Настоящее изобретение относится к биологически активным иуклеозидам, и, в частности, включает антивирусные составы, в которые входит 2-гидроксиметил-5-/5-фторцитозин-1-ил/1,3-оксатиолан /"ФТЦ"/, его физиологически приемлемое производное или физиологически приемлемая соль, а также способ расщепления и использования /-/-β-L и /+/-β-D энантиомеров ФТЦ.
В 1981 г. синдром приобретенного иммунодефицита /СПИД/ был определен как болезнь, которая тяжело поражает иммунную систему человека и которая почти всегда ведет к смерти. В 1983 г. было определено, что этиологической причиной возникновения СПИДа является вирус иммунодефицита человека /ВИЧ/. По оценкам Всемирной организации здравоохранения к декабрю 1990 г. во всем мире ВИЧ-инфицированных людей насчитывалось от 8 до 10 млн., из этого количества от 1 до 1,4 млн. людей проживали в США.
В 1985 г. стало известно, что синтетический нуклеозид 3’-азидо-3’-дезокситимидин /АЗТ/ ингибирует репликацию вируса иммунодефицита человека. С тех пор было доказано, что целый ряд других синтетических нуклеозидов, в том числе 2’,3’-дидезоксиинозин /ДДИ/, 2’,3’-дидезоксицитидин /ДДЦ/, 3’-фтор-3’-дезокситимидин /ФЛТ/ и 2’,3’-дидезокси-2’,3’-дидегидротимидин /Д4Т/, оказался эффективным в борьбе против ВИЧ. Ряд других. 2’,3’-дидезоксинуклеозидов продемонстрировали способность ингибировать рост различных вирусов in vitro. Оказывается, что после клеточного фосфорилирования с помощью клеточной киназы до 5’-трифосфата эти синтетические нуклеозды включаются в растущую нить вирусной ДНК, вызывая обрыв цепи вследствие отсутствия 3’-гидроксильной группы.
Положительные результаты различных 2’,3’-дидеоксинуклеозидов в ингибировании репликации ВИЧ in vivo или in vitro привели ряд ученых к тому, что они занялись созданием и испатанием нуклеозидов, которые замещают гетероатом на атом углерода в положении 3’-нуклеозида. Norbeck et al. обнаружили, что /±/-1-[/2β, 4β/-2-/оксиметил/-4-диоксоланил]тимин (в тексте упоминаемый как (±)-диоксолан-Т) демонстрирует умеренную анти-ВИЧ активность (EC50 20 мкмоль в АТН8 клетках) и не является токсичным по отношению к неинфицированным контрольным клеткам при концентрации 200 мкмоль. Tetrahedren Lettere 30, (46), 6246, (1989). Европейская патентная заявка №0337713 и патент США №5041449, права на которые переданы IАF Biochem International, Inc., описывают 2-замещенный-4-замещенный-1,3-диоксоланы, которые демонстрируют антивирусную активность.
Патент США №5047407 и Европейская патентная заявка №0382526, права на которые также переданы Biochem International, Inc., описывают целый ряд 2-замещенный-5-замещенный-1,3-оксатиолан нуклеозидов, имеющих антивирусную активность, конкретно сообщают, что рацемическая смесь (около С4’-положения/ изомера С1’-β 2-оксиметил-5-/цитозин-1-ил/-1,3-оксотиолана (упоминаемый ниже как (±)-ВСН-189) обладает приблизительно такой же активностью против ВИЧ, как и АЗТ, и не обнаруживают на испытуемых уровнях клеточную токсичность. Было также обнаружено, что (±)-BCH-I89 ингибирует репликацию АЗТ-резистентного изолята ВИЧ in vitro, взятого у пациента, которого лечили АЗТ более 36 недель.
Другим вирусом, вызывающим серьезные проблемы в здоровье человека, является вирус гепатита В (упоминаемый ниже как "ВГВ"). ВГВ является второй после табака причиной рака у человека. Механизм, с помощью которого ВГВ индуцирует рак, неизвестен, хотя не вызывает сомнения, что он может непосредственно вызывать развитие опухоли, или косвенно способствовать ее развитию через хроническое воспаление, цирроз или перерождение клеток вследствие инфекции.
По прошествии инкубационного периода, длящегося от 2 до 6 месяцев, в течение которого носитель заболевания не знает о своей болезни, инфекция ВГВ может привести к острому гепатиту и поражению печени, что вызовет боли в области живота, желтуху и повышение уровней некоторых ферментов в крови. ВГВ может вызвать молниеносный гепатит, быстро прогрессирующую, часто смертельную форму заболевания, при которой разрушаются обширные участки печени.
Заболевшие острым гепатитом пациенты обычно выздоравливают. У некоторых пациентов, однако, высокие уровни вирусного антигена сохраняются в крови в течение продолжительного или неопределенного времени, являясь причиной хронической инфекции. Хроническая инфекция может привести к (хроническому) персистирующему гепатиту. Больные, инфицированные хроническим персистирующим ВГВ, чаще всего встречаются в развивающихся странах. В середине 1991 г. в одной только Азии было приблизительно 225 млн хронических носителей ВГВ, а во всем мире их было почти 300 млн. Хронический персистирующий гепатит может быть причиной усталости, цирроза печени, злокачественной гепатомы, а также первичного рака печени.
В промышленных западных странах группы высокого риска инфицирования ВГВ включают людей, которые находятся в контакте с носителями ВГВ или с образцами их крови. Эпидемиология ВГВ очень схожа с эпидемиологией синдрома приобретенного иммунодефицита, что объясняет, почему инфицирование ВГВ является обычным среди пациентов, больных СПИД или обусловленным СПИД комплексом. Однако ВГВ является более заразным, чем ВИЧ.
Для иммунизации пациентов против ВГВ была разработана человеческая полученная из сыгоротки крови вакцина. Хотя вакцина оказалась эффективной, производство вакцины является затруднительным, поскольку снабжение сывороткой крови, полученной от хронических носителей, ограничено, а процедура очистки является длительной и дорогой, далее каждая партия вакцины, приготовленная из различной сыворотки, для обеспечения безопасности должна быть проверена на шимпанзе. Вакцина была получена также с помощью генетической инженерии. Обещающим оказалось также ежедневное лечение α-интерфероном, полученным с помощью генетической инженерии белком. Однако до настоящего времени не известно фармацевтическое средство, которое эффективно ингибирует репликацию вируса.
Чтобы предложить нуклеозид для использования в фармацевтических целях, он должен быть не только эффективным средством, обладающим низкой токсичностью, но он также должен быть рентабельным в производстве. Значительные работы были проведены по изучению и разработке новых более рентабельных процессов производства нуклеозида в большом масштабе. 2’,3’-дидеоксинуклеозиды в настоящее время получают двумя способами: деривацией ннтактного нуклеозида или конденсацией дериватного фрагмента сахара с гетероциклическим основанием. Хотя существует множество неудобств, связанных с получением новых аналогов нуклеозидов путем модификации интактных нуклеозидов, главное достоинство этого подхода состоит в том, что соответствующая абсолютная стереохимия уже создана природой. Однако этот подход не может быть использован в производстве нуклеозидов, которые содержат не встречающиеся в природе основания или не встречающиеся в природе углеводные фрагменты (которые, следовательно, не были получены из интактных нуклеозидов), например, 1,3-оксатиолановые нуклеозиды и 1,3-диоксолановые нуклеозиды.
В результате конденсации углевод или углеводоподобный фрагмент с гетероциклическим основанием образуют синтетический нуклеозид, полученный нуклеозид имеет два хиральных центра (в положениях С1’ и С4’) и, таким образом, существует как диамтереомерическая пара. Каждый диастереомер является группой энантиомеров. Следовательно, продукт является смесью четырех энантиомеров.
Часто обнаруживается, что нуклеозиды, имеющие не встречающуюся в природе пространственную структуру в положениях С1 или C4, менее активны, чем те же самые нуклеозиды с природной пространственной структурой. Например, Carter et al. сообщили, что концентрация /-/-энантиомера карбовира /2’,3’-дидегидро-2’,3’-дидезоксигуанозин/ в культуре клеток, необходимая для уменьшения активности обратной транскриптазы на 50% (EC50), составляет 0,8 мкмоль, в то время как EC50 для (+)-энантиомера карбовира превышает 60 мкмоль. Antimicrobial Agents and Chemotherapy 34:6, 1297-1300 (июнь 1990).
РСТ международная публикация № WO 91/11186 сообщает, что 1,3-оксатиолановые нуклеозиды могут быть получены с высокой диастетеоселективностью (высокое процентное содержание нуклеозида с β конфигурацией цепи от С1’-углерода до гетероциклического основания) путем тщательного отбора льюисовской кислоты, использовавшейся при конденсации. Было обнаружено, что конденсация 1,3-оксатиоланового нуклеозида с основанием происходит с почти полной β-стереоспецифичностью, когда в качестве катализатора конденсации используется тетрахлорид олова. Другие льюисовские кислоты обеспечивают низкую (или вообще не обеспечивают) C1’-β селективность или просто не способны выступить в качестве катализатора реакций.
В свете того факта, что синдром приобретенного иммунодефицита, обусловленный СПИДом комплекс и вирус гепатита В достигли во всем мире эпидемического уровня и оказывают трагическое воздействие на инфицированных людей, существует настоятельная необходимость в создании новых эффективных фармацевтических средств для лечения этих заболеваний, обладающих низкой токсичностью для носителя.
Существует также потребность в создании рентабельного, коммерчески жизнеспособного способа производства имеющих фармацевтическое значение нуклеозидов и особенно необходимо добиться образования β-стереоспецифичности в С4’-положении синтетических нуклеозидов, полученных путем конденсирования углеводоподобного фрагмента с основанием.
Следовательно, целью настоящего изобретения является создание способа и состава для лечения людей, инфицированных ВИЧ.
Следующей целью настоящего изобретения является создание способа и состава для лечения людей и животных, инфицированных ВГВ.
Еще одной целью настоящего изобретения является создание обогащенных энантиомерами 1,3-оксатиолановых нуклеозидов.
И еще одной целью настоящего изобретения является создание способа расщепления С4’-энантиомеров 1,3-оксатиолановых нуклеозидов.
Краткое изложение существа изобретения
Описывается способ и состав для лечения вызванных ВИЧ и ВГВ инфекций у людей и других животных-носителей, который включает введение эффективного количества 2-оксиметил-5-/5-фторцитозин/-1,3-оксатиолана, его фармацевтически приемлемого производного, 5’ или N4 алкилированное или ацилированное производное или его фармацевтически приемлемой соли в фармацевтически приемлемом носителе.
Было обнаружено, что 2-оксиметил-5-/5-фторцитозин-1-ил/-1,3-оксатиолан ("ФТЦ") демонстрирует неожиданно высокую активность против вируса иммунодефицита человека, причем обнаруживает очень низкую токсичность по отношению к клеткам хозяина. Было также обнаружено, что ФТЦ демонстрирует очень значительную активность против ВГВ и поэтому может быть использован для лечения пациентов, страдающих от различных заболеваний, связанных с инфицированием ВГВ.
Изучение токсичности и фармакокинетических свойств ФТЦ подтверждает ценность его как антивирусного средства для фармацевтического использования. ФТЦ и его энантиомеры не являются токсичным для периферических клеток костного мозга человека при концентрациях до 50 мкмолей и для других клеточных линий при концентрациях до 200 мкмолей. ФТЦ-ТП является главным внутриклеточным метаболитом в МКПК (мононуклеарные клетки периферической крови) и клетках HepG2. ФТЦ-ТП конкретно ингибирует ВИЧ-1 обратной транскриптазы (ОТ) с помощью КI 0,2 мкмоля, используя поли(1)олиго(дц)матрицу-праймер. Используя анализ последовательностей, можно показать, что, когда применяется ВИЧ-ОТ (С-stops), ФТЦ-ТП может быть сильнодействующим терминатором цепи ДНК.
Постоянное лечение ФТЦ не является токсичным для грызунов, даже когда оральные дозы составляют 85 мг/кг в день в течение по крайней мере двух месяцев. Фармакокинетика ФТЦ показывает высокую биологическую доступность на макаках резус при оральном приеме (приблизительно 73±6%) и терминальный период полужизни плазмы приблизительно 1,34±0,18 (среднее орального и внутривенного введения).
Также описывается способ расщепления рацемической смеси энантиомеров нуклеоэида, включая рацемическую смесь ФТЦ, который содержит этап, когда рацемическая смесь подвергается воздействию фермента, который предпочтительно катализирует реакцию в одном из энантиомеров. Способ может быть использован для разделения разнообразных нуклеозидов, включая пиримидиновый и пуриновый нуклеозиды, которые необязательно могут быть замещены в углеводном фрагменте или в основании. Способ может быть также использован для разделения производных нуклеозидов, которые содержат дополнительные гетероатомы в углеводном фрагменте, например, (±)-ФТЦ и (±)-ВСН-189. Разделение нуклеозидов может быть осуществлено в большом масштабе при умеренной стоимости.
Используя описанные здесь способы, ФТЦ был разделен на свои (+)-β-D и (-)-β-L энантиомеры. (-)-β-L-энантиомер представляется более сильнодействующим, чем (+)-β-D-энантиомер, против ВИЧ, ВГВ и ВИО (вирус иммунодефицита обезьян). (+)-энантиомер ФТЦ также активен против ВИЧ, ВГВ и ВИО.
Краткое описание чертежей
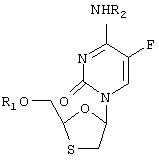
На фиг.1 изображена химическая структура 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана ("ФТЦ").
На фиг.2 схематически изображен способ получения 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана.
На фиг.3 изображена диаграмма специфичности щелочной фосфатазы и фосфдиестеразы яда змей для (+) и (-) энантиомеров ФТЦ.
На фиг.4 представлен график развития катализируемого липазой процесса гидролиза 5’-бутирилового эфира ФТЦ во времени с использованием ферментов Amano РS-800® (-незакрашенный квадрат-) и PLE (-незакрашенный кружок с точкой-).
На фиг.5 представлен график воздействия концентрации (мкмоль) рацемически или энантиомерно обогащенного ФТЦ (полученного способом из примера 4) в сравнении с процентом ингибирования человеческих мононуклеарных клеток периферической крови, инфицированных ВИЧ-1 (-темный кружок-, (±)-ФТЦ), (-незакрашенный кружок-, (-)-ФТЦ), (-закрашенный квадрат-, (+)-ФТЦ).
На фиг.6 представлен график влияния концентрации (мкмоль) рацемически или энантиомерно обогащенного ФТЦ (полученного способом из примера 3) на процент ингибирования человеческих мононуклеарных клеток периферической крови (МКПK), инфицированных ВИЧ-1 (-темный кружок-, (±)-ФТЦ), (-незакрашенный кружок-, (-)-ФТЦ), (-темный квадрат-, (+)-ФТЦ).
На фиг.7 изображен график поглощения меченного тритием (±)-ФТЦ в человеческих мононуклеарных клетках периферической крови (среднее от двух измерений) во времени (часы) и в пикомоль/106 клетки.
Фиг.8 является графиком выхода меченных радиоактивным изотопом (±)-ФТЦ из человеческих мононуклеарных клеток периферической крови (МКПК), измеряемого по часам в пикомоль/106 клетки.
Фиг.9 отражает присутствие [3H]-(±)-ФТЦ и его фосфорилированных производных в человеческих НерG2 клетках (среднее двух измерений), инкубируемых в среде, содержащей 10 мкмолей раствора [3H]-(±)-ФТЦ, измеренного в пикомоль/106 клетки во времени.
На фиг.10 представлен график выхода [3H]-(±)-ФТЦ и его фосфорилированных производных в человеческие клетки НерG2, во времени в пикомоль/106 клетки после того, как клетки были в течение 24 часов сенсибилизированы 10 мкмоль раствора [3H]-(±)-ФТЦ (700 DPM/пикомоль), а также оценка концентрации соединения через 24 часа после удаления.
Фиг.11 иллюстрирует снижение комбинированной концентрации [3H]-(±)ФТЦ и его фосфорилированных производных в человеческих клетках НерG2 после инкубации с 10 мкмолями раствора [3H]-(±)-ФТЦ (700 DРМ/пикомоль) в течение 24 часов, выраженное в пикомоль/106 клетки в зависимости от времени.
Фиг.12 представляет собой график воздействия энантиомеров ФТЦ на образование колонии клеток, предшественников гранулоцитмакрофагов, выраженного в проценте выживания в зависимости от концентрации в мкмолях. (-)-ФТЦ, -незакрашенный кружок; (+)-ФТЦ, -темный кружок; AST - темный квадрат.
Подробное описание изобретения
Используемый здесь термин "энантиомерно обогащенный нуклеозид" относится к составу нуклеозида, который включает по крайней мере 95% одного энантиомера этого нуклеозида.
Используемый здесь термин "ФТЦ" относится к 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолану (рацемической форме или энантиомерам), а также к 2’-дезокси-5-фтор-3’-тиацидину.
Используемый здесь термин "(±)-ФТЦ" относится к (±)-β-D, L-2-оксиметил-5-(5’-фторцитозин-1-ил)-1,3-оксатиолану.
Используемый здесь термин "(-)-ФТЦ" относится к (-)-β-L-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолану.
Используемый здесь термин "(+)-ФТЦ" относится к (+)-β-D-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолану.
Используемые здесь термины "ФТЦ-МФ, ФТЦ-ДФ и ФТЦ-ТФ" относятся к монофосфату, дифосфату и трифосфату ФТЦ соответственно.
Используемый здесь термин "ВСН-189" относится к 2-оксиметил-5-цитозин-1-ил-1,3-оксатиолану.
Используемый здесь термин "предпочтительный ферментативный катализ" относится к катализу ферментом, который предпочтительно способствует одному из субстратов по сравнению с другим.
Используемый здесь термин "отщепляемая группа" обозначает функциональную группу, которая образует начальную карбонизацию, когда отделяется от молекулы, к которой была присоединена.
Описываемое изобретение предлагает способ получения и состав для лечения инфекций, вызванных ВИД и ВГВ, а также другими вирусами, репликация которых происходит аналогичным путем, у людей или других животных-носителей, который включает введение эффективного количества (±)-β-D,L, (-)-β-L или (+)-β-D энантиомера 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана, фармацевтически ("физиологически") приемлемого производного, включая 5’ или N4 алкилированное или ацилированное производное или его фармацевтически ("физиологически") приемлемой соли в фармацевтически приемлемом носителе. Как будет показано ниже, предлагаемые настоящим изобретением соединения сами обладают антиретровирусной активностью, например, анти-ВИЧ-1, анти-ВМ-2 и антивирус иммунодефицита обезьян (анти-ВИО) активностью или в ходе обмена веществ преобразуются в соединение, которое демонстрирует антиретровирусную активность.
ФТЦ и его фармацевтически приемлемые производные или фармацевтически приемлемые составы, содержащие эти соединения, применяются для профилактики и лечения вызванных ВИЧ инфекций и других связанных состояний, например, связанный со СПИДом комплекс (ССК), персистирующая генерализованная лимфеденопатия (ПГЛ), связанные со СПИДом неврологические состояния, состояния, вызванные наличием антител к ВИЧ, и ВИЧ-положительные состояния, саркома Капоши, тромбоцитопения пурпура и условно-патогенные инфекции. Помимо этого эти соединения или составы могут быть использованы профилактически для предотвращения или торможения развития клинической формы заболевания у лиц, имеющих антитела к ВИЧ или антиген ВИЧ или которые были подвергнуты воздействию ВИЧ.
ФТЦ и его фармацевтически приемлемые производные, или соли, или фармацевтически приемлемые составы, содержащие эти соединения, используются также для профилактики и лечения инфекций ВГВ и других связанных состояний, например, состояний, вызванных наличием антител к ВГВ, и ВГВ-положительных состояний, хронического воспаления печени, вызванного ВГВ, цирроза, острого гепатита, молниеносного гепатита, хронического персистируещего гепатита и слабости. Эти соединения или составы могут быть также использованы профилактически с целью предотвращения или торможения развития клинической формы заболевания у лиц, имеющих антитела к ВГВ и антиген ВГВ или которые подверглись воздействию ВГВ.
Резюмируя сказанное, настоящее изобретение включает следующие признаки:
(a) (±)-β-D, L-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан и его фармацевтически приемлемые производные и соли;
(b) (-)-β-L-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан и его фармацевтически приемлемые производные и соли;
(c) (+)-β-D-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан и его фармацевтически приемлемые производные и соли;
(d) (±)-β-D, L-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан, его (-) и (+) энантиомеры и фармацевтически приемлемые производные и соли для использования в медицинских целях, например, для лечения или профилактики ВИЧ или ВГВ инфекций;
(е) использование (±)-β-D, L-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана, его (-) и (+) энантиомеров и фармацевтически приемлемых производных и солей в производстве лекарственного препарата для лечения инфекций ВИЧ или ВГВ;
(f) фармацевтические составы, включающие (±)-β-D, L-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан, его (-) или (+) энантиомер, или фармацевтически приемлемое производное, или соль с фармацевтически приемлемым носителем;
(g) способ получения 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана:
(I) необязательно защищенный 5-фторцитозин вступает в реакцию с 1,3-оксатиоланом формулы А

где R1a является водородом или гидроксильной защитной группой, включая ацильную группу, а L является отщепляемой группой; и необязательно удаляют гидроксилзащитную группу;
(II) соединение формулы В

(где R1a имеет указанное ранее значение, а R1b является аминоэащитной группой) вступает в реакцию с фторирующим реагентом, служащим для введения атома фтора в положение 5 кольца цитозина; или
(III) соединение формулы С

(где R1a имеет указанное ранее значение) вступает в реакцию с реагентом, служащим для преобразования оксогруппы в положении 4 кольца урацила в аминогруппу; и любую остающуюся защитную группу отщепляют для получения целевого продукта;
(f) способ получения (-) или (+) энантиомера 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана, который включает этап, на котором соединение или его производное (например, 5'-сложннй эфир) в виде смеси (-) и (+) энантиомеров помещается в такие условия или вступает в реакцию с таким реагентом, что происходит разделение энантиомеров и, при необходимости, конечное производное преобразуют в исходное соединение.
Что касается способа е) (I), то защитная гидроксильная группа включает защитные группы, подробно описанные ниже, в том числе, апил- (например, ацетил-), арилацил- (например бензоил- или замещенный бензоил-), тритил- или монометокситритил-, бензил- или замещенный бензил-, трехзамещенный силил-, включая триалкилсилил- (например, диметил-Т-бутилсидил-) или дифенилметилсилил-. Соединение 5-фторцитозин необязательно может быть защищено трехзамещенным силилом. Защитные группы могут быть отщеплены обычным образом. Отщепляемая группа L является одной из типичных групп, известных в химии нуклеозидов, например, галоген- (хлор- или бром-), алкокси- (метокси- или этокси-) или ацил- (ацетил- или бензоил-).
Реакция по способу е) (I) может протекать в органическом растворителе (например, 1,2-дихлоратан или ацетонитрил) в присутствии кислоты Льюиса, предпочтительно тетрахлорида олова, или триметилсилилтрифлата.
Соединение формулы А (где L является ацильной группой, например, ацетильной группой) может быть получено путем введения в реакцию соединения формулы Д

(где R1a имеет указанное ранее значение) с восстанавливающим реагентом, например, с алюмогидридом лития, после чего его подвергают воздействию обычного реагента для получения целевого промежуточного соединения, например, ангидрида карбоновой кислоты (ангидрида уксусной кислоты) с целью ацилирования, воздействию хлорирующих или бромирующих реагентов с целью галоидирования, или воздействию алкилирующих реагентов.
Соединение формулы Д можно получить введением в реакцию соединения формулы Е

с HSCH2CO2H при повышенной температуре.
Соединение формулы Е можно получить озонолизом простого или сложного эфира алила формулы CH2=CH-CH2-OR или ди- простого или сложного эфира 2-бутен-1,3-диола формулы ROCH2-CH=CH-CH2OR, в котором R является защитной группой, например, алкил-, силил- или ацил-.
Что касается способа (II), то 5-фтор можно ввести с помощью известных методов (М.Дж. Робина и др. Химия нуклеиновой кислоты, ч.2, Л.В.Тоунсенд и Р.С.Типсон, издатели, Дж.Вилей и Сыновья, Нью-Йорк, 895-900 (19/8) и список литературы там же; Р. Душинский. Химия нуклеиновой кислоты, ч.1, Л.В. Тоунсенд и Р.С. Типсон, издатели, Дж. Вилей и Сыновья, Нью-Йорк, 43-46 (1978) и список литературы там же). Фторирующим реагентом может быть, например, триметилгипофлуорит во фтортрихлорметана.
Что касается способа е) (III), то соединение формулы С можно подвергнуть воздействию 1,2,4-триазола вместе с 4-хлор-фенилдифлофосфатом для получения соответствующего соединения 4-(1,2,4-триазоилил), которое затем преобразуют в целевое соединение 4-амино (цитидин) реакцией, например, с метанолом.
Исходные вещества формул В и С можно получить реакцией, например, соответствующего (необязательно защищенного) основания с соединением формулы А аналогично описанному выше способу е) (I). 5-фторурация и 5-фторцитозин можно закупить у Олдрич Кемикел Ко., Мнлуоки, Висконсин 53233, США.
Раствор (±)-энантиомеров можно выполнить согласно подробному описанию, приведенному ниже в разделе III.
ФТЦ можно преобразовать в фармацевтически приемлемый сложный эфир путем реакции с соответствующим реагентом, например с галогенангидридом или ангидридом кислоты. ФТЦ или его фармацевтически приемлемое производное может быть преобразовано в фармацевтически приемлемую соль обычным способом, например, воздействием соответствующего основания. Сложный эфир или соль ФТЦ могут быть преобразованы в ФТЦ путем, например, гидролиза.
I. Активное соединение, его физиологически приемлемые производные и соли
Описываемым антивирусным активным соединением является 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан (см. фиг.1) в рацемической форме или в виде изолированного энантиомера.
Активное соединение может применяться в виде любого производного, которое после введения реципиенту может дать непосродственно или косвенно исходное ФТЦ соединение или которое само демонстрирует активность. Неограничивающими примерами являются фармацевтически приемлемые соли (или же упоминаемые в тексте как "физиологически приемлемые соли") и 5’ и N4 ацилированные или алкилированные производные активного соединения (или же упоминаемые как "физиологически или фармацевтически активные производные"). В одном варианте осуществления настоящего изобретения ацил является сложным эфиром карбоновой кислоты, в котором некарбонильный фрагмент сложного эфира выбран из прямого, разветвленного или циклического алкила, алкоксиалкила, включая метоксиметил, аралкила, в том числе фенила, необязательно замещенного галогеном, С1-С4 алкила или С1-С4 алкокси сложных эфиров сульфокислот, например, алкил- или аралкил-сульфонил, в том числе метансульфонил, моно, ди или трифосфат сложного эфира, тритил или монометокситритил, замещенный бензил, триал-килсилил (например, диметилбутилсилил) или дифенилметилсилил. Оптимально, чтобы арилом в сложных эфирах был фенил. Алкил может быть прямым, разветвленным или циклическим, оптимально от С1 до С18.
Конкретные примеры фармацевтически приемлемых производных ФТЦ включают, но не ограничиваются ими:
где R1 и R2 независимо выбраны из группы, состоящей из алкила и ацила, в том числе конкретно включая, но не ограничиваясь ими, метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, сек-бутил, t-бутил, изопентил, амил, t-пентил, 3-метилбутирил, гидроген сукцината, 3-хлорбензоат, циклопентил, циклогексил, бензоил, ацетил, пивалоил, мезилат, пропионил, бутирил, валерил, капроновую, каприловую, каприновую, лауриновую, миристиновую, пальмитиновую, стеариновую, олеиновую кислоты, аминокислоты, в том числе, но не ограничиваясь, аланинил, валинил, лейцинил, изолейцинил, пролинил, фенилаланинил, триптофанил, метионинил, глицинил, серинил, треонинил, цистеинил, тирозинил, аспарагинил, глутаминил, аспартоил, глутаоил, лизинил, аргининил и гистидинил и где один из R1 и R2 может быть водородом.
ФТЦ или его производные могут быть предложены в форме фармацевтически приемлемых солей. Используемый здесь термин "фармацевтически приемлемые соли или комплексы" относится к солям или комплексам ФТЦ, которые поддерживают целевую биологическую активность исходного соединения и демонстрируют минимальное, если есть таковое, нежелательное токсикологическое воздействие. Неограничивающими примерами таких солей являются а) соли присоединения кислот, образованные неорганическими кислотами (например, соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, азотная кислота и т.п.), и соли, образованные органическими кислотами, как, например, уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памоевая кислота, альгиновая кислота, полиглютаминовая кислота, нафталинсульфоновые кислоты, нафталиндисульфоновые кислоты и полигалактуроновая кислота;
b) соли присоединения основания, образованные катионами многовалентных металлов, например, цинк, кальций, висмут, барий, магний, алюминий, медь, кобальт, никель, кадмий, натрий, калий и т.п., или органическим катионом, образованным из N,N-дибензилэтилендиамина, аммония или этилендиамина; или с) сочетание а) и b), например, соль дубильной кислоты и цинка и т.п.
Модификации активного соединения, конкретно в положениях N4 и 5’-O, могут влиять на биологическую доступность и скорость метаболизма активных разновидностей, обеспечивая таким образом контроль над их поступлением. Далее модификации могут влиять на антивирусную активность соединения, увеличивая в некоторых случаях его активность по сравнению с исходным соединением. Это можно легко оценить путем получения производного и исследования его антивирусной активности в соответствии с описанными здесь способами или другим способом, известным специалистам в данной области.
II. Получение активных соединений
Рацемическая смесь ФТЦ может быть получена в соответствии с методом, подробно описанным в международной публикации № WО 91/11186 согласно Договору о патентной кооперации, опубликованной 8 августа 1931 г. и зарегистрированной Эмори Юниверсити. В целом, способ включает озонирование простого или сложного эфира алила формулы СН2=СН-СН2-OR или простого или сложного диэфира 2-бутен-1,3-диола формулы ROCH2-CH=CH-CH2OR, где R является защитной группой, как, например, алкил-, силил- или ацил-, для образования гликолевого альдегида формулы OHC-CH2-OR; добавление тиогликолевой кислоты дает лактон формулы 2-(R-окси)-метил-5-оксо-1,3-оксатиолан; восстановление лактона до различных соединений, включающих отщепляемую группу в положении 5 кольца оксатиолана; присоединение этих соединений к силированному 5-фторцитозину в присутствии SnСl4 дает β-изомер ФТЦ; и необязательное отщепление защитных групп.
Пример 1. Получение (±)-β-D, L-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана.
Способ получения рацемической смеси ФТЦ приведен на фиг.2 и подробно описан ниже.
Защита 2-бутен-1,4-диола
В сухой трехгорлой колбе емкостью 2 л, в инертной атмосфере растворили 100 г (93,5 мл = 1,135 моль = 1,00 экв.) 2-бутен-1,4-диола и 15 г (приблизительно 0,1 экв.) ДМАП (4-диметиламинопиридин) в 800 мл обезвоженного пиридина и затем их перемешивали во время охлаждения до 0°С. Затем для предотвращения перегревания медленно добавили хлорид бутирила (260 мл = 2,2 экв.) и перемешивали в течение одного часа. Реакцию охладили небольшим количеством ледяной воды. Жидкость декантировали из соли и выпарили in vitro. Оставшуюся соль растворили в воде и водный раствор дважды экстрагировали простым этиловым эфиром. Другие связанные слои один раз промыли насыщенным CuSO4 и дважды - насыщенным NаНСО3, содержащим Norit®, и затем подвергли вакуум-фильтрации через Celite® томпон .
Концентрированную реакционную смесь растворили в простом эфире и промыли, используя процедуру, описанную выше для соляного раствора. Связанные органические слои выпарили с помощью роторного испарителя и затем поместили в вакуум. Эта реакция обычно очень близка к количественной. В случае необходимости ее масштаб может быть легко увеличен. Продукт, 1,4-дибутирил-2-бутен-1,4-диол, является от бесцветной до желтоватой прозрачной жидкостью.
Озонолиз защищенного диола
1,4-дибутирил-2-бутен-1,4-диол (1,365 моль) растворили в 4 л обезвоженного СН2Сl2 в сухой трехгорлой колбе, емкостью 5 л, оснащенной насадкой с осушителем и открытой трубкой для введения газа. Оптимально трубка не является фриттированной трубкой для барботирования газа, которая будет засоряться при воздействии концентрированного раствора. Раствор перемешивали и охладили до 78°C в то время, как через него барботировали инертный газ. Когда раствор достаточно охладился, газовый клапан закрыли, а колбу и перемешивающее устройство поместили в озоновый генератор. Кислород барботировали через перемешанный раствор, выдерживаемый из ледяной бани в течение по крайней мере 20 минут. Криогенное охлаждение является наилучшим для поддержания низкой температуры во время этой длительной реакции. Затем ввели озон при давлении 8-8,5 фунт/дюйм2. По завершении поток озоне остановили, а кислород барботировали через раствор в течение получаса до того, как добавили 3 эквивалента Me2S. Колбу удалили из охлаждающей бани и перенесли под колпак, где перемешивали в течение 2 дней, чтобы осуществить полное восстановление. Раствор выпарили и поместили в вакуум на несколько часов.
Обычно выход этой реакции составляет приблизительно 95% защищенного альдегида (2-бутирилоксиацетальдегид), от бесцветной до желтой прозрачной жидкости.
Циклизация альдегида меркаптоуксусной, кислотой
Альдегид (0,1 экв.) растворили в толуоле и получили 0,80-0,85 М раствор в колбе, оснащенной насадкой Дина-Старка. Добавили тиогликолевую кислоту и нагрели смесь до обратного холодильника. Воду удалили азеотропной отгонкой. Реакция закончилась через 3 часа и смесь остудили до комнатной температуры. Органический раствор дважды промыли равными объемами насыщенного водного раствора NаНСО3 и один раз водой, высушили над MgSO4 и Norit® профильтровали в вакууме через Celite® перед тем, как выпарили in vacuo. Первый промывочный раствор NаНСО3 экстрагировали один раз простым эфиром; простой эфир один раз промыли водой, высушили над MgSO4 и Norit®, профильтровали в вакууме через Celite® и выпарили вместе другим органическим материалом из раствора толуола. Объединенный материал поместили на всю ночь в вакууме.
Обычно выход реакции составляет 90% 2-(бутирилокси)-метил--5-оксо-1,3-оксатиолана.
Восстановление лактона и получение ацетата
2-бутирилокси-метил-5-оксо-1,3-оксатиолан (1,00 экв.) растворили в обезвоженном тетрагидрофуране (ТГФ) и получили 0,23 М раствор в сухой трехгорлой колбе, оснащенной мешалкой с приводом и находящейся в инертной атмосфере. Раствор перемешали и охладили до 0°С, затем через канюлю добавили 1,1 экв. 1,0 М раствора Li (t-BuO)3AlH в ТГФ. Восстановление закончилось приблизительно через три часа согласно данным тонкослойной хроматографии с использованием системы растворителей простой эфир/гексан в соотношении 2:1 и анисового альдегида для проявления хроматограммы.
Затем добавили приблизительно 10 эквивалентов только что дистиллированного Ас2О и перемешивали в течение 2 дней для получения ацетилированного продукта. Реакционную смесь быстро охладили добавлением насыщенного NaHCO3, затем перемешивали в течение ночи. Затем раствор выпарили и перемешивали в течение ночи еще с некоторым количеством раствора NaHCO3. Смесь экстрагировали добавлением простого эфира, который дважды (осторожно) промыли насыщенным NаНСО3 и один раз водой, высушили над MgSO4 и Norit®, у профильтровали в вакууме через Celite® и выпарили. Продукт является темно-желтой прозрачной жидкостью. Газовая хроматография (начальная температура - 80°С; начальное время = 5 мин; увеличение скорости - 10°С/мин; конечная температура = 240°C) обычно показывает чистоту приблизительно 70%.
Силилирование 5-фторцитозина
5-фторцитозин (1,05 эквивалентов на основе количества ацетилированного лактола, полученного на предыдущем этапе с использованием индикатора чистоты газовой хроматографии) силилировали с обратным холодильником в по крайней мере 10 эквивалентах гексаметилдисилазана, содержащего каталитическое количество чистого сульфата аммония (от 0,05 до 0,10 экв.) в течение двух часов после того, как раствор стал прозрачным. Затем колбу герметично закрыли, а раствор удалили с помощью вакуумного насоса с вспомогательным отделителем. Продукт, белое твердое вещество, оставили на ночь в вакууме, пока он не будет готов для использования в следующей реакции сопряжения.
Реакция сопряжения силированного 5-фторцитозина с ацетилированием лактолом
К силированному 5-фторцитозину (33,86 г, 0,124 моль) в обезвоженном дихлорметане (350 мл) добавили раствор SnCl4 (135,6 мл, 1 М раствора в СН2Сl2) в атмосфере азота. Раствор перемешивали в течение 15 минут при комнатной температуре. Этот раствор через трубочку вливали в раствор ацетата лактола (38 г, 0,113 моль) в дихлорметане (400 мл) в течение 30 минут в атмосфере азота.
Реакционный раствор перемешивали в течение 2 часов, в этот момент окончание реакции определили тонкослойной хроматографией. Затем реакционный раствор разбавили дихлорметаном (500 мл) и быстро охладили раствором гидроксида аммония. Раствор гидроксида аммония (100 мл) добавляли медленно, поддерживая температуру реакции ниже 30°С, в результате чего образовался белый осадок.
Смесь перемешивали в течение еще 30 минут и затем пропустили через колонку с силикагелевой пробкой (диаметр 7 дюймов, высота 5 дюймов). Она вымывалась последовательно дихлорметаном (2 л), этилацетатом (2 л) и этилацетатом:этанолом (9:1) (4 л). Элюенты этилацетата и этилацетата:этанола содержали целевой продукт. Эти растворы соединили и выпарили при пониженном давлении. Оставшееся клейкое твердое вещество промыли обезвоженным простым эфиром (200 мл) и получили белое твердое сухое вещество (25,35 г; 71%), ФТЦ-5’-бутират.
ФТЦ-5'-бутират (8,74 г; 0,26 моль) растворили в 250 мл метилового спирта. Затем добавили метилат натрия (2,85 г; 0,52 г) при комнатной температуре. Реакционную смесь перемешивали в течение 1 часа, в это время завершение реакции подтвердила тонкослойная хроматография. Для быстрого охлаждения добавили раствор NН4Сl (10 мл) и затем при пониженном давлении удалили растворитель. Остаток абсорбировали на селикагеле (5 г) и пропустили через маленькую колонку, используя в качестве элюента смесь этилацетат:этанол (9:1). Содержащие продукт фракции соединили, выпарили и получили клейкое твердое вещество, которое промыли обезвоженным простым эфиром и получили белое сухое вещество ФТЦ (6,00 г, 88%).
(1Н ЯМР: (ДМСО-d6) 8,18 (1Н, d, Н6, J=8,4 Гц), 7,81 и 7,57 (2Н, широкий, NH2), 6,12 (1Н, dd, Н1’, J=5,7 и 4,2 Гц), 5,40 (1Н, t, ОН, J=5,7 Гц), 5,17 (1H, t, 1H4’, J=3-6 Гц), 3,74 (2Н, m, 2Н5’), 3,41 (1H, dd, 1H2’, J=5,7 и 11,7 Гц), 3,11 (1H, dd, 1H2’, J=4,2 и 11,7 Гц); 13C ЯМР: (ДМСО-d6) 157,85 (d, J=13,4 Гц), 153,28, 136,12 (d, J=241 Гц), 126,01 (d, J=32,6 Гц), 86,90, 86,84, 62,48, 37,07; точка плавления 195-196°C.
III. Расщепление энантиомеров нуклеозида
Предлагается способ расщепления рацемических смесей энантиомеров нуклеозида, в том числе, но не ограничиваясь ими, (+) и (-) энантиомеров ФТЦ. Способ может быть также использован для расщепления рацемических смесей углеводородов или углеводородоподобных фрагментов, как, например, производные 1,3-оксатиолан и 1,3-диоксолан. Способ включает использование фермента, который предпочтительно катализирует вступление в реакцию одного энантиомера в рацемической смеси. Прореагировавший энантиомер отделяют от непрореагировавшего энантиомера на основании образовавшегося различия в физической структуре. Пользуясь настоящим описанием, специалист в данной области техники сможет выбрать фермент, обладающий селективностью по отношению к целевому энантиомеру нуклеозида (или являющийся селективным для нецелевого фермента, в качестве способа его удаления), путем выбора одного из обсуждаемых ниже ферментов или с помощью систематической оценки других известных ферментов. Пользусь этим изобретением, специалист в данной области техники будет также знать, как модифицировать субстрат, чтобы обязательно получить целевое разделение. Оптическое обогащение выделенного сложного эфира можно определить с помощью хирального ЯМР смещения реагентов, поляриметрии или хиральной ЖХВД (жидкостная хроматография высокого давления).
Следующие примеры далее показывают использование ферментов для разделения рацемических смесей энантиомеров. В сочетании с описываемым здесь способом разделения рацемических смесей могут быть использованы другие ранее известные способы. Все эти модификации не выходят за пределы объема настоящего изобретения.
Разделение, основанное на гидролизе сложных эфиров С5’-нуклеозидов
В одном варианте осуществления настоящего изобретения С5’-гидроксильная группа смеси рацематов нуклеозида вступает в реакцию с ацильным соединением для образования С5’-сложных эфиров, в которых нуклеозид находится в "карбиноловом" конце сложного эфира. Затем рацемическую смесь С5’-сложных эфиров нуклеозида подвергают воздействию фермента, который предпочтительно разделяет, или гидролизует, один из энантиомеров в данный отрезок времени.
Преимущество этого способа состоит в том, что он может быть использован для разделения широкого круга нуклеозидов, в том числе пиримидиновых и пуриновых нуклеозидов, необязательно замещенных в углеводном фрагменте или основании. Способ может быть также использован для разделения производных нуклеозидов, которые содержат дополнительные гетероатомы в углеводном фрагменте, например, ФТЦ и ВСН-189. Широкая применимость данного способа основывается частично на том, что хотя карбиноловый фрагмент сложного эфира играет роль в способности фермента различать энантиомеры, главным центром распознования этих ферментов является участок карбоновой кислоты эфира. Далее результаты исследования одной системы фермент/субстрат могут быть с успехом экстраполированы на другую, кажущуюся отличной систему, при условии, что фрагменты карбоновой кислоты обоих субстратов одинаковы или по существу одинаковы.
Другое преимущество этого способа заключается в его региоселективности. Ферменты, которые гидролизуют сложные эфиры, обычно не катализуют посторонние реакции в других участках молекулы. Например, фермент липаза катализует гидролиз эфира 2-оксиметил-5-оксо-1,3-оксатиолана, не гидролизуя внутренний лактон. Это очень отличается от "химических" подходов к гидролизу эфиров.
Еще одно преимущество этого способа состоит в том, что отделение негидролизованного энантиомера и гидролизованного энантиомера от реакционной смеси является достаточно простым. Негидролизованный энантиомер является более липофильным, чем гидролизованный, и может быть эффективно выделен путем простого экстрагирования добавлением одного из множества неполярных органических растворителей или смеси растворителей, в том числе гексана и гексана/простого эфира. Затем менее липофильный, более полярный гидролизованный энантиомер может быть экстрагирован добавлением более полярного органического растворителя, например, этилацетата, или путем лиофилизации, вслед за которой осуществляется экстрагирование добавлением этанола или метанола. Во время гидролиза следует избегать спирт, поскольку при определенных условиях он может изменить естественные свойства ферментов.
Ферменты и субстраты
При правильном подборе фермента и субстрата могут быть определены условия изоляции для любого энантиомера нуклеозида. Целевой энантиомер можно изолировать путем воздействия на рацемическую смесь ферментом, который гидролизует его (вслед за этим экстрагирование полярного гидролизата полярным растворителем), или воздействием ферментом, который гидролизует нецелевой энантиомер (вслед за этим удаление нецелевого энантиомера неполярным растворителем).
Ферменты, катализирующие гидролиз сложных эфиров, включают эстеразы, например, эстераза печени свиньи, липазы, в том числе свиная панктеатическая липаза и Amano PS-800 липаза, субстиллин и α-химотрипсин.
На фиг.3 изображена схема последовательности операций специфичности щелочной фосфатазы и экзонуклеазы яда змей для (+) и (-) энантиомеров ФТЦ. Как показано на чертеже, щелочная фосфатаза гидролизует трифосфат обоих энантиомеров ФТЦ, и, следовательно, она не эффективна как средство разделения.
Фосфодиэстераза I предпочтительно гидролизует (+)-изомер ФТЦ до его моноэфира, который затем может быть подвергнут воздействию 5’-нуклеотидазы и дать (+)-ФТЦ.
Наиболее эффективную ацильную группу для эстерификации С5’-положения нуклеозида можно определить без ненужного экспериментирования путем оценки количества гомологов, используя выбранную систему ферментов. Например, при эстерификации 1,3-оксатиолен нуклеозидов масляной кислотой процессы расщепления эстеразой печени свиньи и Amano PS-800 осуществляются с высокой степенью селективности энантиомеров (избыток энантиомера 94-100%) и противоположной селективности. Эстераза печени свиньи предпочтительно гидролизует (+)-энантиомер ФТЦ, a Amano PS-800® предпочтительно гидролизует (-)-энантиомер ФТЦ. Приведенный в таблице 1 выраженный в процентах избыток энантиомера представляет количество очищенного сложного эфира масляной кислоты в обработанной ферментом смеси (т.е. сложный эфир масляной кислоты (-)-ФТЦ в случае применения эстеразы печени свиньи и сложный эфир масляной кислоты (+)-ФТЦ в случае применения Amano РS-800®).
Неограничивающие примеры ацильных групп, которые могут оцениваться с точки зрения использования с определенными энантиометрическими смесями нуклеозидов и определенными ферментами включают алкилы карбоновых кислот и замещенные алкилы карбоновых кислот, в том числе уксусной кислоты, пропионовой кислоты, масляной кислоты и пентановой кислоты. С некоторыми ферментами предпочтительным может быть использование ацильного соединения, сильно оттягивающего электрон, для облегчения гидролиза путем ослабления эфирной связи. Оттягивающие электрон ацильные группы включают α-галоэфиры, как, например, 2-хлорпропионовой кислоты, 2-хлормасляной кислоты и 2-хлорпентановой кислоты, α-галоэфиры являются прекрасными субстратами для липаз.
Условия разделения
Ферментный гидролиз обычно проводится в присутствии достаточного для осуществления роли катализатора количества фермента в водном буферном растворе, который имеет рН, близкий по значению к оптимальному рН для рассматриваемого фермента. По мере развития реакции рН падает вследствие высвобождения карбоновой кислоты. Для поддержания значения рН, близким к оптимальному для данного фермента следует добавить водное основание. За развитием реакции легко следить, наблюдая изменения рН и количество основания, необходимого для поддержания рН. Гидрофобный сложный эфир (негидролизованный энантиомер) и более полярный спирт (гидролизованный энантиомер) могут быть последовательно и избирательно экстрагированы из раствора с помощью рационально выбранных органических растворителей. Или же материал можно расщепить, пропустив его через колонку, которая содержит фермент, неподвижно нанесенный на твердый носитель.
Проводимый в гетерогенных условиях ферментный гидролиз может иметь недостатки вследствие плохой воспроизводимости. Поэтому предпочтительно, чтобы гидролиз выполнялся в гомогенных условиях. Спиртовые растворители не являются предпочтительными, поскольку они могут лишить ферменты их природных свойств. Гомогенность может быть достигнута путем использования неионогенных поверхностно-активных веществ, как, например, Triton Х-100. Однако добавление поверхностно-активных веществ не только способствует расщеплению исходного материала, они также повышают водорастворимость продукта. Поэтому хотя ферментная реакция более эффективно протекает при добавлении неионогенного поверхностно-активного вещества, чем в гетерогенных условиях, изоляцию как выделенного исходного вещества, так и продукта можно усложнить. Продукт можно изолировать, используя соответствующие хроматографические и химические (например, селективное солеобразование) методов. Можно использовать диацилированные нуклеозиды, но часто они бывают вполне липофильными и плохо растворяются в используемой среде.
Пример 2: Энантиоселективный катализируемый липазой гидролиз сложных эфиров ФТЦ
Несколько производных 5’-O-ацил ФТЦ получили путем селективного O-ацилирования соли N-хлоргидрата (см. табл.1 и фиг.4) (±)-ФТЦ. Изучали эффективность гидролиза производных липазами. Как видно из табл.1, эстераза печени свиньи (ЭПС) демонстрирует высокий уровень селективности при гидролизе сложного эфира (+)-энантиомера ФТЦ, по большей части оставляя бутират (-)-ФТЦ в анализируемой ЖХВД смеси. В противоположность этому, PS-800 предпочтительно гидролизует сложный эфир (-)-энантиомера ФТЦ, оставляя по большей части бутират (+)-ФТЦ в анализируемой ЖХВД смеси. Оказалось, что скорость гидролиза также зависит от природы ацильной группы; при ацетильном производном скорость была значительно меньше, чем при бутирильном. Было обнаружено, что скорость гидролиза ФТЦ сложного эфира пропионовой кислоты даже выше, чем наблюдаемая скорость для производного бутирата. Процент выделения и процент избытка энантиомеров определяли с помощью ЖХВД. Хотя при использовании ЭПС энантиоселективность является прекрасной (обычно избыток энантиомера 97% или выше), может быть выполнено дополнительное обогащение путем последовательных реакций ферментного гидролиза, в ходе которых энантиомерически обогащенный бутират, полученный катализированным ЭПС гидролизом, подвергается ферментному гидролизу РS-800.

Пример 3. Методика получения (+)- и (-)-ФТЦ с помощью энантиоселективного катализуемого липазой гидролиза ФТЦ бутирата
5’-O-бутират (±)-ФТЦ (0,47 ммоль, 149 мг) растворили в 16 мл раствора, состоящего из рН 8 буфер:СН3СN в соотношении 4:1. Прозрачный раствор перемешали и подвергли воздействию 26 мг эстеразы печени свиньи (ЭПС-А). За развитием реакции наблюдали с помощью ЖХВД (фиг.4). Через 20 часов (52% преобразования) реакционную смесь экстрагировали добавлением 2×80 мл CHCl3 и 80 мл этилацетата. Органические слои экстрактов соединили, высушили над обезвоженным MgSO4, профильтровали и выпарили с помощью ротора. Полученный остаток элюировали на 2×1000 мкм пластинки тонкослойной хроматографии (pTLC, используя в качестве элюента этилацетат (двойное элюирование) и получили, после изоляции, 53 мг (36% на основе исходного материала) ФТЦ бутирата, который, как определили с помощью ЖХВД, имел 98% энантиомерический избыток (э.и.). Энантиомерически обогащенный бутират подвергли воздействию 1,6 мл метанола, а затем 0,38 ммоль (20 мг) метилата натрия. Полученную смесь перемешивали при комнатной температуре, а за развитием реакции наблюдали с помощью ЖХВД. Реакция завершилась в пределах 30 минут. Растворитель удалили с помощью роторного выпаривания, что дало неочищенное белое сухое вещество (76 мг), которое элюировали на 1000 мкм пластинки тонкослойной хроматографии (pTLC), используя смесь этилацетат:этанол в соотношении 5:1. (-)-ФТЦ изолировали как сухое вещество (33 мг; выход 82%). Анализ ЖХВД ФТЦ как его производного 5’-O-ацетат показал 97% э.и.;
[α] (20,D)-120,5° (с=0,88; чистый этанол).
Добавляя НССl3 к реакционной смеси во время окончания этапа смешивания, можно избежать образования эмульсии (что также лишает фермент его природных свойств), удаляя растворители в вакууме и затем экстрагируя добавлением HCCl3.
Аналогично 1,2 ммоль (375 мг) 5’-O-бутират (±)-ФТЦ растворили в 40 мл смеси 4:1, рН 8, буфер-СН3СN. Прозрачный раствор перемешали и подвергли воздействию 58 мг эстеразы печени свиньи (ЭПС-А). Развитие реакции наблюдали с помощью ЖХВД.
Через 90 минут (38% преобразования) к реакционной смеси добавили 150 мл СНСl3. Слои разделили и водный слой лиофилизировали для удаления растворителя. Полученный в результате лиофилизации белый остаток экстрагировали добавлением 3×10 мл чистого этанола. Экстракты профильтровали, соединили, выпарили в вакууме и получили 179 мг неочищенного масла. Неочищенный материал элюировали на 45×30 мм колонке на силикагеле с использованием 3×75 мл этилацетата, затем смеси 5:1 этилацетат/этанол. (+)-ФТЦ изолировали как сухое белое вещество (109 мг; 37% на основе исходного бутирата). ЖХВД анализ (+)-ФТЦ как его производного 5’-O-ацетат показал 97,4% э.и.;
[a] 0 (20,D)-113,4° (с=2,53; чистый этанол).
Аналогичную реакцию выполнили, используя 0,12 ммоль (37 мг) 5’-O-бутирата ФТЦ и 7 мг РS-800 в 4,0 мл смеси 4:1, рН 8, буфер:СН3СN. Реакция протекала значительно медленнее, чем с ЭПС-А, и потребовалось 74 часа для 59% преобразования. Выделенный бутират (11,4 мг; 31% начального количества) в результате анализа ЗХВД продемонстрировал 94% э.и.
Разделение энантиомеров нуклеозида с помощью цитидин-дезоксицитидиндезаминазы
Как один из вариантов изобретения цитидин-дезоксицитидиндезаминаза используется для разделения рацемических смесей 2-оксиметил-5-(цитозин-1-ил)-1,3-оксатиолана и его производных, в том числе 2-гидроксиметил-5-(фтор-цитозин-1-ил)-1,3-оксатиолана. Фермент катализует дезаминирование части цитозина до урацила. Обнаружили, что один из энантиомеров 1,3-оксатиолан нуклеозидов является предпочтительным субстратом для цитидин-дезоксицидитиндезаминазы. Энантиомер, который не преобразовался в производное урацила (и поэтому является все еще основным), экстрагируется из раствора добавлением кислого раствора. Действовать нужно осторожно, чтобы избежать образования очень кислых растворов (рН ниже 3,0), что может расщепить оксатиолановое кольцо.
Цитидин-дезоксицитидиндезаминазу можно выделить из печени крысы или из печени человека или получить путем экспрессии из рекомбинантных последовательностей в прокариотной системе, как, например, Е.соli.
Способ расщепления энантиомеров цитидинового нуклеозида с использованием цитидин-дезоксицитидиндезаминазы можно использовать как самостоятельный метод или в сочетании с другими способами расщепления, в том числе расщепление с помощью ферментного гидролиза сложных эфиров 5’-O-нуклеозид, описанного выше.
Сочетание ферментного расщепления и классических способов расщепления
Описанный выше способ расщепления рацемических смесей энантиомеров нуклеозида можно сочетать с другими классическими способами энантиомерного расщепления для увеличения оптической чистоты конечного продукта.
Классические способы расщепления включают различные физические и химические методы. Часто наиболее простым и самым эффективным способом является перекристаллизация, основанная на том, что рацематы часто бывают более растворимыми, чем соответствующие отдельные энантиомеры. Перекристаллизацию можно выполнять на любой стадии, в том числе на стадии ацилированных соединений или конечного энантимерного продукта. В случае успеха этот простой способ является лучшим.
Если перекристаллизация не дает материал приемлемой оптической чистоты, то можно рассмотреть другие способы. Если нуклеозид является основным (например, цитидин), то можно использовать хиральные кислоты, образующие диастереомерные смеси, которые могут обладать существенно отличающимися характеристиками растворимости. Неограничивающие примеры хиральных кислот включают яблочную кислоту, миндальную кислоту, дибензоилвинную кислоту, 3-бромкамфора-8-сульфоновую кислоту, 10-камфорсульфоновую кислоту и дипаратолуолвинную кислоту. Аналогично ацилирование свободной гидроксильной группы производным хиральной кислоты также дает образование диастереомерических смесей, физические характеристики которых могут значительно отличаться и разрешить отделение.
Маленькие количества энантиомерно обогащенных нуклеозидов могут быть получены или очищены пропусканием рацемической смеси через колонку ЖХВД, сконструированную для хирального разделения, в том числе колонки со связанным циклодекстрином, продаваемые Рейнин Корпорейшн.
Пример 4: Разделение рацемических смесей нуклеозидов с помощью ЖХВД
Расщепление С4’-энантиомеров (±)-ФТЦ осуществляли с применением хиральной колонки со связанным циклодекстрином (Cyclobond АС-I), полученной от Рейнин Корпорейшн (Уобурн, Массачусетс). Соблюдались следующие условия: изократный раствор 0,5% метанола в воде, скорость потока 1 мл/мин, анализ в ультрафиолетовой области света при 262 им. Метанол для ЖХВД получили от Дж.Т. Вейкер (Филлипсбург, Нью-Джерси). Рацемические смеси ввели в хроматограф, а фракции собрали. Содержащие каждый из энантиомеров фракции поместили в резервуар, заморозили и затем лиофилизировали. Соединения охарактеризовали с помощью ультрафиолетовой спектроскопии и по времени удержания при ЖХВД. Обычно (-)-энантиомеры имеют меньшее время удержания, чем (+)-энантиомеры (см. Журнал жидкостной хроматографии 7:353-376, 1984). Концентрацию соединений определили с помощью ультрафиолетовой спектроскопии, используя исходный раствор с известной концентрацией (15 мкмоль), приготовленный в воде для биологической оценки. Время удержания отделенных энантиомеров приводится в табл.2.

Пример 5: Альтернативные способы разделения энантиомеров ФТЦ с использованием хиральной колонки
При использовании колонки Cyclobond I-Ас (5 мкм, 25×4,6 мм, Рейнин Корпорейшн, Уобурн, Массачусетс, каталог № А Т-41049) со скоростью потока 0,6 мл/мин 0,5% изократного раствора метанола (Фишер Сайентифик, Инк. ЖХВД марка, каталог №А-452-4) в воде и при анализе в ультрафиолетовой области света при 262 нм энантиомеры ФТЦ продемонстрировали время удержания 12,68 мин (-)-ФТЦ и 13,20 мин (+)-ФТЦ.
При использовании колонки Chiralpak AS (10 мкм, 25×4,6 мм, Дж.Т. Вейкер Инк., Филлипсбург, Нью-Джерси, каталог №7406-00, серийный номер 09-29-10320) со скоростью потока 0,8 мл/мин изопропилового спирта (ЖХВД марка, Фишер Сайентифик, Инк., каталог А-451-4) и при анализе в ультрафиолетовой области света при 262 нм ФТЦ энантиомеры продемонстрировали время удержания 5,9 мин (-)-ФТЦ и 9,8 мин (+)-ФТЦ.
IV. Способность 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана ("ФТЦ") ингибировать репликацию ВИЧ
Обычно в качестве предварительного шага желательно проверить ряд рацемических смесей нуклеозидов для определения, какой из них гарантирует в дальнейшем расщепление на энантиомерно обогащенные компоненты и далее оценку антивирусной активности. Способность нуклеозидов ингибировать ВИЧ можно оценить с помощью различных экспериментальных способов. Используемый здесь способ, подробно описанный ниже, оценивает ингибирование вирусной репликации в стимулированных фитогемагглютинином (ФГА) мононуклеарных периферических клетках крови человека (МКПК), инфицированных ВИЧ-1 (штамм ВВЛ, вирус, вызывающий лимфоденопатию). Количество продуцированного вируса определяют, измеряя закодированный вирусом фермент обратная транскриптаза. Количество продуцированного фермента пропорционально количеству продуцированного вируса. В табл. 3 приведены значения ЕС50 (концентрация нуклеозида, ингибирующая репликацию вируса на 50% в МКПК, оцениваемый фактор ошибки 10%) и IC50 (концентрация нуклеозида, которая ингибирует 50% роста митоген-стимулированных неинфицированных человеческих МКПК) некоторых (±)-1,3-оксатиолан и нуклеозидов.
Пример 6: Анти-ВИЧ активность (±)-1,3-оксатиолан нуклеозидов
А. Трехдневные фитогемагглютинин-стимулированные МКПК (106 клетки/мл) здоровых серонегативных на гепатит В и ВИЧ-1 доноров инфицировали ВИЧ-1 (штамм BBЛ) при концентрации, в 100 раз превышающей 50% инфекционную дозу культуры ткани (ИДКТ 50) на 1 л, и культивировали в присутствии и отсутствии различных концентраций антивирусных соединений.
В. Приблизительно через 1 час после инфицирования среду с изучаемым соединением (в 2 раза превышающим конечную концентрацию в среде) или без него добавили в колбы (5 мл; конечный объем 10 мл). АЗТ использовали в качестве позитивного стандарта.
С. Клетки подвергли воздействию вируса (около 2×105 распадов в минуту/мл, как показало испытание обратной транскриптазы) и затем поместили в CO2 инкубатор. ВИЧ-1 (штамм ВВЛ) получили из Центра по контролю заболеваемости, Атланта, Джорджия. Методы культивирования МКПК, сбора вируса и определения активности обратной транскриптазы описаны Mc. Dougal et al. (J.Immun.Meth. 76, 171-163, 1985) и Spira. et al. (J.Clin.Meth. 25, 97-99, 1987), за исключением того, что фунгизон не влючили в среду (см.Schinazi et al., Antimicrob. Agents Chemother. 32, 1784-1787 (1368)); Id., 34:1061-1067 (1990).
D. На 6 день клетки и надосадочную жидкость перенесли в 15 мл пробирку и центрифугировали в течение 10 минут при ускорении 900 g. Удалили 5 мл надосадочной жидкости, а вирус сконцентрировали центрифугированием со скоростью 40 000 оборотов в минуту в течение 30 минут /Beckman 70,1 Ti votor/. Солюбилизированный вирусный осадок в пробирке после центрифугирования обработали с целью определения уровней обратной транскриптазы. Результаты выражены в числе распадов в минуту/мл взятой на пробу надосадочной жидкости. Вирус из меньших объемов надосадочной жидкости (1 мл) также можно концентрировать центрифугированием до солюбилизации и определения уровней обратной транскриптазы.
Среднюю эффективную (ЕС50) концентрацию определили с помощью метода определения среднего эффекта Animicrob. Agents Chemother 30, 491-498 (1966). Кратко, процент ингибирования вируса, определенный из измерений обратной транскриптазы, нанесен на график в сравнении с микромолярной концентрацией соединения. EC50 является концентрацией соединения, при которой происходит 50% ингибирование роста вируса.
Е. Стимулированные мтогеном неинфицированные человеческие МКПК (3,8×105 клетки/мл) культивировали в присутствии и отсутствии лекарства в условиях, подобных описанным выше для антивирусного испытания. Через 6 дней клетки пересчитали с использованием гемоцитометра и метода исключения Tripan blue, описанного Schipanzi et al., Antimicrobial Agents and Chemotherapy 22(3), 433 (1982). IC50 является концентрацией соединения, которая ингибирует 50% роста нормальных клеток.
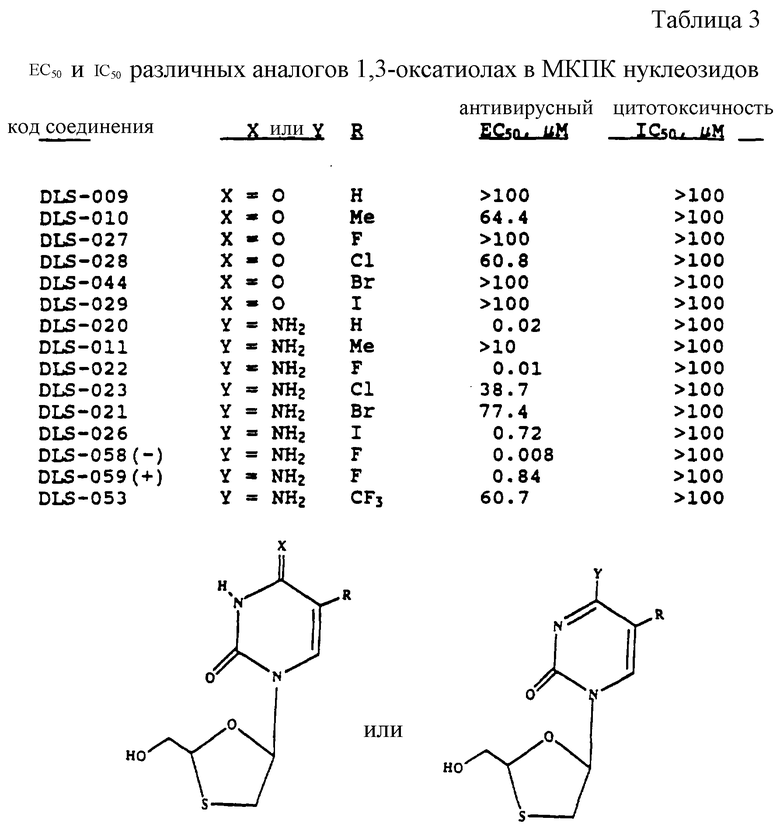
Согласно данным табл.3 замещенные цитозиновые 1,3-оксатиолан нуклеозиды обычно более активны, чем соответствующие урациловые нуклеозиды. Ошибка в измерениях ЕС50 и IC50 оценивается ±10%.
Одно из соединений, (±)-ФТЦ (упоминаемое в тексте как "DLC"-022, соединение 8), не только демонстрирует исключительную активность (приблизительно 10 нм в МКПК), но также достаточно низкую токсичность (>100 мкмоль в МКПК, а также в Vero и CЕМ клетках).
Пример 7: Антивирусная активность энантиомеров ФТЦ, расщепленных с помощью ЖХВД
Энантиомеры ФТЦ изолировали способом, описанным в примере 4, а антивирусную активность оценили методом из примера 6. Результаты приведены в табл.4 и проиллюстрированы на фиг.5.

Из табл.4 видно, что в данном эксперименте (-)-энантиомер ФТЦ оказывается приблизительно на один порядок более сильнодействующим, чем (+)-ФТЦ энантиомер, и обладает приблизительно такой же анти-ВИЧ активностью, как рацемическая смесь. Ни энантиомеры, ни рацемическая смесь не являются токсичными до 100 мкмоль в человеческих МКПК согласно измерениям, выполненным по методу исключения Tripan blue.
Пример 8: Антивирусная активность энантиомеров ФТЦ, расщепленных способом, описанным в примере 3
Энантиомеры (±)-ФТЦ расщепили также с использованием способа из примера 3, а антивирусную активность оценили методом из примера 6. Результаты представлены на фиг.6. Из фиг.6 видно, что EC50 рацемической смеси ФТЦ составила 0,017 мкмоль, ЕС50 (-)-ФТЦ была 0,0077 мкмоль и ЕС50 раствора (+)-ФТЦ была 0,84 мкмоль.
Пример 9: Поглощение (±)-ФТЦ человеческими МКПК
Меченный радиоактивным изотопом ФТЦ использовали для изучения внутриклеточных профилей исходного препарата и метаболитов, обнаруженных внутри клетки. Все исследования дублировались. Человеческие мононуклеарные клетки периферической крови (МКПК) суспендировали в среде указателя числа оборотов в минуту 1640 (RPMI), содержащей 10% фетальной телячьей сыворотки и антибиотики (2×10 клетки/мл, 10 мл/момент времени), и инкубировали с добавлением 10 мкмоль раствора ФТЦ (удельная активность около 700, количество распадов в минуту/пикомоль). Клетки подвергли воздействию препарата в течение 2, 6, 12 и 24 часов.
В эти моменты времени среду удаляли и клетки дважды промывали холодным сбалансированным солевым раствором Хенке. Экстрагирование выполнили добавлением 0,2 мл 60% холодного раствора метанол/вода и оставили на ночь при температуре -70°С. На следующее утро суспензии центрифугировали и экстрагирование провели еще 2 раза по 0,5 часа при температуре -70°С. Все надосадочные жидкости (0,6 мл) лиофилизировали до сухости. Остатки ресуспендировали в 250 мкл воды и аликвотные пробы от 50 до 100 мкмоль проанализировали с помощью ЖХВД. Количественный анализ исходного препарата и метаболических производных провели с помощью ЖХВД. Вследствие потенциальной кислотной неустойчивости некоторых соединений для отделения метаболитов использовали буферную систему, близкую к физиологическому рН.
На фиг.7 представлен график присутствия (поглощения) меченного тритием (±)-ФТЦ в человеческих МКПК (среднее двух измерений) во времени (часы) и в пикомоль/106 клетках. Изучение поглощения показывает, что меченный радиоактивным изотопом ФТЦ быстро поглощается человеческими лимфоцитами, что продуцирует очень большие количества производного 5’-трифосфата ФТЦ.
Пример 10: Антиретровирусная активность ФТЦ в различных клеточных линиях
Антиретровирусную активность ФТЦ оценивали в ряде клеточных линий, используя аналогичные, но не идентичные процедуры, приведенные в примере 6. Клеточные линии получили или от людей-доноров или из Национального института здравоохранения (НИЗ). Справочная программа по использованию реагентов в исследовании СПИДа, Роквилль, Мэриленд, или из Американской коллекции типовых культур, или из Красного Креста. СЕМ клетки с недостаточностью тимидинкиназы получили путем последовательного переноса СЕМ клеток в присутствии 5-бромо-2’-деоксиуридина. Результаты приведены в табл.5.

Пример 11: Выход (±)-ФТЦ из человеческих МКПК
Меченный радиоактивным изотопом ФТЦ использовали для изучения внутриклеточных профилей исходного препарата и метаболитов, обнаруженных внутри клетки после инкубации в среде с препаратом в течение 24 часов и затем после удаления препарата. В ходе этого исследования определяли время, необходимое для снижения внутриклеточных уровней трифосфата. Все эксперименты дублировались. Иеинфицированные клетки (2×106 мл) суспенгировали в соответствующей среде с добавлением сыворотки (10 мл в момент времени) и выдерживали при температуре 37°С в 5% СО2 инкубаторе. Концентрация меченного радиактивным изотопом ФТЦ была 10 мкмоль. После сенсибилизации клеток меченым изотопом в течение 24 часов клетки тщательно промыли и затем повторно наполнили свежей средой без антивирусных препаратов (0 часов). Через 0, 2, 4, 6, 12, 24 и 48 часов (время второй инкубации) клетки удалили и немедлкнно экстрагировали добавлением 60% холодного раствора метанол/вода. Экстракт получили центрифугированием и удалением клеточного осадка после центрифугирования. Экстракты лиофилизировали и затем оставили при температуре -70°С. Материал ресуспендировали в 250 микролитрах буферного раствора ЖВД и сразу же за этим подвергли анализу. Количественный анализ исходного препарата и метаболических производных провели с использованием ЖХВД, применяя "Микромеритикс" или Хьюлетт-Паккард систему ЖХВД, модель 1090 с анионообменной колонкой Партизил 10 САКС (Уэтмен, Инк.), при скорости потока 1 мл/мин, давлении 1 килофунт/кв.дюйм, с ультрафиолетовой спектроскопией при 262 им. Подвижная фаза состояла из деионизированной воды (А), 2 ммоль раствора NаН2РО4/16 ммоль раствора NаОАс (рН 6,6) (В), 15 ммоль раствора NаН2РО4/120,2 ммоль раствора NаОАс (рН 6,6) (С) и 100 ммоль раствора NаН2РО4/800 ммоль раствора NаОАс (рH 6,6) (Д).
Метод сепарации: изократный в течение 5 минут для А, вслед за этим 15-минутным линейным градиентом до 100% В, затем 20-минутным линейным градиентом до 100% С, затем 10-минутным линейным градиентом до 100% Д, затем 30 минут изократным способом для 100% Д.

На фиг.8 представлен график выхода меченного радиоактивным изотопом (±)-ФТЦ из человеческих МКПК, изменение концентрации (пикомоль/106 клетки) в зависимости от времени (часы) после удаления препарата. На чертеже показано, что ФТЦ-трифосфат имеет внутриклеточный период полувыведения приблизительно 12 часов и его можно легко обнаружить внутриклеточно при концентрациях 1-5 мкмоль через 48 часов после удаления внутриклеточного препарата, что значительно превышает ЕС50 соединения. Далее сродство (КI) для трифосфата (±)-ФТЦ при использовании ОТ ВИЧ составляет 0,2 мкмоль, что ниже уровня концентрации через 48 часов.
Пример 12: Анти-ВИЧ активность фармацевтически приемлемых производных (±)-ФТЦ
а. Ряд фармацевтически приемлемых производных (±)-ФТЦ, полученный путем деривации положений 5’ и N4, оценили на ВИЧ-активность в МКПК с использованием методики, аналогичной описанной в примере 6. Получили следующие результаты. Сложный эфир 5’-O-бутирата (±)-ФТЦ продемонстрировал ЕС50 0,0017. Производное N4-ацетил (±)-ФТЦ демонстрирует EC50 0,0028. 5’-O-бутират, N4-сложный эфир (±)-ФТЦ демонстрирует EC50=0,0058.
б. Анти-ВИЧ активность 5"-O-бутират сложного эфира (±)-ФТЦ в системе МТ4 (EC50) была 0,04 мкмоль. В том же самом тесте неацилированная (±)-ФТЦ демонстрировала IC50 при 0,52 мкмоль. IC50 AЗT в этой системе была 0,09 мкмоль.
V. Способность ФТЦ ингибировать репликацию ВГВ
Пример 13: Оценка активности (+) и (-)-энантиомеров ФТЦ в 2.2.15 культурах клеток
Способность энантиомеров ФТЦ ингибировать рост вируса в 2.2.15 культурах клеток (НерG2 клетки, трансформированные вирионом гепатита) подробно описана ниже.
Краткое изложение и описание анализа на антивирусное действие в этой системе культуры клеток, а также анализ ДНК ВГВ содержится в Korba and Milman, 1991, Antiviral. Res. 15:217. Антивирусные оценки выполнили на двух отдельных пассажах клеток. Все гнезда, во всех чашках засеяли плотностью и в одно и то же время.
Параметры анализа
Вследствие врожденных изменений в уровнях внутриклеточной и внеклеточной ДНК ВГВ только депрессии в 3,5 раза (для ДНК вириона ВГВ) или в 3,0 раза превышающие (для промежуточных форм репликации ДНК ВГВ) средние уровни этих форм ДНК ВГВ в необработанных клетках считаются статистически значимыми [р<0,05]. Уровни интегрированной ДНК ВГВ в каждом препарате клеточной ДНК (который имеет в этих экспериментах постоянную клеточную основу) использовали для расчета уровней внутриклеточных форм ДНК ВГВ, обеспечивая таким образом сравнение равных количеств клеточной ДНК при сравнении разных образцов.
Типичные значения для внеклеточной ДНК вирионов ВГВ в необработанных клетках имели размах от 50 до 150 пг/мл культуральной среды (среднее значение приблизительно 76 пг/мл). Уровни промежуточных форм репликации внутриклеточной ДНК ВГВ в необработанных клетках имели размах от 50 до 100 пг/мкг ДНК клетки (среднее значение приблизительно 74 пг/мкг ДНК клетки). Обычно депрессии уровней внутриклеточной ДНК ВГВ вследствие обработки антивирусным соединениями менее явно выражены и происходят медленнее, чем депрессии уровней ДНК вириона ВГВ (Korba and Milman, 1991, Antiviral. Res. 15:217).
Способ осуществления анализа гибридизации для этих экспериментов установил в результате эквивалентность приблизительно 1,0 пг внутриклеточной ДНК ВГВ 2-3 копиям генома на клетку и эквивалентность 1 пг/мл внутриклеточной ДНК ВГВ - 3×105 вирусных частиц/мл.
Анализ токсичности
Анализ токсичности проводили для того, чтобы оценить, является ли любое наблюдаемое антивирусное действие следствием общего воздействия на жизнеспособность клетки. Использованный здесь способ заключается в измерении поглощения нейтрального красного красителя, стандартная и широко используемая проба на жизнеспособность клеток в различных вирус - носитель системах, в том числе вирус простого герпеса и ВИЧ. Для анализа токсичности использовали плоские имеющие дно тканевые культуральные планшеты с 96 ячейками. Клетки для анализа токсичности вырастили и обработали изучаемыми соединениями согласно режиму, описанному ниже для проведения антивирусной оценки. Каждое соединение исследовали при 4 концентрациях, каждая из которых в трех экземплярах культуры (ячейки "А", "В" и "С"). Поглощение нейтрального красного красителя использовали для определения относительного уровня токсичности. Для количественного анализа использовали спектральную поглощательную способность интернализированного красителя при 510 нм (Аsin). Значения представлены как процентное содержание средних Аsin величин в 9 отдельных культурах необработанных клеток, находящихся на том же самом 96-ячеечном планшете, что и испытываемые соединения. Поглощение красителя в 9 контрольных культурах на планшете 5 имело размах от 91,6 до 110,4%, а на планшете 6 - от 96,6 до 109%. Результаты представлены в табл.6.

Оценка токсичности
Из табл. 6 видно, что при концентрациях испытываемых соединений, используемых для антивирусной оценки, не наблюдалось значительной токсичности (в необработанных клетках наблюдалась депрессия более 50% уровней поглощения красителя). Оба испытываемые соединения, (-)-ФТЦ и (+)-ФТЦ, по видимому, являются токсичными при больших концентрациях, используемых для оценки токсичности (330 мкмоль).
Антивирусные оценки
Контрольные
Во время контрольного заражения уровни ДНК вириона ВИЧ и промежуточных форм внутриклеточной репликации ВГВ [ВГВ РПФ) в необработанных клетках остаются постоянными, в пределах нормальных изменений. ДМСО при концентрации 1% не влияет на уровни репликации ВГВ в 2.2.15 культурах клеток.
Испытываемые соединения
Из табл.7 видно, что и (-)-ФТЦ и (+)-ФТЦ значительно ингибировали репликацию ВГВ на испытываемых уровнях. Из табл.8 видно, что (-)-ФТЦ оказывает по-прежнему значительное ингибирующее воздействие на синтез ДНК вириона ВГВ и внутриклеточной ДНК ВГВ при концентрациях 4, 1 и 0,25 мкмоль.
Пример 14: Поглощение (±)-ФТЦ клетками печени человека; связанная с ВГВ активность ФТЦ
Описанную в примере 9 процедуру повторили с клетками печени человека (HepG2 клетки, полученные из Американской коллекции типовых культур) для определения поглощения и метаболизма ФТЦ в этих клетках. На фиг.9 видно, что (±)-ФТЦ поглощается клетками НерG2 в больших количествах. Эти клетки печени человека в ходе обмена веществ преобразуют большой процент (±)-ФТЦ в трифосфат (±)-ФТЦ.
Эти данные в сочетании с другими приведенными здесь данными показывают, что (±)-ФТЦ, как и его (-) и (+) энантиомеры, фосфорилируются в клетках печени. Эти клетки могут быть преобразованы вирусом гепатита В.


Пример 15: Выход ФТЦ из клеток человека НерG2
На фиг.10 показано изменение во времени выхода [3H]-(±)-ФТЦ и его фосфорилированных производных в человеческие клетки НерG2, выраженное в пикомоль/106 клетки, после сенсибилизации клеток 10 мкмоль раствора [3H]-(±)-ФТЦ (700 распадов в минуту/пикомоль) в течение 24 часов и оценке концентрации соединения через 24 часа после удаления.
Фиг.11 иллюстрирует уменьшение с течением времени связанной концентрации [3H]-(±)-ФТЦ н его фосфорилированных производных в человеческих клетках НерG2 после инкубации с 10 мкмоль раствора [3H] -(±)-ФТЦ (700 распадов в минуту/пикомоль) в течение 24 часов, выраженное в пикомоль/106 клетки.
Из иллюстраций видно, что даже через 48 часов в клетках все еще присутствовало более 1 мкмоль раствора активного вещества (что значительно больше, чем ЕС50 соединения).
V. Токсичность в клетках-предшественниках гранулоцит-макрофагов
Пример 16: Воздействие ФТЦ на образование колонии клеток-предшественников гранулоцит-макрофагов
Фиг.12 представляет собой график воздействия (-) и (+) энантиомеров ФТЦ на образование колонии клеток предшественников гранулоцитов-макрофагов, определенную как процент выживания в зависимости от концентрации в мкмолях ((-)-ФТЦ, незакрашенный кружок; (+)-ФТЦ, черный кружок; АЗТ, черный квадрат). На графике видно, что (-)-энантиомер ФТЦ представляется менее токсичным, т.е. он имеет IC50 выше, чем (+)-энантиомер или АЗТ в этой клеточной линии.
VI. Фармакокинетика ФТЦ
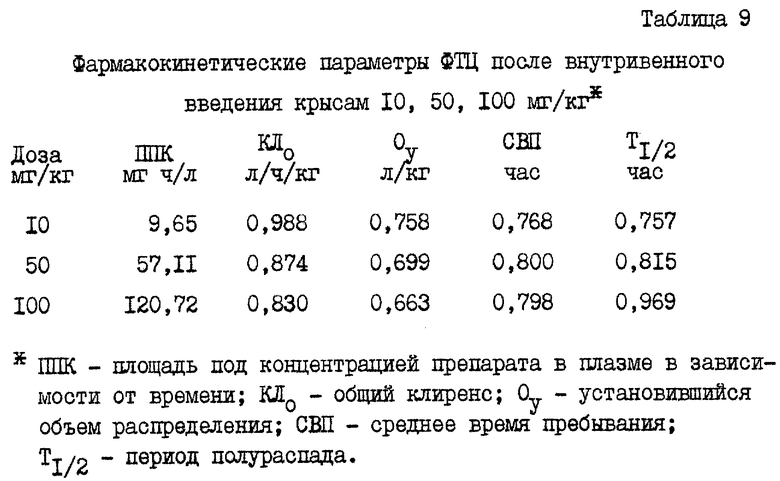
Пример 17: метаболизм ФТЦ при введении его крысам
(±)-ФТЦ ввели крысам внутривенно в дозировках 10, 50 и 100 мг/кг, при этом оценивали площадь под концентрацией препарата в плазме в зависимости от времени (ППК), общий клиренс (KЛО), установившийся объем распределения (Oy), среднее время пребывания (СВП) и период полураспада (Т1/2). Результаты представлены в табл. 9.
Пример 18: Фармакокинетические параметры ФТЦ после внутривенного и орального введения ФТЦ
Не зависящие от модели фармакокинетические параметры (±)-ФТЦ получили путем внутривенного (В.В.) и орального (П.О.) введения 33,3 мг/кг макакам резус. Результаты представлены в табл.10. Следует отметить, что средняя биологическая доступность соединения у макак составила 73% (±6%).


Пример 19: Отношение содержания ФТЦ и его метаболитов в цереброспинальной жидкости и сыворотке крови у макак резус
Способность (±)-ФТЦ преодолевать гематоэнцефалитический барьер оценивали путем введения 33,3 мг/кг активного соединения макакам резус и измерения количества (±)-ФТЦ в цереброспинальной жидкости и сыворотке крови через один час после введения. Результаты приведены в табл.11. Данные показывают, что значительное количество активного соединения проходит через гематоэнцефалический барьер у этих животных.
III. Приготовление Фармацевтических составов
Людей, страдающих от вызванных БИЧ или ВГВ заболеваний, можно лечить введением пациенту эффективного количества (±)-ФТЦ, или его (-) или (+) энантиомера, или его фармацевтически приемлемого производного или соли в присутствии фармацевтически приемлемого носителя или растворителя. Активный материал можно вводить любым подходящим способом, например, орально, парентерально, внутривенно, внутрикожно, подкожно или местно в жидкой или твердой форме.
Активное соединение включают в фармацевтически приемлемый носитель или растворитель в количестве, достаточном для обеспечения больного терапевтически эффективным количеством соединения для ингибирования вирусной репликации in vivo, главным образом репликации ВИЧ и ВГВ, не вызывая серьезных токсических воздействий у проходящего лечение пациента. Под "ингибирующим количеством" имеется в виду количество активного ингредиента, достаточное для оказания давления на ингибирующее действие, определенное, например, с помощью одного из описанных здесь примеров анализа.
Предпочтительная доза (-), (+) или (±)-ФТЦ для всех упомянутых выше состояний будет колебаться от 1 до 50 мг/кг, предпочтительно от 1 до 20 мг/кг веса тела в день, чаще от 0,1 до 100 мг/кг веса реципиента в день. Интервал изменений эффективной дозы фармацевтически приемлемых производных можно рассчитать, исходя из веса исходного нуклеозида, который нужно дать пациенту. Если производное демонстрирует собственную активность, то эффективную дозу можно оценить описанным выше способом, используя вес производного, или другими способами, известными специалистам в данной отрасли.
Соединение удобно вводить в единицах любой подходящей лекарственной формы, в том числе, но не ограничиваясь ею, одной, содержащей от 7 до 3000 мг, предпочтительно от 70 до 1400 мг, активного ингредиента на единицу лекарственной формы. Удобная доза для орального введения составляет 50-1000 мг.
В идеале активный ингредиент следует вводить для достижения максимальных концентраций активного соединения в плазме, составляющих от 0,2 до 70 мкмоль, предпочтительно от 1,0 до 10 мкмоль. Этого можно достичь путем, например, внутривенной инъекции от 0,1 до 5% раствора активного ингредиента, необязательно в физиологическом растворе, или введением большой пилюли с активным ингредиентом.
Концентрация активного соединения в лекарственном составе будет зависеть от скорости поглощения, инактивации и выделения лекарственного препарата, а также от других факторов, известных специалистам в данной отрасли. Следует отметить, что величина дозы будет также меняться в зависимости от тяжести состояния, которое нужно облегчить. Следует понимать далее, что для любого конкретного пациента следует подобрать определенную схему приема лекарственного средства, скорректированную во времени в соответствии с индивидуальной потребностью и профессиональной оценкой лица, назначающего или осуществляющего надзор над введением составов, и что предложенные здесь диапазоны концентраций являются иллюстративными и не ограничивающими объем или применение заявляемого состава. Активный ингредиент можно ввести сразу или разделить на несколько меньших доз для введения через различные промежутки времени.
Предпочтительным способом введения активного соединения является оральный. Составы для орального введения будут обычно включать инертный растворитель или съедобный носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для целей орального терапевтического введения активное соединение можно смешать с наполнителями и использовать в форме таблеток, пастилок или капсул. В состав в качестве его составной части могут быть включены осуществляющие фармацевтически совместимое связывание и/или адъювантные вещества.
Таблетки, пилюли, капсулы, пастилки и тому подобное могут содержать любой из следующих ингредиентов или соединений, обладающих аналогичными основными свойствами: связывающее вещество, как, например, микрокристаллическая целлюлоза, камедь трагаканта или желатин; наполнитель, как, например, крахмал или лактоза, вещество, способствующее распадаемости лекарственной формы, например, альгиновая кислота, Примогель или кукурузный крахмал; смазывающее вещество, например, стеарат магния или Stevotes; скользящее вещество, например, коллоидный кремневый ангидрид; подслащивающее вещество, например, сахароза или сахарин; корригирующее вещество, например, мята перечная, салицилометиловый эфир или цитрусовый корригент. Когда единицей лекарственной формы является капсула, то, помимо веществ вышеперечисленных классов, она может содержать жидкий носитель, например, жирное масло. Помимо этого, единицы лекарственных форм могут содержать различные другие вещества, изменяющие форму лекарственной единицы, например, покрытия из сахара, шеллака или некоторые кишечные агенты.
(±)-ФТЦ, или его (-) или (+)-энантиомер, или фармацевтически приемлемые производные или соли можно применять как компонент элексира, суспензии, сиропа, капсулы, жевательной резинки и тому подобное. Сироп, помимо активных соединений, может содержать в качестве подслащивающего вещества сахарозу, а также некоторые консерванты, красители и красящие вещества, ароматические вещества.
(±)-ФТЦ, его (-) или (+)-энантиомеры или его фармацевтически приемлемые производные или соли можно также смешать с другими активными веществами, которые не ослабят целевое действие, или с веществами, дополнят целевое действие, например, с антибиотками, противогрибковыми, противовоспалительными и другими антивирусными средствами, в том числе с другими содержащими нуклеозиды анти-ВИЧ соединениями.
Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного применения могут включать следующие компоненты: стерильный растворитель, например, вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, например, бензиловый спирт или метилпарабена (methil parablens), антиоксиданты, например, аскорбиновая кислота или бисульфит натрия; хелатные средства, например, этилендиаминтетрауксусная кислота; буферы, например, ацетаты, цитраты или фосфаты, а также вещества для корректировки тонуса, например, хлорид натрия или декстроза. Исходный препарат можно поместить в ампулы, одноразовые шприцы или вмещающие много доз стеклянные или пластмассовые пузырьки.
При внутривенном вводе предпочтительными носителями являются физиологический раствор или фосфатно-буферный раствор (ФБР).
В предпочтительном варианте осуществления настоящего изобретения активные соединения изготовлены с носителями, которые предохранят соединение от быстрого удаления из тела, например, состав с контролируемым выделением, в том числе имплантанты и микрокапсулированные системы доставки. Могут быть использованы биоразлагаемые, биосовместимые полимеры, например, этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры и полимолочная кислота. Способы изготовления таких составов очевидны для специалистов в данной области. Вещества можно также получить коммерческим путем от Альза Корпорейшн и Нова Фармацевтикела, Инк.
Липосомные суспензии (в том числе липосомы, нацеленные на инфицированные клетки с моноклональными антителами к вирусным антигенам) также являются предпочтительным в качестве фармацевтически приемлемых носителей. Они могут быть приготовлены в соответствии со способами, известными специалистам в данной области, например, согласно описанию, содержащемуся в патенте США №4522611 (который включен здесь путем ссылки во всей полноте). Например, липосомные составы могут быть изготовлены путем растворения соответствующего(их) липида(ов) (например, фосфатидилэтаноламин, стеароилфофатидилхолин, араходиолфофатидилхолин и холестерин) в неорганическом растворителе, который затем выпаривают, после чего остается тонкая пленка обезвоженного липида на поверхности контейнера. Затем в контейнер вводят водный раствор активного соединения или его производные монофосфат, дифосфат и/или трифосфат. Затем контейнер крутят вручную чтобы отделить липидный материал от стенок контейнера и распылить липидные совокупности, образуя таким путем липосомную суспензию.
IV. Изготовление фосфатных производных ФТД
Моно-, ди- и трифосфатное производное ФТЦ можно получить описанным ниже способом.
Монофосфат можно изготовить в соответствии с методикой, описанной Imai et al., J.Org.Chem., 34(6), 1547-1550 (июнь 1969). Например, 100 мг ФТЦ и 280 мкл хлорида фосфорида вводят в реакцию при перемешивании в 8 мкл обезвоженного этилацетата при температуре 0°С, которая длится около 4 часов. Реакционную смесь быстро охлаждают льдом. Водную фазу очищают на колонке с активированным древесным углем, затем элюируют 5% гидроокисью аммония в смеси 1:1 этанола и воды. В результате выпаривания элюента получают ФТЦ-5’-монофосфат аммония.
Дифосфат можно получить в соответствии с методикой, описанной Davisson et al., J.Org.Chem., 52(9), 1794-1801 (1987). ФТЦ дифосфат можно получить из соответствующего тозилата, который можно приготовить, например, путем реакции нуклеозида с хлоридом тозила в пиридине при комнатной температуре в течение 24 часов, обработав продукт обычным способом (например, промыванием, высушиванием и кристаллизацией).
Трифосфат можно получить в соответствии с методикой, описанной Hoard et al., J.Org.Chem., 87(8), 1785-1788 (1965). ФТЦ активируют (получением имидазолида в соответствии со способами, известными специалистам в данной области) и подвергают воздействию трибутиламмонийпирофосфата в ДМФ (диметилформамиде). Первоначально в результате реакции получают трифосфат нуклеозида с некоторым количеством непрореагировавшего монофосфата и с некоторым количеством дифосфата. Очищают с помощью анионообменной хроматографии с использованием колонки с диэтиламиноэтанолом (DЕАЕ), после чего выделяют трифосфат, например, как тетранатриевую соль.
Данное изобретение описано на примерах предпочтительных вариантов его осуществления. Специалистам в данной области из приведенного выше подробного описания изобретения очевидны возможные изменения и варианты изобретения. Все эти варианты и изменения изобретения не выходят за пределы его объема, определенного прилагаемой формулой изобретения.


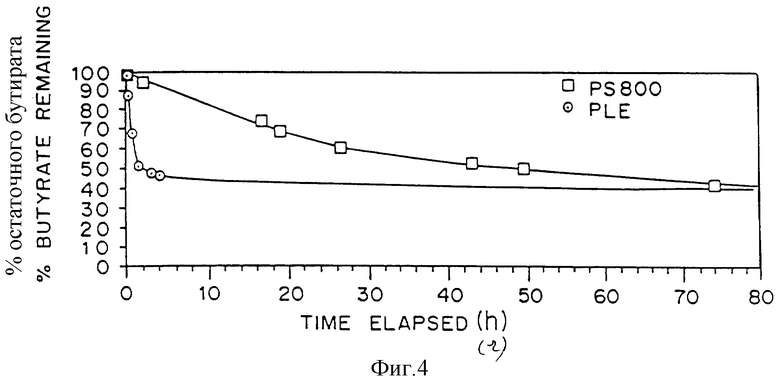





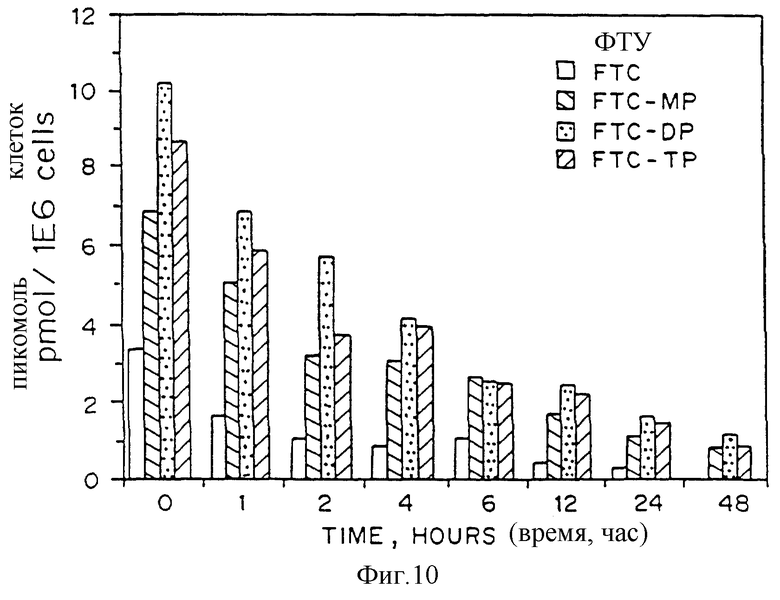


Изобретение относится к способам разделения смеси энантиомеров цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана: посредством пропускания через связанную с циклодекстрином хиральную колонку; селективным гидролизом 5’-O-ацильного производного нуклеозида с помощью ферментов; селективным дезаминированием смеси энантиомеров с помощью цитидиндезаминазы. 4 с. и 8 з.п.ф-лы, 11 табл., 12 ил.

| УЛЬТРАЗВУКОВОЙ СПОСОБ КОНТРОЛЯ состоянияМАТЕРИАЛА | 0 |
|
SU337713A1 |
| WO 8901776 А1, 09.03.1989. | |||
