Данное изобретение относится к новым соединениям, имеющим конденсированное гетероциклическое кольцо, которые обладают превосходной активностью в качестве лекарства, например, антагонистической по отношению к эндотелиновому рецептору активностью, и используются в качестве вазодиляторов и в терапевтической композиции для лечения заболеваний, таких как гипертония, острая почечная недостаточность, инфаркт миокарда, грудная ангина и церебральный ангиоспазм, и способу получения этих соединений.
Описание предшествующего уровня техники
Было предположено, что заболевания взрослых людей, возросшие в последующее время, например, церебральный инфаркт, грудная ангина, инфаркт миокарда и почечная недостаточность, вызванные ишемией, связаны, возможно, с эндотелином. Эндотелин является пептидом, содержащим 21 аминокислоту, выделяемым из эндотелиальных клеток, и получаемым в виде эндотелина-1, эндотелина-2 и эндотелина-3. В дальнейшем, в данном описании, эти эндотелиновые группы объединены понятием "эндотелин". Сообщалось, что среди in vivo или синтетических веществ ранее найденных, эндотелин обладает наиболее сильным и продолжительным ангиоспазматическим действием, прессорной активностью и действием на повышение активности сокращения сердечной мышцы. Считается, что действия этих пептидов осуществляются посредством эндотелинового рецептора, который предполагается существующим на мембране гладкой мышцы кровяных сосудов и т. д. В качестве эндотелиновых рецепторов известны эндотелин-A рецептор и эндотелин-B рецептор (в дальнейшем называемые вместе "эндотелиновый рецептор").
Поэтому соединения, проявляющие сродство к эндотелиновому рецептору, а также проявляющие антагонистическую по отношению к эндотелиновому рецептору активность, обладают профилактическими и терапевтическими действиями против заболеваний, вызываемых ишемией, например церебральный инфаркт, грудная ангина, инфаркт миокарда и почечная недостаточность, таким образом, разработка таких соединений весьма многообещающа.
В качестве веществ, антагонистических по отношению к эндотелиновому рецептору, были получены соединения природного происхождения, как описано в некоторых исследованиях, например, Ishimaru et al. [JPA H4 (1992) - 134048], Fujimoto et al. [Federation of European Biochemical Societies Letters, 305 p. 41 (1992)]. Oh-hata et al. [JPA H3 (1991) - 047163], Miyata et al. [JPA H4 /1992/ - 046127] и Jano et al. [JPA H3 /1991// - 094692].
Далее, сообщения о том, что были получены пептидные соединения, были сделаны Henmi et al. [EP 457195-A2], Ishikawa et al. [EP 460679-A2 и EP 436189-A] , Hashimoto et al. [JPA H3 /1992/ - 130299], Masaki et al. [JPA H3 /1992/ - 024099], G. Hamon et al. [EP 487410-A2], W.L. Cody et al. [J. Med. Chem. /1992/ 35, p. 3303] и Wakimasu et al. [Wo 9113089-A, EP 499266-A1].
Однако, когда принимаются во внимание дозированные формы лекарств, стабильность соединений, длительность фармакологического действия и устойчивость к метаболизму, синтетические антагонисты эндотелинового рецептора, полученные непептидным синтезом этих пептидных соединений, являются весьма необходимыми. При данных обстоятельствах, однако, имеется очень мало сообщений по непептидным синтетическим антагонистам эндотелинового рецептора, например недавнее сообщение K. Bali et al. [EP 510526-A1].
С другой стороны, описаны H.R Haward et al. [WO 92/12979/ производные 2,4/1H, 3H/-диоксопиридо[2,3-d] пиримидин-3-уксусной кислоты, которые имеют относительно похожую структуру как и у соединения по настоящему изобретению. Заместители у указанных соединений, однако, явно отличаются от таковых по настоящему изобретению. Далее, сообщалось, что вышеуказанные соединения используются в терапии в качестве ингибиторов альдозоредуктазы для контролирования некоторых осложнений при хроническом диабете, но не обладают действием антагонистов эндотелинового рецептора.
Объектом настоящего изобретения являются новые и полезные соединения, каждое из которых имеет конденсированное гетероциклическое кольцо, особенно новые соединения, которые могут быть использованы в качестве непептидных синтетических антагонистов эндотелинового рецептора, благодаря которым могут быть решены вышеуказанные проблемы.
Другим объектом настоящего изобретения является новое соединение, имеющее свойства антагониста эндотелинового рецептора, которое стабильно как соединение и имеет длительный фармакологический эффект и метаболическую устойчивость.
Дальнейшим объектом настоящего изобретения является способ получения соединения и композиция антагониста эндотелинового рецептора.
Разработчики настоящего изобретения провели тщательную исследовательскую работу по достижению вышеуказанных целей, в результате чего было найдено, что эти цели могут быть достигнуты новыми производными пиридопиримидина, обладающими антагонистической по отношению к эндотелиновому рецептору активностью, и, таким образом, настоящее изобретение в целом основано на этом найденном факте.
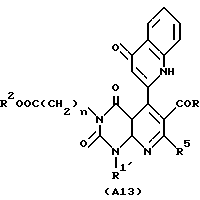
Настоящее изобретение касается производных пиридо-[2,3-d]пиримидина формулы A или их солей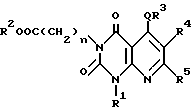
где Q является -(CH2)m - (m = 0, 1, 2), -O-, -S(O)p - (p = 0, 1, 2) или -NH-;
n = 0, 1, 2 3;
R1 и R2 независимо являются атомом водорода, необязательно замещенной C1-6-акильной группой, необязательно замещенной арильной группой или необязательно замещенной аралкильной группой;
R3 является необязательно замещенной циклической углеводородной группой или необязательно замещенной гетероциклической группой;
R4 является атомом водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной арильной группой или необязательно замещенной аралкильной группой, цианогруппой, -COOR6 (R6 является атомом водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной циклической углеводородной группой или необязательно замещенной аралкильной группой) или -CONR7R8 (R7 и R8 независимо являются атомом водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной циклической углеводородной группой или необязательно замещенной аралкильной группой);
R5 является атомов водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной арильной группой, необязательно замещенной аралкильной группой, -X1R9 (X1 является -O-, -NR10- или -S-, R9 и R10 независимо являются атомом водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной арильной группой или необязательно замещенной аралкильной группой).
В качестве особенно предпочтительных примеров производных пиридопиримидина и их солей поэтому изобретению заслуживают внимания производные общей формулы A1 и их соли:
в которой n1 = 1, 2, 3;
R1 является атомом водорода, C1-6-алкильной группой или C7-15-аралкильной группой, необязательно замещенной C1-6-алкоксигруппой или C1-6-алкилтиогруппой;
R2 является атомом водорода или C1-6-алкильной группой;
R3 является C6-16-арильной группой, необязательно замещенной по крайней мере одной группой, выбранной из группы, включающей C1-6-алкильную группу, C3-7-циклоалкильную группу, C1-6-алкоксигруппу, атом галогена, нитрогруппу, цианогруппу и фенильную группу, или 5 - 13-членную ароматическую гетероциклическую группу, содержащую от 1 до 4 гетероатомов, выбранных из атома азота, кислорода и атома серы, необязательно замещенную по крайней мере одной группой, выбранной из группы, включающей C1-6-алкильную группу. C1-6-алкоксигруппу, оксогруппу и гидроксигруппу;
R4 является -COOR6 (R6 является атомом водорода, C1-6-алкильной группой необязательно замещенной карбоксильной группой или 5 - 10-членной гетероциклической группой, содержащей от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода или атома серы, C3-7-циклоалкильной группой или C6-15-аралкильной группой) или -CONR7R8 (R7 и R8 независимо являются атомом водорода, C1-6-алкильной группой или C6-14-арильной группой;
R5 является атомом водорода, C1-6-алкильной группой или C6-14-арильной группой, необязательно замещенной C1-3-алкилендиоксигруппой.
Настоящее изобретение относится к способу получения производного пиридо[2,3-d]пиримидина формулы A или его соли, который заключается в том, что соединение формулы I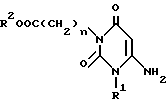
в которой n, R1 и R2 имеют те же значения, что указаны выше,
или его соль подвергают нагреванию с обратным холодильником в растворителе вместе с циклическим углеводородом или гетероциклическим альдегидом формулы R3Q-CHO (Q и R3 имеют те же значения, что указаны выше) и бета-кетоэфиром, или вместе с производным, полученным при дегидратирующей конденсации указанного альдегида и кетоэфира, с получением соединения формулы B
в которой n, R1 - R5 имеют те же значения, что указаны выше,
или его соли.
Соединение B или его соль подвергают окислению окислительным агентом с получением соединения формулы A
в которой n, Q, R1 - R5 имеют те же значения, что указаны выше,
или его соли.
Затем для случая, когда R2 не является атомом водорода, указанное соединение A или его соль могут быть гидролизованы до соединения формулы Aa
в которой n, R1, R3 - R5 и Q имеют те же значения, что указаны выше,
или его соли.
Далее, настоящее изобретение относится к способу получения производного пиридо[2,3-d] пиримидина формулы A или его соли, который заключается в том, что соединение формулы I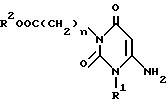
в которой n, R1 и R2 имеют те же значения, что указаны выше,
нагревают вместе с 3,3-бисметилтиопроизводным в диметилформамиде с получением соединения формулы C.
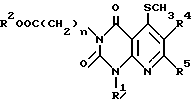
в которой n, R1, R2, R4 и R5 имеют те же значения, что указаны выше,
или его соли. Соединение C или его соль подвергают взаимодействию с нуклеофильным агентом, представленным формулой R3QH (Q и R3 имеют те же значения, что указаны выше) с получением соединения формулы A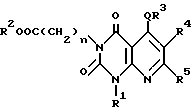
в которой n, Q, R1 - R5 имеют значения, указанные выше,
или его соли. Далее, для случая, когда R2 не является атомом водорода, указанное соединение A или его соль могут быть гидролизованы с получением соединения формулы Aa''
в которой n, R1, R3, R4, R5 и Q имеют те же значения, что указаны выше,
или его соли.
Кроме того, настоящее изобретение касается способа получения производного пиридо[2,3-d] пиримидина формулы A или его соли, который заключается в том, что соединение формулы II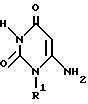
в которой R1 имеет те же значения, что указаны выше,
или его соль подвергают нагреванию с обратным холодильником в растворителе вместе с производным, представленным формулой QR3CHCR4COR5 (Q, R3, R4 и R5 имеют те же значения, что указано выше), полученным путем дегидратной конденсации циклического углеводородного или гетероциклического альдегида, представленного формулой R3Q-CHO (Q и R3 имеют те же значения, что указаны выше) и бета-кетоэфира с получением соединения формулы B
в которой Q, R1, R3 - R5 имеют те же значения, что указаны выше,
или его соли окислением соединения B или его соли с окислительным агентом с получением соединения формулы D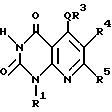
в которой Q, R1, R3 - R5 имеют те же значения, что указаны выше,
затем подвергают соединение D или его соль реакции с галогенированным алкилкарбоксилатным производным формулы X2(CH2)nCOOR2, в которой X2 является атомом галогена, n и R2 имеют те же значения, что указаны выше, в подходящем растворителе в присутствии основания с получением соединения формулы A и его соли. Затем, в случае, когда R2 не является атомом водорода, указанное соединение A или его соль может быть гидролизовано с получением соединения формулы Aa или его соли.
Кроме того, настоящее изобретение касается способа получения производного пиридо[2,3-d] пиримидина общей формулы A или его соли, в которой QR3 является 2-(4-хинолонил)-группой и R4 - карбоксильной группой, который заключается в нагревании соединения формулы E или его соли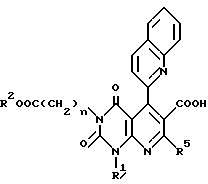
в которой n, Q, R1, R2 и R5 имеют те же значения, что указаны выше,
с обратным холодильником вместе с тионилхлоридом с получением соединения формулы Ab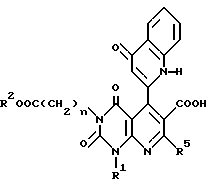
в которой n, Q, R1, R2 и R5 имеют те же значения, что указаны выше,
или его соли.
Затем для случая, когда R2 не является атомом водорода, указанное соединение Ab или его соль могут быть гидролизованы с получением соединения формулы Ac или его соли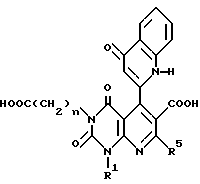
в которой n, R1 и R5 имеют значения, указанные выше.
Кроме того, настоящее изобретение относится к способу получения производного пиридо[2,3-d]пиримидина общей формулы A или его соли, который включает реакцию соединения или его соли;
в которой n, Q, R2 - R5 имеют значения, указанные выше,
галогенированным алкильным, галогенированным арильным или галогенированным аралкильным соединением формулы R1X2, в котором X2 является атомом галогена, R1 имеет те же значения, что указаны выше, в подходящем растворителе в присутствии основания с получением соединения формулы A и его соли. Затем в случае, когда R2 не является атомом водорода, указанное соединение A или его соль могут быть гидролизованы с получением соединения формулы Aa или его соли.
Кроме того, настоящее изобретение относится к способу получения производного пиридо[2,3-d]пиримидина формулы A или его соли, в которой R4 является -COOR6 (R6 является необязательно замещенной C1-6-алкильной группой, необязательно замещенной циклической углеводородной группой или необязательно замещенной аралкильной группой, который включает реакцию соединения формулы C или его соли: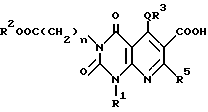
в которой n, Q, R1 - R3 и R5 имеют те же значения, что указаны выше,
с галогенированным алкильным, галогенированным арильным или галогенированным аралкильным соединением, представленным формулой R6'X2, в которой X2 является атомом галогена, R6' имеет те же значения, что указаны выше, в подходящем растворителе в присутствии основания с получением соединения формулы A, в которой R4 является - COOR6 (R6 имеет те же значения, что указаны выше) и его соли. Затем для случая, когда R2 не является атомом водорода, указанное соединение A или его соль могут быть гидролизованы с получением соединения формулы Aa, в которой R4 является - COOR6 (R6 имеет те же значения, что указаны выше) или его соли.
Кроме того, настоящее изобретение относится к способу получения производного пиридо[2,3-d]пиримидина общей формулы A, в которой OR3 является 2-(4-хинолонил)группой, R4 является -COOR6 или (-CONR7R8 (R6, R7 и R8 имеют те же значения, что указаны выше) или его соли, который включает реакцию соединения формулы Ad или его соли: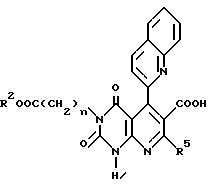
в которой n, R2 и R5 имеют те же значения, что указаны выше,
с тионилхлоридом в подходящем растворителе, затем взаимодействие с галогенированным алкильным, галогенированным арильным или галогенированным аралкильным соединением формулы R1X2, в которой X2 является атомом галогена, R1 имеет те же значения, что указаны выше, в подходящем растворителе в присутствии основания с получением соединения формулы H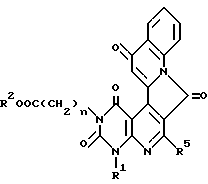
в которой n, R1, R2 и R5 имеют те же значения, что указаны выше,
или его соли и затем взаимодействие соединения H или его соли с нуклеофильным реагентом, который способен ввести заместитель, представленный -QR6 или -NR7R8 (R6, R7 и R8 имеют те же значения, что указаны выше) в подходящем растворителе с получением соединения формулы A, в которой OR3 является 2-(4-хинолонил)группой, R4 является - COOR6 или -CONR7R8 (R6, R7 и R8 имеют те же значения, что указаны выше), и его соли.
Затем для случая, когда R2 не является атомом водорода, указанное соединение A или его соль могут быть гидролизованы с получением соединения формулы Ac, в которой OR3 является 2-(4-хинолонил)группой и R4 является - COOR6 или -CONR7R8 (R6, R7 и R8 имеют те же значения, что указаны выше) или его соли.
Также настоящее изобретение относится к антагонистическому по отношению к эндотелиновому рецептору агенту, содержащему в качестве эффективного компонента производное пиридо-[2,3-d] пиримидина, представленного вышеприведенной формулой A, или его фармацевтически приемлемую соль.
Далее, настоящее изобретение относится к композиции указанного антагониста по отношению к эндотелиновому рецептору в качестве терапевтической композиции для лечения острой почечной недостаточности, инфаркта миокарда, гипертензии, церебрального инфаркта, грудной ангины, артериального склероза, гепатопатии, легочной гипертензии, бронхиальной астмы, органогипофункции, имеющей место при операции или трансплантации органов, особенно терапевтической композиции для лечения острой почечной недостаточности и/или инфаркта миокарда.
Соединение формулы A по данному изобретению (в дальнейшем упоминаемое как соединение A) ниже будет описано подробно.
В формуле A Q означает - (CH2)m (m = 0 или 1, 2), -O-, -S(O)p (p = 0 или 1, 2) или -NH-, предпочтительно Q - -(CH2)-, особенно предпочтительно m = 0 или 1;
n = 0 или 1, 2, 3, предпочтительно 1 или 2, особенно предпочтительно 1.
В формуле A R1 и R2 могут быть одинаковы или отличны друг от друга и соответственно являются атомом водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной арильной группой или необязательно замещенной аралкильной группой.
Примеры C1-6-алкильной группы включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, пентильную группу, изопентильную группу, неопентильную группу, трет-пентильную группу, 1-этилпропильную группу, гексильную группу, изогексильную группу, 1,1-диметилбутильную группу, 2,2-диметилбутильную группу, 3,3-диметилбутильную группу и 2-этилбутильную группу.
Среди них C1-4-алкильные группы, представленные примерами метильной группы, этильной группы, пропильной группы, изопропильной группы, бутильной группы и изобутильной группы, например, являются предпочтительными. Метильная группа является особенно предпочтительной.
C1-6-алкильная группа может иметь от 1 до 3 подходящих заместителей, в качестве примера C3-7-циклоалкильная группа, 5-10-членная ароматическая гетероциклическая группа, содержащая от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода или атома серы, 5-10-членная неароматическая гетероциклическая группа, содержащая от 1 до 4 атомов, выбранных из атома азота, атома кислорода и атома серы, аминогруппа, моно-(C1-6-алкил)аминогруппа, ди(C1-6-алкил)аминогруппа, амидиногруппа, C1-6-алкилкарбонильная группа, C6-14-арилкарбонильная группа, C7-15-аралкилкарбонильная группа, C1-6-алкоксикарбонильная группа, C6-14-арилоксикарбонильная группа, C7-15-аралкилоксикарбонильная группа, карбамоильная группа, моно-(C1-6-алкилкарбамоильная группа, ди-(C1-6)алкилкарбамоильная группа, сульфамоильная группа, моно-(C1-6)алкилсульфамоильная группа, ди-(C1-6)алкилсульфамоильная группа, карбоксильная группа, гидроксильная группа, (C1-6)-алкоксильная группа, C2-6-алкенилоксигруппа, C3-15-аралкилоксигруппа, меркаптогруппа, C1-6-алкилтиогруппа, C6-14-арилтиогруппа, C7-15-аралкилтиогруппа, сульфогруппа, цианогруппа, азидогруппа, нитрогруппа, нитрозогруппа или атом галогена.
Согласно настоящему изобретению арильная группа означает моноциклическую или конденсированную полициклическую ароматическую углеводородные группы, в качестве примеров C6-14-арильной группы, такие, как фенильная группа, нафтильная группа, антрильная группа, фенантрильная группа и аценафтиленильна группа, особенно предпочтительными являются фенильная группа, 1-нафтильная группа и 2-нафтильная группа.
Арильная группа может иметь один или более, предпочтительно от 1 до 3, подходящих заместителей, в качестве примера C1-6-алкильная группа (например, метильная группа, этильная группа и пропильная группа), C2-6-алкенильная группа (например, винильная группа, аллильная группа и 2-бутенильная группа), C2-6-алкинильная группа (например, пропаргильная группа и 2-бутинильная группа), циклоалкильная группа (например, C3-7-циклоалкильная группа, такая как циклопропильная группа, циклобутильная группа, циклопентильная группа и циклогексильная группа), C6-14-арильная группа (например, фенильная группа и нафтильная группа), ароматическая гетероциклическая группа (например, 5-9-членная ароматическая гетероциклическая группа, содержащая от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, в качестве примера фурильная группа, тиенильная группа, пиррольная группа, тиазолильная группа, имидазолильная группа, пиридильная группа, хинолильная группа и хинолонильная группа), неароматическая гетероциклическая группа (например, 5-9-членная неароматическая гетероциклическая группа, содержащая от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, в качестве примера оксиранильная группа, азетидинильная группа, оксетанильная группа, тиетанильная группа, пирролидинильная группа, тетрагидрофурильная группа, тиоланильная группа, пиперидильная группа, тетрагидропиранильная группа, морфонильная группа, тиоморфонильная группа и пиперазинильная группа), аралкильная группа (например, C7-15-аралкильная группа, такая как бензильная группа, фенилэтильная группа, 1-нафтилметил, 1-нафтиэтил), аминогруппа, N-монозамещенная аминогруппа (например, C1-6-моноалкиламиногруппа, такая как метиламиногруппа, этиламиногруппа и пропиламиногруппа), N,N-замещенная аминогруппа (например, N, N-дизамещенная аминогруппа, замещенная C1-6-алкильной группой, в качестве примера диметиламиногруппа и диэтиламиногруппа), ацидиногруппа, ацильная группа (например, C1-8-алкилкарбонильная группа, такая как формильная группа, ацетильная группа, пропионильная группа и бутирильная группа; C6-14-арилкарбонильная группа, такая как бензоильная группа; C7-15-аралкилкарбонильная группа, такая как бензилкарбонильная группа, фенилэтилкарбонильная группа, C1-8-алкоксикарбонильная группа, такая как метоксикарбонильная группа и этоксикарбонильная группа; C6-14-арилоксикарбонильная группа, такая как фенилоксикарбонильная группа, альфа-нафтилкарбонильная группа и C7-15-аралкилоксикарбонильная группа, такая, как бензилоксикарбонильная группа, 1-нафтилоксикарбонильная группа), карбамоильная группа, N-монозамещенная карбонильная группа (например, C1-6-алкилкарбамоильная группа, такая как метилкарбамоильная группа, этилкарбамоильная группа и пропилкарбамоильная группа), N,N-дизамещенная карбамоильная группа (например, N,N-дизамещенная карбамоильная группа, замещенная C1-6-алкильной группой, что подтверждено примерами на диметилкарбамоильную группу и диэтилкарбамоильную группу), сульфамоильная группа, N-монозамещенная сульфамоильная группа (например, N-алкилсульфамоильная группа, имеющая C1-6-алкильную группу, что проиллюстрировано метилсульфамоильной группой, этилсульфамоильной группой и пропилсульфамоильной группой), N,N-дизамещенная сульфамоильная группа (например, N,N-диалкилзамещенная сульфамоильная группа, имеющая C1-6-алкильную группу, в качестве примера диметилсульфамоильная группа и диэтилсульфамоильная группа), карбоксильная группа, гидроксильная группа, C1-6-алкоксильная группа (например, метоксигруппа, этоксигруппа и пропоксигруппа, C2-6-алкенилоксигруппа (например, винилоксигруппа и аллилоксигруппа), циклоалкилоксигруппа (например, C3-7-циклоалкилоксигруппа), такая как циклопропилоксигруппа и циклоэтилоксигруппа), аралкилоксигруппа (например, C7-14-аралкилоксигруппа, такая, как бензилоксигруппа, 1-нафтилоксигруппа), арилоксигруппа (например, C6-14-арилоксигруппа, такая как фенилоксигруппа и нафтилоксигруппа), меркаптогруппа, C1-6алкилтиогруппа (например, метилтиогруппа, этилтиогруппа и пропилтиогруппа), аралкилтиогруппа (например, C7-15-аралкилтиогруппа, такая как бензилтиогруппа, 1-нафтилтиогруппа), арилтиогруппа (например, C6-14-арилтиогруппа, такая как фенилтиогруппа и нафтилтиогруппа), сульфогруппа, цианогруппа, азидогруппа, нитрогруппа, нитрозогруппа, атом галогена (например, атом фтора, атом хлора, атом брома и атом йода), C1-3-алкилендиокси группа (например, метилендиоксигруппа, этилендиоксигруппа) среди прочих.
Упоминавшаяся аралкильная группа означает алкильную группу, имеющую арильную группу в качестве заместителя (арилалкильная группа), и указанная арильная группа является предпочтительной такой же, как вышеуказанная арильная группа, а в качестве предпочтительной алкильной группы подразумевается C1-6-алкильные группы. Предпочтительная аралкильная группа включает C7-15-аралкильную группу, например бензильную группу, фенилэтильную группу, 3-фенилпропильную группу, (1-нафтил)метильную группу и (2-нафтил)метильную группу, особенно предпочтительной является фенил-(C1-3)-алкильная группа, такая как бензильная группа и фенетильная группа.
Арильная группа в аралкильной группе может иметь те же заместители, что может иметь вышеуказанная арильная группа и указанный заместитель предпочтительно является C1-6-алкоксигруппой (например, метоксигруппа, этоксигруппа или пропоксигруппа), C1-6-алкилтиогруппой (например, метилтиогруппой, этилтиогруппой или пропилтиогруппой), особенно предпочтительны заместители, являющиеся C1-3-алкоксигруппой, такой как метоксигруппа.
Предпочтительные примеры R1 включают атом водорода, C1-6-алкильную группу (например, метильную группу, этильную группу), C7-15-аралкильную группу, необязательно замещенную C1-6-алкоксигруппой или C1-6-алкилтиогруппой (особенно предпочтителен заместитель, являющийся фенил-(C1-3)-алкильной группой, необязательно замещенной C1-6-алкоксигруппой или C1-6-алкилтиогруппой).
Предпочтительные примеры R2 включают атом водорода, C1-6-алкильную группу (например, метильную группу, этильную группу, пропильную группу и изопропильную группу).
В формуле A R3 обозначает необязательно замещенную циклическую углеводородную группу или необязательно замещенную гетероциклическую группу, и указанная циклическая углеводородная группа представлена в примерах арильной группой или циклоалкильной группой. Указанная арильная группа обозначает такую же арильную группу, как описано по отношению к R1 и R2, которая может иметь, как и арильная группа радикалов R1 и R2, один или более, предпочтительно от 1 до 3, подходящих заместителей. В качестве указанных заместителей указаны такие же, как описано по отношению к арильной группе радикалов R1 и R2. Особенно предпочтительный заместитель включает C1-6-алкильную группу, C3-7-циклоалкильную группу, C1-6-алкоксигруппу, цианогруппу, нитрогруппу, атом галогена и фенильную группу.
Примеры указанной циклоалкильной группы включают C3-10-циклоалкильную группу или C3-10-бициклоалкильную группу, как показано в примерах циклопропильной группой, циклобутильной группой, циклопептильной группой, циклогексильной группой, циклогептильной группой, циклооктильной группой, бицикло/2,2,1/-гептильной группой, бицикло/2,2,2/октильной группой, бицикло/3,2,1/октильной группой, бицикло/3,2,1/нонильной группой, бицикло/4,2,1/нонильной группой и бицикло/4,3,1/децильной группой. Предпочтительный пример указанной циклоалкильной группы включает C4-7-циклоалкильную группу (например, циклобутильную группу, циклопентильную группу, циклогексильную группу, циклогептильную группу). Указанная циклоалкильная группа может иметь от 1 до 3 подходящих заместителей, таких как C1-6-аклильная группа, C2-6-алкенильная группа, C2-6-алкинильная группа, карбоксильная группа, гидроксильная группа, нитрогруппа или атом галогена.
Примеры указанной гетероциклической группы включают 5-13-членную ароматическую гетероциклическую группу, в качестве атома, содержащего в кольце от 1 до 4 гетероатомов, выбранных из O, S и N, или насыщенную или ненасыщенную неароматическую гетероциклическую группу (алифатическую гетероциклическую группу).
Предпочтительные примеры указанной гетероциклической группы включают ароматические моноциклические гетероциклические группы, такие как фурильная группа, тиенильная группа, пирролильная группа, оксазолильная группа, изоксазолильная группа, тиазолильная группа, изотиазолильная группа, имидазолильная группа, пиразолильная группа, 1,2,3-оксадиазолильная группа, 1,2,4-оксадиазолильная группа, 1,3,4-оксадиазолильная группа, фуразанильная группа, 1,2,3-тиадиазолильная группа, 1,2,4-тиадиазолильная группа, 1,3,4-тиадиазолильная группа, 1,2,3-тиазолильная группа, 1,2,4-тиазолильная группа, тетразолильная группа, пиридильная группа, пиридазинильная группа, пиримидильная группа, пиразинильная группа и триазинильная группа; и ароматические конденсированные гетероциклические группы, такие как бензофуранил, изобензофуранил, бензо/b/тиенил, индолил, изоиндолил, 1H-индазолил, бензоимидазолил, бензоксазолил, 1,2-бензоизоксазолил, бензотиазолил, 1,2-бензоизотиазолил, 1H-бензотриазолил, хинолил, хинолин-N-оксид-2-ил, хинолин-N-оксид-3-ил, изохинолил, циннолинил, хинозолинил, хиноксалинил, фталазинил, нафтиридинил, пуринильная группа, птеридинильная группа, карбазолильная группа, альфа-карболинильная группа, бета-карболинильная группа, гамма-карболинильная группа, акридинильная группа, феноксазинильная группа, фенокситиазинильная группа, тиантренильная группа, фенантридинильная группа, фенантролинильная группа, индолизинильная группа, пирроло/1,2-b/пиридазинильная группа, пиразоло/1,5-a/пиридильная группа, имидазо/1,2-a/пиридильная группа, имидазо/1,2-a/пиридильная группа, имидазо/1,5-a/пиридильная группа, имидазо/1,2-a/пиридазильная группа, имидазо/1,2-a/пиримидильная группа, 1,2,4-триазоло/4,3-a/пиридильная группа, и 1,2,4-триазоло/4,3-b/пиридазинильная группа.
Особенно предпочтительным примером является пиридильная группа, хинолильная группа, хинолин-N-оксид-2-ил, хинолин-N-оксид-3/ил, бензофуранильная группа, бензо/b/тиенильная группа или тиенильная группа.
Предпочтительные примеры указанной гетероциклической группы включают оксиранильную группу, азетидинильную группу, оксетанильную группу, тиетанильную группу, пирролидинильную группу, тетрагидрофуранильную группу, тиоланильную группу, пиперидильную группу, тетрагидропиранильную группу, морфолинильную группу, тиоморфолинильную группу, и пиперазинильную группу.
Также, указанная гетероциклическая группа может иметь один или более, предпочтительно от 1 до 3, подходящих заместителей, которые являются такими же, как указаны для арильной группы радикалов R1 и R2. Среди прочих предпочтительной является C1-6-алкильная группа.
Предпочтительные примеры R3 включают C6-14-арильную группу (например, фенильную группу, нафтильную группу), необязательно замещенную по крайней мере одним заместителем, выбранным из группы, содержащей C1-6-алкильные группы, C3-7-циклоалкильные группы, C1-6-алкоксигруппы, атомы галогена, нитрогруппу, цианогруппу, и фенильную группу; или 5-13-членную ароматическую гетероциклическую группу, содержащую от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы (например, пиридильная группа, хинолильная группа, хинолин-N-оксид-2-ил, хинолин-N-оксид-3-ил группа, бензофуранильная группа, бензо/b/тиенильная группа, тиенильная группа), необязательно замещенную по крайней мере одним заместителем, выбранным из группы, содержащей C1-6-алкильные группы, C1-6-алкоксигрупы, оксогруппу и гидроксильную группу. Среди прочих, фенильная группа, нафтильная группа, пиридильная группа, хинолильная группа, тиенильная группа, хинолин-N-оксид-2-ил, хинолин-N-оксид-3-ил группа, бензофуранильная группа, метил-бензо/b/тиенильная группа и 4-хинолонильная группа (например, 2-(4-хинолонильная группа)) является особенно предпочтительными в качестве R3. 4-хинолонильная группа является хинолонильной группой, замещенной оксогруппой в 4-м положении.
В формуле A R4 является атомом водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной арильной группой, необязательно замещенной аралкильной группой, цианогруппой, -COOR6 (R6 символизирует атом водорода, необязательно замещенную C1-6-алкильную группу, необязательно замещенную циклическую углеводородную группу или аралкильную группу), или -CONR7R8 (R7, R8 независимо обозначают водород, необязательно замещенную C1-6-алкильную группу, необязательно замещенную циклическую углеводородную группу или аралкильную группу).
Указанная C1-6-алкильная группа, арильная группа или аралкильная группа имеют те же значения, что указаны по отношению к R1 и R2, а указанная циклическая углеводородная группа имеет те же значения, что указаны для аналогичной группы по отношению к радикалу R3. Указанные C1-6-алкильная группа, арильная группа, аралкильная группа и циклическая углеводородная группа могут иметь, как и в случае радикалов R1, R2 и R3, один или более, предпочтительно от 1 до 3, подходящих заместителей, которые являются такими же, как описано для R1, R2 и R3.
Предпочтительные примеры R4 включают -COOR6 или -CONR7R8, в которых R6 является предпочтительно C1-6-алкильной группой, C3-7-циклоалкильной группой или C7-15-аралкильной группой, которая может быть необязательно замещена атомом водорода, карбоксильной группой или 5-10-членной гетероциклической группой, содержащей от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы; и R7 и R8 являются, соответственно, атомом водорода, C1-6-алкильной группой или C6-14-арильной группой. Особенно предпочтительным примером R6 является C1-6-алкильная группа, замещенная хинолильной группой.
Предпочтительные примеры - CONR7R8 включают карбамоильную группу или карбамоильную группу, замещенную C1-6-алкильной группой.
В формуле A R5 обозначает атом водорода, необязательно замещенную C1-6-алкильную группу, необязательно замещенную арильную группу, необязательно замещенную аралкильную группу, или -X1R9 (X1 означает -O-, NR10- или -S-; R9 и R10 независимо обозначают атом водорода, необязательно замещенную C1-6-алкильную группу, необязательно замещенную арильную группу или необязательно замещенную аралкильную группу). C1-6-алкильная группа, арильная группа и аралкильная группа в радикалах R5, R9 и R10 являются такими же, как те же группы, указанные по отношению к радикалам R1 и R2, и указанные арильная группа и аралкильная группа могут иметь, как и в случаях для R1, R2 и R3 один или более, предпочтительно, от 1 до 3, подходящих заместителей. В качестве этих заместителей упоминаются такие же заместители, как указаны по отношению к радикалам R1 и R2.
Предпочтительные примеры R5 включают атом водорода, необязательно замещенную C1-6-алкильную группу, необязательно замещенную арильную группу, особенно предпочтительными являются атом водорода, C1-6-алкильная группа (например, метильная группа, этильная группа, пропильная группа, изопропильная группа, и изобутильная группа), или C6-15-арильная группа (например, фенильная группа), необязательно замещенные C1-3-алкилендиоксигруппой.
Предпочтительные примеры соединения A по данному изобретению включают соединение, в котором n = 1, 2, 3; R1 является атомом водорода, C1-6-алкильной группой или C7-15-аралкильной группой, необязательно замещенными C1-6-алкоксигруппой или C1-6-алкилтиогруппой; R2 является атомом водорода или C1-6-алкильной группой; QR3 является C6-14-арильной группой, необязательно замещенной по крайней мере одним заместителем, выбранным из группы, содержащей C1-6-алкильные группы, C3-7-циклоалкильные группы, C1-6-алкоксигруппы, атомы галогена, нитрогруппу, цианогруппу или фенильную группу, или 5-13-членную ароматическую гетероциклическую группу, содержащую от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, необязательно замещенную по крайней мере одним заместителем, выбранным из группы, содержащей C1-6-алкильные группы, C1-6-алкоксигруппы, оксогруппу, и гидроксильную группу; R4 является -COOR6-1 (R6-1 является атомом водорода, C1-6-алкильной группой, необязательно замещенной карбоксильной группой или 5-10-членной гетероциклической группой, содержащей от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы; C3-7-циклоалкильной группой, или 6-15 аралкильной группой) или -CONR71R81 (R71 и R81 - независимо являются атомом водорода, C1-6-алкильной группой, или C6-14-арильной группой); R5 является атомом водорода, C1-6-алкильной группой или C6-14-арильной группой, необязательно замещенной C1-3-алкилендиоксигруппой. Особенно предпочтительным примером является такой, когда 5-13-членная ароматическая гетероциклическая группа, содержащая от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, радикала R3 выбрана из группы, содержащей пиридильную группу, хинолильную группу, хинолин-N-оксид-2-ил, хинолин-N-оксид-3-ил, группу, бензофуранильную группу, бензо/b/тиенильную группу и тиенильную группу, а 5-10-членная гетероциклическая группа, содержащая от 1 до 4 гетероатомов, выбранных из атома азота, атома кислорода и атома серы, радикала R61 является хинолильной группой.
Характерные примеры предпочтительного соединения данного изобретения включают 2,4/1H, 3H/-диоксо-6-этоксикарбонил-/-изопропил-1-/2-метоксибензил/-5- /4-толил/пиридо/2,3-o/пиримидин-3-уксусную кислоту, 2,4/1H,3H/-диоксо-6-этоксикарбонил-7-метил-1-/2-метоксибензил/-5- /4-метоксифенил/пиридо/2,3-o/пиримидин-3-уксусную кислоту, этил/2,4/1H,3H/-диоксо-6-карбокси-7-изопропил-1-/2-метоксибензил/-5- /2-/4-хинолонил//пиридо/2,3-o/пиримидин-3-ацетат, 2,4/1H,3H/-диоксо-6-карбокси-7-изопропил-1-/2-метоксибензил/-5- /2-/4-хинолонил//пиридо/2,3-o/пиримидин-3-уксусную кислоту, 2,4/1H, 3H/-диоксо-6-карбокси-7-изопропил-1-/2-метилтиобензил/-5- /2-/4-хинолонил//пиридо/2,3-o/пиримидин-3-уксусную кислоту и их соли.
В качестве солей соединения A согласно настоящему изобретению соли добавления фармацевтически приемлемой кислоты являются отмеченными как особенно предпочтительные. В качестве таких солей используют полученные, например, соли с неорганической кислотой (например, хлористоводородная кислота, фосфорная кислота, бромистоводородная кислота или серная кислота) или соли с органической кислотой (например, уксусная кислота, муравьиная кислота, малеиновая кислота, янтарная кислота, виноградная кислота, лимонная кислота, малоновая кислота, щавелевая кислота, бензойная кислота, метансульфоновая кислота или бензолсульфоновая кислота). И для случая, когда соединение A настоящего изобретения имеет кислотную группу, такую как -COOH, соединение A может образовывать соль с неорганическим основанием (например, щелочной металл или щелочноземельный металл, такой как натрий, калий, кальций или магний; или аммиак), или с органическим основанием (например, триалкиламин, имеющий C1-8-алкильные группы, такой как триэтиламин).
В качестве солей исходных соединений для получения соединения A согласно настоящему изобретению используют соли, полученные, например, с неорганической кислотой (например, хлористоводородная кислота, фосфорная кислота, бромистоводородная кислота или серная кислота) или соли с органическими кислотами (например, уксусная кислота, муравьиная кислота, пропионовая кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, виноградная кислота, лимонная кислота, малоновая кислота, щавелевая кислота, бензойная кислота, метансульфоновая кислота или бензолсульфоновая кислота). И, для случая, когда исходное соединение имеет кислотную группу, такую как -COOH, оно может образовывать соль с неорганическим основанием (например, щелочной металл или щелочноземельный металл, такой как натрий, калий, кальций или магний, или аммиак), или с органическим основанием (например, триалкиламин, имеющий C1-8-алкильную группу, такой как триэтиламин).
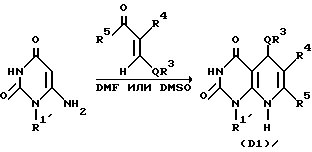
Соединение A согласно настоящему изобретению и его соли могут быть получены известными специалистам способами, и, в качестве типичных, отмечаются следующие семь способов.
Способ получения 1
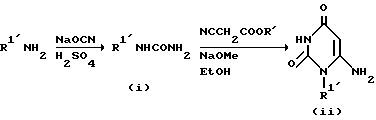
Производное мечены (i), представленное формулой R1'NHCONH2 (R1' означает необязательно замещенную C1-6-алкильную группу, арильную группу или аралкильную группу), которое синтезировано исходя из C1-6-алкиламина, ариламина или аралкиламина, нагревают в подходящем растворителе, таком, как этиловый спирт, в присутствии производного цианоуксусной кислоты в качестве основания, при 80-120oC в течение 2-240 ч, предпочтительно 24-100 ч, с получением 1-/C1-6-алкил/замещенного, 1-арилзамещенного или 1-аралкилзамещенного производного 6-аминопиримидин-2,4/1H,3H/диона (ii).
Это соединение перемешивают вместе с галогенированным производным эфира уксусной кислоты или галогенированным производным эфира пропионовой кислоты в присутствии основания в подходящем растворителе, таком как диметилацетамид или диметилформамид, при 40-70oC обычно в течение 4-96 ч, предпочтительно 12-24 ч, с получением производного эфира 6-амино-2,4/1H,3H/-диоксопиримидин-3-уксусной кислоты или производного эфира-6-амино-2,4/1H,3H/-диоксопиримидин-3-уксусной кислоты или производного эфира -6-амино-2,4/1Н, 3Н/диоксопиримидин-3-пропионовой кислоты, представлены формулой /I'/.
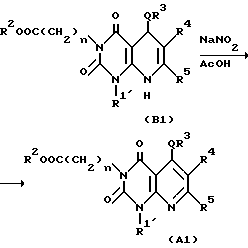
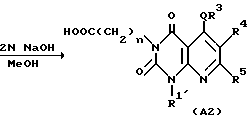
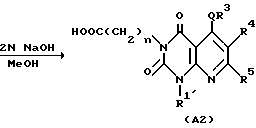
Полученное таким образом производное /I'/ в соответствии с синтетическим методом Ганча [A. Hautzseh, Ann. Chem. 215, 1/1882/] подвергают нагреванию с арилальдегидом и бета-кетоэфиром с обратным холодильником при 80-100oC, обычно в течение 2-240 ч, предпочтительно 12-120 ч, в подходящем растворителе, таком, как этиловый спирт, или предварительно подвергают реакции конденсации Кновенагеля [T. Jamamori, J. Hiramatsu, K. Sakai and I. Adachi, Tetrahedron, 41, 913/1985/] с получением дегидратированного конденсированного производного арилальдегид и бетакетоэфир, затем дегидратированное конденсированное производное и производное /I/ нагревают с обратным холодильником при 100-130oC обычно в течение 2-240 ч, предпочтительно 12-120 ч, в подходящем растворителе, таком как толуол с получением производного 5,8-дигидропиридо/2,3-o/пиримидин-2,4-диона /B1/. Соединение B1 подвергают окислению подходящим окислительным агентом, таким как нитрит натрия в уксусной кислоте с получением соединения A1 по настоящему изобретению. Далее, если необходимо, соединение A1 подвергают в соответствующих условиях, например, щелочному гидролизу в подходящем растворе, таком как метанол, этанол, тетрагидрофуран или диоксан, с получением соединения A2 настоящего изобретения. Реакция щелочного гидролиза может быть проведена перемешиванием реакционной смеси в присутствии подходящего основного катализатора (например, гидроксида натрия, гидроксида лития, гидроксида калия или других), при комнатной температуре или при повышенной температуре (например, 40-100oC) обычно в течение 2-48 ч, предпочтительно в течение 2-24 ч.
Реакции по вышеописанному способу получения сведены в приведенную ниже реакционную схему, в которой каждое обозначение имеет те же значения, что указаны выше.
Способ получения 1.
Способ получения 2.
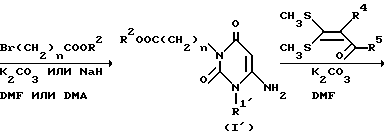
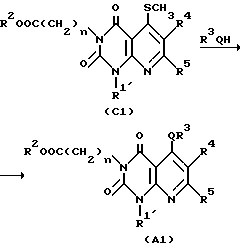
Согласно аналогичному способу, описанному G. Tominaga et al. [Chim. Pharm. Bull. 32, 122 /1984/] производное эфира -6-амино-2,4/1H,3H/-диоксопиримидин-3-уксусной кислоты /I/ нагревают с 3,3-бисметилтиопроизводным в диметилформамиде при 100-150oC обычно в течение 1-12 ч, предпочтительно 2-6 ч с получением производного пиридо/2,3-o/пиримидин-2,4-диона /C1/. Нуклеофильный реагент формулы R3OH (R3 и O имеют значения, указанные выше) подвергают взаимодействию с соединением C1 при 40-100oC обычно в течение 2-120 ч, предпочтительно 2-24 ч, в подходящем растворителе (например, метаноле, этаноле, тетрагидрофуране, диоксане или диметилформамиде), с получением соединения A1 настоящего изобретения.
Затем, если необходимо, соединение A1 подвергают в подходящих условиях, например, щелочному гидролизу в подходящем растворителе, таким как метанол, этанол, тетрагидрофуран или диоксан с получением соединения A2 по настоящему изобретению. Реакция щелочного гидролиза может быть осуществлена перемешиванием реакционной смеси в присутствии подходящего основного катализатора (например, гидроксида натрия, гидроксида лития, гидроксида калия и других), при комнатной температуре или при повышенной температуре (например 40-100oC) обычно в течение 2-48 ч, предпочтительно в течение 2-24 ч. Вышеупомянутые реакции совместно показаны в следующей реакционной схеме, в которой R1 является необязательно замещенной C1-6-алкильной группой, арильной группой или аралкильной группой и каждое обозначение имеет значения, указанные выше /0029/.
Способ получения 2.

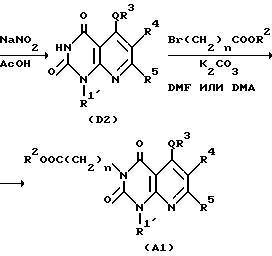
Способ получения 3.
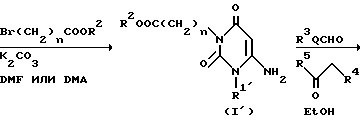
Производное 1-(C1-6-алкил)замещенного, 1-арилзамещенного или 1-аралкилзамещенного 6-аминопиримидан-2,4/1H,3H/диона /ii/ в соответствии с ранее упомянутым синтетическим методом Ганча /A. Hautzseh, Ann. Chem. 215, 1 /1882// подвергают нагреванию с продуктом дегидратной конденсации производного альдегида и бета-кетоэфирного производного при 40-120oC обычно в течение 0,5-4 ч, предпочтительно 0,5-1 ч, в подходящем растворителе (например, диметилацетамиде, диметилсульфоксиде, диметилформамиде) с получением соединения D1.
Продукт дегидратной конденсации является предварительно полученным нагреванием производного альдегида (например, хинолин-2-карбоксальдегид) с бета-кето-эфирным производным (например, этил изобутилацетат, 2-цианоэтил изобутилацетат) с обратным холодильником в подходящем растворителе (например, бензол, толуол) в присутствии подходящего катализатора (например, пирролидин и уксусная кислота, пиперидин с уксусная кислота) при 100-120oC в течение обычно 0,5-1 ч.
Соединение D1 подвергают окислению подходящим окислительным агентом, таким как нитрит натрия в уксусной кислоте, в подходящем растворителе, таком, как уксусная кислота, обычно в течение 0,25-24,0 ч, предпочтительно 1-12 ч, с получением соединения D2. Соединение D2, полученное таким образом, перемешивают с галогенированным производным эфира уксусной кислоты или галогенированным производным эфира пропионовой кислоты в присутствии основания (например, гидрида натрия, карбоната калия) в подходящем растворителе (например, диметилацетамид, диметилформамид) при комнатной температуре или при повышенной температуре, такой, как 40-120oC, в течение обычно 0,5-12,0 ч, предпочтительно 0,5-2,0 ч, с получением соединения A1, производного эфира уксусной кислоты по 3-му положению, или сложного эфира пропионовой кислоты, согласно настоящему изобретению.
Затем, если необходимо, соединение A1 подвергают щелочному гидролизу в подходящем растворителе, таком как метанол, этанол, тетрагидрофуран или диоксан с получением соединения A2 по настоящему изобретению. Реакция щелочного гидролиза может быть проведена перемешиванием реакционной смеси в присутствии подходящего основного катализатора (например, гидроксида натрия, гидроксида лития, гидроксида калия или других), при комнатной температуре или при повышенной температуре (например 40-100oC) обычно в течение 2-12 ч.
Реакции по вышеуказанному способу получения совместно показаны в следующей далее реакционной схеме, в которой каждое обозначение имеет то же значение, что указано выше.
Способ получения 3.

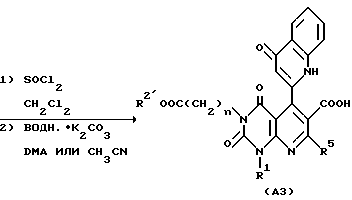
Способ получения 4.
Здесь приведен пример замены заместителя в соединении согласно настоящему изобретению.
5-/2-Хинолил/-6-цианоэтилкарбонильное производное E1 подвергают перемешиванию в подходящем растворителе (например, метанол, этанол, тетрагидрофуран или диоксан) в присутствии подходящего слабого основания (например, водный раствор карбоната натрия или гидрокарбоната натрия) при комнатной температуре или повышенной температуре (например, 40-100oC): обычно в течение 2-12 ч, предпочтительно 0,5-2,0 ч, с получением 6-карбоксильного производного E2.
Соединение E2 растворяют в подходящем растворителе (например, метиленхлорид, четыреххлористый углерод, 1,2-дихлорэтан или толуол) и подвергают нагреванию и реакции с избытком тионилхлорида (например, 10-кратном от эквивалента) с обратным холодильником (например, 41-120oC) в течение 5-120 мин, предпочтительно 5-60 мин. Реакционную смесь затем концентрируют досуха с удалением избытка тионилхлорида. Остаток растворяют в подходящем растворителе (например, ацетонитрил, тетрагидрофуран, диоксан или диметилацетамид). При добавлении подходящего слабого основания (например, водный раствор карбоната натрия или гидрокарбоната натрия) раствор подвергают перемешиванию при комнатной температуре при повышенной температуре (например, 40-100oC) обычно в течение 2-48 ч, предпочтительно 2-24 ч, с получением 4-хинолонильного производного A3.
Затем, если необходимо, соединение A3 подвергают щелочному гидролизу в подходящем растворителе, таком как метанол, этанол, тетрагидрофуран или диоксан с получением соединения A4 настоящего изобретения. Реакция щелочного гидролиза может быть осуществлена перемешиванием реакционной смеси в присутствии подходящего основного катализатора (например, гидроксида натрия, гидроксида лития, гидроксида калия и других) при комнатной температуре или при повышенной температуре (например, 40-100oC) обычно в течение 2-12 ч.
Реакции вышеописанного способа получения совместно показаны в приведенной далее схеме реакции, в которой R2' является необязательно замещенной C1-6-алкильной группой, арильной группой или аралкильной группой, и каждое обозначение, за исключением R2, имеет те же значения, что указаны выше.
Способ получения 4.


Способ получения 5.
1-/2,4-Диметоксибензольное/производное /F1/ подвергают окислению с получением 1-гидропроизводного /F/. Окисление производят следующим образом.
Соединение F1 растворяют в подходящем растворителе (например, водный раствор ацетона или ацетонитрила), к которому добавлен подходящий окислительный агент, такой как церий аммоний нитрат /CAN/, и смесь перемешивают при комнатной температуре или при повышенной температуре (например, 40-60oC) обычно в течение 0,5-2,0 ч, предпочтительно 0,5-1,0 ч, с получением соединения F.
Другой способ окисления включает перемешивание соединения F1 в трифторуксусной кислоте /TFA/ при 40-80oC обычно в течение 1-12 ч, предпочтительно 2-6 ч, с получением соединения F. Соединение F, полученное таким образом, подвергают взаимодействию при перемешивании с галогенированным производным низшего алкила (например, метилиодина) или галогенированным аралкильным производным (например, замещенное производное бензилхлорида или замещенное производное бензилбромида) в подходящем растворителе (например, диметилацетамид, диметилформамид, ацетон или тетрагидрофуран) в присутствии подходящего основания (например, гидрид натрия или карбонат калия) обычно в течение 0,5-4,0 ч, предпочтительно 0,5-2,0 ч, с получением соединения A.
Затем, если необходимо, соединение A подвергают щелочному гидролизу в подходящем растворителе, таком как метанол, этанол, тетрагидрофуран или диоксан с получением соединения Aa настоящего изобретения. Реакция щелочного гидролиза может быть осуществлена перемешиванием реакционной смеси в присутствии подходящего основного катализатора (например, гидроксида натрия, гидроксида лития, гидроксида калия и других) при комнатной температуре или при повышенной температуре (например 40-100oC).
Реакции вышеописанного способа получения совместно показаны в приведенной далее схеме реакции, в которой каждое обозначение имеет те же значения, что указаны выше.
Способ получения 5.
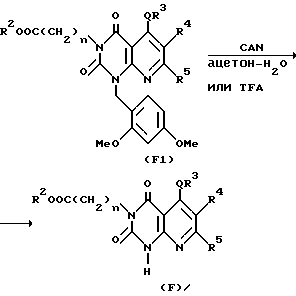

CAN: церий (IV) аммоний нитрат
TFA: Трифторуксусная кислота
Способ получения 6.
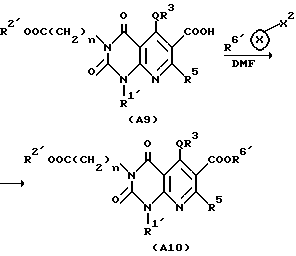
Здесь приведен пример способа получения соединения A настоящего изобретения, в котором заместителем в 6-м положении является - COOR61 (R61 является необязательно замещенной C1-6-алкильной группой, необязательно замещенной циклической углеводородной группой или необязательно замещенной аралкильной группой).
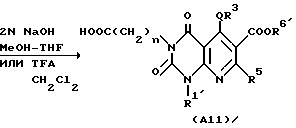
Соединение A9, в котором заместителем в 6-м положении является COOH, подвергают перемешиванию с галогенированным алкильным, галогенированным циклическим углеводородным или галогенированным аралкильным производным (например, этилбромид, изобутилбромид или бензилбромид) в подходящем растворителе (например, диметилацетамид или диметилформамид) в присутствии подходящего слабого основания (например, карбонат калия, триэтиламин или гидрокарбонат натрия) обычно в течение 2-48 ч, предпочтительно 2-24 ч, с получением соединения A10 настоящего изобретения, заместителем в 6-м положении которого является -COOR61.
Затем, если необходимо, соединение A10 подвергают щелочному гидролизу в подходящем растворителе, таком как метанол, этанол, тетрагидрофуран или диоксан, или кислотному гидролизу в подходящем растворителе, таком как метиленхлорид с получением соединения A11 настоящего изобретения.
Реакция щелочного гидролиза может быть осуществлена перемешиванием реакционной смеси в присутствии подходящего основного катализатора (например, гидроксида натрия, гидроксида калия и других) при комнатной температуре или при повышенной температуре (например, 40-100oC) обычно в течение 2-48 ч, предпочтительно 2-24 ч.
Реакция кислотного гидролиза может быть осуществлена перемешиванием реакционной смеси с каталитическим количеством или избыточным количеством подходящей кислоты, такой как трифторуксусная кислота, при комнатной температуре или при повышенной температуре (например, 40-100oC) обычно в течение 2 - 48 ч, предпочтительно 2 - 24 ч.
Реакции вышеописанного способа получения совместно показаны в представленной далее схеме реакции, в которой каждое обозначение имеет значение, указанное выше.
Способ получения 6.
Способ получения 7.
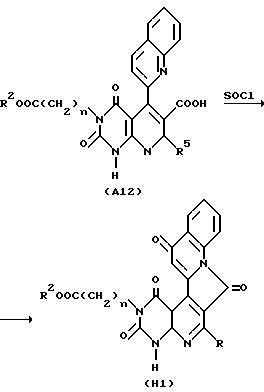
Здесь приведен пример способа получения соединения A настоящего изобретения, в котором заместителем в 5-м положении является 2-/4-хинолонильная/группа, а в 6-м положении - -COOR6 или -CONR7R8 (R6, R7 и R8 являются независимо атомом водорода, необязательно замещенной C1-6-алкильной группой, необязательно замещенной аралкильной группой).
Соединение A12, заместителем которого в 1-м положении является атом водорода, подвергают перемешиванию с избытком тионилхлорида (например, 5-20 предпочтительно 5-10-кратным по отношению к эквиваленту) в подходящем растворителе (например, метиленхлориде или толуоле) при комнатной температуре (например, 40-60oC) обычно в течение 0,5-2,0 ч, предпочтительно 0,5-1,0 ч. Реакционную смесь затем концентрируют досуха при пониженном давлении. Остаток растворяют в подходящем растворителе (например, метиленхлориде). Отфильтровав нерастворимый материал (например, фильтрацией с селлаитом) (магний фторид), фильтрат концентрируют досуха, и, если необходимо, промывают подходящим растворителем (например, этилацетатом) с получением соединения H1.
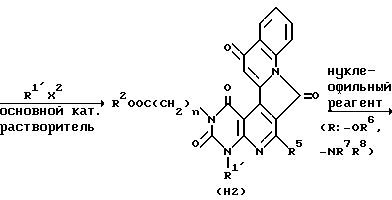
Соединение H1, полученное таким образом, растворяют в подходящем растворителе (например, диметилформамиде, диметилацетамиде или ацетонитриле) и затем подвергают перемешиванию с обычно 1-3-, предпочтительно 1-1,5-кратным по отношению к эквиваленту подходящего галогенированного алкильного производного или галогенированного алкильного производного (например, 2-метилтиобензил хлорид или 2,3-диметоксибензил хлорид и т.п.) в присутствии обычно 1-3-, предпочтительно 1,5-2-кратного по отношению к эквиваленту подходящего основания (например, гидрокарбонат калия, карбонат калия или гидрид натрия) при комнатной температуре или при повышенной температуре (например, обычно 40-100oC, предпочтительно 40-60oC) обычно в течение 2-48 ч, предпочтительно в течение 24-48 ч, с получением соединения H2.
После очистки походящим методом (например, колоночной хроматографией на силикагеле), если необходимо, соединение H2 растворяют в подходящем растворителе (например, ацетонитриле или диметилформамиде) и затем подвергают перемешиванию с избытком (обычно 5-30-, предпочтительно 5-10-кратный по отношению к эквиваленту) нуклеофильного реагента (например, 2 н водный раствор карбоната калия, водного аммиака или соединения амина) при комнатной температуре или при повышенной температуре (например, обычно 40-120oC, предпочтительно 40-80oC) обычно в течение 1-6 ч, предпочтительно 1-2 ч, с получением соединения A13.
Реакции по вышеописанному способу получения совместно показаны в приведенной далее схеме реакции, в которой R является -OR6 или -NR7R8 и каждое обозначение, за исключением R, имеет те же значения, что указаны выше.
Способ получения 7.
В вышеописанных способах получения 1 и 2 соединение A, в котором R1=H, может быть получено, используя соединение, представленное R1''RH2 (R1'' обозначает защитную группу для аммиака) вместо исходного соединения R1'NH2 (R1' имеет те же значения, что указаны выше), с последующим удалением защитной группы, представленной R1'' как в течение, так и после реакции.
В способе получения 3 соединение A, в котором R1 = H, может быть получено с использованием соединения /ii/, имеющего R1'' (R1'' обозначает защитную группу для аммиака) вместо R1' (R1' имеет значения, указанные выше), с последующим удалением защитной группы, показанной R1'' как в процессе, так и после завершения реакции.
Соединение R1''NH2 может быть получено известным специалистам способом или аналогичным способом или оно может быть коммерчески доступным.
Для удаления защитной группы может быть применен известным специалистам способом или его аналогом, например с использованием кислоты, основания, окисляющего агента восстанавливающего агента, ультрафиолетового облучения, гидрозина, фенилгидразина, N-метилдитиокарбамата натрия, тетрабутиламмония фторида, ацетата палладия и т.д.
Исходные соединения C1-6-алкиламин, ариламин и аралкиламин могут быть получены известным специалистам способом или его аналогом.
Соединение A, полученное таким образом, или его соль могут быть выделены и очищены обычным методом, например, перекристаллизацией, дистилляцией или хроматографией. Когда соединение A получают таким образом в качестве свободного соединения, оно может быть преобразовано в соответствующую соль известным специалисту способом или его аналогом и затем соединение A, полученное в виде соли может быть преобразовано в свободную форму или любую другую соль известным специалисту способом или его аналогом.
В случае, когда соединение A или его соль является оптически активным соединением, оно может быть выделено в виде о-изомера и -изомера подходящим методом для оптического разделения.
Соединение A или его соль по настоящему изобретению обладают превосходной антагонистической по отношению к эндотелиновому рецептору активностью, поэтому оно может быть использовано в качестве антагониста эндотелинового рецептора для теплокровных животных (например, крыса, мышь, морская свинка, цыпленок, собака, кошка, свинья, бык, обезьяна, человек и др.).
Более того, ввиду его превосходной антагонистической активности по отношению к эндотелиновому рецептору соединение A или его соль могут быть использованы в качестве профилактической или лечебной композиции против церебрального инфаркта, грудной ангины, инфаркта миокарда и почечной недостаточности.
Соединение A или его соль согласно настоящему изобретению являются безопасными и низкотоксичными.
В то же время соединение A или его соль согласно настоящему изобретению используются в качестве антагониста эндотелинового рецептора или профилактических или терапевтических агентов против острой почечной недостаточности, инфаркта миокарда, гипертензии, церебрального инфаркта, грудной ангины, атеросклероза, гепатопатии, легочной гипертензии, бронхиальной астмы, органогипофункции, связанной с проведением операции или трансплантации органов.
Они могут быть назначены как орально, так и неорально. Обычно они назначаются орально в форме твердого препарата, такого как таблетки, капсулы, гранулы или порошок, или неорально в форме внутривенной, подкожной или внутримышечной инъекции, суппозиториев или сублингвальных таблеток. Количество дозировки варьируется от степени симптомов, возрастов пациентов, пола, веса тела, различия восприимчивости, времени назначения лекарства, интервала, качества медицинских препаратов, препарата, видов: типов эффективных компонентов, помимо прочего, и они никак специально не ограничиваются. Обычно дозировка для взрослого на день колеблется от около 0,1 до 500 мг, предпочтительно, от около 1 до 100 мг, более предпочтительно от 5 до 50 мг, от 1 до 4 разбивочных доз в день.
Соединение согласно настоящему изобретению может быть назначено орально или неорально в составе с фармацевтически приемлемыми носителями, в виде твердого препарата, включающего таблетки, капсулы, гранулы и порошок, или в виде жидкого препарата, такого как сироп или инъекции.
Вышеуказанные фармацевтически приемлемые носители включают общепринятые органические или неорганические носители, используемые при изготовлении фармацевтических препаратов, а именно наполнители, смазки, связующие и разрыхлители для твердых препаратов, растворители, солюбилизаторы, суспендирующие агенты, изотонизаторы, буферные агенты и местные анестетики для жидких препаратов. При необходимости, могут быть также использованы добавки, такие как защитные добавки, антиоксиданты, красители, подсластители или подобные.
Предпочтительные наполнители подтверждены в примерах лактозой, сукрозой, D-маннитолом, крахмалом, кристаллической целлюлозой и легким ангидридом салициловой кислоты.
Предпочтительными смазками в примерах показаны стеарат магния, стеарат кальция, тальк и коллоидный кремнезем.
Предпочтительные связующие проиллюстрированы кристаллической целлюлозой, сукрозой, D-маннитолом, декстрином, гидроксипропилцеллюлозой, гидроксипропил метилцеллюлозой и поливинилпирролидоном.
Предпочтительные разрыхлители показаны крахмалом, карбоксиметилцеллюлозой, кальций карбоксиметилцеллюлозой, натрий клокармеллозой и натрий карбоксиметилкрахмалом.
Предпочтительные растворители показаны примерами на дистиллированную воду для инъекций, спирт, пропиленгликоль, макроголь, кунжутное масло и кукурузное масло.
Предпочтительные солюбилизаторы показаны в примерах на полиэтиленгликоль, пропиленгликоль, D-маннитол, бензилбензоат, этанол, трисаминометан, холестерин, триэтаноламин, карбонат натрия и цитрат натрия.
Предпочтительные примеры суспендирующих агентов включают поверхностно-активные вещества, такие как стеарил триэтаноламин, натрий лаурил сульфат, лауриламинопропионовую кислоту, лецитин, бензалконий хлорид, бензэтоний и хлорид и моностеарат глицерина, гидрофильные полимеры, такие как поливинилы и спирт, поливинилпирролидон, натрий карбоксиметил целлюлозу, метилцеллюлозу, гидроксиметилцеллюлозу, гидроксиэтилцеллюлозу и гидроксипропилцеллюлозу.
Предпочтительные изотонизаторы показаны в примерах как хлорид натрия, глицерин и D-маннитол.
Предпочтительные буферные агенты проиллюстрированы буферными растворами, такими как фосфатный, ацетатный, карбонатный и цитратный.
Предпочтительные местные анестетики проиллюстрированы бензиловым спиртом.
Предпочтительными защитными средствами показаны пара-гидроксибензойные эфиры, хлорбутанол, бензиловый спирт, фенетиловый спирт, дегидроуксусная кислота и сорбитовая кислота.
Предпочтительные антиоксиданты показаны на примерах сернистой и аскорбиновой кислоты.
Добавлением суспендирующей смеси, солюбилизатора, стабилизирующей смеси, изотонизатора, защитного средства или тому подобных к соединению по настоящему изобретению готовят общепринятым способом внутривенные, подкожные и внутримышечные инъекции. При необходимости, эти инъекции могут быть преобразованы в лиофилизующие препараты.
Следующие примеры даны с иллюстративной целью, но не для каких-либо ограничений.
Ссылочный пример 1.
Получение 2-метоксибензилмочевины (химическая формула I)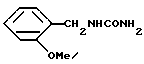
К водному раствору (13 мл), содержащему 2-метоксибензиламин /13,7 г, 0,1 моль/ добавляют по каплям разбавленную серную кислоту (приготовленную из мл 2,8 мл концентрированной серной кислоты и 6,7 мл очищенной воды) при комнатной температуре. После завершения добавления по каплям, к реакционной смеси добавляют при комнатной температуре водный раствор (70 мл), содержащий цианат натрия (7,65 г, 0,12 моль) в течение 15 мин. Полученную суспензию реакционной смеси нагревают при 80oC в течение одного часа. Реакционную смесь охлаждают, полученный кристаллический осадок собирают фильтрацией с последующей перекристаллизацией из этанола с получением 17 г (выход 94%) бесцветных призм.
Ссылочный пример 2.
Получение 6-амино-1-/2-метоксибензил/пиримидин-2,4/1H,3H/диона (химическая формула 2)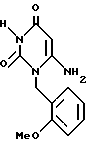
Смесь полученного по ссылочному примеру 1 соединения (10,8 г, 60 ммоль) этилцианоацетат (7,0 г, 62 ммоль) и метоксида натрия (28%-ный раствор в метаноле, 12,0 г, 62 ммоль) нагревают в течение 72 часов в этаноле (100 мл) с обратным холодильником. Реакционную смесь охлаждают, затем концентрируют досуха. В концентрату добавляют насыщенный водный раствор хлорида аммония /50 мл/. Смесь перемешивают, затем устанавливают pH 6 - 7 с помощью 1 н. HCl. Полученный осадок собирают фильтрацией и перекристаллизовывают из смеси этанола и метанола (1:1) в бледно-желтые призмы, т.пл. 278-279oC. Значения элементного анализа показаны в таблице 1.
Ссылочный пример 3.
Способ, описанный в ссылочных примерах 1 и 2 повторяют, с использованием различных замещенных аминосоединений вместо 2-метоксибензиламина. Соединения, представленные химической формулой 3,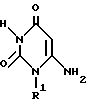
полученные таким образом, совместно показаны в таблице 2.
Ссылочный пример 4.
Получение этил/6-мино-2,4/1H, 3H/-диоксо-1-/2-метоксибензил// пиримидин-3-ацетата (химическая формула 4)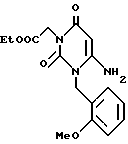
Смесь полученного по ссылочному примеру 2 соединения (6,65 г, 27 ммоль) этилбромацетата (15,0 г, 90 ммоль) и карбоната калия (16,6 г, 120 ммоль) перемешивают в диметилформамиде (500 мл) в течение 72 ч при 60oC. Реакционную смесь после охлаждения концентрируют досуха, затем добавляют насыщенный водный раствор хлорида аммония (30 мл) и этилацетата (50 мл) потом смесь перемешивают. Органический слой отделяют. Водный слой подвергают экстракции этилацетатом (50 мл). Экстракт объединяют с органическим слоем, затем высушивают. Растворитель отгоняют при пониженном давлении. Остаток очищают с помощью хроматографии на колонке с силикагелем с получением 3,5 г (выход 39%) желтого аморфного продукта.
ЯМР-спектр полученного таким образом продукта следующий.
1H-ЯМР (200 МГц, CDCl3) δ ppm: 1,27 (3H, т), 3,93 (3H, с), 4,21 (2H, кв), 4,70 (2H, с), 4,91 (1H, с), 5,12 (2H, с), 5,31 (2H, с), 6,91 - 7,00 (2H, т), 7,27 - 7,40 (1H, м), 7,50 - 7,55 (1H, м).
Ссылочный пример 5.
Получение этил[5,8-дигидро-2,4(1H, 3H)-диоксо-1-этоксикарбонил- 7-изопропил-1-(2-метоксибензил)-5-(4-толил)пиридо/2,3-d] пиримидин] -3-ацетат (химическая формула 5)
Соединение (0,5 г, 1,5 ммоль) полученное по ссылочному примеру 4, нагревают в течение 90 часов с обратным холодильником, в этаноле (10 мл) вместе с п-толуолальдегидом (0,54 г, 4,5 ммоль) и этил изобутилацетатом (0,71 г, 4,5 ммоль). Реакционную смесь охлаждают, затем растворитель отгоняют при пониженном давлении. Остаток очищают с помощью хроматографии на колонке с силикагелем с получением 0,2 г (выход 23%) желтого аморфного продукта. ЯМР спектр полученного таким образом продукта следующий: 1H-ЯМР (200 МГц), CDCl3 δ ppm: 0,80 (3H, д), 1,06 (3H, д), 1,25 (3H, т), 1,26 (3H, с), 2,25 (3H, с), 3,58 (1H, м), 3,96 (3H, с), 4,12 (2H, кв), 4,22 (2H, кв), 4,62 (1H, д), 4,72 (1H, д), 5,07 (1H, с), 5,22 (1H, д), 5,32 (1H, д), 6,40 (1H, с), 6,90 - 7,50 (8H, м).
Ссылочный пример 6.
Способ, описанный как ссылочный пример 5, повторяют, только с использованием различных альдегидов и этил изобутилацетатов. Соединения, представленные химической формулой 6,
полученные таким образом, сведены в таблицу 3.
Ссылочный пример 7.
Получение 5,8-дигидро-2,4-(1H, 3H)-диоксо-6-этоксикарбонил-7- изопропил-1-(2-метоксибензил)-5-(4-толил)пиридо[2,3-d]пиримидин-3- уксусной кислоты (химическая формула 7)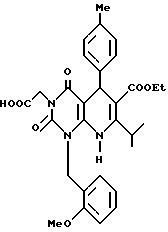
Соединение (0,2 г, 0,35 ммоль) полученное в ссылочном примере 5, растворяют в метаноле (20 мл). К этому раствору добавляют 2 н водный раствор гидроксида натрия (2 мл) и смесь перемешивают 18 часов при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении, при установлении pH в области от 2 до 3 с помощью 1 н. HCl, с последующей экстракцией этилацетатом с получением 0,13 г (выход 68%) желтого маслянистого продукта.
Ссылочный пример 8.
Используя соединения как в ссылочном примере 6, повторяют способ, описанный в ссылочном примере 7. Соединения, представленные химической формулой 8
и полученные таким образом, сведены в таблицу 4.
Ссылочный пример 9.
Получение 2,4-(1H, 3H)-диоксо-(2-цианоэтоксикарбонил)-7- изопропил-1-(2-метоксибензил)-5-(2-хинолил)пиридо/2,3-d//пиримидина (химическая формула 9)
Соединение (3,9 г, 16 ммоль), полученное в ссылочном примере 2, перемешивают в диметилсульфоксиде (23 мл) в течение 25 мин при 125oC вместе с соединением (6,1 г, 19 ммоль) полученным дегидратной конденсацией хинолин-2-карбоксиальдегида и 2-цианоэтил изобутирилацетат. Реакционную смесь охлаждают, затем размешивают в ледяной воде. Полученные коричневатые кристаллы собирают фильтрацией и растворяют в этилацетате. Промывкой раствора водой получают 4,3 г бледно-серого порошка (выход 49%). Соединение, полученное таким образом (4,3 г, 7,8 ммоль), растворяют в уксусной кислоте (50 мл), к которой добавляют нитрит натрия (5,9 г). Смесь перемешивают в течение 15 мин при комнатной температуре. Реакционную смесь размешивают в ледяной воде, затем подвергают экстракции с этилацетатом. Органический слой высушивают и затем растворитель отгоняют при пониженном давлении. Остаток очищают с помощью хроматографии на колонке с силикагелем с получением 4,1 г (выход 47%) желтого аморфного продукта. Значения элементного анализа показаны в таблице 5.
ЯМР-спектр и ИК-спектр соединения такие, как указаны далее.
1H-ЯМР (200 МГц, CDCl3) δ ppm: 1,14 (6H, д), 2,12 (2H, т), 3,15 (1H, м), 3,89 (3H, с), 3,94 (2H, т), 5,61 (2H, с), 6,80 - 6,96 (3H, м), 7,21 (1H, тд), 7,53 (1H, д), 7,59 (1H, тд), 7,72 (1H, тд), 7,88 (1H, д), 8,04 (1H, д), 8,22 (1H, д), 8,77 (1H, с).
ИК (KBr): 3452, 3196, 3068, 2974, 2254, 1736, 1715, 1603, 1572, 1497 см-1.
Ссылочный пример 10.
Используя другие соединения, повторяют способ, описанный в ссылочном примере 9. Соединения, представленные химической формулой 10, полученные таким образом сведены в таблицу 6.
Ссылочный пример 11.
Получение этил /2,4-(1H,3H)-диоксо-5-метилтио-6-этоксикарбонил-7-изопропил-1- (2-метоксибензилпиридо/2,3-d/пиримидин/-3-ацетата (химическая формула 11.
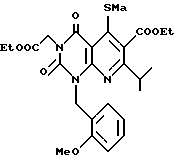
К метанольному раствору (5 мл), содержащему этил изобутилацетат (0,48 г) добавляют метанольный раствор/метоксида натрия (28%, 0,58 г) при охлаждении льдом. После перемешивания в течение 10 мин в тех же условиях к раствору добавляют по каплям дисульфид углерода. После завершения добавления по каплям добавляют диметилсульфат (0,75 мл) к реакционной смеси. Реакционную смесь перемешивают при комнатной температуре в течение 15 мин. Реакционную смесь смешивают с водой (30 мл), перемешивают в течение 5 мин, затем подвергают экстракции с изопропиловым эфиром с получением желтого маслянистого продукта (0,40 г).
Смесь этого маслянистого продукта, соединения, полученного в ссылочном примере 4 (0,50 г, 1,5 ммоль) и карбоната калия (0,31 г) перемешивают в диметилформамиде (10 мл) в течение 5 ч при 150oC. Реакционная смесь после охлаждения концентрируется досуха, затем прибавляется насыщенный водный раствор хлорида аммония (20 мл) и этилацетат (20 мл) и затем смесь перемешивают. Водный слой подвергают экстракции этилацетатом (20 мл). Экстракт объединяют с органическим слоем, который затем высушивают. Растворитель отгоняют при пониженном давлении. Остаток очищают с помощью хроматографии на колонке с силикагелем с получением 0,06 г (выход 8%) желтого аморфного продукта. ЯМР-спектр и масс-спектр полученного таким образом продукта следующие:
1H-ЯМР (200 МГц, CDCl3) δ/ ppm: 1,28 (3H, т), 1,35 (6H, д), 1,41 (3H, т), 2,21 (3H, с), 3,86 (3H, с), 4,20 (2H, кв), 4,40 (2H, кв), 4,40 - 4,60 (1H, м), 4,84 (2H, с), 5,59 (2H, с), 6,7 - 7,0 (3H, м), 7,15 - 7,3 (1H, м).
MS, m/z: 530 (MH).
Ссылочный пример 12.
Получение этил /1,3,7,13-тетраоксо-1,2,3,4,7,13-гексагидро-6- изопропилпиримидо/4'', 5''-: 6',5'/пиридо/3',4':4,3/пирроло/1,2-a/ хинолин/-3-ацетата (химическая формула 12)
Соединение (1,10 г, 2,38 ммоль) полученное в примере 17 (соединение 16) растворяют в дихлорметане (100 мл). К раствору добавляют тионилхлорид (0,87 мл, 11,89 ммоль) и смесь перемешивают в течение 2 ч при комнатной температуре. К реакционной смеси добавляют 20 мл толуола и затем концентрируют при пониженном давлении. Остаток растворяют в дихлорметане, затем фильтруют, с селаитом. Фильтрат концентрируют, и полученный таким образом остаток промывают этилацетатом с получением 0,30 г (выход 28%) желтого порошкообразного продукта. Значения элементного анализа показаны в таблице 7.
ЯМР-спектр, ИК-спектр и масс-спектр соединения следующие:
1H-ЯМР (500 МГц, DMSO-d6) δ ppm: 1,32 - 1,41 (9H, м), 4,28 (2H, кв), 4,37 (1H, м), 4,82 (2H, с), 7,47 (1H, т), 7,78 (1H, т), 8,30 (1H, д), 8,64 (1H, с), 8,78 (1H, с), 9,19 (1H, д).
ИК (KBr): 1738, 1682, 1642, 1576, 1477, 1393 см-1.
FAB-MS, m/x 461.1 (MH)+.
Ссылочный пример 13.
Получение этил[1,3,7,13-тетраоксо-1,2,3,4,7,13-гексагидро-6- изопропил-4-(2-метилтиобензил)пиримидо[4'', 5'': 6', 5'] пиридо[3', 4': 4,3]пирроло[1,2-a]хинолин]-3-ацетата (химическая формула 13).
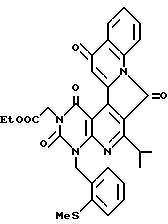
Диметилформамидный (5 мл) раствор, содержащий соединение (0,20 г, 0,43 ммоль), полученное в ссылочном примере 12, 2-метилтиобензилхлорид (0,74 г, 4,30 ммоль) и гидрокарбонат калия (0,07 г, 0,64 ммоль) перемешивают в течение 2 дней при комнатной температуре. К реакционной смеси добавляют насыщенный водный раствор хлорида аммония, затем смесь подвергают разделению добавлением дихлорметана и насыщенного водного раствора хлорида натрия. Органический слой высушивают MgSO4 и затем растворитель отгоняют при пониженном давлении. Остаток очищают путем хроматографии на колонке с силикагелем с получением 0,21 г (выход 80%) желтого порошкообразного продукта.
Значения элементного анализа показаны в таблице 8.
ЯМР-спектр, ИК-спектр и масс-спектр соединения следующие.
1H-ЯМР (200 МГц), (CDCl3) δ ppm: 1,14 (6H, д), 1,34 (3H, т), 2,57 (3H, с), 4,23 - 4,34 (3H, м), 4,93 (2H, с), 5,76 (2H, с), 6,83 (1H, д), 7,03 (1H, т), 7,22 - 7,35 (2H, м), 7,47 (1H, т), 7,77 (1H, т), 8,32 (1H, д), 8,84 (1H, с), 9,18 (1H, с).
ИК (KBr): 1725, 1680, 1640, 1578, 1475, см-1.
FAB-MS m/z: 597,1 (MH+)
Ссылочный пример 14
Используют соединения, упоминаемые в ссылочном примере 12 повторяют способ, описанный в ссылочном примере 13. Соединения, представленные химической формулой 14
полученные таким образом, сведены в таблицу 9.
Пример 1.
Получение этил/2,4(1H, 3H)-диоксо-6-этоксикарбонил-7-метил-1- (2-метоксибензил)-5-(4-метоксифенил)пиридо/2,3-d/пиримидин/-3- ацетата (химическая формула 15)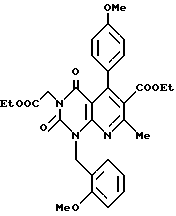
Соединение (2,1 г, 6,3 ммоль), полученное в ссылочном примере 4, нагревают в течение 130 ч с обратным холодильником в этаноле (40 мл) вместе с п-анисальдегидом (0,81 г, 60 ммоль) и этилацетоацетатом (0,79 г, 6,1 ммоль). Реакционную смесь охлаждают, затем отгоняют растворитель при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 2,6 г (выход 70%) желтого аморфного продукта. Аморфное соединение (2,3 г, 4,1 ммоль) растворяют в уксусной кислоте (50 мл), к которой добавляют нитрит натрия (2,0 г). Смесь перемешивают в течение одного часа при комнатной температуре. Реакционную смесь размешивают в ледяной воде, затем подвергают экстракции этилацетатом (100 мл каждый раз, дважды). Органический слой высушивают и затем растворитель отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 1,8 г (выход 77%) желтого маслянистого продукта. ЯМР-спектр этого продукта следующий:
1H-ЯМР (200 МГц, CDCl3) δ/ ppm: 0,97 (3H, т), 1,22 (3H, т), 2,51 (3H, с), 3,84 (3H, с), 3,90 (3H, с), 3,98 (2H, кв), 4,15 (2H, кв), 4,71 (2H, с), 5,65 (2H, с), 6,75 - 7,61 (8H, м).
Пример 2.
Используя подходящий альдегид вместо п-анисальдегида и этил изобутирилацетат или этил ацетоацетат, повторяют способ, описанный в примере 1, с получением соответствующего соединения химической формулы 16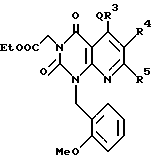
которое представлено в таблице 10.
Пример 3.
Получение трет-бутил /2,4(1H,3H)-диоксо-6-(2-цианоэтокси- карбонил)-7-изопропил-1-(2-метоксибензил)-5-(2-хинолил)пиридо/2,3- d/пиримидин/-3-ацетата (химическая формула (17)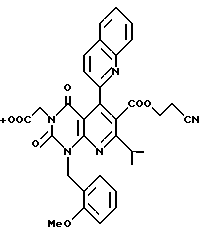
Соединение (1,0 г, 1,9 ммоль) полученное в ссылочном примере 9, перемешивают в течение 40 минут при комнатной температуре в диметилформамиде (5 мл) вместе с трет-бутил бромацетатом (0,88 мл, 5,6 ммоль) и карбонатом калия (0,76 г, 5,6 ммоль). Устанавливают pH реакционной смеси от 1 до 2 с помощью 1 н. HCl, затем подвергают разделению реакционную смесь обработкой этилацетатом и водой. Органический слой высушивают, затем отгоняют растворитель при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле с получением 0,51 г (выход 41%) желтого аморфного продукта. Значения элементного анализа соединения, полученного таким образом, показаны в таблице 11. ЯМР-спектр и ИК-спектр соединения следующие:
1H-ЯМР (200 МГц, CDCl3) δ ppm: 1,15 (6H, д), 1,40 (9H, с), 2,14 (2H, т), 3,15 (1H, м), 3,90 (2H, с), 3,94 (2H, т), 4,59 (2H, с), 5,69 (2H, с), 7,22 (1H, т), 7,54 (1H, д), 7,60 (1H, тд), 7,73 (1H, тд), 7,88 (1H, д), 8,04 (1H, д), 8,22 (1H, д).
ИК (KB): 3452, 2976, 2938, 2364, 2258, 1742, 1721, 1678, 1603, 1574 см-1.
Пример 4.
Используя соответствующее соединение в качестве исходного продукта, повторяют способ, описанный в примере 3, с получением соединений химической формулы 18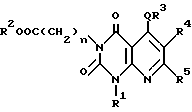
которые сведены в таблицу 12.
Пример 5. Получение этил/2,4(1H,3H)-диоксо-6-/2-цианоэтоксикарбонил/-7-изопропил-5- (2-хинолил/пиридо/2,3-d/пиримидин-/3-ацетата (химическая формула 19)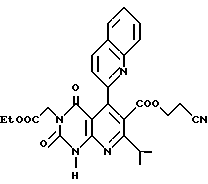
Соединение (соединение 10; 10,0 г, 15,0 ммоль), полученное в примере 4, растворяют в ацетон-эфире (1: 1 по объему, 600 мл). К раствору добавляют церий (IV) аммоний нитрат (24,7 г, 45,0 ммоль) и смесь перемешивают в течение 1 ч при комнатной температуре. Реакционную смесь концентрируют до 1/2 объема и остаток подвергают экстракции этилацетатом.
Органический слой высушивают (над Na2SO4) и растворитель отгоняют при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем с получением 4,59 г (выход 46%) желтого порошкообразного продукта. Данные элементного анализа показаны в таблице 13. ЯМР-спектр и ИК-спектр полученного таким образом соединения приведены ниже.
1H-ЯМР (200 МГц, CDCl3) δ ppm: 1,21 (3H, т), 1,35 (6H, д), 2,17 (2H, т), 3,25 (1H, м), 3,97 (2H, т), 4,15 (2H, кв), 4,61 (2H, с), 7,53 (1H, д), 7,61 (1H, т), 7,74 (1H, т), 7,89 (1H, д), 8,05 (1H, д), 8,23 (1H, д), 8,56 (1H, с).
ИК (KBr): 3424, 3264, 2976, 2362, 1734, 1680, 1620, 1578, 1504 см-1.
Пример 6. Получение этил /2,4(1H,3H)-циоксо-6-(2-цианоэтоксикарбонил)-7-изопропил-5-(2- хинолил)пиридо/2,3-d/пиримидин/-3-ацетата (химическая формула 20)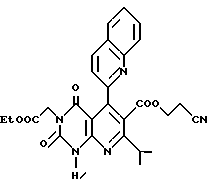
Раствор соединения (соединение 10; 9,1 г, 13,7 ммоль), полученного в примере 4, в трифторуксусной кислоте (50 мл) перемешивают в течение 4 ч при 60oC. Реакционную смесь концентрируют удалением трифторуксусной кислоты и остаток подвергают разделению в этилацетат-водном растворе хлорида натрия. Органический слой высушивают (MgSO4) и растворитель отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 6,47 г (выход 92%) желтого порошкообразного продукта.
Пример 7.
Используя соединение, полученное в примере 4 (соединение N 12), повторяют способ, описанный в примере 5, с получением соединения формулы 21
Пример 8. Получение этил /2,4(1H,3H)-диоксо-6-(2-цианоэтоксикарбонил)-7-изопропил-1-метил-5- (2-хинолил)пиридо/2,3-d/пиримидин/-3-ацетата (химическая формула 22)
Соединение (1,78 г, 3,5 ммоль) полученное в примере 5, метил йодид (0,98 г, 6,9 ммоль) и карбонат калия (1,42 г, 10,4 ммоль) растворяют в диметилформамиде (70 мл). К раствору добавляют 1 н. HCl и подвергают разделению в этилацетате воде. Органический слой высушивают (над Na2SO4) и растворитель отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 1,42 г (выход 78%) желтого пузыристого продукта. Данные элементного анализа показаны в таблице 14. ЯМР-спектр и ИК-спектр полученного таким образом соединения приведены ниже.
1H-ЯМР (200 Гц, CDCl3) δ pp,: 1,21 (3H, т), 1,39 (6H, д), 2,14 (1H, т), 3,30 (1H, м), 3,82 (3H, с), 3,96 (2H, т), 4,15 (2H, кв), 4,65 (2H, с), 7,50 (1H, д), 7,59 (1H, т), 7,73 (1H, т), 7,88 (1H, д), 8,03 (1H, д), 8,21 (1H, д).
IR (KBr): 3452, 2974, 2364, 1742, 1719, 1673, 1620, 1601, 1574, 1506 см-1.
Пример 9.
Используя соответствующие соединения в качестве исходного продукта, повторяют способ, описанный в примере 8, с получением соединения, представленного химической формулой 23.
которое имеет различные заместители в 1-м положении. Соединения, полученные таким образом, сведены в таблицу 15.
Пример 10. Получение 2,4(1H,3H)-диоксо-6-этоксикарбонил-7-изопропил-1-/2-метоксибензил/ -5-(4-толил)пиридо-[2,3-d] пиримидин-3-уксусной кислоты (химическая формула 24)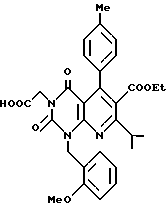
Соединение (0,13 г, 0,24 ммоль), полученное в ссылочном примере 7, растворяют в уксусной кислоте (20 мл). К раствору добавляют нитрит натрия (0,1 г) и смесь перемешивают в течение одного часа при комнатной температуре. Реакционную смесь выливают в ледяную воду и подвергают экстракции этилацетатом (20 мл каждый раз, трижды). Органический слой высушивают и растворитель отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле. Полученные таким образом сырые кристаллы перекристаллизовывают из этилацетат-изопропилового эфира (1:2) с получением 0,06 г (выход 54%). желтого кристаллического порошка, т.пл. 190-192oC (разл. ). Данные элементного анализа показаны в таблице 16.
ЯМР-спектр и ИК-спектр полученного таким образом соединения следующие.
1H-ЯМР (200 МГц, CDCl3) δ/ ppm: 0,89 (3H, т), 1,08 (6H, д), 2,25 (3H, с), 2,9 - 3,1 (1H, м), 3,41 (3H, с), 3,90 (2H, кв), 4,41 (2H, с), 5,53 (2H, с), 6,6 - 7,2 (8H, м).
ИК (KBr): 3456, 1717, 1671, 1562, 1518, 1495, 1466, 1386, 1371, 1305, 1247 см-1.
Пример 11.
Используя соединения, представленные в ссылочном примере 8, повторяют способ, описанный в примере 10. Соединения, полученные таким образом (химическая формула 25)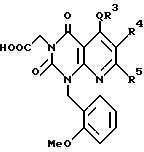
представлены в таблице 17.
Пример 12. Получение 2,4(1H,3H)-диоксо-6-этоксикарбонил-7- метил-1-(2-метоксибензил)-5-(4-метоксифенил)пиридо[2,3-d] пиримидин-3-уксусной кислоты (химическая формула 26)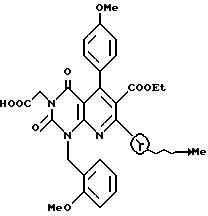
Соединение (соединение N 10; 1,5 г, 2,6 ммоль) полученное в примере 2 растворяют в метаноле (30 мл), к которому добавляют 2 н. водный раствор гидроксида натрия (2,6 мл) и смесь перемешивают в течение 20 ч при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении, pH концентрата устанавливают 2 - 3 с помощью 1 н. HCl и подвергают экстракции этилацетатом. Экстракт очищают с помощью колоночной хроматографии на силикагеле с получением сырых кристаллов, подвергающихся затем перекристаллизации из изопропилового спирта с получением 0,63 г (выход 46%) желтого кристаллического порошка, т.пл. 190-192oC. Элементный анализ продукта показан в табл. 18.
ЯМР-спектр и ИК-спектросоединения следующие:
1H-ЯМР (200 МГц CDCl3) δ ppm: 0,88 (3H, т), 2,39 (3H, с), 3,79 (3H, с), 3,89 (3H, с), 3,92 (2H, кв), 4,18 (2H, с), 5,47 (2H, с), 6,78 (1H, т), 6,68 (1H, д), 6,91 (2H, д), 7,01 (1H, д), 7,06 (2H, д), 7,20 (1H, т).
ИК (KBr): 3450, 1717, 1669, 1609, 1564, 1518, 1483, 1381, 1278, 1247 см-1.
Пример 13.
Используя соединения, представленные в примере 2 (за исключением соединения N 19), примере 7 и примере 9, повторяют способ, описанный в примере 12. Полученные таким образом соединения (химическая формула 27)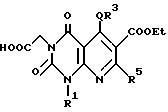
представлены в таблице 19.
Пример 14. Получение 2,4(1H,3H)-диоксо-6-карбокси-7-изопропил-1-(2-метоксибензил)-5- (2-хинолил)пиридо[2,3-d] пиримидин-3-уксусной кислоты (химическая формула 28)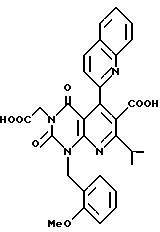
Соединение (соединение N 5; 0,20 г, 0,31 ммоль) полученное в примере 4, растворяют в смеси метанола (1,0 мл) и 1,4-диоксана (2,0 мл) к которой добавляют 1 н. водный раствор гидроксида натрия (0,63 мл, 1,3 ммоль) и смесь перемешивают 30 мин при комнатной температуре. Устанавливают pH реакционной смеси от 1 - 2 с помощью 1 н. HCl, затем разделяют этилацетатом и водой. Органический слой промывают насыщенным водным раствором хлорида натрия и сушат, затем растворитель отгоняют при пониженном давлении. Остаток перекристаллизовывают из изопропилового эфира с получением 0,12 г (выход 71%) бледно-желтого кристаллического порошка, т.пл. выше 300oC. Элементный анализ продукта показан в таблице 20.
ЯМК- и ИК-спектр соединения следующие:
1H-ЯМР (200 МГц, DMSO-d6) δ ppm: 1,05 (6H, д), 3,19 (1H, м), 3,90 (3H, с), 4,47 (2H, с), 6,83 (1H, т), 6,93 (1H, д), 7,05 (1H, д), 7,24 (1H, дд), 7,52 (1H, д), 7,65 (1H, дд), 7,77 (1H, дд), 7,93 (1H, д), 8,02 (1H, дд), 8,32 (1H, д), 12,97 (2H, шир.).
ИК (KBr): 3430, 2980, 1715, 1671, 1576, 1493, 1464 см-1.
Пример 15.
Используя соединения, представленные в примере 4, повторяют способ, описанный в примере 14. Полученные таким образом соединения (химическая формула 29)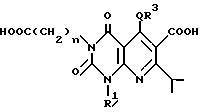
представлены в таблице 21.
Пример 16. Получение трет-бутил [2,4(1H,3H)-диоксо-6-карбокси-7- изопропил-1-(2-метоксибензил)-5-(2-хинолил)пиридо[2,3-d] пиримидин] - 3-ацетат (химическая формула 30)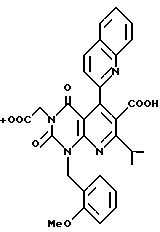
Соединение (2,9 г, 4,3 ммоль), полученное в примере 3, растворяют в смеси метанола (130 мл) и воды (17 мл), к которой добавляют 2 н. водный раствор карбоната калия (8,6 мл, 8,6 ммоль), и смесь перемешивают в течение 1 ч при комнатной температуре. Устанавливают pH реакционной смеси от 2 до 3 с помощью 1 н. HCl и подвергают реакционную смесь разделению этилацетатом и водой. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают, затем растворитель отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 2,3 г (выход 87%) желтого кристаллического порошка, т.пл. выше 300oC. Элементный анализ продукта показан в таблице 22. ЯМР-спектр и ИК-спектр соединения следующие:
1H-ЯМР (200 МГц), DMSO-d6), δ ppm: 1,05 (6H, д), 1,34 (9H, с), 3,19 (1H, м), 3,90 (3H, с), 4,45 (2H, с), 5,54 (2H, с), 6,83 - 6,92 (2H, м), 7,06 (1H, д), 7,24 (1H, тд), 7,53 (1H, д), 7,65 (1H, м), 7,77 (1H, м), 7,94 (1H, д), 8,04 (1H, д), 8,33 (1H, д).
ИК (KBr): 3454, 2972, 2928, 2366, 1719, 1678, 1605, 1572, 1495 см-1.
Пример 17.
Используя соединения, указанные в примере 4, и другие, повторяют способ, описанный в примере 16.
Соединения, полученные таким образом (химическая формула 31)
представлены в таблице 23.
Пример 18. Получение трет-бутил[2,4(1H,3H)-диоксо-6-карбокси- 7-изопропил-1-(2-метоксибензил)-5-[2-(4-хинолонил)]пиридо[2,3-d] пиримидин]-3-ацетата (химическая формула 32)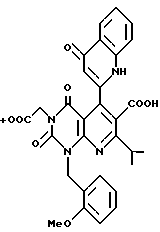
Соединение (500 мг, 0,82 ммоль), полученное в примере 16, растворяют в дихлорметане (20 мл). К раствору добавляют тионилхлорид (0,60 мл, 8,20 ммоль) и смесь нагревают в течение 30 мин с обратным холодильником. Реакционную смесь охлаждают и концентрируют. Остаток растворяют в диметилацетамиде (40 мл) и перемешивают с 2 н. водным раствором карбоната калия в течение 22 ч при 90oC. После охлаждения реакционную смесь выливают в ледяную воду и устанавливают pH от 1 до 2 с помощью 1 н. HCl, затем подвергают экстракции с этилацетатом (150 мл). Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают, затем растворитель отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 180 мг (выход 37%) бледно-коричневого порошка. Полученный таким образом порошок перекристаллизовывают из смешанного растворителя изопропанола и изопропилового эфира с получением 40 мг (выход 8%) бледно-коричневого кристаллического порошка, т.пл. более чем 300oC. Данные элементного анализа показаны в таблице 24. ЯМР-спектр и ИК-спектр полученного таким образом соединения следующие.
1H-ЯМР (200 МГц, DMSO-d6) δ ppm: 0,97 (3H, д), 1,07 (3H, д), 1,33 (9H, с), 3,46 (1H, м), 3,90 (3H, с), 4,48 (2H, с), 5,52 (2H, кв), 5,92 (1H, с), 6,80 (2H, с), 7,04 (1H, д), 7,22 (1H, тд), 7,28 (1H, т), 7,44 (1H, д), 7,58 (1H, тд), 8,10 (1H, д), 11,81 (1H, с).
ИК (KBr): 3452, 2974, 1717, 1673, 1638, 1601, 1570, 1510, 1473 см-1.
Пример 19. Используя соединения, представленные в примере 17, и другие, повторяют способ, описанный в примере 18.
Соединения, полученные таким образом (химическая формула 33)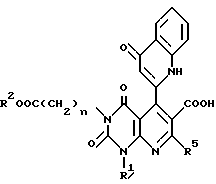
представлены в таблице 25.
Пример 20. Получение 2,4(1H,3H)-диоксо-6-карбокси-7-изопропил- 1-/2-метоксибензил/-5-[2-(4-хинолил)пиридо[2,3-d] пиримидин] -3- уксусной кислоты (химическая формула 34)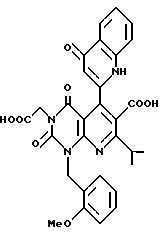
Соединение (соединение N 2: 0,19 г, 0,32 ммоль), полученное в примере 19, растворяют в смеси метанола (2,0 мл) и тетрагидрофурана (2,0 мл), к которому добавляют 1 н. водный раствор гидроксида натрия (0,64 мл, 1,3 ммоль), и смесь перемешивают в течение 5 ч при комнатной температуре. Устанавливают pH реакционной смеси от 1 до 2 с помощью 1 н. HCl, подвергают реакционную смесь экстракции смешанным растворителем из этилацетата и тетрагидрофурана.
Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают, затем растворитель отгоняют при пониженном давлении. Остаток перекриталлизовывают из смешанного растворителя из метанола изопропилового эфира с получением 0,08 г (выход 44%) бесцветного кристаллического порошка, т. пл. выше 300oC. Элементный анализ продукта показан в таблице 26. ЯМР-спектр и ИК-спектр соединения следующие:
1H-ЯМР (200 МГц, DMSO-d6) δ ppm: 1,01 (3H, д), 1,13 (3H, д), 3,18 (1H, м), 3,90 (3H, с), 4,55 (2H, кв), 5,53 (2H, кв), 5,92 (1H, с), 6,81 - 6,94 (2H, м), 7,04 (1H, д), 7,24 (1H, т), 7,35 (1H, т), 7,47 (1H, д), 7,65 (1H, т), 8,12 (1H, д), 11,90 (1H, с), 13,01 (1H, ушир.с).
ИК (KBr): 3444, 2974, 1719, 1676, 1574, 1493 см-1.
Пример 21. Используя соединения, представленные в примере 19, и другие, повторяют способ, описанный в примере 20.
Соединения, полученные таким образом (химическая формула 35)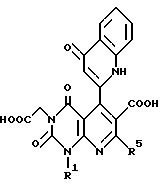
представлены в таблице 27.
Пример 22. Получение трет-бутил[2,4(1H,3H)-диокси-6-этоксикарбонил-7-изопропил-1-/2- метоксибензил/-5-[2-[4-хинолонил)]пиридо[2,3-d]пиримидин]-3- ацетата (химическая формула 36)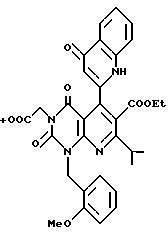
Соединение (0,35 г, 0,56 ммоль), полученное в примере 18, перемешивают с этилиодидом (0,10 г, 0,91 ммоль) в диметилформамиде (5,0 мл) в течение 1 ч при комнатной температуре. Устанавливают pH реакционной смеси от 1 до 2 с помощью 1 н. HCl и затем подвергают разделению в этилацетате и воде. Органический слой высушивают, затем растворитель отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением бледно-коричневого аморфного продукта. Данные элементного анализа соединения, полученного таким образом, показаны в таблице 28.
ЯМР-спектр и ИК-спектр соединения следующие:
1H-ЯМР (200 МГц, CDCl3) δ ppm: 0,98 (3H, т), 1,08 (3H, д), 1,19 (3H, д), 1,42 (9H, с), 3,15 (1H, м), 3,88 (3H, с), 4,09 (2H, т), 4,61 (2H, кв), 5,64 (2H, кв), 6,16 (1H, с), 6,79 - 7,01 (3H, м), 7,18 - 7,35 (3H, м), 7,59 (1H, тд), 8,33 (1H, дд).
ИК (KВr): 3450, 2978, 2936, 1721, 1682, 1638, 1605, 1574, 1510, 1460 см-1.
Пример 23. Получение 2,4(1H,3H)-диоксо-6-этоксикарбонил-7-изопропил-1-/2-метоксибензил/- 5-[2-/4-хинолонил/] пиридо[2,3-d] пиримидин-3-уксусной кислоты (химическая формула 37)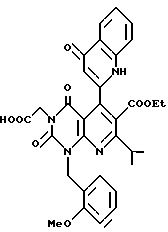
Соединение (0,15 г, 0,23 ммоль), полученное в примере 22 растворяют в дихлорметане и перемешивают с трифторуксусной кислотой в течение 1 ч при комнатной температуре. Реакционную смесь отгоняют при пониженном давлении. Остаток перекристаллизовывают из смешанного растворителя изопропилового спирта и изопропилового эфира с получением 0,12 г (выход 86%) бледно-желтого кристаллического порошка, т. пл. 214-216oC. Элементный анализ продукта показан в таблице 29.
ЯМР-спектр и ИК-спектр соединения следующие.
1H-ЯМР (200 МГц, DMSO-d6) δ ppm: 0,79 (3H, т), 0,89 (3H, д), 1,13 (3H, д), 3,10 (1H, м), 3,89 (3H, с), 4,00 (2H, т), 4,55 (2H, кв), 5,53 (2H, кв), 5,92 (1H, с), 6,79 - 6,92 (2H, м), 7,05 (1H, д), 7,24 (1H, т), 7,37 (1H, т), 7,49 (1H, д), 7,68 (1H, т), 8,13 (1H, д), 12,18 (1H, ушир).
ИК (KBr): 3444, 2972, 2936, 1719, 1680, 1603, 1574, 1492, 1462 см-1.
Пример 24.
Используя соединения, в которых заместителем в 3-м положении является -CH2COO/трет-C4H9, повторяют способ, описанный в примере 23. Соединения, полученные таким образом, представлены в таблице 30.
Пример 25. Получение этил[2,4(1H,3H)-диоксо-6- изобутоксикарбонил-7-изопропил-1-/2-метоксибензил/-5- (2-хинолил)пиридо[2,3-d]пиримидин]-3-ацетата (химическая формула 39)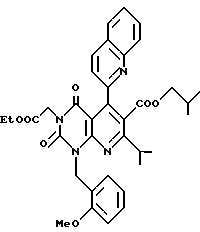
Соединение (соединение N 1 0,26 г, 0,44 ммоль), полученное в примере 17, растворяют в диметилформамиде и перемешивают с изобутилбромидом (0,14 мл, 1,31 ммоль) и карбонатом калия (0,19 г, 1,36 ммоль) в течение 24 ч при комнатной температуре. Устанавливают pH реакционной смеси от 1 до 2 с помощью HCl и подвергают разделению в этилацетате и воде. Органический слой высушивают, затем отгоняют растворитель при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 0,21 г (выход 75%) бесцветного аморфного продукта. Элементный анализ продукта показан в таблице 31. ЯМР-спектр и ИК-спектр соединения следующие.
1H-ЯМР (200 МГц, CDCl3) δ ppm: 0,66 (6H, д), 1,14 (6H, д), 1,23 (3H, т), 1,55 (1H, м), 3,14 (1H, м), 3,53 (2H, д), 3,91 (3H, с), 4,14 (2H, кв), 4,68 (2H, с), 5,69 (2H, с), 6,36 - 7,00 (3H, м), 7,21 (1H, тд), 7,50 (1H, д), 7,56 (1H, т), 7,71 (1H, тд), 7,86 (1H, д), 8,04 (1H, д), 8,19 (1H, д).
ИК (KBr): 3456, 2976, 2968, 1721, 1678, 1574, 1495 см-1.
Пример 26.
Используя соединения, полученные в примерах 16 и 17, повторяют способ, описанный в примере 25. Полученные таким образом соединения представлены в таблице 32.
Пример 27.
Получение 2,4(1H, 3H)-диоксо-6-изобутоксикарбонил-7-изопропил- 1-/2-метоксибензил/-5-(2-хинолил)пиридо[2,3-d] пиримидин-3-уксусной кислоты (химическая формула 41)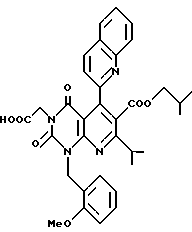
Соединение (0,19 г, 0,30 ммоль), полученное в примере 25, растворяют в смеси растворителей метанола (2,0 мл) и тетрагидрофурана (21,0 мл), к которой добавляют 2 н. водный раствор гидроксида натрия (0,60 мл, 1,2 ммоль), и смесь перемешивают в течение 110 мин при комнатной температуре. Реакционную смесь доводят до pH от 1 до 2 с помощью 1 н. HCl, затем подвергают разделению в этилацетате и воде. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают, после чего отгоняют растворитель при пониженном давлении. Остаток перекристаллизовывают из смешанного растворителя изопропанола и гексана с получением 0,11 г (выход 61%) бесцветного кристаллического порошка, т.пл.) 224-225oC. Элементный анализ продукта показан в таблице 33. ЯМР-спектр и ИК-спектр соединения следующие:
1H-ЯМР (200 МГц, DMSO-d6) δ ppm: 0,57 (6H, д), 1,03 (6H, д), 1,38 (1H, м), 3,10 (1H, м), 3,50 (2H, ушир), 3,80 (3H, с), 4,50 (2H, с), 5,54 (2H, с), 6,83 (1H, т), 6,94 (1H, д), 7,06 (1H, д), 7,24 (1H, т), 7,56 (1H, д), 7,65 (1H, т), 7,78 (1H, т), 7,90 (1H, д), 8,02 (1H, д), 8,34 (1H, д).
ИК (KBr): 3480, 2970, 1717, 1665, 1574, 1466 см-1.
Пример 28.
Используя соединения, полученные в примере 2 (соединение N 27, 28) и примере 26, повторяют способ, описанный в примере 27. Полученные таким образом соединения (химическая формула 42)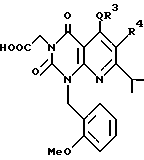
представлены в таблице 34.
Пример 29. Получение 2,4(1H,3H)-диоксо-6-цианоэтоксикарбонил-7- изопропил-1-/2-метоксибензил-/5-(2-хинолил)пиридо/2,3-d/ пиримидин-3-уксусной кислоты (химическая формула 43)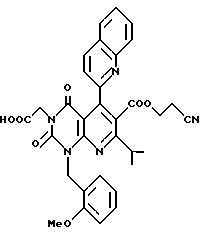
Соединение (0,30 г, 0,45 ммоль), полученное в примере 3, растворяют в дихлорметане (3,0 мл) и перемешивают с трифторуксусной кислотой (1 мл) в течение 4,5 ч при комнатной температуре. Реакционную смесь отгоняют при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 0,31 г бледно-желтого кристаллического порошка, кристаллы перекристаллизовывают из смешанного растворителя - этилацетата и гексан - с получением 0,21 г (выход 78%) бесцветного кристаллического порошка, т.пл. 115-116oC. Элементный анализ продукт показан в таблице 35.
ЯМР-спектр и ИК-спектр соединения следующие:
1H-ЯМР (200 МГц, DMSO-d6) δ/ ppm: 1,06 (6H, д), 2,50 (2H, т), 3,78 (2H, т), 3,13 (1H, м), 3,91 (3H, с), 4,50 (2H, с), 5,55 (2H, с), 6,84 (1H, т), 6,95 (1H, д), 7,05 (1H, д), 7,21 (1H, т), 7,59 (1H, д), 7,67 (1H, т), 7,79 (1H, т), 7,97 (1H, д), 8,04 (1H, д), 8,35 (1H, д).
ИК (KBr): 3438, 2972, 2270, 1719, 1676, 1574, 1495, 1466 см-1.
Пример 30.
Используя соединения, полученные в примере 4 (соединение N1) и примере 26 (соединение N 10-12), повторяют способ, описанный в примере 29. Соединения, полученные таким образом (химическая формула 44)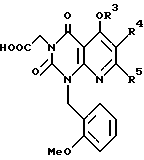
представлены в таблице 36.
Пример 31. Получение этил[2,4(1H,3H)-диоксо-6-карбокси-7- изопропил-1-/2-метилтиобензил/-5-[2-(4-хинолонил)] пиридо[2,3-d] - пиримидин]-3-ацетата (химическая формула 45)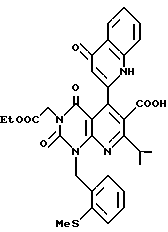
Раствор ацетонитрила (15 мл), содержащий соединение (150 мг, 0,25 ммоль), полученное в ссылочном примере 13, 2 н. водный раствор карбоната калия (1,25 мл, 2,50 ммоль) в воду (1,5 мл), перемешивают в течение 30 мин при 50oC. К реакционной смеси добавляют 1 н. HCl (5 мл) и подвергают разделению этилацетатом и насыщенным водным раствором хлорида натрия. Органический слой высушивают над MoO4 и затем отгоняют при пониженном давлении. Осадок перекристаллизовывают из изопропанола с получением 85 мг (выход 54%) бесцветного порошкообразного продукта, т.пл. 167-170oC. Элементный анализ продукта показан в таблице 37. ЯМР-спектр, ИК-спектр и масс-спектр соединения следующие:
1H-ЯМР (500 МГц, DMSO-d6) δ ppm: 1,03 (6H, д), 1,15 (3H, т), 2,59 (3H, с), 3,16 - 3,24 (1H, м), 4,11 (2H, д), 4,59 (1H, д), 4,69 (1H, д), 5,53 (2H, с), 6,29 (1H, с), 6,86 (1H, д), 7,09 (1H, т), 7,29 (1H, т), 7,41 (1H, д), 7,50 (1H, т), 7,63 (1H, д), 7,79 (1H, т), 8,21 (1H, д), 12,90 (1H, ушир.с).
ИК (KBr): 3428, 1721, 1678, 1574, 1491, 1369 см-1.
FAB-MS m/z: 615.1 (MH+).
Пример 32.
Используя соединения, полученные в ссылочном примере 14, повторяют способ, описанный в примере 31. Соединения, полученные таким образом (химическая формула 46)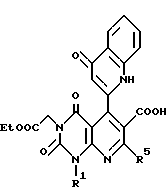
представлены в таблице 38.
Пример 33. Получение трет-бутил[2,4(1H,3H)-диоксо-6-карбамоил- 7-изопропил-1-(2-метоксибензил)-5-[2-(4-хинолонил)] пиридо[2,3-d] пиримидин/-3-ацетата (химическая формула 47)
Соединение (500 мг, 0,82 ммоль), полученное в примере 16, растворяют в дихлорметане (20 мл). В раствор добавляют тионилхлорид (0,60 мл, 8,20 ммоль) и смесь нагревают в течение 30 мин с обратным холодильником. Реакционную смесь охлаждают и концентрируют досуха. Осадок суспендируют в смешанном растворителе - тетрагидрофуран (5 мл) и диметилацетамид (20 мл) - и затем перемешивают с 25%-ным водным аммиаком (10 мл) в течение 15 мин при комнатной температуре. Реакционную смесь выливают в ледяную воду и устанавливают pH реакционной смеси от 1 до 2 с помощью 1 н. HCl, затем подвергают экстракции этилацетатом (150 мл). Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают, затем отгоняют растворитель при пониженном давлении. Остаток очищают с помощью колоночной хроматографии на силикагеле с получением 310 мг (выход 61%) бесцветного порошка. Полученный таким образом порошок перекристаллизовывают из смеси растворителей - изопропанола и изопропилового эфира с получением 170 мг (выход 33%) бесцветного кристаллического порошка, т. пл. более 300oC. Данные элементного анализа показаны в таблице 39. ЯМР-спектр и ИК-спектр полученного таким образом соединения следующие:
1H-ЯМР (200 МГц, DMSO-d6) δ/ ppm: 1,01 (3H, д), 1,13 (3H, д), 1,37 (9H, с), 3,20 (1H, м), 3,90 (3H, с), 4,53 (2H, с), 5,53 (2H, с), 6,06 (1H, с), 6,83 - 6,89 (2H, м), 7,06 (1H, д), 7,23 (1H, тд), 7,32 (1H, т), 7,44 (1H, д), 7,63 (1H, т), 7,75 (1H, ушир), 7,95 (1H, ушир), 8,11 (1H, д), 11,82 (1H, ушир).
ИК (KBr): 3428, 1721, 1678, 1638, 1605, 1572, 1512, 1475 см-1.
Пример 34.
Используя соединения, полученные в примере 16, повторяют способ, описанный в примере 33, но применяя 70%-ный водный раствор этиламина вместо 25%-ного водных раствора аммиака. Соединение, полученное таким образом (химическая формула 48)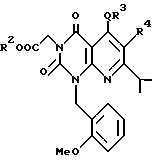
представлено в таблице 40.
Пример 35. Получение трет-бутил[2,4(1H,3H)-диоксо-6-карбокси-7- изопропил-1-(2-метоксибензил)-5-(хинолин-1-оксид-2-ил)- пиридо[2,3-d] пиримидин-3-ацетата (химическая формула 49)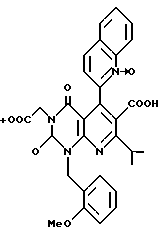
Соединение (300 мг, 0,49 ммоль), полученное в примере 16, растворяют в дихлорметане (20 мл). К раствору добавляют м-хлорбензоил гидропероксид (0,25 г) и смесь перемешивают в течение 21 ч при комнатной температуре. К реакционной смеси добавляют изопропиловый эфир (80 мл) с получением 200 мг (выход 65%) бесцветного кристаллического порошка, т.пл. 176-178oC. Данные элементного анализа показаны в таблице 41. ЯМР-спектр и ИК-спектр полученного таким образом соединения следующие:
1H-ЯМР (200 МГц, DMSO-d6) δ ppm: 1,07 (6H, д), 1,34 (9H, с), 3,33 (1H, м), 3,90 (3H, с), 4,44 (2H, с), 5,53 (2H, с), 6,79 - 6,90 (2H, м), 7,05 (1H, д), 7,24 (1H, тд), 7,43 (1H, д), 7,80 - 7,97 (3H, м), 8,12 (1H, д), 8,49 (1H, д).
ИК (KBr): 3445, 2974, 2372, 1719, 1673, 1572, 1460 см-1.
Пример 36.
Используя соединения, полученные в примере 17 (соединение N 13), повторяют способ, описанный в примере 35. Полученное таким образом соединение (химическая формула 50)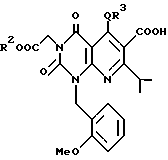
представлено в таблице 42.
Пример 37.
Приготовление таблеток, содержащих соединение по настоящему изобретению в качестве эффективного компонента (1).
Используя соединение 2 примера 19 настоящего изобретения, 165 мг лактозы, 25 мг кукурузного крахмала, 4 мг поливинилового спирта, 5 мг avicel и 1 мг стеарата магния изготовляют таблетки обычным способом.
Пример 38.
Приготовление таблеток, содержащих соединение по изобретению в качестве эффективного компонента (2)
Используя 100 мг соединения по примеру 10 настоящего изобретения, 165 мг лактозы, 25 мг кукурузного крахмала, 4 мг поливинилового спирта, 5 мл avicel и 1 мг стеарата магния изготавливают таблетки общепринятым способом.
Пример 39.
Приготовление таблеток, содержащих соединение по настоящему изобретению в качестве эффективного компонента (3).
Используя 100 мг соединения примера 12 настоящего изобретения, 165 мг лактозы, 25 мг кукурузного крахмала, 4 мг поливинилового спирта, 5 мл avicel и 1 мг стеарата магния, готовят таблетки обычным способом.
Пример 40.
Приготовление таблеток, содержащих соединение настоящего изобретения в качестве эффективного компонента (4).
Используя 100 мг соединения примера 20 настоящего изобретения, 165 мг лактозы, 25 кукурузного крахмала, 4 мг поливинилового спирта, 5 мл avicel и 1 мг стеарата магния, готовят таблетки обычным способом.
Пример 41.
Приготовление таблеток, содержащих соединение настоящего изобретения в качестве эффективного компонента (5).
Используя 100 мг соединения N 9, полученного по примеру 21, 165 мг лактозы, 25 кукурузного крахмала, 4 мг поливинилового спирта, 5 мл avicel и 1 мг стеарата магния, готовили таблетки общепринятым способом.
Пример 42.
Приготовление инъекции, содержащей соединение настоящего изобретения в качестве эффективного компонента (1).
В 15 мг 1 М водного раствора гидроксида натрия растворяют 5 г соединения N 2, полученного в примере 19 настоящего изобретения, pH раствора доводят до 7,6 с помощью 0,1 М HCl, к раствору добавляют воду для инъекции для достижения полного объема 100 мл. Этот раствор подвергают стерильной фильтрации, используя 0,22 микрометровый мембранный фильтр, разделяя в стерилизованные пузырьки по 2 мл каждая порция с последующей лиофилизацией и получением лиофилизованных инъекционных растворов в форме 100 мг/пузырьков.
Пример 43.
Приготовление инъекционного раствора, содержащего соединение настоящего изобретения в качестве эффективного компонента (2).
В 15 мг 1 М водного раствора гидроксида натрия растворяют 5 г соединения примера 10 настоящего изобретения. Устанавливают pH раствора 7,6 с помощью 0,1 М HCl, добавляют воду для инъекции с установлением полного объема в 100 мл. Этот раствор подвергают стерилизующей фильтрации, используя 0,22 микрометровый мембранный фильтр, распределяя в стерильные пузырьки порциями по 2 мл в каждый, последующей лиофилизацией и получением лиофилизованных инъекционных растворов в форме 100 мг/пузырьков.
Пример 44.
Приготовление инъекционного раствора, содержащего соединение настоящего изобретения в качестве эффективного компонента (3).
В 15 мг 1 М водного раствора гидроксида натрия растворяют 5 г соединения примера 12 настоящего изобретения. Устанавливают pH раствора 7,6 с помощью 0,1 М HCl, добавляют воду для инъекции с установлением полного объема 100 мл. Этот раствор подвергают стерилизующей фильтрации, используя 0,22 микрометровый мембранный фильтр, распределяя в стерильные пузырьки порциями по 2 мл в каждый, с последующей лиофилизацией и получением лиофилизованных инъекционных растворов в форме 100 мл/пузырек.
Пример 45.
Приготовление инъекционного раствора, содержащего соединение настоящего изобретения в качестве эффективного компонента (4).
В 15 мл 1 М водного раствора гидроксида натрия растворяют 5 г соединения примера 20 настоящего изобретения. Устанавливают pH раствора 6,7 с помощью 1 М HCl, добавляют воду для инъекции с установлением полного объема в 100 мл. Этот раствор подвергают стерилизующей фильтрации на 0,22 микрометровом мембранном фильтре, распределяя в стерильные пузырьки порциями по 2 мл в каждый, с последующей лиофилизацией и получением лиофилизованных инъекционных растворов в форме 100 мл/пузырек.
Пример 46.
Приготовление инъекции, содержащей соединение настоящего изобретения в качестве активного компонента (5).
В 15 мл 1 М водного раствора гидроксида натрия растворяют 5 г соединения N 9, полученного по примеру 21. Доводя pH раствора до 7,6 0,1 1 М HCl, добавляют воду до получения общего объема инъекции 100 мл.
Раствор подвергают стерильной фильтрации используя 0,22 микрометровый мембранный фильтр, и дополняют им стерильные пузырьки по 2 мл с последующей лиофилизацией обычным методом для получения лиофилизованных инъекционных растворов в форме 100 мг/пузырек.
Фармакологический эксперимент 1.
Эндотелиновый рецептор - исследование.
Эндотелин-A рецептора были получены разбавлением фракции вентрикулярной мышечной мембраны желудочка сердца свиньи аналитическим буфером [200 мМ трис-HCl, 2 мМ ЭГТА (этиленгликоль бис(2-аминоэтиленэфир)тетрауксусной кислоты), 5 мМ ацетата магния, 0,1% БСА (бычий сывороточный альбумин), 0,03% NaN3, 0,5 мМ ФМСФ (фенилметилсульфонилфторид), 20 г/мл лейпептина, 4 мкг/мл Е-64 (продукт из Института Пептидов), 1 мкг/мл пепстатина (pH 7,2)] с получением раствора фракции мембраны желудочка сердца свиньи 12 мкг/мл).
Эндотелиновый-B рецептор был получен разбавлением фракции бычьей церебральной мембраны с тем же самым аналитическим буфером с получением раствора, имеющего концентрацию 180 мкг/мл.
К 100 мкл каждой порции добавляют 5 нМ (125I) эндотелина-1 (2 мкл). Добавляют туда же диметилсульфоксидный раствор образца (3 мл) и инкубируют при 25oC в течение 60 мин.
И для определения максимума связывающего количества (C0) и неспецифически связывающего количества (HCC) также инкубируются образцы, к которым добавлен раствор диметилсульфоксида (3 мкл) или раствор диметилсульфоксида (3 л), содержащий эндотелин-1 (10-5 М).
Эти образцы дополняют 0,05% ХАПС (3-((3-хлорамидопропил) диметиламмонио)-1-пропансульфонат)-аналитическим буфером (1,5 мл), подвергают фильтрации сквозь стеклянный фильтр CF/F (торговое наименование: продукция Wattman Ltd (Англия)) и затем промывают также буфером (1,5 мл).
Радиоактивность фильтрата была измерена гамма-счетчиком для определения процента максимального связывания (ПМС) в соответствии со следующей расчетной формулой.
Концентрация, вызывающая ПМС= 50% указывается как значение IC50. IC50 значения некоторых соединений настоящего изобретения, синтезированных в вышеприведенных примерах, показаны в таблице 43.
ПМС = ((C-HCC)/(C0-HCC)) • 100
Фармацевтический эксперимент 2.
Эндотелиновый рецептор - исследование.
Эндотелиновые (ЭТ) рецепторы были приготовлены разбавлением клеточной (Sf 9) мембраны насекомого, имеющей похожие на человеческие эндотелин-A (ЭТА) рецепторы или на человеческие эндотелин-B (ЭТВ) рецепторы, аналитическим буфером (20 мМ Трис-HCl, 2 мМ ЭГТА (этиленгликоль бис(2-аминоэтиленэфир)тетрауксусной кислоты), 5 мМ ацетата магния, 0,1% БСА (бычий сывороточный альбумин), 0,03% NaN3, 0,5 мМ ФМСФ (фенилметилсульфонилфторид), 20 мкг/мл лейпептина, 4 мкг/мл E-64 (продукт из Института Пептидов), 1 мкг/мл пепстатина, (pH 7,2)), соответственно в концентрации 1,4 мкг/мл в первом случае и 0,7 мкг/мл в последующем случае.
К 100 мл каждой порции добавляют 5 нМ (125I) эндотелина-1 (2 мкл). Добавляют туда же диметилсульфоксидный раствор образца (3 мкл) и инкубируют при 25oC в течение 60 мин.
И, для определения максимума связывающего количества (C0) и неспецифически связывающего количества (HCC), также инкубируют образцы, к которым добавлен раствор диметилсульфоксида (3 мкл) или раствор диметилсульфоксида (3 мкл), содержащий эндотелин-1 (10-5 М).
Эти образцы дополняют 0,05% ХАПС (3-((3-хлорамидопропил)диметиламмонио)-1-пропансульфонат)- аналитическим буфером (1,5 мл), подвергают фильтрации сквозь стеклянный фильтр GF/F (торговое наименование, продукции Wattman Ltd. (Англия)) и затем промывают тем же буфером (1,5 мл).
Радиоактивность фильтрата измеряют гамма-счетчиком для определения процента максимального связывания (ПМС) согласно вышепредставленной формуле. Концентрация, вызывающая ПМС = 50% принимается за значение IC50. Значение IC50 некоторых соединений настоящего изобретения, синтезированных по вышеприведенным примерам, показаны в таблице 44.
Согласно результатам, показанным в таблицах 43 и 44, доказано, что соединение (A) или его соль настоящего изобретения обладают превосходным антагонистическим по отношению к эндотелиновому рецептору действием, как по отношению к эндотелин-A рецептору, так и по отношению к эндотелин-B рецептору.
Соединения настоящего изобретения обладают антагонистическим по отношению к эндотелиновому рецептору действием, и являются эффективными в качестве профилактических и терапевтических агентов при лечении острой почечной недостаточности, инфаркта миокарда, гипертензии, атеросклероза, гепатопатии, легочной гипертензии, бронхиальной астмы, органогипофункции, вызванной операцией или трансплантацией органов.
Ясно, что различные модификации могут быть применены при получении и применении нового соединения по настоящему изобретению без изменения сущности изобретения, как указано в следующей формуле изобретения.
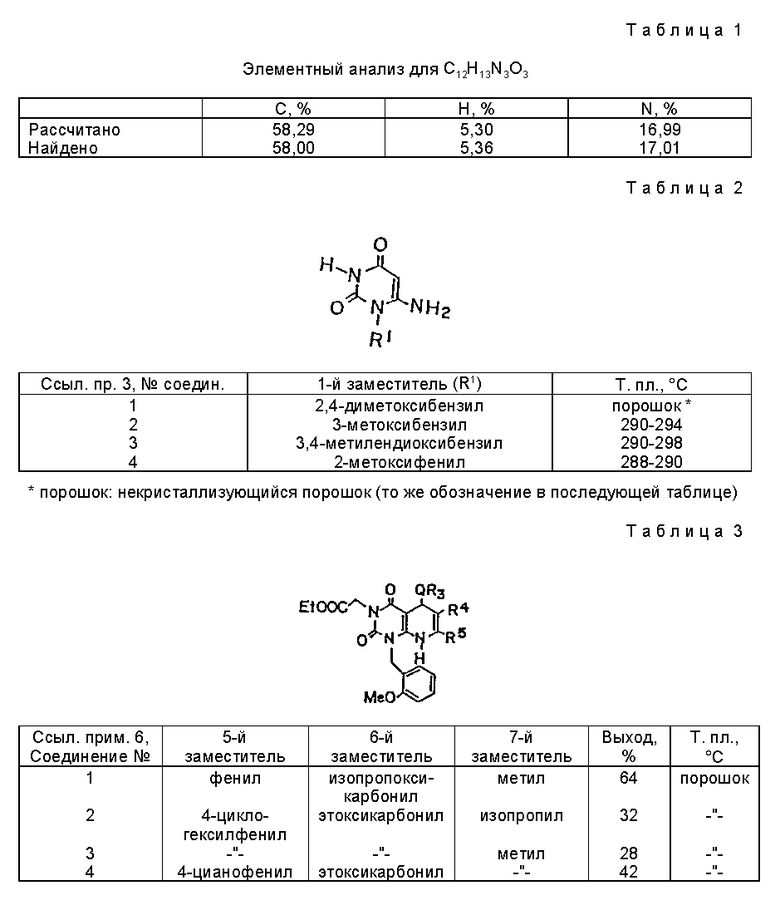


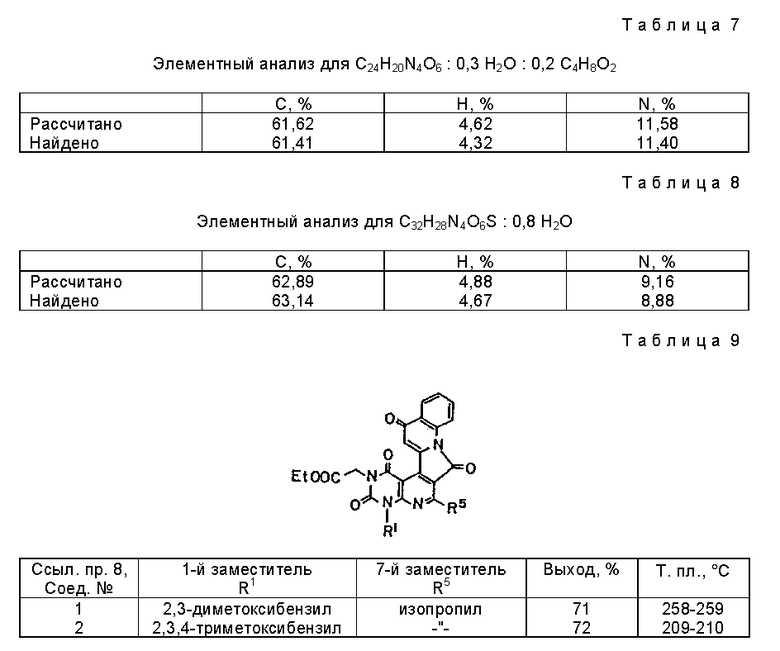


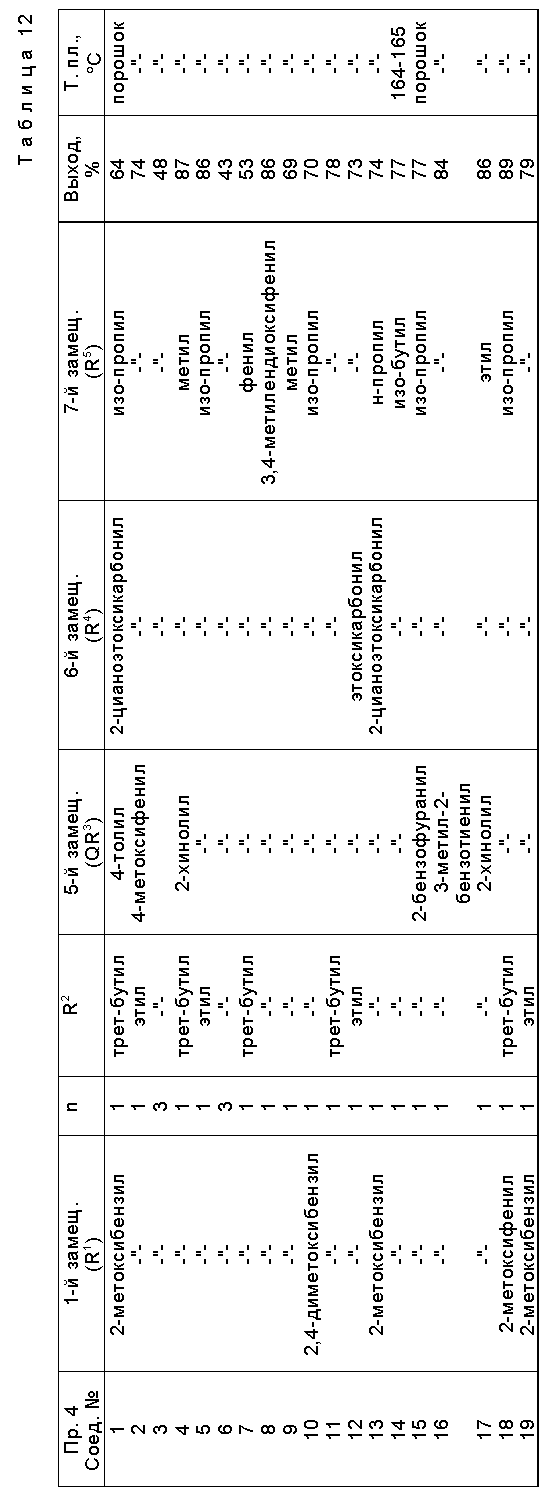


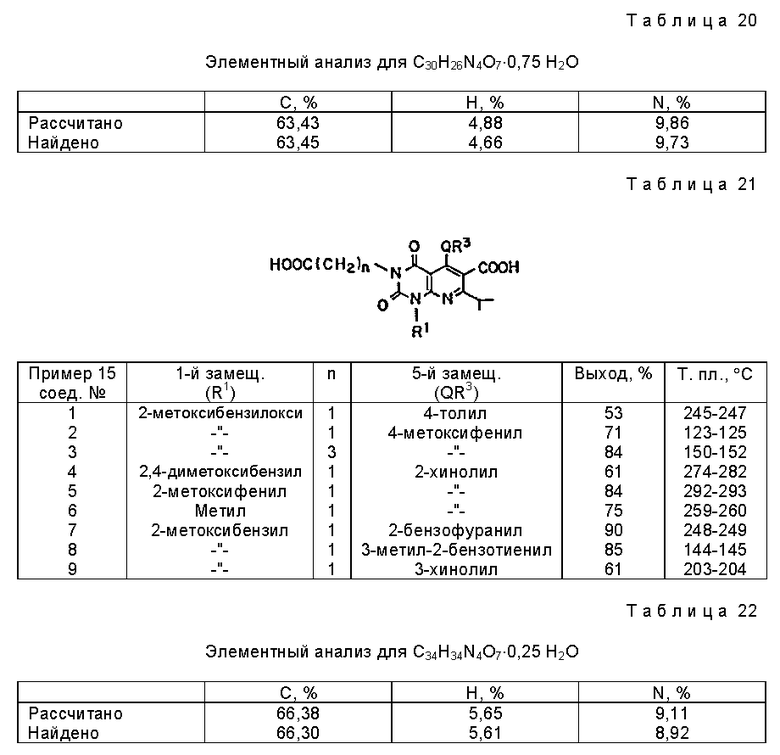
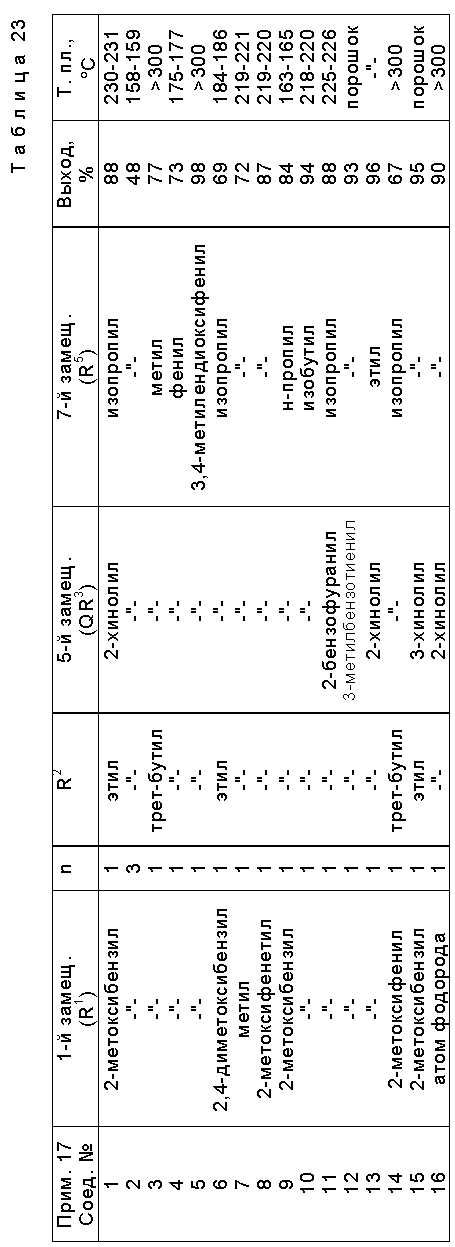
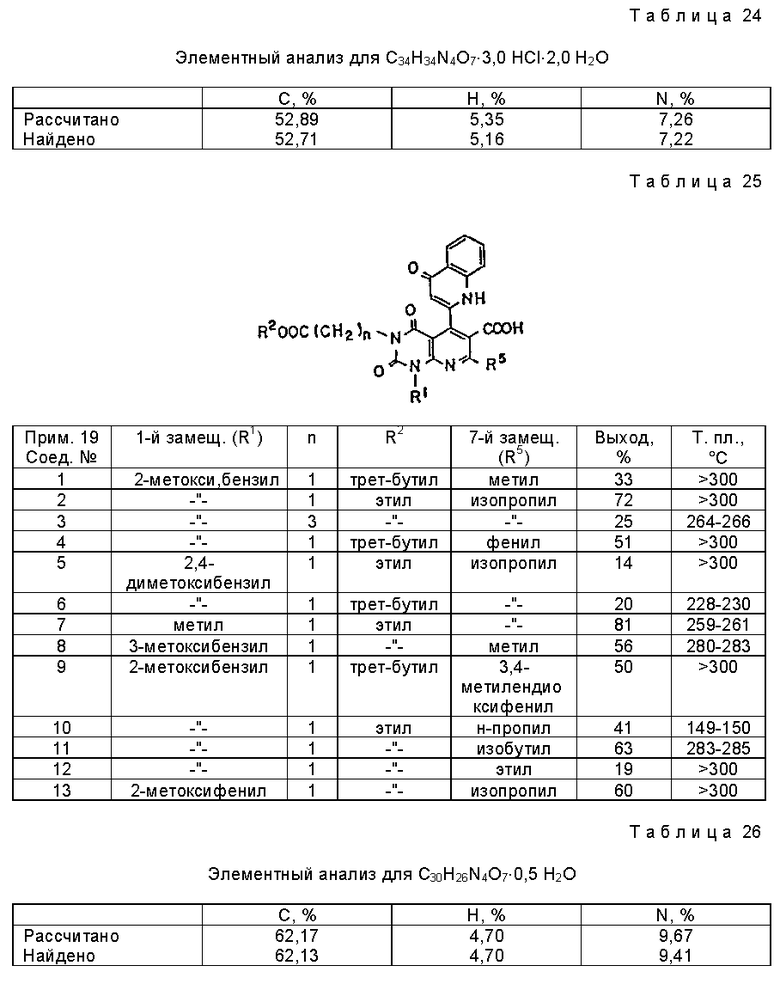
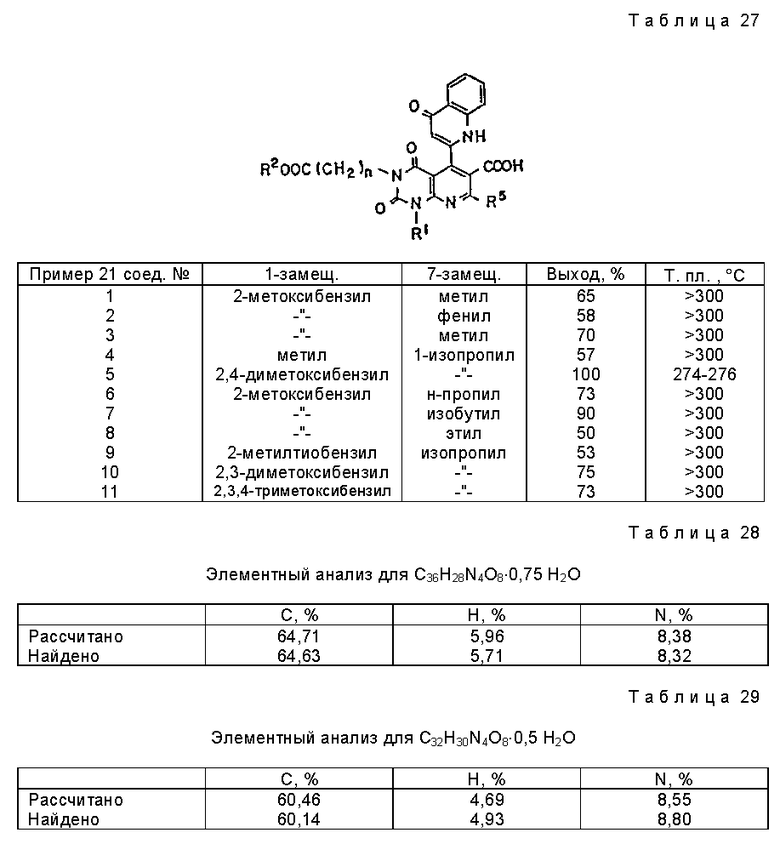
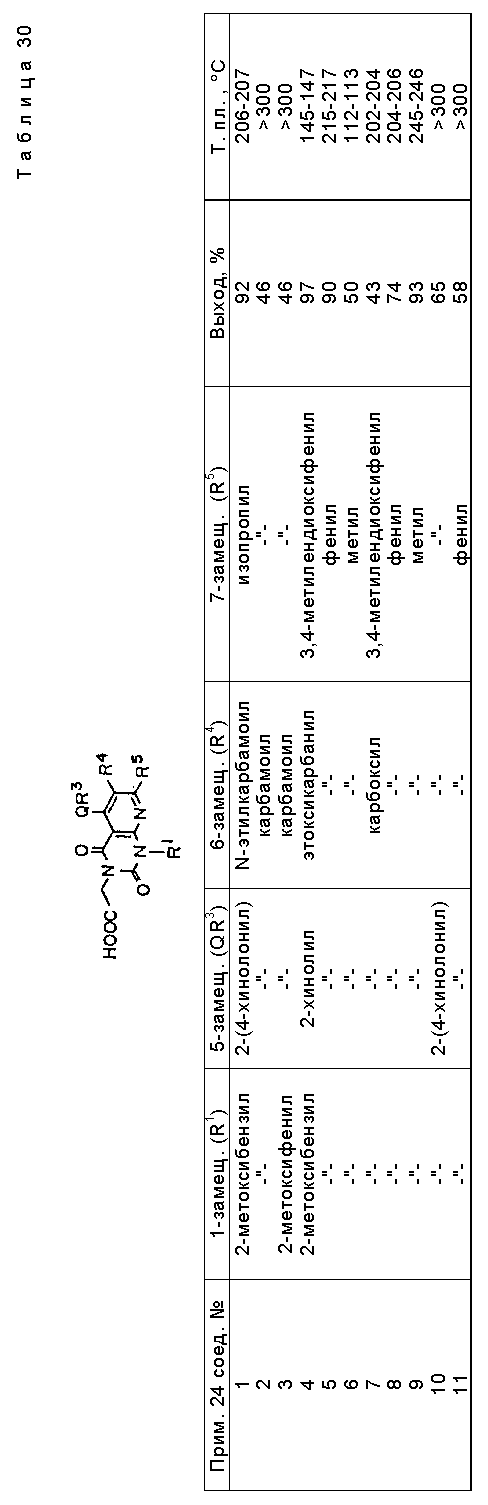

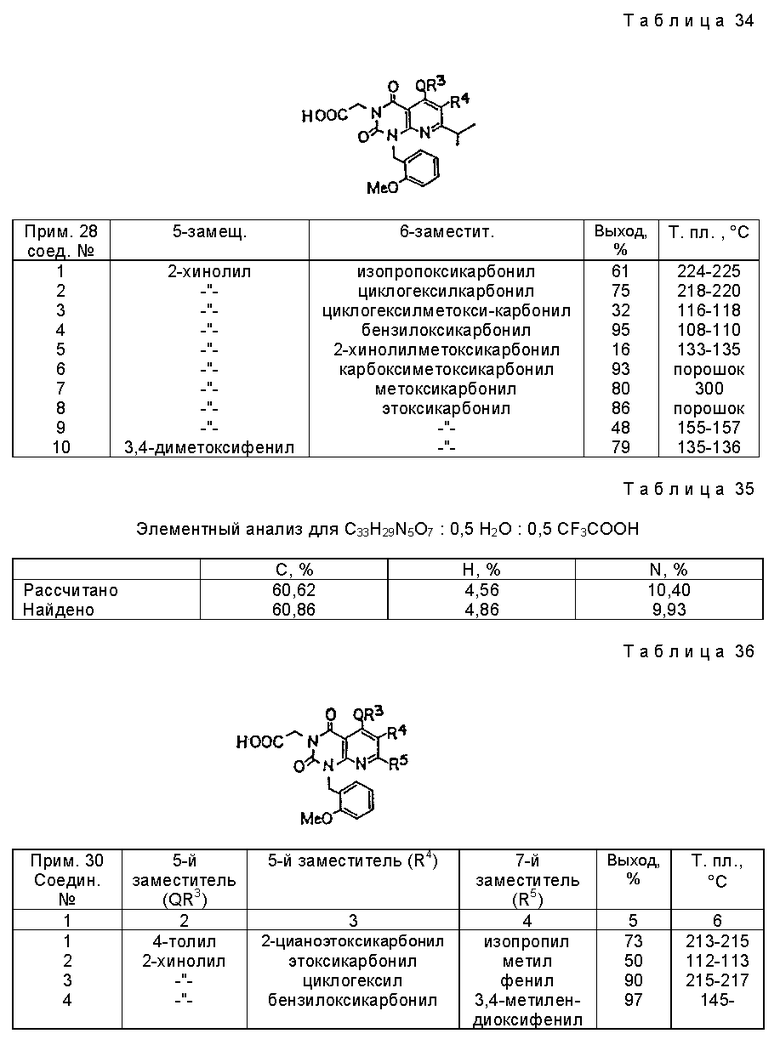
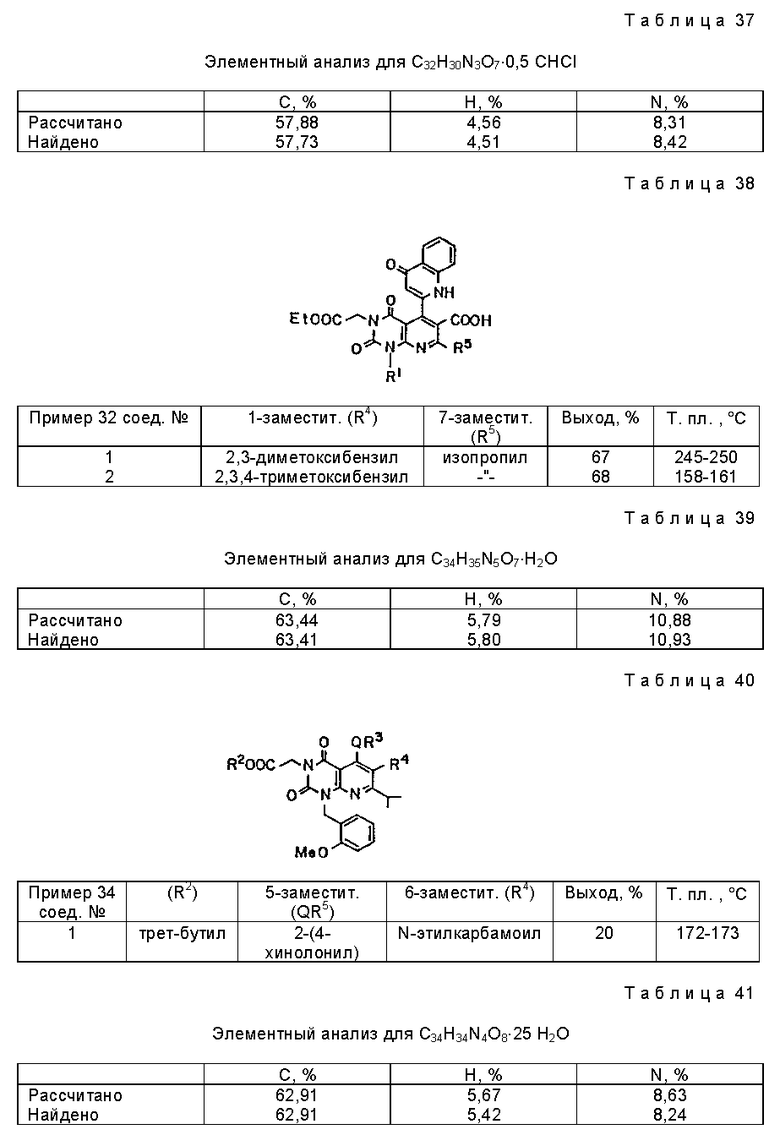
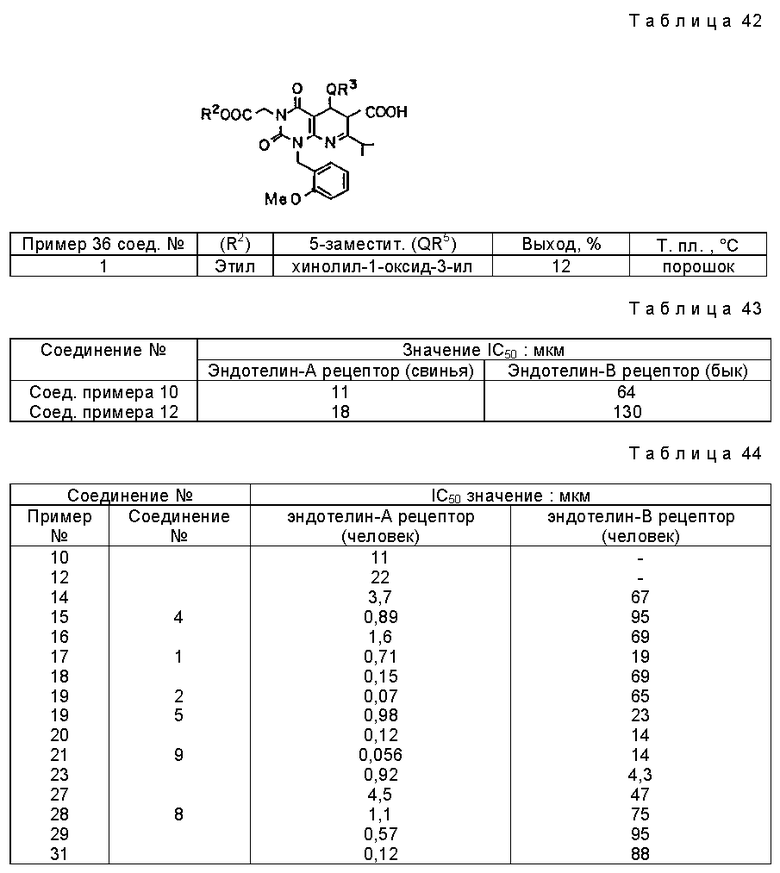
Описаны новые производные пиридопиримидина формулы А, в которой Q представляет -(CH2)m-(m = 0, 1, 2), n = 1, 2, 3; R1 является (1) атомом водорода, (2) C1-6-алкильной группой, которая может быть необязательно замещенной одним из заместителей, выбранных из хинолила или индолида, (3) фенильной группой, которая необязательно может быть замещенной по меньшей мере одной C1-6-алкоксигруппой, или (4) фенил-C1-3-алкильной группой, которая может быть необязательно замещенной по меньшей мере одним заместителем, выбранным из C1-6-алкокси и С1-6-алкилтио; R2 является атомом водорода или C1-6-алкильной группой; R3 является (1) фенильной или нафтильной группой, которая необязательно может быть замещенной по меньшей мере одним заместителем, выбранным из C1-6-алкила, C1-6-алкокси, C1-6-алкилендиокси, атома галогена, нитро, циано, фенила, C3-7-циклоалкила, (2) пиридилом, который необязательно может быть замещенным C1-6-алкилом, (3) тиенилом, который необязательно может быть замещенным C1-6-алкилом, (4) хинолилом, который необязательно может быть замещенным оксогруппой или гидроксильной группой, (5) хинолонилом, (6) бензофурилом или (7) бензотиенилом, который необязательно может быть замещенным C1-6-алкилом; R4 является группой - СООR6, в которой R6 представляет (1) атом водорода, (2) C1-6-алкильную группу, необязательно замещенную по меньшей мере одним заместителем из С3-7-циклоалкильной группы, хинолильной группы, карбоксильной группы, циано и фенильной группы; или (3) С3-7-циклоалкил; или группой -СОNR7R8, в которой R7 и R8 независимо представляют атом водорода или C1-6-алкильную группу; R5 является (1) атомом водорода, (2) С1-6-алкильной группой или (3) фенильной группой, которая может быть необязательно замещенной С1-6-алкилендиоксигруппой, или его соль. Описывается также способ получения соединений формулы А, фармацевтические композиции на основе соединения формулы А и способ антагонистического воздействия на эндотелиновые рецепторы при лечении почечной недостаточности, инфаркта миокарда, гипертензии, церебрального инфаркта, грудной ангины, артериосклероза, гепатопатии, легочной гипертензии, бронхиальной астмы, органогипофункции, вызванной операцией или трансплантацией органов. Новые соединения формулы А и фармкомпозиции на их основе могут найти широкое применение в медицине. 4 с. и 16 з.п.ф-лы, 44 табл.
в которой Q представляет - (CH2)m - (m = 0, 1, 2);
n = 1, 2, 3;
R1 является (1) атомом водорода, (2) C1-6-алкильной группой, которая может быть необязательно замещенной одним из заместителей, выбранных из хинолила или индолила, (3) фенильной группой, которая необязательно может быть замещенной по меньшей мере одной C1-6-алкоксигруппой, или (4) фенил-C1-3-алкильной группой, которая может быть необязательно замещенной по меньшей мере одним заместителем, выбранным из C1-6-алкокси и C1-6-алкилтио;
R2 является атомом водорода или C1-6-алкильной группой;
R3 является (1)фенильной или нафтильной группой, которая необязательно может быть замещенной по меньшей мере одним заместителем, выбранным из C1-6-алкила, C1-6-алкокси, C1-6-алкилендиокси, атома галогена, нитро, циано, фенила, C3-7-циклоалкила, (2) пиридилом, который необязательно может быть замещенным C1-6-алкилом, (3) тиенилом, который необязательно может быть замещенным C1-6-алкилом, (4) хинолилом, который необязательно может быть замещенным оксогруппой или гидроксильной группой; (5) хинолонилом; (6) бензофурилом или (7) бензотиенилом, который необязательно может быть замещенным C1-6-алкилом;
R4 является группой - COOR6, в которой R6 представляет (1) атом водорода, (2) C1-6-алкильную группу, необязательно замещенную по меньшей мере одним заместителем из C3-7-циклоалкильной группы, хинолильной группы, карбоксильной группы, циано и фенильной группы, или (3) C3-7-циклоалкил; или группой -CONR7R8, в которой R7 и R8 независимо представляют атом водорода или C1-6-алкильную группу;
R5 является (1) атомом водорода, (2) C1-6-алкильной группой или (3) фенильной группой, которая может быть необязательно замещенной C1-6-алкилендиоксигруппой,
или его соль.
где n = 0, 1, 2, 3;
R1 является (1) атомом водорода, (2) C1-6-алкильной группой, которая может быть необязательно замещенной одним из заместителей, выбранных из хинолила или индолила, (3) фенильной группой, которая необязательно может быть замещенной по меньшей мере одной C1-6-алкокси группой, или (4) фенил-C1-3-алкильной группой, которая может быть необязательно замещенной по меньшей мере одним заместителем, выбранным из C1-6-алкокси и C1-6-алкилтио;
R2 является атомом водорода или C1-6-алкильной группой;
R5 является (1) атомом водорода, (2) C1-6-алкильной группой или (3) фенильной группой, которая может быть необязательно замещенной C1-6-алкилендиокси группой,
или его соль подвергают реакции с нуклеофильным реагентом, выбранным из группы, состоящей из карбоната калия, водного аммиака и аминосоединения, способным к взаимодействию с заместителем - OR6 или - NR7R8, где значения R6, R7 и R8 указаны выше.
Приоритеты по пунктам:
29.12.92 по пп.1, 4, 10, 11, 16 и 17;
05.11.93 по пп.2, 3, 5 - 8, 9, 12, 13 и 18.
| Способ получения 9-формилпроизводных пиридо @ 1,2- @ пиримидина или их оптически активных антиподов | 1982 |
|
SU1245260A3 |
| Способ получения производных 2,4диоксо-1,2,3,4-тетрагидропиридо (2,3- )пиримидина | 1974 |
|
SU525686A1 |
| АЭРОЗОЛЬНЫЙ ГЕНЕРАТОР | 0 |
|
SU243311A1 |
| ТСХИНЧЕСКАЯ БКБЛИОТЕЯА | 0 |
|
SU251194A1 |
| 0 |
|
SU288048A1 | |
| Машина для просеивания, сортировки и отбора по величине зерна | 1932 |
|
SU37934A1 |
| EP 0457195 A1, 1991 | |||
| EP 0460679 A1, 1991 | |||
| US 3275634 A, 1975 | |||
| US 4981950 A, 1991 | |||
| Способ определения вязкости полимеров | 1988 |
|
SU1670533A1 |
| СПОСОБ И УСТРОЙСТВО РЕГУЛИРОВАНИЯ ДАВЛЕНИЯ В СЕТИ ВОДОСНАБЖЕНИЯ | 2006 |
|
RU2334266C2 |
| Камилов В.Х | |||
| Пиридины и их применение в медицине | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |
