Настоящее изобретение относится к бициклическим производным амидина, к способам их получения, к содержащим их композициям и к использованию их в терапии. Было описано использование некоторых производных амидина в терапевтических целях. Применение производных N- фениламидина для лечения диабетов было описано в патенте США N 3669974 (USV Pharmaceutical Corp.) и в патентной заявке Великобритании 2226562 (Boots). Применение N'N''-дизамещенных амидинов для лечения гипертензии, депрессий и галлюциногенных состояний было описано в международной патентной заявке WO 92/04054 (University of Oregon). Применение некоторых амидинов и симметричных бисамидинов в качестве анальгетиков при лечении воспалений и при лечении гипертензии раскрыто в патенте Бельгии N 71740 и патенте Великобритании N 1180629 (оба Delalande). Применение производных амидина в качестве гербицидов было раскрыто в патентной заявке Германии DE-OS-2321330 (Bayer).
Применение ингибиторов синтетазы окиси азота при лечении заболеваний было также описано, например, в международных патентных заявках WO 94/12163 (Abbott), WO 93/13066 и WO 94/12165 (обе Wellcome) и в Европейских патентных заявках 446699 (Merrell Dow), 547558 и 558468 (обе от Вашингтонского университета). Применение ингибиторов синтетазы окиси азота раскрыто также в WO 95/00505, WO 95/09619, WO 95/09621 (все Wellcome), WO 95/10266 (Otsuka), WO 95/11231 и WO 95/11014 (обе Searle).
Ранее заявитель раскрыл применение производных гуанидина и производных амидина, которые являются ингибиторами синтетазы окиси азота, при лечении, наряду с другими, нейродегенеративного заболевания (WO 094/21621, WO 95/05363).
В настоящее время авторами была обнаружена новая группа бициклических производных амидина, которые обладают полезной фармацевтической активностью.
В соответствии с первым аспектом настоящего изобретения предложено соединение формулы I: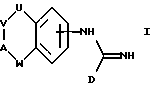
где D представляет пятичленное гетероциклическое ароматическое кольцо, содержащее от 1 до 4 гетероатомов, выбранных из O, N или S, необязательно замещенное по атому углерода галоидом, трифторметилом, алкилом C1-6, нитро или циано, и которое соединено с остальной частью соединения формулы I через атом углерода;
А представляет N(X) или CH(-(CH2)m-NXY);
U представляет NH, O или CH2;
V представляет (CH2)a;
W представляет (CH2)b;
a и b независимо представляют целое число от 0 до 3, при условии, что a+b находится в интервале от 1 до 3;
X и Y независимо представляют водород, алкил C1-6, или группу -(CH2)nQ или -NXY представляет пиперидинил, пирролидинил, морфолинил или тетрагидроизохинолинил;
Q представляет бифенил или фенил, необязательно замещенный одной или более из групп, выбранных из алкил C1-6, акокси C1-6, перфторалкил C1-6, галоида, нитро или циано,
m представляет целое число от 0 до 5;
n представляет целое число от 0 до 6;
или цепочка U-V-A-W имеет указанные ранее значения, за исключением возможности быть ненасыщенной, или цепочка U-V-A-W может представлять -NH-CH2-CH2-O-, замещенную по атому углерода группой -(CH2)m-NXY, где m, X и Y имеют указанные ранее значения, и его фармацевтически приемлемые соли.
Предпочтительная группа соединений формулы I определяется формулой IA: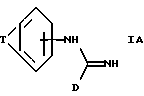
где T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепочку, замещенную -(CH2)m-NXY; -O- (CH2)2-NH-замещенную -(CH2)m-NXY; или -U-(CH2)a-N(X)-(CH2)b-;
X и Y независимо представляют водород, алкил C1-6, или группу - (CH2)nQ, или NXY представляет пиперидинил, пирролидинил, морфолинил или тетрагидроизохинолинил;
Q представляет фенил, необязательно замещенный алкил C1-6, алкокси C1-6, трифторметил, галоид, нитро или циано;
a U, m, n, b и D имеют указанные ранее значения, за исключением того, что если T представляет -U-(CH2)a-N(X)- (CH22)b-, а X представляет -(CH2)nQ, n представляет целое число от 0 до 5, и его фармацевтически приемлемые соли.
Предпочтительно, чтобы D представлял пятичленное гетероциклическое ароматическое кольцо, содержащее один гетероатом, выбранный из O, N или S, необязательно замещенное по атому углерода галоидом. Особенно предпочтительно, чтобы D представлял тиенил, фурил или пирролил, особенно тиенил или фурил, более предпочтительно тиенил, наиболее предпочтителен 2-тиенил.
Предпочтительно, чтобы T представлял C3-5 насыщенную или ненасыщенную алкиленовую цепочку, замещенную -(CH2)m-NXY, особенно C3-5 - насыщенную алкиленовую цепочку, замещенную - (CH2)m-NXY, и особенно, C3-5 насыщенную алкиленовую цепочку, замещенную -(CH2)m-NXY.
Если T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепочку, замещенную -(CH2)m-NXY; или -O- (CH2)2-NH замещенную -(CH2)m-NXY, предпочтительно, чтобы X и Y независимо представляли водород, алкил C1-6 или группу -(CH2)nQ. Особенно предпочтительно, чтобы X и Y независимо представляли водород, метил, этил или группу - (CH2)nQ наиболее предпочтительно, чтобы один из X или Y представлял бы водород, а другой представлял бы водород или группу -(CH2)nQ.
Предпочтительно, чтобы m было равно 0 или 1, особенно 0;
Если T представляет -U-(CH2)a-N(X)- (CH2)b-, предпочтительно, чтобы U представлял CH2,
Если T представляет -U-(CH2)a-N(X)- (CH2)b-, предпочтительно, чтобы a+b было 1 или 2;
Если T представляет -U-(CH2)a-N(X)-(CH2)b-, предпочтительно, чтобы X представлял водород, алкил C1-6 или группу -(CH2)nQ.
Если X и/или Y представляет -(CH2)nQ, предпочтительно, чтобы n представляло 0, 1 или 2, особенно 1.
Предпочтительно, чтобы Q представлял фенил, необязательно замещенный алкил C1-6 или галоидом, хотя наиболее предпочтителен Q, представляющий незамещенный фенил.
В соответствии с изобретением предложен далее способ получения соединений формулы I и их фармацевтически приемлемых солей, который включает:
(а) получение соединения формулы I путем взаимодействия соответствующего соединения формулы II: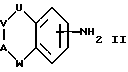
где U, V, A и W имеют указанные ранее значения, с соединением формулы III: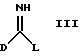
где D имеет указанные ранее значения, a L представляет отщепляемую группу;
(b) получение соединения формулы I путем взаимодействия соответствующего соединения формулы IV: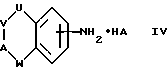
где U, V, A и W имеют указанные ранее значения, а HA представляет кислоту, с соединением формулы V:
где D имеет указанные ранее значения;
(c) получение соединения формулы I, в котором а представляет N(X), а X представляет алкил C1-6 или группу -(CH2)nQ, путем взаимодействия соответствующего соединения формулы I, в котором X представляет водород, с соединением формулы VI:
R9-L
где R9 представляет C1-6 или группу - (CH2)nQ и L представляет отщепляемую группу;
(d) получение соединения формулы I, где A представляет CH(-(CH2)m-NXY), и, по крайней мере, один из X и Y представляет алкил C1-6 или группу - (CH2)nQ, путем взаимодействия соответствующего соединения формулы I, где один из X и Y (или оба) представляют водород, с соединением формулы VI;
(e) получение соединения формулы I, где A представляет CH(-(CH2)m-NXY), a m представляет целое число от 1 до 5, восстановлением соответствующего соединения формулы VII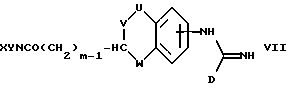
где U, V, W, X, Y и D имеют указанные ранее значения;
(f) получение соединения формулы I, где A представляет CH(-(CH2)m-NXY), и оба X и Y представляют водород, восстановлением соответствующего соединения формулы VIII: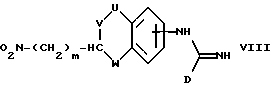
где U, V, W, n и D имеют указанные ранее значения
(g) получение соединения формулы I, где A представляет CH(-(CH2)m-NXY), X представляет водород, a m представляет целое число от 1 до 5, восстановлением соответствующего соединения формулы IX: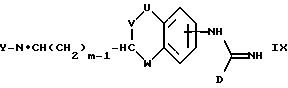
где U, V, W, D и Y имеют указанные ранее значения;
(h) получение соединения формулы I, где A представляет CH(-(CH2)m-NXY), один из X и Y представляет водород, а другой представляет -(CH2)nQ, где n представляет целое число от 1 до 6, за счет восстановления соответствующего соединения формулы X: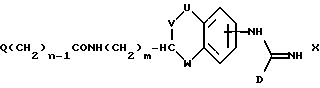
где Q, m, U, V, W и D имеют указанные ранее значения.
(i) получение соединения формулы I, где A представляет CH(-(CH2)m-NXY), один из X и Y представляет водород, а другой представляет -(CH2)nQ, где n представляет целое число от 1 до 6, восстановлением соединения формулы XI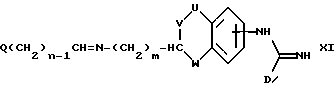
где Q, m, U,V, W и D имеют указанные ранее значения или
(j) получение соединения формулы I, где A представляет CH(-NXY), а X представляет водород, восстановлением соответствующего соединения формулы XII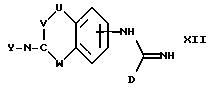
где U, V, W, D и Y имеют указанные ранее значения; и где, при желании или при необходимости, превращают полученное соединение формулы I или другую его соль, в фармацевтически приемлемую соль его или наоборот.
В способе (a) реакцию ведут при перемешивании смеси реагентов в подходящем растворителе, например, в таком низшем алканоле, как этанол, изопропанол или трет. - бутанол, при температуре в интервале от комнатной температуры до температуры кипения растворителя с обратным холодильником. Время реакции (наряду с другим) зависит от растворителя и характера отщепляемой группы, и может составлять вплоть до 48 часов, однако, обычно оно составляет от 1 до 5 часов. Подходящей присутствующей отщепляемой группой L может быть тиоалкил, сульфонил, трифторкарбонилсульфонил, галоид, алкиловый и ариловый спирты, и тозильные группы; другие же указаны в "Advanced Organic Chemistry", J. March (1985) 3 Ed., McGraw- Hill, pp 315, и хорошо известны специалистам.
В способе (b) реакцию, предпочтительно, ведут при температуре кипения с обратным холодильником смеси двух соединений в течение нескольких часов в присутствии подходящего растворителя, за счет чего температура реакции оказывается достаточно высокой, чтобы легко происходила конденсация, но не достаточно высокой, чтобы произошло разложение образующегося амидина. Температура реакции может меняться от комнатной температуры до около 250oC, хотя, предпочтительно, вести реакцию при температуре от около 100oC до 200oC. Обнаружено, что наиболее подходящим растворителем является о-дихлорбензол, и что полезно добавлять в качестве катализатора 4-диметиламинопиридин. При охлаждении образуются два слоя, растворитель можно декантировать, и реакционную смесь обрабатывают, добавляя водное основание. В другом варианте, в случае, если реагенты растворяются в растворителе, растворитель можно выпарить в вакууме, и реакционную смесь обработать, добавляя воду. Кислотой HA может быть органическая или неорганическая кислота, например, соляная, бромистоводородная, йодистоводородная, серная, азотная, фосфорная, уксусная, молочная, янтарная, фумаровая, яблочная, малеиновая, винная, лимонная, бензойная или метансульфоновая кислота.
В способе (c) реакция происходит в стандартных условиях, например, при взаимодействии двух соединений в инертном растворителе в щелочных условиях при комнатной температуре в течение вплоть до 12 часов. Было обнаружено, что часто желательно обрабатывать амин NaOH до того, как осуществляют взаимодействие с соединением формулы VI. Предпочтительно, чтобы L представлял галоид, особенно бромид.
Способ (d) можно осуществить в условиях, аналогичных описанным ранее для способа (c).
В способе (e) восстановление можно осуществить за счет обработки дибораном в инертном растворителе, например, в ТГФ. Альтернативные, хотя менее предпочтительные реагенты, которые можно использовать, включают литийалюминийгидрид и реагенты для каталитического гидрирования, например, H2 на Pd/C. Подробности условий реакции при использовании этих реакций изложены в ссылке на J. March "Advanced Organic Chemistry" на стр. 1099, включая цитированные там ссылки.
В способе (f) реакцию восстановления можно осуществить в различных условиях, например, в тех, которые описаны у J. March "Advanced Organic Chemistry" на стр. 1103-1104. Они включают каталитическое гидрирование, использование Zn, Sn или Fe металлов, AlH3-AlCl3, сульфидов и др. Предпочтительно вести реакцию гидрирования при атмосферном давлении в течение 3-6 часов в присутствии катализатора-палладия на угле.
В способах (g), (i) и (j), восстановление можно осуществить, обрабатывая соединение боргидридом натрия или цианоборгидридом натрия в стандартных условиях.
В способе (h) реакцию можно осуществить в условиях, аналогичных условиям, описанным ранее для способа (e).
Соли соединения формулы I можно получить за счет осуществления взаимодействия свободного основания или его соли, энантиомера, таутомера или защищенного производного, с одним или более эквивалентами соответствующей кислоты. Реакцию можно вести в растворителе, или в среде, в которой соль нерастворима, или в таком растворителе, в котором эта соль растворима, например, в воде, диоксане, этаноле, тетрагидрофуране или в диэтиловом эфире, или в смеси растворителей, которые можно удалить в вакууме или в результате сушки вымораживанием. Такая реакция может быть обменным процессом, или ее можно вести в ионообменной смоле.
Соединения формулы II можно получить восстановлением соответствующего соединения формулы XIII: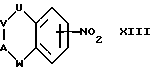
где U, V, A и W имеют указанные ранее значения.
Реакцию восстановления можно вести в условиях, аналогичных условиям, описанным для способа (f).
Некоторые соединения формулы II либо известны, либо их можно получить обычными способами, известными per se. Другие соединения формулы II можно получить из известных соединений с более простыми замещающими группами в соответствии со способами, описанными ранее для способов (c)-(j). Так, например, по аналогии со способом (j), описанным ранее, оказалось удобным получать некоторые соединения формулы XIII, где A представляет CH(-NXY), а X представляет водород, за счет восстановления соответствующего имина, образуемого в реакции соединения формулы NH2Y с нитрированным бициклическим кетоном.
Соединения формулы IV можно получить способами, аналогичными тем, которые описаны для получения соединений формулы II. Соединения формулы IV можно превратить в соответствующие соединения формулы II, обрабатывая их основанием. Соединения формулы II можно превратить в соответствующие соединения формулы IV, обрабатывая протонной кислотой HA, например, одной из тех, которые были перечислены ранее.
Соединения формулы III либо являются известными соединениями, либо их можно получить известными способами. Так, например, соединения формулы II, в которых L представляет тиоалкил, можно получить в результате обработки тиамида формулы XIV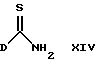
где D имеет указанные ранее значения, алкилиодидом.
Соединения формул VII, VIII, IX, X, XI и XII можно получить способами, аналогичными способам, описанным для получения соединений формулы I. Такие соединения можно легко получить из соединений с более простыми замещающими группами обычными способами, например, используя образование амида (VII, X) путем реакции амина с карбоновой кислотой или ее активированным производным, или образование имина (IX, XI, XII) за счет реакции амина с альдегидом.
Соединения формул V, VI, XIII и XIV либо являются известными соединениями, либо их можно получить обычными способами per se.
Специалистам должно быть понятно, что может оказаться желательным защитить амин или другую реакционно-способную группу, используя защитную группу, как это описано в стандартном тексте "Protecting groups in Organic Synthesis" и 2nd Ed. (1991) Greene and Wuts. Можно упомянуть такие защищающие амин группы, как алкоксикарбонил C2-7, например, трет.-бутоксикарбонил, фенилалкоксикарбонил C8-13, например, бензилоксикарбонил, или, предпочтительно, трифторацетат. Удаляют защиту обычно за счет обработки водным основанием, кислотой, или обрабатывая водородом.
Соединения настоящего изобретения и промежуточные соединения можно выделить из реакционных смесей обычными способами.
Термин "алкил C1-6" включает разветвленные и неразветвленные цепочки, насыщенные, ненасыщенные, алифатические и циклические алкильные группы, содержащие от 1 до 6 атомов углерода.
Соединения формулы I могут существовать в таутомерной, энантиомерной или диастереоизомерной формах, и все они включены в объем настоящего изобретения. Различные оптические изомеры можно выделить, разделяя рацемическую смесь соединений с помощью обычных методов, например, за счет фракционной кристаллизации или с помощью ВЭЖХ. В другом варианте отдельные энантиомеры можно получить за счет реакций соответствующих оптически активных исходных материалов в таких условиях реакции, которые не вызовут рацемизации.
Промежуточные соединения могут существовать в энантиомерных формах, и их можно использовать в виде очищенных энантиомеров, диастереоизомеров, рацематов или смесей.
Соединения формулы I обладают полезной фармакологической активностью в отношении животных. В частности, они обладают полезной ингибирующей синтетазу окиси азота активностью, и ожидается, что они могут быть полезны при лечении или профилактике заболеваний человека или таких состояний, при которых синтез или избыточный синтез окиси азота вносит свой вклад; пример представляет гипоксия, например, в случаях остановки сердца или при ударах, нейродегенеративные заболевания, включая дегенерацию нервов и/или некроз нервов при таких заболеваниях, как гипоксия, гипогликемия, эпилепсия и внешние раны (например, поражения позвоночника или головы), гипербарические кислородные конвульсии и токсичность, слабоумие, например, старческое слабоумие, болезнь Алцгеймера и слабоумие, связанное со СПИДом, хорея Сиденхэма, болезнь Паркинсона, синдром Тоуретта, болезнь Хантингтона, амиотропный латеральный склероз, болезнь Корсакова, имбецильность, связанная с нарушением церебральных сосудов, нарушения сна, шизофрения, депрессия, аутизм, сезонные нарушения, расстройство нормального циркадного ритма, депрессии и другие симптомы, связанные с пременструальным синдромом (PMS), беспокойство и септический шок. Соединения формулы I, как ожидается, могут демонстрировать активность по предотвращению и лечению зависимости от лекарств, облегчению боли и при лечении мигреней и других головных болей сосудистого происхождения. Соединения настоящего изобретения могут также демонстрировать иммуноподавляющую активность, быть полезными при лечении или для профилактики воспалений, нейрогенных воспалений, заболеваний, приводящих к блокаде дыхательных путей, включая астму и респираторный дистресс-синдром взрослых (ARDS), при лечении нарушений моторики желудочно-кишечного тракта, рака, при стимулировании родов, для снижения секреции желудочной кислоты и для повышения силы сокращения скелетных мышц.
Соединения настоящего изобретения формулы I представляют наибольший интерес при лечении нейродегенеративных заболеваний, мигреней или для предотвращения или лечения толерантности к опиатам и диазепинам, или для лечения зависимости от лекарств, и особенно, для лечения нейродегенеративных заболеваний.
Таким образом, в соответствии со следующим аспектом изобретения предложено соединение формулы I или его фармацевтически приемлемая соль для использования в фармацевтике.
В соответствии со следующей отличительной особенностью изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли при получении медикаментов для лечения или профилактики вышеуказанных заболеваний или состояний.
Предложен также способ лечения или профилактики одного из вышеперечисленных заболеваний или состояний, который включает введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, пациенту, страдающему или подверженному такому заболеванию или состоянию.
Для вышеуказанных терапевтических показаний вводимая доза будет, естественно, меняться в зависимости от используемых соединений, способа введения и необходимого лечения. Однако, обычно, удовлетворительных результатов удается достичь в тех случаях, когда эти соединения вводят людям в дневной дозе от 1 мг до 2000 мг (измерения для твердой формы).
Соединения формулы I и их фармацевтически приемлемые соли можно использовать отдельно или в форме соответствующих медицинских препаратов для энтерального или парэнтерального введения.
В соответствии с настоящим изобретение предложена фармацевтическая композиция, содержащая, предпочтительно, менее 80%, и более предпочтительно, менее 50% соединения формулы I или его фармацевтически приемлемой соли, в смеси с фармацевтически приемлемым разбавителем или носителем.
Примеры разбавителей и носителей, которые можно использовать, хорошо известны специалистам.
Энзим синтетаза окиси азота имеет ряд изоформ, и соединения формулы I или их фармацевтически приемлемые соли можно скринировать по активности в отношении синтетазы окиси азота по способам, основанным на способах Bredt and Snyder "Proc. Natl. Acad. Sci. (1990) 87, 682- 685, и Forstermann et.al. , Eur. J. Pharm. (1992) 225, 161-165. Синтетаза окиси азота превращает 3H-L-аргинин в 3H-L- цитруллин, который можно выделить с помощью катионообменной хроматографии, и количественно определить с помощью сцинтилляционного счетчика.
Пример скринирования A
(A) Скринирование по активности синтетазы окиси азота
Энзим выделяют из гиппокампа или мозжечка крыс. Гиппокамп или мозжечок у крыс штамма Spraque-Dawley (250-275 г) удаляют после анестезирования животных CO2 и обезглавливания. Надосадочную жидкость мозжечка или гиппокампа получают в результате гомогенизации в 50 мМ Tris-HCl с 1 мМ EDTA буфере (pH 7,2 при 25oC) и центрифугирования в течение 15 минут при 20000 g. Остаточный L-аргинин удаляют из надосадочной жидкости в результате хроматографической обработки на Dowex AG-50W-X8 (натриевая форма и водородная форма колонок, последовательно), и затем центрифугирования при 1000 g в течение 30 секунд.
Для анализа по 25 мкл конечной надосадочной жидкости добавляют в каждую из 12 тестовых ампул, содержащих по 25 мкл раствора L-аргинина (при концентрации 18 мкМ 1H-L-аргинин, 96 нМ 3H-L- аргинин), и либо по 25 мкл аналитического буфера (50 мМ HEPES, 1 мМ EDTA, 1,5 мМ CaCl2, pH 7,4), либо тестового соединения в буфере при 22oC. В каждую тестовую ампулу добавляют по 75 мкл полного аналитического буфера (50 мМ HEPES, 1 мМ EDTA, 1,5 мМ CaCl2, 1 мМ DTT, 10 мкМ NADPH, 10 мкг/мл калмодулина, pH 7,4) для инициирования реакции, и реакцию останавливают, через 10 минут добавляя 2 мл останавливающего буфера (20 мМ HEPES, 2 мМ EDTA, pH 5,5).
Меченый L-цитруллин отделяют от меченого L-аргинина в результате хроматографической обработки на Dowex AG-50W-X8 200- 400 мешей колонке. По 1 мл каждой из остановленных реакционных смесей вводят в отдельные 1 мл колонки, и элюенты объединяют с двумя промывками по 1 мл дистиллированной водой и 16 мл сцинтилляционного "коктейля". Затем количество L-цитруллина определяют с помощью сцинтилляционного счетчика.
В типичном эксперименте с надосадочной жидкостью мозжечка основная активность повышена на 20000 распадов в минуту/мл по сравнению с контролем, активность для которого составляет 7000 распадов в минуту/мл. Сравнительный стандарт, N-нитро-L-аргинин, который обеспечивает 60% ингибирование синтетазы окиси азота, при концентрации 1 мкМ также тестируют в анализе для контроля за процедурой.
Пример скринирования B
(B) скринирование на активности синтетазы окиси азота макрофагов
Энзим приготавливают, после индуцирования, из культивируемых клеток мышиных макрофагов линии J774A-1 (полученных из лабораторий Imperial Cancer Research Fund). J774A-1 клетки культивируют в Дюльбеко-модифицированной среде Игла (DMEM), дополненной 10% сывороткой плода теленка, 4 мМ L-глутамина и антибиотиками (100 ед/мл пенициллина G, 100 мкг/мл стрептомицина и 0,25 мкг/мл амфотерицина В). Клетки выращивают обычным способом в 225 см2 склянках, содержащих по 35 мл среды, выдерживаемых при 37oC и во влажной атмосфере, содержащей 5% CO2.
Синтетаза окиси азота продуцируется клетками в ответной реакции на интерферон- γ (IFN γ ) и липополисахарид (LPS). Среду из конфлюэнтных культуральных склянок удаляют, и заменяют 25 мл (на склянку) свежей среды, содержащей 1 мкг/мл LPS и 10 ед./мл IFN γ . После 17-20 часов в культуре, сбор клеток осуществляют, снимая слой клеток с поверхности склянки в культуральную среду. Клетки собирают центрифугированием (1000 g в течение 10 минут), и приготавливают лизат, добавляя клеточный осадок в раствор, содержащий 50 мМ Tris-HCl (pH 7,5 при 20oC), 10% (объем/объем) глицерина, 0,1% (объем/объем) Triton-X-100, 0,1 мкМ дитиотреитола и коктейль ингибиторов протеазы, содержащий леупептил (2 мкг/мл), ингибитор трипсина сои (10 мкг/мл), апротинин (5 мкг/мл) и фенилметилсульфонилфторид (50 мкг/мл).
Для анализа 25 мкг коктейля субстрата (50 мМ Tris-HCl (pH 7,5 при 20oC), 400 мкМ NADPH, 20 мкМ флавинадениндинуклеотида, 20 мкМ флавинмононуклеотида, 4 мкМ тетрагидробиоптерина, 12 мкМ L-аргинина и 0,025 мкCi L-[3H] аргинина) добавляют в ячейки фильтровальной пластины с 96 ячейками (0,45 мкМ размер ячеек), содержащие по 25 мкл раствора тестового соединения в 50 мМ Tris-HCl. Реакцию инициируют, добавляя 50 мкл клеточного лизата (полученного как указано ранее), и, после инкубирования в течение 1 часа при комнатной температуре реакцию заканчивают, добавляя 50 мкл водного раствора 3 мМ нитроаргинина и 21 мМ EDTA.
Меченый L-цитруллин отделяют от меченого L-аргинина, используя Dowex AG-50W. 150 мкл 25% водной суспензии Dowex 50W (Na+ форма) добавляют в анализируемый состав, который целиком фильтруют в пластины с 96 ячейками. Отбирают образцы фильтрата по 70 мкл и добавляют в пластины с 96 ячейками, содержащие твердый сцинтиллирующий агент. После того, как образцы оставляют высохнуть, количество L-цитруллина определяют с помощью сцинтилляционного счетчика.
В типичном эксперименте основная активность составляет 300 распадов в минуту для образца в 70 мкл, что повышается до 1900 распадов в минуту для реагентных контролей. Аминогуанидин, который дает ИК50 (50% ингибирующая концентрация) в 10 мкМ, тестируют в качестве стандарта для проверки процедуры.
Пример скринирования C
(C) Скринирование по активности синтетазы окиси азота эндотелия
Энзим можно выделить из эндотелиальных клеток пупочной вены человека (HUVECs) по способу, основанному на способе Pollock et.al (1991), Proc. Nat. Acad. Sci. , 88, 10480-10484. HUVECs закупают у Clonetics Corp. (San Diego, CA, USA) и культивируют до слияния. Клетки можно поддерживать до 35-40 пассажей без заметной потери выхода синтетазы окиси азота. Когда клетки сливаются, их снова суспендируют в Дюльбеко фосфатом буферированном физиологическом растворе, центрифугируют при 8000 об/мин в течение 10 минут, клеточный осадок гомогенизируют в ледяном 50 мМ Tris-HCl, 1 мМ EDTA, 10% глицерина, 1 мМ фенилметилсульфонилфторида, 2 мкМ леупептина при pH 4,2. После центрифугирования при 34000 об/мин в течение 60 минут осадок солюбилизируют в гомогенизирующем буфере, который содержит также 20 мМ CHAPS. После 30 минут инкубирования на льду суспензию центрифугируют при 34000 об/мин в течение 30 минут. Полученную надосадочную жидкость хранят при -80o до использования.
Для анализа 25 мкл полученной надосадочной жидкости добавляют в каждую из 12 тестовых ампул, содержащих 25 мкл раствора L-аргинина (концентрация 12 мкМ 1H-L- аргинина, 64 нМ 3H-L-аргинина) и, либо 25 мкл аналитического буфера (50 нМ HEPES, 1 мМ EDTA, 1,5 мМ CaCl2, pH 7,4), либо 25 мкл тестового соединения в буфере при 22oC. В каждую тестовую ампулу добавляют полный аналитический буфер (50 мМ HEPES, 1 мМ EDTA, 1,5 мМ CaCl2, 1 мМ DTT, 100 мкМ NADPH, 10 мкг/мл калмодулина, 12 мкМ тетрагидробиоптерина, pH 7,4) для инициирования реакции, и реакцию останавливают через 10 минут, добавляя 2 мл останавливающего буфера (20 мМ HEPES, 2 мМ EDTA, pH 5,5).
Меченый L-цитруллин отделяют от меченого L-аргинина в результате хроматографической обработки на Dowex AG-50W-X8 200- 400 мешей колонке. По 1 мл каждого из остановленных реакционных растворов вводят в отдельную 1 мл колонку, и каждый элюент, объединяют с двумя промывками по 1 мл дистиллированной воды и 16 мл сцинтилляционного коктейля. Затем количество L-цитруллина определяют с помощью сцинтилляционного счетчика.
В типичном эксперименте основная активность возрастает на 5000 распадов в минуту/мл для образца по сравнению с реагентным контролем, активность которого составляет 1500 распадов в минуту/мл. Сравнительный стандарт, N-нитро-L-аргинин, который дает 70-90% ингибирования синтетазы окиси азота при концентрации 1 мкМ, тестируют в анализе для проверки процедуры.
Соединения можно также тестировать в экс-виво анализе по определению степени проникновения в мозг.
Пример скринирования D
(D) Экс-виво анализ для определения активности синтетазы окиси азота нейронов
Самцам крыс штамма Spraque-Dawley (250-275 г) вводят внутривенно по 10 мг/кг тестируемого соединения, растворенного в 0,9% физиологическом растворе или в одном только физиологическом растворе в качестве контроля. В определенное заранее время (обычно 2-24 часа) после обработки, животных умерщвляют, удаляют мозжечок, и надосадочную жидкость приготавливают и анализируют на активность синтетазы окиси азота, как описано в примере скринирования A.
В качестве дальнейшего подтверждающего теста, фракцию мозжечковой надосадочной жидкости вводят в колонку 2'-5'-ADP Sepharose (которая связывает синтетазу окиси азота) и затем элюируют за счет NADPH. Элюент тестируют на активность синтетазы окиси азота по способу примера скринирования A.
Соединения, которые проникают в мозг крыс и ингибируют синтетазу окиси азота нейронов, приводят к снижению активности синтетазы окиси азота как в препаратах надосадочной жидкости, так и в элюентах из колонки 2'-5'-ADP Sepharose.
В примерах скринирования по активности ингибирования синтетазы окиси азота активность соединений выражают как ИК50 (концентрация лекарственного вещества, которая обеспечивает 50% ингибирования энзима в анализе). Значения ИК50 для тестовых соединений вначале оценивают из активности ингибирования 1,10 и 100 мкМ растворов соединений. Соединения, которые ингибируют энзим, по крайней мере, на 50% при концентрации 10 мкМ, повторно тестируют, используя соответствующие концентрации для определения величин ИК50.
В примере скринирования A (скринирование по активности против нейронной изоформы синтетазы окиси азота), соединение приводимого далее примера 1 дает ИК50 менее, чем 10 мкМ, что указывает на то, что можно ожидать удовлетворительную терапевтическую активность. В примерах скринирования B и C (скринирование по активности против макрофаговых и эндотелиальных изоформ синтетазы окиси азота) соединение примера 1 демонстрирует ИК50 значения, которые более чем в 10 раз превышают полученные в примере скринирования A значения, демонстрируя нужную активность.
Соединения примеров 2-9, 10(a)-(f), 11-13 и 19-24 также тестируют в примере скринирования A, и они также демонстрируют значения ИК50 менее, чем для 10 мкМ. Таким образом, можно также ожидать, что эти соединения будут обладать удовлетворительной терапевтической активностью.
Соединения формулы I и их фармацевтически приемлемые соли обладают тем преимуществом, что они менее токсичны, более эффективны, более селективны и отличаются более длительным действием, имеют более широкий спектр активности и более эффективны, вызывают меньше побочных эффектов, более легко абсорбируются, или обладают другими полезными фармакологическими свойствами, по сравнению с известными ранее соединениями, и используемыми в указанных ранее областях терапии.
Соединения формулы 1 и их фармацевтически приемлемые соли могут также обладать тем преимуществом, что они более селективны в отношении нейронных изоформ энзима синтетазы окиси азота, и поэтому можно ожидать удовлетворительной терапевтической активности при снижении побочных эффектов, связанных с ингибированием других изоформ.
Далее настоящее изобретение иллюстрируется следующими примерами.
Пример 1
N-((2-(фенилметил)амино)индан-5-ил)-2-тиофенкарбокс- имидамиддиоксалат
(a) 5-нитро-2-инданон
Это соединение получают по способу Heusler, Schief-fer Ber., (1899) 32, 33.
(b) 5-нитро-2-(фенилметил)аминоиндан
5-нитро-2-инданон (1,48 г, 8,36 ммоля), бензиламин (4,40 мл, 41,8 ммоля), уксусную кислоту (15,0 мл), 4 A молекулярные сита (20 мл), ТГФ (15 мл), и MeOH (15 мл) загружают в колбу и охлаждают до 0oC. Затем порциями добавляют цианоборгидрид натрия (1,05 г, 16,7 ммоля) в течение 5 минут. Полученную смесь перемешивают в течение 14 часов, фильтруют через целит и концентрируют до сиропа. Полученную смесь подщелачивают 2н. NaOH и экстрагируют эфиром (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и хроматографически обрабатывают на силикагеле (3% метанол/метиленхлорид) до получения 5-нитро-2-(фенил-метил) аминоиндана: (1,18 г, 53%).
Масс-спектр: (М+Н)+= 269.
(c) 2-(5-нитроинданил)-N-(фенилметил)трифторацетамид
К перемешиваемому раствору 5-нитро-2-(фенилметил)-аминоиндана (1,18 г, 4,40 ммолей) и триэтиламина (0,61 мл, 4,40 ммоля) в 50 мл метиленхлорида добавляют трифторуксусный ангидрид (0,63 мл, 4,40 ммолей) по каплям. После перемешивания в течение 1 минуты раствор сливают в воду и экстрагируют метиленхлоридом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния и фильтруют через короткую пробку силикагеля (20% этилацетат/гексан) до получения 2-(5-нитроинданил)-N- (фенилметил)трифторацетамида: (1,17 г, 73%).
Масс-спектр (М+Н)+=365.
(d) 2-(5-аминоинданил)-N-(фенилметил)трифторацетамид
К перемешиваемому раствору 2-(5-нитроинданил)-N- (фенилметил)трифторацетамида (1,17 г, 3,21 ммоля) в ТГФ/MeOH (100 мл, 1:1) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2) в течение 1 часа, фильтруют через целит и концентрируют до получения 2-(5-аминоинданил)-N-(фенилметил) трифторацетамида, который по данным ТСХ является гомогенным, и используют немедленно на стадии (f).
(e) 3-метил-2-тиофентиокарбоксимидгидроиодид
Раствор 2-тиофенкарбокстиоамида (Maybridge Chemical) (11,1 г) в 60 мл ацетона обрабатывают иодометаном (13,4 г). После 6 часов при 22oC полученное твердое вещество желтого цвета собирают фильтрованием, промывают ацетоном (2 х 25 мл) и сушат до получения 18,45 г S-метил-2-тиофентиокарбоксамидгидроиодида.
Т. плавления 195oC (с разложением).
(f) N-((2-(фенилметил)амино)индан-5-ил)-2-тиофен- карбоксимидамиддиоксалат
К раствору 2-(5-аминоинданил)-N-(фенилметил)-трифторацетамида (1,0 г, 3,0 ммоля) в изопропаноле (6 мл)/ДМФ (0,5 мл) добавляют 3-метил-2-тиофентиокарбоксимидгидрохлорид (0,85 г, 3,0 ммоля). Полученную смесь перемешивают в течение 14 часов, разбавляют метанолом (6 мл) и 2н. NaOH (6 мл) и нагревают до 50oC в течение 0,5 часа. Полученную смесь выливают в воду и экстрагируют этилацетатом (3 х 30 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и обрабатывают хроматографически на силикагеле (20% метанол/метиленхлорид) до получения указанного в заглавии
соединения в виде свободного основания. После обработки IPA/щавелевой кислотой получают N-((2-(фенилметил)-амино)индан-5- ил)-2-тиофенкарбоксимидамиддиоксалат в виде твердого вещества белого цвета (0,47 г, 30%).
Т. плавления 130-135oC.
Пример 2
N-((2-(фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)- 2-тиофенкарбоксимидамид
(a) 7-нитро-3,4-дигидро-2(1H)-нафталинон
Это соединение получают по способу J. Med. Chem. (1989) 32, 2128.
(b) 7-нитро-2-((фенилметил)амино)-1,2,3,4-тетрагидронафталин
7-нитро-3,4-дигидро-2(1H)-нафталинон (1,50 г, 7,85 ммоля), бензиламин (4,30 мл, 39,3 ммоля), уксусную кислоту (8,0 мл), 4  молекулярные сита (20 мл), ТГФ (15 мл) и MeOH (15 мл) загружают в колбу и охлаждают до 0oC. Затем порциями добавляют цианоборгидрид натрия (0,99 г, 15,7 ммоля) в течение 5 минут. Полученную смесь перемешивают в течение 14 часов, фильтруют через целит и концентрируют до сиропа. Полученную смесь подщелачивают 2н. NaOH и экстрагируют эфиром (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и хроматографируют на силикагеле (3% метанол/метиленхлорид) до получения 7-нитро-2- ((фенилметил)амино)-1,2,3,4-тетрагидронафталина: (2, 10 г, 95%).
молекулярные сита (20 мл), ТГФ (15 мл) и MeOH (15 мл) загружают в колбу и охлаждают до 0oC. Затем порциями добавляют цианоборгидрид натрия (0,99 г, 15,7 ммоля) в течение 5 минут. Полученную смесь перемешивают в течение 14 часов, фильтруют через целит и концентрируют до сиропа. Полученную смесь подщелачивают 2н. NaOH и экстрагируют эфиром (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и хроматографируют на силикагеле (3% метанол/метиленхлорид) до получения 7-нитро-2- ((фенилметил)амино)-1,2,3,4-тетрагидронафталина: (2, 10 г, 95%).
Масс-спектр (М+Н)+ 283.
(с) 2-(7-нитро-(1,2,3,4-тетрагидронафтил))-N-(фенилметил) трифторацетамид
К перемешиваемому раствору 7-нитро-2-((фенилметил)амино)-1, 2,3,4-тетрагидронафталина (2,10 г, 7,45 ммоля) и триэтиламина (1,07 мл, 7,45 ммоля) в 50 мл метиленхлорида добавляют трифторуксусный ангидрид (1,05 мл, 7,45 ммоля) по каплям. После перемешивания в течение 1 минуты раствор сливают в воду и экстрагируют метиленхлоридом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния и фильтруют через короткую пробку силикагеля (20% этилацетат/гексан) до получения 2-(7-нитро-(1, 2,3,4-тетрагидронафтил))-N-(фенилметил)трифторацетамида: (2,55 г, 90%).
Масс-спектр (М+Н)+= 379
(d) 2-(7-амино-(1,2,3,4-тетрагидронафтил)-N-(фенилметил) трифторацетамида
К перемешиваемому раствору 2-(7-нитро-(1,2,3,4- тетрагидронафтил)-N-(фенилметил)трифторацетамида (2,55 г, 6,75 ммоля) в ТГФ/MeOH (100 мл, 1:1) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2) в течение 1 часа, фильтруют через целит и концентрируют до получения 2-(7-амино-(1,2,3,4-тетрагидронафтил))-N-(фенилметил)- трифторацетамида, который по данным ТСХ оказывается гомогенным, и немедленно используют на следующей стадии.
(e) N-((2-(фенилметил)амино)-1,2,3,4-тетрагидронафт- 7-ил)-2-тиофенкарбоксимидамид
К раствору 2-(7-амино-(1,2,3,4-тетрагидронафтил))-N-(фенилметил) трифторацетамида (2,11 г, 6,07 ммоля) в 10 мл изопропанола добавляют 3-метил-2-тиофентиокарбоксимидгидрохлорид (1,72 г, 6,07 ммоля). Полученную смесь перемешивают в течение 14 часов, разбавляют метанолом (6 мл) и 2 н. NaOH (6 мл), и нагревают до 50oC в течение 0,5 часа. Полученную смесь выливают в воду и экстрагируют этилацетатом (3 х 30 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до твердого вещества, которое перекристаллизовывают (метиленхлорид/гексан) до получения N-((2-(фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамида в виде белого твердого вещества (0,66 г, 30%).
Т. плавления 119-120oC.
Пример 3
N-((2-амино)-1,2,3,4-тетpaгидpoнaфт-7-ил)-2- тиофенкарбоксимидамиддиоксалат
(a) 7-нитро-2-амино-1,2,3,4-тетрагидронафталингидрохлорид
7-нитро-1-тетралон (1,50 г, 7,85 ммоля), ацетат аммония (6,05 мл, 78,5 ммоля), уксусную кислоту (8,0 мл), 4  молекулярные сита (20 мл), ТГФ (15 мл) и MeOH (15 мл) загружают в колбу и охлаждают до 0oC. Затем добавляют цианоборгидрид натрия (0,99 г, 15,7 ммоля) порционно в течение 5 минут. Полученную смесь перемешивают в течение 1 часа, фильтруют через целит и концентрируют до сиропа. Полученную смесь подщелачивают 2н. NaOH и экстрагируют эфиром (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до получения масла. Это соединение выделяют в виде соли соляной кислоты: 7-нитро-2-амино- 1,2,3,4-тетрагидронафталингидрохлорида (1,00 г, 56%).
молекулярные сита (20 мл), ТГФ (15 мл) и MeOH (15 мл) загружают в колбу и охлаждают до 0oC. Затем добавляют цианоборгидрид натрия (0,99 г, 15,7 ммоля) порционно в течение 5 минут. Полученную смесь перемешивают в течение 1 часа, фильтруют через целит и концентрируют до сиропа. Полученную смесь подщелачивают 2н. NaOH и экстрагируют эфиром (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до получения масла. Это соединение выделяют в виде соли соляной кислоты: 7-нитро-2-амино- 1,2,3,4-тетрагидронафталингидрохлорида (1,00 г, 56%).
Температура плавления более 300oC.
(b) 2-(7-нитро-(1,2,3,4-тетрагидронафтил))-N-трифтор-ацетамид
К перемешиваемому раствору 7-нитро-2-амино-1,2,3,4- тетрагидронафталингидрохлорида (1,00 г, 4,39 ммоля) и триэтиламина (1,22 мл, 8,77 ммоля) в 50 мл метиленхлорида добавляют трифторуксусный ангидрид (0,62 мл, 4,39 ммоля) по каплям. После перемешивания в течение 1 минуты раствор выливают в воду и экстрагируют метиленхлоридом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над безводным сульфатом магния и фильтруют через короткую пробку силикагеля (20% этилацетат/гексан) до получения 2-(7-нитро-(1,2,3,4-тетрагидронафтил))-N-трифторацетамида (0,78 г, 62%).
Масс-спектр (M+H)+ = 289.
(c) 2-(7-амино-(1,2,3,4-тетрагидронафтил))-N-трифторацетамид
К перемешиваемому раствору 2-(7-нитро-(1,2,3,4-тетрагидронафтил))- N-трифторацетамида (0,76 г, 2,21 ммоля) в ТГФ/MeOH (100 мл, 1:1) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2) в течение 1 часа, фильтруют через целит и концентрируют до получения 2-(7-амино(1,2,3,4- тетрагидронафтил))-N-трифторацетамида, который по данным ТСХ оказывается гомогенным, и используют немедленно в следующей реакции.
(d) N-((2-амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамиддигидробромид
К раствору 2-(7-амино-(1,2,3,4-тетрагидронафтил)-N-трифторацетамида (0,70 г, 2,71 ммоля) в 10 мл изопропанола добавляют 3-метил-2- тиофентиокарбоксимидгидроиодид (0,77 г, 2,71 ммоля). Полученную смесь перемешивают в течение 14 часов, разбавляют метанолом (6 мл) и 2н. NaOH (6 мл) и нагревают до 50oC в течение 0,5 часа. Полученную смесь выливают в воду и экстрагируют этилацетатом (3 х 30 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и обрабатывают хроматографически на силикагеле (20% метанол/метиленхлорид) до получения масла, которое превращают в дигидробромид: N-((2-амино)- 1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамиддигидробромид: (0,37 г, 32%).
Т. плавления более 210oC (с разложением).
Пример 4
N-((1-амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамиддиоксала
(a) 7-нитро-1-амино-1,2,3,4-тетрагидронафталин
7-Нитро-1-амино-1,2,3,4-тетрагидронафталин получают так же, как 7-нитро-2-амино-1,2,3,4-тетрагидронафталин. Соединение выделяют в виде соли-гидрохлорида (0,30 г, 12%).
Т. плавления более 300oC.
(b) 1-(7-нитро-(1,2,3,4-тетрагидронафтил))-N-трифтор-ацетамид
1-(7-нитро-(1,2,3,4-тетрагидронафтил))-N-трифторацетамид получают так же, как 2-(7-нитро-(1,2,3,4-тетрагидронафтил))-N-трифторацетамид (0,35 г, 95%).
Масс-спектр (M+H)+ = 289
(c) 1-(7-амино-(1,2,3,4-тетрагидронафтил))-N-трифторацетамид
1-(7-амино-(1,2,3,4-тетрагидронафтил))-N-трифторацетамид получают так же, как 2-(7-амино-(1,2,3,4-тетрагидронафтил))-N-трифторацетамид, и используют немедленно в следующей реакции.
(d) N-((1-амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамиддиоксалат
N-((1-амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамиддиоксалат получают так же, как N-((2-амино) -1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамиддигидробромид за исключением того, что его выделяют в виде диоксалата (0,18 г, 33%).
Температура плавления более 155oC с разложением.
Пример 5
N-((2-амино)индан-5-ил)-2-тиофенкарбоксимидамиддиоксалат
(a) 5-нитро-2-аминоиндангидрохлорид
К 2-аминоиндангидрохлориду (19,11 г, 0,112 моля) при 0oC добавляют серную кислоту (60 мл), затем нитрат калия (11,84 г, 0,117 моля). Полученную смесь оставляют нагреваться до комнатной температуры, перемешивают еще 2 часа, затем выливают в смесь льда/50% NaOH (всего 500 мл). Полученную смесь экстрагируют эфиром (3 х 200 мл) и объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла, которое превращают в соль-гидрохлорид. После перекристаллизации из смеси изопропанол/метанол получают 5-нитро-2-аминоиндангидрохлорид (14,58 г, 60%).
Температура плавления более 300oC.
(b) 2-(5-нитроинданил)-N-трифторацетамид
К перемешиваемому раствору 5-нитро-2-аминоиндангидрохлорида (1,00 г, 5,89 ммоля) и триэтиламина (0,82 мл, 5,89 ммоля) в 50 мл метиленхлорида добавляют трифторуксусный ангидрид (0,83 мл, 5,89 ммоля) по каплям. После перемешивания в течение 1 минуты раствор выливают в воду и экстрагируют метиленхлоридом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния и фильтруют через короткую пробку силикагеля (20% этилацетат/гексан) до получения 2-(5-нитроинданил)-N-трифторацетамида (1,51 г, 93%).
Т. плавления 153-154oC.
(c) 2-(5-аминоинданил)-N-трифторацетамид
К перемешиваемому раствору 2-(5-нитроинданил)-N-трифторацетамида (0,58 г, 2,25 ммоля) в ТГФ/MeOH (100 мл, 1:1) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2), фильтруют через целит и концентрируют до получения 2-(5-аминоинданил)-N-трифторацетамида, который является гомогенным по данным ТСХ, и используют немедленно на следующей стадии.
(d) N-((2-амино)индан-5-ил)-2-тиофенкарбоксимидамиддиоксалат
К раствору 2-(5-аминоинданил)-N-трифторацетамида (0,52 г, 2,25 ммоля) в изопропаноле (6 мл) ДМФ (0,5 мл) добавляют S-метил-2-тиофентиокарбоксимидгидроиодид (0,64 г, 2,23 ммоля). Полученную смесь перемешивают в течение 14 часов, разбавляют метанолом (6 мл) и 2н. NaOH (6 мл) и нагревают до 50oC в течение 0,5 часа. Полученную смесь выливают в воду и экстрагируют этилацетатом (3 х 30 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и обрабатывают хроматографически на силикагеле (20% метанол/метиленхлорид) до получения указанного в заглавии соединения в виде свободного основания. После обработки IPA/щавелевой кислотой получают N-((2-амино)индан-5-ил)-2-тиофенкарбоксимидамиддиоксалат в виде твердого вещества белого цвета (0,60 г, 50%).
Т. плавления 70oC, с разложением.
Пример 6
N-((2-(метил)(фенилметил)амино)индан-5-ил)-2- тиофенкарбоксимидамиддигидробромид
(a) 5-нитро-2-(фенилметил)аминоиндангидрохлорид
К 5-нитро-2-аминоиндангидрохлориду (3,00 г, 14,00 ммолей) в 60 мл ДМФ добавляют триэтиламин (4,07 мл, 29,40 ммоля), а затем бензилбромид (1,74 мл, 14,68 ммоля). Полученную смесь нагревают до комнатной температуры, перемешивают в течение 1 часа, выливают в воду (200 мл) и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют через небольшой слой силикагеля и выпаривают до сиропа. Указанное в подзаглавии соединение выделяют в виде гидрохлоридной соли (2,29 г, 54%).
Т. плавления с разложением 266oC.
(b) 5-нитро-2-(метил)(фенилметил)аминоиндангидрохлорид
К 5-нитро-2-(фенилметил)аминоиндангидрохлориду (2,29 г, 7,52 ммоля) в 100 мл ДМФ добавляют карбонат калия (2,60 г, 18,80 ммоля), а затем метилиодид (0,47 мл, 7,52 ммоля). Полученную смесь нагревают до комнатной температуры, перемешивают в течение 16 часов, выливают в 400 мл воды и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют через небольшой слой силикагеля и выпаривают до сиропа. Указанное в заглавии соединение выделяют в виде соли-гидрохлорида (1,08 г, 45%).
Т. плавления 280oC (с разложением).
(c) 5-амино-2-(метил)(фенилметил)аминоиндандигидрохлорид
К 5-нитро-2-(метил)(фенилметил)аминоиндангидрохлориду (1,08 г, 3,39 ммоля) в 85% уксусной кислоте/воде добавляют порошок цинка (3,0 г). Полученную смесь перемешивают в течение 1 минуты, фильтруют через целит и концентрируют. Полученный концентрат нейтрализуют 2н. NaOH и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния и упаривают до сиропа. Полученное масло обрабатывают IPA/HCl, концентрируют и используют немедленно на следующей стадии.
(d) N-((2-(метил)(фенилметил)амино)индан-5-ил)-2- тиофенкарбоксимидамиддигидробромид
К 5-амино-2-(метил) (фенилметил)аминоиндандигидрохлориду в ДМФ (10 мл) добавляют 3-метил-2-тиофентиокарбоксимидгидроиодид (0,98 г, 3,45 ммоля) и пиридин (0,27 мл, 3,29 ммоля). Полученную смесь перемешивают в течение 14 часов, выливают в смесь воды/2н. NaOH и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и хроматографируют на силикагеле (10% метанол/метиленхлорид) до получения указанного в заглавии соединения в виде свободного основания. В результате обработки IPA/HBr получают N-((2-(метил) (фенилметил)-амино)индан-5-ил)-2-тиофенкарбоксимидамиддигидробромид в виде твердого вещества белого цвета (0,43 г, 25%).
Т. плавления: 196-200oC.
Пример 7
N-((1-амино)индан-6-ил)-2-тиофенкарбоксимидамиддигидрохлорид
(а) 6-нитро-1-аминоиндангидрохлорид
1-аминоиндан (10,0 г, 75,08 ммоля) добавляют к концентрированной серной кислоте (40 мл) при 0oC. Полученную смесь нагревают до комнатной температуры, чтобы помочь растворению, затем охлаждают до 0oC. Затем порционно добавляют нитрат калия (7,60 г, 75,08 ммоля), и полученную смесь оставляют при перемешивании при комнатной температуре в течение 1 часа перед тем, как вылить в смесь лед/50% NaOH. Водный раствор экстрагируют хлороформом (3 х 100 мл). Объединенные экстракты промывают водой, обесцвечивают древесным углем, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HCl получают указанное в подзаглавии соединение (6,90 г, 43%).
Т. плавления 280oC (с разложением).
(b) 6-амино-1-аминоиндангидрохлорид
К раствору 6-нитро-1-аминоиндангидрохлорида (1,00 г 4,66 ммоля) в MeOH (100 мл) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2) в течение 1 часа, фильтруют через целит и концентрируют до получения 6-амино-1-аминоиндангидрохлорида, который по данным ТСХ является гомогенным, и используют немедленно на следующей стадии.
(c) N-((1-амино)индан-6-ил)-2-тиофенкарбоксимидамиддигидрохлорид
К 6-амино-1-аминоиндангидрохлориду (0,74 г, 4,01 ммоля) в ДМФ/IPA (4 мл, 1: 1) добавляют 3-метил-2-тиофентиокарбоксимидгидрохлорид (1,26 г, 4,41 ммоля). Полученную смесь нагревают до 50oC, перемешивают в течение 16 часов, выливают в смесь вода/2н. NaOH и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HCl получают N-((1-амино)-индан-6-ил)-2-тиофенкарбоксимидамиддигидрохлорид в виде твердого вещества белого цвета (0,79 г, 60%).
Т. плавления 200oC с разложением.
Пример 8
N-((1-(фенилметил)амино)индан-6-ил)-2-тиофенкарбоксимидамиддиоксалат
(а) 6-нитро-1-(фенилметил)аминоиндангидрохлорид
К 6-нитро-1-аминоиндангидрохлориду (1,90 г, 8,85 ммоля) в 30 мл ДМФ добавляют триэтиламин (2,50 мл, 18,06 ммоля), а затем бензилбромид (1,07 мл, 9,03 ммоля). Полученную смесь нагревают до комнатной температуры, перемешивают в течение 3 часов, выливают в воду (100 мл) и трижды экстрагируют этилацетатом порциями по 70 мл. Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют через небольшой слой силикагеля и концентрируют до сиропа. Указанное в заглавии соединение выделяют в виде соли-гидрохлорида (1,34 г, 50%).
Т. плавления 234-235oC.
(b) 6-Aмино-1-(фенилметил)аминоиндангидрохлорид
К 6-нитро-1-(фенилметил) аминоиндангидрохлориду (1,34 г, 4,40 моля) в 100 мл MeOH добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см) в течение 1 часа, фильтруют через целит и концентрируют до получения 6-амино-1- (фенилметил)аминоиндангидрохлорида, который по данным ТСХ является гомогенным и используется немедленно на следующей стадии.
(с) N-((1-(фенилметил)амино)индан-6-ил)-2- тиофенкарбоксимидамиддиоксалат
К 6-амино-1-(фенилметил)аминоиндангидрохлориду (1,21 г, 4,40 ммолей) в 20 мл ДМФ добавляют 8-метил-2-тиофентиокарбоксимидгидроиодида (1,38 г, 4,84 ммоля). Полученную смесь нагревают до 50oC в течение 16 часов, затем выливают в смесь воды и 2н. NaOH и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/щавелевой кислотой получают N-((1-(фенилметил)амино)индан-6-ил)-2- тиофенкарбоксимидамиддиоксалат в виде твердого вещества белого цвета (1,06 г, 46%).
Т. плавления более 120oC (с разложением).
Пример 9
N-((2-((3-Хлорфенил)метил)амино)индан-5-ил)-2-тиофенкарбоксамидин
(a) 2-((3-Хлорфенил)карбонил)амино-6-нитроиндан
К 2-амино-6-нитроиндангидрохлориду (1,5 г, 7,0 ммоля) в 50 мл метиленхлорида при 0oC добавляют триэтиламин (2,1 мл, 15,0 ммоля), а затем 3-хлорбензоилхлорид (1,0 мл, 7,5 ммоля). Полученную смесь немедленно выливают в воду и слои разделяются. Водный слой экстрагируют метиленхлоридом (2 x 20 мл), и объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла, которое по данным ТСХ оказывается гомогенным, и его используют немедленно на следующей стадии.
Масс-спектр (М+Н)+= 317.
(b) 2-((3-хлорфенил)метил)амино-6-нитроиндан
К 2-((3-хлорфенил)карбонил)амино-6-нитроиндану (2,2 г, 7,0 ммоля) в 75 мл ТГФ добавляют BH3•ТГФ (1,0 М, 35 мл, 35 ммолей) по каплям. Полученную смесь кипятят с обратным холодильником в течение 12 часов, охлаждают до 0oC, гасят 4н. HCl (60 мл) и кипятят с обратным холодильником в течение 1 часа. Полученный раствор выпаривают до масла, подщелачивают 50% NaOH и экстрагируют метиленхлоридом трижды порциями по 20 мл. Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HCl получают 2-((3-хлорфенил)метил)амино-6-нитроиндан: (2,1 г, 88% за две стадии).
Т. плавления 234-237oC.
(с) 2-((3-хлорфенил)метил)амино-6-аминоиндан
К 2-((3-хлорфенил)метил)амино-6-нитроиндангидрохлориду (2,1 г, 6,13 ммолей) в 40 мл смеси 85% AcOH/H2O добавляют металлический цинк (1,6 г, 24,5 ммоля). Полученную смесь перемешивают в течение 5 минут, фильтруют через целит и выпаривают до масла. Полученное масло выливают в щелочную воду и экстрагируют хлороформом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HCl получают 2-((3-хлорфенил)метил)амино-6-аминоиндан (1,5 г, 70%).
Т. плавления более 270oC
(d) N-((2-((3-хлорфенил)метил)амино)индан-5-ил)-2- тиофенкарбоксамидин
2-((3-хлорфенил)метил)амино-6-аминоиндандигидрохлорид (1,5 г, 4,2 ммоля), 3-метил-2-тиофентиокарбоксимидгидрохлорид (1,3 г, 4,6 ммоля) и пиридин (0,34 мл, 4,2 ммоля) в 10 мл ДМФ перемешивают в течение 24 часов. Полученную смесь выливают в воду, подщелачивают 2н. NaOH и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и обрабатывают хроматографически на силикагеле (12% MeOH/метиленхлорид) до получения бесцветного масла. В результате обработки IPA/HCl получают N-((2-((3-хлорфенил)метил)амино) индан-5-ил)-2-тиофенкарбоксамидин (0,75 г, 40%).
Т. плавления 297-299oC.
Пример 10
По способу примера 9 получают следующие соединения:
(a) N-((2-((2-метилфенил)метил)амино)индан-5-ил)-2- тиофенкарбоксамидин;
Т. плавления 183oC.
(b) N-((2-((3-метилфенил)метил)амино)индан-5-ил) тиофенкарбоксамидин;
Т. плавления 195oC.
(с) N-((2-((4-метилфенил)метил)амино)индан-5-ил)-2- тиофенкарбоксамидин;
Т. плавления 182oC.
(d) N-((2-(этил)амино)индан-5-ил)-2-тиофенкарбоксамидин
Т. плавления 236-238oC.
(o) N-((2-(((4-фенил)фенил)метил)амино)индан-5-ил)- 2-тиофенкарбоксамидин;
Т. плавления 182oC.
(f) N-((2-(((4-гексил)фенил)метил)амино)индан-5-ил)-2- тиофенкарбоксамидин;
Т. плавления 125oC.
(g) N-((2-((3-бромфенил)метил)амино)индан-5-ил)-2- тиофенкарбоксамидин;
Т. плавления 182oC.
Пример 11
N-((2-(((3-Хлорфенил)метил)амино)-1,2,3,4-тетрагидронафт-7-ил)- 2-тиофенкарбоксимидамид
(a) 7-нитро-2-(((3-хлорфенил)метил)амино)-1,2,3,4-тетрагидронафталин
7-Нитро-3,4-дигидро-2(1H)-нафталинон (1,50 г, 7,85 ммоля), 3-хлорбензиламин (4,70 мл, 39,3 ммоля), уксусную кислоту (6,0 мл), 4  молекулярные сита (20 мл), ТТФ (15 мл) и MeOH (15 мл) загружают в колбу и охлаждают до 0oC. Порциями добавляют цианоборгидрид натрия (0,99 г, 15,7 ммоля) в течение 5 минут. Полученную смесь перемешивают в течение 14 часов, фильтруют через целит и концентрируют до сиропа. Полученную смесь подщелачивают 2н. NaOH и экстрагируют эфиром (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и хроматографируют на силикагеле (3% метанол/метиленхлорид). В результате обработки масла IPA/HCl получают 7-нитро-2- (((3-хлорфенил)метил)амино)-1,2,3,4-тетрагидронафталингидрохлорид (1,34 г, 50%).
молекулярные сита (20 мл), ТТФ (15 мл) и MeOH (15 мл) загружают в колбу и охлаждают до 0oC. Порциями добавляют цианоборгидрид натрия (0,99 г, 15,7 ммоля) в течение 5 минут. Полученную смесь перемешивают в течение 14 часов, фильтруют через целит и концентрируют до сиропа. Полученную смесь подщелачивают 2н. NaOH и экстрагируют эфиром (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и хроматографируют на силикагеле (3% метанол/метиленхлорид). В результате обработки масла IPA/HCl получают 7-нитро-2- (((3-хлорфенил)метил)амино)-1,2,3,4-тетрагидронафталингидрохлорид (1,34 г, 50%).
Масс-спектр (М+Н)+ = 317.
(b) 7-Амино-2-(((3-хлорфенил)метил)амино)-1,2,3,4- тетрагидронафталин
К 7-нитро-2-(((3-хлорфенил)метил)амино)-1,2,3,4- тетрагидронафталингидрохлориду (1,34 г, 3,80 ммолей) в 85 AcOH/H2O (75 мл) добавляют металлический цинк (2,48 г, 38,0 ммоля). Полученную смесь перемешивают в течение 5 минут, фильтруют через целит и выпаривают до масла. Полученное масло выливают в щелочную воду и экстрагируют хлороформом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HCl получают 7-амино-2-(((3-хлорфенил)метил)амино)-1,2,3,4-тетрагидронафталин (1,4 г, 99%).
Масс-спектр (M+H)+ = 288.
(c) N-((2-((3-хлорфенил)метил)амино)-1,2,3,4-тетрагидронафт- 7-ил)-2-тиофенкарбоксимидамид
7-Амино-2-(((3-хлорфенил)метил)амино)-1,2,3,4- тетрагидронафталиндигидрохлорид (1,32 г, 3,70 ммолей), 3-метил-2-тиофентиокарбоксимидгидроиодид (1,3 г, 4,6 ммоля) и пиридин (0,30 мл, 3,7 ммоля) в 15 мл ДМФ перемешивают в течение 24 часов. Полученную смесь выливают в воду, подщелачивают 2н. NaOH и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/щавелевой кислотой получают N-((2-((3-хлорфенил)метил)амино)-1,2,3,4- тетрагидронафт-7-ил)-2-тиофенкарбоксимидамиддиоксалат (0,71 г, 33%).
Т. плавления более 100oC с разложением.
Пример 12
N-((2-(фенилметил)(метил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамид
(a) 7-Нитро-2((фенилметил)(метил)амино)-1,2,3,4-тетрагидронафталин
К перемешиваемому раствору 7-нитро-2-((фенилметил)амино)-1, 2,3,4-тетрагидронафталина (1,5 г, 5,4 ммоля) в 30 мл ДМФ добавляют карбонат калия (1,5 г, 10,8 ммоля) и метилиодид (0,36 мл, 5,8 ммоля). Полученную смесь перемешивают в течение 24 часов, выливают в воду и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HCl получают 7-нитро-2-((фенилметил)(метил) амино)-1,2,3,4-тетрагидронафталингидрохлорид (0,89 г, 50%).
Масс-спектр (М+Н)+=297.
b) 7-Амино-2-((фенилметил)(метил)амино)-1,2,3,4- тетрагидронафталин
К 7-нитро-2-((фенилметил)(метил)амино)-1,2,3,4- тетрагидронафталингидрохлориду (0,89 г, 2,7 ммоля) в 85% AcOH/H2O (75 мл) добавляют металлический цинк (3,5 г, 54,0 ммоля). Полученную смесь перемешивают в течение 5 минут, фильтруют через целит и выпаривают до масла. Полученное масло выливают в щелочную воду и экстрагируют хлороформом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HCl получают 7-амино-2-((фенил)метил)(метил)амино)-1,2,3,4-тетрагидронафталин (0,81 г, 88%).
Масс-спектр (М+Н)+= 267
(c) N-((2-(фенилметил)(метил)амино)-1,2,3,4-тетрагидронафт-7- ил)-2-тиофенкарбоксимидамид
7-Амино-2-((фенилметил)(метил)амино)-1,2,3,4- тетрагидронафталиндигидрохлорид (0,81 г, 2,4 ммоля), S-метил-
2-тиофентиокарбоксимидгидрохлорид (0,74 г, 2,6 ммоля) и пиридин (0,19 мл, 2,4 ммоля) в 15 мл ДМФ перемешивают в течение 24 часов. Полученную смесь выливают в воду, подщелачивают 2н. NaOH и экстрагируют этилацетатом (3 х 50 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, концентрируют и хроматографируют на силикагеле (% MeOH/метиленхлорид). В результате концентрирования фракции получают твердое вещество, которое после перекристаллизации из смеси этилацетат/гексан дает N-((2-(фенилметил)(метил)амино)-1,2,3,4-тетрагидронафт-7- ил)-2-тиофенкарбоксимидамиддигидрохлорид (0,14 г,16%).
Температура плавления 176-178oC.
Пример 13
N-((1-Фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамид
(a) 7-Нитро-1-((фенилметил)амино)-1,2,3,4-тетрагидронафталин
7-Нитро-1-тетралон (2,0 г, 10,5 ммоля), бензиламин (1,2 мл, 10,5 ммоля) и изопропоксид титана (3,9 мл, 13,1 ммоля) объединяют и перемешивают в течение 1 часа. Полученную смесь разбавляют абсолютным этанолом (12 мл), обрабатывают цианоборгидридом натрия (0,44 г, 7,0 ммоля) и оставляют при перемешивании на 20 часов. Твердое вещество отфильтровывают и промывают этанолом. Этанол концентрируют, а оставшееся масло используют немедленно в следующей реакции.
Масс-спектр (М+Н)+ = 283.
(b) 1-(7-Нитро-(1,2,3,4-тетрагидронафтил))-N-(фенилметил) трифторацетамид
К перемешиваемому раствору 7-нитро-1-((фенилметил)амино)- 1,2,3,4-тетрагидронафталина (2,96 г, 10,5 ммоля) и триэтиламина (1,46 мл, 10,50 ммоля) в 50 мл метиленхлорида добавляют трифторуксусный ангидрид (1,46 мл, 10,50 ммоля) по каплям. После перемешивания в течение 1 минуты раствор выливают в воду и экстрагируют метилен хлоридом (3 х 20 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния и фильтруют через небольшой слой силикагеля (20% этилацетат/гексан) до получения 1-(7-нитро-(1,2,3,4-тетрагидронафтил))- N-(фенилметил)трифторацетамида (1,90 г, 48%, две стадии).
Масс-спектр (М+Н)+ = 379.
(c) 1-(7-Амино-(1,2,3,4-тетрагидронафтил))-N-(фенилметил) трифторацетамид
К перемешиваемому раствору 1-(7-нитро-(1,2,3,4-тетрагидронафтил))- N-(фенилметил)трифторацетамида (1,91 г, 5,05 ммоля) в ТГФ/MeOH (100 мл, 1:1) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2) в течение 1 часа, фильтруют через целит и концентрируют до получения 1-(7-амино-(1, 2,3,4-тетрагидронафтил))-N-(фенилметил)трифторацетамида, который по данным ТСХ является гомогенным, и немедленно используют на следующей стадии.
(d) N-((1-(Фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)- 2-тиофенкарбоксимидамид
К раствору 1-(7-амино-(1,2,3,4-тетрагидронафтил))-N- (фенилметил)трифторацетамида (1,76 г, 5,05 ммоля) в 10 мл изопропанола добавляют S-метил-2-тиофентиокарбоксимидгидрохлорид (1,44 г, 5,05 ммоля). Полученную смесь перемешивают в течение 14 часов, разбавляют метанолом (6 мл) и 2н. NaOH (6 мл) и нагревают до 50oC в течение 0,5 часа. Полученную смесь выливают в воду и экстрагируют этилацетатом (3 х 30 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. В результате обработки IPA/HBr получают N-((2-фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамиддигидробромид в виде твердого вещества белого цвета (0,53 г, 20%).
Т. плавления 260-262oC.
Пример 14
N-((1-(Фенилметил)амино)индан-5-ил)-N-тиофенкарбоксимидамид
(a) 6-Ацетамидо-(1-((фенил)метил)амино)индан
6-Ацетамидо-1-инданон (5,0 г, 27,6 ммоля), бензиламин (3,1 мл, 27,9 ммоля) и изопропоксид титана (10,2 мл, 34,5 ммоля) объединяют и перемешивают в течение 1 часа. Полученную смесь разбавляют абсолютным этанолом (30 мл), обрабатывают цианоборгидридом натрия (1,2 г, 19,3 ммоля) и оставляют перемешиваться в течение 20 часов. Твердую часть отфильтровывают и промывают этанолом. Этанол концентрируют и оставшееся масло растворяют в этилацетате и экстрагируют 1н. HCl (3 х 50 мл). Водный слой нейтрализуют 2н. NaOH и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла, которое используют без дополнительной очистки на следующей стадии.
(b) 6-Амино-(1-((фенил)метил)амино)индан
6-Ацетамидо-(1-((фенил)метил)амино)индан кипятят с обратным холодильником в 4н. HCl (50 мл) в течение 20 минут, охлаждают и экстрагируют этилацетатом (3 х 50 мл). Водный слой нейтрализуют 2н. NaOH и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. Полученное масло растворяют в IPA и обрабатывают IPA/HCl, в результате чего получают соль-дигидрохлорид (2,0 г, 24%, две стадии).
Т. плавления более 250oC (с разложением).
(c) N-((1-(Фенилметил)амино)индан-5-ил)-2-тиофенкарбоксимидамид
К 6-амино-(1-(фенил)метил)амино)индандигидрохлориду (2,0 г, 6,4 ммоля) в ДМФ (20 мл) добавляют S-метил-2-тиофентиокарбоксимидгидроиодид (2,2 г, 7,7 ммоля) и пиридин (0,57 мл, 7,1 ммоля). Полученную смесь перемешивают при 50oC в течение 20 часов, выливают в подщелаченную воду и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют, концентрируют и хроматографируют на силикагеле (6% метанол/метиленхлорид). Полученные экстракты концентрируют до масла, которое растворяют в метаноле, обрабатывают IPA/HCl и тщательно растирают с эфиром. Твердую часть отфильтровывают и промывают эфиром (1,1 г, 40%).
Температура плавления более 180oC (с разложением).
Пример 15
Нижеследующее соединение получают по способу примера 14:
N-((2-(фенилметил)амино)-1,2,3,4-тетрагидронафт-6-ил)-2- тиофенкарбоксимидамид
Т. плавления более 200oC (с разложением).
Пример 16
N-((2-(фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)- 2-фуранкарбоксимидамид
(а) 2-((Фенил)карбонил)амино-7-нитротетралин
К 2-амино-7-нитротетралину (2,8 г, 14,5 ммоля) в 50 мл ТГФ и 10% K2CO3 (100 мл) добавляют бензоилхлорид (1,7 мл, 15,3 ммоля). После окончания добавления полученную смесь разбавляют водой до объема 250 мл. Выпавшее в осадок твердое вещество отфильтровывают, промывают водой и сушат в вакууме (4,2 г, 98%).
Т. плавления 194-198oC.
(b) 2-((Фенил)метил)амино-7-нитротетралингидрохлорид
К 2-((фенил)карбонил)амино-7-нитротетралину (4,2 г, 14,1 ммоля) в безводном ТГФ (100 мл) добавляют боран-ТГФ (49,3 мл, 1М ТГФ, 49,3 ммоля). Полученную смесь кипятят с обратным холодильником в течение 5 часов, охлаждают до 0oC, и гасят, прикапывая 4н. HCl. Полученную смесь снова доводят до кипения с обратным холодильником в течение 1 часа, концентрируют в вакууме и твердое вещество отфильтровывают (промывают водой) и сушат в вакууме (3,5 г, 78%).
Т. плавления более 300oC.
(c) 2-((Фенил)метил)амино-7-аминотетралингидрохлорид
К перемешиваемому раствору 2-((фенил)метил)амино-7- нитротетралина (2,0 г, 6,3 ммоля) в MeOH (100 мл) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см) в течение 1 часа, фильтруют через целит и концентрируют до получения масла, которое по данным ТСХ оказывается гомогенным, и используют немедленно на следующей стадии.
(d) N-((2-Фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- фуранкарбоксимидамид
К 2-((фенил)метил)амино-7-аминотетралингидрохлориду (1,8 г, 6,3 ммоля) в 20 мл ДМФ добавляют S-метил-2-фурантиокарбоксимидгидроиодид (2,0 г, 7,5 ммоля). Полученную смесь перемешивают в течение 2 часов при 45oC, выливают в подщелаченную воду и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. Это масло растворяют в метаноле, обрабатывают IPA/HCl и тщательно растирают с эфиром. Твердое вещество отфильтровывают и промывают эфиром (2,2 г, 84%).
Температура плавления более 195oC (с разложением).
Получение хиральных промежуточных соединений для примеров 17 и 18
Разделение 2-амино-7-нитротетралина
2-Амино-7-нитротетралин (30 г, 156 ммоля) растворяют в 200 мл ацетона, добавляют к дибензоил-D-винной кислоте (58,7 г, 164 ммоля), также растворенной в 200 мл ацетона. Густую пасту фильтруют и промывают ацетоном. Эту пасту кипятят с обратным холодильником в 3 л смеси вода/этанол/ацетонитрил (1: 1: 1), затем фильтруют в горячем состоянии. Твердое вещество собирают фильтрованием, перекристаллизовывают из этой смеси (3х); получают (5,25 г, 6%) одного изомера (по данным хирального капиллярного зонального электрофореза).
Т. плавления 240-242oC.
Аналогичным образом, дибензоил-L-винную кислоту можно использовать для разделения противоположных энантиомеров, используя ту же самую систему растворителя, что и описанная ранее: (5,3 г, 6%) одного изомера получают (как определено по данным хирального капиллярного зонального электрофореза).
Т. плавления 240-242oC.
Пример 17
(+)-N-((2-((фенил)метил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамид
(a) (+)-2-((фенил)карбонил)амино-1-нитротетралин
К 2-амино-7-нитротетралину (1,8 г, 9,39 ммоля, полученного из дибензоил-D-винной кислоты) в ТГФ (50 мл) и 10% K2CO3 (100 мл) добавляют бензоилхлорид (1,2 мл, 10,1 ммоля). После завершения добавления полученную смесь разбавляют водой до объема 250 мл. Выпавшее в осадок твердое вещество собирают фильтрованием, промывают водой и сушат в вакууме (2,8 г, 100%).
Т. плавления 208-209oC,
[α]D +21,9oC (c 0,33, ДМСО).
(b) (+)-2-((фенил)метил)амино-2-нитротетралингидрохлорид
К (+)-2-((фенил)карбонил)амино-7-нитротетралину (2,8 г, 9,4 ммоля) в безводном ТГФ (100 мл) добавляют боран-ТГФ (32,8 мл, 1М ТГФ, 32,8 ммоля). Полученную смесь кипятят с обратным холодильником в течение 5 часов, охлаждают до 0oC, и гасят, прикапывая 4н. HCl. Полученную смесь доводят до кипения с обратным холодильником в течение 1 часа, концентрируют в вакууме и твердую часть отфильтровывают (промывают водой) и сушат в вакууме (2,8 г, 94%).
Т. плавления более 300oC,
[α]D +51,0oC (c 0,33 ДМСО).
(c) (+)-2-(Фенил)метил)амино-7-аминотетралингидрохлорид
К перемешиваемому раствору (+)-2-((фенил)метил)-амино-7- нитротетралина (2,8 г, 8,7 ммоля) в MeOH (100 мл) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2) в течение 1 часа, фильтруют через целит и концентрируют до получения стеклообразного твердого вещества, которое по данным ТСХ является гомогенным.
[α]D +73,3oC (c 0,87 ДМСО).
(d) (+)-N-((2-)Фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)- 2-тиофенкарбоксимидамид
К (+)-2-((фенил)метил)амино-7-аминотетралингидрохлориду (2,5 г, 8,7 ммоля) в 20 мл ДМФ добавляют S-метил-2- тиофентиокарбоксимидгидрохлорид (3,0 г, 10,4 ммоля). Полученную смесь перемешивают в течение 4 часов при 45oC, выливают в подщелаченную воду и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. Это масло растворяют в метаноле, обрабатывают IPA/HCl и тщательно растирают с эфиром. Твердое вещество собирают фильтрованием и промывают эфиром. После одной перекристаллизации из IPA/MeOH/Et2O получают твердый продукт белого цвета (2,5 г, 66%), т. плавления более 260oC (с разложением) [α]D +44,5oC (с 0,62 ДМСО).
Пример 18
(-)-N-((2-((Фенил)метил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамид
(a) (-)-2-(Фенил)карбонил)амино-7-нитротетралин
К 2-амино-7-нитротетралину (1,8 г, 9,39 ммоля, полученным из дибензоил-L-винной кислоты) в 50 мл ТГФ и 10% K2CO3 (100 мл) добавляют бензоилхлорид (1,2 мл, 10,1 ммоля). После завершения добавления полученную смесь разбавляют водой до объема 250 мл. Выпавший осадок собирают фильтрованием, промывают водой и сушат в вакууме (2,8 г, 100%).
Т. плавления 208-209oC.
[α]D -24,0oC (c 0,87 ДМСО).
(b) (-)-2-((Фенил)метил)амино-7-нитротетралингидрохлорид
К (-)-2-((фенил)карбонил)амино-7-нитротетралину (2,8 г, 9,4 ммоля) в 100 мл безводного ТГФ добавляют боран-ТГФ (32,8 мл, 1М ТГФ, 32,8 ммоля). Полученную смесь кипятят с обратным холодильником в течение 5 часов,
охлаждают до 0oC и гасят, прикапывая 4н. HCl. Полученную смесь снова доводят до кипения с обратным холодильником в течение 1 часа, концентрируют в вакууме и твердое вещество отфильтровывают (промывают водой) и сушат в вакууме (2,8 г, 94%).
Т. плавления более 300oC,
[α]D -59,4oC (с 0,39 ДМСО).
(c) (-)-2-((Фенил)метил)амино-7-аминотетралингидрохлорид
К перемешиваемому раствору (-)-2-((фенил)метил)-амино-7- нитротетралина (2,8 г, 8,7 ммоля) в MeOH (100 мл) добавляют каталитическое количество 10% Pd/C. Полученную смесь гидрируют при 50 пси (3,515 кг/см2) в течение 1 часа, фильтруют через целит и концентрируют до стеклообразного твердого вещества, которое по данным ТСХ является гомогенным; [α]D -74,6oC (с 0,80 ДМСО).
(d) (-)-N-((2-Фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2- тиофенкарбоксимидамид
К (-)-2-((фенил)метил)амино-7-аминотетралингидрохлориду (2,5 г, 8,7 ммоля) в 20 мл ДМФ добавляют S-метил-2-тиофенкарбоксимидгидроиодид (3,0 г, 10,4 ммоля). Полученную смесь перемешивают в течение 4 часов при 45oC, выливают в подщелаченную воду и экстрагируют этилацетатом (3 х 100 мл). Объединенные экстракты промывают водой, сушат над сульфатом магния, фильтруют и концентрируют до масла. Это масло растворяют в метаноле, обрабатывают IPA/HCl и тщательно растирают с эфиром. Твердое вещество отфильтровывают и промывают эфиром. После одной перекристаллизации из IPA/MeOH/Et2O получают твердое вещество белого цвета (2,7 г, 71%).
Т. плавления более 260oC (с разложением).
[α]D -44,5oC (с 0,57 ДМСО).
Пример 19
N-(2,3,4,5-Тетрагидро-1H-3-бензазепин-7-ил)тиофен-2-карбоксимидамид
(a) 2,3,4,5-Тетрагидро-1H-3-бензазепин-7-аминмоногидрохлорид
К раствору 2,3,4,5-тетрагидро-7-нитро-1H-3-бензазепингидрохлорида (1,68 г, 7,35 ммоля) в 100 мл этанола добавляют 5% палладий на угле (0,2 г) и полученный раствор помещают в аппарат Паара для гидрирования, давление доводят до 45 пси (3,164 кг/см2) водорода. После достижения расчетного поглощения водорода (2 часа) катализатор отфильтровывают и промывают водой (25 мл). Полученный фильтрат концентрируют. Добавляют абсолютный этанол и выпаривают до тех пор, пока не выпарится вся вода и не образуется твердый продукт. Твердый продукт растворяют в горячем этаноле (50 мл), и полученный продукт осаждают эфиром (75 мл). Твердое вещество собирают и сушат на воздухе до получения продукта в виде грязно-белого твердого вещества (2,43 г, 94%).
Температура плавления 288-91oC.
(h) N-(2,3,4,5-Тетрагидро-1H-3-бензазепин-7-ил)тиофен-2- карбоксимидамид
Суспензию 2,3,4,5-тетрагидро-1H-3-бензазепин-7- аминмоногидрохлорида (0,60 г, 3,0 ммоля) и 5-метил-2- тиофенкарбоксимидгидроиодида (1,1 г, 3,8 ммоля) в 2,0 мл диметилформамида и 2,0 мл изопропанола перемешивают при комнатной температуре в течение 20 часов. Твердое вещество реакционной смеси отфильтровывают и промывают изопропанолом (5 мл) и этилацетатом (15 мл). Высушенное воздухом твердое вещество весит 1,18 г и представляет смешанную соль. Ее растворяют в воде, подщелачивают и экстрагируют этилацетатом. Растворитель сушат над сульфатом магния и концентрируют до получения свободного основания в виде твердого продукта желтого цвета. Его помещают в 30 мл изопропанола и подкисляют бромистым водородом в изопропаноле до тех пор, пока раствор не становится кислым. Продукт осаждают 35 мл этилацетата. Этот продукт собирают и сушат до получения продукта в виде соли- дигидробромида (0,70 г, 54%).
Т. плавления 281-3oC.
Пример 20
N-(1,2,3,4-Тетрагидроизохинолин-7-ил)тиофен-2-карбоксимидамид
(a) 1,2,3,4-Тетрагидроизохинолин-7-аминмоногидрохлорид
Его получают по способу примера 19, стадии (а). Из 7-нитро- 1, 2, 3, 4-тетрагидроизохинолингидрохлорида (3, 00 г, 14,0 ммоля) и 5% палладия на угле (0,3 г) в 150 мл этанола, выделяют продукт в виде бледно-розового твердого вещества (2,43 г, 94%).
Температура плавления 232-4oC
(b) N-(-1,2,3,4-Тетрагидроизохинолин-7-ил-)тиофен-2-карбоксимидамид
Его получают по способу примера 19, стадии (b). Из 1,2,3,4-тетрагидроизохинолин-7-аминмоногидрохлорида (0,46 г) и 3-метил-2-тиофентиокарбоксимидгидроиодида (0,95 г) в 2,0 мл изопропанола и 2 мл диметилформамида, выделяют после обработки указанное соединение в виде свободного основания (0,60 г, 94%). Его превращают в бисоксалатную соль в растворе метанол/этилацетата до получения продукта в виде грязно-белого твердого вещества (0,59 г, 54%).
Температура плавления 199-200oC (с разложением).
Пример 21
N-(2-Бензил-1,2,3,4-тетрагидроизохинолин-7-ил-тиофен-2- карбоксимидамид
(a) 2-Бензил-7-нитро-1,2,3,4-тетрагидроизохинолинмоногидрохлорид
К раствору 7-нитро-1,2,3,4-тетрагидроизохинолинмоногидрохлорида (2,50 г, 11,6 ммоля) и карбонате калия (2,0 г) в 100 мл ацетонитрила добавляют бензилбромид (2,22 г, 13,0 ммоля) в 10 мл ацетонитрила. Этот раствор перемешивают в течение ночи, и затем твердое вещество удаляют фильтрованием. Растворитель удаляют в вакууме до получения твердого продукта, который разделяют между метиленхлоридом и водой. Высушенную (над сульфатом магния) органическую фазу концентрируют, а полученное масло помещают в этанол (50 мл). Этот раствор подкисляют соляной кислотой в этаноле. Осадок, который образуется, отстаивают и добавляют 150 мл этанола и 50 мл эфира. Твердое вещество собирают и сушат воздухом до получения 2-бензил-7-нитро-1,2,3,4- тетрагидроизохинолингидрохлорида в виде грязно-белого твердого вещества (2,78 г, 79%).
Температура плавления 256-8oC (с разложением).
(b) 2-Бензил-1,2,3,4-тетрагидроизохинолин-7-амингидрохлорид
Это соединение получают по способу примера 19 (стадия а). Из 2-бензил-7-нитро-1,2,3,4-тетрагидроизохинолингидрохлорида (2,00 г, 6,56 ммоля) и 5% Pd/C (0,2 г) в 100 мл этанола. Продукт выделяют в виде твердого вещества желтого цвета (1,05 г, 78%).
Т. плавления 257-9oC (с разложением).
(c) N-(2-Бензил-1,2,3,4-тетрагидроизохинолин-7-ил)тиофен-2- карбоксимидамид
Его получают по способу примера 19 (стадия b). Из 2-бензил-1,2,3,4-тетрагидроизохинолин-7-аминмоногидрохлорида (0,50 г, 1,8 ммоля) и 5-метил-2-тиофентиокарбоксимидгидроиодида (0,67 г, 2,3 ммоля) в изопропаноле (2,0 мл) и диметилформамиде (2,0 мл) выделяют указанное в заглавии соединение в виде твердого вещества желтого цвета (0,53 г, 84%). Его превращают в соль-оксалат в изопропаноле. m/e = 348 (М+Н).
Пример 22
N-(1,2,3,4-тетрагидроизохинолин-5-ил)тиофен-2-карбоксимидамид, диоксалатная соль
Это соединение получают по способу, описанному в примере 20.
Т. плавления 75oC (с разложением).
Пример 23
N-(1,2,3, 4-Тетрагидроизохинолин-6-ил) тиофен-2-карбокс- имидамид
(a) 1,2,3,4-Тетрагидроизохинолин-6-аминмоногидрохлорид
Раствор изохинолин-6-амина (Manske, R.H.F., et.al., J. Am. Chem. Soc., 72, 4997 (1950)) (4,40 г, 30,5 ммоля) и окиси платины (300 мг) в растворе уксусной кислоты (85 мл) и 2,5 М соляной кислоты (30 мл) помещают в аппарат Паара и создают давление водорода 45 пси (3,164 кг/см) в течение 16 часов. Растворитель удаляют в вакууме, а полученную соль разделяют между водным карбонатом калия и 20% изопропанолом в метиленхлориде. Высушенную (сульфат магния) органическую фазу концентрируют, и полученное масло хроматографируют на силикагеле, используя в качестве элюента 2% метанол в хлороформе, до получения 3,08 г (93%) продукта в виде твердого вещества. Этот продукт (3,08 г, 20,8 ммоля) помещают в 200 мл этанола и добавляют 1 эквивалент 0,10 М раствора соляной кислоты. Растворитель удаляют до получения моногидрохлорида в виде твердого вещества
Масс-спектр 149 (М+Н).
(b) N-(1,2,3,4-Тетрагидроизохинолин-6-ил)тиофен- 2-карбоксимидамид
Его получают по способу примера 19 (стадия b). Из 1, 2, 3, 4-тетрагидрохинолин-6-аминмоногидрохлорида (0, 90 г, 4,9 ммоля) и 8-метил-2-тиофентиокарбоксимидгидроиодида (1,80 г, 6,2 ммоля) в изопропаноле (2,0 мл) и диметилформамиде (2,0 мл) выделяют после обработки и хроматографированием на силикагеле, соединение в виде свободного основания (0,74 г, 57%).
Т. плавления 170-5oC.
Пример 24
N-(Изохинолин-7-ил)тиофен-2-карбоксимидамид
(a) 7-Нитроизохинолин
Раствор 7-нитро-3,4-дигидроизохинолина (3,00 г, 17,0 ммоля) и 5% палладия на угле (3,0 г) в 75 мл декалина нагревают с обратным холодильником в течение 3 часов. После охлаждения раствор фильтруют, а катализатор промывают хлороформом (200 мл). Растворитель удаляют в вакууме до получения 7-нитроизохинолина (1,63 г) в виде твердого вещества желто-коричневого цвета.
Масс-спектр 175 (М+Н).
(b) Изохинолин-7-амин
7-Нитроизохинолин (1,62 г, 9,25 ммоля) в 150 мл этанола гидрируют в аппарате Паара над 5% Pd/C (0,2 г) в качестве катализатора в течение 3 часов при 50 пси (3,515 кг/см2). Реакционную смесь фильтруют и растворитель удаляют при пониженном давлении. После перекристаллизации твердого вещества из этанола (3 мл) получают изохинолин-7-амин (0,98 г) в виде твердого вещества желто-коричневого цвета.
Масс-спектр 145 (М+Н),
ЯМР (CDCl3): δ 9,02 (с,1H), 8,29 (д,1H), 7,63 (д,1H), 7,47 (д,1H), 7,13 (дд,H), 7,03 (д,1H), 4,00 (шир.,2H).
(c) N-(Изохинолин-7-ил)тиофен-2-карбоксимидамид
Раствор изохинолин-7-амина (0,96 г, 6,7 ммоля) и S-метил-2-тиофентиокарбоксамида (2,42 г, 8,36 ммоля) в 4 мл изопропанола и 4 мл ДМФ перемешивают в течение 18 часов. Раствор выливают в разбавленный гидроксид натрия и экстрагируют метиленхлоридом. Полученный экстракт сушат над сульфатом магния, и растворитель выпаривают до получения масла, которое отверждается при стоянии. Этот образец очищают на хроматографической колонке с силикагелем (5% метанол в хлороформе, насыщенном газообразным аммиаком) до получения 1,31 г твердого вещества. Это твердое вещество перекристаллизовывают из этилацетата (25 мл) до получения 1,05 г указанного в заглавии соединения в виде твердого вещества грязно-белого цвета.
Т. плавления 177,5-178,5oC.
Описываются бициклические производные амидина формулы I, где D представляет пятичленное гетероциклическое ароматическое кольцо, содержащее от 1 до 4 гетероатомов, выбранных из О, N или S, необязательно замещенное по атому углерода галоидом, трифторметилом, алкил C1-6, нитро или циано, и которое соединено с остальной частью соединения формулы I через атом углерода; А представляет N(Х) или СН (-(СН2)m-NXY), U представляет NH, O или CH2; V представляет (CH2)a; W представляет (CH2)b; а, b, m, X и Y имеют указанные в формуле значения, а также способы их получения и содержащая их фармацевтическая композиция. Соединения формулы I являются ингибиторами синтетазы окиси азота и пригодны для использования в терапии. 5 c. и 15 з.п. ф-лы.
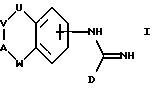
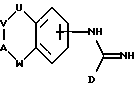
где D представляет пятичленное гетероциклическое кольцо, содержащее гетероатом, выбранный из O или S, и которое связано с остальной частью соединения через атом углерода;
A представляет N(X) или CH (-(CH2)m-NXY);
U представляет NH или CH2;
V представляет (CH2)a;
W представляет (CH2)b;
a и b независимо представляют целое число от 0 до 3 при условии, что a + b представляет целое число от 1 до 3;
X и Y независимо представляют водород, алкил C1-6 или группу -(CH2)nQ;
Q представляет бифенил или фенил, необязательно замещенный одной или более из групп, выбранных из алкил C1-6, галоида, нитро;
m представляет целое число от 0 до 5;
n представляет целое число от 0 до 6;
или цепочка U-V-A-W имеет указанные ранее значения, за исключением того, что она не может быть насыщенной,
и его фармацевтически приемлемые соли и энантиомеры.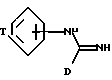
где T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепочку, замещенную (CH2a-NXV, или -U-(CH2)a-N(X)-(CH2)b-;
X и Y независимо представляют водород, алкил C1-6 или группу -(CH2)nQ;
Q представляет фенил, необязательно замещенный алкил C1-6, галоид, нитро;
U, m, n, a, b и D имеют указанные ранее значения, за исключением того, что если T представляет -U(-CH2)a-N(X)-(CH2)b, а X представляет -(CH2)nQ;
n представляет целое число от 0 до 5,
и его фармацевтически приемлемые соли и энантиомеры.
N-((2-(фенилметил)амино)индан-5-ил)-2-тиофенкарбоксимидамидом;
N-((2-(фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамидом;
N-((2-амино)-1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамидом;
N-((1-амино)-1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамидом;
N-((2-амино)-индан-5-ил)-2-тиофенкарбоксимидамидом;
N-((2-метил)(фенилметил)амино)индан-5-ил)-2-тиофенкарбоксимидамидом;
N-((2-амино)-индан-6-ил)-2-тиофенкарбоксимидамидом;
N-((1-(фенилметил)амино)индан-6-ил)-2-тиофенкарбоксимидамидом;
N-((2-((3-хлорфенил)метил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-((2-метилфенил)метил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-((3-метилфенил)метил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-((4-метилфенил)метил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-(этил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-(((4-фенил)метил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-(((4-гексил)фенил))метил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-((3-бромфенил)метил)амино)индан-5-ил)-2-тиофенкарбоксамидином;
N-((2-((3-хлорфенил)метил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамидом;
N-((2-(фенилметил)(метил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамидом;
N-((1-(фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2-тиофенкарбоксимидамидом;
N-((1-(фенилметил)амино)индан-5-ил)-2-тиофенкарбоксимидамидом, N-((1-(фенилметил)амино)-1,2,3,4-тетрагидронафт-6-ил)-2-тиофенкарбоксимидамидом;
N-((2-(фенилметил)амино)-1,2,3,4-тетрагидронафт-7-ил)-2-фуранкарбоксимидамидом;
N-(2,3,4,5-тетрагидро-1H-3-бензазепин-7-ил)тиофен-2-карбоксимидамидом;
N-(1,2,3,4-тетрагидроизохинолин-7-ил)тиофен-2-карбоксимидамидом;
N-(2-бензил-1,2,3,4-тетрагидроизохинолин-7-ил)тиофен-2-карбоксимидамидом;
N-(1,2,3,4-тетрагидроизохинолин-5-ил)тиофен-2-карбоксимидамидом;
N-(1,2,3,4-тетрагидроизохинолин-6-ил)тиофен-2-карбоксимидамидом;
N-(изохинолин-7-ил)тиофен-2-карбоксимидамидом,
или их фармацевтически приемлемыми солями или энантиомерами.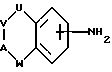
с соединением формулы III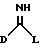
где D имеет указанные в п.1 значения;
L представляет отщепляемую группу,
и, при необходимости, полученное соединение I, в котором X является водородом, дополнительно подвергают взаимодействию с соединением формулы VI
R9-L,
где R9 представляет алкил Cl-6 или группу -(CH2)nQ;
L представляет отщепляемую группу,
с получением соединения формулы I, в котором A представляет группу -N(X), а X представляет алкил C1-6 или группу -(CH2)nQ, или полученное соединение I, в котором X и Y являются водородом, далее подвергают взаимодействию с соединением формулы VI
R9-L,
где R9 представляет алкил C1-6 или группу -(CH2)nQ;
L представляет отщепляемую группу,
с получением соединения формулы I, в которой A представляет группу -CH-((CH2)m-NXY, и по крайней мере, один из X и Y представляет алкил C1-6 или группу -(CH2)nQ.
20 способ получения бициклических производных амидина формулы I по п.1, отличающийся тем, что проводят взаимодействие соединения формулы IV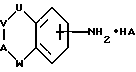
где U, V, A и W имеют указанные в п.1 значения;
HA представляет кислоту,
с соединением формулы V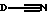
где D имеет указанные в п.1 значения,
и, при необходимости, полученное соединение I, в котором X представляет водород, дополнительно подвергают взаимодействию с соединением VI
R9-L,
где R9 представляет алкил C1-6 или группу -(CH2)nQ;
L представляет отщепляемую группу,
с получением соединения формулы I, в котором A представляет группу -N(X), а X представляет алкил C1-6 или группу -(CH2)nQ, или полученное соединение I, в котором X и Y являются водородом, далее подвергают взаимодействию с соединением формулы VI
R9-L,
где R9 представляет алкил C1-6 или группу -(CH2)nQ;
L представляет отщепляемую группу,
с получением соединения формулы I, в котором A представляет группу -CH-((CH2)m-NXY и, по крайней мере, один из X и Y представляет алкил C1-6 или группу -(CH2)nQ.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| СПОСОБ ВЫВОДА МЫШЬЯКА ИЗ МЫШЬЯКСОДЕРЖАЩИХ МАТЕРИАЛОВ | 2002 |
|
RU2226562C2 |
| 1,3-ЗАМЕЩЕННЫЕ СУЛЬФОНИЛМОЧЕВИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2022963C1 |
