Настоящее изобретение предоставляет усовершенствованные способы ферментативного получения углеводов, особенно фукозилированных углеводов. В нем предложен усовершенствованный синтез гликозил-1- или 2-фосфатов, в результате использования как химических, так и ферментативных средств. Затем эти фосфорилированные гликозиды используют для получения нуклеотидов сахаров, которые в свою очередь используют как донорные сахара для гликозилирования акцепторных углеводов. Наиболее предпочтительным является использование раскрытых способов для фукозилирования.
В настоящем изобретении предложен способ получения фукозилированных углеводов в единой реакционной смеси, включающие стадии: использования фукозилтрансферазы для создания O-гликозидной связи между нуклеозид-5'-дифосфо-фукозой и доступной гидроксильной группой углеводной акцепторной молекулы до получения фукозилированного углевода и нуклеозид-5'-дифосфата; рециркулирование in situ нуклеозид-5'-дифосфата с фукозой до образования соответствующей нуклеозид-5'-дифосфо-фукозы. Предпочтительные способы настоящего изобретения включают использование гуанина в качестве основания для нуклеозида, использование каталитических количеств нуклеозидов, использование N-ацетигликозамина, галактозы, N-ацетилгалактозамина или N-ацетиллактозамина в качестве углеводной акцепторной молекулы, и использование сиалилированной углеводной акцепторной молекулы.
В настоящем изобретении далее рассматривается вышеуказанный способ получения фукозилированной сиалилированной углеводной молекулы через ферментативное образование гликозидных связей в единой реакционной смеси, включающий: создание первой гликозидной связи между дифосфонуклеозид-активированным гликозильным донором, таким, как UDP-Gal, и доступной гидроксильной группой углеводной акцепторной молекулы, такой, как GlcNAc, с использованием первой гликозилтрансферазы, такой, как β-1,4- галактозилтрансфераза, при получении Ga1βI, 4GlcNAc; создание второй гликозидной связи между монофосфонуклеозид-активированным сиалильным донором, таким, как CMP-NeuAc, и доступной гидроксильной группой акцепторной молекулы сахара, такой, как гидроксил в 3-положении Gal Ga1βI, 4GlcNAc, с использованием сиалилтрансферазы, такой, как α-2,3- сиалилтрансфераза; создание третьей гликозидной связи между дифосфонуклеозидактивированным фукозильным донором, таким, как GDP-Fuc, и доступной гидроксильной группой акцепторной молекулы сахара, например, в 3-положении гидроксила GlcNAc Ga1β1,4- GlcNAc, с использованием такой фукозилтрансферазы, как α- 1,3/4-фукозилтрансфераза, где, по крайней мере, одна из стадий (a), (b) или (c) включает далее образование in situ фосфонуклеотид-активированного гликозильного донора из каталитического количества соответствующего монофосфатного и дифосфатного нуклеозида. Наиболее предпочтительны способы настоящего изобретения, в которых продукт с фукозилированным сиалилированным углеводным фрагментом является таким сиалилированным льюисовым лигандом, как сиалил Lex(SLex) или сиалил Lea (SLea), и где фукозу переносят из фукозилированного донора к гидроксильной группе N-ацетилглюкозаминового или галактозного остатка углеводной акцепторной молекулы.
Рассматриваемый способ охватывает многочисленные катализируемые гликозилтрансферазой реакции в единой реакционной смеси, и предпочтительны такие способы, в которых одну гликозилтрансферазу выбирают из группы, состоящей из α-2,3- сиалилтрансферазы, α-2,4- сиалилтрансферазы, α-2,6- сиалилтрансферазы и α-2,8- сиалилтрансферазы. Настоящее изобретение включает фукозилирование олигосахарида, и предпочтительны такие фукозилтрансферазы, которые выбирают из группы, состоящей из: α-1,2- фукозилтрансферазы, α-1,3/4- фукозилтрансферазы, α-1,3- фукозилтрансферазы, α-1,6- фукозилтрансферазы и α-1,4- фукозилтрансферазы. Наиболее предпочтительные фукозилтрансферазы включают β- галактозилазо -α-1,2- фукозилтрансферазу, N-ацетиглюкозамин α-1,3- фукозилтрансферазу, N-ацетилглюкозамин -α-1,4- фукозилтрансферазу и N-ацетил-глюкозамин α-1,6- фукозилтрансферазу.
Углеводные акцепторные молекулы практически неограничены, так как гликозилтрансферазы не селективны относительно положений соседних сахаров. Таким образом, это могут быть любые углеводные замещенные молекулы, в которых углевод является Ga1β1,4G1cNAc молекулой или ее аналогом, или она может оканчиваться Ga1β1,4G1cNAc-X фрагментом, где X представляет органическую молекулу. Дополнительные углеводные акцепторные молекулы, которые являются субстратами для фукозилазы, включают аналоги Ga1β1,4G1cNAc и Ga1β1, 4Glclac-X. Примерами таких молекул являются лактоза, NeuAcα1,6Ga1β1, 4GlcNAc, Ga1β1, 3GlcNAc, Ga1β1,4-G1uca1 (лактал), NeuAcα2,3Ga1β1,4G1uca1, 2-галоидзамещенные продукты реакции вышеуказанных глюкалей, Ga1β1,4/5- Ga1β1,4G1cNAcβ-O- аллил и т.п. Следует также учитывать, что углеводная акцепторная молекула должна содержать доступную гидроксильную группу на сахариде, с которой связана фукозильная или другая донорная группа сахара, а гидроксил, который должен присутствовать, определяется гликозилтрансферазным ферментом, который используют в этой реакции.
Рассматриваемый здесь способ включает далее регенерацию каталитических количеств нуклеотидов, которые используют для создания нуклеозидных сахаров. Предпочтительными основаниями для нуклеотидов являются цитидин, гуанин или уродин. Доноры моносахароидов активируются такими нуклеотидными сахарами, как цитидин-5'-монофосфо-N-ацетилнейраминовая кислота, гуанидин-5'-дифосфофукоза и уродин-5'-дифосфогалактоза.
Помимо вышеуказанных способов, настоящее изобретение включает также in vitro реакционные системы. Такие системы называют инертным или нереакционноспособными контейнерами, или отделениями, сохраняющими реагенты, используемые для проведения вышеуказанных реакций. Более конкретно, эти реакционные системы содержат, как минимум, фукозилтрансферазу и нуклеозиддифосфофукозо-образующие ферменты. Эти реакционные системы могут, кроме того, содержать гуанозиндифосфофукозофосфорилазу в качестве GDP-фукозо-образующих ферментов, киназу, такую, как пируваткиназу или фруктозо-1,6-дифосфаткиназу, ацетилкиназу или фукозокиназу. Другие реагенты могут включать NADPH регенерационную систему гуанозиндифосфатманнозы и гуанозиндифосфоманнозопирофосфорилазу. Если присутствует NADPH система, она может включать каталитическое количество NADP, изопропанол в количестве от около 1 до около 10%, предпочтительно, от около 2 до около 4% вес/объем реакционной системы, и алкоголь-дегидрогеназу.
Ряд химических способов для синтеза олигосахаридов также здесь описан. Один способ включает получение гликозил-1-или 2-фосфата за счет реакции защищенного гликозильного кольца, содержащего гидроксил в аномерном положении (1- или 2-положение) с трехвалентным фосфитилирующим реагентом до получения защищенного гликозил 1- или 2-фосфит-защищенного кольца. Защищенный фосфит окисляют до получения соответствующего фосфата, который используют в ферментативной реакции. Гликозильное кольцо может далее включать галактозил, глюкозил, фукозил, N-ацетилглюкозил и маннозил, также как и другие сахариды. Предпочтительными трехвалентными фосфитилирующими агентами являются такие дибензил-N, N-диалкилфосфорамидиты, как дибензил-N, N-диэтилфосфороамидит. Такие диалкилы являются низшими алкилами, содержащими 1 - 5 атомов углерода включительно, и они могут быть одинаковы или различны. В этом случае далее используют такие защитные реагенты, как ацетил или бензил. Гликозильное кольцо необязательно представляет группу, состоящую из D- или L-альдоз, содержащих четыре, пять или шесть атомов углерода, или образует группу, состоящую из D- или L-кетоз, содержащих четыре, пять или шесть атомов углерода, а также сахаридов, содержащих вплоть до девяти атомов углерода в цепи сахарида.
Далее настоящее изобретение включает новые промежуточные соединения для получения гликозил-1- или 2-фосфатов. Предпочтительным промежуточным соединением является защищенный фосфитилмоносахарид формулы I: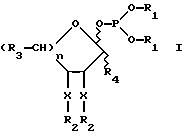
где
R1 представляет арил или низший алкил;
x независимо является кислородом или азотом;
R2 независимо является ацильной, бензильной, силильной или алкильной защитной группой;
R3 независимо представляет -CH3, -OR2, -CH2OR2, -CH(OR2)-CH(OR2) или -CH(OR2)-CH(OR2)-CH(OR2);
R4 представляет водород (H), карбоксил или C1 - C5 или бензилкарбоксилатный сложный эфир; а
n является целым числом 1 или 2.
В предпочтительной группе соединений формулы I R4 представляет водород, так что формула I становится формулой II, приводимой ниже, где R1, R2, R3 и X и n имеют указанные ранее значения.
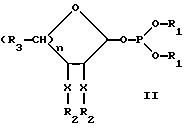
Одной из групп наиболее предпочтительных соединений являются те, в которых моносахарид является шестичленным кольцом, R4 представлен H, а каждый X является кислородом, например, манноза или фукоза. Предпочтительны соединения, в которых R1 представляет бензил, а R2 представляет бензил или ацетил. Примеры предпочтительных промежуточных соединений включают дибензилфосфитил-2,3,4,6-тетра-O-ацетил-D-маннозид или дибензилфосфитил-2,3,4-три-O-ацетил-L-фукозид.
Другой группой наиболее предпочтительных соединений являются те, в которых моносахарид является шестичленным кольцом, R1 и R2 имеют указанные ранее значения, один X представляет азот, а остальные кислород. Примеры соединений этой группы включают GlaNAc, GalNAc и NeuAc. Примерами этих соединений служат дибензилфосфитил-2-ацетамидо-2-деокси-3,4,6-три-O-ацетил-D-глюкозид и 2-ацетамидо-2-деокси-3,4,6-три-O-ацетил-D-галактозид.
Раскрыт также моносахаридный аналог, который является 2,3,4-три-O-бензоил -α-L- фукопиранозилбромидом.
Фраза "доступная гидроксильная группа" относится к гидроксизамещенному углероду, образующему часть кольца углеводной акцепторной молекулы, которая может образовывать гликозидную связь в результате действия гликозилтрансферазы, переносящей моно- или дифосфонуклеозид-активированный гликозильный донор на доступный углерод. "Доступная гидроксильная группа" обычно находится в 3-положении для фукозилирования.
Фраза "защищенное гликозильное кольцо" относится к гликозильным кольцам, в которых доступные амино- или гидрокси-заместители прореагировали с ацильной, бензильной, силильной или алкильной защитными группами. Такие группы описаны в книге Грина Т.В. (Green T.W., "Protective Groups in Organic Synthesis", John Wiley and Sons, 1981).
Термин "углевод(ы)" включает любой органический фрагмент, содержащий углеводы, ковалентно связанные с любыми мономерными сахаридами. Он включает дисахариды, олигосахариды, гликолипиды, гликопротеины и неприродные связи, такие, как связи сахаридов с органическими соединениями, которые в природе не связаны с сахарами.
Фраза "углеводная акцепторная молекула" относится к молекуле, содержащей по крайней мере один моносахарид, где этот моносахарид имеет одну или более доступную гидроксильную группу для образования гликозидных связей с моно- или ди-фосфонуклеозид-активированным гликозильным донором.
Фраза "каталитическое количество" относится к концентрациям реагентов, которые присутствуют в относительно небольших количествах по сравнению с количествами реагентов, которые присутствуют в стехиометрических количествах, и их концентрация в значительной степени не уменьшается в процессе реакции. Те реагенты, которые присутствуют в каталитических количествах, обычно является активирующими реагентами, которые затем регенерируют и возвращают в реакцию за счет побочных реакций.
Фраза "моно- или дифосфонуклеозид-активированный гликозильный донор" или "активированная донорная молекула" относятся к таким нуклеотидным сахарам, как уридин-5'-дифосфо-галактоза. Эти соединения содержат высокоэнергетические связи, которые облегчают образование гликозильной связи с углеводной акцепторной молекулой. Нуклеозид может состоять из любых природных оснований и сахаров, и может также включать незначительное количество производных, таких как метил- или азозамещенные основания, дегидроксилированные или защищенные гидроксильные группы на сахарах, и тиофосфатные аналоги дифосфатного фрагмента.
Фраза "гликозидная связь" относится к кислород/углеродной связи, которая обычно бывает между сахарами. Она может быть либо α, либо β по своей конфигурации, и обычно включает реакцию дегидратации, когда дифосфонуклеозид-активированный гликозильный донор переносят на доступный углерод углеводный акцепторной молекулы, используя гликозилтрансферазу.
Фраза "гликозильное кольцо" относится к сахару или аминосахару, содержащему 5 или 6 атомов углерода в кольце. Сюда входят альдозы, дезоксиальдозы и кетозы, независимо от ориентации или конфигурации связей у асимметричного углерода. Они включают такие сахара, как рибоза, арабиноза, ксилоза, аллоза, альтроза, глюкоза, идоза, галактоза, талоза, рибулоза, ксилулоза, псикоза, N-ацетилгклозамин, N-ацетилгалактозамин, N-ацетилманнозамин, N-ацетилнейраминовая кислота, фруктоза, сорбоза, тагатоза, рамноза и фукоза.
Термин "гликозилтрансфераза" относится к семейству ферментов, которые соединяют дифосфонуклеозид-активированный гликозильный донор с доступным углеродом углеводной акцепторной молекулы за счет гликозидной связи. Эти ферменты включают как выделенные из природных источников, так и из источников генетических модифицированных для выделения таких ферментов. Семейство гликозилтрансфераз включает сиалилтрансферазы, N-ацетилглюкозаминилтрансферазы, N-ацетилгалактозаминилтрансферазы, фукозилтрансферазы, маннозилтрансферазы, галактозилтрансферазы и КДО трансферазы.
Фраза "NADPH регенерационная система" относится к компоненту ферментов, которые рециклизует NADP, образующийся in situ ферментативной реакции обратно в NADPH. Обычно такая система основана на алкогольдегидрогеназном превращении спирта (изопропила) в кетон (ацетон).
Фраза "сиалилированный лиганд Льюиса" относится к молекуле, способной связываться либо с ELAM рецептором, либо с GMP-140 рецептором протеинов. Химически определенные эти лиганды включают природные трисахаридные лиганды SLex и SLea и их производные. Такие производные включают незначительные замещения гидроксильных групп на водород, алкил, алкокси, галоид, гликозил и т. п. , молекулы гликалей, соединения с гликозильным кольцом, где кислород кольца с S или NH и их алкильные, окисленные или ацильные производные, присоединение аномерного углерода к углеводам или органическим молекулам, изменение в ориентации и положении гликозидых связей или замену энантиомеров природных сахаров.
Фраза "стехиометрическое соотношение" относится к количеству исходного продукта, который присутствует в прямом соотношении с продуктами реакции. Реагент существует в стехиометрическом соотношении с конечными продуктами, так как его обычно используют в реакциях получения конечного продукта и не регенерируют в процессе реакции. Стехиометрическое соотношение обычно составляет около 1 : 1 или 2 : 1 (отношение исходного продукта к конечному).
Фраза "трехвалентный фосфитилирующий реагент" относится к реагенту, который реагирует с гидроксильной группой органического соединения с образованием фосфит-содержащего продукта, который можно окислить окисляющим реагентом до получения фосфатного соединения после удаления защитных групп.
Сокращения:
ADP - аденозин-5'-дифосфат,
ATP - аденозин-5'-дифосфат,
CMP, цитидин-5'-монофосфат,
CDP, цитидин-5'-дифосфат,
CTP, цитидин-5'-трифосфат,
CMP, NeuAc цитидин-5'-монофосфо-N-ацетилнейраминовая кислота,
Fuc, фукоза,
Fk, фукокиназа,
Fuc-1-P, фукозо-1-фосфат,
Fuc - T, фукозилтрансфераза,
Ga1, галактоза,
Ga1NAc, N-ацетилгалактозамин,
GTP, гуанозин-5'-трифосфат,
GDP-Fuc, гуанозин-5'-дифосфофукоза,
GDP, гуанозин-5'-дифосфат,
GDP-Man, гуанозин-5'-дифосфоманноза,
GDP-ManPP, GDP-маннозо-пиррофосфорилаза,
GDP-FUCPP, GDP-фукозо-пиррофосфорилаза,
G1c-1-P, глюкозо-1-фосфат,
G1cNAc, N-ацетилглюкозамин,
ManNAc, N-ацетилманнозамин,
NADP(NADPH), никотинамидадениндинуклеотидфосфат,
NeuAc, N-ацетилнейраминовая кислота,
NMK, нуклеозидмонофосфаткиназа,
MK, миокиназа,
PPase, неорганическая пирофосфатаза,
PK, пируваткиназа,
PEP, фосфо(енол)пируват,
Pyr, пируват,
PPi, неорганическая пирофосфатаза,
P, неорганический фосфат,
Rha, раминоза,
UDP, уридин-5'-дифосфат,
UTP, уридин-5'-трифосфат,
UDP-G1c, уридин-5'-дифосфо-глюкоза,
UDP-Ga1, уридин-5'-дифосфо-галактоза.
Во многих использованных здесь структурных формулах имеются только две или три группы, связанные с углеродными атомами кольца. Обычно не изображенные группы представляют собой атомы водорода, и обычно не изображают их связи с атомами углерода, если только их изображение нежелательно, с точки зрения стереометрии. В других формулах зачерненные клинообразные линии изображают связи над плоскостью чертежа, а сужающиеся клиновидные линии используются для обозначения связей расположенных снизу плоскости чертежа. Волнистой линий изображают связи двух типов (и α, и β).
На фиг. 1 представлена типичная временная зависимость превращения GDP-маннозы в GDP-фукозу в результате NADPH окисления с использованием измерений оптической плотности (Abs).
Кривая A: контрольная кювета без GDP-маннозы.
Кривая B: то же, что и для контроля, но с добавлением 1 мкмоль GDP-маннозы.
По оси ординат отложены единицы поглощения на 340 нм, а по оси абсцисс - минуты.
На фиг. 2 представлены три картины A, B и С - хроматограммы ВЖЭХ - элюирование для превращения GDP-маннозы в GDP-фукозу в момент времени 0 (A), спустя 3 ч (2) и спустя 6 ч (C) соответственно после начала реакции.
По оси ординат отложено относительное поглощение на 254 нм, а по оси абсцисс - минуты.
На фиг. 3 представлен график, который иллюстрирует синергическое ингибирование α-1,3- фукозилтрансферазы гуанозин-5'-дифосфата (GDP) с соединением 50 в присутствии 0.2 мМ 14C-GDP-фукозы и 20 мМ MnCl2 при pH 6.2.
Использованы следующие символы: открытый треугольник - нет ингибирования; закрашенный треугольник - 0.05 мМ GDP; пустой квадрат - 34 мМ соединения 50; пустой круг - 0.05 мМ GDP плюс 34 мМ соединения 50.
По оси ординат отложены единицы обратной исходной вязкости образовывающегося продукта (I/v), а по оси абсцисс - обратная величина концентрации N-ацетиллактозамина (LacNAc).
Настоящее изобретение относится к in situ мультиферментативной реакции, в которой углеводная акцепторная молекула подвергается фукозилированию нуклеозид-5'-дифосфофукозой за счет фукозилтрансферазы, где нуклеозид-5'-дифосфофукоза предпочтительно ферментативно образуется из каталитических количеств нуклеотидов (см. общие схемы 1 и 2 далее).
Эти и все последующие схемы приведены в конце описания.
Более конкретно, раскрытое изобретение предлагает улучшенный способ получения предшественников нуклеотидных сахаров, которые действуют как донорские субстраты для реакций гликозилтрансферазы. Эти способы включают как химические, так и ферментативные средства. Химическое усовершенствование относится к повышению выхода и стабильности промежуточных соединений - защищенных сахаров, которые используют для получения гликозил-1 или 2-фосфитов, которые в свою очередь окисляют до фосфатов, которые конденсируют с нуклеозидмонофосфатами до получения нуклеозид-5'-дифосфосахаров или нуклеотидных сахаров.
Другим аспектом настоящего изобретения является усовершенствование мультиферментативной системы, включающей более одной гликозилтрансферазной реакции для синтеза углеводов, где одно усовершенствование состоит в использовании каталитических количеств нуклеотида. Нуклеотиды регенерируются из моно- или дифосфатных форм в трифосфатную форму за счет использования in situ ферментативной реакции одновременно с реакциями гликозилтрансферазы. Используют каталитические количества нуклеотида из-за ингибирующего действия нуклеотидов на гликозилтрансферазы.
Родственные патенты США, содержащие основные материалы, относящиеся к раскрытию изобретения: заявка N 07/670701, поданная 18 марта 1991; N 07/707600, поданная 30 мая 1991; N 07738211, поданная 30 июля 1991 и озаглавленная "Олигосахаридные ферментативные субстраты и ингибиторы: способы и композиции"; N 07/852409, поданная 16 марта 1992, и N 07/889652, поданная 26 мая 1992. Все они включены сюда в качестве ссылок.
A. Фукозилирование.
Один из аспектов настоящего изобретения концентрируется на использования вышеописанной и сравнительной технологии для фукозилирования углеводов. Фукозилирование является обычной терминальной модификацией для многих биологически активных углеводов, таких как антигены Льюиса; как сиалилированные так и несиалилированные.
(1) Фукозилтрансферазы.
Фукозилирование возникает под действием фукозилтрансферазы. Фукозилтрансферазы хорошо известны, и их обзор представлен в Adv. Enzymol, 52 : 44 - 56 (1981). Углерод акцепторного углевода обычно является членом кольца глюкозы, галактозы или N-ацетилглюкозамина или их аналогов. O-гликозидная связь обычно бывает в α- ориентации. Наиболее обычным местоположением являются: 2-, 3- или 6-гидроксилы галактозы; 3-, 4- или 6-гидроксильные группы N-ацетилглюкозамина; или 3- или 4-гидроксилы глюкозы. Глюкаль и (5-тио)-глюкозосодеpжащие сахариды могут также быть акцепторным сахаридным фрагментом акцепторного углевода для фукозилтрансферазных ферментов.
Фукозилтрансферазы можно выделить из природных источников или из рекомбинантных микроорганизмов, которые были генетически изменены для выделения фукозилтрансфераз. Очищенные нативные фукозилтрансферазы были описаны Foster, J. Biol. Chem. 266: 3526 - 3531 (1991); Muramatsu, Eur. J. Biochem., 157: 71 - 75 (1986); и Prieels et al., J. Biol Chem., 256 - 10 456 - 10 463 (1981). Фукозилтрансферазные гены, как сообщалось, были клонированы и экспрессированы Campbell et al. , J. Biol Chem., 259: 11 208 - 11 214 (1984); Larsen, et al. , Proc. Natl. Acad. Sci., U.S.A., 87: 6674 - 6678 (1990); и Kukowska - Latallo, et al., Genes and Devel., 4: 1288 - 1303 (1990); Weston et al., J. Biol. Chem., 267: 4152 (1992).
Вообще фукозилтрансферазы являются мембранносвязанными. Так, целые фукозилтрансферазы обычно не растворимы в водном растворе. Для облегчения их использования в способах и реакционных системах настоящего изобретения предпочтительно использовать растворимые ферменты, где нерастворимый цитоплазмический хвост был бы исключен или сделан более гидрофильным за счет селективной делеции или добавления полярных аминокислот. Однако природные неизменные фукозилтрансферазы можно использовать в настоящем изобретении за счет добавления небольших количеств таких неионных детергентов, как Triton X-100.
Фукозилтрансферазы представляют собой специфический тип гликозилтрансфераз. Активированная донорская молекула представляет обычно нуклеотид-5'-дифосфофукозу. В реакции вырабатывается нуклеотид как уходящая группа и фукоза, содержащая реакционноспособный карбониевый ион, который образует гликозидную связь с доступной гидроксильной группой акцепторной молекулы.
(2) Условия реакции фукозилирования и субстрата.
Фукозилтрансферазы являются типичными гликозилазами и являются относительно устойчивыми ферментами. Условия реакций, подходящие для большинства гликозилтрансфераз, подходят для фукозилтрансфераз. Так, например, подходящие условия реакций включают температурный интервал от около 10 до 40oC, буферы включают органические и неорганические буферы со значениями pH в интервале физиологических значений pH. Приемлемый интервал значений pH составляет от около 4 до около 9. Концентрации солей составляют от около 0 до 200 мМ и от около 0.1 до около 1.0% неионного детергента (например, Triton X-100), который используют, если по-другому фермент не растворяется в водной среде фукозилирования. Часто возникает необходимость в таком двухвалентном катионе, как Mn2+.
Углеводные акцепторные молекулы практически ничем не ограничены. Известные местоположения связывания указаны ранее; однако остальная часть углеводных акцепторных молекул не является критической. Фукозилтрансферазы достаточно толерантны к субстрату, и кроме акцепторного сахара, через который фукоза присоединяется, и сахаров, непосредственно соседствующих с акцепторным сахаром, остальная часть структуры субстрата имеет мало значения. Акцепторные углеводные молекулы можно получить исключительно из остатков сахаров, включая моносахариды, из гликопротеинов, из гликолипидов, или из неприродных соединений, в которых сахар акцептирующий фукозу, связан с такими соединениями, как арил, гетероциклы, циклоалканы и ациклические углеводороды.
Предпочтительная углеводная акцепторная молекула оканчивается GalpI, 4GalcNAc-X фрагментом, где X представляет органическую молекулу. Примеры X групп отмечены далее в тексте. Примеры углеводных акцепторных молекул включают Ga1β1,4G1cNAc, лактозу, NeuAcα2,6Galβ1,4GlcNAc, Ga1β1,3G1cNAc,Ga1β1,4G1ucal/ (лактал), NeuAcα2, 3Galβ1,4Glucal, 2-галоидзамещенные продукты реакций вышеуказанных глюкалей, Galβ1,4(5-тио)Glc,Galβ1,4GlcNAcβ- O-аллил.
SLex или SLea аналог может, таким образом, включать атом галоида вместо одного из кольцевых гидроксилов. Был найден новый способ получения 2- или 3-галоид-моно- или олигосахаридов из их соответствующих гликалей за счет использования хлорпероксидазы. Полученные 2(3)-дезокси-2(3)-галоидсахариды можно затем использовать в обсуждающихся далее синтезах.
В соответствии с этим способом гликаль смешивают с перекисью водорода, выбранным ионом галоида (хлоридом, бромидом или иодидом) и каталитическим количеством хлорпероксидазы (EC 1.11.10) в водном буфере со значением pH около 2.5 - 3.5 до получения реакционной смеси. Полученную реакционную смесь далее поддерживают до тех пор, пока не образуется целевой продукт. Концентрации различных реагентов могут меняться, как это хорошо известно специалистам. Примерные концентрации и синтезы приводятся далее. Таким образом, получают галоидгидрин - продукт и затем его предпочтительно выделяют.
При комнатной температуре обычно время реакции составляет от около 15 мин до 2 - 4 дней. Иодид реагирует быстрее всего, а хлорид реагирует наиболее медленно.
Обычно получают термодинамически образующиеся продукты, за исключением тех случаев, когда 1,3-диаксиальные взаимодействия препятствуют образованию 2-аксиально-замещенных продуктов. Если 1,3-диаксиальные взаимодействия присутствуют в реагирующем гликале, у галоид-гидратированных продуктов наблюдается стереоспецифичность в виде α- или β- ориентации галоидной группы, причем образуются также оба аномера 1- или 2-гидроксильной группы. Примеры синтеза 2- или 3-галоидуглеводных акцепторных молекул и их предшественников проиллюстрированы на схемах 3, 4 и 4a далее.
Бромгидратирование сиалильной Lex молекулы с терминальным гликалем (соединение 36 схемы 4a), который имеет конформацию в растворе, аналогичную конформации сиалил Lex приводят к получению продуктов (соединения 37a и 37b). Продукты, имеющие ту же конформацию, что и соединение 36 и сиалил Lex на участке связывающего домена, состоят из NeuAc-Ga1-Fuc в соответствии с NOE исследованиями.
Бромированные сахариды можно также получить с использованием N-бромсукцинимида (NBS) в растворителе ацетонитрил-вода. Эту процедуру можно использовать для изменения соотношения образующихся продуктов, таких как соединения 32 и 33, которые получаются в равных количествах ферментативной реакции, и в соотношении 1 : 2.5 (32 : 33) при использовании NBS. 3-галоидное соединение 35 и его изомерный галоидгидратированный продукт, соединение 35a, получают в соотношении 2 : 3 (35 : 35a) с использованием NBS реакций по сравнению с отдельным изомером, соединением 35, в тех случаях, когда используют ферментативную реакцию.
Схема 3 иллюстрирует бромгидратирование D-глюкаля, D-галакталя и D-фукаля и образование соответствующих 2-дезокси-2-броммоносахаридов (соединения 20, 21, 22 и 23). Бромгидратирование сиалаля соединения 34 хлорпероксидазой (CPO) до получения соответствующей 3-дезокси-3-бромсиалильной кислоты соединения 35 также представлено в нижней части схемы 3.
Схема 4 иллюстрирует хлоро- и иодогидратацию галакталя до получения соединений 25, 26 и 27. Представлено на схеме 4 бромгидратирование GalI, 3Glucal (соединение 28) и Gal, 4Glucal (соединение 31) до получения соответствующих α- и β-2- дезокси-2-бром соединений, соединений 29 и 30 и соединений 32 и 33 соответственно.
Активированный донор фукозы, нуклеозид-5'-дифосфофукоза, чаще всего содержит гуанозин; однако существуют и альтернативные доноры, такие как нуклеотид, содержащий любой L-сахар, например L-рамноза и L-идоза.
Для того чтобы связать реакцию фукозилтрансферазы со второй реакцией гликозилтрансферазы, можно просто использовать преимущество того факта, что оптимальные условия для реакций большинства гликозилтрансфераз перекрываются. Таким образом, данные условия реакции для любой гликозилтрансферазы обеспечивают функционирование известных фукозилтрансфераз. Используя указанные ранее условия реакции для фукозилтрансферазы и используя обычные эксперименты титрования, можно получить условия реакции, подходящие для синтеза фукозилированного олигосахарида, используя только моносахариды.
Вообще при выборе условий селективной реакции для множественных реакций гликозилтрансферазы в единой реакционной смеси следует принять во внимание температуру, pH, общую концентрацию осмотически активных ионов, как представлено ранее. Если одной из гликозилтрансфераз является фукозилтрансфераза, приемлемые условия реакции включают интервал pH предпочтительно от около 6.0 до около 8.5, и наиболее предпочтительно, между около 7.0 и около 7.5. Такие двухвалентные катионы, как Mn2+, могут быть использованы, но двухвалентные ионы хелатообразующих агентов нежелательны.
Состав буферов не является критическим. Подходят водные буферы, такие как HEPES. Осмомолярность буфера составляет величину от 100 до около 300 мОсм.
Вышеперечисленные условия, при которых функционируют ферменты, здесь и далее называют условиями биологических реакций.
Времена реакций меняются в зависимости от субстратов, ферментов и температур. Обычно время реакции составляет 24 - 96 ч.
В некоторых случаях, если используют галактозилтрансферазу, а моносахаридный акцептор содержит агликон одного положения глюкозы в α- ориентации, условия реакции могут включать лактальбумин, предпочтительно α- лактальбумин.
Так, например, реакция сиалилтрансферазы, описанная Ichikawa et al., J. Amer. Chem. Soc. , 113: 4698 - 4700 (1991) может быть связана с рекомбинантной человеческой α- (1,3/4) фукозилтрансферазой Льюиса, как описано Kukowska - Latallo et al., Genes and Devel., 4: 1288 (1990). Реакционная смесь водного буфера (HEPES) имеет интервал pH 7.0 - 7.5, и концентрацию солей 50 - 200 мМ. Реакция протекает при температуре около 37oC.
В. Специфичность субстрата и исследования ингибирования гликозилтрансфераз.
α- 1,3/4FucT. Фукозилтрансфераза, способная переносить FUC фрагмент из GDP-Fuc на 3- и 4-OH группы GlcNAc с образованием Lexили Lea, является α- 1,3/4FucT (Fukowska-Latallo et al. , Gene and Debelopment 4: 1288 (1990); Dumas et al. , Bioorg. and Med. Chem. Lett. 1: 425 (1990)). Как следует из приводимой далее табл. 1, фермент катализирует фукозилирование Galβ1,3GlcNAc быстрее (Vотн 580), нежели Galβ 4GlcNac (LacNAc) (Vотн 100) (примеры 1 и 4) при концентрации 10 мМ углеводного акцептора. Сиалилированный LacNAc (пример 7) также является субстратом для этого фермента, обеспечивающим синтез (Dumas et al., Bioorg. and Med. Chem. Lett., 1: 425 (1991)) сиалил Lex. Интересно, что Galβ1,4 (5-тио)-Glc (Gautheron - Le Narvor et al., J. Chem. Soc. Chem. Commun., 1130 (1991); Wong et al. J. Am. Chem. Soc., 113: 8137 - 8245 (1991)] является лучшим субстратом, нежели соответствующий дисахарид, лактоза (примеры 2 и 3) в тех же условиях. Каждый из субстратов табл. 1 представляет углеводный акцептор фукозы из фукозильного донора с использованием этого фермента. Однако Galβ1,4 дезоксиноджиримицин (Gautheron - Le Narvor et al., J. Chem. Soc. Chem. Commun., 1130 (1991); Wong et al. J. Am. Chem. Soc. , 113: 8137 - 8245 (1991)) (пример 11) является ингибитором (ИК50 40 мМ). Из-за ограниченного количества α- 1,3/4Fuc, дальнейшие исследования не проводились.
α- 1,3FucT. Этот фермент ответствен за получение сиалил Lex в человеческой плазме типа α- 1,3FucT, который недавно был клонирован, сверхэкспрессирован (Weston et al., J. Biol Chem., 267: 4152 (1992)) и использован в синтезе. В табл. 2 представлена специфичность субстрата, и, как ожидалось, тот фермент более специфичен для LacNAc (V/Km 2.9, пример 1), нежели Gal β-1,3 GlcNAc (V/Km 0.22, пример 5). Аналогично результату для α- 1,3/4FucT (пример 3, табл. 1), Gal β-1,4/5-тио/Glc также является субстратом для α- FucT (пример 3, табл. 2). В отличие от α- 1,3/4 фермента лактал (пример 6) является субстратом для α- 1,3 фермента.
Трисахарид NeuAc α-2,3 Gal β-1,4 GlcNAc (пример 7), предшественник сиалил Lex, является наилучшим субстратом с относительной максимальной скоростью 620% в расчете на LacNAc. α- 2,6-связанный сиалозид (пример 10) примерно в 50 раз менее активен в качестве субстрата, нежели α- 2,3-изомер. Весьма нежелательно, что фермент может также переносить Fuc на глюкальсодержащий сиалилированный трисахарид (Vотн. 330%, пример 9).
Что же касается связывания, этот фермент имеет более высокое сродство и дисахаридами (примеры 1, 3, 4, 6), нежели с трисахаридами. Возрастание сродства наблюдается, когда GlcNAc фрагмент LacNAc заменяют 5-тио-Glc, глюкалем (Haworth et al., J. Chem. Soc. 2644 (1990)) или GlcAc- β-O-аллилом. Лактоза, однако, обладает очень низким сродством, хотя относительная скорость Vмакс достаточно велика (150%). Каждый из субстратов табл. 2 является углеводным акцептором для фукозы из фукозильного донора, использующего этот фермент.
В наших исследованиях ингибирования α 1,3FucT (табл. 3) наблюдение того факта, что 3'-дезокси-Lac-Nac β- O-аллил (Gautheron - Le Narvor et al., J. Chem. Soc. Chem. Commun., 1130 (1991); Wong et al., J. Am. Chem. Soc. 113: 8137 - 8245 (1991)) является слабым ингибитором (пример 5), согласуется с более ранними сообщениями о дезоксигенированных олигосахаридах для гликозилтрансфераз (Hindsgaul et al., J. Biol Chem. 266: 17 858 (1991)). Среди акцепторных углеводных субстратных аналогов, которые были исследованы, Galβ1,4 дезоксиноджиримицин оказался наиболее потенциальным ингибитором (пример 4, ИК50 = 8 мМ).
Два азасахара (Kajimoto et al., J. Am. Chem. Soc. 113: 6679 (1991)), которые, как известно, являются потенциальными ингибиторами α- фукозидазы, были проанализированы в качестве акцепторных аналогов (примеры 8 и 9), и было обнаружено, что они являются умеренными ингибиторами относительно LacNAc для FucT (ИК50 около 34 - 52 мМ). Дезоксиноджиримицин оказался, однако, субстратом для α- 1,4GalT (Wong et al., J. Am. Chem. Soc. 113: 8137 - 8145 (1991)). GDP-Man также является потенциальным ингибитором α- 1,3-FucT (ИК50 2 мМ).
Для исследования продуктов ингибирования наше внимание было сфокусировано на выделенном нуклеозиддифосфате. GDP является побочным продуктом ферментативного фукозилирования и представляет собой очень эффективный неконкурирующий ингибитор по отношению к LacNAc (Kii = 0.13 мМ. Kis = 0.16 мМ, пример 10). Другой нуклеозиддифосфат, UDP выделенный при ферментативном галактозилировании, также весьма эффективный потенциальный ингибитор GalT (Ki = 0.46 мМ). GDP-Fuc является потенциальным ингибитором α- 1,3FucT в концентрациях выше 0.2 мМ в присутствии 10 мМ LacNAc. Однако он не является ингибитором α- 1,4/4 FucT.
В дополнении к азасахарам примеров 8 и 9 в табл. 3, другие азасахара, такие, как соединения 51, 52 и 53 (далее), также ингибируют α- фукозидазную активность, как и соединение 50 (пример 9, табл. 3). Соединения 50 - 53 демонстрируют Ki значения с этим ферментом 4.22, 8 и 1.4 мкМ соответственно (cм. также Dumas et al., Bioorg, and Med. Chem. Lett., 2:33 (1992)).
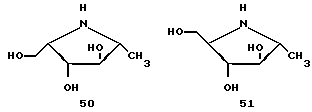
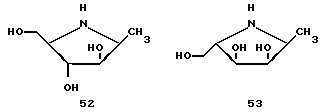
Помимо соединения 50 было обнаружено, что соединение 53 также является конкурирующим ингибитором человеческой плазмы типа α- 1,3-фукозилтрансферазы. ИК50 относительно LacNAc составляет 80 мМ. Кроме того, GDP, который образуется в реакции фукозилирования из GDP-Fuc и является неконкурирующим ингибитором, если присутствует при ИК50, 0.05 мМ, демонстрирует явное синергическое ингибирование в присутствии либо соединения 50, либо 53. Данные исследования ингибирования для соединения 50 представлены на фиг. 3. Этот синергический эффект может быть связан с взаимодействием между GDP и азасахаром в активном сайте фермента до образования комплекса, который имитирует структуру переходного состояния реакции фукозилпереноса.
Вышеуказанные результаты обеспечивают способ ингибирования реакции гликозилтрансферазы, например, реакции фукозилтрансферазы. В соответствии с этим способом такую гликозилтрансферазу, как α-1,3-фукозилтрансферазу человеческой плазмы, углеводную акцепторную молекулу, такую как LacNAc, и активированную гликозильную донорскую молекулу, такую как GDP-Fuc, и ингибирующее количество такого азасахара, как любое из соединений 50 или 53, смешивают в водной среде и поддерживают в условиях биологической реакции в течение промежутка времени, достаточного для ингибирования реакции гликозилтрансферазы.
Более предпочтительно присутствие ингибирующего количества нуклеозиддифосфатного продукта реакции гликозилирования, такого как UDP или GDP, где GDP-Fuc является донором гликозилирования. Ингибирующие количества азасахара и нуклеозиддифосфата, когда они присутствуют, предпочтительно составляют по крайней мере 10% от их индивидуальных ИК50 значений, и более предпочтительно эти количества составляют по крайней мере 50% от их индивидуальных ИК50 значений, измеренных in vitro, как обсуждается далее для конкретных реакций гликозилирования, которые нужно ингибировать. Можно также использовать концентрации, превышающие ИК50. Обычно реакцию гликозилирования ингибируют по крайней мере на 25%, и более предпочтительно по крайней мере на 50%.
Ингибирование гликозилирования происходит как in vitro, так и in vivo. Примеры исследований in vitro ингибирования приводятся далее. Для использования in vivo фермент, гликозильный донор и акцепторные молекулы и GDP присутствуют в животном-хозяине, которое может быть лабораторным животным, например мышью, крысой или кроликом, или это может быть человек. Азасахар вводят хозяину обычно применяемым способом введения лекарственных препаратов, хорошо известным специалистам. При желании можно также вводить дополнительные количества GDP. Условия биологической реакции задаются организмом млекопитающего-хозяина. Добавочный азасахар поддерживают в организме млекопитающего-хозяина до тех пор, пока он не выделяется или не катаболизируется.
C. Химический и ферментативный способы получения GDP-фукозы.
Фукозилтрансферазную циклическую реакцию можно вести, либо добавляя стехиометрические количества соответствующего сахарного нуклеотида, например GDP-фукозу, или предпочтительно сахарный нуклеотид можно создавать за счет каталитических количеств соответствующего нуклеотида и стехиометрических количеств PEP и Man-I-P или Fuc-I-P.
GDP-фуккоза является предпочтительным активированным сахарным донором для известной фукозилтрансферазы. Ее трудно и дорого получать, и для других описываемых далее реакционных циклов предпочтительно, чтобы ее синтез включал in situ регенерацию ее нуклеотидных предшественников. Общая схема представлена в фукозном цикле схемы 1.
(1) Химической синтез фукозо-1-фосфата и GDP-фукозы.
Химический синтез GDP-Fuc основан на соединении фукозо-1-фосфата и такого активированного GMP, как GMP-морфолидат (cм. (a) Кочетков с сотр. Adv. Garbohydr. Chem. Biochem. 28:307 (1973); (b) Moffat, Methods Enzymol., 8:136 (1966); и (c) Roseman et al., Am. Chem. Soc., 83:659 (1961)). За счет относительно высокой лабильности фукозо-1-фосфата и GDP-Fuc известные химические выходы для синтеза Fuc-I-P и реакции присоединения Fuc-I-P и GMP производного были низки. Описанo несколько синтезов фукозо-1-фосфата. Так как только Fuc-I-P среди сахарных нуклеотидов имеет термодинамически нестабильный β- фосфатный фрагмент на аномерном центре фукозы, стереохимию аномерного центра трудно контролировать. Здесь предложены два эффективных способа для GDP-Fuc: один химический, другой - ферментативный.
Первый - химический синтез GDP-Fuc - осуществила группа Barker (Nunez et al. , Can. J. Chem., 59:2086 (1981). Для получения фукозо-1-фосфата они использовали 2,3,4-три-O-ацетил -β-/ L-фукозу, полученную из соответствующего бромпроизводного с последующей кристаллизацией и фосфорилированием полученного β- аномера. Группа Hindsgaul (Gokhale, Can. J. Chem., 68:1063 (1990)) использовала реакцию гликозилирования ацетофукозилбромида и дибензилфосфаттетрабутиламмонийной соли для получения относительно нестабильного гликозилфосфата (менее 10 мин при хроматографии на силикагеле). Schmidt et al., (Schmidt et al., Liebigs Am. Chem. 191:121 (1991)) использовали фукозилимидат и получали продукт гликозилирования без кислотного катализатора Льюиса с высоким выходом. Группа Van Boom (Westerduinn et al., Tetrahedron Lett., 27: 1211 (1986)) использовала трехвалентный фосфитилирующий агент на 2,3,4-тио-O-бензилфукозе и превращение в α- фукозилфосфат.
Здесь раскрыты два усовершенствованных подхода к химическому синтезу GDP-фукозы (см. схемы 5 - 7 далее). В одном из них (схема 5) используют реакцию гликозилирования бензоилированного (Bz) фукозилбромида (соединение 3) и дибензилфосфата. Использование бензоильной группы вместо ацетильной в качестве защитной группы обеспечивает повышенную стабильность фукозильного производного и стереоселективность реакции гликозолирования. Гликозилирование соединения 3 и дибензилфосфата (Linshorst et al., Carbohydr. Rev. 209: 119 (1991)) протекает спокойно, и в результате получают продукт присоединения с 95%-ным выходом. Как и ожидалось, соединение 4 оказывается достаточно стабильным для того, чтобы его можно было очистить на хроматографической колонке с силикагелем (более 3 ч); однако уже очищенный материал оказывается нестабильным. Если очищенное соединение 4 оставить на ночь при комнатной температуре, обнаруживается некоторое количество продуктов разложения и аномеризации. Следует заметить, что очищенное соединение 4 используют на следующей стадии сразу же. Удаление бензильных групп с фосфата, фрагмента и бензольных групп осуществляют поэтапно, как было указано ранее (Gokhale et al., Can. J. Chem., 68: 1063 (1990)).
В другом варианте используют трехвалентный фосфитилирующий агент, такой как дибензил-N,N-диэтилфосфороамидит (DDP), который используют для получения дигидроксиацетилфосфата (DHAP) (см. схему 6 далее (Pederson et al., Tetrahedron, 47: 2643 (1991)). Так, 2,3,4-тио-O-ацетилфукозу, соединение 7, полученную либо химическим, либо ферментативным деацетилированием (Hennen et al. , J. Org. Chem. 53: 4943 (1988)), фосфинируют DDP в присутствии тетразола. Реакция протекает спокойно с 79%-ным выходом соединения 8, соединений формул I и II, которые окисляют до соответствующего фосфатного соединения 9. Удаление защиты у соединения 9 осуществляют аналогично получению соединения 5 из соединения 4.
Реакция фосфитилирования с использованием (BnO2)PNEt2 (DDP), проиллюстрированная на схеме 6, весьма подходит для получения различных фосфитов и соответствующих фосфатов с высокими выходами. Дальнейшие примеры соединений и подробности обсуждаются далее в разделе H.
Fuc-1-P эффективно активируют превращением в триалкиламмониевую соль при взаимодействии с гуанозин-5'-монофосфоморфолидатом (1 : 2) в таком растворителе, как пиридин. Полученный продукт, GDP-Euc, соединение 12, очищают, используя обычную хроматографическую колонку. Эти реакции представлены далее в схеме 7.
(2) Ферментативное получение GDP-фукозы.
Ферментативное получение GDP-фукозы предпочтительно. Ферментативное получение GDP-фукозы описано Schacter et al., Methods of Enzymol., 28: 285 (1972) и проводилось с использованием фукозокиназы и GDP-фукозопирофосфорилазы из печени свиньи.
Как легко видеть, эту мультиферментативную реакцию можно провести, используя сочетание различных ферментов и высокоэнергетических субстратов. Такие циклы фукозных реакций представлены на схемах 1 и 8, представляя примеры такой мультиферментативной системы. В них фукозу добавляют в стехиометрических количествах наряду с PEP. Каталитические количества PK, FK и GDP-FucPP добавляют вместе с ADP и GDP. Условия реакции аналогичны приведенным ранее для трансфераз.
(3) Манноза в качестве исходного материала.
В другом варианте GDP-Fuc можно получить эффективно в результате получения GDP-маннозы и последующего ферментативного превращения в GDP-Fuc. Предшественником GDP-Man является Man-1-P, соединение 18. Man-1-P получают, используя тот же подход, что и для получения фукозо-1-P. Предпочтительно использовать ацетальные защитные группы для получения маннозно-пер-O-ацетата, что представлено на схеме 9 ниже. В другом варианте Man-1-P можно ферментативно получить способом, аналогичным Glc-1-P.
Ферментативный синтез GDP-фукозы из маннозо-1-фосфата in situ с образованием GDP-маннозы достигают в результате комбинации двух ферментативных систем: GDP-маннозо-пирофосфорилазы и GDP-фукозных синтетических ферментов. Регенерация NADPH необходима для образования GDP-фукозы. Такую регенерацию можно осуществить, используя NADPH, зависимую алкоголь-дегидрогеназу из Thermoanaerobacterium brokii в присутствии изопропанола или глюкозофосфат-дегидрогеназы в присутствии глюкозы. GDP-маннозо-пирофосфилазу можно получить из дрожжей, как указано далее; но другие источники, как, например, Arthrobacter (Preiss et al., J. Biol. Chem. 239: 3119 (1964)), Escherichia coli (Lieberman et al. , J. Bact., 101: 965 (1970)), а также из млекопитающих (Smoot et al., Eur. J. Biochem., 148: 83 (1985)) были описаны.
Превращение GDP-фукозы из GDP-маннозы впервые сообщали Ginsburg and Kirkman (Ginsburg et al., J. Am. Chem. Soc., 80: 3481 (1958). Этот фермент частично очищают и используют для демонстрации превращения GDP-маннозы в GDP-фукозу из A. aerogenes (ATCC 12 658), которое в настоящее время переименовано в Klebsiella pneumoniae (Ginsburg, J. Biol. Chem., 235: 2196 (1960)). Эта реакция оказалась NADPH-зависимой. Yamamoto et al., также сообщает о синтезе GDP-фукозы из GDP-маннозы с использованием фермента, полученного из Agrobacterium radiobarter (Yamamoto et al., Agric. Biol. Chem., 48: 823 (1984)).
Далее описано превращение маннозо-1-фосфата в GDP-фукозу in situ с образованием GDP-маннозы. Маннозо-1-фосфат превращают в GDP-фукозу за счет комбинации двух ферментных систем, GDP-маннозопирофосфорилазы и GDP-фукозы - синтетическиx ферментов, с регенерацией NADPH. Хотя начальный результат дает низкий выход, однако ожидают, что, если удастся достичь более высоких активностей ферментов, выход можно существенно повысить. Эти реакции представлены на схемах 10 и 11.
Для синтеза фукозилированного олигосахарида используют кофакторную систему регенерации, в которой выделившийся GDP превращают в GTP с помощью фосфоенолпирувата (PEP) и пируваткиназы (PK), а полученный NADP превращают в NADPH за счет 2-пропанола в присутствии алкоголь-дегидрогеназы. Используя α- 1,3/4 фукозилтрансферазу, Gal-β-1,3 GlcNAc превращают в Gal-β-1,3 (Fuc-α-1,4) GlcNAc (Dumas et al., Biomed. Chem. Lett. 1 : 425 (1991)).
D. Рециркулирование нуклеотидов.
Так как гликозилтрансферазы часто ингибируются нуклеотидами, предпочтительно, чтобы концентрация нуклеотидов поддерживалась минимальной. Регенерация нуклеозидтрифосфатов из нуклеотидных донорных сахаров позволяет использовать каталитические количества нуклеотидов, которые эффективно исключает нежелательное ингибирование активности гликозилтрансферазы. Регенерация нуклеотидов может служить в качестве высокоэнергетичных связей нуклеотидфукозильных донорских молекул, требуемых для того, чтобы условия реакции поддерживали ферментативные реакции как пируваткиназы, так и гуанозин-5'-дифосфофукозо-пирофосфорилазы. Пример системы рециркулирования представлен на схеме 12 далее.
Эти активированные донорские моносахаридные генерационные системы поддерживают реакции гликозилтрансферазы. Системы регенерации включают активированный донорский моносахарид и ферменты для регенерации активированного нуклеотидного сахарного донора из их соответствующих фосфатного донора, нуклеотидного и сахарного донора. Ферменты в регенерационной системе включают такие киназы, как пирувактиназа, ацетилкиназа и 1,6-дифосфофруктокиназа соответственно, и нуклеотид-сахаропирофосфорилазы, такие как GDP-Fuc-PP. Фосфатные доноры включают PEP, ацетилфосфат и D-фруктозо-1,6-дифосфат.
Некоторые фосфатные доноры могут ингибировать активность других ферментов в системе, и доноры следует поэтому выбирать осторожно. Нуклеотид, который фосфорилируется киназой, следует выбирать так, чтобы он функционировал как подходящий субстрат для нуклеотид-сахарофосфорилазы.
Описанная ранее регенерационная система может ингибироваться обратным механизмом, если концентрация неорганического пирофосфата избыточна. Использование каталитических количеств неорганической пирофосфатазы исправляет эту проблему.
E. Сиалильные лиганды Льюиса.
Предпочтительными конечными продуктами, получаемыми и описанными способами, являются фармацевтически активные углеводы. Такие продукты включают сиалильные лиганды Льюиса (см. схемы 1 и 13. Группа R на съеме 13 может быть водородом или органической группой X, как было указано ранее). Сиалильные лиганды Льюиса определяют как любое соединение, которое связывается с селектирующим рецептором, как раскрыто у Polley et al., Proc. Natl. Acad. Sci. USA. 88: 5224 - 6228 (1991). Эти лиганды типифицируют по их концевым структурам (силиловая кислота и фукоза), находящимся на гликопротеинах и гликолипидах. Эти лиганды включают природные лиганды сиалил Lex (SLex) и сиалил Lea (SLea). Далее эти лиганды включают неприродные аналоги, которые связываются аналогичным образом с природными рецепторами лигандов. Так, например, аналоги лигандов можно получить с олигосахаридными аналогами акцепторов для гликозилтрансфераз. Некоторые акцепторные аналоги хорошо известны и включают дезоксигенированные олигосахариды, описанные Hindsgaul et al., J. Biol. Chem., 266: 17 858 - 17 862 (1991).
Аналоги лигандов легко получить, используя вышеуказанные способы, и легко тестировать, используя описываемые далее анализы. Так, например, рецепторы, распознают лиганд, который был модифицирован из природного сайта в результате реакции эпимеризации (из GlcNAc в GalNAc) или изменения ориентации одной из гликозидных связей (с α-2,6 на β-2,6 связь). Примеры таких способов приведены далее.
Галактозилирование.
Были разработаны две мультиферментные системы для синтеза LacNAc с in situ регенерацией кофактора. Одна начинает с Glc-I-P и использует UDP-Glc пирофосфорилазу (EC 2.7.7.9, UDPGP) и UDP-Gal 4 эпимеразу (EC 5.1.3.2, UDPGP) (Wong et al., J. Org. Chem. 47:5416 (1982); Auge et al., Carbohydr. Rev. 151: 147 (1986); Thiem. et al., Angew. Chem. Int. Ed. Engl., 30:1163 (1991); Thiem. et al., Synthesis, 141 (1992)). Это представлено на схеме 14 далее, где NacGlcp O-аллил (соединение 40; х является O-аллилом) используют в качестве иллюстративного акцептора для GalT. UDP-галактозу получают из UDP-Glc с UDPGE; однако такое равновесие благоприятствует образованию UDP-Glc, и Glc-I-P следует получать отдельно. Glc-I-P можно получить, используя реакцию фосфитилирования, которая обсуждается.
Остальные используют Gal вместо Glc-I-P в качестве предшественника донора и UDPGP, галактокиназу (GK; EC 2.7.1.6) и Gal-I-P-Руридил-трансферазу (Gal-I-P UP; EC 2.7.7.12). Это представлено на схеме 15 с использованием I-13C-Gal, что иллюстрируется на схеме заштрихованным кружком в положении I. GK специфична для галактозы и обеспечивает непосредственное получение Gal-I-P, который превращают в UDP-Gal с помощью Gal-I-P UT и UDP-Gal. Последняя система, как было показано, пригодна для получения [Gal-l-13C]-LacNAc.
Мультиферментная система начинается с 113C-Gal (99 атомных % от Isotec Inc. , Miamisburg, OH), GlcNAc-О-аллила (соединение 40) (Lee et al., Carbohydr, Rev., 37:193 (1974)), фосфоенолпирувата (PEP) и каталитических количеств Glc-I-P, ATP и UDP. UDP превращают в UTP с помощью пируваткиназы (PK; EC 2.7.1.40) и PED, и UTP реагирует с Glc-I-P катализируемым UDPGP до получения UDP-Glc. Побочный продукт, неорганический пирофосфат (PPi) разлагают неорганической пирофосфатазой (PPase; EC 3.6.1.1). За счет Gal-I-P UT, UDP-Gal реагирует с 13C-Gal-I-P, получаемой из 13C-Gal и ATP в присутствии GK, до получения UDP-13C-Gal и Glc-I-P. 13C-Cal из UDP-13C-Gal переносят на акцептор (GalcNAc β-О-аллил) с помощью GalT до получения Gal-I-13C-содержащего LacNAc β- аллила (соединение 41). Полученный UDP снова превращают в UDTP в реакции PK и PEP с выделенной Gal-I-P до регенерации UDP-Glc. Используя эту мультиферментную систему, Gal-I-13C-LaNAc β-О-аллил получают с 54%-ным выходом. Тот же способ используют для получения немеченого LaNac и аналогов. Примеры аналогов 41a-c проиллюстрированы на схеме.
Сиалилирование.
Мультиферментативная система для сиалилирования начинается с NeuAc, Gal-I-13C-LacNAc β-О-аллил, PEP, и каталитические количества ATP и CMP, что представлено на схеме 16 далее. CMP превращают в CDP с помощью нуклеозидмонофосфаткиназы (EC 2.7.4.4, NMK) в присутствии ATP, который регенерируют из побочного продукта ADP, катализируемого PK в присутствии PEP, затем CTP за счет PEP с помощью PK. CTP затем подвергают взаимодействию с NeuAc с CMP-NeuAc-синтетазой (EC 2.7.7.42) до получения CMP-NeuAc. Побочный продукт PPi гидролизуют до Pi за счет PPазы. Сиалилирование LacNAc β-О-аллила завершают с помощью CMP-NeuAc и α-2,3-сиалилтрансферазы (α-2,3 SiaT; EC 2.4.99.6). Выделившийся CMP снова превращают в CDP, CTP и, наконец, в CMP-NeuAc. Используя эту систему, получают Gal-I-13C-NeuAc α-2,3 Gal β-1,4GlcNac β-O-аллил (соединение 42), а также немеченые трисахариды.
Интересно, что лактал (Gal β-I, 4Glucal) также является хорошим субстратом для α-2,3 SiaT, что позволяет синтезировать NeuAc α-2,3 Gal β-1,4 Glucal (соединение 43), представлено на схеме 16 с 21%-ным выходом. Лакталь получают либо химически (Haworth et al., J. Chem. Soc., 2644 (1930)), либо ферментативно, используя GalT и глюкаль (Gautheron-Le Narvor et al., J. Chem. Soc. Chem. Commun. 1130 (1991); Wong et al., J. Am. Chem. Soc. 113:8137 - 8245 (1991)) Гликальсодержащий олигосахарид, такой как соединение 43, можно превратить в другие сиалильные Lex производные, используя химические способы, разработанные Данишевским и др. (Griffith et al., J. Am. Chem. Spc. 112: 581 (1990); Halcomb et al., J. Am. Chem. Soc. 111:6661 (1989); Kessler et al. , Angew. Chem. Int. Ed. Engl., 29:425 (1990); Thiem et al., Synthesis, 696 (1978)). Соединение 43 также можно галоидировать до получения 2-гало-2-дезокси-Glc производных.
Способом, аналогичным представленному на схеме 16, используя α- 2,6-сиалилтрансферазу (EC 2.4.99.1) с Gal β-1,4 GlcNAc в качестве акцепторного углевода, получают с выходом 22% NeuAc α-2,6Gal β-1,4GlcNAc после протекания реакции в течениe двух дней при комнатной температуре.
Фукозилирование.
Клонированный человеческий фермент используют для фукозилирования при стехиометрическом количестве GDP-Fuc (99 атомных %, поставка Isotec Inc. Miamisburg, OH), что представлено на схеме 17 ниже.
Таким образом, фукозилирование сиалил LacNAc β-O- аллила (соединение 42) дает сиалил Lex, соединение 44, после очистки на силикагеле и BioGel-P-2. LacNAc β-O- аллил (соединение 41) и сиалилгликаль (соединение 43) также фукозилируют до получения Lex трисахарида (соединение 45) и сиалил Lex гликаля (соединения 46) соответственно, причем два последние соединения представлены на схеме 16. Интересно, что α-1,3-FucT и α- 1,3FucT акцептирует Gal β-1,4/5-тио/Glc до получения (5-тио) Glc-Lex аналога, Gal β-1,4-(Fuc α-1,3)-(5-тио)Glc, как показано далее на схеме 18.
Что же касается in situ регенерации GDP-Fuc превращение Man-I-P в GDP-Fuc, за счет GDP-Man, основанного на биосинтетическом способе GDP-Fuc в микроорганизмах, то она была впервые исследование и представлена на схеме 19. "Акцептор-OH" схем 19 и 20 является гидроксильной группой углеводного акцепторного субстрата, как те, которые перечислены в табл. 2.
Микробные ферменты используют из-за их доступности. Кроме того, эта система позволяет регенерировать GDP-Man. GDP-Man-пирофосфорилаза (GDP-ManPP) была обнаружена в дрожжах (Munch-Peterson, Methods in Ezymol., 5: 171 (1962); Simon et al., J. Org. Chem., 55: 1834 (1990) и, как известно, GDP-Fuc генерирующие ферменты существуют в бактериях (Ginsburg, J. Biol. Chem. 235: 2196 (1960); Ginsburg, Methods in Enzymol., 8: 293 (1966) Klebsiella Pneumonia. В этой регенерации GTP образуется из GDP в присутствии PEP и PK. Man-I-P реагирует с GTP с образованием GTP-Man за счет GTP-Man PP из высушенных клеток дрожжей. GDP-Man трансформируют в GDP-Fuc в присутствии NADPH и GDP-Fuc генерирующих ферментов, частично очищенных из бактерий. Окисленный NADP рециркулирует обратно в NADPH за счет Thermoanaerobium brockii алкоголь-дегидрогеназы (TADH) (EC 1.1.1.1) и изопропанола. Получение GDP-Man и GDP-Fuc подтверждено данными ВЖЭХ, и фукозилирование LacNAc β-O-аллила и соединения 42 до получения соединений 45 и 44 в количестве 5 - 10 мг завершается. Препаративный синтез сиалил Lex с in situ регенерацией GDP-Fuc с использованием очищенных ферментов, находится на стадии разработки.
Альтернативный способ начинают с Fuc-I-P, который превращает в GDP-Fuc, катализируемый GDP-Fuc пирофосфорилазой (GDP-Fuc P), что представлено на схеме 20 далее). (Ishihara et al., J. Biol. Chem., 243: 1103 (1968); Ishihara et al., J. Biol. Chem., 243: 1110 (1968); Schachter et al., Methods in Enzymol, 28: 285 (1972); Richards et al., Biochim. Biophys. Acta, 484: 353 (1977); Kilker et al., Biochim. Biophys. Acta, 570: 271 (1979)). GDP-Fuc P частично очищают из свиной печени (Ishihara et al., J. Biol. Chem. 243: 1110 (1968)) и было показано, что регенерационная система, изображенная на схеме 20, является функциональной в аналитическом масштабе для получения Lex и сиалил Lex.
В дополнениe к сиалильным антигенам Льюиса, SLex и SLea, и им соответствующим аналогам aнтигены группы крови ABH также являются важными олигосахаридами.
В настоящем изобретении предложен быстрый и экономичный способ получения всех этих соединений. Так, например, для получения SLea, который представляет собой NeuAc α-2,3 Gal β-1,3(Fuc α- 1,4)GlcNAc, объединяют следующие три гликозилтрансферазы: β-1,3-галактозилтрансферазу, α- 2,3-сиалилтрансферазу и α-1,4-фукозилтрансферазу. Условия реакции и вспомогательные субстраты ферментов для регенерации сахарных нуклеотидов, как было указано ранее.
Так, для H-активных олигосахаридов O-группы крови антиген, который имеет структуру Fuc α-1,2Gal β-R, где R может быть β-1,3GlcNAc-R1 или β-1,3-GalNAc-R1 и где R1 является рестриктированным олигосахаридом, можно объединить следующие гликозилтрансферазы: β-1,3-галактозилтрансферазу и α-1,2-фукозилтрансферазу с соответствующими вспомогательными компонентами реакции и в указанных ранее условиях для SLex или SLeа до получения Fuc α-1,2Gal β-1,3-GlcNAc-R1. Группа R1 O-группы крови, таким образом, представляет другую X группу, обсуждавшуюся ранее, как и R1 группы для A- и B-групп крови.
Для A-активных олигосахаридов антиген A-группы крови, который имеет структуру GalNac α-1,3(Fuc α-1,2)Gal β-R, где R может быть β1,3 GlcNAc-R1 или β-1,33 GalNAc-R1 и где R1 является рестриктированным олигосахаридом, можно объединить следующие гликозилтрансферазы: β-1,3-галактозилтрансферазу, α- 1,2-фукозилтрансферазу, α-1,3N-ацетилгалактозаминилтрансферазу с соответствующими вспомогательными компонентами реакции и в указанных ранее условиях до получения GAlNAc α/ -1,3(Fuc α-1,2)Gal β-1,3GlcNAc-R1.
Для B-активных олигосахаридов, антиген B-группы крови, который имеет структуру Gal α-1,3(Fuc α-1,2)Gal β-R, где R может быть β-1,3GlcNAc-R1 или β-1,3GalNAc-R1, и где R1 является рестриктированным олигосахаридом, можно объединить следующие гликозилтрансферазы: β-1,3-галактозилтрансферазу α-1,2-фукозилтрансферазу, α-1,3-галактозилтрансферазу с соответствующими вспомогательными компонентами реакции и условиями, которые указаны ранее, до получения Gal α- 1,3(Fuc α-1,2)Gal β-1,3GlcNAc-R1.
Таким образом, предложен катализируемый ферментами постадийный синтез олигосахаридов, включающий фукозилированные и фукозированные сиалилированные углеводные молекулы, в котором продукты каждой из реакций гликозилирования выделяют перед следующей стадией гликозилирования. Такие реакции гликозилирования могут использовать (и предпочтительно используют) обсуждавшиеся ранее стадии рециркулирования.
Также представлены многочисленные гликозилирования в единой реакционной смеси для получения тех же фукозилированных и фукозилированных сиалилированных углеводных молекул. Используют здесь и реакции рециркулирования, обсуждавшиеся ранее. Кроме того, Km и Vотн. данные, представленные в табл. 1 и 2 и опубликованные значения используют для установления концентраций реагентных образцов таким образом, чтобы свести к минимуму побочные реакции. Для контроля за образованием продукта можно также использовать представленные в табл. 3 ингибиторы.
Представлены также мультистадийные реакции гликозилирования в единой реакционной смеси для получения вышеуказанных продуктов, но такие, в которых один фермент или необходимый для гликозилирования реагент добавляют после практического завершения других реакций, так что одна реакция гликозилирования начинается после того, как по крайней мере одна или предпочтительно две другие реакции гликозилирования практически завершены. В одном из примеров синтеза все реагенты и ферменты, представленные на схеме 1, за исключением фукозилтрансферазы (FucT), добавляют к реакционной смеси, и NeuAc α-2,3Gal β-1,4GlcNAc-OR образуется, что видно на схеме 16, для образования соединения 42, где R является аллилом. После образования такого соединения, как соединение 42 схемы 16, добавляют FucT, такую как α-1,3-фукозилтрансфераза или α-1,3/4-фукозилтрансфераза, и образуется такой фукозилированный сиалилированный углевод, как соединение 44 схемы 17. В другом варианте, FucT фермент может присутствовать, и тогда можно опустить такой предшественник фукозильного донора, как фукоза. Аналогично фукоза может присутствовать без фосфорилирующего ее фермента, фукозокиназы.
F. Получение производных фукозилированных продуктов с образованием лигандов.
Описанные ранее фукозилированные продукты являются гаптенами, которые лучше функционируют как лиганды, будучи связаны с более крупными фрагментами. Такие фрагменты включают протеины, гликопротеины, гликолипиды и небиологические аналоги таких молекул. Обычно восстанавливающий конец сахара связан со свободным амином или меркаптаном гликозидной связью. Липосомы пригодны для получения мультивалентной макромолекулы. Различные способы пригодны для получения липосом, как указано, например, в Szoka et al., Ann. Rev. Biophys. Bioeng., 9: 467 (1980), в патенте США N 4235871, N 4501728 и N 4837028.
G. Анализ активности сиалильного лиганда Льюиса.
Один из вариантов настоящего изобретения относится к получению сиалилированных льюисовых антигенов как в природной форме, так и в виде имитаций или аналогов. Эти антигены играют роль в межклеточной адгезии, а также в воспалительных процессах и других болезненных состояниях человека и млекопитающих. Для облегчения продуцирования этих антигенов по способу настоящего изобретения полезно проанализировать полученные продукты по их способности связываться с природными рецепторами сиалилированных льюисовых антигенов, например, ELAM и GMP140 рецепторами. Такие анализы подробно описаны Polley et al., Proc. Natl. Acad. Sci. U.S.A. 88: 6224-6228 (1991) и Phillips et al. , Science, 250: 1130-1132 (1990).
Хотя возможен целый ряд анализов, предпочтительным анализом является тот, в котором определяют способность антигенов блокировать или ингибировать связывание клеток с соответствующими адгезионными рецепторами и клетками, экспрессирующими соответствующий сиалилированный льюисовый антиген. Связывание оценивают визуально под микроскопом. Предпочтительными клетками, экспрессирующими рецепторы, являются активированные тромбоциты или эндотелиальные клетки. Эти рецепторы являются частью семейства, известного как селектины, или LEC-CAM, и включают LEC-CAM-1, ELAM-1, GMP-140 и CD62. Лиганды найдены на нейтрофилах, моноцитах и опухолевых клетках.
В типичном анализе выделяют нейтрофилы, расслаивая обработанную гепарином кровь на Mono-Poly Resolving Medium (разделяющая среда от Ficoll-Hypaque-Flow Laboratories), с последующим центрифугированием в течение 25 мин при 2000 об/мин, а затем еще 25 мин при 2500 об/мин.
Тромбоциты можно выделить таким способом. Кровь берут у здорового донора-человека в шприц, содержащий ACD антикоагулянт (декстроза, 2,0 г, цитрат натрия 2,49 г и лимонная кислота 1,25 г до 100 мл dH2O) в соотношении 6 частей крови на 1 часть антикоагулянта. Эту кровь центрифугируют при 800 об/мин (примерно 90•g) в течение 15 мин при комнатной температуре. Надосадочную жидкость собирают и центрифугируют при 1200 об/мин (примерно 400•g) в течение 6 мин. Надосадочную жидкость удаляют и центрифугируют при 2000 об/мин (примерно 1200•g) в течение 10 мин для осаждения тромбоцитов. Комок тромбоцитов промывают дважды Tyrode-HEPES буфером, pH 6,5 (NaCl 8,0 г, KCl 0,2 г, NaH2PO4 • H2O 0,57 г; MgCl2 • H2O 0,184 г; NaHCO3 0,1 г; декстроза 1,0 г; и HEPES 2,383 г; доводят до 1 л деионизированной водой, pH устанавливают 6,5 1 н NaOH) с последующей одной промывкой в PBS. Тромбоциты суспендируют в концентрации 108/мл в PBS, а затем активируют, инкубируя в течение 20 мин при комнатной температуре с тромбином при конечной концентрации 0,25 ед/мл.
Для анализа 20 мкл суспензии тромбоцитов (2 • 108 мл) помещают в ампулу Эппендорфа для центрифуги. Добавляют равный объем олигосахарида, приготовленного с концентрациями от 200 до 0,3 мкг/мл, или гликолипид-липосомного препарата (полученного описанным ранее способом), с концентрациями от 2 до 0,25 мкг/мл, и ампулы оставляют при комнатной температуре на 20 мин. Затем добавляют 20 мкл препарата нейтрофилов (2 • 106/мл), и ампулы оставляют выстаиваться при комнатной температуре еще 20 мин.
Адгезию активированных тромбоцитов к нейтрофилам оценивают под микроскопом. Обычно оценивают 100 нейтрофилов. Их оценивают как позитивные, если к ним присоединены два или более из тромбоцитов, и как негативные, если связаны менее двух тромбоцитов.
H. Усовершенствованный способ получения глимкозил-1 или 2-P.
Фосфорилированные сахара, содержащие фосфат на аномерном углеводе (1- или 2-положение), представляют ценность в описываемых реакциях и некоторые из них доступны коммерчески. В настоящем изобретении предложены усовершенствованные способы селективного фосфорилирования этого углерода моносахаридорв. Усовершенствование включает использование трехвалентного фосфитилирующего реагента для переноса фоcфитильного фрагмента на нужный углерод. Полученный фосфит используют затем для получения соответствующего фосфата, который используют в ферментативной реакции, описанной здесь.
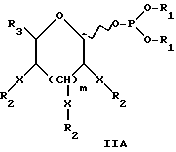
Защищенный фосфитильный моносахарид соответствует приводимой далее формуле I: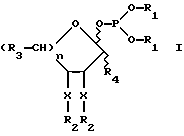
где каждый из R1 может быть одинаковым или различным и представляет такую арильную группу, как фенил или бензил, или C1-C5 низшую алкильную группу;
X независимо является кислородом или азотом,
R2 независимо является ацилом, бензилом, силилом или алкильной защитной группой или X-R2 вместе отсутствуют и заменены водородом;
R3 независимо является водородом (-H), -CH3, -OR2, -CH2OR2, -CH(OR2)-CH(OR2), -CH(OR2)-CH(OR2)-CH-(OR2) , -NH2 или -NHR2;
R4 представляет водород (H), карбоксил или C1-C5-алкил или бензилкарбоксилат; а
n = 1 или 2, предпочтительно 2.
Рассматриваемые защищенные фосфитилмоносахариды включают производные сиалильной кислоты, KDO, KDN или аналогичные соединения, в которых R4 является карбоксилом или сложноэфирной группой кaрбоксилата. В предпочтительной группе соединений формулы I R4 представляет водород. В таком случае формула I превращается в формулу II, где R1, R2, R3, X и n имеют указанные ранее значения: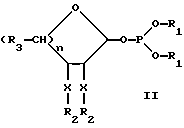
Следует учитывать, что каждая R1 группа может отличаться от других. Это проистекает из того факта, что реагент фосфитилирования можно получить в реакции PCl3 с таким вторичным амином, как диизопропиламин, и двумя молями спирта. Смешивая спиртовую часть реакционной смеси, можно получить реагент фосфитилирования и фосфит, которые будут иметь две различные R1 группы, например бензил и этил. Предпочтительно, чтобы обе R1 группы были одинаковы, и наиболее предпочтительно, чтобы обе были бензильными или фенильными группами, как те группы, которые можно удалить гидрогенолизом.
Следует также учитывать, что каждый X может быть кислородом или азотом, и особенно рассматриваются соединения, в которых есть обе группы, например защищенный сиалилдибензилфосфат, соединение 97. R2 защитные группы включают такие ацильные группы, как C1-C5 ацильные группы, например формил, ацетил, пивалоил и пентаноил, бензоильные и фталоильные группы, алкилзащитные группы и силильные группы. Примеры алкильных групп включают такие C1-C5 алкилы, как метил, этил, изопропил, трет.-бутил, циклопентил и пентил. Ацетали и кетали, полученные из C1-C5 алкилкетонов или альдегидов, таких как наиболее предпочтительный ацетон и формальдегид, могут также образовывать алкилзащитную группу. Бензальдегид также рассматривается как ацетальобразующая защитная группа. Такие кетали и ацетали являются хорошо известными защитными группами в химии сахаридов. Примеры силильных защитных групп включают три-C1-C5 алкилсилильные группы, такие как триметилсилил, трет.-бутилдиметилсилил и т.п. , C1-C5-алкилдифенилсилильные защитные группы, такие как дифенилметилсилильная группа, ди-C1-C5 алкилфенилсилильные защитные группы такие, как фенилдиметилсилильная группа и трифенилсилильная защитная группа.
Обычно предпочтительно, чтобы все защитные группы были одинаковы или, если они различны, чтобы их можно было селективно удалять в различных реакциях. Так, например, бензильную группу можно удалять в присутствии ацетильной группы гидрогенолизом, тогда как ацетильную группу можно удалять в присутствии бензильной группы, обрабатывая таким первичным амином, как бензиламин. Ацетил является наиболее предпочтительной защитной группой такой, как O-ацетильная группа в аномерном положении (1- или 2-положении), и его можно легко удалять в присутствии других O-ацетильных группу в других положениях кольца, обрабатывая первичным амином.
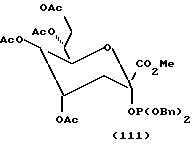
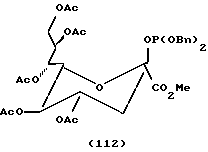
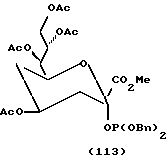
Следует отметить, что X-R2 может отсутствовать и может быть заменен водородом. Как таковой защищенный моносахарид является дезоксимоносахаридом, что видно на примере соединений 97 и 101 - 113.
Следует учитывать, что если n = 2 в формуле I, что предпочтительно для сахаров с шестичленным кольцом, обе из (R3-CH)групп не обязательно должны быть одинаковы, и обычно отличаются. Так, например, для защитного фукозилфосфита соединение 8, обсуждавшееся ранее в отношении схемы 6, одна R3 группа представляет CH3, тогда как другая представляет O-ацетил (OAc). Аналогично для защищенного маннозилфосфита соединения 16, обсуждавшегося в отношении схемы 9, один R3 представляет CH2OAc группу, тогда как другой является OAc группой. Формулу I можно в другом варианте представить как формулу IA далее, где каждый из R1 может быть одинаковым или различным и представляет такие арильные группы, как фенил или бензил, или C1-C5 низшую алкильную группу; X независимо представляет кислород или водород;
R2 независимо представляет ацильную, бензильную, силильную или алкильную защитную группу, или X-R2 вместе отсутствуют и заменены водородом;
R3 независимо представляет водород (-H), -CH3, -OR2, -CH2OR2, -CH(OR2)-CH(OR2), или -CH(OR2)-CH-(OR2)-CH(OR2);
R4 представляет водород (H), карбоксил или C1-C5-алкил, или бензил карбоксилат; а
m = 0 или 1, так что если m = 0, (CH-X-R2) группа отсутствует, и образуется пятичленное кольцо, а если m = 1, тогда присутствует (CH-X-R2) группа, и моносахарид имеет шестичленное кольцо, что является предпочтительным.
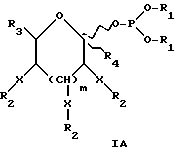
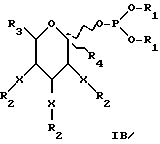
В соответствии с предпочтительностью защищенных моносахаридных фосфитов с шестичленными кольцами формулу IA можно представить как формулу IB, где R1-4 и X имеют значения, указанные для формулы IA.
В соответствии с предпочтительностью соединений, в которых R4 представляет водород, формулу II можно представить как формулу IIA (ниже). В соответствии с предпочтительностью для защищенных моносахаридов с шестичленными кольцами (где m = 1) формулу IIA можно представить как формулу IIB (ниже). R1-3, m и X те же, что и в формуле IA.
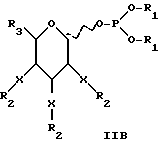
За исключением отмеченных различий в значения R групп в формулах I, II, IA, IB и IIA значения X, ацил, силил, алкил и других групп в этих формулах одинаковы.
Трехвалентные фосфитилирующие реагенты были определены ранее. Доступные трехвалентные фосфитилирующие реагенты включают: дибензил-N,N-диэтилфосфороамидит, 2-цианоэтил-N,N,N',N'-тетраизопропилфосфороамидит или 2-цианоэтил-N,N-диизопропилхлорфосфороамидит.
Способ получения защищенного моносахаридного фосфита из незамещенного (незащищенного) моносахарида представляет собой многостадийный способ, начинающийся с любого моносахарида (альдозы или кетозы без ограничения конформацией или ориентацией). Моносахарид вначале защищают по каждому свободному гидроксилу (или амину), используя стандартные защитные реагенты, такие как ацильные, бензильные, силильные или алкильные группы, как обсуждалось ранее. Затем защитные группы, обычно C1-C5-ацильные или бензильные группы в 1- или 2-положении, селективно удаляют, используя либо свиную липазу поджелудочной железы, либо алкил или бензиламин в таком неводном полярном растворителе, как тетрагидрофуран или дихлорметан. Трехвалентный фосфитилирующий реагент вводят затем в 1- или 2-пoложение в анаэробных условиях, используя ароматический вторичный или третичный амин в качестве конденсирующего агента, например 1,2,4-триазол, тетразол, имидазол или пиридиний-паратолуолсульфонат. В настоящее время предпочтительными конденсирующими агентами являются триазол или тетразол. Затем полученный продукт окисляют, используя такие оксиданты, как перекись водорода или трет.-бутил-гидроксипероксид. У полученной фосфорильной группы удаляют защиту до получения фосфатной соли (например, натриевой), используя восстановление (водород/палладий) для бензильных производных и щелочную обработку для 2-цианоэтильных производных.
Таким образом, используя вышеуказанные реакции получают ряд фосфитов и соответствующих фосфатов. Примерные соединения (в виде фосфатов), которые были получены из 2-ацетамидо-2-дезокси-D-галактозы (CalNAc), соединение 89, 2-ацетамидо-2-дезокси-D-глюкозы (ClcNAc, соединение 90), D-галактозы (Cal, соединение 91), D-глюкозы (Clc, соединение 92), D-маннозы (Man, соединения 18 и 93), L-рамнозы (Rha, соединение 94), L-фукозы (Fuc, соединение 5) и N-ацетилнейраминовой кислоты (NeuAc, соединение 99). На схемах 21 - 23 ниже представлены эти реакции. Фосфит 2-фталимидоил-2-дезокси-D-глюкозо-3,4,6-триацетата (соединение 100) также получают представленным на схеме 23 способом с примерно 90%-ным выходом.
В табл. 4 далее представлены значения различных "R" групп (R1-4), использованных в этих схемах. В табл. 5 представлены выходы и соотношения аномеров для различных соединений схем 21 - 23 и табл. 4.
Было обнаружено, что растворители влияют на аномерное соотношение фосфитилированных продуктов. Так, если соединение 70 фосфитилируют в ТГФ, отношение α:β составляет 1 : 6, тогда как для хлороформа этот соотношение меняется на 1 : 2.
Следует отметить, что соединения на схемах и в таблицах, приведенных ранее, включают производные маннозы и фукозы, которым были присвоены другие номера в более ранних обсуждениях. Эти соединения были перенумерованы здесь для большего удобства представления данных. В последующих примерах приводятся обе нумерации.
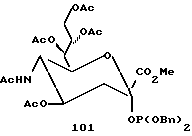
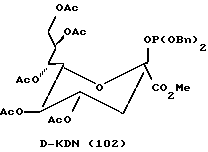
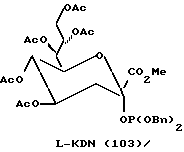
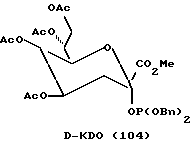
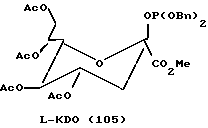
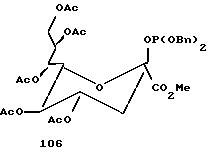
В соответствии с реакциями, изображенными на схеме 23, был проведен синтез еще нескольких специфических моносахаридных карбокислатов формулы I. Эти соединения являются родственными с D-сиалильной кислотой (соединение 101), D- и L-KDN и D- и L-KDO. Cтроение метиловых сложных эфиров (Me) этих соединений, где R2 представляет ацетил, а R1 представляет бензил, показаны далее как соединения 101 - 113. Подчеркнутые моносахаридные карбоксилаты можно получить в реакциях, катализируемых альдолазой сиалильной кислоты, что показано Gautheron-Le Narvor et al., J. Am. Chem. Soc., 113: 7816 (1991) и Sugai et al., J. Am. Chem. Soc., (в печати).
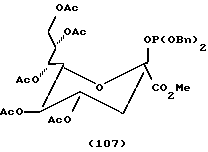
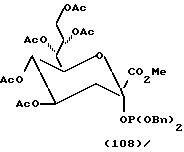
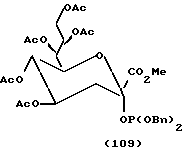
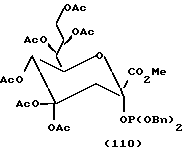
Примеры.
После того как было представлено содержание изобретения в общих чертах и было дано руководство для сочетания реакций фукозилтрансферазы с реакциями с выделением энергии, в которых используют каталитические количества недорогих нуклеотидов и с другими реакциями трансферазы, далее приводятся примеры для более детального описания процессов. Эти примеры представлены исключительно с целью иллюстрации, но не являются ограничительными. Специалистам ясно, что многие параметры не являются критическими, и их можно варьировать.
Пример 1. Химический синтез фукозо-1-фосфата (схемы 5 и 6).
(a) 1,2,3,4-тетра-О-бензоил-L-фукоза (соединение 2)
Бензоилхлорид (21.4 г, 152.3 ммоль, 17.7 мл) прикапывают к охлажденному раствору L-фукозы (5.7 г, 30.5 ммоль) в 100 мл пиридина при 0 - 5oC, и полученную смесь перемешивают в течение 3 ч при комнатной температуре. Полученную смесь выливают в ледяную воду и экстрагируют этилацетатом (EtOAc). Полученные экстракты последовательно промывают ледяной водой и разбавленной HCl, водным бикарбонатом натрия и рассолом, сушат над безводным сульфатом магния и концентрируют. Полученный продукт используют на следующей стадии без дальнейшей очистки.
(b) Дибензилфосфорид-2,3,4-три-О-бензоил β- L-фукозид (соединение 4).
К охлажденному раствору соединения 2 (2.0 г, 3.44 ммоль) в 20 мл CH2Cl2 и Ac2O (2 мл) прикапывают 30% HBr-AcOH (8 мин) при 0 - 5oC, и полученную смесь перемешивают в течение 2 ч при комнатной температуре. Полученную смесь выливают в ледяную воду, и экстрагируют EtOAc. Полученные экстракты последовательно промывают водой, водным бикарбонатом натрия и рассолом, сушат над безводным сульфатом магния и концентрируют до получения соединения 3. Соединение 3 используют на следующей стадии без дополнительной очистки.
1H-ЯМР (CDCl3) δ : 1.36 (3H, д, J = 6.1 Гц, 6-CH3), 4.69 (1H, шир.кв., J = 6.56 Гц, H-5), 5.62 (1H, дд, J = 3.91, 10.5 Гц, H-2), 5.84 (1H, дд, J = 0.97, 3.33 Гц, H-4), 6.01 (1H, дд, J = 3.36, 10.50 Гц, H-3), 6.94 (1H, д, J = 3.92 Гц, H-1);
13C ЯМР (CDCl3) δ 15.8, 68.6, 69.2, 70.4, 89.4, 165.4, 165.6, 165.7 (Здесь и далее в результатах исследования ЯМР и МС встречаются следующие обозначения:
NMR = ЯМР,
S = c (синглет),
d = д (дублет),
t = т (триплет),
q = кв (квартет),
m = м (мультиплет),
br = broad = шир (широкий),
dd = дд (дублет дублетов),
ppm = мд (мил.доли),
Hz = Гц,
HRMS = ВРМС (масс-спектр высокого разрешения),
calcd = рассчитано,
found = найдено.
Ag2CO3 (1.90 г, 6.89 ммоль) добавляют в охлажденный (0 - 5oC) раствор полученного ранее соединения 3, дибензилфосфата (2.88 г, 10.3 ммоль) и МС (молекулярные сита) 3  (6 г) в CH2Cl2-Et2O-CH3CN (20 мл каждого) в круглодонной колбе, закрытой алюминиевой фольгой от света. Полученную смесь перемешивают в течение 10 ч при комнатной температуре, и фильтруют через слой целита, а полученный фильтрат концентрируют. Остаток обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента толуол - EtOAc (2.5 : 1) до получения соединения 4 (2.4 г, 95%) в виде одного продукта. [1H NMR (CDCl3) δ : 1.35 (1H, d, J 6.35 Hz, 6-CH3), 4.225 (1H, br dt, J 5.71, 6.70 Hz, H-5), 4.77 (1H, dd, J 7.07, 11.65 Hz, бензил.), 4.86 (1H, dd, J 6.50, 1.63 Hz, бензил.), 5.11 (1H, dd, J 7.51, 11.71 Hz, бензил. ), 5.14 (1H, dd, J 7.27, 11.70 Hz, бензил.), 5.58 (1H, dd, J 3.48, 10.44 Hz, H-3), 5.69 (1H, dd, J 7.37, 7.89 Hz, H-1), 5.76 (1H, dd, J 8.03, 3.44 Hz, H-4), 5.90 (1H, dd, J 8.03, 10.45 Hz, H-2); 13C NMR (CDCl3) δ : 16.12, 69.31, 69.35, 69.54, 69,58, 69.63, 60.70, 70.57, 70.83, 71.70, 96.97, 90.00, 127.28, 127,40, 127.89, 128.06, 128.13, 128.26, 128.31, 128.43, 128.58, 128.667, 128.83, 129.06, 129.67, 129.72, 129.77, 129.77, 129.93, 133.27, 133.41, 133.51, 165.24, 165.45, 165.79. HRMS рассчитано для C41H37O11PCs (M+Cs+) 869.1128, найдено 869.1138.]
(6 г) в CH2Cl2-Et2O-CH3CN (20 мл каждого) в круглодонной колбе, закрытой алюминиевой фольгой от света. Полученную смесь перемешивают в течение 10 ч при комнатной температуре, и фильтруют через слой целита, а полученный фильтрат концентрируют. Остаток обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента толуол - EtOAc (2.5 : 1) до получения соединения 4 (2.4 г, 95%) в виде одного продукта. [1H NMR (CDCl3) δ : 1.35 (1H, d, J 6.35 Hz, 6-CH3), 4.225 (1H, br dt, J 5.71, 6.70 Hz, H-5), 4.77 (1H, dd, J 7.07, 11.65 Hz, бензил.), 4.86 (1H, dd, J 6.50, 1.63 Hz, бензил.), 5.11 (1H, dd, J 7.51, 11.71 Hz, бензил. ), 5.14 (1H, dd, J 7.27, 11.70 Hz, бензил.), 5.58 (1H, dd, J 3.48, 10.44 Hz, H-3), 5.69 (1H, dd, J 7.37, 7.89 Hz, H-1), 5.76 (1H, dd, J 8.03, 3.44 Hz, H-4), 5.90 (1H, dd, J 8.03, 10.45 Hz, H-2); 13C NMR (CDCl3) δ : 16.12, 69.31, 69.35, 69.54, 69,58, 69.63, 60.70, 70.57, 70.83, 71.70, 96.97, 90.00, 127.28, 127,40, 127.89, 128.06, 128.13, 128.26, 128.31, 128.43, 128.58, 128.667, 128.83, 129.06, 129.67, 129.72, 129.77, 129.77, 129.93, 133.27, 133.41, 133.51, 165.24, 165.45, 165.79. HRMS рассчитано для C41H37O11PCs (M+Cs+) 869.1128, найдено 869.1138.]
(c) β-/ L-фукозо-1-фосфат (соединение 5)
Соединение 4 (2.32 г, 3.15 ммоль) гидрируют над 5% Pd/C (400 мг) в 60 мл EtOH, и 15 мл 1н NaHCO3 в течение 10 ч. Катализатор отфильтровывают на целитном фильтре. К охлажденному раствору остатка в воде (20 мл при 0 - 5oC прикапывают 20 мл 1н NaOH и полученную смесь перемешивают в течение 3 ч при комнатной температуре. Полученную смесь осторожно нейтрализуют, добавляя холодный 1н AcOH до pH 7.5, и нерастворимую часть отфильтровывают на целите. Полученный фильтрат разбавляют до 250 мл и вводят в колонку Dowex 1-X8 (HCO2) (2 • 15 см), и элюируют с поэтапным градиентом NH4OAc2; вода (200 мл), 0,1 М MN4OAc2 (200 мл), 0.2 М NH4OAc2 (200 мл) и 0.3 М NH4-OAc2 (200 мл). Фукозу элюируют водой, а целевое соединение 5 элюируют между 0.2 - 0.3 М NH4OAc2. После удаления соли соединение 5 (700 мг, 82%) получают. Данные 1H 13C ЯМР находятся в хорошем соответствии с данными группы Бейкера (Nunez et al., Can. J. Chem. 59: 2086 (1981)).
(d) 1,2,3,4-тетра-O-ацетил-L-фукоза (соединение 6).
Смесь L-фукозы (3.0 г, 18.2 ммоль) и безводного NaOAc (50 мг, 0.61 ммоль) в уксусном ангидриде (20 мл) перемешивают в течение 2 ч при комнатной температуре, а затем нагревают при 100oC в течение 2 ч. После охлаждения полученную смесь выливают в ледяную воду, перемешивают в течение 2 ч и экстрагируют хлороформом. Полученные экстракты последовательно промывают водным бикарбонатом натрия и водой, сушат над безводным сульфатом магния и концентрируют. Оставшийся сироп обрабатывают на хроматографической колонке с силикагелем, элюируя смесь толуолэтилацетат (10 : 1) до получения соединения 6 (5.92 г, 98%) в виде смеси α и β (1 : 7, по данным 1H ЯМР).
(e) 2,3,4-три-O-ацетил-L-фукоза (соединение 7 или 74).
i) Химический способ:
Раствор соединения 6 или 67 (3.0 г, 9.0 ммоль) и бензиламина (1.45 г, 13.5 ммоль, 1.47 мл) в ТГФ (35 мл) перемешивают в течение дня при комнатной температуре. Полученную смесь разбавляют хлороформом и последовательно промывают охлажденной льдом разбавленной соляной кислотой, водным бикарбонатом натрия и водой, сушат над безводным сульфатом магния и концентрируют. Оставшийся сироп обрабатывают хроматографически на силикагеле смесью толуола и этилацетата (1 : 1) до получения соединения 7 или 74 (2.40 г, 92%). Данные 1H ЯМР соответствуют литературным. (Hennen et al., J. Org. Chem., 53: 4743 (1988)).
ii) Ферментативный способ:
Суспензию соединения 6 или 67 (2.5 г, 7,5 ммоль) и свиную липазу поджелудочной железы (5.6 г) в 13% (объем/объем) ДМФ/(фосфатного буфера (50 мл, pH 7) перемешивают в течение 5 дней при комнатной температуре, поддерживая в это время pH за счет 1н NaOH. Полученную смесь фильтруют, а полученный фильтрат экстрагируют этилацетатом. Полученный экстракт промывают водой и сушат над безводным сульфатом магния. Очистку продукта осуществляют как указано ранее, получая соединение 7 или 74 (1.1. г, 48.4%) в виде смеси α и β (1 : 1).
(f) Дибензилфосфорид-2,3,4-три-О-ацетил-L-фукозид (соединение 9 или 88).
Дибензил-N,N-диэтилфосфороамидат (Pederson et al., Tetrahedron, 47: 2643 (1991) (2.7 г, 8.5 ммоль) прикапывают к раствору соединения 7 или 74 (1.0 г, 3.4 ммоль) и тетразола (1.0 г, 14.5 ммоль) в ТГФ (50 мл) в атмосфере азота при комнатной температуре, и полученную смесь перемешивают в течение часа при комнатной температуре. К этой смеси добавляют эфир (50 мл), и органическую фазу промывают охлажденной льдом соляной кислотой, водным бикарбонатом натрия и водой, сушат над безводным сульфатом магния и концентрируют. Полученный сироп обрабатывают на хроматографической колонке с силикагелем, элюируя смесью гексан-этил-ацетат (4: 1) до получения соединения 8 или 81 (1.43 г, 79%) в виде смеси α и β (1:10).
1H ЯМР (CDCl3) δ : 1.22 (3H, d, J 6.50 Hz, 6-CH3), 1.91, 1.99, 2.19 (3H, каждый, s, 3 x OAc), 3.85 (1H, dg, J 1.00, 6.50 Hz, H-5), 4.82-4.96 (4H, m, бензильные протоны), 5.02-5.08 (2H, m, H-2, 3), 5.25 (1H, dd, J 0.50, 3,50 Hz, H-4), 5.32 (1H, dd, J 8.00, 10.50 Hz, H-1).
К охлажденному раствору соединения 8 или 81 (500 мг, 0.9 ммоль) в 50 мл ТГФ добавляют 30%-ную перекись водорода (7 мл) сразу, и полученную смесь оставляют нагреваться до комнатной температуры, и перемешивают 90 мин. Полученную смесь разбавляют эфиром и промывают ледяным водным тиосульфатом натрия, водным бикарбонатом натрия и водой, сушат над сульфатом магния и концентрируют до получения соединения 9 или 88 (420 мг, 81%). Соединение используют на следующей стадии без дополнительной очистки. Данные 1H ЯМР находятся в хорошем соответствии с литературными. (Schmidt et al., Liebigs Ann. Chem., 191: 121 (1991)).
1H-ЯМР (CDCl3) δ : 1.22 (3H, 3, J 10.0 Hz, 6-CH3), 1.91, 1.99, 2.19 (3H каждый, s, 3 x OAc), 3.90 (1H, dq, J 6.50, 7.50 Hz, H-5), 5.00-5.03 (m, H-3, бензильные протоны), 5.03-5.12 (m, бензильные протоны), 5.26 (1H, dd, J 1.00, 3.50 Hz, H-4), 5.27-5.33 (2H, m, H-1,2). HRMS для C26H31O11PCs: рассчитано для (M+C) + 683.0658, найдено 683.0658.
(g) L-фукозо-1-фосфат (соединение 5 или 95),
Соединение 9 или 88 (5.0 г, 9.1 ммоль) гидрируют над 5% Pd/C (400 мг) в EtOH (70 мл) и бикарбонате натрия (30 мл) в атмосфере водорода в течение 3 ч при комнатной температуре, и катализатор отфильтровывают. К охлажденному фильтрату добавляют 1н NaOH при 0-5oC до тех пор, пока раствор не становится сильно щелочным (pH более 13). Полученную смесь перемешивают в течение 4 ч при комнатной температуре и нейтрализуют, добавляя холодную 1н уксусную кислоту до pH 7.5. Полученную смесь фильтруют, разбавляют до 500 мл, вводят в колонку Dowex 1-X8 (HCOO-) смолы и элюируют с линейным градиентом бикарбоната аммония (0-0.5 М). Соответствующие фракции собирают и лиофилизируют. Избыток бикарбоната аммония удаляют, добавляя к раствору смолу Dowex 50 X8 [H+] к раствору лиофилизированного порошка в воде. Смолу отфильтровывают, и полученный фильтрат лиофилизируют. Раствор продукта пропускают через колонну Dowex W-X8 [Na+] с водой и лиофилизируют до получения соединения 5 или 95 (2.61 г, 99%) в виде смеси α и β (1:10, по данным 1H ЯМР). 1H и 13C ЯМР спектры находятся в хорошем соответствии с литературными данными (Nunez et al., Can. J. Chem., 59: 2086 (1981)).
Пример 2. Химический способ получения GDP-фукозы (соединение 12, схема 7).
Соединение 5 вначале превращают в его триэтиламмонийную соль, пропуская через колонку Dowex 50 W-X8 [Et3NH+ форма] с водой, и лиофилизируют. Лиофилизированную L-фукозо-1-фосфат-триэтиламмонийную соль (соединение 10) (300 мг, 0.83 ммоль) и гуанозин-5'-монофосфатморфолидат (соединение 11, 600 мг, 0.83 ммоль) отдельно сушат, испаряя совместно с пиридином дважды. Затем их объединяют с 20 мл сухого пиридина, и полученную смесь перемешивают в течение 5 дней при комнатной температуре и концентрируют. Полученный продукт очищают на колонке с сефадексом 0 - 25 (суперфайн) (3х65 см), дважды элюируя водой. Соответствующие фракции собирают и пропускают через колонку Dowex 50 W-Xg[Na+] с водой. Фракции собирают и лиофилизируют до получения соединения 12 (300 мг), загрязненного небольшим количеством GMP (по данным 1H ЯМР). 1H ЯМР спектр находится в хорошем соответствии с литературными данными (Gokhale et al., Can. J. Chem., 68 1063 (1990)).
Пример 3. Ферментативное превращение маннозо-I-P в GDP-фукозу и анализы (схемы 9 - 11).
(a) Получение GDP-маннозопирофосфорилазы для превращения Man-I-P в GDP-маннозу.
Ферментом, превращающим GDP-маннозу, является GDP-маннозопирофосфорлаза (GDP-ManPP), которую получают из дрожжей. Большую часть GDP-ManPP из дрожжей выделяют, осаждая сульфатом аммония (около 80% по сравнению с неочищенным, не содержащем клеток экстрактом), со специфической активностью около 0,1 ед/мл раствора фермента.
Дрожжи Saccharomyces cerrevisiae выращивают на среде (г/л): экстракт дрожжей - 5, пептон - 10, pH 6.0. Культуру растят при 30oC в течение ночи. Клетки собирают центрифугированием и промывают 50 мМ трис-буфером (pH 7.5), содержащим 2 мМ MgCl2 и 0.5 мМ DTT. Клетки (около 10 г) разбивают стеклянными шариками, используя шаровую мельницу (Bioseptic Products, ОК) импульсами с одноминутными интервалами пять раз. Затем раствор центрифугируют при 4oC при 23000 g в течение часа. Надосадочную жидкость (экстракт без клеток) собирают и используют для очистки фермента. Для частичной очистки фермента 40 - 80% (при 0oC) осажденного аммонийсульфатом собирают, медленно добавляя порошок сульфата аммония к не содержащему клеток экстракту до 40%-ного насыщения, и центрифугируют при 4oC при 15000 g в течение 30 мин, а затем к надосадочной жидкости добавляют еще сульфат аммония до 80%-ного насыщения. После центрифугирования осадок собирают и снова растворяют в 20 мл 50 мМ tris (pH 7.5) буфера, содержащего 2 мМ MgCl2 и 0.5 мМ DTT, и диализуют в 4 л того же буфера в течение ночи (около 18 ч) при 4oC. Активность этого препарата - около 0.1 ед/мл, по данным анализа активности с помощью ВЖЭХ.
(b) Получение GDP-фукозных синтетических энзимов для превращения GDP-маннозы в GDP-фукозу.
Первоначальные попытки использовать частично очищенный GDP-фукозный синтетический фермент (собранный после осаждения сульфатом аммония) для превращения GDP-маннозы в GDP-фукозу не увенчались успехом из-за сильного внутреннего окисления NADPH. Дальнейшая очистка фермента за счет пропускания через DEAD-Сефарозную CL-6B колонку привела к более высокой активности фермента, а также к снижению MADPH окислительной активности. Раствор фермента на этой стадии оценивают как около 0.05 ед/мл, по данным NADPH окислительного анализа.
Увеличение образования GDP-фукозы наблюдают, используя ВЖЭХ. Спустя шесть часов протекания реакции выход GDP-фукозы оценивают около 9% в расчете на добавленный маннозо-1-фосфат. Ожидается, что более высокого выхода можно достичь, если удастся получить ферментные растворы с более высокими активностями. Во время этой реакции наблюдается разложение GDP-маннозы. Это разложение можно предотвратить, добавляя фторид калия. Это связано с примесями других ферментов в препарате фермента. Если использовать чистый фермент, отпадает необходимость добавлять фторидную соль.
Бактерию Klebsiella pneumonia АТСС 12658 выращивают на 2 л среды, содержащей 10 г казаминовой кислоты (Difco), 5 г экстракта дрожжей, 3 г K2HPO4, 1 г KH2PO4 и 5 г D-глюкозы на литр (pH 7.0). После инкубирования при 37oC в течение 18 ч клетки собирают центрифугированием (10000 х g, 50 мин, 4oC) и снова суспендируют в 50 мМ трис-буфера, содержащего 0.5 мМ DTT (pH 7.5). Клетки разрушают во французской ячейке под давлением 16000 фунт/дюйм (1125 кг/см2). Осколки клеток удаляют центрифугированием при 23000 x g в течение 60 мин, и надосадочную жидкость (экстракт без клеток) используют для очистки фермента. Не содержащий клеток экстракт (50 мл) из 2 л культуры обрабатывают 60 мг сульфата калия, и полученный осадок удаляют после центрифугирования. Затем медленно добавляют сульфат аммония при перемешивании до достижения 70%-ного насыщения (0.436 г/мл) при 0oC. После центрифугировании осадок собирают и повторно суспендируют в 20 мл буфера (50 мМ tris, содержащего 0.5 мМ DTT, pH 7.5), и диализуют в течение ночи (около 18 ч) при 4oC в 4 л того же самого буфера. Полученный раствор (20 мл) пропускают затем через DEAE-Сефарозу CL-6B (колонка Pharmacia) (3х30 см), которую предварительно уравновешивают тем же самым буфером. Фермент элюируют с линейным градиентом от 0 до 1 мМ NaCl в том же буфере (всего 400 мл). Активную фракцию собирают и диализуют в 2 л 50 мМ tris буфера, содержащего 0.5 мМ DTT (pH 7.5). Этот препарат фермента используют в синтезе непосредственно. Активность оценивают около 0.5 ед/мл по данным хроматографии с обращенной фазой и NADH окислительного анализа.
(c) Анализ ферментной активности.
ВЖЭХ систему используют для определения образования GDP-маннозы и GDP-фукозы. Используют колонки партисил 5 SAX (Whatman Co.), 4.6х12.5 см с размером части насадки 5 мкм. Подвижный буфер; 0.1 М фосфатный буфер (pH 3.5) со скоростью потока 0.5 мл/мин (давление 600 пси, 42.19 кг/см2). Соединение детектируют с помощью УФ детектора на 254 нм. Время удерживания для GDP-маннозы составляет около 8.9 мин, а для GDP-фукозы около 13 мин. Активность GDP-маннозопирофосфурилазы оценивают, следя за образованием GDP-маннозы из α- маннозо-1-фосфата и GTP по данным ВЖЭХ анализа. Реакционная среда содержит 10 мкмоль tris-HC1, 1 мкмоль L-маннозо-1-фосфата, 1 мкмоль GTP и частично очищенный фермент в полном объеме 0.5 мл. После инкубирования при 30oC в течение времени, зависящего от активности фермента, реагент (100 мкл) сливают и центрифугируют через фильтр "Ультрафри" (отсекает 100000 МВ, Millipore). Полученный фильтрат (5 мкм) вводят затем в ВЖЭХ для определения образования GDP-маннозы. Количество GDP-маннозы оценивают по GDP-маннозному стандартному раствору, приготовляемому из очищенной GDP-маннозы (Сигма). Одна единица равна 1 мкмоль GDP-маннозы, образующейся в минуту в исследуемых условиях.
После измерения активности превращения GDP-маннозы в GDP-L-фукозу, можно либо провести спектрофотометрическое определение NADPH окисления, либо непосредственно измерить образование GDP-L-фукозы методом ВЖЭХ. Так как ферментные препараты содержат NADPH оксидазную активность, необходимо определить одновременно скорость NADPH окисления в отсутствии субстрата. В двух кюветах, помещается 1 мл 50 мМ tris буфер (pH 7.5), содержащий 0.2 мкмоль NADPH и раствор фермента, а в другую кювету добавляют 0.1 мкмоль GDP-маннозы. Скорость снижения оптической плотности для двух кювет определяют на длине волны 340 нм. Разница в скоростях NADPH окисления и является мерой конверсии. На фиг. 1 представлен типичный анализ, где линия A поглощения соответствует контрольной кювете без GDP-маннозы, а линия B соответствует поглощению того же раствора, содержащего 1 мкмоль GDP-маннозы. Одна единица равна 1 мкмоль NADPH окисления в минуту в условиях анализа.
Для ВЖЭХ анализа в качестве реакционной среды берут 1 мл tris буфера (pH 7.5), содержащего 1 мкмоль GDP-D-маннозы, 0.2 мкмоль NADPH, 2 мкмоль KF, 2 ед. T. brokii алкоголь-дегидрогеназы, 10 мкл изопропанола и соответствующий раствор фермента. После инкубирования в течение определенного промежутка времени (1 ч), 100 мкл реакционного раствора сливают и центрифугируют через Ultrafree фильтр (отсекает 100000 МВ, Millipore). Полученный фильтрат (5 мкл) вводят затем в ВЖЭХ для определения образования GDP-фукозы. Количество GDP-фукозы оценивают по GDP-фукозному стандарту, полученному из очищенной GDP-фукозы (Сигма). Одна единица равна 1 мкмоль GDP-фукозы, образующемуся в минуту в условиях анализа. На фиг. 2 представлены три хроматограммы ВЖЭХ в момент времени 0 (А), спустя три часа (B) и спустя шесть часов (C) после начала реакции. Как было указано ранее, GDP-манноза элюируется около 8.9 мин, тогда как GDP-фукоза выходит примерно через 15 мин.
Пример 4. Ферментативный синтез GDP-фукозы из маннозо-1-фосфата.
К 5 мл реакционного раствора, 5 мкмоль Man-1-фосфата, 1 мкмоль NADPH, 5 мкмоль GTP, 5 мкмоль PEP, 100 ед пируваткиназы, 50 мкл изопропанола, 5 ед. T. brokii алкоголь-дегидрогеназы, 1 мкмоль MgCl2, 100 ед. неорганической фосфатазы, 5 мкмоль KF, 0.1 ед. GDP-маннозопирофосфатазы раствор и 0.05 ед GDP-фукозных-синтетических ферментов добавляют и инкубируют при 30oC в течение 6 ч, а образование GDP-фукозы определяют по данным ВЖЭХ.
Пример 5. Получение Gal β-1.3(Fuc α-1,4) GlcNAc, используя образование GDP-Fuc
Смесь GTP Na соли (6.0 мг, 10 мкмоль), Man-I-P К соли (3.5 мг, 10 мкмоль), Gal β-1,3 GlcNAc (3.8 мг, 10 мкмоль), NaF (0.42 мг, 10 мкмоль), NADPH (9.4 мг, 10 мкмоль), PEP K соли (4.1 мг, 20 мкмоль), MgCl2-6H2O (2.6 мг, 10 мкмоль), MnCl2-4H2O (2 мг, 10 мкмоль), 2-пропанола (50 мкл), ADH (12 ед), PK (200 ед), PPазы (100 ед), неочищенного ферментативного препарата GDP-пирофосфорилазы (1.0 мл) и неочищенный ферментативный препарат GDP-Fuc продуцирующего фермента (1.0 мл) в 100 мМ tris буфера (pH 7.5) и разбавленную до 3 мл α- 1,3/4-фукозилтрансферазу (0.01 ед) добавляют к смеси, и полученную смесь перемешивают в атмосфере аргона в течение трех дней при комнатной температуре. Полученную смесь отфильтровывают, и полученный фильтрат вводят в колонку Dowex 1-X8 (OH-), а затем в колонку Dowex 50W-X8 (H+) и элюируют водой. Собирают фракции и лиофилизируют. Оставшийся материал очищают на колонке Сефадекс G-25 (суперфайн) с водой. Соответствующие фракции собирают и лиофилизируют до получения Gal β-1,3(Fuc α-1,4)GlcNAc. Его 1H-ЯМР спектр находится в хорошем соответствии с литературными данными (Dumas et al., Biomed. Chem. Lett., I: 425 (1991)).
Пример 6. Химический синтез маннозо-1-фосфата (соединение 19 или 93, схема 9).
(a) 1,2,3,4,6-пента-0-ацетил-D-манноза (соединение 14 или 65).
D-маннозу (соединение 13, 5.0 г, 27.8 ммоль) растворяют в безводном пиридине (30 мл) и охлаждают до 0 - 5oC в ледяной бане. Медленно добавляют уксусный ангидрид, и полученную смесь оставляют при перемешивании при комнатной температуре на 8 ч. Полученную смесь выливают в ледяную воду и экстрагируют этилацетатом. Полученный экстракт последовательно промывают холодной соляной кислотой, водой, холодным насыщенным бикарбонатом натрия, водой, насыщенным хлоридом натрия, водой и сушат над безводным сульфатом натрия. Органический слой концентрируют в вакууме и используют на следующей стадии без дальнейшей очистки. Соединение 14 или 65; 10.6 г выход 98% чистого α- изомера.
(b) 2,3,4,6-тетра-O-ацетил-D-манноза (соединение 15 или 72).
Пентаацетат (соединение 14 или 65; 10.0 г, 25.6 ммоль) растворяют в тетрагидрофуране и добавляют 1.5 эквивалентов бензиламина (4.6 мл). Полученную смесь перемешивают при комнатной температуре в течение дня. Затем ее экстрагируют этилацетатом и промывают последовательно холодной соляной кислотой, водой, холодным насыщенным бикарбонатом натрия, водой, холодным насыщенным хлоридом натрия, водой и сушат над безводным сульфатом натрия. Растворитель удаляют в вакууме, а оставшийся сироп обрабатывают на хроматографической колонке с силикагелем, элюируя смесью этилацетат:гексан (2:3 объем/объем) до получения соединения 15 или 72 (7.8 г, 87%-ный выход чистого α- изомера).
1H-NMR (CDCl3) δ : 2.00, 2.05, 2.11, 2.17 (3H, s, 4 x CH3CO), 4.01-4.15, (1H, m, 5-H), 4.21-4.29 (2H, m, 6-H), 5.24 (1H, d, 1-H), 5.26-5.27 (1H, m, 2-H), 5.30-5.34 (1H, d, J=12.03 Hz, 4-H), 5.41-5.45 (1H, dd, J 3.36, 9.99 Hz, 3-H).
(c) Дибензилфосфитил-2,3,4,6-тетра-O-ацетил-D-маннозид (соединение 16 или 79).
Маннозотетраацетат (соединение 15 или 72) (1.5 г, мл) и перемешивают в атмосфере азота при комнатной температуре. 1,2,4-триазол (1.5 г, 21.7 ммоль) добавляют к раствору и перемешивают до растворения. К этому раствору добавляют дибензил-N,N-диэтил-фосфориламидит (10.0 г, 31 ммоль) и полученную смесь перемешивают в течение часа. К раствору добавляют 50 мл эфира, и реакционную смесь последовательно экстрагируют холодным насыщенным бикарбонатом натрия, водой, холодным насыщенным натрий хлоридом и водой, сушат над безводным сульфатом натрия. Затем органический экстракт концентрируют в вакууме, а оставшийся сироп очищают на колонке с силикагелем, элюируя смесью ETOAc-гексан (1:4 объем/объем) (чистый α- изомер) до получения соединения 16 или 79.
1H-NMR (CDCl3) δ : 2.0 - 2.3 (12H, 4s, 4 x CH3CO) 3.92-3.98 (1H, dd, 6-Ha), 4.05-4.1 (1H, m, 5-H), 4.18-4.24 (1H, dd, 6-Hb), 4.85-5.12 (4H m, CH2Ph), 5.22-5.24 (11H, m, 2-H), 5.28-5-32 (1H, t, 4-H), 5.38-5.42 (1H, dd, 3-H), 5.48-5.52 (1H, dd, 1-H).
(d) Дибензилфосфорид-2,3,4,6-тетра-O-ацетил-D-маннозид (соединение 17 или 86).
Соединение 16 или 79 растворяют в безводном тетрагидрофуране и охлаждают до -76oC в бане со смесью сухой лед/ацетон, и к раствору добавляют сразу 7 мл 30% перекиси водорода. Полученному раствору дают нагреться до комнатной температуры и перемешивают в течение 90 мин. Избыток перекиси водорода гасят, добавляя охлажденный льдом тиосульфат натрия. Затем добавляют 100 мл эфира, и экстракцию ведут как указано ранее до получения соединения 17 или 86, которые используют на следующей стадии без дополнительной очистки.
(e) D-маннозо-1-фосфат (соединение 18 или 93).
Соединение 17 или 86 гидрируют над 5% Pd/C (400 мг) в 30 мл этанола и 10 мл бикарбоната натрия в атмосфере водорода в течение двух часов. Катализатор отфильтровывают, и полученный фильтрат концентрируют. Прикапывают гидроксид натрия к остатку при 0 - 5oC до тех пор, пока pH смеси не превышает 12. Полученную смесь перемешивают в течение трех часов при 4oC, а затем нейтрализуют, добавляя холодную уксусную кислоту до pH 7.2. Полученную смесь фильтруют, разбавляют до 400 мл, вводят в колонну Dowex 1-XB (HCOO-форма, 2х28 см), и элюируют с линейным градиентом бикарбоната аммония (0-0.6 М). Фракции, содержащие D-маннозо-1-фосфат, собирают и лиофилизируют. Избыток бикарбоната аммония удаляют, промывая лиофилизированный порошок Dowex 50W-XB (водородная форма) до получения соединения 18 или 93 в виде динатриевой соли.
Пример 7. Ферментативное галоидгидратирование.
А. Общий способ для галоидгидратирования, катализируемого хлорпероксидазой (схемы 3, 4 и 4а)
К реакционной смеси, содержащей 20 мл лимонно/фосфатного буфера (pH 3), 1 ммоль гликаля, 5 ммоль галида калия и 1170 ед. фермента, добавляют 600 мкл H2O2 (30%). Реакцию ведут в течение 30 мин (иодгидратация), трех часов (бромогдиратация) или трех дней (хлоргидратация) при комнатной температуре. Растворитель удаляют при пониженном давлении, и к остатку добавляют метанол. Нерастворимый материал отфильтровывают, а растворитель удаляют при пониженном давлении. Остаток очищают на хроматографической колонке с силикагелем с C8 обращенной фазой до получения 2-деокси-2-галоидсахаров. Полученные продукты превращают в перацетаты стандартными способами (пиридин, каталитическое количество 4-диметиламинопиридина, уксусной ангидрид, один день) и очищают на хроматографической колонке с силикагелем для характеризации.
(1) Перацетат соединения 20:
1H-NMR (CDCl3) δ : α -изомер: 2.04 (3H, s), 2.08 (3H, s), 2.10 (3H, s), 2.21 (3H, s), 4.05 (1H, dd, J = 2.5, 12.5 Hz, H-6), 4.08 (1H, dd, J = 3.5, dd, J = 3.5, 11 Hz, H-2), 4.28 (1H, ddd, J = 2, 4, 10 Hz, H-5), 4.31 (1H, dd, J = 4, 12.5 Hz, H-6), 5.09 (1H, dd, J=9, 10 Hz, H-3), 5.52 (1H, dd, J=9, 11 Hz, H-4), 6.36 (1H, d, J=3.5 Hz, H-1) ppm.
β- изомер: 2.03 (3H, s), 2.09 (3H, s), 2.11 (3H, s), 2.18 (3H, s), 33.88 (1H, ddd, J=2.4, 4.5, 10.5 Hz, H-5), 3.90 (1H, dd, J=9, 10.5 Hz, H-2), 4.11 (1H, dd, J=2.5, 12.5 Hz, H-6), 4.32 (1H, dd, J=4.5, 12.5 Hz, H-6), 5.03 (1H, dd, J=9, 10 Hz, H-3), 5.34 (1H, dd, J=9, 10.5 Hz), 5.81 (1H, d, J=9 Hz, H-1) ppm. 13C-NMR (CDCl3) d: 20.52-20.67 (4 x C), 47.50, 62.10, 68.46, 72.85, 74.36, 93.11, 167.91-172.10 (4 x C) ppm. HRMS (M+Na+): calcd 433.0110/435, found 433.0112/435.
(2) Перацетат соединения 21.
1H-NMR (CDCl3) δ : α -изомер: 2.07 (3H, s), 2.10 (3H, s), 2.11 (3H, s), 2.17 (3H, s), 4.19 (1H, ddd, J=2.5, 4.5, 10.5 Hz, H-5), 4.17 (1H, dd, J=2.5, 10.5 Hz, H-6), 4.23 (1H, dd, J= 4.5, 12.5 Hz, H-6), 4.43 (1H, dd, J=2, 4 Hz, H-2), 5.21 (1H, dd, J= 4, 9.5 Hz, H-4), 5.45 (1H, t, J=10 Hz, H-4), 6.32 (1H, d, J= 2 Hz, H-1) ppm. 13C-NMR (CDCl3) δ : 20.60, 20.66, 20.75, 20.86, 47.77, 61.82, 65.54, 68.75, 71.25, 93.11, 167.20-171.90 (4 x C) ppm.
β- изомер: 2.07 (3H, s), 2.10 (3H, s), 2.12 (3H, s), 2.18 (3H, s), 3.82 (1H, ddd, J=2.5, 5, 9.5 Hz, H-5), 4.13 (1H, dd, J=2.5, 12.5 Hz, H-6), 4.27 (1H, dd, J= 5, 10.5 Hz, H-6), 4.60 (1H, dd, J=3.5, 1.5 Hz, H-2), 5.00 (1H, dd, J= 4, 9.5 Hz, H-3), 5.43 (1H, t, J=9.5 Hz, H-4), 5.74 (1H, d, J=1.5 Hz, H-1) ppm. 13C-NMR (CDCl3) δ : 20.60-20.85 (4 x C), 51.05, 61.82, 65.27, 71.01, 73.04, 90.01, 167.20-171.90 (4 x C) ppm. HRMS (M+Na+): calcd 433.0110/435, found 433.0115/435.
(3) Перацетат соединения 22.
1H-NMR (CDCl3) δ: β- изомер: 2.04 (3H, s), 2.07 (3H, s), 2.16 (3H, s), 2.19 (3H, s), 4.08 (1H, dd, J=9, 11.5 Hz, H-2), 4.10-4.15 (3H, m, 2 x H-6 & H-5), 5.15 (1H, dd, J=3, 11.5 Hz, H-3), 5.35 (1H, d, J=3 Hz, H-4), 5.84 (1H, d, J=9 Hz, H-1) ppm. 13C-NMR (CDCl3) δ : 20.42, 20.52, 20.61, 20.66, 46.35, 60.89, 67.03, 71.94, 72.78, 93.43, 168.31-170.90 (4 x C) ppm. α- изомер: 13C-NMR (CDCl3) δ : 20.42-20.66 (4 xC), 44.39, 67.04, 67.69, 68.64, 69.39, 91.05, 167.01-170.86 (4 x C) ppm. HRMS (M+Na+): calcd 433.0110/435, found 433.0119/435.
(4) Соединение 23.
1H-NMR (CDCl3), β- изомер: 1.12 (3H, d, J=6.5, CH3), 3.45 (1H, dd, J=1, 3 Hz, H-4), 3.52 (1H, dd, J=3, 10.5 Hz, H-3), 3.58 (1H, qd, J=1, 6.5 Hz, H-5), 3.69 (1H, dd, J= 8.5, 10.5 Hz, H-2), 4.45 (1H, d, J=8.5 Hz, H-1). 13C-NMR (CDCl3), β- изомер: 16.71, 58.04, 72.13, 73.56, 76.03, 98.78 ppm. β- изомер: 16.71, 54.71. 67.25, 71.13, 74.55, 94.20 ppm. HRMS (M+Na+): calcd 248.9738/251, found 248.9730/251.
(5) Перацетат соединения 25.
1H-NMR (CDCl3) δ: β--изомер: 2.05 (3H, s), 2.08 (3H, s), 2.16 (3H, s), 2.18 (3H, s), 4.06 (1H, dd, J=9, 11.5 Hz, H-2), 4.07-4.15 (3H, m, 2 x H-6 & H-5), 5.09 (1H, dd, J=3, 11.5 Hz), 5.39 (1H, d, J=3 Hz, H-4), 5.75 (1H, d, J= 9 Hz, H-1) ppm. 13C-NMR (CDCl3), β- изомер: 20.21-22.55 (4 x C), 55.30, 60.87, 66.84, 71.86, 72.74, 93.53, 168.20-180.21 (4 x C) ppm. α- изомер: 20.1-22.55 (4 x C), 53.46, 61.04, 67.44, 68.67, 69.51, 90.94, 168.20-180.21 (4 x C) ppm. HRMS (M+Na+): calcd 389.0615/391, found 389.0610/391.
(6) Перацетат соединения 27.
1H-NMR (CDCl3) δ: β- изомер: 2.04 (3H, s), 2.07 (3H, s), 2.12 (3H, s), 2.17 (3H, s), 4.06-4.17 (4H, m, H-2, 2 x H-6 & H-5), 5.13 (1H, dd, J=3.5, 10 Hz, H-3), 5.25 (1H, d, J=3.5 Hz, H-4), 5.91 (1H, d, J=9.5 Hz, H-1) ppm. 13C-NMR (CDCl3), β- изомер: 20.5-22.50 (4 x C), 24.98, 60.94, 67.10, 72.16, 74.10, 94.15, 168.20-179.36 (4 x C) ppm; α- изомер: 20.5-22.50 (4 x C), 29.87, 60.63, 67.68, 68.31, 69.42, 92.16, 168.20-179.36 (4 x C) ppm. HRMS (M+Na+): calcd 480.9972, found 480.9999.
B. Галоидгидратирование, катализируемое хлорпероксидазой, дисахаридгликалей и сиалаля.
(1) Галоидгидратирование соединения 28 (схема 4).
40 мкл 30% H2O2 добавляют к смеси соединения 28 (20 мг, 0,065 ммоль), KBr (38,6 мг, 0,32 ммоль) и хлорпероксидазы (76 ед), в цитратном буфере (1,4 мл, pH, 3), и реакционную смесь осторожно перемешивают в течение 3 ч при комнатной температуре. Растворитель удаляют при пониженном давлении, и MeOH добавляют к остатку. Нерастворившийся материал отфильтровывают, а полученный фильтрат концентрируют при пониженном давлении. Остаток очищают на хроматографической колонке и силикагелем с C8-обращенной фазой до получения смеси D-галактопиранозио-(1,3)-2-бром-2-деокси-D-глюкопиранозы, соединения 29 (10 мг) и D-галактогпиранозил -β- (1,3)-2-бром-2-дезокси-D-маннопиранозы, соединение 30 (10 мг) с 76%-ным выходом. Полученные продукты ацилируют Ac2O и пиридином в присутствии каталитического количества DMAP для характеризации.
(2) Галоидгидратирование 2,3-дегидро-N-ацетилнейраминовой кислоты катализируемое хлорпероксидазой (соединение 35, схема 3).
100 мкл 30% H2O2 добавляют к смеси соединения 34 (Meindl, Hoppe Seyler's Physiol. Chem. , 1350: 1088 (1969); 50 мг, 0.17 ммоль), KBr (102 мг, 0,857 ммоль) и хлорпероксидазы (200 ед), в цитратном буфере (3,5 мл, pH 3), и реакционную смесь осторожно перемешивают в течение 30 мин при комнатной температуре. Растворитель удаляют при пониженном давлении, и к остатку добавляют MeOH. Нерастворимый материал отфильтровывают, и полученный фильтрат концентрируют при пониженном давлении, а остаток очищают на BioGel P-2 колонке, и затем очищают на колонке с силикагелем с C8-обращенной фазой, элюируя CH3CN/H2O (5 : 1) до получения 2-бром-2-дезокси-N-ацетилнейраминовой кислоты (соединение 35) (43 мг, 65%). Полученный продукт переацилируют Ac2O и пиридином в присутствии каталитического количества DMAP с последующим метилированием Mel и Cs2CO3 для характеризации.
1H-ЯМР (CDCl3) δ : 1.95, 2.04, 2.05, 2.10, 2.19, 2.20, (3H, s, каждый, OAc и NHAC), 3.83 (3H, s, COOCH3), 4.11 (1H, ddd, J=8.7, 10.6, 10.7 Hz, H-5), 4.22 (1H, dd, J=6.4, 12.5 Hz, H-9), 4.32 91H, dd, J-1.8, 10.6 Hz, H-6), 4.57 (1H, dd, J=5/15 (1H, ddd, J=2.4, 5.5, 6.4 Hz, H-8), 5.31 (1H, dd, J= 1.8, 5.5 Hz, H-7), 5.43 (1H, d, J=8.7 Hz, NH), 5.67 (1H, dd, J=3.8, 10.7 Hz, H-4). HRMS: рассчитано для C22H30NO14NrCs (M+Cs+) 743.9904/746, найдено 743.9900/746.
(3) 1,3,6,2', 3',4',6-гептаацетил-D-галактопиранозил- β -(1,4)-2-бром-2-дезоксигликопираноза (соединение 32) и -2-дезоксиманнопираноза (соединение 33)
В соответствии с общей процедурой смесь 1 : 1 соединения 32 и 33 (155 мг, 71%) получают из Gal β (1,4)глюкаля (соединение 31) (96.5 мг). Отношение соединений 32 и 33 определяют из интегрального отношения аномерных протонов соединений 32 и 33. Соединения 32 и 33 получают в виде α:β аномерных смесей: соединение 32 (α:β = 1 : 3), соединение 33 (α:β = 2 : 1).
Масс-спектр высокого разрешения соединений 32 и 33: рассчитано для C26H35O17BrCs (M + C+) 831.0113/833, найдено 831.0112/833.
1H-NMR (CDCl3 ) δ 1.96, 1.97, 1.98, 1.981, 2.03, 2.04, 2.05, 2.06, 2.070, 2.074, 2.075, 2.08, 2.12, 2.13, 2.132, 2.15, 2.16, 2.166 (-OCH3), 3.84 (dd, J=1.6, 9.0, Hz, H-2 Glu ββ- аномер соединения 32), 3.73-4.22 (m), 4.40 (dd, J=2.2, 3.8 Hz. H-2 α- аномер соединения 33), 4.43-4.47 (m), 4.46 (dd, J= 1.0, 7.6 Hz), 4.55 (d, J=8.0 Hz), 4.57 (dd, J=1.6, 4.0 Hz, H-2 α -аномер соединения 33), 4.59 (d, J=8.0 OHz), 4.93 (dd, J=3.5, 5.1 Hz), 4.96 (dd, J=3.5, 4.96 Hz), 4.99 (dd, J=3.5, 10.5 Hz, H-3' β-аномер соединения 32, 5.03 (dd, J=3.8, 8.8 Hz), 4.07-5.12 (m), 5.16 (dd, J=8.0, 10.5 Hz), 5.20-5.26 (m), 5.35-5.38 (m), 5.70 (d, J=1.6, H-1 β-аномер соединения 33), 5.76 (d, J= 9.0 Hz, H-1 β-аномер соединения 32), 6.26 (d, J=2.2 Hz, H-1 α-аномер соединения 33), 6.30 (d, J=3.4 Hz, Р-1 α/ -аномер соединения 32). 13C-NMR (CDCl3) 20.52, 20.61, 20.65, 20.75, 20.80, 20.84, 20.88, 20.93, 46.27, 47.95, 48.13, 51.26, 60.77, 60.91, 61.08, 61.49, 61.67, 61.79, 62.04, 66.56, 66.62, 66.71, 66.75, 68.97, 69.03, 69.09, 69.14, 69.30, 70.68, 70.72, 70.75, 70.78, 70.82, 70.86, 70.91, 70.94, 70.98, 71.16, 71.73, 73.44, 73.68, 73.80, 74.13, 74.29, 76.32, 76.61, 89.77, 90.34, 92.98, 93.13, 100.84, 101.19, 101.45, 168.42, 168.45, 168.53, 168.92, 169.14, 169.22, 169.36, 169.59, 169.65, 170.08, 170.13, 170.16, 170.29, 170.36, 170.46.
(4) 1,3,6,2', 3',4',6'-гептаацетил-D-галактопиранозил β -(1,3)-2-бром-2-дезоксиглюкопираноза (соединение 29) и -2-дезоксиманнопираноза (соединение 30).
В соответствии с общей процедурой смесь 1 : 1 (соединения 29 и 30) (66 мг, 76%) получают из Gal β- (1,3)глюкаля (соединение 28) (38,6 мг). Соединения 29 и 30 выделяют на хроматографической колонке с силикагелем (AcOEt/н-гексан, 5 - 2) в виде α:β- аномерных смесей: соединение 29 (α:β = 1 : 10), соединение 30 (α:β = 12 : 5).
β-аномер соединения 29: 1H-NMR (CDCl3) δ 1.98, 2.04, 2.07, 2.08, 2.09, 2.15, 2.17 (3H, каждый, s, OAc x 7), 3.78 (1H, ddd, J=1.8, 4.6, 9.7 Hz, H-5 Glu), 3.85 (1H, t, J=9.5 Hz, H-2 Glu), 3.89 (1H, t, J=7 Hz, H-5 Gal), 3.96, (1H, t, J=10.0 Hz, H-3 Glu), 4.07 (1H, m, H-6 Gal), 4.09 (1H, dd, J = 1.8, 12.4 Hz, H-6 Glu), 4.13 (1H, dd, J = 7.0, 11.1 Hz. H-6 gal), 4.25 (1H, dd, J = 4.6, 12.4 Hz, H-6 Glu), 4.89 (1H, dd, J = 7.7 Hz, H-1 Gal), 4.97 (1H, t, J = 9.6 Hz, H-4 Glu), 5.03 (1H, dd, J = 3.4, 10.0 Hz, H-3 Gal), 5.13 (1H, dd, J = 7.7, 10.0 Hz, H-2 Gal), 5.36 (1H, d, J = 3.4 Hz, H-4 Gal), 5.75 (1H, d, J = 9.0 Hz, H-1 of Glu).
HRMS: рассчитано для C26H35O17BrCs (M+Cs+)
831.0112/833, найдено 831.0112/833.
13C-NMR (CDCl3) δ 20.55, 20.64, 20.68, 20.71, 20.75, 20.963 50.11, 60.93, 61.62, 66.73, 68.53, 68.96, 70.62, 70.82, 72.87, 77.21, 81.30, 92.86, 101.61, 168.77, 169.03, 169.35, 170.13, 170.20, 170.36, 170.67.
α- аномер соединения 30: 1H-NMR (CDCl3) δ : 4.24 (1H, bd, J = 3.0 Hz, H-2 Man), 4.55 (1H, d, J = 8.0 Hz, H-1 Gal), 5.01 (1H, dd, J = 3.4, 10.5 Hz, H-3 Gal), 5.18 (1H, dd, J = 7.8, 10.5 Hz, H-2 Gal), 5.32 (1H, t, J = 8.7 Hz, H-4 Man), 5.39 (1H, dd, J = 1.0, 3.4 Hz, H-4 Gal), 6.30 (1H, d, J = 3.0 Hz, H-1 Man).
ββ- аномер соединения 30: 1H-NMR (CDCl3) δ : 4.45 (1H, dd, J = 2.0, 3.5 Hz, H-2 Man), 4.55 (1H, d, J = 8.0 Hz, H-1 Gal), 5.01 (1H, dd, J = 3.4, 10.5 Hz, H-3 Gal), 5.20 (1H, dd, J = 7.8, 10.5 Hz, H-2 Gal), 5.30 (1H, t, J = 7.4 Hz, H-4 Man), 5.39 (1H, dd, J = 1.0, 3.4 Hz, H-4 Gal), 5.80 (1H, d, J = 2.0, 3.4 Hz, H-1 Man).
HRMS: рассчитано для C26H35O17BrCs (M+Cs+) 831.0112/833, найдено 831.0110/833.
13C-NMR 20.57, 20.65, 20.76, 20.87, 20.95, 21.01, 21.04, 48.21, 60.98, 61.19, 62.01, 62.61, 66.75, 66.82, 68.43, 68.52, 70.71, 70.88, 71.05, 72.03, 73.36, 74.74, 75.93, 77.23, 90.08, 92.80, 100.10, 100.24, 168.23, 169.13, 169.22, 169.73, 170.18, 170.40.
(5) Сиалил α(1,3)Galβ(1,4)Fucα (1,3)-2-бром-2-дезоксиглюкопираноза (соединение 37a) и -2-бром-2-дезоксиманнопираноза (соединение 37b).
В соответствии с общей процедурой смесь 1 : 1 соединений 37a и 38b (3.5 мг, 56%) получают из NeuAc (2,3)-Gal β-(1,4)Fuc α-(1,3) глюкаля соединение 36 (5.5 мг). Отношение соединений 37a и 37b определяют по отношению интегралов метильных протонов фукозы.
Cмеси соединений 37a и 37b: 1H-NMR (D2O) δ 1.17 (d, J = 6.0 Hz, CH3 Fuc), 1.18 (d, J = 6.0 Hz, CH3 Fuc), 1.80 (t, J = 12.7 Hz, H-3ax NeuAc), 2.02 (dd, J = 5.0, 12.7 Hz, H-3 eq NeuAc), 3.45 - 4.13 (m), 4.48 (d, J = 8.0 Hz, H-1 Gal), 4,49 (d, J = 8.0 Hz, H-1 of Gal), 5.0 - 5.04 (m), 5.18 - 5.22 (m), 5.38 - 4.42 (m).
Пример 8. Общий способ бромгидратирования с помощью NBS.
К раствору 1 ммоль глюкаля в смеси 3.6 мл CH3CN - 1.5 мл H2O добавляют 1 ммоль N-бромсукцинимида (NBS) при комнатной температуре. Реакцию ведут в течение трех часов при той же температуре. Растворитель удаляют при пониженном давлении, а остаток обрабатывают на хроматографической колонке с силикагелем. Полученные продукты превращают в перацетаты с помощью пиридина и уксусного ангидрида в присутствии каталитических количеств 4-диметиламинопиридина, и очищают на хроматогафической колонке с силикагелем для характеризации.
(a) 1,3,6,2', 3',4',6'-гептаацетил-D-галактопиранозил- β (1,4)-2-бром-2-дезоксиглюкопираноза (соединение 32) и -2-дезоксиманнопираноза (соединение 33).
В соответствии с общим способом смесь 1 : 2.5 соединений 32 и 33 (30 мг, 78%) получают из Gal β (1,4)глюкаля (соединение 31) (17 мг). Отношение соединений 32 и 33 определяют из отношения интегралов аномерных протонов. Соединения 32 и 33 получают в виде смеси α:β аномерных смесей: соединение 32 (α:β = 3 : 5), соединение 33 (α:β = 5 : 2).
(b) Метил-5-ацетамидо-2,4,7,8,9-пента-O-ацетил-3-бром-3,5- дидезокси β D-эритро-L-манно-2-нонулопиранозонат (метилперацетил соединения 35, соединение 35a) и метил-5 ацетамидо-2,4,7,8,9-пента-O- ацетил-3-бром-3,5-дидезокси -α- D-эритро-L-глюко-2-нонулопироназонат (соединение 35b).
Химическое бромгидратирование проводят в соответствии с общей процедурой, и полученные продукты превращают в перацетаты с последующей этерификацией метилиодидом в присутствии эквимолярного количества карбоната цезия до получения смеси соединений 35a и 35b (155 мг, 74%). Отношение соединений 35a и 35b определяют из отношения интегралов метильных протонов сложного эфира.
1H-ЯМР спектры соединений 35a и 35b находятся в хорошем соответствии с литературными данными.
Cоединение 35b: 1H-NMR (CDCl3) δ: 1.90, 2.03, 2.08, 2.10, 2.12, 2.15 (3H, s, каждый OAc и NHAc), 3.78 (3H, s, COOCH3), 4.04 (1H, dd, J = 6.0, 12.4 Hz, H-9), 4.09 (1H, d, J = 10.0 Hz, H-3ax), 4.33 (1H, ddd, J = 10.0, 10.6, 10.7 Hz, H-5), 4.36 (1H, dd, J = 2.4, 12.4 Hz, H-9'), 5.10 (1H, ddd, J = 2.4, 6.0, 6.1 Hz, H-8), 5.25 (1H, dd, J = 2.5, 10.7 Hz, H-6), 5.30 (1H, dd, J = 10.0, 10.6 Hz, H-4), 5.38 (1H, dd, J = 2.5, 6.1 Hz, H-7), 5.90 (1H, d, J = 10.0 Hz, NH).
Пример 9. Выделение Galβ1,3/4 GlcNAcα2,3- сиалилтрансферазы.
Высокий выход при выделении растворимой Galβ1,3/4- GlcNAcα2,3- сиалилтрансферазы осуществляют в бакуловирусной системе выделения, используя кДНК, кодирующую протеин слияния преинсулинового сигнального пептида и каталитический домен сиалилтрансферазы. кДНК, кодирующую протеин слияния, конструируют по способу Wen. et al. в плазмидном векторе pGlP199 (Huseh et al., J. Biol. Chem. , 261: 4940 (1986)). Для выделения ДНК-фрагмента, содержащего полную кодирующую последовательность, уникальный Eco RI сайт с 3'-конца chimera вначале переваривают, свисающую часть затупляют, и лигируют синтетические связи, содержащие Nhe сайт. Полученную плазмиду переваривают с Nhe I для высвобождения кДНК протеина слияния, и этот фрагмент клонируют по уникальному Nhe I сайту в pBlueBac, вектор переноса системы выделения бакуловируса, под контролем бакуловирусного полиэдринового промотора (Invitrogen; San Diego, CA). Все манипуляции с рекомбинантным ДНК осуществляют в условиях, рекомендуемых в инструкциях поставщиков ферментов, используя стандартные протоколы (Sambrook et al., Molecular Cloning; A Laboratory Manual, 2nd ed, Cold Spring Harbor Laboratory Press, Plainview, NY (1989)).
Создание рекомбинантных бакуловирусов осуществляют, используя MaxBac выделительной системы (Invitrogen) точно в соответствии с протоколами, рекомендуемыми изготовителем. Короче, плазмиду и дикого типа вирусную ДНК смешивают и используют для трансфектации Sf-9 клеток кальцийфосфатным методом. Рекомбинантные вирусы получают за счет переноса клеток в культуральную среду, и бляшки очищают повторным лимитирующим разбавлением. Некоторые клональные выделенные бляшки анализируют на их способность вызывать секрецию α-2,3 NeuT в инфицированную клеточную среду, тестируя аликвоты среды непосредственно на α-2,3 NeuT активность в сиалилтрансферазном анализе. Изолят, который приводит к наиболее высоким уровням α-2,3 NeuT секреции обозначают rBv 2,3 ST, и доводят до 500 мл, инфицируя свежие Sf-9 клетки. α-2,3 NeuT активность анализируют, используя модификации опубликованного анализа 0.9 мМ. Эти манипуляции проиллюстрированы схематически далее на схеме 24.
Пример 10. Galβ1,3 GlcNAcα1,3 Galβ1,4Glc в качестве акцептора.
Для получения больших количеств α-2,3NeuT, rBv 2,3-ST используют для инфицирования Sf-9 клеток в культуре моносоля, и обычно получают 2 - 3 единицы α- 2,3NeuT активности, секретируемой на 108 инфицированных клеток при выращивании их на среде Excell-400 (JRH Biosciences, Lenexa, KS). Единицы активности (ммоль/мин) определяют, умножая результаты анализа на коэффициент 1,6 для получения активности при Vмакс.. Кондиционную среду из rBv 2,3ST-инфицированных клеток собирают через 72 ч после инфицирования, и рекомбинантные α-2,3NeuT частично очищают на одной стадии хроматографической очистки. 3 л среды, содержащей α-2,3NeuT, фильтруют и кондиционируют до приблизительно 250 мл в Amicon CH2PRS спиральной картриджной системе, снабженной S1Y10 картриджем. Эту установку используют для обессоливания концентрированной надосадочной жидкости тремя объемами 10 мМ какодиловой кислоты, 25 мМ NaCl, 25% глицерина, pH 5,3 (буфер А). Затем образцы вводят в колонку (2,5 х 17 см) S-Sepharose Fast Flow (Pharmacia) уравновешенную буфером А со скоростью 2 мл/мин. После того как все образцы загружены, колонку промывают буфером А до тех пор, пока OD280 не достигает значения базовой линии (1,6 объема колонки).
Затем α-2,3NeuT элюируют из колонки 50 мМ какодиловой кислоты, 1 М NaCl, 25% глицерина, pH 6,5, фракции, содержащие α-2,3NeuT собирают и деализируют в течение в ночи (около 18 ч) против 1 л 50 мМ какодиловой кислоты, 0,5 М NaCl, 50% глицерина, pH 6,0, а затем хранят при -20oC.
Пример 11. Галактозилирование.
(a) LacNac β-O-аллил (схема 14, соединение 41).
Смесь соединения 40 (Lee et al, Carbohydr. Res., 37 (1974) (2,0 г, 7,65 ммоль), Glc-1-Р (2,74 г, 7,65 ммоль), PEP (К соль), (1,6 г, 7,65 мкмоль), NAD+ ( 193 мг, 0,25 ммоль), MnCl2 • 4H2O (79,2 мг, 0,4 ммоль), MgCl2 • 6H2O (162,6 мг, 0,8 ммоль), DTT (306 мг, 2 ммоль), KCl (1,04 г, 15 ммоль) NaN3 (20 мг, 0,31 ммоль) и UDP (90 мг, 0,19 ммоль) в HEPES буфере (100 мМ, pH 7,5; 200 мл) доводят до pH 7,5 с помощью 10 н и н NaOH, и к раствору добавляют энзимы, UDPGE (10 ед), UDPGE ( 20 ед), PK (100 ед), GalT (5 ед) и PPазу (100 ед). Полученную смесь осторожно перемешивают в атмосфере аргона при комнатной температуре (25oC) в течение 5 дней. Полученную смесь концентрируют и хроматографируют на силикагеле, элюируя CHCl3 - EtOAc-MeOH ( 5: 2: 2 до 5: 2:3) до получения дисахарида, который далее очищают на Sephadex G25, элюируя водой, до получения LacNac β-O-аллила (соединение 41) (1.7 г, 50%).
1H-ЯМР (D2O) δ : 2.0 (3H, s, NHAc), 3.49 (1H, dd, J 7.84, 9.97 Hx, H-2 of Gal), 3.52-5.57 (1H, m, H-5 of GlcNAc), 3.63 (1H, dd, J 3.31, 10.04 Hz, H-3 of Gal), 3.65 - 3.75 (8H, m), 3.79 (1H, dd, J 5.10, 12.27 Hz, H-6a of GlcNAc), 3.88 (1H, br d, J 3.22 Hz, H-4 of Gal), 3.95 (1H, dd, J 2.14, 12.27 Hz, H-6b of GlcNAc), 4.43 (1H, d, J 7.81 Hz, H-1 of Gal), 4.55 (1H, d, J 8.28 Hz, H-1 of GlcNAc), 5.21-5.29 (2H, m, allylic), 5.83-5.90 (1H, m, allylic), 13C NMR (D2O) δ : 22.6, 55.5, 60.5, 61,5 69.0, 70.9, 71.4, 72.9, 75.2, 75.8, 78.8, 100.4, 103.3, 118.6, 133.7.
(b) Соединение 1-13C-41 со схемы 15.
Раствор соединения 40 (1.15 г 4.4 ммоль) 1-13C-Gal (99 атомных %, Isotec. Inc. , Miamisburg; 800 мг, 4.4 ммоль), PEP K соль (1.82 г, 8.8 ммоль, 95%), UDP (90 мг, 0.19 ммоль), ATP (100 мг, 0.18 ммоль), цистеина (116 мг, 0.96 ммоль), DTT (183 мг, 1.2 моль) MgCl2 • H2O (244 мг, 1.2 ммоль) MnCl2 • 4H2O (118 мг, 0.6 ммоль), KCl(179 мг, 2.4 ммоль) и Glc-1-Р (77 мг, 0.22 ммоль) в HEPES буфере (100 мМ, pH 7.5; 120 мл) доводят до pH 7.5 с помощью 10 н и н NaOH, и ферменты, GK (10 ед), PK (200 ед), РРазу (10 ед), Gal-1-Р UT (10 ед), UDPGP (10 ед) и GalT (10 ед) добавляют к раствору. Полученную смесь осторожно перемешивают в атмосфере аргона при комнатной температуре (примерно 25oC) в течение 3 дней. Полученную смесь концентрируют в вакууме, а остаток хроматографируют на силикагеле смесью EtOAc-MeOH (2:1) до получения дисахарида, который далее очищают на хроматографической колонке Sephadex G25, элюируя водой, до получения соединения 1-13C-41 (106 г, 57%).
1H-ЯМР (D2O) δ : 2.00 (3H, с, NHAc), 3.48-3.52 (1H, м, H-2 Gal), 4.43 (1H, дд, Jн-1,н-2 8.32, Jн-1,13с-1 162.33 Гц, H-1 Gal), 4.54 (1H, д, J = 8.32 Гц, H-1 GlcNAc). BPCM рассчитано для 12C1613CH29NO11Na (M+Na+) 447.1672, найдено 447.1681.
(c) 2-дезокси-D-галактопиранозил -β (1.4)-2-ацетамидо-2-дезокси-глюкопираноза, соединение 41b.
(36%): Оба спектра 1H-ЯМР: его гептаацетата 13C ЯМР спектр соединения 41a находятся в хорошем соответствии с литературными данными (Thiem el al., Angew. Chem. Int. Ed. Engl., 30: 1163 (1991)).
(d) 2-амино-2-дезокси-D-галактопиранозил -β-(1,4)-2-ацетамидо-2-дезокси-глюкопираноза (соединение 41b).
(12%): 1H-ЯМР для HCl соли (D2O) δ : 2.022, 2.024 (с, NHAc α и β аномеры GlcNAc), 3.17 - 3.23 (1H, м, H-2 GalN), 4.67 (д. J = 7.53 Гц, H-ib GlcNAc), 5.13 (д. J = 1.54 Гц, H-1a GlcNAc). BPMC рассчитано для C14H26N2O10Na (M+Na+) 405.1485, найдено 405.489,
1H ЯМР его ацетатной формы находится в хорошем соответствии в литературными данными (Palcic et al., Glycobioljgy, 1:205 (1991)).
(e) Этил-D-галактопиранозил -β-(1,4)-2-азидо-2-дезокси-D-глюкопиранозид (соединение 41c).
В этом случае DTT исключается, так как 2-азидогруппа восстанавливается до соответствующего амина за счет DTT.
(15%): 1H ЯМР β-аномер (D2O) δ : 1.22 (1H, т, J = 7.80 Гц, OCH2CH3), 3.27 (1H, J = 8.33, 9.64 Гц, H-2 GlcN3), 4.40 (1H, д, J - 7.81 Гц, H-1 Gal), 4.55 (1H, 8.24 Гц, H-1 GlcN3). BPMC рассчитано для C14H25N3O10Na (M+Na+) 418.1438, найдено 418.1438.
Акцептор, этил-2-азидо-2-дезокси-D-глюкопиранозид, получают следующим образом. Триацетил-D-глюкаль азидонитрируют (Lemieux et al., Can. J. Chem., 57: 1244 (1979)) (NaN3 и Ce(NH4)2 (NO3)6 в CH3CN) и ацетолизируют (NaOAc в AcOH) до получения 2-азидо-1,3,4,6-тетра-O-ацетил-2-дезокси -D-глюкопиранозы, которую обрабатывают TiBr4 в CH2Cl2 и EtOAc, получая гликозилбромид, затем гликозилируют EtOH в присутствии AgOTf и МС 4 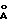 в CH2Cl2 до получения после O-дезацетилирования с помощью NaOMe в MeOH, этил-2-азидо-2-дезокси-D-гликопиранозида (полный выход 22%) в виде смеси α и β 1:1.5.
в CH2Cl2 до получения после O-дезацетилирования с помощью NaOMe в MeOH, этил-2-азидо-2-дезокси-D-гликопиранозида (полный выход 22%) в виде смеси α и β 1:1.5.
1H-ЯМР (D2O) δ : 1.21 (т, J=7.80 Гц OCH2CH3 β аномера), 1.22 (т,J = 7.80 Гц, OCH2CH3), 2.99 (дд, J=7.43, 9.83 Гц, H-2 β- аномера), 5.11 (g, J=3.58 Гц, H-1 α-/ аномера). ВРМС рассчитано для C8H15N3O5Cs (M+Cs) 366.0066, найдено 366.066.
Пример 12. Сиалилирование (схема 16).
(a) соединение 42.
Раствор соединения 1-13C-41 (210 мг, 0.50 мкмоль), NeuAc (160 мг, 0.52 мкмоль), PEP Na3 соль (120 мг, 0.51 ммоль), MgCl2•6H2O (20 мг, 0.10 ммоль), MnCl2•4H2O (4.9 мг, 0.025 ммоль), KCl (7.5 мг, 0.10 ммоль), CMP (16 мг, 0.05 ммоль), ATP (2.7 мг, 0.005 мкмоль) и меркаптоэтанола (0.34 мл) в буфере HEPES (200 мМ, pH 7.5; 3.5 мл) доводят до pH 7.5 с помощью NaOH, и к полученному раствору добавляют NMK (5 ед), PK (100 ед), PPазу (10 ед), CMP-NeuAc синтетазу (0.4 ед) и α 2,3 SiaT (0.1 ед). Полученную смесь осторожно перемешивают при комнатной температуре (25oC) в течение 3 дней. Полученную смесь концентрируют, и остаток обрабатывают хроматографически на силикагеле, элюируя EtOAc-изо PrOH-H2O (2:2:1) до получения трисахарида, который далее очищают BioGel P-2 с водой до получения соединения 42 (88 мг, 24%).
1H NMP (D2O) d 1.81 (1H, br t, J 12.02 Hz, H-3ax NeuAc), 2.04 (6H, s, NHAc GlcNAc и NeuAc), 2.76 (1H, dd, J 4.57, 12.33 Hz, H-1eq NeuAc), 3.96 (1H, br d, J 3.10 Hz, H-4 Gal), 4.13 (1H, dd, J 3.09, 9.94 Hz, H-3 Gal), 4.56 (1H, dd, Jн-1,н-2 7.83, Jн-1,13с 162.78 Hz, H-1 Gal), 4.58 (1H, d, J 8.32 Hz, H-1 GlcNAc). HRMS рассчитано для C27H44N2O19Cs2 (M-H++2Cs+) 980.0759, найдено 980.0720.
(b) NeuAca a2,3'Lactal соединение 43 (82 mg).
1H NMR (D2O, 320oK) δ : 1.84 (1H, br t, J 12.18 Hz, H-3eq NeuAc), 2.08 (3H, s, NHAc NeuAc), 2.82 (1H, dd, J 4.46, 12.32 Hz, H-3eq NeuAc), 4.01 (1H, br d, J 2.50 Hz, H-4 Gal), 4.16 (1H, dd, J 2.50, 9.50 Hz, H-3 Gal), 4.43 (1H, dt, J 1.18, 6.46 Hz, H-3 Glucal), 4.65 (1H, d, J 7.86 Hz, H-1 Gal), 4.88 (1H, dd, J 2.63, 6.07 Hz, H-2 Glucal) и 6.51 (1H, dd, J 1.45, 6.08 Hz, H-1 Glucal). HRMS рассчитано для C23H35NO17NaCs2 (M-H++2Cs+) 864.0092, найдено 864.0066.
Пример 13. Фукозилирование (схема 17).
(a) соединения 44, 45 и 46.
Раствор FucT (0.02 ед., 2 мл) добавляют к раствору соединения 42 (23 мг, 0.031 ммоль) и GDP-Fuc (Ichikawa et al., J. Org. Chem., в печати) (24 мг, 0.036 ммоль) в буфере HEPES (3 мл, 200 мМ, pH 7.5), содержащем 5 мМ АРТ, 20 мМ Mn2+. Полученную смесь осторожно перемешивают в атмосфере аргона в течение 5 дней при комнатной температуре (25oC). Аналогичный результат получают, используя раствор, содержащий соединение 42 (23 мг, 0.031 ммоль) и GDP-Fuc (70 мг, 0.105 ммоль) в буфере HEPES (1 мл, 200 мМ, pH 7.4), содержащем Mn2+ (25 мМ) и α-1,3 FucT раствор (0.01 ед), который обрабатывают аналогичным образом. Полученную смесь концентрируют и хроматографируют на силикагеле, элюируя EtOAc-изо PrOH-H2O (2:2:1), до получения тетрасахарида, который далее очищают BiOGel P-2 с водой. Элюат пропускают через колонку Dowex 50W-XB [H+] , элюируют водой до удаления Mn2+ катионов, нейтрализуют н NaOH и диофилизируют до получения соединения 44 (18 мг), аналогично, соединения 45 (42 мг) и 46 (51 мг) получают таким же способом.
Соединение 44: 1H-NMR (D2O) δ : 1.11 (3H, d, J 6.61 Hz, 6-CH3 Fuc), 1.73 (1H, br t, J 12.04 Hz, H-3ax NeuAc), 1.96 (3H, s, NHAc GlcNAc), 1.97 (3H, s, NHAc NeuAc), 2.69 (1H, dd, J 4.52, 12.38 Hz, H-2eq NeuAc), 3.46 (1H, dt, J 7.00, 9.68 Hz, H-2 Gal), 3.71 (1H br d, J 3.00 Hz, H-4 Fuc), 4.02 (1H, dd, J 2.94, 9.78 Hz, H-3 Gal), 4.46 (1H, dd, J1,2 7.90, J13C,H 162.13 Hz, H-1 Gal), 4.52 (1H, d, J 8.41 Hz, H-1 GlcNAc), 5.04 (1H, d, J 3.98 Hz, H-1 Fuc).
Соединение 45: 1H-NMR (D2O) δ : 1.17 (3H, d, J 6.61 Hz, 6-CH3 Fuc), 2.03 (3H, s, NHAc GlcNAc), 3.50 (1H, ddd, J 6.47, 7.86, 9.86 Hz, H-2 Gal), 3.80 (1H, br d, J 2.88 Hz, H-4 Fuc), 4.46 (1H, dd, J1,2 7.79, J13C,H 161.45 Hz, Hz H-1 Gal), 4.59 (1H, d, J 8.44 Hz, H-1 GlcNAc), 4.84 (1H, br q, J 7.50 Hz, H-5 Fuc), 5.11 (1H, d, J 3.90 Hz, H-1 Fuc).
Соединение 46: 1H-NMR (D2O, 320 K) δ : 1.11 (3H, d, J 6.61 Hz, 6-CH3 Fuc), 1.84 (1H, br t, J 12.00 Hz, H-2ax NeuAc), 2.08 (3H, s, NHAc NeuAc), 2.80 (1H, dd, J 4.52, 12.38 Hz, H-3eq NeuAc), 4.49 (1H, br q, J 7.50 Hz, H-5 Fuc), 4.64 (1H, d, J 8.0 Hz, H-1 Gal), 5.02 (1H, dd, J 2.5, 6.0 Hz, Glucal), 5.09 (1H, d, J 3.98 Hz, H-1 Fuc), 6.51 (1H, dd, J 1.5, 6.0 Hz, H-1 Glucal).
(b) Gal β-1,4Fuc α-1,3/-(5-тио)Glc
Раствор Gal β-1,4/5-тио/Glc (30 мг, 84 ммоль), GDP-Fuc (60 мг, 84 ммоль) и α-1,3/4FucT (0.5 ед.) в Na-кокадилатном буфере (5.4 мл, 50 мМ, pH 6.2), содержащем 5 мМ ATP и 20 мМ MnCl2, перемешивают в течение двух дней при комнатной температуре. Rf значения исходного материала и продукта составляют 0.39 и 0.31, соответственно в EtOAc / AcOH / H2O 3 : 2 : 1 на ТСХ на двуокиси кремния. Реакционную смесь вводят непосредственно в колонку Sephadex G25 Superfine (1.5 x 30 см), и элюируют водой. Фракции, содержащие продукт, собирают и последовательно пропускают через колонки QAE - Sephadeх и Dowex 50-XB [H+] с водой. Элюат собирают и лиофилизируют (21 мг).
1H ЯМР (D2O, 20oC) δ : 1.13 (3H, д, J = 6.7 Гц, 6-CH3 Fuc), 3.40 (1H, дд, J = 6.4 и 11.7 Гц), 3.60 (1H, дд, J = 3.6 и 11.7 Гц), 4.52 (1H, д, J = 7.9 Гц), 4.95 (1H, J = 2.6 Гц), 5.34 (1H, д. J = 3.8 Гц).
Пример 14. Кинетические исследования ферментов.
(a) Для FucT.
Процедура анализа практически та же, что описана ранее (Fukowska - Latallo et al., Gene and Development, 4: 1288 (1990)) с некоторыми модификациями. Приготавливают свежую исходную смесь, состоящую из 0.25 мМ GDP-13C-Fuc (5000 срм/мл), 6.25 мМ ATP, 25 мМ MnCl2 и 62.5 мМ буфера-какодилата натрия, pH 6.2, и хранят на льду. К этому раствору добавляют FucT непосредственно перед использованием, и реакцию начинают, соединяя 16 мкл этой смеси и 4 мкл 100 мМ акцептора (полный объем инкубируемого раствора 20 мкл). Инкубирование ведут при 37oC в течение 30 - 240 мин в зависимости от исследуемого акцептора. Отдельные анализы без акцептора проводят для внесения поправки на фоновый гидролиз GDP-Fuc. После завершения инкубирования добавляют 400 мкл 25% (объем/объем) суспензии QAE-Sephadex. Эти суспензии осторожно перемешивают при комнатной температуре в течение 10 минут перед центрифугированием при 13 000 об/мин в течение 1 мин. Из надосадочной жидкости экстрагируют 200 мкл, и смешивают с 10 мл сцинтилляционной смеси. Радиоактивность определяют на сцинтилляционном счетчике. Принимают предосторожность, чтобы убедиться, что 10% ферментативной реакции прошло за период инкубирования. Этот анализ можно вести в отсутствии ATP.
(b) для GalT.
Начальные скорости ферментативной реакции определяют, измеряя скорость образования LacNAc с небольшой модификацией Pierce et al. (Pierce et al., Anal. Biochem. , 102: 441 (1980)). Все реакции ведут в 100 мМ какодилатном буфере (pH 7.5), с фиксированной концентрацией Mn2+ (9.3 мМ) и UDP-Gal (0.1 мМ; 58.5 срм/пмоль UDP-14C-Gal) в 100 мл раствора. Реакцию инициируют, добавляя GalT (0.05 ед., 120 мг протеина; от Сигмы) и оставляют выстаиваться при 20oC в течение 30 мин. Неспецифический гидролиз UDP-Gal определяют с помощью контрольной реакции без GalT. Реакцию останавливают, пропуская через колонку QAE-Sephadex (700 мл) и элюируя при небольшом давлении воздуха для удаления непрореагировавшего UDP-Ga1. Реакционную ампулу дважды промывают 400 мл воды каждый раз, и реакционную смесь пропускают через колонку со смолой. Полученные фильтраты собирают и непосредственно переносят в сцинтилляционную ампулу. В ампулу добавляют сцинтилляционную жидкость, а затем определяют радиоактивность с помощью счетчика. Полученные данные анализируют методом двойного обратного приближения до получения Km (1.5 мМ для GlcNAc) (величина 1.3 ± 1 мМ сообщалась Palcic et al., Carbohydr. Rev. 159: 315 (1987)), а Ki (0.46 ± 0.06 мМ) для UDP. Аналогично ИК50 величина для GalT была определена с использованием различных концентраций UDP.
Пример 15. GDP-Fuc образующий фермент, используемый в схеме 19.
(a) Получение энзима (GDP-FucS).
Бактерию Klebsiella pneumonia, ATCC 12 658 выращивают в 2 л среды, содержащей 10 г казаминовой кислоты (Дифко), 5 г экстракта дрожжей, 3 г K2HPO4 1 г KH2PO4 и 5 г D-глюкозы на литр (pH 7.0). После инкубирования при 37oC в течение 18 ч клетки собирают центрифугированием (10000 g, 50 мин, 4oC) и снова суспендируют в 50 мМ tris буфера, содержащего 0.5 мМ DTT. Клетки разрушают во французском прессе при 16000 фунт/дюйм (1125 кг/см2). Клеточные осколки удаляют центрифугированием при 23000 g в течение 60 мин, и надосадочную жидкость (экстракт без клеток) используют для очистки фермента. Не содержащий клеток экстракт (50 мл) для 2 л культуры обрабатывают 50 мг протаминсульфата, и образовавшийся осадок удаляют после центрифугирования. Твердый сульфат аммония добавляют при медленном перемешивании до тех пор, пока не достигают 70% насыщения (0.436 г/мл при 0oC). После центрифугирования осадок собирают и снова суспендируют в 20 мл буфера (50 мМ tris, содержащего 0.5 мМ DTT, pH 7.5) и диализуют в течение ночи при 4oC в 4 л того же буфера.
Оставшийся раствор (2 мл) пропускают затем через DEAE-Sepharose CL-6B колонку (Pharmacia) (3 • 30 см), предварительно уравновешенную тем же буфером. Фермент элюируют с линейным градиентом NaCl от 0 до 1 мМ в том же самом буфере (всего 400 мл). Активные фракции собирают и диализуют в 2 л 50 мМ tris буфера, содержащего 0.5 мМ DTT (pH 7.5). Этот препарат GDP-FucS фермента используют для получения GDP-Fuc Активность оценивают в около 0.05 ед/мл на основании см обращенной фазой и NADH окислительного анализа
(b) Ферментативное получение и регенерация GDP-Fuc из Man-I-P, схема 19.
Раствор имидазола (10 мМ), Man-I-P (10 мМ), GDP (10 мМ), PEP (10 мМ), KF (5 мМ), Mg2+ (10 мМ), KC1 (20 мМ), NADP (2 мМ), EDTA (6 мМ), iPrOH (2%), PK (80 ед. ), TBDH (32 ед.), дрожжевые клетки (S, cerevisae 52 мг) высушенного замораживанием из 50 мМ Tris буфера, pH 7.5, GDP-Fuc S вырабатывающий фермент (400 мл) в HEPES буфере (pH 7.5) (полный объем раствора 2 мл) инкубируют при 37oC в атмосфере аргона в течение 18 ч.
ВЖЭХ колонка партисил 5 SAX (Watman Co.), 0.46 • 12,5 см с размером частиц 5 мм была использована. Подвижная фаза представляет собой 0.1 М фосфатный буфер (pH 3.5) со скоростью потока 0.5 мл/мин (давление 600 пси). Соединения определяют с помощью УФ детектора при 254 нм. Время удерживания для GDP-Man и GDP-Fuc составляет 9.92 и 13.4 мин соответственно. GDP-Fuc (5%) и GDP-Man (30%) образуются по данным ВЖЭХ анализа.
Раствор соединения 41 или 42 (10 мМ в 2 мл HEPES буфера, pH 7.5, содержащий 5 мМ ATP и 20 мМ MnCl2) добавляют и полученную смесь перемешивают в течение 5 дней. ТСХ на пластине силикагеля: Rf = 0.28 для соединения 45 и 0.50 для соединения 41 с EtOAc : AcOH : H2O = 4 : 2 : 1 (объем/объем) и 0.56 для соединения 44, и 0.63 для соединения 42 с 1 М NH4OH : iPrOH = 1 : 2 : 4 (объем/объем).
Соединения 45 и 44 (8 и 4 мг каждый) выделяют и очищают как указано ранее.
Пример 16. Очищение GDP-фукозопирофосфорилазы для использования в схеме 20
(a) Получение фермента.
Свиную печень (4 кг) гомогенизируют в охлажденном льдом 10 мМ MOPS, pH 7.5, с 1 мг/мл каждого антипаина, апротинина, химостатина, леупептина и пепстатина в смесителе Уоринга (пять 15-секундных циклов с высокой скоростью). Осколки клеток удаляют центрифугированием при 8000 g в течение 20 мин при 4oC. К надосадочной жидкости добавляют 1 л 2% раствора протаминсульфата. Смесь перемешивают в течение 5 мин, и осадок удаляют центрифугированием, как указано ранее. Твердый сульфат аммония медленно добавляют к надосадочной фракции до 50%-ного насыщения (0.291 г/мл при 0oC). После центрифугирования как указано ранее, осадок собирают и снова суспендируют в 1600 мл 1.2 M сульфата аммония.
Образец смешивают с суспензией фенилсефарозы (250 мл), которую уравновешивают в 1.2 M сульфата аммония. Смолу со связанным ферментом промывают 1.2 M сульфатом аммония (1.5 л), и ферментную активность элюируют 0.4 M сульфатом аммония (750 мл). Этот процесс повторяют в потоке до тех пор, пока основная часть ферментной активности не удается из образца. Часть фенилсефарозного элюата (200 мл) диализуют против 10 мМ MOPS, pH 7.5 и пропускают через колонку DEAE 5PW (15 см x 21.5 мм) уравновешенную в том же самом буфере. Ферментативную активность наблюдают в материале, который протекает через колонку, и концентрируют с помощью Amicon ультрафильтрационного устройства.
Затем образец подвергают гель-фильтрации на колонке TSK Gel 3000 SW9 (30 см x 21.5 мм), уравновешенной и промытой 50 мМ MOPS, pH 7.5 со 150 мМ KCl. Активные фракции собирают и хранят в виде 50%-ной суспензии сульфата аммония. Во время очистки GDP-Fuc пирофосфорилазу оценивают в соответствии со способом Ishihara и Health. (Ishihara et al., J. Biol. Chem., 243: 1110 (1968)). Одну единицу активности определяют как включение 1 ммоль неорганического 32P-пирофосфата в GTP в минуту.
(b) Регенерация GDP-фукозы с использованием GDP-фукозопирофосфорилазы, схема 20, синтез сиалил Льюис-X.
Раствор MOPC, pH 7,5 (50 мМ) Fuc -I-P (10 мМ), GDP (1 мМ), PEP (10 мМ), KF (5 мМ), Mg2+ (10 мМ), Mn2+ (10 мМ), PK (5 ед), сигнал-[3H]-LacNAc β- O-(CH2)6CO2Me (10 мМ) α-1,3 FucT (0,1 ед), неорганической фосфатазы (5 ед) и GDP-Fuc пирофосфорилазы (0,1 ед) смешивают в объеме 100 мл. Реакционную смесь инкубируют во вращающейся ампуле при комнатной температуре в течение 60 ч. Продукты собирают на Sep-Pac C18 колонке и элюируют 50%-ным метанолом. Образец сушат, выпаривая при пониженном давлении, снова суспендируют в воде, и анализируют с помощью тонкослойной хроматографии на пластинах силикагеля со смесью изопропанол/IM ацетат аммония (6:1) в качестве растворителя. Сиалил Льюиса X /получают с выходом около 30%, по данным сцинцилляционного счетчика.
Пример 17. (2R)-метил-(5S)-гидросиметил-(3R, 4R)-дигидроксипирролидин (соединение 50).
A. Цис-2,3-эпокси-1,4-бутан-диол.
Цис-2,3-1,4-бутандиол получают из 1,4-дигидрокси-2-бутена в соответствии с процедурой, описанной Nelson et al., J. Med, Chem., 19: 153 (1976), за исключением того, что реакцию ведут при комнатной температуре в течение 36 ч.
B. 2-азидо-2-дезокси-треитол.
Раствор, содержащий цис-2,3-эпокси-1,4-бутан-диол (1.82 г, 17.50 ммоль), азид натрия (NaN3; 5.68 г, 5 эквивалентов) и хлорид аммония (NH4Cl, 4.68 г, 5 эквивалентов) в 100 мл метанола и 12 мл H2O нагревают при кипячении с обратным холодильником в течение 24 ч. Растворитель удаляют при пониженном давлении, добавляют этанол и осадок отфильтровывают. Процедуру осаждения повторяют несколько раз для удаления избытка NaN3 и NH4Cl, до получения 2-азидо-2-деокси-треитола в виде желтой жидкости (90%: Rf = 0.28), EtOAc (100%); ИК (чистый) 2109 см-1 (-N3);
1H ЯМР (COCD3) δ : 3.49 (1H, м), 3.59 (3H, м), 3.79 (5H, м), 4.03 (1H, т, J = 5.5 Гц), 4.19 (1H, д, J = 5.5 Гц), 4.30 (1H, т, J = 5.5 Гц) мд. BPMC (M + H+) рассчитано 148.0722, найдено 148.072.
C. 5-азидо-5-дезокси-L-ксило-гекслоз-1-фосфат.
Раствор, содержащий 2-азидо-2-дезокси-треитол, полученный ранее (476 мг, 3.24 мл) в 10 мл H2O охлаждают до 0oC и добавляют периодат (NaIO4; 762 мг, 1.1 эквивалент). Спустя 10 мин исходный материал полностью исчезает, и новое пятно появляется в соответствии с данными тонкослойной хроматографии (Rf = 0.5, этилацетат). Хлорид бария (BaCl2 • 2H2O; 870 мг, 1.1 эквивалент) добавляют затем к раствору, и осадок отфильтровывают. Полученный раствор подкисляют до PH I Dowex 50 [H+]. Полученный таким образом рацемический 2-азидо-3-гидроксипропиональдегид не выделяют.
После фильтрования раствор, содержащий рацемат, доводят до pH 7 гидроксидом натрия (NaOH; 10 н). Затем добавляют дигидроксиацетонфосфат (1.5 ммоль), и полученный раствор доводят до pH 7 10 н NaOH. К этому раствору добавляют FDP альдолазу мускула кролика (500 ед), и полученный раствор медленно перемешивают в течение 2 дней.
Ферментативный анализ свидетельствует, что израсходован весь DHAP.
Указанное в заглавии соединение выделяют вначале в виде бариевой соли, добавляя два эквивалента BaCl2 • 2H2O к реакционной смеси. Температуру раствора поддерживают при -20oC в течение ночи (около 18 ч). Осадок выделяют и обрабатывают Dowex 50 [H+] в дистиллированной воде для удаления катионов бария. После фильтрования pH раствора устанавливают 7, и лиофилизируют до получения очищенного указанного в заглавии соединения (75%).
1H-NMR (D2O) δ 3.13 (1H, d, J=9.5 Hz, H-3), 3.14 (1H, ddd, J=9.5, 5, 11 Hz, H-5), 3.20 (1H, t, J=11 Hz, H-6a), 3,31 (1H, t, J=9.5 Hz, H-4), 3.37 (1H, dd, J=6, 11 Hz, H-6e), 3.40-3.44 (2H, m, 2 x H-1) ppm. 13C-NMR (D2O) δ 61.78, 63.36, 67.35, 70.95, 97.67 (d, J=9.5 Hz) ppm. HRMS (M - 4H+ + 5Na+) calcd 395.9540, found 395.9538.
D. Раствор 5-азидо-5-дезокси-L-ксилогксулозо-1-фосфата (100 мг, 0.35 ммоль) в 5 л воды гидрируют 20 мг 10% палладия на угле (Pd-C) при давлении 40 фунтов/дюйм2 (2.812 кг/см2) водорода в течение одного дня. Катализатор удаляют фильтрованием, в полученный фильтрат концентрируют в вакууме. Остаток хроматографируют в колонке с силикагелем (метанол : хлороформ : H2O = 6 : 4 : 2) до получения соединения 50 (40 мг, 78% выход), 2R : 2S ≈ 6 : 1.
1H-NMR (D2O) δ 1.31 (3H, d, J=7 Hz, 2R-CH3), 1.27 (3H, d, J=6.5 Hz, 2S-CH3), 3.36 (1H, m, H-2), 3.66 (1H, m, H-5), 3.74-3.81 (2H, m, 2 x H-5), 3.85 (1H, m, H-3), 4.08 (1H, dd, J=2.5, 4.5 Hz, H-4) ppm; 13C-NMR (D20) δ 16.58 (C-2'), 57.90 (C-5'), 61.50, 63.44, 75.62, 87.09 ppm. HRMS (M + H+) calcd 148.0974, found 148.0974.
Пример 18. Получение FucT ингибитора, соединения 51 - 53.
Ингибирующие соединения 51 - 53 получают обычно как показано на схеме 25 далее.
Для получения соединения 51 - 53 азидо-альдегиды (S)-54 и (R)-54 выбирают в качестве ацепторов для реакций, катализируемых альдолазой, с дигидроксиацетонфосфатом (стадия a), фукозо-1-фосфатальдолазой (стадия b), фруктозо-1,6-дифосфатальдолазой мускула кролика до получения промежуточных фосфатов, которые вначале обрабатывают кислотной фосфатазой, а затем подвергают восстановительному аминированию (стадия c), H2/Pd-C (50 пси) (3.515 кг/см2) до получения окончательных продуктов, которые содержат два хиральных центра в 3- и 4-положениях.
Соединения (S)-54 и (R)-54 получают из 2-бутил-1-ола в реакции с катализатором Линдлара с последующим эпоксидированием и раскрытием азида до получения соответствующих энантиометрных 2- и 3-азидодиолов в соотношении 6 : 1, соответственно. Разделение 2-азиддиола проводят используя липазу из Pseudomonas sp. и винилацетат в качестве ацилирующего агента (Wang et al., J. Org. Chem. , 53: 3127 (1988); Wang et al., J. Am. Chem. Soc., 110: 7200 (1980)) до получения (2R,3S)-2-азидо-3-гидрокси-4-ацетата и (2R,3S)-2-азидо-3,4-диацетата с высокой степенью оптической чистоты по данным 1H ЯМР в присутствии Eu(hfc)3. Очищенные азидогидроксиацетат и азидодиацетат отдельно гидролизуют до получения соответствующих диолов, которые затем окислительно отщепляют периодатом натрия до получения соединений (S)-54 и (R)-54. Физические характеристики соединений 51 - 53 представлены далее.
53: [α]
52: [α]
51: [α]
Пример 19. Синтез и данные соединений в схемах 21 - 23.
Соединения 61 - 99 и их получение обсуждались в отношении схем 21 - 23. В табл. 6 представлены выбранные 1H ЯМР и ВРМС данные для соединений этих схем. Специфические подробности синтеза для примерных соединений в дополнений к тем, которые уже обсуждались ранее, приводятся далее.
Описываемые далее процедуры также применимы к другим гликозил-1-фосфатным соединениям 89 - 95. Единственная модификация наблюдается на стадии очистки соединений 68, 69, 75 и 76. EtOAc используют в качестве элюента для соединений 68 и 69, EtOAc : гексан (2 : 3) для соединения 75, и CHCl3 : EtOAc : MeOH (15 : 0.5 : 0.2) для соединения 76.
A. 2,3,4,6-тетра-O-ацетил-D-глюкоза (соединение 71).
Раствор пентаацетата соединения 64 (5.0 г, 12.9 ммоль) и BnNH2 (19.2 ммоль) в ТГФ (30 мл) поддерживают при комнатной температуре в течение ночи (около 18 ч). Полученную смесь разбавляют холодной водой и экстрагируют CHCl3 (3 • 50 мл). Объединенные органические экстракты последовательно промывают охлажденной льдом разбавленной соляной кислотой, насыщенным бикарбонатом натрия, насыщенным раствором NaCl и водой, сушат над безводным сульфатом натрия и концентрируют в вакууме. Оставшийся сироп очищают хроматографически на силикагеле, элюируя смесью EtOAc/гексан (2 : 3) до получения соединения 71 (3.80 г, 85%) в виде смеси 3 : 1 (α:β) аномеров по данным 1H ЯМР.
B. Дибензилфосфинил-2,3,4,6-тетра-O-ацетил-D-глюкозофосфит (соединение 78).
Дибензил-N, N-диэтилфосфороамидит (0.86, 7.3 ммоль) добавляют к раствору соединения 71 (1.0 г, 2.9 ммоль) и 1,2,4-триазола (0.8 г, 11.5 ммоль) в безводном CH2Cl2 в атмосфере азота при комнатной температуре. Полученную смесь оставляют при перемешивании при комнатной температуре на 1 - 2 ч, перед тем как разбавить эфиром. Полученную смесь последовательно промывают ледяным насыщенным бикарбонатом натрия, насыщенным NaCl и водой, сушат над безводным сульфатом натрия и концентрируют в вакууме. Оставшийся сироп обрабатывают хроматографически на силикагеле, элюируя EtOAc/гексаном (1 : 4) до получения соединения 78 (1.73 г, 97%) в виде смеси (α:β)( 1 : 4, по данным 1H ЯМР (CDCl3).
C. Дибензилфосфорил-2,3,4,6-тетра-O-ацетил-D-глюкоза (соединение 25).
К раствору соединения 78 (1.2 г, 2.2 ммоль) в ТГФ (50 мл), охлажденному до -78oC в бане сухой лед-ацетон, прикапывают 30% H2O2 (10 мл), и полученной смеси дают нагреться до комнатной температуры, и перемешивают в течение 1.5 ч при комнатной температуре. Полученную смесь разбавляют эфиром и последовательно промывают ледяным насыщенным Na2S2O3 насыщенным NaHCO3, насыщенным NaCl и водой. Органическую фазу сушат над безводным сульфатом натрия и концентрируют до получения α:β (1 : 4) смеси соединения 85 (1.36 г, 98%), по данным 1H ЯМР (CDCl3). Этот продут используют на следующей стадии без дальнейшей очистки.
D. Глюкозо-1-фосфат (соединение 92).
Соединение 85 (1.0 г, 1.8 ммоль) гидрируют (14.7 пси, 1.0 кг/см2) над 5% Pd/C (200 мг) в EtOH (30 мл) и 10% NaHCO3 (20 мл) в течение 10 ч при комнатной температуре. Полученную смесь фильтруют, и фильтрат концентрируют. Остаток обрабатывают 1 н NaOH (10 мл) при комнатной температуре в течение 3 ч. Полученную смесь нейтрализуют ледяным 1 и AcOH до pH 7.5, а нерастворимую часть удаляют фильтрованием. В другом варианте вместо NaOH используют раствор MeOH : H2O (1 : 1) (объем/объем) в 10% Et3N, так что последующая стадия нейтрализации исключается. Полученный фильтрат концентрируют, разбавляют водой и пропускают через колонку Dowex 50W-X8 [Na+] (1 • 15 см), элюируя водой. Соответствующие фракции собирают и лиофилизируют до получения соединения 92.
Иногда наблюдаются некоторые количества дефосфорилированного продукта. Его удаляют, пропуская разбавленный фильтрат через колонку Dowex 1W-XB [HCO2-] (1 • 30 см). Колонку вначале элюируют водой для удаления нейтрального продукта, а затем линейный градиент (NH4HCO3 (0.1 М - 0.3 М) вводят для элюирования целевого продукта. Соответствующие фракции собирают, и лиофилизируют. Лиофилизированный порошок растворяют в воде (10 мл), охлаждают до 0oC, и нейтрализуют до pH 7,0 смолой Dowex 50W-XB [H+]. Эту смолу отфильтровывают, а полученный фильтрат снова лиофилизируют до получения соединения 92 (0.30 г, 59%) в виде α:β (1 : 4) смеси по данным 1H ЯМР (D2O).
E. Метил-5-ацетамидо-4,7,8,9-тетра-O-ацетил-3,5-дидеокси -β- D-глицеро-D-галакто-2-нонулопиранозонат (соединение 96).
Это соединение получают по способу Marra et al., Carbohydr. Rev. 190: 317 (1989). В другом варианте, смесь метилд-2-хлор-5-ацетамидо-4,7,8,9-тетра-O-ацетил-3,5-дидеокси -β- D-глицеро-D-галакто-2-нонулопиранозоната (Kuhn et al., Chem. Ber. 99: 611 (1966)) (0.67 г, 1.3 ммоль) и карбоната серебра (0.363 г, 1.3 ммоль) в 5 мл ацетона - 0.5 мл H2O перемешивают в течение 10 ч при комнатной температуре. Полученную суспензию отфильтровывают, пропуская через целитный слой (CeliteTM 545), и полученный фильтрат выпаривают досуха. Остаток разбавляют хлороформом, промывают водой и рассолом, а затем сушат над сульфатом натрия. Растворитель выпаривают в вакууме до получения неочищенного материала, который обрабатывают хроматографически на силикагеле, элюируя смесью хлороформ/метанол 25 : 1, до получения соединения 96 (0.586 г, 88%) в виде белых иголок.
1HNR (CDCl3) δ : 1.90, 2.02, 2.03, 2.10, 2.14 (3H each, s, 4•OAc and NAc), 2.17 (1H dd, J 5.04, 12.72 Hz, H-3eq), 2.29 (1H dd, J 11.52, 12.72 Hz, H-3ax), 3.87 (3H, s, COOMe), 4.02 (1H, dd, J 7.04, 12.4 Hz, H-9), 4.12 (1H, dd, J 2.1, 7.8 Hz, H-6), 4.13 (1H, d, J 7.8 Hz, NH), 4.17 (1H, ddd, 7.8, 9.8, 10.28, Hz, H-5), 4.42 (1H, dd, J 1.92, 12.4 Hz, H-9'), 5.20 - 5.26 (2H, m, H-4 and H-8), 5.32 (1H, dd, J 2.1, 6.50 Hz, H-7), 5.37 (1H, bs, OH).
F. Метил-5-ацетамидо-4,7,8,9-тетра-O-ацетил-2-(дибензилфосфитил) -3,5-дидеокси -β- D-глицеро-D-галакто-2-нонулориганозонат (соединение 97).
Дибензил-N,N-диэтилфосфоамидат (0.25, 0.78 ммоль) прикапывают к раствору соединения 96 (0.166 г, 0.34 ммоль) и 1H-тетразола (0.10 г, 1.43 ммоль) в ТГФ (5 мл) в атмосфере азота, и полученную смесь выдерживают в течение 4 ч при комнатной температуре. К смеси добавляют дихлорметан (10 мл), и органическую фазу промывают ледяной разбавленной HCl, водным NaHCO3 и ледяной водой, сушат над безводным сульфатом натрия. Полученный раствор выпаривают в вакууме до получения неочищенного материала, который обрабатывают на хроматографической колонке с силикагелем, элюируя EtOAc/гексаном (5 : 1) до получения соединения 97 (0.17 г, 68%) в виде бесцветного сиропа.
13C-NMR (CDCl3) δ : 20.7, 20.8, 20.9, 21.00, 23.1, 36.0, 49.5, 53.5, 62.6, 67.3, 67.4, 67.8, 69.3, 70.8, 94.8, 128.0, 128.7, 135.5, 141.8, 153.0, 169.1, 170.2, 170.4, 170.8, 171.0. HRMS: рассчитано для C34H42NO15PCs (M+Cs+) 868.1346, найдено 8768.1346.
G. Метил-5-ацетамидо-4,7,8,9-тетра-O-ацетил-2- (дибензилфосфорил)-3,5-дидеокси -β- D-глицеро-D-галакто-2-нонулопиранозонат (соединение 98).
К охлажденному раствору соединения 97 (0.13 г, 0.17 ммоль) в ТГФ (2 мл) добавляют трет.-BuO2H (0.4 мл) при -10oC, и полученную смесь оставляют нагреваться до комнатной температуры, и перемешивают в течение часа при комнатной температуре. Полученную смесь разбавляют CH2Cl2 и промывают ледяным водным бикарбонатом натрия и водой, затем сушат над безводным сульфатом натрия. Органическую фазу выпаривают в вакууме до получения неочищенного материала, который обрабатывают на хроматографической колонке с силикагелем, элюируя CHCl3/MeOH (25 : 1) до получения соединения 98 (0.126 г, 95%) в виде бесцветного сиропа. ВРМС: рассчитано для C34H42NO16PCs (M + Cs+) 884.1396; найдено 884.1305.
H. Метил-5-ацетамидо-4,7,8,9-тетра-O-ацетил-3,5-дидеокси -β- D-глицеро-D-галакто-2-нонулопиранозаонат-2-фосфорная кислота (соединение 99, водородная форма).
Соединение 98 (0.22 г, 0.25 ммоль) гидрируют (14.7 пси, 1 кг/см2) над 5% Pd/C (10 мг) в атмосфере водорода в течение 7 ч при комнатной температуре. Катализатор отфильтровывают через CeliteTM 545, а полученный фильтрат концентрируют в вакууме. Неочищенный материал обрабатывают на хроматографической колонке с силикагелем с обращенной фазой, элюируя CH3CN/H2O (5 : 1) до получения соединения 99, водородной формы (0.164 г, 99%) в виде бесцветного сиропа. ВРМС: рассчитано для C20H30NO16PCs (M + Cs) 704.0356, найдено 704.0356.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ ФУКОЗИЛИРОВАННЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2584599C2 |
| СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ОПУХОЛЕВОГО РОСТА (ВАРИАНТЫ) | 2001 |
|
RU2276133C2 |
| СПОСОБЫ И КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ИНГИБИРОВАНИЯ αβ-ОПОСРЕДОВАННОГО АНГИОГЕНЕЗА | 1997 |
|
RU2195312C2 |
| СПОСОБЫ ИНГИБИРОВАНИЯ АНГИОГЕНЕЗА И РОСТА ОПУХОЛЕЙ | 2001 |
|
RU2269339C2 |
| ПРОДУКЦИЯ ГЛИКОПРОТЕИНОВ С МОДИФИЦИРОВАННЫМ ФУКОЗИЛИРОВАНИЕМ | 2008 |
|
RU2479629C2 |
| СПОСОБЫ И КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ИНГИБИРОВАНИЯ АНГИОГЕНЕЗА | 1997 |
|
RU2194528C2 |
| СПОСОБ ВЫДЕЛЕНИЯ ФУКОЗИЛИРОВАННЫХ АНТИТЕЛ | 2012 |
|
RU2650873C2 |
| ПРИМЕНЕНИЕ ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ АДЕНИНА ДЛЯ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА | 1993 |
|
RU2130308C1 |
| ПРОДУКЦИЯ ФУКОЗИЛИРОВАННЫХ ОЛИГОСАХАРИДОВ В BACILLUS | 2020 |
|
RU2810729C2 |
| СИСТЕМА ДОСТАВКИ НУКЛЕИНОВЫХ КИСЛОТ | 2002 |
|
RU2294192C2 |
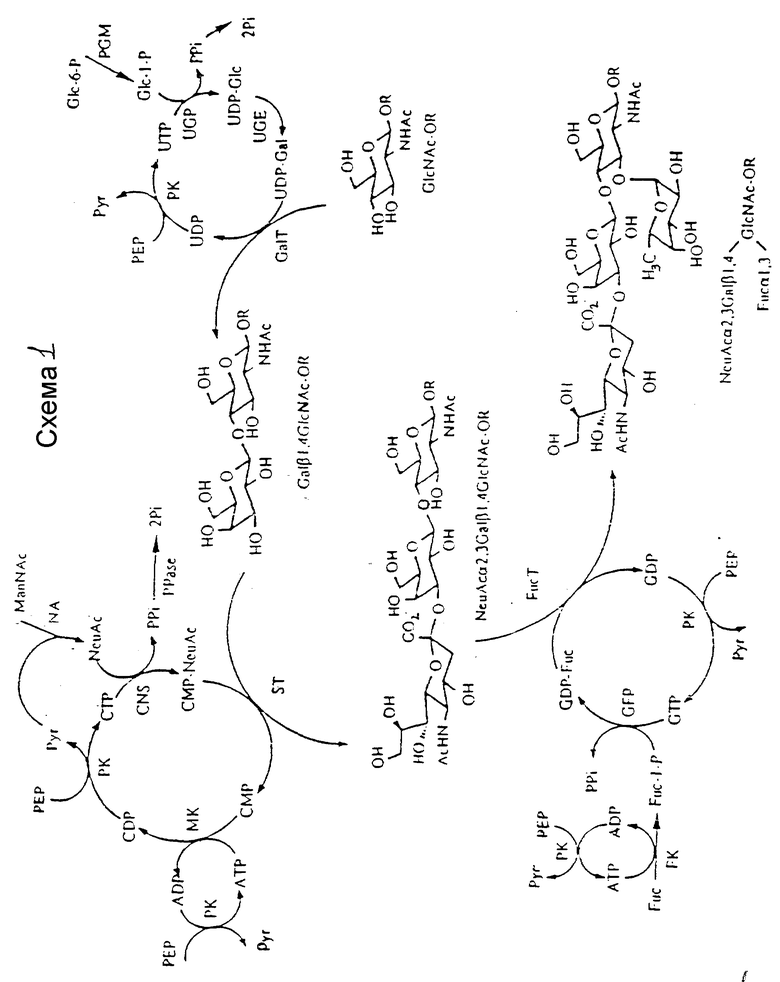
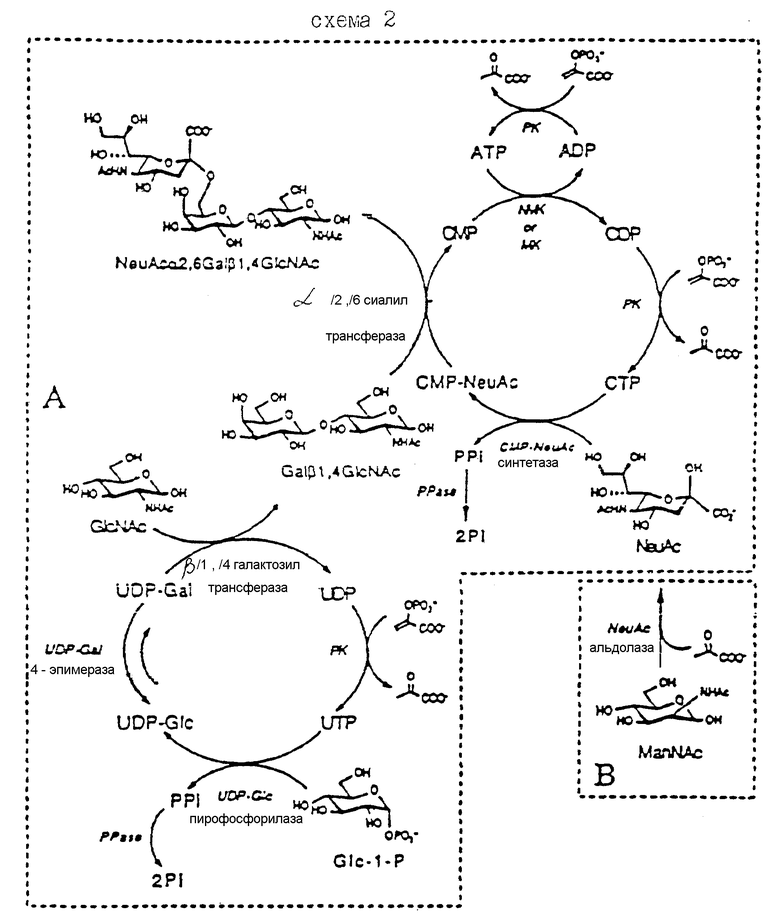
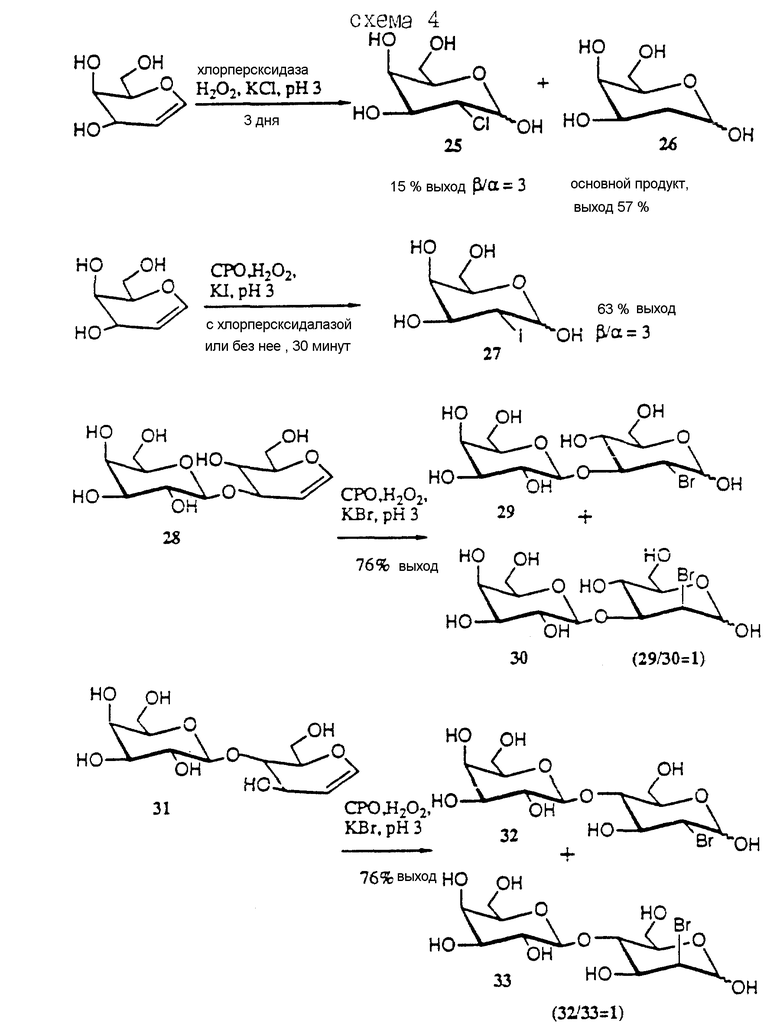
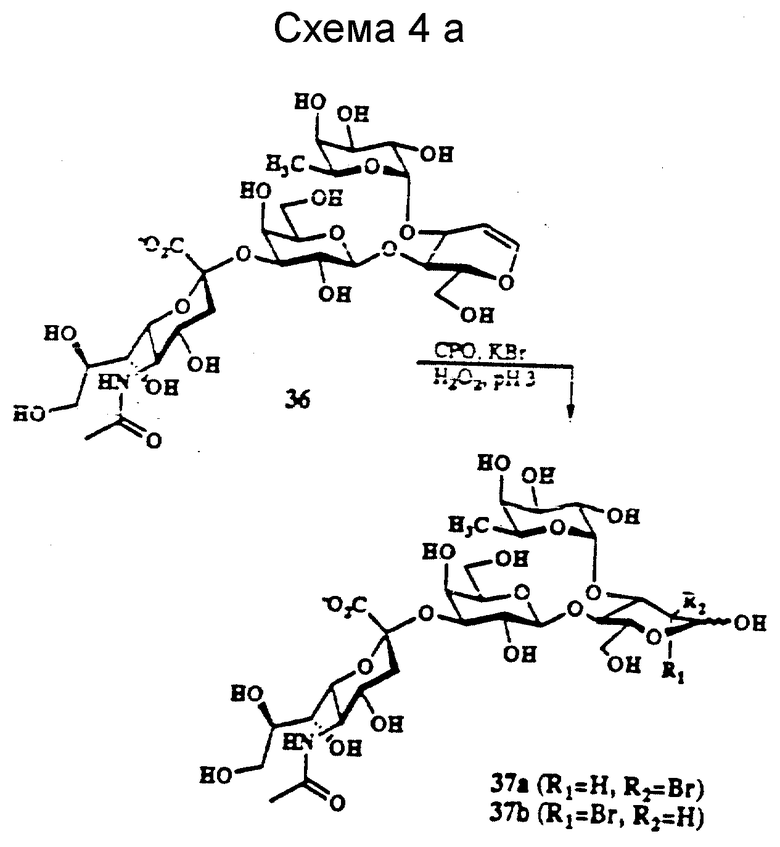
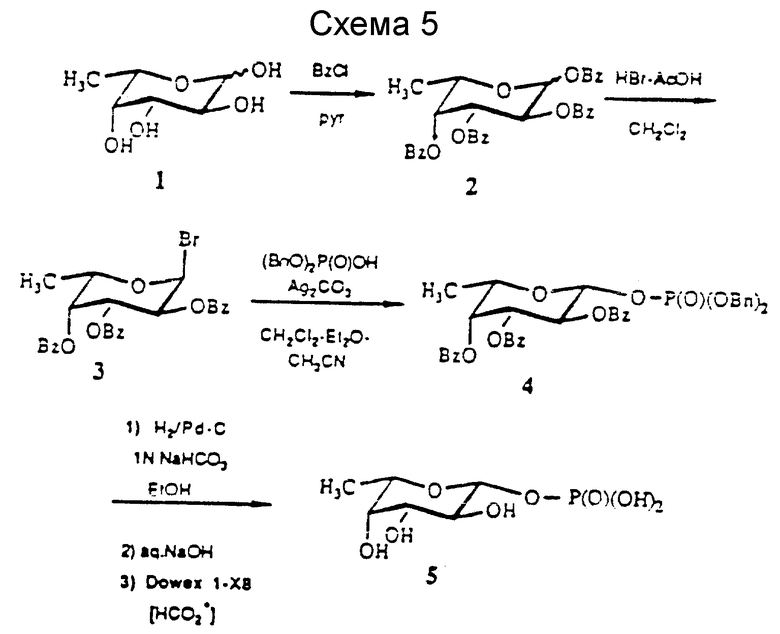
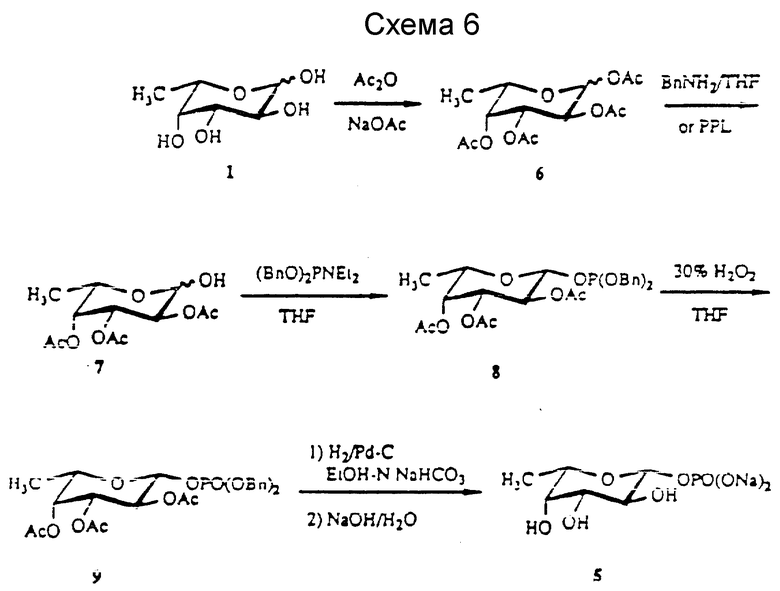
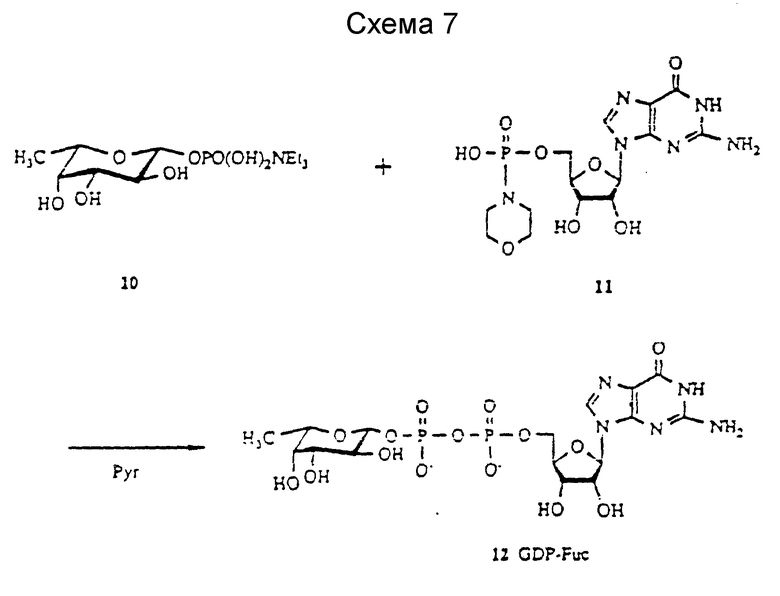
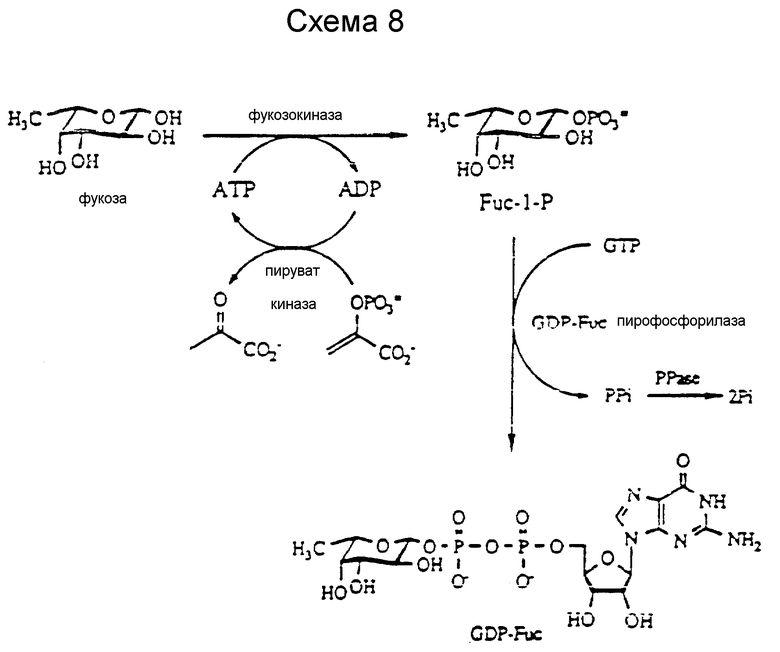
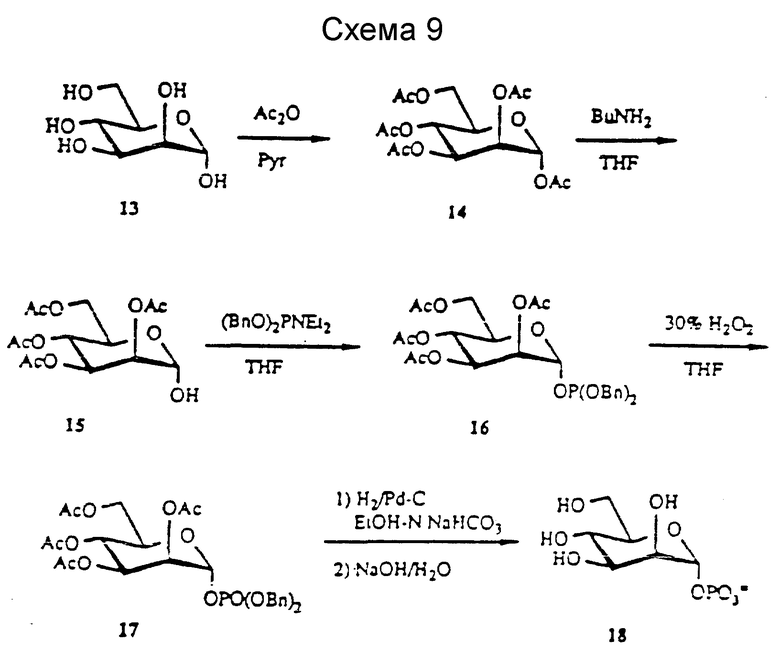
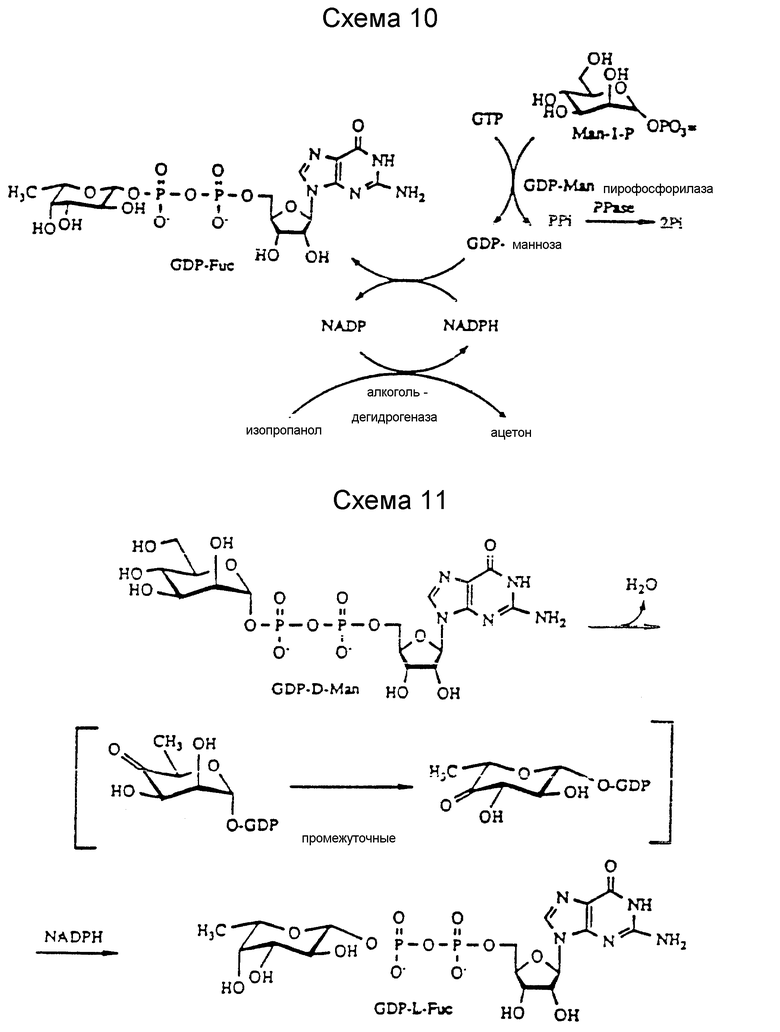
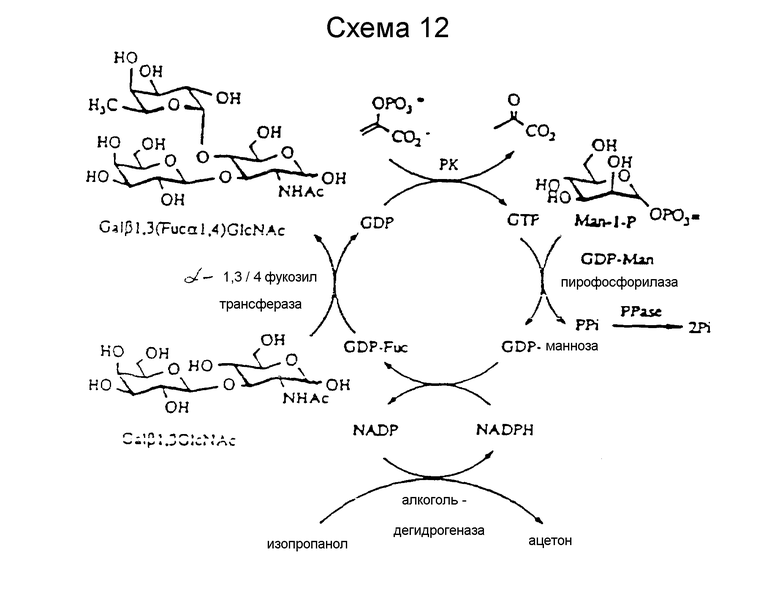
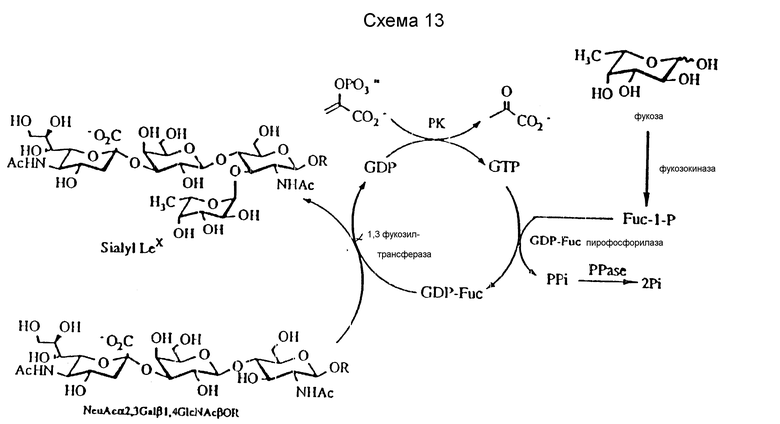
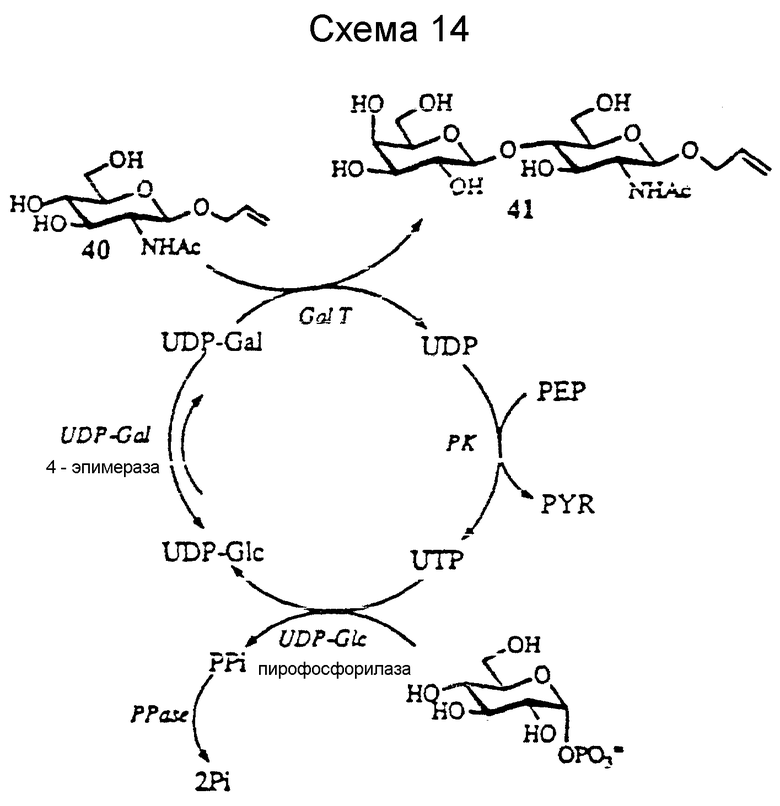
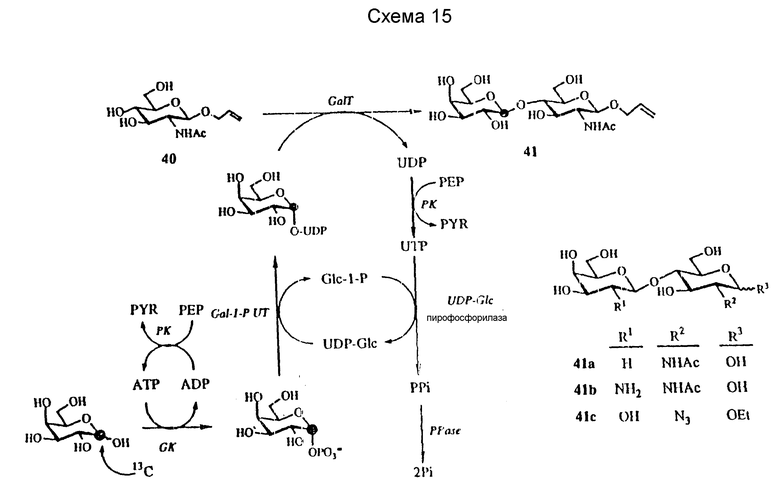
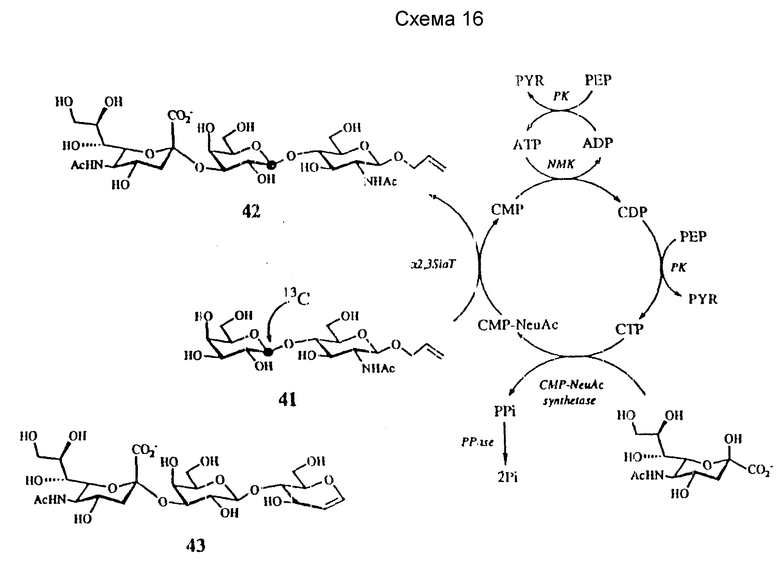
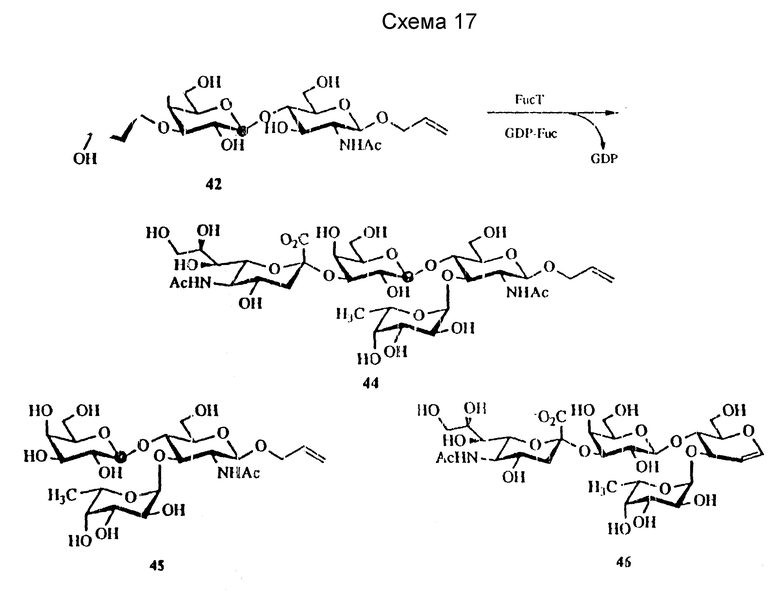
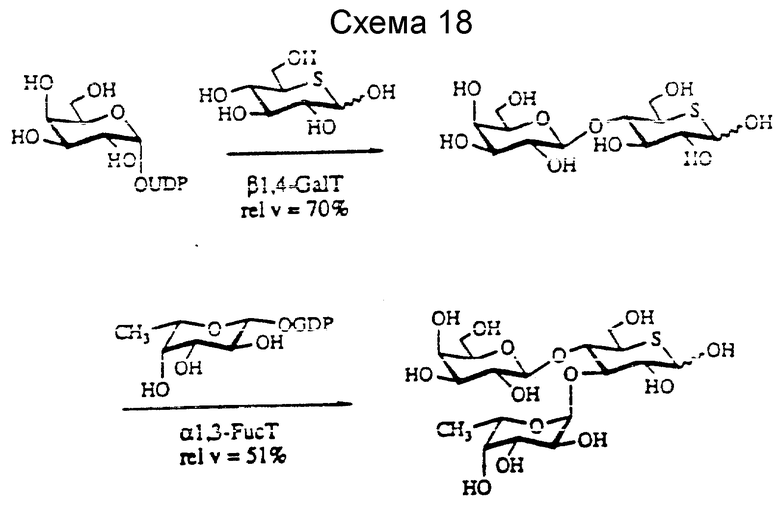
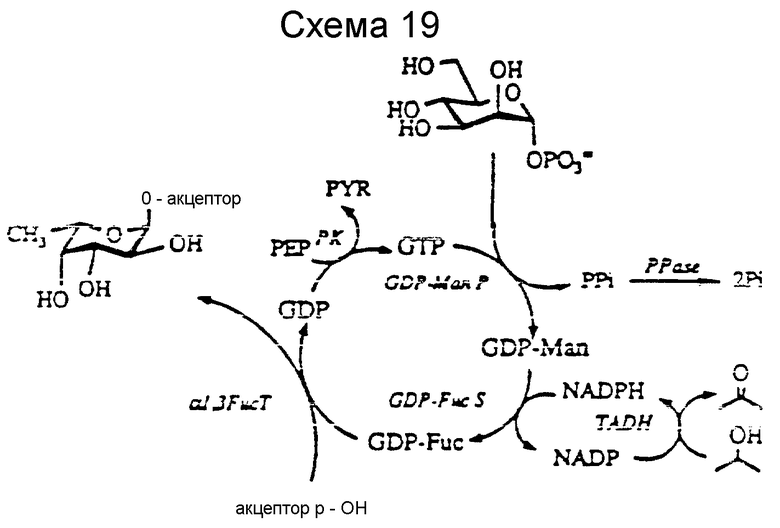
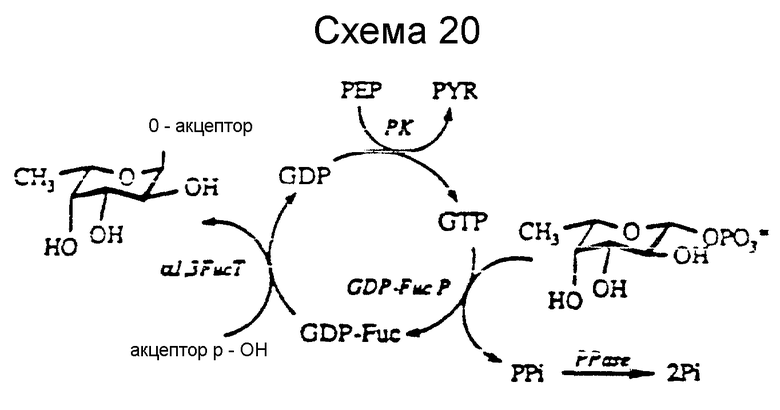
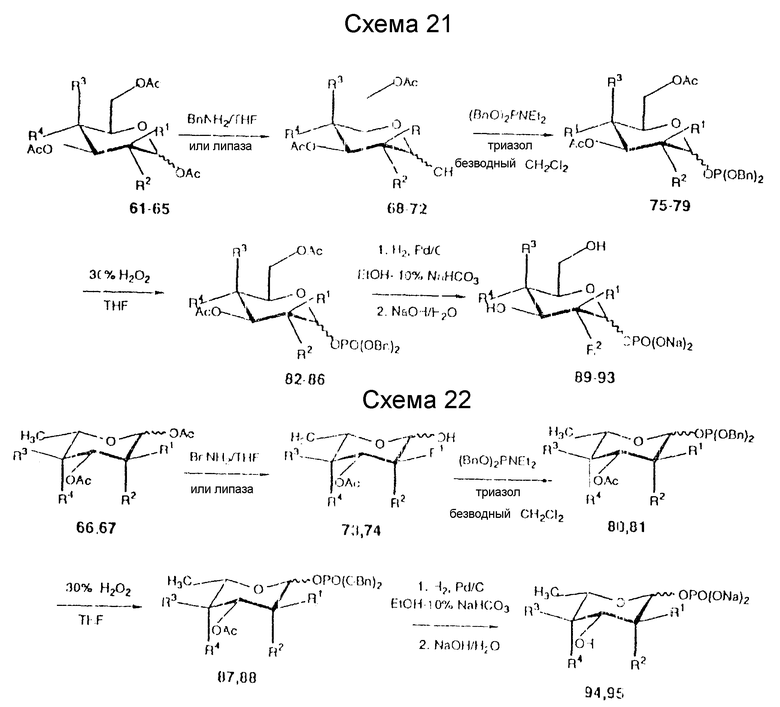
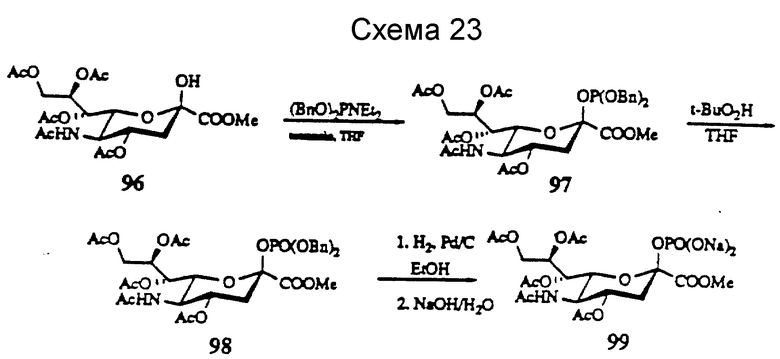
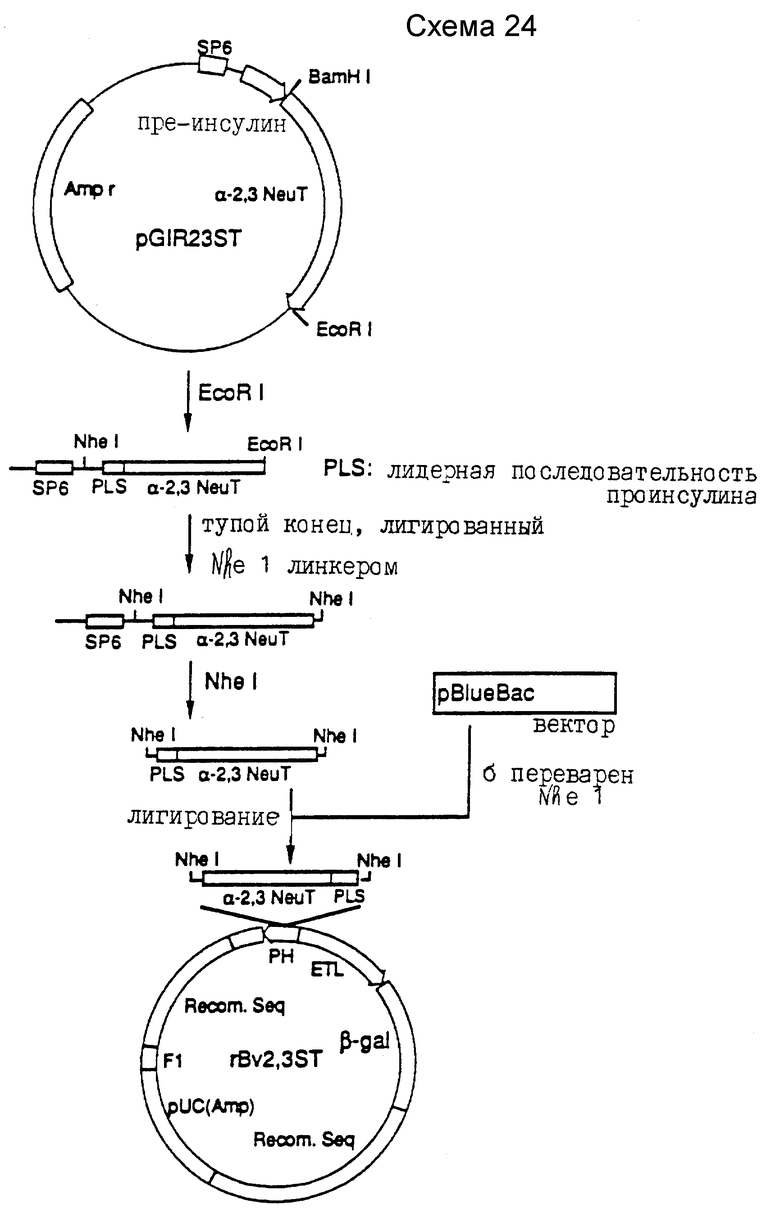
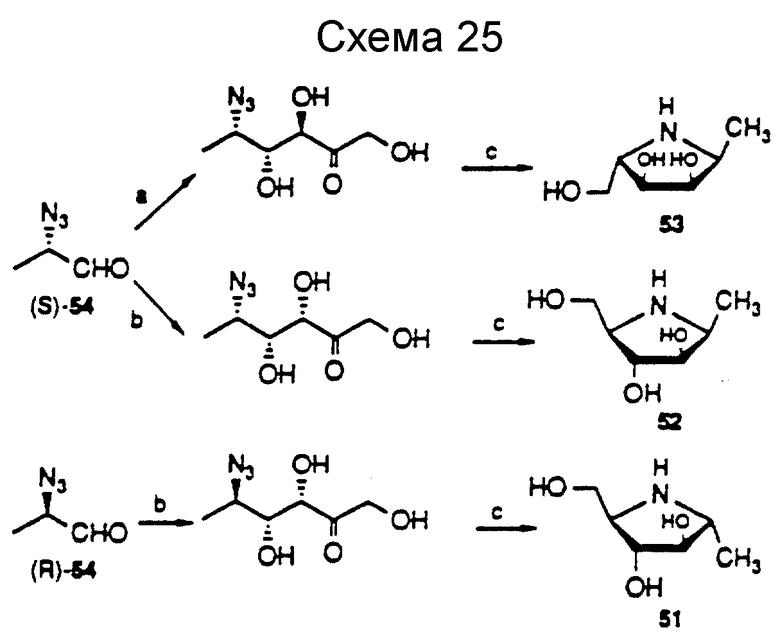


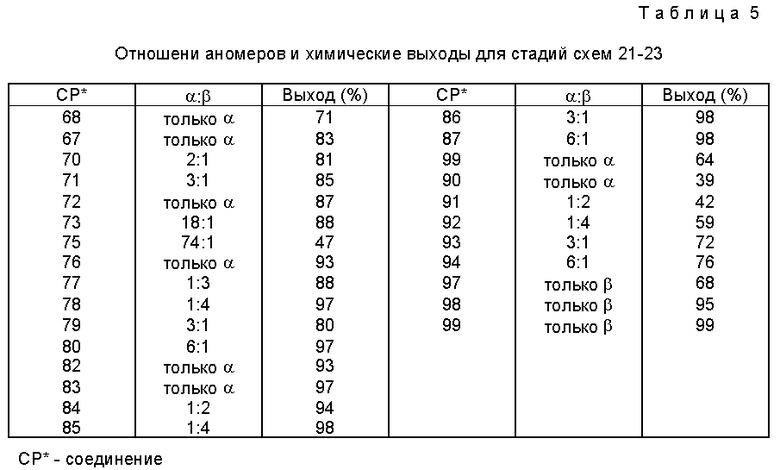
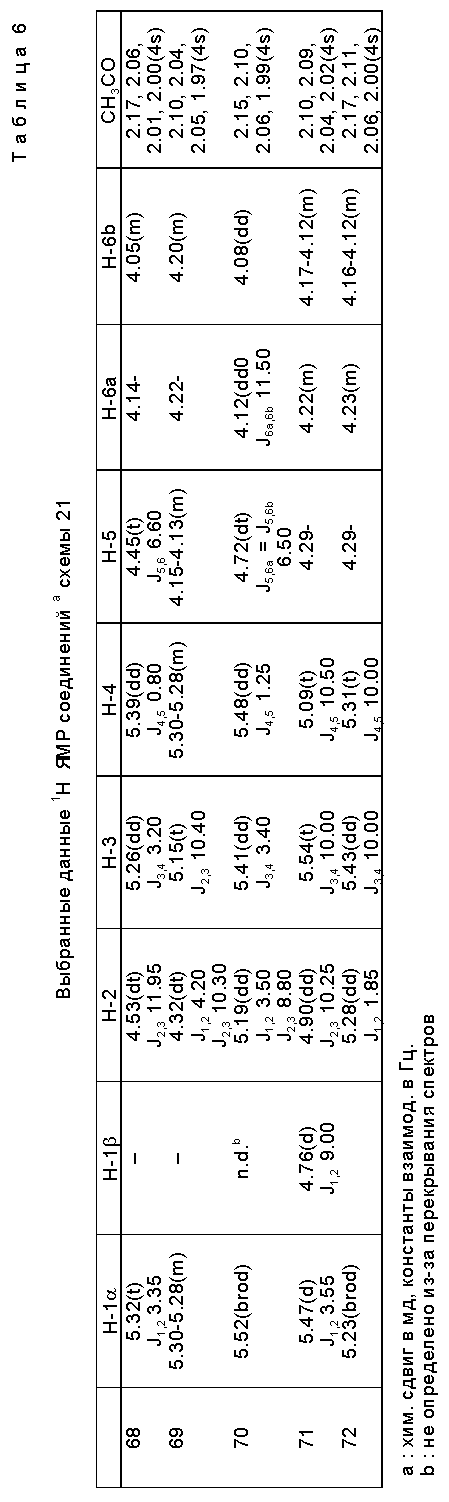

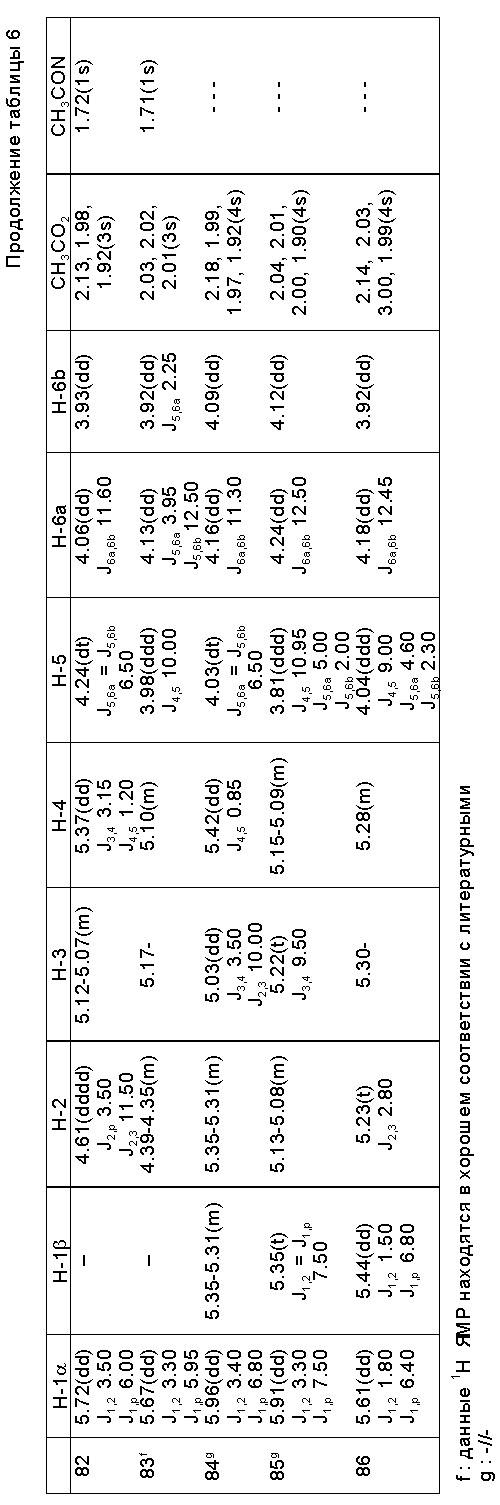
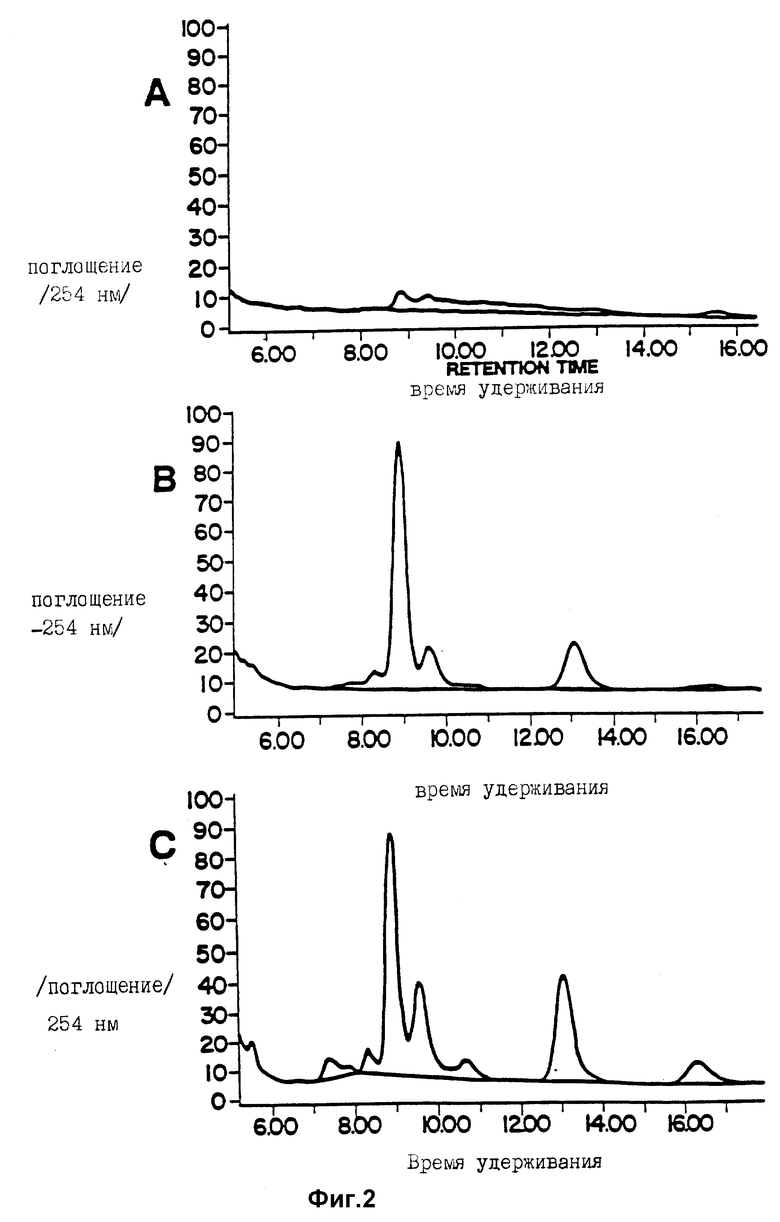
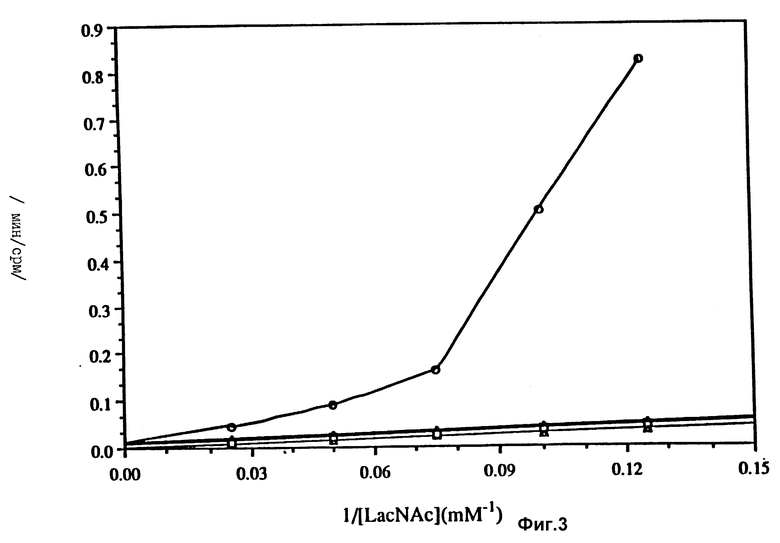
Изобретение относится к биотехнологии и предназначено для получения фукозилированных углеводов. Способ осуществляют в единой реакционной смеси с использованием фукозилтрансферазы для создания С-гликозидной связи между гуанозин 5'-дифосфофукозой и доступной гидроксильной группой углеводной акцепторной молекулы до получения фукозилированного углевода и гуанозин-5'-дифосфата. Гуанозин-5'-дифосфат рециркулируют с образованием гуанозин-5'-дифосфофукозы. Способ позволяет усовершенствовать получение фукозилированных углеводов. 3 с. и 25 з.п. ф-лы, 8 табл., 3 ил.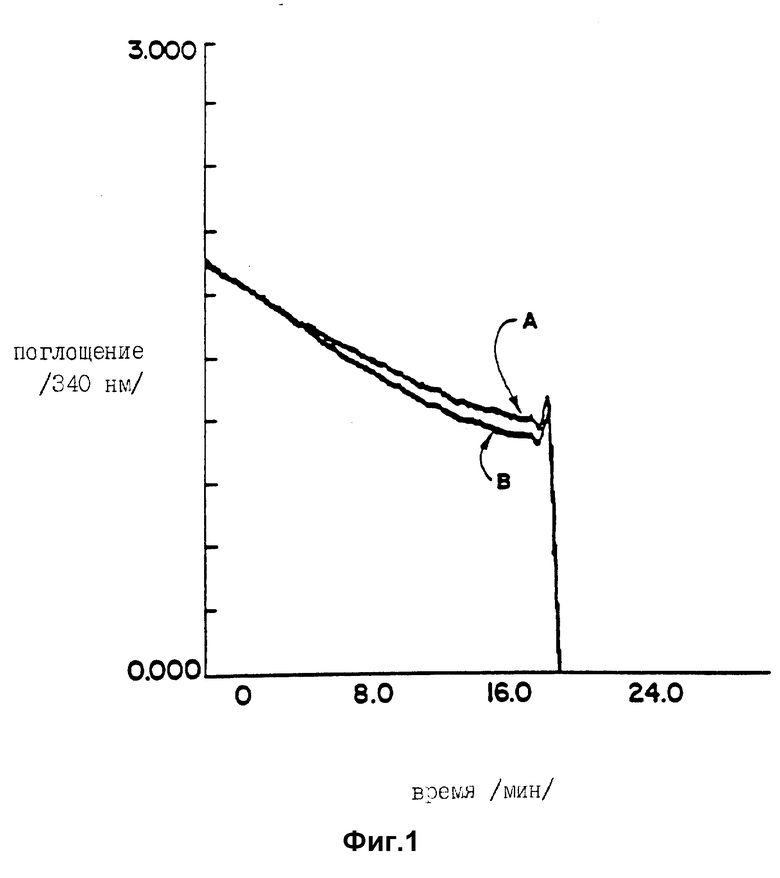
| Adv | |||
| Enzymology | |||
| Устройство для устранения мешающего действия зажигательной электрической системы двигателей внутреннего сгорания на радиоприем | 1922 |
|
SU52A1 |
| Bioch | |||
| Soc | |||
| Transact, v.15, p.396, 1987. | |||
