше всего - 65/35 (о/о).
Хотя в некоторых случаях реакция происходит и при меньших количествах воды, напр. в смеси вода/низший алканол 30/70 или 20/80, обычно скорость протекания реакции очень низкая при соотношении вода/низший алканол ниже, чем 40/60.
Предпочитаемыми низшими алканолами являются (C1-C4)алкильные спирты с линейной или разветвленной цепью, среди которых наиболее предпочтительны n-бутанол, этанол и метанол.
Иногда в отдельных случаях добавляют небольшие количества полярного сорастворителя для того, чтобы полностью растворить исходный материал в ходе реакции, напр. , N, N-диметилформамида, 1,3-диметил-3,4,5,6-тетрагидро-2-(1H)-пиримидона (DMPU), диметилсульфоксида. Иногда добавляют также различные количества диэтилового эфира, чтобы избежать пенообразования.
Наиболее предпочтительным из числа боргидридов щелочных металлов является боргидрид натрия. Количество боргидрида щелочного металла может быть различным в зависимости от типа используемого растворителя и температуры реакции, но рекомендуется использовать боргидрид щелочного металла в большом избытке по отношению к стехиометрическим требованиям, так, чтобы pH реакционной смеси была нейтральным или щелочным, предпочтительно pH от 7 до 10. В общем мольное отношение между боргидридом щелочного металла и исходным материалом для антибиотика составляет от 50 до 300.
Температура реакции может существенно варьироваться в зависимости от типа исходного материала и условий проведения реакции. В общем предпочтительно проводить реакцию при температуре от 0 до 40oC, лучше всего - при комнатной температуре.
Время протекания реакции также может быть различным в зависимости от других параметров реакции, тем не менее время реакции необходимо тщательно контролировать. Обычно реакция завершается приблизительно через 1-4 часа. Если продолжать реакцию в течение свыше 4-х часов, то могут произойти нежелательные побочные реакции, которые приводят к разрыву пептидных связей в ядре молекулы.
В любом случае за ходом реакции наблюдают при помощи тонкослойной хроматографии, предпочтительно ВЭЖХ, используя известные способы. На основе результатов хроматографических анализов любой специалист в состоянии оценить ход реакции и решить, когда ее следует прекратить и приступить к обработке реакционной массы одним из известных способов, который включает, например, экстрагирование растворителями, осаждение путем добавки осадителей и т.п., а также, при необходимости, разделение и очистку колоночной хроматографией.
После завершения реакции избыток боргидрида щелочного металла удаляют путем добавки соответствующего количества кислоты, напр., (C1-C4) алкил-органической кислоты, (C1-C6) алкил-сульфокислоты, арилсульфокислоты и т.п., растворенной в полярном протонном растворителе, таком как, напр., (C1-C4) алкиловый спирт.
В другом варианте соединения формулы (I), где Y представляет собой гидроксиметил, R1, R2 и M имеют значения, указанные в начале описания, X представляет собой аминный остаток
-NR3-alk1-(NR4-alk2)p)- (NR5-alk3)q-W
где
R3, R4, R5, alk1, alk2, alk3, p, q и W имеют значения, указанные в начале описания, а Z представляет собой водород, получают посредством амидирования, которое было описано выше, взаимодействуя соответствующее соединение формулы (I), где Y представляет собой гидроксиметил, X - гидрокси, R1, R2 и M имеют вышеуказанные значения, а Z представляет собой водород, с амином формулы (III), как описано выше.
В этом случае реакцию амидирования можно проводить с использованием соответствующего конденсирующего агента или через промежуточное образование "активированного эфира", как описано выше в отношении получения соединений формулы (I), где Y представляет собой (C1-C4) алкоксикарбонил.
В целом, амидирование производных формул (I), где Y представляет собой гидроксиметил, X - гидрокси и Z - водород, при использовании Py BOP в качестве конденсирующего агента, дает соединения формулы (I), где Z представляет собой водород, даже в тех случаях, когда Py BOP используют в большом молярном избытке по отношению к исходной карбоновой кислоте. Когда реакцию амидирования проводят путем образования активированного эфира" соединения формулы (I), где X - гидрокси, Y - гидроксиметил, а Z - водород, предпочтительно ввели одну из вышеописанных защитных групп в положение N15 аминогруппы указанного соединения.
Далее процесс получения соединения формулы (I), где Y представляет собой (C1-C4) алкоксикарбонил или гидроксиметил, R1, R2, M и Z имеют значения, указанные в начале описания, X представляет собой остаток:
-NR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W
где
R3, R4, R5 каждый независимо представляет собой водород или (C1-C4)алкил, alk1, alk2, alk3 и W имеют значения, указанные в начале описания, p равно 1 и q равно 1 или 0, состоит в том, что производное, амида N63, имеющее защитную группу в N15 (в настоящем описании термин "N63" относится к атому азота в карбоксиамидной группе, которая связана с атомом углерода в молекуле A 40926 под номером 63) формулы (I), где Y, R2, M и Z имеют вышеуказанные значения, а X представляет собой аминный остаток формулы:
-NR3-alk1-(NR5,
где
R3, R4 и alk1 имеют вышеуказанные значения, или -NR3-alk1-NR4-alk2- NR5,
где
R3, R4, R5, alk1 и alk2 имеют вышеуказанные значения,
с аминным реагентом формулы (IV) или (IVa) соответственно;
r-alk2-(NR5-alk3)g-W (IV) и r-alk3-W (IVa),
где
R5, alk2, alk3 и W имеют вышеуказанные значения, q равно 0 или 1, r представляет собой галоген, метансульфонил или тозил, в присутствии акцептора кислоты в инертном растворителе. Производные амида N63 с защитной группой в N15 получают в соответствии с общим способом получения соединений формулы (I) по изобретению. Удаление защитной группы в N15-аминогруппы производят вышеописанным способом.
В этом же случае алкилирование желательно или необходимо применять для того, чтобы ввести защитную группу в аминогруппу (группы) помимо N15-аминогруппы амидного производного N63 формулы (I) и/или аминного реактива (IV) или (IVa), которые не участвуют в реакции алкилирования. Защитную группу амидов N63 можно удалить вышеописанным способом.
Защитные группы, которые можно использовать во всех вышеуказанных реакциях, уже перечислены выше. Однако особое внимание следует уделить удалению защитной группы из производного формулы (I), где Y является гидроксиметилом. Для этих соединений, когда защитная группа в 15 положении удаляется под воздействием кислоты, этап удаления защитной группы очень важен, поскольку происходит относительно быстрое замещение соответствующего ацилглюкозаминного фрагмента в 56 при обработке, например, сухой трифторуксусной кислоты (TFA). В любом случае эти нежелательные побочные реакции можно легко свести к минимуму. Например, когда в качестве защитной группы используют трет-бутилоксикарбонил (t-ВОС), можно использовать следующий способ: обработку сухой TFA в течение одной минуты при комнатной температуре или в течение 10-30 мин при температуре от 0 до 5oC с последующим быстрым осаждением продукта реакции диэтиловым эфиром или смесью метанол/диэтиловый эфир при 0-5oC. Наоборот, в отношении соединений формулы (I), где Y представляет собой карбокси или метоксикарбонил, замечено, что фрагмент ациламиноглюкуроновой кислоты в 56 является значительно более устойчивым к TFA. Фактически образование следов соответствующих деглюкуронильных псевдоагликонов отмечалось только через 1 час течения реакции. Однако в данном случае удаление защитной группы t-BOC осуществляется в течение 30 мин.
Другой походящий способ удаления защитной группы t-BOC без существенного воздействия на другие части молекулы состоит в обработке сухой TFA в дихлорметане при 0-10oC в течение 1-2 часов с последующим осаждением продукта реакции осадителем.
Соединения формулы (I), где R1, R2, M, X и Z имеют значения, указанные в начале описания, а Y представляет собой карбокси, получают из соответствующих соединений формулы (I), где Y представляет собой (C1-C4) алкоксикарбонил, предпочтительно метоксикарбонил, а все другие радикалы имеют вышеуказанные значения, посредством обработки водным раствором гидроксидов щелочных металлов (напр. NaOH или KOH) при температуре от 0 до 30oC (более высокие температуры недопустимы, поскольку может произойти эпимеризация на атоме углерода в 3 положении молекулы), в инертном органическом растворителе, например ди-(низший алкил) эфире этиленгликоля или тетрагидрофуране.
Соединения формулы (I), где R1, R2, M, X и Z имеют значения, указанные в начале описания, а Y представляет собой аминокарбонил, (C1-C4) алкиламинокарбонил, ди (C1-C4) алкиламинокарбонил, где алкильный фрагмент может иметь заместитель, выбираемый из гидрокси, амино, (C1-C4) алкиламино и ди (C1-C4) алкиламино, можно получить следующим способом:
i) Получение производных, где радикал Y и фрагмент COX в C63 представляют одну и ту же группу (C1-C4) алкиламинокарбонил или ди (C1-C4) алкиламинокарбонил, где алкильный фрагмент может иметь заместитель, выбираемый из амино, (C1-C4) алкиламино и ди (C1-C4) алкиламино;
(a) Амидирование антибиотического комплекса A 40926, его деманнозильного производного или его фактора (формулы (II), X' = гидрокси, Y' = карбокси, R'1, R'2 и M' имеют те же значения, что R1, R2 и M) большим избытком соответствующего амина формулы (III), где радикалы R3, R4, R5, alk1, alk2, alk3, p, q и W имеют значения, совместимые с вышеуказанными карбоксиамидными остатками Y и COX. Эту реакцию амидирования осуществляют в тех же условиях, которые описаны выше.
ii) Получение производных, где радикал Y и фрагмент COX в C63 представляют собой различные карбоксамидные остатки, значение Y выбирают из аминокарбонила, (C1-C4) алкиламинокарбонил, ди(C1-C4) алкиламинокарбонил, причем алкильный фрагмент может иметь заместитель, выбираемый из амино, гидрокси, (C1-C4) алкиламино и ди (C1-C4) алкиламино, а X представляет собой аминный остаток, описанный в начале описания;
Способ A: Амидирование соответствующего соединения формулы (I), где R1, R2, M и Z имеют значения, указанные в начале описания, X представляет собой аминный остаток формулы I, NR3-alk1-(NR4-alk2)p- (NR5-alk3)q-W, где все радикалы имеют значения, указанные в начале описания, а Y представляет собой карбокси, посредством реакции с соответствующим амином до образования вышеуказанного карбоксамидного остатка Y в присутствии конденсирующего агента (напр., Py BOP или DPPA) при вышеописанных условиях;
Способ (B): (a) в исходное соединение (такое же, как в Способе A) вводят защитную группу в положение N15 аминогруппы (например, группу t-BOC или CBz); (b) получают "активированный эфир" карбоксильной группы в положении 6b (например, путем взаимодействия с хлорацетонитрилом); (c) взаимодействуют фрагмент "активированного эфира" указанного соединения с соответствующим амином до получения вышеуказанного карбоксамидного остатка Y в вышеописанных условиях, (d) при желании удаляют защитную группу в N15 вышеописанным способом (напр., ацидолизом или гидрогенолизом).
Соединения формулы (I), где M представляет собой водород, получают вышеописанным способом при использовании соответствующих исходных молекул формулы (II), где M' представляет собой водород.
Кроме того, альтернативный способ получения соединения формулы (I), где M представляет водород, состоит в преобразовании соединения формулы (I), где M представляет собой α-D- маннопиранозил или 6-O-ацетил -α-D- маннопиранозил, в соответствующее соединение, где M представляет собой водород, путем селективного кислотного гидролиза в соответствии с условиями, описанными в EP-A-240609.
Как описано выше, соединения формулы (I) могут состоять из отдельных соединений, соответствующих индивидуальным факторам предшественника антибиотика A 40926 или их смеси в различных пропорциях. Поскольку во многих случаях биологическая активность смесей аналогична биологической активности индивидуальных факторов, нет смысла отделять индивидуальные компоненты после получения смеси. Однако, в тех случаях, когда нужды чистые факторы формулы (I), их можно отделить из смесей с помощью колоночной хроматографии с обращенной фазой по способу, описанному в EP 177882. В другом варианте их можно получить, используя исходные материалы формулы (II), содержащие отдельные соединения, соответствующие индивидуальным факторам антибиотического комплекса A 40926.
Согласно общему способу и условиям протекания описанных здесь реакций можно использовать предшественник комплекса A 40926, содержащий один из индивидуальных факторов (напр., фактор B0) в преобладающей пропорции к остальным компонентам смеси (напр., 60% по ВЭЖХ). Соответственно, соединения формулы (I), получаемые из такого предшественника посредством процесса по изобретению, если эти соединения не подвергают вышеописанному разделению, то они состоят из смесей, преобладающий компонент которых соответствует тому фактору, который преобладал в указанном предшественнике комплекса A 40926.
Способ получения комплекса A-40926, насыщенного факторами A и/или B0, или PA и/или PB, описан, например, в EP-A-259781.
Соединения по изобретению, обладающие щелочными свойствами, могут образовывать соли с органическими и неорганическими кислотами обычными способами.
Примеры подходящих солей соединений по изобретению включают соли, полученные с помощью стандартных реакций с органическими и неорганическими кислотами, такими, как, например, соляная, бромистоводородная, серная, фосфорная, уксусная, трифторуксусная, трихлоруксусная, янтарная, лимонная, аскорбиновая, молочная, малеиновая, фумаровая, пальмитиновая, холевая, памовая, слизевая, глутаминовая, камфорная, глутаровая, гликолевая, фталиевая, винная, лауриновая, стеариновая, салициловая, метансульфоновая, бензолсульфоновая, сорбиновая, пикриновая, бензойная, коричная и т.п. кислоты.
Соединения формулы (I), где X представляет собой гидрокси, а Y - гидроксиметил, и соединения, где Y представляет собой карбокси, также обладают кислотной группой, которая может образовывать соли с органическими и неорганическими основаниями.
Примерами оснований, которые могут образовывать соли с соединениями по изобретению, содержащими кислотные группы, являются: гидроксиды щелочных или щелочноземельных металлов, таких как натрий, калий, кальций, магний, гидроксид бария, аммиак и алифатические, ациклические или ароматические органические амины, такие как метиламин, диметиламин, триэтиламин, этаноламин и пиколин.
Преобразование соединений по изобретению в соответствующие соли и обратный процесс, т. е. преобразование соответствующих солей в соединения по изобретению, осуществляются обычными способами и входят в рамки настоящего изобретения.
Например, соединение формулы (I) может быть преобразовано в соответствующие соли с помощью кислот или оснований путем растворения или суспендирования соединения формулы (I) в водном растворителе и добавки выбранной кислоты или основания в небольшом молярном избытке. Полученный раствор или суспензию затем лиофилизируют, чтобы восстановить искомую соль.
В случае, когда конечная соль нерастворима в растворителе, в котором растворимо само соединение по изобретению, эту соль можно восстановить фильтрацией из раствора соединения по изобретению после добавки стехиометрического количества или небольшого молярного избытка выбранной кислоты или основания.
Соединение по изобретению можно получить из соответствующей соли, растворенное в водном растворителе, который затем нейтрализуют для высвобождения соединения. Затем соединение восстанавливают, например, экстрагированием органическим растворителем или преобразуют в другую соль путем добавки выбранной кислоты или основания и проведения вышеописанной обработки.
В тех случаях, когда проводят нейтрализацию, необходимо обессоливание, которое осуществляют обычными способами. Например, с помощью колоночной хроматографии на полидекстрановых смолах с заданной пористостью (таких, как "Sephadex LH 20) или на силанизированном силикагеле. После элюирования нежелательных солей водным раствором, искомый продукт элюируя линейным градиентом или ступенчатым градиентом смеси воды с полярным или неполярным органическим растворителем, такой смеси, как ацетонитрил - вода в соотношении от 50:50 до 100% ацетонитрила.
Известно, что образование солей при использовании либо фармацевтически-приемлемых кислот и оснований, либо фармацевтически-неприемлемых кислот и оснований можно использовать в качестве способа очистки. После образования и выделения соль соединения формулы (I) можно преобразовать в соответствующее соединение не в форме соли или фармацевтически-приемлемую соль.
Ввиду того, что соединения формулы I их соли обладают схожими свойствами, то все, что относится к биологической активности соединений формулы (I), можно отнести также к фармацевтически-приемлемым солям этих соединений.
В нижеследующей табл. 1 представлена серия представителей соединений по настоящему изобретению.
Производные антибиотика A 40926 проявляют активность главным образом против грамположительных бактерий.
В частности, соединения по изобретению проявляют высокую активность против устойчивых к гликопептидам энтерококков и стафилококков.
Антимикробная активность производные формулы (I) антибиотика A 40926, выражаемая минимальной ингибирующей концентрацией (MIC), против выбранных штаммов грамположительных бактерий была сопоставлена с антимикробной активностью тейкопланина и антибиотического комплекса A 40926. Для этого использовали способ разбавления микрокультуральной среды в бульоне Мюллера-Хинтона в присутствии 0,01% (м/о) сыворотки бычьего альбумина (фракция сигма), конечный инокулят составлял около 105 колониеобразующих единиц/мл, а минимальная ингибирующая концентрация (MIC) была на самом низком уровне (мкг/мл), что говорило об отсутствии видимого роста через 18-24 часов инкубации при 37oC.
На нижеследующей табл. 2 спектр антимикробной активности репрезентативных соединений по изобретению.
В нижеследующей табл. 3 представлены данные по активности in vitro некоторых репрезентативных соединений по настоящему изобретению в сравнении с тейкопланином и ванкомицином, показывающие, что соединения по изобретению проявляют активность in vitro против штаммов энтерококков, обладающих высокой устойчивостью к гликопептидам в обычной терапии.
В нижеследующей табл. 4 показаны результаты экспериментального использования некоторых репрезентативных соединений по изобретению против стрептококкового сепсиса мышей.
Эксперименты проводились по методике, описанной а работе В.Ариоли и др., "Journal of Antibiotics" 29. 511 (1976)
Вышеприведенные данные показывают, что несмотря на то, что соединения по изобретению в целом менее активны против Neisseria gonorrheae, чем предшественник A 40926, эти соединения обладают более высокой активностью против культур Staphylococci и Enterococci по сравнению с другими соединениями. В частности, эти соединения:
a) проявляют заметно более высокую активность in vitro, чем тейкопланин и A 40926, против стафилококков, являющихся интермедиатами гликопептидов или обладающих устойчивостью к гликопептидам;
b) проявляют активность in vitro против энтерококков, обладающих устойчивостью к гликопептидам, которые обладают также высокой устойчивостью к тейкопланину и ванкомицину и проявляют некоторую устойчивость к A 40926 (MIC ≥ 64 мкг/мл);
c) проявляют большую эффективность in vivo, чем тейкопланин и A 40926, при стрептококковом сепсисе мышей.
Благодаря такой антимикробной активности соединения по изобретению можно эффективно применять в качестве активных ингредиентов антимикробных препаратов, используемых для лечения людей и в ветеринарной практике для предотвращения и лечения инфекционных заболеваний, вызываемых патогенными бактериями, в частности, для лечения инфекционных заболеваний, вызываемых энтерококками, стрептококками и стафилококками, которые обладают низкой чувствительностью к гликопептидным антибиотикам.
Соединения по настоящему изобретению могут назначаться перорально, местно или парентерально, парентеральное введение является предпочтительным.
В зависимости от способа введения соединения по изобретению входят в состав препаратов в различных дозах. Препараты для перорального приема могут быть изготовлены в виде капсул, таблеток, жидких растворов или суспензий. Согласно обычной практике капсулы и таблетки могут содержать, помимо активного ингредиента, обычные носители, такие как разбавители, например лактозу, фосфат кальция, сорбит и т.п., смазывающие вещества, например стеарат магния, тальк, полиэтиленгликоль, связующие вещества, например поливинилпирролидон, желатин, сорбит, трагакант, акацию, ароматизаторы и приемлемые расщепляющие и смачивающие агенты. Жидкие препараты, обычно в виде водных или масляных растворов или суспензий, могут содержать обычные добавки, такие как суспендирующие агенты.
Для местного применения соединения по изобретению вводят в состав препаратов, пригодных для поглощения через слизистые носоглотки или бронхиальные ткани, такие препараты обычно изготавливают в виде жидких аэрозолей или ингаляционных препаратов, таблеток или препаратов, предназначенных для смазывания горла.
Для лечения заболеваний глаз или уха препараты могут иметь жидкую или полужидкую форму. Для местного применения препараты изготавливают на гидрофобных или гидрофильных основах, таких как мази, кремы, лосьоны или порошки.
Для ректального применения соединения по изобретению назначают в виде свечей, содержащих обычные носители, такие как например, кокосовое масло, воск, спермацеты или полиэтиленгликоли и их производные.
Композиции для инъекций могут иметь вид суспензий, растворов или эмульсий в масляных или водных носителях и могут содержать дополнительные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты.
В другом варианте активный ингредиент может быть в виде порошка, который перед инъекцией смешивают с подходящим носителем, например водой.
Количество активного ингредиента, подлежащего введению, зависит от различных факторов, таких как масса и состояние пациента, способ и периодичность введения, тип возбудителя болезни.
Соединения по изобретению обычно эффективны в дозах от около 1 до около 40 мг активного ингредиента на 1 кг массы тела пациента. В зависимости от характеристик конкретного соединения, типа инфекции и состояния пациента эффективную дозу вводят один раз в день или делят ее на 2-4 приема в день. Предпочтительными являются композиции, приготовленные в виде отдельных доз по 30-500 мг на дозу.
Пример 1
Получение исходного материала (МА) (соединения формулы (II), где Y' представляет собой - COOCH3, X' представляет собой - OH, R'1 является - H, R'2 является (C9-C12) алкил, соответствующий факторам комплекса A 40926, M' является α-D- маннопиранозилом и Z является - H).
Антибиотический комплекс A 40926 (150 мг, 0,0866 ммоль) полученный способом, описанным в EP-A-177882, растворяют в метаноле (30 мл) и с помощью концентрированной серной кислоты доводят pH до 2. Смесь перемешивают при комнатной температуре в течение 26 часов. В тот момент, когда pH доводят до 6 с помощью 0,15 мл триэтиламина (ТЕА), появляется осадок. После добавки диэтилового эфира осадок собирают, тщательно промывают диэтиловым эфиром и высушивают. Выход: 150 мл (99%).
Пример 2
Получение соединения 1 (RA) (соединения формулы (I), где Y является - CH2OH, X является - OH, R1 является - H, R2 является (C9-C12) алкилом, соответствующим факторам комплекса A 40926, M представляет собой α-D- маннопиранозил, а Z является - H).
Этап a: получение N15-(t-BOC)-MA
При перемешивании в раствор 1,8 г соединения из примера 1 (МА) и 1 г бикарбоната натрия в 50 мл смеси диоксан/вода 1/1, добавляют раствор 0,25 г ди-трет-бутил-дикарбоната в 5 мл диоксана при 5oC, добавку производят по капле в течение 15 мин. Через 1 час при комнатной температуре pH реакционной смеси доводят до 4 с помощью 1Н HCL. После этого добавляют 150 мл воды и полученную смесь экстрагируют n-бутанолом (2 х 100 мл). Органический слой отделяют, промывают 100 мл воды и затем концентрируют до малого объема (около 25 мл) при 40oC под пониженным давлением. Осадок, выпавший после добавки диэтилового эфира (100 мл) собирают и высушивают под вакуумом при комнатной температуре в течение ночи до получения 1,6 г искомого соединения N15-(t-BOC)-MA, достаточно чистого для использовании на последующем этапе.
Этап b: получение N15-(t-BOC)-RA
При перемешивании в суспензию 0,9 г соединения, полученного на этапе a, в 50 мл воды добавляют 30 мл смеси n-бутанол/диэтиловый эфир 1/1, после чего добавляют 0,9 г боргидрида натрия. В течение 30 мин при комнатной температуре производят добавку восстановителя (порциями) затем реакционную смесь перемешивают при комнатной температуре в течение 1 часа. После этого реакционную смесь охлаждают до 5oC и добавляют 1,5 мл ледяной уксусной кислоты, а затем 50 мл воды. Полученную смесь экстрагируют н-бутанолом (100 мл) и органический слой обрабатывают вышеописанным способом до получения 0,8 г искомого соединения N15-(t-BOC)-RA, достаточно чистого для использования на последующем заключительном этапе.
Этап c: Раствор 0,5 соединения N15-(t-BOC)-RA, полученного на этапе B выше, в 5 мл сухой трифторуксусной кислоты (TFA) перемешивают при комнатной температуре в течение 1 минуты (или, по другому варианту, при 0-5oC в течение 20-30 мин), затем выливают в 10 мл смеси метанол/диэтиловый эфир 1/4 при 0-5oC. Искомое соединение RA собирают фильтрацией до получения (после промывки диэтиловым эфиром, и высушивания при комнатной температуре под вакуумом в течение ночи) 0,35 г, продукта. Образец в 0,15 г чистого соединения RA получают с помощью колоночной хроматографии с обращенной фазой на силанизированном силикагеле, соединив все фракции, содержащие чистые индивидуальные факторы, как описано ниже.
Пример 3
Получение соединения 2 (MA-A-1/B0) и соединения 6 (MA-A-1). (соединение формулы (I), где Y представляет собой -COOCH3, X представляет собой - NH-(CH2)3- N(CH3)2, R1 является - H, R2 представляет собой 9-метилдецил (MA-A-1/B0) или (C9-C12)-алкил, соответствующий факторам комплекса A 40926 (MA-A-1), M представляет собой L-D-маннопиранозил, и Z является - H).
Способ A.
Этап a: получение N15-(t-BOC)-MA-A-1.
При перемещении в раствор 1,3 г N15-(t-BOC)-MA- в 30 мл диметилсульфоксида (полученный на этапе a в примере 2 выше) добавили 0,2 мл 3,3-диметиламино-1-пропиламина и 0,3 мл дифенилфосфоразидата (DPPA). Через 4 часа перемешивания при комнатной температуре добавили еще 0,15 мл DPPA и продолжали перемешивание при комнатной температуре еще в течение 20 часов. Твердый осадок, выпавший после добавки 170 мл диэтилового эфира, собирали до получения 1,3 г искомого соединения N15-(t-BOC)-MA-A-1.
Этап b: Продукт вышеописанной реакции растворяли в 10 мл TFA. Полученный раствор перемешивали при комнатной температуре в течение 20 мин, затем добавили 90 мл диэтилового эфира. Выпавший осадок собрали, дважды промыли 50 мл диэтилового эфира, а затем высушивали при комнатной температуре под вакуумом в течение ночи, получив 0,9 г сырого неочищенного искомого соединения (MA-A-1), которое подвергали хроматографии с обращенной фазой на колонке с силанизированным силикагелем (соединяя фракции, содержащие чистый искомый фактор) до получения 0,15 г, чистого MA-A-1/B0.
Способ B.
При перемешивании в раствор 1,8 г (около 1 ммоль) соединения из примера 1 (MA) в 30 мл диметилформамида добавили 0,14 мл (около 1,15 ммоль) 3,3-диметиламино-1-пропиламина и 600 мг (около 1,2 моль) PуBOP, добавку производили при комнатной температуре. После перемешивания при 20-25oC в течение 3 часов добавили 150 мл диэтилового эфира. Выпавший осадок собрали и очистили колоночной хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы), получив 1,15 г соединения MA-A-I.
Пример 4
Получение соединения 7 (Py MA-A-I). (Соединение формулы (I), где Y представляет собой - COOCH3, X является -NH-(CH2)3- N(CH3)2, R1 является - H, R2 представляет собой (C9-C12) алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D- маннопиранозил и Z является P⊕(NC4H8)3CH3COO⊖.
При перемешивании в раствор 1,8 г (около 2 ммоль) соединения MA (полученного в примере 1) в 40 мл диметилформамида добавили 2 мл (около 16 ммоль) 3,3-диметиламино-1-пропиламина и 3,12 г (около 6 ммоль) Py BOP, добавку производили при комнатной температуре. Спустя 30 мин реакционную смесь подвергали обработке, описанной в примере 3, способ B, получив 1,5 г искомого соединения Py MA-A-I.
Пример 5
Получение соединения 3 (RA-A-1/B0) и 8 (RA-A-1). (Соединение формулы (I), где Y является - CH2OH, X является - NH-(CH2)3-N(CH3)2, R1 является - H, R2 представляет собой 9-метилдецил (RA-A-1B0) или (C9-C12) алкил, соответствующий факторам комплекса A 40926 (RA-A-1), M представляет собой α-D- маннопиранозил и Z является - H).
Способ A
Этап a: получение N15-(t-BOC)-RA-A-1.
Используя способ по примеру 3, способ A, этап a, из 2 г N15-(t-BOC)-RA (пример 2, этап b) получили 1,7 г искомого соединения N15-(t-BOC)-RA-A-1.
Этап b. Используя способ, описанный в примере 2, этап c, из 1,7 г вышеуказанного соединения N15-(t-BOC)-RA-1 получили 0,22 г чистого соединения RA-A-1.
Фактор RA-A-1/B0 получили, используя указанный способ, за исключением того, что очистку хроматографией с обращенной фазой производили в отношении только тех фракций, которые содержали чистый искомый индивидуальный фактор (объединяя эти фракции).
Способ B.
При перемешивании в раствор 50 г (около 27 ммоль) соединения из примера 2 (RA) в 200 мл диметилформамида добавили 11 мл (около 90 ммоль) 3,3-(N,N-диметиламино)-1-пропиламина и 18 г (около 35 ммоль) Py BOP, добавку производили при комнатной температуре. После перемешивания в течение 15 минут добавили 1 литр этилацетата и выпавший осадок (около 63 г) собрали и подвергали очистке колоночной хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы), получив 25 г соединения RA-A-1.
Пример 6
Получение соединения 4 (MA-A-2/B0) (Соединение формулы (I), где Y является - COOCH3, X является - NH-(CH2)3-[NH-(CH2)3]2-NH2, R1 является - H, R2 представляет собой 9-метилдецил, M представляет собой L-D-маннопиранозил и Z является - H).
Этап a: получение N15-(t-BOC)-MA, цианометилового эфира.
Раствор 2,5 г соединения из примера 2, этап a (N15-(t-BOC)-MA), 0,25 мл TEA и 2,5 мл хлорацетонитрила в 10 мл диметилсульфоксида (DMSO) перемешивают при комнатной температуре в течение 4 часов. После этого добавляют 90 мл этилацетата и выпавший осадок собирают, получая 2,8 г неочищенного искомого соединения N15-(t-BOC)-MA, цианометиловый эфир.
Этап b: получение N15-(t-BOC)-MA-A-2.
Вышеописанный цианометиловый эфир (неочищенный) растворяют в 30 мл DMSO. В полученный раствор добавляют 2,8 мл N,N'-бис(3-аминопропил)-1,3-пропандиамина и перемешивают реакционную смесь при комнатной температуре в течение 4 часов. После этого добавляют 200 мл этилацетата и выпавший твердый осадок собирают, получая 3 г неочищенного искомого соединения N15-(t-BOC)-MA-A-2.
Этап c: Вышеуказанное неочищенное соединение обрабатывают TFA, как описано выше в примере 3, способ A, этап b, до получения (после колоночной хроматографии с обращенной фазой (соединяя только фракции, содержащие чистый искомый индивидуальный фактор)) 0,45 г чистого соединения MA-A-2/Bo.
Пример 7
Получение соединения 5 (MA-A-3/B0). (Соединение формулы (I), где Y является COOH3, X является - NH-(CH2)3-N[(CH2)3-NH2]2, R1 является - H, R2 представляет собой 9-метилдецил, M представляет собой α-D-маннопиранозил и Z является - H).
Этап a: получение N',N''-ди(t-BOC)-трис(3-аминопропил)амина.
N',N''-защищенный полиамин получают способом, описанным в Заявке на международный патент N WO 90/11300.
Этап b: конденсация MA с помощью N',N"-ди(t-BOC)-трис (3-аминопропил)амина.
Раствор 18 г (около 10 ммоль) соединения из примера 1 (MA), 14 г (около 36 ммоль) защищенного амина, 3 мл (около 22 ммоль) TEA и 6 мл (около 28 ммоль) DPPA в 150 DMSO перемешивают при комнатной температуре в течение 2 часов, затем добавляют 500 мл этилацетата. Выпавшее в осадок твердое вещество собирают (около 22 г) и используют на следующем этапе без дальнейшей очистки.
Этап с: удаление защитных групп t-BOC:
Неочищенный продукт реакции этапа b растворяют в 150 мл сухого TFA, предварительно охлажденного до 0oC и полученный раствор перемешивают при 0-5oC в течение 20 минут. Затем добавляют 150 мл метанола и 300 мл диэтилового эфира. Выпавший осадок собирают, несколько раз промывают диэтиловым эфиром, затем очищают колоночной хроматографией с обращенной фазой (соединяя только фракции, содержащие чистый искомый индивидуальный фактор) до получения 9 г соединений МА-А-3/B0.
Пример 8
Получение соединения 9 (RA-A-2). (Соединения формулы (I), где Y является - CH2OH, X является - NH-(CH2)3- [NH(CH2)3] 2-NH2, R1 является - H, R2 представляет собой (C9-C12) алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D- маннопиранозил и Z является - H).
Этап а: получение N15-(t-BOC)-RA, цианометилового эфира.
Раствор 8 г (около 4 ммоль) соединения из примера 2, этап b (N15-(tBOC)-RA), 0,75 мл (около 5,5 моль) TEA и 8 мл хлорацетонитрила в 40 мл DMSO перемешивают при комнатной температуре в течение 5 часов. Затем добавляют 200 мл этилацетата и выпавший твердый осадок собирают, получая 8,2 искомого неочищенного цианометилового эфира. Этапы b и c: Конденсация с помощью N', N''-бис-(3-аминопропил)-1,3- пропандиамина и ацидолиз защитной группы t-BOC:
Неочищенный цианометиловый эфир из этапа а растворяют в 80 мл DMCO и добавляют 9 г N,N'-бис-(3-аминопропил)-1,3-пропандиамина. После перемешивания при комнатной температуре в течение 20 часов добавляют этилацетат. Выпавший твердый осадок собирают и повторно растворяют в 70 мл ледяного сухого TFA. Полученный раствор перемешивают при 0oC в течение 10 минут, затем добавляют 230 мл охлажденного диэтилового эфира. Выпавший твердый осадок собирают и быстро повторно растворяют в 200 мл воды. pH раствора доводят до 5,5 с помощью 1Н NaOH и очищают хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы), получая 1,3 чистого искомого соединения RA-A-2.
Пример 9
Получение соединения 10 (RA-A-3). (Соединение формулы (I), где Y является - CH2OH, X является - NH-(CH2)3- N[(CH2)3NH2]2, R1 является H, R2 представляет собой (C9 - C12) алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D- маннопиранозил и Z является - H).
При перемешивании в раствор 9 г (около 5 ммоль) в 100 мл DMSO добавили 7 г (около 18 ммоль) N',N''-ди(-tBOC)-трис-(3-аминопропил)амина (пример 7, этап а), 1,5 мл TEA и 3 мл DPPA при 10oC. После перемешивания при 10oC в течение 1 часа и при комнатной температуре в течение 4 часов добавили 400 мл этилацетата. Выпавший твердый осадок (около 12 г) повторно растворили в 80 мл ледяного TFA и полученный раствор перемешивали при 0-5oC в течение 10 минут. Затем добавили смесь метанол/диэтиловый эфир 1/1 (около 300 мл), предварительно охлажденную до - 10oC. Твердый осадок собрали и быстро повторно растворили в 200 мл воды. pH полученного раствора довели до 4 с помощью 1Н NaOH и провели очистку хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы) до получения 1,8 г чистого искомого соединения RA-A-3.
Пример 10
Получение соединения II (A-A-1). (Соединение формулы (I), где Y является - COOH, X является - NH-(CH2)3-N(CH3)2, R1 является - H, R2 представляет собой (C9-C12) алкил, соответствующий факторам комплекса A 40926, M представляет собой - маннопиранозил и Z является - H).
При перемешивании в суспензию 5 г (около 2,5 ммоль) соединение 6 (MA-A-1), полученного как описано в примере 3, способ B, в 60 мл тетрагидрофурана (THF) добавили 10 мл воды и 20 мл 1H NaOH при комнатной температуре. Спустя 30 мин pH полученного раствора довели до 7 с помощью 1Н HCl, добавили 150 мл n-бутанола и концентрировали смесь до малого объема (около 20 мл) при 40oC под пониженным давлением. Твердый осадок, выпавший после добавки диэтилового эфира (около 200 мл) собрали (5,2 г) и очищали хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы), получив 2,1 искомого соединения A-A-1.
Пример 11
Получение соединений 12 (Py A-A-1) (Соединение формулы (I), где Y является - COO⊖ , X является - NH-(CH2)3- N(CH3)2, R1 является - H, R2 представляет собой (C9-C12) алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D- маннопиранозил и Z является P⊕ (NC4H8)3).
Соединение 12 (Py A-A-1) получают из соединения 7 (Py MA-A-1) из примера 4 способом, описанным в примере 10 для получения соединения II (A-A-1) из соединения 6 (MA-А-1), выход - 35%.
Пример 12
Получение соединения 13 (A-A-3/B0). (Соединение формулы (I), где Y является - COOH, X является - NH - (CH2)3 - N[(CH2)3NH2]2, R1 является - H, R2 представляет собой 9-метилдецил, M представляет собой α-D-манопиранозио и Z является - H).
Соединение 13 (A-A-3/B0) получают из соединения (MA-A-3/B0) из примера 7 способом, описанным в примере 10 для получения соединения II (A-A-1) из соединения 6 (MA-A-1), выход - 41%.
Пример 13
Получение соединения 14 (ABA-A-1). (Соединение формулы (I), где Y является - CONHCH3, X является - NH-(CH2)3- N(CH3)2, R1 является - H, R2 представляет собой (C9-C12) алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D- маннопиранозил и Z является - H).
Этап a: получение N15-(t-BOC)-A-A-1, 6b-цианометиловый эфир.
При перемешивании в раствор 22 г (около 11 ммоль) соединения II (A-A-1) из примера 10 и 3 г NaHCO3 в 220 мл смеси вода/диоксан 1/1 добавили раствор 5 г ди-трет-бутил-дикарбоната в 20 мл сухого диоксана, добавку производили по капле при комнатной температуре в течение 10 мин. После перемешивания в течение 2 часов при комнатной температуре добавили 200 мл воды, затем pH полученного раствора довели до 3 с помощью 1Н HCl и экстрагировали раствор 300 мл n-бутанола. Органический слой отделяли и концентрировали при 35oC под пониженным давлением до малого объема (около 45 мл). Твердый осадок, выпавший после добавки диэтилового эфира (около 250 мл) собрали (около 20 г неочищенного N15-(t-BOC)- A-A-1) и повторно растворили в 150 мл DMSO. После добавки 3 мл TFA и 20 мл хлорацетонитрила полученный раствор перемешивали при комнатной температуре в течение 5 часов, затем добавили 500 мл этилацетата. Выпавший твердый осадок (около 18 г. цианометилового эфира) был достаточно чистым для использования на следующем этапе.
Этап b: Взаимодействие полученного выше 6b-цианометлового эфира с метиламином и удаление защитной группы t-BOC.
Раствор 5 г вышеуказанного продукта в 75 мл 250 (м/о) метиламина в этаноле перемешивали при комнатной температуре в течение 3 часов, затем добавляли 300 мл диэтилового эфира. Выпавший твердый осадок (около 5,1 г) собирали и повторно растворяли в 35 мл TFA при 0oC. Полученный раствор перемешивали при 0oC в течение 15 мин, затем добавляли 50 мл смеси метанол/диэтиловый эфир 1/1 до выпадения в осадок 4,5 г неочищенного продукта, который очищали колоночной хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы) до получения 1,7 г искомого соединения 14 (ABA-A-1).
Пример 14
Получение соединения 15 (ADA-A-1). (Соединение формулы (I), где Y является - CONH-(CH2)3-N(CH3)2, X является - NH-(CH2)3-N(CH3)2, R1 является - H, R2 представляет собой (C9-C12)алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D- маннопиранозил и Z является - H).
Раствор 7 г (около 4 ммоль) антибиотического комплекса A 40929, 2,5 мл (около 20 ммоль) 3,3-диметиламино-1-пропиламина и 5,2 г (около 10 ммоль) Py BOP в 70 мл DMF перемешивали при комнатной температуре в течение 1 часа, затем добавляли 400 мл этилацетата. Выпавший твердый осадок собирали и очищали хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы), получая 2,1 г искомого соединения 15 (ADA-A-1).
Пример 15
Получение соединения 16 (Py Ra-A-A-1). (Соединение формулы (I), где Y является - CH2OH, X является - NH-(CH2)3- N(CH3)2, R1 является - H, R2 является (C9-C12)алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D- маннопиранозил и Z является P⊕(NC4H8)3CH3COO⊖.
При перемешивании в раствор 400 мг (около 0,2 ммоль) соединения 7 (Py MA-A-1), полученного способом, описанным в примере 4, в 20 мл воды добавили 4 мл n-бутанола и 200 мг NaBH4, добавку производили при комнатной температуре. После перемешивания при комнатной температуре в течение ночи pH реакционной смеси доводили до pH 4,5 с помощью ледяной уксусной кислоты и экстрагировали смесь 50 мл n-бутанола. Органический слой отделяли и растворитель выпаривали при 45oC под пониженным давлением. Твердый осадок очищали хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные факторы) до получения 175 мг чистого искомого соединения 16 (Py RA-A-1).
Пример 16
Получение соединения 25 (MA-A-4). (Соединение формулы (I), где Y является - COOCH3, X представляет собой:
R1 является - H, R2 представляет собой (C9-C12)алкил, соответствующий факторам комплекса A 40926, M является α-D- маннопиранозилом и Z является - H).
При перемешивании в раствор 5 г соединения из примера 1 (MA) в 60 мл смеси DMF/DMSO 5/1, добавляли 0,3 мл N-метил-пиперазина и 1,7 г Py BOP, добавку производили при комнатной температуре. Через 1 час протекания реакции добавили 140 мл этилацетата и выпавший твердый осадок собрали и очистили колоночной хроматографией с обращенной фазой (соединяя все фракции, содержащие чистые индивидуальные компоненты), получив 1,9 г искомого соединения MA-A-4.
Пример 17
Получение соединения 24 (RA-A-4). (Соединение формулы (I), где Y является - CH2OH, X представляет собой: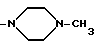
R1 является - H, R2 представляет собой (C9-C12)алкил, соответствующий факторам комплекса A 40926, M представляет собой α-D-маннопиранозил и Z является - H).
Применив способ, описанный в примере 16 выше и в тех же условиях, из 5 г RA получили 2,7 г чистого искомого соединения RA-A-4.
Колоночная хроматография с обращенной фазой
Чистые образцы вышеуказанных соединений получали с помощью колоночной хроматографии с обращенной фазой на силанизированном силикагеле (0,063 - 0,2 мм; "Merck"). Неочищенный продукт (напр., 0,5 г) растворяли в минимальном количестве смеси ацетонитрил/вода 1/1, затем pH раствора доводили до 7 с помощью 1H NaOH и разбавляли раствор водой до образования мутного раствора. После этого добавляли несколько капель ацетонитрила при энергичном перемешивании. Как только раствор становился прозрачным, его помещали на колонку силанизированного силикагеля (100 г) в воде. Элюирование проводили линейным градиентом с 10 до 60% ацетонитрила в 0,1 н. уксусной кислоты в течение 10 часов при скорости потока около 250 мл/час, при этом собирая 20 мл фракции, которые подвергали ВЭЖХ анализу. Фракции, содержащие чистые соединения формулы (I), выбирали и, в тех случаях, когда искомым соединением были соединения формулы (I), где R2 представляло собой (C9-C12)-алкил, соответствующий фракциям комплекса A 40926, то все фракции, содержащие чистые факторы, соединяли и растворители выпаривали при 40oC под пониженным давлением в присутствии n-бутанола для того, чтобы избежать пенообразования.
В тех случаях, когда процесс получения соединения формулы (I) антибиотического комплекса A 40926 использовался для получения предшественника и искомым соединением являлся индивидуальный фактор амидного соединения формулы (I), где R2 соответствует одному из значений, характеризующих индивидуальные факторы комплекса A 40926 (напр., R2 = 9-метилдецил), то соединяли и подвергали вышеописанной обработке только те фракции, которые согласно ВЭЖХ содержали искомый чистый фактор.
Идентичность и структуру каждого отдельного фактора соединений по изобретению определяли, проводя ВЭЖХ анализ каждого продукта реакции. Соответственно, предварительную идентификацию искомого фактора производили путем сравнения отпечатков ВЭЖХ комплекса A 40926 с отпечатками неочищенных продуктов реакции. (см. напр., спектрограммы в работе Л.П.Церилли и др. "Rapid Communications in Mass Spectrometry", том 6, 109, 1992) (в этой работе фактор B0 комплекса A 40926 назван фактором B).
ВЭЖХ - анализ проводили на колонке "Hibar" (125 • 4 мм; "Merck"), предварительно насаженной реактивом "Li-Chrospher RP-8" (5 мкм), используя жидкостной хроматограф "Varian Model 5500, оснащенный переменным УФ-детектором. Хроматограммы записывали в 254 нм. Элюирование производили с помощью линейного ступенчатого градиента от 20 до 60% ацетонитрила в 0,2% водном формиате аммония в течение 30 мин при скорости потока в 1,5 мл/мин.
Поскольку в целом все комплексные соединения по изобретению дают типичные отпечатки ВЭЖХ, аналогичные отпечатку соответствующего предшественника комплекса A 40926, индивидуальные факторы соединения по изобретению, соответствующие факторам предшественника комплекса A 40926, можно легко идентифицировать посредством корреляции двух спектрограмм ВЭЖХ. Элюированные фракции хроматограмм с обращенной фазой, которые содержат указанные чистые факторы, можно отделить и подвергнуть обработке, как описано выше. Чтобы подтвердить идентичность (C9-C12) алкильных цепей, образец каждой фракции можно выпарить вышеописанным способом до получения продукта, который можно подвергнуть газ-хроматографии/масс-спектрометрическому анализу согласно способу, описанному в работе Л.Ф. Церилли и др., упомянутой выше.
В табл. 5 приведены значения времени удерживания (tR) чистого фактора каждого из соединений формулы (I) по изобретению, где R2 представляет собой 9-метилдецил, т.е. соответствующего фактору B0 комплекса A 40926 (эти значения использовались в ходе очистки с помощью хроматографии с обращенной фазой).
В табл. 5 приведены также значения tR фактора B0 предшественника комплекса A 40926 и соответствующего эфирного исходного материала (MA), полученные в вышеописанных условиях.
Далее показан наиболее значительный химический сдвиг (дельта ммд) у некоторых репрезентативных соединений.
Соединение 1 (RA):
0,85, 1,13, 1,42, 1,98 (ацильная цепь); 3,72 (CH2OH), 4,05-6,22 (пептидные CH); 6,43-8,52 (ароматические протоны и пептидные NH).
Соединение 2 (MA-A-1/B0):
0,83, 1,14, 1,38, 1,99 (ацильная цепь); 1,83, 2,83 (CH2 - боковая цепь), 2,73 (N(CH3)2); 4,11 - 6,10 (пептидные CH); 6,48 - 9,50 (ароматические протоны и пептидные NH).
Соединение 3 (RA-A-1/B0):
0,84, 1,14, 1,38, 1,92 (ацильная цепь); 1,72, 2,75 (CH2 - боковая цепь); 2,53 (N(CH3)2); 3,69 (CH2-OH); 4,09 - 6,11 (пептидные CH); 6,41 - 9,18 (ароматические протоны и пептидные NH)
Соединение 4 (MA-A-2/B0):
0,84, 1,15, 1,39 ,1,98 (ацильная цепь); 1,96, 2,86 (CH2 - боковая цепь); 4,08 - 6,15 (пептидные CH); 6,42 - 9,61 (ароматические протоны и NH).
Соединение 5 (MA-A-3/B0):
0,85, 1,13, 1,42, 2,02 (ацильная цепь); 1,73, 2,82 (алкиламинные цепи); 2,42 (-N-CH3); 3,63 (COOCH3); 3,10 - 3,80 (сахара); 4,10 - 6,10 (пептидные CH); 6,41 - 8,52 (ароматические протоны и пептидные NH).
Соединение 7 (Py MA-A-1);
0,84, 1,13, 1,42, 2,01 (ацильная цепь); 1,83, 2,16 (диметилпропил-амид); 2,32 (HN-CH3); 1,70, 3,23 (пирролидин); 3,10 - 3,80 (сахара); 4,10 - 6,20 (пептидные CH); 6,38 - 8,40 (ароматические протоны и пептидные NH).
Соединение 9 (RA-A-2):
0,84, 1,13, 1,39, 1,98 (ацильная цепь); 1,88, 2,91 (алкиламинные цепи); 2,41 (HN-CH3); 3,10 - 3,80 (сахара); 4,10 - 6,10 (пептидные CH); 6,38 - 8,49 (ароматические протоны и пептидные NH).
Соединение 10 (RA-A-3):
0,84, 1,13, 1,39, 1,98 (ацильная цепь); 1,73, 2,82 (алкиламинные цепи); 2,47 (HN-CH3); 3,10 - 3,80 (сахара); 4,10 - 6,10 (пептидные CH); 6,37 - 8,70 (ароматические протоны и пептидные NH); 9,2 - 1,04 (фенольные OH).
Соединение 11 (A-A-1):
0,84, 1,13, 1,39, 2,00 (ацильные цепи); 1,74 - 2,79 (алкиламинные цепи); 2,37 (NH-CH3); 3,10 - 3,80 (сахара); 4,10 - 6,10 (пептидные CH); 6,39 - 8,50 (ароматические протоны и пептидные NH).
Соединение 12 (Py A-A-1):
0,84, 1,13, 1,42, 2,02 (ацильные цепи); 1,87, 2,73, 3,00 (диметилпропиламид); 2,48 (NH-CH3); 1,71, 3,30 (пирролидин); 3,10 - 3,80 (сахара); 4,10 - 6,25 (пептидные CH); 6,38 - 8,55 (ароматические протоны и пептидные NH).
Соединение 13 (A-A-3/B0):
0,84, 1,13, 1,42, 2,02 (ацильные цепи); 2,33 (NH-CH3); 1,71, 2,80 (алкиламинные цепи); 3,10 - 3,80 (сахара); 4,10 - 6,10 (пептидные CH); 6,37 - 8,50 (ароматические протоны и пептидные NH).
Соединение 14 (ABA-A-1):
0,84, 1,13, 1,42, 1,96 (ацильные цепи); 2,35 [(CH-NH-)-CH3); 1,78, 2,70 (алкиламинные цепи); 3,10 - 3,80 (сахара); 4,10 - 6,10 (пептидные CH); 6,37 - 8,50 (ароматические протоны и пептидные NH).
Соединение 15 (ADA-A-1):
0,82, 1,13, 1,40, 1,98 (ацильные цепи); 2,50 (NH-CH3); 1,72, 1,85, 2,73, 3,00 (алкиламинные цепи); 3,10 - 3,80 (сахара); 4,10 - 6,10 (пептидные CH); 6,40 - 8,55 (ароматические протоны и пептидные NH).
Соединение 16 (Py RA-A-1): 0,84, 1,13, 1,41, 2,00 (ацильные цепи); 2,33 (NH-CH3); 1,82, 2,16 (диметилпропиламид); 1,71, 3,23 (пирролидин); 3,10 - 3,80 (сахара); 4,10 - 6,20 (пептидные CH), 6,38 - 8,40 (ароматические протоны и пептидные NH).
Соединение 24 (RA-A-4):
0,84, 1,13 1,40, 1,97 (ацильные цепи); 2,10 (пиперазин CH3); 2,38 (NH-CH3); 3,10 - 3,80 (сахара); 4,05 - 6,07 (пептидные CH); 6,38 - 8,49 (ароматические протоны и пептидные NH).
Соединение 25 (MA-A-4):
0,84, 1,13 1,40, 2,00 (ацильные цепи); 2,13 (пиперазин CH3); 2,43 (NH-CH3); 3,10 - 3,80 (сахара); 3,63 (COOCH3); 4,05 - 6,09 (пептидные CH); 6,38 - 8,49 (ароматич. протоны и пептидные NH).
31P-ЯМР - спектры брали при 161,98 МГц (соединение 12) или при 202,46 МГц (соединения 7 и 16) в DMSO-d6 (растворе) с 85% H3PO4 в качестве внутренней справки.
Соединение 7 (Py MA-A-1) (31P): один сигнал при дельта 24,12 ммд
Соединение 12 (Py A-A-1) (31P): один сигнал при дельта 23,50 ммд
Соединение 16 (Py PA-A-1) (31P): один сигнал при дельта 24,11 ммд
Композиция для парентерального введения.
Композицию готовят растворением 100 мг соединения 3 (табл. 1) в стерильной воде для инъекций (5 мл), доведенной до pH 4,5 - 5,5. Формы для парентерального введения для соединений 5, 6, 8, 10, 13, 21 25, 26 и 31 готовят аналогичным образом с использованием того же количества активного вещества и носителя.
Мазь для местного применения
Соединение 33 - 200 мг
Полиэтиленгликоль 4000 - 3-6 г
Полиэтиленгликоль 400 - 6,2 г,




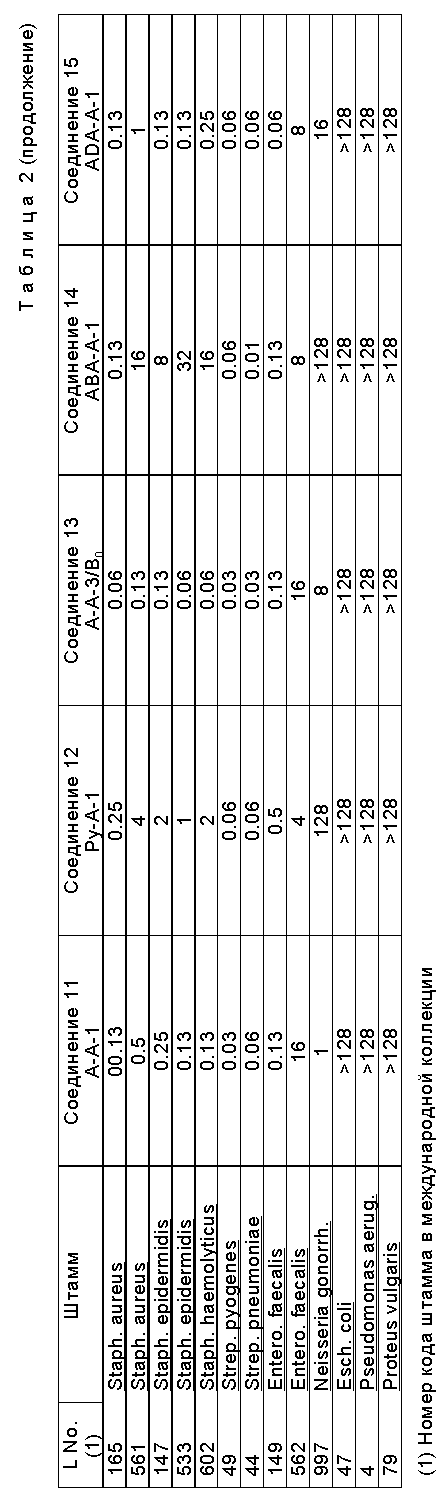



Настоящее изобретение относится к новым производным антибиотика А 40926 общей формулы 1, которые имеют карбоксильный, (C1-C4) алкоксикарбонильный, аминокарбонильный, (C1-C4) алкиламинокарбонильный, ди (C1-C4) алкиламинокарбонильный или гидроксиметильный заместитель на N -ациламиноглюкуронильном фрагменте и гидроксильный или полиаминный заместитель в положении 63 молекулы. Соединения по изобретению проявляют высокую активность in vitro против энтерококков и стафилококков, устойчивых к гликопептидам. Синтез производных А 40926 ведут с использованием реакций амидирования, восстановления аминогруппы. 3 с. и 19 з.п. ф-лы, 5 табл.
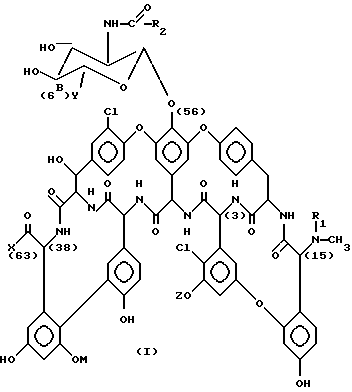

R1 - водород, или защитная группа аминогруппы;
R2 - С9-С12 алкил;
М - водород, α-D-маннопиранозил;
Y - карбокси, - С1-С4 алкоксикарбонил, аминокарбонил, гидроксиметил;
Х - гидрокси- или аминный остаток формулы
NR3-alK1-(NR4- alK2)p-(NR5- alK3)q-W,
где R3 - водород или С1-С4 алкил; alK1, alK2 и alK3 каждый независимо представляет собой линейный или разветвленный алкилен С2-С10; p и q каждый = 0 или 1;
R4 и R5 каждый независимо водород, С1-С4 алкил;
W - водород, С1-С4 алкил, амино, С1-С4-алкиламино, ди(С1-С4)-алкиламино, аминогруппа, замещенная одним или двумя амино(С2-С4)-алкильными группами, и если p и q равны 0, то W вместе с фрагментом -NR3- alK1- представляет собой пиперазино- или 4-метилпиперазинорадикал при условии, что когда Х-гидрокси, то Y-гидроксиметил, Z-водород или группа формулы
где A⊖ анион минеральной или органической кислоты, или их фармацевтически приемлемые соли присоединения.
NR3-alK1-(NR4- alK2)p-(NR5- alK3)q-W,
где R3, R4 и R5-водороды, alK1, alK2 и alK3 каждый независимо представляют собой линейный или разветвленный алкилен С2-С4 p и q = 0 или 1, W - водород, С1-С4 алкил, амино, С1-С4алкиламино, ди(С1-С4)-алкиламино, аминогруппа, замещенная одним или двумя амино(С2-С4)-алкильными группами и, если p и q равны 0, то W вместе с фрагментом - NR3- alK1- представляет собой пиперазино- или 4-метилпиперазинорадикал, при условии, что когда Х - гидрокси, то Y - гидроксиметил, Z - водород или группа формулы
где А- анион минеральной или органической кислоты или его фармацевтически приемлемые соли присоединения.
NR3-alK1-(NH-alK2)p-(NH-alK3)q-W,
где R3 - водород, alK1, alK2 и alK3 каждый независимо линейный алкилен С2-С4, p и q каждый независимо равны 0 или 1, W-амино, С1-С4 алкиламино, ди С1-С4 алкиламино, аминогруппа, замещенная одним или двумя амино (С2-С4)алкильными группами, или, когда оба p и q равны 0, то W вместе с фрагментом -NR3-alK1- представляет пиперазино- или 4-метилпиперазино, Z-водород или его фармацевтически приемлемые соли присоединения. Z - водород или его фармацевтически приемлемые соли присоединения.
Z - водород или его фармацевтически приемлемые соли присоединения.
R1 - водород или защитная группа аминогруппы;
R2 - С10-С11 алкил;
M - водород, α-D-маннопиранозил;
Y - гидроксиметил;
Х - гидрокси или аминный остаток -NR3-alK1-(NR4- alK2)p-(NR5- alK3)q-W, где R3-водород или C1-C4алкил, alK1, alK2 и alK3 каждый независимо линейный или разветвленный алкилен С2-С10;
p и q независимо = 0 или 1;
R4 и R5 независимо водород, С1-С4 алкил;
W - водород, С1-С4 алкил, амино, С1-С4 алкиламино, ди(С1-С4)алкиламино, аминогруппа, замещенная одним или двумя амино (С2-С4) алкильными группами, и при условии, что когда Х - гидрокси, Y - гидроксиметил,
или его фармацевтически приемлемые соли присоединения.
а) в случае, когда Х - аминный остаток -NR3-alK1-(NR4- alK2)p-(NR5- alK3)q-W, соединение общей формулы II
где R'1, R'2, M' имеют значения, указанные для R1, R2 и M;
Y'-C1-C4 алкоксикарбонил;
Х' - гидрокси,
подвергают реакции амидирования с аминным реагентом общей формулы III
-NHR3-alK1-(NR4- alK2)p-(NR5- alK3)q-W,
где R3, R4, R5, alK1, alK2, alK3, p, q, и W имеют вышеуказанные значения,
в присутствии конденсирующего агента или путем образования активированного эфира в С63-карбоновой кислоты и
i) необязательно полученное производное формулы I, где Y'-C1-C4 алкоксикарбонил, а R1 - защитная группа аминогруппы, а все другие радикалы имеют вышеуказанные значения, подвергают восстановлению с помощью боргидрида щелочного металла и возможно защитную группу аминогруппы удаляют с получением соединения I, где Y - гидроксиметил, а R1 - водород;
ii) необязательно полученное производное формулы I, где Y-С1-С4 алкоксикарбонил или гидроксиметил, а R1 - защитная группа для N15 аминогруппы, R2, M, Z имеют вышеуказанные значения, X - аминный остаток -NR3-alK1-NHR4, где R3 и R4 каждый независимо водород или C1-C4-алкил, alK1 имеет вышеуказанные значения; или является остатком -NR3-alK1- alK2-NHR5, где R3, R4 и R5 каждый независимо водород или (C1-C4) алкил, alK1 и alK2 имеет вышеуказанные значения, алкилируют с аминным реагентом общих формулы IY или IYа:
r-alK2-(NR5-alK3)q-W, IY
r-alK2-W, IYа
где R5, alK2, alK3 и W имеют вышеуказанные значения,
q = 0 или 1;
r - галоид, метансульфонил или тозил,
в присутствии акцептора кислоты в инертном растворителе и, возможно, защитную группу в N15 удаляют;
iii) необязательно полученное производное формулы I, где где Y-С1-С4 алкоксикарбонил и все остальные радикалы имеют вышеуказанные значения, обрабатывают водным гидроксидом щелочного металла до получения соответствующего соединения I, где Y - карбокси;
d) в тех случаях, когда целевым соединением является производное формулы I, где R1, R2, M, Z имеют значения, указанные в начале данного пункта, а Y и COX представляют различные карбоксиамидные остатки, причем Y - аминокарбонил, X - аминный остаток -NR3-alK1-(NR4-alK2)p-(NR5-alK3)-W, где R3, R4, R5, alK1, alK2, alK3, p, g и W имеют вышеуказанные значения, производное формулы I, где R1, R2, M и Z имеют вышеуказанные значения, а X - аминный остаток -NR3-alK1-(NR4-alK2)p-(NR5-alK3)q-W, где R3, R4, R5, alK1, alK2, alK3, p, g и W имеют вышеуказанные значения, а Y - карбокси, подвергают амидированию соответствующим амином до образования вышеуказанного карбоксиамидного остатка Y в присутствии конденсирующего агента.
X - гидрокси;
Y' -С1-С4-алкоксикарбонил;
R1 1 - соответствующая защитная группа аминогруппы,
восстанавливают боргидридом щелочного металла, предпочтительно боргидридом натрия, при температуре 0 - 40oC в водно-спиртовой среде с последующим удалением защитной группы.
Приоритет по пунктам:
29.07.91 - по пп.1 - 22, кроме значений R2 - C9-C12-алкил, Y - карбокси, аминокарбонил, Z - группа  W - вместе с фрагментом NR3-alK1-пиперазино или 4-метилпиперазино, когда p и q равны 0;
W - вместе с фрагментом NR3-alK1-пиперазино или 4-метилпиперазино, когда p и q равны 0;
12.06.92 - по пп.1 - 22.
| 0 |
|
SU178152A1 | |
| Способ получения полипропилена | 1975 |
|
SU538002A1 |
| СПОСОБ УПАКОВКИ ПОРОШКООБРАЗНОГО МАТЕРИАЛА | 0 |
|
SU240609A1 |

