Данное изобретение частично профинансировано грантом United States National Institutes of Health №1R01-A1-41980-01. Правитель сто США имеет определенные права на данное изобретение.
В соответствии с данной заявкой испрашивается приоритет по предварительной заявке на выдачу патента США №60/116773, поданной 22 января 1999 года.
Предпосылки изобретения:
В 1983 году в качестве этиологического фактора СПИД был определен вирус иммунодефицита человека (ВИЧ). В 1985 году появились сообщения о том, что синтетический нуклеозид 3'-азидо-3'-дезокситимидин (AZT) ингибирует репликацию вируса иммунодефицита человека. С тех пор было доказано, что ряд других синтетических нуклеозидов, включая 2',3'-дидезоксиинозин (DDI), 2',3'-дидезоксицитидин (DDC), 2',3'-дидезокси-2',3'-дидегидротимидин (D4T), цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан (FTC), (-)-цис-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолан (3ТС), эффективны против ВИЧ. После клеточного фосфорилирования 5'-трифосфатом клеточными киназами данные синтетические нуклеозиды включаются в растущую цепь вирусной ДНК, вызывая терминацию цепи из-за отсутствия 3'-гидроксильной группы. Они также способны ингибировать вирусный фермент обратную транскриптазу.
Было выявлено, что устойчивые к лекарственным средствам варианты ВИЧ могут возникать после продолжительного лечения противовирусным средством. Устойчивость к лекарственным средствам обычно возникает в результате мутации гена, который кодирует фермент, используемый при репликации вируса, и, как правило, в случае ВИЧ обратной транскриптазы, протеазы или ДНК-полимеразы. Недавно было показано, что эффективность лекарственного средства против ВИЧ-инфекции может быть продлена, улучшена или восстановлена путем введения соединения в сочетании или чередовании со вторым и, возможно, третьим противовирусным средством, которое индуцирует мутацию, отличную от таковой, индуцированной основным лекарственным средством. Альтернативно, с помощью подобной сочетанной или чередующейся терапией могут быть изменены фармакокинетика, биологическое распределение или иной параметр лекарственного средства. Вообще, сочетанная терапия, как правило, является предпочтительной по сравнению с чередующейся терапией, поскольку она оказывает множественное одновременное давление на вирус. Однако, нельзя предсказать, какие мутации будут индуцированы в геноме ВИЧ-1 введенным лекарственным средством, является ли мутация постоянной или временной, или как инфицированная ВИЧ-1 с мутантной последовательностью будет реагировать на сочетаную или чередующуюся терапию другими средствами. Это осложняется тем фактом, что имеется мало данных по кинетике устойчивости к лекарственным средствам в долгоживущих клеточных культурах, обрабатываемых современными противоретровирусными средствами.
Варианты ВИЧ-1, устойчивые к 3'-азидо-3'-дезокситимидину (AZT), 2',3'-дидезоксиинозину (DDI) или 2',3'-дидезоксицитидину (DDC), были выделены из организма пациентов, получавших длительную монотерапию данными лекарственными средствами (Larder BA, Darby G, Richman DD, Science, 1989; 243:1731-4; St.Clair MH, Martin JL, Tudor WG, et al., Science, 1991; 253:1557-9; St.Clair MH, Martin JL, Tudor WG, et al., Science, 1991; 253:1557-9; and Fitzgibbon JE, Howell RM, Haberzettl CA, Sperber SJ, Gocke DJ, Dubin DT, Antimicrob Agents Chemother, 1992; 36:153-7). Растущие клинические данные свидетельствуют о том, что устойчивость к AZT является предвестником неблагоприятного клинического исхода как у детей, так и у взрослых (Mayers DL, Lecture at the Thirty-second Interscience Conference on Antimicrobial Agents and Chemotherapy (Anaheim, CA. 1992); Tudor-Williams G, St.Clair MH, McKinney RE, et al., Lancet, 1992; 339:15-9; Ogino MT, Dankner WM, Spector SA, J.Pediatr., 1993; 123:1-8; Crumpacker CS, D'Aquila RT, Johnson VA, et al., Third Workshop on Viral Resistance, (Gaithersburg, MD. 1993); and Mayers D, and the RV43 Study Group, Third Workshop on Viral Resistance, (Gaithersburg, MD. 1993)). Также сообщалось о быстром развитии устойчивости ВИЧ-1 к ненуклеозидным ингибиторам обратной транскриптазы (ННИОТ) как в клеточной культуре, так и в ходе клинических испытаний на людях (Nunberg JH, Schleif WA, Boots EJ, et al., J.Virol., 1991; 65(9):4887-92; Richman D, Shin CK, Lowy I, et al., Proc.Natl.Acad.Sci.(USA), 1991; 88:11241-5; Mellors JW, Dutschman GE, Im GJ, Tramontane E, Winkler SR, Cheng YC, Mol.Pharm., 1992; 41:446-51; Richman DD and the ACTG 164/168 Study Team, Second International HIV-1 Drug Resistance Workshop, (Noordwijk, the Netherlands, 1993); and Saag MS, Emini EA, Laskin OL, et al., N. Engi. J.Med., 1993; 329:1065-1072). В случае ННИОТ L'697661, устойчивый к лекарственному средству ВИЧ-1 возник в течение 2-6 недель от начала терапии в сочетании с возвратом виремии к уровням, существовавшим до лечения (Saag MS, Emini EA, Laskin OL, et al., N.Engl.J.Med., 1993; 329:1065-1072). Внезапно развивающаяся виремия, связанная с присутствием устойчивых к лекарственным средствам штаммов, также отмечалась и в случае с другими классами ингибиторов ВИЧ-1, включая ингибиторы протеазы (Jacobsen H, Craig CJ, Duncan IB, Haenggi M, Yasargil К, Mous J, Third Workshop on Viral Resistance, (Gaithersburg, MD, 1993)). Данный опыт привел к осознанию того, что потенциал к развитию устойчивости к лекарственным средствам ВИЧ-1 должен быть оценен в ходе ранних доклинических исследований всех новых способов лечения ВИЧ-1.
2',3'-Дидезокси-2',3'-дидегидро-5-фторцитидин (D4FC) является известным соединением. В публикации заявки на выдачу европейского патента №0 409 227 А2, поданной Ajinomoto Co., Inc., описан β-D-D4FC (пример 2) и его применение для лечения гепатита В. В патенте Нидерландов №8901258, выданном Stichting Rega V.Z.W., описаны, главным образом, производные 5-галоген-2',3'-дидезокси-2',3'-дидегидроцитидина для применения в лечении ВИЧ-инфекции и гепатита В (HBV). В патенте США №5703058 описан способ лечения ВИЧ- и HBV-инфекций, который предусматривает введение эффективного количества β-L-D4FC в сочетании или чередовании с цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиоланом, цис-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиоланом, 9-[4-(гидроксиметил)-2-циклопентен-1-ил)гуанином (карбовиром), 9-[(2-гидроксиэтокси)-метил]гуанином (ацикловиром), интерфероном, 3'-дезокси-3'-азидотимидином (AZT), 2',3'-дидезоксиинозином (DDI), 2',3'-дидезоксицитидином (DDC), (-)-2'-фтор-5-метил-β-L-арауридином (L-FMAU) или 2',3'-дидегидро-2',3'-дидезокситимидином (D4T). В патенте США №5905070 описан способ лечения ВИЧ- и HBV-инфекций, который предусматривает введение эффективного количества β-D-D4FC в сочетании или чередовании с цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиоланом, цис-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиоланом, 9-[4-(гидроксиметил)-2-циклопентен-1-ил)гуанином (карбовиром), 9-[(2-гидроксиэтокси)метил]гуанином (ацикловиром), интерфероном, 3'-дезокси-3'-азидотимидином (AZT), 2',3'-дидезоксиинозином (DDI), 2',3'-дидезоксицитидином (DDC), (-)-2'-фтор-5-метил-β-L-арауридином (L-FMAU) или 2',3'-дидегидро-2',3'-дидезокситимидином (D4T).
Объектом настоящего изобретения является определение оптимального способа введения β-D-D4FC для лечения ВИЧ-инфекции.
Другими объектами настоящего изобретения являются способ и композиция, которая включает β-D-D4FC, для лечения пациентов, инфицированных ВИЧ, которая проявляет преимущественные или улучшенные фармакокинетические параметры, параметры биологического распределения, метаболические параметры, параметры устойчивости и другие параметры по сравнению с введением одного β-D-D4FC.
Другими объектами настоящего изобретения являются способ и композиция для лечения пациентов, инфицированных ВИЧ, в которой β-D-D4FC вводят в сочетании или чередовании со вторым соединением, которое действует синергически β-D-D4FC против вируса.
Другими объектами настоящего изобретения являются способ и композиция для лечения пациентов, инфицированных устойчивой к лекарственным средствам форме ВИЧ.
Другими объектами настоящего изобретения являются способ и набор для оценки наилучшего способа введения β-D-D4FC.
Сущность изобретения:
Было выявлено, что β-D-D4FC индуцирует мутации в ВИЧ-1 в 70 (К на N), 90 и 172 кодонах обратно-транскриптазного участка генома вируса. На основании данного открытия разработан способ лечения ВИЧ-инфекции, который предусматривает введение β-D-D4FC или его фармацевтически приемлемой соли или пролекарства человеку, нуждающемуся в таком лечении, в сочетании или чередовании с лекарственным средством, которое индуцирует мутацию ВИЧ-1 в положении, отличном от 70 (К на N), 90 и 172 кодонов обратно-транскриптазного участка. Данное изобретение может быть осуществлено с использованием опубликованных профилей мутаций, индуцируемых известными анти-ВИЧ лекарственными средствами, или с помощью определения профиля мутаций, индуцируемых новым лекарственным средством.
На основании данного изобретения также предоставляется способ применения β-D-D4FC в качестве "терапии спасения" для пациентов, у которых наблюдается устойчивость к другим анти-ВИЧ средствам. Было обнаружено, что β-D-D4FC не проявляет существенной перекрестной устойчивости с AZT, DDC, DDI, D4T, 3ТС, (-)-FTC или β-L-D4FC. Напротив, β-L-D4FC быстро индуцирует мутацию в кодоне 184 (метионин на валин), что приводит к высокому уровню устойчивости 3ТС и FTC. β-D-D4FC может быть, как правило, использован как терапия спасения для любого пациента, у которого наблюдается устойчивость к лекарственному средству, которое индуцирует мутацию кодонов, отличных от 70 (К на N), 90 или 172 кодонов.
Изобретение, описанное здесь, в более общем смысле охватывает, по крайней мере, следующие осуществления:
(i) Способ лечения ВИЧ-инфекции у человека, предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли человеку, необязательно в фармацевтически приемлемом носителе, в сочетании или чередовании с лекарственным средством, которое индуцирует мутацию ВИЧ-1 в положении, отличном от 70 (К на N), 90 или 172 кодонов обратно-транскриптазного участка, и которое отлично от цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана, цис-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолана, 9-[4-(гидроксиметил)-2-циклопентен-1-ил)гуанина (карбовира), 9-[(2-гидроксиэтокси)метил]гуанина (ацикловира), интерферона, 3'-дезокси-3'-азидотимидина (AZT), 2',3'-дидезоксиинозина (DDI), 2',3'-дидезоксицитидина (DDC), (-)-2'-фтор-5-метил-β-L-арауридина (L-FMAU) или 2',3'-дидегидро-2',3'-дидезокситимидина (D4T).
(ii) Способ лечения ВИЧ-инфекции у человека, предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемой соли человеку, необязательно в фармацевтически приемлемом носителе, в сочетании или чередовании с лекарственным средством, которое индуцирует мутацию ВИЧ-1 кодона 70 с К на N (т.е. с лизина на аспарагин), мутацию кодона 90 с V на I (т.е. с валина на изолейцин) и мутацию кодона 172 с R на К (т.е. с аргинина на лизин) обратно-транскриптазного участка и которое отлично от цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолана, цис-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолана, 9-[4-(гидроксиметил) -2-циклопентен-1-ил)гуанина (карбовира), 9-[(2-гидроксиэтокси)метил]гуанина (ацикловира), интерферона, 3'-дезокси-3'-азидотимидина (AZT), 2',3'-дидезоксиинозина (DDI), 2',3'-дидезоксицитидина (DDC), (-)-2'-фтор-5-метил-β-L-арауридина (L-FMAU) или 2',3'-дидегидро-2',3'-дидезокситимидина (D4T).
(iii) Способ лечения пациента, инфицированного штаммом ВИЧ, который устойчив к ЗТС, предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли пациенту, необязательно в фармацевтически приемлемом носителе.
(iv) Способ лечения пациента, инфицированного штаммом ВИЧ, который устойчив к AZT, предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли пациенту, необязательно в фармацевтически приемлемом носителе.
(v) Способ лечения пациента, инфицированного штаммом ВИЧ, который устойчив к цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолану, предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли пациенту, необязательно в фармацевтически приемлемом носителе.
(vi) Способ лечения пациента, инфицированного штаммом ВИЧ, который устойчив к цис-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолану, предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли пациенту, необязательно в фармацевтически приемлемом носителе.
(vii) Способ лечения пациента, инфицированного штаммом ВИЧ, который устойчив к 2',3'-дидегидро-2',3'-дидезокситимидину (D4T), предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли пациенту, необязательно в фармацевтически приемлемом носителе.
(viii) Способ лечения пациента, инфицированного штаммом ВИЧ, который устойчив к 2',3'-дидезоксиинозину (DDI), предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли пациенту, необязательно в фармацевтически приемлемом носителе.
(ix) Способ лечения пациента, инфицированного штаммом ВИЧ, который устойчив к 2',3'-дидезоксицитидину (DDC), предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли пациенту, необязательно в фармацевтически приемлемом носителе.
(х) Способ лечения пациента, инфицированного ВИЧ, предусматривающий введение эффективного количества β-D-D4FC или его фармацевтически приемлемого пролекарства или соли в сочетании или чередовании с эффективным количеством (S)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он (SUSTIVA, смотри патент США 5519021).
Описанные схемы сочетания, чередования и спасения применимы для профилактики и лечения ВИЧ-инфекции и других ассоциированных состояний, таких как СПИД-ассоциированный комплекс (ARC), персистирующая генерализованная лимфаденопатия (PGL), СПИД-ассоциированные неврологические состояния, состояния, связанные с наличием анти-ВИЧ антителами и ВИЧ, саркома Калоши, тромбоцитопеническая пурпура и оппортунистические инфекции. Кроме того, данные соединения или композиции могут применяться профилактически для предотвращения или замедления прогрессирования клинических проявлений заболевания у субъектов, положительных по анти-ВИЧ антителам или антигенам ВИЧ или контактировавших с ВИЧ.
Краткое описание чертежей
На фиг.1 представлена формула β-D-D4FC.
Фиг.2 представляет собой график зависимости концентрации β-D-D4FC, выраженной в микромоль/л, от процентного ингибирования продукции антигена р24. Фиг.2 иллюстрирует отбор вируса с пониженной чувствительностью к β-D-D4FC.
Подробное описание изобретения:
I. Определения
Используемый здесь термин "устойчивый вирус" относится к вирусу, который проявляет трех-, а обычно пятикратное или большее увеличение значения EC50 по сравнению с интактным вирусом в непрерывной клеточной линии, включающей, не ограничиваясь, мононуклеарные клетки периферической крови (РВМС) или клетки МТ2 или МТ4.
Термин "D-D4FC" используется ниже взаимозаменяемо с термином "β-D-D4FC".
Используемый здесь термин "по существу, чистый" или "по существу, в виде одного оптического изомера" относится к нуклеозидной композиции, которая содержит по крайней мере 95-98% и более предпочтительно 99-100%, одного энантиомера данного нуклеозида. В предпочтительном осуществлении β-D-D4FC вводят по существу в чистом виде по любому из указанных показаний.
Используемый здесь термин "пролекарство" относится к 5'- и N4-ацилированным, алкилированным или фосфорилированным (включая моно-, ди- и трифосфатные эфиры, равно как и стабилизированные фосфатные и фосфолипидные) производные D-D4FC. В одном осуществлении ацильная группа представляет собой эфир карбоновой кислоты, в котором некарбонильный радикал сложноэфирной группы выбран из неразветвленного, разветвленного или циклического алкила, алкоксиалкила, включая метоксиметил, аралкила, включая бензил, арилоксиалкила, включая феноксиметил, арила, включая фенил, необязательно замещенный галогеном, алкилом или алкокси, эфиры сульфоната, такие как алкил- или аралкилсульфонил, включая метансульфонил, тритил или монометокситритил, замещенного бензила, триалкилсилила или дифенилметилсилила. Арильные группы в сложных эфирах предпочтительно содержат фенильную группу. Алкильная группа может быть неразветвленной, разветвленной или циклической и предпочтительно представляет собой C1-C18.
Используемый здесь термин "фармацевтически приемлемые соли" относится к фарцевтически приемлемым солям, которые, будучи введены реципиенту, способны непосредственно или опосредованно образовывать β-D-D4FC или которые проявляют активность сами по себе.
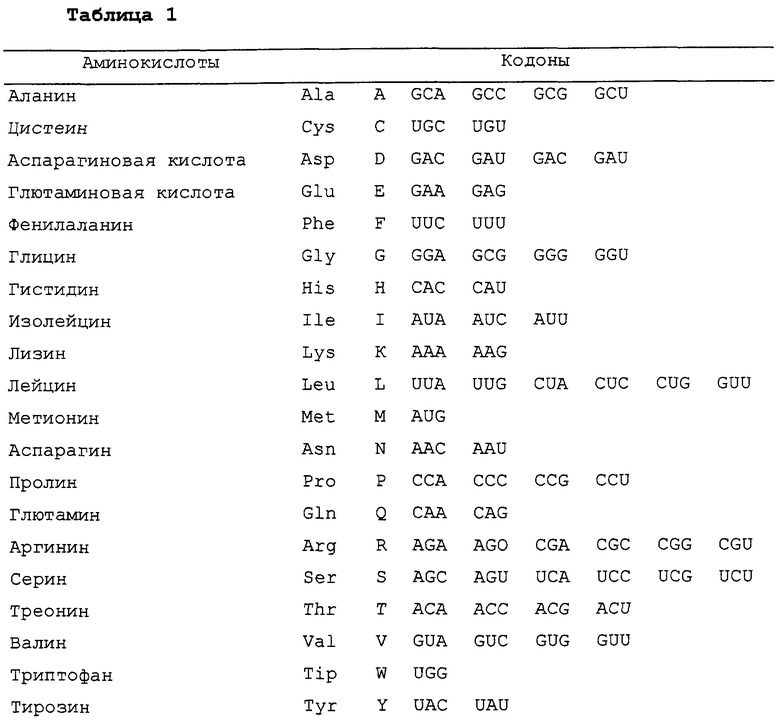
Сокращения наименований аминокислот, используемые здесь, пояснены в таблице 1.
II. Мутации обратной транскриптазы ВИЧ-1, отбираемые с помощью β-D-D4FC
Как D-, так и L-энантиомеры β-2',3'-дидегидро-2',3'-дидезокси-5-фторцитидина (D4FC) являются мощными и селективными ингибиторами ВИЧ-1, хотя D-энантиомер является более селективным. Развитие устойчивости к D-D4FC in vitro оценивали путем серийных посевов ВИЧ-1LAI в клетках МТ-2 и мононуклеарных клетках периферической крови (РВМС) в присутствии растущих концентраций лекарственного средства. Варианты, устойчивые к D-D4FC, возникли лишь после продолжительного контактирования с лекарственным средством. Вирус, полученный после 20 посевов в клетках МТ-2, проявил 5,3-кратную устойчивость к D-D4FC. Устойчивый вирус не удалось выделить из РВМС, несмотря на многочисленные попытки. Секвенирование ДНК ОТ вируса, отобранного в клетках МТ-2, выявило две мутации: K65R и V179D. Отбор устойчивого к D-D4FC вируса повторяли в клетках МТ-2, и варианты, проявляющие 19,3-кратную устойчивость, кодировали три новые мутации ОТ: K70N, V90I и R172K. Рекомбинантный K65R вич-1LAI проявлял 3,9-кратную устойчивость к D-D4FC. V179D, мутация, придающая устойчивость к ненуклеозидным ингибиторам ОТ, наиболее вероятно, компенсирует K65R. Роль других мутаций в формировании устойчивости к D-D4FC также оценивали путем конструирования рекомбинантного вируса с одной или множественными мутациями, однако, ни один из протестированных рекомбинантов не проявлял более чем 2-кратную устойчивость.
Материалы и методы.
Химикалии. D-D4FC синтезировали в одной из лабораторий авторов настоящего изобретения, как описано ранее (Shi et al., 1999). Его получали в виде 10 мМ маточных растворов в стерильной воде и хранили при -20°С. Раствор оттаивали и разбавляли до желаемой концентрации непосредственно перед использованием.
Клетки. Клетки МТ-2 (AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health при содействии D. Richman) культивировали в RPMI 1640 (Whittaker M.A., Byproducts, Walkersville, MD), обогащенной 10% околоплодной сывороткой теленка, 10 мМ буфером HEPES, пенициллином (50 МЕ/мл) и стрептомицином (50 мкг/мл).
Вирусы. ВИЧ-1LAI, молекулярно клонированный клинический изолят, применяли в качестве исходного вируса как для отбора устойчивости, так и для получения рекомбинантных мутантов. Маточные препараты ВИЧ-1LAI получали путем электропорации 5-10 мкг провирусной плазмидной ДНК в 1,3×107 клеток МТ-2. На пике цитопатического эффекта вируса (обычно через 7 суток после трансфекции) супернатант отбирали из инфицированных культур, разделяли на аликвоты и хранили при -80°С до использования. Препараты вируса титровали до трехкратного конечного разведения в клетках МТ-2 и значение TCID50 рассчитывали по уравнению Reed и Muench.
Отбор устойчивых вирусов. До начала отбора вирусов, устойчивых к D-D4FC, исходный вирус (ВИЧ-1LAI) высевали в виде бесклеточного вируса 10 циклов в клетках МТ-2 в отсутствие лекарственного средства. Устойчивый к D-D4FC вирус отбирали путем серийного высева ВИЧ-lLAI в клетках МТ-2 в присутствии постепенно растущих концентраций D-D4FC. Отбор устойчивых к D-D4FC вирусов проводили дважды. Отбор начинали путем инокуляции 1×106 клеток МТ-2 0,01 моль вируса. На пике цитопатического эффекта вируса (4-7 суток после инфекции), супернатант отбирали из инфицированных культур и 0,1-0,3 мл использовали далее для начала нового цикла инфекции. Супернатант также разделяли на аликвоты и хранили при -80°С для охарактеризования отобранного вируса. Вирус высевали по крайней мере трижды при каждой концентрации, причем число циклов при каждой конкретной концентрации лекарственного средства зависело от способности вируса к размножению при конкретной концентрации D-D4FC. В ходе первой процедуры отбора вирус высевали один раз в отсутствие лекарственного средства до повышения концентрации лекарственного средства (таблица 2). В качестве контроля параллельно также высевали вирус в отсутствие лекарственного средства. Первую процедуру отбора начинали при 0,75 мкМ и постепенно повышали концентрацию до 4,0 мкМ в течение 37 циклов бесклеточных посевов. Вторую процедуру отбора начинали при 0,2 мкМ и постепенно повышали концентрацию до 6,2 мкМ в течение 27 циклов бесклеточных посевов (таблица 2).
Анализы чувствительности вируса. Чувствительность вируса к D-D4FC измеряли путем измерения процентного ингибирования продукции антигена р24. Вкратце, клетки МТ-2 (1×105 клеток/мл) инфицировали вирусом при moi 0,01 в присутствии серийных разведений D-D4FC. Каждое разведение тестировали трижды. Культуральные супернатанты собирали на 7 сутки после инфекции и исследовали на предмет продукции антигена р24, используя коммерчески доступную тест-систему (DuPont, NEN Products, Wilmington, Del.). Чувствительность вируса выражали как концентрацию лекарственного средства, требующуюся для ингибирования продукции антигена р24 на 50% (EC50).
Секвенирование ДНК отобранной вирусной обратной транскриптазы. Вирусную РНК (вРНК) из отобранного вируса выделяли, используя реагент TRIzol (GibcoBRL, Grand Island, NY). Полноразмерную кодирующую ОТ область амплифицировали с помощью ОТ-ПЦР. Продукт ПЦР далее очищали, используя наборы для ПЦР Wizard (Promega, Madison, WI), и секвенировали.
Получение мутантного рекомбинантного ВИЧ-1. Мутантную ОТ получали, используя систему мутагенеза in vitro Altered SitesII (Promega, Madison, WI). Мутагенез проводили на ВИЧ-lLAI, ОТ клонировали в вектор мутагенеза (PALTER, Promega). Наличие желаемой мутации определяли путем прямого секвенирования гена ОТ. Мутантную ОТ далее лигировали в вектор pxxHIV-1LAI. Маточные растворы вируса получали путем электропорирования 5-10 мкг ДНК в 1,3×107 клеток МТ-2, как описано выше.
Результаты и обсуждение
Фенотипирование устойчивого вируса. Вирус, устойчивый к D-D4FC, отбирали после 37 (отбор №1) и 20 (отбор №2) циклов инфекции в клетках МТ-2. Оценка чувствительности к D-D4FC вируса из посева 37 отбора №1 (р37 D-D4FC#1) показала, что р37 D-D4FC#1 в 19,4 раза менее чувствителен к D-D4FC, чем дикий тип, что выражается в увеличении значения EC50 с 0,21 мкМ до 4,07 мкМ (фиг.2, таблица 3). Оценка чувствительности к D-D4FC вируса из посева 20 отбора №2 (р20 D4FC#2) показала, что р20 D-D4FC#2 в 5,3 раза менее чувствителен к D-D4FC, чем дикий тип, что выражается в увеличении значения EC50 с 0,21 мкМ до 1,1 мкМ (фиг.2, таблица 3).
Генотипирование устойчивого вируса. вРНК из р37 D-D4FC#1 и р20 D-D4FC#2 выделяли и подвергали амплификации с помощью ОТ-ПЦР. Секвенирование продукта ПЦР выявило наличие трех новых мутаций в р37 D-D4FC#1: K70N (ААА→4ААТ), V901 (GTT→ATT) и R172K (AGA→AAA) (таблица 2). Две различные мутации были обнаружены в р20 D-D4FC#2: K65R (AAA→AGA) и V179D (GTT→GAT) (таблица 3). Других мутаций генов ОТ данных вирусов обнаружено не было (по аминокислотам 8-330). Кроме того, мутации не были обнаружены в контрольных вирусах, высеваемых параллельно в отсутствие лекарственного средства.
Выделение устойчивых к D-D4FC вирусов с различными ассоциированными мутациями может являться результатом различных методик отбора, применяемых для выделения устойчивого к D-D4FC вируса. В отборе №1 давление отбора снимали на один цикл инфекции до повышения концентрации лекарственного средства. Этого не было сделано в ходе второй процедуры отбора. Кроме того, исходная концентрация D-D4FC, примененная в каждой из процедур отбора, варьировалась приблизительно в 3 раза. Гораздо более высокая исходная концентрация лекарственного средства применялась для отбора №1 (0,75 мкМ). Данные два различия, вероятно, вызвали различные мутации, наблюдаемые в отобранных вирусах.
Мутантный рекомбинантный ВИЧ-1. Мутантный рекомбинантный ВИЧ-1, содержащий мутации, обнаруженные в отобранных вирусах, получали с помощью сайт-направленного мутагенеза. В таблице 4 перечислен каждый полученный мутантный рекомбинантный вирус, равно как и соответствующее значение EC50. В то время, как вирус xxHIV-1LAI, K65R проявил 3,9-кратное снижение чувствительности к D-D4FC, ни один другой вирус не проявил более чем 3,0-кратной устойчивости. Следует отметить, однако, что тройной мутант (xxHIV-1LAI, K70N/V90I/R172K) размножался плохо, и это могло быть причиной неспособности воспроизводить устойчивость, наблюдаемую в отобранном in vitro вирусе.
Чувствительность и ассоциированные мутации отобранного с помощью D-D4FC вируса в клетках МТ-2
V90I
R172K
Чувствительность к D-D4FC рекомбинантного ВИЧ-1 в клетках МТ-2
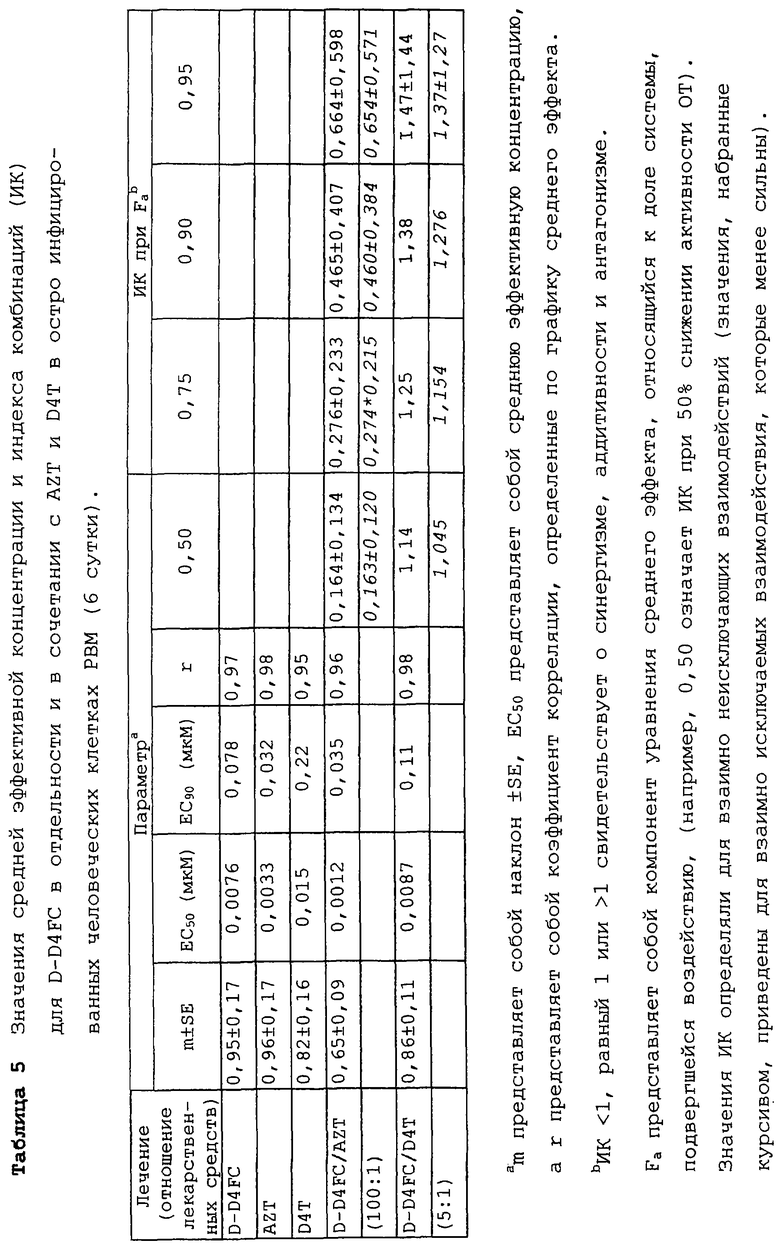
В таблице 5 приведены значения средней эффективной концентрации и индекса комбинаций (ИК) для D-D4FC в отдельности и в сочетании с AZT и D4T в остро инфицированных человеческих клетках РВМ (6 сутки). В таблице 6 описан эффект β-D и β-L-D4FC против ВИЧ-1 и клонируемых вирусов в человеческих клетках РВМ.

III. Сочетание или чередование анти-ВИЧ средств
Как правило, при чередующейся терапии эффективную дозу каждого агента вводят серийно, тогда как при сочетанной терапии эффективную дозу двух или более агентов вводят совместно. При чередующейся терапии, например, один или несколько первых агентов могут вводиться в эффективном количестве в течение эффективного периода времени для лечения вирусной инфекции, а затем могут вводиться один или несколько вторых агентов, замещающих указанные первые агенты в традиционных схемах лечения и аналогичным образом вводимых в эффективном количестве в течение эффективного периода времени.
Дозировки будут зависеть от таких факторов, как скорость всасывания, биологическое распределение, метаболизм и выведение каждого лекарственного средства, равно как и от других факторов, известных специалистам в данной области. Следует также отметить, что значение дозировки будет также варьироваться в зависимости от тяжести подлежащего лечению состояния. Следует также понимать, что для каждого конкретного субъекта должны быть разработаны особые временные режимы и схемы введения в зависимости от индивидуальной потребности и профессионального мнения лица, осуществляющего или наблюдающего за введением композиций. Примеры подходящих интервалов дозировок анти-ВИЧ соединений, включая нуклеозидные производные (например, AZT, D4T, DDI и 3ТС) или ингибиторы протеаз, например нелфинавир и индинавир, можно найти в научной литературе и в справочниках терапевта. Множество примеров подходящих интервалов дозировок других соединений также можно найти в опубликованной литературе или установить в соответствии с известными методиками. Данные интервалы дозировок могут быть модифицированы по желанию для достижения желаемого результата.
В одном предпочтительном осуществлении D-D4FC вводят в сочетании с ингибитором протеаз. В конкретных осуществлениях D-D4FC вводят в сочетании или чередовании с индинавиром (Crixivan), нелфинавиром ([3S-[2(2S*,3S*), 3-альфа,4-а-бета, 8а-бета]]-N-(1,1-диметилэтил)декагидро-2-)2-гидрокси-3-[(3-гидрокси-2-метилбензоил) амино]-4-(фенилтио)бутил]-3-изохинолинкарбоксамида монометансульфонатом) (Viracept), саквинавиром (Invirase) или 141W94 (ампренавиром; (S)-тетрагидрофуран-3-ил-N-[(1S,2R)-3-[N-[(4-аминофенил)сульфонил]-N-изобутиламино]-1-бензил-2-гидроксипропил] карбаматом); или эфавиренцем (efavirenz) ((S)-6-хлор-4-(циклопропилэтинил)-1,4-дигидро-4-(трифторметил)-2Н-3,1-бензоксазин-2-оном).
В другом предпочтительном осуществлении D-D4FC вводят в сочетании или чередовании с нуклеозидным аналогом, включая абакавир (1592U89), который представляет собой (1S,4R)-4-[2-амино-6-циклопропиламино)-9Н-пурин-9-ил]-2-циклопентен-1-метанола сукцинат.
В другом осуществлении D-D4FC вводят в сочетании с ненуклеозидным ингибитором обратной транскриптазы, таким как DMP-266 ((S)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2Н-3,1-бензоксазин-2-он) (SUSTIVA, смотри патент США №5519021); делавирдин (1-[3-(1-метилэтил)амино]-2-пиридинил-4-[[5-[(метилсульфонил)амино]-1Н-индол-2-ил]карбонил]моноэтансульфонат), невирапин или деларвирдин.
В других осуществлениях D-D4FC вводят в сочетании или чередовании с ингибитором ВИЧ-интегразы или ингибитором хемокина.
II. Анализ индуцированной D-D4FC мутации генома ВИЧ
Для анализа присутствия мутаций, индуцированных D-D4FC, могут быть использованы способы и наборы, сходные с таковыми, описанными в патенте США №5409810, выданном Larder et al., для AZT, но основанные на профиле мутаций для D-D4FC, и, таким образом, они образуют часть настоящего изобретения, представленного здесь. В дополнение к включению патента Larder в качестве ссылки во всей полноте, данные методики изложены ниже.
В одном аспекте настоящее изобретение относится к способу оценки чувствительности образца ВИЧ-1 к D-D4FC, который предусматривает:
(i) выделение нуклеиновой кислоты из образца,
(ii) гибридизацию олигонуклеотида с нуклеиновой кислотой, причем олигонуклеотид комплементарен области последовательности ДНК дикого типа (или соответствующей ей РНК) или области мутантной последовательности ДНК (или соответствующей ей РНК);
(iii) попытку полимеризации нуклеиновой кислоты с 3'-конца олигонуклеотида,
(iv) проверку факта присутствия удлиненного продукта олигонуклеотидного праймера.
В данной методике может быть использована геномная ДНК или РНК, выделенная из образцов ВИЧ-1. Подходящие клетки для поддержания роста изолята ВИЧ-1 инкубируют в течение некоторого периода времени. Клетки выделяют путем центрифугирования. ДНК может быть затем выделена путем разрушения клеток протеиназой К в присутствии ЭДТА и детергента, такого как ДСН, с последующей экстракцией добавлением фенола.
Для выделения ДНК из образца доступны широко известные методики экстракции и очистки. РНК может быть выделена с использованием следующей методики. Подходящие клетки инфицируют и инкубируют в течение некоторого периода времени. Клетки выделяют путем центрифугирования. Клетки ресуспендируют в буфере для экстракции РНК с последующим разрушением с использованием буфера для протеиназного разрушения и разрушением протеиназой К. Белки удаляют в присутствии смеси фенол/хлороформ. РНК может быть затем выделена с помощью следующих стадий центрифугирования (Maniatis, Т., et al., Molecular Cloning, A Laboratory Manual, 2nd Edition, Cold Spring Harbor Laboratory Press, (1989)).
Хотя можно применять неамплифицированную нуклеиновую кислоту, вследствие относительной редкости нуклеиновой кислоты в образце ВИЧ-1 предпочтительно ее амплифицировать. Нуклеиновая кислота может быть селективно амплифицирована с использованием технологии полимеразной цепной реакции (ПЦР), которая представляет собой in vitro методику получения больших количеств конкретного фрагмента нуклеиновой кислоты определенной длины и последовательности с малых количеств матрицы.
ПЦР состоит из стандартных условий реакции, включая концентрацию Mg2+, олигонуклеотидные праймеры и температурные условия проведения циклов. Выбирают такие праймеры, которые амплифицируют полноразмерный ген ОТ или выбранную последовательность, которая включает нуклеотиды, соответствующие области ДНК ВИЧ-1 дикого типа, которая включает мутированный кодон.
РНК не может подвергаться непосредственной амплификации путем ПЦР. Должна быть синтезирована соответствующая ей кДНК. Синтез кДНК, как правило, проводят путем праймированной обратной транскрипции с применением олиго-dT праймеров. Преимущественно, выбирают такие праймеры, которые упрощают последовательность нуклеиновой кислоты для ОТ или выбранную последовательность, которая включает нуклеотиды, соответствующие области РНК, соответствующей последовательности ДНК дикого типа или области мутантной последовательности ДНК, соответствующей 70-му (К на N), 90-му или 172-му кодону области обратной транскриптазы. Это может быть достигнуто путем получения олигонуклеотидного праймера, который комплементарен области цепи РНК, которая находится выше соответствующей последовательности ДНК дикого типа. кДНК, полученная в соответствии с данной методикой (смотри Maniatis, Т., et al., supra), может быть далее применена так же, как и уже обсуждавшаяся ДНК.
Следующей стадией методики является гибридизация с нуклеиновой кислотой олигонуклеотида, который комплементарен области последовательности ДНК дикого типа (или соответствующей ей РНК) или области мутантной последовательности ДНК (или соответствующей ей РНК).
Далее предоставляются условия и реагенты, обеспечивающие протекание полимеризации нуклеиновой кислоты с 3'-конца олигонуклеотидного праймера. Такие реакции полимеризации широко известны в данной области.
Если олигонуклеотидный праймер содержит на своем 3'-конце нуклеотид, который комплементарен мутантному генотипу, который представляет собой генотип, который содержит нуклеотидную замену по 70-му (К на N), 90-му или 172-му кодону области ОТ, полимеризация последовательности нуклеиновой кислоты происходит лишь в случае, если нуклеиновая кислота образца такая же, что и мутантный генотип. Полимеризация последовательности нуклеиновой кислоты дикого типа не будет происходить или по крайней мере не будет происходить в значительной степени вследствие несовпадения нуклеотидов на 3'-конце олигонуклеотидного праймера и последовательности нуклеиновой кислоты из образца.
Если олигонуклеотидный праймер содержит на своем 3'-конце нуклеотид, который комплементарен генотипу дикого типа, который представляет собой генотип, который содержит нуклеотид дикого типа в 70-м (К на N), 90-м или 172-м кодоне области ОТ, будет иметь место полимеризация последовательности нуклеиновой кислоты, которая соответствует дикому типу по данному положению. Полимеризация не будет происходить с нуклеиновой кислотой, которая содержит мутантный нуклеотид в 3'-положении.
Предпочтительная длина каждого нуклеотида составляет 15-20 нуклеотидов. Олигонуклеотид может быть получен в соответствии с методикой, хорошо известной специалистам в данной области (Koster, H., Drug Research, 30, р.548 (1980); Koster, H., Tetrahedron Letters, p.1527 (1972); Caruthers, Tetrahedron Letters, p.719, (1980); Tetrahedron Letters, p.1859, (1981); Tetrahedron Letters, 24, р.245, (1983); Gate, M. Nucleic Acid Research, 8, p. 1081, (1980)), и, как правило, его получают с применением автоматизированного ДНК-синтезатора, такого как синтезатор Applied Biosystems 381A.
Существует удобный способ определять наличие удлиненного продукта олигонуклеотидного праймера. Инструментом для проведения определения является подходящая метка.
Метка может быть подходящим образом прикреплена к олигонуклеотидному праймеру или к какой-либо другой молекуле, которая будет связываться с удлиненным продуктом олигонуклеотидного праймера.
Метка может представлять собой, например, фермент, радиоизотоп или флуорохром. Предпочтительной меткой может являться биотин, который может быть впоследствии определен с помощью стрептавидина, конъюгированного с ферментом, таким как пероксидаза или щелочная фосфатаза. Присутствие удлиненного продукта олигонуклеотидного праймера может быть определено, например, путем разгона реакции полимеризации в агарозном геле и наблюдения конкретного фрагмента ДНК, меченного бромидом этидия, или путем Саузерн-блоттинга и авторадиографии для определения наличия или отсутствия полос, соответствующих полимеризованному продукту. Если имеется преобладающая полоса, которая соответствует только меченому олигонуклеотиду, это свидетельствует о том, что полимеризация произошла. Если полосы имеют точно предсказанный размер, это будет указывать на то, что полимеризация произошла.
Например, ДНК, выделенную из лимфоцитов пациентов, как здесь описано, применяют в качестве матрицы для ПЦР-амплификации с применением синтетических олигонуклеотидных праймеров, которые либо совпадают, либо не совпадают с амплифицированными последовательностями. Применимость ПЦР для определения таких мутаций уже была продемонстрирована. ПЦР с помощью системы Amplification Refractory Mutation ("ARMS") (Newton, C.R., et al., Nucleic Acids Research, 17, p.2503, (1989)). Получают такие синтетические олигонуклеотиды, которые гибридизуются с областями, смежными с таковой, включающей специфичные мутации, что 3'-конец олигонуклеотида либо совпадает, либо не совпадает с мутантной последовательностью или последовательностью дикого типа. Проводят ПЦР, которая приводит к идентификации фрагмента ДНК (с применением электрофореза в геле), в котором произошло совпадение, или к неопределению фрагмента, в котором произошло несовпадение.
ДНК выделяют из инфицированных ВИЧ-1 Т-клеток, как здесь описано, и подвергают ПЦР-анализу с помощью "ARMS" с применением данных праймеров.
Наличие фрагмента определяют путем использования олигонуклеотидного праймера, как описано выше, например путем попытки проведения полимеризации с применением олигонуклеотидного праймера, который может быть помечен, с получением фрагмента ДНК, амплифицированного в жестких условиях, которые обеспечивают гибридизацию лишь комплементарной ДНК (единственным отличием является то, что дифференциальная гибридизация не должна проводиться, поскольку фрагменты ДНК, амплифицированные по методу "ARMS", полученные как из мутантной ДНК, так и из ДНК дикого типа, будут одинаковы, так что общий олигонуклеотид может быть применен для определения наличия данных фрагментов. Последовательность такого олигонуклеотида происходит из последовательности ДНК из фрагмента ДНК, который консервативен среди штаммов ВИЧ-1).
Описанный выше ПЦР-анализ может быть приспособлен для обеспечения прямого определения мутаций, связанных с устойчивостью к D-D4FC, в ДНК из образцов PBL из инфицированных субъектов, которые не культивировали для получения вируса. Поскольку данный материал, как правило, содержит значительно меньше ДНК ВИЧ-1, чем таковой в инфицированных лимфоидных культурах, может быть использован протокол "двойной ПЦР" (или уплотненного набора) (Simmonds, P., Balfe, P, Peutherer, J.F., Ludlam, С. A., Bishop, J.O. and Leigh Brown, A.J., J. Virol., 64, 864-872, (1990)) для увеличения количества сигнала целевой ДНК ОТ ВИЧ-1 в образцах. Двойная ПЦР позволяет преодолеть проблему ограниченной амплификации редкой матричной последовательности. Малое количество предварительно амплифицированного материала может быть использовано во второй ПЦР с парами праймеров, сконструированных для обеспечения дискриминации остатков дикого типа и мутантных остатков.
Подходящий набор для теста для применения в анализе для определения статуса устойчивости образца ВИЧ-1 к D-D4FC, который использует методику в соответствии с первым аспектом настоящего изобретения, содержит олигонуклеотид, комплементарный области последовательности ДНК дикого типа (или соответствующей ей РНК) или области последовательности мутантной ДНК, как описано здесь, другие материалы, необходимые для полимеризации нуклеиновой кислоты с 3'-конца олигонуклеотида, и средства для определения присутствия удлиненного продукта олигонуклеотидного праймера. Такие другие материалы включают подходящие ферменты, буферы и промывающие растворы, и метку и субстрат для метки, если необходимо. Если для амплификации нуклеиновой кислоты применяют ПЦР, должны быть также включены дополнительные материалы, такие как подходящие олигонуклеотидные праймеры, которые амплифицируют область последовательности ДНК дикого типа (или соответствующую ей РНК) или область последовательности мутантной ДНК (или соответствующую ей РНК) и dNTP.
Во втором аспекте настоящее изобретение относится к способу определения чувствительности образца ВИЧ-1 к D-D4FC, который предусматривает:
(i) выделение нуклеиновой кислоты из образца,
(ii) гибридизацию нуклеиновой кислоты с олигонуклеотидом, комплементарным области последовательности ДНК дикого типа (или соответствующей ей РНК) или области мутантной последовательности ДНК, указанной на фиг.1 (или соответствующей ей РНК), содержащей один или несколько нуклеотидов в области 70-го (К на N), 90-го или 172-го кодона в области ОТ; и
(III) проверку факта, что полученные гибриды олигонуклеотида и нуклеиновой кислоты содержат комплементарные нуклеотиды в одном из данных положений.
Предпочтительно, олигонуклеотид сконструирован таким образом, что образует высококомплементарный гибрид со своим комплементом.
Нуклеиновую кислоту (ДНК или РНК) выделяют из образца в соответствии с указанными выше методиками, описанными в первом аспекте настоящего изобретения.
Аналогично, ПЦР может применяться для амплификации ДНК ОТ (или соответствующей ей РНК) или, предпочтительно, для амплификации области ДНК ОТ (или соответствующей ей РНК), которая включает ДНК (или соответствующую ей РНК), содержащую один или несколько нуклеотидов в желаемом положении.
На второй стадии данной методики далее используется нуклеиновая кислота для гибридизации с олигонуклеотидами, комплементарными области последовательности ДНК дикого типа (или соответствующей ей РНК) или области мутантной последовательности ДНК.
Олигонуклеотид может иметь любую длину в зависимости от числа интересующих исследуемых нуклеотидных положений. Если олигонуклеотид сконструирован таким образом, чтобы включать нуклеотид только в одном интересующем положении, данный нуклеотид, предпочтительно, находится в непосредственной близости или рядом с центральным положением олигонуклеотида.
Для проверки факта образования комплементарного гибрида между олигонуклеотидом и последовательностью нуклеиновой кислоты специфичные условия гибридизации устанавливаются таким образом, что гибрид образуется только в том случае, если нуклеотид или нуклеотиды в 70-м (К на N), 90-м или 172-м кодоне области обратной транскриптазы комплементарны соответствующему нуклеотиду или нуклеотидам олигонуклеотида, что либо обеспечивает гибридизацию, либо нет. Важным, например, является установить температуру реакции и концентрацию соли в растворе до осуществления стадии гибридизации для определения условий, которые являются достаточно жесткими для обеспечения специфичности (Maniatis, Т., et al., Molecular Cloning, A Laboratory Manual, 2nd edition, Cold Spring Harbour Laboratory Press, (1989)). Если олигонуклеотидный зонд имеет последовательность ДНК, которая комплементарна последовательности нуклеиновой кислоты дикого типа по одному или нескольким ее нуклеотидам, соответствующим 70-му (К на N), 90-му или 172-му кодону в области обратной транскриптазы, данный олигонуклеотид высококомплементарно гибридизуется с нуклеиновой кислотой дикого типа. Если гибридизации не происходит, это позволяет предположить, что нуклеиновая кислота, выделенная из образца, содержит одну или несколько мутаций.
Если олигонуклеотидный зонд имеет последовательность ДНК, которая комплементарна последовательности мутантной нуклеиновой кислоты, данный олигонуклеотид гибридизуется с мутантной нуклеиновой кислотой. Если гибридизации не происходит, это позволяет предположить, что нуклеиновая кислота, выделенная из образца, не содержит такой мутации или мутаций. Олигонуклеотидные зонды могут быть помечены как инструмент определения, как описано в отношении первого аспекта настоящего изобретения.
Гибридизацию и последующее удаление негибридизованных нуклеиновых кислот проводят в жестких условиях, которые обеспечивают гибридизацию только комплементарной ДНК, но не олигонуклеотида, содержащего несовпадение (т.е. олигонуклеотид-специфичная мутация, как описано для определения мутации при серповидноклеточной анемии, затрагивающей ген β-глобина, или HLA-DQα (Saikt, R.К., et al., Nature, 324, р.163, (1986)), активированный ген Ras (Ver Laande, Vries, M., et al., Gene, 50, 313, (1986)) и при β-талассемии (Wong, С., et al., Nature, 330, р.384, (1987)).
Гибридизация может быть проведена путем иммобилизации последовательности нуклеиновой кислоты ОТ на нитроцеллюлозе, найлоне или другом твердом носителе (например, дот-блот). Удобно определять присутствие гибрида, используя в качестве инструмента метку. Например, химически синтезированные олигонуклеотидные зонды могут быть подходящим образом помечены ферментом, радиоизотопом или флуорохромом. Предпочтительной меткой может являться биотин, который может быть впоследствии определен с помощью стрептавидина, конъюгированного с ферментом, таким как пероксидаза или щелочная фосфатаза.
Альтернативно, гибридизация может быть проведена путем иммобилизации химически синтезированных олигонуклеотидов, на которые ссылались выше, которые не являются мечеными, на твердой подложке, на которую ссылались выше, и последующей гибридизацией с помеченной последовательностью нуклеиновой кислоты ОТ, описанной ранее.
В обеих описанных выше для гибридизации ситуациях будут включены подходящие контрольные реакции для определения эффективности гибридизации (например, гибридизация олигонуклеотидов с комплементарным нуклеотидом).
Результаты могут быть легко интерпретированы, поскольку выделенная нуклеиновая кислота гибридизуется либо с олигонуклеотидом дикого типа, либо с мутантным олигонуклеотидом.
Подходящий набор для теста для применения в анализе для определения чувствительности образца ВИЧ-1 к D-D4FC, который использует методику в соответствии со вторым аспектом настоящего изобретения, содержит олигонуклеотид, комплементарный области последовательности ДНК дикого типа (или соответствующей ей РНК) или соответствующей области последовательности мутантной ДНК, а также другие материалы, необходимые для обеспечения гибридизации. Такие материалы включают подходящие буферы и промывающие растворы и метку и субстрат для метки, если это необходимо. Как правило, олигонуклеотид помечен. Если ПЦР применяют для амплификации нуклеиновой кислоты до гибридизации, должны быть также включены дополнительные материалы, такие как подходящие олигонуклеотидные праймеры, которые амплифицируют область последовательности ДНК дикого типа (или соответствующую ей РНК) или область последовательности мутантной ДНК (или соответствующую ей РНК), подходящие ферменты и dNTP (дезоксинуклеотидтрифосфаты).
В одном альтернативном формате анализа dNTP при амплификации могут быть или не быть помечены детекторной молекулой, такой как радиоизотоп, биотин, флуорохром или фермент.
Также возможно определить мутации устойчивости к зидовудину РНК ОТ ВИЧ-1, выделенной из клинических образцов, с применением системы амплификации РНК. С использованием методики, описанной Guatelli et al. (Proc.Natl.Acad.Sci, (USA), 8/7, 1874-1878, (March 1990)), целевая последовательность нуклеиновой кислоты может быть реплицирована (амплифицирована) экспоненциально in vitro в изотермальных условиях путем применения трех ферментативных активностей, необходимых для репликации ретровируса: обратной транскриптазы, РНКазы Н и ДНК-зависимой РНК-полимеразы. Такая методика может быть использована с последующей стадией гибридизации для отличения мутантных нуклеотидов от нуклеотидов дикого типа, как обсуждалось ранее.
Получение фармацевтических композиций
Люди, страдающие от эффектов, вызванных любым заболеванием, описанным здесь, и, в частности, ВИЧ-инфекцией, могут получать лечение путем введения пациенту эффективного количества D-D4FC или его фармацевтически приемлемой соли или пролекарства в присутствии фармацевтически приемлемого носителя или разбавителя по любому показанию или в соответствии с любым способом введения, подробно описанными здесь. Активные вещества могут вводиться любым подходящим путем, например, перорально, парентерально, энтерально, внутривенно, внутрикожно, подкожно, чрескожно, интраназально или местно в жидкой или твердой форме.
Активное(ые) соединение(я) включают в фармацевтически приемлемый носитель или разбавитель в количестве, достаточном для доставки пациенту терапевтически эффективного количества соединения для ингибирования репликации вируса in vivo, особенно репликации ВИЧ, без вызывания серьезных токсических эффектов у получающего лечение пациента. Под "ингибирующим количеством" подразумевается количество активного ингредиента, достаточное для оказания ингибирующего эффекта, измеренного, например, с помощью анализа, такого как описанные здесь.
Предпочтительная доза соединения для всех указанных выше состояний составляет приблизительно от 1 до 75 мг/кг, предпочтительно от 1 до 20 мг/кг массы тела в сутки, вообще от 0,1 приблизительно до 100 мг/кг массы тела реципиента в сутки. Эффективный интервал дозировки фармацевтически приемлемых производных может быть рассчитан, исходя из массы исходного нуклеозида, подлежащего доставке. Если производное проявляет активность само по себе, эффективная дозировка может быть установлена, как описано выше, исходя из массы производного, или другими способами, известными специалистам в данной области.
Соединения удобно вводить в виде любой подходящей дозированной формы, включая, но не ограничиваясь, формой, содержащей от 7 до 3000 мг, предпочтительно от 70 до 1400 мг активного ингредиента на разовую дозированную форму. Обычно удобной является пероральная дозировка от 50 до 1000 мг.
В идеале, активный ингредиент должен вводиться для достижения пика концентрации активного соединения в плазме, составляющего приблизительно от 0,2 до 70 микромоль/л, предпочтительно приблизительно от 0,5 до 10 мМ. Это может быть достигнуто, например, с помощью внутривенной инъекции 0,1-25% раствора активного ингредиента, необязательно в солевом растворе, или введено в виде болюса активного ингредиента.
Концентрация активного соединения в композиции лекарственного средства будет зависеть от скорости всасывания, распределения, метаболизма и выведения лекарственного средства, равно как и от других факторов, известных специалистам в данной области. Следует отметить, что значения дозировки также будут варьироваться в зависимости от тяжести подлежащего излечению состояния. Следует также понимать, что для каждого конкретного субъекта должны быть разработаны особые временные режимы и схемы введения в зависимости от индивидуальной потребности и профессионального мнения лица, осуществляющего или наблюдающего за введением композиций, и что интервалы концентраций, приведенные здесь, являются лишь иллюстративными и не предназначены для ограничения объема притязаний или осуществления заявленной композиции. Активный ингредиент может вводиться сразу или может быть разделен на ряд меньших доз, подлежащих введению через варьирующиеся интервалы времени.
Предпочтительным способом введения активного соединения является пероральный. Пероральные композиции, как правило, включают инертный разбавитель или съедобный носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. В целях перорального терапевтического введения активное соединение может быть включено в наполнители и применено в форме таблеток, пастилок или капсул. В качестве части композиции могут быть включены фармацевтически совместимые связующие агенты и/или адъюванты.
Таблетки, пилюли, капсулы, пастилки и т.п. могут содержать любой из следующих ингредиентов или соединения схожей природы: связующее вещество, такое как микрокристаллическая целлюлоза, смола трагаканта или желатин; наполнитель, такой как крахмал или лактоза; дезинтегратор, такой как альгиновая кислота, Primogel или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или Sterotes; скользящее вещество, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; и корригент, такой как перечная мята, метилсалицилат или апельсиновый корригент. Когда разовая дозированная форма представляет собой капсулу, она может содержать, помимо веществ, описанных выше, жидкий носитель, такой как нелетучее масло. Кроме того, разовые дозированные формы могут содержать различные другие вещества, которые модифицируют физическую форму дозированной единицы, например, покрытия из сахара, шеллака или других энтеросолюбильных агентов.
Соединения могут вводиться в виде компонента эликсира, суспензии, сиропа, вафли, жевательной резинки и тому подобного. Сироп может содержать, помимо активных соединений, сахарозу в качестве подсластителя и определенные консерванты, красители и корригенты.
Соединения или их фармацевтически приемлемые производные или их соли также могут быть смешаны с другими активными веществами, которые не ослабляют желаемого воздействия, или с веществами, которые усиливают желаемое воздействие, такими как антибиотики, противогрибковые средства, противовоспалительные средства, ингибиторы протеаз или другие нуклеозидные или ненуклеозидные противовирусные средства, более подробно обсуждавшиеся выше. Растворы и суспензии, применяемые для парентерального, внутрикожного, подкожного или местного введения, могут содержать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота и бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие ацетаты, цитраты или фосфаты, и агенты для доводки тоничности, такие как хлорид натрия или декстроза. Парентеральный препарат может быть заключен в ампулы, одноразовые шприцы и сосуды на много доз, изготовленные из стекла или пластика.
При внутривенном введении предпочтительными носителями являются физиологический раствор или фосфатно-буферный раствор (ФБР).
Суспензии липосом (включая липосомы, нацеленные на инфицированные клетки моноклональными антителами против вирусных антигенов) также предпочтительны в качестве фармацевтически приемлемых носителей, они могут быть приготовлены в соответствии с методиками, известными в данной области, например, как описано в патенте США №4522811 (который включен сюда полностью в качестве ссылки). Например, липосомные композиции могут быть приготовлены путем растворения подходящего(их) липида(ов), (такого(их) как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, арахадоилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем упаривают, оставляя тонкую пленку высушенного липида на поверхности контейнера. Водный раствор активного соединения или его монофосфатных, дифосфатных и/или трифосфатных производных вносят затем в контейнер. Контейнер затем взбалтывают вручную для высвобождения липидного материала со стенок контейнера и для диспергирования липидных агрегатов, в результате чего образуется суспензия липосом.
Композиции с контролируемым высвобождением
Все патенты США, процитированные в данном разделе о композициях с контролируемым высвобождением, включены сюда полностью в качестве ссылки.
Область биологически разрушаемых полимеров развивалась стремительно с тех пор, как о синтезе и биологической разрушаемости полимолочной кислоты сообщили Kulkarni et al., в 1966 ("Polylactic acid for surgical implants," Arch.Surg., 93:839). Примеры других полимеров, о применимости которых в качестве матриксного вещества для устройств доставки сообщалось, включают полиангидриды, сложные полиэфиры, такие как полигликолиды и полилактид-со-гликолиды, полиаминокислоты, такие как полилизин, полимеры и сополимеры полиэтиленоксида, терминированный акрилом полиэтиленоксид, полиамиды, полиуретаны, полиортоэфиры, полиакрилонитрилы и полифосфазены. Смотри, например, патенты США №4891225 и 4906474, выданные Langer (полиангидриды); 4767628, выданный Hutchinson (полилактид, полилактид-со-гликолидовая кислота), и 4530840, выданный Tice, et al. (полилактид, полигликолид и сополимеры). Также смотри патент США №5626863, выданный Hubbell, et al, в котором описаны фотополимеризуемые биологически разрушаемые гидрогели в качестве материалов, контактирующих с тканью, и носителей с контролируемым высвобождением (гидрогели полимеризованных и поперечно сшитых макромеров, содержащие гидрофильные олигомеры, содержащие биологически разрушаемые мономерные или олигомерные удлинения, которые представляют собой кэпированные по концам мономеры или олигомеры, способные к полимеризации и поперечному сшиванию); и РСТ WO 97/05185, поданную Focal, Inc., направленную на многозвенные биологически разрушаемые гидрогели для применения в качестве агентов с контролируемым высвобождением для доставки лекарственных средств и агентов для лечения тканей.
Хорошо известны разрушаемые материалы биологического происхождения, например, поперечно сшитый желатин. Гиалуроновую кислоту поперечно сшивали и применяли в качестве разрушаемого набухающего полимера для биомедицинских применений (патент США 4957744, выданный Delia Valle et al.; (1991) "Surface modification of polymeric biomaterials for reduced thrombogenicity," Polym.Mater.Sci.Eng., 62:731-735).
Множество биологических систем ныне используются либо разрабатывались для применения в качестве носителей веществ, особенно биологически активных соединений. Дисперсионные системы, применяемые в фармацевтических и косметических композициях, могут быть подразделены на суспензии или эмульсии. Суспензии определяют как твердые частицы размером от нескольких нанометров до сотен микрон, диспергированные в жидкой среде с использованием суспендирующих агентов. Твердые частицы включают микросферы, микрокапсулы и наносферы. Эмульсии определяют как дисперсии одной жидкости в другой, стабилизированные межповерхностной пленкой эмульгаторов, таких как поверхностно-активные вещества и липиды. Эмульсионные композиции включают эмульсии "вода-в-масле" и "масло-в-воде", многокомпонентные эмульсии, микроэмульсии, микрокапли и липосомы. Микрокапли представляют собой однослойные фосфолипидные пузырьки, которые состоят из сферического липидного слоя с масляной фазой внутри, как определено в патентах США №4622219 и 4725442, выданных Haynes. Липосомы представляют собой фосфолипидные пузырьки, полученные путем смешивания водонерастворимых полярных липидов с водным раствором. Неблагоприятная энтропия, вызываемая смешиванием нерастворимого липида с водой, приводит к образованию высоко упорядоченного ансамбля концентрических закрытых мембран фосфолипида с заключенным в них водным раствором.
В патенте США №4938763, выданном Dunn, et al, описан способ образования имплантата in situ путем растворения нереакционноспособного водонерастворимого термопластического полимера в биологически совместимом водорастворимом растворителе с образованием жидкости, помещения жидкости внутрь организма и рассеивания растворителя с получением твердого имплантата. Раствор полимера может быть введен в организм посредством шприца. Имплантат способен принимать форму окружающей его полости. В альтернативном осуществлении имплантат образуется из реакционноспособных жидких олигомерных полимеров, которые не содержат растворителя и которые образуют твердое вещество непосредственно на месте, обычно с добавлением катализатора затвердевания.
В ряде патентов описаны системы доставки лекарственных средств, которые могут применяться для введения D-D4FC или нуклеотида или любого их определенного пролекарства. В патенте США №5749847 описан способ доставки нуклеотидов в организм методом электрофореза. В патенте США №5718921 описаны микросферы, содержащие полимер и диспергированное в нем лекарственное средство. В патенте США №5629009 описана система доставки для контролируемого высвобождения биологически активных факторов. В патенте США №5578325 описаны наночастицы и микрочастицы из нелинейных гидрофильных гидрофобных многозвенных сополимеров. В патенте США №5545409 описана система доставки для контролируемого высвобождения биологически активных факторов. В патенте США №5494682 описаны поперечно сшитые ионными связями полимерные микрокапсулы.
В патенте США №5728402, выданном Andrx Pharmaceuticals, Inc., описана композиция с контролируемым высвобождением, которая включает внутреннюю фазу, которая содержит активное лекарственное средство, его соль или пролекарство, в смеси с гидрогель-образующим агентом, и внешнюю фазу, которая содержит покрытие, которое препятствует растворению в желудке. В патентах США №№5736159 и 5558879, выданных Andrx Pharmaceuticals, Inc., описана композиция с контролируемым высвобождением для лекарственных средств с низкой водорастворимостью, в которой проход формируется in situ. В патенте США №5567441, выданном Andrx Pharmaceuticals, Inc., описана ежесуточно вводимая композиция с контролируемым высвобождением. В патенте США №5508040 описана мультикорпускулярная система с пульсирующей доставкой лекарственного средства. В патенте США №5472708 описана корпускулярная система с пульсирующей доставкой лекарственного средства. В патенте США №5458888 описана таблетированная композиция с контролируемым высвобождением, которая может быть изготовлена с использованием смеси, содержащей внутреннюю содержащую лекарственное средство фазу и внешнюю фазу, которая содержит полиэтиленгликолевый полимер, который обладает средней молекулярной массой от 3000 до 10000 Да. В патенте США №5419917 описаны способы модификации скорости высвобождения лекарственного средства из гидрогеля, которые основаны на применении эффективного количества фармацевтически приемлемого ионизируемого соединения, которое способно обеспечивать, по существу, нулевого порядка скорость высвобождения лекарственного средства из гидрогеля. В патенте США №5458888 описана таблетированная композиция с контролируемым высвобождением.
В патенте США №5641745, выданном Elan Corporation, plc, описана фармацевтическая композиция с контролируемым высвобождением, которая содержит активное лекарственное средство в биологически разрушаемом полимере с образованием микросфер или наносфер. Биологически разрушаемый полимер предпочтительно представляет собой поли-D,L-лактид или смесь поли-D,L-лактида и поли-D,L-лактид-со-гликолида. В патенте США №5616345, выданном Elan Corporation, plc, описана композиция с контролируемым всасыванием для ежесуточного введения, которая содержит активное соединение в сочетании с органической кислотой и многослойную мембрану, окружающую ядро и содержащую основную долю фармацевтически приемлемого пленкообразующего водонерастворимого синтетического полимера и малую долю фармацевтически приемлемого пленкообразующего водорастворимого синтетического полимера. В патенте США №5641515 описана композиция с контролируемым высвобождением на основе биологически разрушаемых наночастиц. В патенте США №5637320 описана композиция с контролируемым всасыванием для ежесуточного введения. Патенты США №№5580580 и 5540938 относятся к композициям и их применению в лечении неврологических заболеваний. Патент США №5533995 относится к пассивному чрескожному устройству с контролируемой доставкой лекарственного средства. В патенте США №5505962 описана фармацевтическая композиция с контролируемым высвобождением.
Композиции пролекарства
D-D4FC или любой нуклеозид или другое соединение, которое описано здесь для применения в сочетанной или чередующейся терапии D-D4FC или родственными ему соединениями, может вводиться в виде ацилированного пролекарства или нуклеотидного пролекарства, как подробно описано ниже.
Любой описанный здесь нуклеозид или другое соединение, которое содержит гидроксильную или аминовую функциональную группу, может вводиться в виде пролекарства нуклеотида для повышения активности, биологической доступности, стабильности или иного изменения свойств нуклеозида. Известен ряд лигандов пролекарств нуклеотидов. Как правило, алкилирование, ацилирование или иная липофильная модификация гидроксильной группы соединения или моно-, ди- или трифосфата нуклеозида приводит к увеличению стабильности нуклеотида. Примеры замещающих групп, которые способны замещать один или несколько водородов фосфатного радикала или гидроксила, представляют собой алкил, арил, стероиды, углеводы, включая сахара, 1,2-диацилглицерол и спирты. Многие описаны в R. Jones and N. Bischofberger, Antiviral Research, 27 (1995) 1-17. Любое из них может быть использовано в сочетании с описанными нуклеозидами или другими соединениями для достижения желаемого эффекта.
Активное нуклеозидное или иное гидроксилсодержащее соединение также может быть получено в виде эфирного липида (и, в частности, 5'-эфирного липида нуклеозида), как описано в следующих ссылках, которые включены сюда в качестве ссылок: Kucera, L.S., N. Iyer, E. Leake, A. Raben, Modest E.K., D.L.W. and C. Piantadosi, 1990, "Novel membrane-interactive ether lipid analogs that inhibit infectious HIV-1 production and induce defective virus formation", AIDS Res.Hum.Retro Viruses, 6:491-501; Piantadosi, C., J. Marasco C.J., S.L. Morris-Natschke, K.L. Meyer, F. Gumus, J.R. Surles, K.S. Ishaq, L.S. Kucera, N. lyer, C.A. Wallen, S. Piantadosi and E.J. Modest 1991, "Synthesis and evaluation of novel ether lipid nucleoside conjugates for anti-HTV activity", J.Med.Chem., 34:1408-1414; Hosteller, K.Y., D.D. Richman, D.A. Carson, L.M. Stuhmiller, G.M. T. van Wijk, and H. van den Bosch, 1992, "Greatly enhanced inhibition of human immunodeficiency virus type 1 replication in СЕМ and HT4-6C cells by 3'-deoxythymidine diphosphate dimyristoylglycerol, a lipid prodrug of 3'-deoxythymidine", Antimicrob.Agents Chemother., 36:2025-2029; Hostetler, K.Y., L.M. Stuhmiller, H.B. Lenting, H. van den Bosch and D.D. Richman, 1990, "Synthesis and antiretroviral activity of phospholipid analogs of azidothymidine and other antiviral nucleosides", J.Biol.Chem., 265:61127.
Неограничивающие примеры патентов США, в которых описаны подходящие липофильные заместители, которые могут быть ковалентно включены в нуклеозид или другое гидроксил- или аминосодержащее соединение, предпочтительно в 5'-ОН положении нуклеозидных или липофильных препаратов, включают патенты США №№5149794 (22 сентября 1992 года, Yatvin et al.); 5194654 (16 марта 1993 года, Hostetler et al.); 5223263 (29 июня 1993 года, Hostetler et al.); 5256641 (26 октября 1993 года, Yatvin et al.); 5411947 (2 мая 1995 года, Hostetler et al.); 5463092 (31 октября 1995 года, Hostetler et al.); 5543389 (6 августа 1996 года, Yatvin et al.); 5543390 (6 августа 1996 года, Yatvin et al.); 5543391 (6 августа 1996 года, Yatvin et al.) и 5554728 (10 сентября 1996 года; Basava et al.), каждый из которых включен сюда в качестве ссылки. Зарубежные заявки на выдачу патента, в которых описаны липофильные заместители, которые могут быть присоединены к нуклеозидам по настоящему изобретению, или липофильные препараты, включают WO 89/02733, WO 90/00555, WO 91/16920, WO 91/18914, WO 93/00910, WO 94/26273, WO 96/15132, ЕР 0 350 287, ЕР 93917054.4 и WO 91/19721.
Неограничивающие примеры нуклеотидных пролекарств описаны в следующих ссылках: Но, D.H.W. (1973) "Distribution of Kinase and deaminase of 1β-D-arabinofuranosylcytosine in tissues of man and mouse". Cancer Res., 33, 2816-2820; Holy, A. (1993) Isopolar phosphorous-modified nucleotide analogues," In: De Clercq (Ed.), Advances in Antiviral Drug Design, Vol. 1, JAI Press, pp. 179-231; Hong, C.I., Nechaev, A. and West, C.R. (1979a) "Synthesis and antitumor activity of 1β-D-arabinofuranosylcytosine conjugates of cortisol and cortisone", Biochem.Biophys.Res.Commun., 88, 1223-1229; Hong, C.I., Nechaev, A., Kirisits, A.J. Buchheit, D.J. and West, C.R. (1980) "Nucleoside conjugates as potential antitumor agents. 3. Synthesis and antitumor activity of 1-(β-D-arabinofuranosyl)cytosine conjugates of corticosteroids and selected lipophilic alcohols", J.Med.Chem., 28, 171-177; Hosteller, K.Y., Stuhmiller, L.M., Lenting, H.B.M. van den Bosch, H. and Richman, J.Biol.Chem., 265, 6112-6117; Hosteller, K.Y., Carson, D.A. and Richman, D.D. (1991); "Phosphatidylazidothymidine: mechanism of antiretroviral action in СЕМ cells", J.Biol.Chem., 266, 11714-11717; Hosteller, K.Y., Korba, B. Sridhar, C., Gardener, M. (1994a) "Antiviral activity of phosphatidyl-dideoxycytidine in hepatitis B-infected cells and enhanced hepatic uptake in mice". Antiviral Res., 24, 59-67; Hosteller, K.Y., Richman, D.D., Sridhar. C.N. Feigner, P.L. Feigner, J., Ricci, J., Gardener, M.F. Seileseth, D.W. and Ellis, M.N. (1994b) "Phosphatidylazidothymidine and phosphatidyl-ddC: Assessment of uptake in mouse lymphoid tissues and antiviral activities in human immunodeficiency virus-infected cells and in rauscher leukemia virus-infected mice", Antimicrobial Agents Chemother., 38, 2792-2797; Hunston, R.N., Jones, A.A. McGuigan, C., Walker, R.T., Balzarini, J., and DeClercq, E. (1984) "Synthesis and biological properties of some cyclic phosphotriesters derived from 2'-deoxy-5-fluorouridine", J.Med.Chem., 27,440-444; Ji, Y.H., Мооg, С., Schmitt, G., Bischoff, P. and Luu, B. (1990); "Monophosphoric acid esters of 7-β-hydroxycholesterol and of pyrimidine nucleoside as potential antitumor agents: synthesis and preliminary evaluation of antitumor activity", J. Med.Chem., 33 2264-2270; Jones, A.S., McGuigan, C., Walker, R.T., Balzarini, J. and DeClercq, E. (1984) "Synthesis, properties, and biological activity of some nucleoside cyclic phosphoramidates", J.Chem.Soc., Perkin Trans. I, 1471-1474; Juodka, B.A. and Smart, J. (1974) "Synthesis of diribonucleoside phosph (P→N) amino acid derivatives". Coil.Czech.Chem. Comm., 39, 363-968; Kataoka, S., Imai, J., Yamaji, N., Kato, M., Saito, M., Kawada, T. and Imai, S. (1989) "Alkylated cAMP derivatives: selective synthesis and biological activities", Nucleic Acids Res.Sym.Ser., 21, 1-2; Kataoka, S., Uchida, "(cAMP) benzyl and methyl triesters", Heterocycles, 32, 1351-1356; Kinchington, D., Harvey, J.J., O'Connor, T.J., Jones, B.C.N.M., Devine, K.G., Taylor-Robinson D., Jeffries, D.J. and McGuigan, C. (1992) "Comparison of antiviral effects of zidovudine phosphoramidate and phosphorodiamidate derivatives against HIV and ULV in vitro". Antiviral Chem.Chemother., 3, 107-112; Kodama, К., Morozumi, M., Saithoh, K.I., Kuninaka, H., Yosino, H. and Saneyoshi, M. (1989) "Antitumor activity and pharmacology of 1-β-D-arabinofuranosylcytosine-5'-stearylphosphate; an orally active derivative of 1-β-D-arabinofuranosylcytosine", Jpn.J.Cancer Res., 80, 679-685; Korty, M. and Engels, J. (1979) "The effects of adenosine- and guanosine 3',5' phosphoric and acid benzyl esters on guinea-pig ventricular myocardium", Naunyn-Schmiedeberg's Arch. Pharmacol., 310, 103-111; Kumar, A., Goe, P.L., Jones, A.S. Walker, R.T.Balzarini, J. and DeClercq, E. (1990) "Synthesis and biological evaluation of some cyclic phosphoramidate nucleoside derivatives", J.Med.Chem., 33, 2368-2375; LeBec, С., and Huynh-Dinh, T. (1991) "Synthesis of lipophilic phosphate triester derivatives of 5-fluorouridine an arabinocytidine as anticancer prodrugs". Tetrahedron Lett., 32, 6553-6556; Lichtenstein, J., Bamer, H.D. and Cohen, S.S. (1960) "The metabolism of exogenously supplied nucleotides by Escherichia coli", J.Biol.Chem., 235, 457-465; Lucthy, J., Von Daeniken, A., Friederich, J. Manthey, В., Zweifel, J., Schlatter, C. and Benn, M.H. (1981) "Synthesis and toxicological properties of three naturally occurring cyanoepithioalkanes", Mitt.Geg. Lebensmittelunters.Hyg., 72, 131-133 (Chem.Abstr. 95, 127093); McGigan, С. Tollerfield, S.M. and Riley, P.A. (1989) "Synthesis and biological evaluation of some phosphate triester derivatives of the anti-viral drug Ara", Nucleic Acids Res., 17, 6065-6075; McGuigan, С., Devine, K.G., O'Connor, T.J., Galpin, S.A., Jeffries, D.J. and Kinchington, D. (1990a) "Synthesis and evaluation of some novel phosphoramidate derivatives of 3'-azido-3'-deoxythymidine (AZT) as anti-HIV compounds". Antiviral Chem.Chemother., 1 107-113; McGuigan, С., O'Connor, T.J., Nicholls, S.R. Nickson, C. and Kinchington, D. (1990b) "Synthesis and anti-HIV activity of some novel substituted dialkyl phosphate derivatives of AZT and ddCyd", Antiviral Chem.Chemother., 1, 355-360; McGuigan, С., Nicholls, S.R., O'Connor, T.J., and Kinchington, D. (1990с) "Synthesis of some novel dialkyl phosphate derivative of 3'-modified nucleosides as potential anti-AIDS drugs". Antiviral Chem.Chemother., 1, 25-33; McGuigan, С., Devin, K.G., O'Connor, T.J., and Kinchington, D. (1991) "Synthesis and anti-HIV activity of some haloalkyl phosphoramidate derivatives of 3'-azido-3' deoxythylmidine (AZT); potent activity of the trichloroethyl methoxyalaninyl compound". Antiviral Res., 15, 255-263; McGuigan, С., Pathirana, R.N., Balzarini, J. and DeClercq, E. (1993b) "Intracellular delivery of bioactive AZT nucleotides by aryl phosphate derivatives of AZT", J.Med.Chem., 36, 1048-1052.
Алкилгидрофосфатные производные анти-ВИЧ средства AZT могут быть менее токсичными, чем исходные нуклеозидные аналоги. Antiviral Chem. Chemother., 5, 271-277; Meyer, R.B., Jr., Shuman, D.A. and Robins, R.K. (1973) "Synthesis of purine nucleoside 3',5'-cyclic phosphoramidates". Tetrahedron Lett., 269-272; Nagyvary, J. Gohil, R.N., Kirchner, C.R. and Stevens, J.D. (1973) "Studies on neutral esters of cyclic AMP", Biochem.Biophys.Res.Commun., 55, 1072-1077; Namane, A. Gouyette, C., Pillion, M.P., Pillion, G. and Huynh-Dinh, T. (1992) "Improved brain delivery of AZT using a glycosyl phosphotriester prodrug", J.Med.Chem., 35, 3039-3044; Nargeot, J. Nerbonne, J.M. Engels, J. and Leser, H.A. (1983) Natl.Acad. Sci.U.S.A., 80, 2395-2399; Nelson, К.A., Bentrude, W.G. Stser, W.N. and Hutchinson, J.P. (1987) "The question of chair-twist equilibria for the phosphate rings of nucleoside cyclic 3',5' monophosphates. 1HNMR and x-ray crystallographic study of the diastereomers of thymidine phenyl cyclic 3',5'-monophosphate", J.Am.Chem.Soc., 109, 4058-4064; Nerbonne, J.M., Richard, S., Nargeot, J. and Lester, H.A. (1984) "New photoactivatable cyclic nucleotides produce intracellular jumps in cyclic AMP and cyclic GMP concentrations". Nature, 301, 74-76; Neumann, J.M., Herv, M., Debouzy, J.C., Guerra, F.I., Gouyette, C., Dupraz, B. and Huyny-Dinh, T. (1989) "Synthesis and transmembrane transport studies by NMR of a glucosyl phospholipid of thymidine", J.Am.Chem.Soc., 111, 4270-4277; Ohno, R., Tatsumi, N., Hirano, M., Imai, K. Mizoguchi, H., Nakamura, Т., Kosaka, M., Takatuski, K., Yamaya. Т., Toyama K., Yoshida, Т., Masaoka, Т., Hashimoto, S., Ohshima, Т., Kimura, I., Yamada, K. and Kimura, J. (1991) "Treatment of myelodysplastic syndromes with orally administered 1-(3-D-arabmouranosylcytosine-5' stearylphosphate". Oncology, 48, 451-455. Palomino, E., Kessle, D. and Horwitz, J.P. (1989) "A dihydropyridine carrier system for sustained delivery of 2',3' dideoxynucleosides to the brain", J.Med. Chem., 32, 22-625; Perkins, R.M., Barney, S. Wittrock, R., Clark, P.H., Levin, R. Lambert, D.M., Petteway, S.R., Serafinowska, H.T., Bailey, S.M., Jackson, S., Hamden, M.R. Ashton, R., Sutton, D., Harvey, J.J. and Brown, A.G. (1993) "Activity of BRL47923 and its oral prodrug, SB203657A against a rauscher murine leukemia virus infection in mice". Antiviral Res., 20 (Suppl. I). 84; Piantadosi, C., Marasco, С J., Jr., Norris-Natschke, S.L., Meyer, K.L., Gumus, F., Surles, J.R., Ishaq, K.S., Kucera, L.S. lyer, N., Wallen, C.A., Piantadosi, S. and Modest, E.J. (1991) "Synthesis and evaluation of novel ether lipid nucleoside conjugates for anti-HFV-1 activity", J.Med.Chem., 34, 1408-1414; Pompon, A., Lefebvre, I., Imbach, J.L., Kahn, S. and Farquhar, D. (1994), "Decomposition pathways of the mono- and bis(pivaloyloxymethyl) esters of azidothymidine-5'-monophosphate in cell extract and in tissue culture medium: an application of the on-line ISRP-cleaning HPLC technique". Antiviral Chem.Chemother., 5, 91-98; Postemark, Т. (1974) "Cyclic AMP and cyclic GMP", Annu.Rev.Pharmacol., 14, 23-33; Prisbe, E.J., Martin, J.C.M., McGhee, D.P.C., Barker, M.F., Smee, D.F. Duke, A.E., Matthews, T.R. and Verheyden, J.P.J. (1986) "Synthesis and antiherpes virus activity of phosphate an phosphonate derivatives of 9-[(1,3-dihydroxy-2-propoxy)methyl]guanine", J.Med.Chem., 29, 671-675; Pucch, F., Gosselin, G., Lefebvre, I., Pompon, A., Aubertin, A.M. Dim, and Imbach, J.L. (1993) "Intracellular delivery of nucleoside monophosphate through a reductasemediated activation process", Antiviral Res., 22, 155-174; Pugaeva, V.P., Klochkeva, S.I., Mashbits, F.D. and Eizengart, R.S. (1969), "Toxicological assessment and health standard ratings for ethylene sulfide in the industrial atmosphere", Gig.Trf.Prof.Zabol., 14, 47-48 (Chem.Abstr. 72, 212); Robins, R.K. (1984) "The potential of nucleotide analogs as inhibitors of Retro viruses and tumors", Pharm.Res., 11-18; Rosowsky. A., Kim S.H., Ross and J. Wick, M.M. (1982) "Lipophilic 5'-(alkylphosphate) esters of 1-β-D-arabinofuranosylcytosine and its N4-acyl and 2,2'-anhydro-3'-O-acyl derivatives as potential prodrugs", J.Med.Chem., 25, 171-178; Ross, W. (1961) "Increased sensitivity of the walker turnout towards aromatic nitrogen mustards carrying basic side chains following glucose pretreatment", Biochem. Pharm., 8, 235-240; Ryu, E.K., Ross, R.J. Matsushita, Т., MacCoss, M, Hong, C.I. and West, C.R. (1982) "Phospholipid-nucleoside conjugates. 3. Synthesis and preliminary biological evaluation of 1-β-D-arabinofuranosylcytosine 5' diphosphate [-],2-diacylglycerols", J.Med. Chem., 25, 1322-1329; Saffhill, R. and Hume, W.J. (1986) "The degradation of 5-iododeoxyuridine and 5-bromoethoxyuridine by serum from different sources and its consequences for the use of these compounds for incorporation into DNA", Chem.Biol. Interact., 57, 347-355; Saneyoshi, M., Morozumi, M., Kodama, K., Machida, J., Kuninaka, A. and Yoshino, H. (1980) "Synthetic nucleosides and nucleotides. XVI. Synthesis and biological evaluations of a series of 1-β-D-arabinofuranosylcytosine 5'-alkyl or arylphosphates", Chem.Pharm.Bull., 28, 2915-2923; Sastry, J.K., Nehete, P.N., Khan, S., Nowak, B.J., Plunkett, W., Arlinghaus, R.B. and Farquhar, D. (1992) "Membrane-permeable dideoxyuridine 5'-monophosphate analogue inhibits human immunodeficiency virus infection", Mol.Pharmacol., 41, 441-445; Shaw, J.P., Jones, R.J. Arimilli, M.N., Louie, M.S., Lee, W.A. and Cundy, K.C. (1994) "Oral bioavailability of PMEA from PMEA prodrugs in male Sprague-Dawley rats", 9th Annual AAPS Meeting. San Diego, CA (Abstract); Shuto, S., Ueda, S., Imamura, S., Fukukawa, K. Matsuda, A. and Ueda, T. (1987) "A facile one-step synthesis of 5' phosphatidiylnucleosides by an enzymatic two-phase reaction", Tetrahedron Lett., 28, 199-202; Shuto, S. Itoh, H., Ueda, S., Imamura, S., Kukukawa, K., Tsujino, M., Matsuda, A. and Ueda, T. (1988) Pharm.Bull., 36, 209-217. Примером применимой фосфатной пролекарственной группы является S-aцил-2-тиoэтильнaя группа, также называемая SATE.
Данное изобретение описано в свете его предпочтительных осуществлений. Вариации и модификации настоящего изобретения будут очевидны специалистам в данной области из предшествующего подробного описания настоящего изобретения. Подразумевается, что все данные вариации и модификации входят в объем данного изобретения.
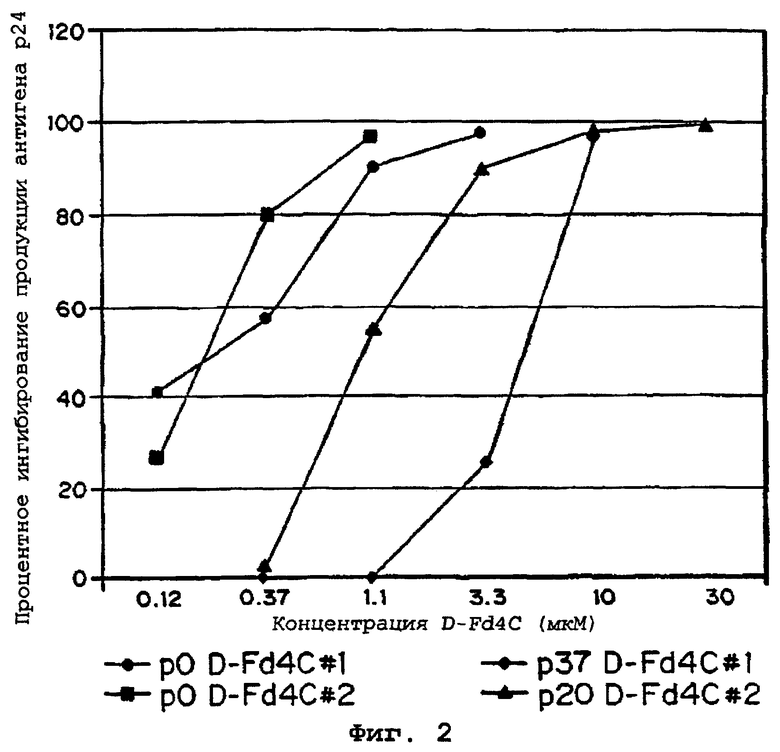
Изобретение относится к фармацевтической композиции, используемой для лечения или профилактики ВИЧ-инфекции у человека, содержащей эффективное количество β-D-D4FC или его фармацевтически приемлемой соли или пролекарства, необязательно в фармацевтически приемлемом носителе, в сочетании с эффективным количеством соединения, необязательно в фармацевтически приемлемом носителе, которое выбрано из группы, состоящей из индинавира, нелфинавира, саквинавира, ампренавира, эфаверенца, делавирдина, невирапина и абакавира в виде единого препарата, а также к способу лечения или профилактики ВИЧ-инфекции у человека, предусматривающему введение эффективного количества этой композиции. Технический результат: фармацевтическая композиция согласно изобретению проявляет преимущественные или улучшенные фармакокинетические параметры, параметры биологического распределения, метаболические параметры, параметры устойчивости и другие параметры по сравнению с введением одного β-D-D4FC. 2 н. и 15 з.п. ф-лы, 6 табл., 2 ил.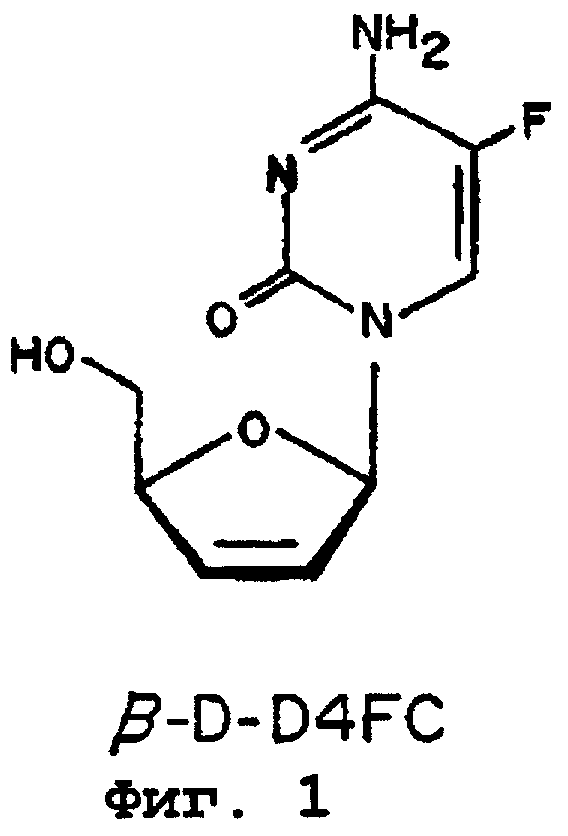
| WO 9622778 A1, 01.08.1996 | |||
| US 5703058 А, 30.12.1997 | |||
| WO 9628170 A1, 19.09.1996 | |||
| CHEN SH et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Bioorganis & Medical Chemistry Letters | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ДЕЙСТВИЕМ ПРОТИВ ИНФЕКЦИОННЫХ АГЕНТОВ | 1994 |
|
RU2092177C1 |
| Способ получения хлоргидрата 2"2 -ангидро-1- - -арабинофуранозил - 5-фторцитозина | 1978 |
|
SU707246A1 |

