Настоящее изобретение относится к способу лечения инфекций, вызываемых вирусом гепатита В (обозначаемым также "HBV"), заключающемуся во введении эффективного количества одного или нескольких активных соединений, раскрываемых в настоящей заявке, либо фармацевтически приемлемого производного или пролекарственного предшественника одного из этих активных соединений.
HBV является второй после курения причиной, вызывающей раковые заболевания у человека. Механизм индуцирования рака вирусом гепатита В пока неизвестен, хотя было высказано предположение, что этот вирус может стимулировать развитие опухоли как непосредственно, так и посредством хронического воспаления, цирроза и регенерации клеток, ассоциированной с данной инфекцией.
Инфекции, вызываемые вирусом гепатита В, широко распространены во всем мире и достигают уровня эпидемий. После 2-6 месячного инкубационного периода, в течение которого инфицированный не подозревает о наличии у него HBV-инфекции, эта инфекция может индуцировать явления острого гепатита и поражения печени, вызывая боли в животе, желтуху, и повышение уровня некоторых ферментов в крови. HBV может также вызывать молниеносный гепатит, который быстро прогрессирует и часто переходит в летальную форму заболевания с обширными участками поражения печени.
Пациенты, страдающие острым вирусным гепатитом, как правило, выздоравливают. Однако у некоторых пациентов в течение длительного или неопределенного периода времени сохраняются высокие уровни вирусного антигена в крови, вызывая хронические инфекции. Эти инфекции могут приводить к хроническому персистирующему гепатиту. Заболевания хроническим персистирующим гепатитом наиболее широко распространены в развивающихся странах. Например, к середине 1991 года только в Азии было зарегистрировано около 225 миллионов носителей HBV, а во всем мире их насчитывается почти 300 миллионов. Хронический персистирующий гепатит может вызвать усталость, цирроз печени и злокачественную гепатому, т.е. первичный рак печени.
В западных индустриальных странах, высокий риск HBV - инфицирования обусловлен контактами с HBV - носителями или непосредственно с их образцами крови. Фактически, эпидемиология HBV-инфекций очень схожа с эпидемиологией синдрома приобретенного иммунодефицита, что вполне объясняет факт широкого распространения HBV - инфекции среди пациентов, страдающих СПИДом, или среди ВИЧ-инфицированных пациентов. Однако HBV является более контагиозным вирусом, чем ВИЧ.
Для иммунизации пациентов против HBV была разработана вакцина на основе сыворотки человека. Эта вакцина была продуцирована посредством генной инженерии, и хотя указанная вакцина оказалась весьма эффективной, однако, при ее продуцировании последователи столкнулись с рядом серьезных проблем, связанных с ограниченными поставками человеческой сыворотки от носителей хронической HBV-инфекции, а также с продолжительностью и трудоемкостью процедуры очистки. Кроме того, каждая партия вакцины, полученной из сыворотки различных источников, должна быть проверена на шимпанзе для гарантии ее безопасности. Более того, эта вакцина неэффективна для пациентов, уже инфицированных вирусом.
Хорошие результаты были также получены при ежедневном введении α-интерферона, генетически сконструированного белка. Однако в настоящее время еще не найдено такого фармацевтического средства, которое могло бы эффективно ингибировать репликацию вируса гепатита В у человека.
Поскольку распространение HBV - инфекции во всем мире достигает уровня эпидемий и часто влечет за собой трагические последствия для инфицированных пациентов, то получение нового эффективного фармацевтического средства для лечения гепатита у человека, которое обладало бы низкой токсичностью, остается крайне актуальной проблемой.
Поэтому другой целью настоящего изобретения является разработка способа и композиции для лечения человека или других животных, инфицированных HBV.
В предпочтительном варианте своего осуществления настоящее изобретение относится к способу лечения людей, инфицированных HBV, заключающемуся во введении эффективного количества анантиомерно чистого β-D - диоксоланилпурин-нуклеозида формулы:
где R представляет собой OH, Cl, NH2 или H, либо его фармацевтически приемлемые соли или производное, необязательно в сочетании с фармацевтически приемлемым носителем или разбавителем. Соединение, в котором R является хлором, представляет собой (-)-(2R, 4R)-2-амино-6-хлоро-9-[(2-гидроксиметил)-1,3-диоксолан-4-ил) пурин. Соединение, в котором R является гидрокси, представляет собой (-)-(2R, 4R)-9-[(2-гидроксиметил)-1,3-диоксолан-4-ил) гуанин. Соединение, в котором R является амино, представляет собой (-)-(2R, 4R)-2 амино-9-[(2-гидроксиметил)-1,3-диоксолан-4-ил]аденин.
Соединение, в котором R является водородом, представляет собой (-)-(2R, 4R)-2-амино-9-[(2-гидроксиметил)-1,3-диоксолан-4-ил] пурин. Абсолютная конфигурация указанных соединений не была определена методами кристаллографии. Вышеуказанные обозначения соединений были даны на основании сравнения их структуры с конфигурацией исходного сахара, используемого для получения данного соединения. В другом варианте настоящего изобретения пациенту вводят эффективное количество энантиомера β- L-диоксоланилпуриннуклеозида, или рацемической смеси β- L- и β- D-диоксоланилпуриннуклеозида.
Было установлено, что в случае ACPD ((-)-(2R,4R)-2-амино-6-хлоро-9-[(2-гидроксиметил)-1,3-диоксолан-4-ил)пурин) и DAPD ((-(2R-4R)-2-амино-9-[(2-гидроксиметил)-1,3- диоксолан-4-ил] аденин)) EC50 для ингибирования промежуточных продуктов репликации HBV - ДНК для синтеза вириона HBV составляет около 0,1 мКм. При испытании соединений DAPD, ACPD или DG((-(-(2R, 4R)-9-[(2-гидроксиметил)-1,3-диоксолан-4-ил] гуанин) в концентрации до 300 мкм в клетках 2.2.15 заметной цитотоксичности не наблюдалось. В клоногенных анализах указанные три пуринкуклеозида оказались в значительной степени нетоксичными для миелоидных и эритроидных клеток (IC50 = от 50 до более 100 по сравнению с AZT, для которого IC50 составляет 1 мкм).
Было также обнаружено, что DG, DAPD, ACPD и APD) ((-)-(2R, 4R)-2-амино- 9-[(2-гидроксиметил)-1,3-диоксолан-4-ил] пурин), взятые в концентрации до 1 мм, не ингибируют ферменты, участвующие в биосинтезе пурина и пиримидина, например, также как аденозиндезаминаза, пурин-нуклеозидфосфорилаза, гипоксантингуанозин-фосфорибозилтрансфероза, аденозинкиназа, инозинкиназа, цитидинкиназа, ксантиноксидаза, альдегидоксидаза и ксантиндегидрогеназа.
Раскрытые в настоящей заявке β- -диоксаланпуриннуклеозиды, их фармацевтически приемлемые производные или соли, либо фармацевтически приемлемые композиции, содержащие указанные соединения, могут быть использованы для предупреждения и лечения HBV -инфекций и других связанных с ними состояний, таких как состояния с положительной реакцией на антитела против HBV и с HBV - положительной реакцией, хроническое воспаление печени, вызванное вирусом HBV, цирроз печени, острый гепатит, молниеносный гепатит, хронический персистирующий гепатит и усталость. Указанные соединения или композиции могут быть также использованы в профилактических целях для предупреждения или замедления прогрессирования заболевания у индивидуумов, имеющих положительную реакцию на HBV -антиген или на антитело против HBV, или у индивидуумов, экспонированных вирусом HBV.
В одном из вариантов осуществления настоящего изобретения, в целях проведения эффективного лечения HBV-инфекций, пациентам вводят одно или несколько активных соединений настоящего изобретения поочередно с одним или несколькими другими средствами против HBV. Примерами таких средств против HBV, которые могут быть использованы в альтернирующей терапии, являются, но не ограничиваются ими, энантиомер или рацемическая смесь 2-гидроксиметил 5-(5-фтороцитозин-1-ил)-1-3-оксатиолана (FTC, см. WO 92/147443), его физиологически приемлемое производное или физиологически приемлемая соль, (-)-энантиомер или радемическая смесь 2 гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолана (именуемого также BCH-189 или ЗТС, см. публикацию ЕРА N 0382526 и WO 91/17159 соответственно) его физиологически приемлемое производное, или физиологически приемлемая соль, энантиомер или рацемическая смесь 2'-фтор-5-иод-арабинозилурацила (FIAU) энантиомер или рацемическая смесь 2'-фтор-5-этил-арабинозил-урацила (FEAU), карбовир, или интерферон.
Для лечения пациентов может быть использован любой метод альтернирующей терапии. Примерами таких методов (которые, однако, не ограничивают объема настоящего изобретения) являются следующие схемы введения лекарственных средств, в течение 1-6 недель вводят эффективное количество одного лекарственного средства, а затем в течение 1-6 недель вводят эффективное количество второго лекарственного средства. Схема поочередного введения лекарственных средств может также включать периоды, в течение которых введение лекарственных средств не проводится.
В другом варианте осуществления настоящего изобретения активное соединение, его производное или его соль могут быть введены в сочетании с другим агентом против вируса HBV, включая вышеперечисленные средства. В основном, при чередующейся терапии эффективные дозы каждого из агентов против вируса HBV вводят периодически, а при комплексной терапии, сниженные дозы двух или нескольких противовирусных агентов вводят вместе, в комбинации друг с другом. Дозы вводимых противовирусных агентов зависят от скорости их абсорбции, инактивации и экскреции, а также от других факторов, хорошо известных специалистам. Например, дозы лекарственных средств могут также варьироваться в зависимости от тяжести состояния пациента. Кроме того, следует отметить, что для каждого конкретного пациента должна быть установлена индивидуальная схема приема лекарственного средства, скорректированная в соответствии с потребностями данного пациента, и эта индивидуальная схема должна проводиться под наблюдением лечащего врача.
Краткое описание рисунков.
Фиг. 1 иллюстрирует способ получения энантиомерно чистых β- D-диоксоланилпуриннуклеозидов.
Фиг.2 представляет собой график, иллюстрирующий влияние пуриндиоксоланов и AZT на образование колоний эритроидных предшественников (BFU-E) человека, измеренных как % от контрольных клеток, в зависимости от логарифма концентрации испытуемого лекарственного средства (AZT, 3-азидодезокси-тимидин, APD, (-)-(2R, 4R)-2-амино-9-[(2-гидроксиметил)-1,3- диоксолан-4-ил] пурин, ACPD, (-)-(2R, 4R)-2-амино-6-хлоро-9-[(2- гидроксиметил)-1,3-диоксолан-4-ил]пурин, DG, (-)-(2R, 4R)-9-(2- гидроксиметил)-1,3-диоксолан-4-ил]гуанин, DAPD, (-)(-(2R,4R)- 2-амино-9-[(2-гидроксиметил)-1,3-диоксолан-4-ил]аденин).
Фиг.3 представляет собой график, иллюстрирующий влияние пуриндиоксоланов и AZ на образование колоний клеток предшественников гранулоцитов и макрофагов, измеренных как % от контрольных клеток, в зависимости от логарифма концентрации испытуемого лекарственного средства. Аббревиатуры, используемые на этом рисунке, определены выше для фиг.2.
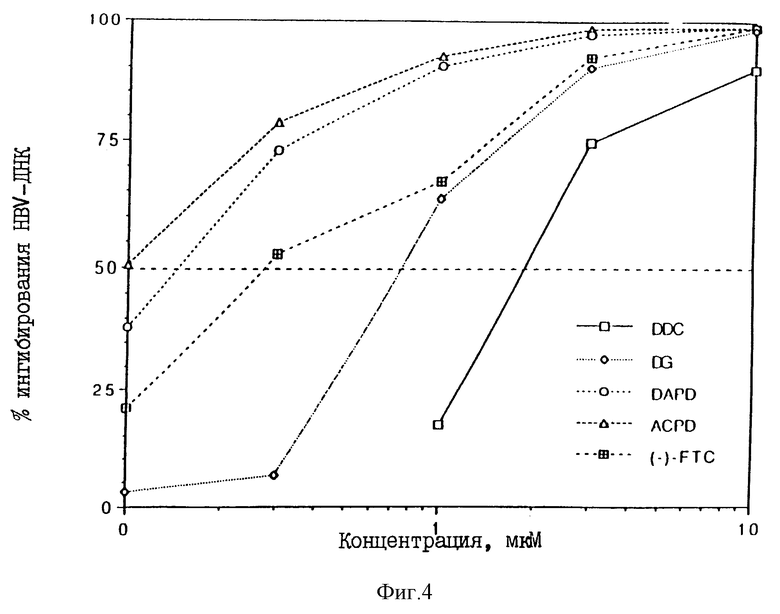
Фиг. 4 представляет собой график, иллюстрирующий процент ингибирования репликации ДНК HBV в клетках 2.2.15 на 9 день в зависимости от различных концентраций испытуемых соединений. Аббревиатуры, используемые на этом рисунке, определены выше для фиг.2 ((-)-PTC означает (-)-2-гидроксиметил-5-(5-фтороцитозин-1-ил)-1,3 оксатиолан). См. таблицу для соответствующих данных.
Фиг.5 представляет собой график, иллюстрирующий поглощение 5 мкм меченного тритием (-)-(2R, 4R)-2-амино-9-(2-гидроксиметил)-1,3-диоксолан-4-ил)адекина (ДАРД) в клетках Hep2G. Экстракт получали через 4 часа после DAPD экспонирования клеток. 1000 число распадов/мин, инъекция 80 мкл.
Фиг. 6 представляет собой график, иллюстрирующий поглощение 5 мкм меченного тритием (-)-(2R,4R)-2-амино-9-[(2-гидроксиметил)-1,3- диоксолан-4-ил] аденина (DAPD) в клетках Нер2C. Экстракт был получен через 12 часов после DAPD-экспонирования клеток. 1000 число распадов/мин, инъекция 145 мкл.
Используемый в настоящем описании термин "энантиомерно чистый" относится к нуклеозидной композиции, содержащей, по крайней мере, около 95%, а предпочтительно 97%, одного энантиомера данного нуклеозида.
Изобретение, раскрываемое в настоящей заявке, относится к способу и композиции для лечения HBV инфекции у человека и других животных, при этом указанный способ заключается во введении эффективного количества одного или нескольких вышеупомянутых соединений либо их физиологически приемлемых производных, включая 5' - и/или N6 алкилированных или адилированных производных, либо их фармацевтически приемлемой соли, необязательно в фармацевтически приемлемом носителе. Соединения настоящего изобретения либо обладают активностью против вируса HBV, либо они являются метаболическими предшественниками указанных соединений и могут превращаться в соединения, обладающие противовирусной активностью.
В другом варианте своего осуществления настоящее изобретение относится к способу лечения пациентов, инфицированных вирусом HBV, заключающемуся в том, что этим пациентам вводят эффективное для лечения HBV - инфекций количество пролекарственного предшественника вышеописанных энантиомерно чистых β- D-диоксоланил- пуриннуклеозидов. Используемый в настоящем описании термин "пролекарственный предшественник" относится к фармацевтически приемлемому производному вышеописанного нуклеозида, то есть указанный лекарственный предшественник может превращаться в конкретный нуклеозид настоящего изобретения после введения in vivo либо этот предшественник может сам по себе обладать противовирусной активностью. В качестве примеров, не ограничивающих объема настоящего изобретения, могут служить фармацевтически приемлемые соли (иногда именуемые "физиологически приемлемые соли"), 5' и N6-ацилированные или алкилированные производные активного соединения (обозначаемые также "физиологически или фармацевтически приемлемые производные"). В одном из вариантов настоящего изобретения ацильная группа представляет собой сложный эфир карбоновой кислоты, где некарбонильную часть сложноэфирной группы выбирают из прямого, разветвленного или циклического C1-C20 алкила, алкоксиалкила, например метоксиметила, аралкила, например бензила, арилоксиалкила, например феноксиметила, арила, например фенила, необязательно замещенного галогеном, C1-C4-алкилом или C1-C4-алкокси, дикарбоновой кислоты, например янтарной кислоты, сложных эфиров сульфоновой кислоты, таких как алкил- или аралкилсульфонил, например метансульфонил, и сложных эфиров моно, ди- и трифосфорной кислоты.
Используемый в настоящем описании термин "алкил" означает, но не ограничивается ими: метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил, трет-бутил, изопентил, амил, трет-пентил, циклопентил и циклогексил.
Используемый в настоящем описании термин "ацил" означает, но не ограничивается ими: ацетил, пропионил, бутирил, пентаноил, 3-метилбутирил, бисукцинат, 3-хлоробензоат, бензоил, ацетил, пивалоил, мезилат, пропионил, валорил и группы капроновой, каприловой, каприновой, лауриновой, мириетиновой, пальмитиновой, стеариновой и олеиновой кислот. Указанные нуклеозиды могут быть также получены в виде 5'-этерифицированных липидов, описанных в нижеследующих работах: Kucera, L.A., M. Lyer, E.Leake, A. Raben, Modest E.J., D.L.W., H.C. Piantadosi. 1990. Novel membrane - interactive ether lipid analogs thah inhibit infections HIV-1 production and induce defective virus formation. AIDS Res Hum Retroviruses. 6-491-501; Piantadosi, C.,T. Marasco C. . J., S.L. morres-Natschke, K.L.Meyer, F. Gumus, J.R. Surles, K.S. Ashaq, L. S. Kucera, N. Lyer, C. A. Wallen, S. Piantadosi и E.J. Modest 1991 - Synthesis and evaluation of novel ether lipid nucleoside conjugates for anti - HIV activity. J.Med. Chim 34:1408 - 1414; Hostetler, K.Y.D.D. Richman, D. A. Carson, L.M. Stuhmiller, G.M.T. van Wijk и H. van den Bosch. 1992. Greatly enhanced inhibition of human immunodeficiency virus type 1 replication in CEM and HT4-6C cells bu 31-deoxythymidine diphosphate dimyristoylglycerol, a lipid prodrug of 31-deoxythymidine. Antimicrob Agents Chemother. 36: 2025-2029; Hostetler, K.Y., L.M. Stuhmiller, H.B. Lenting, H. van den Bosch и D.D. Richman. 1990. Synthesis and antiretroviral activity of phospholipid analogs of agidothymidine and other antiviral nucleosides. J.Biol. Chem. 265:6112-6117.
- Диоксиланилпуриннуклеозид может быть превращен в фармацевтически приемлемый сложный эфир посредством реакции с соответствующим этирифицированным агентом, например галогенангидридом или ангидридом.
Нуклеозид или его фармацевтически приемлемое производное могут быть превращены в их фармацевтически приемлемую соль стандартным способом, например путем обработки соответствующим основанием. Сложный эфир или соль могут быть, в свою очередь, превращены в исходный нуклеозид, например, путем гидролиза.
Активное соединение может быть получено в виде фармацевтически приемлемой соли. Используемый в настоящем описании термин "фармацевтически приемлемые соли или комплексы" относится к солям или комплексам нуклеозидов, сохраняющим желательную биологическую активность исходного соединения и не имеющим либо имеющим минимальное токсическое действие. В качестве примеров таких солей (не ограничивающих, однако, объем настоящего изобретения) могут служить: (а) кислые аддитивные соли, образованные неорганическими кислотами (например, соляной кислотой, бромистводородной кислотой, серной кислотой, фосфорной кислотой, азотной кислотой и т.п.), и соли, образованные органическими кислотами, такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновая кислота и полигалактуроновая кислота, (b) основные аддитивные соли, образованные катионами, такими как натрий, калий, цинк, кальций, висмут, барий, магний, алюминий, медь, кобальт, никель, кадмий и т.п., либо органическими катионами, такими как N,N-дибензил-этилен-диамин, аммоний, или этилендиамин, или (с) комбинации солей (а) и (b), например, таннат цинка и т.п.
Модификации активного соединения, а в частности, в N6 - и 5'-O-положениях могут влиять на биологическую доступность и скорость метаболизма активного соединения и тем самым обеспечивать регулируемую доставку активного ингредиента к нужным тканям или органам.
Активное соединение, или его фармацевтически приемлемое производное, или соль могут быть также смешаны с другими активными материалами, не оказывающими неблагоприятное воздействие на активность указанного соединения, либо с материалами, оказывающими дополнительно желательное действие, например, такими как антибиотики, противогрибковые средства, противовоспалительные средства или другие противовирусные средства, например средства против вирусов HBV или ВИЧ.
Энантиомерно чистые β-D -диоксолан-нуклеозиды могут быть получены способом, подробно описанным ниже, а также описанным в PCT /US 91/09124. Этот способ предусматривает сначала получение (2R,4R)- - и (2R,4S)-4-ацетокси-2-(защищенный оксиметил)-диоксолана из 1,6-ангидроманнозы, т.е. сахара, который содержит все необходимые стереоизомеры для получения энантиомерно чистого конечного продукта, включая диастеомер с правильной конфигурацией в 1- положении сахара (которое становится 4'-положением в образуемом затем нуклеозиде).
После этого (2R, 4R) и (2R,4S)-4-ацетокси-2-(защищенный оксиметил)-диоксолан конденсируют с соответствующим гетероциклическим основанием в присутствии SnCl4, другой кислоты Льюиса, или триметилсилилтрифторацетата в органическом растворителе, таком как дихлорэтан, ацетонитрил или метиленкхлорид, в результате чего получают стереохимически чистый диоксоланнуклеозид.
Однако, следует иметь в виду, что при получении энантиомерно чистых диоксоланнуклеозидов необходимо следить за тем, чтобы условия реакции не были сильнокислотными, поскольку сильная кислота может способствовать размыканию диоксоланового кольца. Вышеуказанные реакции должны проводитьея, если это возможно, в основной или нейтральной среде, но, если условия реакции предусматривают использование кислоты, то время реакции должно быть минимизировано.
Рацемические смеси диоксоланпуриннуклеозидов могут быть получены как описано в публикации EPA 0382526, β- L-энантиомер может быть выделен из рацемической смеси известными методами, например методом, предусматривающим использование хиральной ВЭЖХ-колонки.
Способ получения активных соединений проиллюстрирован на фиг.1 и в примере 1. Исходный материал (соединение 1) был получен в соответствии с описанием в PCT/US 91/09124 (в данной заявке соединение 8). 2,6-дизамещенные пуриновые производные были синтезированы путем конденсации ацетата 1 с силилированным 6-хлор- 2-фторпурином, в результате чего получали смесь α/β = 1/1,3 изомеров 2 и 3. Первоначально полученный N7-изомер снова превращали в N9-изомер, размешивая в течение ночи при комнатной температуре. Аналитический образец получали в результате разделения α,β- смеси на отдельные изомеры 2 и 3 с помощью, препаративной TCX с использованием CH2Cl2- ацетона (19:1) в качестве проявляющего растворителя. Однако, для получения конечных продуктов 10-15 смесь изомеров 2 и 3 обрабатывали аммиаком в DME (Robins M.I; Ugnanski, B. Nucleic acid related - compounds. 34. Non aqueous Diasotisation with tert - butyl nitrite. Introduction of fluorine, chlorine - and bromine at C-2 of purine nucleosides - Can. J. Chemistry,
1981, 2608), в результате чего получали смесь из 10-13, которая была разделена на отдельные изомеры 4 (24%) 5 (18,6%), 6 (25,89%) и 7 (16%) и производные гуанина 8 и 2,6-диамино 9 были получены путем обработки изомера 4. 2 меркаптоэтанолом NaOme и аммиаком в этаноле, соответственно. Свободные нуклеозиды 10-15 были получены с хорошим выходом после обработки соответствующих 5'-силилированных нуклеозидов н-Bu4NF
α- изомеры 12 и 13 были получены таким же способом, как и β- изомеры.
Пример 1.
Получение энантиомерно чистых β- D-диоксоланилпуриннуклеозидов
(2R, 4R, ) и (2R,4S)-9-[(2-(трет-бутилдифенилсилил)оксиметил]- 1,3 диоксолан-4-ил]-6-хлор-2-фторпурин (2 и 3)
Смесь 2-фтор-6-хлорпурина (4,05 г, 23,47 мм) и сульфата аммония (каталитическое количество) в гексаметилдисилазане (940 мл) нагревали с обратным холодильником в течение 2 часов. Полученный раствор концентрировали в безводных условиях, в результате чего получали 2 фтор-6-хлорпурин в виде белого твердого вещества. Затем к охлажденному (0oC) и размещенному раствору силилированного 2-фтор-6-хлорпурина (5,69 г, 23,69 мм) и соединению 1 (7,84 г, 19,57 мм) в сухом метиленхлориде (175 мл) добавляли TMS OTF (4,41 мл, 23,44 мм).
Реакционную смесь нагревали до комнатной температуры и размешивали в течение 16 часов, по истечении которых весь первоначально полученный конденсированный N7-продукт превращался в N9-изомер. Затем реакцию гасили насыщенным раствором NaHCO3 (50 мл) и размешивали еще 20 минут при комнатной температуре, после чего смесь выпаривали досуха при пониженном давлении. Остаток растворяли в этилацетате (200 мл), промывали водой и солевым раствором, осушали безводным сульфатом натрия, фильтровали и выпаривали, в результате чего получали твердый остаток, после очистки которого с помощью колоночной хроматографии на силикагеле (20% E ОЛс в гексане) получали смесь β- аномера 8 и α- аномера 9 (1,3:1, β/α ) в виде белых твердых кристаллов (6,30 г, 62,8%).
Аналитический образец очищали с помощью препартивной TCX, используя CH2Cl2 - ацетон (19:1) в качестве проявляющей системы, и получали соединения 2 (Rf = 0,50) и 3 (Rf = 0,55) для ЯМР-характеризации: УФ (МеОН) μmax 269,0 нм.
(-)-(2R, 4R)-2-Амино-9-[[2-[(трет-бутилдифенилсилил)оксиметил] - 1,3-диоксолан-4-ил] -6-хлорпурин (4), (-)-(2R, 4R)-9[[2- [(трет-бутилдифенилсилил)окси] метил)-1,3-диоксолан-4-ил] -2- фтораденин (5), (+)-(2R,4S)-2-амино-9-[[2-[(трет-бутилдифенил- силил)оксиметил]-1,3-диоксолан-4-ил]-6-хлорпурин (6), и (+)- (2R,4S) -9[[2-[(трет-бутилдифенилсилил)оксиметил]-1,3-диоксолан-4- ил]-2-фтораденин (7)
Сухой газообразный аммиак барботировали в размешанный раствор соединенный 2 и 3 (6,25 г 12,18 мм) в ME (125 мл) в течение ночи. Растворитель выпаривали при пониженном давлении, а остаток подвергали хроматографическому разделению на колонке с силикагелем (20-30% в этилацетат в CH2Cl2), в результате чего получали четыре соединения: 4 (Rf=0,35, 1,49 г, 24%), белое кристаллическое твердое вещество. УФ (МеОН) λmax= 309,5 нм. Анализ (C25H28ClN5O3Si), C, H, Cl, N. 5 (Rf= 0,21 1,12 г, 18,6%), бесцветные игольчатые кристаллы, УФ (МеОН)A λmax= 261,0 268,0 (сд) нм.
Анализ (C25H26FN5O3Si) C, H, F, N, 6 (Rf=0,43, 1,60 г 25,76%), белые кристаллы, УФ (МеОН) λmax= 261,0, 269,0 (сд). нм. Анализ (C25 H28FN5O3Si) C, H, F, N. 7 (Rf=0,12, 0,96 г, 16%), микрокристаллическое твердое вещество, УФ (метанол) λmax= 261,0, 269,0 (сд) нм.
Анализ C25H28FN5O3Si) C, H, F, N.
(-)-(2R, 4R)-2-Амино-6-хлор-9-[(2-гндроксиметил)-1,3- диоксолан-4-ил] пурин (10)
Раствор соединения 4 (0,47 г, 0,91 мм) в ТГФ (20 мл) обрабатывали IM н-Bu4NF/ТГФ (1,1 мл, 1,1 мМ), в результате чего получали 10 (Rf = 0,50 0,21 г, 84%) в виде кристаллического твердого вещества, которое перекристаллизовывали из МеОН: УФ (H2O)=307,0 нм ( ε 8,370) (pH 7), 307,5 (ε 8,590) (pH 2), 307,0 (ε 8,800) (pH 11).
Анализ (C9H10ClN5O3) C, H, Cl, N
(-)-(2R,4R)-2-фтор-9-[(2-гидроксиметил)-1,3-диоксолан-4- ил]-аденин (II)
Раствор соединения 5 (0,56 г, 1,12 мм) в ТГФ (20 мл) обрабатывали IM н-Bu4NF/ТГФ (1,35 мл, 1,35 мм), в результате чего получали соединение 22 (0,24 г, 85%) в виде белого кристаллического твердого вещества, которое перекристаллизовывали из МеОН: УФ (H2O) λmax= 260,8 нм (ε 17,010), 268,5 (сд.) нм ( ε 13,510) (рН 7), 261,0 (ε 16,390), 268,5 (сд) (ε 13.300) (pH 2), 260,8 (ε 16,700), 268,5 (сд) (ε 13,200) (pH 11).
Анализ (C9H10FN5O3) C, H, F, N.
(-)-(2R,4R)-9-[(2-Гндроксиметил)-1,3-диоксолан-4-ил]гуанин (14)
Смесь соединения 4 (0,29 г, 0,57 мм), H, CH2CH2OH, (0,51 мл) и 1,0 NaOMe/MeOH (11,6 мл) в МеОН (20 мл) нагревали с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали и нейтрализовали ледяной уксусной кислотой.
Затем раствор выпаривали досуха, остаток растворяли с CHCl3, фильтровали, фильтрат концентрировали досуха и получали неочищенное соединение 8 (0,21 г 75%), которое затем без дополнительной очистки подвергали десилилированию с получением соединения 3 (0,07 г, 61%) в виде микрокристаллического твердого вещества, которое перекристаллизировали из МеОН: УФ (H2O) λmax= 252,0 ( ε 8,730) (pH 7), 254,4 ( ε 12,130), 277,5 (сд.) ( ε 8,070) (pH 2), 264,3 ( ε 10,800) (pH 11).
Анализ (C9H11N5O4 G, Н, N.
(-)-(2R, 4R)-2-Амино-9-[(2 гидроксиметил)-1,3-диоксолан-4- ил] -аденин (15).
В стальной баллон загружали соединение 4 (0,28 г 0,55 мм) безводный этанол (20 мл), насыщенный аммиаком, и нагревали при 90oC в течение 6 часов. После охлаждения растворитель выпаривали в вакууме и полученное соединение 9 (0,26 г, 95%) десилилировали в соответствии с процедурой, описанной для получения соединения 12, в результате чего получали соединение 15 (0,10 г. 75%) в виде белых игольчатых микрокристаллов, которые перекристаллизовывали из MeOH: УФ (H2O) λmax= 279,0 нм ( ε 8,040) (pH 7) 290,0 ( ε 7,070) (pH 2), 278,8 ( ε 7,580) (pH 11).
Анализ ( C9H12N6O3 C, H, N.
(-(-(2-R, 4R, )-2-Амино-9-[(2-гидроксиметил)-1,3-диоксолан-4 ил] пурин может быть получен путем восстановления соединения 10 с использованием ряда восстанавливающих агентов, включая палладированный уголь и газообразный водород или гидрид трибутилолова и азабисизобутиронитрил.
Способность β- D-диоксолан-нуклеозидов ингибировать HBV может быть измерена с помощью различной экспериментальной техники. Используемый в данном эксперименте анализ для оценки способности соединений настоящего изобретения ингибировать репликацию HBV подробно описан в работе Korba and Gerin, Antiviral Res 19: 55-70 (1992). Ниже, в целях иллюстрации, не ограничивающей объема настоящего изобретения, приводятся результаты оценки токсичности и анти-HBV -вирусной активности (-)-(2R,4R,)-2-амино-6-хлор-9-[2- гидроксиметил)-1,3-диоксолан-4-ил] -пурина, (-)-(2R, 4R)-2-амино-9-] 2- гидроксиметил)-1,3-диоксолан-4-ил] -аденин, и (-)-(2R,4R)-9- [(2-гидроксиметил)-1,3-диоксолан-4-ил]гуанина. Другие соединения настоящего изобретения были оценены аналогичным образом.
Оценки противовирусной активности осуществляли на двух отдельных пассажах клеток, где на один пассаж приходилось две культуры (всего 4 культуры). Все лунки в планшетах были засеяны с одинаковой плотностью и в одно и то же время.
Из-за естественных различий в уровнях внутриклеточной и внеклеточной ДНК HBV статистически значимыми оценками P менее 0,05 считались лишь величины ингибирования, которые в 3 раза (для ДНК вириона HBV) или в 2,5 раза (для промежуточных продуктов репликации ДНК HBV) превышали средние уровни для указанных форм HBV-ДНК в необработанных клетках (Korba & Gerin, Antiviral Res. 19: 55-70, 1992). Уровни интегрированной HBV ДНК в каждом клеточном ДНК-препарате (которые в этих экспериментах остаются постоянными в отношении клеточной массы) использовали для вычисления уровней внутриклеточных форм ДНК HBV, что позволило устранить экспериментальные отклонения в блок-гибридизационных анализах.
Типичные значения для уровней ДНК HBV-вириона в необработанных клетках составляет в пределах от 50 до 150 пг на миллилитр культуральной среды (в среднем, около 76 пг/мл). Уровень промежуточных продуктов репликации внутриклеточной ДНК HBV в необработанных клетках составляет в пределах от 50 до 100 пг/мкг клеточной ДНК (в среднем, около 74 пг/мкг).
В основном, ингибирование уровней внутриклеточной HBV-ДНК в результате обработки противовирусными соединениями менее явно выражено и происходит более медленно, чем ингибирование уровней ДНК вириона HBV.
Например, результаты гибридизационных анализов, проведенных для этих экспериментов выявили эквивалентность приблизительно 1,0 пг внутриклеточной HBV - ДНК/мкг клеточной ДНК для 23 гепомных копий на клетку и 1,0 пг внеклеточной HBV ДНК/мл культуральной среды для 3 • 105 вирусных частиц/мл.
Были осуществлены анализы на токсичность для того, чтобы определить являются ли наблюдаемые противовирусные эффекты результатом общего действия на жизнеспособность клетки. Используемый для этой цели способ и был основан на поглощении красителя (нейтрального красного) и представляет собой стандартный и широко используемый анализ на жизнеспособность клеток в различных системах вирус-хозяин, включая вирусы HBV (вирус простого герпеса) и ВИЧ. Процедура этого анализа более подробно проиллюстрирована в нижеприведенной таблице.
Испытуемые соединения использовали в виде 40 мм маточных растворов в ДМСО (замороженных на сухом льду). Для этого были изготовлены суточные аликвоты испытуемых образцов и заморожены при -20oC так, чтобы каждая отдельная аликвота подвергалась лишь одному циклу замораживания-оттаивания. Затем суточные испытуемые аликвоты размораживали, суспендировали в культуральной среде при комнатной температуре и сразу добавляли к клеточным культурам. В тестах на противовирусную активность соединения испытывали при концентрации 0,0 и 1 мкм. В тестах на токсичность, соединения испытывали в пяти кондентрациях, составляющих до 300 мкм.
В нижеприведенных таблицах использовали следующие сокращения: ACPD: (-)-(2R, 4R)-2-амино-6-хлор-9-[(2-гидроксиметил)-1,3- диоксолан-4-ил] пурин, DAPD: (-)-(2R,4R)-2-амино-9-[(2- гидроксиметил)-1,3-диоксолан-4-ил]аденин, и Диоксолан-C: (2R,4R)-9-[(2-гидроксимпетил)-1,3-диоксолан-4-ил]гуанин.
Пример 2.
Токсичность соединений
Оценивали способность энантиомеров ACPD, DAPD и диоксолана -C ингибировать рост вируса в культурах клеток 2.2.15 (клетки HepC2, трансформированные вирионом гепатита). Как показано в таблице 1, для испытуемых соединений, взятых в концентрациях, используемых для оценки противовирусной активности, значительной токсичности не наблюдалось (для необработанных клеток наблюдалось более, чем 50%-ное ингибирование поглощения окраски). Испытуемые соединения оказались нетоксичными для клеток 2.2.15 при концентрации 100 мкм. При концентрации 300 мкм соединения обнаруживали умеренную токсичность, однако все три соединения оказались менее токсичными при данной концентрации, чем ddC.
Анализ на токсичность осуществляли в 96-луночных плоскодонных планшетах для культивирования тканей. Клетки, используемые в анализе на токсичность, культивировали и обрабатывали испытуемыми соединениями в соответствии со схемой, использованной для анализа на противовирусную активность. Каждое соединение испытывали при 4 концентрациях и культуру для каждой концентрации использовали в трех копиях. Для определения относительного уровня токсичности оценивали степень поглощения нейтрального красного. Для количественного анализа использовали оптическую плотность интернализованной окраски при 510 нМ (A510). Полученные величины выражали как процент от средних значений A50 (+ среднеквадратическое отклонение), полученных для 9 отдельных культур необработанных клеток, культивированных в тех же самых 96- луночных планшетах, что и испытуемые соединения. Процент поглощения окраски для 9 контрольных культур в планшете 40 составлял 100 + 3. В этих анализах для соединения 2', 3'- ddC, взятого в концентрации 150-190 мкм, наблюдалось 2-кратное снижение поглощения окраски (по сравнению с уровнями, наблюдаемыми для необработанных культур (Korba & Gerin, Antiviral Res. 19:55-70, 1992) (см. табл. 1).
Пример 3.
Активность против вируса гепатита В
Как показано в таблице 2, уровни ДНК вириона HBV и уровни промежуточных продуктов репликации внутриклеточного HBV (HBV R1) в пределах нормальных отклонений остаются постоянными для необработанных клеток на протяжении всего периода экспериментального заражения. Позитивный контроль (2,3 - дидезоксицитозин 2',3 '- ddC) индуцировал значительное подавление репликации HBV - ДНК при используемой концентрации. Предварительные исследования показали, что в данной аналитической системе (Korba & Gerin, Antiviral, Res 19:55 70, 1992), при концентрации 9-12 мкм 2', 3' - ddC обычно наблюдается 90%-ное подавление HBV - R1 (по отношению к средним уровням в необработанных клетках).
Все три испытуемые соединения являются сильными ингибиторами репликации HBV, и степень подавления ими ДНК вириона HBV и HBV - R1 сравнима или превышает степень подавления, которая наблюдалась при использовании соединения 2', 3' - ddC.
Пример III
Токсичность для эритроидных клеток предшественников (BF U -Е) человека
Фиг. 2 представляет собой график, иллюстрирующий влияние ряда выбранных пуриндиоксоланов и AZT на образование колоний эритроидных клеток-предшественников (BF U-E) человека, измеренных как % от контрольных клеток, в зависимости от концентрации (мкм). Как видно из рисунка, четыре исследуемых диоксоланилнуклеозидов, а именно APD, (-)-(2R,4R)-2-амино-9-[(2-гидроксиметил)-1,3- диоксолан-4-ил]пурин, ACPD,
(-)-(2R, 4R)-2-амино-6-хлор-9-[2-гидроксиметил)-1,3- диоксолан-4-ил]пурин, DG (-)-(2R,4R,)-9-[(2-гидроксиметил- 1,3-диоксолан-4ил]гуанин, и DAPD, (-)-(2R, 4R)-2-амино-9-[(2- гидроксиметил)-1,3-диоксолан-4-ил]аденин, обнаруживали значительно меньшую токсичность, чем AZT для этой линии клеток.
Пример 4.
Влияние на образование колоний клеток-предшественников гранулоцитов и макрофагов человека
Фиг. 3 представляет собой график, иллюстрирующий влияние ACPD, DG, DAPD, APD и AZT на образование колоний клеток- предшественников гранулоцитов и макрофагов человека, измеренных как % от контрольных клеток, в зависимости от логарифма концентрации испытуемого лекарственного средства. Как видно из рисунка, пуриндиоксоланилнуклеозиды обнаруживали значительно меньшую токсичность, т.е. имели более высокую IC50, чем AZT для этой линии клеток.
Пример 5.
Ингибирование репликации HBV - ДНК
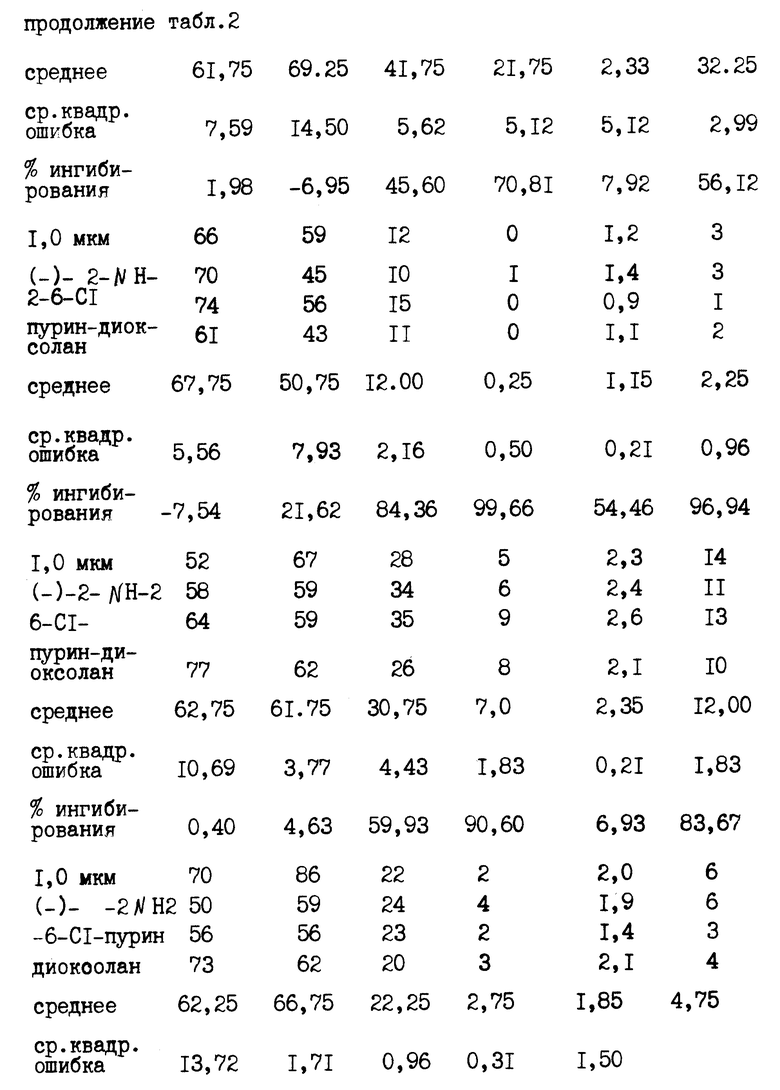
Фиг. 4 представляет собой график, иллюстрирующий процент ингибирования репликации ДНК НВV в клетках 2.2.15 на 9-й день в зависимости от различных концентраций испытуемых соединений с использованием более узкого диапазона концентраций, чем это было использовано в примере 1. В таблице 3 приводятся EC50 и EC90 для вириона HBV и HBV - RI, значения цитотоксичности и индекс селективности для DG, DAPG, ACPD, FTC и DDC.
Пример 6
Фиг. 5 представляет собой график, иллюстрирующий поглощение 5 мкм меченного тритием (-)-(2R,4R)-2-амино-9-[(2-гидроксиметил)- 1,3-диоксолан-4ил] аденина (DAPD) в клетках Hep G2. Экстракт получали через 4 часа после обработки клеток DAPD (1000 число распадов в мин-пМ, инъекция 80 мкл.) Полученные данные показали, что в ходе обмена веществ внутри клетки указанное соединение преобразуется, главным образом, в трифосфатную форму.
Пример 7.
Фиг. 6 представляет собой график, иллюстрирующий поглощение 5 мкм меченного тритием (-)-(2R,4R)-2-амино-9[(2-гидроксиметил)- 1,3-диоксолан-4-ил] аденина (DAPD) в клетках Hep G2. Экстракт получали через 12 часов после обработки клеток соединением DAPD (1000 число распадов в мин-пМ, иньекция 145 мкл. ). Полученные данные показали, что после 4-часового инкубирования с соединением, меченным тритием, наблюдались высокие внутриклеточные уровни трифосфата.
Соединения настоящего изобретения, а также их фармацевтически приемлемые соли, пролекарственные предшественники и производные могут быть использованы для предупреждения и лечения HBV инфекций, и других связанных с ними состояний, таких как состояния, характеризующиеся положительной реакцией на антитела против HBV и положительной реакцией на HBV, хроническое воспаление печени, вызванное вирусом HBV, цирроз печени, острый гепатит, молниеносный гепатит, хронические персистирующий гепатит и усталость. Указанные соединения или композиции могут быть также использованы в профилактических целях для предупреждения или замедления прогрессирования заболевания у индивидуумов, имеющих положительную реакцию на антитело против НВV или НВV - антиген, либо подвергшихся экспонированию вирусом НВУ.
Индивидуумам страдающим любым из вышеуказанных состояний, может быть введено эффективное количество (-)-(2R,4R)-2-амино-6-хлор-9-[(2- гидроксиметил)-1,3-диоксолан-4-ил] пурина, (-)-(2R, 4R)-9-[(2- гидроксиметил)-1,3-диоксолан-4-ил] -гуанина, (-)-(2R,4R)-2-амино- 9-[(2 гидроксиметил)-1,3-диоксолан-4-ил] -аденина или (-)- (2R,2R)-2-амино-9-[2-гидроксиметил)-1,3-диоксолан-4-ил] пурина либо их фармацевтически приемлемой соли или производного, необязательно в фармацевтически приемлемом носителе или разбавителе. Указанные активные материалы могут быть введены любым подходящим способом, например, перрорально, парентерально, внутривенно, внутрикожно, подкожно или путем местного применения в жидкой или твердой форме.
Активное соединение может быть введено в фармацевтически приемлемом носителе или разбавителе в количестве, достаточном для обеспечения нужного терапевтического эффекта и в то же время не оказывающем серьезного токсического действия на пациента.
Предпочтительная доза активного соединения для всех вышеупомянутых состояний или заболеваний варьируется в пределах от около 1 до 60 мг/кг, более предпочтительно 1-20 мг/кг, а в более широком диапазоне от 0,1 до около 100 мг на один килограмм веса тела пациента в день. Диапазон эффективных доз фармацевтически приемлемых производных может быть вычислен на основании массы исходного нуклеозида, необходимого для введения. Если это производное обладает собственной активностью, то эффективные дозы могут быть определены, как указано выше, исходя из массы производного либо другими способами, известными специалистам.
Соединение настоящего изобретения может быть введено в виде любой подходящей унифицированной лекарственной формы, например, содержащей 7-3000 мг, а предпочтительно 70-1400 мг активного ингредиента на одну лекарственную форму. Пероральный препарат обычно содержит 50-1000 мг активного ингредиента.
В идеальном случае, если после введения препарата максимальные концентрации активного соединения в плазме крови составляют от около 0,2 до 70 мкм, а предпочтительно от около 1,0 до 10 мкм. Такие концентрации могут быть достигнуты, например, путем внутривенной инъекции 0,1-5%-ного раствора активного ингредиента необязательно в физиологическом растворе либо путем введения активного ингредиента в виде болюса. Концентрация активного соединения в лекарственной композиции зависит от скорости абсорбции, инактивации и экскреции лекарственного средства, а также от других факторов, хорошо известных специалистам. Кроме того, дозы активного соединения могут также варьироваться в зависимости от тяжести состояния пациента. При этом следует отметить, что для каждого конкретного пациента должна быть установлена индивидуальная схема введения лекарственного средства, которая назначается в соответствии с потребностями данного пациента и проводится под наблюдением лечащего врача, а указанные выше диапазоны концентраций представлены лишь в иллюстративных целях и не должны рассматриваться как некое ограничение объема или применения заявленной композиции. Активный ингредиент может быть введен в виде разовой дозы, либо он может быть разделен на несколько доз меньшего количества и введен через различные интервалы времени.
Предпочтительно, если активное соединение вводят перорально. В основном, пероральные композиции содержат инертный разбавитель или пищевой носитель. Эти композиции могут быть включены в мелатиновые капсулы или спрессованы в таблетки. Для перорального введения в терапевтических целях активное соединение может быть смешано с наполнителями и использовано в форме таблеток, пастилок, или капсул.
Лекарственная композиция может также включать в себя фармацевтически приемлемые связующие агенты и/или адъюванты.
Указанные таблетки, пилюли, капсулы, пастилки, и т.п. могут содержать следующие ингредиенты (или соединения аналогичной природы): связующее соединение, такое как микрокристаллическая целлюлоза, трагекантовая камедь или желатин, наполнитель, такой как крахмал или лактоза, дезинтегрирующий агент, такой как альгиновая кислота. Примогель или кукурузный крахмал, смазывающее, такое как стеарат магния или Sterotes, усилитель скольжения, такой как коллоидальный диоксид кремния, подслащивающий агент, такой как сахароза, или ароматизирующий агент, такой как перечная мята, метилсалицилат, или апельсиновая отдушка. Если в качестве лекарственной формы используется капсула, то помимо вышеуказанных ингредиентов, эта капсула может содержать жидкий носитель, например, жирное масло. Кроме того, унифицированные лекарственные формы могут содержать и другие материалы, которые модифицируют физическую форму лекарственного препарата, например, также, как покрытия, изготовленные из сахара, шеллака или других энтеросолюбильных агентов.
Активное соединение, или его фармацевтически приемлемая соль, или фармацевтически приемлемое производное могут быть введены в качестве компонента, входящего в состав эликсира, суспензии, сиропа, облатки, жевательной резинки и т.п. Сироп, помимо активного соединения, может также содержать сахарозу в качестве подслащивающего агента, консерванты, красители и ароматические вещества.
Активное соединение или его фармацевтически приемлемая соль или фармацевтически приемлемое производное могут быть также совмещены с другими активными материалами, не оказывающими неблагоприятного воздействия на активность соединений настоящего изобретения либо с материалами, оказывающими дополнительное желательное действие, например, такими как антибиотики, противогрибковые средства, противовоспалительные средства или другие противовирусные средства, например, средства против вируса HBV, цитомегаловируса или ВИЧ.
Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного применения, могут содержать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, жирное масло, полиэтиленгликоль, глицерин, пропиленгликоль или другие синтетические растворители, антибактриальные агенты, также, как бензиловый спирт или метилкарабены, антиоксиданты, также, как аскорбиновая кислота или бисульфит натрия, хелатообразующие агенты, также, как этилендиаминтетрауксусная кислота, буферы, также, как ацетаты, цитраты, или фосфаты, и агенты, корректирующие тоничность, также, как хлорид натрия или декстроза. Парентеральная композиция может быть помещена в ампулы, одноразовые шприцы, либо в стеклянные или пластиковые флаконы для мнокократного использования.
В случае внутривенного введения предпочтительными носителями являются физиологический раствор или забуференный фосфатом физиологический раствор (PBS).
В предпочтительном варианте осуществления настоящего изобретения, во избежание быстрого удаления активного соединения из организма, получают композицию с регулируемым высвобождением, содержащую активное соединение вместе с носителем, например, такую, как имплантат, и микрокапсулированные системы доставки лекарственного средства. Для этих целей могут быть использованы биологически разлагаемые и биологически совместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры, и полимолочная кислота. Способы получения таких композиций хорошо известны специалистам. Указанные материалы могут быть также получены в готовом виде от Alga Corporation и Nova Pharmaceutical Inc. В качестве фармацевтически приемлемых носителей, предпочтительно, являются также липосомные суспензии (включая липосомы, направленные на инфицированные клетки и конъюгированные с моноклональными антителами против вирусных антигенов). Такие липосомные суспензии могут быть получены методами, хорошо известными специалистам и описанными, например, в патенте США N 4522811. Так, например, липосомные композиции могут быть получены путем растворения соответствующих липидов (таких, как стеароилфосфатицилэтаноламин, стеароилфосфатидилхолин, арахадоилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем выпаривают, в результате чего на поверхности контейнера остается тонкая пленка осушенного липида. Затем в контейнер вводят водный раствор активного соединения или его монофосфатных, дифосфатных и/или трифосфатных производных. После этого содержимое контейнера перемешивают вручную в целях отделения липидного материала от стенок контейнера и диспергирования липидных агрегатов, в результате чего получают липосомную суспензию.
Получение фосфатных производных β- D-диоксоланнуклеозидов
Моно-, ди-, и трифосфатные производные β- D-диоксоланнуклеозидов могут быть получены в соответствии с процедурой, описанной выше.
Монофосфат может быть получен способом, описанным Imai и др. J.Org.Chem. 34 (6), 1547-1550 (June 1868). Например, около 100 мг β- D-диоксолан-нуклеозида и около 280 мкл фосфорилхлорида, размешивая, подвергали реакции приблизительно в 8 мл безводного этилацетата при около 0oC в течение примерно 4 часов. Затем реакцию гасили с использованием льда. Видную фазу очищали на колонке с активированным углем, элюируя 5% гидроксидом аммония в смеси этанола и воды (1:1). После выпаривания элюата получали аммоний-( β- D-диоксалан-нуклеозид)-5'-монофосфат.
Дифосфат может быть получен в соответствии с процедурой, описанной Davisson и др. J. Org Chem., 52(9), 1794-1801 (1987). β- D- -Диоксолан-нуклеозиды могут быть получены из соответствующего тозилата, который может быть получен, например, посредством реакции нуклеозида с тозилхлоридом в пиридные при комнатной температуре в течение примерно 24 часов с последующей обработкой полученного продукта стандартными способами (например, путем промывания, осушки и кристаллизации).
Трифосфат может быть получен в соответствии с процедурой, описанной Hoard и др. J.Am.Сhem. Soc., 87 (8), 1785-1788 (1965). Например, β- D-диоксоланнуклеозид активируют (путем обработки имидазолидом в соответствии с известными процедурами), а затем обрабатывают пирофосфатом трибутиламмония в ДМФ. В результате этой реакции образуется, главным образом, трифосфат с некоторым количеством непрореагировавшего монофосфата и некоторым количеством дифосфата. После очистки с помощью анионообменной хроматографии на DEAE-колонне получают трифосфат, например, в виде тетранатриевой соли.
Настоящее изобретение было описано на предпочтительных примерах его осуществления. Однако, специалистам в данной области ясно, что возможны различные изменения и модификации изобретения, которые могут быть внесены, исходя из вышеприведенного подробного описания изобретения. При этом следует отметить, что указанные модификации и изменения не должны выходить за рамки объема нижеследующей формулы изобретения.










Изобретение относится к медицине. Предложен способ лечения HBV-инфекции, заключающийся во введении эффективного количества энантиомерно чистого β-D-диоксоланилпуриннуклеозида форомулы 1.

где R является OH, Cl, NH2 или H, а X выбирают из группы, включающей алкил, ацил, монофосфат, дифосфат и трифосфат. Способ ингибирует репликацию ДНК HBV-вириона. 18 с. и 7 з.п. ф-лы, 3 табл., 6 ил.

где R является OH, а X выбирают из группы, включающей водород, алкил, ацил, монофосфат, дифосфат, и трифосфат,
или его фармацевтически приемлемой соли, причем указанное соединение, по крайней мере на 95% не содержит соответствующего β-L - энантиомера.
где R представляет NH2, Х выбирают из группы, состоящей из водорода, алкила, ацила, монофосфата, дифосфата и трифосфата,
или его фармацевтически приемлемой соли, причем указанное соединение, по крайней мере, на 95% свободно от соответствующего β-L - энантиомера.
где R представляет водород или хлор, а X выбирают из группы, состоящей из водорода, алкила, ацила, монофосфата, дифосфата и трифосфата,
или его фармацевтически приемлемой соли, причем указанное соединение, по крайней мере, на 95% свободна от соответствующего β-L - энантиомера.
где R представляет собой OH, а X выбирают из группы, состоящей из водорода, алкила, ацила, монофосфата, дифосфата и трифосфата;
или его фармацевтически приемлемой соли.
где R представляет собой NH2, а Х выбирают из группы, состоящей из водорода, алкила, ацила, монофосфата, дифосфата и трифосфата, или его фармацевтически приемлемой соли.
где R представляет собой H, или Cl, а X выбирают из группы, состоящей из водорода, алкила, ацила, монофосфата, дифосфата и трифосфата,
или его фармацевтически приемлемой соли.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| УЛЬТРАЗВУКОВОЙ СПОСОБ КОНТРОЛЯ состоянияМАТЕРИАЛА | 0 |
|
SU337713A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO A 9210497, 25.06.1992 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| WO A 9208717, 29.05.1992. | |||

