Изобретение относится к способу выделения диацетилреина фармацевтически пригодной чистоты с остаточным содержанием нежелательных производных алоэ-эмодина в целом менее чем 200 ppm, полученному по этому способу диацетилреину и фармацевтическому средству, которое содержит это соединение.
Диацетилреин формулы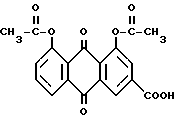
представляет собой лекарственное биологически активное вещество, которое обладает антиаритмической, противовоспалительной, жаропонижающей и анальгетической активностью. Диацетилреин поэтому используется для лечения артритных заболеваний, например патент ФРГ А-27 11493 и патент США А-4 244968.
Диацетилреин можно получать, например, путем ацетилирования барбалоина и окисления полученного надацетилированного барбалоина с помощью триоксида хрома. Кроме того, диацетилреин можно получать путем ацетилирования реина, который можно получать, например, из высушенного лекарственного растения сенны.
В полученном по этому способу диацетилреине в качестве нежелательных примесей содержатся производные алоэ-эмодина, которые происходят от неполного окисления с помощью триоксида хрома или совместно экстрагируются при экстракции высушенного лекарственного растения сенны. Эти примеси содержатся в относительно незначительных количествах и поэтому только с другом могут отделяться/ руководствуясь классическими операциями очистки. Кроме того, при указанном выше сначала способе образуются остатки хрома, которые пригодным образом нужно удалять.
Поэтому в основу настоящего изобретения положена задача разработки способа получения диацетилреина, который прост и осуществляется с высоким выходом, при котором диацетилреин образуется фармацевтически пригодной чистоты с остаточным содержанием нежелательных производных алоэ-эмодина в целом менее чем 20 ppm.
Эта задача решается благодаря предлагаемому в изобретении способу, который отличается, тем, что:
A/ содержащий алоэ-эмодиновые компоненты /т.е. алоэ-эмодин и/или его производные/ реин-9-антрон-8-глюкозид подвергают жидкостно-жидкостному диспергированию между только частично смешивающимися с водой полярным органическим растворителем и водной фазой;
Б/ содержащийся после диспергирования в водной фазе реин-9-антрон-8-глюкозид окисляют от реин-8-глюкозида;
В/ глюкозный остаток и 8-положении реин-8-глюкозида отщепляют в кислой среде и
Г/ полученный реин ацетилируют и получают диацетилреин.
Важным источником реин-9-антрон-8-глюкозида являются содержащиеся в лекарственном растительном сырье сенны сеннозиды. Предпочтительным вариантом осуществления изобретения поэтому является способ получения диацетилреина, который в основном свободен от производных алоэ-эмодина, причем
А/ смесь сеннозидов подвергают восстановлению до соответствующего реин-9-антрон-8-глюкозида и алоэ-эмодин-9-антрон-8-глюкозидных соединений
Б/ жидкостно-жидкостное диспергирование полученных соединений осуществляют между только частично смешивающимся с водой полярным органическим растворителем и водной фазой
В/ содержащиеся после экстракции в водной фазе реин-9-антрон-8-глюкозидные соединения окисляют до соответствующего антрахинонового соединения.
Г/ глюкозный остаток в 8-положении атрахинонового соединения отщепляют в кислой среде и
Д/ полученное 1,8-диоксиантрахиновое соединение ацетилируют и получают диацетилреин.
Отдельные стадии способа поясняются ниже:
Восстановление сеннозидов:
Служащую в качестве исходного материала смесь сеннозидов можно получать, например, из лекарственного растительного сырья сенны.
Лекарственное растительное сырье сенны состоит из высушенных листьев и плодов сенны, например индийской сенны /Cassia angusti folia/ и египетской сенны /Cassia acutifolia/.
Лекарственное растительное сырье сенны содержит диантронглюкозиды реина и алоэ-эмодина. Важнейшими являются сеннозиды A, B, A, I, C, D и D 1. Сеннозиды соответствуют формуле а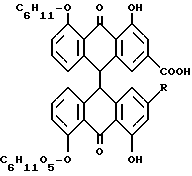
В случае сеннозидов A, B и A I, R обозначает COOH, а в случае сеннозидов C, D и D I. R обозначает CH2OH. Сеннозиды A, B и A I, соответственно C D и D I представляют собой стереизомеры и отличаются друг от друга конфигурацией C-атомов в положениях 10 и 10'.
Выделение сеннозидов из лекарственного растительного сырья сенны описано, например, в патенте ФРГ А-3200131, на который этим ссылаются полностью. Сообразно с этим сначала лекарственное растительное сырье сенны экстрагируют водным метанолом. Остающийся после полного удаления метанола концентрат содержит сеннозиды в форме калиевой соли. Этот концентрат пригоден в качестве исходного материала для предлагаемого в изобретении способа. Концентрат можно еще очищать путем жидкостной экстракции с помощью частично растворимых в воде спиртов или кетонов /например, бутанол-2-2-бутанон/ /рафинат/. Рафинат подкисляют до pH-значения примерно 1,5 - 2 и сеннозиды доводят до кристаллизации при внесении затравки. Полученная смесь сырых сеннозидов также пригодна в качестве исходного продукта для предлагаемого в изобретении способа. В желательном случае смесь сырых сеннозидов также можно еще перекристаллизовывать.
Альтернативно можно применять в качестве исходного продукта смешанный с частично растворимым в воде спиртом или кетоном, в особенности бутанолом-2, концентрат.
При экстракции лекарственного растительного сырья сенны соотношение лекарственного высушенного растения к экстрагирующему растворителю составляет предпочтительно 1:4 - 1:15, в особенности 1:4 - 1:10.
Экстракцию осуществляют предпочтительно в присутствии буфера, например, как тринатрийцитрат, глицин, бикарбонат натрия или сахароза.
Согласно предлагаемому в изобретении способу, эти исходные материалы подвергают полному восстановлению до соответствующего реин-9-антрон-8-клюкозида /R = COOH/ и соответствующего алоэ-эмодин-9-антрон-8-глюкозида /R = CH2H/ формулы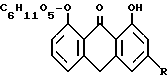
R = COOH, CH2 OH
Восстановителем с пригодным восстановительным потенциалом являются, например, хлорид цинка-II, диоксид серы, бороводороды щелочных металлов и предпочтительно дитиониты щелочных металлов, в особенности дитионит натрия.
Для проведения восстановления исходный материал можно использовать в водной растворе или суспензии, а восстановитель добавляют в твердой форме или растворенным в воде. Также можно работать в двухфазной смеси тем, что добавляют частично смешивающийся с водой, полярный органический растворитель, в особенности 2-бутанол или ацетон.
Можно восстанавливать при температуре окружающей среды или при повышенной температуре. Восстановление целесообразнее осуществлять при 40 - 60oC, в особенности при 50 - 55oC. Работают при слабокислом, вплоть до слабощелочного pH-значения раствора, соответственно суспензии, исходных сеннозидов, предпочтительно при pH 7-9. В желательном случае можно осуществлять восстановление многократно, в особенности 2 - 10 раз.
Образовавшиеся 9-антрон-8-глюкозиды выделяются за счет добавки кислоты, например серной кислоты, вплоть до pH 4 - 4,5. Температура при этом целесообразнее не должна составлять больше чем 40oC. Целесообразнее работать при осаждении 9-антрон-8-глюкозидов и при их выделении /например, путем отфильтрования/ в атмосфере азота, чтобы избежать неконтролируемого окисления этих соединений.
Существенно, чтобы восстановление протекало полностью. Целесообразнее применять поэтому восстановитель в большом избытке. При применении дитионита натрия используют в общем 1-4-кратное весовое количество дитионита натрия, в расчете на содержание в исходном материала сеннозидов. Кроме того, восстановителем можно воздействовать минимально 2 ч, предпочтительно минимально 3 ч. В общем восстановление осуществляют не более чем 10 ч. Предпочтительно осуществляют дополнительное восстановление в указанных условиях.
Полученный продукт перед его использованием предпочтительно в самой ближайшей стадии переосаждают тем, что растворяют в водном растворе путем добавки основания /NaOH, KOH/ примерно вплоть до pH 6 - 7, водный раствор экстрагируют 2-бутанолом, ацетоном или 2-бутаноном и продукт осаждают снова путем добавки кислоты до pH-значения примерно 2 - 4.
Жидкостно-жидкостное диспергирование.
В этой стадии алоэ-эмодиновые компоненты, в особенности алоэ-эмодин-9-антрон-8-глюкозид удаляют. Для этой цели осуществляют жидкостно-жидкостное диспергирование полученного продукта в только частично смешивающемся с водой полярном органическом растворителе и водной фазе. Пригодными полярными органическими растворителями являются C4-C5-алканолы и ди-C1-C3-алкилкетоны, как ацетон, 1-бутанол, 2-бутанол и 2-бутанон. Предпочтительно применяют 2-бутанол или ацетон.
Предпочтительно к водной фазе добавляют восстановитель, чтобы придать водной фазе во время всей жидкостно-жидкостного диспергирования редокс-потенциал-210 mV или отрицательнее. Целесообразнее применять такой же восстановитель, как и в стадии A. При применении дитионита щелочного металла в качестве восстановителя и общем достаточно 2 - 4 вес.%-ного раствора при pH-значения 7 - 11, чтобы соблюдать упомянутые условия в отношении потенциала.
Объемное соотношение водной фазы (тяжелой фаза) к органической фазе (легкая фаза) составляет в общем величину в пределах 1:5 - 1:40.
Предпочтительно жидкостно-жидкостную экстракцию осуществляют в противотоке. Смесь антроновых соединений при этом подают в виде полученного после восстановления раствора или, если выделяются антроновые соединения, в форме 3 - 15 вес.%-ного раствора.
После экстракции желательный реин-9-антрон-8-глюкозид находится в водной фазе. Его осаждают путем добавки кислоты вплоть до pH - значения примерно 2 - 4 и получают соединение обычным образом.
Окисление реинантрон-8-глюкозида
Полученный реин-9-антрон-8-глюкозид теперь окисляют до реин-8-глюкозида формулы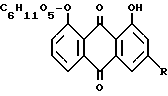
R = COOH, CH2OH
Пригодными окислителями для этой цели являются, например, кислород, пероксидные соединения (пероксид водорода), соединения марганца, хрома или железа в высоких стадиях окисления. Предпочтительно применяют соль железа-III, в особенности сульфат железа-III. Целесообразнее работать при повышенной температуре, однако, ниже 60oC. Благодаря этому избегают образования нежелательных и неопределяемых продуктов окисления. По окончании окисления образовавшийся реин-8-глюкозид выделяют обычным образом.
Отщепление остатка глюкозы
Остаток глюкозы в 8-положении отщепляют в кислом растворе. Целесообразнее работать примерно при 85 - 95oC. Полученный продукт выделяют обычным образом.
Известно переведение сеннозидов после кислотного гидролиза прямо в реин путем введения во взаимодействие с хлоридом железа-III, см., например, патент ФРГ А-2711493. При этом выход, однако, составляет примерно 10% и, кроме того, образовавшийся реин трудно отделять.
В случае предлагаемого в изобретении способа осуществляется восстановительное расщепление сеннозидов, окисление образовавшихся антроновых соединений до соответствующих антрахиноновых соединений и отщепление остатка глюкозы в 8-положении антрахиноновых соединений, смотря по обстоятельствам, в отдельных стадиях. После восстановительного расщепления все соединения, которые в дальнейшем ходе могут приводить к образованию алоэ-эмодина или его производных, количественно разделяют с помощью жидкостно-жидкостной экстракции. Кроме того, можно осуществлять окисление при невысоких температурах, так что избегают образования нежелательных и неопределяемых продуктов окисления. Помимо этого, при этом осуществлении реакции используемую соль железа можно почти количественно регенерировать и после вторичного окисления можно снова использовать. Разделение стадий окисления и гидролиза на основании более высокой растворимости в воде антронглюкозидов по сравнению с соответствующими аглюконами позволяет осуществлять окисление осторожно при комнатной температуре ниже 60oC, благодаря чему избегают иначе неизбежного образования неопределяемых побочных продуктов.
Ацетилирование 1,8-диоксиантрахинонового соединения
Ацетилирование полученных 1,8-диоксиантрахиноновых соединений осуществляют обычным образом. Например, можно ацетилировать с помощью ацетангидрида в присутствии ацетата натрия, как описано в Arch Pharm. 241, 607 (1903). Ацетилирование, однако, также можно осуществлять другими, известными специалисту способами, например, путем введения во взаимодействие с ацетилхлоридом и т.д.
Полученный таким образом диацетилреин по существу лишен алоэ-эмодина и его производных. Содержание в нем примесей при этом составляет еще примерно 50 ppm (определено согласно описанному в примерах методу анализа). Содержание в нем примесей можно снижать далее, если полученный диацетилреин перекристаллизуют следующим образом.
Диацетилреин переводят в соль щелочного металла тем, что его обрабатывают пригодным основанием. Пригодным основанием является, например, ацетат щелочного металла, предпочтительно ацетат калия. Предпочтительно применяют эквимолярные количества основания и водного C1 - C3 - спирта, например 80 - 90%-ного этанола, в качестве реакционной среды. Можно выкристаллизовывать соль щелочного металла диацетилреина на холоду, растворять ее в водном C1 - C3 - спирте и осаждать ее за счет добавки кислоты вплоть до pH-значения примерно 3. Осаждавшийся диацетилреин затем выделяют обычным образом и обрабатывают.
Таким образом полученный продукт содержит менее чем 20 ppm вышеупомянутых примесей. Кроме того, продукт находится в форме иглообразных кристаллов, которые особенно пригодны для галеновой формулировки.
Продукт можно обычным образом высушивать. Целесообразнее прежде всего осуществлять высушивание в вакууме при относительно низкой температуре, например, не выше чем 40oC, до тех пор, пока содержание воды в продукте не снизится примерно до 3% или менее. Затем температуру можно повышать до 70 - 110oC.
Изобретение относится также к получаемому согласно изобретению, по существу чистому диацетилреину, а также к фармацевтическому средству, которое содержит это соединение. Область применения, вводимая доза и пригодные дозировочные формы известны, см. патент США А-4244968, патент США А-4346103, А-4950687, патент ФРГ А-2711493, а также Drugs ExptI. CIi п. Res. 6 (1) 53 - 64 (1980).
Нижеследующие примеры поясняют изобретение.
Пример 1. Получение используемой в качестве исходного материала смеси синнозидов: в соединенные в ряд два перколятора объемом 250 л помещают смотря по обстоятельством 40 кг лекарственного растительного сырья сенны /содержание сеннозидов примерно 1,5%/ и покрывают их перфорированной стальной плитой. В качестве растворителя для экстракции применяют 70%-ный метанол, который подают на лекарственное растительное сырье в первый перколятор. Образовавшийся в первом перколяторе раствор направляют на лекарственное растительное сырье, которое находится во втором перколяторе. При этом растворитель может свободно протекать через первый перколятор.
Для экстракции 40 кг лекарственного растительного сырья сенны применяют в целом 160 л растворителя. После того, как этот объем 70%-ного метанола пропустят через оба перколятора и получают соответствующее количество перколята, выпускной шланг перколятора соединяют с дополнительной емкостью для перколята и через перколяторы пропускают еще дополнительно 60 л 70%-ного метанола. После этого остаточный свободный растворитель из первого перколятора вводят в верхнюю часть второго перколятора и собирают дополнительный перколят до тех пор, пока в целом он не составит 120 л. Затем опорожняют первый перколятор, заполняют его снова 40 кг высушенного лекарственного сырья сенны и дополнительный перколят перекачивают на высушенные лекарственные растения, причем достаточно 120 л дополнительного перколята, чтобы покрыть высушенное лекарственное сырье в перколяторе. Затем доводят температуру раствора до +30oC.
Соединяют этот перколятор с прежде используемым для экстракции перколятором и осуществляют экстракцию, как описано выше.
Смотря по обстоятельствам, из 40 кг высушенных лекарственных растений получают 160 л перколята, из которого удаляют метанол на ротационном вакуумном испарителе, который снабжен колонкой с наполнителем. Получают примерно 30 л продукта. Этот концентрат экстрагируют с помощью такого же объема насыщенного водой бутанола-2.
Стадия А: восстановление сеннозидов до реин-9-антрон-8-глюкозидов.
1,0 л Проэкстрагированного концентрата доводят pH-значения 7,5 с помощью 48%-ного раствора гидроксида натрия. Нагревают до 60oC и при перемешивании в течение получаса в раствор добавляют 90 г дитионита натрия в твердой форме. После окончания добавления перемешивают следующий час. Затем при перемешивании добавляют концентрированную серную кислоту вплоть до pH-значения 2. В течение двух часов охлаждают до температуры окружающей среды, отфильтровывают выделившийся кристаллический осадок и промывают его содержащей диоксид серы водой.
В случае необходимости сырой реин-9-антрон-8-глюкозид переосаждают. Еще влажный осадок на фильтре растворяют в смеси 15 об.ч. 2-бутанола и 85 об.ч. воды, которая содержит 0,5 вес.% пиросульфита натрия, так, чтобы благодаря добавке 48%-ного раствора гидроксида натрия вплоть до pH-значения 7 получают 10%-ный раствор /вес/объем/. Раствор подкисляют с помощью концентрированной соляной кислоты до pH-значения 2,8 или ниже и оставляют стоять в течение 2 ч. Выпавший осадок отфильтровывают и промывают содержащий диоксид серы или пиросульфит натрия водой и высушивают.
Выход: 90%.
Для полученного таким образом продукта проводят новое восстановление /дополнительное восстановление/ следующим образом:
3,0 г Сырого высушенного реин-9-антрон-8-глюкозида или соответствующее количество влажного продукта вместе с 1,4 г дитионита натрия и 2,3 мл 5 н. NaOH растворяют в 15 мл воды. Затем доливают водой до 24 мл и нагревают раствор в течение 20 мин при 55oC. После этого добавляют следующие 1,5 г дитионита натрия в раствор и нагревают 20 мин при 55oC. Затем добавляют 0,8 мл 5 н. NaOH и 1,5 г дитионита натрия. После нагревания в течение 20 мин при 55oC добавляют еще раз 0,9 мл 5н NaOH, полученный раствор вводят прямо в последующую жидкостно-жидкостную экстракцию.
Стадия Б: отделение алоэ-эмодиновых компонентов.
Отделение алоэ-эмодиновых компонентов осуществляют путем жидкостно-жидкостного диспергирования 9-антрон-8-глюкозидов в противотоке с помощью аппаратуры из 60 единиц смеситель-сепаратор (Mixer - Settler-аппаратура). В качестве водной, более тяжелой фазы применяют раствор 3,0 г дитионита натрия в 3,5 мл 5 н. NaOH и 96 мл воды. В качестве органической, более легкой фазы применяют (насыщенный водой) 2-битанол или ацетон. Обе фазы пропускают через аппаратуру так, чтобы объемное соотношение более тяжелой фазы к более легкой фазе составляло 1:10.
Разделяемую смесь вводят в аппаратуру в виде свежевосстановленного раствора или в форме раствора с соответствующим pH-значением и соответствующей концентрации, которые содержат полученные из стадии А 9-антрон-8-глюкозиды, а именно, что на об.ч. разделяемой смеси используются 30 объемн. частей органической фазы.
pH-Значение содержащего смесь раствора поддерживают с помощью глицинового буфера при 9 - 9,5. Буфер из 3 об. ч. 7,5%-ного раствора глицина, 1 об. ч. 1н. NaOH добавляют в количестве 240 мл буферного раствора на 150 г сырого реин-9-антрон-8-глюкозида. Нежелательные алоэ-эмодиновые соединения обогащают органическую фазу, в то время как реин-9-антрон-8-глюкозид остается в водной фазе. Водную фазу подкисляют серной кислотой вплоть до pH-значения, равного 2,8; образовавшийся осадок отфильтровывают и промывают водой и ацетоном и высушивают на воздухе при температуре окружающей среды. Получают таким образом реин-9-антрон-8-глюкозид с содержанием алоэ-эмодиновых компонентов 41 ppm, определенных в виде алоэ-эмодина по способу, который описывается в конце этой заявки на патент.
Выход: 97%, в расчете на реин-9-антрон-8-глюкозид.
Стадия B: окисление до реин-8-глюкозида.
Продукт из стадии Б / в расчете на содержание 3,0 кг сеннозидов A, A1 и B) суспендируют в растворе из 184 л деминерализованной воды и 75,5 кг гидрата сульфата железа-III (22% Fe+3). Суспензию нагревают до 55 - 62oC и окисляют в течение 14 ч при применении быстро движущегося диспергатора. По окончании окисления образовавшийся реин-8-глюкозид отфильтровывают и промывают 50 л деминерализованной воды, в которой установлено значение pH 2 с помощью серной кислоты.
Стадия Г: гликолиз до реина.
Влажный осадок на фильтре из стадии B суспендируют в 200 кг 20 вес.%-ной серной кислоты и перемешивают 8 ч при 88 - 92oC. Образовавшийся реин отфильтровывают и для хранения его можно высушивать в течение 48 ч при 40oC и под давлением 1 мбар или также его тотчас используют во влажном состоянии для ацетилирования в стадии Г.
Общий выход для стадий А - Г: 79%, в расчете на используемые в стадии А сеннозиды A, A1 и B.
Стадия Д: ацетилирование до диацетилреина.
6,5 кг Реина из стадии B суспендируют в 100 л уксусного ангидрида в течение 10 мин, смешивают с 2 кг ацетата калия, при перемешивании нагревают до 95oC, смешиваются с 0,65 кг активного угля и перемешивают 0,5 ч при 90 - 95oC. Активный уголь отфильтровывают от горячего раствора и фильтрат при 90oC смешивают с 2,1 кг 96 - 98 вес.%-ной серной кислоты. После этого при перемешивании по возможности быстро охлаждают до 20oC. Образовавшуюся суспензию отфильтровывают,остаток промывают деминерализованной водой до полного удаления сульфатов.
Выход: 83%.
Стадия E: перекристаллизация, высушивание, размалывание.
При быстром перемешивании 7,5 кг диацетилреина из стадии D (в расчете на высушенное вещество) суспендируют в 375 л 90 об. %-ного этанола. Суспензию нагревают до 70oC и затем смешивают с 3,75 кг ацетата калия. При охлаждении до 0 - 2oC из промежуточного образующегося, прозрачного раствора выкристаллизовывается чистая калиевая соль диацетилреина.
Калиевую соль отфильтровывают и растворяют в 300 л 40 об. %-ного этанола при добавке 3 кг ацетата калия при 20 - 30oC. Прозрачный раствор доводят до pH-значения 3,0 с помощью 10 вес.%-ной серной кислоты. Выкристаллизовавшийся диацетилреин отфильтровывают и промывают деминерализованной водой до полного удаления сульфатов.
Высушивание продукта осуществляют сначала в вакууме при 40oC и под давлением 1 мбар в течение 24 ч. Если содержание остаточной воды снижается до величины ниже 3%, то материал грубо размельчают и дополнительно высушивают в течение 24 ч под давлением 1 мбар и при 70oC.
Затем размалывают при использовании сита до размера 0,5 мм и для удаления остатков растворителя дополнительно высушивают в вакууме 1 мбар и при 70oC. Выход: 95%.
Пример 2. Повторяют описанную в примере 1 экстракцию лекарственного растительного сырья сенны и восстановление сеннозидов. Дополнительное восстановление затем осуществляют следующим образом:
14,0 г Сахарозы, 4,5 г дитионита натрия (85%) и 13,3 г ацетата калия растворяют в 133 мл воды и добавляют 1,3 мл 48%-ного раствора гидроксида натрия и 17,3 г карбоната калия. После этого смешивают с 293 мл ацетона и 50 мл воды. Встряхивают смесь в делительной воронке и разделяют фазы, причем получают 375 мл верхней фазы (ацетоновая фаза) и 130 мл нижней фазы.
В 98 мл нижней фазы растворяют 1,4 мл 48%-ного раствора гидроксида натрия и 10 г реин-9-антрон-8-глюкозида. Нагревают до 45 - 50oC и выдерживают 20 - 30 мин при этой температуре. После этого добавляют 1,0 мл 48%-ного раствора гидроксида натрия и 3,4 г дитионита натрия и нагревают следующие 20 - 30 мин при 45 - 50oC. Затем снова добавляют 1,0 мл 48%-ного раствора гидроксида натрия и 3,4 г дитионита натрия и нагревают 20 - 30 мин при 45 - 50oC.
Отделение алоэ-амодиновых компонентов осуществляют путем жидкостно-жидкостного диспергирования восстановленного раствора противотоком к вышеупомянутой верхней фазе (ацетоновая фаза). Вытекающую, содержащую реин-9-антрон-8-глюкозид рафинатную фазу испаряют до объема 400 мл и смешивают с 20 мл бутанола-2. Добавляют соляную или серную кислоту до pH-значения 4,0 - 4,2. Образовавшийся осадок отфильтровывают, промывают 40 мл воды и 30 мл ацетона и затем высушивают. Последующее окисление проводят, как описано в примере 1.
Пример 3. Полученный после экстракции высушенных лекарственных растений сенны концентрат смешивают примерно с 2 л бутанола-2. Восстановление смеси концентрат плодов сенны-бутанол-2- затем осуществляют в 7 стадий в атмосфере азота в качестве защитного газа. После стадии восстановления 1 осуществляют осаждение сырого реин-9-антро-8-глюкозида.
Стадия восстановления I
100 мл Смеси концентрат плодов сенны/бутанол-2, содержащей примерно 4 кг сеннозидов, помещают в сосуд с перемешиванием и устанавливают атмосферу азота. При перемешивании последовательно добавляют 6 л 20%-ного /по весу/ раствора гидроксида натрия, затем 350 л насыщенного водой бутанола-2 /например, из стадии II/ и перемешивают в течение 15 мин. Смесь нагревают до 42 - 50oC и смешивают с 7 кг дитионита натрия. Перемешивают еще 45 мин. pH-Значение поддерживают равным 7,5 - 8 с помощью 20 вес.%-ного раствора гидроксида натрия. Восстановительный потенциал (против Ag/AgCl - электрода) в случае необходимости поддерживают ниже -630 mV за счет добавки дитионита натрия. После охлаждения до 30 - 35oC осаждают с помощью 10 вес.%-ного раствора серной кислоты вплоть до pH < 4 в течение 1,5 ч. Образовавшуюся суспензию перемешивают при маленькой скорости мешалки примерно в течение 10 ч при 25oC. Образовавшийся осадок отфильтровывают. Осадок суспендируют в 60 л 15%-ного /по весу/ бутанола-2, перемешивают 30 мин при 50 - 60oC и затем отфильтровывают. Остаток промывают с помощью 100 л деминерализованной воды. Сырой выход реин-9-антрон-8-глюкозида, в расчете на используемые сеннозиды, составляют более 82%.
Стадия восстановления II
3,3, кг Сырого реин-9-антрон-8-глюкозида из стадии 1 суспендируют в смеси из 42 л деминерализованной воды и 7,4 л бутанола-2. С помощью 2 л 20 вес. %-ного раствора гидроксида натрия и 9,9 кг тринатрийцитрата суспензию переводят в раствор и затем смешивают с 3,3 кг дитионита натрия и 250 л насыщенного водой бутанола-2 (например, из стадии III). Смесь нагревают до 42 - 45oC, pH-значение поддерживают при 8,5 - 9 с помощью 20 вес.%-ного раствора гидроксида натрия. Восстановительный потенциал /против Ag/AgCl-электрода/ в случае необходимости поддерживают ниже - 750 mV за счет добавки дитионита натрия.
После времени выдерживания 30 мин верхнюю фазу удаляют, а нижнюю фазу перерабатывают далее в стадии III.
Стадия восстановления III
При использовании нижней фазы из стадии II при добавке химикалиев повторяют описанный в стадии II восстановительно/ экстракционный способ: 1,65 кг дитионита натрия, 0,8 л 20 вес.%-ного раствора гидроксида натрия, 350 л насыщенного водой бутанола-2 (например, из стадии IV).
Стадии восстановления IV-VII
При использовании нижней фазы из, смотря по обстоятельствам, предыдущей стадии при добавке следующих химикалиев повторяют описанный в стадии II восстановительно/экстракционный способ: 0,825 кг дитионита натрия, 0,4 л 20 вес. %-ного раствора гидроксида натрия, 350 л насыщенного водой бутанола-2 (например, из одной из последующих стадий - принцип противотока).
Отделенную в стадии VII нижнюю фазу охлаждают до 30 - 35oC и осаждают реин-9-антро-8-глюкозид, как описано в стадии I. Образовавшийся осадок отфильтровывают и промывают с помощью 100 л деминерализованной воды. Затем заливают 10 л раствора сульфата железа-III /получение см. стадию Б, пример 1/.
Реин-9-антрон-8-глюкозид затем, как описано в примере 1 или 2, переводят в сеннозиды.
Фармакологические исследования
Определяют эффективность диацетилреина в случае моделей с хроническим воспалением после орального приема. Используют следующие опытные модели: гранулема за счет комочка ваты в случае крыс и вызванный внутриартикулярным введением витамина А артроз в случае кроликов.
а) Гранулема за счет комочка ваты, в случае крыс
Молодые, половозрелые крысы (п = 10) получают 25, 50 или 100 мг диацетилреина, соответственно 5 мг индометацина/кг или 100 мг ацетилсалициловой кислоты/кг ежедневно в течение 5 дней. Также имеется обработанная только водой контрольная группа. Имплантацию комочка осуществляют в первый день лечения. Свежие и сухие веса препарированных в конце опыта гранулем показывают значительное и отчетливо зависящее от дозы уменьшение по сравнению с контрольной группой. При этом действие 100 мг диацетилреина/кг соответствует примерно действию 5 мг индометацина или 100 мг ацетилсалициловой кислоты. Веса вилочной железы и надпочечников не изменяются во время обработки.
б) Артроз за счет витамина А
За счет трех внутриартикулярных инъекций 30000 IE витамина A в течение 9 дней у двух групп по 10 индивидумов кроликов /белые новозеландские/ вызывают артрозоподобное изменение суставов. Спустя 56 дней 10 животных обрабатывают с помощью 3 мг диацетилреина/кг/день в течение 8 недель. По сравнению с контрольной группой макроскопически и микроскопически определяемые изменения суставов значительно уменьшаются в обработанной группе.
Лечебное действие диацетилреина далее сравнивают с таковым ацетилсалациловой кислоты на каждом из 7 кроликов, которые после шестидневной предобработки с помощью трехкратно 10000 IE витамина A и через интервал без обработки в 26 дней в течение 8 недель получают либо 5 мг диацетилреина/кг/день /подопытная группа/, 15 мг ацетилсалициловой кислоты/кг/день (с положительная контрольная группа) или остаются необработанными (отрицательная контрольная группа). Во всех трех группах спустя 24 дня после последней инъекции витамина A появляются сравнимые нарушения движений в форме волочащихся задних лап. В отрицательной контрольной группе во время следующих 8 недель усиливаются клинические симптомы проявляющегося артроза.
В подопытной группе и положительной контрольной группе эти симптомы значительно улучшаются при 8-недельной обработке.
Изменения слизистой оболочки желудка
В то время, как одноразовый прием 400 мл диацетилреина/кг или растворителя в случае крысы не вызывает никаких эрозий слизистой оболочки желудка, после приема ибупрофена (200 мг/кг) или индометацина (20 мг/кг) наблюдаются отчетливые повреждения слизистой оболочки от точечнообразных (диаметром 1 мм) до крупных (диаметром 3 мм) эрозий. Также двукратный в день прием 100 100 мг диацетилреина /кг в течение 3-х дней не вызывает никаких повреждений слизистой оболочки, пожалуй, однако, и соответствующее применение 10 мг индометацина/кг. При этом речь идет об эрозиях диаметром 1 - 3 мм.
Токсикология
Острая токсичность ЛД50 в зависимости от исследуемого вида (крыса, мышь, кошка), после орального применения составляет 1,9 - 7,9 г/кг. При этом крыса оказывается менее всего чувствительной. После парентерального введения (внутривенно, интраперитонеально) ЛД50 - значения у этих видов составляют 119 - 339 мг/кг.
Клинические исследования
1. При изучении двойным слепым методом по отношению к напроксену и последующему дополнительному лечению плацебо исследуют действие диацетилреина в случае коксартроза и гонита на 95 /49/46/ пациентах. Используемая доза оставляет 50 мг диацетилреина 2 раза в день, соответственно 750 мг напроксена ежедневно. Длительность лечения составляет 60 дней после 7-дневной фазы промывки. Последующее лечение плацебо распространяется на время более 60 дней.
Испытуемыми величинами являются симптоматика болей и движения по оценочной шале, ограничение функций и совместимость.
В обеих, подвергающихся лечению группах /диацетилреин/напроксен/ в отношении всех испытуемых параметров установлена статистически заметная доля улучшения /P < 0,01, соответственно P < 0,05/ по сравнению с исходным значениями. После прекращения лечения и последующего приема плацебо, однако, для диацетилреин/плацебо-группы в отношении параметра спонтанной боли, активной и пассивной боли передвижения, оказалось значительное превосходство /P < 0,01/ по сравнению с напроксен-/плацебо- коллективом. Это различие на уровне 5% обеспечивается также для переменных ночных болей и болей при надавливании спустя 30 дней после прекращения приема диацетилреина.
2. В раскрытом изучении протекания процессов с контролем исследуют действие диацетилреина против остеоартроза позвоночника и колен у 70 пациентов /35/35/. Используемая доза составляет 100 мг диацетилреина в день. Длительность лечения составляет 60 дней, продолжительность наблюдения - 75 дней. Испытуемыми величинами являются ограничения болей и движения. Величины определяются по оценочной системе.
Контрольная группа охватывает 35 пациентов, в случае которых осуществляются исключительно физико-терапевтические меры. В группе, которую лечат диацетилреином, также проводят физиотерапию.
Оценка результатов относительно всех параметров показывает статистически заметное превосходство излечиваемой группы по отношению к контрольной. Также после прекращения лечения для группы, которую лечат диацетилреином, смогли установить удерживающийся терапевтический эффект /"переживающий эффект"/.
3. При осуществляемом по одинарному слепому методу перекрестном изучении по отношению к напроксену исследуют действие диацетилреина при локализованном артрозе на 20 пациентах. Их разделяют на две группы, причем первой группе сначала дают 2 раза по 50 мг диацетилреина в течение 20 дней. Затем в течение трех дней осуществляют фазу промывки и дальнейшее лечение проводят с помощью дачи 2 раза в день 250 мг напроксена в следующие 20 дней. Во второй группе устанавливают обратную последовательность. Длительность лечения составляет в целом 43 дня. Испытуемыми величинами являются боли, боль от нажатия, пассивная боль при движении, ограничение функций и припухание по оценочной шкале.
Оценка результатов показывает превосходство лечения с помощью диацетилреина по сравнению с лечением напроксеном. Не наблюдаются никакие достойные упоминания побочные действия, также нет никаких изменений клинических лабораторных параметров.
4. При изучении двойным слепым методом, по случаю, в "двойном холостом способе" по отношению к напроксену исследуют действие диацетилреина на 23 пациентах /12/11/ с остеоартрозом /изучение совместимости/. Исследуемая доза составляет 2 раза в день 50 мг диацетилреина и 3 раза в день 250 мг напроксена. Длительность лечения составляет 4 недели. Испытуемыми величинами являются эзофагогастродуоденоскопически полученные данные до и после терапии. Для изучения отбираются только пациенты с нормальными результатами исследования слизистой оболочки, соответственно с легкими повреждениями слизистой оболочки /степень 1/.
Спустя 4 недели эндоскопические результаты в одном случае /10%/ в подвергаемой лечению диацетилреином группе показывают повреждения слизистой оболочки степени 2, в то время как в группе, которую лечат напроксеном, 5 пациентов /50%/ показывают повреждения слизистой оболочки степени 2,3 и 4. Во всех случаях отмечается нормальный результат приема.
Аналитическое определение алоэ-эмодина
50 мг Диацетилреина растворяют в 25,3 мл 0,5 М NaOH в делительной воронке и встряхивают в течение 10 мин. Затем добавляют 74,6 мл раствора, который содержит 0,5 М глицина и 0,5 М NaCl. При этом достигается pH 9,5.
Этот раствор экстрагируют 3 раза с помощью 25 мл хлороформа. Объединенные органические фазы экстрагируют 1 раз с помощью 10 мл 0,5 М буфера с pH 9,5 /глицин, NaOH, NaCl/ и 1 раз с помощью 10 мл 0,01 М серной кислоты. Удаляют растворитель органической фазы и остаток растворяют в 1 мл метанола.
Для стандартного раствора 2 мг алоэ-эмодина растворяют в 20 мл N,N-диметилацетамида и разбавляют метанолом вплоть до концентрации 2 мкг/мл/ соответственно 40 ppm.
Содержание растворов исследуют с помощью жидкостной хроматографии высокого давления /ЖХВД/. Линейность метода ЖХВД со стандартным раствором алоэ-эмодина обнаруживается в области от 0,11 мкг/мл/соответственно 2,2 ppm/до 53,6 мкг/мл/соответственно 1072 ppm/. Определения содержания осуществляют с помощью ЖХВД-колонки Merck 1 250 - 4, заполненной Li - Chrospher-100 RP-18, 5 мкм, при 40oC, с подвижной фазой из 1%-ной уксусной кислоты в метаноле /по объему/, 1%-ной уксусной кислоты в воде /по объему/ и ацетонитрила (в соотношении 49:46:5).
Аналитическое определение продукта стадии Б, а именно реин-9-антрон-8-глюкозида с содержанием алоэ-эмодиновых компонентов 41 ppm, определенных в виде алоэ-эмодина
Исследуемое вещество путем окисления с помощью хлорида железа-III при одновременном гидролизе с помощью соляной кислоты в двухфазной смеси из водного раствора и четыреххлористого углерода переводят в реин и алоэ-эмодин. Реин переводят в соль, так что ее можно отделять от алоэ-эмодина путем жидкостно-жидкостной экстракции. Находящийся в органической фазе алоэ-эмодин определяют с помощью ЖХВД.
Изобретение относится к области медицины и касается способа получения диацетилреина и фармацевтического средства, содержащего диацетилреин. Задача изобретения заключается в разработке способа получения диацетилреина с низким содержанием алоэмодиновых примесей. Сущность изобретения состоит в экстракции реин-9-антрон-8- глюкозида или смеси сеннозидов, восстановленных до соединений реин-9-антрон-8-глюкозида и алоээмодинатрон-8-глюкозида 2-бутанолом или ацетоном в водной среде, содержащей дитионит щелочного металла и восстановительно-окислительный потенциал, который является отрицательным и составляет не менее 210 mV, подкислении водной фазы кислотой, далее полученный реин-9-антрон-8-глюкозид окисляют солью железа /Fe III/ до реин-8-глюкозида, остаток глюкозы отщепляют в кислой среде, полученный реин ацетилируют и получают диацетилреин. Фармацевтическое средство для лечения артритных заболеваний содержит полученный таким образом диацетилреин и фармацевтически пригодную основу и вспомогательные вещества. Преимущество изобретения заключается в разработке препарата фармацевтически пригодной степени чистоты. 3 с. и 4 з.п. ф-лы.
| СПОСОБ ПРИГОТОВЛЕНИЯ КОНСЕРВИРОВАННОГО ПРОДУКТА "КОТЛЕТЫ РУБЛЕНЫЕ ИЗ ИНДЕЙКИ С ГАРНИРОМ И СОУСОМ БЕЛЫМ С ОВОЩАМИ" | 2013 |
|
RU2508798C1 |
| SU 757519 A, 1980 | |||
| US 3998966 A, 1976 | |||
| ЖИДКОСТНОЕ ИЛИ ГИДРАВЛИЧЕСКОЕ ФОРМОВАНИЕ С РАЗДУВОМ | 2011 |
|
RU2566772C2 |
| US 4244968 A, 1981. | |||

