Это изобретение относится к производным амидина, способам их получения, содержащим их композициям и к их применению в терапии.
В качестве нейрозащитных средств были описаны определенные азотсодержащие соединения. В международной заявке WO 91/12797 (Штат Оригон) в качестве нейрозащитных средств представлены три-тетразамещенные гуанидины. В патенте США N 5266594 (Dawson et al.) (опубликованном после самой ранней даты приоритета этой заявки) описано применение производных аргинина при лечении удара и других нейродегенеративных заболеваний. Кроме того, в заявке на Европейский патент 547558 (Вашингтонский университет) описано применение аминогуанидина при лечении иммунологических и других расстройств.
Применение ингибиторов синтетазы оксида азота (V) при лечении заболеваний описано также, например, в международных заявках на патент WO 94/12163 (Абботт) и WO 94/12165 (Велкам) (обе опубликованы после самой ранней даты приоритета этой заявки) и в заявке на Европейский патент 4446699 (Мерелл Дау).
Производные амидина при использовании в качестве гербицидов описаны в заявке на патент Германии DE-OS-2321330 (Байер).
В патенте США N 3669974 (Фармацетикал Корп. USV) и заявке на патент Великобритании 2226562 (Бутс) описаны также производные N-фениламидина, используемые при лечении диабета. В заявке на международный патент WO 92/04054 (Университет Оригоны) описаны N',N''-дизамещенные амидины, используемые для лечения гипертензии, депрессивных синдромов и галюциногенных состояний. В патенте UK N 1180629 (Делаланд) описано использование определенных симметрических бис-амидинов в качестве аналгезирующих средств при лечении воспалений и гипертензии.
В ряде патентных документов описаны способы получения амидинов или применение амидинов в качестве промежуточных продуктов без раскрытия фармацевтического применения этих соединений. Простые производные амидина в качестве промежуточных продуктов при получении пригодных производных бензимидазола описаны в патенте UK 1088095 (Мерк). Способы получения других простых N-арил- и N-гетероарильных амидинов описаны в патенте США 3299081 (Мерк), а фторосодержащие производные амидина в качестве химических промежуточных продуктов описаны в заявке на патент Японии N 58057357 (Дейкин).
Мы обнаружили новую группу производных амидина, которые обладают пригодной фармацевтической активностью.
В соответствии с первым аспектом изобретения мы обеспечили соединение формулы I: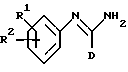
в которой D представляет фенил, пиридинил или 5-членное гетероциклическое ароматическое кольцо, содержащее от 1 до 4 гетероатомов, выбранных из O, S и N, три группы которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила, C1-C6-алкокси, галогена и C1-C6-перфторалкила; или C1-C6-перфторалкил;
R1 представляет водород, C1-C6-алкил или галоген;
R2 представляет -X(CH2)n ZCONR3R4, -X(CH2)n NHCO(CH2)3NR3R4, -X(CH2)pNR3R4, -X(CH2)nNHCOR5 или -(CH2)q NHC(NH)R6;
R3 и R4 независимо представляют водород, C1-C6-алкил, (CH2)rA, -(CH2)mOA или CH(CH3)(CH2)tA; или NR3R4 вместе представляют 1-инданил, пиперониламино-, пиперидинил, морфолинил, пирролидинил, 1,2,3,4-тетагидроизохинолинил; или пиперазинил, необязательно 4-замещенный C1-C6-алкилом;
R6 представляет C1-C6-алкил, C1-C6-перфторалкил, -(CH2)rA или -O(CH2)wA; A представляет фенил, пиридинил, пиримидинил или 5-членное гетероциклическое ароматическое кольцо, содержащее от 1 до 4-х гетероатомов, выбранных из O, S и N, четыре группы которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила, галогена, нитро, циано и трифторметила;
R6 представляет фенил, пиридинил или 5-членное гетероциклическое ароматическое кольцо, содержащее от 1 до 4-х гетероатомов, выбранных из O, S и N, три группы которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила, C1-C6-алкокси, галогена и C1-C6-перфторалкила; или C1-C6-перфторалкил,
n и r независимо представляют целое число от 0 до 6 включительно;
p и w независимо представляют целое число в диапазоне от 1 до 5 включительно;
m представляет целое число в диапазоне от 2 до 5 включительно;
g и t независимо представляют целое число в диапазоне от 0 до 5 включительно;
S представляет целое число от 1 до 3 включительно;
X представляет O или связь;
Z представляет O, NR7 или связь;
R7 представляет водород или C1-C6-алкил; при условии, что:
(a) когда D содержит гетероатом, он не связан с остатком соединения формулы I через гетероатом;
(b) когда R2 представляет -X(CH2)nZCONR3R4 и ни X, ни Z не представляют связь, тогда n представляет целое число в диапазоне от 2 до 6 включительно;
(c) когда R2 представляет -X(CH2)nNHCO(CH2)sR3R4 или X(CH2)nNHCOR5, и X представляет O, тогда n представляет целое число в диапазоне от 2 до 6 включительно;
(d) когда R2 представляет -X(CH2)pNR3R4 и X представляет O, тогда p представляет целое число в диапазоне от 2 до 5 включительно;
(e) когда R2 представляет -(CH2)gNHC(NH)R6, R1 представляет водород и D и R6 имеют одинаковое определение и представляют фенил, необязательно замещенный C1-C4-алкилом, или одной или несколькими группами C1-C3-алкокси или одним или несколькими атомами галогена; или пиридинил, тогда q не равно 0;
или его фармацевтически приемлемую соль.
Предпочтительно, чтобы D представлял фенил, пиридинил или 5-членное гетероциклическое ароматическое кольцо, содержащее от 1 до 4-х гетероатомов, выбранных из O, S и N, три группы которого необязательно замещены одной или несколькими группами, выбранных из C1-C6-алкила, C1-C6 -алкокси, галогена или C1-C6-перфторалкила.
Более предпочтительно, чтобы D представлял фенил, тиофен, фуран, пиррол или тиазол, пять групп которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила, C1-C6-алкокси, галогена или C1-C6-перфторалкила.
Еще в большей степени предпочтительно, чтобы D представлял тиофен, пиррол, фуран или тиазол, четыре группы которого необязательно замещены C1-C6-алкилом или галогеном.
Главным образом предпочтительно, чтобы D представлял тиофен, фуран или пиррол, наиболее предпочтительно тиофен, D представлял 2-тиофен, R1 представлял водород.
Когда R2 представляет -X(CH2)nZCONR3R4, -X(CH2)n NHCO(CH2)sNR3R4, или -X(CH2)pNR3R4, мы предпочитаем, чтобы -NR3R4 представлял пиперидинил, морфолинил, пирролидинил, 1,2,3,4-тетрагидроизохинолинил или 1-инданил, или, чтобы по крайней мере один из R3 и R4 представлял (CH2)rA или -(CH2)mOA. Мы в особенности предпочитаем, чтобы -NR3R4 представлял 1,2,3,4-тетрагидроизохинолинил или 1-инданил, или чтобы один из R3 и R4 представлял -(CH2)rA и другой представлял водород или метил. Мы, главным образом, предпочитаем, чтобы один из R3 и R4 представлял -(CH2)rA и другой представлял водород или метил.
Когда R2 представляет -X(CH2)nNHCOR5, мы предпочитаем, чтобы R5 представлял -(CH2)rA.
Когда R2 представляет -X(CH2)nZCONR3R4, -X(CH2)nNHCO(CH2)sNR3R4,
-X(CH2)pNR3R4 или -X(CH2)nNHCOR5, мы предпочитаем, чтобы X представлял связь.
Когда R2 представляет -X(CH2)nZCONR3R4 и Z представляет NR7, мы предпочитаем, чтобы R7 представлял водород.
Когда R2 представляет -X(CH2)nZCONR3R4, мы предпочитаем, чтобы Z представлял связь.
Когда R2 представляет -(CH2)gNHC(NH)R6, мы предпочитаем, чтобы R6 представлял фенил или 5-членное гетероциклическое ароматическое кольцо, содержащее от 1 до 4 гетероатомов, выбранных из O, S и N, две группы которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила, C1-C6-алкокси и галогена.
Когда R2 представляет -(CH2)gNHC(NH)R6, мы в особенности предпочитаем, чтобы R6 представлял фенил или тиофен, две группы которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила и галогена.
Когда R2 представляет -(CH2)qNHC(NH)R6, мы предпочитаем, чтобы q представлял 0, 1 или 2.
Мы в особенности предпочитаем, чтобы q представлял 0 или 2, главным образом 0.
Когда R2 представляет -(CH2)gNHC(NH)R6, g представляет O и R6 представляет фенил, необязательно замещенный галогеном, C1-C6-алкилом или C1-C6-алкокси, или R6 представляет пиридинил, тогда мы предпочитаем, чтобы D не имел такое же определение, как К6.
Когда R2 представляет -(CH2)qNHC (NH)R6 и g представляет 0, тогда мы обычно предпочитаем, чтобы R6 не имел такое же определение, как D.
Когда R2 представляет -X(CH2)pNR3R4, мы предпочитаем, чтобы p представляло целое число в диапазоне от 1 до 4 включительно, в особенности 1, 2 или 3, главным образом 1 или 2.
Когда R2 представляет -X(CH2)nZCOHR3R4, -X(CH2)nNHCO(CH2)sNR3R4 или -X(CH2)nNHCOR5, мы предпочитаем, чтобы n представляло 1, 2 или 3, главным образом 2 или 3.
Когда R3, R4 или R5 представляют -(CH2)rA, мы предпочитаем, чтобы r представляло целое число в диапазоне от 0 до 4 включительно, в особенности 0, 1 или 2, более предпочтительно 1 или 2, главным образом 1.
Когда R3 или R4 представляют -(CH2)m OA, мы предпочитаем, чтобы m представляло 2, 3 или 4.
Когда R5 представляет -O(CH2)wA, мы предпочитаем, чтобы w представляло 2, 3 или 4.
Когда R3 или R4 представляет -CHMe(CH2)tA, мы предпочитаем, чтобы t представлял 0, 1 или 2, главным образом 0 или 1.
Мы предпочитаем, чтобы A представляло фенил, пиридинил, пиримидинил, тиофенил или фуранил, пять групп которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила и галогена. Мы в особенности предпочитаем, чтобы A представляло фенил, необязательно замещенный одной или несколькими группами, выбранными из C1-C6-алкила и галогена.
Когда D или R6 представляют C1-C6-перфторалкил, мы предпочитаем, чтобы они представляли пентафторэтил или трифторметил, главным образом трифторметил.
Мы предпочитаем, чтобы R2 представлял -X(CH2)pNR3R4 или -(CH2)gNHC(NH)R6.
Мы предпочитаем, чтобы R2 был ориентирован в мета- или параположении по отношению к атому азота амидиновой составляющей.
В соответствии с изобретением мы, кроме того, обеспечиваем способ получения соединений формулы I и его фармацевтически приемлемых солей, который включает:
(a) получение соединения формулы I путем взаимодействия соответствующего соединения формулы II: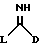
в которой D является таким, как он определен выше, и L является отщепляемой группой, с соединением формулы (III),
в которой R1 и R2 являются такими, как они определены выше;
(b) получение соединения формулы I путем взаимодействия соответствующего соединения формулы IV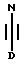
в которой D является таким, как он определен выше, с соединением формулы V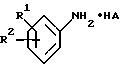
в которой R1 и R2 являются такими, как они определены выше, и HA является кислотой;
(c) получение соединения формулы I, в которой R2 представляет -X(CH2)nZCONR3R4, -X(CH2)nNHCO(CH2)sNR3R4 или -X(CH2)pNR3R4 и по крайней мере один из R3 и R4 представляет C1-C6-алкил, -(CH2)rA, -(CH2)mOA или -CH(CH3)(CH2)tA, путем взаимодействия соответствующего соединения формулы I, в которой один или оба из R3 и R4 представляют водород, с соединением формулы VI
R8 - L,
в которой R8 представляет C1-C6-алкил, -(CH2)rA, -(CH2)mOA или -CH(CH3)-(CH2)tA, и L является отщепляемой группой;
(d) получение соединения формулы I, в которой R2 представляет -(CH2)gNHC(NH)R6, путем взаимодействия соответствующего соединения формулы VII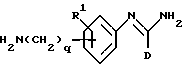
в которой D, R1 и O являются такими, как они определены выше,
с соединением формулы VIII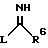
в которой R6 является таким, как он определен выше, и L является отщепляемой группой;
(e) получение соединения формулы I, в которой R2 представляет -(CH2)gNHC(NH)R6, путем взаимодействия соответствующего соединения формулы IX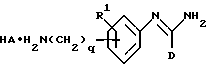
в которой D, R1, q и HA являются такими, как они определены выше, с соединением формулы X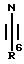
в которой R6 является таким, как он определен выше;
(f) получение соединения формулы I, в которой R2 представляет -X(CH2)nZCONR3R4, путем взаимодействия соответствующего соединения формулы XI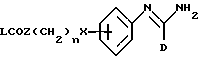
в которой D, R1, X, n, Z и L являются такими, как они были определены выше, с соединением формулы XII,
R3R4NH,
в которой R3 и R4 являются такими, как они определены выше;
(q) получение соединения формулы I, в которой R2 представляет -X(CH2)nNHCO(CH2)sNR3R4, путем взаимодействия соединения формулы XIII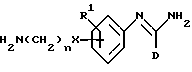
в которой D, R1, X и n являются такими, как они определены выше, с соединением формулы XIV
R3R4N(CH2)sCOL,
в которой R3, R4 и S являются такими, как они определены выше, и L является отщепляемой группой;
(h) получение соединения формулы I, в которой R2 представляет -X(CH2)nNHCOR5, путем взаимодействия соединения формулы XIII с соединением формулы XV
R5COL,
в которой R5 является таким, как он определен выше, и L является отщепляемой группой;
(i) получение соединения формулы I, в которой R2 представляет -X(CH2)nZCONR3R4 и Z представляет NR7, путем взаимодействия соответствующего соединения формулы I, в которой R2 представляет -X(CH2)nZCONR3R4 и Z представляет -NH, с соединением формулы XVI
R7 - L,
в которой R7 является таким, как он определен выше, и L является отщепляемой группой;
(j) получение соединения формулы I, в которой R2 представляет -X(CH2)pNR3R4 и p не менее чем 2, путем восстановления соединения формулы XVII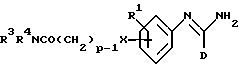
в которой D, X, R1, R3, R4 и p являются такими, как они определен выше;
(k) получение соединения формулы I, в которой R2 представляет -X(CH2)pNR3R4 и оба R3 и R4 представляют водород, путем восстановления соединения формулы XVIII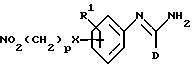
в которой R1, D, p и X представляются такими, как они определены выше;
(l) получение соединения формулы I, в которой R2 представляет -X(CH2)sZCONR3R4, Z представляет O или NR7 и R3 представляет водород, путем взаимодействия соединения формулы XIX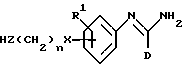
в которой R1, D, X и n являются такими, как они определены выше, и Z представляет O или NR7 с соединением формулы XX
R4 - N = C = O,
в которой R4 является таким, как он определен выше;
(m) получение соединения формулы I, в которой R2 представляет -X(CH2)nNHCOR5 и R5 представляет -O(CH2)wA, путем взаимодействия соединения формулы XXI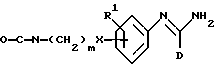
в которой R1, D, X и n являются такими, как они определены выше,
с соединением формулы XXII
A(CH2)wOH,
в которой A и w являются такими, как они определены выше;
(n) получение соединения формулы I, в которой R2 представляет -X(CH2)nZCONR3R4 и Z представляет O или NR7, путем взаимодействия соединения формулы XIX с соединением формулы XXIII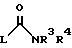
в которой R3 и R4 являются такими, как они определены выше;
(o) получение соединения формулы I, в которой R2 представляет -X(CH2)pNR3R4, R3 представляет водород и p представляет целое число от 2 до 5, путем восстановления соединения формулы XXIV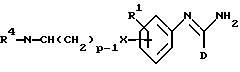
в которой R1, R4, D, X и p являются такими, как они определены выше;
(p) получение соединения формулы I, в которой R2 представляет -X(CH2)pNR3R4, один из R3 и R4 представляет водород и другой представляет -(CH2)rA, где r представляет целое число от 2 до 6, путем восстановления соединения формулы XXV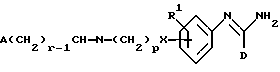
в которой R1, A, D, r и p являются такими, как они определены выше;
(q) получение соединения формулы I, в которой R2 представляет -X(CH2)pNR3R4, один из R3 и R4 представляет водород, а другой представляет -(CH2)mOA, путем восстановления соединения формулы XXVI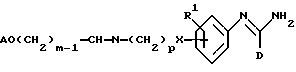
в которой R1, A, D, p и m являются такими, как они определены выше,
(r) получение соединения формулы I, в которой R2 представляет -X(CH2)pNR3R4, один из R3 и R4 представляет водород, и другой представляет -(CH2)rA, где r представляет целое число от 2 до 6, путем восстановления соединения формулы XXVII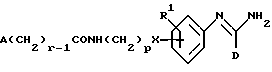
в которой R1, A, D, p и r являются такими, как они определены выше;
(s) получение соединения формулы I, в которой R2 представляет -X(CH2)pNR3R4, один из R3 и R4 представляет водород, и другой представляет -(CH2)mOA, путем восстановления соединения формулы XXVIII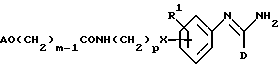
в которой R1, A, D, p и m являются такими, как они определены выше, и когда желательно или необходимо превращение полученного соединения формулы I или его соли в его фармацевтически приемлемую соль или наоборот.
В способе (a) реакция проходит при перемешивании смеси реагента в соответствующем растворителе, например низшем спирте, например этаноле, изопропаноле или третичном бутаноле, при температуре между комнатной температурой и температурой флегмы растворителя. Время реакции будет зависеть, между прочим, и от природы отщепляемой группы и может составить до 48 часов, однако, оно обычно составляет от 1 часа до 5 часов. Подходящие отщепляемые группы, которые может представлять 1, включают группы тиоалкила, сульфоновой кислоты, трифторуглеродсульфокислоты, галогенида, алкильных и арильных спиртов и тозильные группы; другие перечислены в "Advanced Organic Chemistry", J. Merch (1985) 3rd Edition, McGrow-Hill на стр. 315, и являются хорошо известными в данной области.
В способе (b) реакцию предпочтительно осуществляют путем нагревания с обратным холодильником смеси двух соединений в течение нескольких часов в присутствии соответствующего растворителя, вследствие чего температура реакции является достаточно высокой для того, чтобы быстро происходила конденсация, но недостаточно высокой для разложения образованного амидина. Температура реакции может изменяться от комнатной температуры до около 250oC, хотя предпочтительно осуществлять реакцию при температурах от около 100oC до 200oC. Мы обнаружили, что о-дихлорбензол является в особенности подходящим растворителем и что полезно добавить 4-диметиламинопиридин в качестве катализатора. При охлаждении образуются 2 слоя, растворитель может быть декантирован, и реакция может быть завершена путем добавления водного основания. Альтернативно, когда реагенты растворимы в растворителе, растворитель может быть выпарен под вакуумом, и реакционной смеси придают законченный вид путем добавления воды.
Кислота HA может быть органической или неорганической кислотой, например соляной, бромистоводородной, иодистоводородной, серной, азотной, фосфорной, уксусной, молочной, янтарной, фумаровой, яблочной, малеиновой, винной, лимонной, бензойной или метансульфокислотой.
В способе (c) реакция происходит при стандартных условиях, например при взаимодействии двух материалов в инертном растворителе в основной среде при комнатной температуре в течение времени до 12 часов. Мы многократно обнаруживали, что перед взаимодействием с соединением формулы II амин желательно обработать NaH. Мы предпочитаем, чтобы L представлял галоген, в особенности бромид.
Способ (d) можно осуществлять в условиях, аналогичных тем, которые описаны выше для способа (a).
Способ (e) можно осуществлять в условиях, аналогичных тем, которые описаны выше для способа (b).
Способы (f), (q) и (h) можно осуществлять при стандартных условиях, хорошо известных в данной области для конденсации амина и карбоновой кислоты или активированной карбоновой кислоты для образования амида. Реакция соединения для образования амида может быть осуществлена, например, при перемешивании реагентов в течение 12-24 часов при температуре между 0oC и 25oC в воде или в смеси воды и менее полярного растворителя, например диоксана, тетрагидрофурана или этанола. Мы предпочитаем осуществлять реакцию в основной среде, например, в присутствии водного раствора карбоната натрия или бикарбоната натрия.
Способ (i) можно осуществить при стандартных условиях, аналогичных тем, которые указаны выше для способа (c).
В способе (j) восстановление можно осуществлять путем обработки дибораном в инертном растворителе, например THF.
Альтернативные, хотя менее предпочтительные реагенты, которые могут быть подходящими, включают алюмогидрид лития и реагенты для каталитической гидрогенизации, например H2 на Pd/C. Дополнительные сведения, касающиеся реакционных условий для этих реакций, могут быть получены при обращении к ссылке J. March "Advanced Organic Chemistry", стр. 1099, включая ссылки, цитированные в этом источнике.
В способе (k) реакцию восстановления можно осуществлять при различных условиях, например при тех, которые описаны в J. March "Advanced Organic Chemistry", на стр. 1103 - 1104. Эти условия включают каталитическую гидрогенизацию, применение Zn, Sn или Fe, AlH3-AlCl3, сульфидов и других веществ. Мы предпочитаем осуществлять реакцию путем гидрогенизации при атмосферном давлении в течение 3-6 часов в присутствии палладиевого и углеродного катализатора.
В способе (l) и (m) реакцию можно осуществлять путем перемешивания реагентов в присутствии инертного растворителя при температуре между комнатной температурой и температурой флегмы растворителя в течение времени до 24-х часов.
Способ (n) можно осуществлять при условиях, аналогичных тем, которые описаны выше для способов (f), (q) и (h).
В способах (o), (p) и (q) восстановление можно осуществлять путем обработки соединения боргидридом натрия при стандартных условиях.
В способах (r) и (s) реакцию можно осуществлять при условиях, аналогичных тем, которые описаны выше для способа (j).
Соли соединений формулы I могут быть образованы путем взаимодействия свободной кислоты, основания или соли, энантиомера, таутомера или их защищенной производной с одним или несколькими эквивалентами соответствующего основания или кислоты. Реакцию можно осуществлять в растворителе или среде, в которой соль нерастворима, например в воде, диоксане, этаноле, тетрагидрофуране или диэтиловом эфире, или в смеси растворителей, которые могут быть удалены в вакууме или сушкой вымораживанием. Реакция может представлять обменный процесс или ее можно осуществлять на ионообменной смоле.
Для специалистов в данной области очевидно, что может быть желательным защитить гидроксильную группу, аминогруппу или другую реакционноспособную группу с использованием защитной группы, которая описана в руководстве "Protecting groups in Organic Synthesis", 2-nd Edition (1991) Greene and Wuts. Защитные группы для аминогруппы, которые могут быть упомянуты, включают C2-C7-алкилоксикарбонил, например трет-бутилоксикарбонил, C8-C13-фенилалкилоксикарбонил, например бензилоксикарбонил, или предпочтительно трифторацетат. Защитную группу обычно удаляют при обработке водным раствором основания.
Соединения формулы II являются или известными или могут быть получены известными методами. Например, соединения формулы II, в которых L представляет тиоалкил, могут быть получены путем обработки соответствующего тиамида формулы XXIX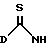
в которой D является таким, как он определен выше,
с алкилиодидом.
Соединения формулы (III) могут быть получены путем восстановления соответствующего соединения формулы XXX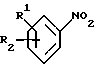
в которой R1 и R2 являются такими, как они определены выше.
Реакцию восстановления можно осуществлять при условиях, аналогичных тем, которые описаны выше для способа (k).
Определенные соединения формулы XXX являются или известными или могут быть получены обычными способами, по существу известными. Другие соединения формулы XXX могут быть получены из известных соединений с более простыми боковыми цепями методами, аналогичными тем, которые описаны для способов (c) - (s).
Соединения формулы V могут быть получены способами, аналогичными тем, которые описаны для получения соединений формулы III. Соединения формулы V могут быть превращены в соответствующие соединения формулы III путем обработки основанием. Соединения формулы III могут быть превращены в соответствующие соединения формулы V обработкой протонной кислотой HA, например одной из вышеперечисленных.
Соединения формулы VII, IX, XI, XIII; XVII, XXVIII, XIX, XXI, XXIV, XXV, XXVI, XXVII и XXVIII могут быть получены способами, аналогичными тем, которые описаны для получения соединений формулы I.
Соединения формулы VIII являются или известными или могут быть получены способом, аналогичным тому, который описан выше для получения соединений формулы II.
Соединения формулы IV, VI, X, XII, XVI, XX, XXII, XXIII и XXIX являются известными или могут быть получены обычными способами, по существу известными.
Соединения формулы XIV или XV являются или известными или могут быть легко получены из соответствующей карбоновой кислоты, которая или является известной или может быть получена обычными способами, по существу, известными.
Когда необходимо, гидроксильная группа, аминогруппа или другие реакционноспособные группы в промежуточных соединениях могут быть защищены с использованием защитной группы, которая описана в руководстве "Protecting groups in Organic Synthesis", 2-nd Edittion (1991 ) Greene and Wuts.
Соединения изобретения и промежуточные химические соединения могут быть выделены из их реакционных смесей стандартными методиками.
Термин "C1-C6-алкил" включает прямую цепь, разветвленные, насыщенные, ненасыщенные, алифатические и циклические алкильные группы, содержащие от 1 до 6 углеродных атомов.
Соединения формулы I могут существовать в таутомерных, энантиомерных или диастереомерных формах, все из которых включены в область изобретения. Путем разделения рацемической смеси соединений при использовании общепринятых методик, например фракционированной кристаллизации или HPLC (высокоэффективной жидкостной хроматографии), могут быть выделены различные оптические изомеры. Альтернативно отдельные энантиомеры могут быть получены путем взаимодействия соответствующих оптически активных исходных материалов при реакционных условиях, которые не вызывают рацемизацию.
Промежуточные химические соединения могут также существовать в энантиомерных формах и могут быть использованы в виде очищенных энантиомеров, диастереомеров, рацематов или смесей.
Соединения общей формулы I обладают пригодной фармакологической активностью по отношению к животным. В частности, они обладают пригодной ингибирующей активностью против синтетазы оксида азота (V) и ожидается, что они будут пригодны при лечении или профилактике заболеваний человека или состояний, в которых значительную часть составляет синтез или избыточный синтез оксида азота (V), например, гипоксии, например, в случаях остановки сердца и ударов, нейродегенеративных расстройств, включая дегенерацию и/или отмирание нервных клеток при таких расстройствах, как гипоксия, гипогликемия, эпилепсия, и наружных ран (например, спинного мозга и травм головы), судорог, связанных с гипербаризмом и токсичностью, слабоумия, например преждевременного слабоумия, болезни Альцгеймера и синдромов приобретенного иммунного дефицита (СПИДа), родственного со слабоумием, хореи Сиденгама, болезни Хантингтона, бокового амиотрофического склероза, полиневротического психоза, психической неполноценности, связанной с церебральными сосудистыми расстройствами, сонных болезней, шизофрении, депрессии, эмоциональных расстройств, связанных с переменой погоды, депрессии или других симптомов, связанных с предменструальным синдромом (PMS), тревоги и септического шока. Можно ожидать, что соединения формулы I проявляют активность также при предотвращении и аннулировании устойчивости к наркотикам и диазепинам, при лечении привыкания к чрезмерному употреблению лекарственных средств, ослаблении боли и лечении мигрени и других сосудистых заболеваний головного мозга. Соединения настоящего изобретения могут также проявлять пригодную иммунодепрессивную активность, быть пригодными при лечении или профилактике воспалений, при лечении желудочно-кишечных расстройств и при стимуляции родов.
Ожидается, что соединения формулы I в особенности пригодны при лечении нейродегенеративных расстройств при мигрени или для предотвращения и аннулирования устойчивости к наркотикам и диазепинам или для лечения привыкания к чрезмерному употреблению лекарственных средств и, главным образом, при лечении нейродегенеративных расстройств.
Таким образом, в соответствии с еще одним аспектом изобретения мы обеспечили соединение формулы I или его фармацевтически приемлемую соль для применения в качестве фармацевтического препарата.
В соответствии с еще одной особенностью изобретения мы обеспечили применение соединения формулы I, без условия (e), или его фармацевтически приемлемой соли при производстве лекарственного препарата для лечения вышеназванных заболеваний или состояний.
Для вышеупомянутых терапевтических показаний вводимая доза будет, конечно, изменяться в зависимости от используемого соединения, способа введения лекарственного средства и от желательного лечения. Однако, когда соединения вводят человеку при суточной дозе между 1 мг и 2000 мг (в пересчете на твердую форму) в день, получают удовлетворительные результаты.
Соединения формулы I и их фармацевтически приемлемые соли могут быть использованы сами по себе или в форме соответствующих медицинских препаратов для тонкокишечного или парентерального введения.
В соответствии с изобретением обеспечена технология приготовления лекарственного средства, предпочтительно включающая менее чем 80% и, более предпочтительно менее чем 50% соединения формулы I или его фармацевтически приемлемой соли, в смеси с фармацевтически приемлемым разбавителем или носителем.
Обеспечен также способ лечения из вышеупомянутых заболеваний или состояний, который включает введение терапевтически эффективного количества соединения формулы I, без условия (e), или его фармацевтически приемлемой соли, человеку, страдающему от такого заболевания или состояния.
Примерами таких разбавителей и носителей являются для таблеток и драже: лактоза, крахмал, стеариновая кислота; для капсул: винная кислота или лактоза; для инъецируемых растворов: вода, спирты, глицерин, растительные масла; для суппозиториев: природные или отвержденные масла или воски.
Композиции в форме, подходящие для орального введения, т.е. введения в пищевод, включают: таблетки, капсулы и драже; композиции для длительного облегчения боли включают такие, в которых активный ингредиент связан с ионообменной смолой, которую необязательно покрывают диффузионным барьером для изменения выделяющих свойств смолы.
Фермент синтеза оксида азота (V) имеет ряд изоформ и соединения формулы I или их фармацевтически приемлемые соли могут быть проверены на ингибирующую активность в отношении синтетазы оксида азота (V) методиками, основанными на тех, которые представлены в Bredt and Snyder in Proc. Natl. Acad. Sci. (1990), 87, 682-685 и Forstermann et al. (1992 ) Fur J. Pharm., 225, 161-165, как следует далее. Синтетаза оксида азота (V) превращает 3H-L-аргинин в 3H-L-цитрулин, который может быть отделен катионообменной хроматографией и определен количественно посредством сцинтиляционного счетчика жидкости.
Проверка A.
(A) Проверка на ингибирующую активность против нейронной синтетазы оксида азота (V).
Из гиппокампа или мозжечка крысы выделили фермент. Мозжечок или гиппокамп крысы мужского пола Sprague-Dawley (250-275 г) удалили, осуществив анестезию животного CO2 и декапитацию. Посредством гомогенизации в 50 мМ трис-HCl и декапитацию. Посредством гомогенизации в 50 мМ трис-HCl с 1 мМ буфера EDTA этилендиаминтетрауксусной кислоты (pH 7,2 при 25oC) и центрифугирования в течение 15 минут при 20000 q приготовили надосадочную жидкость мозжечка или гиппокампа.
Посредством последовательной хроматографии через колонки Dowex AG-50W-X8 в натриевой форме и водородной форме и центрифугирования при 10000 q в течение 30 с удалили остаточный L-аргинин из надосадочной жидкости.
Для анализа 25 мкл полученной надосадочной жидкости добавили в каждую из 12 пробирок, содержащих 25 мкл раствора L-аргинина (концентрации 18 мкл 1H-аргинина, 96 нм 3H-L-аргинина), или 25 мкл анализируемого буфера (50 мМ HEPES, 1 мМ EDTA, 1,5 мМ CaCl2, pH 7,4) или 25 мкл испытуемого соединения в буфере при 22oC. В каждую пробирку добавили 75 мкл конечного анализируемого буфера (50 мМ HEPES, 1 мМ EDTA, 1,5 мМ CaCl2, 1 мМ DTT, 100 мкм NaDPH, 10 мкг/мл калмодулина, pH 7,4) для начала реакции и через 10 минут реакцию остановили путем добавления 2 мл буферного раствора для завершения реакции (20 мМ HEPES, 2 мМ EDTA, pH 5,5).
Меченый L-цитрулин отделили от меченого L-аргинина хроматографией через колонку с Dowex AG-50W-X8 200-400. В отдельную 1 мл колонку добавили 1 мл буферного раствора, который использовали для завершения реакции, и элюент соединили с элюентом от двух промывок 1 мл дистиллированной воды и с 16 мл сцинтилляционной смеси. Затем с помощью сцинтилляционного счетчика количественно определили L-цитрулин.
В типичном эксперименте, в котором использовали надосадочную жидкость мозжечка, основная активность увеличилась на 20000 распадов в минуту/мл пробы относительно реагентного бланка, который имеет активность 7000 распадов в минуту/мл. Для проверки методики при анализе подвергали испытанию сравнительный эталон, N-нитро-L-аринин, который дает 60% ингибирования синтетазы оксида азота (V) при концентрации 1 мкм.
Проверка B.
(B) Проверка на ингибирующую активность против макрофаговой синтетазы оксида азота (V).
Из культивируемой солянокислой макрофаговой линии клеток, J1774A-1 (полученной из лабораторий Империал Кансер Рисерч Фанд) после индукции приготовили фермент. Клетки J774A-1 культивировали в Dulbecco's Modified Eagles Medium (DMEM), дополненной 10%-ной плодной бычьей сывороткой 4 мМ L-глутамина и антибиотиками (100 ед./мл пенициллина G, 100 мкг/мл стрептомицина и 0,25 мкг/мл амфотерицина B). Клетки выращивали в установленном режиме в 225 см3 колбах, содержащих 35 мл среды, поддерживаемой при 37oC, и в увлажненной атмосфере, содержащей 5% CO2. Синтетазу оксида азота (V) продуцировали посредством клеток в ответ на интерферон -γ(IFN-γ) и липополисахарид (LPS).
Из колб с культурой удалили среду и заменили ее 25 мл (на колбу) свежей среды, содержащей 1 мкг/мл LPS и 10 ед./мл IFN-γ. Через 17-20 часов осуществляли сбор клеток, выросших в культуре, посредством соскоба пласта клеток с поверхности колбы в культуральную среду. Клетки собрали центрифугированием (1000 г в течение 10 минут) и путем добавления к осадку клеток после центрифугирования раствора, содержащего 50 мМ трис-HCl (pH 7,5 при 20oC). 10% (об. /об. ) глицерина, 0,1% об. /об. Triton-X-100, 0,1 мкм дитиотреита и смеси ингибиторов протеазы, содержащей лейпетин (2 мкг/мл), ингибитор соевого трипсина (10 мкг/мл), апротинин (5 мкг/мл) и фенилметилсульфонилфторид (50 мкг/мл). Для анализа в ячейки фильтровальной пластины с 96-ю ячейками (размер пор 0,45 мкМ), содержащей 25 мкл раствора испытуемого соединения в 50 мМ трис-HCl, добавили 25 мкл питательной смеси (50 мМ трис-HCl, pH 7,5 при 20oC), 400 мкМ NaDPH, 20 мкМ флавинадениндинуклеотида, 20 мкМ флавинмононуклеотида, 4 мкМ тетрагидробиоптерина, 12 мкМ L-аргинина и 0,025 мкМ Cl L-[3H] аргинина). Реакцию начали путем добавления 50 мкл клеточного лизата (полученного, как указывалось выше) и после инкубации в течение 1 часа при комнатной температуре реакцию завершили путем добавления 50 мкл водного раствора, 3 мМ нитроаргинина и 21 мМ EDTA.
Меченый L-цитрулин отделили от меченого L-аргинина с использованием Dowex AG-50W. К анализируемому добавили 150 мкл 25%-ной водной суспензии Dowex 50W (Na+ форма), после чего полученное отфильтровали на пластинах, содержащих 96 ячеек. Отобрали 70 мкл фильтрата и добавили в ячейки пластин, имеющих 96 ячеек, которые содержали твердый сцинтиллянт. После сушки проб L-цитрулин определили количественно посредством сцинтилляционного счетчика.
В типичном эксперименте основная активность составляла 300 распадов в минуту/на 70 мкл пробы, которая возросла до 1900 распадов в минуту в контрольных реагентных группах. Для проверки методики в качестве стандарта испытывали аминогуанидин, который дает IC50 (концентрацию, дающую ингибирование фермента на 50%), равную 10 мкМ.
Проверка C.
(C) Проверка на ингибирующую активность против эндотелиальной синтетазы оксида азота (V).
Фермент может быть выделен из умбиликальных венных эндотелиальных клеток человека (HUVECs) методикой, основанной на той, которая представлена в Pollock et al., (1991) Proc., Nat. Acad. Sci., 88, 10480-10484, HUVEC приобретали от Клонетикс Корп. (Сан Диего, СА, США) и культивировали до слияния. Клетки можно сохранять без значительных потерь выхода синтетазы оксида азота (V). Когда клетки достигли слияния, их повторно суспендировали в фосфатной буферной соли Dulbecco, центрифугировали при 800 оборотах в минуту в течение 10 минут, осадок клеток после центрифугирования раствора гомогенизировали в 50 мМ трис-HCl, охлажденной льдом, 1 мМ EDTA, 10% глицерина, 1 мМ фенилметилсульфонилфторида, 2 мкМ лейптина при pH 4,2. После центрифугирования при 34000 оборотах в минуту в течение 60 минут осадок солюбилизировали в буфере, содержащем также 20 мкМ CHAPS. Через 30 минут инкубации на льду суспензию центрифугировали при 34000 оборотах в минуту в течение 30 минут.
Полученную надосадочную жидкость хранили до использования при -80oC.
Для анализа в каждую из 12 пробирок, содержащих 25 мкл раствора L-аргинина (концентрации 12 мкМ 1H-L-аргинина, 64 нМ 3H-L-аргинина) и/или 25 мкл анализируемого буфера (50 мМ HEPES, 1 мМ EDTA, 1,5 мМ CaCl2, pH 7,4) или 25 мкл испытуемого соединения в буферном растворе при 22oC, добавили 25 мкл конечного анализируемого буфера (50 мМ HEPES 1 мМ EDTA, 1,5 мМ CaCl2, 1 мМ DTT, 100 мкМ NaDPH, 10 мкг/мл калмодулина, 12 мМ тетрагидробиоптерина, pH 7,4), чтобы вызвать реакцию, которую через 10 минут закончили путем добавления 2 мл буфера для завершения реакции (20 мМ HEPES, 2 мМ EDTA, pH 5,5).
Меченый L-цитрулин отделили от меченого L-аргинина посредством хроматографии через колонку с Dowex AG-50 X8 200-400. В отдельную 1 мл колонку добавили 1 мл каждого реагента, завершающего реакцию, и элюент соединили с элюентом от двух промывок 1 мл дистиллированной воды и с 16 мл сцинтилляционной смеси. Затем с помощью сцинтилляционного счетчика количественно определили L-цитрулин.
В типичном эксперименте основная активность увеличилась на 5000 распадов в минуту/мл пробы относительно реагентного бланка, который имеет активность 1500 распадов в минуту/мл. Для проверки методики при анализе подвергали испытанию сравнительный эталон, N-нитро-L-аргинин, который дает 70% - 90% ингибирования синтетазы оксида азота (V) при концентрации 1 мкМ.
Для определения степени проникновения внутрь мозга соединения могут быть также испытаны в анализе ex-vivo.
Проверка D.
(D) Анализ ex-vivo на ингибирующую активность против нейронной синтетазы оксида азота (V).
Крысам мужского пола Aprague-Dawley (250 - 275 г) дозировали внутривенно 10 мг/кг испытуемого соединения, растворенного в 0,9%-ном солевом растворе, или в качестве контроля только соль. В предварительно определенное время (обычно 2-24 часа) после обработки животных умерщвили, удалили мозжечок, приготовили надосадочную жидкость и анализировали на активность против синтетазы оксида азота (V), как описано в проверке A.
В качестве дополнительного подтверждающего испытания в 2'-5'-ADP Sephorose колонку (которая связывает синтетазу оксида азота (V)) подали фракцию надосадочной жидкости мозжечка и потом элюировали NADPH. Элюент испытывали на активность против синтатетазы оксида азота (V), следуя методике проверки A.
Соединения, которые проникают внутрь мозга крысы и ингибируют нейронную синтетазу оксида азота (V), приводят к уменьшению активности синтетазы оксида азота (V) как в надосадочной жидкости, так и в элюенте из 2'-5'-ADP Sephorose колонки.
В проверках на ингибирующую активность против синтетазы оксида азота (V) активность соединения выражали в виде IC50 (концентрация лекарственного вещества, которая дает при анализе ингибирование фермента на 50%). Значения IC50 для испытуемых соединений первоначально оценивали, исходя из ингибирующей активности 1,10 и 100 мкМ растворов соединений. Соединения, которые ингибировали фермент по крайней мере на 50% при 10 мкМ, испытывали повторно, используя более походящие концентрации с тем, чтобы можно было определить IC50.
В проверке A, приведенной выше (проверка на активность против нейронной изоформы синтетазы оксида азота (V)) соединения примера 1, представленное ниже, дает IC50 менее, чем 10 мкМ, что указывает на то, что предполагается пригодная терапевтическая активность.
В проверках B и C (проверка на активность против макрофаговой и эндотелиальной изоформ синтетазы оксида азота (V)) соединение примера 1 дает значения IC50 более чем в 10 раз выше, чем те, которые были получены в проверке A, что указывает на то, что оно показывает желательную селективность.
Соединения примеров 2-20, 21(a) - (n), 22(a) - (e), 23(a) - (f), 24-26, 27(a), (b), 28-47 и 49-71 испытывали в проверке A, и они также давали значения IC50 менее, чем 10 мкМ. Соединение примера 48 испытывали в проверке A и получили значение IC50 менее чем 100 мкМ. Соединение примера 72 испытывали в проверке A и получили ингибирование на 17% при концентрации 10 мкМ. Таким образом, предполагается, что эти соединения обладают пригодной терапевтической активностью.
Соединения формулы I и их фармацевтически приемлемые соли имеют преимущество, состоящее в том, что они являются менее токсичными, более эффективными, более селективными, являются соединениями более длительного действия, обладают более широким диапазоном активности, являются более сильнодействующими, дают меньшее количество побочных воздействий, более легко всасываются или имеют другие пригодные фармакологические свойства, чем ранее известные соединения, использованные в вышеупомянутых областях терапии.
Соединения формулы I и их фармацевтически приемлемые соли могут также иметь преимущество, состоящее в том, что они являются более селективными для нейронной изоформы фермента синтетазы оксида азота (V) и, следовательно, ожидается, что они имеют пригодную терапевтическую активность с профилем уменьшенного побочного воздействия, связанным с ингибированием других изоформ.
Изобретение иллюстрировано посредством следующих примеров.
Пример 1
N-(4-(2-((фенилметил)амино)этил)фенил-2-тиофенилкарбоксимидамид.
(a) N-(2-(4-(4-нитрофенил)этил)трифторацетамид.
К перемешанному раствору 4-нитрофенетиламингидрохлорида (1,84 г, 9,10 ммоль) и триэтиламина (3,03 мл, 21,70 ммоль) в метаноле (12 мл) добавили по каплям трифторуксусный ангидрид (1,51 мл, 20,66 ммоль). После перемешивания в течение 1 минуты при пониженном давлении удалили растворитель, и оставшийся остаток смешали с водой и экстрагировали метиленхлоридом (3 • 20 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения твердого вещества, которое перекристаллизовали из метиленхлорида/гексана до получения N-(2-(4-нитрофенил)этил)трифторацетамида в виде белого твердого вещества: 1,92 г (выход 80%); точка плавления 103-104oC.
(b) N-(2-(4-нитрофенил)этил)-N-(фенилметил)трифторацетамид.
К перемешанному раствору продукта стадии (a) (0,89 г, 3,40 ммоль) в THF ((5 мл) при 0oC добавили NaH (60%, 0,18 г, 4,42 ммоль), затем бензилбромид (0,50 мл, 4,10 ммоль). Смесь перемешивали при комнатной температуре в течение 6 часов, резко охладили водой и экстрагировали этилацетатом (3 • 30 мл).
Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (18% этилацетат/гексан) для получения N-(2-(4-нитрофенил)этил-N-(фенилметил)трифторацетамида в виде бесцветного масла (0,52 г, 44%); M.S. (M+H)+ = 353.
(c) N-(2-(4-аминофенил)этил-N-(фенилметил)трифторацетамид.
К перемешанному раствору продукта стадии (b) (0,52 г, 1,48 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали при давлении 3,5155 кг/см2 в течение 1 часа; фильтровали через целит и концентрировали до получения N-(2-(4-аминофенил)этил)-N-(фенилметил)трифторацетамида, который стал однородным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(d) S-метил-2-тиофентиокарбоксимидгидроиодид.
Раствор 2-тиофенкарбокситиоамида (Мэйбридж Кемикал) (11,1 г) в 60 мл ацетона обработали иодидометаном (13,4 г). Через 6 часов при 22oC полученные желтые частицы собрали фильтрацией, дважды промыли 25 мл ацетона и сушили для обеспечения 18,45 г S-метил-2-тиофентиокарбоксимидгидроиодида, точка плавления 195oC (разложения).
(e) N-(4-(2-((фенилметил)амино)этил)фенил-2-тиофен-карбоксимидамид.
К раствору N-(2-(4-аминофенил)этил-N-(фенилметил)трифторацетамида (0,48 г, 1,48 ммоль) в изопропаноле (6 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (0,42 г, 1,48 ммоль). Смесь перемешали в течение 4 часов, разбавили метанолом (5 мл) и 2N NaOH (6 мл) и нагрели до 70oC в течение 1 часа. Пои пониженном давлении удалили растворители, и остаток сбросили в воду и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения твердого вещества, которое перекристаллизовывали (этилацетат/гексан) до получения N-(4-(2-((фенилметил)амино)этил)фенил-2-тиофенкарбоксимидамида в виде белого твердого вещества: (0,17 г, 34%); точка плавления 116-118oC.
Пример 2
N-(4-(1-((фенилметил)амино)метил)фенил-2-тиофенкарбоксимидамид.
(a) N-((4-нитрофенил)метил)трифторацетамид.
К перемешанному раствору 4-нитробензиламиногидрохлорида (4,06 г, 21,5 ммоль) и триэтиламина (6,60 мл, 47,4 ммоль) в метиленхлориде (30 мл) добавили по каплям трифторуксусный ангидрид (3,34 мл, 23,7 ммоль). После перемешивания в течение 1 минуты добавили воду и разделили слои. Затем водный слой экстрагировали метилехлоридом (3 • 20 мл), и смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения твердого вещества, которое перекристаллизовали из метиленхлорида/гексана до N-(4-((нитрофенил)метил)трифторацетамида в виде белого твердого вещества: 3,9 г (выход 73%); точка плавления 97-98oC.
(b) N-(4-((нитрофенил)метил)-N-(фенилметил)трифторацетамид.
К перемешанному раствору продукта стадии (a) (1,0 г, 4,03 ммоль) в THF (10 мл) при 0oC добавили NaH (60%, 0,21
г, 5,24 ммоль), затем бензилбромид (0,72 мл, 4,84 ммоль). Смесь перемешали при комнатной температуре в течение 12 часов, резко охладили водой и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промывали водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (16% этилацетата/гексана) до получения N-((4-нитрофенил)метил-N-(фенилметил)трифторацетамида в виде белого бесцветного масла (0,50 г, 40%); M.S. (M+H)+ = 339.
(c) N-((4-аминофенил)метил-N-(фенилметил)трифторацетамид.
К перемешанному раствору продукта стадии (b) (1,76 г, 5,16 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали при давлении 3,51 кг/см2 в течение 0,5 часа, фильтровали через целит и концентрировали до получения N-((4-аминофенил)метил-N-(фенилметил)трифторацетамида, который стал гомогенным посредством тонкослойной хроматографии, и использовали тотчас же в следующей реакции.
(d) N-(4-(1-фенилметил)амино)метил)фенил-2-тиофенкарбоксимидамид.
К раствору продукта стадии (c) (1,60 г, 5,16 ммоль) в изопропаноле (6 мл) добавили S-метил-2-S-тиофентиокарбоксимидгидрохлорид (продукт примера 1, стадия (d) (1,47 г, 5,16 ммоль). Смесь перемешивали в течение 24 часов при 40oC, разбавили метанолом (5 мл) в 2 NaOH (15 мл) и нагрели до 70oC в течение 1 часа. При пониженном давлении удалили растворители, и остаток сбросили в воду и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промывали водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (8% метанола/метиленхлорида) до получения твердого вещества, которое перекристализовывали (этилацетат/гексан) до выхода N-(4-(1-((фенилметил)амино)метил)фенил)-2-тиофенкарбоксимидамида в виде белого твердого вещества (60 мг, 4%); точка плавления 73-74oC.
Пример 3 N-(4-(1-((фенетил)амино)метил)фенил)-2-тиофенкарбоксиамид.
(a) N-(2-фенилэтил)трифторацетамид.
К перемешанному раствору фенетиламина (4,91 г, 40,5 ммоль) и триэтиламина (6,50 мл, 46,6 ммоль) в метиленхлориде (30 мл) добавили по каплям трифторуксусный ангидрид (6,3 мл, 44,6 ммоль). После перемешивания в течение 1 минуты добавили воду и слои разделили. Затем водный слой экстрагировали метиленхлоридом (3 • 40 мл), и смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения твердого вещества, которое перекристаллизовывали из метиленхлорида/гексана до получения N-(2-фенилэтил)трифторацетамида в виде белого твердого вещества: (60 г, выход 69%); точка плавления 50-52oC.
(b) N-(2-фенилэтил)-N-(4-нитрофенил)метил)трифторацетамид.
К перемешанному раствору продукта стадии (a) (2,0 г, 9,26 ммоль) в THF (10 мл) при 0oC добавили NaH (60%, 0,37 г, 9,26 ммоль) затем 4-нитробензилбромид (1,0 мл, 4,63 ммоль). Смесь перемешали при комнатной температуре в течение 1 часа, резко охладили водой и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промывали водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (16% этилацетата/гексана) до получения N-(2-фенилэтил)-N-((4-нитрофенил)метил)трифторацетамида в виде белого бесцветного масла: (1,60 г, 98%); M.S. (M+H)+ = 353.
(c) N-(2-фенилэтил)-N-((4-аминофенил)метил)трифторацетамид.
К перемешанному раствору продукта стадии (b) (1,60 г, 4,54 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали при давлении 3,5155 кг/см2 в течение 0,75 часа, фильтровали через целит и концентрировали до получения N-(2-фенилэтил)-N-((4-аминофенил)метил)трифторацетамида, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(d) N-(4-(1-((2-фенилэтил)амино)метил)фенил-2-тиофенкарбоксимидамид.
К раствору продукта стадии (c) (1,17 г, 4,54 ммоль) в изопропаноле (5 мл) добавили S-метил-2-тиофентиокарбоксимидгидрохлорид (продукт примера 1, стадия (a) (1,30 г, 4,54 ммоль). Смесь перемешали в течение 24-х часов при 40oC, разбавили метанолом (5 мл) и 2N NaOH (10 мл) и нагрели в течение 1 часа до 70oC. При пониженном давлении удалили растворители, остаток сбросили в воду и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промывали водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (10% метанола/метиленхлорида) до получения твердого вещества, которое перекристализовывали (этилацетат/гексан) до получения N-(4-(1-((2-фенилэтил)амино)метил)фенил)-2-тиофенкарбоксимидамида в виде белого твердого вещества: (20 мг, 2%); M.S. (M+H)+ = 336.
Пример 4 N-(4-(2-((2-хлорфенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид
(a) N-(4-нитрофенил)этил-N-((2-хлорфенил)метил) трифторацетамид.
К перемешанному раствору N-(2-(4-нитрофенил)этил)трифторацетамида (продукт примера 1 стадии (a) (2,0 г, 7,63 ммоль) и каталитическому количеству 15-круан-5- в THF (10 мл) при 0oC добавили NaH (60%, 0,18 г, 4,42 ммоль), затем 2-хлорбензилбромид (1,49 мл, 11,45 ммоль). Смесь перемешивали при комнатной температуре в течение 2-х часов, резко охладили водой и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (18% этилацета/гексана) до получения N-(2-(4-нитрофенил)этил-N-((2-хлорфенил)метил)трифторацетамида в виде бесцветного масла: (2,31 г, 78%); M.S. (M+H)+ = 353.
(b) N-(2-аминофенил)этил)-N-((2-хлорфенил)метил)трифторацетамид.
К перемешанному раствору продукта стадии (a) (231 г, 5,96 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C.
Смесь гидрировали под давлением 3,5155 кг/см3 в течение 1 часа, фильтровали через целит и концентрировали до получения N-(2-(4-аминофенил)этил)-N-(2-хлорфенил)метил)трифторацетамида, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(c) N-(4-((2-хлорфенилметил)амио)этил)фенил-2-тиокарбоксимидамид.
К раствору продукта стадии (b) (2,1 г, 5,96 ммоль) в изопропаноле (10 мл) добавили S-метил-2-тиофентиокарбоксимидгидрохлорид (продукт примера 1, стадия (d) (1,7 г, 5,96 ммоль). Смесь перемешали в течение 24-х часов, разбавили метанолом (10 мл) и 2N NaOH (6 мл) и нагрели в течение 1 часа до 70oC. При пониженном давлении удалили растворители, остаток сбросили в воду и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промывали водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (10% метанола/метиленхлорид) до получения твердого вещества, которое перекристаллизовывали (метиленхлорид/гексан) до получения N-(4-(2-(2-хлорфенилметил)амино)этил)фенол)-2-тиофенкарбоксимидамида в виде белого твердого вещества: (0,21 г, 10%); точка плавления 81-82oC.
Пример 5 N-(4-(2-((3-фторфенилметил)амино)этил)фенил-2-тиофенкарбоксимидамид
(a) N-((3-фторфенил)метил-N-(2-(4-нитрофенил)этил) трифторацетамид.
К перемешанному раствору N-(2-(4-нитрофенил)этил)трифторацетамида (продукт примера 1, стадии (a) (1,5 г, 5,75 ммоль) и каталитическому количеству 15-краун-5- в THF (10 мл) при 0oC добавили NaH (60%, 0,25 г, 6,34 ммоль), затем 3-фторбензилбромид (1,40 мл, 11,45 ммоль). Смесь перемешали при комнатной температуре в течение 4 часов, резко охладили водой и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (18% этилацетата/гексана) до получения N-((3-фторфенил)метил-N-(2-(4-нитрофенил)этил)трифторацетамида в виде бесцветного масла: (1,63 г, 77%); M.S. (M+H)+ = 371.
(b) N-(2-(4-аминофенил)этил)-N-(3-фторфенил)метил)трифторацетамид.
К перемешанному раствору продукта стадии (a) (1,63 г, 4,40 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали при давлении 3,5155 кг/см3 в течение 1 часа, фильтровали через целит и концентрировали до получения N-(2-(4-аминофенил)этил)-N-(3-фторфенил)метил)трифторацетамида, который стал гомогенным посредством тонкослойной хроматографии и тотчас же использовали в следующей реакции.
(c) N-(4-(2-(3-фторфенил)метил)амино)этил)фенил-2-тиофенкарбоксимидамид.
К раствору продукта стадии (b) (1,5 г, 4,40 ммоль) в метаноле (10 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадия (d) (1,3 г, 4,40 ммоль). Смесь перемешали в течение 2 часов, разбавили метанолом (5 мл) и 2N NaOH (8 мл) и нагрели в течение 1 часа до 70oC. При пониженном давлении удалили растворители, остаток сбросили в воду и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения твердого вещества, которое перекристаллизовывали (метиленхлорид/гексан) до получения N-(4-(2-((3-фторфенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамида в виде белого твердого вещества: (0,14 г, 8%); точка плавления 130-131oC.
Пример 6 N-(4-(2-(((2-метилфенил)метил)амино)этил)фенил-2-тиофенекарбоксимидамид.
(a) N-((2-метилфенил)метил-N-(2-(4-нитрофенил)этил) трифторацетамид.
К перемешанному раствору N-(2-(4-нитрофенил)этил)трифторацетамида (продукт примера 1, стадии (a) (1,5 г, 5,75 ммоль) и каталитическому количеству 15-краун-5- в THF (10 мл) при 0oC добавили NaH (60%, 0,25 г, 6,34 ммоль), затем 2-метилбензилбромид (1,53 мл, 22,45 ммоль).
Смесь перемешали при комнатной температуре в течение 2 часов, резко охладили водой и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (18% этилацетата/гексана) до получения N-((2-метилфенил)метил-N-(2-(4-нитрофенил)этил)трифторацетамида в виде белого бесцветного масла: (1,76 г, 84%); M.S. (M+H)+ = 367.
(b) N-(2-(4-аминофенил)этил)-N-((2-метилфенил)метил)трифторацетамид.
К перемешанному раствору продукта стадии (a) (1,76 г, 4,82 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали при давлении 3,5155 кг/см3 в течение 1 часа, фильтровали через целит и концентрировали до получения N-(2-(4-аминофенил)этил)-N-((2-метилфенил)метил)трифторацетамида, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(c) N-(4-(2-(((2-метилфенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид.
К раствору продукта стадии (b) (1,62 г, 4,82 ммоль) в метаноле (10 мл) добавили S-метил-2-тиофентиокарбоксимидгидрохлорид (продукт примера 1, стадия (d) (1,37 г, 4,82 ммоль). Смесь перемешали в течение 2-х часов, разбавили 2N NaOH (8 мл) и нагрели в течение 1 часа до 70oC. При пониженном давлении удалили растворители, остаток сбросили в воду и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали до получения твердого вещества, которое перекристаллизовали (метиленхлорид/гексан) до получения N-4-(((2-метилфенил)метил)амино)этил)фенил-2-тиофенкарбоксимидамида в виде белого твердого вещества: (0,46 г, 28%); точка плавления 105-106oC.
Пример 7
N-(4-(2-(метиламино)этил)фенил)-2-тиофенкарбоксимидамид.
(a) N-метил-N-(2-(4-нитрофенил)этил)трифторацетамид.
К перемешанному раствору N-(2-(4-нитрофенил)этил)трифторацетамида (продукт примера 1, стадии (a)) (1,5 г, 5,75 ммоль) и каталитическому количеству 15-краун-5 в THF (10 мл) при 0oC добавили NaF (60%, 0,25 г, 6,43 ммоль), затем метилиодид (0,71 мл, 11,45 ммоль). Смесь перемешали при комнатной температуре в течение 4 часов, резко охладили водой и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения N-метил-N-(2-(4-аминофенил)этил)трифторацетамида в виде белого бесцветного масла: (1,40 г, 88%); M.S. (M+H)+ = 277.
(b) N-метил-N-(2-аминофенил)этил)трифторацетамид.
К перемешанному раствору продукта стадии (a) (1,45 г, 5,25 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали при давлении 3,5155 кг/см3 в течение 1 часа, фильтровали через целит и концентрировали до получения N-метил-N-(2-(4-аминофенил)этил)трифторацетамида, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(c) N-(4-(2-(метиламино)этил)фенил)-2-тиофенкарбоксимидамид.
К раствору продукта стадии (b) (1,32 г, 5,37 ммоль) в метаноле (10 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d) (1,53 г, 5,37 ммоль). Смесь перемешали в течение 2-х часов, разбавили 2N NaOH (8 мл) и нагрели в течение 1 часа до 70oC. При пониженном давлении удалили растворители, остаток сбросили в воду и экстрагировали этилацетатом (3 • 30). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали до получения твердого вещества, которое перекристаллизовали (метиленхлорид/гексан) до получения N-(4-(2-((2-метилфенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамида в виде белого твердого вещества: (0,43 г, 31%); M.S. (M+H)+ = 260.
Пример 8 N-(4-(2-аминоэтил)фенил)-2-тиофенкарбоксимидамид.
(a) N-(2-аминофенил)этил)трифторацетамид.
К перемешанному раствору N-(2-(4-нитрофенил)этил-N-(фенилметил)трифторацетамида (продукт примера 1, стадии (a)) (1,00 г, 3,81 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали при давлении 3,5155 кг/см2 в течение 1 часа, фильтровали через целит и концентрировали до получения N-(2-(4-аминофенил)этил)трифторацетамида, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(c) N-(4-(2-аминоэтил)фенил)-2-тиофенкарбоксимидамид.
К раствору продукта стадии (a) (0,88 г, 3,81 ммоль) в метаноле (10 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)) (1,09 г, 3,81 ммоль). Смесь перемешали в течение 12 часов, разбавили 2N NaOH (8 мл) и нагрели в течение 1 часа до 70oC. При пониженном давлении удалили растворители, остаток сбросили в воду и экстрагировали этилацетатом (3 • 30). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали до получения твердого вещества, которое перекристаллизовали (этилацетат/метанол) до получения N-(4-(2-аминоэтил)фенил)-2-тиофенкарбоксимидамида в виде белого твердого вещества: (70 мг, 8%); точка плавления 134-137oC.
Пример 9 N-((4-морфолинилметил)фенил-2-тиофенкарбоксимидамид.
(a) 4-(4-нитробензил)морфолин.
К перемешанному раствору (2,00 г, 0,0093 моль) 4-нитробензолбромида (Aldrich) и (0,736 г, 0,011 моль) безводного карбоната калия (Aldrich) в 20,00 мл DMF добавили морфолин (0,796 мл, 0,0093 моль). Реакционную смесь нагревали до 50oC и перемешали в течение 30 минут, после чего добавили дополнительно 0,1 эквивалента морфолина и карбоната калия. Через 30 минут реакционную смесь резко охладили 100 мл воды и экстрагировали (4 • 100 мл) этилацетатом. Органические слои собрали и сушили над сульфатом магния, и выпарили растворитель. Полученное твердое перекристаллизовали из этилацетата и гексана для получения 1,90 г 4-(4-нитробензил)морфолина.
(b) 4-морфолинилметил)анилин.
A пробу (1,00 г; 0,0045 моль) 4-(4-нитробензил)морфолина растворили в 25 мл THF и 25 мл метанола в толстостенной склянке для реакций под давлением. Добавили каталитическое количество 10% палладия на углероде, и реакционную смесь гидрировали. Когда поглощение водорода прекратилось, катализатор удалили фильтрацией, и растворитель выпарили. Твердое растворили в 30 мл этилацетата и 30 мл воды и 30 мл 2N гидроксида натрия. Водные слои экстрагировали этилацетатом (4 • 75 мл). Органические слои собрали, сушили над сульфатом магния и растворитель выпарили под вакуумом. Полученное твердое перекристаллизовали из этилацетата и гексана до получения 0,68 г (4-морфолинилметил)анилина.
(c) N-((4-морфолинилметил)фенил-2-тиофенкарбоксимидамид.
К перемешанному раствору продукта стадии (b) (0,68 г, 0,0035 моль) и 150 мл изопропилового спирта добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)) (0,99 г; 0,0035 моль). Смесь перемешали при 35oC. К этой смеси вместе с 2М соляной кислотой в изопропиловом спирте, добавленной по каплям, добавляли 10,0 мл метанола до тех пор, пока все реагенты были в растворе. Обеспечили перемешивание реакционной смеси в течение 48 часов. Затем реакционную смесь разбавили в 50 мл насыщенного хлорида натрия и экстрагировали этилацетатом (3 • 75 мл). Органические слои собрали, сушили над сульфатом магния, и растворитель выпарили. Сырой продукт отделили на силикагельной колонке при элюировании 10%-ным метанолом в метиленхлориде. Растворитель выпарили и сырое твердое вещество дважды перекристаллизовали из этилацетата и гексана до получения 60 мг N-((4-морфолинилметил)фенил-2-тиофенкарбоксимидамида, точка плавления 148150oC.
Пример 10
N-(3-(((фенилметил)амино)метил)фенил)-2-тиофенкарбоксимидамидбис-оксалат.
(a) N-(3-нитробензил)бензамид.
К раствору 3-нитробензиламингидрохлорида (2,45 г, 0,013 моль) в растворе 50 мл метиленхлорида и 50 мл полунасыщенного водного раствора карбоната калия при 0oC добавили по каплям раствор бензоилхлорида (2,1 г, 0,0149 моль) в 10 мл метиленхлорида. После завершения добавления реакционную смесь перемешали в течение 2 часов при 0oC и затем нагревали до окружающей температуры всю ночь. Органический слой отделили и последовательно промыли разбавленной соляной кислотой и водой. Высушенную органическую фазу (MgSO4) концентрировали в вакууме до получения 2,92 г (88%) названного продукта, точка плавления 136-138oC.
b) N-бензил-2,2,2-трифтор-N-(3-нитробензил)ацетамид.
К раствору продукта стадии (a) (2,85 г, 11,1 ммоль) в 50 мл безводного тетрагидрофурана при 0oC под азотом добавили 18,6 мл 1,0 М борана в растворе THF (18,6 ммоль). Затем реакционную смесь в течение 5,5 часа нагревали в колбе с обратным холодильником. Раствор охлаждали в течение всей ночи и затем резко охладили путем последовательного добавления 2 мл метанола и 10 мл 6М соляной кислоты. Реакционную смесь опять нагрели в колбе с обратным холодильником в течение 1 часа. После охлаждения до окружающей температуры повысили основность реакционной смеси и экстрагировали в простой эфир, сушили (MgSO4) и концентрировали до получения масла. Масло подвергали хроматографии на силикагеле при использовании в качестве элюента метиленхлорида до получения 1,95 г (72%) бензил-3-(нитробензил)амина в виде масла К раствору сырого бензил-3/(нитробензил)амина (1,95 г, 8,05 ммоль) и триэтиламина (2,6 мл, 18 ммоль) в 20 мл метиленхлорида под азотом при 0oC добавили по каплям трифторуксусный ангидрид (3,4 г, 16 ммоль). Перед сливом в воду реакционную смесь перемешали в течение 10 минут. Отделили органический слой и сушили над сульфатом магния. Раствор фильтровали и концентрировали до получения масла. Хроматографией на силикагеле при использовании 20% этилацетата в гексане в качестве элюента получили 1,46 г (54%) продукта в виде масла, масс-спектр m/e 339 (100%, M+H).
(c) N-(3-аминобензил)-N-бензил-2,2,2-трифторацетамид.
К раствору продукта стадии (b) (1,21 г, 3,58 ммоль), растворенного в растворе 100 мл метанола, добавили 20 мл насыщенного раствора хлористого водорода в изопропаноле и 0,1 г 5% Pd/C. Полученный раствор гидрировали в течение 1 часа при давлении 3,5155 кг/см2. Удалили катализатор фильтрацией, и фильтрат концентрировали в вакууме до получения твердого вещества. При растирании в порошок твердого вещества с простым эфиром получили 1,15 г (93%) названного соединения в виде гидрохлоридной соли, тока плавления 169-174oC.
(d) N-3-(((фенилметил)амино)метил)фенил-2-тиофенкарбоксимидамид.
К раствору 0,25 г (0,94 ммоль) S-метил-2-тиофентиокарбоксимидгидроиодида (продукт примера 1, стадии (d)) в 4 мл изопропанола добавили 0,41 г (1,3 ммоль) N-(3-аминобензил)-N-бензил-2,2,2-трифторацетамида (полученного посредством гидрохлоридной соли, нейтрализации 2,5 м NaOH и экстрагирования в метиленхлорид). Реакционную смесь перемешали в течение 5 часов. Добавили 2,5М раствора гидроксида натрия (2 мл) и около 5 капель метанола, и полученный раствор нагревали в течение 1 часа в колбе с обратным холодильником. Раствор концентрировали, и продукт экстрагировали в этилацетат. Раствор сушили и концентрировали до получения твердого вещества. Твердое вещество растворили в этаноле и добавили дигидрат щавелевой кислоты (0,16 г, 1,3 ммоль). Полученную соль собрали и сушили до получения 0,26 г (55%) названного соединения в виде бисоксалатной соли, точка плавления 178-183oC.
Пример 11
Альтернативный синтез соединения примера 2.
N-(4-(((фенилметил)амино)метил)фенил)-2-тиофенкарбоксимидамид.
(a) N-(4-нитробензил)бензамид).
Это соединение получили, следуя методике примера 10, стадия (a). Из 4-нитробензиламина (2,45 г, 0,031 моль) и бензохлорида (2,1 г, 0,0149 моль) выделили 2,56 г (77%) названного продукта, точка плавления 150-153oC.
(b) N-бензил-2,2,2-трифтор-N-(4-нитробензил)ацетамид.
Это соединение бензил-(4-нитробензил)амин получили, используя методику, описанную в примере 10, стадия (b), для получения бензил-3-(нитробензил)амина. Из 2,49 г (9,36 ммоль) N-(4-нитробензил)бензамида и 18,6 мл 1,0М борана в THF получили 3,12 г сырого бензил-(4-нитробензил)амина, который использовали без дополнительной очистки. Этот сырой продукт смешали с 4,3 мл триэтиламина в 40 мл метиленхлорида при 0oC под азотом. К этому раствору по каплям добавили 3,6 мл трифторуксусного ангидрида. Этот раствор перемешали в течение 10 минут, влили в воду и отделили. Органическую фазу сушили (MgSO4) и концентрировали до получения 3,1 г (94%) названного соединения в виде масла.
(c) N-(4-аминобензил)-N-бензил-2,2,2-трифторацетамид.
Это соединение получили с использованием методики, описанной в примере 10, стадии (c), для получения N-(3-аминобензил)-N-бензил-2,2,2-трифторацетамида.
Из N-бензил-2,2,2-трифтор-N-(4-нитробензил)ацетамида (3,1 г, 9,2 ммоль) после гидрирования получили 2,46 г (78%) названного соединения в виде гидрохлоридной соли. Перекристаллизацией из изопропанола и простого эфира получили 1,74 г чистого вещества, точка плавления 115-119oC.
(d) N-(4-(((фенилметиламино)метил)фенил)-2-тиофенкарбоксимидамид.
Из 0,60 г (1,9 ммоль) свободного основания N-(4-аминобензил)-N-бензил-2,2,2-трифторацетамида и 0,42 г (1,6 ммоль) S-метил-2-тиофентиокарбоксимида (полученного посредством гидрохлоридной соли, полученной при следовании методике, аналогичной той, которая представлена в примере 1, стадия (d), и нейтрализации 2,5 м NaOH и экстракции в метиленхлорид) в 4 мл изопропанола. Реакционную смесь перемешали в течение 5 часов. Добавили раствор (2 мл) 2,5М гидроксида натрия и около 5 капель метанола, и полученный раствор нагревали в колбе с обратным холодильником в течение 1 часа. Раствор концентрировали, и продукт экстрагировали в этилацетат. Раствор сушили и концентрировали до получения твердого вещества. Твердое вещество превратили в бис-оксалат в изопропаноле и затем перекристаллизовали из 95% этанола до получения 110 мг (10%) названного соединения, точка плавления 209-213oC.
Пример 12
Следуя методике примера 1, получили следующие соединения:
(a) N-(4-(2-(((2,6-дихлорфенил)метил)амино)этил)фенил)- 2-тиофенкарбоксимидамид, точка плавления 104-105oC.
(b) N-(4-(2-(((2-бромфенил)метил)амино)этил)фенил)- 2-тиофенкарбоксимидамид, точка плавления 81-82oC.
(с) N-(3-(2-((фенилметил)амино)этил)фенил)-3- тиофенкарбоксимидамиддигидрохлорид, точка плавления 145-147oC.
(d) Свободное основание N-(4-(2-((2,6-дихлорфенил)метил)амино)этил)фенил)-3- тиофенкарбоксимидамид, точка плавления 109-110oC.
(e) N-(4-(2-аминоэтил)-3-тиофенкарбоксимидамиддигидробромид, точка плавления 158-170oC (разлож.).
(f) Свободное основание N-(4-(2-((2,6-дихлорфенил)метил)амино)этил)фенил)-2- фуранкарбоксимидамид, точка плавления 101-104oC.
(q) Свободное основание N-(3-(3-(1-пиролидинил)пропил)фенил)-2- тиофенкарбоксимидамид, точка плавления 110-111oC.
(h) N-(4-(2-аминоэтил)фенил)-2-фурокарбоксимидамиддиоксилат, точка плавления 162oC (разлож.).
Пример 13
Следуя методике примера 9, получили следующие соединения:
(a) N-(4-((1-пиперидинил)метил)фенил)-2- тиофенкарбоксимиддигидробромид, точка плавления 277-278oC.
(b) N-(4-((1-пирролидинил)метил)фенил)-2- тиофенкарбоксимиддигидробромид, точка плавления 248-250oC.
Пример 14
Следуя методике примера 10, получили следующее соединение:
N-(3-(((фенилметил)амино)метил)фенил)-2- тиофенкарбоксимиддималеат, точка плавления 171-173oC.
Пример 15
Следуя общей методике примера 1, осуществляя стадию (c) взаимодействием S-метил-2-тиофентиокарбоксимидгидроиодида с 3-(метиламино)фениламином, получили следующее соединение:
N-(3-((амино)метил)фенил)-2- тиофенкарбоксимиддималеат, точка плавления 145-148oC.
Пример 16
Следуя методике примера 10, получили следующее соединение:
N-(3-(2-((фенилметил)амино)этил)фенил)-2- тиофенкарбоксамидиндиоксалат, точка плавления 132-134oC.
Пример 17
N-(3-(2-(этиламино)этилфенил)-2-тиофенкарбоксимидамид.
(a) (3-нитрофенил)ацетилхлорид.
Перемешанный раствор 3-нитрофенилуксусной кислоты (10,0 г, 55,2 ммоль) в тионилхлориде (100 мл, 1,37 моль) нагрели в колбе с обратным холодильником в течение 2-х часов, затем концентрировали до выхода 11,1 г (3-нитрофенил)ацетилхлорида в виде рыжевато-коричневого твердого вещества.
(b) N-этил-(2-нитрофенил)ацетамид.
К перемешанному раствору 70 вес.% этиламина в воде (35 мл), охлажденного в бане со льдом, добавили одной порцией (3-нитрофенил)ацетилхлорид (3 г, 15,0 ммоль).
Полученную смесь нагрели до достижения прозрачного раствора, который охладили. Полученный осадок отфильтровали, получив N-этил-2-(3-нитрофенил)ацетамид в виде желтого твердого вещества: (2,2 г, 71%; точка плавления 115-117oC).
(c) Этил-(2-(3-нитрофенил)этил)амингидрохлорид.
К перемешанному раствору продукта стадии (b) (2,2 г, 10,6 ммоль) в тетрагидрофуране под азотом добавили по каплям 1,0М боран-тетрагидрофуран (42 мл, 42 ммоль). Реакционную смесь нагревали в колбе с обратным холодильником в течение 1,5 часов, охладили в бане со льдом, затем добавили по каплям водный раствор 6N HCl (75 мл). Полученную смесь нагревали в колбе с обратным холодильником в течение 1 часа, повысили основность до pH 11 20%-ным водным раствором гидроксида натрия, экстрагировали дважды простым эфиром. Смешанные экстракты сушили над сульфатом магния, фильтровали, концентрировали. Сырую гидрохлоридную соль получили из изопропанола и этилацетата, перекристаллизовали до выхода этил-(2-(3-нитрофенил)этил)амингидрохлорида в виде светло-желтого твердого вещества: (1,7 г, 70%); точка плавления 186-188oC.
(d) 3-(2-этиламиноэтил)фениламингидрохлорид.
К раствору продукта стадии (c) (1,7 г, 7,0 ммоль) в метаноле (30 мл) добавили каталитическое количество 10% палладия на углероде. Смесь гидрировали в течение 30 минут при давлении 3,5155 кг/см2, фильтровали через целит, концентрировали до выхода 3-(2-этиламиноэтил)фениламингидрохлорида в виде не совсем белого твердого вещества: (1,4 г, 100%); точка плавления 192-194oC.
(e) N-(3-(2-этиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид.
К раствору продукта стадии (d) (1,4 г, 7,0 ммоль) в изопропаноле (20 мл) и диметилформамиде (20 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодидом (2,5 г, 8,8 ммоль). Смесь перемешали в течение 16 часов, разбавили 20%-ным водным раствором гидроксида натрия и экстрагировали дважды этилацетатом. Смешанные экстракты дважды промывали водой, сушили над сульфатом магния, фильтровали и концентрировали до получения 2,7 г масла. Дигидробромидную соль получили из изопропанола и этилацетата, перекристаллизовали из изопропанола, метанола и этилацетата о выхода N-(3-(2-этиламино)этил)-фенил)-2-тиофенкарбоксимидамиддигидробромида в виде рыжевато-коричневого твердого вещества: (1,72 г, 49%); точка плавления 192-194oC (разлож.).
Пример 18
N-(3-(3-((фенилэтил)амино)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
(a) 3-(3-фенилэтиламинопропил)фениламиндигидрохлорид.
Его получили, следуя методике, аналогичной той, которая описана в примере 17, стадии (a) - (o).
(b) N-(3-(3-((фенилэтил)амино)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
К раствору продукта стадии (a) (3,0 г, 9,17 ммоль) и S-метил-2-тиофентиокарбоксимидгидроиодида (3,3 г, 11,5 ммоль) в пропаноле (25 мл) и диметилформамиде (25 мл) добавили одной порцией пиридин (0,74 мл, 9,17 ммоль). Смесь перемешали в течение 16 часов, разбавили 20%-ным водным раствором гидроксида натрия и экстрагировали дважды этилацетатом. Смешанные экстракты дважды промывали водой, сушили над сульфатом магния, фильтровали и концентрировали. Сырую диоксалатную соль получили из этанола и простого эфира, перекристаллизовали из этанола до выхода N-(3-(3-фенилэтил)амино)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалата в виде белого твердого вещества: (2,3 г, 44%); точка плавления 102-105oC.
Пример 19 N-(3-((2-(((2-бромфенил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
(a) N-(2-бромбензил)-2-(3-нитрофенил)ацетамид.
Его получили, следуя методике, аналогичной той, которая представлена в примере 17, стадии (a) - (b).
(b) N-(2-бромбензил)-2-(3-аминофенил)ацетамид.
К раствору продукта стадии (a) (5,45 г, 15,6 ммоль) в 85%-ным ледяной уксусной кислоте (400 мл) добавили одной порцией цинковую пыль (10,2 г, 156 ммоль). Реакционную смесь перемешали в течение 30 минут, фильтровали и концентрировали. Остаток отделили 20%-ным водным раствором гидроксида натрия и дихлорметана, органический слой сушили над сульфатом магния, фильтровали, концентрировали до выхода N-(2-бромбензил)-2-(3-аминофенил)ацетамида в виде белого твердого вещества: (4,7 г, 94%); точка плавления 110-112oC.
(c) 3-(2-(2-бромбензиламино)этил)фениламингидрохлорид.
Его получили, следуя методике, аналогичной той, которая представлена в примере 17, стадия (c).
(d) N-(3-(2-(((2-бромфенил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
Его получили, следуя методике, аналогичной той, которая представлена в примере 18, стадия (b), точка плавления 175-178oC (разл.).
Пример 20
N-(3/(2-фениламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид.
(a) (2-(3-нитрофенил)этил)фениламин.
Его получили, следуя методике, аналогичной той, которая представлена в примере 17, стадии (a) - (c).
(b) 2,2,2-трифтор-N-(2-(3-нитрофенил)этил)-N-фенилацетамид.
Его получили, следуя методике, аналогичной той, которая представлена в примере 1, стадия (a).
(c) 2,2,2-трифтор-N-(2-(3-аминофенил)этил-N-фенилацетамид.
Его получили, следуя методике, аналогичной той, которая представлена в примере 1, стадия (c).
(d) N-(3-(2-(фениламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид.
Его получили, следуя методике, аналогичной той, которая приведена в примере 1, стадия (c). Точка плавления 235-240oC (разлож.).
Пример 21
Следуя методике, аналогичной той, которая приведена в примере 17, получили следующие соединения:
(a) N-(4-(2-этиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 176-178oC.
(b) N-(4-(2-(2-пропиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 240-242oC.
(c) N-(4-(2-(1-пропиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 233-235oC.
(d) N-(4-(2-трет-бутиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 241-242oC.
(e) N-(4-(2-(н-бутиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 238-240oC.
(f) N-(3-(2-(метиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 219-223oC.
(g) N-(3-(2-(-пропиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 72-75oC (размягч.).
(h) N-(3-(2-трет-бутиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 232-235oC (разлож.).
(i) N-(3-(2-пропиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 206-210oC (разлож.).
(j) N-(3-(2-аминоэтил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 194-199oC (разлож.).
(k) N-(3-(2-диметиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 232-233oC (разлож.).
(l) N-(3-(2-(диэтиламино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 75-80oC (размягч.).
(m) N-(3-(2-(2-(1,2,3,4-тетрагидро)изохинолинил)этил)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления 172-175oC (разлож.).
(n) N-(4-(3-(2-(1,2,3,4-тетрагидро)изохинолинил)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления 138-142oC.
(o) Свободное основание N-(4-(2-(3,5-бис-трифторметилфенилметил)амино)этил)фенил)-2- тиофенкарбоксимидамид, точка плавления 98-100oC.
(p) Свободное основание N-(4-(2-диэтиламино)этил)фенил)-2- тиофенкарбоксимидамид, точка плавления 113-115oC.
(q) N-(4-(2-((3-хлорфенилметил)амино)этил)фенил) бензолкарбоксимидамиддигидрохлорид, точка плавления 253-254oC.
(r) N-(4-(2-((3-хлорфенилметил)амино)этил)фенил)-3- хлортиофен-2-карбоксимидамиддигидрохлорид, 257oC.
(s) N-(4-(2-((4-метилфенилметил)амино)этил)фенил)-2- тиофенкарбоксимидиндигидрохлорид, 218-219oC.
(t) N-(4-(2-(пиперониламино)этил)фенил)-2- тиофенкарбоксамидиндигидрохлорид, 205-206oC.
Пример 22
Используя методику, аналогичную той, которая представлена в примере 18, получили следующие соединения:
(a) N-(3-(2-(((2-хлорфенил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления 155-157oC (разлож.).
(b) N-(3-(3-((фенилметил)амино)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления 138-141oC (разлож.).
(c) N-(4-(2-(((3-хлорфенил)метил)амино)этил)фенил)-2- тиофенкарбоксамидиндиоксалат, точка плавления 216-217oC.
(d) N-(4-(2-(((4-хлорфенил)метил)амино)этил)фенил)-2- тиофенкарбоксамидиндиоксалат, точка плавления 203-204oC.
(e) N-(4-(2-(((3-хлорфенил)метил)амино)этил)фенил)-3- хлортиофен-2-карбоксимидамиддигидрохлорид, точка плавления 257-258oC.
(f) N-(4-(3-этиламино)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления 98-100oC.
Пример 23
Используя методику, аналогичной той, которая представлена в примере 19, получили следующие соединения:
(a) Свободное основание N-(3-(2-((N-фенилметил-N-метил)амино) этил)фенил)-2-тиофенкарбоксимидамид, точка плавления 85-87oC.
(b) Свободное основание N-(4-(2-((N-фенилметил-N-метил)амино) этил)фенил)-2-тиофенкарбоксимидамид, точка плавления 110-112oC.
(c) N-(3-(2-(((3-хлорфенил)метил)амино) этил)фенил)-2-тиофенкарбоксимидамиддиоксалат, точка плавления 185-188oC (разлож.).
(d) N-(3-(2-(((фторфенил)метил)амино) этил)фенил)-2-тиофенкарбоксимидамиддиоксалат, точка плавления 183-184oC.
(e) N-(4-(3-(((3-хлорфенил)метил)амино) пропил)фенил)-2-тиофенкарбоксимидамиддиоксалат, точка плавления 212-215oC.
(f) N-(4-(3-(((3-фенилметил-N-метил)амино) пропил)фенил)-2-тиофенкарбоксимидамиддигидробромид, точка плавления 228-232oC (разлож.).
(g) Свободное основание N-(4-(2-((этил)(фенилметил)амино) этил)фенил)-2-тиофенкарбоксимидамид, точка плавления 87-89oC.
(n) Свободное основание N-(4-(2-((пропил)(фенилметил)амино)этил) фенил)-тиофенкарбоксимидамид, точка плавления 100-102oC.
(i) Свободное основание N-(4-(2-((1,1-диметилэтил)(фенилметил)амино)этил) фенил-2-тиофенкарбоксимидамид, точка плавления 145-148oC.
(j) Свободное основание N-(4-(2-(((3,4-дихлорфенил)метил)амино)этил) фенил)-2-тиофенкарбоксимидамид, точка плавления 111-114oC.
Пример 24 N-(4-(2-(3-(фениламино)карбонил)пропил)фенил) -2-тиофенкарбоксимидамид.
(a) 4-(3-((фениламино)карбонил)пропил)анилин.
Перемешанный раствор 4-(4-нитрофенил)масляной кислоты (5,0 г, 0,023 моль) в 20 мл тионилхлорида нагревали в колбе с обратным холодильником в течение 4-х часов. Растворитель выпаривали и к перемешанному раствору анилина (2,0 г, 0,02 моль) в 30 мл тетрагидрофурана и 10 мл триэтиламина по каплям добавили 2,5 г сырого хлорангидрида, и затем реакционную смесь перемешивали в течение 18 часов. Фильтрацией удалили триэтиламингидрохлорид, и к органической фазе добавили 50 мл этилацетата. Органическую фазу промыли 1 • 100 мл 1N соляной кислоты и сушили над сульфатом магния. Растворитель выпарили до получения желтого твердого вещества. Твердое вещество растворили в 100 мл метанола и добавили 5200 мг 10% палладия на углероде, реакционную смесь гидрировали в течение 4-х часов. Было обнаружено, что продукт не восстановился и затем его растворили в 100 мл метанола. Добавили 10 мл насыщенного раствора HCl в изопропаноле, затем 250 мг 10% палладия на углероде. Смесь гидрировали в течение 4-х часов. Удалили катализатор фильтрацией, и выпарили растворитель. Остаток растворили в 100 мл горячей воды с минимальным количеством метанола и затем 50% раствором гидроксида натрия повысили основность раствора. Смесь экстрагировали 150 мл этилацетата, экстракт сушили сульфатом магния и выпаривали, получив твердый 4-(3-(((фенил)амино)карбонил)пропиланилин, выход 1,0 г, одно пятно при тонкослойной хроматографии.
(b) N-(4-(3-((фениламино)карбонил)пропил)фенил)-2- тиофенкрабоксимидамид.
К перемешанной суспензии продукта стадии (a) (1,00 г, 0,0037 моль) приблизительно в 5 мл изопропанола добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадия (d) (1,01 г, 0,0035 моль).
Эту смесь нагревали в течение 1 часа в колбе с обратным холодильником и затем охладили до получения твердых частиц. Продукт отфильтровали, сушили под вакуумом всю ночь, получив при этом N-(4-(3-((фениламино)карбонил)пропил)фенил)-2- тиофенкрабоксимидамид, выход 1,76 г, точка плавления 229-231oC.
Пример 25
Используя методику, аналогичную той, которая представлена в примере 24, получили следующие соединения:
(a) N-(4-(3-((фенилметиламино)карбонил)пропил)фенил)-2- тиофенкарбоксимидамид, точка плавления 169-171oC.
(b) N-(4-(3-((1-пирролидил)карбонил)пропилокси)фенил)-2- тиофенкарбоксимидамид, точка плавления 191-194oC.
(c) N-(4-(3-морфолинил)карбонил)пропилокси)фенил)-2- тиофенкарбоксимидамид, точка плавления 136-138oC.
(d) N-(4-(2-((фенилметиламино)карбонил)этил)фенил)-2- тиофенкарбоксимидамид, точка плавления 63-65oC.
(e) N-(3-(2-((фениламино)карбонил)этил)фенил)-2- тиофенкарбоксимидамид, точка плавления 203-205oC.
(f) N-(4-(3-((фениламино)карбонил)пропил)фенил)-2- пирролкарбоксимидамид, точка плавления 195-196oC.
(g) N-(4-(3-((фениламино)карбонил)пропил)фенил)-2- фурокарбоксимидамид, точка плавления 197-199oC.
(h) N-(4-(3-((фениламино)карбонил)пропил)фенил)-3- хлор-2-тиофенкарбоксимидамид, точка плавления 141-144oC.
(i) N-((3-((фениламино)карбонил)пропил)фенил)-1- метилпиррол-2-карбоксимидамид, точка плавления 154-155oC.
(j) N-(4-(3-(1-(4-метилпиперазинил)карбонил)пропил)фенил)-2- тиофенкарбоксимидамид, точка плавления 132-134oC.
Пример 26
N-(4-(3-(1-пирролидил)карбонил)пропил)фенил)тиофен-2- карбоксимидамидгидроиодид
(a) N-(4-(3-((1-пирролидинил)карбонил)пропил)анилин.
4-(4-нитрофенил)масляную кислоту (2,25 г, 0,01076 моль) растворили в 40 мл дихлорметана и охладили до -5oC в бане со смесью льда с ацетоном. Добавили триэтиламин (1,09 г, 0,01076 моль) и этилхлорформиат (1,17 г, 0,01076 моль), смесь перемешали в течение 10 минут и по каплям при поддержании температуры ниже 0oC добавили пирролидин (0,92 г, 0,01291 моль). Через 10 минут холодную баню удалили, и реакционную смесь перемешали в течение 16 часов при комнатной температуре. Раствор дихлорметана промыли насыщенным раствором бикарбоната натрия, 2 • 75 мл, и водой, 2 • 75 мл. Слой дихлорметана сушили над сульфатом магния и под вакуумом выпарили растворитель, получив при этом 2,19 г светло-коричневого масла. Светло-коричневое масло восстановили под давлением 3,5155 кг/см2, растворителем был этанол, а в качестве катализатора использовали 10% палладий на углероде. Через 4 часа катализатор отфильтровали и под вакуумом выпарили растворитель, обеспечив при этом 4-(3-((1-пирролидинил)карбонил)пропил)анилин, масло, которое отверждалось при стоянии (его использовали в том виде, как оно есть).
(b) N-(4-(3-((1-пирролидил)карбонил)пропил)фенил)-2- тиофенкарбоксимидамидгидроиодид.
К продукту стадии (a) (1,50 г, 0,00646 моль) в 6 мл изопропанола добавили S-метил-2-тиофенкарбоксимидгидроиоидид (продукт примера 1, стадии (d)) (1,53 г, 0,00538 моль) и перемешали в течение 16 часов при комнатной температуре. Полученную суспензию твердых частиц разбавили 50 мл изопропанола, и твердые частицы собрали фильтрацией, получив при этом N-(4-(3-((1-пирролидил)карбонил)пропил)фенил)-2- тиофенкарбоксимидамидиодид, точка плавления 213-216oC.
Пример 27
Используя методику, аналогичную той, которая представлена в примере 26, получили следующие соединения:
(a) N-(4-(3-((4-морфолинил)карбонил)пропил)фенил)-2- тиофенкарбоксимидамидгидроиодид, точка плавления 189-192oC.
(b) N-(4-((фениламино)карбонил)фенил)-2- тиофенкарбоксимидамид, точка плавления 200-201oC.
(c) N-(4-(2-(((4-морфолинил)карбонил)этил)фенил)-2-метилтиазол- 4-карбоксимидамид, точка плавления 280-281oC.
Пример 28 N-(4-(2-(((4-морфолинил)карбонил)амино)этил)фенил) тиофен-2-карбоксимидамидгидроиодид.
(a) 4-(2-(((4-морфолинил)карбонил)амино)этил)аналингидрохлорид.
К перемешанному раствору 4-нитрофенетиламингидрохлорида (4,0 г, 0,24 моль) в 50 мл тетрагидрофурана добавили 10 мл триэтиламина. К этой смеси по каплям добавили 4-морфолинкарбонилхлорид (3,6 г, 0,0024 моль) в 20 мл тетрагидрофурана, и реакционную смесь перемешали в течение 6 часов. Фильтрацией удалили триэтиламиновую соль, и органическую фазу промыли 1 • 100 мл 1N соляной кислоты. Органическую фазу сушили над сульфатом магния; после выпаривания растворителя получили сырое масло. Затем сырое масло растворили в 250 мл метанола, добавили 250 мг 10% палладия на углероде, и реакционную смесь гидрировали в течение 4-х часов. Фильтрацией удалили катализатор и выпарили растворитель. К остатку добавили 150 мл этилацетата, и для получения соли затем добавили хлористоводородный газ. При охлаждении кристаллизовался 4-(2-(((морфолинил)карбонил)амино)этил)анилингидрохлорид, твердое вещество 4,4 г, которое собрали фильтрацией.
(b) 4-(2-(((4-морфолинил)карбонил)амино)этил)анилин.
Продукт стадии (a), 4,4 г растворили в 200 мл воды 50%-ным раствором гидроксида натрия повысили основность смеси и экстрагировали 2 • 50 мл этилацетата. Экстракты этилацетата смешали, сушили над сульфатом магния и выпаривали в вакууме до получения твердого вещества. Твердое вещество растворили в горячем этилацетате и гексане, и при охлажднении образовались кристаллы 4-(2-(((4-морфолинил)карбонил)амино)этил)анилина. Анализ: вычислено: C 62,63, H 7,68, N 16,85; найдено: C 62,43, H 7,65, N 16,59.
(c) N-(4-(2-(((4-морфолинил)карбонил)амино)этил)фенил-2- тиофенкарбоксимидамидгидроиодид.
Продукт стадии (b), 1,0 г, и S-метил-2-тиофенкарбоксимидгидроиодид (продукт примера 1, стадии (d)), 1,09 г смешали с минимальным количеством изопропанола. Смесь нагревали в колбе с обратным холодильником в течение 1 часа. Полученный раствор охладили, осадили твердые частицы и собрали их фильтрацией, получив при этом 0,95 г N-(4-(2-(((4-морфолинил)карбонил)амино)этил)фенил-2- тиофенкарбоксимидамидгидрохлорида, точка плавления 209-211oC.
Пример 29 N-(4-(3-(((фенил)амино)карбонил)пропилокси)фенил-2- тиофенкарбоксимидамидгидроиодид.
(a) 4-(4-нитрофенокси)масляная кислота.
Этил-4-бромбутират, 9,9 г, 4-нитрофенол, 7,0 г, и карбонат натрия, 6,0 г, смешали в 50 мл DMF, и смесь нагрели на 100oC плитке в течение четырех часов. Фильтрацией удалили твердые частицы и промыли 20 мл ацетона. Фильтрат разбавили до 400 мл холодной водой и экстрагировали смесью из 50 мл этилацетата и 50 мл гексана. Полученный органический слой промыли 3 • 200 мл 0,2М карбоната калия для удаления непрореагировавшего нитрофенола. Полученный органический слой выпарили в вакууме до получения 11 г желтого масла. Сырое желтое масло разбавили 200 мл метанола, обработали 25 мл 2М гидроксида натрия и перемешивали всю ночь при комнатной температуре. Смесь выпарили в вакууме и разбавили водой до 250 мл. Раствор остветлили целитом и затем подкислили 20 мл 4М соляной кислоты. Полученные твердые частицы собрали фильтрацией, промыли водой и сушили в вакууме до получения 4-(4-нитрофенокси)масляной кислоты, точка плавления 116-118oC.
(b) N-(4-(3-(((фенил)амино)карбонил)пропилокси)анилин.
Раствор 4-(4-нитрфоенокси)масляной кислоты, 4,0 г в 20 мл тионилхлорида, нагревали в колбе с обратным холодильником в течение 4-х часов, и затем избыток тионилхлорида выпарили в вакууме. Сырой хлорангидрид добавили к раствору 1,68 г анилина и 10 мл триэтиламина в 30 мл THF, и реакционную смесь перемешали в течение 18 часов. Фильтрацией удалили твердые частицы, и фильтрат разбавили 50 мл этилацетата. Раствор промыли 100 мл 1N соляной кислоты, сушили сульфатом магния и выпарили до получения твердого вещества. Твердое растворили в 100 мл метанола и гидрировали в присутствии 10% Pd/C в течение 40 часов, получив при этом 1,25 г 4-(3-(((фенил)амино)карбонил)пропилокси)анилина, белого твердого вещества. M.S. (M+H)+ = 271.
(c) N-4-(3-(((фенил)амино)карбонил)пропилокси)фенил-2- тиофенкарбоксимидамидгидродиодид.
Продукт (b) 1,0 и S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)), 1,01 г смешали в минимальном количестве изопропанола, и смесь нагрели в колбе с обратным холодильником в течение 1 часа. Полученный прозрачный раствор охладили, твердое осадили и собрали фильтрацией, получив при этом 1,76 г N-(4-(3-(((фенил)амино)карбонил)пропилокси)фенил)-2- тиофенкарбоксимидамидгидроиодида, точка плавления 229-231oC.
Пример 30 N-(4-(2-(((трифторфенил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимид.
(a) 4-(2-(((трифторфенил)карбонил)амино)этил)нитробензол.
К перемешанному раствору 4-нитрофенетиламингидрохлорида (1,84 г, 9,10 ммоль) и триэтиламина (3,03 мл, 21,70 ммоль) в метаноле (12 мл) добавили по каплям трифторуксусный ангидрид (1,51 мл, 10,66 ммоль). После перемешивания в течение 1 минуты при пониженном давлении удалили растворитель, и оставшийся остаток смешали с водой и экстрагировали метиленхлоридом (3 • 20 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до выхода твердого вещества, которое перекристаллизовали из метиленхлорида/гексана до получения 4-(2-(((трифторметил)карбонил)амино)этил)нитробензола в виде белого твердого вещества; 1,92 г (выход 80%); точка плавления 103-104oC.
(b) 4-(2-(((трифторметил)карбонил)амино)этил)анилин.
К перемешанному раствору продукта стадии (a) (0,52 г, 1,98 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали в течение 1 часа при давлении 3,5155 кг/см2, фильтровали через целит и концентрировали до получения 4-(2-(((трифторметил)карбонил)амино)этил)анилина, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(c) N-4-(2-(((трифторметил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
К раствору продукта стадии (b) (0,30 г, 1,29 ммоль) в изопропаноле (6 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1 (стадия d) (0,37 г, 1,29 ммоль). Смесь перемешали в течение 4-х часов, перевели в раствор насыщенного NaCl (50 мл) и 50% NaOH (4 мл) и экстрагировали этилацетатом (3 • 20 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до твердого вещества, которое перекристаллизовали из гексана/этилацетата до выхода N-4-(2-(((трифторметил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамида в виде желтоватого твердого вещества: 0,19 г (выход 43%), точка плавления 181-182oC.
Пример 31 N-4-(2-(((метил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
(a) 4-(2-(((метил)карбонил)амино)этил)нитробензол.
Вышеуказанное соединение получили способом примера 30 стадии (a), за исключением того, что трифторуксусный ангидрид заменили уксусным ангидридом. Получили 1,28 бледно-желтого твердого вещества, которое тотчас же использовали в следующей реакции.
(b) 4-(2-(((метил)карбонил)амино)этил)анилин.
К перемешанному раствору продукта стадии (a) (0,82 г, 3,94 ммоль) в THF/MeOH (100 мл, 1:1) добавили 4 мл 1N HCl и каталитическое количество 10% Pd/C. Смесь гидрировали в течение 4-х часов при давлении 3,5155 кг/см2, фильтровали через целит и концентрировали до получения твердого вещества. Твердое вещество перевели в раствор насыщенного NaCl (50 мл) и 50% NaOH (4 мл) и экстрагировали этилацетатом (3 • 20 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до твердого вещества, которое тотчас же использовали в следующей реакции.
(c) N-4-(2-(((метил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
Вышеуказанное соединение получили, следуя методике примера 30, стадия (c). После перекристаллизации из этилацетата/метанола получили 0,56 г рыжевато-коричневого твердого вещества, точка плавления 186-187oC.
Пример 32
N-(4-(2-(((фенилметил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
(a) 4-(2-(((метил)карбонил)амино)этил)нитробензол.
Вышеуказанное соединение получили, следуя методике примера 30, стадия (a), за исключением того, что трифторуксусный ангидрид заменили фенилацетилхлоридом. Получили 1,42 г бледно-желтого твердого вещества, которое тотчас же использовали в следующей реакции.
(b) 4-(2-(((фенилметил)карбонил)амино)этил)анилин.
Вышеуказанное соединение получили способом, аналогичным тому, который описан в примере 8, стадия (b). Полученное масло тотчас же использовали в следующей реакции.
(c) N-(4-(2-(((фенилметил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
Вышеуказанное соединение получили, следуя методике примера 30, стадия (c). После перекристаллизации из этилацетата/метанола получили 0,45 г рыжевато-коричневого твердого вещества, точка плавления 210-211oC.
Пример 33 N-(4-(2-((фенил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид
(a) 4-(2-(((фенил)карбонил)амино)этил)нитробензол.
Вышеуказанное соединение получили, следуя методике примера 30, стадия (a), за исключением того, что трифторуксусный ангидрид заменили бензоилхлоридом. Получили 1,77 г бледно-желтого твердого вещества, которое тотчас же использовали в следующей реакции.
(b) 4-(2-(((фенил)карбонил)амино)этил)анилин.
Вышеуказанное соединение получили способом, аналогичным тому, который описан в примере 8, стадия (b). Полученное масло тотчас же использовали в следующей реакции.
(c) N-(4-(2-((фенил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
Вышеуказанное соединение получили, следуя методике примера 30, стадия (c). После перекристаллизации из этилацетата/метанола получили 1,10 г рыжевато-коричневого твердого вещества, точка плавления 196-197oC.
Пример 34 N-(4-(((фенил)иминокарбонил)амино)этил)фенил)-2- тиофенкарбоксимидамид.
(a) N-(4-нитрофенил)бензолкарбоксимидамид.
К перемешанному раствору бензонитрила (25 мл) добавили каталитическое количество 4-диметиламинопиридина. К этой смеси добавили 4-нитроанилингидробромид (10,0 г, 0,57 моль), и реакционную смесь нагрели в течение шести часов до 190oC. Реакционную смесь охладили и добавили 25 мл изопропанола. Твердое вещество собрали фильтрацией, получив при этом 8,6 г N-(4-нитрофенил)бензокарбоксимидамида, точка плавления 240-241oC.
(b) N-(4-аминофенил)бензолкарбоксимидамид.
В толстостенную склянку для реакций под давлением, загруженную продуктом стадии (a) (8,6 г, 0,024 моль) в 200 мл метанола, добавили 20 мл изопропанола/HCl и 0,5 г 10% палладия на углероде. Реакционную смесь гидрировали в течение шести часов; фильтрацией удалили катализатор и выпарили растворитель. К остатку добавили 50 мл изопропанола и 100 мл этилацетата, твердое суспендировали и затем собрали фильтрацией, получив при этом 10,6 г N-(4-аминофенил)бензолкарбоксимидамидгидрохлорида. N-(4-аминофенил)бензолкарбоксимидамидгидрохлорид растворили в 100 мл воды и 20 мл 50% гидроксида натрия. Водную фазу экстрагировали этилацетатом (3 • 100 мл) и сушили над сульфатом магния. После выпаривания растворителя получили твердый продукт N-(4-аминофенил)бензолкарбоксимидамид, выход 7,6 г.
(c) N-(4-(((фенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамид.
К перемешанной суспензии продукта стадии (b) (1,1 г, 0,0052 моль) в 5 мл изопропанола добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадия (o) (1,5 г, 0,0054 моль), и реакционную смесь перемешивали в течение 48 часов. Твердое собрали фильтрацией, растворили в растворе, содержащем 100 мл воды и 10 мл 50% гидроксида натрия, и водную фазу экстрагировали три раза этилацетатом (100 мл). Органическую фазу сушили над сульфатом магния и при последующем испарении растворителя получили сырое масло. Сырое масло растворили в 20 мл метанола и добавили хлористоводородный газ; при стоянии выкристаллизовывалось твердое вещество, которое собрали фильтрацией. Продукт, N-(4-(((фенил)иминокарбонил)амино)фенил)-2-тиофенкарбоксимидамид, сушили в течение 24-х часов над сульфатом магния, точка плавления 317-318oC.
Пример 35
Используя методику, аналогичную той, которая описана в примере 34, получили следующие соединения:
(a) N-N''-(1,4-фенилен)бис-2-тиофенкарбоксимидамидгидроиодид, точка плавления 278-279oC.
(b) N-N''-(1,3-фенил)бис-2-тиофенкарбоксимидамидгидроиодид, точка плавления 219-220oC.
(c) N-N'-(1,3-фенилен)бис-2-хлорфенил-карбоксимидамиддималеат, точка плавления 200-201oC.
(d) Свободное основание N-N-(1,4-фенилен)бис-3-хлортиофен-2-карбоксимидамид, точка плавления 247-248oC.
(e) Свободное основание N-(4-(((2-метоксифенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид, точка плавления 187-188oC.
(f) Свободное основание N-4-(((фенил)иминокарбонил)амино)фенил)-3-хлортиофен-2- карбоксимидамид, точка плавления 213-214oC.
(q) N-(4-(((фенил)иминокарбонил)амино)фенил)-3- тиофенкарбоксимидамиддигидрохлорид, точка плавления 323-324oC.
(h) N-(3-(((фенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамид, точка плавления 295-296oC.
(i) N-(4-(((4-хлорфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамидаминдигидрохлорид, точка плавления 296-297oC.
(j) N-(4-(((2-хлорфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамидамиддиоксалат, точка плавления 166-167oC.
(k) N-(4-(((4-бромфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамидамиддиоксалат, точка плавления 236-237oC.
(l) N-(4-(((3-хлор-4-метилфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамиддигидрохлорид, точка плавления 294oC.
(m) N-(4-(((3,5-диметоксифенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления 226-227oC.
(n) N-(4-(((3,5-дихлорфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления 237-238oC.
(o) N-(4-(((фенил)иминокарбонил)амино)фенил)-2-фуран- карбоксимидамиддиоксалат, точка плавления 210-211oC.
(p) Свободное основание N-(4-(((3-метилфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамид, точка плавления 205-206oC.
(q) Свободное основание N-(4-(((3-метоксифенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамид, точка плавления 194-195oC.
(r) N-(4-(((3-бромфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 293-294oC.
(s) N-(4-(((3-хлорфенил)иминокарбонил)амино)фенил)-2- тиофенкарбоксимидамиддигидрохлорид, точка плавления 310-311oC.
(t) N-(4-(((3-метилфенил)иминокарбонил)амино)фенил)-2- пирролкарбоксимидамиддигидробромид, точка плавления 210-211oC.
(u) N-(4-(((4-хлорфенил)иминокарбонил)амино)фенил)-2- пирролкарбоксимидамиддифумарат, точка плавления 228-229oC.
Пример 36 N-(4-(2-((фениламино)карбонил)амино)этил)фенил)-2- тиофенкарбоксамидингидроиодид.
(a) N-(2-(4-нитрофенил)этил-N'-фенилмочевина.
До начала реакции пробу 3,09 г 4-нитрофениламингидрохлорида растворили в 20 мл воды и обработали 30 мл 2N NaOH. Свободное основание экстрагировали 2 • 75 мл диэтилового эфира. Слой эфира сушили над сульфатом магния и объем эфира уменьшали под вакуумом до ~60 мл. К этому раствору добавили по каплям 1,81 г фенилизоцианата. Белые твердые частицы, которые осадились при добавлении, перемешали в течение 3-х часов, затем собрали фильтрацией и промывали простым эфиром. Воздушной сушкой получили 3,67 г N-(2-(4-нитрофенил)этил)-N'-фенилмочевины, точка плавления 170-172oC.
(b) N-(2-(4-аминофенил)этил)-N'-фенилмочевина/
В толстостенную склянку для реакций под высоким давлением, загруженную 3,67 г N-(2-(4-нитрофенил)этил)-N'-фенилмочевиной в 100 мл смеси метанола (THF 50/50 об.%), добавили каталитическое количество 5% Pd/C. Смесь гидрировали под давлением водорода 3,5155 кг/см2 в течение 24-х часов, катализатор отфильтровывали, и тонкослойная хроматография показала исходный материал и два пятна Rf.
Растворители удалили выпариванием в вакууме, и полученное твердое вещество соединили с метанолом и добавили избыток щавелевой кислоты. Раствор резко охладили простым эфиром, и осажденные белые твердые частицы собрали фильтрацией. Твердое обработали 100 мл 2N NaOH, и этилацетатом экстрагировали свободное основание; органический слой сушили сульфатом магния и выпарили, получив при этом желтое твердое вещество, N-(2-(4-аминофенил)этил-N'-фенилмочевину (430 мг).
(c) N-(2-(((фениламино)карбонил)амино)этил)фенил)-2- тиофенкарбоксамидингидроиодид.
К раствору 430 мг N-(2-(4-аминофенил)этил-N'-фенилмочевины, суспеднированной в 3 мл изопропилового спирта, добавили 435 мг S-метил-2-тиофентиокарбоксамиддигидроиодида (продукт примера 1, стадия (d)). Смесь перемешивали в течение 16 часов при комнатной температуре. Суспензию разбавили в 25 мл изопропилового спирта, и твердые частицы собрали фильтрацией, получив при этом желто-рыжевато-коричневое твердое вещество. Твердое перекристаллизовали из метанола/простого эфира. Две порции собрали и соединили для получения 470 г N-(4-(2-(((фениламино)карбонил)амино)этил)фенил)-2- тиофенкарбоксамидингидроиодида, точка плавления 216-219oC.
Пример 37
Способом, аналогичным тому, который представлен в примере 36, получили следующее соединение:
N-(4-(2-(((фениламино)карбонил)окси)этил)фенил)-2- тиофенкарбоксамид, точка плавления 222-224oC.
Пример 38
N-(4-((бис(фенилметил)амино)метил)фенил)-2- тиофенкарбоксимидамид.
(a) 4-((бис(фенилметил)амино)метил)нитробензол.
К 4-нитробензиламину (1,61 г, 10,60 ммоль) в DMF (25 мл) добавили карбонат калия (3,32 г, 23,30 ммоль), затем бензилбромид (2,64 мл, 22,30 ммоль). Смесь перемешали в течение 2-х дней, сбросили в воду и экстрагировали этилацетатом (3 • 50 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (20% этилацетата/гексана) до выхода 4-((бис(фенилметил)амино)метил)нитробензола: (1,64 г, 47%); M.S. (M+H)+ = 333.
(b) 4-((бис(фенилметил)амино)метил)анилин.
К продукту стадии (a) (0,56 г, 1,69 ммоль) в AcOH (15 мл) добавили дигидрат хлорида олова (II) (2,00 г, 19,03 ммоль), затем концентрировали HCl (5 мл). Смесь перемешали в течение 20 часов, охладили до 0oC, резко охладили 50% NaOH и экстрагировали этилацетатом (3 • 50 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали и подвергали хроматографии на силикагеле (30% этилацетата/гексана) до выхода 4-((бис(фенилметил)амино)метил)анилина: (0,29 г, 57%); M.S. (M+H)+ = 303.
(c) N-(4-((бис(фенилметил)амино)метил)фенил)-2- тиофенкарбоксимидамид.
К раствору продукта стадии (b) (0,28 г, 0,93 ммоль) в изопропаноле (6 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадия (d)) (0,26 г, 0,93 ммоль). Смесь перемешали в течение 14 часов, резко охладили 2N NaOH (2 мл) и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали и до получения твердого вещества, которое перекристаллизовали из этилацетата/гексана до выхода N-(4-((бис(фенилметил)амино)метил)фенил)-2- тиофенкарбоксимидамида в виде белого твердого вещества (86 мг, 22%); точка плавления 127-128oC.
Пример 39
Следуя способу, аналогичному тому, который представлен в примере 8, получили следующее соединение: N-(4-(2-аминометил)фенил)-2- тиофенкарбоксимидамидгидробромид, точка плавления 188-189oC.
Пример 40
N-(3-(1,2,3,4-тетрагидроизохинолин-2-илметил)фенил-2- тиофенкарбоксимидамиддигидробромид.
(a) 3-(1,2,3,4-тетрагидроизохинолин-2-ил-метил)нитробензол.
К 3-нитробензилхлориду (2,00 г, 11,66 ммоль) в DMF (25 мл) добавили карбонат калия (1,93 г, 13,96 ммоль), затем тетрагидроизохинолин (1,55 г, 11,66 ммоль). Смесь перемешали в течение 4 часов, сбросили в воду и экстрагировали этилацетатом (3 • 50 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения масла. Масло растворили в простом эфире и обработали 1PA/HCl для получения 3-(1,2,3,4-тетрагидроизохинолин-2-ил-метил)нитробензолгидрохлорида: (1,96 г, 55%); точка плавления 196-197oC.
(b) 3-(1,2,3,4-тетрагидроизохинолин-2-ил-метил)анилингидрохлорид.
К перемешанному раствору продукта стадии (a) (1,00 г, 3,29 ммоль) в THF/MeOH (100 мл, 1:1) добавили каталитическое количество 10% Pd/C. Смесь гидрировали в течение 0,5 часа при давлении 3,5155 кг/см2, фильтровали через целит и концентрировали до получения 3-(1,2,3,4-тетрагидроизохинолин-2-ил-метил)анилингидрохлорида, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(c) N-(3-(1,2,3,4-тетрагидроизохинолин-2-ил-метил)фенил)-2- тиофенкарбоксимидамид.
К раствору продукта стадии (b) (0,90 г, 3,29 ммоль) в изопропаноле (3 мл)/DMF (1 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)) (0,94 г, 3,29 ммоль). Смесь перемешали в течение 14 часов, резко охладили 2N NaOH (2 мл) и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и подвергали хроматографии на силикагеле (8% метанола/метиленхлорида) для выхода названного соединения в виде свободного основания. Обработкой 1PA/HBr получили N-(3-(1,2,3,4-тетрагидроизохинолин-2-ил-метил)фенил)-2- тиофенкарбоксимидамид в виде белого твердого вещества: (0,26 г, 16%); точка плавления (разлож.) > 179oC.
Пример 41
N-(4-(4-((фенилметил)амино)бутил)фенил)-2- тиофенкарбоксимидамиддималеат.
(a) 4-(4-нитрофенил)-N-(фенилметил)бутиламид.
Пробу 4-(4-нитрофенил)масляной кислоты, 2,09 г, растворенной в 20 мл дихлорметана, обработали бензиламином, 1,07 г, получив суспендированные твердые частицы. Смесь обработали дифенилфосфорилазидом, 2,75 г, и 20 мл диоксана, и после перемешивания в течение 4-х часов она стала прозрачной. Смесь разбавили 100 мл этилацетата и дважды промыли 100 мл 2М карбоната калия, затем 100 мл 1М соляной кислоты. Органический слой сушили сульфатом магния и выпарили до получения твердых частиц. Твердые частицы растворили в 150 мл циклогексана и 50 мл этилацетата и охладили до получения белых твердый частиц; эти твердые частицы собрали фильтрацией и сушили на воздухе до получения 4-(4-нитрофенил)-N-(фенилметил)бутиламида, точка плавления 133-135oC.
(b) 4-(4-аминофенил)-N-(фенилметил)бутиламидгидрохлорид.
Пробу продукта стадии (a), 0,80 г, в 20 мл этанола и 20 мл этилацетата обработали 0,4 г 5% палладия на углероде и подвергли давлению водорода 3,5155 кг/см2. Через 1 час тонкослойная хроматография показала новое пятно, P 0,2 с 15% ацетона в метиленхлориде. Смесь выпарили досуха и затем обработали 30 мл толуола. Остаток растворили в 10 мл ТНГ и обработали 4 мл 1М алюмогидрида лития в ТНГ, получив при этом прозрачный раствор. Через 1 час при комнатной температуре по каплям добавили 1 мл 2М гидроксида натрия, затем 10 г безводного сульфата натрия. После перемешивания в течение 30 минут смесь фильтровали и обработали хлористоводородным газом, образовалось масло. Смесь обработали 3 мл изопропанола и затем хлористоводородным газом, при этом получили твердые частицы. Смесь охладили до -20oC в течение 2-х часов, затем фильтровали и сушили на воздухе до получения 4-(4-аминофенил)-(фенилметил)бутиламидгидрохлорида.
Анализ хлорида: вычислено 20,78; найдено 20,64.
(c) 4-(4-((фенилметил)амино)бутил)анилин)дигидрохлорид.
Пробу продукта стадии (b) суспендировали в 5 мл ТНГ (Aldrich), получив при этом прозрачный раствор. Смесь нагрели в колбе с обратным холодильником в течение 5 часов, затем охладили. Полученную твердую массу разбавили диэтиловым эфиром до 20 мл и затем по каплям добавили 1 мл 2М гидроксида натрия, затем 4 см2 безводного сульфата натрия. После перемешивания в течение 15 минут смесь фильтровали, и твердые частицы промыли 20 мл диэтилового эфира. Смешанные фильтраты обработали хлористоводородным газом и выдержали при комнатной температуре. Образовавшиеся твердые частицы собрали фильтрацией и сушили в вакууме до получения 4-(4-((фенилметил)амино)бутил)анилин)дигидрохлорида; анализ хлорида: вычислено: 21,66, найдено 21,63.
(d) N-(4-(4-((фенилметил)амино)бутил)фенил)-2- тиофенкарбоксимидамиддималеат.
Пробу продукта стадии (c) 0,50 г и S-метил-2-тиофенкарбоксимидгидроиодид (продукт примера 1, стадии (d)) 0,44 г смешали в 4 мл изопропанола и нагрели до 60oC. Через 2 часа тонкослойная хроматография при использовании 15% метанола в хлороформе на диоксиде кремния показала, что исходный анилин был большей частью израсходован и что появилось новое пятно низшего Rf. Смесь разбавили 20 мл 1М карбоната калия и экстрагировали этилацетатом. Экстракт этилацетата сушили 10 г карбоната калия и обработали 0,150 г малеиновой кислоты, получив при этом клейкий осадок. Тонкослойная хроматография осадка в сравнении с надосадочной жидкостью показала, что осадок представлял смесь исходного амина и продукта и что надосадочная жидкость содержала главным образом продукт. Затем надосадочную жидкость обработали опять 0,150 г малеиновой кислоты, получив при этом клейкий осадок, и оставшуюся надосадочную жидкость слили. Твердые частицы растворили в 5 мл метанола и осадили 100 мл диэтилового эфира. Полученный клейкий осадок обработали 1 мл воды, разбавили ацетоном до 200 мл, получив при этом прозрачный раствор, разбавили диэтиловым эфиром до 275 мл и охладили до -20oC. Фильтрацией собрали твердые частицы, промыли 20 мл диэтилового эфира и сушили в вакууме для получения N-(4-((фенилметил)амино)бутил)фенил)-2- тиофенкарбоксимидамиддималеата, точка плавления 104-106oC.
Пример 42 N-(4-(((2-тиофенил)иминометил)амино)метил)фенил)-2- тиофенкарбоксимидамиддифумарат.
(a) 4-аминометиланилин.
К раствору 4-нитробензиламингидрохлорида (9,0 г, 0,0477) в метаноле (200 мл) добавили 20 мл 1PA/HCl и каталитическое количество 10% палладия на углероде.
Смесь гидрировали в течение 4-х часов при давлении 3,5155 кг/см2, фильтровали через целит, концентрировали до твердого вещества. Затем твердое вещество растворили в 300 мл воды и 20 мл 2N гидроксида натрия и экстрагировали в метиленхлорид (3 • 100 мл). Смешанные экстракты сушили над сульфатом магния, фильтровали и концентрировали до получения 4-аминометиланилина в виде масла (6,1 г).
(b) N-(4-(((2-тиофенил)иминометил)амино)метил)фенил)-2- тиофенкарбоксимидамиддифумарат.
К перемешанному раствору 4-аминометиланилина (1,6 г, 0,0013 ммоль) в диметилформамиде (10 мл) и изопропаноле (10 мл) добавили N-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)) (4,4 г, 0,015 ммоль). Смесь нагрели в течение 72 часов до 40oC. Реакционную смесь разбавили 20%-ным водным раствором гидроксида натрия и фильтрацией собрали твердые частицы (2,5 г). Из изопропанола и метанола получили соль фумаровой кислоты, при этом выход составил 2,0 г N-(4-(((2-тиофенил)иминометил)амино)метил)фенил)-2- тиофенкарбоксимидамиддифумарата, точка плавления 200-201oC.
Пример 43
Способом, аналогичным тому, который представлен в примере 17, получили следующие соединения:
(a) N-(3-(3-(1-пирролидинил)пропил)фенил)-фенилкарбоксимидамиддиоксалат, точка плавления 138-139oC.
(b) Свободное основание N-(4-(2-((4-метоксифенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид, точка плавления 144-145oC.
(c) N-(4-(2-((4-метилфенилметил)амино)этил)фенил)-2- тиофенкарбоксимидамидмоногидрохлорид, точка плавления 225-226oC.
(d) N-(3-(2-((3-фенилпропил)амино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 183-186oC.
(e) Свободное основание N-(3-(2-(2-метилфенилметил)амино) этил)фенил)-2-тиофенкрабоксимидамид, точка плавления 114-116oC.
(f) N-(4-(2-((1-инданил)этил)амино) фенил)-2-тиофенкарбоксимиддиоксалат, точка плавления 95oC (разлож.).
(g) N-(4-(2-(((4-пиридил)метил)амино)этил) фенил)-2-тиофенкарбоксимидамидтригидрохлорид, точка плавления > 250oC.
(h) N-(4-(2-(((2-тиенил)метил)амино)этил) фенил)-2-тиофенкарбоксимиддиоксалат, точка плавления 226-227oC.
Пример 44 N-(3-(2-((2-фенилэтил)амино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид.
(a) N-(2-фенилэтил)-3-нитрофенилацетамид.
2,50 г (0,0138 моль) 3-нитрофенилуксусной кислоты нагрели в колбе с обратным холодильником в 25 мл тионилхлорида. Растворители выпарили, и остаток перед добавлением по каплям при 0oC 3,51 г (0,290 моль) фенетиламина поместили в 50 мл THF. Смесь перемешивали в течение 48 часов и отфильтровали гидрохлоридную соль амина в виде белого твердого вещества. Выпарили промывочную воду, оставив при этом N-(2-фенилэтил)-3-нитрофенилацетамид, тусклое оранжевое масло, 4,48 г. Продукт анализировали M.S. (масс-спектрометрией) и NMR (ядерным магнитным резонансом).
(b) N-(2-фенилэтил)-2-(3-нитрофенил)этиламин.
К перемешанному раствору продукта стадии (a), 4,48 г (0,0158 моль) в 80 мл сухого THF добавили 47,7 мл 1М борана/THF. Смесь нагрели в колбе с обратным холодильником в течение 3-х часов, охладили и осторожно добавили 10 мл метанола, затем 20 мл 4N HCl. Раствор концентрировали выпариванием под вакуумом, при этом получили красноватую жидкость. 2М NaOH увеличили основность масла, и продукт экстрагировали 3 • 50 мл EtOAc. Органические слои соединили, сушили над сульфатом магния и выпарили, оставив при этом масло. Масло растворили в растворе HCl/изопропанола. Образовались белые твердые частицы, которые собрали фильтрацией, получив при этом N-(2-фенилэтил)-2-(3-нитрофенил)этиламин, точка плавления 196-200oC.
(c) N-(2-фенилэтил)-N-(2-(3-нитрофенил)этил)трифторацетамид.
К суспензии продукта стадии (b), 2,63 г (0,00857 моль) в 40 мл дихлорметана добавили 1,99 г (0,0197 моль) триэтиламина и до добавления по каплям 21,34 г (0,111 моль трифторуксусного ангидрида смесь охладили. Через 45 минут смесь резко охладили 50 мл воды, и продукт экстрагировали 3 • 50 мл дихлорметана. Органические слои смешали, сушили над сульфатом магния и выпарили, оставив при этом 3,3 г N-(2-фенилэтил)-N-(2-(3-нитрофенил)этил)трифторацетамида в виде масла.
(d) N-(2-фенилэтил)-N-(2-(3-аминофенил)этил)трифторацетамид.
К раствору продукта стадии (c), 3,3 г, в 75 мл THF и 75 мл метанола добавили каталитическое количество 10% Pd на углероде. Через 1 час под давлением 3,5155 кг/см2 реакцию завершили. Отфильтровали катализатор, и выпарили растворители, оставив при этом 2,88 г N-(2-фенилэтил)-N-(2-(3-аминофенил)этил) трифторацетамида в виде масла.
(e) N-(3-(2-((2-фенилэтил)амино)этил)фенил)-2- тиофенкарбкосимидамиддигидробромид.
К раствору продукта (d) 2,88 г (0,00857 моль) в 15 мл изопропанола добавили 2,94 г S-метил-2-тиофентиокарбоксимидгидроиодида (продукт примера 1, стадии (d)). Смесь перемешали при комнатной температуре в течение 16 часов, затем твердый желтый осадок отфильтровали и выбросили. Промывочную воду выпарили, и остаток растворили в минимальном количестве метанола; 2М NaOH повысили основность раствора и его нагрева в течение 30 минут до 50oC. Продукт экстрагировали 3 • 50 мл этилацетата, органические слои соединили, промыли 2 • 50 мл воды, сушили над сульфатом магния и выпаривали, оставив при этом масло. Свободное основание растворили в изопропаноловом растворе HBr. Твердые частицы, образованные при добавлении этилацетата и охлаждении, собрали фильтрацией, получив при этом 102 мг N-(3-(2-((2-фенилэтил)амино)этил)фенил)- 2-тиофенкарбоксимидамиддигидробромидной соли, точка плавления 137-139oC.
Пример 45
Следуя методике, аналогичной той, которая представлена в примере 44, получили следующие соединения:
(a) N-(4-(2-(аминоэтил)фенил)-2- пирролкарбоксимидамиддиоксалат, точка плавления 145oC (разлож.).
(b) (S)-N-(4-(2-((1-фенилэтил)амино)этил)фенил)-2- тиофенкарбоксимидамиддигидрохлорид, точка плавления 197oC (разлож.).
(c) Свободное основание (R)-N-(4-(2-((1-фенилэтил)амино)этил)фенил)-2- тиофенкарбоксимидамид, точка плавления 92-94oC.
(d) N-(3-(2-((4-фенилбутил)амино)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид, точка плавления 136-139oC.
Пример 46
N-(4-(((фенилметокси)карбонил)аминометил)фенил)-2- тиофенкарбоксимидамидоксалат.
(a) 4-(((фенилметокси)карбонил)аминометил)нитробензол.
Пробу 4-нитробензиламингидрохлорида, 5 г, и 200 мл воды обработали 10 г бикарбоната натрия. Смесь обработали 50 мл этилацетата и затем 4 мл бензилхлорформиата. Через 4 часа смесь обработали 200 мл гексана, и осажденные твердые частицы собрали фильтрацией. Сырой продукт растворили в 150 мл горячего метанола, фильтровали, разбавили 250 мл воды и охладили. Полученные твердые частицы собрали, получив при этом 4-(((фенилметокси)карбонил)аминометил)нитробензол, точка плавления 92-93oC.
(b) 4-(((фенилметокси)карбонил)аминометил)анилин.
Продукт стадии (a), 6,0 г, обработали 10 мл уксусной кислоты и 100 мл метанола. Смесь обработали сульфидом платины на углероде, 0,97 г, при давлении водорода 3,5155 кг/см2; через 20 часов смесь фильтровали и выпарили в вакууме. Остаток растворили в 50 мл простого эфира и разбавили гексаном до 200 мл. Затем смесь перемешали в течение пяти дней и фильтровали до получения 4-(((фенилметокси)карбонил)аминометил)анилина, точка плавления 60-64oC.
(с) N-4-(((фенилметокси)карбонил)аминометил)фенил)-2- тиофенкарбоксимидамидоксалат.
Продукт стадии (b), 0,89 г, и S-метил-2- тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)), 1,0 г, смешали в 6 мл изопропанола и перемешали при 30oC. Через 4 часа смесь осадили простым эфиром, при этом получили клейкие твердые частицы, которые поместили в 100 мл горячей воды, обработали целитом и фильтровали. Смесь охладили, получив клейкие твердые частицы, которые обработали 50 мл этилацетата и 5 г бикарбоната натрия. Слой этилацетата сушили сульфатом магния и охладили до -20oC. Затем смесь обработали гексаном. Смесь выпарили в вакууме, и сырое свободное основание, 0,7 г, растворили в 40 мл теплого изопропанола и обработали 0,26 г дигидрата щавелевой кислоты. При охлаждении и обработке 150 мл простого эфира образовался липкий осадок. После перемешивания в течение всей ночи при комнатной температуре полученные твердые частицы собирали фильтрацией, получив при этом N-((4-((фенилметокси)карбонил)аминометил)фенил)-2- тиофенкарбоксимидамидоксалат, точка плавления 150-160oC.
Пример 47
N-(4-((2-(фенилметил)амино)этокси)фенил)-2- тиофенкарбоксимидамидгидроиодид.
(a) 4-(нитрофенокси-N-(фенилметил)ацетамид.
Пробу 4-(нитрофенокси-N-(фенилметил)ацетамида уксусной кислоты (Ланкастер), 4,22 г, обработали 20 мл 1М водного раствора HCl и 200 мл этилацетата. Смесь охладили до 10oC и затем в течение 2-х минут добавили 1,38 г нитрата натрия в 20 мл воды. Смесь перемешали в течение 5 минут, соли разделили, и слой этилацетата сушили сульфатом натрия. Раствор этилацетата обработали 5 мл бензиламина, что привело к быстрому образованию осадка. Через 20 минут смесь промыли 100 мл насыщенного карбоната натрия, затем 100 мл 1М HCl (водного раствора). Затем выпарили слой этилацетата. Твердые частицы растворили в 50 мл ацетона и осадили водой. Твердые частицы собрали фильтрацией, при этом получили 3,76 г 4-(нитрофенокси-N-(фенилметил)ацетамида, точка плавления 125-126oC.
(b) 4-(аминофенокси-N-(фенилметил)ацетамид.
Пробу продукта стадии (a), 3,74 г, поместили в 100 мл метанола и 100 мл этилацетата. Смесь обработали 0,4 г 10% палладия на углероде и подвергли давлению водорода 3,5155 кг/см2. Через 1 час смесь фильтровали и концентрировали в вакууме, обеспечив при этом сырой 4-(аминофенокси-N-(фенилметил)ацетамид; вычислено: C 70,29; H 6,29; N 10,93; найдено: C 69,97; H 6,3; N 10,90.
(c) 4-(2-((фенилметил)амино)этокси)анилин.
Раствор 3,2 г продукта стадии (b) в 40 мл сухого THF под азотом обработали 40 мл 1М диборана в THF. Смесь нагрели в колбе с обратным холодильником в течение 3-х часов, обработали 40 мл 6М водного раствора HCl и нагревали в колбе с обратным холодильником в течение 2-х часов. Фильтрат концентрировали под вакуумом до 150 мл. Мутную смесь обработали 100 мл измельченного льда и нейтрализовали 50% NaOH, и полученные тонкоизмельченные твердые частицы собрали, промыли водой и сушили инфракрасным излучением до получения 4-(2-((фенилметил)амино)этокси)анилина, MS = 243,98%, как было найдено посредством капиллярного электрофореза.
(d) N-(4-(2-((фенилметил)амино)этокси)фенил)-2- тиофенкарбоксимидгидроиодид.
Пробу 4-(2-((фенилметил)амино)этокси)анилина, 0,81 г, и S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)), 1,42 г, смешали в 100 мл изопропанола и перемешали в течение 7 часов при комнатной температуре. Полученные белые твердые частицы собрали фильтрацией, промыли 10 мл изопропанола и сушили в вакууме, обеспечив при этом белое твердое вещество N-(4-(2-((фенилметил)амино)этокси)фенил-2- тиофенкарбоксимидамидгидроиодид, точка плавления 193-195oC.
Пример 48
N-[4-(((дифениламино)карбонил)амино)фенил] -2- тиофенкарбоксамидингидрохлорид.
(a) 4-[дифениламино(карбонил)амино]анилин.
К перемешанному раствору 1,4-фенилендиамина (1,00 г, 9,25 ммоль) и триэтиламина (1,29 мл, 9,25 ммоль) в метиленхлориде (50 мл) добавили дифенилкарбамилхлорид (2,14 г, 9,25 ммоль). После перемешивания в течение 14 часов смесь сбросили в воду и экстрагировали метиленхлоридом (3 • 20 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали, концентрировали и повергали хроматографии на силикагеле (80% этилацетата/гексан) до выхода 4-[дифениламино(карбонил)амино]анилина: (0,49 г, 17%); M.S. (M+H)+ = 304.
(b) N-[4-(((дифениламино)карбонил)амино)фенил] -2- тиофенкарбоксамидингидрохлорид.
К раствору продукта стадии (a) (0,49 г, 1,62 ммоль) в изопропаноле (10 мл) добавили S-метил-2- тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)) (0,46 г, 1,62 ммоль).
Смесь перемешали в течение 48 часов, сбросили в щелочную воду и экстрагировали этилацетатом (3 • 30 мл). Смешанные экстракты промыли водой, сушили над сульфатом магния, фильтровали и концентрировали до получения масла. Обработкой 1PA/HCl получили N-[4-(((дифениламино)карбонил)амино)фенил]-2- тиофенкарбоксамидингидрохлорид в виде белого твердого вещества: (24 мг, 3,3%); точка плавления 210-211oC.
Пример 49
N-(3-((бензоил)амино)фенил)-2-тиофенкарбоксимидамидоксалат.
(a) N-(3-нитрофенил)бензамид.
К раствору 7,5 г (54 ммоль) 3-нитроанилина в двухфазном растворе, состоящем из 100 мл метиленхлорида и 100 мл 20% карбоната калия, добавили по каплям 6,0 мл (52 ммоль) бензоилхлорида в 25 мл метиленхлорида. Реакционная смесь простояла всю ночь, и органическую фазу отделили и промыли разбавленной соляной кислотой. Растворитель концентрировали до получения 3,83 г (32%) названного соединения, MS 243 (M+H).
(b) N-(3-аминофенил)бензамид.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 30, стадия (b), MS 213 (M+H).
(c) N-(3-бензоил)амино)фенил)-2- тиофенкарбоксимидамидоксалат.
Это соединение получили, следуя методике примера 30, стадия (b). Свободное основание превратили в оксалатную соль в изопропаноле, MS 323 (M+H).
Пример 50
N-(4-((бензоил)амино)фенил)-2-тиофенкарбоксимидамидгидроиодид.
(a) N-(3-нитрофенил)бензамид.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 49, стадия (a), MS 243 (M+H).
(b) N-(4-аминофенил)бензамид.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 30, стадия (b), MS 213 (M+H).
(c) N-(4-((бензоил)амино)фенил)-2- тиофенкарбоксимидамидгидроиодид.
Это соединение получили, следуя методике примера 26, стадия (b) и перекристаллизовали из воды, точка плавления 234-235oC.
Пример 51
N-(3-(((фениламино)карбонил)амино)фенил)-2- тиофенкарбоксимидамидоксалат.
(a) N-фенил-N'-(3-нитрофенил)мочевина.
К раствору 54,0 г (36 ммоль) m-нитроанилина в 40 мл простого эфира добавили 5,0 мл (47 ммоль) фенилизоцианата. Раствор перемешали в течение 6-ти часов. Продукт фильтровали до получения 9,2 г (99%) названного соединения, MS 258 (M+H).
(b) N-фенил-N'-(3-аминофенил)мочевина.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 30, стадия (b). Суспендирование выделенного продукта в простом эфире привело к получению твердого продукта, точка плавления 199-202oC.
(c) N-(3-(((фениламино)карбонил)амино)фенил)-2- тиофенкарбоксимидамидоксалат.
Это соединение получили, следуя методике примера 30, стадия (c). Свободное основание превратили в оксалатную соль в изопропаноле, 208-210oC.
Пример 52 N-(3-(((4-феноксибутил)амино)карбонил)фенил)-2- тиофенкарбоксимидамидоксалат.
(a) 3-нитро-N-(4-феноксибутил)бензамид.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 49, стадия (a), MS 315 (M+H).
(b) N-((3-(4-феноксибутил)амино)карбонил)анилингидрохлорид.
Раствор 7,8 г (25 ммоль) N-4-феноксибутил-3-нитробензамида и 1 г 5% палладия на углероде в 120 мл изопропанола при добавлении хлористого водорода гидрировали в течение 3-х часов при давлении 3,164 кг/см2. Катализатор отфильтровали, и растворитель концентрировали, получив при этом 6,7 г (84%) названного соединения, MS 285 (M+H).
(c) N-(3-(((4-феноксибутил)амино)карбонил)-2- тиофенкарбоксимидамидоксалат.
Вышеуказанное соединение сначала превратили в свободное основание и, используя методику примера 30, стадия (c), получили названное соединение. Затем свободное основание названного соединения превратили в оксалатную соль в изопропаноле, MS 349 (M+H), точка плавления 154-156oC.
Пример 53
N-(3-(((4-фенилбутил)амино)карбонил)фенил)-2- тиофенкарбоксимидамидоксалат.
(a) 3-нитро-N-(4-фенилбутил)бензамид.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 49, стадия (a), MS 299 (M+H).
(b) 3-амино-N-(4-фенилбутил)бензамидгидрохлорид.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 52, стадия (b), MS 273 (M+H).
(c) N-3-(((4-фенилбутил)амино)карбонил)фенил)-2- тиофенкарбоксимидамидоксалат.
Это соединение получили, следуя методике примера 30, стадия (c), за исключением того, что к тому же добавили эквивалент триэтиламина. Свободное основание превратили в оксалатную соль в изопропаноле, MS 376 (M+H), точка плавления 118-120oC.
Пример 54
N-4-(((бензил)амино)карбонил)метил)фенил)-2- тиофенкарбоксимидамид.
(a) N-бензил-(4-нитро)фенилацетамид.
Это соединение получили, следуя методике, аналогичной той, которая представлена в примере 49, стадия (a), точка плавления 172-182oC.
(b) N-бензил-(4-амино)фенилацетамид.
Это соединение получили, следуя методике примера 17, стадия (d), точка плавления 137-140oC.
(c) N-(4-((((бензил)амино)карбонил)метил)фенил)-2- тиофенкарбоксимидамид.
Это соединение получили, следуя методике примера 30; стадия (c), точка плавления 257-161oC.
Пример 55
N-(4-(2-(1-пирролидинил)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид.
(a) N-пирролидинил-(4-нитрофенил)уксусная кислота.
К раствору 3,12 г (43,9 ммоль) пирролидина в двухфазном растворе, состоящем из 100 мл метиленхлорида и 100 мл 20% карбоната калия, добавили по каплям 7,3 г (36,5 ммоль) 4-нитрофенилацетилихлорида в 25 мл метиленхлорида. Реакционная смесь простояла всю ночь, и органическую фазу отделили и промыли разбавленной соляной кислотой. Растворитель концентрировали до получения 6,26 г (73%) названного соединения, точка плавления 103-105oC.
(b) 4-(2-(1-пирролидинил)этил)нитробензол.
Это соединение получили, следуя методике примера 17, стадия (c), MS 221 (M+H).
(c) 4-(2-(1-пирролидинил)этил)анилин.
Это соединение получили, следуя методике примера 34, стадия (c), MS 191 (M+H).
(d) N-(4-(2-(1-пирролидинил)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид.
Это соединение получили, следуя методике примера 18, стадия (b). Дигидробромидную соль выкристаллизовали из изопропанола и простого эфира, MS 300 (M+H).
Пример 56
N-(4-(2-(1-пиперидинил)этил)фенил)-2- тиофенкарбоксимидамиддигидрохлорид.
(a) N-пиперидинил-(4-нитрофенил)уксусная кислота.
Это соединение получили, следуя методике примера 55, стадия (a), точка плавления 105-107oC.
(b) N-(4-(2-(1-пиперидинил)этил)нитробензол.
Это соединение получили, следуя методике примера 17, стадия (c), MS 235 (M+H).
(c) N-(4-(2-(1-пиперидинил)этил)анилин.
Это соединение получили, следуя методике примера 34, стадия (c). Гидрохлоридную соль превратили в свободное основание в виде масла, MS 205 (M+H).
(d) N-(4-(2-(1-пиперидинил)этил)фенил)-2- тиофенкарбоксимидамиддигидрохлорид.
Это соединение получили, следуя методике примера 34, стадия (d). Дигидрохлоридную соль выкристаллизовали из пропанола и простого эфира, точка плавления 256-261oC.
Пример 57
N-(4-(3-(1-пирролидинил)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
(a) N-пирролидинил-(4-нитрофенил)пропенамид.
Это соединение получили, следуя методике примера 55, стадия (a), MS 247 (M+H).
(b) 4-(2-(1-((пирролидинил)карбонил)этил)анилин.
Это соединение получили, следуя методике примера 34, стадия (c), MS 219 (M+H).
(c) 4-(3-(пирролидинил)пропил)анилин)дигидрохлорид.
Это соединение получили, следуя методике примера 17, стадия (c). Дигидрохлоридную соль выкристаллизовали из этанола, точка плавления 262-265oC.
(d) N-(4-(3-(1-пирролидинил)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
Это соединение получили, следуя методике примера 18, стадия (b). Диоксалатную соль получили из простого эфира, точка плавления 86-92oC.
Пример 58
N-(4-(3-(1-пиперидинил)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
(a) N-пиперидинил-(4-нитрофенил)пропенамид.
Это соединение получили, следуя методике примера 55, стадия (a). Точка плавления 168-171oC.
(b) 4-(2-(1-((пиперидинил)карбонил)этил)анилин.
Это соединение получили, следуя методике примера 34, стадия (c), MS 233 (M+H).
(c) 4-(2-1-((пиперидинил)пропил)анилин.
Это соединение получили, следуя методике примера 17, стадия (c), точка плавления 180-185oC.
(d) N-(4-(3-(1-((пиперидинил)пропил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
Это соединение получили, следуя методике примера 17, стадия (e). Из этанола и этилацетата получили диоксалатную соль, MS 328 (M+H).
Пример 59
N-(4-(2-(2-(1,2,3,4-тетрагидро)изохинолил)этил)фенил-2- тиофенкарбоксимидамиддигидробромид.
(a) 4-нитро-N-(2-изохинолил)фенилацетамид.
К раствору 4-нитрофенилуксусной кислоты (5,43 г, 930 ммоль) и 1,2,3,4-тетрагидроизохинолина (5,6 г, 42 ммоль) в метиленхлориде (200 мл) добавили 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (6,13 г, 32 ммоль). Реакционную смесь перемешали в течение 18 часов. Реакционную смесь промыли разбавленной соляной кислотой и разбавленным гидроксидом натрия, сушили, и растворитель концентрировали до получения твердого вещества. При растирании в порошок с простым эфиром получили названное соединение, точка плавления 137-139oC.
(b) N-(4-(2-(2-(1,2,3,4-тетрагидро)изохинолил)этил)нитробензол.
Это соединение получили, следуя методике примера 17, стадия (c), точка плавления 97-102oC.
(c) N-(4-(2-(2-(1,2,3,4-тетрагидро)изохинолил)этиланилин.
Это соединение получили, следуя методике примера 34, стадия (c), точка плавления 300oC (разлож.).
(d) N-(4-(2-(2-(1,2,3,4-тетрагидро)изохинолил)этил)фенил)-2- тиофенкарбоксимидамиддигидробромид
Это соединение получили, следуя методике примера 18, стадия (b). Из этанола и этилацетата получили дигидробромидную соль, MS 362 (M+H).
Пример 60
Свободное основание N-(4-((((фенилметил)амино)метил- карбонил)амино)фенил)-2-тиофенкарбоксимидамид.
(a) N-(4-нитроофенил)-2-хлорацетамид.
4-нитроанилин (13,8 г) в этилацетате (200 мл) обработали триэтиламином (15 мл) и затем порциями обработали хлорацетилхлоридом (8 мл). Полученную смесь перемешали в течение 10 минут. Затем смесь обработали водой (200 мл) и этилацетатом (100 мл). Смесь нагревали до тех пор, пока растворились все твердые частицы, и слои разделили. Затем слой ацетилацетата концентрировали до 100 мл при нагреве и охладили до комнатной температуры. На следующий день смесь отфильтровали, и твердые частицы промыли этилацетатом и сушили на воздухе до получения 14,83 г N-(4-нитрофенил)-2-хлорацетамида, точка плавления 183-185oC.
(b) 4-((((фенилметил)амино)метилкарбонил)амино)нитробензол.
Соединение стадии (a) (4,28 г) и бензиламин (2,5 мл) смешали в DMF с карбонатом калия (3,2 г), перемешали при комнатной температуре в течение 3-х часов. Отфильтровывали твердые частицы и промыли метанолом (2 • 10 мл). Смешанные фильтры медленно разбавили водой до 150 мл, получив при этом желтые твердые частицы, которые собрали фильтрацией и сушили на воздухе, получив при этом 4-((((фенилметил)амино)метилкарбонил)амино)нитробензол, MS 286 (M + H).
(c) 4-((((фенилметил)трифторметилкарбонил)амино)метилкарбонил)амино) нитробензол.
Соединение стадии (b) (4,9 г), трифторуксусный ангидрид (2,5 мл) и триэтиламин (2,5 мл) смешали в этилацетате (50 мл), и смесь с суспендированными частицами нагревали всю ночь до 50oC. Затем смесь промыли водой (50 мл), фильтровали для удаления твердых частиц, и выпарили слой этилацетата. Остаток поместили в простой эфир (151 мл) и охлаждали до -20oC всю ночь. Твердые частицы собрали фильтрацией, получив при этом 4-((((фенилметил)трифторметил)карбонил)амино)метилкарбонил)амино) нитробензол, MS (M+H) = 282.
(d) 4-((((фенилметил)трифторметилкарбонил)амино) метилкарбонил)амино)анилин.
Соединение стадии (c) (3,8 г) растворили в этилацетате (50 мл) и этаноле (50 мл). Смесь гидрировали при давлении 3,5155 кг/см2 в течение 4 часов палладием на углероде. Смесь фильтровали и выпарили в вакууме, получив при этом белые твердые частицы 4-((((фенилметил)трифторметил)карбонил)амино)метилкарбонил)амино) анилина, MS (M+H) = 352.
(e) Свободное основание N-4-((((фенилметил)амино)метилкарбонил)амино)фенил)-2- тиофенкарбоксимидамид.
Соединение стадии (d) (1,05 г) и S-метил-2- тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (o)) (0,85 г) обработали метанолом (2 мл). Через 15 минут твердые частицы растворились, и смесь продули азотом для удаления метантиола. Тонкослойная хроматография с использованием 10% изопропанола в хлороформе на диоксиде кремния показала новое пятно низшего Rf и, что исходный амин израсходован.
Смесь растворили в метаноле (6 мл) и обработали карбонатом калия (1,1 г). Тонкослойная хроматография с использованием 15% метанола в хлороформе на диоксиде кремния показала, что гидролиз не завершен, поэтому добавили дополнительное количество карбоната калия, 1,1 г. Через 2 часа превращение завершилось, и смесь для удаления твердых частиц отфильтровали. На следующий день фильтрат обработали 0,65 г малеиновой кислоты, разбавили простым эфиром и перемешивали всю ночь. Собранные твердые частицы не представляли желательный продукт. Затем фильтрат разбавили гексаном и промыли водой. Водный слой обработали 1М карбонатом калия и экстрагировали этилацетатом. Этилацетат выпарили в вакууме, и остаток поместили в 20 мл метанола.
Раствор медленно обработали водой до тех пор, пока растворились твердые частицы. Твердые частицы собрали фильтрацией и сушили под вакуумом при 40oC до получения свободного основания N-4-((((фенилметил)амино)метилкарбонил)амино)фенил)-2- тиофенкарбоксимидамида, точка плавления 161-163oC.
Пример 61
N-(4-(2-(((2-фуранил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
(a) 4-нитрофенилацетилхлорид.
К 4-нитрофенилуксусной кислоте (30 г) добавили тионилхлорид (100 мл). Смесь нагрели в колбе с обратным холодильником под азотом и перемешивали в течение 2-х часов. Избыток тионилхлорида выпарили под вакуумом, и оставшееся масло азеотропно сушили толуолом. При этом получили 4-нитрофенилацетилхлорид (35 г).
(b) 4-(2-(((2-фуранил)метил)амино)этил)нитробензолгидрохлорид.
К перемешанному раствору фурфуриламина (1,32 г) в метиленхлориде (125 мл) добавили триэтиламин (2,36 мл), затем по каплям добавили раствор соединения стадии (a) (3,0 г) в метиленхлориде (10 мл). Смесь перемешали при 0oC в течение 15 минут. Смесь влили в воду (150 мл), и метиленхлоридом (2 • 100 мл) экстрагировали сырые продукты. Органические слои собрали, сушили (MgSO4), фильтровали и концентрировали. Полученные твердые частицы поместили в раствор 7% метанола/метиленхлорида и очистили на силикагельной колонке при элюировании тем же самым растворителем. Продукт собрали и концентрировали. Твердое поместили в THF (50 мл) и обработали 1М бораном/THF (50 мл). Раствор нагревали в колбе с обратным холодильником в течение 15 часов. Смесь охладили до 0oC и медленно добавили 4N соляную кислоту. Смесь повторно нагревали в колбе с обратным холодильником и перемешали в течение 4-х часов. Избыток кислоты и THF выпарили под вакуумом и оставшуюся суспензию поместили в воду (100 мл) и этилацетат (100 мл), 50% гидроксидом натрия увеличили основность и экстрагировали этилацетатом (3 • 125 мл). Органические слои собрали, сушили (MgSO4), фильтровали и концентрировали. Сырой продукт сушили на силикагельной колонке при элюировании 10% метанолом/метиленхлоридом. Продукт собрали и концентрировали. Оставшиеся твердые частицы поместили в изопропиловый спирт (25 мл) и обработали насыщенным 1PA/HCl (10 мл). Белые частицы отфильтровали и промыли изопропиловым спиртом, получив при этом 4-(2-(((2-фуранил)метил)амино)этил)нитробензолгидрохлорид (2,9 г).
(c) 4-(2-(((2-фуранил)метил)амино)этил)анилингидрохлорид.
К перемешанному раствору соединения стадии (b) (2,46 г) в уксусной кислоте (100 мл) добавили одной порцией (3,3 г) цинковую пыль. Смесь перемешали в течение 10 минут. Цинк отфильтровали, и избыток кислоты выпарили под вакуумом. Оставшиеся твердые частицы поместили в воду (100 мл) и этилацетат (100 мл), 50% гидроксидом натрия повысили основность и экстрагировали этилацетатом (3 • 125 мл). Органические слои собрали, сушили (MgSO4), фильтровали и концентрировали. Оставшееся масло поместили в изопропиловый спирт (25 мл) и обработали насыщенным 1PA/HCl (10 мл). Белые твердые частицы отфильтровали и промыли изопропиловым спиртом, получив при этом 4-(2-(((2-фуранил)метил)амино)этил) анилиндигидрохлорид (1,50 г).
(d) 4-(2-(((2-фуранил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамиддиоксалат.
К перемешанной суспензии соединения стадии (c) (1,5 г) в DMF (15 мл) добавили пиридин (0,42 мл), затем S-метил-2- тиофентиокарбоксадигидроиодид (продукт примера 1, стадии (d)) (1,51 г). Смесь нагрели до 50oC и перемешали в течение 48 часов. Затем смесь разбавили в 100 мл воды и избытком 50% гидроксида натрия повысили основность. Сырой продукт экстрагировали этилацетатом (3 • 100 мл). Органические слои собрали и промыли водой. Органический слой сушили (MgSO4), фильтровали и концентрировали. Сырой продукт очистили на силикагельной колонке при элюировании 20% метанолом/метиленхлоридом. Продукт собрали и концентрировали до получения масла, которое поместили в изопропиловый спирт и обработали 2,5 эквивалентами щавелевой кислоты. Белые твердые частицы отфильтровали и промыли простым эфиром, получив при этом N-(4-(2-(((2-фуранил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамиддиоксалат (580 г), точка плавления (разлож.) > 220oC.
Пример 62
Следуя методике, аналогичной той, которую использовали в примере 61, получили следующие соединения:
(a) N-(4-(2-(((2-пиридил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамидтригидрохлорид, точка плавления (разлож.) > 250oC.
(b) N-(4-(2-(((2-тиофенил)метил)амино)этил)фенил)-2- тиофенкарбоксимидамиддиоксалат, точка плавления (разлож.) > 226oC.
Пример 63
N-(4-((амино)карбонил)фенил)-2-тиофенкарбоксимидамидгидроиодид.
Это соединение получили, следуя методике примера 26, стадия (b). Соль перекристаллизовали из 30% изопропанола в воде, точка плавления 236oC (разлож.).
Пример 64
N-(4-(2-(((2-тиенил)карбонил)амино)этил)фенил)-2- тиофенкарбоксимидамидоксалат.
(a) N-(4-нитрофенил)-2-тиофенкарбоксамид.
Это соединение получили, следуя методике примера 26, стадия (a), MS 249 (M+H).
(b) N-(4-амино)анилин-2-тиофенкарбоксамид.
Это соединение получили, следуя методике примера 17, стадия (d), MS 219 (M+H).
(c) N-(4-(((2-тиенил)карбонил)амино)фенил-2- тиофенкарбоксимидамидоксалат.
Это соединение получили, следуя методике примера 7, стадия (c). Свободное основание превратили в оксалатную соль в изопропаноле, точка плавления 231-233oC.
Пример 65
N-[4-((((дифениламино)карбонил)амино)метил)фенил] -2- тиофенкарбоксамидиноксалат.
(a) 4-((((дифениламино)карбонил)амино)метил)нитробензол.
К перемешанному раствору 4-нитробензиламингидрохлорида (1,04 г, 5,51 ммоль) и триэтиламина (1,56 мл, 11,22 ммоль) в метиленхлориде (10 мл) добавили дифенилкарбамилхлорид (1,40 г, 6,07 ммоль). После перемешивания в течение 5 часов смесь сбросили в воду и слои разделили. Затем водный слой экстрагировали метиленхлоридом (3 • 20 мл). Смешанные экстракты промыли водой, сушили (MgSO4), фильтровали и концентрировали до выхода твердого вещества, которое перекристаллизовали иэ этилацетата/гексана/метанола для получения 4-((((дифениламино)карбонил)амино)метил)нитробензола в виде белого твердого вещества: 1,37 г (выход 72%); точка плавления 137-138oC.
(b) 4-((((дифениламино)карбонил)амино)метил)анилин.
К перемешанному раствору соединения стадии (a) (1,37 г, 3,94 ммоль) в THF/MeOH (100 мл, 1:1)) добавили каталитическое количество 10% Po/C. Смесь гидрировали при давлении 3,5155 кг/см2 в течение 1 часа, фильтровали через целит и конденсировали до получения 4-((((дифениламино)карбонил)амино)метил)анилина, который стал гомогенным посредством тонкослойной хроматографии, и тотчас же использовали в следующей реакции.
(c) N-[4-((((дифениламино)карбонил)амино)метил)фенил]-2- тиофенкарбоксамидиноксалат.
К раствору соединения стадии (b) (1,24 г, 3,90 ммоль) в изопропаноле (10 мл) добавили S-метил-2- тиофентиокарбоксимидгидроиодид (1,06 г, 3,70 ммоль). Смесь перемешали в течение 18 часов, сбросили в щелочную воду и экстрагировали хлороформом (3 • 30 мл). Смешанные экстракты промыли водой, сушили (MgSO4), фильтровали, концентрировали и подвергали хроматографии на силикагеле (6% метанола/метиленхлорида) до получения масла, которое отвердилось при стоянии. Маленькое количество было выделено в виде оксалатной соли: (48 мг); точка плавления (разлож.) 150oC.
Пример 66
N-[4-((((2-тиофенил)иминометил)амино)метил)фенил] -2- тиофенкарбоксамидиндифумарат.
(a) 4-(аминометил)анилин.
К раствору 4-нитробензиламингидрохлорида (9,0 г, 4,7 ммоль) в метаноле (200 мл) добавили каталитическое количество 10% палладия на углероде. Смесь гидрировали в течение 4-х часов при давлении 3,5155 кг/см2, фильтровали через целит и концентрировали до получения сырого масла. Масло растворили в воде (100 мл) и 20% гидроксиде натрия (20 мл), дважды экстрагировали дихлорметаном, органический слой сушили (MgSO4), фильтровали и концентрировали до выхода (6,1 г) 4-(аминометил)анилина.
(b) N-(4-((((2-тиофенил)иминометил)амино)метил)фенил)-2- тиофенкарбоксамидиндифумарат.
Смесь соединения стадии (a) (1,6 г, 1,3 ммоль) и S-метил-2-тиофентиокарбоксамидгидроиодид (продукт примера 1, стадии (d)) (4,4 г, 1,5 ммоль) в HF (10 мл) нагрели до 40oC в течение 72 часов. Затем смесь разбавили 20% водным раствором гидроксида натрия, и твердое собрали фильтрацией, получив при этом 2,5 г сырого N-((((2-тиофенил)иминометил)амино)метил)фенил)-2- тиофенкарбоксамидина. Из метанола и изопропанола получили дифумаратную соль, точка плавления 200-201oC.
Пример 67
Следуя методике, аналогичной той, которая представлена в примере 66, получили следующее соединение:
(a) N-(4-((((2-тиофенил)иминометил)амино)этил)фенил)-2- тиофенкарбоксамидиндифумарат, точка плавления 200-201oC.
Пример 68
N-(4-((((3-метилфенил)иминометил)амино)этил)фенил)-2- тиофенкарбоксамидиндифумарат.
(a) N-(4-(2-аминоэтил)фенил)-2-тиофенкарбоксамидин.
К раствору 4-аминоэтиланилиндигидрохлорида (1,4 г, 6,6 ммоль) и S-метил-2-тиофентиокарбоксимидгидроиодида (продукт примера 1, стадии (d)) (2,2 н, 7,6 ммоль) в DMF (10 мл) добавили пиридин (0,52 г, 6,6 ммоль). Смесь перемешали при 40oC в течение 24-х часов, разбавили 20% водным раствором гидроксида натрия, дважды экстрагировали дихлорметаном, сушили (MgSO4), фильтровали и концентрировали до получения N-(4-(2-аминоэтил)фенил)-2-тиофенкарбоксамидина (4,1 г) в виде масла.
(b) N-(4-(((3-метилфенил)иминометил)амино)этил)фенил- 2-тиофенкарбоксамидиндифумарат.
К раствору соединения стадии (a) (2,0 г, 8,2 ммоль) добавили S-метил-(3-метилфенил)тиокарбоксимидигидроиодид (2,8 г, 9,2 ммоль) в изопропаноле (10 мл). Смесь перемешали при 40oC в течение 18 часов, разбавили 20% водным раствором гидроксида натрия и дважды экстрагировали этилацетатом. Органический слой промыли водой (100 мл), сушили (MgSO4), фильтровали и концентрировали до получения масла. Из метанола, изопропанола и этилацетата получили дифумаратную соль, N-(4-((((3-метилфенил)иминометил)амино)этил)фенил)-2- тиофенкарбоксамидиндифумарат: точка плавления 200-201oC.
Пример 69
Следуя методике, аналогично той, которая представлена в примере 68, получили следующее соединение:
(a) N-(3-((((2-тиофенил)иминометил)амино)этил)фенил)- -2-тиофенкарбоксамидиндифумарат, точка плавления 211-212oC.
Пример 70
N-(4-(2-(пиримидин-2-ил)амино)этил)фенил)-2- тиофенкарбоксамиддигидрохлорид.
(a) [2-(4-нитрофенил)этил]пиримиди-2-иламин.
К раствору 4-нитрофенетиламингидрохлорида (2,0 г, 9,8 ммоль) в диметилформамиде (20 мл) добавили карбонат калия (10 г) и 2-хлорпиримидин (1,6 г, 1,4 ммоль). Смесь нагревали в течение 24 часов до 100oC, разбавили водой (300 мл), дважды экстрагировали этилацетатом, сушили (MgSO4), фильтровали и концентрировали до получения сырого твердого вещества. Из этилацетата, изопропанола получили моногидрохлоридную соль [2-(4-нитрофенил)этил]пиримидин-2-иламингидрохлорид (2,4 г).
(b) [2-(4-аминофенил)этил]пиримидин-2-иламин.
К раствору соединения стадии (a) в уксусной кислоте (100 мл) добавили цинковую пыль (3,0 г). Реакционную смесь перемешали в течение 30 минут, фильтровали и концентрировали. Остаток разделили 20% водным раствором гидроксида натрия и дихлорметаном и органический слой сушили (MgSO4), фильтровали и концентрировали до выхода 1,3 г [2-(4-аминофенил)этил]пиримиди-2-иламина.
(c) N-(4-(2-((пиримидин-2-ил)амино)этил)фенил)-2- тиофенкарбоксамиддигидрохлорид.
К раствору соединения (b) (1,3 г, 6,6 ммоль) в диметилформамиде (10 мл) добавили S-метил-2-тиофентиокарбоксимидгидроиодид (продукт примера 1, стадии (d)). Реакционную смесь перемешали в течение 72 часов, разбавили 20% раствором гидроксида натрия и дважды экстрагировали этилацетатом. Смешанные экстракты дважды промыли водой, сушили (MgSO4), фильтровали и концентрировали.
Из метанола и изопропанола получили гидрохлоридную соль N-(4-(2-((пиримидин-2-ил)амино)этил)фенил)-2- тиофенкарбоксамиддигидрохлорид, точка плавления 191-192oC.
Пример 71
N-(4-(2-((фенилметил)амино)этокси)-2-фторфенил)-2- тиофенкарбоксимидамид.
(a) 3-фтор-4-нитрофеноксиуксусная кислота.
Пробу 3-фтор-4-нитрофенола (5,18 г) и карбоната калия (10 г) обработали DMF (20 мл). Затем добавили бромацетат (5 мл), и смесь перемешали при 22oC. Через 2 часа смесь обработали метанолом (20 мл) и водой (40 мл) и перемешали. Еще через 2 часа смесь разбавили водой до 200 мл и отфильтровали твердые частицы. Фильтрат подкислили, собрали твердые частицы, получив при этом 3-фтор-4-нитрофеноксиуксусную кислоту, точка плавления 90-92oC.
(b) N-(фенилметил)-3-фтор-4-нитрофеноксиацетамид.
Пробу соединения стадии (a) (2,84 г) растворили в сухом THF (100 мл), затем добавили метилморфолин (1,45 мл), и смесь перемешали при 0oC. Добавили этилхлорформиат (1,26 мл), и смесь перемешали в течение 2-х минут. Добавили бензиламин (1,45 мл), и смесь перемешали при 0oC. Через 15 минут смесь медленно разбавили водой до 22 мл и перемешали при 0oC. Полученные твердые частицы собрали и промыли водой, затем сушили на воздухе до получения N-(фенилметил)-3-фтор-4-нитрофеноксиацетамида, точка плавления 128,5-130oC.
(c) N-(4-(2-((фенилметил)амино)этокси)-2-фторфенил)-2- тиофенкарбоксимидамид.
Продукт стадии (b) превратили в свободное основание N-(4-(2-((фенилметил)амино)этокси)-2-фторфенил)-2- тиофенкароксимидамид, следуя методике, аналогичной той, которая представлена в примере 19, стадии (c) и (d)), точка плавления 105-107oC.
Пример 72
N-(4-((2-(фенилметил)(метил)амино)этил)фенил)трифторацетимидамид.
4-(2-((фенилметил)(метил)амино)этил)анилиндигидрохлорид (полученный в соответствии с методикой примера 19, стадии (a) - (c)) (1,03 г), растворили в воде (25 мл), обработали 50% водным раствором гидроксида натрия (5 мл) и экстрагировали этиловым эфиром. Экстракт сушили (NaOH) и выпаривали до обеспечения свободного основания, 0,72 г. Затем свободное основание обрабатывали трифторацетамидамидом и нагрели до 100oC. Через 1 час для улучшения перемешивания добавили толуол (5 мл). Через 2 часа смесь охладили, добавили воду (30 мл), и смесь перемешали. Через 15 минут с образованного полутвердого рыжевато-коричневого остатка слили воду. Остаток перекристаллизовали из метанола и водной смеси (125 мл), получив при этом N-(4-(2-((фенилметил)(метил)амино)этил)фенил)-трифторацетамид в виде рыжевато-коричневого твердого вещества, точка плавления 105-107oC.
Предложены производные амидина общей формулы I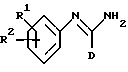 ,
,
в которой D представляет фенил и 5-членное гетероциклическое ароматическое кольцо, содержащее от 1 до 2-х гетероатомов, выбранных из O, S и N, одна группа которого необязательно замещена группой, выбранной из C1-C6-алкила, галогена, или C1-C6-перфторалкил;
R1 представляет водород, галоген;
R2 представляет группу - X(CH2)nZCONR3R4, -X(CH2)nNHCO(CH2)sNR3R4;
-X(CH2)pNR3R4, -X(CH2)nNHCOR5 или -(CH2)qNHC(NH)R6;
R3 и R4 представляют водород, C1-C4-алкил, -(CH2)r-A, -(CH2)m-OA или CH(CH3)/CH2)t-A; или NR3R4 вместе представляют пиперониламино, пиперидинил, морфолинил и др.;
R5 представляет собой C1-C6 -алкил, C1-C6-перфторалкил, -(CH2)r-A или -O(CH2)w-A;
A представляет фенил, пиридинил, пиримидинил или 5-членное гетероциклическое ароматическое кольцо, содержащее I гетероатом, выбранный из O, S и N;
R6 представляет фенил или 5-членное гетероциклическое ароматическое кольцо, содержащее 1 гетероатом, выбранный из O, S и N;
n и r = 0-4; p; w = 1-4;
m = 2-4; q и t = 0-4; S = 1-3;
X представляет O или связь;
Z представляет O, NR7 или связь;
R7 представляет водород или C1-C6 -алкил,
или его фармацевтически приемлемые соли. Способ получения соединения формулы I или его фармацевтически приемлемой соли взаимодействием соединения формулы II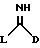 ,
,
где L является отцепляемой группой и D определен выше,
с соединением формулы III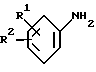 ,
,
где R1 и R2 определены выше.
Лекарственное средство обладает свойством ингибировать активность синтетазы оксида азота (Y). 3 с. и 9 з.п. ф-лы.
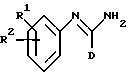
в которой D представляет фенил, 5-членное гетероциклическое ароматическое кольцо, содержащее от 1 до 2 гетероатомов, выбранных из O, S, и N, одна группа которого необязательно замещена группой, выбранной из C1-C6-алкила, галогена; или C1-C6-перфторалкил;
R1 представляет водород или галоген;
R2 представляет X(CH2)nZCONR3R4, -X(CH2)nNHCO(CH2)sNR3R4, -X(CH2)pNR3R4, -X(CH2)nNHCOR5 или -(CH2)qNHC(NH)R6;
R3 и R4 независимо представляют водород, C1-C4-алкил, -(CH2)rA, -(CH2)mOA или CH(CH3)(CH2)t-A; или NR3R4 вместе представляют пиперониламино-, пиперидинил, морфолинил, пирролидинил, 1,2,3,4-тетрагидроизохинолинил; или пиперазинил, необязательно 4-замещенный C1-C6-алкилом;
R5 представляет C1-C6-алкил, C1-C6-перфторалкил, -(CH2)rA или -O(CH2)wA; A представляет фенил, пиридинил, пиримидинил, или 5-членное гетероциклическое ароматическое кольцо, содержащее 1 гетероатом, выбранный из O, S, и N, две группы которого необязательно замещены одной или несколькими группами, выбранными из C1-C6-алкила, галогена, и трифторметила;
R6 представляет фенил или 5-членное гетероциклическое ароматическое кольцо, содержащее 1 гетероатом, выбранный из O, S, и N, две группы которого необязательно замещены одной или двумя группами, выбранными из C1-C6-алкила, C1-C6-алкокси, галогена;
n и r независимо представляют целое число от 0 до 4 включительно;
p и w независимо представляют целое число в диапазоне 1 - 4 включительно;
m представляет целое число в диапазоне 2 - 4 включительно;
q и t независимо представляют целое число в диапазоне 0 - 4 включительно;
s представляет целое число 1 - 3 включительно;
X представляет O или связь;
Z представляет O, NR7 или связь;
R7 представляет водород или C1-C6-алкил при условии, что (а) когда D содержит гетероатом, он не связан с остатком соединения формулы (1) через гетероатом; (в) когда R2 представляет -X(CH2)nZCONR3R4 и ни X, ни Z не представляют связь, тогда n представляет целое число в диапазоне 2 - 4 включительно; (с) когда R2 представляет -X(CH2)nNHCO(CH2)sNR3R4 или -X(CH2)nNHCOR5, X представляет O, тогда n представляет целое число в диапазоне от 2 до 4 включительно; (d) когда R2 представляет -X(CH2)pNR3R4 и X представляет O, тогда p представляет целое число в диапазоне от 2 до 4 включительно; (е) когда R2 представляет -(CH2)qNHC(NH)R6, R1 представляет водород и D и R6 имеют одинаковое определение и представляют фенил, необязательно замещенный C1-C4-алкилом, или одной или несколькими группами C1-C2-алкокси или одним или несколькими атомами галогена, или пиридинил, тогда q не равно 0;
или его фармацевтически приемлемая соль.
N-(4-(2-((фенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(1-((фенилметил)амино)метил)фенил)-2-тиофенкарбоксимидамид,
N-4-(1-((фенилэтил)амино)метил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((2-хлорфенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((3-фторфенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((2-метилфенил)метил)амино)этил)-фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((метил)амино)этил)-фенил)2-тиофенкарбоксимидамид,
N-(4-(2-аминоэтил)фенил)-2-тиофенкарбоксимидамид,
N-((4-морфолинилметил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(((фенилметил)амино)метил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((2,6-дихлорфенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((2-бромфенил)метил)амино)этил)фенил)-2-тиофенилкарбоксимидамид,
N-(3-(2-((фенилметил)амино)этил)фенил)-3-тиофенкарбоксимидамид,
N-(4-(2-((2,6-дихлорфенилметил)амино)этил)фенил)-3-тиофенкарбоксимидамид,
N-(4-(2-аминоэтил)фенил)-3-тиофенкарбоксимидамиддигидробромид,
N-(4-(2-((2,6-дихлорфенилметил)амино)этил)фенил)-2-фуранкарбоксимидамид,
N-(3-(3-(1-пирролидинил)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-аминоэтил)фенил-2-фурокарбоксимидамиддиоксалат,
N-(4-((1-пиперидинил)метил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((1-пирролидинил)метил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(((фенилметил)амино)метил)фенил)-2-тиофенкарбоксимидамид,
N-(3-((амино)метил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-((фенилметил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(3-(2-(этиламино)этилфенил)-2-тиофенкарбоксимидамид,
N-(3-(3-((фенилэтил)амино)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(((2-бромфенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-фениламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-этиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(2-пропиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(1-пропиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-трет-бутиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(н-бутиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(метиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(1-пропиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(трет-бутиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(2-пропиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-аминоэтил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(диметиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-диметиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(1,2,3,4-тетрагидро)изохинолинил)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-(2-(1,2,3,4-тетрагидро)изохинолил)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(((2-хлорфенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(3-((фенилметил)амино)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((3-хлорфенил)метил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(4-(2-(((4-хлорфенил)метил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(4-(2-(((3-хлорфенил)метил)амино)этил)фенил)-3-хлортиофен-2-карбоксамидин,
N-(3-(2-((N-фенилметил-N-метил)амино)этил)фенил-2-тиофенкарбоксимидамид,
N-(4-(2-((N-фенилметил-N-метил)амино)этил)фенил-2-тиофенкарбоксимидамид,
N-(3-(2-(((3-хлорфенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-(((3-фторфенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-(((3-хлорфенил)метил)амино)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-((фенилметил-N-метил)амино)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-((фениламино)карбонил)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-((фенилметиламино)карбонил)пропил)фенил-2-тиофенкарбоксимидамид,
N-(4-(3-((1-пирролидил)карбонил)пропил)окси)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-((4-морфолинил)карбонил)пропокси)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((фенилметиламино)карбонил)этил)фенил)-2-тиофенкарбоксимидамид,
N-3-(2-((фениламино)карбонил)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-((фениламино)карбонил)пропил)фенил)-2-пирролкарбоксимидамид,
N-(4-(3-((фениламино)карбонил)пропил)фенил)-2-фурокарбоксимидамид,
N-(4-(3-((фениламино)карбонил)пропил)фенил)-3-хлор-2-тиофенкарбоксимидамид,
N-((3-((фениламино)карбонил)-пропил)фенил)-1-метилпиррол-2-карбоксимидамид,
N-(4-(3-(1-(4-метилпиперазинил)карбонил)пропил)фенил-2-тиофенкарбоксимидамид,
N-(4-(3-((1-пирролидил)карбонил)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-((4-морфолинил)карбонил)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((фениламино)карбонил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((4-морфолинил)карбонил)этил)фенил)-2-метилтиазол-4-карбоксимидамид,
N-(4-(2-(((4-морфолинил)карбонил)амино)этил)фенил)тиофен-2-карбоксимидамид,
N-(4-(3-(((фенил)амино)карбонил)пропилокси)фенил-2-тиофенкарбоксимидамид,
N-(4-(2-(((трифторметил)карбонил)амино)этил)фенил-2-тиофенкарбоксимидамид,
N-(4-(2-((метил)карбонил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((фенилметил)карбонил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((фенил)карбонил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((фенилиминокарбонил)амино)фенил)-2-тиофенкарбоксимидамид,
N-N''-(1,4-фенилен)бис-2-тиофенкарбоксимидамид,
N-N''-(1,3-фенилен)бис-2-тиофенкарбоксимидамид,
N-N'-(1,3-фенилен)бис-2-хлорфенил-карбоксимидамид,
N-N'-(1,4-фенилен)бис-3-хлортиофен-2-карбоксимидамид,
N-(4-(((2-метоксифенил)иминокарбонил)амино)фенил)-2-тиофенкарбоксимидамид,
N-(4-(((фенил)иминокарбонил)амино)фенил)-3-хлортиофен-2-карбоксимидамид,
N-(4-(((фенил)иминокарбонил)амино)фенил)-3-тиофенкарбоксимидамид,
N-(3-(((фенил)иминокарбонил)амино)фенил)-2-тиофенкарбоксимидамид,
N-(4-(((4-хлорфенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамин,
N-(4-(((2-хлорфенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид,
N-(4-(((4-бромфенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид,
N-(4-(((3-хлор-4-метилфенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид,
N-(4-(((3,5-диметилоксифенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид,
N-(4-((3,5-дихлорфенил)иминокарбонил)амино)фенил)-2-тиофенкарбоксимидамид,
N-(4-(((фенил)иминокарбонил)амино)фенил)-2-фуранкарбоксимидамид,
N-(4-(((3-метилфенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид,
N-(4-(((3-метоксифенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид,
N-(4-(((3-бромфенил)иминокарбонил)амино)фенил-2-тиофенкарбоксимидамид,
N-(4-(((3-хлорфенил)иминокарбонил)амино)фенил)-2-тиофенкарбоксимидамид,
N-(4-(((3-метилфенил)иминокарбонил)амино)фенил)-2-пирролкарбоксимидамид,
N-(4-(((4-хлорфенил)иминокарбонил)амино)фенил-2-пирролкарбоксимидамид,
N-(4-(2-((фениламино)карбонил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(4-(2-((фениламино)карбонил)окси)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((бис(фенилметил)амино)метил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-аминометил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(1,2,3,4-тетрагидроизохинолин-2-илметил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(4-((фенилметил)амино)бутил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((((-2-тиофенил)иминометил)амино)метил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(3-(1-пирролидин)пропил)фенил)-фенилкарбоксомидамид,
N-(4-(2-((4-метоксифенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((4-метилфенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-((3-фенилпропил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-((2-метилфенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((1-инданил)этил)амино)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((4-пиридил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((2-тиенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-((2-фенилэтил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-аминоэтил)фенил)-2-пирролкарбоксимидамид,
(S)-N-(4-(2-((1-фенилэтил)амино)этил)фенил-2-тиофенкарбоксимидамид,
(R)-N-(4-(2-((1-фенилэтил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(3-(2-((4-фенилбутил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(((фенилметокси)карбонил)аминометил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((фенилметил)амино)этокси)фенил)-2-тиофенкарбоксимидамид,
N-[4-(((дифениламино)карбонил)амино)фенил]-2-тиофенкарбоксамидин,
N-(3-((бензоил)амино)фенил-2-тиофенкарбоксидамид,
N-(4-((бензоил)амино)фенил-2-тиофенкарбоксимидамид,
N-(3-((((фениламино)карбонил)амино)фенил)-2-тиофенкарбоксимидамид,
N-(3-(((4-феноксибутил)амино)карбонил)фенил-2-тиофенкарбоксимидамид,
N-(3-(((4-фенилбутил)амино)карбонил)фенил-2-тиофенкарбоксимидамид,
N-(4-(((бензил)амино)карбонил)метил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(1-пирролидининил)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(1-пиперидинил)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-(1-пирролидинил)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(3-(1-пиперидинил)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(2-(1,2,3,4-тетрагидро)изохинолил)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(((фенилметил)амино)метилкарбонил)амино)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((2-фуранил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(2-пиридил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((2-тиофенил)метил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((амино)карбонил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-(((2-тиенил)карбонил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-((((дифениламино)карбонил)амино)метил)фенил)-2-тиофенкарбоксамидин,
N-(4-((((2-тиофенил)иминометил)амино)метил)фенил)-2-тиофенкарбоксамидин,
N-(4-((((2-тиофенил)иминометил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(4-((((3-метилфенил)иминометил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(3-((((2-тиофенил)иминометил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(4-(2-((пиримидин-2-ил)амино)этил)фенил)-2-тиофенкарбоксамид,
N-(4-(2-((фенилметил)амино)этокси)-2-фторфенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((фенилметил)(метил)амино)этил)фенил)-трифторацетимидамид,
N-(4-(2-((этил)(фенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((пропил)(фенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((1,1-диметилэтил)фенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((3,4-дихлорфенилметил)амино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((3,5-бис-трифторметилфенилметил)амино)этил)фенил-2-тиофенкарбоксимидамид,
N-(4-(3-((этиламино)пропил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((диэтиламино)этил)фенил)-2-тиофенкарбоксимидамид,
N-(4-(2-((3-хлорфенилметил)амино)этил)фенил)-бензолкарбоксимидамид,
N-(4-(2-((4-метилфенилметил)амино)этил)фенил)-2-тиофенкарбоксамидин,
N-(4-(2-((2-пиперониламино)этил)фенил)-2-тиофенкарбоксамидин,
или фармацевтически приемлемую соль одного любого из этих соединений.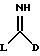
в которой D является таким, как он определен в п.1;
L является отщепляемой группой,
с соединением формулы III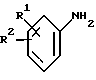
в которой R1 и R2 являются такими, как они определены в п.1.
Приоритет по пунктам:
12.08.93 - по пп.2, 3, 9 (соединения 51, 52, 62, 63, 63-71), 10, 11, 12;
25.09.93 - по п.9 (соединения 72-74);
11.12.93 - по пп. 1 и 4 (где R2 = -X(CH2)pNR3R4), 5, 6, 9 (соединения 1-12, 19-21);
27.01.94 - по пп.1 и 4 (где R2 = -(CH2)qNHC(NH)R4), 7, 8, 9 (соединения 75-79);
10.06.94 - по п.9 (соединения 22-38, 40-43, 45-46).
| EP 0 446 699 A, 18.09.91 | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
