Изобретение касается способа получения диацетилреина с фармацевтически приемлемой чистотой с остаточным содержанием нежелательных производных алоэ-эмодина в количестве менее чем 20 ppm (частей на миллион), содержащего диацетилреин, который можно получить этим способом, и фармацевтического средства, включающего это соединение.
Диацетилреин формулы: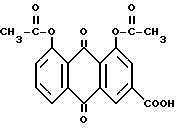
представляет собой лекарственное вещество, обладающее противоартритным, противовоспалительным, антипиретическим и обезболивающим действием.
Диацетилреин используется для лечения артритных заболеваний, (см., например, выложенную заявку ФРГ N 2711493 и выложенную заявку США N 4244968).
Диацетилреин можно получить, например, путем ацетилирования барбалоина и окисления полученного перацетилированного барбалоина трехокисью хрома. Кроме того, диацетилреин можно изготовить путем ацетилирования реина, который можно выделить, например, из лекарственного растительного сырья сенны (Sennadroge).
В диацетилреине, полученном таким способом, содержатся нежелательные примеси производных алоэ-эмодина, которые образуются от неполного окисления трехокисью хрома или извлекаются при экстрагировании лекарственного растительного сырья из сенны. Эти примеси содержатся в относительно небольшом количестве и поэтому могут быть отделены лишь с большим трудом с помощью классической очистительной операции. Кроме того, при вышеуказанном первом способе образуются осадки хрома, которые необходимо удалить соответствующим способом.
В соответствии с этим в основу данного изобретения положена задача создания способа получения диацетилреина, имеющего простое осуществление и высокий выход продукта, и образующего диацетилреин с фармацевтически приемлемой чистотой с остаточным содержанием нежелательных производных алоэ-эмодина в количестве менее чем 20 ppm (частей на миллион).
Эта задача решается предложенным в изобретении способом, отличающимся тем, что диацетилреин, содержащий компоненты алоэ-эмодина (т.е. алоэ-эмодин и/или его производные), подвергают диспергированию жидкость-жидкость в частично смешиваемым с водой полярном органическом растворителе и водной фазе при pH от 6,5 до 7,5 и получают диацетилреин, и в случае необходимости перекристаллизовывают его.
В предложенном согласно изобретению способе может использоваться диацетилреин, содержащий алоэ-эмодин. Значительным источником диацетилреина являются сеннозиды, содержащиеся в лекарственном растительном сырье из сенны, а также получаемый из сеннозидов реин-9-антрон-8-глюкозид.
Предпочтительным вариантом осуществления изобретения является способ получения диацетилреина в значительной степени свободного от производных алоэ-эмодина, причем:
A) реин-9-антрон-8-глюкозид, содержащий компоненты алоэ-эмодина, окисляют в соответствующие антрахиноновые соединения,
B) глюкозный остаток отщепляют в 8 положение антрахиновых соединений в кислой среде,
C) ацетилируют полученные 1,8-дигидроксиантрахиноновые соединения и
D) проводят диспергирование жидкость-жидкость полученного продукта между только частично смешиваемым с водой полярным органическим растворителем и водной фазой при pH от 6,5 до 7,5 и выделяют диацетилреин и в случае необходимости перекристаллизовывают.
Другим предпочтительным вариантом осуществления изобретения является способ получения диацетилреина в значительной степени свободного от производных алоэ-эмодина, причем:
A) смесь сеннозидов подвергают восстановлению до соответствующих антроновых соединений,
B) полученные антроновые соединения окисляют до соответствующих антрохиноновых соединений,
C) глюкозный остаток отщепляют в 8 положение антрахиноновых соединений в кислой среде,
D) полученные соединения ацетилируют 1,8-дигидроксиантрахиноновыми соединениями и
E) проводят диспергирование жидкость-жидкость полученного продукта между частично смешиваемым с водой полярным органическим растворителем и водной фазой при pH от 6,5 до 7,5 и выделяют диацетилреин, и в случае необходимости перекристаллизовывают.
Ниже поясняются отдельные этапы предложенного в изобретении способа.
Восстановление смеси сеннозидов в соответствующие антроновые соединения
Смесь сеннозидов, используемую в качестве исходного материала, можно получить, например, из лекарственного растительного сырья сенны (Sennadroge).
Лекарственное растительное сырье из сенны состоит из высушенных листьев и фруктов сенны, например, индийской сенны (Cassia angustifolia) и египетской сенны (Cassia acutifolia). Лекарственное растительное сырье из сенны содержит диантронглюкозиды реина и алоэ-эмодин. Важнейшими являются сеннозиды A, B, A1, C, D, D1. Сеннозиды соответствуют формуле: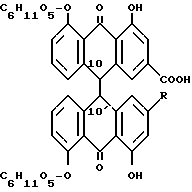
При сеннозидах A, B и A1 R обозначает COOH и при сеннозидах C, D и D1 R обозначает CH2OH. Сеннозиды A, B и A1 или C, D и D1 являются стереоизомерами и отличаются друг от друга конфигурацией по атомам C 10 и 10'.
Выделение сеннозидов из лекарственного растительного сырья из сенны описано, например, в выложенной заявке ФРГ N 3200131, которая принимается для данного описания в полном объеме. В соответствии с этой выкладкой сначала экстрагируют лекарственное растительное сырье из сенны водным метанолом. Концентрат, оставшийся после полного удаления метанола, содержит сеннозиды в виде соли щелочного металла, предпочтительно в виде соли калия. Концентрат очищают путем экстракции жидкость-жидкость частично растворимыми в воде спиртами или кетонами (например, бутанол-2,2-бутанон) (рафинит). Рафинит подкисляют примерно до pH от 1,5 до 2,0 и сеннозиды с помощью затравки кристаллизуют. Полученную смесь неочищенных сеннозидов пригодна в качестве исходного продукта для предложенного в изобретении способа. При желании можно также неочищенную смесь сеннозидов перекристаллизовать.
Как альтернативный вариант в качестве исходного продукта для предложенного в изобретении способа можно использовать концентрат, смешанный с частично растворимым в воде спиртом или кетоном, в частности, бутанолом-2.
При экстракции лекарственного растительного сырья из сенны соотношение лекарственного растительного сырья к экстрагирующему растворителю предпочтительно 1:4 до 1:10, в частности, 1:4 до 1:10.
Экстракцию осуществляют, предпочтительно, в присутствии буферного раствора, например, тринатрийцитрата, глицина, бикарбоната натрия или сахарозы.
По предложенному в изобретении способу эти исходные продукты подвергают восстановлению в реин-9-антрон-8-глюкозид и алоэ-эмодин-9-антрон-8-глюкозид (R=CH2OH) формулы: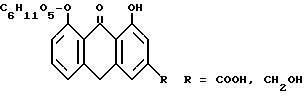
Восстановителями с приемлемым восстановительным потенциалом являются хлорид II олова, двуокись серы, бороводороды щелочных металлов, предпочтительно дитионит щелочного металла, в частности дитионит натрия. Восстановитель применяют с большим избытком. Дитионит, в частности дитионит натрия, применяют обычно в 1 - 4 кратном весовом количестве в расчете на содержание исходного материала по сеннозидам.
Для проведения восстановления можно брать исходный материал в водном растворе или суспензии и к нему добавить восстановитель в твердом виде или растворенный в воде. Предпочтительной является работа в двухфазной смеси, в которую добавляют, в крайнем случае, частично смешиваемый с водой полярный органический растворитель, в частности 2-бутанол.
Восстановление проводят, в частности, при температуре 40 - 60oC, в частности, при температуре 50 - 55oC и при pH от 7 до 9. Предпочтительно проводят многократное восстановление, в частности, от 2 до 10 раз.
Образующиеся 9-антрон-8-глюкозиды осаждают путем добавления кислоты, например, серной кислоты до pH от 4 до 4,5. При этом целесообразно, чтобы температура не превышала 40oC. При осаждении антроновых глюкозидов и при их выделении (например, путем фильтрации) целесообразно работать с азотом во избежание неконтролируемого окисления этого соединения.
Окисление антроновых соединений до антрахиноновых соединений.
Полученные антроновые соединения окисляют до соответствующих антрахиноновых соединений формулы: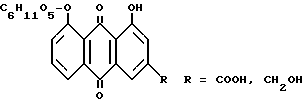
Подходящими окислителями для этой цели являются, например, кислород, перекисные соединения (перекись водорода), соединения марганца, хрома или железа с высокой степенью окисления. Предпочтительно используют соль железа (III), в частности сульфат железа (III). Целесообразно работать при повышенной температуре, тем не менее поддерживают 60oC. Благодаря этому обеспечивается отсутствие нежелательных и не поддающихся определению продуктов окисления. По окончанию окисления образуемые антрахинон-8-глюкозиды выделяют обычным способом.
Отщепление глюкозного остатка.
Глюкозный остаток в 8 позиции антрахиноновых соединений отщепляют в кислом растворе. Целесообразно работать при температуре примерно 85 - 95oC. Полученный продукт выделяют обычным способом.
Известно превращение сеннозида после гидролиза в кислой среде путем реакции обмена с хлоридом железа (III) непосредственно в реин (см., например, выложенную заявку ФРГ N 2711493). Однако, при этом выход продукта достигает только примерно 10% и, кроме того, полученный реин отделяется с трудом.
В способе согласно изобретению восстановительное расщепление сеннозидов, окисление образованных антроновых соединений и отщепление глюкозного остатка в 8 положение антрахиноновых соединений проводят соответственно по отдельным этапам. Неожиданно по этому способу был получен реин с выходом продукта 89%. К тому же возможно проведение окисления при щадящих температурах во избежание образования нежелательных и не поддающихся определению продуктов окисления. Кроме того, при проведении этой реакции можно снова выделить примерно в том же количестве использованную соль железа и после повторного окисления вновь использовать. Разделение этапов окисления и гидролиза позволяет на основании повышенной водорастворимости антроновых глюкозидов по сравнению с соответствующими агликами проводить окисление в щадящем режиме при комнатной температуре ниже 60oC, благодаря чему предотвращается обычно неизбежное образование не поддающихся определению побочных продуктов.
Ацетилирование 1,8-дигидроксиантрахинонового соединения
Ацетилирование полученных 1,8-дигидроксиантрахиноновых соединений осуществляют обычным способом. Например, можно ацетилировать ацетангидридом в присутствии ацетата натрия, как описано в Arch. Pharm, 241, 607 (1903). Однако, ацетилирование может осуществлять также другими, известными специалисту данной области методами, например, путем преобразования ацетилхлоридом и т. д.
Диспергирование жидкость-жидкость.
Проводят диспергирование жидкость-жидкость полученного продукта в полярном органическом растворителе, по крайней мере, частично смешиваемым с водой, и водной фазе при pH от 6,5 до 7,5. Подходящими полярными органическими растворителями являются C4-C5-алканол и C1-C3-диалкилкетон, как например, 1-бутанол, 2-бутанол, изобутанол и 2-бутанон. Предпочтителен последний.
Соотношение объемов тяжелой и легкой фаз находится обычно в пределах от 1:2 до 2:1. Более легкой фазой является раствор диацетилантрахиноновых соединений в полярном органическом растворителе. В качестве более тяжелой фазы служит водная фаза с pH 6,5 - 7,5, которая образуется буферным раствором, в частности, ацетатным буферным раствором.
Предпочтительно экстрагирование жидкость-жидкость осуществляют в противотоке. При этом диацетилреин добавляют в органической фазе в концентрации примерно 0,01 M.
После диспергирования необходимый диацетилреин находится в более тяжелой фазе. Диацетилреин путем подкисления доводят примерно до pH 5,2 и получают обычным способом, и перекристаллизовывают в виде соли щелочных металлов, предпочтительно соли калия, причем полученную соль превращают в нерастворимую свободную кислоту. Как вариант можно применять прямую перекристаллизацию этиллактатом.
Полученный этим способ диацетилреин в основном свободен от алоэ-эмодина и его производных. Содержание примесей достигает при этом еще примерно 50 частей на миллион (ppm) (определено по результатам анализов, описанных в примерах). Содержание этих примесей можно еще снизить, если полученный диацетилреин перекристаллизовать следующим образом. Диацетилреин превращают в соль щелочных металлов, причем обрабатывают подходящим основанием. Подходящим основанием является, например, ацетат щелочных металлов, предпочтительно, ацетат калия. Используют, предпочтительно, эквимолярные количества основания и водный C1-C3-спирт, например, 80 - 90% этанола в качестве реакционной среды. Соль щелочных металлов диацетилреина выкристаллизовывают на холоде, берут в водном C1-C3-спирте и путем добавления кислоты доводят до величины pH примерно 3. Осажденный диацетилреин выделяют обычным способом и обрабатывают. Как вариант можно использовать прямую перекристаллизацию этиллактатом.
Полученный таким образом продукт содержит менее чем 20 ppm (частей на миллион) вышеупомянутых примесей. Кроме того, продукт имеет форму иглообразных кристаллов, которые особенно подходят для галенового состава.
Продукт может высушиваться обычным способом. Целесообразно проводить сначала высушивание в вакууме при относительно низкой температуре, например, не более чем 40oC до тех пор, пока содержание воды в продукте упадет примерно на 3% или ниже. Затем можно повысить температуру до 70 - 110oC.
Изобретение касается также получения согласно изобретению практически чистого диацетилреина, а также фармацевтического средства, которое содержит это соединение. Области применения, отпускаемые дозы и приемлемые формы дозирования известны, см. выложенную заявку США N 4244968, 4346103, 4950687, выложенную заявку ФРГ N 2711493, а также лекарственные растительные средства Expt1. C1in. Res. 6(I) 53 - 64 (1980).
Нижеследующие примеры поясняют изобретение.
Пример 1. Выделение смеси сеннозидов в качестве исходного материала.
Всякий раз подают 40 кг лекарственного растительного сырья из сенны (содержание сеннозидов примерно 1,5%) в перколяторы, расположенные по два в ряду, с объемом 250 л и накрывают его перфорированной стальной пластиной. В качестве растворителя для экстрагирования используют 70%-ный метанол, который направляют к лекарственному сырью в первом перколяторе. Раствор, образовавшийся в первом перколяторе, направляют к лекарственному сырью, находящемуся во втором перколяторе. При этом растворитель свободно протекает первый перколятор.
Для экстрагирования 40 кг лекарственного растительного сырья из сенны используют всего 160 л растворителя. После того как эти объемы 70%-го метанола проведены через оба перколятора и получено соответствующее количество перколятора, сливной шланг перколятора присоединяют к резервуару соседнего перколятора и дополнительно направляют еще 60 л 70%-го метанола через перколяторы. Затем остаточный свободный растворитель направляют из первого перколятора в верхнюю часть второго перколятора и собирают вторичный перколят, пока он не составит 120 л. Затем освобождают первый перколятор, снова заполняют его 40 кг лекарственного растительного сырья из сенны, закачивают вторичный перколят к лекарственному сырью, причем 120 л вторичного перколята достаточно, чтобы покрыть лекарственное сырье в перколяторе. Затем температуру раствора доводят до +30oC.
Этот перколятор соединяют с перколятором, в котором раньше проводилось экстрагирование, и проводят экстрагирование как описано выше.
Всякий раз для 40 кг лекарственного сырья собирают 150 л перколята из которого в вакуумном ротационном испарителе, снабженном насадочной колонкой, удаляют метанол. Получают примерно 30 л осажденного продукта (концентрат), который экстрагируют в аппарате "смеситель-осадитель" (10 ступеней) с 40 л бутанола-2, насыщенного водой. Получают примерно 38-40 л водного очищенного продукта и примерно 30-32 л экстракта бутанола-2.
Водный очищенный продукт подкисляют 93% серной кислотой при помешивании в течение 20 ч, причем используют 1,6 об.% (относительно подкисляемого объема жидкости). Тогда подкисленный раствор имеет pH от 1,5 до 2,0. Перемешивают следующие 6 дн., оставляют отстаиваться на ночь, фильтруют его, промывают водой до бесцветного состояния воды, промывают метанолом и высушивают в потоке воздуха при комнатной температуре. Выход продукта на каждые 40 кг сырого материала достигает 760-790 г (высушенное вещество) сырого сеннозида с содержанием сеннозидов от 90 до 94%. Таким образом, выход продукта составляет примерно 70% количества сеннозидов, в пересчете на сырой материал.
Этап A: Восстановление сеннозидов в реин-9-антрон-8-глюкозид.
9 кг дитионита натрия растворяют в 100 л деминерализированной воды. К этому раствору добавляют при помешивании полученные сырые сеннозиды, содержащие примерно 3,0 кг сеннозидов A, A1 и B. Гомогенный раствор перемешивают 2 ч при температуре 55-58oC, затем охлаждают до 50-55oC. Затем с помощью 96-98% серной кислоты доводят до pH примерно 4,2. Полученную суспензию перемешивают далее в течение 1,5 ч при макс. температуре 25oC и затем фильтруется в атмосфере азота. Остаток промывают 50 л деминерализированной воды, которая с помощью серной кислоты доводится до pH 2. Затем сверху наливают раствор сульфата железа (III). (Получение см. этап B).
Этап B: Окисление до реин-8-глюкозид.
Продукт предшествующего этапа суспендируют в растворе из 184 л деминерализованной воды и 75,5 кг сульфатгидрата железа (III) (22% Fe3+). Суспензию нагревают до 55-62oC и окисляют 14 ч при использовании быстро протекающего диспергатора. После окончания окисления образованный реин-8-глюкозид отфильтровывают и промывают 50 л деминерализированной воды, pH которой доведена серной кислотой до 2.
Этап C: Гидролиз в реин.
Влажный остаток на фильтре из этапа B суспендируют в 200 кг 20 вес.% серной кислоты и перемешивают 8 ч при температуре 88-92oC. Образованный реин отфильтровывают и он может быть высушен для хранения при вакууме 1 мбар в течение 48 ч при температуре 40oC или также во влажном состоянии вводят сразу же для ацетилирования в этап D.
Общий выход продукта для этапов A-C: 89% в пересчете на сеннозиды A, A1 и B, введенные в этапе A.
Этап D: Ацетилирование в диацетилреин.
6,5 кг реина из этапа C суспендируют в 100 л уксусного ангидрида в течение 10 мин, смешивают с 2 кг ацетата калия, нагревают при помешивании до 95oC, смешивают с 0,65 кг активированного угля и перемешивают 1/2 ч при температуре 90-95oC. Активированный уголь отфильтровывают из горячего раствора и фильтрат смешивают с 2,1 кг 96-98%-ой по весу серной кислоты при температуре 90oC. Затем по возможности быстро охлаждают при помешивании до 20oC. Полученную суспензию фильтруют. Остаток промывают деминерализированной водой до состояния, свободного от сульфата.
Выход продукта: 83%.
Этап E: Удаление свободного и ацетилированного алоэ-эмодина.
Части алоэ-эмодина удаляют путем противоточного экстрагирования на пульсационной экстракционной колонне с по меньшей мере 15 теоретическими тарелками. Соотношение объемов тяжелой фазы к легкой равно 1:1. В качестве тяжелой фазы служит бутанон-2-насыщенный 0,1 молярным, водным раствором ацетата калия. В легкой фазе, состоящей из насыщенного водой бутанон-2, растворяют подлежащий очистке 0,01 молярный диацетилреин. Из протекающей тяжелой фазы с помощью 10 процентной по весу серной кислоты при pH 5,2 выпадает диацетилреин. Осадок отфильтровывают и промывают деминерализованной водой до свободного от сульфата состояния.
Выход продукта: 88%, в пересчете на введенный сырой диацетилреин из этапа D.
Этап F: Перекристаллизация, сушка, измельчение.
Вариант A
7,5 кг диацетилреина из этапа E (в пересчете на высушенное вещество) суспендируют при быстром помешивании в 250 л 90%-го по объему этанола. Суспензию нагревают до 70oC и затем смешивают с 3,75 кг ацетата калия. При охлаждении на 0-2oC из периодически образующегося прозрачного раствора выкристаллизовывается чистая соль калия диацетилреина.
Соль калия отфильтровывают и растворяют в 300 л 40%-го по объему этанола при добавлении 3 кг ацетата калия при температуре 20-30oC. Прозрачный раствор доводят 10%-ой по весу серной кислотой до pH 3,0. Выкристаллизованный диацетилреин отфильтровывают и промывают деминерализированной водой до состояния, свободного от сульфата.
Вариант B.
7,5 кг диацетилреина суспендируют в 275 л этиллактата, путем нагрева вводят в раствор, фильтруют и кристаллизуют при помешивании до 20-25oC. Выкристаллизованный диацетилреин отфильтровывают и промывают деминераллизированной водой.
Высушивание продукта осуществляют сначала в вакууме при 1 мбар и 40oC в течение 24 ч. Если содержание остаточной воды падает ниже 3%, материал крупно измельчают и при вакууме 1 мбар и 70oC повторно сушат в течение 24 ч.
Затем измельченный материал просеивают через просеивающее устройство с ячейками 0,5 мм и для удаления остатков растворителя повторно сушат при вакууме 1 мбар и 70oC.
Выход продукта: 95%.
Пример 2. Повторяют способ, описанный в примере 1, причем предпринимают следующие модификации.
При экстрагировании лекарственного растительного сырья из сенны применяют тринатрийцитрат, причем 2,85 кг тринатрийцитрата перед подачей растворителя добавляют всякий раз к 40 кг лекарственного растительного сырья. При этом в качестве растворителя используют нагретый до 60oC 70% метанол. После удаления метанола до остаточного объема 11,4 л концентрат разбавляют примерно 2 л бутанола-2.
Восстановление смеси концентрата плодового сока сенны/бутанола-2 осуществляется затем в 7 этапов в атмосфере азота в качестве защитного газа. После первого этапа восстановления происходит осаждение сырого реин-9-антрон-8-глюкозида. Этапы восстановления II-VII служат для частичного удаления производных алоэ-амодина. Эти этапы проводят без осаждения. Окончательное осаждение реин-9-антрон-8-глюкозида происходит после последнего этапа восстановления.
Этап восстановления I.
100 л смеси концентрата плодового сока сенны/бутанола-2, содержащей примерной 4 кг сеннозидов, подают в резервуар с мешалкой, с подачей азота. При помешивании поочередно добавляют 6 л 20 вес.% раствора едкого натра, затем добавляют 350 л насыщенного водой бутанола-2 (например, из этапа II) и перемешивают 15 мин. Исходную смесь нагревают до 42-50oC и разбавляют 7 кг дитионата натрия. Перемешивают еще 45 мин. Величина pH поддерживается 20 вес. % раствором едкого натра около 7,5-8. Восстановительный потенциал (относительно электродов Ag/AgCl), при необходимости поддерживается подачей дитионита натрия ниже - 630 мВ. После охлаждения на 30-35oC осаждают 10 вес. % серной кислотой в течение 1,5 ч до величины pH < 4. Образовавшуюся суспензию перемешивают при невысокой скорости вращения при температуре < 25oC в течение примерно 10 ч. Полученный осадок отфильтровывают. Осадок суспендируют в 60 л 15 вес.% бутанола-2, в течение 30 мин при температуре 50-60oC перемешивают и затем фильтруют. Осадок промывают 100 л деминерализированной воды. Сырой выход реина-9-антрон-8-глюкозида, в пересчете на введенные сеннозиды, выше 82%.
Восстановительный этап II.
3,3 кг сырого реин-9-антроглюкозида из этапа I суспендируют в смеси из 42 л деминерализированной воды и 7,4 л бутанола-2. Суспензия вместе с 2 л 20 вес. % раствора едкого натра и 9,9 кг тринатрийцитрата подается в раствор и затем разбавляется 3,3 кг дитионата натрия и 350 л бутанола-2, насыщенного водой (например, из этапа III). Исходную смесь нагревают до 42-45oC. Величина pH поддерживается 20 вес.% раствором едкого натра около 8,5-9. Восстановительный потенциал (относительно электродов Ag/AgCl) поддерживается при необходимости путем подачи дитионита натрия ниже -750 мВ.
После выдерживания продолжительностью 30 мин верхнюю фазу отбирают и продолжают обработку нижней фазы в этапе III.
Восстановительный этап III.
С нижней фазой из этапа II при добавлении следующих химических веществ повторяют способ восстановления/экстрагирования, описанный в этапе II:
1,65 кг дитионит натрия,
0,8 л 20 вес.% раствор едкого натра,
350 л насыщенного водой бутанола-2,
(например, из этапа IV).
Восстановительные этапы IV-VII.
С нижней фазой из соответственно предшествующих этапов при добавлении следующих химических веществ повторяют способ восстановления/экстрагирования, описанный в этапе II:
0,825 кг дитионит натрия,
0,4 л 20 вес.% раствор едкого натра,
350 л насыщенного водой бутанола-2,
(например, из соответственно нижеследующих этапов).
Нижнюю фазу, отделенную на этапе VII, охлаждают до температуры 30 - 35oC и реин-9-антрон-8-глюкозид выпадает в осадок, как описано на этапе I. Полученный осадок фильтруют и промывают 100 л деминерализированной воды. Затем сверху наливают 10 л раствора сульфата железа (III). (Получение см. этап В, пример 1).
Затем реин-9-антрог-8-глюкозид, как описано в примере 1, превращают в диацетилреин.
Фармакологические исследования
Действие диацетилреина установлено при хронических воспалительных заболеваниях при оральном применении.
Применялись следующие экспериментальные модели:
а) Cotton-Pellet-гранулема у крысы и
б) вызванный интраартикулярным применением витамина A артроз у кролика.
а) Cotton-Pellet-гранулема у крысы.
Молодые бесполые крысы (н = 10) получали 25, 50 или 100 мг диацетилреина/кг или 5 мг индометацина/кг или 100 мг ацетилсалициловой кислоты/кг ежедневно в течение 5 дн. Одновременно действовала контрольная группа, обрабатываемая только водой. Имплантация шариков осуществлялась в первый день лечения. Сырой и сухой вес препарированный в конце испытания гранул показали значительное и отчетливое зависимое от дозы изменение по сравнению с контрольной группой. При этом действие 100 мг диацетилреина/кг соответствовало примерно действию 5 мг индометацина или 100 мг ацетилсалициловой кислоты. Вес тимуса и надпочечников во время обработки не изменялся.
б) От витамина A-артроз.
В результате трех интраартикулярных инъекций 30000 ед. витамина A в течение 9 дн. в двух группах, в каждой по 10 кроликов (белая Новозеландская порода) было вызвано артрозоподобное суставное изменение. 56 дн. спустя 10 животных лечили 3 мг диацетилреина/кг/день в течение 8 недель. По сравнению с контрольной группой макроскопически и микроскопически распознаваемые суставные изменения были значительно ниже в группе, подвергавшейся лечению.
Далее лечебное действие диацетилреина сравнили с действием ацетилсалициловой кислоты на каждого из 7 кроликов, которые после 6 дневного предварительного лечения с трехразовым введением инъекций по 10000 ед. витамина A и после 26 дневного интервала в лечение в течение 8 недель получали либо 5 мг диацетилреина/кг/день (опытная группа), 15 мг ацетилсалициловой кислоты/кг/день (позитивная контрольная группа), либо оставались без лечения (негативная контрольная группа). Во всех трех группах через 24 дн. после последней инъекции витамина A возникало расстройство движений, выражавшееся в волочении задних лапок. В негативной контрольной группе в течение следующих 8 недель усиливались клинические признаки явного артроза. В опытной группе и позитивной контрольной группе эти симптомы значительно улучшились при 8 недельном лечении.
Изменения слизистой оболочки желудка.
В то время как одноразовый прием 400 мл диацетилреина/кг или растворителя не вызывал у крысы никаких эрозий слизистой оболочки желудка, после приема ибупрофена (200 мг/кг) или индометацина (20 мг/кг) обнаруживались очевидные повреждения слизистой оболочки от точечных (диаметром 1 мм) до больших (диаметром 3 мм) эрозий. Также двухразовый ежедневный прием 100 мг диацетилреина/кг более 3 дн. не вызвал никаких повреждений слизистой оболочки желудка, однако, возможно соответствующее применение 10 мг индометацина/кг. При этом отмечались эрозии диаметром 1 - 3 мм.
Токсикология.
Острая токсичность ЛД50 в зависимости от исследуемого вида (крыса, мышь, кошка) при оральном применении достигает 1,9 - 7,9 кг. При этом крыса проявила наименьшую чувствительность. При парентеральном введении (внутривенно, внутрипарентерально) величины ЛД50 у этих видов находятся между 119 и 339 мг/кг.
Клинические исследования.
1. В двойном слепом опыте против напроксена и последующей обработки с помощью плацебо исследовалось действие диацетилреина при коксартрозе и гонартрозе у 95 (49/46) пациентов. Доза приема была 50 мг диацетилреина 2 раза в день или 750 мг напроксена ежедневно. Длительность обработки 60 дн. после 7-дневной подачи вымывающей фазы. Последующая обработка с помощью плацебо продолжалось свыше 60 дн.
Испытуемыми величинами являлись болевые и двигательные симптомы по шкале отсчета, функциональные ограничения и переносимость.
В обеих лечебных группах (диацетилреин/напроксен) относительно всех проверочных параметров была установлена статистически значимая скорость улучшения (P < 0,01 или P < 0,05) по сравнению с исходными величинами. Однако, после прерывания обработки и последующего введения плацебо в группе диацетилреин/плацебо на 90 и 120 дн. относительно таких параметров как спонтанная боль, активная и пассивная двигательная боль, обнаружилось статистически значимое превосходство (P < 0,01) по сравнению с группой напроксен/плацебо. Это различие обеспечивалось на уровне 5% также для переменных параметров, отражающих ночную боль, боль при надавливании через 30 дн. после прекращения введения диацетилреина.
2. В открытом длительном опыте с контролем исследовалось действие диацетилреина против остеоартроза позвоночника и колена у 70 пациентов (35/35). Принимаемая доза - 100 мг диацетилреина ежедневно. Длительность обработки достигала 60 дн., длительность наблюдения 75 дн. Параметры определялись по системе подсчета.
Контрольная группа включала 35 пациентов, в которой проводились исключительно физио-терапевтические мероприятия. В группе лечения диацетилреином также проводилась физиотерапия.
Анализ результатов показал по всем параметрам статистически значительное превосходство по сравнению с контрольной группой. Также после прерывания лечения для группы лечения диацетилреином мог установится продолжительный терапевтический эффект ("подвешенный эффект").
3. В слепом одиночном сквозном опыте относительно напроксена исследовалось действие диацетилреина при локализованном артрозе у 20 пациентов. Пациенты были разделены на две группы, причем в первой группе сначала выдавали два раза в день по 50 мг диацетилреина в течение 20 дн. Затем в течение 3 дн. проходила вымывающая фаза и дальнейшее лечение с приемом 250 мг напроксена два раза в день, ежедневно, в течение следующих 20 дн. Во второй группе соблюдалась обратная последовательность. Длительность лечения достигла в целом 43 дн. Проверочными параметрами были боль, компрессионная боль, пассивная двигательная боль, функциональное ограничение и опухание по системе подсчета.
Анализ результатов показывает превосходство лечения диацетилреином по сравнению с лечением напроксеном. Не наблюдались никакие достойные внимания побочные явления, а также никакие изменения клинических лабораторных параметров.
4. При произвольном выборе двойного слепого опыта в "технологии двойного плацебо" относительно напроксена исследовалось действие диацетилреина на 23 пациентах (12/11) с остеоартрозом (опыт на переносимость). Принималась доза 50 мг диацетилреина два раза в день, ежедневно и 250 мг напроксена 3 раза в день, ежедневно. Лечение продолжалось 4 недели. Проверочными параметрами были эзофагогастродуоденоскопические результаты, собранные до и после терапии. На исследование принимались только пациенты с нормальными результатами слизистой оболочки или с легкими поражениями слизистой оболочки (степень 1).
Через 4 недели эндоскопические результаты показали в одном случае (10%) в группе лечения диацетилреином поражения слизистой оболочки второй степени, в то время как в группе лечения напроксеном у 5 пациентов (50%) поражения слизистой оболочки второй, третьей и четвертой степени. Во всех случаях при поступлении имел место нормальный диагноз.
Аналитическое определение алоэ-эмодина.
50 мг диацетилреина растворяют в 25,3 мл 0,5 М NaOH в делительной воронке и встряхивают 10 мин. Затем добавляют 74,6 мл раствора, содержащего 0,5 М глицина и 0,5 NaCl. При этом величина pH равнялась 9,5.
Этот раствор экстрагируют трижды с 25 мл хлороформа. Соединенные органические фазы экстрагируют 1 раз с 10 мл 0,5 М буферного раствора с величиной pH 9,5 (глицин, NaOH, NaCl) и один раз 10 мл 0,01 М серной кислоты. Удаляют растворитель органической фазы и остаток растворяют в 1 мл метанола. Для стандартного раствора 2 мг алоэ-эмодина растворяют в 20 мл N,N-диметилацетамида и разбавляют метанолом до концентрации 2 мкм/мл (соответственно 40 частей на миллион).
Содержание растворов исследуется с помощью жидкостной хроматографии высокого разрешения (HPLC). Линейность метода HPLC подтверждается стандартным раствором алоэ-эмодина в диапазоне от 0,11 мкм/мл (соответственно 2,2 ppm) до 53,6 мкм/мл (соответственно 1072 ppm). Определение содержания осуществляется колонками HPLC-Lichrocart 250-4 фирмы МЕРК, заполненными Li-Chrospher-100 RP - 18,5 мкм при 40oC с подвижной фазой из 1%-го раствора уксусной кислоты в метаноле (объем/объем), 1% раствора уксусной кислоты в воде (объем/объем) и ацетонитрила в соотношении 49:46:5.
Использование: очистка диацетилреина от примесей алоэ-эмодин-компонентов с получением фармацевтически чистого диацетилреина. Сущность изобретения: исходный диацетилреин, включающий примеси алоэ-эмодин-компонентов, подвергают диспергирования в системе жидкость-жидкость с ограниченно смешивающимся с водой полярным органическим растворителем, например 2-бутаноном, и водной фазой с величиной pH 6,5-7,5, при этом выделяют диацетилреин, который при необходимости перекристаллизовывают. Исходный диацетилреин получают из смеси сеннозидов через антроновые соединения, которые окисляют до антрахиноновых соединений и затем полученный продукт ацетилируют. В качестве окислителя предпочтительно используют соль железа III. В результате получают диацетилреин с остаточным содержанием производных алоэ-эмодина в количестве 20 ppm, который используют в качестве активного начала в фармацевтической композиции, обладающей противоартритным, противовоспалительным, антипиритическим и анальгетическим действием. 2 с. и 13 з.п.ф-лы.
жидкость и выделяют диацетилреин, который при необходимости перекристаллизовывают.
| US, патент, 4950687, кл | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US, патент, 4244968, кл | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

