Область техники, к которой относится изобретение
Изобретение относится, в целом, к медицине и конкретно относится к способам и композициям для ингибирования ангиогенеза тканей с использованием антагонистов витронектинового рецептора αvβ3.
Предпосылки изобретения
Интегрины представляют собой класс клеточных рецепторов, которые, как известно, связывают внеклеточные белки матрикса, и поэтому опосредуют взаимодействия клетка-клетка и клетка-внеклеточный матрикс, именуемые как события адгезии клетки. Однако несмотря на то, что многие интегрины и лиганды, которые связывают интегрин, и описаны в литературе, биологическая функция многих интегринов остается смутной. Интегриновые рецепторы составляют семейство белков с совпадающими структурными характеристиками нековалентных гетеродимерных гликопротеиновых комплексов, образованных α- и β-субъединицами.
Витронектиновый рецептор, названный по его исходной характеристике предпочтительного связывания с витронектином, известен в настоящее время как относящийся к трем разным интегринам, обозначенным αvβ1, αvβ3 и αvβ5. Horton, Int. J. Exp. Patnol., 71:741-759. αvβ1 связывает фибронектин и витронектин. αvβ3 связывает большое множество лигандов, в том числе фибрин, фибриноген, ламинин, тромбоспондин, витронектин, фактор фон Вилленбранда, остеопонтин и костный сиалопротеин I. αvβ5 связывает витронектин. Эти три интегрина важны для осуществления специфической клеточной адгезии во многих клеточных взаимодействиях в тканях и исследуются до сих пор, но очевидно, что существуют разные интегрины с разными биологическими функциями.
Существенный участок узнавания в лиганде для многих интегринов представляет собой трипептидную последовательность аргинин-глицин-аспарагиновая кислота (RGD). RGD обнаружена во всех вышеуказанных лигандах для витронектиновых рецепторных интегринов. Этот RGD-узнающий участок можно имитировать с помощью полипептидов ("пептидов"), которые содержат данную RGD-последовательность и такие RGD-пептиды представляют собой известные ингибиторы с функцией интегрина. Важно, однако, отметить, что в зависимости от последовательности и структуры RGD-пептида специфичность ингибирования может изменяться для мишенно-специфичных интегринов.
Обсуждение RGD-узнающего участка смотрите у Nature, 309:30-33 (1984) и Pierschbacher с соавт., Proc. Natl. Acad. Sci. USA, 81:5985-5988 (1984). Различные RGD-полипептиды с варьирующей интегриновой специфичностью описаны у Grant с соавт., Cell, 58:933-943 (1989), Cheresh с соавт., Cell, 58-945-953 (1989), Aumailley с соавт., FEBS Letts., 291-50-54 (1991) и Pfaff с соавт., J. Biol. Chem. 269:20233-20238 (1994) в Патентах Соединенных штатов 4517686, 4578079, 4589881, 4614517, 4661111, 4792525, 4683291, 4879237, 4988621, 5041380 и 5061693.
Ангиогенез представляет собой васкуляризацию тканей, в которой происходит рост вновь образующихся сосудов, и представляет собой также так называемую реваскуляризацию. Данный процесс опосредован с помощью инфильтрации эндотелиальных клеток и клеток гладкой мускулатуры. Данный процесс осуществляется любым одним из трех путей: сосуды могут вырастать из ранее существовавших сосудов, создание сосудов de novo может осуществляться из предшественников клеток (васкулогенез) или существующие маленькие сосуды могут увеличиваться в диаметре. Blood с соавт., Biochem. Biophys. Acta, 1032:89-118 (1990). Известно, что в сосудистых эндотелиальных клетках содержится как минимум пять RDG-зависимых интегринов, в том числе рецептор витронектина (αvβ3 и αvβ5), рецептор коллагена типа I или IV (α1β1), рецептор ламинина (α2β1), фибронектиновый (ламининовый) коллагеновый рецептор (α3β1) рецептор фибронектина (α5β1). Davis с соавт., J. Cell. Biochem., 51:206-218 (1993). Клетки гладкой мускулатуры, как известно, содержат, по крайней мере, шесть RGD-зависимых интегринов, в том числе α5β1, αvβ3 и αvβ5.
Ангиогенез является важным процессом в неонатальном росте, и также важен в заживлении ран и в разнообразных клинических заболеваниях, в том числе воспалении ткани, артритах, опухолевом росте, диабетической ретинопатии, дегенерации желтого пятна при образовании новых сосудов в ретине и подобных состояниях. Эти клинические проявления ассоциируются с ангиогенезом и именуются сосудистными заболеваниями. Folkman с соавт., Science, 235-442-447 (1987). Как правило, ангиогенез отсутствует во взрослой или зрелой тканях, хотя он может происходить при заживлении раны и в ростовом цикле corpeus leuteum. Смотрите, например, Moses с соавт., Science, 248:1408-1410 (1990).
Было предположено, что ингибирование ангиогенеза, должно быть, представляет собой неплохую терапию по ограничению опухолевого роста. Было предложено ингибировать ангиогенез путем (1) ингибирования высвобождения "ангиогенных молекул", таких как bFGF (фактор роста базисных фибробластов), (2) нейтрализации ангиогенных молекул, таких как используемые в качестве антител к анти-βbFGF и (3) подавления ответа эндотелиальной клетки на ангиогенные стимулы. Этот последний подход привлек внимание и Folkman с соавт. , Cancer Biology, 3:89-96 (1992) описали несколько ингибиторов ответа эндотелиальной клетки, в том числе ингибитор коллагеназы, базальной мембраны оборотные ингибиторы, ангиостатические стероиды, ингибиторы ангиогенеза, производимые грибами, фактор 4 тромбоцитов, тромбоспондин, артритные лекарственные средства, такие как D-пеницилламин и тиомалат золота, аналоги витамина D3, альфа-интерферон и другие, которые могли бы использоваться для подавления ангиогенеза. Дополнительно предлагаемые ингибиторы ангиогенеза смотрите у Blood с соавт., Bioch. Biophys. Acta., 1032:89-118 (1990), Moses с соавт. , Science, 248-1408-1410 (1990), Ingber с соавт., Lab. Invest., 59-44-51 (1988) и Патенты Соединенных Штатов 5092885, 5112946, 5192744 и 5202352. Ни один из ингибиторов в вышеприведенных ссылках не приводит к ингибированию αvβ3.
Описаны также RGD-содержащие пептиды, которые ингибируют витронектиновый рецептор αvβ3. Aumailley с соавт., FEBS Letts., 291:50-54 (1991), Choi с соавт. , J. Vasс. Surg., 19:125-134 (1994), Smith с соавт., J. Biol. Chem., 265: 12267-12271 (1990), и Pfaff с соавт., J. Biol. Chem., 269:20233-20238 (1994). Однако роль интегрина αvβ3 в ангиогенезе никогда не предполагалась и не была установлена до настоящего времени.
Например, Hammes с соавт. , Nature Med., 2:529-53 (1996) подтвердил факты, обнаруженные в настоящем изобретении. В частности, в его статье показано, что циклические пептиды, включающие циклический RGDfV, структура и функция которого была описана раньше в приоритетных заявках, на которых основана настоящая заявка, ингибирует ретинальную реваскуляризацию в мышиной модели ретинальной реваскуляризации, индуцированной гипоксией. В специальном исследовании, которое также подтверждает настоящее изобретение, а также приоритетные заявки, Luna с соавт., Lab. Invest., 75:563-573 (1996) описал два отдельных циклических метилированных RGD-содержащих пептида, которые были частично эффективны в ингибировании ретинальной реваскуляризации в мышиной модели, индуцированной кислородом ишемической ретинопатии. Напротив, пептиды настоящего изобретения почти полностью ингибируют реваскуляризацию в модельных системах, описанных здесь.
Ингибирование клеточной адгезии in vitro с использованием моноклональных антител, иммуноспецифичных к разным субъединицам интегрина, α или β, вовлекают αvβ3 в клеточную адгезию разных типов клеток, в том числе эндотелиальные клетки капилляров. Davis с соавт., J. Cell. Biol., 51:206-218 (1993). Кроме того, Nicosia с соавт., Am. J. Pathol., 138:829-833 (1991), описал применение RGD-пептида GRGDS для подавления in vitro образования "микрососудов" из крысиной аорты, культивируемой в коллагеновом геле. Однако подавление образования "микрососудов" in vitro в культурах коллагенового геля не является моделью ингибирования ангиогенеза в тканях, поскольку она не показала, что микрососудистые структуры не представляют собой то же самое, что и капиллярные отростки, или что образование микрососуда в культуре коллагенового геля представляет собой то же, что и неоваскулярный рост в интактной ткани, такой как артритная ткань, опухолевая ткань или заболевание ткани, где ингибирование ангиогенеза представляется желательным.
Для осуществления ангиогенеза эндотелиальные клетки должны вначале деградировать и пересечь кровеносный сосуд базальной мембраны аналогичным способом, используемым опухолевыми клетками во время инвазии и образования метастазов.
Авторы настоящего изобретения ранее сообщали, что ангиогенез зависит от взаимодействия между интегринами сосудов и белками внеклеточного матрикса. Brooks с соавт., и Science, 264:569-571 (1994). Кроме того, он сообщил, что программируемая клеточная смерть (апоптоз) ангиогенных сосудистых клеток инициируется путем взаимодействия, которое должно быть подавлено некоторыми антагонистами васкулярного интегрина αvβ3. Brooks с соавт., Cell, 79:1157-1164 (1994). Совсем недавно авторы настоящего изобретения сообщили, что связывание металлопротеиназы-2 (ММР-2) матрикса с витронектиновым рецептором (αvβ5) можно подавить, используя антагонисты αvβ5 и, таким образом, ингибировать ферментативную функцию протеиназы. Brooks с соавт., Cell, 85:683-693 (1996).
Помимо исследований, сообщенных здесь, заявители не знают какой-либо другой аргументации, что ангиогенез мог бы быть ингибирован в ткани с использованием ингибиторов клеточной адгезии. В частности никогда прежде не доказывалось другими, что αvβ3-функция необходима для ангиогенеза в ткани или что антагонисты αvβ3 могут подавлять ангиогенез в ткани.
Краткое описание настоящего изобретения
В настоящем изобретении описаны доказательства того, что ангиогенез в ткани нуждается в интегрине αvβ3 и что ингибиторы αvβ3 могут подавлять ангиогенез. Представленное описание также аргументирует, что антагонисты других интегринов, таких как αIIbβ3 или αvβ1, не подавляют ангиогенез, главным образом потому, что эти другие интегрины не существенны для осуществления ангиогенеза.
Поэтому в настоящем изобретении описаны способы подавления ангионеза в ткани, включающие введение в определенную ткань композиции, включающей количество αvβ3-антагониста, подавляющего ангиогенез.
Обрабатываемая ткань может представлять собой любую ткань, в которой желательно осуществить ингибирование ангиогенеза, такую как больную ткань, где наблюдается реваскуляризация. Иллюстративные ткани включают воспаленные ткани, твердые опухоли, метастазы, ткани, подвергающиеся рестенозу и им подобные ткани.
αvβ3-антагонист, используемый в настоящих способах, обладает способностью к связыванию с αvβ3 и к конкурентному ингибированию способности αvβ3 связываться с природным лигандом. Данный антагонист предпочтительно проявляет специфичность по αvβ3 перед другими интегринами. В конкретном варианте осуществления настоящего изобретения указанный αvβ3-антагонист подавляет связывание фибриногена или других RGD-содержащих лигандов с αvβ3, но практически не подавляет связывание фибриногена с αIIbβ3. Предпочтительный αvβ3-антагонист может быть слитым полипептидом, циклическим или линейным полипептидом, производным полипептида, моноклональным антителом, иммунно реагирует с αvβ3, органическим имитатором αvβ3 или его функциональным фрагментом.
Краткое описание графических материалов
В чертежах, составляющих часть данного описания:
Фигуры 1A-1D иллюстрируют тканевое распределение субъединиц интегрина, β3 и β1, в нормальной коже и в коже, подвергнувшейся раневому заживлению, обозначаемой в качестве гранулированной ткани. Иммунохимию с антителами к β3 и β1 осуществляли, как описано в примере 3А. Фигуры 1А и 1В иллюстрируют соответственно иммунореактивность анти-β3 в нормальной коже и в гранулированной ткани. Фигуры 1С и 1D иллюстрируют соответственно иммунореактивность анти-β1 в нормальной коже и в гранулированной ткани.
Фигуры 2A-2D иллюстрируют тканевое распределение фактора фон Виллебранда и лигандов ламинина, которые соответственно связывают субъединицы интегрина β3 и β1 в нормальной коже и в коже, подвергнувшейся раневому заживлению, обозначаемой в качестве гранулированной ткани.
Иммуногистохимию с антителами к фактору фон Виллебранда (анти-vWF) и к ламинину (анти-ламинин) осуществляли, как описано в примере 3В. Фигуры 2А и 2В иллюстрируют соответственно иммунореактивность анти-vWF в нормальной коже и в гранулированной ткани. Фигуры 2С и 2D иллюстрируют соответственно иммунореактивность анти-ламинина в нормальной коже и в гранулированной ткани.
Фигуры 3A-3D иллюстрируют тканевое распределение витронектинового интегринового рецептора αvβ3 в тканевых биоптатах соответственно рака мочевого пузыря, рака толстой кишки, рака молочной железы и рака легкого. Иммуногистохимию с LМ609-антителами к αvβ3 осуществляли, как описано в примере 3С.
Фигура 4 иллюстрирует типичную микрофотографию САМ настоящего изобретения без кровеносных сосудов в необработанном 10-дневном эмбрионе цыпленка. Данный препарат описан в примере 5В.
Фигуры 5А-5С иллюстрируют тканевое распределение интегринов β1 и αvβ3 в САМ-препарате настоящего изобретения. Фигура 5А показывает распределение субъединицы β1 в необработанном 10-дневном САМ-препарате, которую детектировали путем иммунофлуоресцентной иммунореактивности с CSAT, антителом к анти-β1. Фигура 5В показывает распределение рецептора αvβ3 в необработанном 10-дневном САМ-препарате, который детектировали путем иммунофлуоресцентной иммунореактивности с LM609, антителом к анти-αvβ3. Фигура 5С показывает распределение рецептора αvβ3 в bFGF-обработанном 10-дневном САМ-препарате, который детектировали путем иммунофлуоресцентной иммунореактивности с LM609, антителом к анти-αvβ3. Эти обработки и результаты описаны в примере 5С.
Фигура 6 иллюстрирует количественный анализ гистограммы, касающейся экспрессии αvβ3 и β1 в необработанных и bFGF-обработанных 10-дневных CAMs, как описано в примере 6А. Средняя интенсивность флуоресценции отложена на оси Y, а профили интегрина отложены на оси X.
Фигуры 7А и 7С иллюстрируют внешний вид соответственно необработанных 10-дневных САМ, bFGF-обработанных САМ и САМ, обработанных TNFα, методы и результаты которых описаны в примере 6А.
Фигуры 8А-8Е иллюстрируют эффект местной обработки антителами ангиогенеза, индуцированного bFGF, у 10-дневного САМ, как описано в примере 7А1). Фигура 8А показывает необработанный САМ-препарат, который избавлен от кровеносных сосудов. Фигура 8В показывает инфильтрацию новой сосудистой сети в область, ранее избавленную от сосудистой сети, вызванной обработкой bFGF. Фигуры 8С, 8D и 8Е показывают соответственно эффекты антител к β1 (анти-β1; CSAT), αvβ5 (анти-αvβ5; P3G2) и αvβ3 (анти-αvβ3; LM609).
Фигуры 9А-9С иллюстрируют эффект внутривенной инъекции синтетического пептида 66203 на ангиогенез, индуцированный опухолями, как описано в примере 7Е2). Фигура 9 показывает отсутствие ингибирующего эффекта внутривенной обработки контрольным пептидом (контрольный опухолевый пептид) на ангиогенез, полученного в результате индукции опухоли. Ингибирование такого ангиогенеза внутривенной инъекцией пептида 66203 (циклический RGD опухоли) показано на фигуре 9В. Отсутствие ингибирующих эффектов или цитотоксичности на зрелые предсуществующие сосуды после внутривенной инфузии пептида 66203 в области, примыкающей к данной обработанной опухолевой области, показано на фигуре 9С (циклический RGD прилегающего САМ).
Фигуры 10А-10С иллюстрируют эффект внутривенного применения моноклональных антител к фактору роста, индуцирующего ангиогенез, как описано в примере 7В1). Фигура 10А показывает ангиогенез, индуцированный bFGF, не подвергнутый обработке антителом (контроль). Ингибирования ангиогенеза не происходило, когда аналогичный препарат обрабатывали Р3G2-антителом к анти-αvβ5, как показано на фигуре 10В. Ингибирование ангиогенеза происходило при обработке LM609-антителом к анти-αvβ3, как показано на фигуре 10С.
Фигуры 11А-11С иллюстрируют эффект на эмбриональный ангиогенез после местного применения антител к антиинтегрину, как описано в примере 7С. Ангиогенез не ингибировался обработкой 6-дневного САМ соответственно антителами к анти-β1 и к анти-αvβ5, показанной на фигурах 11А и 11В. Напротив, обработка LМ609-антителом к анти-αvβ3 давала ингибирование образования кровеносного сосуда, как показано на фигуре 11С.
Фигура 12 иллюстрирует количественный анализ числа сосудов, пронизывающих опухоль в САМ-препарате, как описано в примере 7D1). График показывает число сосудов, графически отложенных на оси Y, возникающих после местного применения CSAT (анти-β1), LM609 (анти-αvβ3) или P3G2 (анти-αvβ3).
Фигуры 13A-13D иллюстрируют сравнение между сырым весом опухолей на 7-й день после обработки и исходными весами, как описано в Примере 9А1а). Каждый столбик соответствует средней
± S. E. для группы из 5-10 опухолей. Опухоли были получены от САМ-препаратов и внутривенно обработанных препаратов человеческой меланомы (M21-L) (фигура 13А), карциномы поджелудочной железы (Fg) (фигура 13В), карциномы легкого (UCLAP-3) (фигура 13С) и от карциномы гортани (НЕр3) (фигура 13D) с помощью PBS, CSAT (анти-β1) или LM609 (анти-αvβ3). Построенные графики показывают вес опухоли, отложенный на оси Y, после внутривенного применения CSAT (анти-β1), LM609 (анти-αvβ3) или PBS, что указано на оси X.
Фигуры 14А и 14В иллюстрируют гистологические срезы опухолей, обработанных с помощью P3G2 (анти-αvβ5) (фигура 14А) и LM609 (анти-αvβ3) (фигура 14В) и окрашенных гематоксилином и эозином, как описано в примере 9А1)b. Как показано на фигуре 14А, опухоли, обработанные с помощью контрольного антитела (P3G2), показали множество жизнеспособных активно делящихся опухолевых клеток, как показано на мистических фигурах (стрелка-указатель), а также множество кровеносных сосудов (линейные стрелки), пронизывающих всю опухолевую строму. Напротив, на фигуре 14В если и есть, то в опухолях, обработанных с помощью LM609 (анти-αvβ3), если и были детектированы, то только несколько опухолевых клеток или кровеносных сосудов.
Фигуры 15А-15Е относятся к М21L-опухолям, обработанным с помощью M21L, как описано в примере 9А2), и представляют собой следующее: фигура 15А, контрольный циклический RAD-пептид (69601); фигура 15В, циклический RGD-пептид (66203); фигура 15С, прилегающая САМ-ткань, взятая от одних и тех же эмбрионов, обработанных циклическим RGD-пептидом (66203) и сильно увеличенные опухоли на фигуре 15D, обработанные с помощью контрольного RAD (69601) или циклическим RGD-пептидом (66203) на фигуре 15Е. Фигура 15D изображает нормальные сосуды опухоли, обработанной контрольным пептидом RAD (69601). Фигура 15Е изображает примеры разорванных кровеносных сосудов опухоли, обработанной циклическим RGD-пептидом (66203) (линейные стрелки).
Фигуры 16А-16Е изображают ингибирование ангиогенеза с помощью антагонистов ангиогенеза в глазной кроличьей модели in vivo, как описано в примере 10. Фигура 16А и 16В изображает ангиогенез глаза кролика в присутствии bFGF и mAb P1F6 (анти-αvβ5). Фигура 16С, 16D и 16Е изображают ингибирование ангиогенеза в глазу кролика в присутствии bFGF и mAb LM609 (анти-αvβ3).
Фигура 17 изображает схему последовательности операций получения in vivo мышиной модели химеры мышь: человек, как описано в примере 11. Лоскут кожи SCID-мыши заменяли неонатанальной крайней плотью человека и давали приживляться в течение 4-х недель. После приживления трансплантата человеческую крайнюю плоть инокулировали опухолевыми клетками человека. В течение последующего 4-недельного периода определяли заметную опухоль, которая включает человеческую опухоль с человеческой сосудистой сетью, прорастающей из человеческой кожи в человеческую опухоль.
Фигура 18 иллюстрирует процентную долю одиночных клеток, полученных из СAМs, обработанных mAb и пептидом и окрашенных с помощью Арор Tag, как установлено FACS-анализом и описано в примере 12. Затемненные и заштрихованные столбики соответствуют клеткам эмбрионов, обработанных соответственно за 24 часа и 48 часов до анализа. Каждый столбик соответствует средней ± S.E. трех повторов. CAMs обрабатывали mAb LM609 (анти-αvβ3) или CSAT (анти-β1) или PBS. CAMs обрабатывали также циклическим пептидом 66203 (цикло-RGDfV, указанным в качестве Пептида 203) или контрольным циклическим пептидом 69601 (цикло-RADfV, указанным в качестве Пептида 601).
Фигуры 19А и 19В иллюстрируют объединенные результаты по суспензиям одиночных клеток CAMs эмбрионов, обработанных CSAT (анти-β1) (фигура 19А) или LM609 (анти-αvβ3) (фигура 19В), окрашенных с помощью Арор Tag и пропидиййодидом и проанализированных с помощью FACS, как описано в примере 12С. На оси Y представлены численности клеток (Апоптоз), окрашенных Арор Tag, на оси Х представлены значения окраски пропидиййодидом (содержание ДНК). Горизонтальная линия представляет ячейку для негативного окрашивания. Левая и правая секции представляют САМ-клетки эмбрионов, обработанных соответственно CSAT (анти-β1) (фигура 19А) и LM609 (анти-αvβ3) (фигура 19В). Анализ клеточного цикла осуществляли путем анализа приблизительно 8000 событий на состояние.
Фигуры 20А-20С представляют САМ-ткань CSAT-(анти-β1) обработанных эмбрионов, а фигуры 20D-20E представляют САМ-ткань LM609-(анти-αvβ3) обработанных эмбрионов, полученных, как описано в примере 12С. Фигуры 20А и 20D изображают ткани, окрашенные с помощью Арор Tag и визуализированные путем флуоресцентного (FITC) наложения на D.I.С. изображение. Фигуры 20В и 20Е изображают те же ткани, окрашенные с помощью mAb LM609 (анти-αvβ3) и визуализированные с помощью флуоресценции (родамин). Фигуры 20С и 20F представляют слитые изображения одних и тех же тканей, окрашенных с помощью Арор Tag и LM609, где солокализация представлена желтым окрашиванием. Полоса соответствует 15 и 50 мкм соответственно на левой и правой панелях.
Фигура 21 показывает результат ингибирования клеточного прикрепления при испытании с пептидом 85189, как описано в примере 4А. Эффекты пептидного антагониста оценивали в диапазоне доз от 0,001 до 100 мкМ, как отложено на оси X. Прикрепление клеток откладывали на оси Y, измеряемой оптической плотностью (O.D.) при 600 нм. Прикрепление клеток измеряли для поверхностей, покрытых витронектином (прерывистая линия) по отношению к ламинину (непрерывная линия).
Фигуры 22А и 22В показывают представленные ступенчато последовательность кДНК цыпленка ММР-2 вместе с выведенной для нее во второй линии аминокислотной последовательностью. Третья и четвертая линии показывают соответственно выведенные аминокислотную последовательность ММР-2 человека и мыши, как описано в примере 4А. кДНК-последовательность цыпленка представлена в списке SEQ ID NO 29 вместе с кодируемой аминокислотной последовательностью, которая также представлена отдельно в виде SEQ ID NO 30. Нумерация первого нуклеотида 5'-нетранслируемой области и области, кодирующей проферментную последовательность, показана на фигуре 22А в виде отрицательного числа, которое в действительности представлено в качестве номера 1 в Списке Последовательностей, создавая впечатление более длинной последней, чем на фигуре; однако данная нуклеотидная последовательность на данной фигуре идентична по длине и последовательности последовательности, которая представлена в списке последовательностей, за исключением нумерации. В соответствии с этим ссылки на позицию нуклеотида куриного или человеческого ММР-2, таких как в праймерах, для использования в амплифицируемых фрагментах ММР-2 основаны на позиции нуклеотида, как указано на фигуре, а не так, как приведено в Списке Последовательностей.
Фигура 23 показывает результаты в форме гистограммы анализа твердофазного связывания рецептора йодированного ММР-2 по связыванию с αvβ3 при наличии и без ингибиторов, как описано далее в примере 4В. Эти данные представлены графически в виде связанного СРМ на оси Y по отношению к разным потенциальным ингибиторам и контролю.
Фигура 24 показывает специфичность ММР-2-композиций, полученных от цыпленка по интегриновым рецепторам αvβ3 и αIIbβ3 в присутствии ингибиторов ММР-2, как описано далее в примере 4В. Эти данные представлены, как описано в объяснении на фигуре 23.
Фигура 25 показывает эффект куриного ММР-2 (410-637) GST-слитого белка на ангиогенез, индуцированный bFGF, как описано в примере 7А3). Фигуры 25А-В и 25C-D показывают соответственно контроль (фрагмент ММР-2, не содержащий слитый белок) и эффекты фрагмента ММР-2 GST-слитого белка.
Фигуры 26 и 27, обе иллюстрируют гистограмму индекса ангиогенеза (измерение точек ветвления) действия куриного ММР-2 (410-637) GST-слитого белка (помеченного СТММР-2) против контроля (RAP-GST или GST-RAP) на bFGF-обработанные CAMs, как описано в примере 7А3). Индекс ангиогенеза отложен на оси Y, а отдельные обработки - на оси X.
Фигура 28 показывает действие пептидов и органических соединений на ангиогенез, индуцированный bFGF, по измерению действия на точки ветвления, отложенные на оси Y, и различные обработки, отложенные на оси X, в том числе только bFGF и bFGF, обработанные CAMs пептидами 69601 или 66203 и органическими соединениями 96112, 96113 и 96229, как описано в примерах 7В и 14.
Фигура 29 графически показывает дозовый ответ пептида 85189 на ингибирование ангиогенеза, индуцированного bFGF, как описано дополнительно в примере 7В2), где число точек ветвления отложено на оси Y, а количество пептида, введенного в эмбрион, - на оси X.
Фигура 30 показывает ингибирующую активность пептидов 66203 (помеченный 203) и 85189 (помеченный 189) на ангиогенез, индуцированный bFGF, в САМ-анализе, как описано в примере 7В2). Контроли не включают пептида в bFGF-обработанных CAMS и пептида 69601 (помеченный 601). Данные представлены графически, как описано в пояснении к фигуре 27.
Фигуры 31A-L показывают эффект разных обработок против необработанных САМ-препаратов во времени, начиная от 24 ч и заканчивая 72 ч, как дополнительно описано в примере 7В3). Фотографии необработанных классов, помеченные bFGF, bFGF + MAID (bFGF-обработанные с последующим воздействием куриного ММР-2 (2-4) GST слитым белком) и соответственно bFGF + контроль (bFGF-обработка с последующим воздействием с ММР-2 (10-1), показаны на фигурах 31А-С, 31D-F, 31G-I и 31J-L.
Фигуры 32, 33 и 34 показывают соответственно снижение веса опухоли для UCLAP-3, M21-L и FgM-опухолей после внутривенного воздействия контрольного пептида 69601 и антагониста 85189, как описано дополнительно в примере 9А. Эти данные представлены графически для веса опухоли на оси Y, а пептидные обработки - на оси X.
Фигура 35 иллюстрирует эффект пептидов и антител на рост меланомной опухоли в химерной модели мышь: человек, что описано, кроме того, в примере 11В. Испытываемые пептиды включают контроль 69601 (помеченный 601) и антагонист 85189 (помеченный 189). Испытываемым антителом являлось LM609. Объем опухоли в мм3 откладывали на оси Y, а разные обработки - на оси X.
Фигуры 36А и В показывают соответственно эффект антагониста 85189 (помеченный 189) в сравнении с контрольным пептидом 69601 (помеченный 601), на уменьшение объема и сырого веса М21L-опухолей в диапазоне от 10, 50 и 250 мкг/инъекцию, как дополнительно описано в примере 11С.
Фигуры 37А и 37В показывают эффективности пептида-антагониста 85189 (обозначенный 189 и представленный непрерывной линией с зачерненными кружками) и контрольного пептида 69601 (обозначенный 601, и представленный линией точек и чистыми квадратами) ингибирования объема опухоли M21L в модели мышь: человек при двух разных режимах обработки, как дополнительно описано в примере 11С. Объем опухоли в мм3 откладывали на оси Y, а дни - на оси X.
Фигуры 38-42 схематически иллюстрируют разные химические синтезы органической молекулы αvβ3-антагонистов, как дополнительно описано в примере 13.
Фигуры 43 и 44 показывают эффекты разных органических молекул на ангиогенез, индуцированный bFGF, в САМ-анализе, как описано дополнительно в примере 14. Точки ветвления откладывали на оси Y, а разные соединения, используемые при 250 мкг/мл, - на оси X на фигуре 43, и при 100 мкг/мл - на фигуре 44.
Подробное описание настоящего изобретения
А. Определения
Аминокислотный остаток: аминокислота, полученная при химическом расщеплении (гидролизе) полипептида по его пептидным связям. Аминокислотные остатки, описанные здесь, представлены предпочтительно в "L"-изомерной форме. Однако остатки в "D"-изомерной форме могут замещать любой L-аминокислотный остаток, пока нужное функциональное свойство сохраняется данным полипептидом. NH2 относится к свободной аминогруппе, представленной на N-конце полипептида. СООН относится к свободной карбоксигруппе, представленной на С-конце полипептида. В соответствии со стандартной номенклатурой полипептидов (описанной в J. Biol. Chem., 243:3552-59 (1969) и принятой в 37 CFR 1.822(b) (2)), аббревиатура аминокислотных остатков показана в Таблице Соответствия (см. в конце описания).
Следует отметить, что все последовательности аминокислотных остатков представлены здесь формулами, чьи левая и правая ориентация находятся в обычном направлении от амино-конца к карбокси-концу. Кроме того, следует отметить, что тире в начале или в конце последовательности аминокислотных остатков указывает на пептидную связь с дополнительной последовательностью из одного или нескольких аминокислотных остатков.
Полипептид: относится к линейным группам аминокислотных остатков, соединенных с другой группой пептидными связями между альфа-аминогруппой и карбоксигруппой смежных аминокислотных остатков.
Пептид: как он используется здесь, относится к последовательным линейным соединениям не более чем около 50 аминокислотных остатков, соединенные друг с другом в виде полипептида.
Циклический пептид: относится к соединению, обладающему гетероатомной кольцевой структурой, которая включает несколько амидных связей, как в обычном пептиде. Данный циклический пептид может представлять собой замкнутый в кольцо, "голова к хвосту", линейный полипептид, в котором n-концы линейного пептида образуют амидную связь с концевым карбоксилатом линейного пептида, или он может содержать циклическую структуру, в которой данный полимер представляет собой homodetic или heterodetic и включает амидные связи и/или иные связи, чтобы закрыть данный цикл, такие как дисульфидные мостики, тиоэфиры, тиоамиды, гуанидино-группу и подобные связи.
Белок: относится к линейным последовательным соединениям свыше чем 50 аминокислотных остатков, соединенных друг с другом в полипептид.
Слитый белок: относится к полипептиду, содержащему как минимум два отличающихся домена, соединенных путем сшивки обычной пептидной связью ("слитый"), где эти два домена соответствуют пептидам, не обнаруживаемым в природе слитыми.
Синтетический пептид: относится к химически полученной цепи аминокислотных остатков, соединенных вместе пептидными связями, которые представляют собой естественно встречаемые белки и их фрагменты.
В. Общие положения
Настоящее изобретение относится в основном к обнаружению того, что ангиогенез опосредован специфическим витронектиновым рецептором αvβ3 и что ингибирующая αvβ3-функция подавляет ангиогенез. Данное открытие представляется важным вследствие той роли, которую ангиогенез играет в разных болезненных процессах. Подавляя ангиогенез, оно может вмешиваться в заболевание, улучшая данные симптомы, и в некоторых случаях излечивать данное заболевание.
Когда рост новых кровеносных сосудов вызывается или обусловлен патологией, ассоциированной с болезнью, ингибирование ангиогенеза будет снижать повреждающие эффекты данного заболевания. Примеры включают ревматоидный артрит, диабетическую ретинопатию, воспалительные заболевания, рестеноз и тому подобное. Когда рост новых кровеносных сосудов необходим для поддержания роста поврежденной ткани, ингибирование ангиогенеза будет снижать снабжение кровью данной ткани и, таким образом, содействовать уменьшению массы ткани, основанной на потребностях в снабжении кровью. Примеры включают рост опухолей, где необходима потребность в непрерывной реваскуляризации, для того чтобы данная опухоль росла на несколько миллиметров в толщину и для создания метастазов твердой опухоли.
Способы настоящего изобретения представляются эффективными отчасти потому, что данная терапия является высокоселективной для ангиогенеза, а не для других биологических процессов. Как показано в примерах, только новый сосудистый рост содержит значительный αvβ3 и по этой причине терапевтические методы не оказывают неблагоприятного действия на зрелые сосуды. Кроме того, αvβ3 не имеет широкого распространения в нормальных тканях, но скорее обнаруживаются в новых сосудах, убеждая, таким образом, что данная терапия может быть избирательно нацелена на рост нового сосуда.
Обнаружение того факта, что ингибирование одного только αvβ3 будет эффективно ингибировать ангиогенез, позволяет создать терапевтические композиции с потенциально высокой специфичностью и по этой причине с низкой токсичностью. Поэтому несмотря на то, что в настоящем изобретении описывается использование реагентов, основанных на пептиде, который обладает способностью ингибировать один или несколько интегринов, он может создавать другие реагенты, которые более селективно ингибируют αvβ3, и по этой причине не обладает побочным эффектом ингибирования иных биологических процессов, тех иных, которые не опосредованы αvβ3.
Например, как показано настоящими отличительными особенностями, возможно получение моноклональных антител, высокоселективных для иммунной реакции с αvβ3, а также селективных для ингибирования αvβ3-функции. Кроме того, RGD-содержащие пептиды могут предназначаться для селективного ингибирования αvβ3, как далее описывается здесь.
До открытий настоящего изобретения оставалось неизвестным, что ангиогенез и любой из процессов, зависимых от ангиогенеза, мог бы быть ингибирован in vivo при использовании реагентов, которые антагонистичны биологической функции αvβ3.
С. Способы ингибирования ангиогенеза
В настоящем изобретении создали способ подавления ангиогенеза в ткани и, таким образом, подавления событий в данной ткани, которые зависят от ангиогенеза. В основном данный способ включает введение в определенную ткань композиции, включающей количество антагониста αvβ3, подавляющего ангиогенез.
Как описано раньше, ангиогенез включает целый ряд процессов, вовлекающих реваскуляризацию ткани, в том числе "прорастание", образование и развитие сосудов или увеличение сосуда, все процессы ангиогенеэа, которые опосредованы и зависят от экспрессии αvβ3. За исключением заживления травматической раны, образование corpus leuteum и эмбриогенеза, полагают, что большинство процессов ангиогенеза ассоциированы с процессами заболевания, и по этой причине использование настоящих терапевтических способов являются избирательными для данного заболевания и не привносят побочных повреждающих эффектов.
Существует целый ряд заболеваний, отнесенных к так называемым ангиогенным заболеваниям, для которых ангиогенез считается важным, включающие, но не ограничиваемые воспалительными заболеваниями, такие как неиммунное воспаление, хронический суставной ревматизм и псориаз, заболевания, ассоциированные с неподходящей или несвоевременной инвазией сосудов, такие как диабетическая ретинопатия, неоваскулярная глаукома, рестеноз, пролиферация капилляров в атеросклеротические бляшки и остеопороз, и заболевания, ассоциированные с канцером, такие как солидные опухоли, метастазы солидной опухоли, ангиофибромы, ретролентальная фиброплазия, гемангиома, саркома Капоши и подобные канцеры, которые нуждаются в реваскуляризации для поддержки опухолевого роста.
Поэтому способы, которые подавляют ангиогенез в больной ткани, улучшают симптомы данного заболевания и в зависимости от данного заболевания могут способствовать излечению данного заболевания. В первом варианте осуществления настоящего изобретения предполагается подавить ангиогенез, per se, в ткани. Объем ангиогенеза в ткани и, следовательно, объем ингибирования, осуществляемый с помощью настоящих способов, можно оценить разными способами, такими, которые описаны в примерах по детектированию с помощью иммунохимии αvβ3-иммунопозитивно зрелых или нарождающихся сосудистых структур.
Как описано здесь, любая из разнообразных тканей или органов, включающих организованную ткань, может поддерживать ангиогенез при состояниях заболевания, включающих кожную, мышечную, кишечную, соединительную ткань, суставы, кости и подобную ткань, в которую при ангиогенных стимулах могут проникать кровеносные сосуды.
Поэтому в близком варианте осуществления настоящего изобретения обрабатываемая ткань представляет собой воспаленную ткань, а подавляемый ангиогенез представляет собой ангиогенез воспаленной ткани, где имеется реваскуляризация воспаленной ткани. Для этого класса способа предполагается подавление ангиогенеза в артритных тканях, таких как у пациента с хроническим суставным ревматизмом, в тканях иммунно или неиммунно воспаленных, в псориатической ткани и тому подобное.
Пациент, лечимый в настоящем изобретении, во многих вариантах его осуществления, желательно, представляет собой человека, хотя должно быть очевидно, что принципы настоящего изобретения свидетельствуют, что настоящее изобретение эффективно в отношении всех млекопитающих, которые должны быть включены в данный термин "пациент". В данном контексте подразумевается млекопитающее, которое включает любые виды млекопитающих, особенно сельскохозяйственные и домашние виды, для которых лечение болезни, ассоциированной с ангиогенезом, является желательным.
В другом близком варианте осуществления настоящего изобретения ткань для лечения представляет собой ретинальную ткань пациента с ретинальным заболеванием, таким как диабетическая ретинопатия, дегенерация желтого пятна или неоваскулярная глаукома, и ангиогенез подавляется в ретинальной ткани, где осуществляется реваскуляризация ретинальной ткани.
В дополнительном родственном варианте осуществления настоящего изобретения ткань, подвергаемая лечению, представляет собой опухолевую ткань пациента с солидной опухолью, метастазами, раком кожи, раком молочной железы, гемангиомой или ангиофибромой и подобным канцером, а подавляемый ангиогенез представляет собой ангиогенез опухолевой ткани, в которой происходит реваскуляризация опухолевой ткани. Типичные опухолевые ткани, подвергаемые лечению настоящими способами, включают легкие, поджелудочную железу, молочную железу, толстую кишку, гортань, яичники и подобные ткани. Типичный ангиогенез опухолевой ткани и его подавление описаны в нижеследующих примерах.
Подавление ангиогенеза опухолевой ткани является особенно предпочтительным вариантом осуществления настоящего изобретения, потому что реваскуляризация играет важную роль в росте опухоли. В отсутствие реваскуляризации опухолевой ткани опухолевая ткань не получает нужных питательных веществ, замедляет в росте, прекращает дополнительный рост, регрессирует и одновременно становится некротической в результате гибели опухоли.
Иными словами, в настоящем изобретении разработали способ ингибирования опухолевой реваскуляризации путем подавления опухолевого ангиогенеза в соответствии с настоящими способами. В настоящем изобретении создали также способ ингибирования опухолевого роста путем применения способов подавления ангиогенеза.
Данные способы особенно эффективны против образования метастазов, потому что (1) их образование требует реваскуляризации первичной опухоли для того, чтобы метастатические раковые клетки могли существовать в первичной опухоли и (2) их образование во вторичном участке требует реваскуляризации для поддержания роста этих метастазов.
В родственном варианте осуществления настоящего изобретения предполагается применение способа в соединении с другими терапиями, такими как традиционная химиотерапия, направленная против солидных опухолей и для контроля за образованием метастазов. Введение ингибитора ангиогенеза обычно проводят в течение или после химиотерапии, хотя предпочтительно ингибировать ангиогенез после режима химиотерапии в периоды, когда опухолевая ткань будет отвечать на токсическую атаку индукцией ангиогенеза для восстановления временного кровоснабжения и снабжения питательных веществ опухолевой ткани. Кроме того, предпочтительно применять способы ингибирования ангиогенеза после хирургии, когда солидная опухоль была удалена, в качестве профилактики против метастазов.
Поскольку настоящие способы применимы для ингибирования опухолевой реваскуляризации, данные способы можно также применить для ингибирования роста опухолевой ткани, для ингибирования образования опухолевых метастазов и для регрессии образовавшихся опухолей. В нижеследующих примерах демонстрируется регрессия образовавшейся опухоли после однократного введения αvβ3-антагониста настоящего изобретения.
Рестеноз представляет собой процесс миграции клетки гладкой мускулатуры и пролиферацию на подкожном участке просвечивающей ангиопластики, что затрудняет успешную ангиопластику. Эта миграция и пролиферация SMC's во время рестеноза может считаться процессом ангиогенеза, который подавлен настоящими способами. По этой причине в настоящем изобретении в соответствии с настоящими способами подавление рестеноза рассматривается также через подавление ангиогенеза у пациента после процедур по ангиопластике. Для ингибирования рестеноза αvβ3-антагонист обычно вводят после операции ангиопластики в течение от около 2 до около 28 дней, но более обычно в течение первых 14 дней после данной операции.
Настоящий способ по ингибированию ангиогенеза в ткани и, следовательно, применения также способов лечения заболеваний, связанных с ангиогенезом, включает контактирование ткани, в которой происходит ангиогенез, или существует риск его осуществления, с композицией, включающей терапевтически эффективное количество αvβ3-антагониста, способного ингибировать связывание αvβ3 с его естественным лигандом. Поэтому данный способ включает введение пациенту терапевтически эффективного количества физиологически толерантной композиции, содержащей αvβ3-антагонист настоящего изобретения.
Диапазон доз для введения αvβ3-антагониста зависит от формы данного антагониста и его силы, как описано здесь далее, и представляет собой достаточно большие количества для получения требуемого эффекта, в котором ангиогенез и симптомы болезни, опосредованные ангиогенезом, уменьшены. Данная дозировка не должна быть больше той, которая не вызывает неблагоприятные побочные эффекты, такие как синдром повышенной вязкости, отек легких, застойная сердечная недостаточность и тому подобное. Вообще, данная дозировка должна варьировать у пациента с возрастом, состоянием, полом и продолжительностью заболевания и может быть определена специалистом в данной области. Эту дозировку может откорректировать отдельный врач для случая любой сложности.
Терапевтически эффективное количество представляет собой количество αvβ3-антагониста, достаточное для получения измеряемого подавления ангиогеназа в ткани, которую лечат, т.е. количество, подавляющее ангиогенез. Ингибирование ангиогенеза может быть измерено in situ с помощью иммуногистохимии, как описано здесь, или с помощью других методов, известных специалисту в данной области.
Поскольку αvβ3-антагонист может имитировать форму αvβ3, RGD-содержащего пептида, моноклонального антитела к анти-αvβ3 или его фрагмента, следует принять во внимание, что сила и, следовательно, проявление "терапевтического эффективного" количества может варьировать. Однако, как показано настоящими испытываемыми способами, специалист в данной области может легко оценить потенцию αvβ3-антагониста - кандидата настоящего изобретения.
Потенция αvβ3-антагониста может быть измерена различными средствами, включающими ингибирование ангиогенеза в САМ-опыте, в опыте in vivo кроличьего глаза, в анализе in vivo химеры мышь: человек и путем измерения ингибирования связывания естественного лиганда с αvβ3, которые все описаны здесь, и в подобных опытах.
Предпочтительный αvβ3-антагонист обладает способностью фактически ингибировать связывание естественного лиганда, такого как фибриноген или витронектин, с αvβ3 в растворе при концентрациях антагониста менее чем 0,5 микромолей (мкМ) предпочтительно менее чем 0,1 мкМ и более предпочтительно менее чем 0,05 мкМ. "Фактически" означает, что как минимум 50 процентов снижения связывания фибриногена наблюдается при ингибировании в присутствии αvβ3-антагониста, а 50% ингибирования рассматривается здесь в качестве значения IC50.
Более предпочтительный αvβ3-антагонист проявляет селективность в отношении αvβ3, помимо других интегринов. Поэтому предпочтительный αvβ3-антагонист фактически подавляет связывание фибриногена с αvβ3, но практически не подавляет связывания фибриногена с другим интегрином, таким как αvβ1, αvβ5 или αIIbβ3. Особенно предпочтительным является αvβ3-антагонист, который проявляет активность в 10 раз - 100 раз ниже IC50-aктивности при подавлении связывания фибриногена с αvβ3 в сравнении с IC50-активностью при ингибировании связывания фибриногена с другим интегрином. Типичные опыты по измерению IC50-активности в подавлении связывания фибриногена с интегрином описаны в нижеследующих примерах.
Терапевтически эффективное количество αvβ3-антагониста настоящего изобретения в форме моноклонального антитела обычно представляет собой такое количество, что при введении в физиологически толерантную композицию является достаточным для достижения в плазме концентрации от около 0,01 микрограмма (мкг) на миллилитр (мл) до около 100 мкг/мл, предпочтительно от около 1 мкг/мл до около 5 мкг/мл, но обычно около 5 мкг/мл. По-разному установленная, данная дозировка может варьировать от около 0,1 мг/кг до около 300 мг/кг, предпочтительно от около 0,2 мг/кг до около 200 мг/кг, наиболее предпочтительно от около 0,5 мг/кг до около 20 мг/кг в однократной или большей ежедневной дозе для введения, в течение одного или нескольких дней.
Когда данный антагонист представлен в форме фрагмента моноклонального антитела, данное количество можно легко адаптировать на основе массы данного фрагмента относительно массы целого антитела. Предпочтительная концентрация в плазме в молярности составляет от около 2 микромолей (мкМ) до около 5 миллимолей (мМ), но предпочтительно около 100 мкМ до 1 мМ антитела-антагониста.
Терапевтически эффективное количество αvβ3-антагониста данного изобретения в форме полипептида или другой подобной по размеру небольшой молекулы, имитирующей αvβ3, является обычно количеством полипептида, такого, который при введении в физиологически толерантную композицию достаточен для достижения в плазме концентрации от около 0,1 мкг/мл до около 200 мкг/мл, предпочтительно от около 1 мкг/мл до около 150 мкг/мл. На основе полипептида, обладающего массой около 500 граммов на моль, предпочтительная концентрация в плазме, в молярности, равна от около 2 мкМ до около 5 мМ, но предпочтительна около 100 мкМ до 1 мМ полипептидного антагониста. Различно установленная, дозировка на вес тела может варьировать от около 0,1 мг/кг до около 300 мг/кг, но предпочтительно от около 0,2 мг/кг до около 200 мг/кг в однократной или более ежедневной дозе введения в течение одного или нескольких дней.
Моноклональные антитела или полипептиды настоящего изобретения могут быть введены парентерально с помощью инъекции или постепенно во времени инфузии. Хотя данная подвергаемая лечению ткань обычно может быть доступной в данном организме в результате системного введения и, следовательно, более обычно подвергается лечению путем внутривенного введения терапевтических композиций, предполагаются другие ткани и средства доставки, где существует вероятность того, что данная мишенированная ткань содержит данную молекулу-мишень. Поэтому моноклональные антитела или полипептиды настоящего изобретения могут быть введены внутривенно, внутрибрюшинно, внутримышечно, подкожно, внутриполостно, трансдермально и могут быть доставлены с помощью перистальтических средств.
Терапевтические композиции, содержащие моноклональное антитело или полипептид данного изобретения, традиционно вводят внутривенно, например в виде инъекции стандартной дозы. Термин "стандартная доза", используемый в отношении терапевтической композиции в настоящем изобретении, относится к физически дискретным единицам, пригодным для субъекта в качестве унифицированной дозировки, содержащие в каждой единице заранее определенное количество активного вещества, вычисленного для получения требуемого терапевтического эффекта в сочетании с требуемым разбавителем; т.е., переносчиком или носителем.
Как показано в примерах, в первом предпочтительном варианте осуществления настоящего изобретения αvβ3-антагонист вводили внутривенно однократной дозой.
Данные композиции вводили способом, сопоставимым с дозировкой препарата, и в терапевтически эффективном количестве. Количество и время введения зависит от пациента, подвергаемого лечению, системной вместимости пациента для использования данного активного ингредиента и уровня требуемого терапевтического эффекта. Точные количества активного ингредиента, необходимые для введения, зависят от мнения практикующего врача и специфики каждого индивидуума. Однако подходящие дозовые диапазоны для системного применения, описанные здесь, зависят от пути введения. Подходящие режимы для введения также изменяются, но типируются в результате первоначального введения с последующими повторными дозами с интервалами в один или несколько часов в последней инъекции или иного введения. Альтернативно, непрерывная внутривенная инфузия достаточна для поддержания концентраций в крови в ряду предполагаемых точно указанных терапий in vivo.
Как показано в представленных примерах, ингибирование ангиогенеза и опухолевой регрессии происходит уже через 7 дней после первоначального контактирования с антагонистом. Дополнительное или пролонгированное воздействие антагониста предпочтительно в течение 7 дней - 6 недель, предпочтительно около 14-28 дней.
В родственном варианте осуществления настоящего изобретения, представленные примеры показывают взаимосвязь между ингибированием αvβ3 и индукцией апоптоза в реваскуляризованных клетках, несущих αvβ3. Поэтому в настоящем изобретении рассматриваются также способы ингибирования апоптоза в несосудистой системе ткани. Данный способ применяли фактически, как описано здесь для ингибирования ангиогенеза во всех тканях и состояниях. Имеется только заметное различие эффекта во времени, то есть, что апоптоз проявляется быстро, обычно около 48 часов после контактирования антагониста, а ингибирование ангиогенеза и опухолевая регрессия проявляются более медленно, как описано здесь. Это различие сказывается на терапевтическом режиме во временных сроках введения и требуемом эффекте. Обычно управление апоптоза новой сосудистой сетью может осуществляться в течение от 24 часов до около 4 недель, хотя срок 48 часов - 7 дней является предпочтительным.
D. Терапевтические композиции
Представленное изобретение рассматривает терпевтические композиции, полезные для практического применения терапевтических способов, описанных здесь. Терапевтические композиции представленного изобретения содержат физиологически толерантный переносчик вместе, как описано здесь, с αvβ3-антагонистом, растворенным или диспергированным в нем в качестве активного ингредиента. В предпочтительном варианте осуществления настоящего изобретения терапевтическая композиция αvβ3-антагониста при введении млекопитающему или человеку в терапевтических целях не является иммуногенной.
Термины "фармацевтически приемлемый", "физиологически толерантный", которые используются здесь, и их грамматические вариации, поскольку они относятся к композициям, носителям, разбавителям и реагентам, используются взаимозаменяемо и представляют собой вещества, которые при введении млекопитающему не способны производить нежелательные физиологические эффекты, такие как тошнота, головокружение, расстройство желудка и тому подобное.
Получение фармакологической композиции, которая содержит активные ингредиенты, растворенные или диспергированные в ней, хорошо известны в данной области и не ограничиваются составом. Обычно такие композиции получают в виде инъецируемых жидких растворов или суспензий, однако твердые формы подходят для растворения или суспендирования и перед использованием могут быть получены в жидкой форме. Данный препарат может быть также эмульгированным.
Данный активный ингредиент может быть смешан с наполнителями, которые фармакологически приемлемы и совместимы с данным активным ингредиентом в количествах, подходящих для использования в описанных здесь терапевтических способах. Подходящими наполнителями, например, являются вода, солевой раствор, декстроза, глицерол, этанол или им подобные и их сочетания. Кроме того, данная композиция может содержать, по желанию, минорные количества вспомогательного вещества, такие как увлажняющие или эмульгирующие агенты, буферные агенты и тому подобное, которые усиливают эффективность данного активного ингредиента.
Терапевтическая композиция настоящего изобретения может включать фармацевтически приемлемые соли ее компонентов. Фармацевтически приемлемые соли включают кислые аддитивные соли (образованные свободными аминогруппами определенного полипептида), которые образуются с неорганическими солями, такими как, например, хлористоводородная или фосфорная кислоты, или такими органическими кислотами, как уксусная, виннокаменная, миндальная и тому подобное. Соли, образованные с помощью свободных карбоксильных групп, могут быть произведены из неорганических оснований, таких как, например, натриевый, калиевый, аммониевый, кальциевый гидроксиды или гидроксид трехвалентного железа, и таких органических оснований, как изопропиламин, триметиламин, 2-этиламиноэтанол, гистидин, прокаин и тому подобное.
Особенно предпочтительными являются соли TFA и НСl при использовании получения циклических полипептидных αvβ3-антагонистов. Типичные соли пептидов описаны в нижеследующих примерах.
Физиологически толерантные переносчики хорошо известны в данной области. Типичные водные переносчики представляют собой стерильные водные растворы, которые не содержат веществ в дополнение к активным ингредиентам и воде или содержат буфер, такой как натрийфосфатный при физиологическом значении рН, физиологический солевой раствор или оба, такой как фосфатно-солевой буферный раствор. Более того, водные переносчики могут содержать более чем одну буферную соль, а также такие соли, как натрий и калийхлориды, декстрозу, полиэтиленгликоль и другие растворенные вещества.
Жидкие композиции могут также содержать дополнительные жидкие фазы и исключающие воду. Типичным представителем таких дополнительных жидких фаз является глицерин, растительные масла, такие как хлопковое масло, и водно-масляные эмульсии.
Терапевтическая композиция содержит ингибирующее ангиогенез количество αvβ3-антагониста настоящего изобретения, составленного обычно из антагониста в количестве менее 0,1 вес.% на вес общей терапевтической композиции. Весовой процент представляет собой отношение веса ингибитора к общей композиции. Поэтому, например, 0,1 весовой процент представляет собой 0,1 г ингибитора на 100 г тотальной композиции.
Е. Антагонисты интегрина αvβ3
αvβ3-антагонисты используются в настоящих способах для ингибирования ангиогенеза в тканях и могут принимать разнообразные формы, которые включают соединения, которые взаимодействуют с αvβ3 таким образом, чтобы вмешиваться в функциональные взаимодействия с естественными αvβ3-лигандами. Типичные
представители антагонистов включают аналоги αvβ3, полученные из лигандсвязывающего участка в αvβ3, миметики либо из αvβ3, либо из естественного лиганда αvβ3, которые имитируют структурную область, участвующую в αvβ3-лигандсвязывающих взаимодействиях, пептиды, обладающие последовательностью, соответствующей функциональному связывающему домену естественного лиганда, специфичного для αvβ3, соответствующего, в частности, RGD-содержащему домену естественного лиганда αvβ3 и антитела, которые иммунно реагируют либо с αvβ3, либо с естественным лигандом, которые все, как здесь указано, проявляют антагонистическую активность.
1. Полипептиды
В первом варианте осуществления настоящего изобретения рассматриваются αvβ3-антагонисты в форме полипептидов. Полипептидный (пептидный) αvβ3-антагонист может обладать характеристиками последовательности либо естественного лиганда αvβ3, либо самого αvβ3 в области, участвующей в αvβ3-лигандном взаимодействии, и проявляет, как описано здесь, активность αvβ3-антагониста. Предпочтительный пептидный αvβ3-антагонист содержит RGD-трипептид и соответствует последовательности естественного лиганда по RGD-содержащей области.
Предпочтительные RGD-содержащие полипептиды обладают последовательностью, соответствующей последовательности аминокислотных остатков из RGD-содержащей области естественного лиганда αvβ3, такого как фибриноген, витронектин, фактор фон Виллебранда, ламинин, тромбоспондин и подобных лигандов. Последовательности этих αvβ3-лигандов хорошо известны. Поэтому пептидный αvβ3-антагонист можно получить из любого естественного лиганда, хотя предпочтительными являются фибриноген и витронектин.
Особенно предпочтительный пептидный αvβ3-антагонист предпочтительно ингибирует связывание αvβ3 с его естественным лигандом(ами) при сравнении с другими интегринами, которые описаны раньше. Эти αvβ3-специфичные пептиды особенно предпочтительны, по крайней мере, потому, что специфичность по αvβ3 снижает появление нежелательных побочных эффектов, таких как ингибирование других интегринов. Выявление предпочтительных пептидных αvβ3-антагонистов, обладающих селективностью по αvβ3, можно легко осуществить в обычном ингибирующем анализе связывания, таком как ИФА, описанный в нижеследующих примерах.
Полипептид настоящего изобретения обычно включает не более чем около 100 аминокислотных остатков, предпочтительно не более чем около 60 остатков, более предпочтительно - не более чем около 30 остатков. Пептиды могут быть линейными или циклическими, но особенно предпочтительными пептидами являются циклические.
Когда данный полипептид равен более чем 100 остаткам, он обычно создается в форме слитого белка или белкового фрагмента, как описано здесь.
Предпочтительные циклические и линейные пептиды и их обозначения показаны в таблице 1 в нижеследующих примерах.
Следует иметь в виду, что данный оригинальный полипептид не нуждается в идентичности с последовательностью аминокислотных остатков αvβ3-естественного лиганда, поскольку он включает данную требуемую последовательность и способен функционировать в качестве αvβ3-антагониста в опыте, таком как опыты, описанные здесь.
Данный оригинальный полипептид включает любой аналог, фрагмент или химическое производное полипептида, чья последовательность аминокислотных остатков показана здесь, поскольку данный полипептид представляет собой αvβ3-антагонист. По этой причине настоящий полипептид может быть подвергнут различным изменениям, замещениям, вставкам и делениям, когда такие изменения создают определенные преимущества для его использования. В этом отношении полипептидный αvβ3-антагонист настоящего изобретения соответствует, а не идентичен последовательности изложенного пептида, в котором осуществили одно или несколько изменений и он сохраняет способность к функционированию в качестве αvβ3-антагониста в одном или нескольких опытах, которые указаны здесь.
Поэтому полипептид может находиться в любой из различных форм пептидных производных, которые включают амиды, конъюгаты с белками, циклические пептиды, полимеризованные пептиды, аналоги, фрагменты, химически модифицированные пептиды и подобные производные.
Термин "аналог" включает полипептид, обладающий последовательностью аминокислотных остатков, фактически идентичных последовательности, конкретно показанной здесь, в которой один или несколько остатков были консервативно замещены на функционально подобный остаток, который проявляет активность αvβ3-антагониста, как описано здесь. Примеры консервативных замен включают замену неполярного (гидрофобного) остатка, такого как изолейцин, валин, лейцин или метионин на другой, замену полярного (гидрофильного) остатка на другой, такую как между аргинином и лизином, между глутамином и аспарагином, между глицином и серином, замену одного основного остатка, такого как лизин, аргинин или гистидин, такого как кислотный остаток, такой как аспарагиновая кислота или глутаминовая кислота, на другой.
Выражение "консервативная замена" включает также применение химически произведенного остатка вместо непроизведенного остатка, созданного так, чтобы такой полипептид проявлял требующуюся ингибирующую активность.
"Химически производный" относится к оригинальному полипептиду, обладающему одним или несколькими остатками, химически произведенными в реакции с функциональной боковой группой. Кроме того, для производных боковой группы химическое производное может обладать одной или несколькими модификациями основной цепи, включающими α-аминозамены, такие как N-метил, N-этил, N-пропил и тому подобное, и α-карбонильные замены, такие как тиоэфир, тиоамид, гуанидино и тому подобное. Такие производные молекулы включают, например, производные молекулы, в которых были произведены свободные аминогруппы для образования аминных гидрохлоридов, p-толуолсульфонильных групп, карбобензокси-групп, трет-бутилоксикарбонильных групп, хлороацетильных групп или формильных групп. Свободные карбонильные группы могут быть произведены для образования солей, метилового и этилового эфиров или эфиров другого вида или гидразидов. Свободные гидроксильные группы могут быть произведены для образования O-ацил- или O-алкил-производных. Имидазольный азот гистидина может быть произведен для образования N-имбензилгистидина. В качестве производных включены также пептидные производные, которые содержат один или несколько естественно встречаемых аминокислотных производных двадцати обычных аминокислот. Например, 4-гидроксипролин может быть заменен на пролин; 5-гидроксилизин может быть заменен на лизин; 3-метилгистидин может быть заменен на гистидин; гомосерин может быть заменен на серин; и орнитин может быть аменен на лизин. Полипептиды настоящего изобретения включают также любой полипептид, обладающий одной или несколькими добавками и/или делениями или остатками, связанными с последовательностью полипептида, последовательность которого показана здесь, поскольку поддерживается нужная активность.
Особенно предпочтительным производным является циклический пептид в соответствии с формулой цикло(Arg-Gly-Asp-D-NMeVal), сокращенно с(RGDf-NMeV), в которой имеется N-метилзамещенная α-аминогруппа по валиновому остатку данного пептида, а циклизация соединила первоначальные амино- и карбоксиконцы данного пептида.
Термин "фрагмент" относится к любому оригинальному полипептиду, обладающему последовательностью аминокислотных остатков более короткой, чем последовательность аминокислотных остатков, которая показана здесь.
Когда полипептид настоящего изобретения обладает последовательностью, которая не идентична последовательности αvβ3 естественного лиганда, это обычно происходит потому, что осуществляются одна или несколько консервативных или неконсервативных замен, обычно не более чем около 30 числовых процентов и предпочтительно не более чем 10 числовых процентов замен аминокислотных остатков. Дополнительные остатки могут быть также добавлены на конце полипептида с целью создания "линкера", с помощью которого полипептиды данного изобретения могут быть традиционно прикреплены к метке, или твердой, матрице или носителю.
Метки, твердые матрицы и носители, которые могут использоваться с полипептидами данного изобретения описаны здесь ниже.
Аминокислотные остатки-линкеры обладают, как правило, хотя бы одним остатком и могут быть представлены 40 или более остатками, чаще всего 1-10 остатками, но не образуют αvβ3-лигандных эпитопов. Обычными аминокислотными остатками, используемыми для связывания, являются тирозин, цистеин, лизин, глутаминовая и аспарагиновая кислоты или подобные. Кроме того, оригинальный полипептид может отличаться, если не оговорено особо, от естественной последовательности αvβ3-лиганда вследствие модификации последовательности, которая ацилирована по NН2-концу, например, ацетилированием или амидированием тиогликолевой кислоты, путем концевого карбоксиламидирования, например, с помощью аммиака, метиламина и подобными концевыми модификациями. Как хорошо известно, концевые модификации полезны для снижения чувствительности к протеиназному расщеплению и, следовательно, служат для пролонгирования времени полужизни данных полипептидов в растворах, особенно в биологических жидкостях, где могут присутствовать протеазы. В этом отношении полипептидная циклизация также полезна для концевой модификации, но особенно предпочтительна также потому, что с помощью циклизации образуются стабильные структуры и с точки зрения наблюдаемых биологических активностей таких циклических пептидов, которые описаны здесь.
Любой пептид настоящего изобретения может использоваться в форме фармацевтически приемлемой соли. Подходящие кислоты, которые способны образовывать соли с данными пептидами настоящего изобретения, включают неорганические кислоты, такие как трифторуксусная кислота (TFA), хлористоводородная кислота (НСl), бромистоводородная кислота, хлорная кислота, азотная кислота, тиоциановая кислота, серная кислота, метансульфокислота, уксусная кислота, фосфорноуксусная кислота, пропионовая кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, малоновая кислота, янтарная кислота, малеиноая кислота, фумаровая кислота, антраниловая кислота, коричная кислота, нафтилсульфоновая кислота, сульфаниловая кислота и тому подобное. Особенно предпочтительными являются соли НСl и TFA.
Подходящие основания, способные образовывать соли с пептидами настоящего изобретения, включают неорганические основания, такие как гидроксид натрия, гидроксид аммония, гидроксид калия и тому подобное; и органические основания, такие как моно-, ди- и триалкил и ариламины (например, триэтиламин, диизопропиламин, метиламин, диметиламин и тому подобное) и факультативно замещенные этаноламины (например, этаноламин, диэтаноламин и тому подобное).
Кроме того, пептид данного изобретения может быть получен, как описано в нижеследующих примерах, без включения свободной ионной соли, в которой заряженные кислотная или основная группы, присутствующие в боковых группах аминокислотного остатка (например, Arg, Asp и тому подобное), ассоциируют и нейтрализуют друг друга с образованием соединения "внутренняя соль".
Пептид настоящего изобретения, также указанный здесь в качестве оригинального полипептида, может быть синтезирован любым методом, который известен специалистам в области полипептидов, включая методы рекомбинантной ДНК. Методы синтетической химии, такие как твердофазный метод синтеза Merrifield, являются предпочтительными по причинам очистки, антигенной специфичности, отсутствия нежелательных побочных продуктов, легкостью получения и тому подобное. Превосходное доступное краткое изложение многих методов можно найти у Steward с соавт., "Solid Phase Peptide Synthesis", W.H. Freeman Co., San Francisco, 1969; Bodanszky с соавт., "Peptide Synthesis", John Wiley & Sons, Second Edition, 1976; J. Meienhofer, "Hormonal Proteins and Peptides", Vol. 2, p. 46, Academic Press (New York), 1983; Merrifield, Adv. Enzymol., 32: 221-96, 1969; Fields с соавт., Int. J. Peptide Protein Res., 35:161-214, 1990; патент Соединенных Штатов 4244946 по твердофазному пептидному синтезу; Schroder с соавт., "The Peptides", Vol. 1, Academic Press (New York), 1965 по классическому синтезу в растворе, каждый из которых включен здесь путем ссылки. Подходящие защитные группы, полезные для такого синтеза, описаны в вышеприведенных текстах и в J.F.W. McOmie "Protective Groups in Organic Chemistry", Plenum Press, New York, 1973, который включен здесь путем ссылки.
Вообще, рассматриваемые методы твердофазного синтеза включают последовательное добавление одного или нескольких аминокислотных остатков или подходящих защищенных аминокислотных остатков к растущей пептидной цепи. Обычно амино- или карбоксильная группа первого аминокислотного остатка защищается подходящей селективно удаляемой блокирующей группой. Другая селективно удаляемая защитная группа используется для аминокислот, содержащих реакционную боковую группу, такую как лизин.
Используя в качестве типичного твердофазного синтеза, защищенную или произведенную аминокислоту присоединяют к инертной твердой подложке через ее незащищенную карбоксильную или аминогруппу. Затем защитная группа из амино- или карбоксильной группы селективно удаляется и добавляется следующая аминокислота в данной последовательности, обладающая комплементарной (амино- или карбоксильной) группой, соответственно защищенной, и реагирует в условиях, подходящих для образования амидной связи с остатком, уже присоединенным к твердой подложке. Затем данная защитная группа из амино- или карбоксильной группы удаляется из этого вновь добавленного аминокислотного остатка и затем добавляется следующая аминокислота (соответственно защищенная) и так далее. После соединения всех требуемых аминокислот в желаемую последовательность все остающиеся концевые и боковые защитные группы (и твердая подложка), последовательно или одновременно удаляются для получения конечного линейного полипептида.
Результирующие линейные полипептиды, полученные, например, как описано выше, могут реагировать с образованием соответствующих им циклических пептидов. Типичный метод получения циклического пептида описан у Zimmer с соавт. , Peptides 1992, pp. 393-394, ESCOM Science Publishers, B.V., 1993. Обычно метиловый эфир пептида, защищенный третбутоксикарбонилом, растворяли в метаноле и добавляли раствор гидроксида натрия и данную смесь инкубировали при 20oС для гидролитического удаления метилэфирной защитной группы. После выпаривания растворителя пептид, защищенный третбутоксикарбонилом, экстрагировали этилацетатом из подкисленного водного растворителя. Затем данную третбутоксикарбонильную защитную группу удаляли в мягких кислотных условиях в диоксановом растворителе. Полученный незащищенный линейный пептид со свободными амино- и карбоксиконцами преобразовывали в соответствующий циклический пептид путем взаимодействия разбавленного раствора линейного пептида в смеси из дихлорметана и диметилформамида с дициклогексилкарбодиимидом в присутствии 1-гидроксибензотриазола и N-метилморфолина. Затем этот полученный циклический пептид выделяли очисткой с помощью хроматографии.
Альтернативные методы для циклического пептидного синтеза описаны у Gurrath с соавт. , Eur. J. Biochem., 210-911-921 (1992) и описаны в нижеследующих примерах.
Кроме того, αvβ3-антагонист может быть создан в форме слитого белка. Слитые белки представляют собой белки, получаемые с помощью методов рекомбинантной ДНК, как описано здесь, в которых оригинальный полипептид экспрессируется в виде слитого со вторым белком-носителем, таким как глутатионсульфгидрилтрансфераза (GST), или с другим хорошо известным носителем. Предпочтительные слитые белки включают описанный здесь полипептид ММР-2. Получение ММР-2-слитого белка описано в нижеследующих примерах.
Особенно предпочтительные пептиды и производные пептиды для использования в настоящих способах включают c-(GrGDFV) (SEQ ID NO 4), c-(RGDfV) (SEQ ID NO 5), c-(RADfV) (SEQ ID NO 6), c-(RGDFv) (SEQ ID NO 7), с-(RGDf-NMeV) (SEQ ID NO 15) и линейный пептид YTAECKPQVTRGDVF (SEQ ID NO 8), где "с" указывает циклический пептид, заглавные буквы представляют собой одиночный буквенный код для L-аминокислоты, а строчные буквы представляют собой одиночный буквенный код для D-аминокислоты. Последовательность аминокислотных остатков этих пептидов показана соответственно также в SEQ ID NOs 4, 5, 6, 7, 15 и 8.
Предпочтительными также являются полипептиды, произведенные из ММР-2, описанного здесь, обладающего последовательностями, показанными в SEQ ID NОs 17-28 и 45.
2. Моноклональные антитела
Настоящее изобретение описывает в первом варианте осуществления αvβ3-антагонисты в форме моноклональных антител, которые иммунно взаимодействуют с αvβ3 и подавляют, как описано здесь, связывание αvβ3 с его естественным лигандом. В настоящем изобретении описаны также клеточные линии, которые продуцируют антитела, способы получения клеточных линий и способы получения моноклональных антител.
Моноклональное антитело настоящего изобретения включает молекулы антитела, которые 1) иммунно реагируют с выделенным αvβ3 и 2) ингибируют связывание фибриногена с αvβ3. Предпочтительные моноклональные антитела, которые предпочтительно связываются с αvβ3, включают моноклональное антитело, обладающее иммунореактивными характеристиками mAb LM609, продуцируемого гибридомной клеточной линией АТСС НВ 9537. Данная гибридомная клеточная линия АТСС НВ 9537 была депонирована в соответствии с требованиями Будапештского Договора в Американской Коллекции Типовых Культур (АТСС), 1301 Parklawn Drive, Rockville, MD, США, 15 сентября 1987 г.
Термин "антитело или антительная молекула" используется здесь в разных грамматических формах в качестве собирательного имени существительного, которое относится к популяции иммуноглобулиновых молекул и/или иммунологически активным частям иммуноглобулиновых молекул, т.е. молекулам, которые содержат антительный объединяющий сайт или паратоп.
"Участок антитела для связывания" представляет собой структурную часть антительной молекулы, включающей тяжелую и легкую цепь вариабельного и гипервариабельного участков, которые специфически связывают антиген.
Типичными представителями антител, используемыми в настоящем изобретении, являются интактные иммуноглобулиновые молекулы, в значительной степени интактные иммуноглобулиновые молекулы и те части иммуноглобулиновой молекулы, которые содержат паратоп, в том числе те части, которые известны в данной области в качестве Fab, Fab', F(аb')2 и F(v), а также отнесенные к антительным фрагментам.
Во втором предпочтительном варианте осуществления настоящего изобретения рассматривается усеченная иммуноглобулиновая молекула, включающая Fab-фрагмент, полученный из моноклонального антитела данного изобретения. Fab-фрагмент, не имеющий Fc-рецептора, является растворимым и дает терапевтические преимущества для времени полужизни в сыворотке и диагностическое преимущество способам, использующим растворимый Fab-фрагмент. Получение растворимого Fab-фрагмента хорошо известно в области иммунологии и может быть выполнено разными способами.
Например, Fab и F(ab')2 части (фрагменты) антител получали с помощью протеолитической реакции соответственно папаина и пепсина, по существу интактных антител, хорошо известными способами. Смотрите, например, патент США 4342566 Theofilopolous и Dixon. Fаb'-антительная часть тоже хорошо известна и ее получают из F(ab')2 частей с последующим восстановлением дисульфидных связей, соединяющих две части тяжелой цепи, с помощью меркаптоэтанола и с последующим алкилированием полученного белкового меркаптана с таким реагентом, как йодацетамид. Антитела, содержащие интактные иммуноглобулиновые молекулы, являются предпочтительными и используются здесь в качестве иллюстративных.
Выражение "моноклональное антитело" и его различные грамматические формы относятся к популяции антительных молекул, которые содержат только один из видов участка антитела для связывания, способного к иммунному взаимодействию с отдельным эпитопом. Поэтому моноклональное антитело обычно проявляет одиночное связывание по сродству для любого эпитопа, с которым оно иммунно взаимодействует. Моноклональное антитело может поэтому содержать антительную молекулу, обладающую множеством участков для связывания, каждое иммуноспецифическое для отдельного эпитопа, например, диспецифичное моноклональное антитело.
Моноклональное антитело обычно составлено из антител, полученных в клонах единственной клетки, называемой гибридомой, которая секретирует (продуцирует) только один вид антительной молекулы. Гибридомная клетка образуется путем слияния клетки, продуцирующей антитело, и миеломы или другой бессменной клеточной линии. Получение таких антител впервые было описано Kohler и Milstein, Nature 256:495-497 (1975), описание которого включено здесь путем ссылки. Дополнительные методы описаны у Zola, Monoclonal Antibodies: A Manual of Techniques, CRC Press, Inc. (1987). Гибридомные супернатанты, полученные таким образом, можно проскринировать в присутствии антительных молекул, которые иммунно реагируют с αvβ3, и для ингибирования связывания αvβ3 с естественными лигандами.
Вкратце, для образования гибридомы, из которой получают композицию моноклонального антитела, миелому или другую бессменную клеточную линию сливали с лимфоцитами, полученными из селезенки млекопитающего, гипериммунизированного источником αvβ3, таким как αvβ3, выделенным из меланомных клеток М21 человека, как описано у Cheresh с соавт., J. Biol. Chem., 262:17703-17711 (1987).
Предпочтительно, чтобы данная миеломная клеточная линия, используемая для получения гибридомы, была бы от тех же видов, что и лимфоциты. Обычно предпочтительным млекопитающим является мышь штамма 129 GIX+. Подходящие мышиные миеломы, используемые в настоящем изобретении, включают гипоксантинаминоптерин-тимидин-чувствительные (HAT) клеточные линии Р3Х63-Аg8.653 и Sр2/0-Аg14, которые доступны от Американской Коллекции Типовых Культур, Rockville, MD соответственно под обозначениями CRL 1580 и CRL 1581.
Спленоциты обычно сливали с миеломными клетками, используя полиэтиленгликоль (PEG) 1500. Гибриды слияния отбирали по их чувствительности к HAT. Гибридомы, продуцирующие моноклональное антитело данного изобретения, идентифицировали с использованием твердофазного иммуноферментного анализа (ELISA), описанного в нижеследующих примерах.
Моноклональное антитело настоящего изобретения можно также получить в результате инициации моноклональной гибридомной культуры, включающей питательную среду, содержащую гибридому, которая выделяет антительные молекулы соответствующей специфичности. Полученную культуру поддерживают в условиях и в течение периода времени, достаточных для того, чтобы данная гибридома выделяла антительные молекулы в данную среду. Затем эту среду, содержащую антитело, собирали. В дальнейшем, полученные антительные молекулы можно выделить с помощью хорошо известных методик.
Среда, пригодная для приготовления этих композиций, хорошо известна в данной области, коммерчески доступна и включает синтетическую культуральную среду, инбредных мышей и тому подобное. Образцовая синтетическая среда представляет собой основную минимальную среду Дульбекко (DMEM; Dulbecco с соавт. , Virol. 8:396, 1959) с добавлением 4,5 г/л глюкозы, 20 мМ глутамина и 20% фетальной сыворотки теленка. Типичный инбредный мышиный штамм представляет собой Balb/c.
Другие способы получения моноклонального антитела гибридомной клетки или гибридомной клеточной культуры также хорошо известны. Смотрите, например, способ выделения моноклональных антител из иммунологического ассортимента, как описано у Sastry с соавт., Proc. Natl. Acad. Sci. USA, 86:5728-5732 (1989); и Нusе с соавт., Science, 246:1275-1281 (1989).
В настоящем изобретении рассматривается также гибридомная клетка и культуры, содержащие гибридомную клетку, которая продуцирует моноклональное антитело данного изобретения. Особенно предпочтительной является гибридомная клеточная линия, которая выделяет моноклональное антитело mAb LM609, обозначенное АТСС НВ 9537. mAb LM609 получали, как описано у Cheresh с соавт., J. Biol. Chem. , 262: 17703-17711 (1987), и его получение описано также в нижеследующих примерах.
В настоящем изобретении рассматривается, в первом варианте осуществления настоящего изобретения, моноклональное антитело, которое обладает иммунореакционными характеристиками mAb LM609.
Можно также определить без чрезмерного экспериментирования, когда моноклональное антитело обладает одной и той же (т.е. эквивалентной) специфичностью (иммунореакционные характеристики), как моноклональное антитело данного изобретения, путем выяснения, действительно ли первое предохраняет последнее от связывания с заранее отобранной молекулой-мишенью. Если определенное моноклональное антитело, которое испытывают, конкурирует с моноклональным антителом настоящего изобретения, как показано при уменьшении связывания с моноклональным антителом настоящего изобретения в обычных анализах по конкурентному связыванию с данной молекулой-мишенью, при наличии в твердой фазе, тогда вероятно, что два моноклональные антитела связаны с одним и тем же или близкородственным эпитопом.
Еще один способ, чтобы определить, действительно ли моноклональное антитело обладает специфичностью моноклонального антитела настоящего изобретения, заключается в предварительной инкубации моноклонального антитела настоящего изобретения с молекулой-мишенью, с которой оно нормально реагирует, а затем добавить моноклональное антитело, которое испытывают, чтобы определить, когда ингибируется моноклональное антитело, которое испытывают, что касается его способности связываться с данной молекулой-мишенью. Если моноклональное антитело, которое испытывают, ингибируется после, то, по всей вероятности, оно обладает той же или функционально эквивалентной эпитопической специфичностью в качестве моноклонального антитела настоящего изобретения.
Дополнительный способ определения, действительно ли моноклональное антитело обладает указанной специфичностью моноклонального антитела настоящего изобретения, заключается в определении последовательности аминокислотных остатков участков CDR антител, о которых идет речь. Антительные молекулы, обладающие идентичностью или функциональной эквивалентностью последовательностей аминокислотных остатков по своим участкам CDR, обладают одной и той же специфичностью связывания. Методы секвенирования полипептидов хорошо известны в данной области.
Иммуноспецифичность антитела, связывающая способность его молекул-мишеней и сопутствующее сродство данного антитела, проявляемое по данному эпитопу, определяли по эпитопу, с которым данное антитело иммунологически взаимодействует. Специфичность данного эпитопа определяли как минимум по части последовательности аминокислотных остатков вариабельной области тяжелой цепи данного иммуноглобулина данного антитела, и по части последовательности аминокислотных остатков вариабельной области легкой цепи.
Применение термина "обладающий специфичностью по связыванию" указывает, что данное эквивалентное моноклональное антитело проявляет такие же или аналогичные иммунореакционные (связывание) характеристики и конкурирует за связывание с ранее выбранной молекулой-мишенью.
Человеческие моноклональные антитела обладают особыми преимуществами перед мышиными моноклональными антителами, в частности, поскольку они могут применяться терапевтически на людях. А именно, человеческие антитела не устраняются из кровотока так быстро, как "чужие" антигены, и не активируют данную иммунную систему таким же способом, как чужие антигены и чужие антитела. Способы получения "человеческих" антител в целом хорошо известны в данной области и могут быть легко перенесены на антитела настоящего изобретения.
Таким образом, настоящее изобретение рассматривает в первом варианте осуществления настоящего изобретения моноклональное антитело данного изобретения, которое гуманизируют путем трансплантации для интродуцирования компонентов человеческой иммунной системы без фактического вмешательства в способность антитела связывать антиген.
3. αvβ3-специфические миметики
В настоящем изобретении продемонстрировали, что αvβ3-антагонисты могут в целом использоваться в настоящем изобретении, если антагонисты могут включать полипептиды, антитела и другие молекулы, называемые "миметиками", которые обладают способностью препятствовать αvβ3-функции. Особенно предпочтительными являются антагонисты, которые специфически препятствуют αvβ3-функции и не вмешиваются в функцию других интегринов.
В данном контексте следует иметь в виду, что целый ряд реагентов может быть пригоден для применения в настоящих способах, поскольку эти реагенты обладают необходимой биологической активностью. Эти реагенты относятся, главным образом, к миметикам, так как они обладают способностью "имитировать" связывающий домен либо в αvβ3, либо в αvβ3-лиганде, участвующих в функциональном взаимодействии данного рецептора и лиганда и, таким образом, вмешиваться в (т.е. подавлять) нормальную функцию.
αvβ3-миметик представляет собой любую молекулу, иную, чем антитело или лиганд, полученный из пептида, которая проявляет вышеописанные свойства. Она может быть синтетическим пептидом, аналогом или производным пептида, соединением, которое напоминает форму связывающего кармана вышеописанного связывающего домена, такого как органическая миметик-молекула, или другой молекулой.
Предпочтительный миметик данного изобретения представляет собой миметик, основанный на органической молекуле и отнесенный поэтому к органическому миметику. Особенно предпочтительными органическими молекулами-миметиками, которые функционируют в качестве αvβ3-антагонистов путем присоединения миметика к лиганду αvβ3, являются соединения 7, 9, 10, 12, 14, 15, 16, 17 и 18, как описано в примере 10.
Создание αvβ3-миметика может быть осуществлено любым из различных способов структурного анализа, известного в данной области при создании лекарственного средства, включая молекулярное моделирование, анализ двумерным ядерным магнитным резонансом (2-D NMR), рентгеновской кристаллографией, случайным скринингом библиотек пептида, пептидного аналога или иного химического полимера или соединения и подобными подходами при создании лекарственного средства.
Ввиду обширности структурного доказательства, представленного в настоящем описании, которое показывает, что αvβ3-антагонист может быть слитым белком (например, слитый белок ММР-2), небольшим полипептидом, циклическим полипептидом, производным пептида, органической молекулой-миметик или моноклональным антителом, которые различным образом отличаются химической структурой, которая совпадает с функциональным свойством избирательного ингибирования αvβ3, структура оригинального αvβ3-антагониста, пригодного для настоящих способов, не нуждается в ограничении, но включает любой αvβ3-миметик, как здесь указано.
F. Методы идентификации антагонистов αvβ3
В настоящем изобретении описаны также аналитические методы идентификации кандидата в αvβ3-антагонисты для использования в соответствии с настоящими методами. В этих аналитических методах молекулы-кандидаты оценивали по их способности ингибировать связывание αvβ3 с естественными лигандами и к тому же оценивали их способность ингибировать ангиогенез в ткани.
В первом анализе измеряли ингибирование прямого связывания естественного лиганда с αvβ3, и предпочтительный вариант подробно описан в нижеследующих примерах. В данном анализе обычно измеряют степень ингибирования связывания естественного лиганда, такого как фибриноген, с выделяемым αvβ3 в твердофазном ELISA.
Данный анализ можно также использовать для идентификации соединений, которые проявляют специфичность по αvβ3 и не ингибируют связывание естественных лигандов другими интегринами. Анализ специфичности осуществляли проведением параллельных ELISA-анализов, в которых и αvβ3 и другие интегрины скринировали совместно в отдельных аналитических камерах на их соответствующие способности связывать естественный лиганд и на кандидатное соединение, которое ингибирует соответствующие способности данных интегринов связывать ранее отобранный лиганд. Предпочтительные форматы аналитического скринирования описаны в нижеследующих примерах.
Во втором анализе измеряли ангиогенез в хориоаллантоидной мембране цыпленка (САМ) и он назван САМ-анализ. САМ-анализ подробно был описан другими и впоследствии использовался для измерения ангиогенеза и реваскуляризации опухолевых тканей. Смотрите Ausprunk с соавт., Am. J. Pathol., 79:597-618 (1975) и Ossonski с соавт., Cancer Res., 40:2300-2309 (1980).
САМ-анализ хорошо распознает аналитическую модель ангиогенеза in vivo из-за того, что происходит реваскуляризация целой ткани, а кровеносные сосуды эмбриона цыпленка практически прорастают в САМ или в ткань, выросшую в САМ.
Как показано здесь, САМ-анализ иллюстрирует ингибирование реваскуляризации на основе количества и протяженности нового сосудистого роста. Кроме того, легко отслеживать рост любой ткани, трансплантированной в САМ, такой как опухолевая ткань. Наконец, данный анализ особенно полезен, потому что что имеется внешний контроль за токсичностью в данной аналитической системе. На эмбрион цыпленка воздействовали любым испытуемым реагентом, и поэтому жизнеспособность данного эмбриона является критерием токсичности.
Третий анализ, который измеряет ангиогенез, представляет собой in vivo модель кроличьего глаза и относится к так называемому анализу глаза кролика. Анализ глаза кролика был подробно описан другими и впоследствии использовался для измерения ангиогенеза и реваскуляризации в присутствии ангиогенных ингибиторов, таких как талидомид. Смотрите D'Amato, с соавт., Proc. Natl. Acad. Sci. USA, 91:4082-4085 (1994).
Анализ кроличьего глаза является хорошо узнаваемой моделью для ангиогенеза in vivo из-за процесса реваскуляризации, иллюстрируемого кроличьими кровеносными сосудами, растущими от края роговой оболочки глаза в роговицу через естественно прозрачную роговицу кроличьего глаза. Кроме того, можно легко контролировать во времени и продолжительность и итог стимуляции или ингибирования реваскуляризации или регрессии реваскуляризации.
Наконец, на кролика воздействуют любым испытываемым реагентом, и поэтому жизнеспособность данного кролика является критерием токсичности данного испытываемого реагента.
Четвертый анализ измеряет ангиогенез в химерной мышиной модели мышь: человек и относится к так называемому анализу химерной мыши. Этот анализ подробно описан другими и дополнительно описан здесь для измерения ангиогенеза, реваскуляризации и регрессии опухолевых тканей. Смотрите Yan с соавт., J. Clin. Invest. , 91: 986-996 (1993). Анализ химерной мыши является полезной аналитической моделью ангиогенеза in vivo, поскольку трансплантируемые кожные трансплантаты весьма гистологически близки нормальной человеческой коже, реваскуляризация целой ткани происходит там, где действительно человеческие кровеносные сосуды проращиваются из трансплантированной человеческой кожи в человеческую опухолевую ткань на поверхности трансплантированной человеческой кожи. Начало реваскуляризации в человеческом трансплантате можно показать с помощью иммуногистохимического окрашивания новой сосудистой сети специфичными для человека маркерами эндотелиальной клетки.
Как показано здесь, анализ химерной мыши демонстрирует регрессию реваскуляризации, основанную и на степени и на протяженности регрессии роста нового сосуда. Более того, легко контролировать эффекты роста любой ткани, трансплантированной на пересаженную кожу, такой как опухолевая ткань. Наконец, данный анализ полезен потому, что существует внешний контроль токсичности данной аналитической системы. Полученную химерную мышь подвергали воздействию любого испытываемого реагента, и поэтому жизнеспособность данной мыши является критерием токсичности.
G. Изделие
В настоящем изобретении рассматривается также изделие, которое представляет собой маркированный контейнер для создаваемого в настоящем изобретении антагониста αvβ3. Изделие включает упаковывающий материал и фармацевтический агент, содержащийся в упаковывающем материале.
Фармацевтический агент в изделии представляет собой αvβ3-антагонист настоящего изобретения, созданный в фармацевтически приемлемой форме, как описано здесь в соответствии с описанными указаниями. Изделие содержит любое количество фармацевтического агента, достаточного для применения в лечении состояния, указанного здесь, либо в виде однократной или многократной доз.
Упаковывающий материал включает этикетку, которая указывает на применение данного фармацевтического агента, представленного в ней, например, для лечения состояний, связанных с ингибированием ангиогенеза, и подобных состояний, описанных здесь. Данная этикетка может дополнительно включать инструкции по применению и связанную с этим информацию, необходимую для продажи. Данный упаковывающий материал может включать контейнер(ы) для хранения данного фармацевтического агента.
Термин упаковывающий материал, как он используется здесь, относится к материалу, такому как стекло, пластик, бумага, фольга и тому подобное, способному удерживать в фиксированном состоянии любой фармацевтический агент. Так, например, данный упаковывающий материал может представлять собой пластиковый или стеклянный флаконы, клееный сосуд и подобные контейнеры, используемые для содержания фармацевтической композиции, включающей данный фармацевтический агент.
В предпочтительных вариантах осуществления настоящего изобретения данный упаковывающий материал включает этикетку, которая реально выражает описание данного содержимого изделия и применение данного фармацевтического агента, представленного на ней.
Примеры
Нижеследующие примеры, относящиеся к данному изобретению, являются иллюстративными и конечно не должны интерпретироваться в качестве специфически ограничивающих настоящее изобретение. Кроме того, такие изменения настоящего изобретения, известные в настоящее время или созданные позже, которые должны находиться в компетенции специалиста в данной области, рассматриваются для того, чтобы они могли попасть в сущность настоящего изобретения, изложенного в нижеследующей формуле изобретения.
1. Получение синтетических пептидов
а. Методика синтеза
Линейные и циклические полипептиды, представленные в таблице 1, были синтезированы с использованием стандартных методик по твердофазному синтезу, как, например, описанные у Merrifield, Adv. Enzymol., 32:221-96 (1969) и у Fields G.B. and Noble R.L., Int. J. Peptide Protein Res., 35:161-214 (1990).
Два грамма (г) BOC-Gly-D-Arg-Gly-Asp-Phe-Val-OMe (SEQ ID NO 1) вначале растворяли в 60 миллилитрах (мл) метанола, к которому добавляли 1,5 мл 2 N раствора гидроксида натрия с образованием смеси. Затем полученную смесь перемешивали в течение 3 часов при 20oС. После выпаривания остаток помещали в воду, подкисляли до рН 3,0 разбавленной НСl и экстрагировали этилацетатом. Полученный экстракт сушили над Na2SO4, вновь выпаривали и этот полученный BOC-Gly-D-Arg-Gly-Asp-Phe-Val-OH (SEQ ID NO 2) перемешивали при 20oС в течение 2-х часов с 20 мл 2 N НСl в диоксане. Данную полученную смесь выпаривали до получения H-Gly-D-Arg-Gly-Asp-Phe-Val-OH (SEQ ID NO 3), который впоследствии растворяли в смеси из 1800 мл дихлорметана и 200 мл диметилформамида (DMF) с последующим охлаждением до 0oС. После этого последовательно с перемешиванием добавляли 0,5 г дициклогексилкарбодиимида (DCCI), 0,3 г 1-гидроксибензотриазола (HOBt) и 0,23 мл N-метилморфолина.
Данную полученную смесь перемешивали в последующие 24 часа при 0oС, а затем при 20oС в следующие 48 часов. Полученный раствор концентрировали и пропускали через слой смешанной ионообменной смолы для освобождения его от солей. После использования смолу удаляли путем фильтрации, осветленный раствор выпаривали, а остаток очищали с помощью хроматографии, получая цикло(-Gly-D-Arg-Gly-Asp-Phe-Val) (SEQ ID NO 4). Нижеследующие пептиды, приведенные в таблице 1 с использованием однобуквенного кода аббревиатур аминокислотных остатков и идентифицированные с помощью пептидного численного обозначения, были получены аналогично: цикло(Arg-Gly-Asp-D-Phe-Val) (SEQ ID NO 5); цикло(Arg-Ala-Asp-D-Phe-Val) (SEQ ID NO 6); цикло(Arg-D-Ala-Asp-Phe-Val) (SEQ ID NO 9); цикло (Arg-Gly-Asp-Phe-D-Val) (SEQ ID NO 7) и цикло(Arg-Gly-Asp-D-Phe-NMeVal) (метилирование по азоту альфа-амино амидной связи остатка валина) (SEQ ID NO 15).
Пептид, обозначенный как 66203, обладающий последовательностью, идентичной с последовательностью пептида 62184, отличается от последнего лишь содержанием соли НСl, но не солью TFA, присутствующей в 62184. Это же справедливо для пептидов 69601 и 62185 и для 85189 и 121974.
b. Методика альтернативного синтеза
i. Синтез цикло-(Arg-Gly-Asp-DPhe-NMeVal), TFA-соль
Fmoc-Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-ONa синтезировали с использованием твердофазного метода по Merrifield путем последовательного добавления NMeVal, DPhe, Asp (OBut), Gly и Fmoc-Arg(Mtr) в поэтапном способе с 4-гидроксиметил-феноксиметил-полистироловой смолой (Wang type resin) (применяли обычные методы пептидного синтеза по методу Merrifield, как описано у Houben-Weyl, 1.с., Том 15/II, Страницы 1-806 (1974). Данная полистироловая смола и предшественники аминокислотных остатков коммерчески доступны от химических компаний Aldrich, Sigma или Fluka). По окончании последовательного добавления указанных аминокислотных остатков эту смолу отторгали от полученной пептидной цепи с использованием смеси TFA/дихлорметана 1:1, которые создают продукт Fmoc-Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-OH. Затем группу Fmoc удаляли смесью пиперидин/DMF 1:1, которая создает неочищенный предшественник Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-OH, который затем выделяли очисткой в HPLC обычным способом.
Для образования кольца раствор из 0,6 г Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-OH (синтезированный выше) в 15 мл DMF (диметилформамид; Aldrich) разбавляли 85 мл дихлорметана (Аldrich) и добавляли 50 мг NaHCO3. После охлаждения в смеси сухой лед/ацетон, добавляли 40 мкл дифенилфосфорилазида (Aldrich). После инкубации при комнатной температуре в течение 16 часов этот раствор концентрировали. Полученный концентрат подвергали гель-фильтрации (колонка с Sephadex G10 изопропанол/вода 8:2), а затем очищали HPLC обычным методом. Обработка с TFA (трифторуксусная кислота)/Н2О (98:2) дает цикло-(Arg-Gly-Asp-DPhe-NMeVal)хTFA, который затем очищали на HPLC обычным методом; RT=19,5; FAB-MS (М+Н): 589.
ii. Синтез "внутренней соли"
Из вышеполученного циклического пептида удаляли TFA-соль путем суспендирования цикло-(Arg-Gly-Asp-DPhe-NmeVal)хTFA в воде с последующим упариванием под вакуумом для удаления данной TFA. Образованный циклический пептид относится к так называемой "внутренней соли" и обозначается цикло-(Arg-Gly-Asp-DPhe-NmeVal). Термин "внутренняя соль" используется потому, что этот циклический пептид содержит два противоположно заряженных остатка, которые внутриэлектронно уравновешивают
друг друга с образованием незаряженной, в целом, молекулы. Один из заряженных остатков содержит кислотную составляющую, а другой заряженный остаток содержит амино-составляющую. Когда кислотная составляющая и амино-составляющая находятся в непосредственной близости друг к другу, кислотная половина может депротонироваться амино-составляющей, которая образует карбоксилат/аммонийные виды соли, в целом, с нейтральным зарядом.
iii. HCl-обработка, которая дает цикло-(Arg-Gly-Asp-DPhe-NMeVal)хHCl
80 мг цикло-(Arg-Gly-Asp-DPhe-NMeVal) растворяли в 0,01 М HCl пять-шесть раз и высушивали замораживанием после каждой операции растворения. Последующая очистка HPLC дает цикло-(Arg-Gly-Asp-DPhe-NMeVal)хHCl; FAB-MS (М+Н): 589.
iv. Обработка метансульфоновой кислотой, которая дает цикло-(Arg-Gly-Asp-DPhe-NMeVal)хМеSО3Н
80 мг цикло-(Arg-Gly-Asp-DPhe-NMeVal) растворяли в 0,01 М МеSО3Н (метансульфоновая кислота) пять-шесть раз и высушивали замораживанием после каждой операции растворения. Последующая очистка HPLC дает цикло-(Arg-Gly-Asp-DPhe-NMeVal)хМеSО3Н; RT=17,8; FAB-MS (М+Н): 589.
Альтернативные способы циклизации включают получение цепей боковых групп ациклического предшественника с сульфгидрильными составляющими и при выдерживании в условиях с несколько более высоким, чем нормальное значением рН (рН 7,5) внутримолекулярно образуют дисульфидные связи с другими сульфгидрильными группами, представленные в данной молекуле, с образованием циклического пептида. Кроме того, С-концевая карбоксилатная составляющая из предшественника ациклического пептида может взаимодействовать со свободной сульфгидрильной составляющей, представленной в данной молекуле с получением тиоэфир-циклизованных пептидов.
В анализах по ингибированию ангиогенеза, как описано в примере 7, где используется синтетический пептид, пептид 66203 в НСl был несколько более эффективен в ингибировании ангиогенеза, чем идентичный пептид - в TFA.
2. Моноклональные антитела
Моноклональные антитела LM609, продуцируемые гибридомой АТСС НВ 9537, получали с использованием стандартных гибридомных методов путем иммунизации выделенного αvβ3, адсорбированного на сефарозно-чечевичных лектиновых шариках. αvβ3 выделяли из человеческих меланомных клеток, обозначенных М21, а антитело получали, как описано у Cheresh с соавт., J. Biol. Chem., 262: 17703-17711 (1987). Клетки М21 были созданы Dr. D. L. Morton (University of California в Los Angeles, CA) и росли в суспензионных культурах в культуральной среде RPMI 1640, содержащей 2 мМ L-глутамина, 50 мг/мл гентамицин-сульфата и 10% фетальной сыворотки теленка.
Моноклональные антитела LM609 показывают специфическое иммунное взаимодействие с комплексом αvβ3, но иммунно не взаимодействует с αv-субъединицей, с β3-субъединицей или с другими интегринами.
3. Характеристика тканевого распределения экспрессии αvβ3
А. Иммунофлуоресценция с анти-интегриновыми рецепторными антителами
Во время заживления раны базальные мембраны кровеносных сосудов экспрессируют несколько адгезивных белков, в том числе фактор фон Виллебранда, фибронектин и фибрин. Кроме того, несколько представителей интегринового семейства из адгезионных рецепторов экспрессируются на поверхности культивируемых клеток гладкой мускулатуры и эндотелиальных клеток. Смотрите, Cheresh, Proc. Natl. Acad. Sci. USA, 84:6471 (1987); Janat с соавт., J. Cell. Physiol. , 151-588 (1992), и Cheng с соавт., J. Cell Physiol., 139-275 (1989). Среди этих интегринов имеется αvβ3 эндотелиальноклеточный рецептор для фактора фон Виллебранда, фибриногена (фибрина) и фибронектина, как описано у Cheresh, Proc. Natl. Acad. Sci. USA, 84:6471 (1987). Этот интегрин инициирует кальцийзависимый сигнальный путь, приводящий к миграции эндотелиальных клеток, и поэтому выступает в ключевой роли в клеточной биологии сосудов, как описано у Leavelsey с соавт., J. Cell Biol., 121:163 (1993).
Чтобы исследовать экспрессию αvβ3 во время ангиогенеза, от пациентов, выразивших согласие, получали гранулированную раневую ткань человека или примыкающей нормальной кожи, промывали 1 мл фосфатно-солевого буфера и помещали в среду О.Т.С. (Tissue Tek). Эти помещенные ткани резко замораживали в жидком азоте в течение приблизительно 30-45 секунд. Из замороженных блоков на криостатном микротоме получали срезы толщиной шесть микрон для последующего иммунопероксидазного окрашивания с антителами, специфичными либо по β3-интегринам (αvβ3 или αIIbβ3), либо по β1-интегриновому подсемейству.
Результаты окрашивания нормальной человеческой кожи и гранулированной ткани раны показаны на фигурах 1A-1D. Моноклональные антитела АР3 и LM534 соответственно к интегринам β3 и β1, использовали для иммуногистохимического анализа замороженных срезов. Эксперименты с тканью из четырех разных сосудов человеческих доноров давали идентичные результаты. Представлены микрофотографии с увеличением 300х.
Интегрин αvβ3 в большом количестве экспрессировался в кровеносных сосудах в гранулированной ткани (фигура 1В), но не обнаруживался в дерме и эпителии нормальной кожи от того же донора (фигура 1А). А противоположность этому интегрины β1 экспрессировались в большом количестве в кровеносных сосудах и клетках стромы в нормальной коже (фигура 1С) и в гранулированной ткани (фигура ID) и, как показано прежде, описано у Adams с соавт., Cell, 63:425 (1991), на базальных клетках в эпителии.
В. Иммунофлуоресценция с анти-лигандными антителами
Исследовали также дополнительные срезы вышеполученных человеческой кожи и гранулированных тканей на наличие лигандов соответственно для интегринов β3 и β1, фактора фон Виллебранда и ламинина. Фактор фон Виллебранда локализовали в кровеносных сосудах нормальной кожи (фигура 2А) и гранулированной ткани (фигура 2В), а ламинин локализовали во всех кровеносных сосудах, а также в эпителиальной базальной мембране в обоих тканевых препаратах (фигуры 2С и 2D).
С. Распределение антител к анти-αvβ3 в раковой ткани
Помимо вышеуказанных анализов исследовали также биоптаты раковой ткани от человеческих пациентов на наличие и распределение αvβ3. Данные ткани получали, как описано в примере 1А, за тем исключением, что их окрашивали с помощью моноклональных антител LM609, полученных в примере 2, которое специфично для данного интегринового рецепторного комплекса αvβ3. Кроме того, опухоли получали для микроскопического гистологического анализа путем фиксации соответствующих образцов опухолей в Bulins Fixative в течение 8 часов, получали последовательные срезы и окрашивали Н&Е.
Результаты иммунопероксидазного окрашивания раковых тканей мочевого пузыря, толстой кишки, молочной железы и легкого показаны соответственно на фигурах 3А и 3D. В большом количестве αvβ3 экспрессируется лишь в кровеносных сосудах, представленных в четырех проанализированных раковых биоптатах, но не с какими-либо другими клетками в данной ткани.
Таким образом, результаты, описанные здесь, показывают, что рецептор αvβ3-интегрина избирательно экспрессируется в специфических типах тканей, а именно в гранулированной, в метастазных тканях и в других тканях, в которых проявляется ангиогенез, но не в нормальных тканях, где образование новых кровеносных сосудов остановлено. Эти ткани поэтому создают идеальную мишень для терапевтических аспектов данного изобретения.
4. Идентификация αvβ3-специфических синтетических пептидов, детектируемых путем ингибирования прикрепления клеток и в анализе связывания лиганд-рецептор
А. Ингибирование прикрепления клеток.
В качестве первого пути для определения специфичности антагонистов интегринового рецептора в данном изобретении анализ ингибирования прикрепления клеток осуществляли, как описано ниже.
Вкратце, меланомные клетки CS-1 хомяка, не имеющие экспрессии αvβ3 и αvβ5, вначале трансфецировали плазмидой для экспрессии β3-субъединицы, как описано ранее у Filardo с соавт., J. Cell. Biol., 130:441-450 (1995). Специфичность потенциальных антагонистов определяли по их способности блокировать связывание клеток CS-1, экспрессирующих αvβ3, с планшетами, покрытыми VN или ламинином. В качестве примера типичного анализа вначале лунки покрывали субстратом, 10 мкг/мл, в течение ночи. После промывки и блокирования 1% термоденатурированным BSA в PBS при комнатной температуре в течение 30 минут пептид 85189 (SEQ ID NO 15) с концентрацией в диапазоне от 0,0001 мкМ до 100 мкМ смешивали отдельно с клетками CS-1 для нанесения в лунки с числом клеток, равным 50000 клеток/лунку. После 10-15 минут инкубации при 37oС данный раствор, содержащий клетки и пептиды, удаляли. Затем после окрашивания 1% кристаллвиолетом определяли число присоединившихся клеток. Клетки, ассоциированные с кристаллвиолетом, элюировали путем добавления 100 микролитров (мкл) 10% уксусной кислоты. Клеточную адгезию определяли количественно путем измерения оптической плотности элюированного кристаллвиолета при длине волны 600 нм.
Фигура 21 показывает результат типичного анализа антагониста αvβ3, в данном случае пептида 85189. Ингибирование не детектировали для пептида на поверхностях, покрытых ламинином. Напротив, полное ингибирование связывания получили на поверхностях, покрытых VN, для пептидной концентрации от 10 мкМ и больше, как показано с помощью дозозависимой кривой.
Аналогичные анализы осуществляли со слитыми белками, содержащими разные области белка ММР-2. Полипептиды, производные ММР-2, включают области из С-конца ММР-2, активного в связывающем взаимодействии с αvβ3 и, таким образом, способного ингибировать активацию ММР-2, и ассоциированные активности. Эти полипептиды получали либо в виде синтетических полипептидов, обладающих последовательностью, полученной из С-концевого домена ММР-2, как описано в примере 1, либо в виде слитых белков, включающих все или часть С-концевого домена ММР-2, полученных как описано ниже. С-концевые молекулы ММР-2 представлены для специфичных последовательностей и цыпленка и человека.
С-концевой домен, полученный для ММР-2 цыпленка, относящийся также к гемопексин-домену, непосредственно примыкающему к шарнирной области, включает аминокислотные остатки 445-637 из ММР-2. Полная нуклеотидная и кодируемая аминокислотная последовательность ММР-2 цыпленка описаны ниже. Нуклеотидная и кодируемая аминокислотная последовательность ММР-2 человека также описаны ниже. С-концевой домен в ММР-2 человека, который соответствует области цыпленка 445-637, начинающийся с аминокислотного остатка 439 и заканчивающийся на 631, связан с шестью недостающими остатками из человеческой последовательности, как показано на фигурах 22А и 22В. С-концевые синтетические пептиды, полученные из ММР-2 цыпленка и человека, используются на практике в методах данного изобретения и приведены в таблице 1. Последовательности аминокислотных остатков данных синтетических пептидов являются теми же, что и полученные с помощью рекомбинантных аналогов слитого белка, но без компонента слияния GST. С-концевые слитые белки, произведенные и из цыпленка и из человека, получали, как описано ниже.
Слитый белок ММР-2 представляет собой химерный полипептид, обладающий последовательностью С-концевого домена ММР-2 или его части, слитой (присоединенной путем сшивки ковалентной пептидной связью) с носителем (слияние) белка, таким как глутатионсульфгидрилтрансфераза (GST).
Чтобы амплифицировать разные области ММР-2 цыпленка и человека, создали праймерные последовательности на основе соответствующих последовательностей кДНК из ММР-2 цыпленка и человека. Полная верхняя цепь нуклеотидной последовательности кДНК непроцессируемого ММР-2 цыпленка, отнесенная также к так называемой прожелатиназе, показана на фигурах 22А и 22В вместе с выведенной аминокислотной последовательностью, показанной на второй линии (Aimes с соавт. , Biochem J., 300:729-736, 1994). Третья и четвертая линии данной фигуры показывает соответственно выведенную аминокислотную последовательность человека (Collier с соавт., J. Biol. Chem., 263:6579-6587 (1988)) и ММР-2 мыши (Reponen с соавт., J. Biol. Chem., 267:7856-7862 (1992)). Идентичные остатки отмечены точками, а отличающиеся остатки даны в их однобуквенном написании IUPAC. Недостающие остатки отмечены прочерком. Нумерация аминокислотных остатков начинается от первого остатка профермента вместе с остатками сигнального пептида, представленного отрицательными числами. Данная нуклеотидная последовательность пронумерована в соответствии с этим. Предполагаемая инициация трансляции (ATG) отмечена тремя направленными вперед стрелками-указателями, а сигнал терминации трансляции (TGA) указан звездочкой. Данные аминокислотные последовательности профермента и активного фермента цыпленка ограничены ромбовидными и одиночными стрелками-указателями. Последовательности нуклеотидных и аминокислотных остатков прожелатиназы цыпленка представлены вместе в виде SEQ ID NO 29, а кодируемая последовательность аминокислотных остатков представлена отдельно в виде SEQ ID NO 30.
Матрицы для получения амплифицированных областей ММР-2 цыпленка представляют собой либо кДНК, кодирующую полноразмерный зрелый полипептид ММР-2 цыпленка, созданный Dr. J. Р. Quigley из the State University of New York at Stoney Brook, New York, либо кДНК, полученную из тотальной клеточной РНК-матрицы, произведенной с помощью стандартных методик, из вырезанного образца хориоаллантоидной мембранной ткани цыпленка. В последнем случае кДНК получали с помощью обратной транскриптазы MuLV и нижележащими праймер-специфичными по 3'-концу нуклеотидами, 5' ATTGAATTCTTCTACAGTTCA3' (SEQ ID NO 31), 5' и 3'-концы которых комплементарны соответственно нуклеотидам 1932-1912 из опубликованной последовательности ММР-2 цыпленка. Полимеразную целую реакцию с обратной трансферазой (RT-PCR) осуществляли в соответствии с техническими условиями изготовителей для набора GeneAmp RNA PCR (Perkin Elmer). Данный праймер был также сконструирован так, чтобы содержать внешний сайт рестрикции EcoRI.
Из любых вышеописанных кДНК-матриц с помощью PCR вместе с 3'-праймером, представленным выше (SEQ ID NO 31), спаренным вместе с одним из ряда 5'-праймеров, представленных ниже, были получены ряд С-концевых областей ММР-2 цыпленка, каждая, обладающая естественным цистеиновым остатком в позиции 637 на карбокси-конце. Эти амплифицированные области кодируют нижеследующие слитые белки ММР-2, обладающие последовательностями, соответствующими с позициями аминокислотных остатков, как показано на фигурах 22А и 22В и представленные также в SEQ ID NO 30: 1) 203-637; 2) 274-637; 3) 292-637; 4) 410-637; 5) 445-637. Вышележащие или 5'-праймеры для амплификации каждой из нуклеотидных областей для кодирования представленных выше слитых белков ММР-2 были предназначены для кодирования полипептидных биотехнологических стартовых сайтов 3', т.е., PCR-интродуцированного внешнего сайта рестрикции BamHI, чтобы получить возможность прямого лигирования в любом экспрессионном векторе, pGEX-1λT или pGEX-3Х. Эти 5'-праймеры включают нижеследующие последовательности, 5' и 3'-концы которых соответствуют указанным 5' и 3' нуклеотидным положениям последовательности ММР-2 цыпленка, как показано на фигурах 22А и 22В (для каждого праймера указана также позиция аминокислотных остатков стартовых сайтов): 1) Нуклеотиды 599-619, кодирующие 203-й стартовый сайт 5' ATGGGATCCACTGCAAATTTC3' (SEQ ID NO 32); 2) Нуклеотиды 809-830, кодирующие 274-й стартовый сайт 5' GCCGGATCCATGACCAGTGTA3' (SEQ ID NO 33); 3) Нуклеотиды 863-883, кодирующие 292-й стартовый сайт 5' GTGGG-ATCCCTGAAGACTATG3' (SEQ ID NO 34); 4) Нуклеотиды 1217-1237, кодирующие 410-й старт 5' AGGGGATCCTTAAGGGGATTC3' (SEQ ID NO 35); и 5) Нуклеотиды 1325-1345, кодирующие 445 стартовый сайт 5' CTCGGATCCTCTGCAAGCACG3' (SEQ ID NO 36).
Указанные нуклеотидные области матричной кДНК были впоследствии амплифицированы в течение 35 циклов (температура отжига 55oС), в соответствии с инструкциями изготовителя для Expand High Fidelity PCR System (Boehringer Mannheim). Полученные в PCR продукты были выделены очисткой в геле, обработаны с помощью ферментов рестрикции BamHI и EcoRI и повторно выделены очисткой перед лигированием в любом векторе, pGEX-1λT или pGEX-3Х (Pharmacia Biotech, Uppsala, Sweden), которые были аналогично обработаны, а также дефосфорилированы перед реакцией лигирования. Выбор плазмиды определялся требуемой рамкой считывания данного продукта амплификации. Компетентные клетки штамма Е. coli BSJ72 или BL21 трансформировали разными конструкциями с помощью теплового шока. Полученные колонии скринировали на включение соответствующего ММР-2 слитого белка-кодирующей плазмиды с помощью PCR перед дидезокси-секвенированием позитивных клонов, чтобы проверить интегрированность интродуцированной кодирующей последовательности. Кроме того, проверку инкорпорирования плазмиды подтверждали экспрессией слитого белка GST-MMP-2 соответствующего размера.
Выделение очисткой каждого из ре комбинантных слитых белков GST-MMP-2 осуществляли с использованием индуцированных IPTG культур в log-фазе, по существу, как описано у изготовителя GST Gene Fusion System (Pharmacia Biotech). Вкратце, извлеченные бактерии лизировали путем ультразвуковой обработки и инкубировали с детергентом перед осветлением и иммобилизацией данного рекомбинантного белка на сефарозе 4В с пришитым глутатионом (Pharmacia Biotech). После экстенсивной промывки данные иммобилизованные слитые белки были по отдельности элюированы из этой матрицы по сродству 10 мМ восстановленным глутатионом в 50 мМ Трис-HCl, рН 8,0, и экстенсивно диализовали против PBS для удаления остаточного глутатиона перед использованием.
В ранних попытках получить слитые белки между остатками 445 и 637 ММР-2 цыпленка, который обладает только одним кодируемым цистеиновым остатком, получали нерастворимые продукты. По этой причине, для того чтобы получить дополнительную растворимость слитых белков ММР-2, произведенных из С-концевой области, которая не включает эндогенный концевой цистеиновый остаток, как представлено в слитом белке, описанном прежде, нуклеотидные последовательности интродуцировали при необходимости в амплифицируемые области ММР-2 для кодирования цистеинового остатка в зависимости от конкретного слитого белка. Цистеиновый остаток естественно присутствует в последовательности ММР-2 цыпленка в позиции 446 и в позиции 637. В данной последовательности человека эти позиции соответствуют 440 и 631. Поэтому слитые белки были сконструированы так, чтобы содержать введенные концевые цистеиновые остатки по амино- или карбокси-концам представляющих интерес последовательностей ММР-2 цыпленка для того, чтобы создать дисульфидную связь с природным цистеином на другом конце, как необходимо для данной конструкции.
В соответствии с этим олигонуклеотидные праймеры были сконструированы так, чтобы дать возможность амплифицироваться С-концевым областям ММР-2 цыпленка для экспрессии растворимых слитых белков MMP-2/GST. Амплифицируемые С-концевые области ММР-2 цыпленка включают области кодирования аминокислотных остатков в позициях 445-518, 445-552, 516-637 и 549-637. В слитых белках, содержащих остаток 517, естественно кодируемый тирозиновый остаток заменяли на цистеин, чтобы обеспечить дисульфидную связь с любым цистеиновым остатком в позиции 446 или 637. В слитых белках остаток 551, естественно представленный триптофаном, заменяли цистеином, чтобы обеспечить дисульфидную связь с природным цистеином в позиции 446 или 637.
Вкратце, конструкцию плазмиды pGEX-3Х, кодирующую вышеполученный рекомбинантный слитый белок GST/MMP-2 (410-637), использовали в качестве матрицы для амплификации в соответствии с протоколом изготовителя в Expand High Fidelity PCR Kit (Boerhringer Mannheim) с использованием набора олигонуклеотидных праймеров, которые были сконструированы на основе опубликованной последовательности ММР-2 цыпленка (показано также на фигурах 22А и 22В). Один вышележащий праймер, сконструированный для кодирования стартового сайта белка ММР-2 цыпленка в позиции 445 после биотехнологического внешнего эндонуклеазного сайта рестрикции BamHI для инсерции в GST-вектор pGEX-3Х, обладает нуклеотидной последовательностью (5' CTCGGATCCTCTGCAAGCACG3' (SEQ ID NO 37)). На данной фигуре 5' и 3'-концы данного праймера соответствуют позициям 1325-1345 последовательности ММР-2 цыпленка. Другой вышележащий праймер, сконструированный для кодирования стартового сайта белка ММР-2 цыпленка в позиции 516 после биотехнологического внешнего сайта рестрикции BamHI, для инсерции в GST-вектор pGEX-1λT и для кодирования остатка в позиции 517, обладает нуклеотидной последовательностью (5'GCAGGATCCGAGTGCTGGGTTTATAC3' (SEQ ID NO 38)). 5' и 3'-концы данного праймера соответствуют позициям 1537-1562 последовательности ММР-2 цыпленка. Третий вышележащий праймер, сконструированный для кодирования стартового сайта белка ММР-2 цыпленка в позиции 549 после биотехнологического внешнего эндонуклеазного сайта рестрикции EcoRI для инсерции в GST-вектор pGEX-1λT и для кодирования цистеинового остатка в позиции 551 обладает нуклеотидной последовательностью (5' GCAGAATTCAACTGTGGCAGAAACAAG3' (SEQ ID NO 39)). 5' и 3'-концы данного праймера соответствуют позициям 1639-1665 последовательности ММР-2 цыпленка.
Эти предлежащие праймеры использовали отдельно с одним из нижележащих праймеров, приведенных ниже, для получения вышеуказанных областей из С-концевого домена ММР-2 цыпленка. Первый нижележащий праймер (антисмысловой), сконструированный для кодирования сайта терминации белка в позиции 518, для кодирования цистеинового остатка в позиции 517 и для введения внешнего эндонуклеазного сайта рестрикции EcoRI для инсерции в GST-вектор, обладает нижеследующей нуклеотидной последовательностью (5' GTAGAATTCCAGCACTCATTTCCTGC3' (SEQ ID NO 40)). 5' и 3'-концы данного праймера, написанные в 5'-3'-направлении, были частично комплементарны соответственно позициям 1562-1537 последовательности ММР-2 цыпленка. Второй нижележащий праймер, сконструированный для кодирования сайта терминации белка ММР-2 цыпленка в позиции 552 для кодирования цистеинового остатка в позиции 551 и для введения внешнего эндонуклеазного сайта рестрикции EcoRI для инсерции в GST-вектор, обладает нижеследующей нуклеотидной последовательностью (5' TCTGAATTCTGCCACAGTTGAAGG3' (SEQ ID NO 41)). 5' и 3'-концы данного праймера, написанные в 5'-3'-направлении, были частично комплементарны соответственно позициям 1666-1643 последовательности ММР-2 цыпленка. Третий нижележащий праймер, сконструированный для кодирования сайта терминации белка ММР-2 цыпленка в позиции 637 и для введения внешнего эндонуклеазного сайта рестрикции EcoRI для инсерции в GST-вектор обладает нижеследующей нуклеотидной последовательностью (5' ATTGAATTCTTCTACAGTTCA3' (SEQ ID NO 42)). 5' и 3'-концы данного праймера, написанные в 5'-3'-направлении, были частично комплементарны соответственно позициям 1932-1912 последовательности ММР-2 цыпленка.
Области карбокси-концов ММР-2 цыпленка, соединенные с вышележащим и нижележащим праймерами, используемые в определенных сочетаниях для получения слитых белков, содержащих как минимум один биотехнологический цистеиновый остаток, как описано выше, были по отдельности амплифицированы в течение 30 циклов с температурой отжига 55oС в соответствии с инструкциями изготовителя для Expand High Fidelity PCR System (Boehringer Mannheim). Полученные продукты амплификации выделяли очисткой, обрабатывали по необходимости ферментами рестрикции BamHI и/или EcoRI и повторно выделяли очисткой перед лигированием в надлежащий GST-вектор слитого белка, pGEX-3Х либо pGEX-1λT, как указано выше, благодаря открытой рамке считывания вышележащего олигонуклеотидного праймера. Для лигирования амплифицированных продуктов ММР-2 векторы обрабатывали аналогично, а также дефосфорилировали перед реакцией лигирования. Затем путем теплового шока отдельно трансформировали компетентные клетки Е. coli штамма BL21 с получением векторных конструкций, содержащих ММР-2. Затем полученные колонии скринировали для инкорпорирования с помощью PCR-плазмиды, кодирующей слитый белок, и получали надлежащего размера GST-слитый белок перед дидезокси-секвенированием позитивных клонов для проверки интегрируемости интродуцированной кодирующей последовательности. Затем осуществляли выделение очисткой рекомбинантных GST слитых белков с использованием индуцированных IPTG культур в log-фазе, по существу как описано выше при получении других слитых белков GST-MMP-2.
Результаты ингибирования клеточного прикрепления при анализе с различными белками ММР-2 цыпленка, а также с другими пептидами, свидетельствуют, что интактный ММР-2, слитый белок СТММР-2(2-4) из остатков 445-637 и пептида 66203 (SEQ ID NO 5), но не ММР-2 (1-445) и контрольного пептида 69601, ингибирует адгезию β3-экспрессирующих клеток CS-1 с витронектином, но не с ламинином и тем самым ингибирует витронектиновый рецептор (αvβ3), связывающийся с витронектином, путем препятствования нормальной активности связывания αvβ3. Другие проанализированные СТММР-2-слитые белки - 7-1, из остатков 274-637, 10-1, из остатков 292-637, и 4-3, из остатков 274-400, обладают меньшим воздействием на клеточную адгезию по сравнению с 2-4.
Кроме GST-слитых белков ММР-2 цыпленка, описанных выше, были получены два GST-слитых белка ММР-2 человека для экспрессии аминокислотных областей 203-631 и 439-631 из зрелого проферментного полипептида ММР-2 человека. Указанные области соответственно соответствуют областям 203-637 и 445-637 ММР-2 цыпленка. ММР-2-GSТ-слитые белки человека были получены с помощью PCR, как описано выше для ММР-2-GSТ-слитых белков с использованием кДНК-матрицы, которая кодирует полную открытую рамку считывания ММР-2, созданную Dr. W. G. Stetler-Stevenson в National Cancer Institute, Bethesda, MD. Последовательности вышележащего 5'-праймера сконструировали на основе ранее опубликованной последовательности ММР-2 человека (Collier с соавт., J. Biol. Chem. , 263-6579-6587 (1988) и для введения внешнего сайта рестрикции EcoRI, чтобы обеспечить возможность клонирования амплифицированных продуктов в надлежащий ЭКС-прессионный вектор.
Первый вышележащий праймер, сконструированный для кодирования стартового сайта белка, в позиции 203 после биотехнологического внешнего эндонуклеазного сайта рестрикции EcoRI для клонирования в GST-вектор pGEX-1λT обладает нижеследующей последовательностью (5' GATGAATTCTACTGCAAGTT3' (SEQ ID NO 43)). 5' и 3'-концы данного праймера соответствуют позициям 685-704 последовательности открытой рамки считывания ММР-2 человека. Второй вышележащий праймер, сконструированный для кодирования стартового сайта белка ММР-2 человека в позиции 439 после биотехнологического внешнего сайта рестрикции EcoRI для клонирования в GST-вектор pGEX-1λT, обладает нижеследующей нуклеотидной последовательностью (5' CACTGAATTCATCTGCAAACA3' (SEQ ID NO 44)). 5'и 3'-концы данного праймера соответствуют позициям 1392 и 1412 последовательности открытой рамки считывания ММР-2 человека.
Каждый из вышеуказанных праймеров использовали отдельно с нижележащим праймеров, обладающим 5' и 3'-концами, комплементарными соответственно основаниям 1998 и 1978 последовательности ММР-2 человека, концы которой находятся снаружи открытой рамки считывания и указывают терминацию белка после аминокислотного остатка 631. Полученные амплифицированные продукты экспрессировали слитые белки, содержащие аминокислотные остатки 203-631 ММР-2 человека (SEQ ID NO 45) и 439-631 (SEQ ID NO 18).
Эти полученные PCR-продукты выделяли очисткой, обрабатывали EcoRI и повторно выделяли очисткой для лигирования в плазмиду pGEX-1λT, которую обрабатывали аналогично и дефосфорилировали перед данной реакцией лигирования. Клетки трансформировали, как описано выше.
Другие слитые белки ММР-2 человека, содержащие аминокислотные остатки 410-631 (SEQ ID NO 17), 439-512 (SEQ TD NO 19), 439-546 (SEQ ID NO 20), 510-631 (SEQ ID NO 21) и 543-631 (SEQ ID NO 22) получали так же, как описано выше, для использования в способах данного изобретения.
В. Анализ связывания лиганд-рецептор
Синтетические пептиды, полученные в примере вместе с описанными выше слитыми белками ММР-2, скринировали дополнительно, чтобы измерить их способность находиться в антагонизме с рецептор-связывающей активностью αvβ3 и αIIbβ3 в анализах по связыванию выделенного очисткой лиганд-рецептора. Метод изучения этих связываний описаны у Barbas с соавт., Proc. Natl. Acad. Sci. USA, 90:10003-10007 (1993), Smith с соавт., J. Biol. Chem., 265:11008-11013 (1990), и Pfaff с соавт., J. Diol. Chem., 269:20233-20230 (1994), описания которых включены, таким образом, путем ссылки.
Здесь описан метод идентификации антагонистов в анализе по связыванию лиганд-рецептор, в котором рецептор иммобилизован на твердой подложке, а лиганд и антагонист - растворимы. Описан также анализ по связыванию лиганд-рецептора, в котором лиганд иммобилизован на твердой подложке, а рецептор и антагонист - растворимы.
Вкратце, выбранные очищенные интегрины были по отдельности иммобилизованы в лунках планшета для микротитрования Titertek в концентрации для покрытия 50 нанограмм (нг) на лунку. Очищенные рецепторы, используемые для анализов по связыванию лиганд-рецептора, хорошо известны в данной области и легко получаемы методами, хорошо знакомыми для рядового специалиста в данной области. После инкубации в течение 48 часов при 4oС, неспецифические связывающие сайты на данном планшете блокировали с помощью бычьего сывороточного альбумина (ВСА), 10 мг/мл, в Трис-солевом буфере. В исследованиях по ингибированию тестировали различные концентрации выбранных пептидов из таблицы 1, чтобы блокировать связывание 125I-витронектина или 125I-фибриногена с интегриновыми рецепторами αvβ3 и αIIbβ3. Хотя эти лиганды проявляют оптимальное связывание по конкретному интегрину, витронектин - по αvβ3, а фибриноген - по αIIbβ3, исследования по ингибированию связывания с использованием пептидов для блокирования связывания фибриногена с любым рецептором позволяют точно определять количество в микромолях (мкМ) пептида, необходимого для полумаксимального ингибирования связывания рецептора с лигандом. Лиганды с радиоактивной меткой использовали в концентрациях 1 нМ, а связывание контролировали отдельно с немечеными синтетическими пептидами.
После трехчасовой инкубации свободный лиганд удаляли промывкой, а связанный лиганд детектировали гамма-счетчиком. Данные из этих анализов, в которых использовали выбранные циклические пептиды, представленные в таблице 1 для ингибирования связывания рецепторов и фибриногена с радиоактивной меткой, чтобы отдельно иммобилизовать рецепторы αvβ3 и αIIbβ3, были высоковоспроизводимыми с ошибкой между частными значениями, обычно ниже 11%. IC50-данные в микромолях (IC50 мкМ) выражены в виде средней повторенных частных значений ± стандартное отклонение, как показано в таблице 2.
Следовательно, RGD-содержащие или RGD-полученные циклические пептиды 62181, 62184, 62185 и 62187, обладающие, каждый, одним D-аминокислотным остатком, проявляют предпочтительное ингибирование связывания фибриногена с рецептором αvβ3 по измерению нижней концентрации пептида, необходимого для полумаксимального ингибирования по сравнению с ингибированием по рецептору αIIbβ3. В противоположность этому другие RGD-содержащие или RGD-полученные циклические пептиды 62186, 62175 и 62179 были либо не настолько эффективны в блокировании связывания фибриногена с αvβ3, либо не проявляли предпочтительного ингибирования связывания фибриногена с αIIbβ3 в сравнении с αvβ3. Эти результаты согласуются с результатами, недавно опубликованными Pfaff с соавт., J. Biol. Chem., 269:20233-20238 (1994), в которых циклический пептид 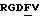 (где
(где  указывает D-аминокислотный остаток) специфически ингибирует связывание фибриногена с αvβ3-интегрином, а не с интегринами αIIbβ3 или α5β1. Подобные анализы по ингибированию связывания осуществляли с линеаризованными пептидами, обладающими или не обладающими RGD-мотивом, последовательностями, которые получены из αv-субъединицы рецептора, αIIb-субъединицы рецептора или из последовательностей аминокислотных остатков витронектинового лиганда. Последовательности линеаризованных пептидов, 62880 (аминокислотные остатки 35-49, производные VN), 62411 (аминокислотные остатки 676-687, производные αv); 62503 (аминокислотные остатки 655-667, производные αv) и 62502 (аминокислотные остатки 296-306, производные αIIb), представлены в таблице 1. Каждый из этих пептидов использовали в отдельных анализах, чтобы ингибировать связывание витронектина (VN) или фибриногена (FG) либо с αIIbβ3, либо с αvβ3. Данные по IC50 в микромолях (IC50 мкМ) в индивидуальном анализе для каждого эксперимента показаны в таблице 3.
указывает D-аминокислотный остаток) специфически ингибирует связывание фибриногена с αvβ3-интегрином, а не с интегринами αIIbβ3 или α5β1. Подобные анализы по ингибированию связывания осуществляли с линеаризованными пептидами, обладающими или не обладающими RGD-мотивом, последовательностями, которые получены из αv-субъединицы рецептора, αIIb-субъединицы рецептора или из последовательностей аминокислотных остатков витронектинового лиганда. Последовательности линеаризованных пептидов, 62880 (аминокислотные остатки 35-49, производные VN), 62411 (аминокислотные остатки 676-687, производные αv); 62503 (аминокислотные остатки 655-667, производные αv) и 62502 (аминокислотные остатки 296-306, производные αIIb), представлены в таблице 1. Каждый из этих пептидов использовали в отдельных анализах, чтобы ингибировать связывание витронектина (VN) или фибриногена (FG) либо с αIIbβ3, либо с αvβ3. Данные по IC50 в микромолях (IC50 мкМ) в индивидуальном анализе для каждого эксперимента показаны в таблице 3.
Результаты опытов по ингибированию связывания лиганда с выбранными интегриновыми рецепторами для линеаризованных пептидов показывают, что только пептид 62880 был эффективен в ингибировании полумаксимального связывания FG или VN с αvβ3, как измерено по наименьшей концентрации пептида, необходимого для полумаксимального ингибирования, по сравнению с рецептором αIIbβ3. Ни один из других линеаризованных пептидов не был эффективен в блокировании связывания лиганда с αvβ3, хотя пептид 62502 был эффективен в блокировании связывания VN с αIIbβ3.
В других опытах по связыванию рецептора с лигандом, осуществленных как описано выше, за исключением того, что детектирование связывания или его ингибирование осуществляли в ELISA и с конъюгированными с пероксидазой антителами козы к анти-IgG кролика, лиганды VN, ММР-2 и фибронектин в диапазоне 5-50 нг/лунку, и приведенных в порядке эффективности, показали связывание с иммобилизованным рецептором αvβ3, а коллаген - не показал. Кроме того, способность пептидов ингибировать связывание ММР-2 или VN с иммобилизованным αvβ3 испытывали с пептидами 69601 (SEQ ID NO 6) и 66203 (SEQ ID NO 5). Только пептид 66203 был эффективен в ингибировании связывания любого субстрата с рецептором αvβ3, а контрольный пептид 69601 не обладает эффектом с любым лигандом.
Специфичность связывания ММР-2 с интегриновыми рецепторами была подтверждена твердофазным методом связывания рецептора, в котором полированный ММР-2 связывался с αvβ3 и не связывался с αIIbβ3, который был иммобилизован на твердой фазе (300 cpm по связыванию против приблизительно 10 СРМ по связыванию). Способность пептида, произведенного ММР-2 или слитого белка ингибировать специфическое связывание ММР-2 с αvβ3 было продемонстрировано в сопоставимом опыте, результаты которого показаны на фигуре 23. GST-CTMMP-2 (445-637) (именуемый также как СТММР-2(2-4)) слитый белок, полученный как описано выше, меченный GST-MAID, ингибирует связывание йодированного ММР-2 с αvβ3, тогда как один GST не обладает эффектом с уровнями связывания СРМ, соизмеримого со всеми лунками, не получившими ингибитора (помеченный NT). ММР-2-слитый белок, именуемый как СТММР-2 (274-637), именуемый также как СТММР-2 (10-1), не ингибирует связывание меченого ММР-2 с
αvβ3.
Специфичность взаимодействия рецептора с ММР-2-производными антагонистов подтверждали прямым и конкурентным твердо-фазным методом. СТММР-2 (2-4), отмеченный на фигуре в виде [1251]GST2-4, связывается с αvβ3, но не с αIIbβ3, тогда как СТММР-2 (10-1), отмеченный на фигуре в виде [1251]GST10-1, не связывается с любым рецептором в твердофазном методе in vitro. Кроме того, связывание меченого GST2-4 конкурирует с немеченым GST2-4.
Поэтому лиганд-рецепторный опыт, описанный здесь, может использоваться для скринирования и циклических и линеаризованных синтетических пептидов, которые проявляют избирательную специфичность по конкретному интегриновому рецептору, особенно αvβ3, который используется в качестве антагониста витронектинового рецептора (αvβ3) в практике данного изобретения.
5. Характеристика необработанной хориоаллантоидной мембраны цыпленка (САМ)
А. Получение САМ
Ангиогенез, который можно индуцировать в хориоаллантоидной мембране цыпленка (САМ), после нормального эмбрионального ангиогенеза получали для образования зрелых кровеносных сосудов. Было показано, что ангиогенез индуцируется в ответ на специфические цитокины или фрагменты опухоли, как описано у Leibovich с соавт., Nature, 329:630 (1987) и Ausprunk с соавт., Am. J. Pathol. , 79: 597 (1975). САМs получали из эмбрионов цыплят для последующей индукции ангиогенеза и его ингибирования, как описано соответственно в примерах 6 и 7. 10-дневные эмбрионы цыплят получали от McIntyre Poultry (Lakeside, СА) и инкубировали при 37oС и 60% влажности. В скорлупе на оконечности яйца высверливали отверстие непосредственно над воздушным пузырем с использованием небольшой профессиональной дрели (Dremel, Division of Emerson Electris Co. Racine WI). Второе отверстие сверлили на широкой оконечности яйца в области, не содержащей эмбриональных кровеносных сосудов, определенных заранее просвечиванием яйца. К первому отверстию прикладывали отрицательное давление, которым вытягивали САМ (хориоаллантоидная мембрана) из скорлуповой мембраны и создавали ложный мешок над САМ. Квадратное окно 1,0 сантиметр (см) • 1,0 см вырезали в скорлупе над отброшенным САМ с использованием небольшой модели шлифовального круга (Dremel). Это небольшое окно дает непосредственный доступ к подлежащей САМ.
Затем полученный препарат САМ использовали либо на 6-й день эмбриогенеза, по отмеченной стадии активной реваскуляризации, без дополнительной обработки САМ, отражающего модель, используемую для оценки эффектов на эмбриональную реваскуляризацию, либо использовали на 10-й день эмбриогенеза, когда ангиогенез убывает. Этот последний препарат, таким образом, использовали в данном изобретении для индукции возобновленного ангиогенеза в ответ на цитокиновую обработку или на контакт с опухолью, как описано в примере 6.
В. Гистология САМ
Чтобы проанализировать микроскопическую структуру CAMs эмбрионов цыплят и/или опухоли человека, которую удаляли из эмбриона цыпленка, как описано в примере 8, CAMs и опухоли получали в виде замороженных срезов, как описано в примере 3А. Замороженные блоки нарезали на срезы толщиной шесть микрон (мкм) на замораживающем микротоме для иммунофлуоресцентного анализа.
Фигура 4 показывает типичную микрофотографию области без кровеносных сосудов в необработанном 10-дневном САМ. Поскольку на этой стадии эмбриогенеза ангиогенез в САМ-системе убывает, эта система полезна для данного изобретения для стимуляции образования новой сосудистой системы из существующих сосудов примыкающих областей в область САМ, не имеющей в данное время каких-либо сосудов.
С. Интегриновые профили, детектируемые в САМ с помощью иммунофлуоресценции
Чтобы проанализировать тканевое распределение интегриновых рецепторов, присутствующих в САМ-тканях, 6 микронные замороженные срезы опухолевой ткани и САМ-ткани эмбриона цыпленка фиксировали в ацетоне в течение 30 секунд и окрашивали для иммунофлуоресценции 10 микрограммами/миллилитр (мкг/мл) mAb CSAT, моноклональным антителом, специфичным по интегриновой субъединице β1, как описано у Buck с соавт., J. Cell Biol., 107:2351 (1988), и, таким образом, использовали для контролей, или LM609, как получали в примере 2. Первичное окрашивание с последующим окрашиванием при разведении 1:250, родамин-меченными вторичными антителами козы к антимышиным антителам (Таgо), обеспечивает детекцию первичного продукта иммунной реакции. Эти срезы анализировали затем в Цейсовском иммунофлуоресцентном микроскопе.
Результаты анализа иммунофлуоресценции показали, что зрелые кровеносные сосуды, присутствующие в необработанном 10-дневном эмбрионе цыпленка, экспрессируют интегриновую субъединицу β1 (фигура 5А). Напротив, в серийном срезе ткани, показанном на фигуре 5А, не обнаруживается иммунная реактивность с LM609 (фигура 5В). Следовательно, интегрин αvβ3, детектируемый с помощью антитела LM609, активно не экспрессируемый в зрелых кровеносных сосудах, присутствует у 10-дневного необработанного эмбриона цыпленка. Как показано в СДМ-модели и в последующих примерах, пока кровеносные сосуды подвергаются новому росту в нормальном эмбриогенезе или индуцированы каким-либо цитокином или опухолью, эти кровеносные сосуды экспрессируют αvβ3. Однако после активной реваскуляризации, когда развитие кровеносных сосудов прекратилось, экспрессия αvβ3 уменьшается до уровней, не детектируемых в иммунофлуоресцентном анализе. Данная регуляция экспрессии αvβ3 в кровеносных сосудах, подвергающихся ангиогенезу, в противоположность отсутствию экспрессии в зрелых сосудах создает уникальную способность данного изобретения по контролю и ингибированию ангиогенеза, как показано в нижеследующих примерах с использованием аналитической системы ангиогенеза в САМ.
В других профилях в 10-дневной САМ-модели в эндотелиальных клетках, подвергающихся ангиогенезу через 3 дня после индукции bFGF, солокализовали металлопротеиназу ММР-2 и αvβ3. ММР-2 лишь минимально экспрессировался в сосудах, в которых отсутствует рецептор αvβ3. Кроме того, ММР-2 солокализовали с αvβ3 в ангиогенных кровеносных сосудах, ассоциированных in vivo с опухолью M21-L (опухоли получали от инъецированных меланомными клетками человека M21-L кожных трансплантатов человека, выращенных на SCID-мыши, как описано в примере 11), но не с предсуществующими кровеносными сосудами, не ассоциированными с опухолью. Аналогичные результаты избирательной ассоциации ММР-2 и αvβ3 были также получены в САМ-модели с меланомными опухолями CS-1α, несущими αvβ3, но не с клетками CS-1 без αvβ3.
6. Анализ ангиогенеза в САМ
А. Ангиогенез, индуцированный факторами роста
Было показано, что ангиогенез индуцируется цитокинами или факторами роста, как рассматривается в примере 5А. В описанных здесь экспериментах ангиогенез в САМ-препарате, описанный в примере 5, индуцировали факторами роста, которые, как описано здесь, наносили местно на кровеносные сосуды САМ.
Ангиогенез индуцировали путем помещения 5 мм • 5 мм Ватмановского фильтровального диска (Whatman Filter paper No. 1), насыщенного сбалансированным солевым раствором Хэнкса (HBSS, GIBCO, Grand Island, NY) или HBSS, содержащим 150 нг/мл рекомбинантного основного фактора роста фибробластов (bFGF) (Genzyme, Cambridge, МА), на САМ 10-дневного эмбриона цыпленка в области без кровеносных сосудов и позднее это окно заклеивали пластырем. В других опытах 125 нг/мл bFGF был также эффективен в индукции роста кровеносного сосуда. Для опытов, где ингибирование ангиогенеза оценивали с помощью внутривенных инъекций антагонистов, ангиогенез вначале индуцировали с помощью 1-2 мкг/мл bFGF в фибробластной ростовой среде. Ангиогенез контролировали с помощью фотомикроскопии через 72 часа. CAMs резко замораживали и 6-микронные замороженные срезы фиксировали в ацетоне и окрашивали для иммунофлуоресценции, как описано в примере 5С, с 10 мкг/мл моноклонального антитела CSAT или LM609 к анти-β1.
Полученная иммунофлуоресцентная микрофотография на фигуре 5С показывает усиление экспрессии αvβ3 в САМ цыпленка во время ангиогенеза, индуцированного bFGF, в противоположность отсутствию экспрессии αvβ3 в необработанном САМ цыпленка, как показано на фигуре 5В. αvβ3 было легко детектировать во многих (75%-80%) сосудах CAMs, обработанных bFGF. Кроме того, экспрессия интегрина β1 не изменялась, поскольку β1 в необработанном САМ был также легко детектируем в стимулированных кровеносных сосудах.
Затем количественно выражали относительную экспрессию интегринов αvβ3 и β1 во время ангиогенеза, индуцированного bFGF, с помощью анализа лазерного софокального изображения замороженных срезов САМ. Затем окрашенные срезы анализировали с помощью Цейсовского лазерного софокального микроскопа. Двадцать пять сосудов, окрашенных с помощью LM609, и 15 - окрашенных с помощью CSAT (средний размер ~1200 мм2, в диапазоне 350-3500 мм2) были отобраны из случайных полей, а среднюю родаминовую флуоресценцию для каждого сосуда на единицу области измеряли в произвольных единицах с помощью анализа лазерного софокального изображения. Данные выражены в виде средней интенсивности флуоресценции в произвольных единицах сосудов ± стандартная ошибка (SE).
Графические результаты на фигуре 6 показывают, что окрашивание αvβ3 было существенно усилено (выше в четыре раза) в CAMs, обработанных bFGF, как установлено в критерии ранговых сумм Вилкоксона (Р<0,0001), хотя окрашивание β1 с bFGF-обработкой существенно не отличалось.
Опыт с САМ дополнительно использовали для проверки эффекта другого сильного индуктора ангиогенеза, фактора некроза опухолей альфа (TNFα), на экспрессию интегринов β1 и β3. Обнаружили, что фильтровальные диски, импрегнированные либо с bFGF, либо с TNFα и помещенные на САМs 10-дневных эмбрионов, способствуют локальному ангиогенезу через 72 часа.
Полученные результаты показаны на микрофотографиях CAMs необработанных (фигура 7А), обработанных bFGF (фигура 7В) или обработанных TNFα (фигура 7С). Кровеносные сосуды легко различимы на препаратах, обработанных и bFGF и TNFα, но не на необработанных САМ. Следовательно, местное применение ростового фактора/цитокина приводит к индукции ангиогенеза от зрелых сосудов примыкающей области в область исходно без кровеносных сосудов. Принимая во внимание индукцию кровеносных сосудов bFGF и сопутствующую экспрессию αvβ3, как показано на фигуре 5С, обработка TNFα приводит к сравнимым активностям.
Эти факты свидетельствуют, что кровеносные сосуды и человека и цыпленка, участвующие в ангиогенезе, показывают усиление экспрессии αvβ3. В соответствии с этим экспрессия αvβ3 в культивируемых эндотелиальных клетках может быть индуцирована различными цитокинами in vitro, как описано у Janat с соавт. , J. Cell Physiol., 151:588 (1992); Enenstein с соавт., Exp. Cell Res., 203:499 (1992) и Swerlick с соавт., J. Invest. Derm., 99:715 (1993).
Эффект фактора роста, индуцирующего ангиогенез с помощью антител и пептидных ингибиторов, представлен в примерах 7А и 7В.
В. Эмбриональный ангиогенез
САМ-препарат для оценки эффекта ингибиторов ангиогенеза на естественное образование эмбриональной новой сосудистой системы представлял собой, как описано ранее, 6-дневный находящийся в зачаточном состоянии эмбрион цыпленка. На этой стадии развития кровеносные сосуды подвергали росту de novo и, таким образом, создавали пригодную систему для определения участия αvβ3 в эмбриональном ангиогенезе. САМ-систему получали, как описано выше, за исключением того, что данный опыт осуществляли на 6-й день зародышевого развития, а не на 10-й день. Эффект на эмбриональный ангиогенез путем обработки антителами и пептидами данного изобретения представлен в примере 7С.
С. Ангиогенез, индуцированный опухолями
Чтобы исследовать роль αvβ3 в ангиогенезе, индуцированном опухолью, в САМ-анализе применяли различные фрагменты карциномы и меланомы человека, негативные по αvβ3, ранее выращенные и выделенные из САМ 17-дневного эмбриона цыпленка, как описано у Brooks с соавт., J. Cell Biol., 122:1351 (1993) и как описано здесь. Данные фрагменты индуцировали обширную реваскуляризацию в присутствии одного буфера.
Ангиогенез индуцировали в САМ-аналитической системе прямым наложением опухолевого фрагмента на САМ. Получение САМ эмбриона цыпленка было идентично методике, описанной выше. Вместо фильтровального бумажного диска, 50 миллиграмм (мг) - 55 мг фрагмента одной из меланомной опухоли M21-L человека, карциномной опухоли UCLAP-3 легкого человека, панкреатической карциномной клеточной линии FG человека (Cheresh с соавт., Cell 58:945-953, 1989) или карциномной клеточной линии НЕр3 гортани человека, все они являются негативными опухолями по αvβ3, помещали на САМ в области исходно без кровеносных сосудов.
Меланомная клеточная линия M21-L человека, карциномная клеточная линия легкого человека UCLAP-3, панкреатическая карциномная клеточная линия FG или карциномная клеточная линия гортани человека НЕр3, все негативные по αvβ3, использовали для роста твердых опухолей человека на САМs эмбрионов цыпленка. Суспензию из единственной клетки, 8•106 M21-L, UCLAP-3 и FB или 5•105 НЕр3-клеток вначале наносили на CAMs в общем объеме 30 мкл стерильного HBSS. Окна заклеивали пластырем и данные эмбрионы инкубировали в течение 7 дней, чтобы дать вырасти опухолевым поражениям. В конце 7-го дня, теперь 17-дневного эмбриона, полученные опухоли удаляли из CAMs и подравнивали свободную ткань, окружающую САМ. Эти опухоли резали на опухолевые фрагменты 50 мг - 55 мг для использования либо в ангиогенезе, либо в опытах по опухолевому росту. Эти опухолевые фрагменты помещали в новую группу CAMs 10-дневных эмбрионов цыпленка, как описано в примере 6А, в область без кровеносных сосудов.
Опухоли, выращенные in vivo в CAMs эмбрионов цыпленка, окрашивали с помощью mАb LM609, как описано в примере 3А, для экспрессии αvβ3. Наблюдаемое неспецифическое окрашивание опухолевых клеток свидетельствует об отсутствии экспрессии αvβ3.
Эти опухолевые препараты САМ были затем последовательно обработаны, как описано в примерах 7D и 7Е, для измерения эффекта антител и пептидов на ангиогенез, индуцированный опухолью. Опухолевые препараты САМ обрабатывали так же, как описано в примере 8, 9 и 12, для измерения эффекта антител и пептидов на регрессию опухолей и апоптоза ангиогенных кровеносных сосудов и сосудистых клеток.
7. Ингибирование ангиогенеза по измерению в САМ-анализе
А. Ингибирование ангиогенеза, индуцированного фактором роста путем местного применения ингибиторов
1) Обработка моноклональными антителами
Чтобы определить, действительно ли αvβ3 играет активную роль в ангиогенезе, фильтровальные диски, насыщенные bFGF или TNFa помещали на CAMs, затем к данному препарату добавляли моноклональные антитела (именуемые также mAb), LM609 (специфичными по αvβ3 CSAT (специфичный β1) или P3G2 или также P1F6 (оба специфичные по αvβ5.
Ангиогенез индуцировали в CAMs от 10-дневных эмбрионов цыплят путем насыщения фильтровальных дисков bFGF. Затем диски обрабатывали 50 мл HBSS, содержащего 25 мг mAb в общем объеме 25 мкл стерильного HBSS в 0, 24 и 48 часов. Через 72 часа CAMs собирали и помещали в 35 мм чашку Петри и однократно промывали 1 мл фосфатно-солевого буфера. Затем данную сторону фильтровальной бумаги и САМ-ткани анализировали под стереомикроскопом Olympus с двумя наблюдениями в дважды слепом методе. Ингибирование ангиогенеза считалось существенным, если CAMs проявляли >50% снижения в инфильтрации кровеносного сосуда из САМ непосредственно ниже на диск. Эксперименты повторяли четыре раза на антитело с 6-7 эмбрионами на состояние.
Результаты воздействия mАb-обработки на ангиогенез, индуцированный bFGF, показаны на фигурах 8А-8В. Необработанный САМ-препарат без кровеносных сосудов показан на фигуре 8А для сравнения с индуцированным bFGF кровеносным сосудом, показанным на фигуре 8В, и после воздействия на него mAb - на фигурах 8С-8Е. Почти 75% этих CAMs, обработанных с помощью mAb LM609, проявляют >50% ингибирования ангиогенеза, как показано на фигуре 8Е и многие из них появляются без сосудистой инфильтрации. Напротив, контроль с буфером (фигура 8А) и диски, обработанные с помощью mAb CSAT (фигура 8С) и P3G2 (фигура 8D) последовательно проявляют васкуляризацию.
Идентичные результаты были получены, когда ангиогенез индуцировали с помощью TNFα. Чтобы проверить влияние этих одних и тех же антител на предсуществующие зрелые кровеносные сосуды, присутствующие в нормальной ткани, обрабатывали прилежащие области без кровеносных сосудов, фильтровальные диски, насыщенные mAbs помещали на васкуляризованные области CAMs 10-дневных эмбрионов, которые не получали местного применения цитокина. Ни один из этих трех mAbs не воздействовал на предсуществующие сосуды, как установили визуализацией под стереомикроскопом. Следовательно, mAb LM609 избирательно ингибирует только рост новых кровеносных сосудов и не влияет на зрелые кровеносные сосуды, присутствующие в прилегающих областях. Этот же эффект был виден при применении синтетических пептидов, либо нанесенных местно, либо введенных внутривенно, как описано соответственно в примерах 7А2) и 7Е2).
2) Обработка синтетическими пептидами
САМ-анализы осуществили также с синтетическими пептидами данного изобретения, чтобы определить эффект циклического и линеаризованного пептидов в ростовом факторе, индуцирующем ангиогенез. Эти пептиды получали, как описано в примере 1, и 80 мкг присутствовало в общем объеме 25 мкл стерильного HBSS. Этот пептидный раствор наносили непосредственно на САМ-препарат и нанесение повторяли через 24 и 48 часов. Через 72 часа эту фильтровальную бумагу и окружающую САМ ткань иссекали и просматривали, как описано выше.
Обнаруженные результаты данного анализа были аналогичны результатам, показанным на фигурах 9А-9С, как описано в примере 7Е2), где синтетические пептиды внутривенно инъецировали в опухоль, индуцирующую кровеносные сосуды. В данном случае, что касается контрольного пептида 62186, кровеносные сосуды, индуцированные bFGF, оставались ненарушенными, как показано на фигуре 9А. В противоположность этому при нанесении на фильтр циклического RGD-пептида 62814 образование кровеносных сосудов ингибировалось в данной области без образования новой сосудистой системы. Данный эффект внешне был аналогичен эффекту, который показан на фигуре 9В, как описано ниже в примере 7Е2). Кроме того, как показано также на фигуре 9С для внутривенно инъецируемых пептидов, в областях, в которых присутствуют зрелые кровеносные сосуды, отдаленное размещение фильтра, насыщенного фактором роста, не оказывает видимого влияния с обработанными местно синтетическими пептидами на эти находящиеся на расстоянии сосуды. Следовательно, ингибирующая активность данных пептидов на ангиогенез ограничена областями ангиогенеза, индуцируемого факторами роста и не оказывает влияния на прилегающие предсуществующие зрелые сосуды или не приводит к пагубной цитотоксичности на окружающую область.
Аналогичные опыты осуществляли с другими синтетическими пептидами, полученными в примере 1 и приведенными в таблице 1.
3) Обработка ММР-2-пептидными фрагментами
Чтобы продемонстрировать биологические эффекты пептидных фрагментов ММР-2 на ангиогенез, осуществляли опыты с САМ, как описано выше, за исключением того, что ангиогенез индуцировали диск-фильтрами, насыщенными в течение 10 минут bFGF в концентрации 1,0 мкг/мл HBSS. Затем эти диски позиционировали на САМ в области, в которой численность предсуществующих сосудов снижена. С-концевой слитый белок СТММР-2 (410-637), полученный как описано раньше, или слитый белок, ассоциированный с контрольным GST-рецептором (RAP) (1,5 мкг в 30 мкл HBSS), наносили затем местно однократным фильтровальным диском в день в течение трех дней. В конце инкубационного периода прекращали инкубацию этих эмбрионов и данный фильтровальный диск и подлежащую САМ-ткань удаляли и анализировали на ангиогенез под стереомикроскопом. Ангиогенез выражали количественно путем подсчета числа точек ветвления кровеносных сосудов в границах фильтровальных дисков. Ветвленые кровеносные сосуды рассматривали по отношению к исходным как вновь проросшие ангиогенные кровеносные сосуды.
Количественный анализ осуществляли дважды слепым способом как минимум в двух независимых наблюдениях. Полученные результаты выражали в виде чисел точек ветвления (стимулируемых bFGF) минус число точек ветвления (нестимулируемый контроль) на фильтровальный диск. Эксперименты, как положено, имели 6-10 эмбрионов на состояние.
Результаты опыта по ангиогенезу САМ показаны на фигурах 25A-D, 26 и 27. На фигуре 25 серии фотографий, разделенные на четыре фигуры. Фигуры 25A-D иллюстрируют в сравнении ингибирование ангиогенеза в присутствии слитого белка СТММР-2 (СТММР-2 (410-637)) (фигуры 25C-D) и неингибирование в присутствии контрольного GST-слитого белка (фигуры 25 А-В). Фигуры 26 и 27 представляют собой гистограммы, иллюстрирующие индекс ангиогенеза в опыте по ангиогенезу САМ с СТММР-2, тот же слитый белок, что и выше, в сравнении с контролями (только bFGF или слитый белок GST-RAP). На Фигуре 27 показаны результаты двух отдельных экспериментов (#1 & #2) с использованием слитого белка СТММР-2 (410-637).
Эти результаты показывают, что на всех трех фигурах слитый белок СТММР-2 или полипептид, содержащий С-концевой домен из ММР-2, является пригодной композицией для ингибирования ангиогенеза, опосредованного bFGF, через ингибирование αvβ3.
В. Ингибирование ангиогенеза, индуцируемого фактором роста, при внутривенном введении ингибиторов
1) Обработка моноклональными антителами
Влияние на ангиогенез, индуцируемый ростовым фактором, моноклональных антител, внутривенно инъецируемых в САМ-препарат, использовали для оценки в данном изобретении.
Получение CAMs эмбрионов цыплят для внутривенных инъекций было, в сущности, таким, как описано в примере 7А с некоторыми модификациями. Во время просвечивания в проходящем свете выбирали кровеносные сосуды, а метки ставили на яичной скорлупе с указанием их позиций. В скорлупе сверлили отверстия и вываливали CAMs, а фильтровальные бумажки, насыщенные bFGF, помещали на CAMs, как описано выше. Окна заклеивали стерильным пластырем и эмбрионов помещали в инкубатор. Через двадцать четыре часа небольшое второе окно вырезали на латеральной стороне скорлупы яйца прямо над заранее выбранными просвеченными кровеносными сосудами. Наружную яичную скорлупу аккуратно удаляли от краевых интактных эмбриональных мембран. Оболочечную мембрану делали прозрачной с помощью небольшой капли минерального масла (Perkin-Elmer Corp. Norwalk, CT), что позволяло легко визуализировать кровеносные сосуды. Выделенные очисткой стерильные mAb, или синтетические пептиды, последние описаны ниже, однократно инокулировали в кровеносные сосуды с помощью иглы 30-го размера в дозе 200 мкг IgG в общем объеме 100 мкл стерильного PBS. Полученные окна заклеивали пластырем и эмбрионам позволяли инкубироваться в течение 72 часов. Полученные фильтровальные диски и окружающие САМ ткани анализировали, как описано раньше.
Чтобы определить локализацию mAb LM609 в САМ-тканях или в опухолевых тканях, как показано здесь и в последующих примерах, которые были заранее инокулированы внутривенно LM609, фиксированные срезы блокировали с помощью 2,5% BSA в HBSS в течение 1 часа при комнатной температуре с последующим окрашиванием при разведении 1:250, родамин-меченным вторичным антителом козы к анти-мышиному антителу (Таgо). Эти срезы анализировали затем в Цейсовском иммунофлуоресцентном микроскопе.
Результаты внутривенной антительной обработки, индуцированного bFGF, кровеносного сосуда САМ-препарата, показаны на фигурах 10А-10С. На фигуре 10А показан ангиогенез как результат обработки bFGF. Изменения в присутствии bFGF, индуцирующего образование сосудистой системы, не было видно при внутривенном выдерживании с mAb P3G2, антителом к анти-αvβ5, как показано на фигуре 10В. Напротив, обработка индуцируемого bFGF ангиогенеза САМ-препарата с помощью LM609, антителом к анти-αvβ3, приводила к полному ингибированию роста новых сосудов в области фильтра, как показано на фигуре 10С. Ингибирующий эффект ангиогенеза происходит, следовательно, из ингибирующей активности рецептора αvβ3 с помощью LM609 антитела, специфичного к анти-αvβ3. Поскольку блокирование αvβ5 не ингибирует образование вновь образованной сосудистой системы в CAMs на участке фильтра, следовательно αvβ5 по сравнению с αvβ3 не существенен для роста новых сосудов.
2) Обработка синтетическими пептидами
В САМ-препаратах, в которых ангиогенез индуцировали с помощью 1-2 мкг/мл bFGF, как описано прежде, синтетические пептиды 69601 (контроль) и 66203 (SEQ ID NO 5) инъецировали внутривенно отдельно в 18-часовые САМ-препараты после bFGF-индукции ангиогенеза. Эти препараты поддерживали в дополнительные 36-40 часов, после чего, как описано прежде, определяли число точек ветвления.
Полученные результаты показаны на фигуре 28, где пептид 66203 полностью ингибировал ангиогенез, индуцированный bFGF, в противоположность отсутствию ингибирования контрольным пептидом.
В других анализах оценивали вклад пептида 85189 (SEQ ID NO 15) в ингибирование ангиогенеза, индуцированного bFGF в САМ-анализе с диапазоном дозы от 10 мкг/эмбрион до 300 мкг/эмбрион. Данный анализ осуществляли, как описано ранее. Полученные результаты показаны на фигуре 29, где наименьшая эффективная доза равнялась 30 мкг при том, что 100 и 300 мкг почти полностью ингибировали ангиогенез.
В последующих анализах пептид 85189 еще сравнивали с пептидами 69601 и 66203 на анти-ангиогенную активность. Данный анализ осуществляли, как описано выше, за тем исключением, что использовали 50 мкг пептида. Полученные результаты, графически изображенные на фигуре 30, показывают, что пептиды 66203 (маркированный 203) и 85189 (маркированный 189) являлись эффективными ингибиторами ангиогенеза, опосредованного bFGF по сравнению с bFGF-обработанным (маркированный bFGF) и 69601-обработанным (маркированный 601) контролями.
Эффективность пептида 85189 различных солевых составов также оценивали в аналогичном bFGF-индуцированном САМ-анализе. Эти пептиды использовали при 100 мкг/эмбрион. Одна и та же пептидная последовательность в НСl (пептид 85189) и в TFA (пептид 121974) ингибирует bFGF-индуцированный ангиогенез с HCL-составным пептидом несколько более эффективно, чем с TFA-составным пептидом (соответствующие числа точек ветвления для пептида 85189 в сравнении с 121974 составляет 30 против 60). Необработанные САМs, маркированные "без цитокина", имели вдвое меньшее число точек ветвления, как это можно видеть при обработке с помощью bFGF, соответственно 70 против 190. Обработка контрольным пептидом 69601 не имела эффекта на ингибирование ангиогенеза (230 точек ветвления).
Другие синтетические пептиды, полученные в примере 1, по отдельности инъецировали внутривенно в кровеносные сосуды САМ-препарата, индуцированного фактором роста, как описано выше. Эффект пептидов на жизненность сосудов оценивали аналогичным образом.
3) Обработка с помощью фрагментов ММР-2
С помощью вышеописанного протокола оценивали также эффект ММР-2-слитых белков, СТММР-2 (2-4), именуемого также в качестве СТММР-2 (445-467) и СТММР-2 (10-1), именуемого также в качестве СТММР-2 (274-637). Данный анализ осуществляли, как описано прежде, за тем исключением, что 50 мкг слитого белка вводили в эмбрионы, обработанные bFGF. Эффект обработки слитым белком оценивали через 24 часа, 48 часов и 72 часа.
Полученные результаты показаны в эти выбранные периоды времени на фигурах 31A-L, где ангиогенез оценивали фотографически при анализе состояний без обработки, без bFGF-обработки, без bFGF-обработки с последующей обработкой СТММР-2 (2-4), маркированного в качестве bFGF + MAID (MAID = ММР-2-домену, ингибирующему ангиогенез) и без bFGF-обработки с последующей обработкой СТММР-2 (10-1), маркированного в качестве bFGF + Контроль. Существенная индукция ангиогенеза после 48 часов и 72 часов, после обработки bFGF, была почти полностью ингибирована лишь при выдерживании с СТММР-2 (2-4). Протяженность ингибирования с помощью СТММР-2 (2-4) была выше, чем это можно видеть для СТММР-2 (10-1), который проявляет некоторую анти-ангиогенную активность in vivo.
Другие ММР-2-композиции, целый ММР-2, фрагменты и слитые белки, полученные как описано прежде, также по отдельности, внутривенно инъецированные в кровеносные сосуды САМ-препарата, индуцированного фактором роста, как описано выше. Эффект пептидов на жизненность сосудов оценивали аналогичным образом.
С. Ингибирование эмбрионального ангиогенеза путем местного применения
1) Обработка моноклональными антителами
Чтобы определить, действительно ли αvβ3 участвует в эмбриональном ангиогенезе, эффект LM609 на рост кровеносных сосудов de novo в CAMs проверяли у 6-дневных эмбрионов на стадии, отмеченной активной реваскуляризацией, как описано в примере 5А. САМ-анализ готовили, как описано в примере 6С, с последующей местной аппликацией дисков, насыщенных mAbs и помещенных в CAMs 6-дневных эмбрионов в отсутствие цитокинов. Через 3 дня CAMs вырезали и фотографировали. Каждый эксперимент включал 6 эмбрионов в группе и был повторен 2 раза.
Антитело LM609 (фигура 11С), но не CSAT (фигура НА) или P3G2 (фигура 11В) предотвращали рост сосудов в этих состояниях; это свидетельствует, что αvβ3 играет существенную роль в эмбриональной реваскуляризации, которая не зависела от добавляемых факторов роста для индукции ангиогенеза.
2) Обработка с помощью синтетических пептидов
Синтетические пептиды, полученные в примере 1 добавляли по отдельности, к эмбриональному САМ-препарату, полученному выше и как описано в примере 5А2) либо путем местной аппликации на САМ, либо внутривенным введением в кровеносные сосуды. Эффект пептидов на жизненность сосудов оценивали аналогичным образом.
D. Ингибирование ангиогенеза, индуцированного опухолью, при местном применении
1) Обработка моноклональными антителами
Кроме опытов по ангиогенезу, описанных выше, где оценивали эффекты антагонистов анти-αvβ3, LM609 и различных пептидов на эмбриональный ангиогенез, исследовали также роль αvβ3 в ангиогенезе, индуцированном опухолью. В качестве индуктора использовали заранее выращенные αvβ3-негативные фрагменты меланомы M21-L человека и выделенные из 17-дневного эмбриона цыпленка. Данные фрагменты получали, как описано в примере 6С.
Как описано выше в примере 7А1), mAbs по отдельности накладывали местно на фрагменты опухоли в концентрации 25 мкг в 25 мкл HBSS и затем окна заклеивали пластырем. Таким же образом mAbs вновь добавляли через 24 часа и 48 часов. Через 72 часа опухоли и окружающие САМ ткани анализировали, как описано выше в примере 7А1).
Как описано в примере 6С, опухоли вначале получали с помощью трансплантированных культивируемых клеток M21-L, которые не экспрессируют интегрин αvβ3, как описано у Felding-Habermann с соавт., J. Clin. Invest., 89:2018 (1992) в CAMs 10-дневных эмбрионов. Эти αvβ3-негативные фрагменты индуцировали обширную реваскуляризацию в присутствии одного буфера или - mAbs CSAT (анти-β1) или P3G2 (анти-αvβ5). В противоположность этому mAb LM609 (анти-αvβ3) упраздняют инфильтрацию большинства сосудов в опухолевую массу и окружающий САМ.
Для того чтобы количественно выразить эффект mAbs на ангиогенез, индуцированный опухолью, кровеносные сосуды, проникающие в опухоль в пределах фокальной плоскости САМ, подсчитывали под стереомикроскопом в двух наблюдениях дважды слепым способом. Каждые данные, представленные столбиком на фигуре 12, соответствуют среднему числу сосудов ± SE из 12 CAMs в каждой группе соответствующих повторных экспериментов.
Данный количественный анализ обнаружил трехкратное снижение в числе сосудов, проникающих в опухоли, обработанных mAb LM609 в сравнении с опухолями, обработанными буфером или другими mAbs, P3G2 или CSAT (Р<0,0001), как определено в критерии ранговых сумм Вилкоксона. Тот факт, что опухоли M21-L не экспрессируют αvβ3, свидетельствует о том, что mAb LM609 ингибирует ангиогенез скорее путем прямого воздействия на кровеносные сосуды, чем на опухолевые клетки. Эти результаты согласуются с гистологическим распределением αvβ3 в раковых тканевых биоптатах, показанных на фигуре 3A-3D, где распределение αvβ3 ограничено кровеносными сосудами в опухоли, но не в самих опухолевых клетках.
2) Обработка синтетическими пептидами
Синтетические пептиды, полученные в примере 1, включающие пептиды, полученные из ММР-2 и слитых белков, применяли местно в индуцированной опухолью ангиогенной САМ-аналитической системе, как описано выше. Эффект пептидов на жизненность сосудов оценивали аналогичным образом.
Е. Ингибирование ангиогенеза, индуцированного опухолью путем внутривенного введения
1) Обработка моноклональными антителами
Индуцированные опухолью кровеносные сосуды, полученные, как описано в примере 7Е1), обрабатывали также с помощью mAbs, применяемых путем внутривенной инъекции. Опухоли помещали на CAMs, как описано в примере 7D1), а окна заклеивали пластырем и через 24 часа 200 мкг выделенных очисткой mAbs однократно инокулировали внутривенно в кровеносные сосуды эмбриона цыпленка, как описано раньше. После этого эмбрионы цыплят позволяли высиживать в течение 7 дней. Затем наблюдали величину ангиогенеза, как описано выше. Как описано ниже в примере 8, после этого периода времени опухоли удаляли и анализировали их вес, чтобы определить эффект воздействия антитела на рост опухоли и супрессию.
2) Обработка синтетическими пептидами
Эффекты пептидов, воздействующих на сосудистую систему, индуцированную опухолью, также оценивали в САМ-аналитической системе. Использовали опухолевый САМ-препарат, как описано выше, за тем исключением, что вместо внутривенной инъекции mAb синтетические пептиды, полученные, как описано в примере 1 и в примере 7А2), по отдельности инъецировали внутривенно в видимые кровеносные сосуды.
Полученные результаты с циклическим пептидом 66203, содержащимся в виде HCl-соли, и контрольный пептид 62186, показаны на фигурах 9А-9С. На фигуре 9А обработка контрольным пептидом не влияет на наличие в изобилии больших кровеносных сосудов, которые были индуцированы путем обработки опухолью, чтобы врасти в область САМ, исходно без кровеносных сосудов. Напротив, когда данный циклический пептид 66203, антагонист по отношению к αvβ3, наносили на фильтр, образование кровеносных сосудов было ингибировано, исключая область без новой сосудистой системы, как показано на фигуре 9В. Ингибирующий эффект RGD-содержащего пептида был специфичным и локализован, о чем свидетельствует отсутствие каких-либо повреждающих эффектов на сосуды, прилегающие к месту локализации опухоли. Следовательно, на фигуре 9С, когда ингибирующие пептиды инъецировали внутривенно в САМ-аналитическую систему, не наблюдали эффекта в предсуществующих зрелых сосудах, представленных в САМ-областях, прилегающих еще далее от местоположения данной опухоли. Эти предсуществующие сосуды в данном месте не находились под воздействием ингибирующего пептида, который истекал в границах этих сосудов, хотя образование новых сосудов из этих предсуществующих сосудов в опухолевую массу было ингибировано. Следовательно, синтетические пептиды, в том числе 66203 и 62184, ранее показанные в лиганд-рецепторных анализах в примере 4 антагонистами αvβ3, теперь продемонстрировали подавление ангиогенеза, которое ограничено сосудами, подвергающиеся развитию, но не зрелыми предсуществующими сосудами. Кроме того, внутривенная инфузия пептидов не ведет к какой-либо вредной цитоксичности в окружающей области, о чем свидетельствует интактная сосудистая система на фигуре 9С.
Аналогичные опыты осуществляли с другими синтетическими пептидами, полученными в примере 1 и приведенными в таблице 1 вместе с ММР-2-композициями данного изобретения.
3) Обработка фрагментами
ММР-2 Опухоль CS-1 (β3-негативная) получали в САМ, как описано выше. Через 24 часа опухолевого роста композиция из ММР-2-фрагмента, сконструированная в виде СТММР-2 (2-4) и полученная, как описано в примере 4А, вводили внутривенно в виде 50 мкг фрагмента в 100 мкл PBS. Через 6 дней опухоль оценивали по массе. Опухоли, обработанные с помощью СТММР-2 (2-4), снижали в скорости роста почти на 50% при сравнении со скоростью роста контрольных опухолей, обработанных с помощью СТММР-2 (10-1) или с помощью PBS. Следовательно, антагонист αvβ3 ингибирует опухолевый рост.
8. Ингибирование роста опухолевой ткани с помощью αvβ3-антагонистов по измерению в САМ-анализе
Как описано в примере 7Е1) в дополнение к визуально
определенному эффекту анти-αvβ3-антагонистов на ангиогенез, индуцированный ростовым фактором или опухолью, оценивали также эффект антагонистов, после воздействия, путем измерения изменений в опухолевой массе. Для данного анализа получали САМ-аналитическую систему ангиогенеза, индуцированного опухолью, как описано в примере 6С и 7D. В конце 7-дневного периода инкубации полученные опухоли удаляли из CAMs и подрезали любую остаточную САМ-ткань, промывали 1 мл фосфатно-солевого буфера и определяли сырой вес каждой опухоли.
Кроме того, получение опухоли для микроскопического гистологического анализа включает фиксирование соответствующих образцов опухолей в фиксаторе Bulins в течение 8 часов и заключение в парафин. Последовательно получали срезы и окрашивали гематоксилином и эозином (Н&Е). Gladson с соавт., J. Clin. Invest. , 88: 1924 (1991). Срезы фотографировали под компаундным (compound) микроскопом Olympus при 250х.
А. Местное применение
Результаты взвешиваний обычной меланомной опухоли человека (M21L), полученной при местном применении контрольного буфера (HBSS), P2G2 (анти-αvβ5) или LM609 (анти-αvβ3), приведены в таблице 4. Число эмбрионов определяли для каждой обработки со средним весом для каждой опухоли в миллиграммах (мг), который вычисляли вместе со SE средней, как показано внизу табл.4.
Выдерживание массы αvβ3-негативной опухоли меланомы человека в САМ-аналитической системе с LM609 вызывает снижение среднего веса необработанной опухоли от 172±26 мг до 52±13 мг. Антитела P3G2 не влияют на опухолевую массу. Поэтому блокирование αvβ3-рецептора при местном применении αvβ3-специфичных антител LM609 приводит к регрессии опухолевой массы вместе с ингибированием ангиогенеза, как показано в предыдущих примерах. Измерение диаметра опухолевой массы, полученной при воздействии P3G2, составило приблизительно 8 миллиметров на 1 сантиметр в среднем. Напротив, опухоли, обработанные LM609, имели в среднем диаметр, равный 2-3 миллиметрам.
Замороженные срезы этих опухолей обнаруживают интактную опухолевую цитоархитектуру для опухоли, подвергнутой воздействию P3G2 в противоположность отсутствию организованной клеточной структуры у опухолей, подвергнутых воздействию LM609. По этой причине αvβ3-рецепторная активность является существенной для опухоли αvβ3-негативной для поддержки питания ее массы при развитии новой сосудистой системы, экспрессирующей αvβ3. Блокирование αvβ3 при помощи антагонистов αvβ3 данного изобретения приводит к ингибированию ангиогенеза в данной опухоли с одновременным уменьшением опухолевой массы.
В. Внутривенное применение
Результаты взвешиваний типичной раковой опухоли (UCLAP-3), полученной после внутривенного применения контрольного буфера (фосфатно-солевой буфер, PBS), CSAT (анти-β1) или LM609 (анти-αvβ3), приведены в таблице 5. Число эмбрионов определяли для каждой обработки со средним весом для каждой опухоли, который вычисляли вместе со SE средней, как показано внизу табл.5.
Выдерживание массы αvβ3-негативной раковой опухоли человека в САМ-аналитической системе с LM609 вызывает уменьшение среднего веса необработанной опухоли от 85±7 мг до 30±6 мг. Антитела CSAT существенно не влияют на вес данной опухолевой массы. Поэтому блокирование αvβ3-рецептора при внутривенном введении антител LM609, специфичных к αv β3, приводит к регрессии раковой опухоли, которую они осуществляет в отношении меланомной опухолевой массы вместе с ингибированием ангиогенеза, как показано в предыдущих примерах. Кроме того, рост меланомной опухоли человека сходным образом ингибируется при внутривенной инъекции LM609.
9. Регрессия роста опухолевой ткани при помощи αvβ3-антагонистов по измерению в САМ-анализе
Чтобы дополнительно оценить эффекты αvβ3-антагонистов на рост и жизнеспособность опухоли, в CAMS 10-дневные эмбрионы помещали фрагменты меланомы человека и фрагменты рака легкого, поджелудочной железы и гортани, как описано в примере 5А.
А. Внутривенное применение
1) Обработка моноклональными антителами
а. Обработка с помощью LM609 (анти-αvβ3) и CSAT (анти-β1)
Через двадцать четыре часа после имплантации в САМ с фрагментами раковой опухоли αvβ3-негативной меланомы человека M21-L, панкреатической раковой опухоли FG, рака легкого человека UCLAP-3 или рака гортани НЕр3, эмбрионам внутривенно инъецировали лишь один PBS или однократную дозу (300 мкг/100 мкл) либо mAb LM609 (анти-αvβ3), либо CSAT (анти-β1). Опухолям позволяли размножаться в течение шести дополнительных дней. В конце инкубационного периода эти опухоли аккуратно удаляли и вырезали свободную окружающую САМ-ткань. Резекцию опухолей осуществляли с помощью двух независимых исследователей, удаляющих только легко определяемую массу твердой опухоли. Данные опухоли имели хорошо определяемые края, поэтому тонкую полупрозрачную мембрану (САМ), которая легко отличалась от массы твердой опухоли, удаляли без повреждения самой опухолевой массы. Эти вырезанные опухоли взвешивали и проверяли на морфологию и гистологию.
Как показано на фигуре 13, определяли сырые веса опухолей в конце 7-го дня и сравнивали с первоначальными опухолевыми весами до обработок. Каждый столбик соответствует средней ± SE из 5-10 опухолей в группе. mAb LM609 существенно ингибировал опухолевый рост (р<0,001) в сравнении с контролями у всех тестируемых опухолей. Опухоли, обработанные PBS или CSAT, пролиферировали во всех случаях. Напротив, mAb LM609 не только предотвращали рост этих опухолей, но и индуцировали в большинстве случаев обширную регрессию. Важно, что эти опухолевые клетки не экспрессируют интегрин αvβ3, демонстрируя, что данное ингибирование роста связано с анти-ангиогенными эффектами этого антитела на реваскуляризацию, а не непосредственно на опухолевые клетки.
b. Обработка с помощью LM609 (анти-αvβ3) и P3G2 (анти-αvβ5)
Опухолевые фрагменты меланомы M21-L человека (50 мг) имплантировали в CAMs 10-дневных эмбрионов, как описано в примере 5А. Двадцать четыре часа спустя эмбрионам внутривенно инъецировали один PBS или однократную дозу (300 мкг/100 мкл) mAb LM609 (анти-αvβ3) или P3G2 (анти-αvβ5). Опухолям позволяли размножаться, как описано выше в примере 9А1), и проверяли на морфологию и гистологию, как здесь описано.
Соответствующие образцы опухолей M21-L, обработанных mAbs P3G2 (aнти-αvβ5) или LM609 (анти-αvβ3), проверяли на морфологию. Опухоли, обработанные P3G2, были больше (8 мм в диаметре) и хорошо васкуляризованы, а опухоли, обработанные mAb LM609, были намного меньше (3 мм в диаметре) и без детектируемых кровеносных сосудов.
Эти опухоли дополнительно проверяли с помощью препаратов гистологических срезов и окраски гематоксилином и эозином, как описано в примере 9А1)а. Как показано на фигуре 14 (верхняя группа), опухоли, обработанные с помощью mAb P3G2 (анти-αvβ3), показали множество жизнеспособных и активно делящихся клеток, как указано на митотических фигурах (стрелки-указатели), а также множество кровеносных сосудов (размерные стрелки) во всей опухолевой строме. Напротив, немногие, если вообще жизнеспособные опухолевые клетки или кровеносные сосуды, были детектированы в опухолях, обработанных с помощью mAb LM609 (анти-αvβ3) (фигура 14, нижняя группа). Эти результаты демонстрируют, что антагонисты интегрина αvβ3 ингибируют ангиогенез, индуцированный опухолью, приводя к остановке роста и регрессии in vivo различных опухолей человека. Важно отметить, что эмбрионы, обследованные после семи дней опухолевого роста (17-й день развития), являются нормальными при обследовании в целом, были ли они обработаны или нет антагонистом αvβ3. Эти данные свидетельствуют, что антагонисты данного интегрина нетоксичны для развития эмбрионов.
2) Обработка синтетическими пептидами
Опухолевые фрагменты меланомы M21-L человека (50 мг) имплантировали в САМs 10-дневных эмбрионов, как описано в примере 5А. Двадцать четыре часа спустя эмбрионы получали однократную внутривенную инъекцию либо из 300 мкг/100 мкл цикло-RADfV (69601) и либо цикло-RGDfV (66203). Через общие 72 часа опухоли удаляли, проверяли на морфологию и фотографировали под стереомикроскопом, как описано в примере 9А1).
Панели, показанные на фигурах 15А-15Е, соответствуют следующему: фигура 15А, дублированные образцы обрабатывали с помощью пептида цикло-RADfV (69601); фигура 15В, дублированные образцы обрабатывали с помощью пептида цикло-RGDfV (66203); фигура 15С, прилегающая САМ-ткань, взятая от одних и тех же эмбрионов, обработанная с помощью пептида цикло-RGDfV (66203) и фигуры 15D и 15Е, опухоли под большим увеличением (13х), обработанные пептидом. Фигура 15D изображает нормальные кровеносные сосуды из опухоли, обработанной контрольным пептидом (69601). Фигура 15Е изображает примеры поврежденных кровеносных сосудов из опухолей (размерные стрелки), обработанные пептидом цикло-RGDfV (66203).
Эти результаты поясняют, что только пептид 66203 в противоположность контрольному пептиду 69601 подавляет образование сосудов и, кроме того, что сосуды в САМ-ткани, прилегающей к данной опухоли, не подвержены воздействию.
Дополнительно осуществляли анализ регрессии опухоли с помощью αvβ3-реагирующего пептида 85189 (SEQ ID NO 15) против 69601 в качестве контроля. Эти анализы осуществляли, как описано выше, за исключением того, что 100 мкг пептида инъецировали внутривенно в эту САМ через 18 часов после имплантации. Более чем через 48 часов данные опухоли удаляли и измеряли их сырой вес.
Фигуры 32, 33 и 34 показывают соответственно редукцию веса опухоли для UCLAP-3, M21-L и FgM-опухолей после внутривенного воздействия пептида 85189 в противоположность отсутствию эффекта с PBS либо с пептидом 69601.
10. Регрессия роста опухолевой ткани под влиянием αvβ3-антагонистов по измерению в опыте с кроличьей моделью глаза in vivo
Эффект анти-αmβ3-антагонистов на ангиогенез, индуцированный ростовым фактором, можно получить в естественно прозрачных структурах, что проиллюстрировано на примере роговицы глаза. Новые кровеносные сосуды росли от края роговицы, которая обладает богатым кровоснабжением, в направлении к центру роговицы, которая обычно не обладает кровоснабжением. Стимуляторы ангиогенеза, такие как bFGF, при нанесении на роговицу индуцируют рост новых кровеносных сосудов из обода роговицы. Антагонисты роговицы, нанесенные на данную роговицу, ингибируют рост новых кровеносных сосудов из обода роговицы. Следовательно, роговица подвергается ангиогенезу через инвазию эндотелиальных клеток от края роговицы в плотную упакованную в коллаген роговичную ткань, которая легко просматривается. Анализ модели глаза кролика создает по этой причине модель in vivo для прямого наблюдения стимулирования и ингибирования ангиогенеза после имплантации соединений прямо в роговицу глаза.
А. Анализ модели глаза кролика in vivo
1) Ангиогенез, индуцированный ростовыми факторами
Ангиогенез, индуцированный в аналитической in vivo - модели глаза кролика при помощи ростового фактора bFGF, описан в нижеследующих разделах.
а. Получение hydron-осадков, содержащих ростовой фактор и моноклональные антитела
Осадки hydron-полимера, содержащие ростовой фактор и mAbs, были получены, как описано у D'Amato с соавт., Рrос. Natl. Acad. Sci. USA, 91:4082-4085 (1994). Отдельные гранулы, содержащие 650 нг ростового фактора bFGF, соединяли с sucraflate (Carafet, Marion Merrell Dow Corporation), чтобы стабилизировать bFGF и обеспечить его медленное высвобождение в окружающую ткань. Кроме того, были получены hydron-гранулы, которые содержали либо 40 мкг mAb LM609 (анти-αvβ3), либо mAb P1F6 (анти-αvβ5) в PBS. Эти гранулы формовали в специально полученном тефлоновом штифте, который имел 2,5 мм высверленный керн в их поверхностях. Приблизительно 12 мкл выбранного материала помещали в каждый штифт и полимеризовали в течение ночи в стерильном колпачке. Затем гранулы стерилизовали ультрафиолетовым облучением.
b. Обработка моноклональными антителами
Каждый эксперимент состоял из трех кроликов, у которых один глаз получал гранулу, содержащую bFGF и LM609, а другой глаз получал гранулу, содержащую bFGF и мышиное mAb P1F6 (анти-αvβ5). Применяя парное тестирование глаза для сравнения LM609 (анти-αvβ3) с другим mAb и PBS-контролями, создавали средние строгого тестирования для демонстрации существенных различий между испытываемыми mAbs.
mAb P1F6 иммунно реагирует с интегрином αvβ5, который обнаружен на поверхности васкулярных эндотелиальных клеток, но по преимуществу не участвует в ангиогенезе. Чтобы определить, действительно ли mAb P1F6 участвует в ангиогенезе, получали гранулы, содержащие только данное mAb, и анализировали, как описано ниже, для подтверждения того, что mAb не индуцирует ангиогенез.
Все тестируемые mAbs выделяли из асцитных жидкостей с помощью аффинной хроматографии на протеин-А-сефарозе CL-4B в соответствии с хорошо известными методами. Затем элюированные иммуноглобулины диализовали против PBS и обрабатывали Detoxi-гелем (Pierce Chemicals) для удаления эндотоксина. Было показано, что эндотоксин является сильным ангиогенным и воспалительным стимулятором. Поэтому mAbs тестировали в присутствии эндотоксина в опыте с хромогенным лизатом амебоцита мечехвоста (Bio-Whittaker) и в аналитической модели глаза кролика использовали только mAbs, в препаратах которых эндотоксин не детектировался.
Hydron-гранула, включающая bFGF и mAb LM609 (анти-αvβ3) или P1F6 (анти-αvβ5), внедряли в роговичный карман, формируемый в глазу кроликов. Hydron-гранула содержит также sucralfate для стабилизации bFGF во время опыта. Индивидуальные гранулы имплантировали в хирургически создаваемые "карманы", сформированные в срединной строме роговицы кроликов. Данную хирургическую операцию осуществляли в стерильных условиях с использованием операционного микроскопа Wild model М691, снабженного светоделителем, с которым смонтировали камеру для фотографической регистрации индивидуальных роговиц. "Карман" размером 3-5 мм создавали в роговичной строме путем создания 3 мм надреза до половины толщины роговицы с помощью лезвия Beaver 69. Эту строму рассекали периферически с использованием шпателя для радужной оболочки и указанную гранулу имлантировали в ее периферический край в 2 мм от лимба роговицы.
Во время последующих 14 дней bFGF и mAb диффундировали из имплантированной гранулы в окружающую ткань и тем самым осуществляли ангиогенез от края в роговицу.
Соответствующие результаты по каждой обработке изображены на фигурах 16А-16Е. Количество присутствующих сосудов определяли количественно и описывали в пределах циферблата часов, которые определяли следующим образом. Данный глаз делили на 12 равных секторов таким же образом, как циферблат делят на часы. "Первый циферблатный час сосудов" относится к такому количеству сосудов, которые заполняют область глаза, эквивалентную первому часу на часах. Пять кроликов, которые получили только bFGF, проявляли флоридный ангиогенез, в котором новые кровеносные сосуды росли от края роговицы в направлении к центру данной роговицы, которая обычно не имеет кровеносных сосудов. Один из этих кроликов обладал только 1 циферблатным часом сосудов на указанную гранулу. Два кролика, которые получали bFGF и mAb LM609 абсолютно не проявляли ангиогенеза до 14 дней после операции. Один из этих кроликов имел на 14-й день 3 геморрагических очага и прорастание сосудов. Двое из этих кроликов, которые получали bFGF и mAb P3G2 (анти-αvβ5), показали обширную васкуляризацию, в которой новые кровеносные сосуды росли от края роговицы в роговицу. Один из этих кроликов имел только 1-2 часа сосудов к указанной грануле.
Как установлено анализом кроличьей глазной модели, у кроликов, которые получали mAb LM609 (анти-αvβ3), в присутствии ростового фактора bFGF, в нормальных паралимбальных (paralimbal) сосудах ангиогенный эффект не наблюдался. Напротив, ангиогенез наблюдали в паралимбальных сосудах в присутствии ростового фактора bFGF у кроликов, которые получали mAb P3G2 (анти-αvβ3). Полное ингибирование роговичного ангиогенеза с помощью mAb LM609 существенно выше, чем у любого прежде сообщавшегося анти-ангиогенного реагента.
с. Обработка полипептидами
Каждый эксперимент состоял из восьми кроликов, у которых один глаз получал гранулу, содержащую 100 нанограмм (нг) bFGF, а другой глаз получал гранулу, содержащую 1 микрограмм (мкг) VEGF. Эти гранулы внедряли в роговичный карман, как описано выше, и данные цитокины последовательно стимулировали рост новых кровеносных сосудов в данной роговице. Пептиды вводили подкожно (s. q. ) в 1 мл PBS в начальной дозе 50 мкг на кг веса кролика в день внедрения гранулы, а после этого давали ежедневные дозы s.q. в концентрации 20 мкг/кг. Через 7 дней оценивали роговицу, как описано выше.
Кролики, получающие контрольные пептиды 69601, показали практический рост роговичных кровеносных сосудов на 7-й день в обоих глазах, стимулированных bFGF и VEGF. Кролики, получающие пептид 85189, показали менее чем 50% количества от роста роговичных кровеносных сосудов в сравнении с контролями для глаз, стимулированных bFGF и почти 100% ингибирования для глаз, стимулированных VEGF.
11. Регрессия с помощью αvβ3-антагонистов роста in vivo опухолевой ткани по измерению в опыте химерная мышь: человек
Химерную модель in vivo мышь: человек получали путем замещения части кожи от SCID-мыши на неонатальную крайнюю плоть человека (фигура 17). После акклиматизации кожного имплантата в крайнюю плоть человека инокулировали карциномные клетки. После акклиматизации измеримой опухоли, в хвостовую вену мыши инъецировали либо mAb LM609 (анти-αvβ3), либо PBS. После 2-3-недельного периода данную опухоль вырезали и анализировали на вес и гистологию.
А. Анализ химеры in vivo мышь: человек
Химерную модель in vivo мышь: человек получали, в сущности, как описано у Yan с соавт., J. Clin. Invest., 91:986-996 (1993). Вкратце, из SCID-мыши (возраст 6-8 недель) хирургически удаляли квадратную область кожи 2 см2 и заменяли на крайнюю плоть человека. Данную мышь анестезировали, волосы сбривали с области 5 см2, с каждой боковой стороны брюшной области. Два круглых имплантата поддерживающей ткани в 2 см2 получали удалением всего слоя кожи на глубину до фасции. Полный слой имплантатов кожи человека того же размера, полученных от неонатальной крайней плоти человека, помещали на раневое ложе и накладывали шов. Данный имплантат покрывали Band-Aid, который пришивали к данной коже. Данную рану укрывали марлевой повязкой.
Для образования солидных опухолей человека в кожных имплантатах человека на SCID-мыше использовали клеточную линию меланомы M21-L человека или клеточную линию карциномы молочной железы 23.1 MDA (АТСС НТВ 26; αvβ3-негативная по иммунной реакции тканевых срезов с mAb LM609). Суспензию, полученную из единственной клетки, из 5•106 M21-L или клеток MDA 23.1 инъецировали внутрикожно в кожный имплантат человека. Затем данных мышей обследовали в течение 2-4 недель, наблюдая за ростом заметных опухолей человека.
В. Внутривенное введение
1) Обработка моноклональными антителами
После роста заметных опухолей SCID-мыши, которым были инъецированы опухолевыми клетками M21L, инъецировали внутривенно в хвостовую вену 250 мкг mAb LM609 (анти-αvβ3) или PBS, дважды в неделю в течение 2-3 недель. После этого времени данные опухоли удаляли из кожи и подравнивали свободные края окружающей ткани. В каждой обработке несколько мышей оценивали по среднему весу опухоли из каждой обработки (см. таблицу 6).
Воздействие αvβ3-негативной раковой опухолевой массой на химерную аналитическую систему мышь: человек с LM609 (анти-αvβ3) является причиной уменьшения среднего веса опухоли, обработанной PBS, со 198 до 113 мг.
Соответствующие образцы опухолей M21L, обработанных mAb LM609 (анти-αvβ3) и PBS исследовали на морфологию. Опухоли, обработанные PBS, были большими (8-10 мм в диаметре) и хорошо васкуляризованы, тогда как опухоли, обработанные с помощью mАb LM609 (анти-αvβ3) были намного меньше (3-4 мм в диаметре) и не обнаруживали кровеносных сосудов.
В других экспериментах с опухолевыми клетками меланомы M21-L в химерной аналитической системе мышь: человек ответ на mAb LM609 сравнивали с ответом, полученным на синтетический пептид 85189 (SEQ ID NO 15), в сравнении с контрольным синтетическим пептидом 69601 (SEQ ID NO 6). Эти опыты осуществляли, как описано выше. Полученные результаты, показанные на фигуре 35, демонстрируют, что синтетический пептид 85189 редуцирует объем опухоли до менее чем 25 мм3 в сравнении с контрольным пептидом, где объем опухоли составлял, приблизительно 360 мм3. mAb LM609 также существенно редуцировал объем опухоли до приблизительно 60 мм3.
Опухоли, сформированные в кожных трансплантатах, которые инъецировали с помощью клеток MDA 23.1, детектировали и измеряли. Морфологическое исследование созданных опухолей обнаружило, что реваскуляризация происходит из трансплантированной ткани человека в опухолевые клетки MDA 23.1.
Следовательно, блокирование рецептора αvβ3 в результате внутривенного введения αvβ3-специфичного антитела LM609 и пептидов ведет к регрессии раковой опухоли в данной модельной системе таким же образом, как и в модельных системах САМ и кроличьего глаза, как описано соответственно в примерах 9 и 10.
2) Обработка синтетическими пептидами
В методике, аналогичной той, которая описана выше для моноклональных антител, пептидные антагонисты αvβ3 были внутривенно инъецированы в хвостовую вену мышей SCID, обладающих заметными опухолями M21-L. В предварительном анализе для пептида 69601 (контроль) и 85189 (опыт), инъецированных в диапазоне концентраций от 10 до 250 мкг/мл, строили кривую зависимости "доза-эффект". Результаты определения средних объемов и веса удаляемых опухолей после обработки приведены соответственно на фигурах 36А и 36В. Пептид 85189, эффективный в ингибировании роста опухоли M21-L в тестируемом диапазоне концентраций, сравнивали с обработкой контрольным пептидом с наиболее эффективной дозой, равной 250 мкг/мл.
Для анализа эффективности обработки пептидом 85189 во времени устанавливали два режима обработки на одной и той же модели опухоли SCID. В первом опыте обработку любым пептидом, 85189 или 69601, начинали на 6-й день, от дня 0, дня, в который в кожу мыши подкожно инъецировали 3•106 клеток опухоли M21-L с интраперитонеальными инъекциями 250 мкг/мл пептида 85189 или контроля 69601, через день вплоть до 29-го дня. Второй опыт осуществляли идентично, за тем исключением, что обработку начинали на 20-й день. В конце данных опытов удаляли опухоли и определяли средний объем опухоли в мм3. Значения полученных данных представляли графически ± стандартная ошибка средней.
Результаты этих опытов, показанные соответственно на фигурах 37А и 37В, свидетельствуют, что пептид 85189, но не 69601, ингибирует опухолевый рост в разные дни после начатой обработки в зависимости от конкретного режима обработки. Следовательно, пептид 85189 является эффективным антагонистом αvβ3 в ангиогенезе и поэтому в опухолевом росте.
12. Стимуляция сосудистых клеток с полным клеточным циклом и подверженных апоптозу в присутствии антагонистов интегрина αvβ3, измеренная в САМ-опыте
Ангиогенный процесс четко зависит от способности цитокинов, таких как bFGF и VEGF, стимулировать пролиферацию сосудистых клеток. Mignatti с соавт. , J. Cell. Biochem., 471:201 (1991); Takeshita с соавт., J. Clin. Invest., 93: 662 (1994); и Koyama с соавт., J. Cell. Physiol., 158:1 (1994). Вместе с тем очевидно, что сигнальные события могут регулировать дифференциацию этих сосудистых клеток в зрелые кровеносные сосуды. Поэтому понятно, что вмешательство сигналов, связанных либо с ростом, либо с дифференциацией сосудистых клеток, подвергающихся новому росту или ангиогенезу, может приводить к осложнению ангиогенеза.
Было показано, что события, связанные с интегрином, принимают участие как в клеточной пролиферации, так и в апоптозе или запрограммированной клеточной смерти in vitro. Schwartz, Cancer Res., 51:1503 (1993); Meredith с соавт., Mol. Biol. Cell., 4:953 (1993); Frisch с соавт., J. Cell Biol., 124: 619 (1994); и Ruoslahti с соавт., Cell, 77:477 (1994). При близком рассмотрении влияния антагонистов αvβ3 на ангиогенез обнаружили наличие разорванных и поврежденных кровеносных сосудов, ассоциированных с опухолью. По этой причине, возможно, что утрата непрерывности кровеносного сосуда может быть связана с избирательным некрозом или апоптозом сосудистых клеток.
Чтобы выяснить эту возможность, исследовали САМs после индукции ангиогенеза с помощью ростового фактора bFGF и обработки с помощью mAb и циклических пептидов данного изобретения.
А. Обработка моноклональными антителами
Апоптоз можно выявить разными способами, которые включают прямое исследование ДНК, выделенной из ткани, обнаружением фрагментации ДНК и детекции 3'ОН в интактной ткани с помощью определенного антитела, которое специфически детектирует свободные 3'ОН-группы фрагментированной ДНК.
1) Анализ фрагментации ДНК
Ангиогенез индуцировали путем помещения фильтровальных дисков, насыщенных bFGF, на CAMs 10-дневных эмбрионов, как описано в примере 6А. Иммуногистологический анализ CAMs с помощью LM609 (анти-αvβ3) обнаруживает пик экспрессии αvβ3 в кровеносных сосудах через 12-14 часов после инициации ангиогенеза с помощью bFGF. Поэтому через 24 часа после стимуляции с помощью bFGF в эмбрионы внутривенно инокулировали 100 мкл одного PBS или PBS, содержащего 300 мкг mAb CSAT (анти-β1) или LM609 (анти-αvβ3).
Фрагментацию ДНК детектировали путем удаления САМ-ткани непосредственно ниже фильтровальных дисков, насыщенных bFGF, через 24 или 48 часов после внутривенных инокуляций mAb LM609 (анти-αvβ3), CSAT (анти-β1) или PBS. Вырезанные ткани трижды промывали стерильным PBS и в конце споласкивали, ресуспендировали в 0,25% бактериальной коллагеназе (Worthington Biochemical; Freehold, NJ) и инкубировали в течение 90 минут при 37oС с нерегулярным встряхиванием. ДНК экстрагировали из САМ-клеток одинаковой численности из одноклеточной суспензии, как описано прежде. Bissonette с соавт., Nature, 359:552 (1992). Вкратце, одинаковые численности САМ-клеток лизировали в 10 мМ Трис-HCl, рН 8,0, 10 мМ EDTA в 0,5 (о/о) Тритоне Х-100 (Sigma, St. Louis, МО). Клеточные лизаты центрифугировали при 16000•g в течение 15 минут при 4oС для разделения растворимой фрагментированной ДНК и интактного хроматинового осадка. Фрагментированную ДНК промывали, осаждали и анализировали в 1,2% (в/о) агарозном геле.
Растворимую фрагментированную ДНК выделяли из САМ-клеток одинаковой численности в каждой обработке, разделяли электрофоретически в агарозном геле и визуализировали окрашиванием с этидийбромидом. Через 24 часа после обработки различий в относительном количестве фрагментированной ДНК, полученной в трех различных обработках, не обнаружили. Однако через 48 часов после обработки mАb LM609 (анти-αvβ3) наблюдали существенное увеличение в фрагментации ДНК при сравнении с эмбрионами, обработанными mAb CSAT (анти-β1) или одним PBS.
2) Стимуляция сосудистых клеток с полным клеточным циклом
Чтобы экспериментально исследовать роль αvβ3 в этих процессах, клетки, полученные из CAMs, обработанные или не обработанные bFGF, окрашивали пропидиййодидом и подвергали иммунному взаимодействию с mAb LM609 (анти-αvβ3).
CAMs, выделенные из эмбрионов, обработанные через 24 и 48 часов с помощью mAb LM609 (анти-αvβ3), CSAT (анти-β1) или PBS, диссоциировали в одноклеточные суспензии путем инкубации с бактериальной коллагеназой, как описано выше. Затем одиночные клетки пермеабилизировали и окрашивали с помощью Арор Tag Insitu Detection Kit в соответствии с инструкциями изготовителя (Oncor, Gaithersburg, MD). Арор Tag представляет собой антитело, которое специфически детектирует свободные 3'ОН-группы фрагментированной ДНК. Детекция таких свободных 3'ОН-групп представляет собой установленный метод для детекции апоптозных клеток. Gavrieli с соавт., J. Cell Biol., 119:493 (1993).
Затем клетки, окрашенные Арор Tag, промывали в 0,1% (о/о) Тритоне Х-100 в PBS и ресуспендировали в FACS-буфере, содержащем 0,5% (в/о) BSA, 0,02% (в/о) натрийазида и 200 мкг/мл РНКазы А в PBS. Клетки инкубировали в течение 1,5 часа, промывали и анализировали в активируемом флуоресценцией активаторе клеток. Клеточную флуоресценцию измеряли с использованием проточного цитометра FACScan и данные анализировали, как описано ниже.
Клеточную флуоресценцию измеряли в проточном цитометре FACScan (Becton Dickinson, Mountain View, CA). Боковое рассеяние (SSC) и рассеяние в прямом направлении (FSC) определяли одновременно и все данные собирали с помощью компьютера Hewlet Packard (HP9000), снабженного исследовательской программой FACScan (Becton Dickinson, Mountain View, CA). Полученные данные анализировали с помощью программы Р.С. Lysis version I (Becton Dickinson, Mountain View, CA). Негативные контрольные ячейки регулировали с использованием клеточных суспензий без добавления первичных антител из набора Арор Tag. Идентичное просеивание применяли к обеим клеточным популяциям, полученным в анализах, приблизительно из 8000 клеток на отдельную клеточную обработку.
Процентная доля одиночных клеток, полученных из CAMs, обработанных mAb и окрашенных с помощью Арор Tag, которую определили в FACS-аналиэе, показана на фигуре 18. Черный столбик представляет клетки эмбрионов, обработанных за 24 часа до анализа. Заштрихованный столбик представляет клетки эмбрионов, обработанных за 48 часов до анализа. Каждый столбик соответствует средней ± S.E. из трех повторностей.
Как показано на фигуре 18, CAMs, обработанные двумя днями раньше с помощью mAb LM609 (анти-αvβ3), показали 3-4-кратное увеличение в окрашивании Арор Tag в сравнении с CAMs, обработанными либо одним PBS, либо CSAT (анти-β1).
В. Обработка синтетическими пептидами
САМ-опыты с ангиогенезом, индуцированным ростовым фактором, как описано в примере 6А, осуществляли также с синтетическими пептидами данного изобретения, чтобы выяснить эффект циклических пептидов на апоптоз. Пептиды цикло-RGDfV (66203) и цикло-RADfV (69601) получали, как описано в примере 1. Эти пептидные растворы или PBS инъецировали в САМ-препарат в концентрации 300 мкг/мл. Через 24 и 48 часов иссекали фильтровальную
бумагу и ткань, окружающую САМ, и окрашивали с помощью Арор Tag, чтобы детектировать апоптоз, как описано в примере 12А2).
Как показано на фигуре 18, CAMs, обработанные двумя днями раньше с помощью пептида 69203, показали 3-4-кратное увеличение в окраске с помощью Арор Tag по сравнению с CAMs, обработанными либо одним PBS, либо контрольным циклическим пептидом 69601 (цикло-RADfV).
С. Влияние обработки моноклональными антителами на апоптоз и клеточный цикл
Одноклеточные суспензии исследовали на число копий хромосомной ДНК путем окрашивания с помощью пропидиййодида для определения эффекта обработки моноклональными антителами на клеточный цикл и на апоптоз при окрашивании с помощью Арор Tag.
Одноклеточные суспензии из CAMs, обработанные за 24 или 48 часов с помощью mАb LM609 (анти-αvβ3), или CSAT (анти-β1), или PBS, были получены, как описано в примере 12А1).
Для окраски клеток с помощью Арор Tag клеточные суспензии трижды промывали буфером, содержащим 2,5% (в/о) BSA и 0,25% (в/о) натрийазида в PBS. Затем клетки фиксировали в 1% (в/о) параформальдегиде в PBS в течение 15 минут с последующими тремя промывками, как описано выше. Чтобы предотвратить неспецифическое связывание, одноклеточные суспензии блокировали с помощью 5% (в/о) BSA в PBS в течение ночи при 4oС. Затем, как и раньше, клетки промывали, окрашивали с помощью Арор Tag, и измеряли клеточную флуоресценцию с помощью FACScan, как описано выше в примере 12А.
Клетки из каждого экспериментального состояния окрашивали с помощью пропидиййодида (Sigma, St. Louis, МО) в 10 мкг/мл в PBS в течение 1 часа, промывали два раза с помощью PBS и анализировали ядерные характеристики, типичные для апоптоза, в том числе хроматиновую конденсацию и сегментацию. В морфологическом анализе клеток оценивали процент апоптозных клеток как минимум из 10-15 случайно выбранных микроскопных полей.
Объединенные результаты по одноклеточных суспензиям САМs из эмбрионов, обработанных либо с помощью CSAT (анти-β1), либо LM609 (анти-αvβ3), окрашенных с помощью Арор Tag и пропидиййодида и проанализированных в FACS, даны на фигуре 19. На оси Y представлены результаты окрашивания с помощью Арор Tag (апоптоз), на оси Х представлены результаты окрашивания с помощью пропидиййодида (ДНК-содержание). Горизонтальная линия соответствует негативному просеиванию при окрашивании Арор Tag. Левая и правая панели указывают САМ-клетки эмбрионов, обработанных соответственно CSAT и LM609. Анализ клеточного цикла осуществляли анализом приблизительно из 8000 событий на состояние и данные представлены на контурном графике.
Образцы одиночных клеток, окрашенных с помощью ДНКового красителя пропидиййодида, обнаруживают, что 25-30% САМ-клеток, обработанных LM609 (анти-αvβ3), через 48 часов после обработки дают доказательство ядерной конденсации и/или сегментации. Эти процессы характеризуют клетки, подвергающиеся апоптозу. Это находится в противоречии с CAMs, обработанными с помощью CSAT (анти-β1), где 90-95% данных клеток показывают нормальное ядерное окрашивание.
Как показано на фигуре 19, наблюдали согласованность индуцированного апоптоза с помощью LM609, со значительной численностью клеток в пике, содержащем менее чем одну копию ДНК (АО). Ранее было показано, что данный пик представляет фрагментированную ДНК в клетках на последней стадии апоптоза. Telford с соавт., Cytometry, 13:137 (1992). Более того, эти АО-клетки легко окрашиваются с помощью Арор Tag, подтверждая способность данного реагента детектировать апоптозные клетки. Однако помимо окрашивания клеток в АО значительная численность клеток, содержащих более чем одну копию ДНК, также окрашиваются с помощью Арор Tag (фигура 19). Эти результаты демонстрируют, что LM609 обладают способностью содействовать апоптозу сосудистых клеток, которые уже вошли в клеточный цикл. В противоположность этому клетки, полученные из контрольных CAMs, которые вошли в клеточный цикл, показывают минимальное окрашивание с помощью Арор Tag, что согласуется с меньшинством апоптозных клеток, детектированных в контрольных обработанных CAMs.
У других клеток в стимулированных bFGF CAMs, которые вошли в клеточный цикл (фаза S и G2/M), 70% показывают позитивное окрашивание с помощью LM609 (анти-αvβ3). Этот показатель сравнивали с 10%, окрашенных LM609, в клетках, проходящих цикл из CAMs, не обработанных bFGF. Эти данные указывают, что после стимуляции bFGF большинство клеток, несущих αvβ3, показывают активную пролиферацию.
Взятые вместе, эти данные свидетельствуют, что внутривенная инъекция mAb LM609 циклически пептидным антагонистом αvβ3 содействуют апоптозу в границах САМ цыпленка после индукции ангиогенеза.
CAMs исследовали также гистологически на экспрессию αvβ3 путем иммунного взаимодействия с LM609 и на клетки, которые подвергались апоптозу, путем иммунного взаимодействия с Арор Tag. САМ-срезы, вырезанные из эмбрионов, обработанные 48 часами ранее с помощью LM609 (анти-αvβ3), CSAT (анти-β1), или PBS, полученного в примере 5А, промывали, заливали в ОТС (Baxter) и резко замораживали в жидком азоте. Из САМ-тканей нарезали шестимикронные срезы, фиксировали в ацетоне в течение 30 секунд и хранили при -70oС до использования. Тканевые срезы получали в течение окрашивания в результате краткого споласкивания в 70% (о/о) этаноле (ЕТОН) с последующей трехкратной промывкой в PBS. Далее срезы блокировали с помощью 5% (в/о) BSA в PBS в течение 2 часов, с последующей инкубацией с 10 мкг/мл mAb LM609 в течение 2 часов. Данные срезы затем промывали и инкубировали при разведении 1:50 с конъюгированными с родамином антителами козы к анти-IgG мыши (Fisher Scientific, Pittsburg, PA) в течение 2 часов. Наконец, эти срезы промывали и окрашивали с помощью Арор Tag, как описано в примере 12А2). Окрашенные тканевые срезы монтировали на предметном стекле и анализировали под софокальным иммунофлуоресцентным микроскопом.
На фигуре 20, панели А-С представлена САМ-ткань из эмбрионов, обработанных CSAT (анти-β1), а панели D-F представляют САМ-ткань из эмбрионов, обработанных LM609 (анти-αvβ3). На панелях А и D изображены ткани, окрашенные с помощью Арор Tag и визуализированные с помощью флуоресценции (FITC), наложенные на изображение D.I.С. Панели В и Е изображают те же ткани, окрашенные с помощью mAb LM609 (анти-αvβ3) и визуализированные флуоресценцией (родамин). Панели С и F представляют сливающиеся изображения тех же тканей, окрашенных и с Арор Tag, и с LM609, где желтое окрашивание отражает солокализацию. Столбики представляют соответственно 15 и 50 мкм на левой и правой панелях.
Как показано на фигуре 20 (А-С), после внутривенной инъекции CSAT или PBS-контроля окрашивание с помощью Арор Tag проявляется минимально и случайно, указывая на минимальный уровень апоптоза в данной ткани. Напротив, CAMs из эмбрионов, ранее обработанных с помощью LM609 или циклическим пептидом 203, показывают большинство сосудов, окрашенных интенсивно с Арор Tag, а минимальная реактивность наблюдалась у окружающих несосудистых клеток (фигура 20 D-F). Более того, когда для окраски этих тканей (19С и 19F) использовали и Арор Tag и LM609, существенную солокализацию в CAMs, полученных из эмбрионов, обработанных αvβ3-антагонистами (фигура 20F), наблюдали только между этими маркерами. Эти данные демонстрируют, что после индукции ангиогенеза in vivo ингибиторы интегрина αvβ3 избирательно содействуют апоптозу кровеносных сосудов, несущих αvβ3.
Поскольку ангиогенез представляет собой сложный процесс с участием многих молекулярных и клеточных биологических событий, некоторые особенности доказательно подтверждают, что интегрин αvβ3 сосудистой клетки играет относительно последнюю роль в данном процессе. Во-первых, в иммуногистологическом анализе обнаружили, что экспрессия αvβ3 в сосудистых клетках достигает максимума через 12-24 часа после индукции ангиогенеза при помощи bFGF. Во-вторых, антагонисты αvβ3, осложняющие ангиогенез, индуцированный множеством активаторов, позволяют предполагать, что данный рецептор участвует в обычном пути, может быть, позже всех первичных сигнальных событий, ведущих к ангиогенезу. В-третьих, CAMs, обработанные mAb LM609 или циклическим пептидом, не показывают существенного увеличения апоптоза, как определено путем лидерной последовательности нуклеотидов ДНК, даже после 48 часов обработки с помощью этих антагонистов. Наконец, антагонисты αvβ3 содействуют апоптозу сосудистых клеток, в которых уже был индуцирован полный клеточный цикл.
Результаты, представленные здесь, дают первое прямое доказательство того, что интегрин-лигирующие события могут регулировать клеточную выживаемость in vivo. Поэтому предполагается, что как только начинается ангиогенез, индивидуальные сосудистые клетки делятся и начинают двигаться в направлении к ангиогенному источнику, после которого αvβ3-лигирование создает сигнал, позволяющий клетке непрерывно выживать, что приводит к дифференциации и образованию зрелых кровеносных сосудов. Однако, если αvβ3-лигирование предотвратить, тогда в клетках отсутствует получение данного молекулярного сигнала и клетки входят в апоптоз вследствие пассивности. Данная гипотеза должна была бы также предсказать, что после дифференциации получившихся зрелых кровеносных сосудов более не требуется αvβ3-сигнализация для выживаемости, и поэтому они невосприимчивы к антагонистам данного интегрина.
Наконец, результаты, представленные здесь, дают доказательство того, что антагонисты интегрина αvβ3 могут создавать сильный терапевтический подход к лечению неоплазии или других болезней, характерных для ангиогенеза. Во-первых, антагонисты αvβ3 разрывают вновь формирующиеся кровеносные сосуды без воздействия предсуществующей сосудистой системы. Во-вторых, эти антагонисты обладают существенным эффектом на жизнеспособность эмбриона цыпленка, что дает основание полагать, что они не токсичны. В-третьих, ангиогенез был существенно блокирован независимо от ангиогенных стимулов. Наконец, системное введение αvβ3-антагонистов является причиной драматической регрессии различных гистологически отличающихся опухолей человека.
13. Получение органической молекулы αvβ3-антагонистов
Синтез органических соединений αvβ3-антагониста 7 (96112), 9 (99799), 10 (96229), 12 (112854), 14 (96113), 15 (79959), 16 (81218), 17 (87292) и 18 (87293) описан ниже и показан также на отмеченных фигурах. Органические антагонисты указаны числами в скобках. Полученные органические молекулы, указанные в качестве органических миметиков в данном изобретении, охарактеризованы раньше, а затем использовались в способах по ингибированию ангиогенеза, опосредованного αvβ3, как описано в примере 11.
Для каждого синтеза, описанного ниже, определяли оптическое вращение в УФ-спектрофотометре Perkin-Elmer 241, а видимые спектры записывали на спектрометре Beckmann DU-70. Спектры ЯМР 1H и 13С записывали при 400 и 500 MГц на спектрометре АМХ-400 и АМХ-500. Высокоразрешающие масс-спектры (HRMS) записывали на масс-спектрометре VG ZAB-ZSE в условиях быстрой бомбардировки атомами. Колоночную хроматографию осуществляли на силикагеле 70-230-меш. Препаративную TLC осуществляли на Merck Art. 5744 (0,5 мм). Точки плавления регистрировали на аппарате Thomas Hoover.
А. Соединение 1: t-Boc-L-тирозин, бензиловый эфир, как показано на фигуре 38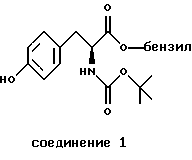
Для растворения N-(трет-бутоксикарбонил)-L-тирозин(t-Boc-L-тирозина) (1,0 эквивалентов; Aldrich) в 0,10 М (М) метиленхлориде добавляли дициклогексилкарбодиимид (DCC) (1,5 эквивалентов) при 25oС и перемешивали в течение 1 часа. Затем добавляли 1,5 эквивалентов бензилового спирта и полученную смесь перемешивали в последующие 12 часов при 25oС. Затем реакционную смесь разбавляли этилацетатом (0,10 М) и дважды (2Х) промывали водой, однократно (1X) - солевым раствором и сушили над сульфатом магния. Затем данный растворитель удаляли при отрицательном давлении и затем полученный неочищенный продукт очищали хроматографией на силикагеле. Соединение 1, t-Boc-L-тирозин, бензиловый эфир можно также купить у Sigma.
В. Соединение 2: (S)-3-(4-(4-бромбутилокси)фенил-2-N-трет-бутилоксикарбонил-пропионовой кислоты бензиловый эфир, как показано на фигуре 38, стадия i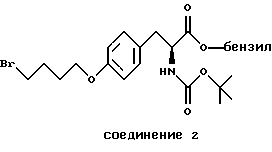
Смесь из t-Boc-L-тирозина, бензиловый эфир, (2 г, 5,38 ммоль; синтезированного как описано выше), 1,4-дибромбутана (1,9 мл, 16,2 ммоль; Aldrich), углекислого калия (5 г) и 18-крон-6 (0,1 г; Aldrich) нагревали при 80oС в течение 12 часов. После охлаждения полученный осадок отфильтровывали, а реакционную смесь выпаривали досуха под отрицательным давлении. Затем неочищенный продукт выделяли очисткой путем кристаллизации с использованием 100% гексана с выходом 2,5 г (92%) соединения 2.
С. Соединение 3: (S)-3-(4-(4-азидбутокси)фенил-2-N-трет-бутилоксикарбонил-пропионовой кислоты бензиловый эфир, как показано на фигуре 38, стадия ii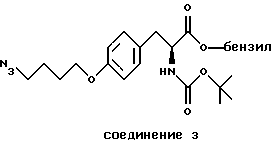
Соединение 2 (2,5 г, 4,9 ммоль) перемешивали с азидом натрия (1,6 г, 25 ммоль) в диметилформамиде (DMF) (20 мл) при 25oС в течение 12 часов. Затем данный растворитель выпаривали, а полученный остаток обрабатывали водой (приблиз. 10 мл) и дважды экстрагировали этилацетатом. Полученные органические слои объединяли, сушили над сульфатом магния и выпаривали с выходом 2,0 г (90%) соединения 3 в виде неокрашенного сиропа (FAB-MS: 469 (М+Н+).
D. Соединение 4: (S)-3-(4-(4-азидбутилокси)фенил-2-амино-пропионовой кислоты бензиловый эфир, как показано на фигуре 38, стадия iii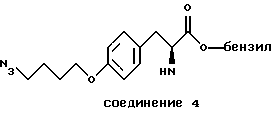
Соединение 3 (2,0 г (4,4 ммоль)) растворяли в трифторуксусной кислоте (TFA; 2 мл) и перемешивали в течение 3-х часов при комнатной температуре. Выпаривали при отрицательном давлении с выходом 1,6 г (количественно) соединения 4 в виде неокрашенного сиропа, который в дальнейшем использовали без дополнительной очистки на следующей стадии. FAB-MS: 369 (M+Н+).
Е. Соединение 5: (S)-3-(4-(4-азидбутилокси)фенил-2-бутилсульфонамидо-пропионовой кислоты бензиловый эфир, как показано на фигуре 38, стадия iv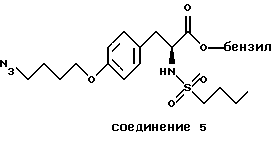
Смесь из соединения 4 (1,6 г; 4,3 ммоль), хлорида бутансульфоновой кислоты (0,84 мл; 6,6 ммоль) и триэтиламина (1,5 эквивалентов) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем данную реакционную смесь выпаривали, а полученный остаток растворяли в этилацетате, промывали разбавленной НСl, водным натрийбикарбонатом и водой. После выпаривания досуха полученный неочищенный продукт выделяли очисткой с помощью флэш-хроматографии (силикагель, толуол/этилацетат 15:1) с выходом 1,4 г (67%) соединения 5 в виде аморфного твердого вещества.
F. Соединение 6: (S)-3-(4-(4-азидбутилокси)фенил-2-бутилсульфонамидо-пропионовой кислоты бензиловый эфир, как показано на фигуре 38, стадия v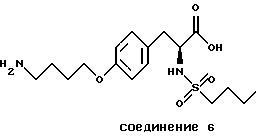
Соединение 5 (1,3 г (2,6 ммоль)) растворяли в 20 мл этилацетат/метанол/вода 5/3/1/ и 0,2 мл трифторуксусной кислоты (TFA) и гидрировали под водородом (1 атмосфера; аппарат Parr Shaker) при 25oС в присутствии 100 мг палладия (10% на угле). Через 3 часа данный катализатор отфильтровывали, а данный растворитель выпаривали с выходом соединения 6 в виде масляного остатка. После лиофилизации из воды получали 1,0 г (количественно) соединения 6 в виде белого порошка. FAB-MS: 373 (М+Н+).
G. Соединение 7: (S)-3-(4-(4-гуанидинбутилокси)фенил-2-бутилсульфонамидо-пропионовой кислоты, как показано на фигуре 38, стадия vi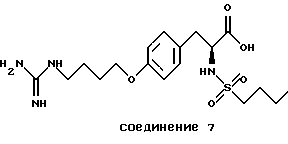
Соединение 6 (200 мг; 0,5 ммоль), 3,5-диметилпиразол-1-карбоксамидиннитрат (DPFN) (170 мг; 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения данный растворитель выпаривали при отрицательном давлении, а полученный остаток очищали с помощью HPLC (Lichrocart RP-18, градиент ацетонитрил/вода + 0,3% TFA 99:1-1:99) с выходом после лиофилизации 50 мг (25%) соединения 7 в виде белого, аморфного порошка. FAB-MS: 415 (М+Н+), т.пл.: 70oС.
Н. Соединение 8: (S)-3-(4-(4-аминобутилокси)фенил-2-N-трет-бутилоксикарбонил-пропионовой кислоты, как показано на фигуре 38, стадия iii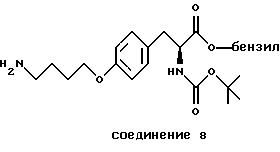
Соединение 3 (0,5 г (1,07 ммоль)) растворяли в 10 мл этилацетат/метанол/вода 5/3/1 и 0,1 мл трифторуксусной кислоты (TFA) и гидрировали под водородом (1 атмосфера; аппарат Parr Shaker) при 25oС в присутствии 30 мг палладия (10% на угле). Через 3 часа данный катализатор отфильтровывали, а данный растворитель выпаривали с выходом соединения 8 в виде масляного остатка. После лиофилизации из воды получали 370 мг (количественно) соединения 8 в виде белого порошка. FAB-MS: 353 (М+Н+).
I. Соединение 9: (S)-3-(4-(4-гуанидинобутилокси)фенил-2-N-трет-бутилоксикарбонил-пропионовой кислоты, как показано на фигуре 38, стадия iv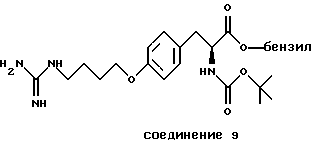
Соединение 8 (200 мг; 0,5 ммоль), 3,5-диметилпиразол-1-карбоксамидиннитрат (DPFN) (170 мг; 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения данный растворитель выпаривали при отрицательном давлении, а полученный остаток очищали с помощью HPLC (Lichrocart RP-18, градиент ацетонитрил/вода + 0,3% TFA 99:1-1:99) с выходом после лиофилизации 160 мг (90%) соединения 9 в виде белого аморфного порошка. FAB-MS: 395 (М+Н+).
J. Соединение 10: (R)-3-(4-(4-гуанидинобутилокси)фенил-2-бутилсульфонамидо-пропионовой кислоты, как показано на фигуре 38, стадия i-vi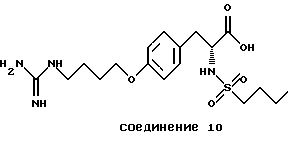
Для получения 205 мг D-тирозин - аналога 10 в виде белого аморфного материала использовали реакционную последовательность, идентичную синтезу соединения 7, FAB-MS: 415 (М+Н+), как следует ниже, с использованием промежуточных соединений 100-600 с образованием соединения 10.
1) Соединение 100: t-Boc-D-тирозин, бензиловый эфир, как показано на фигуре 40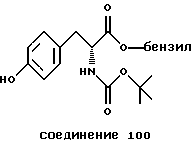
К раствору N-(трет-бутоксикарбонил)D-тирозин(t-Boc-L-тирозин) (1,0 эквивалентов; Aldrich) в 0,10 М метиленхлориде добавляли дициклогексилкарбодиимид (DCC) (1,5 эквивалентов) при 25oС и оставляли перемешиваться в течение 1 часа. Затем добавляли 1,5 эквивалентов бензилового спирта и полученную смесь перемешивали в последующие 12 часов при 25oС. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем удаляли данный растворитель при отрицательном давлении, а затем полученный неочищенный продукт очищали колоночной хроматографией на силикагеле.
2) Соединение 200: (R)-3-(4-(4-бромбутилокси)-фенил-2-N-трет-бутилоксикарбонил-пропионовой кислоты бензиловый эфир, как показано на фигуре 40, стадия i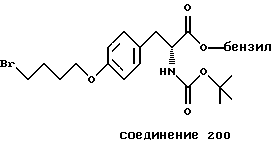
Смесь из t-BOC-D-тирозина, бензиловый эфир, (2 г, 5,38 ммоль; синтезированный, как описано выше), 1,4-дибромбутана (1,9 мл, 16,2 ммоль; Aldrich), углекислого калия (5 г) и 18-крон-6 (0,1 г; Aldrich), нагревали при 80oС в течение 12 часов. После охлаждения отфильтровывали полученный осадок, а данную реакционную смесь выпаривали досуха при отрицательном давлении. Затем полученный неочищенный продукт очищали кристаллизацией с использованием 100% гексана с выходом 2,5 г (92%) соединения 200.
3) Соединение 300: (R)-3-(4-(4-азидбутилокси)фенил-2-N-трет-бутилоксикарбонил-пропионовой кислоты бензиловый эфир, как показано на фигуре 40, стадия ii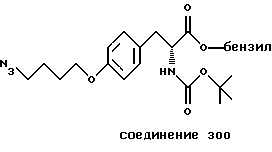
Соединение 200 (2,5 г, 4,9 ммоль) перемешивали с азидом натрия (1,6 г, 25 ммоль) в диметилформамиде (DMF) (20 мл) при 25oС в течение 12 часов. Затем данный растворитель выпаривали, а полученный остаток обрабатывали водой (приблиз. 10 мл) и дважды экстрагировали этилацетатом. Полученные органические слои объединяли, сушили над сульфатом магния и выпаривали с выходом 2,0 г (90%) соединения 300 в виде неокрашенного сиропа (FAB-MS: 469 (М+Н+).
4) Соединение 400: (R)-3-(4-(4-азидбутилокси)-фенил-2-амино-пропионовой кислоты бензиловый эфир, как показано на фигуре 40, стадия iii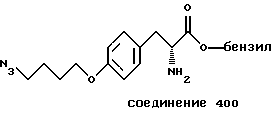
Соединение 300 (2,0 г (4,4 ммоль)) растворяли в трифторуксусной кислоте (TFA; 2 мл) и перемешивали в течение 3 часов при комнатной температуре. Выпаривание осуществляли при отрицательном давлении с выходом 1,6 г (количественно) соединения 400 в виде неокрашенного сиропа, который использовали без дальнейшей очистки на следующей стадии. FAB-MS: 369 (М+Н+).
5) Соединение 500: (R)-3-(4-(4-азидбутилокси)-фенил-2-бутилсульфонамидо-пропионовой кислоты бензиловый эфир, как показано на фигуре 40, стадия iv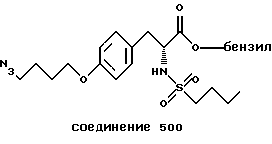
Смесь из соединения 400 (1,6 г; 4,3 ммоль), хлорида бутансульфоновой кислоты (0,84 мл; 6,6 ммоль) и триэтиламина (1,5 эквивалентов) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем данную реакционную смесь выпаривали, а полученный остаток растворяли в этилацетате, промывали разбавленной НСl, водным бикарбонатом натрия и водой. После выпаривания досуха полученный неочищенный продукт очищали флэш-хроматографией (силикагель, толуол/этилацетат 15:1) с выходом 1,4 г (67%) соединения 500 в виде аморфного твердого вещества.
6) Соединение 600: (R)-3-(4-(4-аминобутилокси)-фенил-2-бутилсульфонамидо-пропионовой кислоты, как показано на фигуре 40, стадия v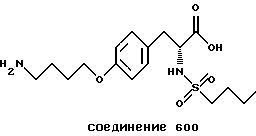
Соединение 500 (1,3 г (2,6 ммоль)) растворяли в 20 мл этилацетат/метанол/вода 5/3/1 и 0,2 мл трифторуксусной кислоты (TFA) и гидрировали под водородом (1 атмосфера; аппарат Parr Shaker) при 25oС в присутствии 100 мг палладия (10% на угле). Через 3 часа отфильтровывали данный катализатор, а данный растворитель выпаривали с выходом соединения 600 в виде масляного остатка. После лиофилизации из воды получали 1,0 г (количественно) соединения 600 в виде белого порошка. FAB-MS: 373 (М+Н+).
7) Соединение 10: (R)-3-(4-(4-гуанидинобутилокси)фенил-2-бутилсульфонил-амидо-пропионовой кислоты, как показано на фигуре 40, стадия vi
Стадия 600 (200 мг; 0,5 ммоль), 3,5-диметилпиразолкарбоксамидиннитрат (DPFN) (170 мг; 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения данный растворитель выпаривали при отрицательном давлении, а полученный остаток очищали с помощью HPLC (Lichrocart RP-18, градиент ацетонитрил/вода + 0,3% TFA 99:1-1:99) с выходом, после морфолизации, 50 мг (25%) соединения 10 в виде белого аморфного порошка. EAB-MS: 415 (М+Н+), т.пл.: 70oС.
К. Соединение 11: (S)-3-(4-(4-азидбутилокси)фенил-2-(10-камфорсудьфонамидо)-пропионовой кислоты бензиловый эфир, как показано на фигуре 4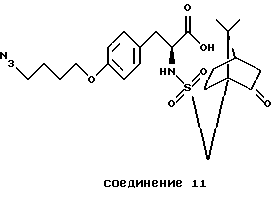
Смесь из соединения 4 (1,0 г; 2,7 ммоль), хлорида 10-камфорсульфоновой кислоты (6,6 ммоль; Aldrich Chemical Company) и триэтиламина (1,5 эквивалентов) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем данную реакционную смесь выпаривали, а полученный остаток растворяли в этилацетате, промывали разбавленной НС1, водным бикарбонатом натрия и водой. После выпаривания досуха полученный неочищенный продукт очищали флэш-хроматографией (силикагель, толуол/этилацетат 15:1) с выходом 1,4 г (67%) соединения 11 в виде аморфного твердого вещества.
L. Соединение 12: (S)-3-(4-(4-гуанидинобутилокси)фенил-2-(10-камфорсульфонамидо)-пропионовой кислоты, как показано на фигуре 41, стадии i-ii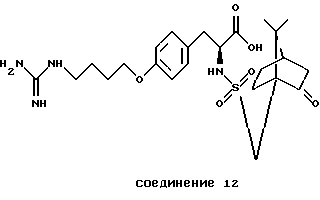
Соединение 12 получали после гидрирования и гуанилирования соединения 11 в соответствии с нижеследующими условиями.
Стадия i: Соединение 11 (1,3 г, 2,6 ммоль)) растворяли в 20 мл этилацетат/метанол/вода 5/3/1 и 0,2 мл трифторуксусной кислоты (TFA) и гидрировали под водородом (1 атмосфера; аппарат Parr Shaker) при 25oС в присутствии 100 мг палладия (10% на угле). Через 3 часа данный катализатор отфильтровывали, а данный растворитель выпаривали с выходом промежуточного амина в виде масляного остатка. После лиофилизации из воды получали 1,0 г (количественно) этого промежуточного амина в виде белого порошка, что осуществляли следующим образом.
Стадия ii: Вышеобразованное соединение промежуточного амина (200 мг; 0,5 ммоль), 3,5-диметилпиразол-1-карбоксамидиннитрат (DPFN) (170 мг; 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения данный растворитель выпаривали при отрицательном давлении, а полученный остаток очищали с помощью HPLC (Lichrocart RP-18, градиент ацетонитрил/вода + 0,3% TFA 99:1-1:99) с выходом после лиофилизации 50 мг (25%) соединения 12 в виде белого аморфного порошка. FAB-MS: 509,6 (М+Н+).
М. Соединение 13: (S)-3-(4-(4-бромпентилокси)фенил-2-2-N-трет-бутилоксикарбонил-пропионовой кислоты бензиловый эфир, как показано на фигуре 41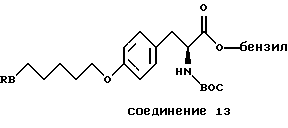
Смесь из t-Boc-L-тирозина, бензиловый эфир (4,5 г, 12,1 ммоль; соединение 1 синтезировали, как описано выше), 1,5-дибромбутана (5 мл, 36,7 ммоль; Aldrich), углекислого калия (10 г) и 18-крон-6 (0,25 г; Aldrich) нагревали при 80oС в течение 12 часов. После охлаждения отфильтровывали полученный преципитат, а данную реакционную смесь выпаривали досуха при отрицательном давлении. Затем полученный неочищенный продукт очищали кристаллизацией с использованием 100% гексана с выходом 5,35 г (85%) соединения 13.
N. Соединение 14: (S)-3-(4-(5-гуанидинопентилокси)-фенил-2-бутилсульфонамидо-пропионовой кислоты, как показано на фигуре 40, стадии i-v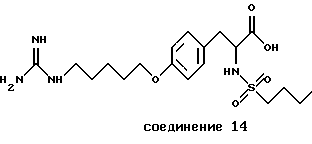
Последовательность реакции на стадии 5 - бромин-азидобмен, Вос-гидролиз, сульфонилирование с помощью хлорида бутансульфоновой кислоты, гидрирование и гуанилирование с помощью DPFN осуществляли идентично вышеприведенным методикам с использованием промежуточных соединений 1-6 с образованием соединения 7 или методикам с использованием соединений 100-600 с образованием соединения 10, как описано выше. Соединение 14 получали в виде белого порошка FAB-MS: 429 (М+Н+).
О. Соединение 15: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-амино-этил)фенокси)метил-2-оксазолидинон, дигидрохлорид, как показано на фигуре 42
1) Синтез стартового материала 2-N-ВОС-амино-3-(4-гидроксифенил)пропионата для соединения 15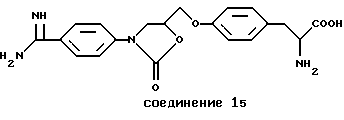
Стартовый материал 2-N-ВОС-амино-3-(4-гидрокси-фенил)-пропионат, получали путем этерификации (D или L), N-(трет-бутоксикарбонил)-L(D)-тирозина (t-Boc-L(D)-тирозин) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавляли 1%-ной НСl. Данную реакционную смесь перемешивали при 25oС в течение 12 часов, а затем нейтрализовали с помощью углекислого калия, а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли данный растворитель, а затем полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 2-N-BOC-амино-3-(4-гидрокси-фенил)пропионата.
2) Синтез стартового материала 3-р-N-ВОС-амидино-фенил-5-метансульфонилокси-метил-2-оксазолидинона для соединения 15; с ниже следующей 3-стадийной методикой
р-Амино-бензонитрил (1,0 эквивалент; Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалент; Aldrich) в течение 12 часов при 25oС. Затем этот растворитель удаляли при отрицательном давлении, а неочищенный 4-(2,3-дигидроксипропиламино)бензонитрил передавали на следующую стадию;
4-(2,3-дигидроксипропиламино)бензонитрил (1,0 эквивалент, как описано выше), в диметилформамиде (0,10 М), при 25oС перемешивали с диэтилкарбонатом (1,1 эквивалента; Aldrich) и трет-бутилатом калия (1,1 эквивалента; Aldrich) при 110oС в течение 6 часов. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем удаляли данный растворитель при отрицательном давлении и полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 3-(4-циано-фенил)-5-гидроксиметил-2-оксазолидин и передавали на следующую стадию;
3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин (1,0 эквивалент, как описано выше) в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента сероводорода, 1,1 эквивалента метилйодида и 1,1 эквивалента ацетата аммония. Данную реакционную смесь перемешивали в течение 6 часов, а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем данный растворитель удаляли при отрицательном давлении, а полученный неочищенный продукт впоследствии очищали колоночной хроматографией на силикагеле с получением указанного амидина, который передавали на следующую стадию;
1,0 эквивалент амидина, синтезированного, как описано выше, защищали 1,1 эквивалента BOC-ON (2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldrich) в метиленхлориде (0,10 М) при 25oС и перемешивали в течение 6 часов. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем удаляли данный растворитель при отрицательном давлении, а полученный неочищенный продукт впоследствии этерифицировали в 0,10 М метиленхлориде и 1,1 эквивалентном метансульфонилхлориде. Эту реакционную смесь перемешивали при 0oС в течение 6 часов и затем останавливали реакцию водой (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем данный растворитель удаляли при отрицательном давлении, а затем очищали полученный неочищенный продукт колоночной хроматографией на силикагеле с получением 3-p-N-BOC-амидино-фенил-5-метансульфонилокси-метил-2-оксазолидинона.
3) Связывание промежуточных соединений 2-N-BOC-амино-3-(4-гидрокси-фенил)пропионата и 3-p-N-ВОС-амидино-фенил-5-метансульфонилокси-метил-2-оксазолидиноном с образованием защищенной формы соединения 15, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2N-ВОС-аминоэтил)феноксиметил-2-оксазолидинона
Смесь из 1,9 г 2-N-ВOС-амино-3-(4-гидроксифенил)-пропионата (как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент), перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-р-N-ВОС-амидино-фенил-5-метансульфонилокси-метил-2-оксазолидинона (как описано выше) в 10 мл диметилформамида (DMF) и вновь перемешивали в течение 15 минут при комнатной температуре. Полученную реакционную смесь разбавляли затем этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем данный растворитель удаляли при отрицательном давлении, а полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением защищенной формы соединения 15 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2N-ВОС-аминоэтил)феноксиметил-2-оксазолидинона, который передавали на следующую стадию.
4) Снятие защиты с защищенной формы соединения 15 с образованием соединения 15: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-аминоэтил)фенокси)-метил-2-оксазолидинон, дигидрохдорида, фигура 42
Обрабатывали защищенную форму соединения 15 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2N-ВОС-аминоэтил)феноксиметил-2-оксазолидинона (1,0 эквивалент, синтезированного, как описано выше) с помощью 4 мл 2N NaOH в течение 4 часов при комнатной температуре. Затем в полученную смесь добавляли последовательно по каплям при 0-25oС в течение 3 часов 40 мл 2N HCl-раствора. Затем данную реакцию резко останавливали с помощью натрия бикарбоната (5 эквивалентов), а после разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали хроматографией на колонке с силикагелем для получения соединения 15: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-амино-этил)фенокси)метил-2-оксазолидинона дигидрохлорида; т.пл. 165oС (d).
Р. Соединение 16: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-бутилсульфониламиноэтил)фенокси)метил-2-оксазолидинон, как показано на фигуре 42,
1) Синтез стартового материала 2-N-бутилсульфонил-амино-3-(4-гидрокси-фенил)пропионата для соединения 16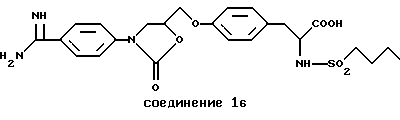
Стартовый материал 2-N-бутилсульфониламино-3-(4-гидроксифенил)пропионат получали путем этерификации ((D или L) тирозин) (1,0 эквивалентов; Sigma) в 0,10 М метаноле и разбавляли 1% НСl. Данную реакционную смесь перемешивали при 25oС в течение 12 часов и затем нейтрализовали с помощью углекислого калия, а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем растворитель удаляли при отрицательном давлении, а полученный неочищенный продукт передавали на следующую операцию:
Смесь из вышеуказанного соединения (4,3 ммоль), хлорид бутансульфоновой кислоты (6,6 ммоль) и триэтиламин (1,5 эквивалентов) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Эту реакционную смесь выпаривали, а полученный остаток растворяли в этилацетате, промывали разбавленной НСl, водным бикарбонатом натрия и водой. После выпаривания досуха полученный неочищенный продукт очищали флэш-хроматографией (силикагель, толуол/этил-ацетат 15:1) с выходом названного соединения.
2) Синтез стартового материала 3-р-N-ВОС-амидинофенил-5-метансудьфонидокси-метил-2-оксазолидинон для соединения 16; следующей 3-стадийной методикой
р-Амино-бензонитрил (1,0 эквивалентов; Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалентов; Aldrich) в течение 12 часов при 25oС. Затем растворитель удаляли при отрицательном давлении, а полученный неочищенный 4-(2,3-дигидроксипропиламино)бензонитрил передавали в нижеследующую стадию;
4-(2,3-дигидроксипропиламино)бензонитрил (1,0 эквивалент, как описано выше), в диметилформамиде (0,10 М), при 25oС, перемешивали с диэтилкарбонатом (1,1 эквивалента; Aldrich) и трет-бутилатом калия (1,1 эквивалента; Aldrich) при 110oС в течение 6 часов. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем данный растворитель удаляли при отрицательном давлении, а полученный неочищенный продукт затем очищали хроматографией на колонке с силикагелем с получением 3-(4-цианофенил)-5-гидроксиметил-2-оксазолидинона и передавали в нижеследующую стадию;
3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин (1,0 эквивалент, как описано выше), в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента сероводорода, 1,1 эквивалента метилйодида и 1,1 эквивалента аммонийацетата. Данную реакционную смесь перемешивали в течение 6 часов, а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем удаляли растворитель при отрицательном давлении, а полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением амидина, который передавали в нижеследующую стадию;
1,0 эквивалент амидина, синтезированного, как описано выше, защищали с помощью 1,1 эквивалента BOC-ON (2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldrich) в метиленхлориде (0,10 М) при 25oС и перемешивали в течение 6 часов. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1Х насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт этерифицировали затем в 0,10 М метиленхлориде и 1,1 эквивалента метансульфонилхлорида. Данную реакционную смесь перемешивали при ОoС в течение 6 часов и реакцию резко останавливали водой (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали колоночной хроматографией на силикагеле с получением 3-р-N-ВОС-амидино-фенил-5-метансульфонилокси-метил-2-оксазолидинона.
3) Связывание промежуточных соединений 2-N-бутилсульфониламино-3-(4-гидрокси-фенил)пропионата и 3-р-N-ВОС-амидино-фенил-3-метансульфонилоксиметил-2-оксазолидинона с образованием защищенной формы соединения 16, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-бутилсульфониламиноэтил)фениоксилметил-2-оксазолидинона
Смесь из 1,9 г 2-N-бутилсульфониламино-3-(4-гидрокси-фенил) пропионата (как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-р-N-ВОС-амидино-фенил-5-метансульфонилоксиметил-2-оксазолидинона (как описано выше) в 10 мл диметилформамида (DMF) и вновь перемешивали в течение 15 минут при комнатной температуре. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель и затем полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением защищенной формы соединения 16, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2-N-бутилсульфониламиноэтил)фениоксилметил-2-оксазолидинона, который передавали на следующую стадию.
4) Снятие защиты с защищенной формы соединения 16 с образованием соединения 16: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-бутилсульфониламиноэтил)фенокси)метил-2-оксазолидинона, фигура 42
Обрабатывали защищенную форму соединения 16, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-бутилсульфониламиноэтил)фениоксилметил-2-оксазолидинона (1,0 эквивалент, суспендированных, как описано выше) с 4 мл 2N NaOH в течение 4 часов при комнатной температуре. Затем, к данной смеси приливали капля за каплей при 0-25oС в течение 3 часов, 40 мл 2N раствора НСl в диоксане. Затем в данной реакционной смеси резко останавливали протекающую реакцию с помощью бикарбоната натрия (5 эквивалентов), а затем разбавляли этил-ацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением соединения 16: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-бутилсульфониламиноэтил)фенокси)метил-2-оксазолидинона; т. пл. 236-237oС.
Q. Соединение 17: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-пропил-сульфониламиноэтил)фенокси)метил-2-оксазолидинон, как показано на фигуре 42
1) Синтез стартового материала 2-N-пропил-сульфонил-амино-3-(4-гидроксифенил)пропионат для соединения 17: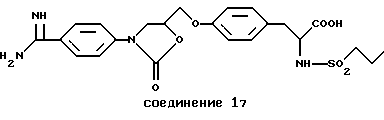
Стартовый материал 2-N-пропил-сульфониламино-3-(4-гидрокси-фенил) пропионат получали путем этерификации ((D или L) тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавляли 1% НСl. Данную реакционную смесь перемешивали при 25oС в течение 12 часов, а затем нейтрализовали с помощью углекислого калия и затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт передавали ниже.
Смесь из вышеуказанного соединения (4,3 ммоль), хлорида пропилсульфоновой кислоты (6,6 ммоль; Aldrich) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем данную реакционную смесь выпаривали, а полученный остаток растворяли в этилацетате, промывали разбавленной НСl, водным бикарбонатом натрия и водой. После выпаривания досуха полученный неочищенный продукт очищали флэш-хроматографией (силикагель, толуол/этилацетат 15:1) с выходом вышеназванного соединения.
2) Синтез стартового материала 3-р-N-ВОС-амидинофенил-5-метансульфонилокси-метил-2-оксазолидинона для соединения 17: нижеследующая 3-стадийная методика
р-Амино-бензонитрил (1,0 эквивалент; Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалентов; Aldrich) в течение 12 часов при 25oС. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный 4-(2,3-дигидроксипропиламино)бензонитрил передавали в нижеследующую стадию;
4-(2,3-дигидроксипропиламино)бензонитрил (1,0 эквивалент, как описано выше), в диметилформамиде (0,10 М), при 25oС, перемешивали с диэтилкарбонатом (1,1 эквивалента; Aldrich) и трет-бутилатом калия (1,1 эквивалента; Aldrich) при 110oС в течение 6 часов. Затем, данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали затем хроматографией на колонке с силикагелем с получением 3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин и передавали в нижеследующую стадию,
3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин (1,0 эквивалент, как описано выше), в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента сероводорода, с 1,1 эквивалента метилйодида и с 1,1 эквивалента аммонийацетата. Данную реакционную смесь перемешивали в течение 6 часов и затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали затем хроматографией на колонке с силикагелем с получением амидина, который передавали в нижеследующую стадию;
1,0 эквивалент амидина, синтезированного, как описано выше, защищали с помощью 1,1 эквивалента BOC-ON (2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldrich) в метиленхлориде (0,10 М) при 25oС и перемешивали в течение 6 часов. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт этерифицировали затем в 0,10 М метиленхлориде и 1,1 эквивалентном метансульфонилхлориде. Данную реакционную смесь перемешивали при 0oС в течение 6 часов, а затем реакцию останавливали водой (5 эквивалентов) и затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением 3-p-N-BOC-амидино-фенил-5-метансульфонилокси-метил-2-оксазолидинона.
3) Связывание промежуточных соединений 2-N-пропил-сульфониламино-3-(4-гидроксифенил)пропионата и 3-р-N-ВОС-амидино-фенил-5-метансульфонилокси-метил-2-оксазолидинона с образованием защищенной формы соединения 17, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2-N-пропил-сульфониламиноэтил)фениоксиметил-2-оксазолидинона
Смесь из 1,9 г 2-N-пропил-сульфониламино-3-(4-гидрокси-фенил) пропионата (как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент), перемешивали в течение 30 минут при комнатной температуре. После перемешивания, добавляли 1,8 г 3-р-N-ВОС-амидино-фенил-5-метансульфонил-окси-метил-2-оксазолидинона (как описано выше) в (10 мл) диметилформамиде (DMF) и вновь перемешивали в течение 15 минут при комнатной температуре. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали затем хроматографией на колонке с силикагелем с получением защищенной формы соединения 17, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2-N-пропил-сульфониламиноэтил)-фениоксилметил-2-оксазолидинона, который передавали в нижеследующую стадию.
4) Снятие защиты с защищенной формы соединения 17 с образованием соединения 17: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-пропил-сульфониламиноэтил)-фенокси)метил-2-оксазолидинона, фигура 42
Защищенную форму соединения 17, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2-N-пропил-сульфониламиноэтил)-фениоксиметил-2-оксазолидинона (1,0 эквивалент, синтезированный, как описано выше) обрабатывали с помощью 4 мл 2N NaOH в течение 4 часов при комнатной температуре. Затем, в данную смесь добавляли, при 0-25oС в течение 3 часов, капля за каплей, 40 мл 2N раствора НСl в диоксане. Затем в данной реакционной смеси протекание реакции резко останавливали с помощью бикарбоната натрия (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением соединения 17: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-пропил-сульфониламиноэтил)-фенокси)-метил-2-оксазолидинона; т.пл. 200oС (d).
R. Соединение 18: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-этил-сульфониламиноэтил)-фенокси)метил-2-оксазолидинон, как показано на фигуре 42
1) Синтез стартового материала 2-N-этил-сульфонил-амино-3-(4-гидрокси-фенил)пропионата для соединения 18: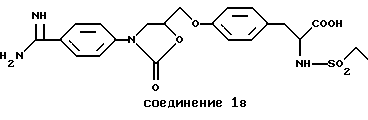
Стартовый материал 2-N-этил-сульфониламино-3-(4-гидрокси-фенил)пропионата получали путем этерификации ((D или L) тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавляли в 1% НСl. Данную реакционную смесь перемешивали при 25oС в течение 12 часов и затем нейтрализовали с помощью углекислого калия, а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт передавали в нижеследующую операцию.
Смесь из вышеуказанного соединения (4,3 ммоль), хлорида этилсульфоновой кислоты (6,6 ммоль; Aldrich) и триэтиламина (1,5 эквивалентов) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем данную реакционную смесь выпаривали, а полученный остаток растворяли в этилацетате, промывали разбавленной НСl, водным бикарбонатом натрия и водой. После выпаривания досуха полученный неочищенный продукт очищали флэш-хроматографией (силикагель, толуол/этилацетат 15:1) с выходом вышеназванного соединения.
2) Синтез стартового материала 3-р-N-ВОС-амидинофенил-5-метансульфонилокси-метил-2-оксазолидинона для соединения 18: нижеследующая 3-стадийная методика
р-Амино-бензонитрил (1,0 эквивалентов; Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалентов; Aldrich) в течение 12 часов при 25oС. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный 4-(2,3-дигидроксипропиламино)бензонитрил передавали в нижеследующую стадию;
4-(2,3-дигидроксипропиламино)бензонитрил (1,0 эквивалент, как описано выше), в диметилформамиде (0,10 М), при 25oС, перемешивали с диэтилкарбонатом (1,1 эквивалентов; Aldrich) и трет-бутилатом калия (1,1 эквивалентов; Aldrich) при 110oС в течение 6 часов. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали затем хроматографией на колонке с силикагелем с получением 3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин и передавали в нижеследующую стадию;
3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин (1,0 эквивалент, как описано выше), в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента сероводорода, с 1,1 эквивалента метилйодида и с 1,1 эквивалента аммонийацетата. Данную реакционную смесь перемешивали в течение 6 часов и затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали затем хроматографией на колонке с силикагелем с получением амидина, который передавали в нижеследующую стадию;
1,0 эквивалент амидина, синтезированного, как описано выше, защищали с помощью 1,1 эквивалентов BOC-ON (2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldrich) в метиленхлориде (0,10 М) при 25oС и перемешивали в течение 6 часов. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт этерифицировали затем в 0,10 М метиленхлориде и 1,1 эквивалентном метансульфонилхлориде. Данную реакционную смесь перемешивали при 0oС в течение 6 часов, а затем реакцию останавливали водой (5 эквивалентов) и затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением 3-р-М-ВОС-амидинофенил-5-метансульфонилокси-метил-2-оксазолидинона.
3) Связывание промежуточных соединений 2-N-этил-сульфониламино-3-(4-гидрокси-фенил)пропионата и 3-р-N-ВОС-амидино-фенил-5-метансульфонилоксиметил-2-оксазолидинона с образованием защищенной формы соединения 18, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2-N-этил-сульфониламиноэтил)-фениоксилметил-2-оксазолидинона
Смесь из 1,9 г 2-N-этил-сульфониламино-3-(4-гидроксифенил)пропионата (как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент), перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-р-N-ВОС-амидино-фенил-5-метансульфонилоксиметил-2-оксазолидинона (как описано выше) в (10 мл) диметилформамиде (DMF) и вновь перемешивали в течение 15 минут при комнатной температуре. Затем данную реакционную смесь разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением защищенной формы соединения 18, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2-N-этил-сульфониламиноэтил)-фениоксилметил-2-оксазолидинона, который передавали в нижеследующую стадию.
4) Снятие защиты с защищенной формы соединения 18 с образованием соединения 18; 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-этилсульфониламиноэтил)-фенокси)метил-2-оксазолидинона, фигура 42
Защищенную форму соединения 18, 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2-N-этил-сульфониламиноэтил)-фениоксилметил-2-оксазолидинона (1,0 эквивалент, синтезированный, как описано выше) обрабатывали с помощью 4 мл 2N NaOH в течение 4 часов при комнатной температуре. Затем в данную смесь добавляли при 0-25oС в течение 3 часов, капля за каплей, 40 мл 2N раствора НСl в диоксане. Затем в данной реакционной смеси протекание реакции резко останавливали с помощью бикарбоната натрия (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М) и промывали 2Х водой, 1X насыщенным раствором соли и сушили над сульфатом магния. Затем при отрицательном давлении удаляли растворитель, а полученный неочищенный продукт очищали хроматографией на колонке с силикагелем с получением соединения 18: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-этилсульфониламиноэтил)-фенокси)метил-2-оксазолидинона; т. пл. 212oС (d).
14. Ингибирование ангиогенеза, индуцированного ростовым фактором по измерению в САМ-анализе, с помощью внутривенного введения органических миметиков αvβ3-лиганда
Также определяли влияние органических миметиков αvβ3-лиганда, инъецированных внутривенно в САМ-препарат, на ангиогенез, индуцированный ростовым фактором, для использования в данном изобретении.
Использовали 10-дневный САМ-препарат, как описано раньше в примере 5А. Через двадцать четыре часа после начала ангиогенеза, индуцированного bFGF, органические миметики, именуемые в качестве соединений 16 (81218), 17 (87292) и 18 (87293) по отдельности инъецировали внутривенно в САМ-препарат в объеме 100 мкл в концентрации 1 мг/мл (100 мкг/эмбрион) в 20% тетрагликоль-PBS при рН 7,0. В параллельных анализах аналогичным образом определяли соединение 7 (96112), 9 (99799), 10 (96229), 12 (112854) и 14 (96113). Влияние органических миметиков анализировали через 48 часов, когда осуществляли количественный анализ путем подсчета числа точек ветвления кровеносного сосуда в области данного фильтровального диска с помощью дважды слепого метода.
Полученные результаты показаны соответственно на фигурах 43 и 44. На фигуре 43 соединения 14 (96113), 10 (96229), 9 (99799) и 12 (112854), расположенные в порядке уменьшения ингибирования, эффективно снижали число точек ветвления новых кровеносных сосудов по сравнению с контрольной индукцией bFGF и с соединением 7 (96112). На фигуре 44 соединения 17 (87292) и 18 (87293) проявляют анти-ангиогенные свойства по сравнению с необработанным bFGF-контролем и обработанным с помощью соединения 16 (81218).
В третьем анализе оценивали органические соединения 7 (96112), 10 (96229) и 14 (96113) в качестве ингибиторов ангиогенеза, индуцированного bFGF вместе с пептидами 69601 и 66203. В этом анализе через 18 часов после bFGF-обработки вводили 250 мкг/мл органического соединения, как описано в примере 7В. Полученные результаты показаны выше, на фигуре 28 соединения 14 (96113) и 10 (96229) почти полностью ингибируют образование новых кровеносных сосудов, индуцируемых с помощью bFGF.
Следовательно, вышеприведенные примеры демонстрируют, что интегрин αvβ3 играет ключевую роль в ангиогенезе, индуцированном с помощью различных стимулов и в качестве такого αvβ3 представляет собой ценную терапевтическую мишень для αvβ3-антагонистов данного изобретения при заболеваниях, характеризующихся реваскуляризацией.
Вышеизложенное письменное описание сочтено достаточным, чтобы дать возможность специалисту в данной области осуществить на практике данное изобретение. Объем настоящего изобретения не ограничен депонированной клеточной линией, поскольку депонирование осуществляли с намерением лишь проиллюстрировать один из аспектов данного изобретения и какую-либо клеточную линию, которая является функционально эквивалентной в рамках объема данного изобретения. Депонирование материала не означает допущение того, что изложенное здесь письменное описание не отвечает требованию для применения на практике какого-либо аспекта настоящего изобретения, включая наилучший его способ, и не является ограничением объема формулы изобретения, которое она представляет. В действительности, различные модификации настоящего изобретения, кроме тех, которые показаны и описаны здесь, очевидны специалистам в данной области из предшествующего описания и попадают в объем прилагаемой формулы изобретения.





















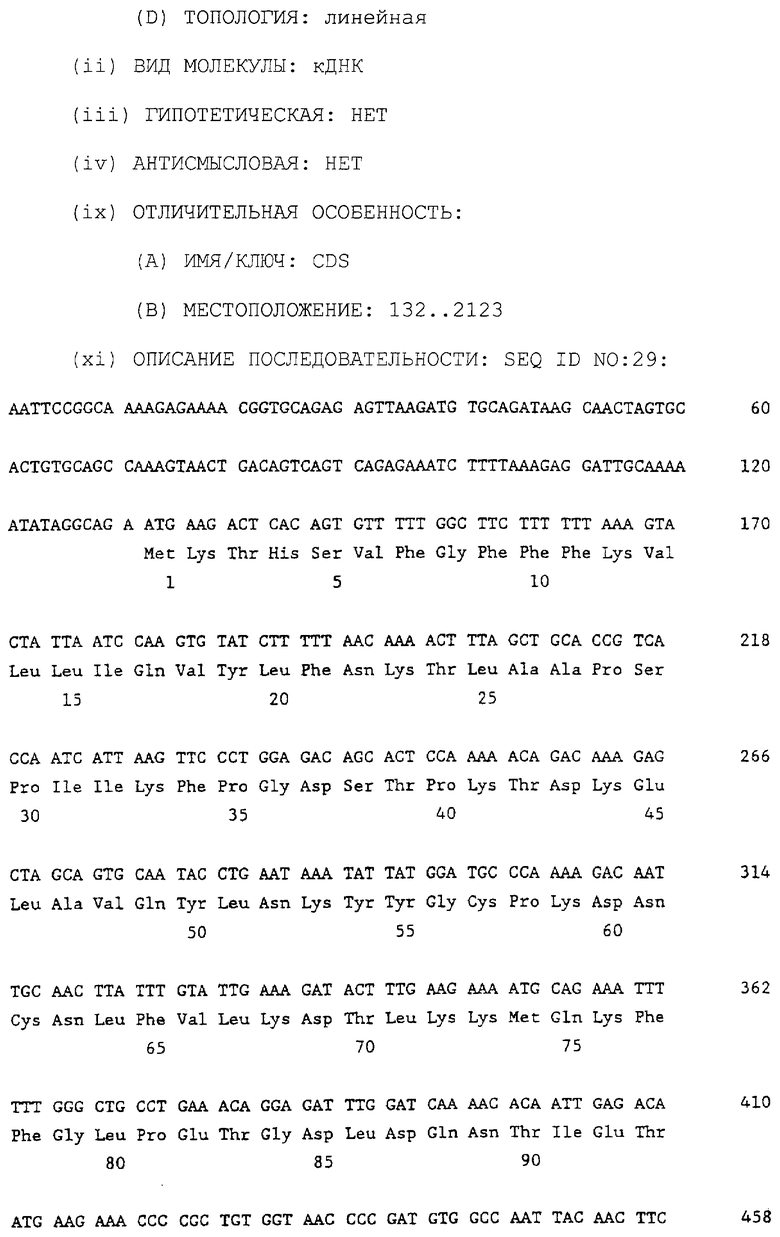


















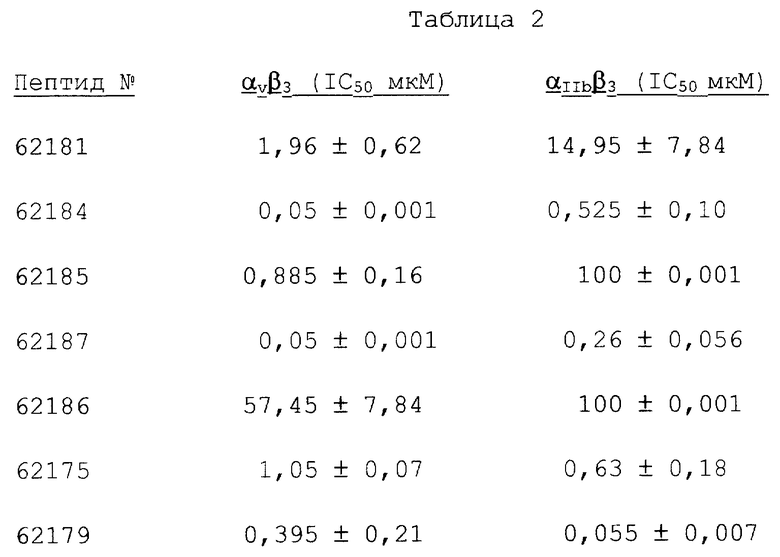
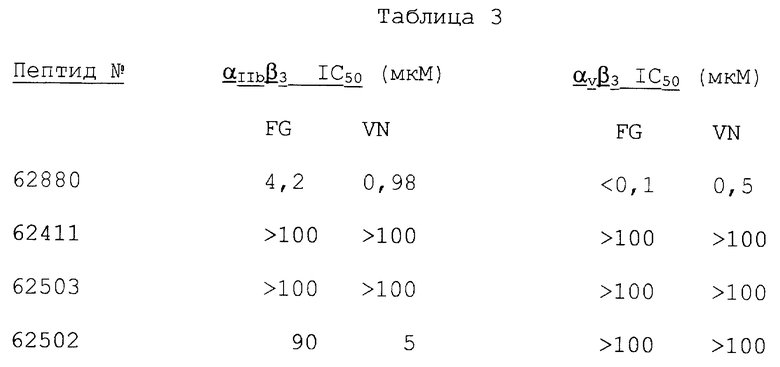






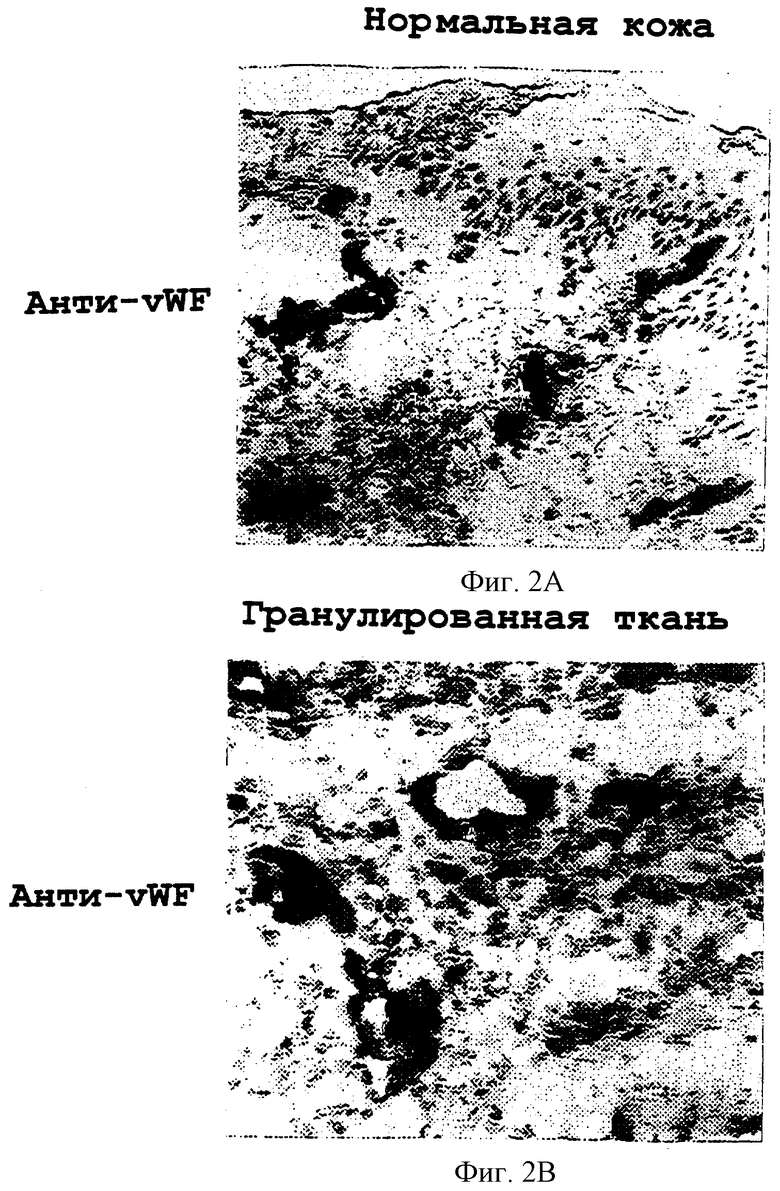



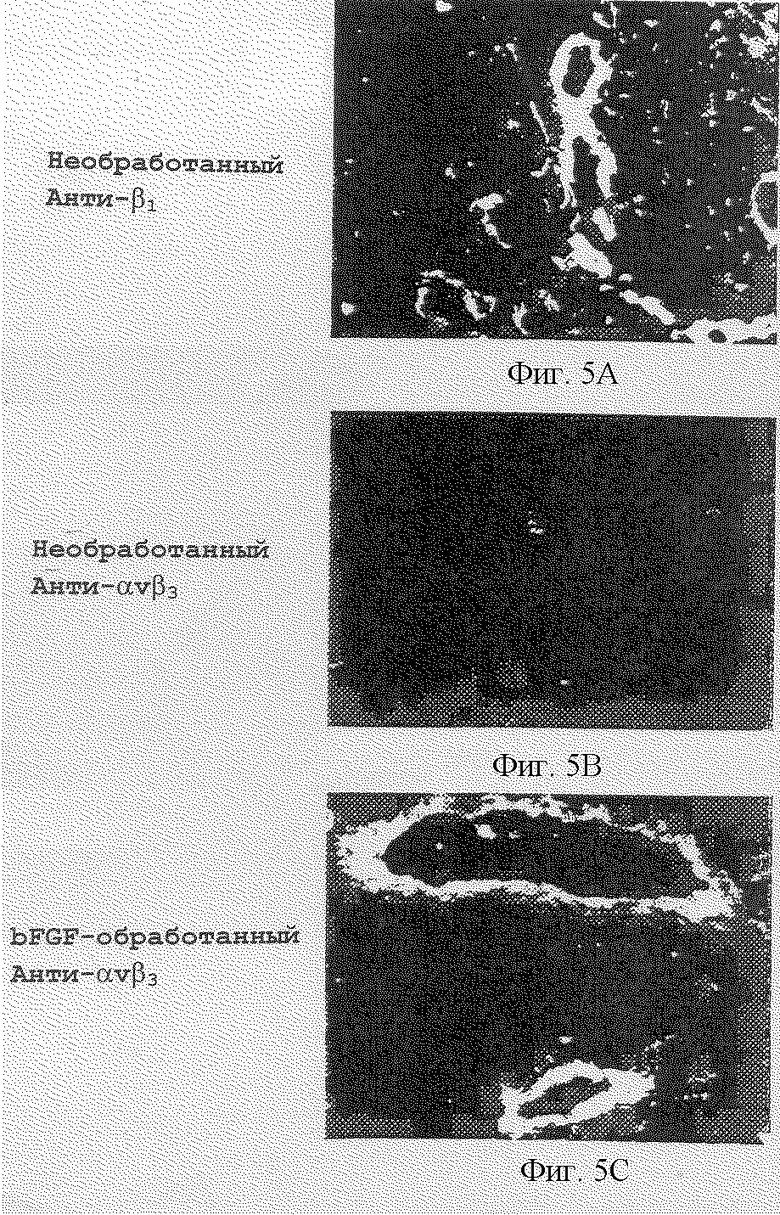
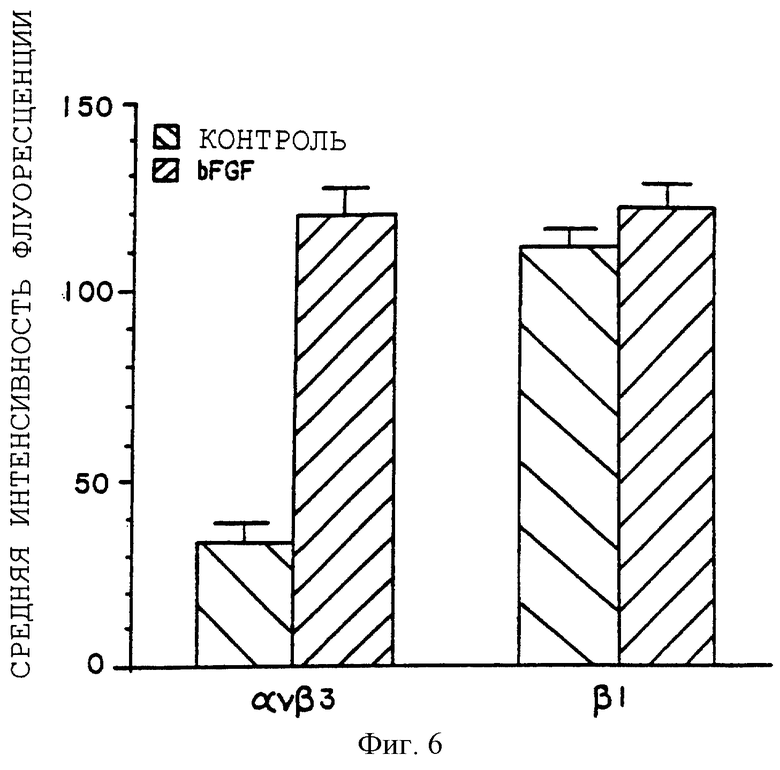
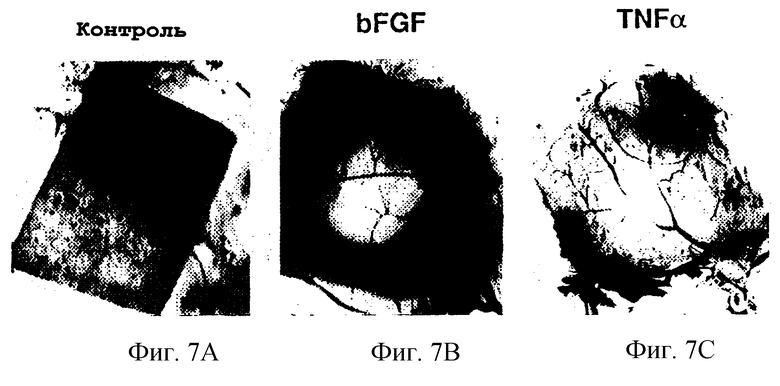
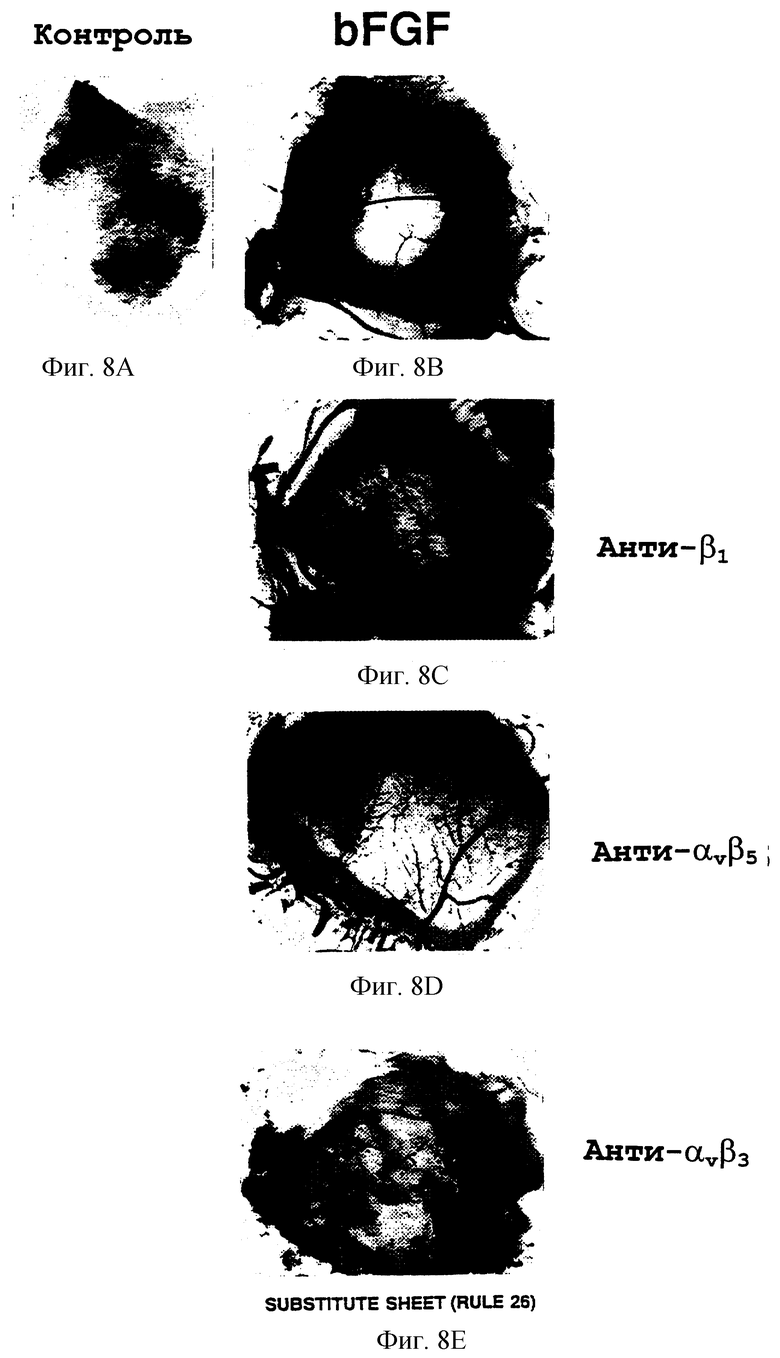
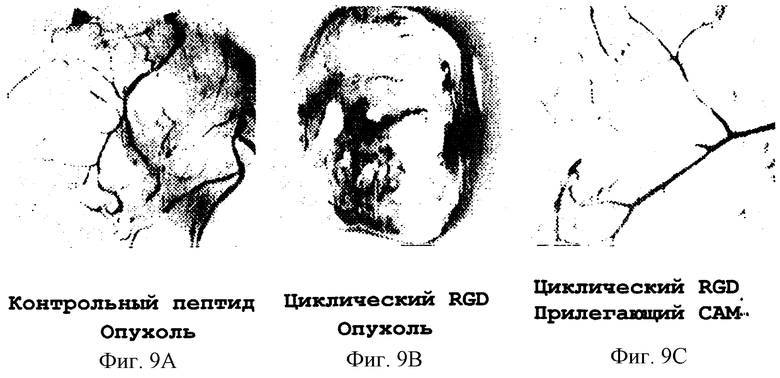
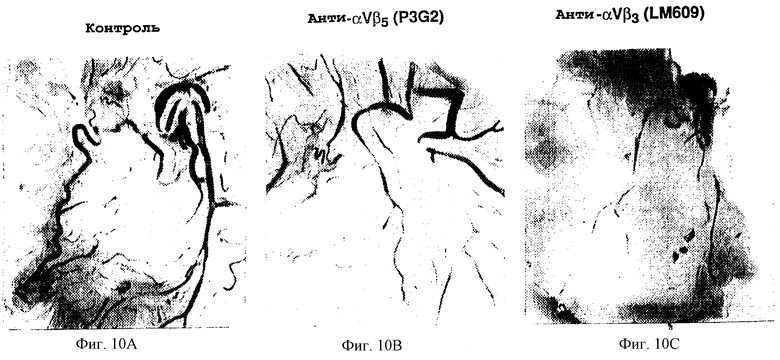
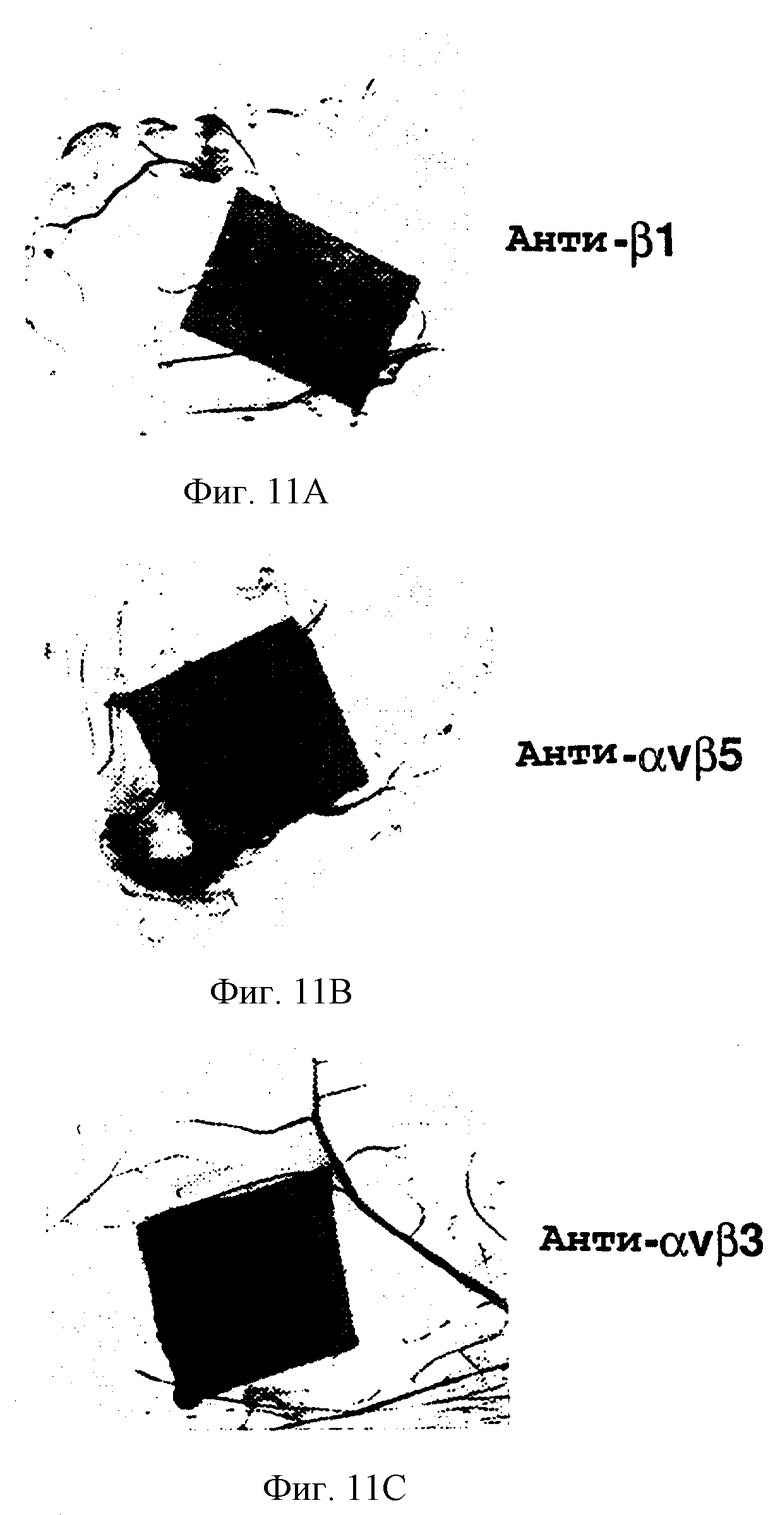
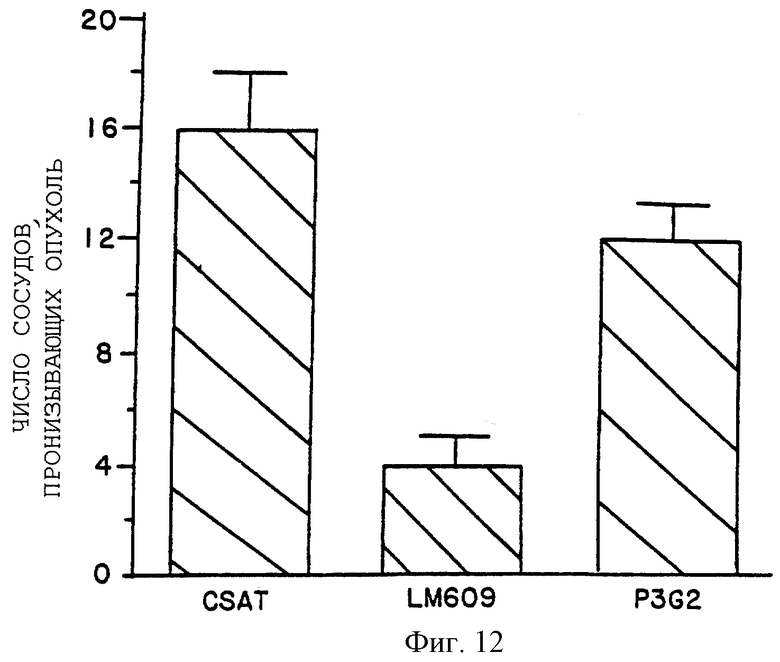
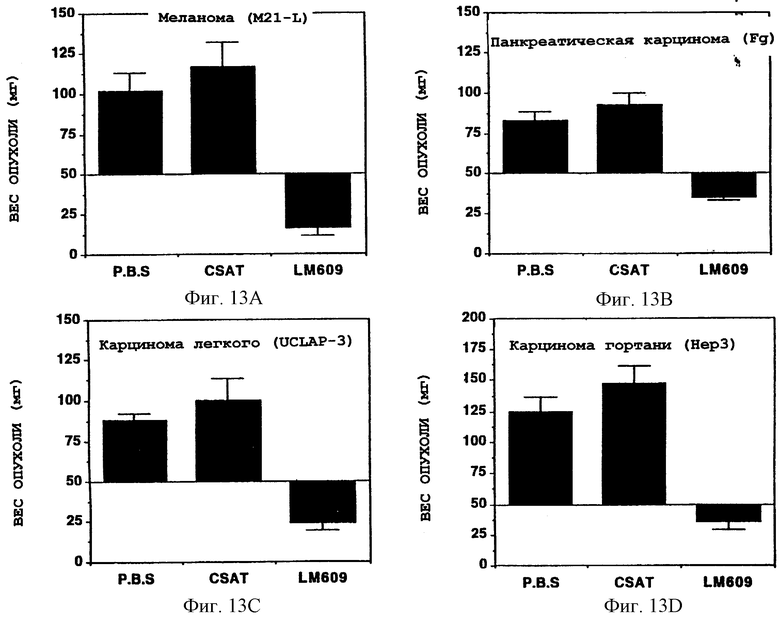
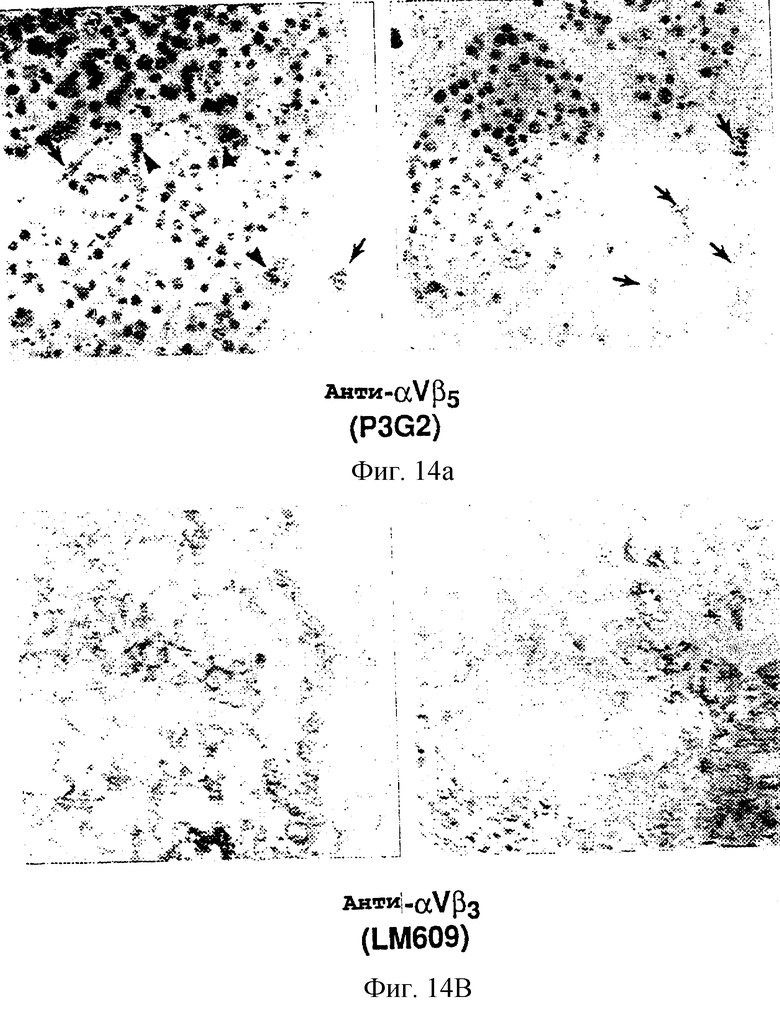
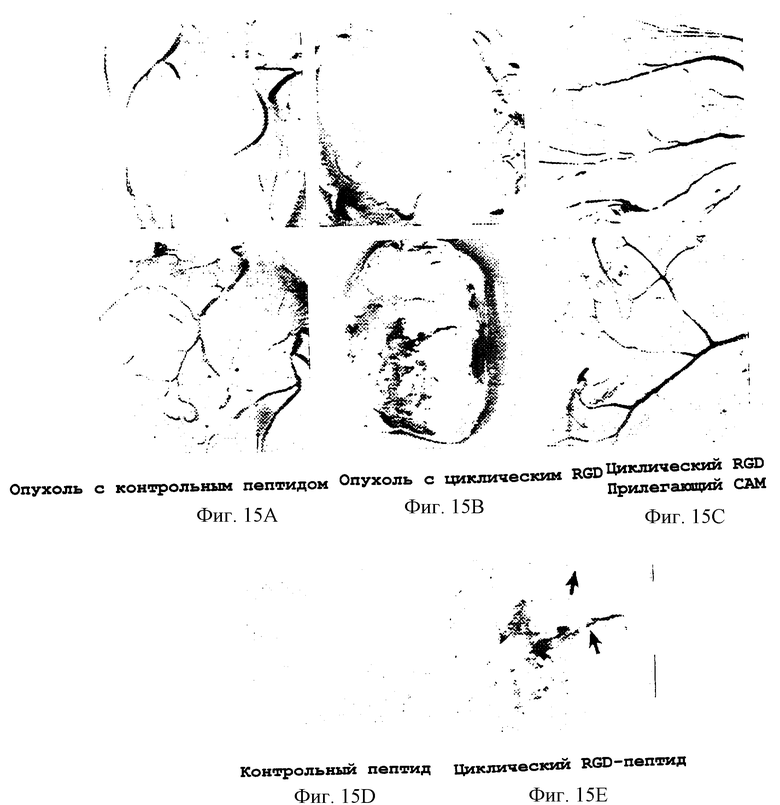
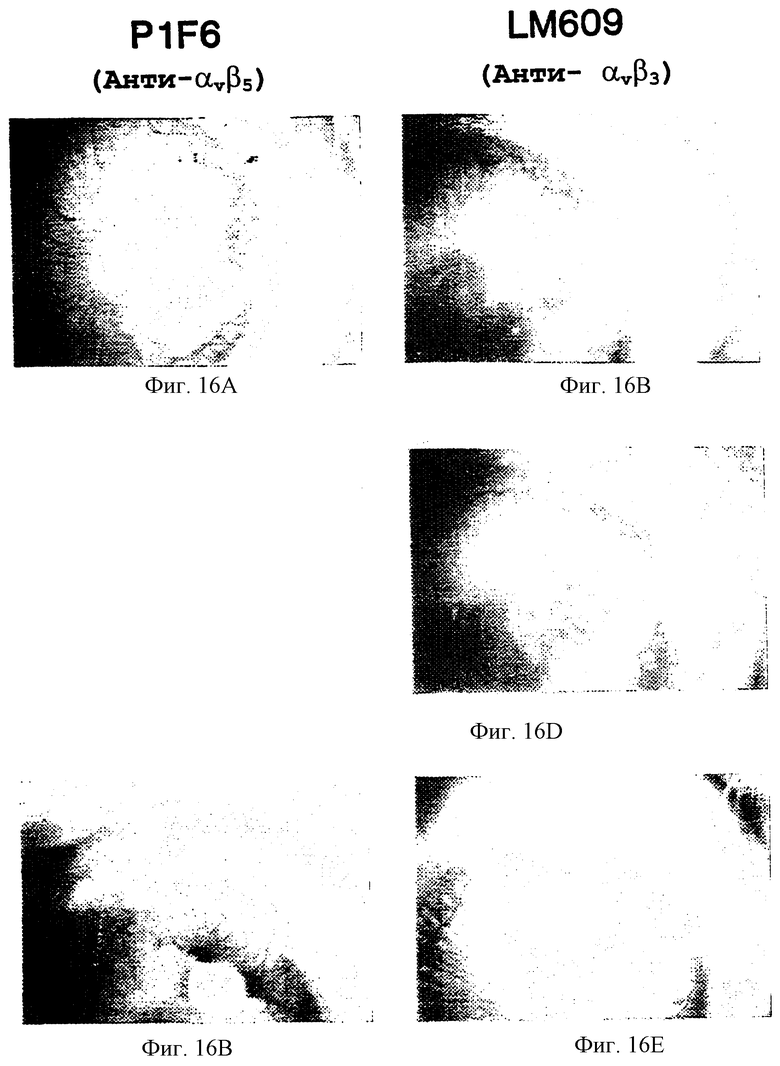
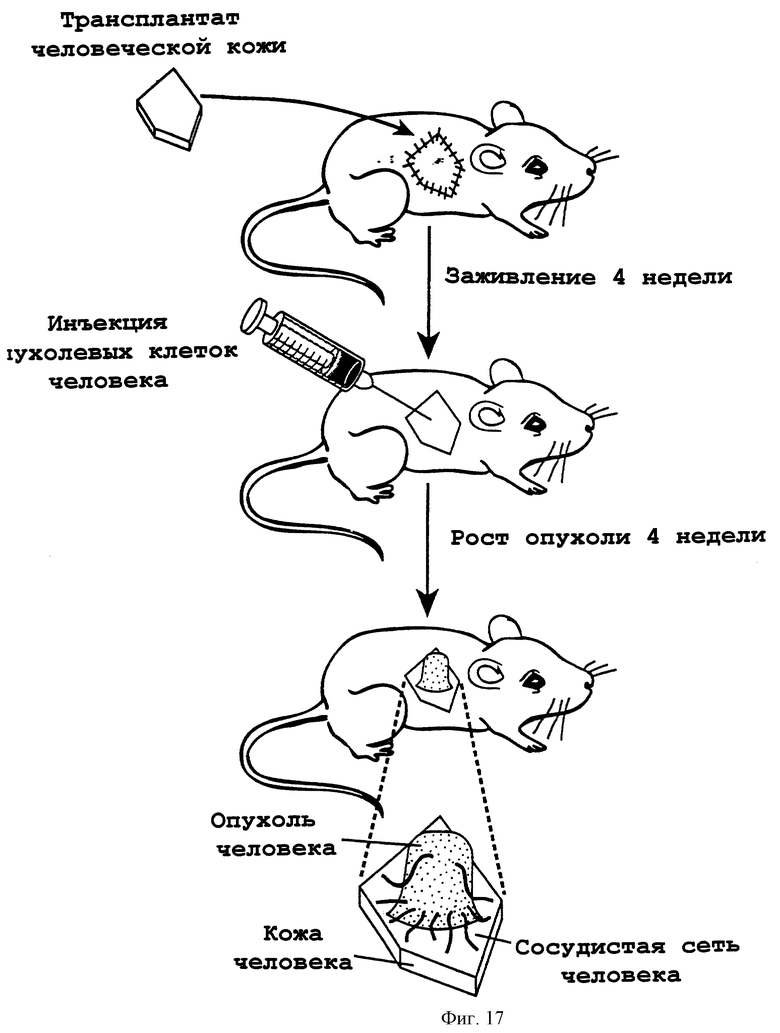
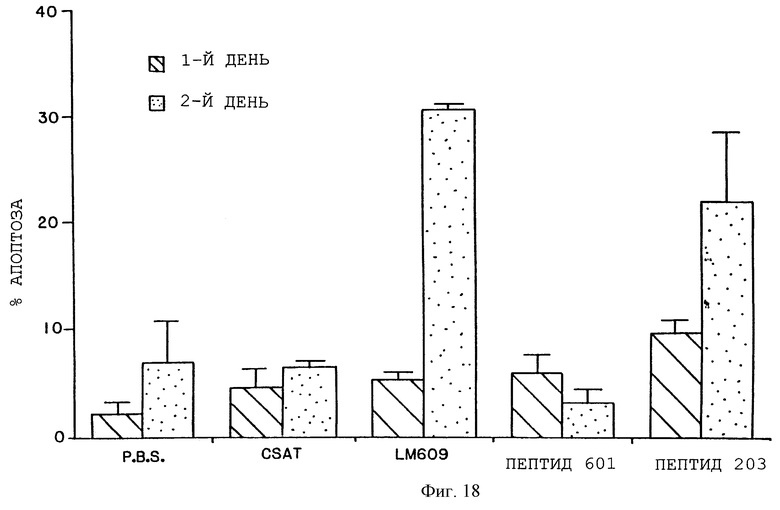
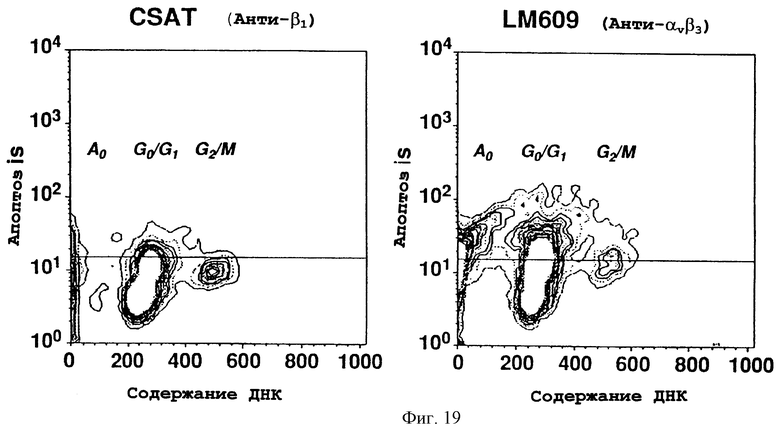
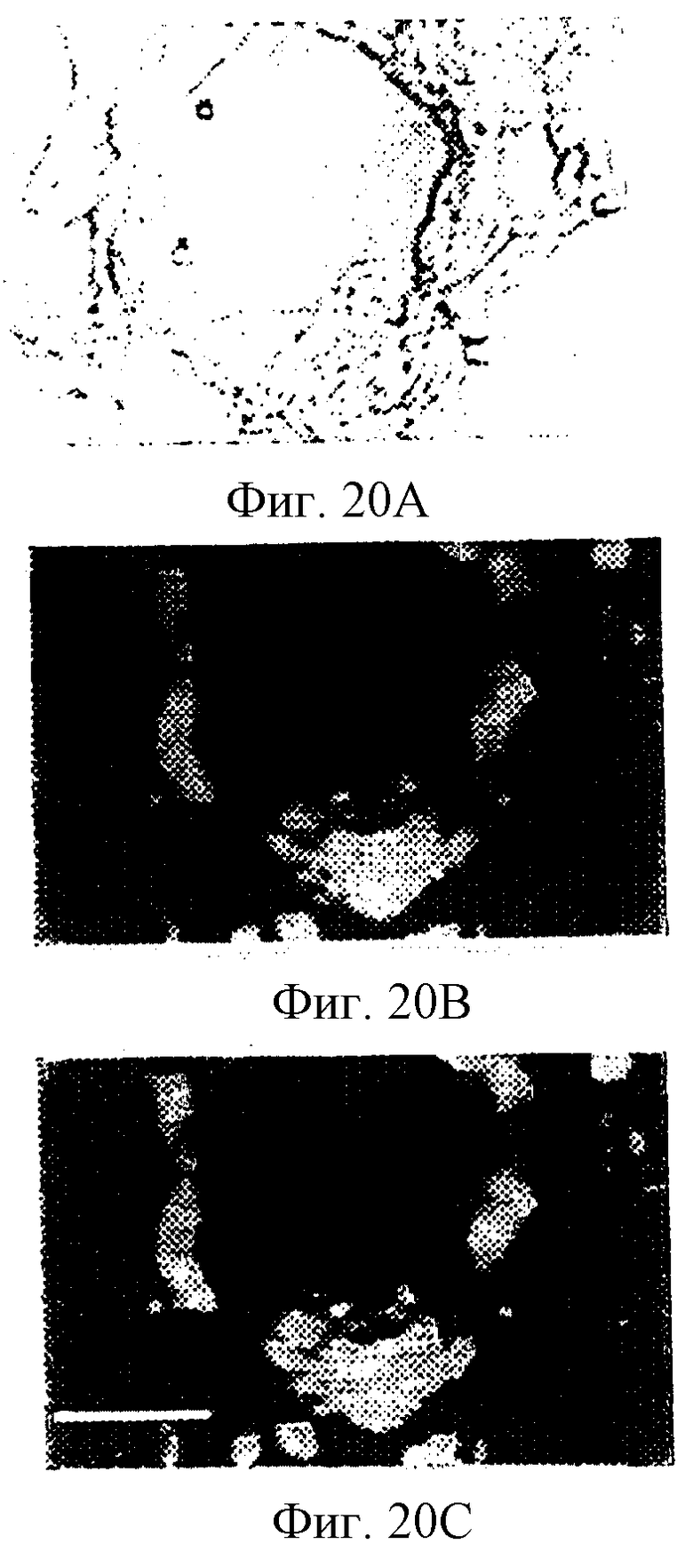

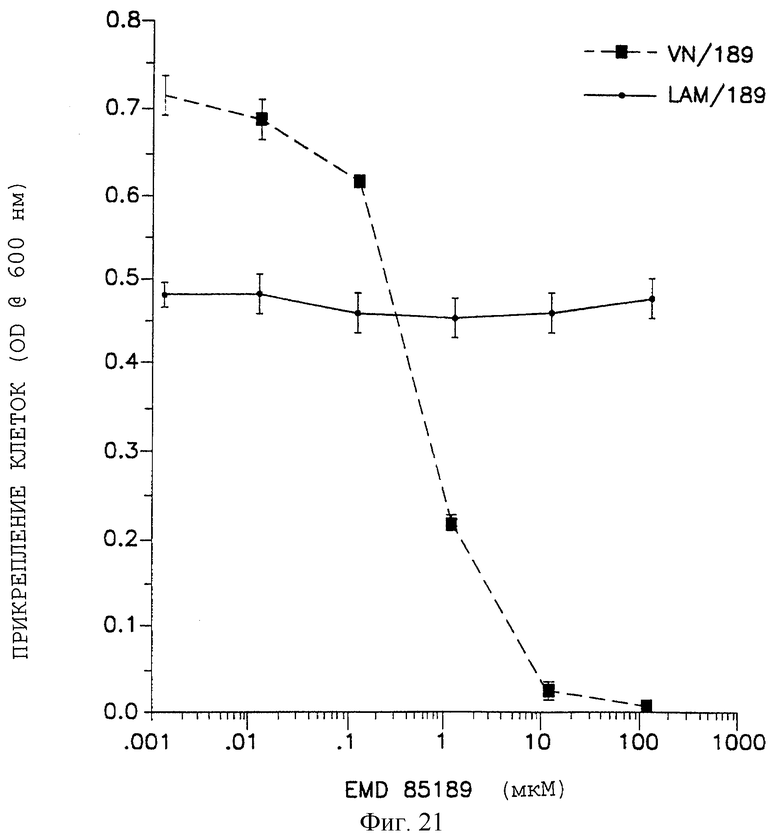
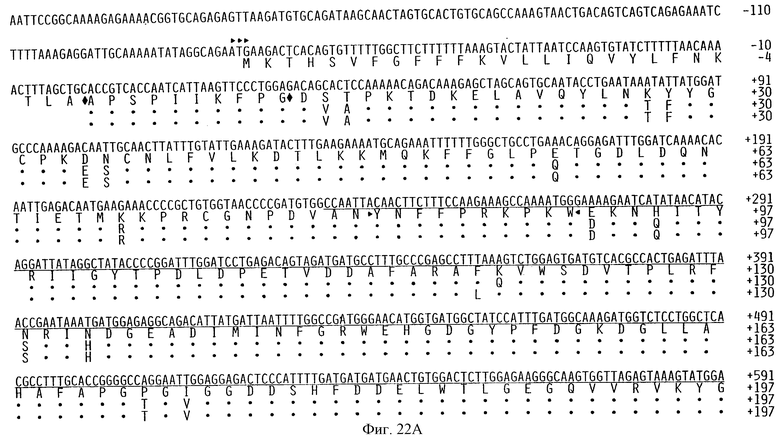

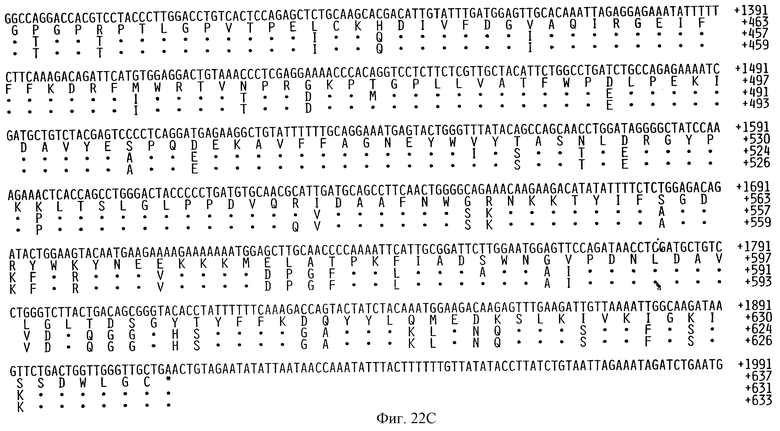

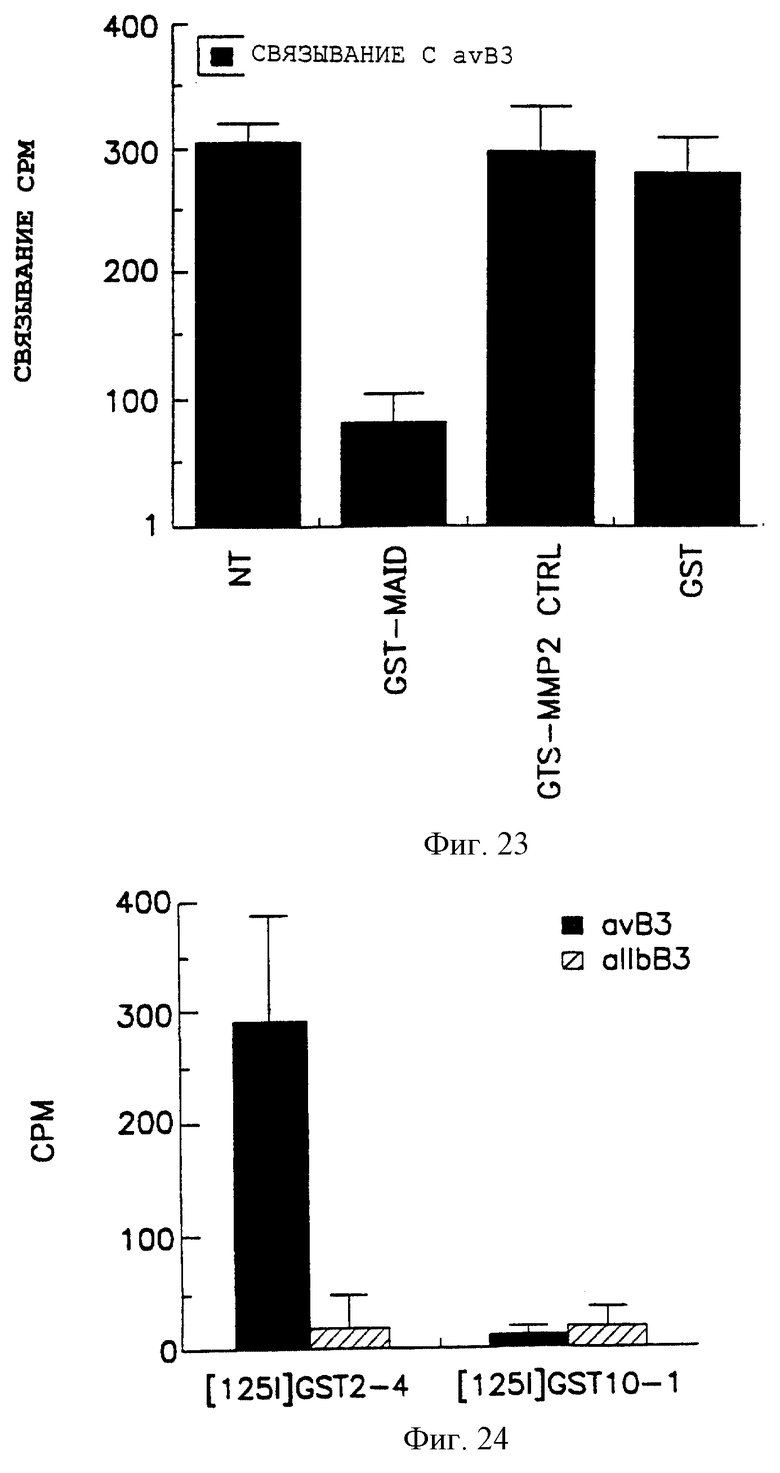
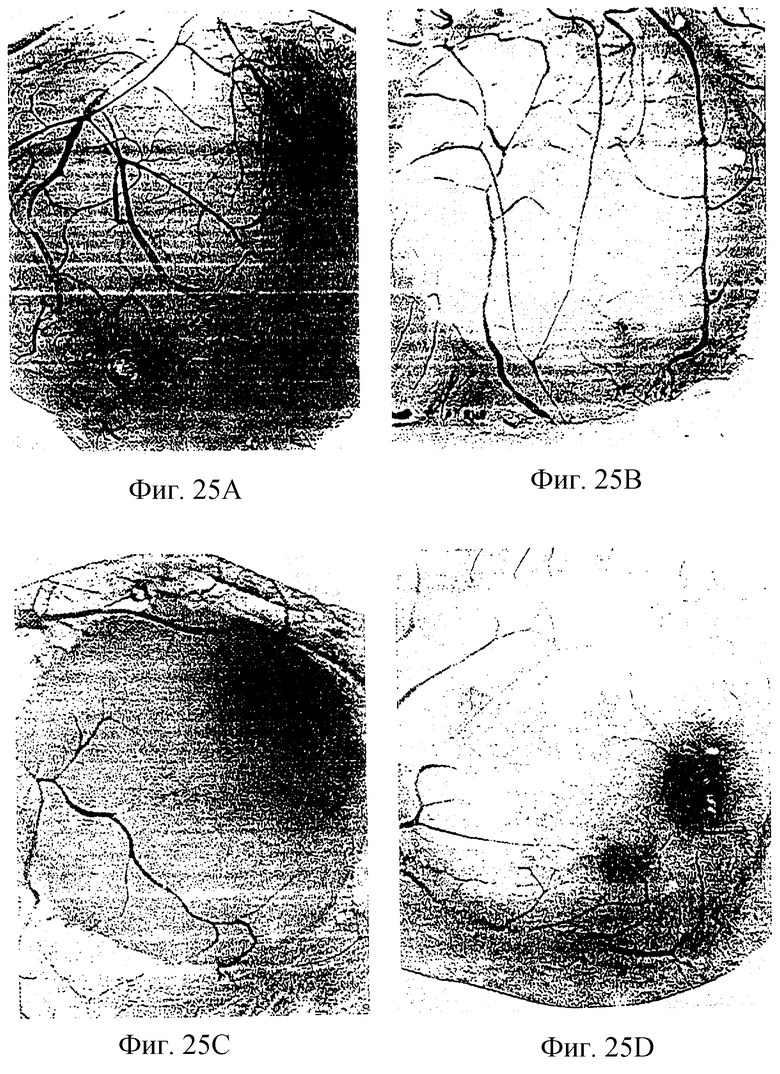
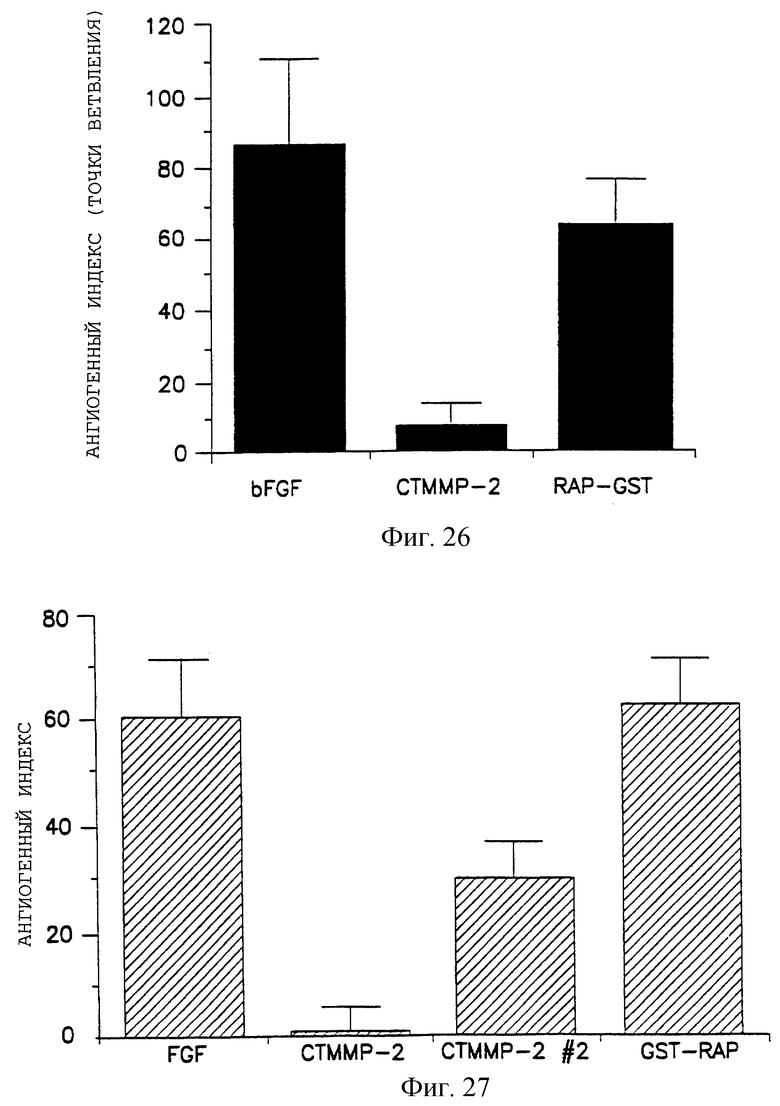
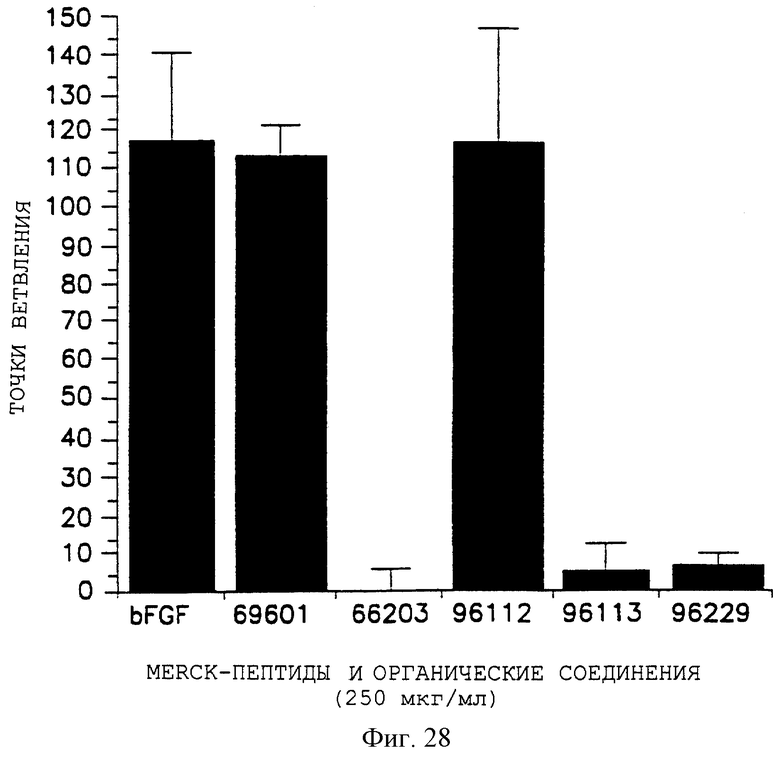
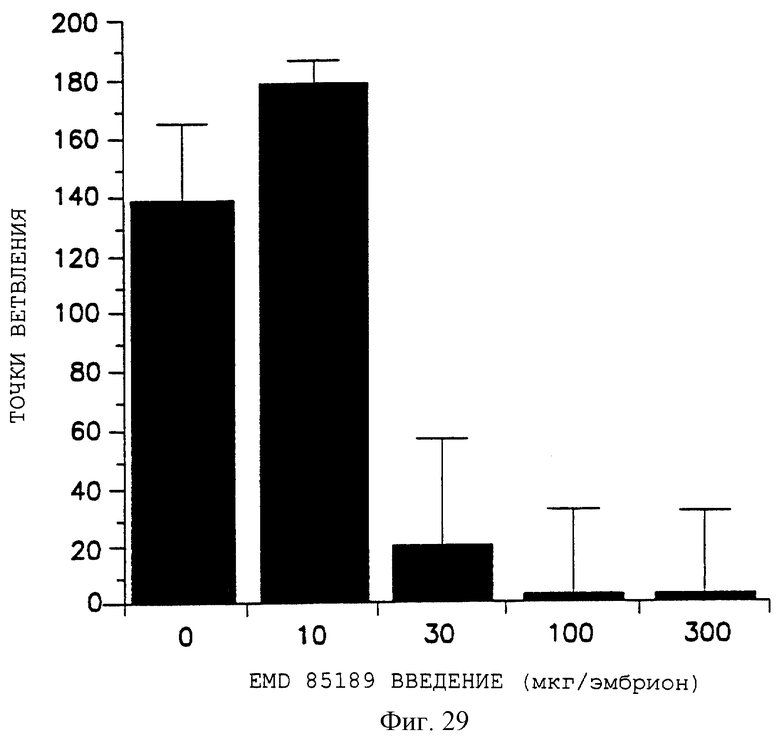
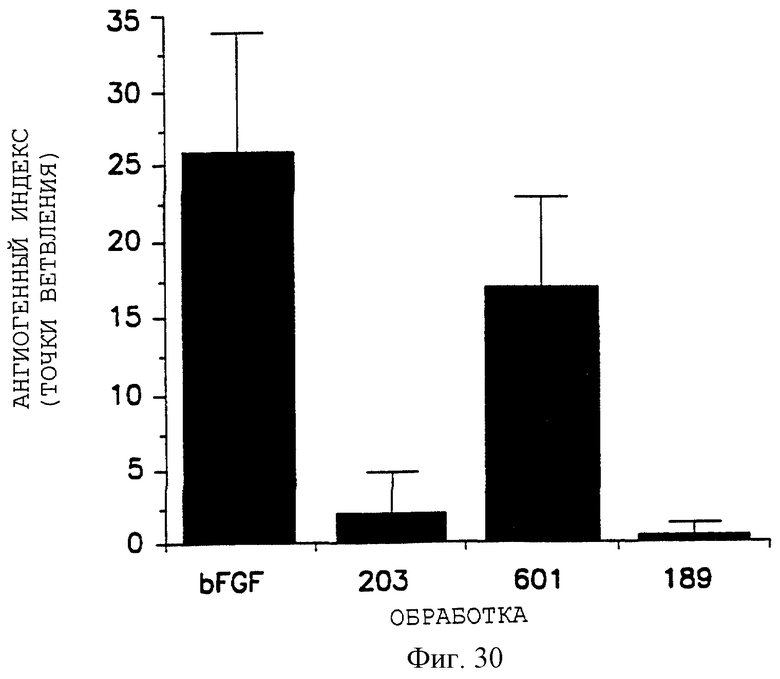
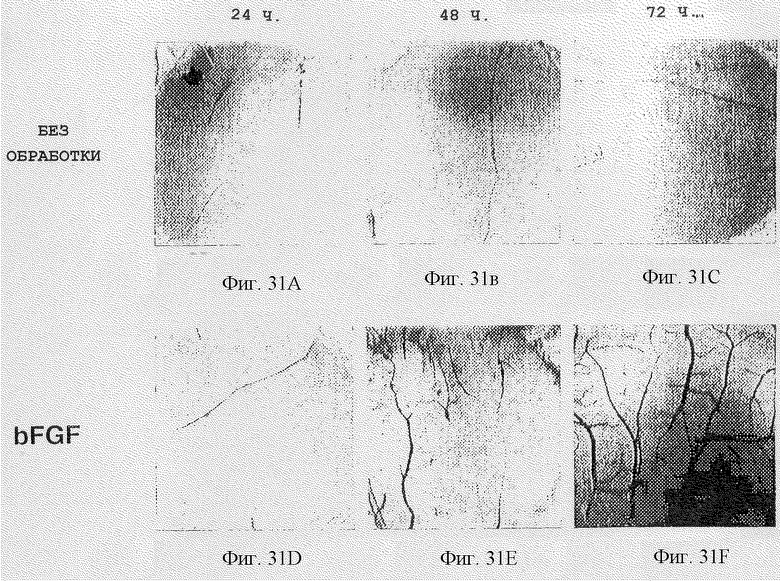
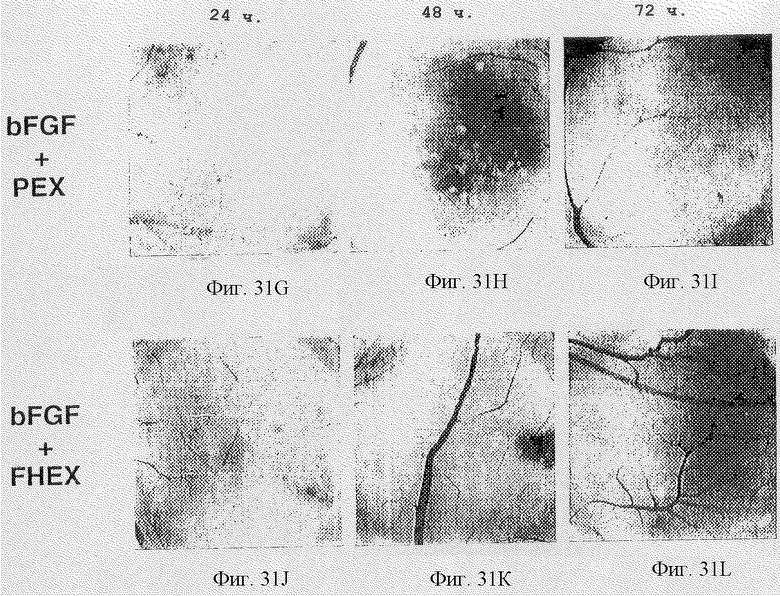
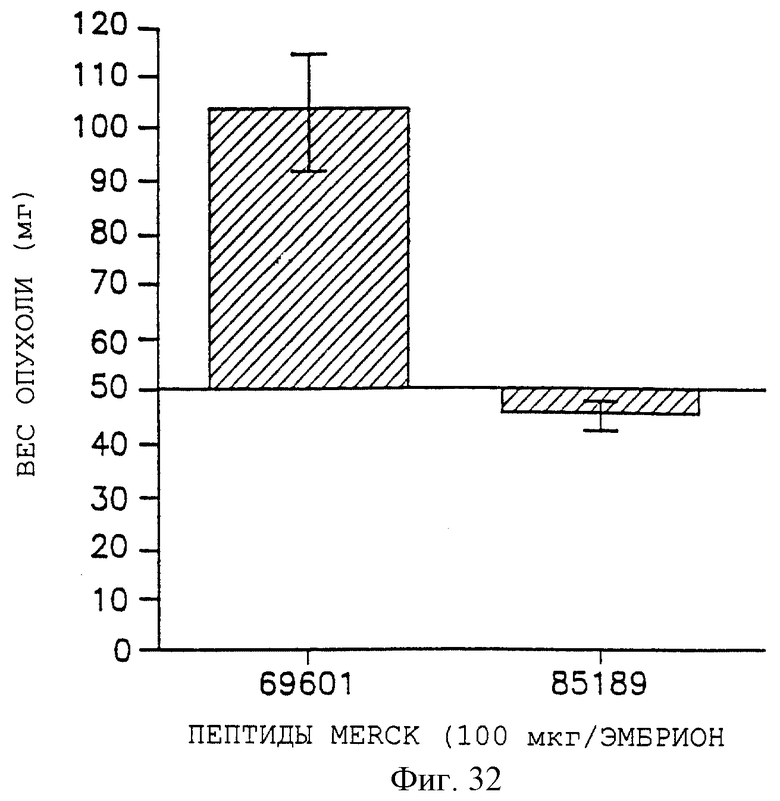
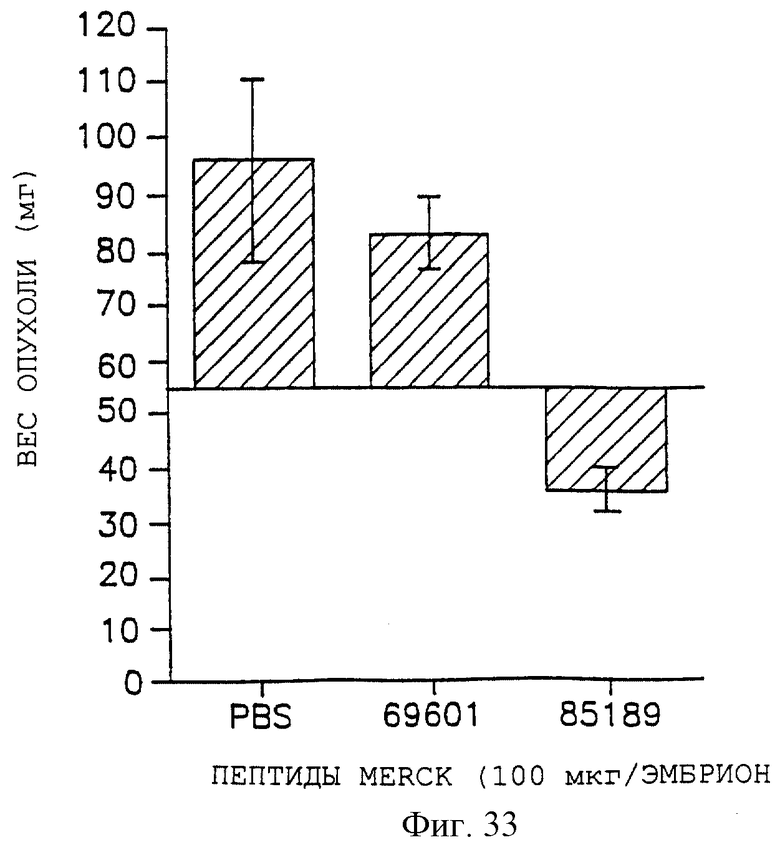
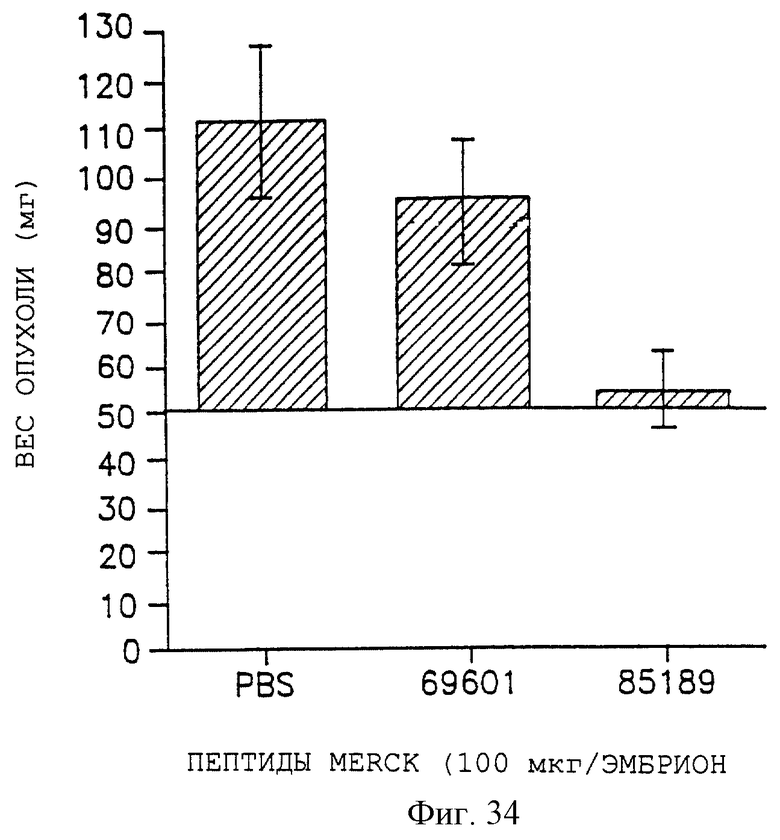
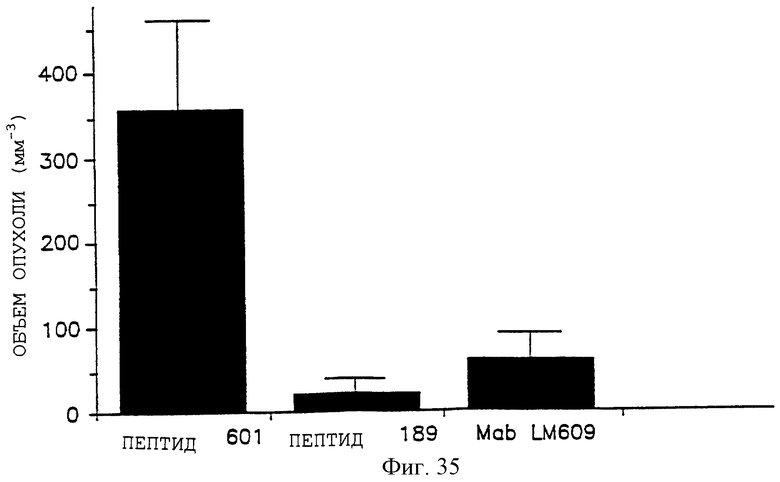
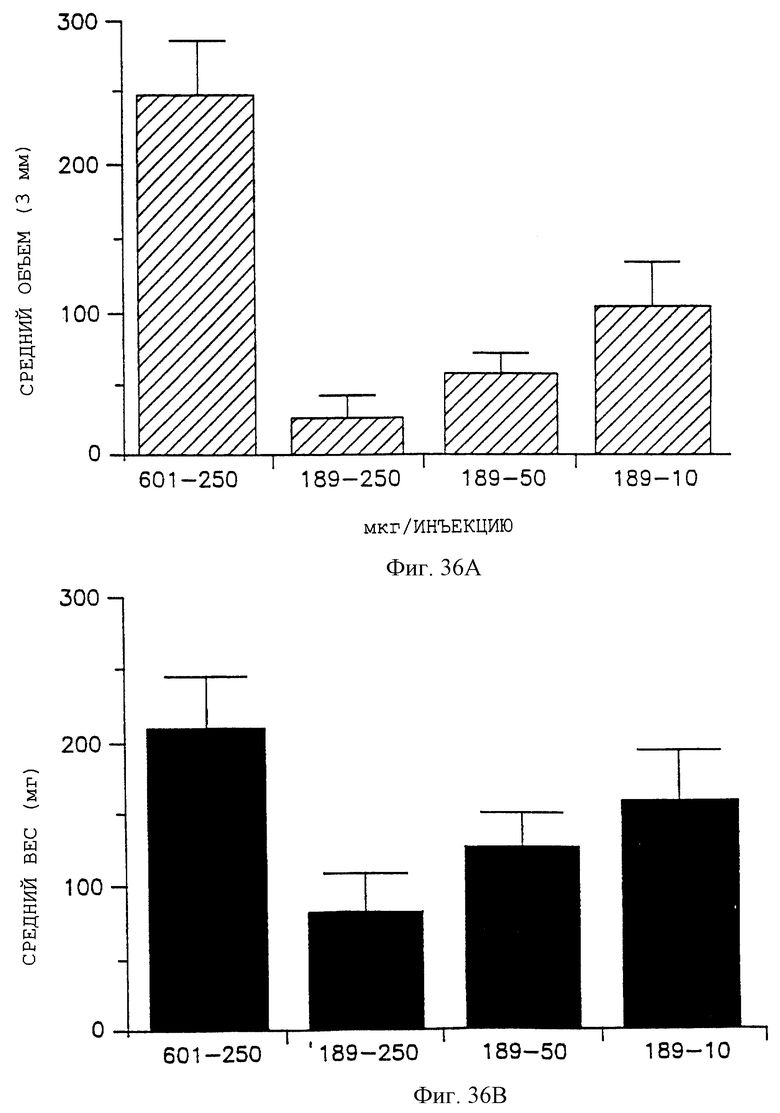
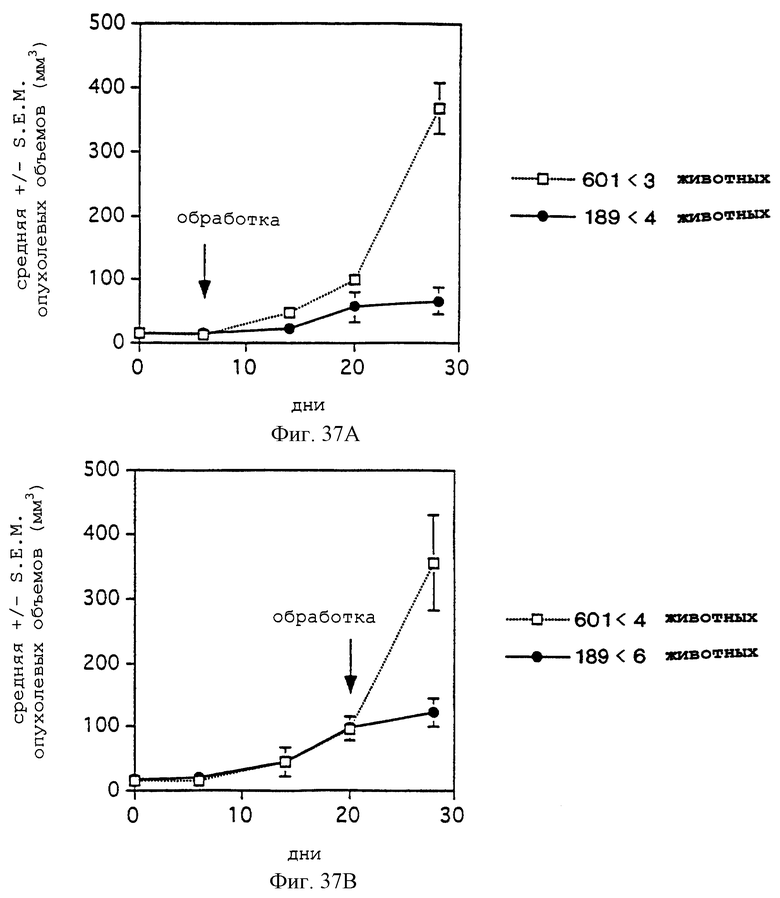
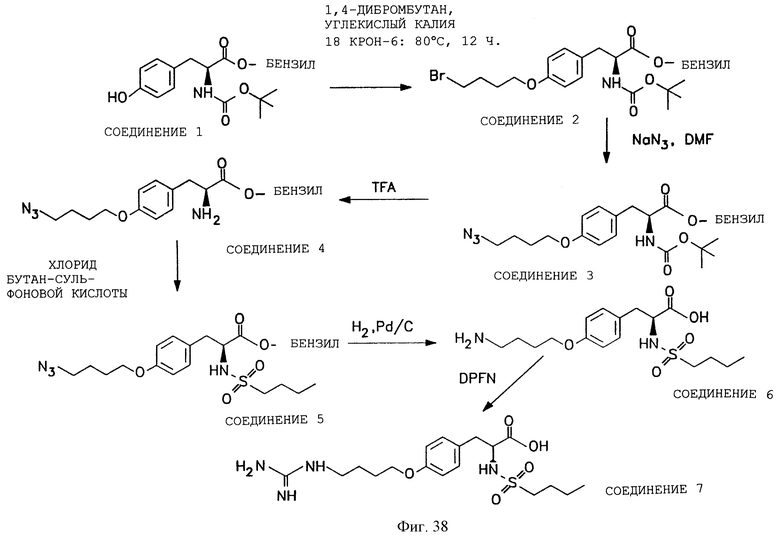
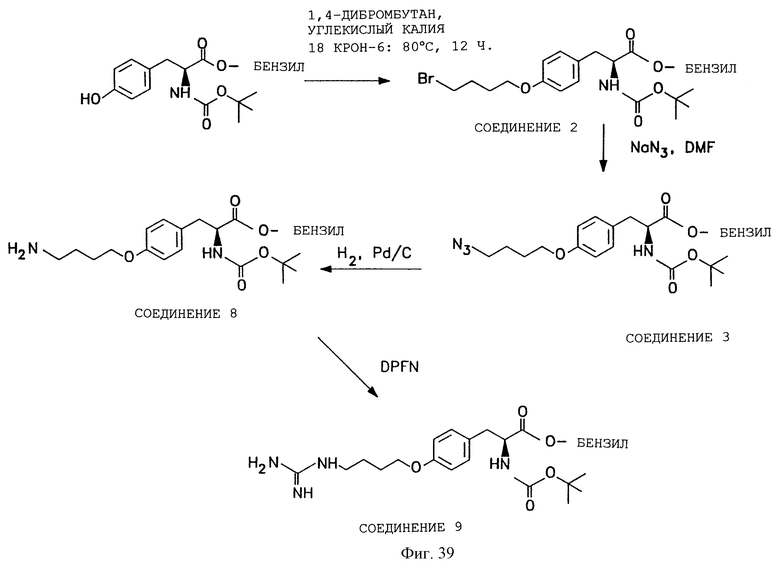
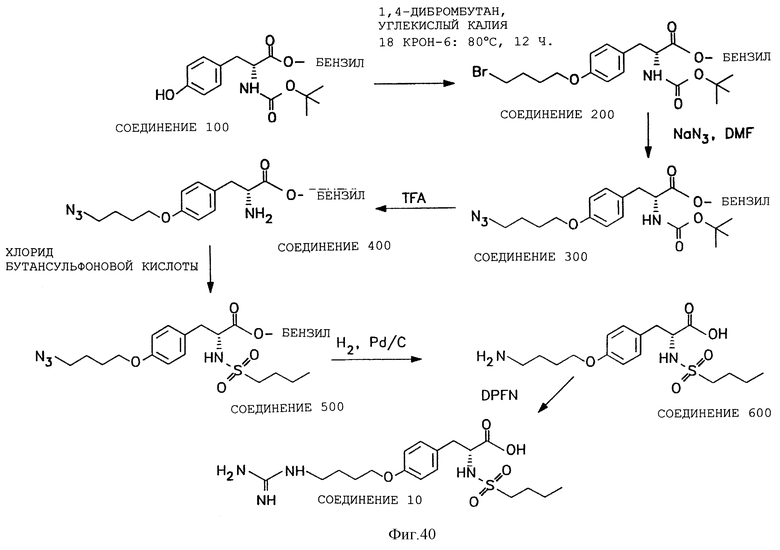
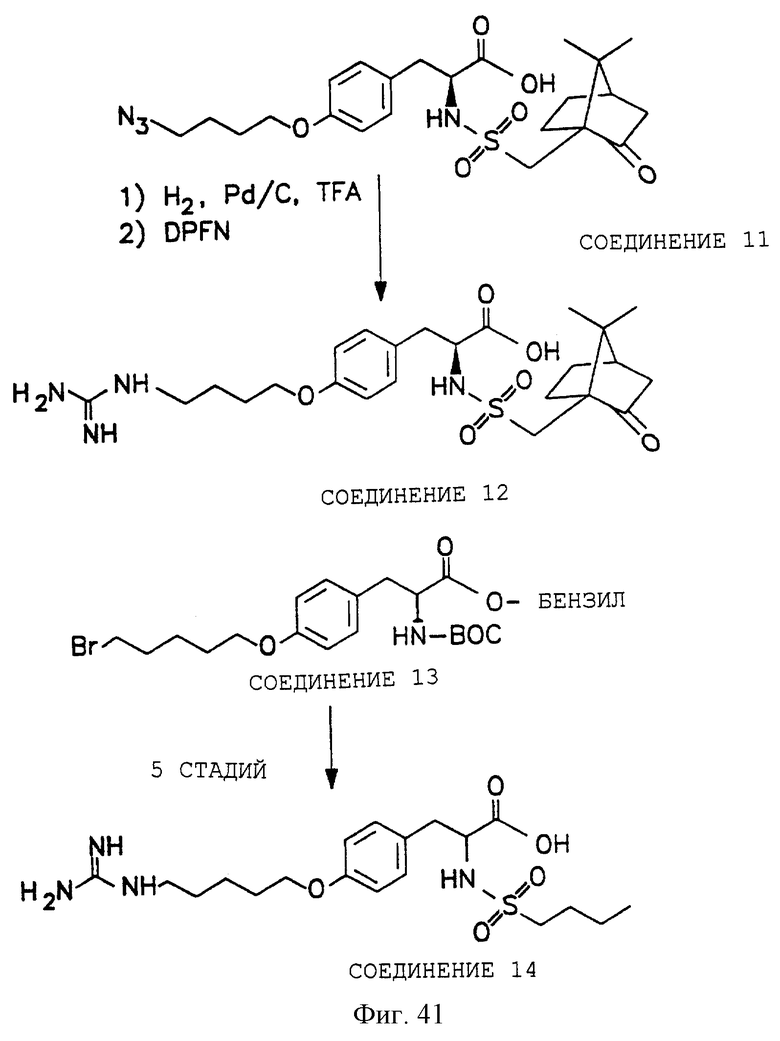
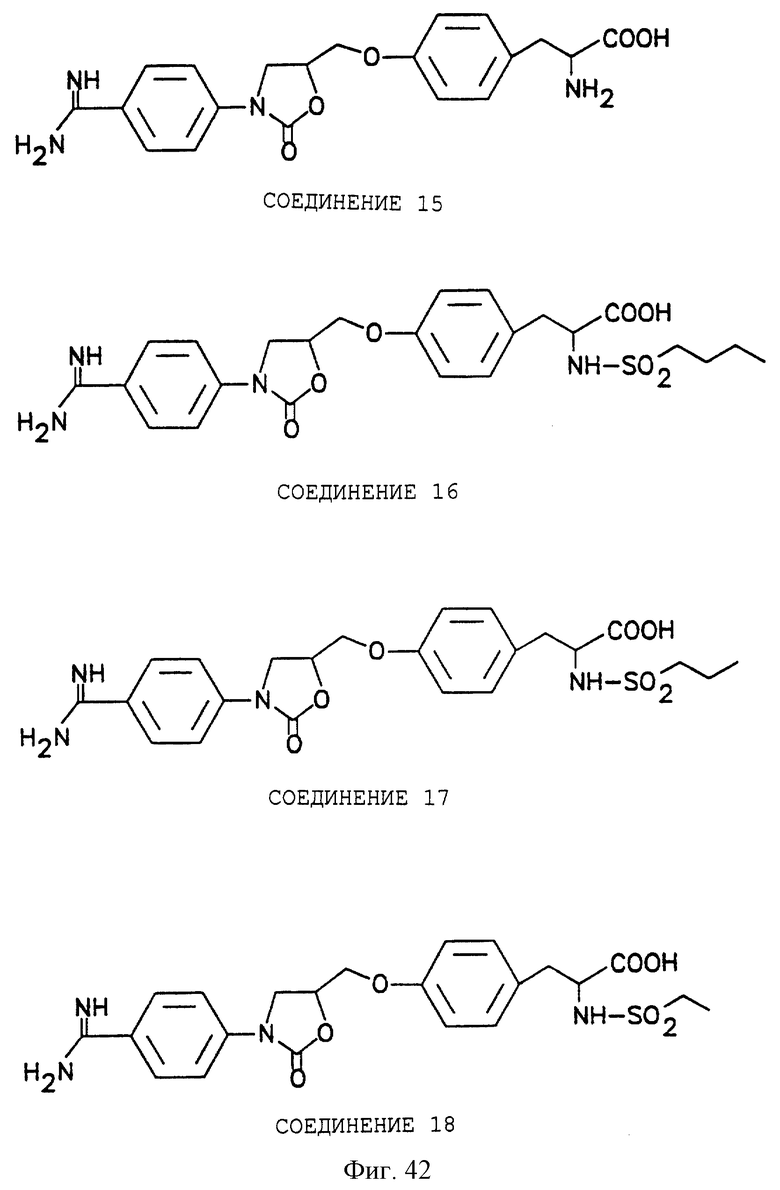
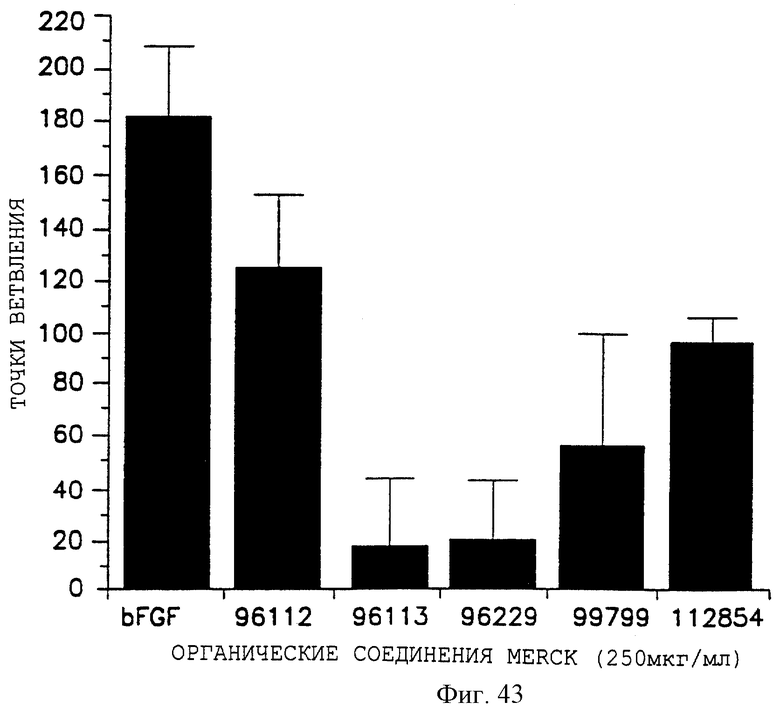
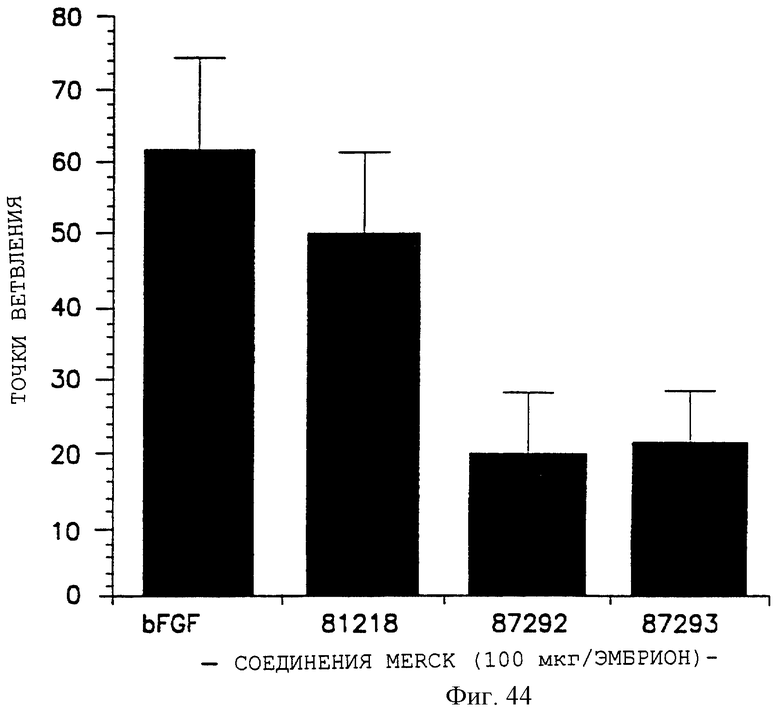
Изобретение относится к медицине. В изобретении описаны способы ингибирования ангиогенеза в тканях с использованием витронектиновых антагонистов αvβ3 и, в частности, для ингибирования ангиогенеза в воспалительных тканях и в опухолевых тканях и в метастазах с использованием терапевтических композиций, содержащих αvβ3-антагонисты. Изобретение обеспечивает возможность лечения некоторых воспалительных, опухолевых заболеваний. 7 с. и 33 з.п. ф-лы, 44 ил., 7 табл.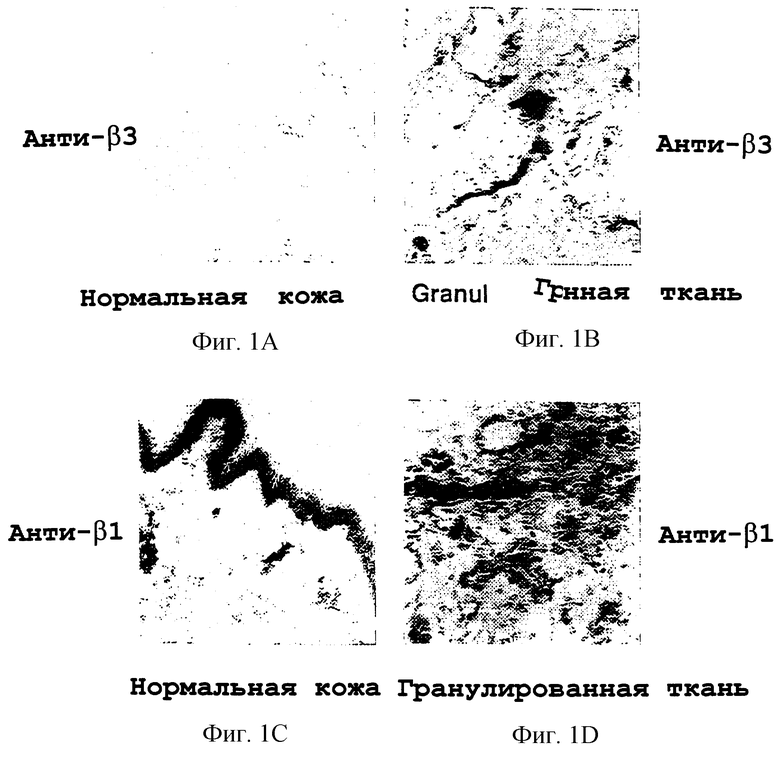
21. Способ по любому из пп. 13-19, отличающийся тем, что указанный αvβ3-антагонист включает последовательность аминокислотных остатков, показанную в SEQ ID NO 15, 17, 18, 19, 20, 21, 22, 23, 25, 26, 27 или 28.
Приоритет по пунктам:
по пп. 1-9, 11-40 31.05.1996 по заявке 60/018.773 и 31.05.1996 по заявке 60/015.869; по п. 10 31.05.1996 по заявке 60/015.869.
| BROOKS et al, А, Cell 1996, 85:683-693 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Технология лекарственных форм | |||
| /Под ред | |||
| Т.С | |||
| Кондратьевой | |||
| - М.: Медицина, 1991, т.1, с | |||
| Разборное приспособление для накатки на рельсы сошедших с них колес подвижного состава | 1920 |
|
SU65A1 |

