Способ получения 2',3'-дидезокси-3'-тиацитидина или 2',3'-дидезокси-3-'тиа-5-фторцитидина, обогащенной энантиомером нуклеозид, способ получения 1,3-оксатиолана, способ получения обогащенного энантиомером 2-ацилокси-5-ацилокси-1,3-оксатиолана, энантиомера, способ расщепления энантиомеров нуклеозида.
Настоящее изобретение касается способов и составов для приготовления противовирусных нуклеозидных аналогов, в частности, ВСН-189 (2',3'-дидезокси-3'-тиа-цитидин). Более подробно, изобретение относится к избирательному синтезу β-изомера ВСН-189 и родственных соединений, а также избирательному синтезу ВСН-189 и родственных соединений, обогащенных энантиомером.
Предшествующий уровень техники
С 1981 г. началась документация заболевания, которое стало известно как Синдром Приобретенного Иммунного Дефицита /СПИД/, а также его предшественника, - связанного со СПИДом Комплекса /ПСК/. В 1983 г. была установлена причина заболевания СПИДом, и вирус был назван как Вирус Иммунодефицита человека, тип 1 /ВИЧ-1/. Обычно, у лица, зараженного вирусом, развивается СПИД, во всех случаях СПИД, а это всегда приводит к смерти.
Болезнь СПИД представляет собой конечный результат после прохождения вирусом ВИЧ-1 его собственного жизненного цикла. Жизненный цикл вириона начинается с прикрепления самого вириона с иммунной клеткой-хозяином, лимфоцитом Т-4 человека посредством связывания гликопротеина на поверхности защитной оболочки вириона с С 4-гликопротеином лимфоцитной клетки. Однажды прикрепившись, вирион сбрасывает собственную гликопротеиновую оболочку, проникает через мембрану клетки-хозяина и обнажает ее РНК. Фермент вириона, обратная транскриптаза, руководит процессом транскрибирования РНК в однонитевую ДНК. Вирусная РНК разрушается, и вторая нить ДНК синтезируется. Теперь двухнитевая ДНК интегрируется в гены клеток человека, и эти гены используются для воспроизведения клеток.
С этого момента клетки человека осуществляют процесс собственной репродукции, используя свою РНК-полимеразу для транскрипции ДНК, интегрированной в вирусную РНК. Вирусная РНК транслируется в гликопротеины, структурные белки и вирусные ферменты, которые компонуются с интактной вирусной РНК. Когда клетка-хозяин заканчивает репродуктивную стадию, новая клетка-вирион, не лимфоцит Т-4, дает начало новому процессу. Число вирусных клеток ВИЧ-1, таким образом, растет, в то время как число лимфоцитов Т-4 снижается.
Типичная для человека иммунная система ответной реакции, уничтожающая вторгшиеся вирионы, подвергается испытанию, потому что большая часть жизненного цикла вириона проходит в латентном состоянии в иммунной клетке. Кроме того, вирусная обратная транскриптаза - фермент, используемый для создания новых вирионных клеток, не является очень специфичным и допускает ошибки при транскрипции, что приводит к появлению непрерывно измененных гликопротеинов на поверхности вирусной защитной оболочки. Это отсутствие специфичности снижает эффективность иммунной системы, так как антитела, специфично вырабатываемые против одного гликопротеина, могут быть бесполезны против другого, и это приводит в итоге к снижению числа антител, способных бороться с вирусом. Вирус продолжает увеличиваться, в то время как защитная иммунная система продолжает ослабевать. В итоге ВИЧ устанавливает беспредельный контроль над иммунной системой организма, что позволяет начать наступление условно-патогенных инфекций, что без введения противовирусных средств и/или иммуномодуляторов приводит в конце концов к смерти.
В процессе жизненного цикла вируса есть три критических момента, которые были идентифицированы как "мишени" для действия противовирусных лекарств: (1) начальное прикрепление вириона к лимфоциту Т-4 или к участку макрофага; (2) транскрипция вирусной РНК в вирусную ДНК; (3) сборка новых вирионных клеток в процессе репродукции.
Ингибирование вируса на второй критической стадии, то есть в процессе транскрипции вирусной РНК в вирусную ДНК привело к созданию большого числа терапевтических средств, используемых для лечения больных СПИДом. Эта транскрипция должна осуществляться для вириона в целях репродукции, потому что гены вириона закодированы в РНК; клетка-хозяин считывает только ДНК. Вводя лекарства, которые блокируют обратную транскриптазу от полного образования вирусной ДНК, можно остановить репликацию ВИЧ-1.
Аналоги нуклеозидов, такие как 3'-азидо-3'-дизокситимидин (AZT), 2', 3'-дидезоксицитидин (DDC), 2',3'-дидезокситимидинен (D4T), 2',3'-дидезоксиинозин (DD1), и различные фтор-производные этих нуклеозидов являются относительно эффективными для остановки репликации ВИЧ на стадии обратной транскриптазы. Другой обещающий ингибитор транскриптазы - 2',3'-дидезокси-3'-тиацитидин (BCH-189), который содержит кольцо окситиолана, замещающее остаток сахара в нуклеозиде.
AZT успешно применяется как анти-ВИЧ лекарство, потому что нарушает образование вирусной ДНК внутри клетки-хозяина, лимфоцитов T-4. Когда AZT поступает в клетку, клеточные киназы активируют его посредством фосфолирования до AZT-трифосфата. Затем AZT-трифосфат вступает в конкуренцию с природными тимидиновыми нуклеозидами за рецепторный участок на ферменте, обратной транскриптазе ВИЧ. Природные нуклеозиды обладают двумя реакционно-активными концами: первый - для прикрепления к предыдущему нуклеозиду и второй для связывания с последующим нуклеозидом. Молекула AZT содержит только первый реакционно-активный конец; попав на участок ВИЧ-фермента, азидная группа AZT останавливает образование вирусной ДНК, так как азид не может образовывать 3',5'-фосфодиэфирную связь с рибозой последующего нуклеозида.
Преимущества AZT в клиническом аспекте заключаются в длительности существования, сниженной частоте и опасности условно-патогенных инфекций и повышенном количестве периферических CD 4-лимфоцитов.
При иммунносорбентном способе определения вирусного (белка) p24, антигена, применяемого для контроля над активностью ВИЧ-1, отмечается значительное снижение его, когда применяют AZT. Однако преимущества AZT должны быть оценены в отношении острых неблагоприятных реакций подавления костного мозга, рвоты, миалгий, бессонницы, сильных головных болей, анемии, периферической нейропатии и судорог. Более того, эти нежелательные побочные эффекты наблюдаются сразу же после начала лечения, в то время как не менее шести недель терапии необходимо для реализации преимуществ с использованием AZT.
Как DDC, так и D4T являются мощными ингибиторами репликации ВИЧ, активность которых сравнима с AZT (D 4T) или превосходит AZT (DDC). Однако как DDC, так и D4T превращаются в их 5'-трифосфаты менее эффективно, чем природные аналоги и обладают устойчивостью к дезаминазам и фосфорилазам. Клинически, оба соединения являются токсичными. В настоящее время DD1 применяется в сочетании с AZT для лечения СПИДа. Однако побочные эффекты DD1 включают спорадически возникающие панкреатиты и периферическую нейропатию. Начальные тесты на 3'-фтор-2'-3'-дидезокситимидин показывают, что его антивирусная активность сравнима с таковой для AZT.
Недавно проведенные тесты на BCH-189 показали, что он обладает анти-ВИЧ активностью, аналогичной с AZT и DDC, но без клеточной токсичности, которая вызывает ослабление побочных эффектов AZT и DDC. Для проведения клинических испытаний и лечения требуется достаточное количество BCH-189.
Обычно применяемые химические подходы для синтеза нуклеозидов или аналогов нуклеозидов можно классифицировать на две широкие категории: (1) модификация интактных нуклеозидов путем изменения их углеводного компонента, азотистого основания, или обоих и (2) модификация углеводов и основания или их синтетического предшественника на подходящей стадии синтеза. Поскольку в структуре BCH-189 атом углерода замещен на атом серы в углеводном кольце, второй подход представляется более осуществимым. Наиболее важным фактором в осуществлении этой последней стратегии является помещение основания из β-стороны углеводородного кольца в реакции гликозилирования, поскольку только β-изомеры обладают полезной биологической активностью.
В этой области техники хорошо известно, что стериоизбирательное включение оснований в аномерные центры углеводов можно контролировать посредством усиления эффекта по соседней группе в положении 2-заместителя по углеводному кольцу (Chem.Ber.114: 1234 (1981)). Однако BCH-189 и его аналоги не имеют 2-заместителя и, следовательно, не могут быть использованы в данной методике, пока не будут осуществлены дополнительные стадии для введения функциональной группы, что позволяет их использовать для синтеза. Эти дополнительные стадии снижают общую эффективность синтеза.
В этой области техники хорошо известно, что "значительные количества нежелательных α-нуклеозидов всегда образуются при синтезе 2'-дезоксирибозидов" (Chem. Ber. 114:1234, 1244 (1981)). Более того, эта ссылка учит, что использование простых катализаторов Friedel-Crafts, подобно SnCl4, при синтезе нуклеозидов ведет к образованию нежелательных эмульсий при обработке реакционной смеси, образуются сложные смеси α- и β-изомеров и стабильные комплексы между SnCl4 и более основными силицированными гетероциклами, такими как силицированный цитозин. Эти комплексы удлиняют время реакции, снижают выходы и ведут к образованию нежелательных неприродных N-3-нуклеозидов. Таким образом, известный уровень техники предусматривает использование триметилсилилтрифталата или триметилсилилперхлората в качестве катализатора при соединении пиримидиновых оснований с углеводным кольцом, что позволяет достичь высоких выходов биологически активных β-изомеров. Однако использование этих катализаторов для синтеза BCH-189 или аналогов BCH-189 не дает преимущественного получения β-изомера; результатом этих реакций является соотношение изомеров, равное приблизительно 50:50.
Таким образом, существует необходимость в эффективном синтетическом способе синтеза BCH-189 и его аналогов. Также существует необходимость пути синтеза биологически активного изомера этих соединений, β-BCH-189 и родственных β-аналогов.
Более того, есть необходимость в стереоселективном пути синтеза β-BCH-189, обогащенного энантиомером, поскольку другой энантиомер является неактивным и, следовательно, представляет 50% нежелательной примеси.
Раскрытие изобретения
Настоящее изобретение связано с открытием исключительно эффективного пути синтеза соединения BCH-189 и различных аналогов BCH-189 из недорогих предшественников с выбором нужной функциональности. Этот путь синтеза позволяет осуществить стереоселективное получение биологически активного изомера этих соединений, β-BCH-189 и родственных соединений. Более того, стереохимия в 4'-положении нуклеозида может контролироваться, чтобы получать обогащенный энантиомером β-BCH-189 и его аналоги.
Термин "аналоги BCH-189" обозначает ссылку на нуклеозиды, которые образуются из пиримидиновых оснований, замещенных в положении 5 и которые соединяются с замещенными 1,3-оксатиоланами.
Способ настоящего изобретения включает озонирование аллилового эфира или эфира с формулой CH2= CH-CH2-OR, в которой R - защитная группа, такая как алкил, силил или ацил, что приводит к образованию гликоальдегида с формулой OCH-CH2-OR; добавление тиогликолевой кислоты к гликоальдегиду образует лактон с формулой 2-(R-окси)-метил-5-оксо-1,3-оксатиолан; превращение лактона в соответствующий ему карбоксилат в положении 5 оксатиоланового кольца; соединение ацетата с силицированным пиримидиновым основанием в присутствии SnCl4 с образованием β-изомера аналога 5'-(R-окси)-2',3'-дидезокси-3'-тиа-нуклеозида; и замещение защитной группы R на водород с образованием BCH-189 или аналога BCH-189.
Изобретение может быть использовано для получения BCH-189 или аналогов BCH-189, которые обогащены энантиомером по положению 4' посредством избирательного подбора защитной группы R, чтобы дать возможность ферменту осуществлять стереоселективный выбор. Например, защитная группа R может быть подобрана таким образом, чтобы заместитель в положении 2 оксатиоланового лактона будет бутирилокси-группа, а это дает возможность для стереоселективного ферментативного гидролиза эстеразой из печени свиньи. В итоге оптически активный гидролизованный лактон можно затем превратить в соответствующий ему диацетат и соединить с силицированным пиримидиновым основанием, как сказано выше.
Соответственно, одной из целей данного изобретения является создание эффективного способа для приготовления β-изомера BCH-189 и аналогов BCH-189 с высокими выходами. Более того, целью данного изобретения является создание способа синтеза для изготовления только одного оптического изомера, а не рацематной смеси BCH-189 и аналогов BCH-189. Еще одной целью этого изобретения является разработка пути синтеза для получения  BCH-189, обогащенного энантиомером.
BCH-189, обогащенного энантиомером.
В дополнение, целью этого изобретения является получение промежуточных веществ, из которых BCH-189 или аналоги BCH-189 могут быть синтезированы, и соответствующие формуле 2-(R-ок-симетил)-5-ацилокси-1,3-оксатиолан, где R представляет собой защитную группу, такую как алкил, силил или ацил, а также способ получения этих соединений. Более того, целью этого изобретения является получение обогащенного энантиомером 2-ацетоксиметил-5-ацетокси-1,3-оксатиолана и 2-бутоксиметил-оксо-1,3-оксатиолана, а также разработка способов получения этих соединений.
Другой целью этого изобретения является получение промежуточных соединений, из которых BCH-189 или аналоги BCH-189 могут быть синтезированы, следующей формулы:
где R - защитная группа, такая как алкил, силил или ацил;
Y - может быть водород, метил, галоид, алкил, алкенил, алкинил, гидроксиалкил, карбоксиалкил, тиоалкил, селеноалкил, фенил, циклоалкил, циклоалкенил, тиоарил и селеноарил,
а также разработка способов получения этих соединений.
Более того, это изобретение обеспечивает получение промежуточных соединений, из которых BCH-189 или аналоги BCH-189 могут быть синтезированы следующей формулы:
где R - защитная группа, такая как алкил, силил или ацил;
Y - может быть водород, метил, галоид, алкил, алкенил, алкинил, гидроксиалкил, карбоксиалкил, тиоалкил, селеноалкил, фенил, циклоалкил, циклоалкенил, тиоарил и селеноарил,
а также разработка способов получения этих соединений.
Фиг. 1 иллюстрирует один из вариантов синтеза BCH-189 и аналогов BCH-189 согласно настоящему изобретению;
фиг.2 иллюстрирует один из вариантов синтеза BCH-189 согласно настоящему изобретению;
фиг. 3 иллюстрирует один из вариантов синтеза 5-метил-цитидиновых и тимидиновых производных BCH-189 согласно настоящему изобретению;
фиг. 4 иллюстрирует один из вариантов синтеза BCH-189, обогащенного энантиометром согласно настоящему изобретению.
Лучший вариант осуществления изобретения.
BCH-189 является соединением следующей формулы:
Процесс получения BCH-189 и аналогов BCH-189 по данному изобретению представлен на фиг. 1. Аллиловый эфир или эфир  подвергают озонированию с образованием альдегида
подвергают озонированию с образованием альдегида  , который реагирует с тиогликолевой кислотой с образованием лактона
, который реагирует с тиогликолевой кислотой с образованием лактона  . Лактон
. Лактон  обрабатывают восстанавливающим агентом: например, диизобутилалюминий-гидридом (DIBAL), бис(2-метоксиэтокси)алюминий-гидридом натрия (который можно приобрести в виде 3,4 молярного раствора в толуоле под торговым названием "Red-AI"TM), и NaBH4, с последующей обработкой карбоксильным ангидридом, получая карбоксилат β- Этот карбоксилат сочетают с силицированным пиримидиновым основанием в присутствии кислоты Льюиса, которая может катализировать стереоспецифическое сочетание, например с SnCl4, с образованием β изомера замещенного нуклеозида 5 с важным соотношением
обрабатывают восстанавливающим агентом: например, диизобутилалюминий-гидридом (DIBAL), бис(2-метоксиэтокси)алюминий-гидридом натрия (который можно приобрести в виде 3,4 молярного раствора в толуоле под торговым названием "Red-AI"TM), и NaBH4, с последующей обработкой карбоксильным ангидридом, получая карбоксилат β- Этот карбоксилат сочетают с силицированным пиримидиновым основанием в присутствии кислоты Льюиса, которая может катализировать стереоспецифическое сочетание, например с SnCl4, с образованием β изомера замещенного нуклеозида 5 с важным соотношением  : d - изомеров, равным 100:0. С замещенного нуклеозида
: d - изомеров, равным 100:0. С замещенного нуклеозида  удаляют защитную группу, получая BCH-189 или аналог
удаляют защитную группу, получая BCH-189 или аналог  BCH-189.
BCH-189.
Эта методика может быть изменена для получения BCH-189 или аналогов BCH-189, обогащенных энантимером в положении 4', посредством подбора подходящей защитной группы R, что делает возможным стереоселективный ферментативный гидролиз 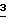 эстеразой из печени свиньи, липазой из поджелудочной железа свиньи или субтилизином, или другими ферментами, которые гидролизуют
эстеразой из печени свиньи, липазой из поджелудочной железа свиньи или субтилизином, или другими ферментами, которые гидролизуют  в соответствии со стереоселективным механизмом. Образующийся оптически активный
в соответствии со стереоселективным механизмом. Образующийся оптически активный  может быть превращен в обогащенный энантиомером карбоксилат и подвергнут сочетанию с силицированным пиримидиновым основанием, как было отмечено выше при получении обогащенного энантиомером BCH-189 или аналогов BCH-189.
может быть превращен в обогащенный энантиомером карбоксилат и подвергнут сочетанию с силицированным пиримидиновым основанием, как было отмечено выше при получении обогащенного энантиомером BCH-189 или аналогов BCH-189.
Защитная группа R в  может быть выбрана таким образом, чтобы обеспечить защиту соответствующего спирта до проведения заключительной стадии синтеза (снятие защиты с
может быть выбрана таким образом, чтобы обеспечить защиту соответствующего спирта до проведения заключительной стадии синтеза (снятие защиты с  с образованием
с образованием  ). Кроме того, защитную группу можно выбрать, по требованию, для обеспечения дополнительного участка узнавания для фермента, который применяют позже в реакции энантио-избирательного гидролиза. Можно использовать любую группу в этом способе. Например, можно использовать алкил, силил и ацил-защитные группы или группы, обладающие по существу такими же свойствами, как и отмеченные выше.
). Кроме того, защитную группу можно выбрать, по требованию, для обеспечения дополнительного участка узнавания для фермента, который применяют позже в реакции энантио-избирательного гидролиза. Можно использовать любую группу в этом способе. Например, можно использовать алкил, силил и ацил-защитные группы или группы, обладающие по существу такими же свойствами, как и отмеченные выше.
Примененная алкильная защитная группа представляет собой трифенилметильную или алкильную группу, обладающую по существу такими же защитными свойствами, как трифенилметильная группа. Силил-защитная группа, примененная здесь, представляет собой три-замещенную силильную группу следующей формулы:
где R1, R2 и R3 могут быть низшими алкилами, например, метилом, этилом, бутилом и алкилом с 5 углеродными атомами или меньше, или фенилом, более того, R1 может быть одинаков с R2; R1, R2 и R3 могут быть все идентичными.
Примеры силил-защитных групп включают триметилсилил и t-бутилдифенилсилил, но не ограничиваются ими.
Примененная ацильная группа приводится для описания ацил-защитной группы (как в  ) или для описания карбоксилата (как в
) или для описания карбоксилата (как в  ), и имеет следующую формулу:
), и имеет следующую формулу:
где R' - низший алкил, например, метил, этил, бутил и алкил с 5 углеродными атомами или меньше; замещенный низший алкил, где алкил содержит один, два или более простых заместителей, включая, но не ограничивая, амино, карбоксил, гидроксил, фенил, низший алкокси-заместитель, например, метокси и этокси; фенил; замещенный фенил, где фенил содержит один, два или более простых заместителей, включая, но не ограничивая, низший алкил, галоид, например, хлор и бром, сульфато, сульфонилокси, карбоксил, карбо-низший-алкокси-заместитель, например, карбометокси и карбэтокси, амино, моно- и ди-низшую алкиламино-группу, например, метиламино, амидо, гидрокси, низшую алкокси-группу, например, метокси и этокси, низшую алканоилокси-группу, например, ацетокси-группу.
Примененное здесь силицированное пиримидиновое основание представляет собой соединение следующей формулы:
где X - или три-замещенная силилокси- или три-замещенная силиламино-группа;
Z - три-замещенная силил-группа;
Y - группа, описанная ниже.
Примененная здесь три-замещенная силильная группа имеет следующую формулу:
где R1, R2 и R3 могут быть низшим алкилом, например, метилом, этилом, бутилом и алкилом с 5 углеродными атомами или менее, или фенилом. Более того, R1 может быть идентична с R2; R1, R2 и R3 могут быть все идентичны. Примеры три-замещенных силильных групп включают триметилсилил- и t-бутилдифенилсилил-группы, но лимитированы ими.
Силицированное пиримидиновое основание может быть замещено различными Y-заместителями, включая, но не лимитируя их, водород, метил, галоид, алкил, алкенил, алкинил, гидроксиалкил, карбоксиалкил, тиоалкил, селеноалкил, фенил, циклоалкил, циклоалкенил, тиоарил и селеноарил-группу, в положении 5 силицированного пиримидинового основания (Y-заместитель на фиг.1) для модификации свойств, таких как транспортные свойства или скорость метаболизма аналога BCH-189.
Иллюстративные примеры синтеза BCH-189 или аналогов BCH-189 согласно настоящему изобретению приведены на фиг.2 - 4 и в следующих описаниях.
На фиг. 2 представлен синтез BCH-189, исходя из аллилового спирта  . Масляную суспензию NaH (4,5 г, 60%, 110 ммоль) промывают дважды THF (тетрагидрофураном) (100 мл•2) и полученное твердое вещество суспендируют в THF (300 мл). Суспензию охлаждают до 0oC, и по каплям добавляют аллиловый спирт
. Масляную суспензию NaH (4,5 г, 60%, 110 ммоль) промывают дважды THF (тетрагидрофураном) (100 мл•2) и полученное твердое вещество суспендируют в THF (300 мл). Суспензию охлаждают до 0oC, и по каплям добавляют аллиловый спирт  (6,8 мл, 100 ммоль), и смесь перемешивают в течение 30 минут при 0oC. t-бутил-дифенилсилил-хлорид (25,8 мл, 100,8 ммоль) добавляют по каплям при 0oC и реакционную смесь перемешивают 1 час при 0oC. Раствор "тушат" водой (100 мл) и экстрагируют диэтиловым эфиром (200 мл•2). Объединенные экстракты промывают водой, сушат над MgSO4, фильтруют, концентрируют и остаток перегоняют под вакуумом (90-100oC при 0,5 - 0,6 мм рт.ст.), получая бесцветную жидкость
(6,8 мл, 100 ммоль), и смесь перемешивают в течение 30 минут при 0oC. t-бутил-дифенилсилил-хлорид (25,8 мл, 100,8 ммоль) добавляют по каплям при 0oC и реакционную смесь перемешивают 1 час при 0oC. Раствор "тушат" водой (100 мл) и экстрагируют диэтиловым эфиром (200 мл•2). Объединенные экстракты промывают водой, сушат над MgSO4, фильтруют, концентрируют и остаток перегоняют под вакуумом (90-100oC при 0,5 - 0,6 мм рт.ст.), получая бесцветную жидкость 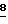 (28 г, 94 ммоль, 94%). (1H-ЯМР: 7,70-7,35 (10H, м, ароматический -H); 5,93 (1H, м, H2); 5,37 (1H, д, H1) J=1,4 и 14,4 Hz; 5,07 (1H, д, H1) J=1,4 и 8,7 Hz; 4,21 (2H, м, H3); 1,07 (9H, с, t-Bu).
(28 г, 94 ммоль, 94%). (1H-ЯМР: 7,70-7,35 (10H, м, ароматический -H); 5,93 (1H, м, H2); 5,37 (1H, д, H1) J=1,4 и 14,4 Hz; 5,07 (1H, д, H1) J=1,4 и 8,7 Hz; 4,21 (2H, м, H3); 1,07 (9H, с, t-Bu).
Силилаллиловый эфир  (15,5 г, 52,3 ммоль) растворяют в CH2Cl2 (400 мл) и озонируют при -78oC. По достижении полного озонолиза добавляют при -78oC DMS (диметилсульфоксид) (15 мл, 204 ммоль, 3,9 экв), и смесь нагревают до комнатной температуры и перемешивают в течение ночи. Раствор промывают водой (100 мл•2), сушат над MgSO4, фильтруют, концентрируют и перегоняют под вакуумом (100-110oC при 0,5 - 0,6 мм рт.ст), получая бесцветную жидкость
(15,5 г, 52,3 ммоль) растворяют в CH2Cl2 (400 мл) и озонируют при -78oC. По достижении полного озонолиза добавляют при -78oC DMS (диметилсульфоксид) (15 мл, 204 ммоль, 3,9 экв), и смесь нагревают до комнатной температуры и перемешивают в течение ночи. Раствор промывают водой (100 мл•2), сушат над MgSO4, фильтруют, концентрируют и перегоняют под вакуумом (100-110oC при 0,5 - 0,6 мм рт.ст), получая бесцветную жидкость  (15,0 г, 50,3 ммоль, 96%). 1H-ЯМР: 9,74 (1H, с, H-CO); 7,70 - 7,35 (10H, м, ароматический H); 4,21 (2H, с, -CH2); 1,22 (9H, с,t-Bu)).
(15,0 г, 50,3 ммоль, 96%). 1H-ЯМР: 9,74 (1H, с, H-CO); 7,70 - 7,35 (10H, м, ароматический H); 4,21 (2H, с, -CH2); 1,22 (9H, с,t-Bu)).
Силированный гликоальдегид  (15,0 г, 50,3 ммоль) растворяют в толуоле (200 мл) и одной порцией добавляют тиогликолевую кислоту (3,50 мл, 50,3 ммоль). Раствор отгоняют в течение 2 часов и образующуюся воду удаляют с помощью ловушки Dean-Stark'a. Раствор охлаждают до комнатной температуры и промывают насыщенным раствором NaHCO3, водные промывные воды экстрагируют диэтиловым эфиром (200 мл•2). Объединенные экстракты промывают водой (100 мл•2), сушат над MgSO4, фильтруют и концентрируют, получая бесцветное масло
(15,0 г, 50,3 ммоль) растворяют в толуоле (200 мл) и одной порцией добавляют тиогликолевую кислоту (3,50 мл, 50,3 ммоль). Раствор отгоняют в течение 2 часов и образующуюся воду удаляют с помощью ловушки Dean-Stark'a. Раствор охлаждают до комнатной температуры и промывают насыщенным раствором NaHCO3, водные промывные воды экстрагируют диэтиловым эфиром (200 мл•2). Объединенные экстракты промывают водой (100 мл•2), сушат над MgSO4, фильтруют и концентрируют, получая бесцветное масло  (16,5 г, 44,3 ммоль, 88%), которое под вакуумом постепенно затвердевает.
(16,5 г, 44,3 ммоль, 88%), которое под вакуумом постепенно затвердевает.
Рекристаллизация из гексана дает белый твердый  (15,8 г, 84%). (1H-ЯМР: 7,72-7,38 (10H, м, ароматический H); 5,53 (1H, т, H2) J = 2,7 Hz; 3,93 (1H, дд, -CH2O) J = 9,3 Hz; 3,81 (1H, д, 1H4) J = 13,8 Hz; 3,79 (1H, дд, -CH2O); 3,58 (1H, д, 1H4); 1,02 (9H, с, t-Bu)).
(15,8 г, 84%). (1H-ЯМР: 7,72-7,38 (10H, м, ароматический H); 5,53 (1H, т, H2) J = 2,7 Hz; 3,93 (1H, дд, -CH2O) J = 9,3 Hz; 3,81 (1H, д, 1H4) J = 13,8 Hz; 3,79 (1H, дд, -CH2O); 3,58 (1H, д, 1H4); 1,02 (9H, с, t-Bu)).
2-(t-Бутил-дифенилсилилокси)-метил-5-оксо-1,2-оксатиолан 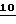 (5,0 г, 13,42 ммоль) растворяют в толуоле (150 мл) и раствор охлаждают до -78oC. Раствор Dibal (14 мл, 1,0 М в гексанах, 14 ммоль) добавляют по каплям, в то время как внутреннюю температуру поддерживают ниже -70oC в течение всего времени. По окончании добавления смесь перемешивают в течение 30 минут при -78oC. Добавляют уксусный ангидрид (5 мл, 53 ммоль) и смесь нагревают до комнатной температуры и перемешивают в течение ночи. В смесь добавляют воду (5 мл) и полученную смесь перемешивают 1 час при комнатной температуре. Смесь разбавляют диэтиловым эфиром (300 мл), добавляют MgSO4 (40 г) и смесь интенсивно перемешивают 1 ч при комнатной температуре. Смесь фильтруют, концентрируют и остаток подвергают мгновенной хроматографии с 20% EtOAc в гексанах, получая бесцветную жидкость
(5,0 г, 13,42 ммоль) растворяют в толуоле (150 мл) и раствор охлаждают до -78oC. Раствор Dibal (14 мл, 1,0 М в гексанах, 14 ммоль) добавляют по каплям, в то время как внутреннюю температуру поддерживают ниже -70oC в течение всего времени. По окончании добавления смесь перемешивают в течение 30 минут при -78oC. Добавляют уксусный ангидрид (5 мл, 53 ммоль) и смесь нагревают до комнатной температуры и перемешивают в течение ночи. В смесь добавляют воду (5 мл) и полученную смесь перемешивают 1 час при комнатной температуре. Смесь разбавляют диэтиловым эфиром (300 мл), добавляют MgSO4 (40 г) и смесь интенсивно перемешивают 1 ч при комнатной температуре. Смесь фильтруют, концентрируют и остаток подвергают мгновенной хроматографии с 20% EtOAc в гексанах, получая бесцветную жидкость  (3,60 г, 8,64 ммоль, 64%), которая представляет смесь аномеров в соотношении 6:1. (1H-ЯМР главного изомера: 7,70-7,35 (10H, м, ароматический H); 6,63 (1H, д, H5) J = 4,4 Hz; 5,47 (1H, т, H2); 4,20-3,60 (2H, м, -CH2O); 3,27 (1H, дд, 1H4) J = 4,4 и 11,4 Hz; 3,09 (1H, д, 1H4) J = 11,4 Hz; 2,02 (3H, с, CH3CO); 1,05 (9H, с, t-Bu); 1H-ЯМР минорного изомера: 7,70-7,35 (10H, м, ароматический H); 6,55 (1H, д, H5) J = 3,9 Hz; 5,45 (1H, т, H2); 4,20-3,60 (2H, м, -CH2O); 3,25 (1H, дд, 1H4) J = 3,9 и 11,4 Hz; 3,11 (1H, д, 1H4) J = 11,4 Hz; 2,04 (3H, с, CH3CO); 1,04 (9H, с, t-Bu)).
(3,60 г, 8,64 ммоль, 64%), которая представляет смесь аномеров в соотношении 6:1. (1H-ЯМР главного изомера: 7,70-7,35 (10H, м, ароматический H); 6,63 (1H, д, H5) J = 4,4 Hz; 5,47 (1H, т, H2); 4,20-3,60 (2H, м, -CH2O); 3,27 (1H, дд, 1H4) J = 4,4 и 11,4 Hz; 3,09 (1H, д, 1H4) J = 11,4 Hz; 2,02 (3H, с, CH3CO); 1,05 (9H, с, t-Bu); 1H-ЯМР минорного изомера: 7,70-7,35 (10H, м, ароматический H); 6,55 (1H, д, H5) J = 3,9 Hz; 5,45 (1H, т, H2); 4,20-3,60 (2H, м, -CH2O); 3,25 (1H, дд, 1H4) J = 3,9 и 11,4 Hz; 3,11 (1H, д, 1H4) J = 11,4 Hz; 2,04 (3H, с, CH3CO); 1,04 (9H, с, t-Bu)).
2-(t-Бутил-дифенилсилилокси)-метил-5-ацетокси-1,3-оксатиолан  (0,28 г, 0,67 ммоль) растворяют в 1,2-дихлорэтане (20 мл) и добавляют силилированный цитозин
(0,28 г, 0,67 ммоль) растворяют в 1,2-дихлорэтане (20 мл) и добавляют силилированный цитозин  (0,20 г, 0,78 ммоль) одной порцией при комнатной температуре. Смесь перемешивают в течение 10 минут и к ней прибавляют раствор SnCl4 (0,80 мл, 1,0 М раствор в CH2Cl2, 0,80 ммоль), по каплям при комнатной температуре. Цитозин
(0,20 г, 0,78 ммоль) одной порцией при комнатной температуре. Смесь перемешивают в течение 10 минут и к ней прибавляют раствор SnCl4 (0,80 мл, 1,0 М раствор в CH2Cl2, 0,80 ммоль), по каплям при комнатной температуре. Цитозин  (0,10 г, 0,39 ммоль) и раствор SnCl4 (0,60 мл) прибавляют таким же образом часом позже. По завершении реакции в течение 2 часов раствор концентрируют и остаток тритируют триэтиламином (2 мл) и подвергают мгновенной хроматографии (сначала с чистым EtOAc и затем с 20% этанолом в EtOAc); получая твердый
(0,10 г, 0,39 ммоль) и раствор SnCl4 (0,60 мл) прибавляют таким же образом часом позже. По завершении реакции в течение 2 часов раствор концентрируют и остаток тритируют триэтиламином (2 мл) и подвергают мгновенной хроматографии (сначала с чистым EtOAc и затем с 20% этанолом в EtOAc); получая твердый  красновато-коричневого цвета (100% в β-конфигурации) (0,25 г, 0,54 ммоль, 80%). (1H-ЯМР (DMSO-d6): 7,75 (1H, д, H6) J = 7,5 Hz; 7,65-7,35 (10H, м, ароматический H); 7,21 и 7,14 (2H, широкий, -NH2); 6,19 (1H, т, H5); 5,57 (1H, д, H5); 5,25 (1H, т, H2); 3,97 (1H, дд, -CH2O) J = 3,9 и 11,1 Hz; 3,87 (1H, дд, -CH2O); 3,41 (1H, дд, 1H4) J = 4,5 и 11,7 Hz; 3,03 (1H, дд, 1H4); 0,97 (9H, с, t-Bu)).
красновато-коричневого цвета (100% в β-конфигурации) (0,25 г, 0,54 ммоль, 80%). (1H-ЯМР (DMSO-d6): 7,75 (1H, д, H6) J = 7,5 Hz; 7,65-7,35 (10H, м, ароматический H); 7,21 и 7,14 (2H, широкий, -NH2); 6,19 (1H, т, H5); 5,57 (1H, д, H5); 5,25 (1H, т, H2); 3,97 (1H, дд, -CH2O) J = 3,9 и 11,1 Hz; 3,87 (1H, дд, -CH2O); 3,41 (1H, дд, 1H4) J = 4,5 и 11,7 Hz; 3,03 (1H, дд, 1H4); 0,97 (9H, с, t-Bu)).
Силиэфир  (0,23 г, 0,49 ммоль) растворяют в THF (30 мл) и к нему добавляют раствор H-Bu4NF (0,50 мл, 1,0 М раствор в THF, 0,50 ммоль), по каплям при комнатной температуре. Смесь перемешивают в течение 1 часа и концентрируют под вакуумом. Остаток отбирают смесью этанол-триэтиламин (2 мл/1 мл) и подвергают мгновенной хроматографии (сначала с EtOAc, затем с 20%-ным этанолом в EtOAc), получая белый твердый
(0,23 г, 0,49 ммоль) растворяют в THF (30 мл) и к нему добавляют раствор H-Bu4NF (0,50 мл, 1,0 М раствор в THF, 0,50 ммоль), по каплям при комнатной температуре. Смесь перемешивают в течение 1 часа и концентрируют под вакуумом. Остаток отбирают смесью этанол-триэтиламин (2 мл/1 мл) и подвергают мгновенной хроматографии (сначала с EtOAc, затем с 20%-ным этанолом в EtOAc), получая белый твердый  100%-й чистотой аномера (BCH-189; 0,11 г, 0,48 ммоль, 98%), который далее перекристаллизовывают из смеси этанол/CHCl3/гексаны. (1H-ЯМР (DMSO-d6): 7,91 (1H, д, H6) J=7,6 Hz; 7,76 и 7,45 (2H, широкий, -NH2); 6,19 (1H, т, H5); 5,80 (1H, д, H5) J = 7,6 Hz; 5,34 (1H, широкий, -OH); 5,17 (1H, т, H2); 3,74 (2H, м, -CH2O); 3,42 (1H, дд, 1H4) J = 5,6 и 11,5 Hz; 3,09 (1H, дд, 1H4) J = 4,5 и 11,5 Hz)).
100%-й чистотой аномера (BCH-189; 0,11 г, 0,48 ммоль, 98%), который далее перекристаллизовывают из смеси этанол/CHCl3/гексаны. (1H-ЯМР (DMSO-d6): 7,91 (1H, д, H6) J=7,6 Hz; 7,76 и 7,45 (2H, широкий, -NH2); 6,19 (1H, т, H5); 5,80 (1H, д, H5) J = 7,6 Hz; 5,34 (1H, широкий, -OH); 5,17 (1H, т, H2); 3,74 (2H, м, -CH2O); 3,42 (1H, дд, 1H4) J = 5,6 и 11,5 Hz; 3,09 (1H, дд, 1H4) J = 4,5 и 11,5 Hz)).
BCH-189 и его аналоги можно также синтезировать сочетанием силилированного производного урацила с  Силилированное производное урацила
Силилированное производное урацила  (1,80 г, 7,02 ммоль) сочетают с
(1,80 г, 7,02 ммоль) сочетают с  (1,72 г, 4,13 ммоль) в 1,2-дихлорэтане (50 мл) в присутствии SnCl4 (5,0 мл), как описано выше, при получении производного цитозина 13. Реакция завершается полностью после 5 часов. Методом мгновенной хроматографии, сначала с 40% EtOAc в гексане и затем с EtOAc получают
(1,72 г, 4,13 ммоль) в 1,2-дихлорэтане (50 мл) в присутствии SnCl4 (5,0 мл), как описано выше, при получении производного цитозина 13. Реакция завершается полностью после 5 часов. Методом мгновенной хроматографии, сначала с 40% EtOAc в гексане и затем с EtOAc получают  в виде белой пены (1,60 г, 3,43 ммоль, 83%). (1H-ЯМР: 9,39 (1H, широкий, -NH) 7,90 (1H, д, H6) J = 7,9 Hz; 7,75-7,35 (10H, м, ароматический H); 6,33 (1H, дд, H5); 5,51 (1H, д, H5) J = 7,9 Hz; 5,23 (1H, т, H5); 4,11 (1H, дд, -CH2O) J = 3,2 и 11,7 Hz; 3,93 (1H, дд. -CH2O); 3,48 (1H, дд, 1H4) J = 5,4 и 12,2 Hz; 3,13 (1H, дд, 1H4) J = 3,2 и 12,2 Hz)).
в виде белой пены (1,60 г, 3,43 ммоль, 83%). (1H-ЯМР: 9,39 (1H, широкий, -NH) 7,90 (1H, д, H6) J = 7,9 Hz; 7,75-7,35 (10H, м, ароматический H); 6,33 (1H, дд, H5); 5,51 (1H, д, H5) J = 7,9 Hz; 5,23 (1H, т, H5); 4,11 (1H, дд, -CH2O) J = 3,2 и 11,7 Hz; 3,93 (1H, дд. -CH2O); 3,48 (1H, дд, 1H4) J = 5,4 и 12,2 Hz; 3,13 (1H, дд, 1H4) J = 3,2 и 12,2 Hz)).
Производное урацила  может быть превращено в производное цитозина 13. Производное урацила 16 (0,20 г, 0,43 ммоль) растворяют в смеси пиридин/дихлорэтан (2 мл/10 мл) и раствор охлаждают до 0oC. Трифторуксусный ангидрид (72 мкл, 0,43 ммоль) добавляют по каплям при 0oC и смесь нагревают до комнатной температуры и перемешивают в течение 1 часа. Дополнительно добавляют трифторуксусный ангидрид (0,50 мкл, 0,30 ммоль) и смесь перемешивают в течение 1 часа. Методом ТСХ не выявили подвижности с EtOAc. Реакционную смесь затем отбирают трубочкой и переводят в насыщенный аммиаком раствор метанола (30 мл), и смесь перемешивают в течение 12 часов при комнатной температуре. Раствор концентрируют и остаток подвергают мгновенной хроматографии, получая красновато-коричневую пену
может быть превращено в производное цитозина 13. Производное урацила 16 (0,20 г, 0,43 ммоль) растворяют в смеси пиридин/дихлорэтан (2 мл/10 мл) и раствор охлаждают до 0oC. Трифторуксусный ангидрид (72 мкл, 0,43 ммоль) добавляют по каплям при 0oC и смесь нагревают до комнатной температуры и перемешивают в течение 1 часа. Дополнительно добавляют трифторуксусный ангидрид (0,50 мкл, 0,30 ммоль) и смесь перемешивают в течение 1 часа. Методом ТСХ не выявили подвижности с EtOAc. Реакционную смесь затем отбирают трубочкой и переводят в насыщенный аммиаком раствор метанола (30 мл), и смесь перемешивают в течение 12 часов при комнатной температуре. Раствор концентрируют и остаток подвергают мгновенной хроматографии, получая красновато-коричневую пену  (0,18 г, 0,39 ммоль, 91%), которая была идентична с соединением, полученным при реакции сочетания цитозина.
(0,18 г, 0,39 ммоль, 91%), которая была идентична с соединением, полученным при реакции сочетания цитозина.
На фиг. 3 представлен синтез 5-метилцитидиновых и тимидиновых производных ВСН-189. Ацетат 11 (0,93 г, 2,23 ммоль) в 1,2-дихлорэтане (50 мл) реагирует с силилированным производным тимина 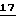 (1,0 г, 3,70 ммоль) и раствором SnCl4 (4,0 мл), как описано при приготовлении производного цитозина
(1,0 г, 3,70 ммоль) и раствором SnCl4 (4,0 мл), как описано при приготовлении производного цитозина  (1H-ЯМР: 8,10 (1H, широкий, NH); 7,75-7,30 (11H, м, 10 ароматических H и 1H6); 6,32 (1H, т, H1) J = 5,4 Hz; 5,25 (1H, т, H4) J = 4,2 Hz; 4,01 (1H, дд, 1H5) J = 3,9 и 11,4 Hz; 3,93 (1H, дд, 1H5) J = 4,5 и 11,4 Hz; 3,41 (1H, дд, 1H2) J = 5,4 и 11,7 Hz; 3,04 (1H, дд, 1H2) J= 5,7 и 11,7 Hz; 1,75 (3H, с, CH3); 1,07 (9H, с, t-Bu)).
(1H-ЯМР: 8,10 (1H, широкий, NH); 7,75-7,30 (11H, м, 10 ароматических H и 1H6); 6,32 (1H, т, H1) J = 5,4 Hz; 5,25 (1H, т, H4) J = 4,2 Hz; 4,01 (1H, дд, 1H5) J = 3,9 и 11,4 Hz; 3,93 (1H, дд, 1H5) J = 4,5 и 11,4 Hz; 3,41 (1H, дд, 1H2) J = 5,4 и 11,7 Hz; 3,04 (1H, дд, 1H2) J= 5,7 и 11,7 Hz; 1,75 (3H, с, CH3); 1,07 (9H, с, t-Bu)).
Производное тимина 18 (0,20 г, 0,42 ммоль) растворяют в смеси пиридин/дихлорэтан (2 мл/10 мл), и раствор охлаждают до 0oC. К нему добавляют трифторуксусный ангидрид (100 мкл, 0,60 ммоль) по каплям при 0oC, и смесь отставляют при постоянном перемешивании, давая ей нагреться до комнатной температуры. По достижении комнатной температуры ее перемешивают в течение 1 часа. Методом ТСХ показано отсутствие подвижности с EtOAc. Затем реакционную смесь с помощью трубочки переводят в насыщенный аммиаком раствор метанола (20 мл), и смесь перемешивают в течение 12 часов при комнатной температуре. Раствор концентрируют и остаток подвергают мгновенной хроматографии, получая красновато-коричневую пену  (0,18 г, 0,38 ммоль, 90%). (1Н-ЯМР: 7,07-7,30 (12H, м, 10 ароматических H, 1NH и H6); 6,0 (1H, широкий, 1NH); 6,34 (1H, т, H1) J = 4,5 Hz; 5,25 (1H, т, H4) J = 3,6 Hz; 4,08 (1H, дд, 1H5) J = 3,6 и 11,4 Hz; 3,96 (1H, дд, 1H5) J = 3,6 и 11,4 Hz; 3,52 (1H, дд, 1H2) J = 5,4 и 12,3 Hz; 3,09 (1H, дд, 1H2) J = 3,9 и 12,3 Hz; 1,72 (3H, с, CH3); 1,07 (9H, с, t-Bu)).
(0,18 г, 0,38 ммоль, 90%). (1Н-ЯМР: 7,07-7,30 (12H, м, 10 ароматических H, 1NH и H6); 6,0 (1H, широкий, 1NH); 6,34 (1H, т, H1) J = 4,5 Hz; 5,25 (1H, т, H4) J = 3,6 Hz; 4,08 (1H, дд, 1H5) J = 3,6 и 11,4 Hz; 3,96 (1H, дд, 1H5) J = 3,6 и 11,4 Hz; 3,52 (1H, дд, 1H2) J = 5,4 и 12,3 Hz; 3,09 (1H, дд, 1H2) J = 3,9 и 12,3 Hz; 1,72 (3H, с, CH3); 1,07 (9H, с, t-Bu)).
Силиловый эфир 19 (0,18 г, 0,38 ммоль) растворяют в THF (20 мл) и прибавляют раствор H-Bu4NF (0,50 мл, 1,0 М раствор в THF, 0,50 ммоль), по каплям, при комнатной температуре. Смесь перемешивают 1 час и концентрируют под вакуумом. Остаток отбирают с помощью смеси этанол/триэтиламин (2 мл/1 мл) и подвергают мгновенной хроматографии (сначала с EtOAc, затем с 20%-ным этанолом в EtOAc), получая белый твердый 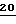 (0,09 г, 0,37 ммоль, 97%), который далее перекристаллизовывают из смеси этанол/CHCl3/гексаны, получая 82 мг чистого соединения (89%). (1H-ЯМР: (в d6-DMSO): 7,70 (1H, с, H6); 7,48 и 7,10 (2H, широкий, NH2); 6,19 (1H, т, H1) J = 6,5 Hz; 5,31 (1H, т, OH); 5,16 (1H, т, 1H4) J = 5,4 Hz; 3,72 (2H, м, 2H5) 3,36 (1H, дд, 1H2) J = 6,5 и 14,0 Hz; 3,05 (1H, дд, 1H2) J = 6,5 и 14,0 Hz; 1,85 (3H, с, CH3)).
(0,09 г, 0,37 ммоль, 97%), который далее перекристаллизовывают из смеси этанол/CHCl3/гексаны, получая 82 мг чистого соединения (89%). (1H-ЯМР: (в d6-DMSO): 7,70 (1H, с, H6); 7,48 и 7,10 (2H, широкий, NH2); 6,19 (1H, т, H1) J = 6,5 Hz; 5,31 (1H, т, OH); 5,16 (1H, т, 1H4) J = 5,4 Hz; 3,72 (2H, м, 2H5) 3,36 (1H, дд, 1H2) J = 6,5 и 14,0 Hz; 3,05 (1H, дд, 1H2) J = 6,5 и 14,0 Hz; 1,85 (3H, с, CH3)).
Силиловый эфир  (0,70 г, 1,46 ммоль) растворяли в THF (50 мл), и раствор H-Bu4NF (2 мл, 1,0 М раствор в THF, 2 ммоль) добавляют по каплям при комнатной температуре. Смесь перемешивают 1 час и концентрируют под вакуумом. Остаток отбирают смесью этанол/триэтиламин (2 мл/1 мл) и подвергают мгновенной хроматографии, получая белый твердый 21 (0,33 г, 1,35 ммоль. 92%). (1H-ЯМР: (в d6-ацетоне): 9,98 (1H, широкий, NH); 7,76 (1H, д, H6) J = 1,2 Hz; 6,25 (1H, т, H4) J = 5,7 Hz; 5,24 (1H, т, H1) J = 4,2 Hz; 4,39 (1H, т, OH) J = 5,7 Hz; 3,85 (1H, дд, 2H5) J = 4,2 и 5,7 Hz; 3,41 (1H, дд, 1H2) J = 5,7 и 12,0 Hz; 3,19 (1H, дд, 1H2) J = 5,4 и 12,0 Hz; 1,80 (3H, с, CH3)).
(0,70 г, 1,46 ммоль) растворяли в THF (50 мл), и раствор H-Bu4NF (2 мл, 1,0 М раствор в THF, 2 ммоль) добавляют по каплям при комнатной температуре. Смесь перемешивают 1 час и концентрируют под вакуумом. Остаток отбирают смесью этанол/триэтиламин (2 мл/1 мл) и подвергают мгновенной хроматографии, получая белый твердый 21 (0,33 г, 1,35 ммоль. 92%). (1H-ЯМР: (в d6-ацетоне): 9,98 (1H, широкий, NH); 7,76 (1H, д, H6) J = 1,2 Hz; 6,25 (1H, т, H4) J = 5,7 Hz; 5,24 (1H, т, H1) J = 4,2 Hz; 4,39 (1H, т, OH) J = 5,7 Hz; 3,85 (1H, дд, 2H5) J = 4,2 и 5,7 Hz; 3,41 (1H, дд, 1H2) J = 5,7 и 12,0 Hz; 3,19 (1H, дд, 1H2) J = 5,4 и 12,0 Hz; 1,80 (3H, с, CH3)).
На фиг. 4 представлен синтез обогащенного энантиомером BCH-189 и его аналогов. Аллилбутират  (19,0 г, 148 ммоль) растворяют в CH2Cl2 (400 мл) и озонируют при -78oC. По завершении озонолиза добавляют диметилсульфид (20 мл, 270 ммоль, 1,8 экв) при -78oC, смесь нагревают до комнатной температуры и перемешивают в течение ночи. Раствор промывают водой (100 мл•2), сушат над MgSO4, фильтруют, концентрируют и перегоняют под вакуумом (70-80oC при 0,5-0,6 мм Нд), получая бесцветную жидкость
(19,0 г, 148 ммоль) растворяют в CH2Cl2 (400 мл) и озонируют при -78oC. По завершении озонолиза добавляют диметилсульфид (20 мл, 270 ммоль, 1,8 экв) при -78oC, смесь нагревают до комнатной температуры и перемешивают в течение ночи. Раствор промывают водой (100 мл•2), сушат над MgSO4, фильтруют, концентрируют и перегоняют под вакуумом (70-80oC при 0,5-0,6 мм Нд), получая бесцветную жидкость  (17,0 г, 131 ммоль, 88%). (1H-ЯМР: 9,59 (1H, с, H-CO); 4,66 (2H, с, -CH2O); 2,42 (2H, т, CH2CO) J = 7,2 Hz; 1,71 (2H, секстет, -CH2); 0,97 (3H, т, CH3) J = 7,2 Hz; (ИК (чистый): 2990, 2960, 2900, 1750, 1740, 1460, 1420, 1390, 1280, 1190, 1110, 1060, 1020, 990, 880, 760).
(17,0 г, 131 ммоль, 88%). (1H-ЯМР: 9,59 (1H, с, H-CO); 4,66 (2H, с, -CH2O); 2,42 (2H, т, CH2CO) J = 7,2 Hz; 1,71 (2H, секстет, -CH2); 0,97 (3H, т, CH3) J = 7,2 Hz; (ИК (чистый): 2990, 2960, 2900, 1750, 1740, 1460, 1420, 1390, 1280, 1190, 1110, 1060, 1020, 990, 880, 760).
Бутирилоксиацетальдегид  (15,0 г, 115 ммоль) растворяют в толуоле (200 мл) и смешивают с тиогликолевой кислотой (8,0 мл, 115 ммоль). Раствор перегоняют в течение 5 часов, и образующуюся воду удаляют с помощью ловушки Dean-Stark'a. Раствор охлаждают до комнатной температуры и переносят в делительную воронку объемом 500 мл. Затем раствор промывают насыщенным раствором NaHCO3. Водные смывы экстрагируют диэтиловым эфиром (200 мл • 2) с целью рекуперации любого неочищенного продукта из водного слоя. Эфирные экстракты добавляют к толуольному слою и полученную смесь промывают водой (100 мл • 2), сушат над MgSO4, фильтруют, концентрируют и отгоняют под вакуумом (70-80oC при 0,5-0,6 мм Нд), получая бесцветное масло
(15,0 г, 115 ммоль) растворяют в толуоле (200 мл) и смешивают с тиогликолевой кислотой (8,0 мл, 115 ммоль). Раствор перегоняют в течение 5 часов, и образующуюся воду удаляют с помощью ловушки Dean-Stark'a. Раствор охлаждают до комнатной температуры и переносят в делительную воронку объемом 500 мл. Затем раствор промывают насыщенным раствором NaHCO3. Водные смывы экстрагируют диэтиловым эфиром (200 мл • 2) с целью рекуперации любого неочищенного продукта из водного слоя. Эфирные экстракты добавляют к толуольному слою и полученную смесь промывают водой (100 мл • 2), сушат над MgSO4, фильтруют, концентрируют и отгоняют под вакуумом (70-80oC при 0,5-0,6 мм Нд), получая бесцветное масло  (19 г, 93 ммоль, 81%). (1H-ЯМР: 5,65 (1H, дд, H5) J = 5,0 и 1,4 Hz; 4,35 (1H, дд, -CH2O) J = 3,2 и 12,2 Hz; 4,29 (1H, дд, -CH2O) J = 5,7 и 12,2 Hz; 3,72 (1H, д, -CH2S) J = 16,2 Hz; 3,64 (1H, д, -CH2S); 2,34 (2H, т, -CH2CO) J = 7,2 Hz; 1,66 (2H, секстет, -CH2); 0,95 (3H, т, CH3) J = 7,2 Hz; (ИК (чистый): 2980, 2960, 2900, 1780, 1740, 1460, 1410, 1390, 1350, 1300, 1290, 1260, 1220, 1170, 1110, 1080, 1070, 1000, 950, 910, 830, 820, 800, 760).
(19 г, 93 ммоль, 81%). (1H-ЯМР: 5,65 (1H, дд, H5) J = 5,0 и 1,4 Hz; 4,35 (1H, дд, -CH2O) J = 3,2 и 12,2 Hz; 4,29 (1H, дд, -CH2O) J = 5,7 и 12,2 Hz; 3,72 (1H, д, -CH2S) J = 16,2 Hz; 3,64 (1H, д, -CH2S); 2,34 (2H, т, -CH2CO) J = 7,2 Hz; 1,66 (2H, секстет, -CH2); 0,95 (3H, т, CH3) J = 7,2 Hz; (ИК (чистый): 2980, 2960, 2900, 1780, 1740, 1460, 1410, 1390, 1350, 1300, 1290, 1260, 1220, 1170, 1110, 1080, 1070, 1000, 950, 910, 830, 820, 800, 760).
Раствор эстеразы из печени свиньи (90 мкл) добавляют к буферному раствору (pH 7, 100 мл) при комнатной температуре, и смесь интенсивно перемешивают в течение 5 минут. Бутират  (2,8 г, 13,7 ммоль) добавляют одной порцией к раствору фермент/буфер, и смесь энергично перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь выливают в делительную воронку. Реакционную колбу промывают эфиром (10 мл) и смывы объединяют с реакционной смесью в воронке. Объединенную смесь экстрагируют гексанами трижды (100 мл • 3). Три гексановых экстракта объединяют и сушат над MgSO4, фильтруют и концентрируют, получая оптически активный бутират
(2,8 г, 13,7 ммоль) добавляют одной порцией к раствору фермент/буфер, и смесь энергично перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь выливают в делительную воронку. Реакционную колбу промывают эфиром (10 мл) и смывы объединяют с реакционной смесью в воронке. Объединенную смесь экстрагируют гексанами трижды (100 мл • 3). Три гексановых экстракта объединяют и сушат над MgSO4, фильтруют и концентрируют, получая оптически активный бутират  (1,12 г, 5,48 ммоль, 40%). Избыток энантиомера определяют методом ЯМР, используя производное Трис[3-гептафторпропил-гидроксиметилен)-(+)-камфорато]европия (III) в качестве реактива для химического сдвига; по этой методике обогащение для одного энантиомера составило примерно 40%. Оставшийся от реакции водный слой подвергают непрерывной экстракции с помощью CH2Cl2 в течение 20 часов. Органический слой из экстрактора удаляют, сушат над MgSO4, фильтруют и концентрируют, получая масло (1,24 г), которое, по данным ЯМР-анализа, состоит преимущественно из 2-гидроксиметил-оксо-1,3-оксатиолана
(1,12 г, 5,48 ммоль, 40%). Избыток энантиомера определяют методом ЯМР, используя производное Трис[3-гептафторпропил-гидроксиметилен)-(+)-камфорато]европия (III) в качестве реактива для химического сдвига; по этой методике обогащение для одного энантиомера составило примерно 40%. Оставшийся от реакции водный слой подвергают непрерывной экстракции с помощью CH2Cl2 в течение 20 часов. Органический слой из экстрактора удаляют, сушат над MgSO4, фильтруют и концентрируют, получая масло (1,24 г), которое, по данным ЯМР-анализа, состоит преимущественно из 2-гидроксиметил-оксо-1,3-оксатиолана 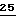 с небольшим количеством масляной кислоты и бутирата 24.
с небольшим количеством масляной кислоты и бутирата 24.
Лактон 25 (0,85 г, 4,16 ммоль) растворяют в толуоле (30 мл), и раствор охлаждают до -78oC. Раствор Dibal-H (9 мл, 1,0 М в гексанах, 9 ммоль) добавляют по каплям, в то время как внутреннюю температуру поддерживают ниже -70oC в процессе добавления. По окончании добавления смесь перемешивают в течение 0,5 часа при -78oC. Уксусный ангидрид (5 мл, 53 ммоль) добавляют в смесь при непрерывном перемешивании и оставляют на ночь для достижения комнатной температуры. К реакционной смеси прибавляют воду (5 мл) и полученную смесь перемешивают 1 час. Затем вносят MgSO4 (40 г), и смесь энергично перемешивают при комнатной температуре в течение 1 часа. Смесь фильтруют, концентрируют и остаток подвергают мгновенной хроматографии с 20% EtOAc в гексанах, получая бесцветную жидкость 26 (0,41 г, 1,86 ммоль, 45%), которая представляет смесь аномеров по положению C-4.
2-Ацетоксиметил-5-ацетокси-1,3-оксатиолан  (0,40 г, 1,82 ммоль) растворяют в 1,2-дихлорэтане (40 мл) и к ней добавляют силилированный цитозин 12 (0,70 г. 2,74 ммоль) одной порцией при комнатной температуре. Смесь перемешивают в течение 10 минут, и к ней добавляют раствор SnCl4 (3,0 мл, 1,0 М раствор в CH2Cl2, 3,0 ммоль), по каплям, при комнатной температуре. Дополнительно раствор SnCl4 (1,0 мл) добавляют через 1 час. Реакцию контролируют методом ТСХ. По завершении реакции сочетания раствор концентрируют, остаток тритируют триэтиламином (2 мл) и подвергают мгновенной хроматографии (сначала в чистом EtOAc и затем с 20%-ным этанолом в EtOAc), получая красновато-коричневый твердый 27 (0,42 г, 1,55 ммоль, 86%). (1H-ЯМР: 7,73 (1H, д, H6) J = 7,5 Hz; 6,33 (1H, т, H4) J = 4,8 Hz; 5,80 (1H, д, 5) J = 7,5 Hz; 4,52 (1H, дд, 1H5) J = 5,7 и 12,3; 4,37 (1H, дд, 1H5) J = 3,3 и 12,3 Hz; 3,54 (1H, дд, H2) J = 5,4 и 12,0 Hz; 3,10 (1H, дд, 1H3); 2,11 (3H, с, CH3)).
(0,40 г, 1,82 ммоль) растворяют в 1,2-дихлорэтане (40 мл) и к ней добавляют силилированный цитозин 12 (0,70 г. 2,74 ммоль) одной порцией при комнатной температуре. Смесь перемешивают в течение 10 минут, и к ней добавляют раствор SnCl4 (3,0 мл, 1,0 М раствор в CH2Cl2, 3,0 ммоль), по каплям, при комнатной температуре. Дополнительно раствор SnCl4 (1,0 мл) добавляют через 1 час. Реакцию контролируют методом ТСХ. По завершении реакции сочетания раствор концентрируют, остаток тритируют триэтиламином (2 мл) и подвергают мгновенной хроматографии (сначала в чистом EtOAc и затем с 20%-ным этанолом в EtOAc), получая красновато-коричневый твердый 27 (0,42 г, 1,55 ммоль, 86%). (1H-ЯМР: 7,73 (1H, д, H6) J = 7,5 Hz; 6,33 (1H, т, H4) J = 4,8 Hz; 5,80 (1H, д, 5) J = 7,5 Hz; 4,52 (1H, дд, 1H5) J = 5,7 и 12,3; 4,37 (1H, дд, 1H5) J = 3,3 и 12,3 Hz; 3,54 (1H, дд, H2) J = 5,4 и 12,0 Hz; 3,10 (1H, дд, 1H3); 2,11 (3H, с, CH3)).
5'-Ацетат BCH-189  (140 мг, 0,52 ммоль) растворяют в безводном метаноле (10 мл) и к нему добавляют метоксил натрия (110 мг, 2,0 ммоль) одной порцией. Смесь перемешивают при комнатной температуре до полного завершения гидролиза. Гидролиз длится примерно 1 час, и реакцию контролируют методом ТСХ. По завершении реакции смесь концентрируют, и остаток отбирают с помощью этанола (2 мл). Спиртовой раствор подвергают колоночной хроматографии, сначала применяя этилацетат и затем 20% этанол в EtOAc, получая белую пену (110 мн, 92%), ЯМР-спектр которой идентичен со спектром стандартного BCH-189, 14.
(140 мг, 0,52 ммоль) растворяют в безводном метаноле (10 мл) и к нему добавляют метоксил натрия (110 мг, 2,0 ммоль) одной порцией. Смесь перемешивают при комнатной температуре до полного завершения гидролиза. Гидролиз длится примерно 1 час, и реакцию контролируют методом ТСХ. По завершении реакции смесь концентрируют, и остаток отбирают с помощью этанола (2 мл). Спиртовой раствор подвергают колоночной хроматографии, сначала применяя этилацетат и затем 20% этанол в EtOAc, получая белую пену (110 мн, 92%), ЯМР-спектр которой идентичен со спектром стандартного BCH-189, 14.




Изобретение касается способов и составов для приготовления противовирусных нуклеозидных аналогов, в частности 2',3'-дидезокси-3'-тиа-цитидин (ВСН-189). ВСН-189 или 2',3'-дидезокси-3'-тиа-5-фторцитидин преимущественно в виде β-изомеров получают взаимодействием 1,3-оксатиолана формулы I, где R - гидрозащитная группа, R' - ацильная группа, с основанием, выбранным из группы, включающий силилированный цитозин или 5-фторцитозин в присутствии SnCl4. Этот путь синтеза дает возможность для стереоизбирательного получения биологически активного изомера β-ВСН-189 и родственных соединений. Более того, стереохимия в положении 4 нуклеозида может быть проконтролирована для получения обогащенного энантиомером β-ВСН-189 и его аналогов. 15 с. и 12 з. п.ф-лы, 4 ил.


где R - гидроксизащитная группа;
R' - ацильная группа,
с основанием, выбранным из группы, включающей силилированный цитозин или 5-фторцитозин в присутствии SnCl4.
где X - триалкилсилилоксигруппа;
Y - водород, метил, фтор;
Z - триалкилсилильная группа.
R - водород, алкил, силил, ацил;
Y - водород.
R - водород или ацил;
Y - водород.
где R - гидроксизащитная группа;
R' - ацильная группа,
отличающийся тем, что проводят стадии: а) азонирования соединения общей формулы CH2 CHCH2OR, где R имеет указанные значения, с образованием гликоальдегида общей формулы OHC CH2OR, где R - гидроксизащитная группа; в) добавления тиогликолевой кислоты к указанному гликоальдегиду с образованием лактона общей формулы
и с) восстановления лактона с получением целевого 1,3-оксатиолана.
где R - гидроксизащитная группа,
с образованием 2-гидроксиметил-5-оксо-1,3-оксатиолана, обогащенного энантиомером; в) восстановления и ацилирования указанного 2-гидроксиметил-5-оксо-1,3-оксатиолана, обогащенного энантиомером, с образованием 2-ацилоксиметил-5-ацилокси-1,3-оксатиолана, обогащенного энантиомером.
где Y - водород, фтор;
R - ацил,
отличающийся тем, что нуклеозид обрабатывают ферментом, выбранным из группы, включающей эстеразу из печени свиньи.
| J.of Med | |||
| Chem | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
