Изобретение относится к оптически активному пиранобензоксадиазольному соединению, которое является важным интермедиатом при синтезе оптически активного производного пиранобензоксади- азола, полезного при лечении гипертензии (артериальной гипертонии) и астмы, и к способам оптического раз- деления пиранобензоксадиазольного соединения в форме рацемической модификации.
В форме рацемической смеси, как это описано в Выкладке патента Японии N 49788/1990 и в патенте США N 4900752, получается производное пиранобензоксадиазола, представленное формулой (III)
O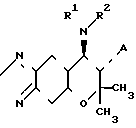 в которой А представляет гидроксильную группу или группу ОС(О)СH3-nХn, в которой Х представляет атом фтора, атом хлора, атом брома, метильную группу или метокси-группу, а n представляет 0 или целое число 1-3;
в которой А представляет гидроксильную группу или группу ОС(О)СH3-nХn, в которой Х представляет атом фтора, атом хлора, атом брома, метильную группу или метокси-группу, а n представляет 0 или целое число 1-3;
когда R1 представляет атом водорода, R2 представляет атом водорода, группу С(Z)СH3-nХn, в которой Z представляет атом кислорода или атом серы, а Х и n являются такими, как это определено выше, или группу С(Z)NHCH3-nХn, в которой Z и n являются такими, как это определено выше; и когда RI не представляет атом водорода, R1 и R2 вместе представляют группу (СH2)m-1C(Z), в которой m представляет целое число 4 или 5, а Z является таким, как это определено выше, группу (СН2)m-2NHC(Z) или группу (СH2)m-2OC(Z), в которой Z и m являются такими, как это определено выше. Соединение III проявляет интенсивные сосудорасширяющие и гипотензивные (понижающие артериальное давление) активности и таким образом, как это ожидается, являются полезными в качестве лекарственного средства для лечения гипертензии, стенокардии, аритмии, нарушений мозгового кровообращения и астмы.
Как это описано в выкладке патента Японии N 49788/1990, соединение III может быть синтезировано следующим образом. Схема реакции I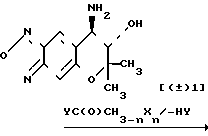
Cхема реакции 2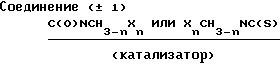
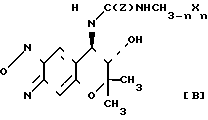
Cхема реакции 3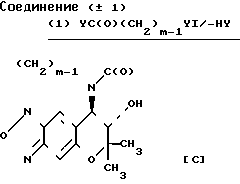
Cхема реакции 4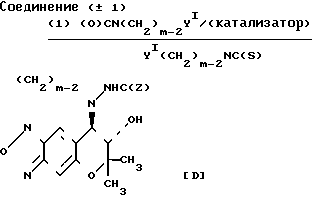
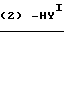
Cхема реакции 5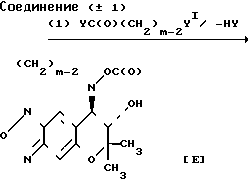
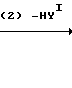
В приведенных схемах реакции, Y представляет уходящую группу, такую как атом галогена (например, атом хлора, брома или йода), ацетокси-группа или трифторацетокси-группа;
YI представляет атом хлора, атом брома, атом йода, орто- или п-толуолсульфонатную группу или метансульфонатную группу; и m, n и Х являются такими, как это определено выше.
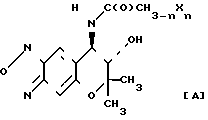
Соединение (А), которое является соединением III, в котором RIпредставляет атом водорода, может быть получено путем взаимодействия пиранобензоксадиазольного соединения, которое получается в форме рацемической смеси (вкратце называется как соединение (± 1) в дальнейшем), с ацилирующим реагентом YC(O)CH3-nХn, в котором Х, Y и n являются такими, как это определено выше, не обязательно в присутствии основания (относится к Схеме реакции I).
Соединение (В), которое является соединением (III), в котором RIпредставляет атом водорода, может быть получено путем взаимодействия соединения (± 1) c изоцианатом С(О)NCH3-nХn или изотиоцианатом ХnCH3-nNC(S), в котором Х, Z и n являются такими, как это определено выше (относится к схеме реакции 2).
Соединение С, которое является соединением III, в котором R1 и R2вместе представляют группу (СH2)m-1C(O), может быть получено путем взаимодействия соединения (± 1) с ацилирующим реагентом УС(О)(СН2)m-1Y1, в котором Y, Y1 и m являются такими, как это определено выше, необязательно в присутствии основания, а затем циклизацией продукта реакции необязательно в присутствии основания (относится к Схеме реакции 3).
Соединение Д, которое соединением III, в котором R1 и R2 вместе представляют группу (СH2)m-2NHC(Z), в которой Z и m являются такими, как это определено выше, может быть получено путем взаимодействия соединения (± 1) с изоцианатом (О)CN(CH2)m-2Y1 или изотиоцианатом (S)CN(CH2)m-2YI,в которой YI и m являются такими, как это определено выше, а затем циклизацией продукта реакции необязательно в присутствии основания (относится к схеме реакции 4).
Соединение (Е), которое является соединением (III), в котором R1 и R2 вместе представляют группу (СН2)m-2OC(O), в которой m является таким, как это определено выше, может быть получено путем взаимодействия соединения (± 1) с галоген-карбонатом YC(О)О(СH2)m-2Y1, в котором Y, Y1и m являются такими, как это определено выше, необязательно в присутствии основания, а затем циклизацией продукта реакции необязательно в присутствии основания (относится к схеме реакции 5).
В указанных схемах реакции, соединение III, в котором Z является атомом серы, может быть получено сульфурированием соответствующего соединения, в котором Z является атомом кислорода, с помощью реагента Ловиссона.
Как описывается в выкладке патента Японии N 49788/1990, соединение (± 1) может быть получено следующим образом: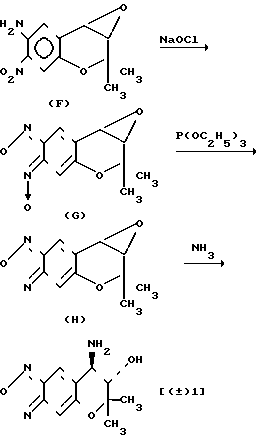
Соединение (± 1) может быть получено путем обработки известного соединения F с гипохлоритом натрия, восстановлением N-оксидной группы соединения G, полученного таким образом, с помощью восстановителя, такого как триэтилфосфат, а затем взаимодействием полученного таким образом соединения (Н) с аммиаком в инертном растворителе.
Однако нигде не сообщено об оптическом разделении соединения (± 1).
Кроме того, упомянутое рацемическое соединение III, которое обладает асимметрическими атомами углерода в положениях 3 и 4 пиранового цикла, имеет два оптических изомера (соединения III* и III**). Однако упомянутая выкладка патента Японии N 49788/1990 не описывает ни эти оптически активные производные пиранобензоксадиазола, ни какой-либо способ получения этих оптически активных производных.
В области медицины часто наблюдается, что оптические изомеры отличаются друг от друга по фармакологической активности и безопасности (безвредности). Поэтому является желательным оптически разделять эти изомеры для того, чтобы разрабатывать лучшие лекарственные средства.
Авторы изобретения обнаружили, что оптически активное производное пиранобензоксадиазола, соответствующее соединению III*, которое синтезируется через оптически активное пиранобензоксадиазольное соединение, показывающее правое вращение в этаноле, соответствующее соединению (± 1), которое будет описано ниже, заметно является превосходным по сравнению с оптически активным производным пиранобензоксадиазола, соответствующим соединению (III**), которое синтезируется через энантиомер, соответствующий соединению (-) I, которое будет описано ниже с точки зрения лекарственной активности.
Краткое описание
Таким образом, изобретение относится к способам оптического разделения соединения (± I), который включает реакцию взаимодействия пиранобензоксадиазольного соединения формулы ( I)
O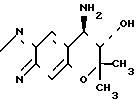 [(±)1] в форме рацемической смеси с оптически активной карбоновой кислотой формулы II
[(±)1] в форме рацемической смеси с оптически активной карбоновой кислотой формулы II
HO O
O O2H а затем разделение полученной таким образом диастереомерной соли. Кроме того, изобретение относится к оптически активному пиранобензоксадиазольному соединению (+) I, показывающему правое вращение в этаноле, соответствующему указанному соединению [+] I, из двух оптических изомеров, полученных по упомянутому способу.
O2H а затем разделение полученной таким образом диастереомерной соли. Кроме того, изобретение относится к оптически активному пиранобензоксадиазольному соединению (+) I, показывающему правое вращение в этаноле, соответствующему указанному соединению [+] I, из двух оптических изомеров, полученных по упомянутому способу.
Соединение II, которое является оптическим активным разделяющим реагентом и встречается в виде двух оптических изомеров соединения [+] II и [-] II, может быть синтезировано по методу, описанному в выкладке патента Японии N 83144/1986.
Теперь способ оптического разделения соединения (± I) на соединение ([+] 1) и его антипод, а именно, соединение ([-] I), будет описан подробно.
Схема реакции I
Стадия А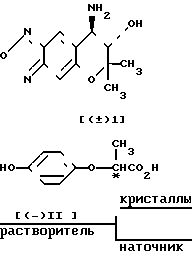


Стадия Б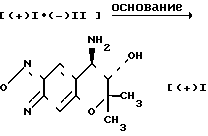

В cтадии А соединение (± I) подвергается взаимодействию с соединением [-] II, которое используется в качестве оптически активного разделяющего реагента, и кристаллизуется. Таким образом диастереомерная соль [+] I˙ [-] II может быть легко получена в форме кристаллов.
Однако должны здесь отметить, что полученные кристаллы могут быть сольватированы в зависимости от используемого растворителя (см. примеры).
Подобным образом, диастереомерная соль [-] I˙ [+] II) может быть легко получена при использовании соединения ([+] II) в качестве оптически активного разделяющего реагента.
Таким образом соответствующий оптический изомер соединения (± I) может быть легко получен, выбирая соответствующим образом разделяющий реагент.
В качестве растворителя, который используется в стадии А, предпочтительным являются кетоны, такие как ацетон и метил-изобутилкетон, хотя изобретение не ограничивает выбора растворителя. В этом случае диастереомерная соль кристаллизуется в форме сольвата.
Температура реакции обычно может варьировать от -20 до 100оС, предпочтительно от 10 до 30оС.
Температура кристаллизации обычно может варьировать в пределах от -20 до 50оС, предпочтительно от -10 до 20оC.
Выкристаллизованная таким образом диастереомерная соль может быть дополнительно перекристаллизована, например, из ацетона, тем самым получая кристаллическую диастереомерную соль с высокой чистотой.
В стадии Б, кристаллическая диастереомерная соль ([+] I˙ [-] II) или ее сольват подвергается реакции с основанием, выбираемым из следующих бикарбоната натрия, бикарбоната калия, карбоната натрия, карбоната калия, гидроокиси натрия и гидроокиси калия. Таким образом целевое соединение ([+] I), показывающее правое вращение в этаноле, может быть легко получено.
Подобным образом, соединение ([-] I) может быть легко получено из диастереомерной соли ([-] I ˙ [+] II) или ее сольвата.
Оптическая чистота соединения ([+] I) может быть определена путем взаимодействия указанного соединения с метил-изоцианатом, получая тем самым соединение мочевины формулы [+] IV
O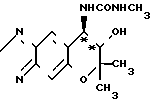 а затем анализируя полученное соединение с использованием оптически активной жидкостной хроматографической колонки (Чиралсел ОС, производимой фирмой Дэйсел Кемикал Индастриз Лимитед).
а затем анализируя полученное соединение с использованием оптически активной жидкостной хроматографической колонки (Чиралсел ОС, производимой фирмой Дэйсел Кемикал Индастриз Лимитед).
Оптическая чистота соединения [-] I) может быть определена подобным путем.
Как будет видно ниже в примере испытаний, соединение III**, полученное из соединения [+] I в соответствии с методом, описанным в упомянутой выкладке патента Японии N 49788/1990, показывает исключительно высокую активность при понижении кровяного давления, по сравнению с энантиомером соединение (III**), полученным из соединения [-] I.
Поэтому очевидно, что применение соединения III* при лечении, например, гипертензии является более эффективным, чем применение соединения III в форме рацемической модификации.
Анализ с использованием оптически активной жидкостной хроматографической колонки (Чиралсел ОС, производимый фирмой Дэйсел Кемикал Индастриз Лимитед) доказывает, что не происходит рацемизации во время получения соединения (III*) из соединения [+] I или получения соединения (III**) из соединения [-] I.
Пример испытаний (гипотензивная активность).
Соединения III* и III** были каждый по отдельности растворены или суспендированы в 0,5%-ном водном растворе метилцеллюлозы и принудительно внесены пероральным путем трем самцам спонтанно гипертензивных крыс II- недельного возраста с использованием желудочного зонда.
Животные были предварительно обогреты в обогреваемом ящике при 50оС в течение 3-5 мин, а затем перенесены в ограничительную клетку при 37оС, чтобы измерить систолическое кровяное давление методом хвостовой манжетки (KN-210-I, производимой Натсуме Сейсакушо Ко. Лимитед). Табл.1 приводит проценты понижения кровяного давления спустя 1 ч после введения лекарственного средства. Каждая величина представляет среднее значение от трех животных, подвергнутых испытанию.
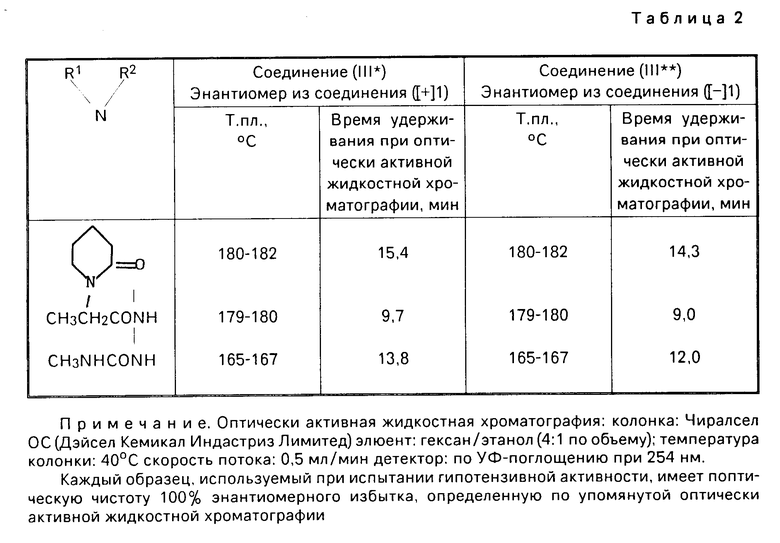
Табл.2 дает аналитические данные соединения III* и III**.
П р и м е р 1. (а) Стадия А. Разделение диастереомерной соли (ацетонового сольвата ([+] I ˙ [-] II и диастереомерной соли (ацетонового сольвата [-] I ˙ [+] II).
В 1000 г ацетона растворяют 117,6 г (0,500 моля) [ ±]-7,8-дигидро-6,6-диметил-7-окси-8-амино-6Н-пирано [2,3-f]бенз-2,1,3-оксадиазола (соединение [± I и 92,9 г (0,510 моля) (-)-2-(4-оксифенокси)-пропионовой кислоты соединение [-] I и смесь перемешивают в течение 3 ч при охлаждении со льдом.
Выпадающие таким образом в осадок кристаллы фильтруют путем отсасывания, промывают с помощью 500 мл охлажденного льдом ацетона и высушивают при пониженном давлении. Таким образом получают 64,6 г диастереомерной соли (ацетоновый сольват [+] I ˙ [-] II в форме светло-желтых кристаллов (выход: 27,2% оптическая чистота: 95,7% энантиомерного избытка).
Эту диастереомерную соль (ацетоновый сольват ([+] I˙ [-] II нагревают при кипячении с обратным холодильником в 270 г ацетона, а затем отгоняют 77 г ацетона. Остаток кристаллизуют при охлаждении со льдом в течение 2 ч. Таким образом оптическая чистота диастереомерной соли (ацетонового сольвата [+] I ˙ [-] II повышается до 100% энантиомерного избытка (выход: 80%).
При измерении т. пл. этого вещества оно начинает медленно разлагаться примерно при 102оС. В результате неводного титрования с помощью хлорной кислоты в уксусной кислоте в качестве растворителя было подтверждено, что одна молекула ацетона находится в сольватной форме.
С другой стороны фильтраты объединяют и ацетон удаляют из фильтратов. Затем к остатку добавляют 1500 мл этилацетата, 1000 мл воды, 32,8 г (0,39 моля) бикарбоната натрия и 200 г хлористого натрия и смесь встряхивают.
Полученный раствор оставляют стоять, чтобы тем самым вызвать разделение фаз. Этилацетатную фазу собирают и к ней добавляют 200 мл воды, 6,56 г (0,078 моля) бикарбоната натрия и 40 г хлористого натрия. Полученную смесь снова встряхивают и оставляют стоять, чтобы тем самым вызвать разделение фаз.
Полученную таким образом этилацетатную фазу высушивают путем добавления безводного сульфата натрия и фильтруют, а затем из нее отгоняют этилацетат. Таким образом получают 94,7 г коричневого твердого вещества.
Это коричневое твердое вещество и 73,4 г (0,403 моля) [+]-2-(4-оксифенокси)-пропионовой кислоты (соединение ([+] II) растворяют в 700 г ацетона и перемешивают в течение трех часов при охлаждении со льдом.
Выпадающие в осадок таким образом кристаллы фильтруют путем отсасывания, промывают с помощью 280 мл охлажденного льдом ацетона, а затем высушивают при пониженном давлении. Таким образом получают 75,79 г диастереомерной соли ([-] I [+] II) ацетоновый сольват) в форме светло-желтых кристаллов (выход: 31,9% оптическая чистота: 100% энантиомерного избытка).
При измерении т.пл. этого продукта реакции он начинает медленно разлагаться примерно при 102оС. В результате неводного титрования с помощью хлорной кислоты в уксусной кислоте подтверждено, что одна молекула ацетона сольватирована.
(б) Стадия Б: разделение соединений ([+] I) и ([-] I)
O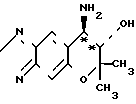
К 66,7 г (0,140 моля) диастереомерной соли (ацетоновый сольват ([+] I ˙ [-] II) добавляют 1000 мл этилацетата, 700 мл воды, 17,0 г (0,160 моля) карбоната натрия и 140 г хлористого натрия, за которым следует встряхивание, и оставляют стоять, чтобы тем самым вызвать разделение фаз.
Этилацетатную фазу собирают и промывают с помощью 200 мл воды, 2,1 г (0,020 моля) карбоната натрия и 40 г хлористого натрия. Кроме того, эту фазу промывают водным раствором хлористого натрия (40 г/200 мл воды). Затем этилацетатную фазу высушивают добавлением безводного сульфата натрия и фильтруют. После отгонки этилацетата получают 31,85 г соединения ([+] I) (выход 96%).
Отдельно, диастереомерную соль (ацетоновый сольват [-] I ˙ [+] II) обрабатывают подобным образом, как это описано выше. Таким образом получают соединение ([-] I).
Аналитические данные т. пл. 145-146оС (для обоих соединений ([+] I) и ([-] I). Оптическое вращение:
Соединение ([+] I): [d]D25 + 189о (с 0,50, этанол)
Соединение ([-] I): [d]D25 189о (с 0,50, этанол)
Оптическая чистота (определяется при условиях, указанных в табл.2).
Каждое испытуемое соединение подвергают взаимодействию с метилизоцианатом. Мочевиновое соединение, полученное таким образом, анализируют с использованием оптически активной жидкостной хроматографической колонки (Чиралсел ОС, производимой фирмой Дэйсел Лимитед).
Каждое из соединений ([+] I) и ([-] I) показывает оптическую чистоту 100% энантиомерного избытка.
ЯМР cпектр:
Каждое из обоих соединений ([+] I) и ([-] I) показывает спектр, идентичный со спектром соединения ([± I), т.е. рацемической модификации.
ЯМР (CDCl3 + DMCO-d6) м.д.
1,26 (3Н), 1,49 (3Н), 2,80-3,30 (5Н), 3,33 (1Н), 3,78 (1Н), 6,82 (1Н) и 7,98 (1Н).
П р и м е р 2. (а) Cтадия А: разделение диастереомерной соли (метил-изобутил кетоновый сольват [+] I ˙ [-] II) и диастереомерной соли (метил-изобутилкетоновый сольват [-] I ˙ [+] II).
В 27,8 г метилизобутилкетона растворяют 4,70 г (20 ммолей) (±)-7,8-дигидро-6,6-диметил-7-окси-8-амино-6Н -пирано[2,3-] бенз-2,1,3-оксадиазола (соединение ([± I)) и 3,70 г (20,3 ммолей) (-)-2-(4-оксифенокси)-пропионовой кислоты (соединение ([-] II) и раствор перемешивают при 21оС в течение 15 мин.
К полученному таким образом раствору добавляют 10 мг затравочного кристалла (метил-изобутилкетонового сольвата [+] I ˙ [-] II). Перемешивание продолжают в течение дополнительных 2 ч, чтобы тем самым выкристаллизовать продукт реакции. Затем прекращают перемешивание и реакционную смесь оставляют стоять в холодильнике в течение ночи.
Выпадающие таким образом кристаллы собирают отсасыванием, промывают с помощью 7,1 г холодного метилизобутилкетона и высушивают при пониженном давлении. Таким образом получают 4,59 г диастереомерной соли (метил-изобутилкетонового сольвата [+] I ˙ [-] II) в форме светло-желтых кристаллов (выход: 44,4%).
При измерении т.пл. этого продукта реакции он начинает медленно разлагаться примерно при 95оС. В результате неводного титрования с помощью хлорной кислоты в уксусной кислоте в качестве растворителя подтверждено, что одна молекула метилизобутилкетона сольватирована.
С другой стороны фильтраты объединяют и к нему добавляют 28,2 г 20%-ного водного раствора хлористого натрия и 1,22 г (11,5 ммолей) карбоната натрия. Полученную смесь встряхивают, оставляют стоять, чтобы тем самым вызвать разделение фаз. Метилизобутилкетоновую фазу собирают и встряхивают снова вместе с 9,4 г 20%-ного водного раствора хлористого натрия. Затем ее оставляют стоять, чтобы тем самым вызвать разделение фаз.
К полученной таким образом метил-изобутилкетоновой фазе добавляют 2,07 г (11,4 молей) (+)-2-(4-оксифенокси)-пропионовой кислоты (соединение [+] II)). После растворения соединения путем перемешивания при комнатной температуре к этому раствору добавляют 10 мг затравочного кристалла (метил-изобутилкетонового сольвата [-] I ˙ [+] II), чтобы тем самым выкристаллизовать продукт реакции. Затем реакционную смесь оставляют стоять в холодильнике в течение ночи.
Выпадающие таким образом кристаллы фильтруют путем отсасывания, промывают с помощью 7,1 г охлажденного метилизобутилкетона и высушивают при пониженном давлении. Таким образом получают 4,25 г диастереомерной соли (метил-изобутилкетонового сольвата [+] I ˙ [-] II) в виде светло-желтых кристаллов (выход: 41,1%).
При измерении т.пл. этого продукта реакции он начинает медленно разлагаться примерно при 95оС. В результате неводного титрования с помощью хлорной кислоты в уксусной кислоте в качестве растворителя подтверждено, что одна молекула метил-изобутилкетона сольватирована.
(б) Стадия Б: разделение соединений ([+] I) и ([-] I).
К 4,26 г (8,23 ммолей) диастереомерной соли (метил-изобутилкетонового сольвата ([+] I ˙ [-] II), полученной в приведенной стадии А, добавляют 53,4 г этилацетата, 42,7 г воды, 0,873 г (8,23 ммолей) карбоната натрия и 10,7 г хлористого натрия, за которым следует встряхивание и оставление стоять, чтобы тем самым вызвать разделение фаз. Этилацетатную фазу собирают и промывают с 14,2 г воды и 3,6 г хлористого натрия.
Этилацетатную фазу высушивают добавлением в нее безводного сульфата натрия и фильтруют. После отгонки 48,6 г этилацетата к остатку добавляют 7,3 г гексана, за которым следует кристаллизация при охлаждении со льдом в течение 3 ч. Затем выпадающие таким образом кристаллы собирают. Таким образом получают 1,84 г соединения ([+] I) (выход: 95%).
Диастереомерную соль (метил-изобутилкетоновый сольват [-] I, [+] II) обрабатывают таким же образом, что и описанный выше. Таким образом получают соединение ([-] I).
Соединения [+] I и [-] I показывают (каждое из них) оптическую чистоту 100% энантиомерного избытка.
Рекомендуемый пример 1. (а) Синтез (+)-7,8-дигидро-6,6- диметил-7-окси-8-(н-(I-оксо-5-хлор)-пентил)-амино-6Н- пирано[2,3-f] бенз-2,1,3-оксадиазола (интермедиат)
формула (+)-формы
O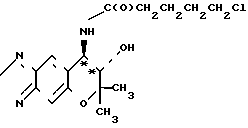
715 мг (3,04 ммоля)(+)-7,8-дигидро-6,6-диметил-7-окси-8-амино- 6Н-пирано[2,3-f]бенз-2,1,3-оксадиазола (соединение [+] I), 470 мкл триэтиламина и 70 мл хлористого метилена перемешивают при комнатной температуре. К полученному раствору добавляют 430 мкл (3,34 ммоля) хлорангидрида 5-хлорвалериановой кислоты. После реакции в течение 2 ч реакционную смесь промывают водой трижды. Метиленхлористую фазу высушивают над безводным сульфатом натрия и фильтруют. После отгонки растворителя получают названное соединение. Этот продукт реакции далее не очищают каким-либо образом, но как таковой подвергают следующей реакции.
(б) Cинтез (+)-7,8-дигидро-6,6-диметил-7-окси-8-(2-оксо- пиперидил)-6Н-пирано [2,3-f] бенз-2,1,3-оксадиазола соответствующего соединению III*
O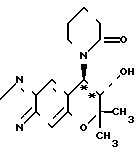
В 200 мл ацетона суспендируют 1,08 г (+)-7,8-дигидро-6,6- диметил-7-окси-8-(н-(1-оксо-5-хлор)-пентил)-амино-6Н-пирано [2,3-f]-бенз-2,1,3-оксадиазола, 8,40 г (60,8 ммолей) карбоната калия и 1,01 г (6,08 ммолей) йодистого калия и смесь нагревают при кипячении с обратным холодильником в течение 9 ч в атмосфере азота.
После охлаждения нерастворимые вещества отфильтровывают и фильтрат разбавляют этилацетатом, промывают водой дважды и рассолом (насыщенным солевым раствором) один раз и высушивают над безводным сульфатом натрия.
После отгонки растворителя остаток обрабатывают на препаративной cиликагелевой тонкослойной хроматографии (проявляющий растворитель: этилацетат). Таким образом получают 40 мг названного соединения (выход: 4%). Некоторую часть этого продукта реакции кристаллизуют затем из этилацетата, чтобы тем самым получить светло-желтые кристаллы.
[Аналитические данные] т.пл. 180-182оC.
Оптическая чистота 100% энантиомерного избытка приведена в табл.2.
Рекомендуемый пример 2. (а) Cинтез (-)-7,8-дигидро-6,6-диметил-7- окси-8-(n-1-оксо-5-хлор)-пентил)-амино-6Н-пирано [2,3-f]-бенз-2,1,3-оксадиазола (интермедиат)
O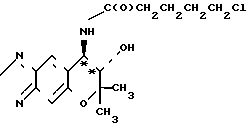
При комнатной температуре перемешивают 769 мг (3,27 ммоля) (-)-7,8-дигидро-6,6-диметил-7-окси-8-амино-6Н-пирано [2,3-f] -бенз-2,1,3-оксадиазола (соединение [-] I), 500 мкл (3,60 ммолей) триэтиламина и 70 мл хлористого метилена. К полученному раствору добавляют 465 мкл (3,60 ммолей) хлорангидрида 5-хлор-валериановой кислоты. После реакции взаимодействия в течение 2 ч, реакционную смесь промывают водой трижды. Метиленхлористую фазу высушивают над безводным сульфатом натрия и фильтруют. После отгонки растворителя получают названное соединение. Этот продукт реакции далее не очищают каким-либо образом, но как таковой используют в следующей реакции.
(б) Синтез (-)-7,8-дигидро-6,6-диметил-7-окси-8-(2-оксо-1- пиперидинил)-6Н-пирано-[2,3-f] -бенз-2,1,3-оксадиазола (соответ- ствует соединению III**
O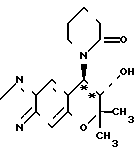
В 200 мл ацетона суспендируют 1,16 г (-)-7,8-дигидро-6,6-диметил-7-окси-8-(п-(1- оксо-5-хлор)-пентил) -амино-6Н-пирано-[2,3-т]-бенз-2,1,3-оксадиазола, 9,04 г (65,4 ммолей) карбоната калия и 1,09 г (6,54 ммолей) йодистого калия и смесь нагревают при кипячении с обратным холодильником в течение девяти часов в атмосфере азота.
После охлаждения нерастворимые вещества отфильтровывают и фильтрат разбавляют этилацетатом, промывают насыщенным водным раствором обычной соли один раз и высушивают над безводной глауберовой солью.
После отгонки растворителя остаток обрабатывают с помощью препаративной тонкослойной хроматографии на силикагеле (проявляющий растворитель: этилацетат). Таким образом получают 47 мг названного соединения (выход: 5%). Некоторую часть этого продукта реакции кристаллизуют затем из этилацетата, чтобы тем самым получить светло-желтые кристаллы.
[Аналитические данные] т.пл. 180-182оС.
Оптическая чистота 100% энантиомерного избытка приведена в табл.2.
Рекомендуемый пример 3. Синтез (+)-7,8-дигидро-6,6-диметил-7-окси-8-пропиониламино-6Н-пирано- [2,3-f] -бенз-2,1,3-оксадиазола (соответствует соединению III*
O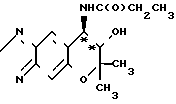
При комнатной температуре перемешивают 1,29 г (5,48 ммолей) (+)-7,8-дигидро-6,6-диметил-7-окси-8-амино-6Н-пирано- [2,3-f]-бенз-2,1,3-оксадиазола (соединение [+] 1), 690 мг (6,8 ммолей) триэтиламина и 40 мл хлористого метилена, добавляя при этом к смеси 610 мг (6,6 ммолей) пропионил-хлорида (хлорангидрид пропионовой кислоты). Смесь перемешивают при комнатной температуре в течение 4 ч. Реакционную смесь экстрагируют с помощью 600 мл этилацетата и 300 мл воды. Органическую фазу собирают и высушивают над безводным сульфатом натрия. Фильтрат и остаток, полученный после отгонки растворителя, кристаллизуют из смеси растворителей, содержащей 10 г этилацетата и 5 г гексана, оставляют стоять в холодильнике в течение ночи, а затем фильтруют путем отсасывания. Полученные кристаллы промывают с помощью 3 л порций смеси этилацетата/гексана (2:1) дважды и высушивают при пониженном давлении, чтобы тем самым получить названное выше соединение в виде бесцветного вещества.
[Аналитические данные] т.пл. 179-180оС.
Оптическая чистота: 100% э.и. приведен в табл.2.
Рекомендуемый пример 4. Синтез (-)-7,8-дигидро-6,6-диметил-7- окси-8-пропиониламино-6Н-пирано-[2,3-f] -бенз-2,1,3-окса- диазола (соединению III**).
O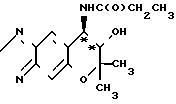
При комнатной температуре перемешивают 52 мг (0,22 ммоля) (-)-7,8-дигидро-6,6-диметил-7-окси-8-(н-(1-оксо-5-хлор)-пентил) -амино-6Н-пирано-[2,3-f] -бенз-2,1,3-окса- диазола (соединение (-) I), 34 мкл (0,24 ммоля) триэтиламина и 5 мл хлористого метилена, добавляя при этом к смеси 21 мкл (0,24 ммоля) пропионил-хлорида. Смесь перемешивают при комнатной температуре в течение шести часов.
После завершения реакции реакционную смесь промывают водой трижды и высушивают над безводным сульфатом магния. После отгонки растворителя остаток перекристаллизовывают из этанола, чтобы тем самым получить 15 мг названного выше чистого соединения (выход: 23%).
[Аналитические данные] т.пл. 179-180оС.
Оптическая чистота: 100% э.и. приведена в табл.2.
Рекомендуемый пример 5. Синтез (+)-7,8-дигидро-6,6-диметил-7-окси -8-метилуреидо-6Н-пирано-[2,3-f] -бенз-2,1,3-оксади- азола (соответствующего соединению (+)-IV).
O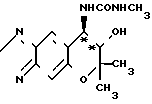
При комнатной температуре перемешивают 300 мг (1,28 ммоля) (+)-7,8-дигидро-6,6-диметил-7-окси-8-амино-6Н-пирано- [2,3-f]-бенз-2,1,3-оксадиазола (соединение (+) I) и 15 мл хлористого метилена. К полученному раствору добавляют 120 мг (2,10 ммоля) метилизоцианата. Смесь перемешивают при комнатной температуре (20оС) в течение 3 ч.
Реакционную смесь кристаллизуют в холодильнике и кристаллы, выпадающие в осадок таким образом, фильтруют. Таким образом получают 214 мг названного соединения в виде бесцветных кристаллов (выход: 58%).
Аналитические данные: т.пл. 165-167оС.
Оптическая чистота: 100% э.и. приведена в табл.2.
Рекомендуемый пример 6. Синтез (-)-7,8-дигидро-6,6-диметил-7-окси -8-метилуреидо-6Н-пирано-[2,3-f] -бенз-2,1,3-оксадиазола (соответствующего соединению (-) IV).
O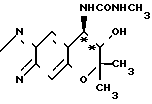
При комнатной температуре перемешивают 300 мг (1,28 ммоля) (-)-7,8-дигидро-6,6-диметил-7-окси-8-амино-6Н-пирано-[2,3-f] -бенз-2,1,3- оксодиазола (соединение (-) 1) и 20 мл хлористого метилена. К полученному раствору добавляют 120 мг (2,10 ммоля) метилизоцианата. Смесь перемешивают при комнатной температуре (20оС) в течение 5 ч.
Реакционную смесь кристаллизуют в холодильнике и выпадающие таким образом кристаллы фильтруют. Таким образом получают 195 мг названного выше соединения в виде бесцветных кристаллов (выход: 52%).
Аналитические данные: т.пл. 165-167оС.
Оптическая чистота: 100% э.и. приведена в табл.2.
Рекомендуемый пример 7. Синтез 7,8-дигидро-6,6-диметил-7,8-эпокси -6Н-пирано-[2,3-f]-бенз-2,1,3-оксадиазол-3-оксида (соединение G)
O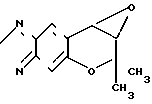
При комнатной температуре перемешивают 4,41 г (18,9 ммолей) 6-амино-3,4-дигидро-2,2-диметил-3,4-эпокси-7-нитро-2Н-бен- зо-[b]-пирана (соединение (F)), 1,29 г (32 ммолей) гидроокиси натрия, 400 мл этанола и 40 мл воды, добавляя медленно по каплям в эту смесь 32,3 г (26 ммолей) 6%-ного водного раствора гипохлорита натрия. Затем полученную смесь перемешивают в течение 1 ч.
После завершения реакции добавляют 1 л водного раствора обычной соли и смесь экстрагируют этилацетатом трижды. Этилацетатные фазы объединяют, промывают насыщенным солевым раствором и высушивают над безводным сульфатом натрия.
После отгонки растворителя остаток обрабатывают для очистки с помощью колочной хроматографии на силикагеле (проявляющий растворитель: этилацетат/гексан 1: 2 по объему). Таким образом получают 4,00 г названного выше соединения в виде желтых кристаллов (выход: 92%).
Аналитические данные: т.пл. 144-145оС
Рекомендуемый пример 8. Синтез 7,8-дигидро-6,6-диметил-7,8-эпокси -6Н-пирано-[2,3-f]-бенз-2,1,3-оксадиазола (соединение Н).
O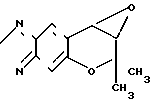
При 60оС перемешивают 1,00 г (4,27 ммоля) 7,8-дигидро-6,6- диметил-7,8-эпокси-6Н-пирано-[2,3-f] -бенз-2,1,3- оксадиазол-3-оксида (соединение G) и 6 мл бензола, добавляя при этом по каплям в пределах 15 мин 0,80 мл (4,70 ммоля) триэтилфосфита. Затем полученную смесь перемешивают в течение 3 ч.
После отгонки растворителя остаток подвергают очистке с помощью хроматографии на силикагелевой колонке (проявляющий растворитель: этилацетат/гексан 1: 1 по объему). Таким образом получают 0,82 г названного выше соединения (выход: 88%).
Некоторую часть этого вещества перекристаллизовывают из гексана, чтобы тем самым получить желтые кристаллы.
Аналитические данные: т.пл. 97-99оС
Рекомендуемый пример 9. Синтез 7,8-дигидро-6,6-диметил-7- окси-8-амино-6Н-пирано-[2,3-f]-бенз-2,1,3-оксадиазола (соединение ± 1)
O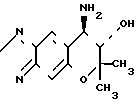
В 25 мл 16,7% -ного раствора аммиака в этаноле растворяют 0,82 г (3,8 ммоля) 7,8-дигидро-6,6-диметил-7,8-эпокси-6Н-пирано-[2,3-f] -бенз-2,1,3-оксадиазола (соединение Н), а затем смеси позволяют реагировать в толстостенной стеклянной трубке под давлением при 60оС в течение 48 ч.
Растворитель реакции отгоняют и остаток подвергают хроматографии на силикагелевой колонке (проявляющий растворитель: смесь этилацетата и метанола 5: 1 по объему), чтобы тем самым получить 0,77 г названного выше соединения в виде коричневого твердого вещества (выход: 87%).
Некоторую часть этого вещества перекристаллизовывают из этанола, чтобы тем самым получить чистое названное выше соединение в виде бесцветных кристаллов.
Аналитические данные: т.пл. 159-162оС.
ЯМР (СDCl3 + DMCO-d6) м.д. 1,26 (3Н), 1,49 (3Н), 2,80-3,30 (5Н), 3,33 (1Н), 3,78 (1Н), 6,82 (1Н) и 7,98 (1Н).
Масс-спектр:
133 (50%), 163 (100%) и 235 (M+, 3%).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПТИЧЕСКОГО РАЗДЕЛЕНИЯ ПИРАНОБЕНЗОКСАДИАЗОЛЬНОГО СОЕДИНЕНИЯ | 1990 |
|
RU2026297C1 |
| ПРОИЗВОДНЫЕ ПИРАНОБЕНЗОКСАДИАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ. | 1989 |
|
RU2054007C1 |
| ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ | 1994 |
|
RU2128043C1 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО(3,4-B)ПИРИДИНА | 1989 |
|
RU2022964C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕВАЛОНОЛАКТОНОВЫХ ПРОИЗВОДНЫХ | 1989 |
|
RU2045529C1 |
| СЕРУСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СОСТАВ, ОБЛАДАЮЩИЙ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ НА КОСТНУЮ РЕЗОРБАЦИЮ | 1989 |
|
RU2041875C1 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА, ОПТИЧЕСКИ ИНЕРТНЫЕ ИЛИ ИМЕЮЩИЕ R- ИЛИ S-КОНФИГУРАЦИЮ C-2 И C-3 АСИММЕТРИЧНЫХ ЦЕНТРОВ, ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2039050C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ АНТАГОНИСТА АНГИОТЕНЗИНА II И СПОСОБ АНТАГОНИЗИРОВАНИЯ АНГИОТЕНЗИНА II У МЛЕКОПИТАЮЩИХ | 1992 |
|
RU2104276C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХИНОЛИНКАРБАЛЬДЕГИДА | 1999 |
|
RU2217423C2 |
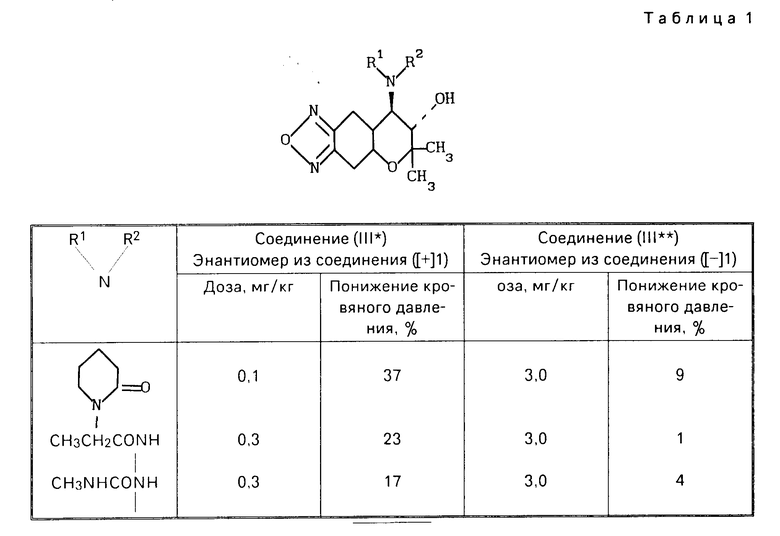
Использование: в медицине, в частности в синтезе препарата. полезного при лечении гипертензии. Сущность изобретения: продукт (+)форма 7,8-дигидро -6,6-диметил -7-окси-8- амино-6Н- пирано(2,3 f)бенз- 2, 1,3-оксадиазол, угол вращения при 25°С +189, выход 96% т. пл. 145 - 146°С. 2 табл.
(+) Форма 7,8-дигидро-6,6- диметил-7-окси-8- амино-6Н-пирано (2,3-f)бенз-2,1,3-оксадиазола формулы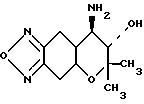
показывающая правое вращение в этаноле.
| Chem.Rev., 1972, 72, 627. |
