Настоящее изобретение относится к применению фармацевтически активных производных бензопирана в качестве лекарственных средств для лечения сердечной недостаточности млекопитающих, включая людей.
В выложенных заявках на патенты Японии N Hei 2-4791, N Hei 2-49788, N Hei 2-152974 и N Hei 5-43432, патенте США N 4900752, Европейском патенте N 327 127 и EP-A492 391 описана возможность применения производных бензопирана в качестве лекарственного средства для лечения заболеваний сердечно-сосудистой системы, например гипертензии, стенокардии, аритмии и т.д., а также в качестве стимуляторов роста волос для лечения облысения. Однако в них не сообщается о возможности применения этих производных в качестве лекарственного средства для лечения заболеваний, связанных с сердечной недостаточностью.
Авторы настоящего изобретения интенсивно изучали и исследовали различные производные бензопирана, чтобы получить производные, обладающие слабой способностью открывать калиевые каналы, не обладающие кардиоподавляющей активностью, но скорее обладающие способностью усиливать сокращение сердца, и в результате нашли, что соединения следующей формулы (I) обладают сильной кардиотонической активностью.
Конкретно, настоящее изобретение касается соединений следующей формулы (I), их оптических изомеров, их стереоизомеров и их фармацевтически приемлемых солей, когда они могут образовать соли.
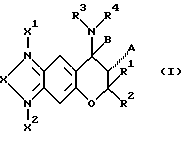
где X1 и X2 отсутствуют или представляют собой атом кислорода;
X представляет собой атом кислорода, атом серы, атом азота (этот атом азота незамещен или замещен атомом водорода или C1-C4-алкилом), C(O), C(S) или C(N-CN);
A представляет собой атом водорода, гидроксигруппу или OC(O)R5 (R5 представляет собой C1-C4-алкил) или A вместе с B образуют одинарную (дополнительную) связь;
B представляет собой атом водорода или вместе с A образует одинарную связь;
R1 и R2, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или C1-C4-алкил или R1 и R2 вместе образуют 1,4-бутиленовую или 1,5-пентиленовую группу, которая незамещена или замещена одним или несколькими C1-C4-алкилами;
R3 и R4, одинаковые или отличающиеся друг от друга, представляют собой атом водорода, C1-C6-алкил (этот алкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C2-C6-алкенил (этот алкенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C2-C6-алкинил (этот алкинил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C3-C6-циклоалкил (этот циклоалкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), фенил (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы) или C(=Y)ZR6 [Y представляет собой атом кислорода, атом серы или NR7 представляет собой атом водорода, цианогруппу, нитрогруппу, C1-C4-алкил, C1-C4-алкоксигруппу или CO2R8 (R8 представляет собой C1-C4-алкил)]; Z представляет собой атом кислорода, атом серы или NR9 (R9 представляет собой атом водорода, C1-C6-алкил (алкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-С4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C2-С6-алкенил (этот алкенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-С5-алкоксикарбонила, гидроксигруппы, C1-С4-алкоксигруппы, CH(OR)2 (R представляет собой C1-С4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C2-C6-алкинила (алкинил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C3-C6-циклоалкил (этот циклоалкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы) или фенил (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-С4-алкоксигруппы)); и R6 представляет собой атом водорода, C1-С8-алкил (этот алкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-С5-алкоксикарбонила, гидроксигруппы, C1-С4-алкоксигруппы, CH(OR)2 (R представляет собой C1-С4-алкил), фенила (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-С4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C2-С6-алкенил (этот алкенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), C2-C6-алкинил (этот алкинил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы, формила, цианогруппы и нитрогруппы), C3-C6-циклоалкил (этот циклоалкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-aлкил), фенила (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы), или фенил (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы) или
R3 и R4 вместе образуют 1,4-бутиленовую или 1,5-пентиленовую группу (эти 1,4-бутиленовая и 1,5-пентиленовая группы незамещены или замещены одним или несколькими заместителями, выбранными из C1-C4-алкила, фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), атома галогена, OR10 (R10 представляет собой атом водорода, C1-C4-алкил, COR11 (R11 представляет собой C1-C4-алкил), нитрогруппу, SO3H или PO3H2)}, или
R3 и R4 вместе образуют (CH3)mX4(CH2)I [m и I представляют собой числа 1, 2 или 3, причем сумма их равна 3, 4 или 5, X4 представляет собой атом кислорода, атом серы или NR12 { R12 представляет собой атом водорода, C1-C4-алкил или фенил (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы)}], или
R3 и R4 вместе образуют группу (CH2)nZC(=Y) (n обозначает число 2, 3 или 4 и Z и Y имеют значения, указанные выше).
Настоящее изобретение относится к лекарственным композициям для лечения сердечной недостаточности, содержащим эти соединения в качестве активных компонентов.
Соединения, показанные в формуле (1) настоящего изобретения, усиливают сокращения сердечных мышц, они пригодны для улучшения сердечной функции. Поэтому они пригодны в качестве лекарственного средства для лечения сердечной недостаточности. Эти соединения не только имеют кардиотоническую активность, но также обладают сильной активностью по снижению скорости сердечного сокращения.
Заместители в соединениях формулы (1) будут объясняться более детально ниже.
В описании настоящего изобретения "n-" обозначает нормальный "i-"; обозначает изо-; "sec" - обозначает вторичный; "t-"обозначает третичный; "c-" обозначает цикло-; "Me" обозначает метил; "Et" обозначает этил; "Pr" обозначает пропил; "Bu" обозначает бутил; "Pen" обозначает пентил; "Hex" обозначает гексил и "Ph" обозначает фенил.
Примеры R1 и R2 включают атом водорода. Me, Et, n-Pr, i-Pr, n-Bu, i-Bu, sec-Bu и t-Bu. R1 и R2 могут вместе образовать (CH2)4 или (CH2)5, в результате чего образуется спиросоединенные ядра.
Примеры A включают OH, OC(O)Me, OC(O)Et, OC(O)-n-, Pr, OC(O)-i-Pr, OC(O)-n-Bu, OC(O)-i-Bu, OC(O)-Sec-Bu, OC(O)-t-Bu и т.д.
A и B могут вместе образовать одинарную (дополнительную) связь.
Примеры X включают атом кислорода, атом серы, C(O), C(S), NH, NMe, NEt, N-n-Pr, N-i-Pr, N-n-Bu, N-i-Bu, N-sec-Bu, N-t-Bu и т.д.
Примеры R3 и R4 включают атом водорода. Me, Et, n-Pr, c-Pr, n-Bu, i-Bu, sec-Bu, t-Bu, n-Pen, c-Pen, бензил, пара-хлорфенилметил, пара-фторфенилметил, пара-бромфенилметил, фенилэтил, пара-хлорфенилэтил, парафторфенилэтил, парабромфенилэтил,
CH2CO2H, CH2CO2Me, CH2CO2Et, (CH2)2CO2Me, (CH2)2CO2Et, (CH2)2CH(OMe)2, (CH2)2CH(OEt)2, (CH2)3OH, (CH2)3OMe, (CH2)3OEt, (CH2)3Cl, (CH2)3Br, (CH2)3F, (CH2)3CO2H, (CH2)3CO2Me, (CH2)3CO2Et, (CH2)3CH(Me)2,
(CH2)3CH(Et)2, C(O)OMe, C(O)OEt, C(O)O-n-Pr, C(O)O-i-Pr, C(O)O-c-Pr, C(O)O-n-Bu, C(O)O-i-Bu, C(O)O-sec-Bu, C(O)O-t-Bu, C(O)O-n-Pen, C(O)O-c-Pen, C(O)O-n-Hex, C(O)O-c-Hex, C(O)O(CH2)2Cl, C(O)O(CH2)2Br, C(O)O(CH2)3Cl, C(O)O(CH2)3Br, C(O)OPh, C(O)OCH2Ph, C(O)NHMe, C(O)NHEt, C(O)NH-n-Pr, C(O)NH-i-Pr, C(O)NH-c-Pr, C(O)NH-n-Bu, C(O)NH-i-Bu, C(O)NH-sec-Bu, C(O)NH-t-Bu, C(O)NH-n-Pen, C(O)NH-c-Pen, C(O)NH-n-Hex, C(O)NH-c-Hex, C(O)NH(CH2)2Cl, C(O)NH(CH2)2Br, C(O)NH(CH2)3Cl, C(O)NH(CH2)3Br, C(O)NHPh,
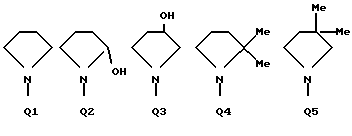
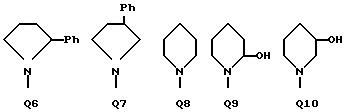
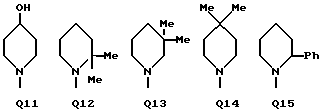
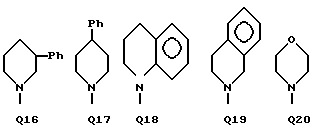
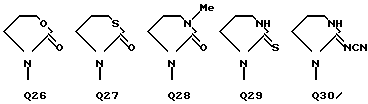
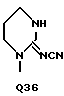
C(O)NHCH2Ph, C(O)NHC(O)CCl3, C(S)NHMe, C(S)NHEt, C(S)NH-n-Pr, C(S)NH-i-Pr, C(S)NH-c-Pr, C(S)NH-n-Bu, C(S)NH-i-Bu, C(S)NH-sec-Bu, C(S)NH-t-Bu, C(S)NH-n-Pen, C(S)NH-c-Pen, C(S)NH-n-Hex, C(S)NH-c-Hex, C(S)NH(CH2)2Cl, C(S)NH(CH2)2Br, C(S)NH(CH2)3Cl, C(S)NH(CH2)3Br, C(S)NHPh, C(S)NHCH2Ph, C(N-CN)NHMe, C(N-CN)NHEt, C(N-CN)NH-n-Pr, C(N-CN)NH-i-Pr, C(N-CN)NH-c-Pr, C(N-CN)NH-n-Bu, C(N-CN)NH-i-Bu, C(N-CN)NH-sec-Bu, C(N-CN)NH-t-Bu, C(N-CN)NH-n-Pen, C(N-CN)NH-c-Pen, C(N-CN)NH-n-Hex, C(N-CN)NH-c-Hex, C(N-CN)NH(CH2)2Cl, C(N-CN)NH(CH2)2Br, C(N-CN)NH(CH2)3Cl, C(N-CN)NH(CH2)3Br, C(N-CN)NHPh, C(N-CN)NHCH2Ph, вместе с атомом азота, к которому они присоединены, образуют радикал формулы Q1-Q36.


Среди лекарственных препаратов для лечения сердечной недостаточности настоящего изобретения предпочтителен лекарственный препарат для лечения сердечной недостаточности, содержащий в качестве активного компонента по меньшей мере одно из соединений формулы (II), их оптических изомеров, их стереоизомеров или их фармакологически приемлемых солей, если они могут образовать соли: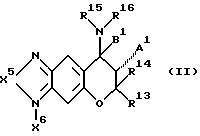
где X6 отсутствует или представляет собой атом кислорода, X5- представляет собой атом кислорода, атом серы или атом азота (этот атом азота незамещен или замещен атомом водорода или C1-C4-алкилом);
A1 представляет собой атом водорода, гидроксигруппу или OC(O)R17, (R17 представляет собой C1-C4-алкил);
B1 - представляет собой атом водорода;
R13 и R14, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или C1-C4-алкил;
R15 и R16, одинаковые или отличающиеся друг от друга, представляют собой атом водорода, C1-C6-алкил { этот алкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C6-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы} C3-C6-циклоалкил { этот циклоалкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы}, фенил (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы) или C(=Y1)Z1R18 [Y1 представляет собой атом кислорода,
атом серы или NR19 (R19 представляет собой атом водорода, цианогруппу, нитрогруппу, C1-C4-алкил или C1-C4-алкоксигруппу); Z1 представляет собой атом кислорода, атом серы или NR20 (R20 представляет собой атом водорода, C1-C6-алкил { этот алкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы}, C3-C6-циклоалкил { этот циклоалкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы} или фенил (этот фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы); и R18 представляет собой атом водорода, C1-C8-алкил {этот алкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы}, C3-C6-циклоалкил { этот циклоалкил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, карбоксигруппы, C2-C5-алкоксикарбонила, гидроксигруппы, C1-C4-алкоксигруппы, CH(OR)2 (R представляет собой C1-C4-алкил), фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), формила, цианогруппы и нитрогруппы} , или фенил (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы)] или R15 и R16 вместе образуют 1,4-бутиленовую или 1,5-пентиленовую группу {эти 1,4-бутиленовая и 1,5-пентиленовая группы незамещены или замещены одним или несколькими заместителями, выбранными из C1-C4алкила, фенила (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы), атома галогена и OR21 (R21 представляет собой атом водорода, C1-C4-алкил, COR22 (R22 представляет собой C1-C4-алкил), нитрогруппу, SO3H или PO3H2)}, или R15 и R16 вместе образуют ((CH2)o X7(CH2)p [o и p представляют собой число 1, 2 или 3, причем сумма их равна 3, 4 или 5; X7 представляет собой атом кислорода, атом серы или NR23 (R23 представляет собой атом водорода, C1-C4-алкил или фенил (фенил незамещен или замещен одним или несколькими заместителями, выбранными из атома галогена, гидроксигруппы и C1-C4-алкоксигруппы)}] ; или
R15 и R16 вместе образуют группу (CH2)q Z1C(=Y1) (q обозначает число 2, 3 или 4 и Z1 и Y1 имеют значения, указанные выше).
Предпочтительны также указанные выше лекарственные композиции для лечения сердечной недостаточности, у которых R15 в формуле (II) представляет собой атом водорода и R16 представляет собой группу C(=Y2)NHR24 (в которой Y2 представляет собой атом кислорода, атом серы или N-CN; и R24 представляет собой фенил, бензил или C1-C8-алкил, который может быть разветвлен).
Предпочтительны также лекарственные композиции для лечения сердечной недостаточности, у которых R15 и R16 в формуле (II) вместе образуют группу (CH2)kNHC(= Y3) (в которой к представляет собой число 2, 3 или 4 и Y3 представляет собой атом кислорода, атом серы или N-CN).
Предпочтительны также лекарственные композиции для лечения сердечной недостаточности, у которых R15 и R16 в формуле (II) одновременно представляют собой C1-C6-алкилы.
Предпочтительны также лекарственные композиции для лечения сердечной недостаточности, у которых R15 и R16 в формуле (II) вместе образуют группу (CH2)4 или (CH2)5.
Ниже, в табл/ 1, приведены примеры соединений формулы (I) настоящего изобретения. В таблице знак "-" обозначает отсутствие X1 и/или X2. Qn имеет указанное выше значение. (Qn=(R3-)(R4-)N-).
Соединения формулы (1) настоящего изобретения имеют асимметричные атомы углерода в положениях 3 и 4 ядра пирана и поэтому включают оптически активные соединения на основе асимметричных атомов углерода. Такие оптически активные соединения можно также применять в настоящем изобретении, подобно рацемическим модификациям. Кроме того, можно также применять стереоизомеры по положениям 3 и 4 ядра пирана. Если соединения могут образовать соли, их фармакологически приемлемые соли можно также применять в качестве активных компонентов композиций настоящего изобретения.
Способы получения соединений формулы (1) настоящего изобретения будут указаны ниже.
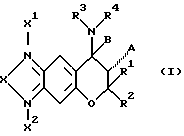
Соединений формулы (1), у которых X представляет собой атом кислорода и одновременно R3 и R4 не образуют группу C(=Y)ZR6, получают, как указано в следующей реакционной схеме, реакцией соединения формулы (3) с соединением формулы (4) в инертном растворителе. Соединения, у которых A представляет собой ОН, показаны в формуле (5). Соединения, у которых A образует одинарную (дополнительную) связь с B, показаны формулой (6).
Растворители, пригодные для проведения реакции соединения (3) и соединения (4), включают например сульфоксидные растворители, например диметилсульфоксид, амидные растворители, например диметилформамид и диметилацетамид, растворители типа простых эфиров, например диэтиловый эфир, диметоксиэтан и тетрагидрофуран, и спиртовые растворители, например, метанол, этанол и изопропанол. Из этих растворителей предпочтительны спиртовые растворители.
Температура реакции соединений (3) и (4) может быть от температуры, создаваемой охлаждением льдом, до температуры кипения применяемого растворителя. Реакцию предпочтительно проводят при температуре кипения применяемого растворителя. При необходимости реакцию можно проводить под давлением.
Исходные соединения (4)/(3) применяют в молярном соотношении в пределах от 0,5 до 2,0, предпочтительно от 1,0 до 1,1.
В зависимости от условий реакции или условий обработки реакционной смеси после реакции, которые будут подробно указаны ниже, получают соединение формулы (5) или соединение формулы (6). Соединение формулы (6) получают реакцией соединения формулы (3) с соединением формулы (4) в тетрагидрофуране в присутствии гидрида натрия. Однако, соединение формулы (5) можно также получить той же реакцией, но проводимой в других условиях (например время реакции, температура реакции и т.д.).
В некоторых случаях, которые зависят от условий последующей обработки, полученное соединение формулы (5) может дегидратироваться. Например, если реакционный раствор содержит кислоту или щелочь и когда его сразу нагревают или концентрируют без удаления кислоты или щелочи промыванием, образованное соединение в реакционном растворе часто дегидратируется.
Однако, на дегидратацию оказывают влияние тип образованного соединения, условия реакции получения и условия последующей обработки.
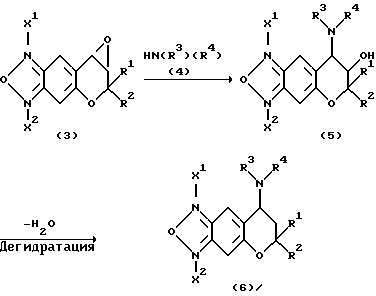
Соединения формулы (1) настоящего изобретения, у которых X является атомом кислорода и R3 или R4 представляет собой группу C(=Y)ZR6, получают по указанной ниже реакционной схеме, в которой соединение формулы (3) обрабатывают соединением формулы (7) в инертном растворителе для получения соединения формулы (8) и соединение формулы (8) обрабатывают соединением R6NCY(R6NCO, R6NCS) или ClCO2R6 для получения соединения формулы (9) или (10)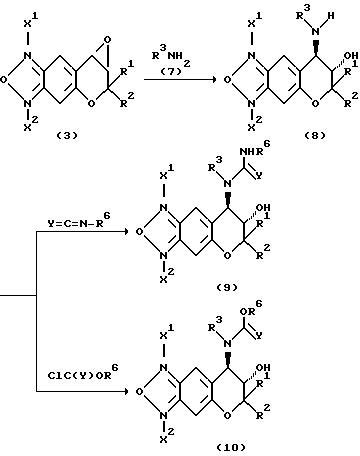

В реакционных схемах Z представляет собой N(R9) или атом кислорода или атом серы; W представляет собой атом хлора, атом брома, атом йода, низшую алкилсульфонатную, бензолсульфонатную или толуолсульфонатную группу.
По указанной выше схеме соединение, у которого R3 и R4 вместе образуют ядро, получают циклизацией соединения формулы (11).
Исходные соединения формулы (3):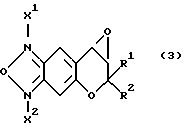
где R1 и R2 имеют значения, указанные для формулы (1), можно получить по указанной выше реакционной схеме. Полная схема получения их приводится ниже (см. в конце описания).
Соединение формулы (13):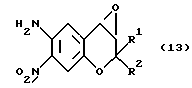
где R1 и R2 имеют значения, указанные для формулы (1), обрабатывают гипохлоридом натрия (NaOCl) для получения соединения, которое представлено формулой (3) и у которого X1 отсутствует и X2 представляет собой атом кислорода (соединение 3 (X1 =-, X2=0)), полученное соединение затем обрабатывают восстановителем, обладающим способностью удалять кислород типа N-оксида, например, азидом натрия (NaN3) или триэтилфосфитом (P(OEt)3), получая соединение, которое представлено формулой (3) и у которого X1 и X2 отсутствуют (соединение 3 (X= X2= -)). Когда соединение 3(X1=X2=-) обрабатывают близким к одному эквиваленту количеством пригодной перкислоты (например, м-хлорпербензойной кислоты, пероксида водорода, перуксусной кислоты, причем эти же кислоты будут применяться далее), то получают соединение, которое представлено формулой (3) и у которого X1 представляет собой атом кислорода и X2 отсутствует (соединение 3 (X1=0, X2=-). Если в реакции применяют более одного эквивалента перкислоты, то получают соединение формулы (3), у которого X1 и X2 представляют собой атомы кислорода (соединение 3 (X1=X2=0)). Соединение формулы (13) можно получить известными методами (например, описанными в Med. Chem., 27, 1127 (1987)).
Соединение 3(X1=0, X2=-) можно также получить обработкой соединения формулы (14):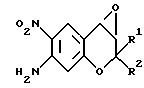 (14)
(14)
где R1 и R2 имеют значения, указанные для формулы (1), гипохлоритом натрия. Соединение формулы (14) можно получить известными методами (например, описанными в указанной выше литературе). Растворители, пригодные для проведения реакции соединения (8) и соединения Y=C=N-R6 или ClC(O)OR6, включают, например, сульфоксидные растворители, например диметилсульфоксид, амидные растворители, например диметилформамид или диметилацетамид, растворители типа простых эфиров, например диэтиловый эфир, диметоксиэтан или тетрагидрофуран, и растворители типа галогенированных соединений, например хлористый метилен или хлороформ. Из этих растворителей предпочтительны галогенированные соединения.
Температура реакции может быть от температуры, создаваемой при охлаждении льдом, до температуры кипения применяемого растворителя, предпочтительна температура кипения применяемого растворителя. При необходимости реакцию можно проводить под давлением.
Что касается молярного соотношения исходных соединений, то соединение (8) и соединение Y=C=N-R6 или ClC(O)OR6 применяют в молярном соотношении в пределах от 0,5 до 2,0, предпочтительно от 1,0 до 1,1.
Соединения формулы (1) настоящего изобретения, у которых X представляет собой атом серы или атом азота (который незамещен или замещен атомом водорода или C1-C9-алкилом), можно получить из соединения формулы (15) синтезом из трех или четырех стадий. Превращение соединения формулы (15) в соединение формулы (17) осуществляют, например, известными методами, описанными в выложенных заявках на патент Японии N Sho 56-57785 и N Sho 56-122380. Соединение формулы (17) затем обычным образом диазотируют, например обработкой его нитритом натрия в водном растворе в присутствии неорганической кислоты, например соляной кислоты или серной кислоты, или органической кислоты, например уксусной кислоты, и затем циклизуют при нагревании при 5-100oC, предпочтительно при 50-100oC, для получения соединения формулы (18), где X представляет собой атом азота.
Соединение формулы (1), у которого A и B вместе образуют одинарную связь, часто образуется просто при нагревании соединения формулы (18). Следовательно, соединение формулы (1) можно получать во время реакции синтеза соединения формулы (18) или в процессе его последующей обработки. При желании его можно синтезировать дегидратацией соединения формулы (18) путем обработки ангидридом, например бензойным ангидридом или уксусным ангидридом. , или основанием, например карбонатом калия.
Соединение формулы (1), у которого X представляет собой алкиламиногруппу, можно получить реакцией соединения формулы (18) или продукта его дегидратации диазометаном или алкилгалогенидом в присутствии карбоната калия.
Соединение формулы (17) можно реакцией с тиониланилином в инертном растворителе, например бензоле, толуоле, ксилоле или дихлорбензоле, превратить в соединение формулы (20), у которого X представляет собой атом серы.
Температура реакций может быть от 5 до 120oC, предпочтительно от 50 до 100oC.
Соединение формулы (1), у которого A и B вместе образуют одинарную связь, часто получают просто нагреванием соединения формулы (20). Следовательно, первое соединение можно получать в процессе реакции синтеза соединения формулы (20) или в процессе его последующей обработки. При желании его можно также синтезировать дегидратацией соединения формулы (20) ангидридом, например бензойным ангидридом или уксусным ангидридом, или основанием, например карбонатом калия.
В этих реакционных схемах, представленных ниже, Z представляет собой N(R9) или атом кислорода или серы и W представляет собой атом хлора, атом брома, атом иода, низшую алкилсульфонатную, бензолсульфонатную или толуолсульфонатную группу.
Из соединений формулы (15) соединение формулы (24), у которого R3 и R4 не являются группой C(=Y)ZR6 (R3 и R4 в этом случае не включают одновременно C(= Y)ZR6, можно получить реакцией соединения формулы (23) с соединением формулы (4) в инертном растворителе.
Растворители, применяемые для реакции соединения формулы (23) и соединения формулы (4), включают, например, сульфоксидные растворители, например диметилсульфоксид, амидные растворители, например диметилформамид или диметилацетамид, растворители типа простых эфиров, например диэтиловый эфир, диметоксиэтан или тетрагидрофуран, и спиртовые растворители, например метанол, этанол или изопропанол. Из этих растворителей предпочтительны спиртовые растворители.
Температура реакции может быть от температуры, создаваемой при охлаждении льдом, до температуры кипения применяемого растворителя реакции. Предпочтительной температурой реакции является температура кипения применяемого растворителя. При необходимости реакцию можно проводить под давлением.
Что касается молярного соотношения исходных соединений, то соединение (4) и соединение (23) применяют в молярном соотношении в пределах от 0,5 до 2,0, предпочтительно от 1,0 до 1,1.
Из соединений формулы (15) соединения формулы (26) и (27), у которых R3 или R4 представляет собой группу C(=Y)ZR6, можно получить по указанной выше реакционной схеме, в которой соединение формулы (23) реагирует с соединением формулы (7) в инертном растворителе с образованием соединения формулы (25), реакцией которого с R6NCY (R6NCO, R6NCS) или ClCO2R6 получают соединение формулы (26) или (27).
Соединение формулы (29), у которого R3 и R4 вместе образуют ядро, можно получить циклизацией соединения формулы (28) по указанной ниже реакционной схеме.
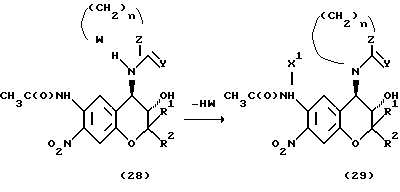
Растворители, применяемые для реакции соединения формулы (25) и соединения Y= C= N-R6 или ClC(Y)OR6, включают, например, сульфоксидные растворители, например диметилсульфоксид, амидные растворители, например, диметилформамид или диметилацетамид, растворители типа простых эфиров, например диэтиловый эфир, диметоксиэтан или тетрагидрофуран, и растворители типа галогенированных соединений, например хлористый метилен или хлороформ. Из этих растворителей предпочтительны растворители типа галогенированных соединений.
Температура реакции может быть от температуры, создаваемой при охлаждении льдом, до температуры кипения применяемого растворителя реакции. Предпочтительной температурой реакции является температура кипения применяемого растворителя. При необходимости реакцию можно проводить под давлением.
Что касается молярного соотношения исходных соединений, то соединение (25) и Y=C=N=R6 или ClC(Y)OR6 применяют в молярном соотношении в пределах от 0,5 до 2,0, предпочтительно от 1,0 до 1,1.
Соединения формулы (1) настоящего изобретения, у которых A представляет собой ацил, можно получить реакцией соединения формулы (30) с ацелирующим реагентом в инертном растворителе в присутствии пригодного основания по указанной ниже реакционной схеме.
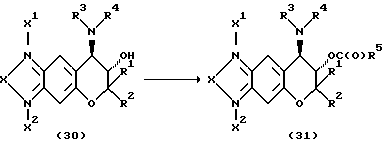
Растворители, применяемые для этой реакции, включают например сульфоксидные растворители, например диметилсульфоксид, амидные растворители, например диметилформамид или диметилацетамид, растворители типа простых эфиров, например диэтиловый эфир, диметоксиэтан или тетрагидрофуран, и растворители типа галогенированных соединений, например дихлорметан, хлороформ или дихлорэтан. Реакцию можно проводить в отсутствие растворителя. Основания, применяемые для этой реакции, включают например триэтиламин, пиридин, диизопропилэтиламин и DBU (диазабициклоундецен). Ацилирующие реагенты включают галогенангидриды, например хлорангидриды, бромангидриды, и ангидриды. Температура реакции может быть от температуры, создаваемой охлаждением льдом, до температуры кипения применяемого растворителя.
Что касается молярного соотношения исходных соединений, то соединение (30) и ацилирующий реагент применяют в молярном соотношении в пределах от 0,5 до 2,0, предпочтительно от 1,0 до 1,1.
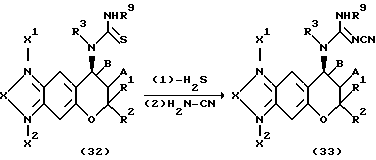
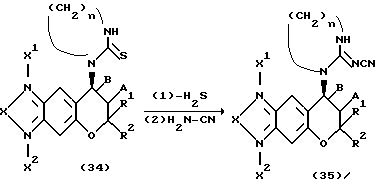
Соединения формулы (1) настоящего изобретения, у которых R4 представляет собой группу NC(N-CN)NHR9, можно получать по указанной ниже реакционной схеме, в которой соединение формулы (32) обрабатывают карбодиимидом в инертном растворителе, затем удаляют сероводород и после этого обрабатывают цианамидом, получая соединение формулы (33).Соединение формулы (34), у которого R3 и R4 вместе образуют остаток циклического тиокарбамида, можно также превратить в соединение формулы (35) тем же самым способом.
Оптически активные изомеры соединений формулы (1) настоящего изобретения можно получить например способами разделения (расщепления) рацемических модификаций на оптические антиподы (смотри выложенную заявку на патент Японии N 3-141286, патент США N 5097037, Европейский патент N 409165) и способами асимметричного синтеза (смотри выложенную заявку на патент Японии N 5-301878, Европейский патент N 535377).
Как указано выше, заявители настоящего изобретения обнаружили, что соединения формулы (1) обладают сильной активностью по усилению сокращения сердечных мышц и обладают также сильной активностью по снижению скорости сердечного сокращения. Поскольку соединения настоящего изобретения не обладают активностью по торможению сердечной функции, но скорее обладают активностью по усилению сокращения сердечных мышц, они могут быть активными по снижению скорости сердечного сокращения, даже при введении их в том же количестве, которое требуется для экспрессии кардиотонической активности, на основе активности соединении настоящего изобретения полагают, что они могут снижать количество кислорода, потребляемого сердечными мышцами, что приводит к снижению нагрузки на сократительную функцию сердечных мышц и способствует антистенокардической активности. Кроме того, установлено также, что эти соединения не обладают активностью по пролонгированию эффективного рефракторного периода, что способствует антиаритмической активности. Следовательно, предполагается, что соединения настоящего изобретения пригодны для лечения сердечно-сосудистых заболеваний вследствие потребления кислорода, потребления энергии или метаболизма, вызванного сердечной сократительной способностью, а также для лечения других сердечных нарушений, вследствие активности соединений по снижению скорости сердечного сокращения. Соединения настоящего изобретения например пригодны в качестве лекарственных средств для лечения сердечной недостаточности у млекопитающих, включая людей, а также лекарственных средств для лечения сердечно-сосудистых нарушений, вызванных сердечной недостаточностью, например в качестве лекарственных средств для лечения ишемической кардиопатии, лекарственных средств для лечения гипертензии, лекарственных средств для лечения задержки сердечной жидкости, лекарственных средств для лечения легочной гипертензии, лекарственных средств для лечения вальвулита, лекарственных средств для лечения застойных сердечных нарушений, лекарственных средств для лечения сердечно-мышечных нарушений, лекарственных средств для лечения отека легких, лекарственных средств для лечения стенокардии напряжения, лекарственных средств для лечения инфаркта миокарда, лекарственных средств для лечения аритмии и лекарственных средств для лечения фибрилляции предсердий.
Настоящее изобретение предлагает фармацевтические композиции, содержащие эффективное количество соединений формулы (1) и предназначенные для лечения этих заболеваний.
В качестве способов введения соединений настоящего изобретения можно указать парентеральное введение инъекциями (подкожными, внутривенными, внутримышечными или внутрибрюшинными инъекциями), мазями, суппозиториями или аэрозолями или пероральное введение в форме таблеток, капсул, гранул, пилюль, сиропов, жидкостей, эмульсий или суспензий.
Указанные выше фармакологические или ветеринарные композиции настоящего изобретения содержат указанные выше соединения настоящего изобретения в количестве от около 0,01 до 99,5 масс.%, предпочтительно от около 0,1 до 30 масс.%, считая на общую массу композиции.
В соединения настоящего изобретения или композиции, содержащие настоящие соединения, можно вводить другие фармакологически или ветеринарно активные соединения. Кроме того, композиции настоящего изобретения могут содержать несколько соединений настоящего изобретения.
Клиническая доза соединений настоящего изобретения изменяется в зависимости от возраста, массы тела, восприимчивости или симптомов и т.д. пациента. В общем, однако, эффективная суточная доза обычно составляет от около 0,003 до 1,5 г, предпочтительно от около 0,01 до 0,6 г для взрослого пациента. Однако, если необходимо, можно применять количество, выходящее из этого предела.
Соединения настоящего изобретения можно приготовлять в виде различных пригодных готовых препаративных форм (в зависимости от способа введения) в соответствии с обычными методиками, обычно применяемыми для получения фармацевтических готовых препаративных форм.
Таблетки, капсулы, гранулы или пилюли для перорального введения можно получить с применением наполнителей, например белого сахара, лактозы, глюкозы, крахмала или маннита; связующих, например гидроксипропилцеллюлозы, сиропов, аравийской камеди, желатина, сорбита, трагакантовой камеди, метилцеллюлозы или поливинилпирролидона; дезинтеграторов, например крахмала, карбоксиметилцеллюлозы или ее кальциевой соли, кристаллического порошка целлюлозы или полиэтиленгликоля; смазывающих веществ, например, талька, стеарината магния или кальция, диоксида кремния; и средств, придающих однородность композиции, например лаурината натрия, глицерина и т.д.
Инъекционные композиции, растворы (жидкости), эмульсии, суспензии, сиропы или аэрозоли можно получить с применением растворителя для активного компонента, например воды, этилового спирта, изопропилового спирта, пропиленгликоля, 1,3-бутиленгликоля или полиэтиленгликоля; поверхностно-активных веществ, например эфиров жирных кислот и ангидросорбита, полиоксиэтилированных эфиров жирных кислот и ангидросорбита, полиоксиэтилированных эфиров жирных кислот, полиоксиэтилированного гидрогенизированного касторового масла, лецитина; суспендирующих средств, таких как производные целлюлозы, например натриевой соли карбоксиметилцеллюлозы, или природные смолы, например трагакантовой камеди или аравийской камеди; или консервантов, например пара-гидроксибензойной кислоты, хлористого бензалкония, солей сорбиновой кислоты и т.д.
Мази, которые являются чрескожными препаратами, можно получать с применением например белого вазелина, жидкого парафина, высших спиртов, мази Макроголя, гидрофильной основы мази или гидрогеля и т.д.
Суппозитории можно получать с применением например, какао-масла, полиэтиленгликоля, ланолина, триглицеридов жирных кислот, кокосового масла, полисорбата и т.д.
Наилучший способ осуществления изобретения.
Далее настоящее изобретение будет объясняться при помощи примеров, но оно не ограничивается этими примерами.
Синтетические примеры.
Синтетические примеры получения соединений, которые пригодны в качестве лекарственных средств настоящего изобретения, приведены ниже.
Синтез оптически активного 7,8-дигидро-6,6-диметил-7,8-эпокси-6H-пирано [2,3-f]бензо- 2,1,3-оксадиазола:
В 300 мл раствора 40 г (198 ммоля) 6,6-диметил-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола в хлористом метилене добавляли 2,44 г (3,7 ммоля) ацетата (R, R) - [1,2-бис (3,5-дитрет-бутилсалицилидамино) циклогексан] марганца (III). (Получение этого ацетата описано в выложенной заявке на патент Японии N Hei 5-507645 и Европейском патенте N 521 099). В смесь добавляли 1,2 л водного раствора гипохлорита натрия (содержание активного хлора: 5%). Для установления pH реакционной смеси 11,3 в нее добавляли 0,5N водный раствор едкого натра. Полученный раствор перемешивали при комнатной температуре в течение 10 час. После прекращения перемешивания раствор выдерживали при комнатной температуре в течение ночи. Реакционный раствор экстрагировали хлороформом (300 мл х 1; 200 мл х 1; 50 мл х 1) и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Остаток хроматографировали на колонке с силикагелем (элюирование смесью бензола и этилацетата 5: 1) и снова на колонке с силикагелем (элюирование бензолом и смесью бензола и этилацетата 5:1) и полученные кристаллы перекристаллизовали из этанола (60 мл), получая 15,7 г целевого соединения (выход: 36%, оптическая чистота выше 99% ее-конформера).
Колонка: Chirallcell OJ (Daisel Chemical Industries, Ltd.).
Подвижная фаза: гексан и изопропанол в соотношении 4:1.
Детектирование: по УФ-спектру у полосы 254 нм.
Скорость потока: 1 мл/мин.
Температура колонки: 40oC.
Время удерживания: 9,2 мин.
Ссылочный пример 2
Синтез энантиомера соединения ссылочного примера 1
Энантиомер соединения ссылочного примера 1 синтезировали таким же образом, как в ссылочном примере 1, применяя ацетат (S, S)- [1,2-бис(3,5-ди-трет-бутилсалицилидамино)циклогексан] -марганца (III). Выход продукта был 20,8 г (48%).
Оптическая чистота: выше 99% ее-конформера время удерживания: 12,5 мин.
Пример синтеза 1
Синтез 3-оксида 7,8-дигидро-6,6-диметил-7-гидрокси-8-диэтиламино-6H-пирано[2,3-f] бензо-2,1,3-оксадиазола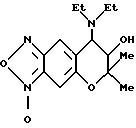
465 мг (1,50 ммоля) 3,4-дигидро-2,2-диметил-3-гидрокси-4- диэтиламино-6-амино-7-нитро-2H-бензо[b] пирана, 102 мг (2,56 ммоля) едкого натра, 32 мл этанола, 6 мл воды и 0,1 мл полиэтиленгликоля перемешивали при 40oC и в полученный раствор добавляли 2,59 г (2,10 ммоля) 6%-ного водного раствора NaOCl, и перемешивали в течение 15 мин. Реакционную жидкость выливали в воду и экстрагировали три раза этилацетатом. Слои этилацетата объединяли и затем промывали водой и насыщенным солевым раствором и сушили над безводным сульфатом натрия. Растворитель отгоняли и остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и гексана, 1:1 (объем/объем)), получая 189 мг целевого соединения (выход 41%). Часть таким образом полученного соединения растворяли в этаноле и в раствор добавляли раствор HCl-EtOH и абсолютный простой эфир для получения гидрохлорида этого соединения в виде желтых кристаллов. Соль имела следующие свойства:
т.пл.: 160-164oC (разложение)
ЯМР-спектр (CDCl) δ (млн. ч); 1,15 (6H), 1,26(3H), 1,52(3H), 2,64-3,14(5H), 3,56(1H), 3,85 (1H), 6,57(1H), 7,30(1H).
Пример синтеза 2
Синтез 3-оксида 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пиперидинил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
924 мг (2,88 ммоля) 3,4-дигидро-2,2 -диметил-3-гидрокси-4- (1-пиперидинил)-6-амино-7-нитро-2H-бензо[b] пирана, 0,7 мл 50%-ного раствора едкого натра, 4 мл дихлорэтана и 10 мг Bu4 N+Br- перемешивали при комнатной температуре и затем в смесь добавляли 4,97 г (4,03 ммоля) водного 6%-ного водного раствора NaOCl и реакцию проводили в течение 9 час при комнатной температуре и при перемешивании. Органический слой отделяли и водный слой экстрагировали два раза хлористым метиленом. Слои хлористого метилена объединяли, промывали водой и сушили над безводным сульфатом натрия. После отгонки растворителя остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и гексана, 1:3, объем/объем), получая 297 мг целевого соединения в виде масла (выход: 43%). Часть полученного таким образом соединения растворяли в этаноле и в раствор добавляли HCl-EtOH и сухой простой эфир для получения гидрохлорида целевого соединения в виде желтых кристаллов.
Т.пл.: 210-213oC.
Пример синтеза 3
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пиперидинил)- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола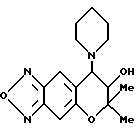
297 мг 3-оксида (0,93 ммоля) 7,8-дигидро-6,6-диметил-7-гидрокси- 8-(1-пиперидинил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола, полученного в синтетическом примере 2,6 мл этиленгликоля и 60 мг (0,93 ммоля) NaN3 нагревали при 140oC в течение 1,2 часа. После охлаждения реакционную жидкость выливали в воду и экстрагировали три раза хлороформом. Слои хлороформа объединяли и сушили над безводным сульфатом натрия. После удаления растворителя отгонкой остаток хроматографировали на колонке (элюент: смесь этилацетата и гексана, 1:3, объем/объем), получая 84 мг целевого соединения (выход 30%). Часть таким образом полученного соединения растворяли в смеси этанола и диэтилового эфира и в раствор добавляли HCl-EtOH для получения гидрохлорида целевого соединения в виде бледно-желтых кристаллов.
Т.пл. 202-205oC.
Пример синтеза 4
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пирролидинил)- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола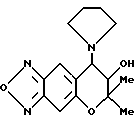
150 мг (0,687 ммоля) 7,8-дигидро-6,6-диметил-7,8-эпокси-6H- пирано[2,3-f] бензо-2,1,3-оксадиазола, 63 мкл (0,756 ммоля) пирролидина и 2 мл этанола кипятили с обратным холодильником при перемешивании в течение 31 час. Растворитель удаляли отгонкой и остаток очищали распределительной тонкослойной хроматографией (элюент: смесь этилацетата и гексана, 1:1, объем/объем), получая 120 мг целевого соединения (выход 60%). Часть таким образом полученного соединения растворяли в сухом простом эфире и в раствор добавляли HCl-EtOH для получения гидрохлорида этого соединения в виде бледно-желтых кристаллов.
Т.пл. 208-209oC.
Пример синтеза 5.
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-метил амино-6-пирано[2,3-f] бензо-2,1,3-оксадиазола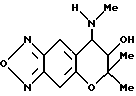
300 мг (1,37 ммоля) 7,8-дигидро-6,6-диметил-7,8-эпокси-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола, 0,53 г водного 40%-ного раствора метиламина и 15 мл этанола перемешивали при 60oC 3 дня, применяя трубку для высокого давления. После окончания реакции растворитель отгоняли и остаток чистили распределительной тонкослойной хроматографией (элюент: смесь этилацетата и метанола, 10: 1), получая 263 мг целевого соединения (выход 77%). Часть полученного таким образом соединения растворяли в сухом простом эфире и добавляли в раствор HCl-EtOH для получения гидрохлорида целевого соединения в виде бесцветных кристаллов.
Т.пл.: 244,5-260oC
Пример синтеза 6
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(4-фтор бензил)амино-6H-пирано[2,3-f] бензо-2,1,3-оксадиазола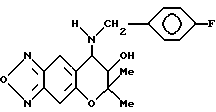
150 мг (0,687 ммоля) 7,8-дигидро-6,6-диметил-7,8-эпокси-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола, 86 мкл (0,756 ммоля)4-фторбензиламина и 2 мл этанола кипятили с обратным холодильником при перемешивании в течение 20 час. Растворитель удаляли отгонкой и остаток очищали распределительной тонкослойной хроматографией (элюент: смесь этилацетата и гексана, 1:2), получая 204 мг целевого соединения в виде масла (выход: 86%). Часть полученного таким образом соединения растворяли в сухом простом эфире и добавили HCl-EtOH для получения гидрохлорида целевого соединения в виде бесцветных кристаллов.
Т.пл. 207-210oC.
Пример синтеза 7
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-бензиламино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола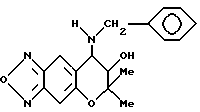
Таким же образом, как описано в синтетическом примере 6, с применением бензиламина получали целевое соединение.
ЯМР-спектр (60 МГц, CDCl3, δ/ млн. д. ): 7,77(1H), 7,37-6.92(5H), 6,81(1H), 3,9-3,8(4H), 2,73(2H), 1,51(3H), 1,25(3H).
Пример синтеза 8
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(N-бензил-N -метил)амино-6H-пирано[2,3-f] бензо-2,1,3-оксадиазола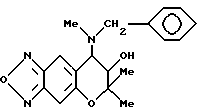
Таким же образом, как описано в синтетическом примере 6, с применением N-метилбензиламина получали целевое соединение. Гидрохлорид целевого соединения имел температуру плавления 148-150oC.
Пример синтеза 9
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-циклогексилмино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола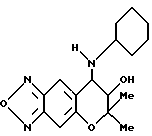
Таким же образом, как описано в синтетическом примере 6, с применением циклогексиламина получали целевое соединение. Гидрохлорид целевого соединения имел температуру плавления 208-210oC.
Пример синтеза 10
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(4-метил -1-пиперазинил)-6H-пирано [2,3-f] бензо-2,1, 3-оксадиазола
Таким же образом, как описано в синтетическом примере 6, с применением N-метилпиперазина получали целевое соединение в виде желтых кристаллов (выход 75%).
Т.пл.: 225-226oC
Масс-спектр: 70(100%), 246(56%), 318 (M+, 13%).
Пример синтеза 11
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пиперазинил) -6H-пирано [2,3-f] бензо-2,1,3-оксадиазола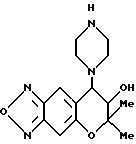
Таким же образом, как описано в синтетическом примере 6, с применением пиперазина получали целевое соединение в виде желтых кристаллов (выход 72%).
Т.пл. 245-246oC
Масс-спектр: 56(67%), 232(100%), 304(M+, 8%).
Пример синтеза 12
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(4-фенил-1- пиперазинил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола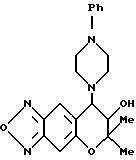
Таким же образом, как описано в синтетическом примере 6, с применением N-фенилпиперазина получали целевое соединение (выход 75%).
Масс-спектр: 132(100%), 308(36%), 380(M+, 32%)
Гидрохлорид целевого соединения получали в виде бесцветных кристаллов
Т.пл. 198-201oC
Пример синтеза 13
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(4-фенил-1- пиперидинил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола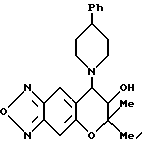
Таким же образом, как описано в синтетическом примере 6, с применением 4-фенилпиперазина получали целевое соединение (выход 87%)
Масс-спектр: 186(21%), 307(100%), 379(M+, 4%).
Гидрохлорид целевого соединения получали в виде бесцветных кристаллов
Т.пл.: 195-197oC
Пример синтеза 14
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1,2,3,4- тетрагидроизохинолин-2-ил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола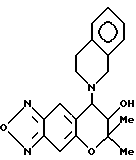
Таким же образом, как описано в примере 6, с применением 1,2,3,4-тетрагидроизохинолина получали целевое соединение (выход 80%).
Масс-спектр: 262(32%), 279(100%), 351(M+, 4%).
Гидрохлорид целевого соединения получали в виде бесцветных кристаллов
Т.пл. 188,5-190oC
Пример синтеза 15
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(4-морфолинил)- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола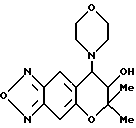
Таким же образом, как описано в примере 6, с применением морфолина получали целевое соединение (выход 11%).
Т. пл.: 185-186,5oC.
Пример синтеза 16
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(3-метокпропиламино) -6H-пирано [2,3-f] бензо-2,1,3-оксадиазола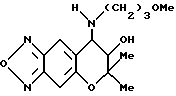
Таким же образом, как описано в синтетическом примере 6, с применением 3-метоксипропиламина получали целевое соединение (выход 60%).
Масс-спектр: 177(100%), 235(100%), 289(M+-18,1%).
Т.пл. 175,5-178oC
Пример синтеза 17
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(3- этоксикарбонилпропиламино)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола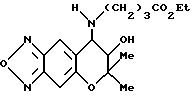
Таким же образом, как описано в синтетическом примере 6, с применением этил-4-аминобутирата получали целевое соединение (выход 38%).
Гидрохлорид целевого соединения получали в виде бесцветных кристаллов.
Т.пл.: 189-191oC
Пример синтеза 18
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- этоксикарбонилметиламино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола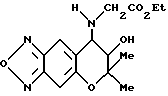
Таким же образом, как описано в синтетическом примере 6, с применением этилового эфира глицина получали целевое соединение (выход 5%).
Гидрохлорид целевого соединения получали в виде оранжевого масла.
Пример синтеза 19
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(3-хлорпропиламино)- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола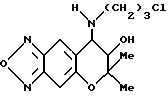
Таким же образом, как описано в синтетическом примере 6, с применением 3-хлорпропиламина получали целевое соединение (выход 20%).
Гидрохлорид целевого соединения получали в виде бесцветных кристаллов.
Т.пл.: 216-220oC
Синтетический пример 20
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (2-гидроксиэтиламино)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола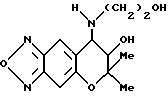
Таким же образом, как описано в синтетическом примере 6, с применением этаноламина получали целевое соединение (выход 89%).
Гидрохлорид целевого соединения получали в виде бесцветных кристаллов.
Т.пл. 200-204oC.
Пример синтеза 21
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
0,82 г (3,8 ммоля) 7,8-дигидро-6,6-диметил-7,8-эпокси-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола растворяли в 25 мл 16,7% H3-EtOH и реакцию проводили при 60oC в течение 48 час в стеклянной трубке для высокого давления. Растворитель удаляли отгонкой и остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и метанола, 5:1), получая 0,77 г целевого соединения в виде коричневого твердого вещества (выход 87%). Часть продукта перекристаллизовали из этанола, получая чистые бесцветные кристаллы целевого соединения.
Т.пл.: 223-225oC.
ЯМР-спектр (CDCl3 + DMSO-d6) δ/ (млн.д.): 1,26(3H), 1,49(3H), 2,80-3,30(5H), 3,33(1H), 3,78(1H), 6,82(1H), 7,98(1H).
Масс-спектр: 133(50%), 163(100%), 235(M+, 3%).
Пример синтеза 22
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-метилуреидо- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
200 мг (0,850 ммоля)7,8-дигидро-6,6-диметил-7-гидрокси-8-амино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола и 20 мл дихлорметана перемешивали при комнатной температуре и в раствор добавляли 55 мкл (0,935 ммоля) метилизоцианата и смесь перемешивали 23 часа. Осажденные кристаллы отделяли фильтрованием, получая 227 мг целевого соединения в виде бесцветных кристаллов (выход 92%).
Т.пл. 213-215oC
Масс-спектр: 44,202(30%), 274(M+-H2O, 6%).
Пример синтеза 23
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-метилтиоуреидо- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
200 мг (0,850 ммоля) 7,8-дигидро-6,6-диметил-7-гидрокси-8-амино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола и 20 мл дихлорметана перемешивали при комнатной температуре и в раствор добавляли 68 мг (0,935 ммоля) метилизотиоцианата и смесь перемешивали 23 часа. Осажденные кристаллы отделяли фильтрованием, получая 122 мг целевого соединения в виде бесцветных кристаллов (выход 47%).
Т.пл. 213-215oC.
Масс-спектр: 91(62%), 202(67%), 290,308(M+, 27%).
Пример синтеза 24
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-фенилуреидо- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола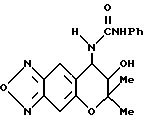
200 мг (0,850 ммоля)7,8-дигидро-6,6-диметил-7- гидрокси-8-амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола и 20 мл дихлорметана перемешивали при комнатной температуре и в раствор добавляли 102 мкл (0,935 ммоля) фенилизоцианата и смесь перемешивали 4 часа. Осажденные кристаллы отделяли фильтрованием, получая 203 мг целевого соединения в виде бесцветных кристаллов (выход 67%).
Т.пл. 215-217oC
Масс-спектр: 93,163(56%), 321(20%), 354(M+, 10%).
Пример синтеза 25
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-трихлорацетилуреидо- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола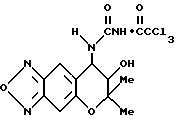
200 мг (0,850 ммоля) 7,8-дигидро-6,6-диметил-7-гидрокси-8-амино- 6H-пирано [2,3-f] бензо-2,1, 3-оксадиазола и 20 мл дихлорметана перемешивали при комнатной температуре и в раствор добавляли 100 мкл (0,935 ммоля) трихлорацетилизоцианата и смесь перемешивали 5 час. Осажденные кристаллы отделяли фильтрованием, получая 90 мг целевого соединения в виде бесцветных кристаллов (выход 25%).
Т.пл. 248-250oC
Масс-спектр: 44,163(43%), 422(M+, 2%).
Пример синтеза 26
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (3-хлорпропилуреидо)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола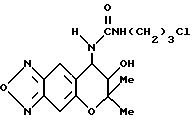
400 мг (1,70 ммоля) 7,8-дигидро-6,6-диметил-7-гидрокси-8-амино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола и 40 мл дихлорметана перемешивали при комнатной температуре и в раствор добавляли 192 мкл (1,87 ммоля) 3-хлорпропилизоцианата и смесь перемешивали 5 час. Осажденные кристаллы отделяли фильтрованием, получая 250 мг целевого соединения в виде бледно-желтых кристаллов (выход 41%).
Т.пл. 83-85oC
Масс-спектр: 41(53%), 163,318(93%), 354(M+, 5%).
Пример синтеза 27
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(2-хлорэтилуреидо)- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола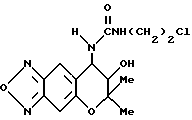
400 мг (1,70 ммоля) 7,8-дигидро-6,6-диметил-7-гидрокси-8-амино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола и 40 мл дихлорметана перемешивали при комнатной температуре и в раствор добавляли 200 мкл (1,87 ммоля)2-хлорэтилизоцианата и смесь перемешивали 6 час. Осажденные кристаллы отделяли фильтрованием, получая 480 мг целевого соединения в виде бесцветных кристаллов (выход 83%).
Т.пл. 178-180oC.
Масс-спектр: 87(57%), 163,304(78%), 340(M+, 8%)
Пример синтеза 28
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-изопропилуреидо-6H- пирано [2,3-f] бензо-2,1,3-оксадиазола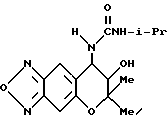
200 мг (0,850 ммоля)7,8-дигидро-6,6-диметил-7-гидрокси- 8-амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола и 20 мл дихлорметана перемешивали при комнатной температуре и в раствор добавляли 92 мкл (0,935 ммоля) изопропилизоцианата и смесь перемешивали 6 час. Осажденные кристаллы отделяли фильтрованием, получая 120 мг целевого соединения в виде бесцветных кристаллов (выход 44%).
Т.пл. 201-203oC
Масс-спектр: 43(40%), 202,302(20%), 320(M+, 12%).
Пример синтеза 29
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-этоксикарбониламино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола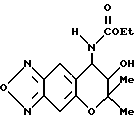
200 мг (0,850 ммоля)7,8-дигидро-6,6-диметил-7-гидрокси-8-амино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола, 166 мкл (1,19 ммоля) триэтиламина и 20 мг дихлорметана перемешивали при комнатной температуре, в смесь добавляли 114 мкл (1,19 ммоля) этилхлорформиата и перемешивали 21 час. Реакционную жидкость промывали три раза водой и затем сушили над безводным сульфатом натрия. Растворитель удалили отгонкой и остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и метанола, 20:1, объем/объем), получая 227 мг целевого соединения в виде желтого масла (выход 87%).
Масс-спектр 133(48%), 235,307 (M+, 25%).
Пример синтеза 30
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (2-хлорэтоксикарбониламино)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола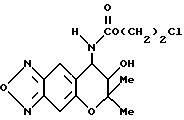
400 мг (1,70 ммоля) 7,8-дигидро-6,6-диметил-7-гидрокси-8-амино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола, 260 мкл (1,87 ммоля) триэтиламина и 40 мг дихлорметана перемешивали при комнатной температуре, в смесь добавляли 193 мкл (1,87 ммоля)2- хлорэтилхлорформиата и перемешивали 21 час. Реакционную жидкость промывали три раза водой и затем сушили над безводным сульфатом натрия. Растворитель удалили отгонкой и остаток перекристаллизовали из хлороформа, получая 507 мг целевого соединения в виде бледно-желтых кристаллов (выход 87%).
Т.пл. 164-166oC
Масс-спектр: 133(48%), 235,307 (M+, 25%).
Пример синтеза 31
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (2-оксо-3-оксазолин-1-ил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола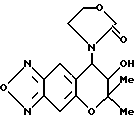
400 мг (1,17 ммоля) соединения, полученного в синтетическом примере 30, 3,24 г (23,4 ммоля) карбоната калия, 388 мг (2,34 ммоля) йодистого калия и 50 мл абсолютного ацетона нагревали с обратным холодильником при комнатной температуре в течение 26 час. После охлаждения смеси до комнатной температуры нерастворимую часть отделяли фильтрованием, добавляли в полученный фильтрат этилацетат и промывали три раза водой. Полученное таким образом соединение сушили над безводным сульфатом магния. Растворитель удаляли отгонкой и остаток очищали колоночной хроматографией на силикагеле (элюент: смесь этилацетата и метанола, 10:1, объем/объем), получая 339 мг целевого соединения в виде коричневого масла (выход 94%). Часть продукта перекристаллизовали из этилацетата, получая желтые кристаллы, имеющие следующие физические свойства:
Т.пл.: 177,5-180oC
Масс-спектр: 43(25%), 272,287(65%), 305(M+, 8%).
Пример синтеза 32
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(2-оксо-3- мидазолин-1-ил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола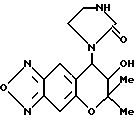
Соединение, полученное в синтетическом примере 27, обрабатывали таким же образом, как в синтетическом примере 31, для получения целевого соединения в виде бесцветных кристаллов (выход 34%).
Т.пл. 251-252,5oC.
Пример синтеза 33
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-циклогексилуреидо- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
Целевое соединение в виде бесцветных кристаллов получали таким же образом, как в синтетическом примере 22, применяя циклогексилизоцианат (выход 28%).
Т.пл. 203-206oC
Пример синтеза 34
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (трет-бутилуреидо)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
Целевое соединение в виде бесцветных кристаллов получали таким же образом, как в синтетическом примере 22, применяя трет-бутилизоцианат (выход 52%).
Т.пл. 203-205oC.
Пример синтеза 35
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- метоксикарбониламино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола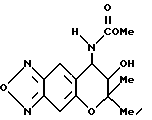
Целевое соединение в виде желтого масла получали таким же образом, как в синтетическом примере 29, применяя метилхлорформиат (выход 19%).
Пример синтеза 36
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(трет- бутилтиоуреидо)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола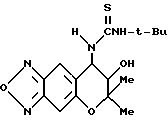
Целевое соединение получали таким же образом, как в синтетическом примере 23, применяя трет-бутилизотиоцианат (выход 57%).
Пример синтеза 37
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (2-оксогексагидропиримидин-1-ил)-6H-пирано [2,3-f] бензо-2,1,3- оксадиазола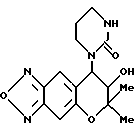
Соединение, полученное в синтетическом примере 26, обрабатывали таким же образом, как в синтетическом примере 31, получая целевое соединение в виде бесцветных кристаллов (выход 39%).
Т.пл. 233-234oC.
Пример синтеза 38
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (3-хлорпропоксикарбониламино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола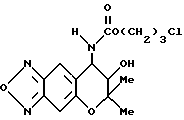
Целевое соединение в виде желтого масла получали таким же образом, как в синтетическом примере 30, применяя 3-хлорпропилхлорформиат (выход 100%).
Пример синтеза 39
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (2-оксотетрагидрооксазин-3-ил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
Соединение, полученное в синтетическом примере 38, обрабатывали таким же образом, как в синтетическом примере 31, получая целевое соединение в виде бесцветных кристаллов (выход 34%).
Т.пл. 220-234oC.
Пример синтеза 40
Синтез 3-оксида-7,8-дигидро-6,6-диметил-7-ацетокси-8- диэтиламино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола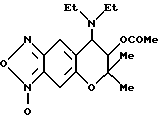
1 мл пиридина и 130 мг (1,27 ммоля) уксусного ангидрида добавляли в 103 мг (0,34 ммоля) соединения, полученного в синтетическом примере 1, и смесь перемешивали при 80-90oC в течение 2 час. После охлаждения растворитель удаляли отгонкой при пониженном давлении и остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и н-гексана, 1:3, объем/объем, Rf= 0,3), получая 90,4 мг целевого соединения в виде желтого твердого вещества (выход 77%).
Пример синтеза 41
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-диэтил амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола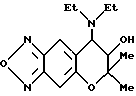
310 мг (1,01 ммоля) соединения, полученного в синтетическом примере 1, растворяли в 3 г бензола, в раствор добавляли 0,20 г (1,2 ммоля) триэтилфосфита и смесь нагревали при 60oC в течение 1 часа. После охлаждения до комнатной температуры полученное таким образом соединение перемешивали в течение ночи. Растворитель удаляли отгонкой при пониженном давлении и остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и н-гексана, 1: 3, объем/объем, Rf=0,3), получая 267 мг целевого соединения в виде желтого твердого вещества (выход 91%).
Пример синтеза 42
Синтез 7,8-дигидро-6,6-диметил-7-ацетокси-8-диэтиламино-6H- пирано [2,3-f] бензо-2,1,3-оксадиазола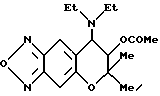
2 мл пиридина и 0,29 г (2,8 ммоля) уксусного ангидрида добавляли в 220 мг (0,75 ммоля) соединения, полученного в синтетическом примере 41, и смесь нагревали при 80-90oC в течение 2 час. После охлаждения растворитель удаляли отгонкой при пониженном давлении и остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и н-гексана, 1:3, объем/объем, Rf= 0,3), получая 209,1 мг целевого соединения в виде бледно-желтого твердого вещества (выход 83%).
Пример синтеза 43
Синтез 7,8-дигидро-6,6-диметил-7-ацетокси-8-(1-пиперидинил)-6H- пирано [2,3-f] бензо-2,1,3-оксадиазола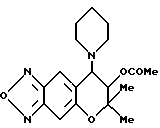
Соединение, полученное в синтетическом примере 3, обрабатывали таким же образом, как в синтетическом примере 42, получая целевое соединение в виде желтых кристаллов (выход 100%).
Т.пл. 158-160oC.
Синтетический пример 44
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (4-диэтоксибутиламино)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола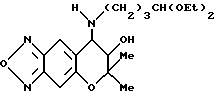
Целевое соединение в виде коричневого масла получали таким же образом, как в синтетическом примере 6, применяя диэтилацеталь-4-аминомасляного альдегида (выход 93%).
Пример синтеза 45
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (2-гидроксипирролидин-1-ил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола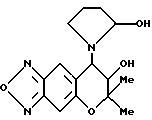
1 мл 2,5N соляной кислоты и 2 мл 1,4-диоксана добавляли в 316 мг (0,832 ммоля) соединения, полученного в синтетическом примере 44, и реакцию проводили при комнатной температуре в течение 3 час. В реакционную смесь добавляли водный раствор карбоната натрия, чтобы гидратировать продукт и образованный гидрат затем экстрагировали диэтиловым эфиром. Раствор в эфире сушили над безводным сульфатом натрия. Растворитель удаляли отгонкой при пониженном давлении. Остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и н-гексана, 1:1, объем/объем, Rf=0.5), получая целевое соединение в виде желтого масла (выход 40%).
Пример синтеза 46
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (3-карбоксипропиламино)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола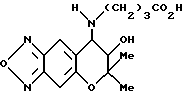
Раствор 18,4 мг (0,460 ммоля) едкого натра, 1 мл воды и 4 мл этанола добавляли в 80,6 мг (0,209 ммоля) гидрохлорида, полученного в синтетическом примере 17, и реакцию проводили 3 часа при комнатной температуре. Реакционную смесь подкисляли добавлением в нее 1N соляной кислоты и затем экстрагировали диэтиловым эфиром. Экстракт сушили над безводным сульфатом натрия. Растворитель удаляли отгонкой при пониженном давлении, получая целевое соединение в виде бесцветного масла.
Пример синтеза 47
Синтез 7,8-дигидро-6,6-диметил-7-ацетокси-8-амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола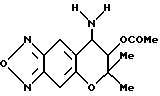
240 мл (1,10 ммоля)-7,8-дигидро-6,6-диметил-7,8-эпокси-6H- пирано [2,3-f] бензо-2,1,3-оксадиазола растворяли в 5 мл дихлорметана и в раствор добавляли 0,5 мл комплекса BF3-диэтиловый эфир. Реакцию проводили 30 мин при комнатной температуре. Затем в реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия и реакционную жидкость экстрагировали хлороформом. После высушивания экстракта над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и н-гексана, 1:1, объем/объем, Rf= 0,3), получая 152 мг целевого соединения в виде желтого масла (выход 50%).
Масс-спектр: 188 (100%), 277 (M+, 4%).
Пример синтеза 48
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пиперидинил)-6H- пирано [2,3-f] бензо-2,1,3-оксадиазола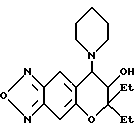
87,7 г (0,36 ммоля) 7,8-дигидро-6,6-диэтил-7,8-эпокси-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола, 61 мг (0,72 ммоля) пиперидина и 2 мл этанола кипятили с обратным холодильником в течение 20 час при перемешивании. После отгонки растворителя остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и н-гексана, 1:1, объем/объем, Rf=0,3), получая 30 мг целевого соединения в виде желтого масла (выход 25%).
Пример синтеза 49
Синтез 7,8-дигидро-6,6-диэтил-7-гидрокси-8-амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола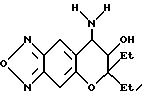
Целевое соединение в виде бесцветных кристаллов получали таким же образом, как в синтетическом примере 21, применяя 7,8-дигидро-6,6-диэтил-7,8-эпокси-6H-пирано [2,3-f] бензо-2,1,3-оксадиазол (выход 44%).
Т.пл. 122-124oC.
Пример синтеза 50
Синтез 7,8-дигидро-6,6-диэтил-7-гидрокси-8-метил-уреидо-6H- пирано [2,3-f] бензо-2,1,3-оксадиазола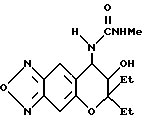
Соединение, полученное в синтетическом примере 49, обрабатывали таким же образом, как в синтетическом примере 22, получая целевое соединение в виде коричневого масла (выход 89%).
Пример синтеза 51
Синтез 7,8-дигидро-6,6-диэтил-7-ацетокси-8-амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола
Целевое соединение в виде бледно-желтых кристаллов получали таким же образом, как в синтетическом примере 47, применяя 7,8-дигидро-6,6-диэтил-7,8-эпокси-6H-пирано [2,3-f] бензо-2,1,3-оксадиазол (выход 62%).
Т.пл. 92-95oC.
Пример синтеза 52
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (2-циано-3-трет-бутил-1-гуанидино)-6H-пирано [2,3-f] бензо-2,1,3- оксадиазола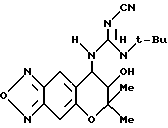
Раствор, содержащий 100 мг (0,29 ммоля) соединения, полученного в синтетическом примере 36, 97 мг (0,37 ммоля) трифенилфосфина, 40 мкл четыреххлористого углерода, 40 мкл (0,29 ммоля) триэтиламина и 1 мл дихлорметана нагревали с обратным холодильником в течение 4 час. После удаления растворителя отгонкой при пониженном давлении остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и н-гексана, 1:2, объем/объем, Rf=0,1), получая 77 мг бледно-желтых кристаллов.
Раствор, содержащий 122 мг (0,39 ммоля) полученного таким образом соединения, 21 мг (0,50 ммоля) цианамида, 2 мл тетрагидрофурана и 20 мг диизопропилэтиламина, перемешивали 14 час при комнатной температуре. Осажденные кристаллы отделяли фильтрованием, получая 112 мг целевого соединения в виде желтых кристаллов (выход 80%).
Т.пл. 146-148oC.
Пример синтеза 53
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пиперидинил) -6H-пирано [2,3-f] бензо-1,2,3-триазола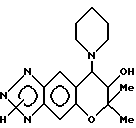
0,14 г (0,44 ммоля)3,4-дигидро-2,2-диметил-3-гидрокси-4- (1-пиперидинил)-6амино-7-нитро-2H-бензо[b] пирана растворяли в 23,7 г этанола и через раствор продували газообразный водород в присутствии применяемого в качестве катализатора 0,10 г 5% палладия на угле. Продувание проводили в течение 3 час при комнатной температуре и атмосферном давлении при перемешивании. Реакционную жидкость фильтровали с отсасыванием для удаления из нее катализатора и из фильтрата затем отгоняли растворитель, получая 0,12 г (выход 95%) 3,4-дигидро-2,2-диметил-3-гидрокси-4-(1-пиперидинил)-6,7- диамино-2H-бензо[b] пирана в виде темно-красного масла. Поскольку это соединение нестабильно, его применяли сразу для последующего диазотирования.
Все количество диаминосоединения, полученного в предыдущей стадии, растворяли в смеси 0,13 г уксусной кислоты и 0,23 г воды и в раствор при комнатной температуре добавляли раствор, полученный растворением 35 мг (0,51 ммоля) нитрита натрия в 0,15 г воды. После обнаружения выделения тепла реакционную смесь нагревали на водяной бане при 80o в течение 1 мин. В реакционную смесь добавляли 20 мл воды, 0,13 г едкого натра и 4,0 г хлористого натрия и затем экстрагировали ее три раза 40 мл этилацетата в каждом случае. Слои этилацетата объединяли, сушили над безводным сульфатом натрия и фильтровали. После удаления растворителя отгонкой получали 0,10 г желтовато-красного порошка 90 мг порошка очищали хроматографией на колонке с силикагелем (элюент: смесь этилацетата и этанола, 5:1), получая 80 мг целевого соединения в виде бледного желтовато-коричневого порошка. Общий выход после двух стадий был 72%.
Масс-спектр: 284(M+-H2O, 18%), 230(M+-72, 100%), 84(5%).
Пример синтеза 54
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пирролидинил) -6-пирано [2,3-f] бензо-1,2,3-триазола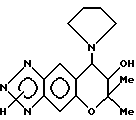
0,20 г (0,65 ммоля)3,4-дигидро-2,2-диметил-3-гидрокси-4- (1-пирроилинил)-6-амино-7-нитро-2H-бензо[b] пирана растворяли в 34,9 г этанола и через раствор продували газообразный водород в присутствии применяемого в качестве катализатора 0,15 г 5% палладия на угле. Продувание проводили в течение 3 час при комнатной температуре и атмосферном давлении при перемешивании. Реакционную жидкость фильтровали с отсасыванием для удаления из нее катализатора и из фильтрата затем отгоняли растворитель, получая 170 мг (выход 94%) 3,4-дигидро-2,2-диметил-3-гидрокси-4-(1-пирролидинил)-6,7- диамино-2H-бензо[b]пирана в виде темно-красного масла.
Все количество диаминосоединения, полученного в предыдущей стадии, растворяли в смеси 0,19 г уксусной кислоты и 0,34 г воды и в раствор при комнатной температуре добавляли раствор, полученный растворением 52 мг (0,75 ммоля) нитрита натрия в 0,22 г воды. После обнаружения выделения тепла реакционную смесь нагревали на водяной бане при 80o в течение 3 мин. Соединение затем обрабатывали таким же образом, как в синтетическом примере 53, получая 160 мг целевого соединения в виде желтовато-коричневого порошка. Общий выход после двух стадий был 85%.
Масс-спектр: 288 (M+, 3%), 270(M+-H2O, 3%), 216(M+-72,88%), 188(M+-100, 100%), 70(22%).
Пример синтеза 55
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-диэтиламино-6-пирано [2,3-f] бензо-1,2,3-триазола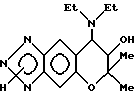
0,20 г (0,65 ммоля)3,4-дигидро-2,2-диметил-З-гидрокси- 4-диэтиламино-6-амино-7-нитро-2H-бензо[b]пирана растворяли в 34,9 г этанола и через раствор продували газообразный водород в присутствии применяемого в качестве катализатора 0,15 г 5% палладия на угле. Продувание проводили в течение 2,5 часа при комнатной температуре и атмосферном давлении при перемешивании. Реакционную жидкость фильтровали с отсасыванием для удаления из нее катализатора и из фильтрата затем отгоняли растворитель, получая 0,15 г (выход 83%)3,4- дигидро-2,2-диметил-3-гидрокси-4-диэтиламино-6,7-диамино-2H- бензо[b]пирана в виде темно-коричневого масла.
Все количество диаминосоединения, полученного в предыдущей стадии, растворяли в смеси 0,19 г уксусной кислоты и 0,34 г воды и в раствор при комнатной температуре добавляли раствор, полученный растворением 52 мг (0,75 ммоля) нитрита натрия в 0,22 г воды. После обнаружения выделения тепла реакционную смесь нагревали на водяной бане при 80o в течение 3 мин. Соединение затем обрабатывали таким же образом, как в синтетическом примере 53, получая 70 мг целевого соединения в виде бледно-коричневого порошка. Общий выход после двух стадий был 37%.
Масс-спектр (FAB): 291 [(M+H)+]
Пример синтеза 56
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пиперидинил)-6- пирано [2,3-f] бензо-2,1,3-тиадиазола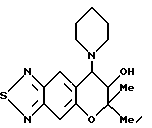
0,28 г (0,87 ммоля)3,4-дигидро-2,2-диметил-3-гидрокси-4- (1-пиперидинил)-6-амино-7-нитро-2H-бензо[b] пирана растворяли в 44,8 г этанола и через раствор продували газообразный водород в присутствии применяемого в качестве катализатора 0,20 г 5% палладия на угле. Продувание проводили в течение 3 час при комнатной температуре и атмосферном давлении при перемешивании. Реакционную жидкость фильтровали с отсасыванием для удаления из нее катализатора и из фильтрата затем отгоняли растворитель, получая 0,24 г (выход 95%) 3,4-дигидро-2,2-диметил-3-гидрокси-4-(1-пиперидинил)-6,7- диамино-2H-бензо[b]пирана в виде темно-красного масла. В полученное масло добавляли 0,12 г (0,86 ммоля) тиониланилина и 4 г бензола и смесь нагревали с обратным холодильником в течение 2,5 час. После отгонки растворителя при пониженном давлении остаток хроматографировали на колонке с силикагелем (элюент: смесь этилацетата и этанола, 1:3), получая 60 мг (выход 23%) целевого соединения в виде желтого твердого вещества.
Масс-спектр: 84(85%), 247(100%), 319 (M+, 2%).
Синтетический пример 57
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(1-пирролидинил)- 6H-пирано [2,3-f] бензо-2,1,3-тиадиазола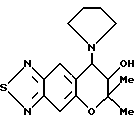
3,4-дигидро-2,2-диметил-3-гидрокси-4-(1-пирролидинил)-6-амино- 7-нитро-2H-бензо[b] пиран обрабатывали таким же образом, как в синтетическом примере 56, получая целевое соединение в виде коричневых кристаллов (выход 25%).
Пример синтеза 58
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-диэтиламино-6H-пирано [2,3-f] бензо-2,1,3-тиадиазола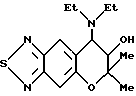
3,4-дигидро-2,2-диметил-3-гидрокси-4-диэтиламино-6-амино-7- нитро-2H-бензо[b]пиран обрабатывали таким же образом, как в синтетическом примере 56, получая целевое соединение в виде коричневого масла (выход 17%).
Пример синтеза 59
Синтез (-)-7,8-дигидро-6,6-диметил-7-гидрокси-8- (1-пирролидинил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола (оптически активного (-)-изомера соединения синтетического примера 4)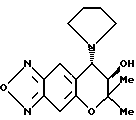
14,7 г (206,2 ммоля) пирролидина добавляли в 60 мл раствора 15,0 г (68,7 ммоля) соединения ссылочного примера 1 в этаноле и смесь нагревали с обратным холодильником 2 часа. После удаления растворителя отгонкой при пониженном давлении в остаток добавляли 100 мл воды и смесь экстрагировали хлороформом (100 мл х 1,30 мл х 2). После высушивания раствора в хлороформе над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток хроматографировали на колонке с силикагелем (элюент: смесь бензола и этилацетата, 5:1 - 4:1) и полученные кристаллы перекристаллизовали из смеси бензола и гексана (1:2), получая 13,8 г целевого соединения в виде желтых кристаллов (выход 69%).
240 мл смеси соляная кислота : метанол (10%) добавляли в 240 мл раствора 13 г (44,9 ммоля) целевого соединения в метаноле и смесь перемешивали 3 часа при комнатной температуре. Затем растворитель отгоняли при пониженном давлении. Остаток кристаллизовали в 250 мл 2-пропанола, получая 11,5 г гидрохлорида целевого соединения в виде бесцветных кристаллов (выход: 78%).
Т.пл.: выше 200oC (разложение)
Пример синтеза 60
Синтез (+)-7,8-дигидро-6,6-диметил-7-гидрокси-8- (1-пирролидинил)-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола (оптически активного (+)-изомера соединения синтетического примера 4)
Таким же образом, как в синтетическом примере 59, получали 15,0 г (выход 58%) целевого соединения в виде желтых кристаллов. Таким же образом получали также 12,9 г (выход 82%) гидрохлорида целевого соединения в виде бесцветных кристаллов.
Т.пл.: выше 200oC (разложение).
Пример синтеза 61
Синтез 7,8-дигидро-6,6--диметил-7-гидрокси-8-ди-н-пропиламино- 6H-пирано [2,3-f] бензо-2,1,3-оксадиазола: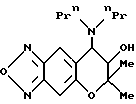
219 мг (1 ммоль) 7,8-дигидро-6,6-диметил-7,8-эпокси-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола, 304 мг (3 ммоль) ди-н-пропиламина и 5 мл этанола перемешивают при 150oC в течение 4 часов с использованием трубы высокого давления. После завершения реакции отгоняют растворитель и остаток подвергают хроматографии на колонке с силикагелем (элемент: гексанэтилацетат = 9: 1) с получением 112 мг целевого соединения (выход составляет 35%). Часть полученного таким образом соединения растворяют в сухом этаноле и добавляют HCl-этанол, после чего образуется гидрохлоридная соль соединения в виде бесцветного порошка.
Т.пл.: от 178 до 183oC.
Пример синтеза 62:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-н-пропиламино-6H- пирано [2,3-f] бензо-2,1,3-оксадиазола: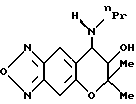
Целевое соединение получают с использованием н-пропиламина-по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 248 до 250oC (выход: 56%).
Пример синтеза 63:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(3'-этоксипропил) амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола: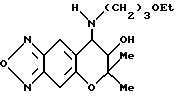
Целевое соединение получают с использованием 3-этоксипропиламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 175 до 178oC (выход: 56%).
Пример синтеза 64:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(N-этил-N-н-пропил) амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола: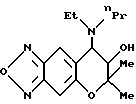
Целевое соединение получают с использованием N-этил- н-пропиламина по методу, приведенному в Примере синтеза 61 Гидрохлорид целевого соединения имеет точку плавления от 222 до 225oC (выход, 35%).
Пример синтеза 65:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(N-этил-N-н-бутил) амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола;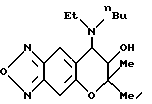
Целевое соединение получают с использованием N-этил- н-бупиламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 180 до 182oC (выход: 28%).
Пример синтеза 66:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (N,N-диаллил) -амино-6H-пирано [2,3-f] бензо-2,1,3- оксадиазола: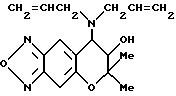
Целевое соединение получают с использованием диаллиламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 172 до 174oC (выход: 35%).
Пример синтеза 67:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(4'-гидроксибутил) амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола:
Целевое соединение получают с использованием 4-гидроксибутиламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 220 до 223oC (выход: 49%).
Пример синтеза 68:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(ди-2'-гидроксиэтил) амино-6H-пирано [2,3-f] бензо-2,1,3- оксадиазола:
Целевое соединение получают с использованием диэтаноламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 108 до 110oC (выход: 43%).
Пример синтеза 69:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(N-метил-N-фенэтил) амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола:
Целевое соединение получают с использованием N-метилфенэтиламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 190 до 193oC (выход: 79%).
Пример синтеза 70:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8-(N-метил-N-изобутил) амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола:
Целевое соединение получают с использованием метил-изобутиламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 178 до 181oC (выход: 26%).
Пример синтеза 71:
Синтез 7,8-дигидро-6,6-диметил-7-гидрокси-8- (1', 1'-диметилфенэтил)амино-6H-пирано [2,3-f] бензо-2,1,3-оксадиазола:
Целевое соединение получают с использованием 1,1-диметилфенэтиламина по методу, приведенному в Примере синтеза 61. Гидрохлорид целевого соединения имеет точку плавления от 190 до 192oC (выход: 34%).
Примеры составления готовых лекарственных форм.
Ниже приведены примеры составления готовых лекарственных форм таблеток, твердых капсул, эластичных капсул, суппозиториев, средств для инъекций и гранулированных средств.
1. Таблетка содержит 5 мас.% соединения N 59, 60 мас.% лактозы, 20 мас.% кукурузного крахмала, 5 мас.% гидроксипропилметилцеллюлозы, 9 мас.% КМЦ-Ca соли и 1 мас.% стеарата магния.
2. Твердая капсула содержит 10 мас.% соединения N 59, 30 мас.% лактозы, 59 мас.% микрокристаллической целлюлозы и 1 мас.% стеарата магния.
3. Эластичная капсула содержит 6,7 мас.% соединения N 59, 92,6 мас.% триглицерида C4-10 алифатической кислоты и 0,7 мас.% Полисорбата 80 (т.е. полиоксиэтилен 20-сорбитана моноолеата, определенного в Фармакопее Японии).
4. Суппозитория содержит 1,0 мас.% соединения N 59, 98,5 мас.% твердого жира и 0,5 мас.% Полисорбата 80 ("Твердый жир" представляет собой триглицерид C6-18 алифатической кислоты, определенный в Фармакопее Японии).
5. Средство для инъекций содержит 0,01 мас.% соединения N 59 и 99,99 мас.% дистиллированной воды для инъекций.
6. Гранулированное средство содержит 5 мас.% соединения N 59, 60 мас.% лактозы, 20 мас.% кукурузного крахмала, 5 мас.% гидроксипропилметилцеллюлозы и 10 мас.% КМЦ-Ca соли.
ПРИМЕРЫ ПРИГОТОВЛЕНИЯ ПРЕПАРАТИВНЫХ ФОРМ
Пример 1 готовой препаративной формы.
Готовая препаративная форма в виде таблеток:
Соединение (получено по примеру синтеза 4) - 10 г
Лактоза - 260 г
Порошок кристаллической целлюлозы - 600 г
Кукурузный крахмал - 350 г
Гидроксипропилцеллюлоза - 100 г
Кальциевая соль карбоксиметилцеллюлозы - 150 г
Стеаринат магния - 30 г
Всего - 1500 г
Указанные выше компоненты смешивали обычным образом и смесь затем таблетировали, получая 10000 покрытых сахарной оболочкой таблеток, причем каждая из них содержала 1 мг активного компонента.
Пример 2 готовой препаративной формы:
Готовая препаративная форма в виде капсул:
Соединение (получено по примера синтеза 4) - 10 г
Лактоза - 440 г
Порошок кристаллической целлюлозы - 1000 г
Стеаринат магния - 50 г
Всего - 1500 г
Указанные выше компоненты смешивали обычным способом и затем смесью заполняли желатиновые капсулы, получая 10000 капсул, причем каждя из них содержала 1 мг активного компонента.
Пример 3 готовой препаративной формы
Готовая препаративная форма в виде мягких капсул:
Соединение (получено в примере 4) - 10 г
PEG-400 - 479 г
Триглицериды насыщенных жирных кислот - 1500 г
Мятное масло - 1 г
Полисорбат 80 - 10 г
Всего - 2000 г
Указанные выше компоненты смешивали и затем смесью наполняли мягкие желатиновые капсулы N 3 обычной методикой, получая 10000 мягких капсул, причем каждая из них содержала 1 мг активного компонента.
Пример 4 готовой препаративной формы
Готовая препаративная форма в виде мази:
Соединение (получено по примеру синтеза 4) - 1,0 г
Жидкий парафин - 10,0 г
Цетанол - 20,0 г
Белый вазелин - 68,4 г
Этилпарабен - 0,1 г
L-Ментол - 0,5 г
Всего - 100,0 г
Указанные выше компоненты смешивали обычным методом, получая 1%-ную мазь.
Пример 5 готовой препаративной формы
Готовая препаративная форма в виде суппозиториев:
Соединение (получено по примеру синтеза 4) - 1 г
Witepsol H15x - 478 г
Witepsol W 35x - 520 г
Полисорбат 80 - 1 г
Всего - 1000 г
(x обозначает фирменное название триглицеридного соединения).
Указанные выше компоненты смешивали в расплаве обычным методом и наливали в контейнеры суппозиториев, после чего охлаждали для отверждения, получая 1000 суппозиториев по 1 г, причем каждый из них содержит 1 мг активного компонента.
Пример 6 готовой препаративной формы
Готовая препаративная форма в виде инъекции;
Соединение (получено в примере 4) - 1 мг
Дистиллированная вода для инъекции - 5 мл
Готовую препаративную форму получают, как только требуется, растворением соединения в дистиллированной воде.
ПРИМЕРЫ ФАРМАЦЕВТИЧЕСКИХ ИСПЫТАНИЙ
Влияние на сократительную способность сердечных мышц
Методика испытания:
Сердце вынимали у самца морской свинки Harley и в жидкости Krebs Henseleit аэрированной газовой смесью (95% 02//5% CO2), от него отделяли левое предсердие. Образец выдерживали при напряжении 0,5 г в ванне для органов, заполненной питательной жидкостью, которую выдерживали при 31oC. Для определения сократительной способности сердечных мышц левого предсердия образец трансмурально подвергали электрическому стимулированию при помощи платиновых диполярных электродов и регистрировали напряжение, генерированное сокращением сердечных мышц образца. Условия для электрического стимулирования были следующие:
Напряжение: двухразовое пороговое напряжение для достижения сокращения (B)
Время: 3 (мсек)
Частота: 1 (Гц)
После уравновешивания образца при замене питательной жидкости на образец акумулятивно наносили изопротеренол, чтобы получить максимальную сократительную реакцию образца. После промывания добавленного изопротеренола образец снова уравновешивали в течение 60 мин при замене питательной жидкости. Затем на образец наносили указанные ниже испытываемые соединения и наблюдали их действие.
Действие, вызванное нанесением 100 мкМ и 300 мкМ каждого соединения, выражены как степень изменения (в %), считая от максимального сокращения (100%), ранее полученного при применении изопротеренола.
Результаты:
Результаты испытания, приведенные в табл. 1а, подтверждают, что соединения настоящего изобретения обладают сильной активностью по усилению сокращения сердечных мышц и что активность зависит от концентрации применяемого соединения.
Результаты испытания 1 - влияние на сократительную способность сердечных мышц даны в табл. 1а.
Влияние на скорость сердечного сокращения
Методика испытания:
Сердце вынимали у самца морской свинки Harley и в жидкости Krebs Henseleit аэрированной газовой смесью (95% O2/5% CO2), от него отделяли правое предсердие. Образец выдерживали при напряжении 1 г в ванне для органов, заполненной питательной жидкостью, которую выдерживали при 31o.
После уравновешивания образца при замене питательной жидкости на образец аккумулятивно наносили изопротеренол, чтобы получить максимальную реакцию образца. После промывания добавленного изопротеренола образец снова уравновешивали в течение 60 мин при замене питательной жидкости. Затем на образец наносили указанные ниже испытываемые соединения и наблюдали их действие.
Получали относительное изменение (%) скорости сердечного сокращения образца, обусловленное добавлением испытываемого соединения (100 мкМ или 300 мкМ), от максимальной реакции (100%), ранее полученной при применении изопротеренола.
Результаты:
Результаты испытаний, показанные в табл. 2, подтверждают, что соединения настоящего изобретения обладают активностью по снижению скорости сердечных сокращений и что эта активность зависит от концентрации применяемого соединения.
Результаты испытания 2 - влияние на скорость сердечных сокращений представлены в табл. 2.
Влияние на сердечную функцию анестезированных собак
Методика испытания:
Самок и самцов собак-дворняжек анестезировали пентобарбиталом натрия и выдерживали в условиях искусственного дыхания. В брюшную аорту через правую бедренную артерию вставляли канюлю и кровяное давление у животного измеряли с применением усилителя кровяного давления и датчика давления. Скорость сердечного сокращения животного измеряли на основе моментальной волны сердечного сокращения, применяя волну кровяного давления в качестве триггера. Катетер (датчик кровяного давления) вставляли в левый желудочек из левой сонной артерии и внутреннее давление в левом желудочке измеряли с применением усилителя кровяного давления. Внутреннее давление в левом желудочке дифференцировали дифференциальным устройством для получения первичной дифференцированной величины внутреннего давления в левом желудочке (эта величина соответствует основной степени систолического давления левого желудочка, LV dp/dlmax). Испытываемые соединения, указанные ниже, растворяли в смешанном растворителе из полиэтиленгликоля-400, этанола и воды (2:3:5) и вводили как ударную дозу через канюлю, вставленную в левую головную вену животного. До испытания было установлено, что сам смешанный растворитель не оказывает влияние на сердечную функцию и кровяное давление животного.
Результаты:
Результаты испытания, показанные в табл. 3 подтверждают, что соединения настоящего изобретения обладают сильной активностью по усилению сокращения сердечных мышц и сильной активностью по снижению скорости сердечных сокращений и не оказывают влияния на кровяное давление.
Результаты испытания 3:
(Влияние соединения примера синтеза 59 (-)-изомер соединения синтетического примера 4), которое внутривенно вводили анестезированным собакам, - как изменение (в %) (получено как средняя величина трех определений) величины, полученной до добавления испытываемого соединения.
Испытание 1. Действие на сократительную способность сердечной мышцы представлено в табл. 4.
Испытание 2. Действие на частоту сердечных сокращений представлено в табл. 5.
МЕТОДЫ
Исследованы действия тестируемого соединения на β1- и β2-адренергические рецепторы. Для проведения экспериментов по конкурентному связыванию с 1251-меченым йодопиндололом (специфическая активность, 2200 Ci/ммоль; конечная концентрация 0,2 мН) β1- и β2-неселективными блокаторами в присутствии тестируемого соединения при указанных в таблице концентрациях использовали микросомальные мембраны, полученные из коры головного мозга крыс. Реакции проводили в 50 мМ Трис-HCl (pH 7,5), содержащем 150 мМ NaCl, 2,5 мМ MgCl2 и 0,5 мМ аскорбате в течение 30 минут при 37oC. Заканчивали реакцию посредством быстрой вакуумной фильтрации на фильтрах из стеклянных волокон. При помощи счетчика гамма-излучения определяли радиоактивность, оставшуюся на фильтрах. Селективность связывания для β1- и β2-адренергических рецепторов определяли одновременно в присутствии избытка ICI-118.551 ( β2-селективный блокатор) или ICI-89.406 ( β1-селективный блокатор), соответственно. Неспецифическое связывание определяли, добавляя к пробе избыток немеченного изопротеренола (100 мкм). Специфическое связывание рассчитывали как разницу: общее связывание минус неспецифическое связывание. Специфическое связывание составляет от 80 до 85% общего связывания.
Результаты представлены в виде процентов максимального специфического связывания 1251-меченого йодопиндолола в отсутствие тестируемого соединения.
Тестируемое соединение = Соединение Примера Синтеза 59. Результаты.
Как показано в табл. 6, тестируемое соединение не связывает ни β1- и β2-адренергические рецепторы и не влияет на них. Эти результаты показывают, что механизм брадикардического действия тестированного соединения отличен от механизма β-блокатора.
Действие, тестируемого соединения на связывание β1- и β2-адренергических рецепторов представлено в табл. 6.
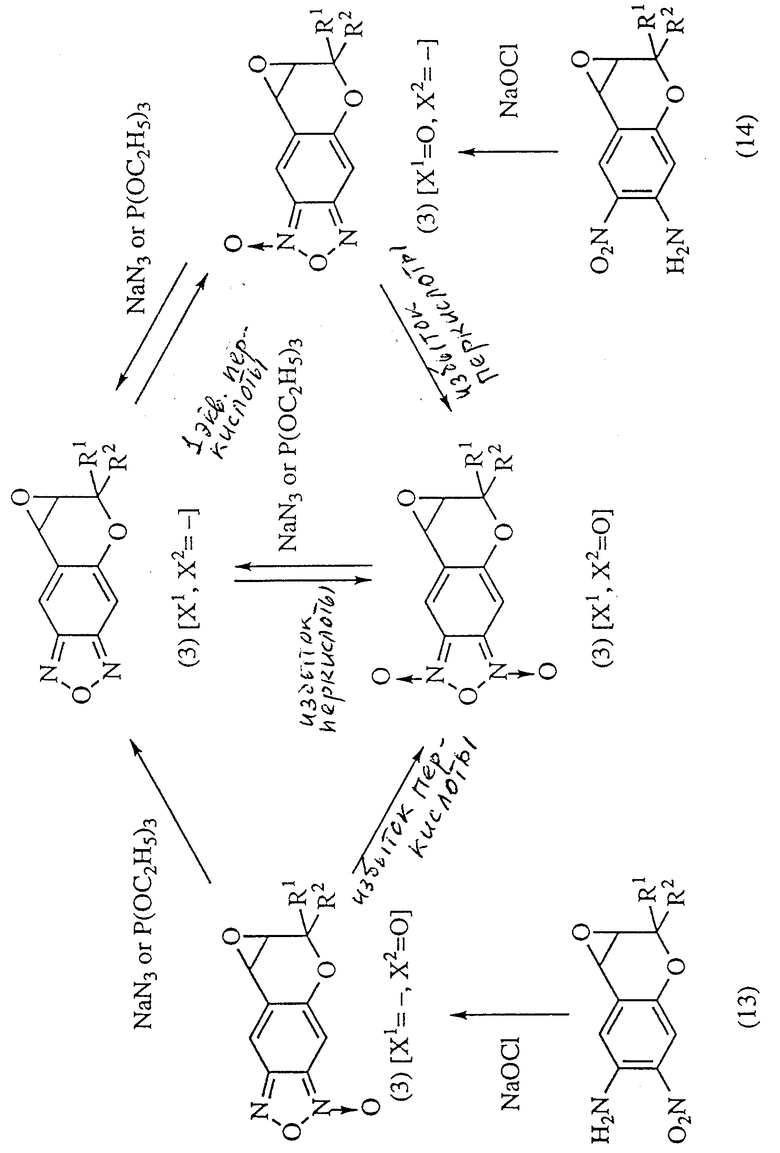
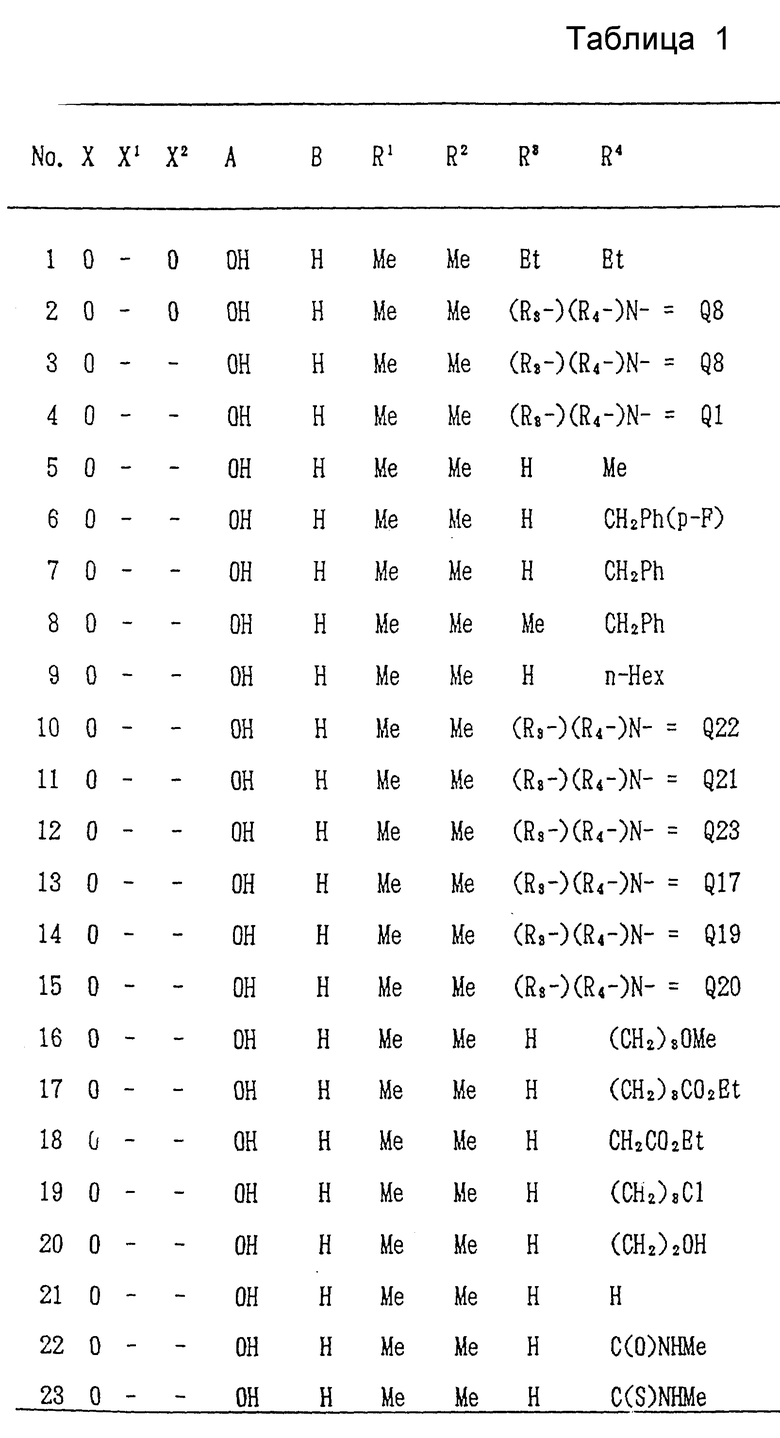
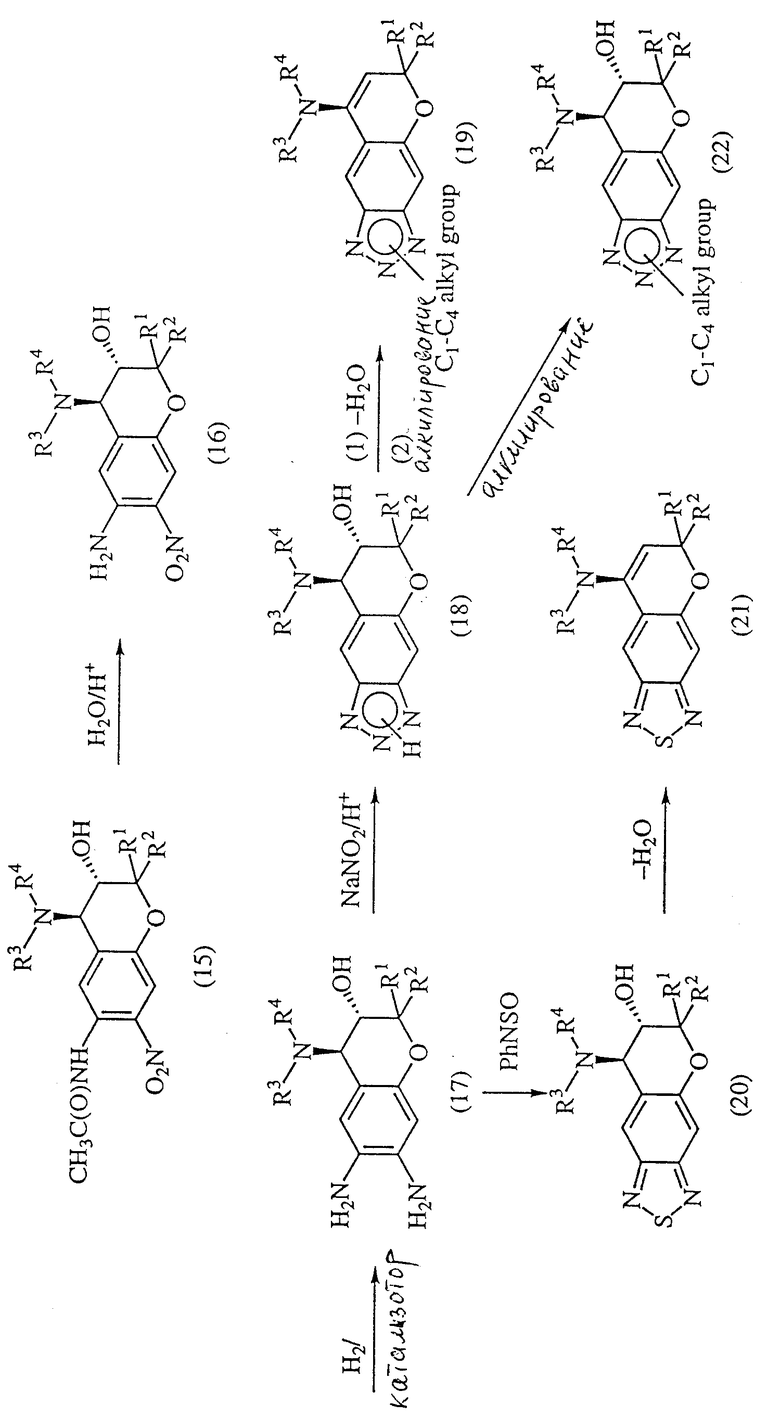
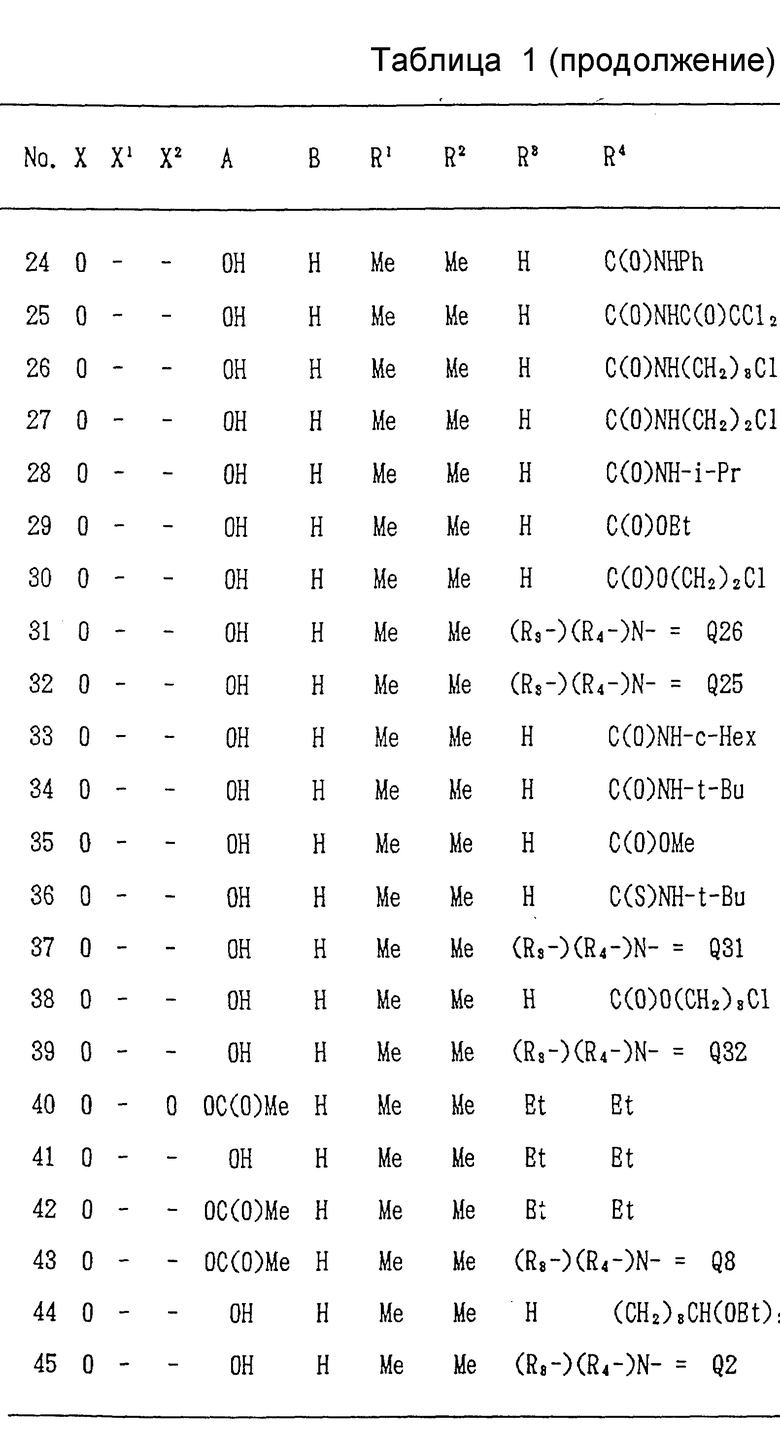
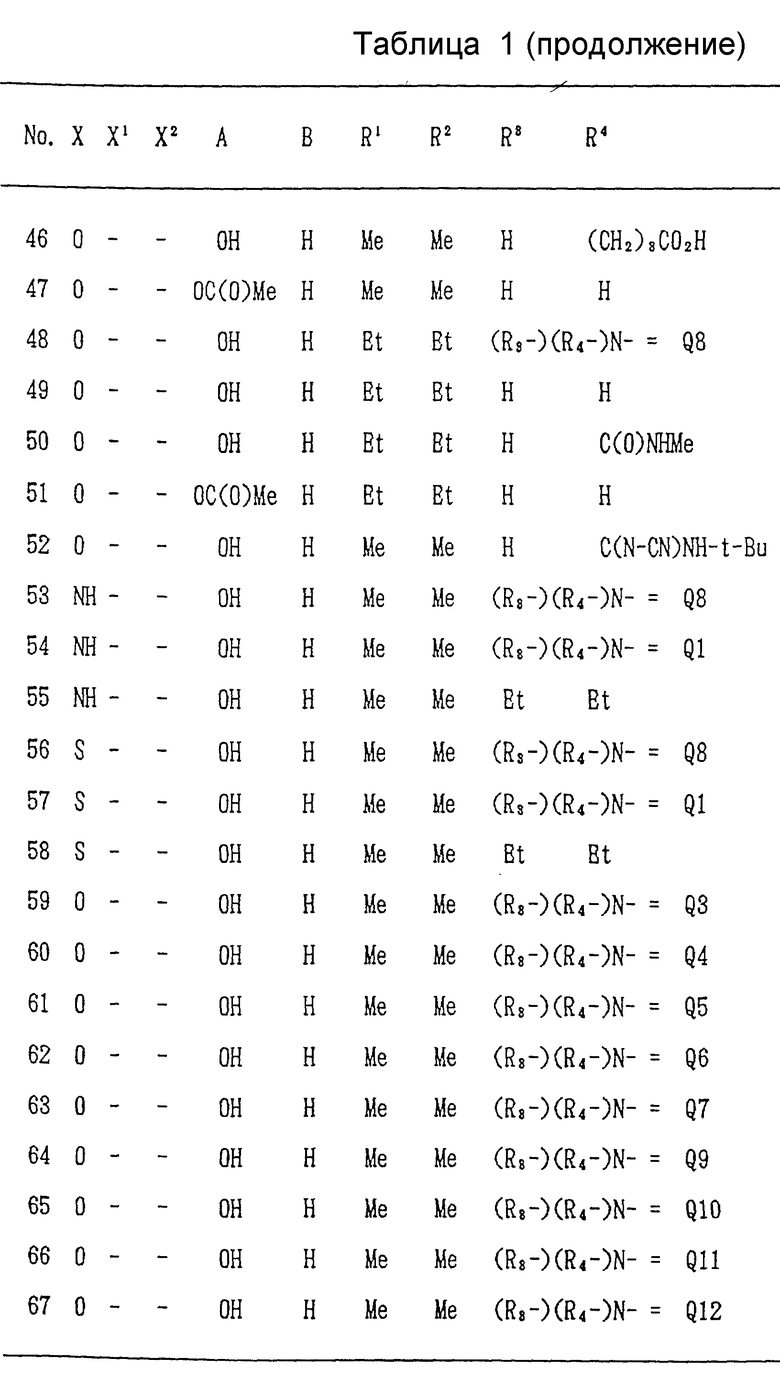
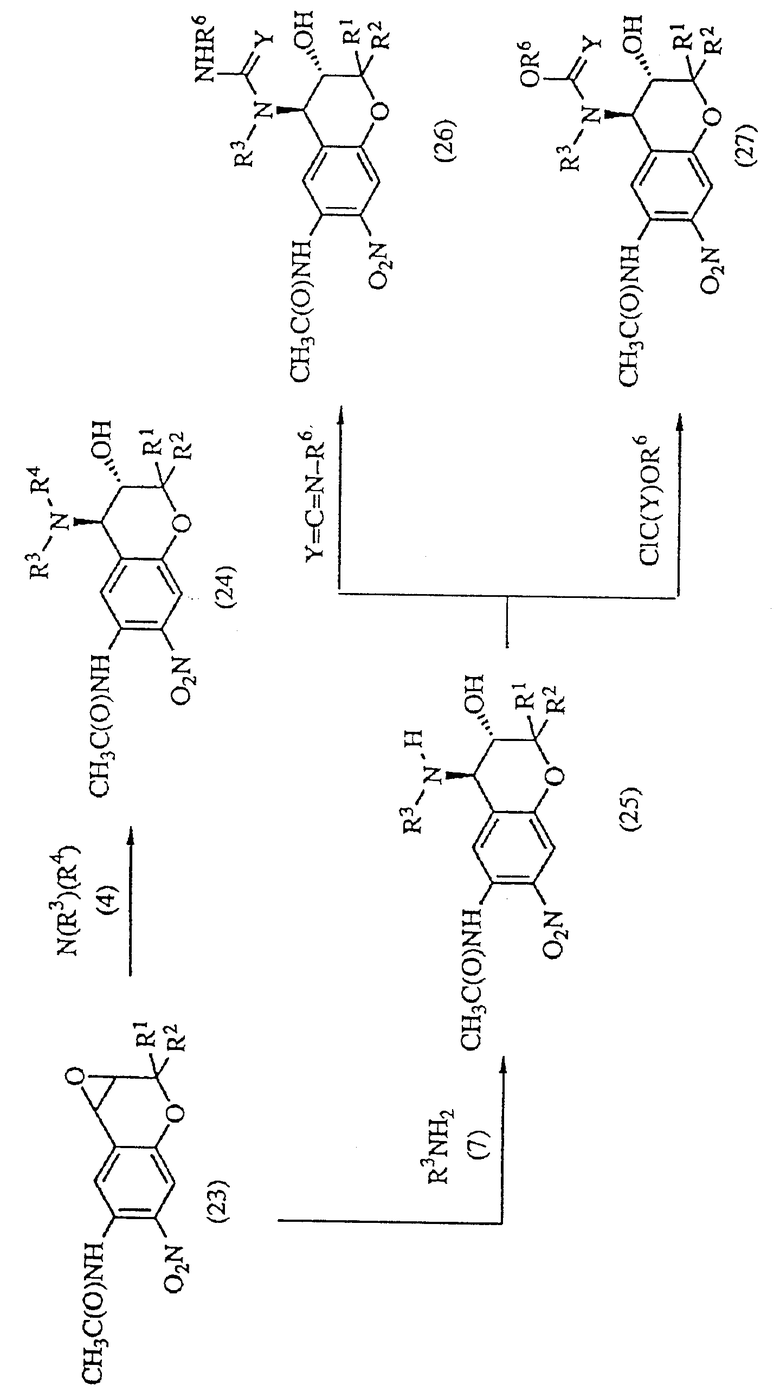
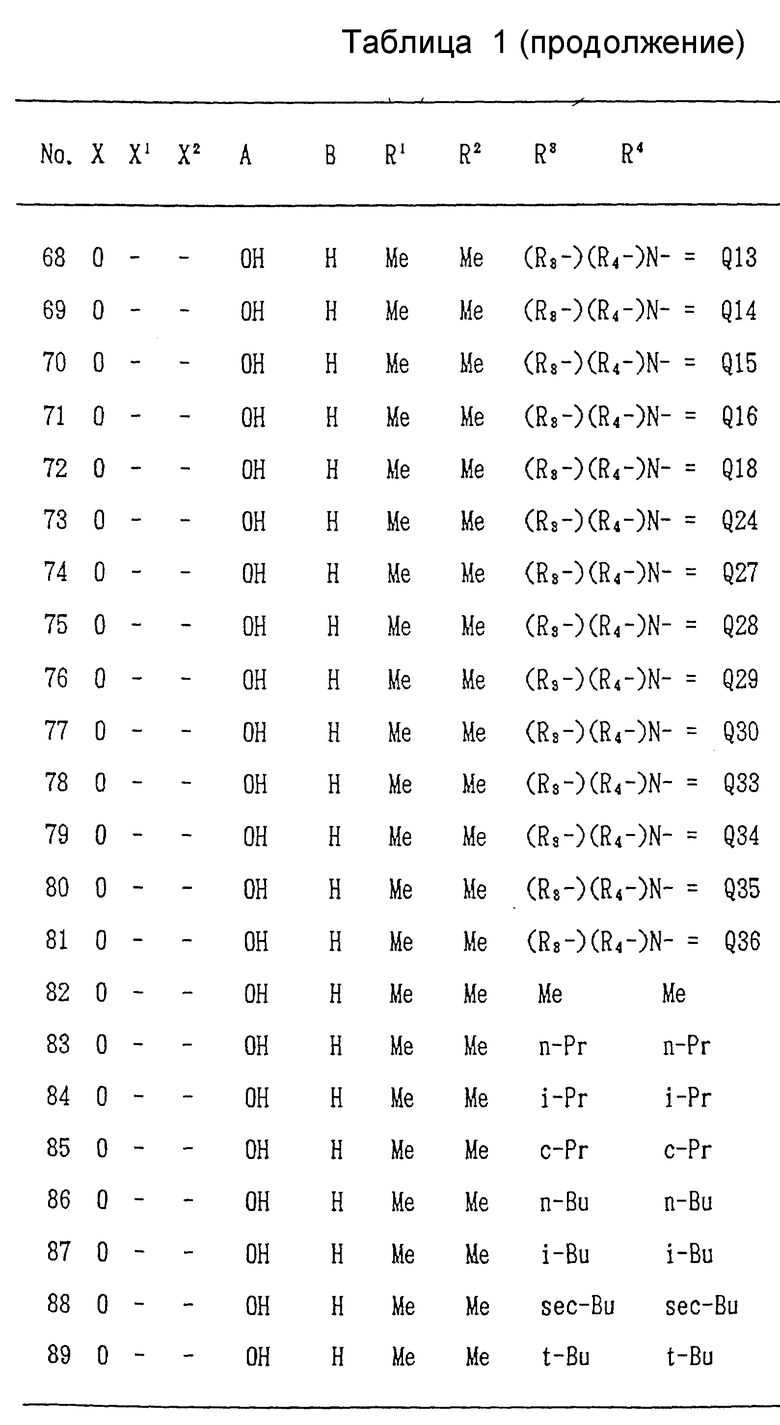




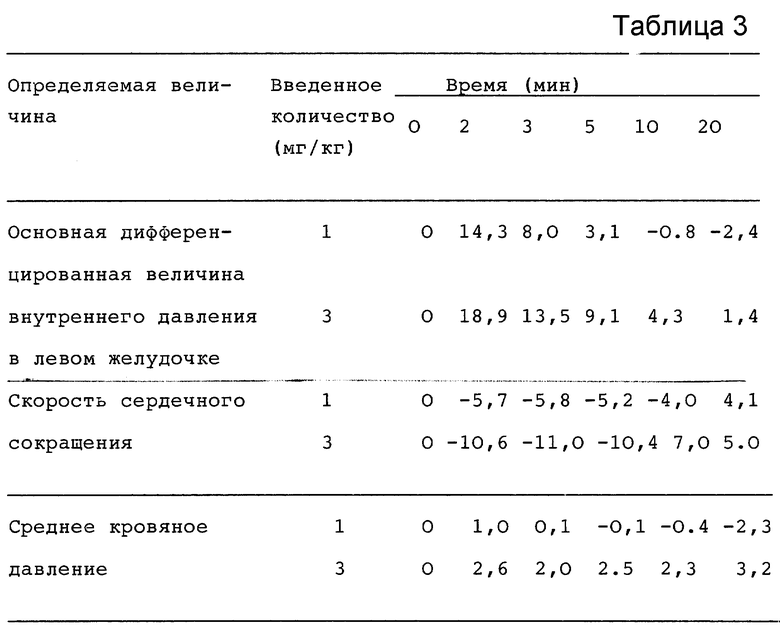
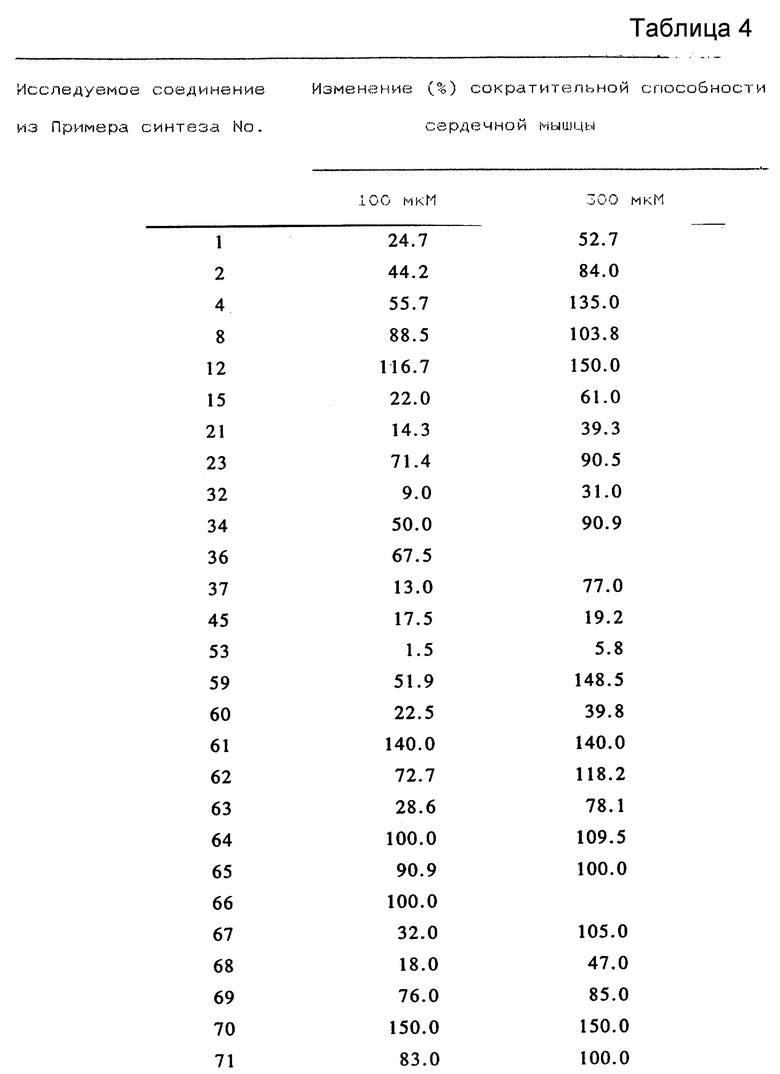
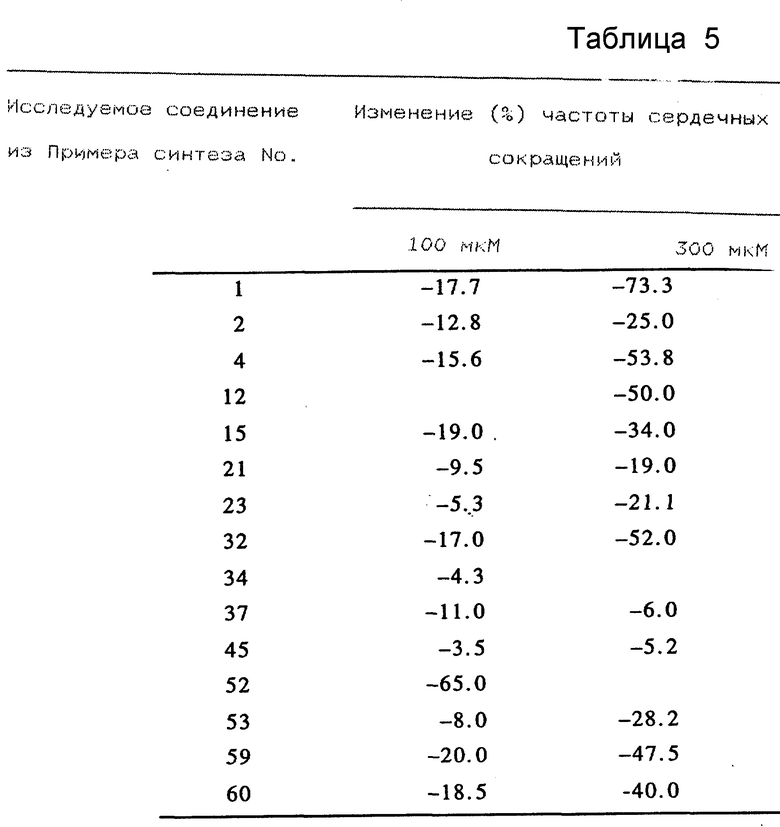
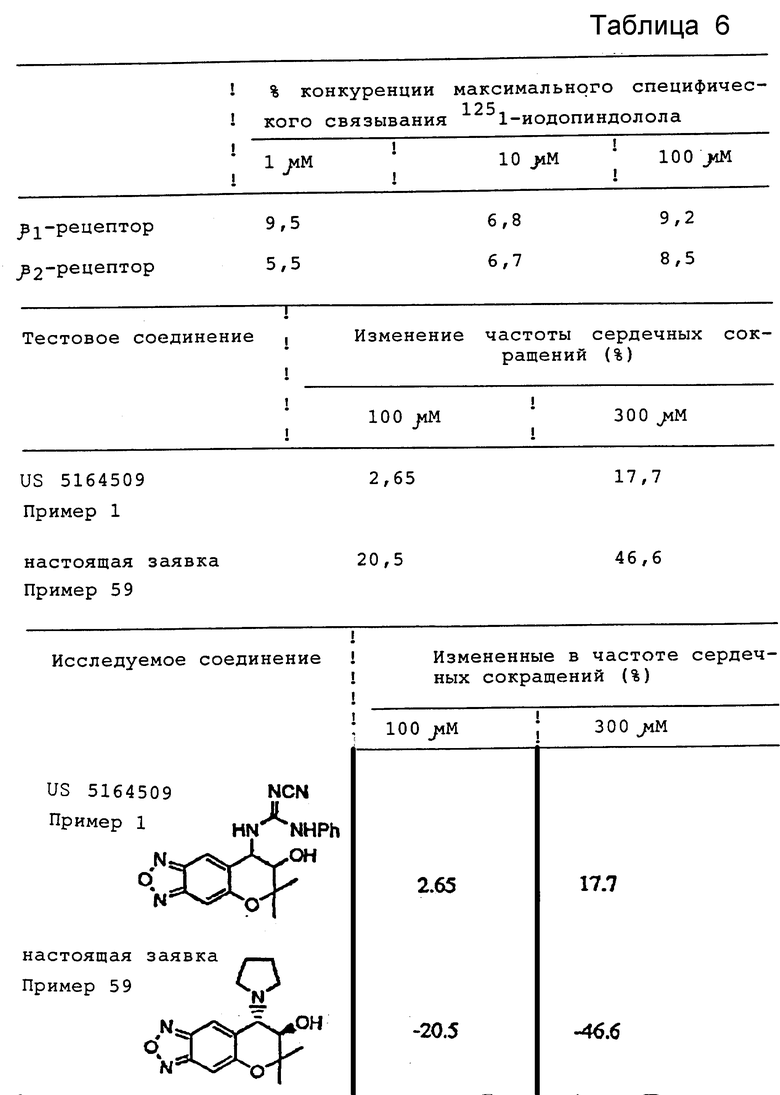
Кардиотонические лекарственные средства, используемые в медицине для лечения сердечной недостаточности, содержат в качестве активного компонента по меньшей мере одно из соединений формулы I или их фармакологически приемлемых солей.
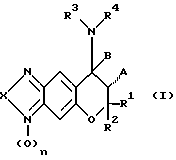
где n = 0 или 1; А представляет собой OH или Н; В представляет собой атом водорода; X представляет собой атом кислорода, атом азота; R1 и R2 представляют собой атом водорода, C1-C4-алкил; R3 и R4 представляют собой атом водорода, C1-C6-алкил, C2-C6-алкенил, или С(=Y)ZR6, где Z=NH; Y=О,S, N-C≡N, или они вместе образуют 1,4-бутилен или 1,5-пентилен. Эти соединения обладают повышенной активностью по усилению сокращения сердечных мышц и сильной активностью по снижению скорости сердечного сокращения. 4 з.п.ф-лы, 6 табл.
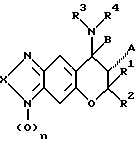
где n - 0 или 1;
Х представляет атом кислорода или атом азота (указанный атом азота незамещен или замещен атомом водорода);
А представляет атом водорода или гидроксильную группу;
В представляет атом водорода;
R1 и R2 одинаковые или отличающиеся друг от друга, обозначают атом водорода или C1-C4-алкильную группу;
R3 и R4 одинаковые или отличающиеся друг от друга, представляют атом водорода, C1-C6-алкильную группу (указанная алкильная группа незамещена или замещена одним или более заместителями, выбранными из гидроксильной группы, C1-C4-алкокси группы или фенильной группы), C2-C6-алкенильная группа или C(= Y)ZR6, где Y обозначает атом кислорода, атом серы или группу NR7, в которой R7 представляет цианогруппу, Z представляет группу NR9, в которой R9 обозначает атом водорода, R6 обозначает C1-C8-алкильную группу; или R3 и R4 вместе образуют 1,4-бутиленовую или 1,5-пентиленовую группу (указанные 1,4-бутиленовая и 1,5-пентиленовая группы не замещены или замещены гидроксильной группой); или R3 и R4 образуют вместе группу -(CH2)m X4(CH2)I-, в которой m и I каждый принимает значение 1, 2 или 3, при этом их сумма должна составлять 3, 4 или 5; X4 представляет атом кислорода или группу NR12, в которой R12 обозначает атом водорода или фенильную группу; или R3 и R4 образуют вместе -(CH2)2 ZC(=Y)- или -(CH2)3 ZC(=Y)-, где значения Z указаны выше;
их оптические изомеры, их стереоизомеры или их фармацевтически приемлемые соли в количестве от 0,01 до 99,5 мас.%.
Приоритеты по пунктам:
02.04.93 и 08.03.94 - по всем пунктам формулы.
| US 5164509 A, 17.11.92 | |||
| US 5026712 A, 25.06.91 | |||
| US 4900752 A, 13.02.90 | |||
| Стенд для определения сил упругости резино-металлических опор | 1973 |
|
SU488107A1 |
