Изобретение относится к полипептидам и их получению. Эти полипептиды были выделены из пиявки Hirudinaria manillensis. Указанные полипептиды обладают антитромбинными свойствами.
Наиболее известные антикоагулирующие пептиды, очевидно, принадлежат к семейству гирудинов. Гирудин, который был выделен из медицинской пиявки Hirudo medicinalis, хорошо известен специалистам в качестве полипептидного ингибитора тромбина 1,2. В частности, гирудин связывает тромбин посредством ионных взаимодействий, предупреждая тем самым превращение фибриогена в фибрин и последующее образование фибриновых сгустков. При исследованиях на животных гирудин показал эффективность в предупреждении венозного тромбоза, окклюзии анастоматических сосудов и тромбин-индуцированного диссеминированного внутрисосудистого свертывания. Кроме того, гирудин обнаруживает низкую токсичность, невысокую антигенность, или полное ее отсутствие, и короткое время выведения из кровообращения 3.
Полипептиды с антикоагулирующими свойствами были выделены из различных видов пиявок Hirudinaria manillensis (ЕР-A-0347376 и WO 90/05143). Этот вид пиявки является эволюционно более продвинутым, чем Hirudo medicinalis и поэтому может синтезировать антикоагулирующие пептиды, аминокислотные последовательности которых могут отличаться от аминокислотных последовательностей гирудина и других известных его вариантов.
Авторами настоящей заявки были проведены исследования по получению препаратов из пиявок Hirudinaria manillensis. В результате этих исследований были обнаружены три новых полипептида, обладающих антитромбиновой активностью. В соответствии c этим настоящее изобретение относится к полипептиду, содержащему аминокислотную последовательность (см. в конце описания), и к его фармацевтически приемлемым солям.
Аминокислотные остатки представлены в соответствии с их стандартным трехбуквенным кодом (Eur. J. Biochem. 138. 9-37, 1984). Остаток Yaa представляет собой Asp или Tyr-остатки, а Zaa представляет собой -OH, -NH2 или Gly-OH. Указанные соли могут быть кислыми аддитивными солями. Эти соли могут быть образованы неорганическими кислотами, такими как галогенводородные кислоты, например соляная кислота, серная кислота, фосфорная кислота, или пирофосфорная кислота, или органическими кислотами, такими как бензолсульфоновая, п-толуолсульфоновая, метансульфоновая, уксусная, молочная, пальмитиновая, стеариновая, яблочная, винная, аскорбиновая, или лимонная кислота. Указанные полипептиды содержат также свободные гидроксильные группы и поэтому могут образовывать натриевые, калиевые, кальциевые, магниевые или аммониевые соли, или эти соли могут быть образованы о физиологически приемлемыми органическими азотсодержащими основаниями. Полипептиды могут быть также в виде внутренних солей.
Полипептиды настоящего изобретения состоят в основном из вышеуказанных аминокислотных последовательностей (i) - (iii). Природные полипептиды, выделенные из Hirudinaria manillensis, имеют аминокислотную последовательность (i) или (ii), где Yaa является Asp, a Zaa является -OH или частичной аминокислотной последовательностью (iii). Полипептиды настоящего изобретения могут быть выделены и очищены в целях их использования в качестве антагонистов тромбина.
Полипептиды могут быть получены с использованием предшествующей всей или части лидерной последовательности. Эта лидерная последовательность может быть нативной либо чужеродной лидерной последовательностью по отношению к клетке, из которой происходит данный полипептид. Лидерная последовательность обладает способностью направлять секрецию полипептида из клетки. Оба натуральных полипептида настоящего изобретения экспрессируются лидерной последовательностью, которая затем отщепляется. Поэтому может присутствовать полная или часть лидерной последовательности, которой, в частности, является следующая последовательность: Met Phe Ser Leu Lys Leu Phe Val Val Phe Leu Ala Val Cys Ile Cys Val Ser Gln Ala.
Натуральный полипептид настоящего изобретения или его соли могут быть получены путем выделения указанного полипептида или его фармацевтически приемлемой соли из ткани или секретов пиявки вида Hirudinaria manillensis. В частности, полипептид настоящего изобретения может быть получен путем продуцирования в соответствии с WO 90/05143 и очистки с помощью жидкостной хроматографии высокого давления.
Полипептид настоящего изобретения или его соль могут быть также получены путем
а) использования хозяина, трансформированного вектором экспрессии, содержащим ДНК-последовательность, кодирующую указанный полипептид в условиях, обеспечивающих экспрессию этого полипептида в хозяине;
b) выделения полученного таким образом полипептида или его фармацевтически приемлемой соли.
Этот способ, в основном, заключается в получении нуклеотидной последовательности, кодирующей нужный полипептид, и экспрессии указанного полипептида в рекомбинантных организмах. Культивирование генетически модифицированных организмов приводит к продуцированию нужного продукта, обладающего полной биологической активностью. Кроме того, настоящее изобретение относится
- к экспрессирующему вектору, содержащему ДНК-последовательность, кодирующую полипептид настоящего изобретения;
- к хозяину, трансформированному с использованием совместимого вектора экспрессии в соответствии с настоящим изобретением;
- к ДНК-последовательности, кодирующей полипептид настоящего изобретения.
Хозяин, предназначенный для экспрессии полипептида настоящего изобретения, может быть получен путем трансформации хозяйской клетки с использованием совместимого вектора экспрессии настоящего изобретения. Этот вектор экспрессии может быть получен путем:
а) химического синтеза ДНК-последовательности, кодирующей полипептид настоящего изобретения;
b) инсерции указанной ДНК в вектор экспрессии.
Альтернативно, вектор экспрессии может быть получен путем
a) продуцирования и выделения кДНК, кодирующей полипептид настоящего изобретения, из мРНК пиявки вида Hirudinaria manillensis; и
b) инсерции выделенной кДНК в вектор экспрессии.
Таким образом, согласно настоящему изобретению полипептид продуцируют путем получения трансформированного хозяина в условиях, обеспечивающих экспрессию полипептида в этом хозяине. Если хозяином является эукариотическая клетка, то полученный полипептид может быть гликосилированным. Полученный полипептид может быть выделен как таковой, или в виде его фармацевтически приемлемой соли. Таким образом, полипептид настоящего изобретения или его соль могут быть получены в чистом виде.
Полипептиды настоящего изобретения могут быть модифицированными посредством аминокислотных удлиняющих вставок у одного или обоих концов. Полипептид, состоящий из такой удлиненной последовательности, должен, разумеется, еще обладать антитромбиновой активностью. Например, короткая последовательность до 30 аминокислотных остатков может быть введена у любого или у обоих концов.
Полипептиды настоящего изобретения могут быть подвергнуты одной или нескольким посттрансляционным модификациям, таким как сульфатирование, COOH-амидирование, ацилирование или химическое изменение полипептидной цепи. Например, полипептид, имеющий на своем карбоксильном конце глициновый остаток, может быть подвергнут ферментному амидированию с использованием пептидил-глицин-α-амидирующей монооксигеназы (РАМ-фермента).
Для продуцирования полипептида с противотромбиновой активностью путем использования техники рекомбинантных ДНК получают ген, кодирующий полипептид настоящего изобретения. Кодирующая последовательность ДНК обычно не содержит интроны. Эту ДНК выделяют и очищают. Ген вставляют в вектор экспрессии, способный к продуцированию рекомбинантного продукта. ДНК-последовательности могут быть кДНК-последовательностями. Они также могут быть синтетическими ДНК-последовательностями. Синтетический ген обычно получают путем химического синтеза олигонуклеотидов, которые в целом соответствуют нужному гену. Затем для получения гена осуществляют сборку этих олигонуклеотидов.
Указанный ген может быть сконструирован из шести химически синтезированных олигонуклеотидов, каждый из которых представляет около 1/3 нити двухнитевой генной ДНК. Для получения нужного гена олигонуклеотиды сшивают и отжигают. По желанию, генная последовательность может быть модифицирована с помощью сайт-направленного мутагенеза путем замены одного или нескольких кодонов. Обычно, ген конструируют с помощью рестриктирущих сайтов у каждого конца с целью облегчения последующей манипуляции.
Как указывалось выше, может быть получена ДНК-последовательность, которая кодирует лидерный пептид. Указанный лидерный пептид обладает способностью к направлению секреции полипептида из клеток, в которых этот пептид был экспрессирован. Последовательность, кодирующую лидерный пептид, обычно, лигируют с 5'-концом ДНК-последовательности, кодирующий полипептид.
Таким лидерным пептидом может быть OmpA-лидерный пептид, в том случае, если необходима экспрессия в бактериальном хозяине, таком как Е. coli. При экспрессии в клетках насекомого лидерным пептидом может быть лидерный пептид белка вируса везикулярного стоматита G (VSV-G-белка). Соответствующие ДНК-последовательности, кодирующие лидерные последовательности OmpA и VSV-G-белка, приведены ниже:
В соответствии с настоящим изобретением может быть получена ДНК-последовательность, кодирующая слитый белок, который расщепляется с высвобождением нужного полипептида. При этом может быть использована ДНК-последовательность, которая кодирует несущую полипептид последовательность, сшитую посредством отщепляемой связи с N-концом полипептида настоящего изобретения. Указанная смесь может расщепляться цианистым бромидом.
Для экспрессии полипептида конструируют вектор экспрессии, который содержит ДНК-последовательность, кодирующую полипептид, и который способен к экспрессии полипептида в соответствующем хозяине. Могут быть использованы транскрипционные и трансляционные регуляторные элементы, включая промотор для ДНК-последовательности, сайт терминации транскрипции, и кодоны инициации и терминации трансляции. ДНК-последовательность получают в правильной рамке считывания так, чтобы экспрессия полипептида происходила в хозяине, совместимом с вектором.
Вектор экспрессии обычно включает в себя область начала репликации и при необходимости ген селектируемого маркера, такой как ген резистентности к антибиотику. Промотор сшивают с ДНК-последовательностью, кодирующей полипептид. Указанный вектор экспрессии может быть плазмидой. В этом случае промотор, предпочтительно выбранный из Ptrp- и Plcc/lac-промоторов, сшивают с ДНК-последовательностью. Альтернативно, вектор экспрессии может быть вирусом. Таким вирусом может быть рекомбинантный бакуловирус, в котором промотор полиэдрина оперативно связывают с ДНК-последовательностью, кодирующей полипептид.
Вектор экспрессии, обладающий способностью к экспрессии полипептида, может быть получен с использованием стандартной техники. ДНК-фрагмент, кодирующий полипептид, может быть вставлен в соответствующий рестрикционный сайт вектора экспрессии, например, плазмидного вектора. Рекомбинантный бакуловирус может быть получен путем
(i) клонирования гена, кодирующего полипептид, в бакуловирусном векторе-переносчике в рестриктирующем сайте, расположенном за (в направлении 5′ __→ 3′) полиэдриновым промотором; и
(ii) совместной трансфекции клеток насекомого, восприимчивых к бакуловирусной инфекции, с рекомбинантным вектором, определенным в стадии (i), и интактной бакуловирусной ДНК дикого типа.
В результате происходит гомологичная рекомбинация, позволяющая получить рекомбинантный бакуловирус, содержащий полипептидный ген за (5′ __→ 3′) полиэдриновым промотором. Бакуловирусный вектор-переносчик может иметь уникальный сайт клонирования, расположенный за (5′ __→ 3′) инициирующим ATG-кодоном полиэдрина. Продукт, экспрессированный полученным рекомбинантным бакуловирусом, представляет собой гибридный белок, в котором N-концевая часть полиэдринового белка сплавлена с N-концом полипептида. Как указывалось выше, расщепляемая связь может находиться в месте лигирования.
В стадии (ii) в качестве клеток насекомых обычно используются клетки Spodoptera frugiperda. В качестве бакуловируса дикого типа обычно используют вирус ядерного полиэдроза Autographa californica (AcNPV).
Вектор экспрессии, кодирующий полипептид, получают в соответствующем хозяине. Клетки трансформируют с помощью полипептидного гена. Трансформированные хозяйские клетки получают в условиях, обеспечивающих экспрессию в них полипептида. Трансформированные клетки, например, культивируют таким образом, чтобы создать возможность для осуществления указанной экспрессии. В данном случае может быть использована любая система совместимых хозяина и вектора.
Трансформированным хозяином может быть прокариот или эукариот. Может быть использован бактериальный или дрожжевой хозяин, например E.coli или S.cerevisiae. В частности, может быть использована грамм-положительная бактерия. Предпочтительным бактериальным хозяином является штамм E.coli, типа B. Альтернативно, могут быть использованы клетки насекомого, если бакуловирусная экспрессирующая система является в данном случае подходящей, Такими клетками насекомого являются клетки Spodoptera frugiperda. В другом альтернативном варианте могут быть трансформированы клетки, принадлежащие к клеточной линии млекопитающего. В этом случае могут быть использованы не относящиеся к человеку трансгенные животные, в которых продуцируется нужный полипептид.
Экспрессированный полипептид может быть выделен и очищен. При этом может быть получен полипептид, имеющий одну из вышеуказанных аминокислотных последовательностей (i), (ii) или (iii), которым предшествует остаток Met, кодируемый кодоном инициации трансляции. Альтернативно, как указывалось выше, может быть получен гибридный белок, содержащий аминокислотную последовательность полипептида настоящего изобретения, т.е. последовательность (i), (ii) или (iii), лигированную с последовательностью-носителем. В случае, если в указанном гибридном белке между аминокислотной последовательностью (i) и (ii) или (iii) и последовательностью-носителем имеется соответствующая связь, то полипептид, имеющий вышеуказанную аминокислотную последовательность (i), (ii) или (iii) может быть выделен путем расщепления этой связи соответствующим агентом.
Полипептид настоящего изобретения или его фармацевтически приемлемая соль могут быть также получены путем
(a) химического синтеза указанного полипептида; и
(b) выделения полученного таким образом полипептида или его фармацевтически приемлемой соли.
Таким образом, указанный полипептид может быть сконструирован посредством химического синтеза из отдельных аминокислот и/или из пептидов, предварительно сформированных из двух или нескольких аминокислот в порядке, соответствующем последовательности нужного полипептида. При этом могут быть использованы твердофазный или растворительный способы. Полученный в результате полипептид может быть по необходимости превращен в его фармацевтически приемлемую соль.
В твердофазном синтезе аминокислотную последовательность нужного полипептида последовательно конструируют из C-концевой аминокислоты, связанной с нерастворимой смолой. После продуцирования нужного полипептида он может быть выделен из смолы. При использовании синтеза в жидкой фазе нужный полипептид может быть снова сконструирован из C-концевой аминокислоты. Карбоксильную группу указанных аминокислотных остатков, блокированных соответствующей защитной группой, в конце синтеза удаляют.
Независимо от того, используется ли твердофазная или жидкофазная техника, каждая аминокислота, добавленная в реакционную систему, обычно имеет защищенную аминогруппу и активированную карбоксигруппу. Функциональные боковые группы также являются защищенными. После каждой стадии синтеза, аминозащитные группы удаляют. Функциональные группы боковой цепи обычно удаляют в конце синтеза.
Полипептид может быть превращен в его фармацевтически приемлемую соль. Он может быть превращен в кислую аддитивную соль с использованием органической или неорганической кислоты. Такой кислотой является уксусная, янтарная и соляная кислота. Альтернативно, указанный пептид может быть превращен в соль карбоновой кислоты, такую как аммониевая соль или соль щелочного металла, например натриевая или калиевая соль.
Полипептид или его фармацевтически приемлемая соль могут быть использованы в фармацевтической композиции в сочетании с фармацевтически приемлемым носителем или наполнителем. Такой композицией может быть, например, препарат для внутривенного введения (в этом случае носителем обычно является стерильный солевой раствор или вода с приемлемой степенью чистоты). Полипептид настоящего изобретения является антагонистом тромбина и может быть использован при лечении тромбоэмболии, например при свертывании крови человека. Полипептид может быть также использован для лечения и профилактики тромбоза и тромбоэмболии, включая профилактику постоперационного тромбоза, при лечении шоковых состояний (например, при септическом шоке или политравматическом шоке), при лечении коагулопатии потребления, при гемодиализе, гемосепарации и при искусственном кровообращении. В одном из вариантов осуществления настоящего изобретения полипептид или его соль могут быть введены вместе с активатором плазминогена, например тканевым плазменным активатором.
Доза полипептида, в основном, зависит от конкретных форм введения и целей лечения или профилактики. Размер отдельной дозы и схема введения может быть определена специалистом в зависимости от заболевания, поскольку каждому специалисту известны способы определения факторов крови, необходимых для назначения соответствующего лечения, В основном, в случае введения инъекции, терапевтически эффективное количество соединений настоящего изобретения составляет в пределах приблизительно от 0,005 до 0,1 мг на кг веса тела. Предпочтительной является доза приблизительно в пределах от 0,01 до 0,05 мг/кг веса тела. Эффективным способом введения являются внутривенные, внутримышечные или подкожные инъекции. В соответствии с этим одноразовая доза композиции для парентерального введения зависит от способа введения и составляет приблизительно от 0,4 до 7,5 мг соединения настоящего изобретения. Помимо активного ингредиента указанная фармацевтическая композиция также содержит буфер (например, фосфатный буфер) который используется для поддержания pH в пределах приблизительно от 3,5 до 7, и другие компоненты, такие как хлорид натрия, маннит или сорбит для регулирования изотоничности раствора. Указанные композиции могут быть в лиофилизованной форме или в виде растворов, предпочтительно содержащих антибактериальные активные консерванты, например от 0,2 до 0,3% метилового или этилового сложного эфира 4-гидроксибензойной кислоты.
Композиция для наружного применения может быть получена в виде водного раствора, лосьона или геля, масляного раствора или суспензии, или жиросодержащей, или, в частности, эмульгируемой мази. Композицию в виде водного раствора получают, например, путем растворения активных ингредиентов настоящего изобретения, или его терапевтически приемлемой соли, в водном буферном растворе (напр., pH 4-6,5), в который, по желанию, кроме активного ингредиента может быть добавлено противовоспалительное средство, и/или полимерное связующее, например поливинилпирролидон, и/или консервант. Концентрация активного ингредиента составляет приблизительно от 0,1 до 1,5 кг, а предпочтительно от 0,25 до 1,0 мг на 10 мл раствора или на 10 г геля.
Масляная форма для наружного применения может быть получена, например, путем суспендирования активного ингредиента настоящего изобретения или его терапевтически приемлемой соли в масле, по желанию, с добавлением агента, вызывающего набухание, например, такого как стеарат алюминия, и/или поверхностно-активных веществ (тензиды), имеющих значение HLB ("гидрофильно-липофильного баланса") ниже 10, например, таких как жирнокислые моноэфиры многоатомных спиртов, например глицеринмоностеарат, сорбитанмонолаурат, сорбитанмоностеарат или сорбитанмоноолеат. Жиросодержащие мази могут быть получены, например, путем суспендирования активного ингредиента настоящего изобретения или его соли в подходящем для нанесения жирном основании, и по желанию, с добавлением ПАВ, имеющего значение HLB ниже 10. Эмульгируемую мазь получают путем размешивания водного раствора активного ингредиента настоящего изобретения или его соли в мягком жирном основании, способном к размазыванию, и с добавлением тензида, имеющего значение HLB ниже 10. Все формы, предназначенные для наружного употребления, могут также содержать консерванты. Концентрация активного ингредиента составляет приблизительно от 0,1 до 1,5 мг, а предпочтительно от 0,25 до 1,0 мг на приблизительно 10 г основы.
Помимо описанных выше композиций и их фармацевтических аналогов, предназначенных для введения непосредственно в организм человека или животных, настоящее изобретение также относится к фармацевтическим композициям и препаратам для медицинского применения вне организма человека или животного. Такие композиции и препараты обычно используют в качестве противосвертывающего средства, добавляемого в кровь, подвергаемую циркуляции или обработке вне организма (например, при искусственном кровообращении или диализе искусственной почки), консервации или модификации (например, гемосепарации). Указанные препараты, такие как маточные растворы или альтернативные препараты в одноразовых дозах, по своему составу аналогичны описанным выше препаратам для инъекций; однако количество или концентрация активного ингредиента зависит преимущественно от объема обрабатываемой крови, а более точно от содержания в ней тромбина. В связи с этим следует учесть, что активный ингредиент настоящего изобретения в свободной форме полностью дезактивирует приблизительно 5-кратное количество по массе тромбина и является при этом физиологически безвредным даже в относительно больших количествах и быстро выводится из кровообращения даже при высоких концентрациях, поэтому риск передозировки практически отсутствует, например, даже при гемотрансфузии. В зависимости от конкретных целей подходящая доза активного ингредиента составляет приблизительно от 0,01 до 1,0 мг на литр крови, хотя даже превышение верхнего предела является практически безопасным.
Ниже приводятся примеры, иллюстрирующие настоящее изобретение вместе с сопровождающими их фигурами.
На фиг. 1 изображена хроматограмма, представляющая результаты ВЭЖХ-анализа примера 1, P1 - P3 указывают на три различных пика, полученных в соответствии о описанием в примере 1. FT соответствует потоку, а 4-AB соответствует 4-аминобензамидину.
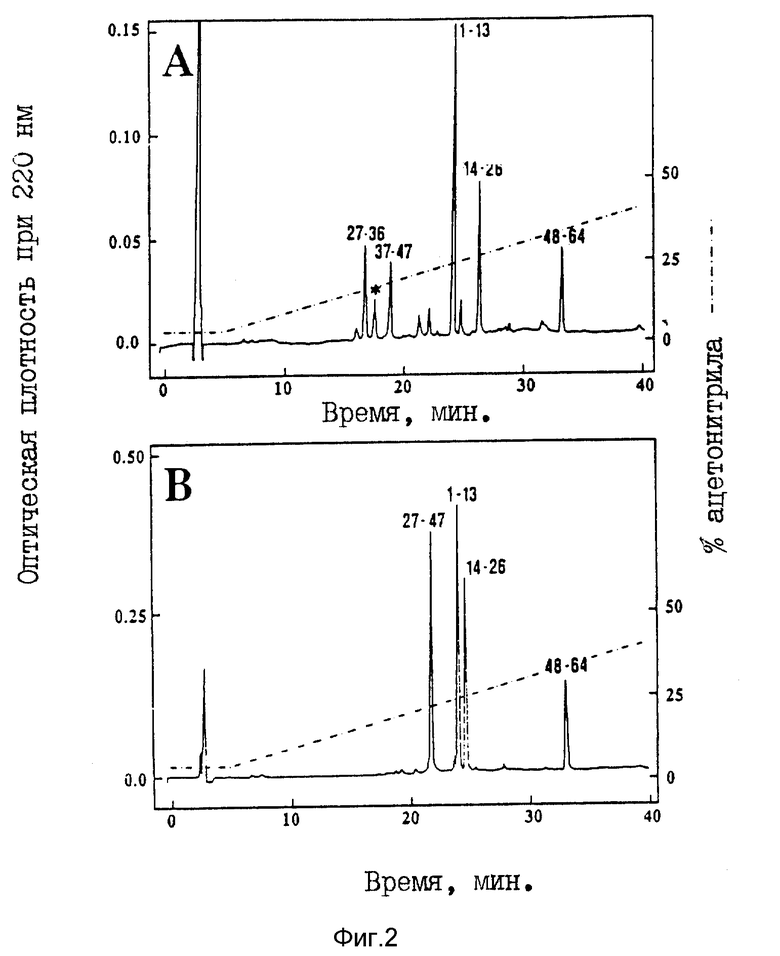
На фиг. 2 показаны профили элюирования, полученные в примере 2 (b) для трипсин-переваренного PE-P1 (A) и PE-P2 (B).
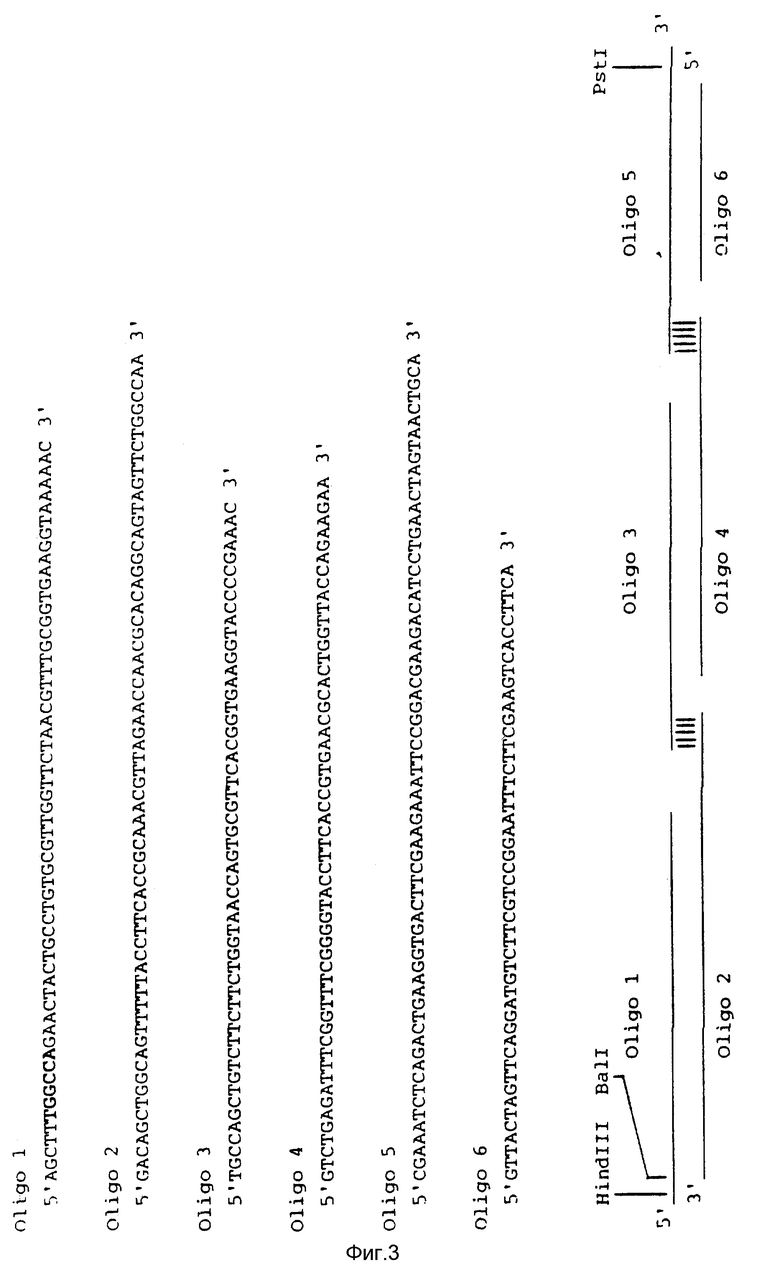
На фиг. 3 показана нуклеотидная последовательность шести олигонуклеотидов, кодирующих большую часть белка, соответствующего пику 2 (P2), в котором аминокислотным остатком в положении 61 является Asp, а последней аминокислотой полипептидной цепи является Asn64. Жирным шрифтом показан BalI-сайт, который был использован для дальнейших конструкций. В нижней части фигуры показан способ сборки шести олигонуклеотидов. Для последующих манипуляций были использованы Hind III и PstI-сайты.
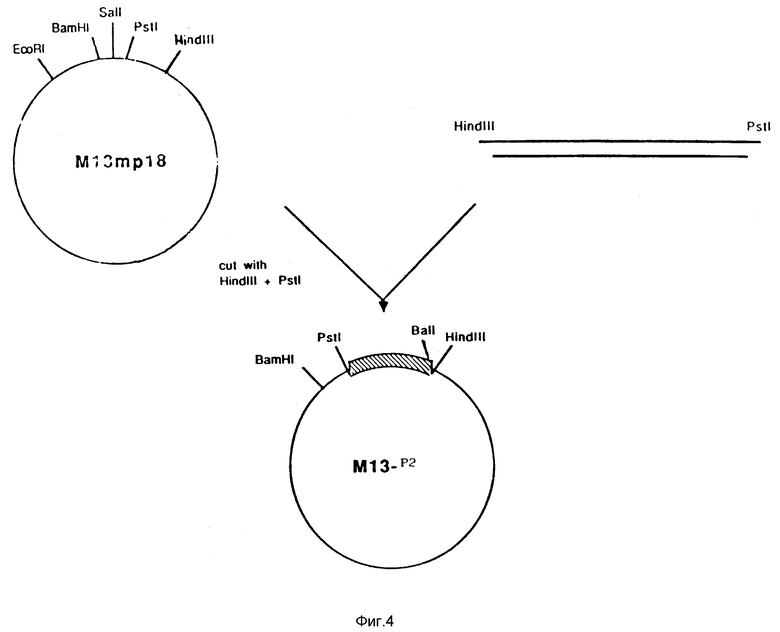
На фиг. 4 показана схема конструирования промежуточной плазмиды M13-P2, которая является источником BalI-BamHI-фрагмента ДНК для всех последующих конструкций P2.
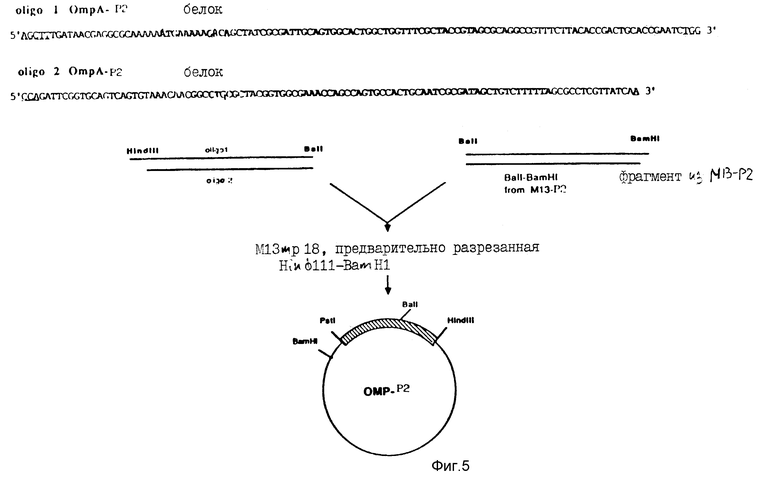
На фиг. 5 схематически показано конструирование нового рекомбинанта M13, обозначенного OMP-P2, который несет полный ген P2, связанный с лидерным пептидом OmpA. Лидерная пептидная последовательность показана жирным шрифтом, а тупой конец BalI и липкий конец Hind III подчеркнуты.
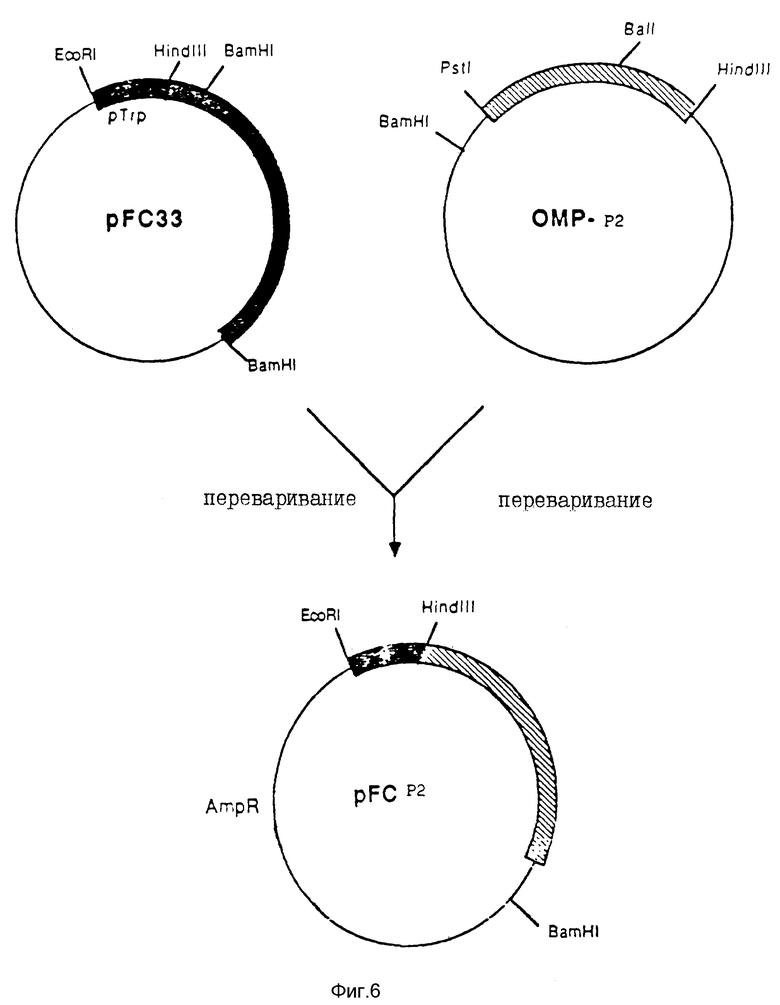
На фиг. 6 схематически показано конструирование pFC-P2, которая является плазмидой, используемой для продуцирования белка P2 в E.coli.
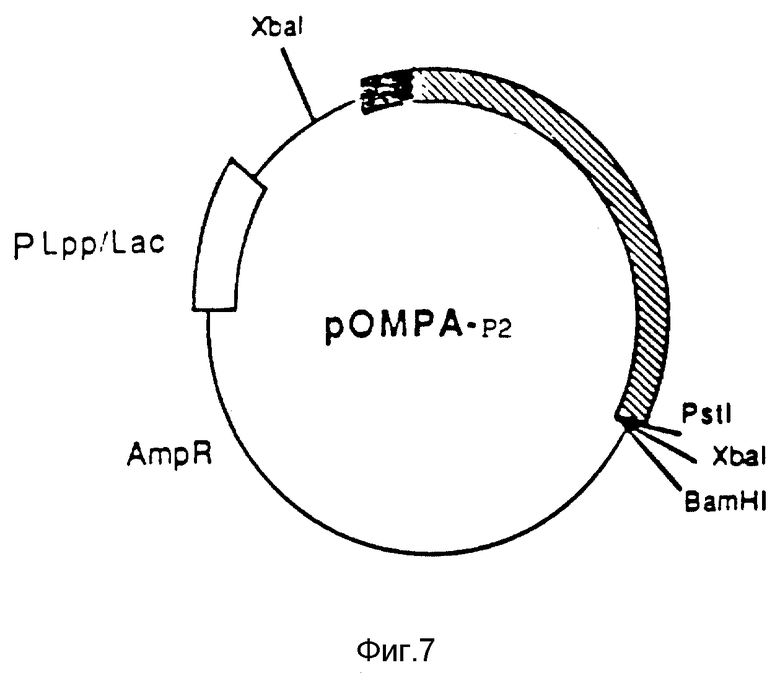
На фиг. 7 показана в общих чертах структура плазмиды pOMPA-P2, использованная для продуцирования P2 в E.coli. Для получения этой новой плазмиды, где P2-ген находится под транскрипционным контролем гибридного промотора Plpp/lac, была использована традиционная техника генной рекомбинации. Даже в этом случае лидерный пептид OmpA направляет секрецию P2 в периплазму E.coli.
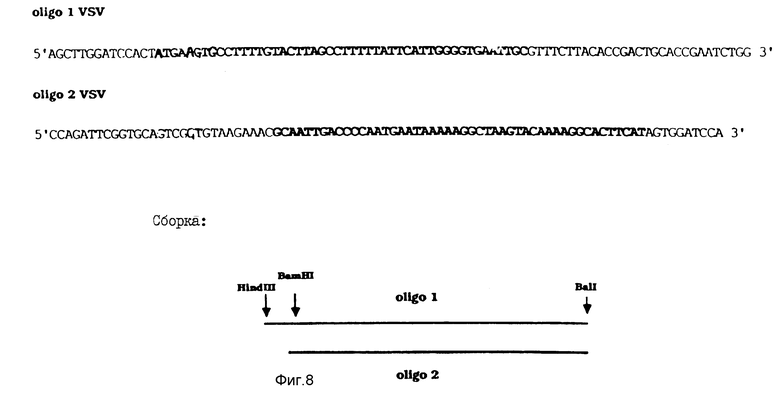
На фиг. 8 показана нуклеотидная последовательность и сборка синтетических олигонуклеотидов, использованных для секреции P2 из клеток насекомых. Последовательность, показанная жирным шрифтом, относится к лидерному пептиду белка VSV-G.
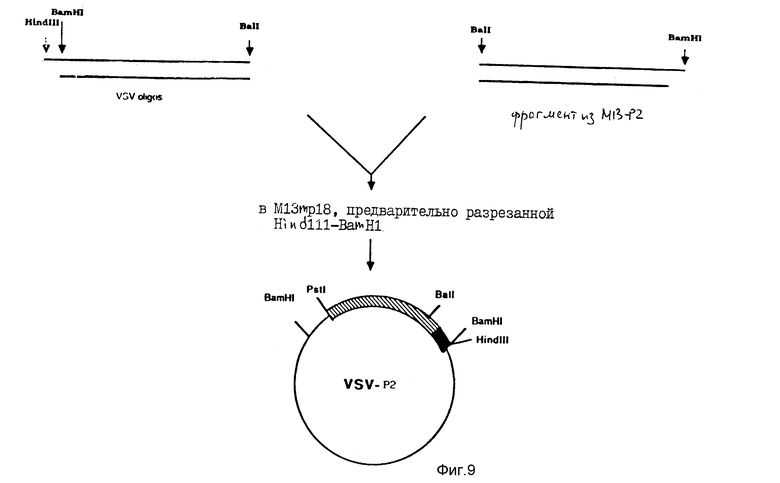
На фиг. 9 схематически показано конструирование нового рекомбинанта M13, обозначенного VSV-P2, где полный P2-ген является связанным с лидерным пептидом белка VSV-G.
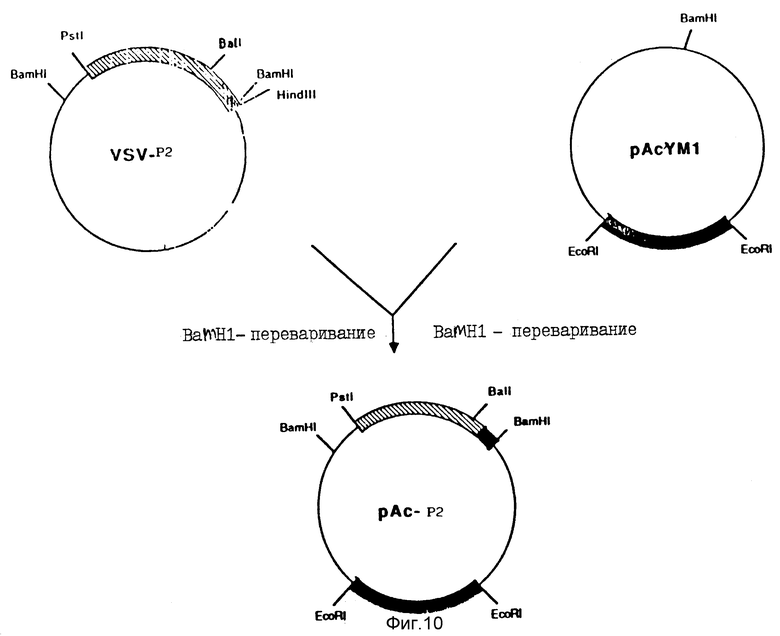
На фиг. 10 схематически показано конструирование pAc-P2, которая была использована в качестве вектора-переносчика в бакуловирусный геном. pAcYM1 является исходной плазмидой, широко используемой в качестве акцептора гетерологичной последовательности, переносимой в вирус.
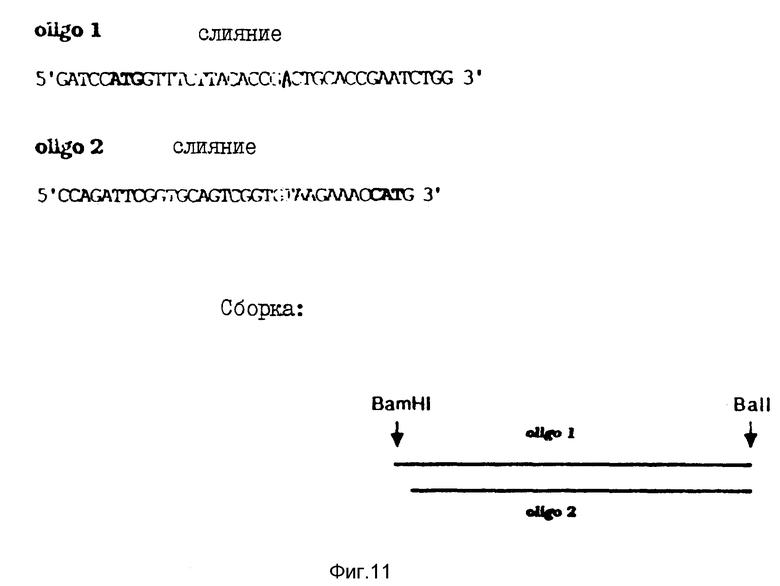
На фиг. 11 показана нуклеотидная последовательность и сборка синтетических олигонуклеотидов, кодирующих начало P2-цепи. Жирным шрифтом показан ATG-кодон, кодирующий дополнительный метиониновый остаток.
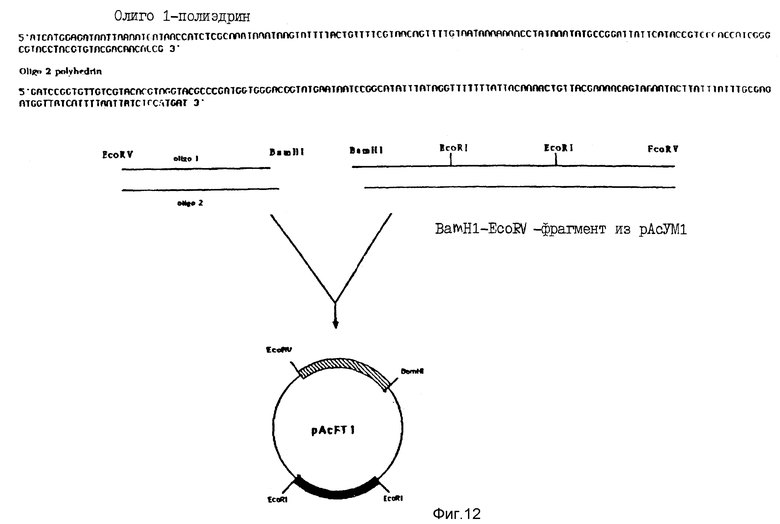
На фиг. 12 схематически показано конструирование pAcFT1, которая используется для внутриклеточной экспрессии.
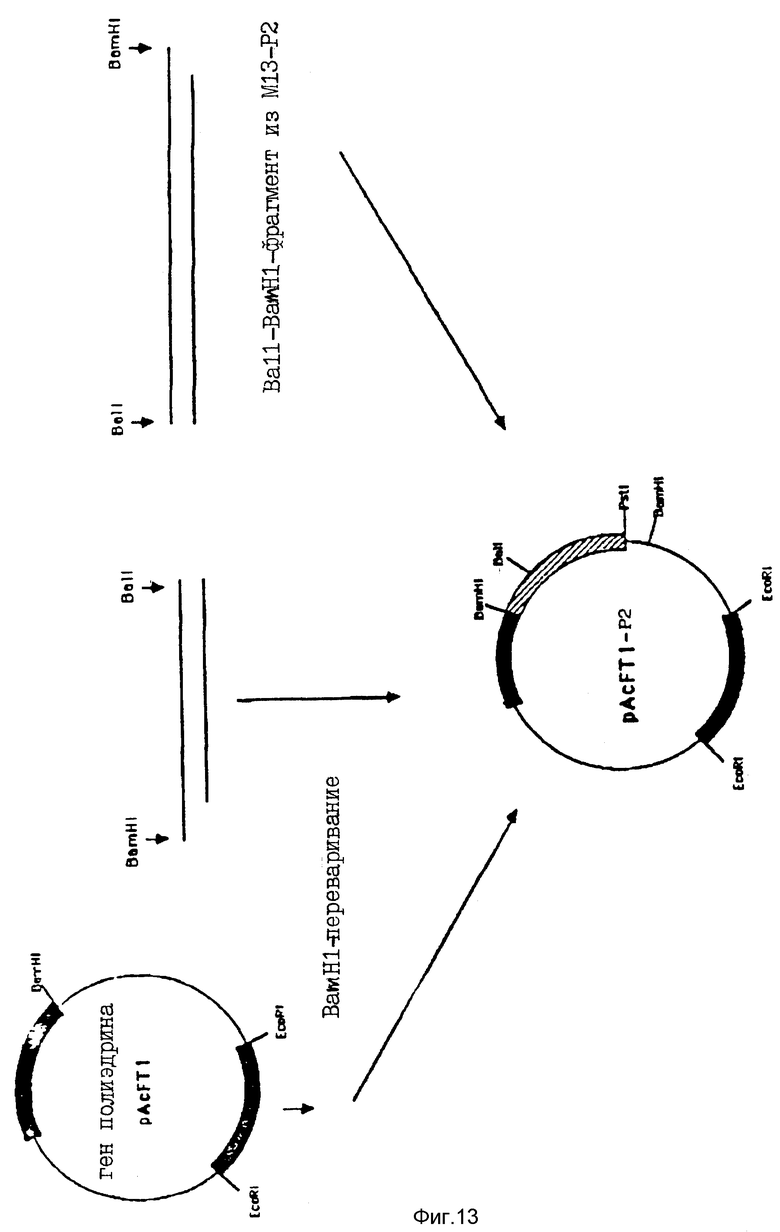
На фиг. 13 схематически показана новая транспортная плазмида, обозначенная pAcFT1-P2, которая содержит полную P2-последовательность, связанную с первыми 18 аминокислотами полиэдрина. Эта плазмида была использована для переноса гетерологичной последовательности в бакуловирусный геном.
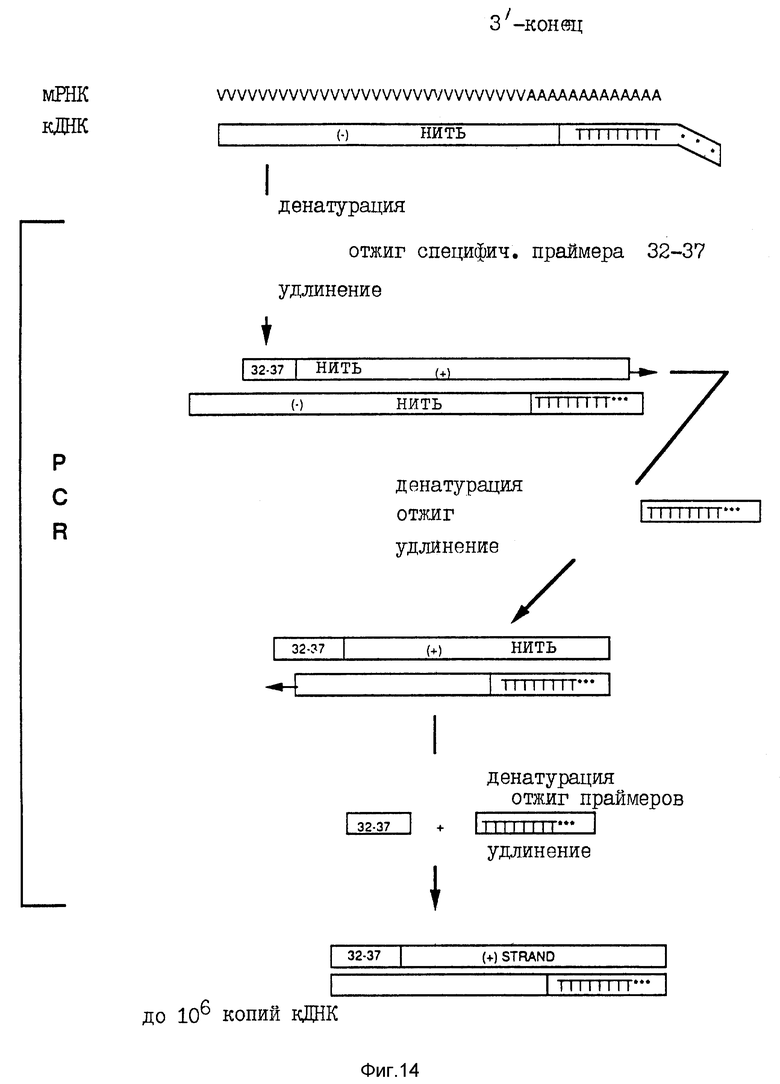
На фиг. 14 схематически представлен протокол RACE для амплификации 3'-концов. На этой фигуре "*** TTTT..." обозначает адапторный праймер dT17. В каждой стадии диаграммы для упрощения показано только, каким образом используется новый продукт, полученный в предыдущей стадии.
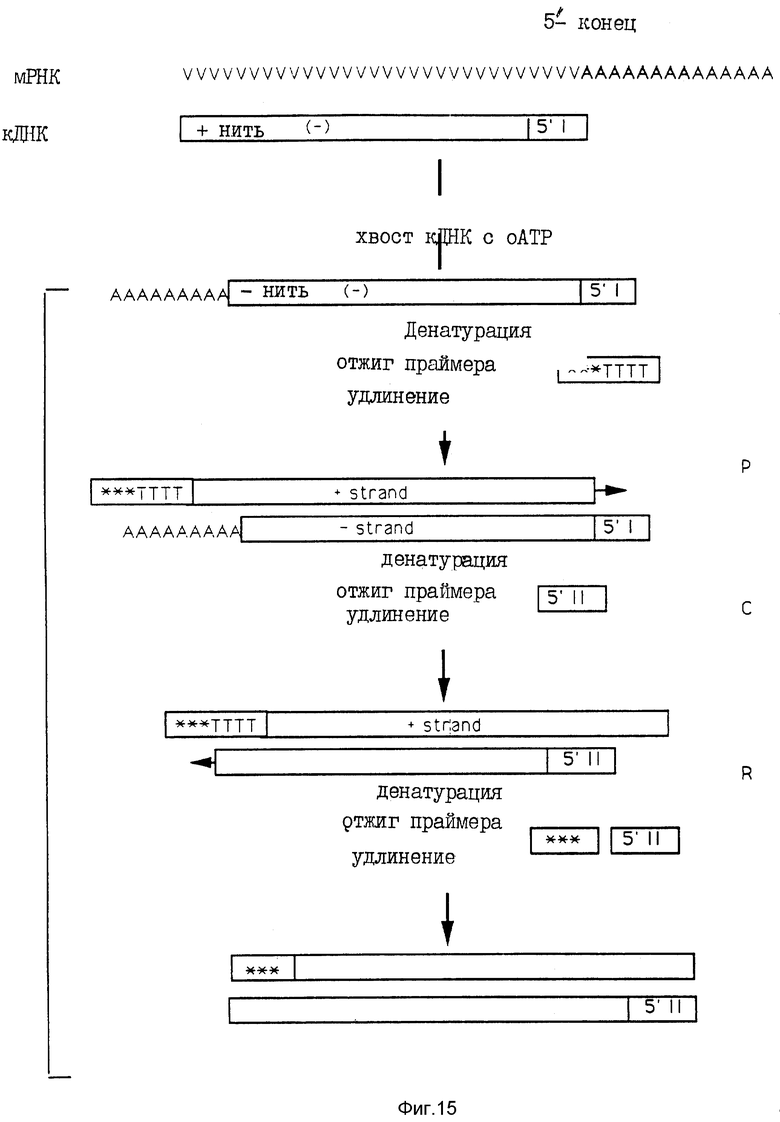
На фиг. 15 схематически представлен протокол RACE для амплификации 5'-концов. На этой фигуре "*** TTTT..." и "***" обозначают адапторный праймер dT17 и адапторный праймер соответственно. Для упрощения в каждой стадии диаграммы показано только, каким образом используется новый продукт, полученный в предыдущей стадии.
Пример 1
Антитромбиновый препарат получали из пиявки Hirudinaria manillensis в соответствии с WO 90/05143, согласно описанным ниже процедурам (а) и (b).
a) Экстракция ацетоном
Осушенные этанолом головы пиявок (2920 г) мелко измельчали и обрабатывали смесью (40:60) ацетона и воды (7,5 л). После гомогенизации путем размешивания при комнатной температуре смесь центрифугировали в течение 15 мин при 2700 об/мин, супернатант сливали, а полученный осадок ресуспендировали в смеси (40: 60) ацетона и воды, размешивали в течение 30 минут и полученную смесь центрифугировали в течение 15 минут при 2700 об/мин. Супернатант собирали и объединяли, а затем подкисляли ледяной уксусной кислотой до pH 4,5 (объем 8,5 л). Полученную смесь центрифугировали 15 минут при 2700 об/мин, после чего супернатант декантировали, и pH раствора доводили до pH 6,0 путем добавления 30% аммиака. После роторного выпаривания при 35oC pH концентрированного раствора понижали до 1,8, осажденные примеси удаляли путем цетрифугирования и сырой антитромбиновый материал осаждали из смеси с использованием 9-кратного избытка ацетона. Затем смесь центрифугировали, осадок ресуспендировали в ацетоне и снова центрифугировали. После этого осажденный материал лиофилизовали.
b) Ионообменная очистка
Сырой антитромбинный материал восстанавливали в воде, диализовали против 10 мМ аммонийацетатного буфера при pH 4,0 и загружали в колонку о карбоксиметиловой сефарозой (СМ Сефароза, Pharmacia, 2,6 х 30 см), предварительно уравновешенную тем же буфером. Затем 100 мл промывали исходным буфером, антитромбин-активные фракции элюировали 20 мМ ацетатом аммония (pH 4,5), собирали и объединяли (1,3 л). Для последующих стадий очистки объединенные фракции концентрировали до 0,5 л в аппарате Минитана (Millipore); концентрированный раствор нейтрализовали NaOH, а затем загружали в колонку с Q-Сефарозой, уравновешенную 20 мМ Трис-HCl (pH 7,0). Связанный материал элюировали линейным градиентом 0 - 1 М NaCl в исходном буфере. Фракции, обладающие антитромбинной активностью, объединяли, концентрировали и обессоливали на колонке Superdex S-200, элюируя 20 мМ Трис-HCl (pH 7,5) при расходе 4 мл/мин. Активный пул из гель-фильтрации концентрировали в аппарате Минитана, а затем очищали посредством слабой ионообменной хроматографии (DEAE FPLC). Активный материал затем загружали в колонку с Protein Pak DEAE-5PW (Waters) и элюировали градиентом 0 - 1 M NaCl в 20 мМ Трио-HCl (pH 6,5), при скорости потока 1,0 мл/мин. Активные фракции объединяли, анализировали на содержание белка и активность (специфическую активность: 800 ATU/мг), и лиофилизовали в концентраторе (Speed Vac, Savants).
Полученный таким образом частично очищенный материал (специфическая активность 800 ATU/мг) затем подвергали двум дополнительным стадиям хроматографии для получения гомогенных полипептидов в соответствии с приведенными ниже описаниями (с) и (d).
с) Тромбин-Сефароза
Коммерческий бычий тромбин (Sigma) был очищен в соответствии с процедурой, описанной Lundblad (*), а затем подвергнут связыванию с активированной Сефарозой CL 6B (Pharmacia) согласно инструкции производителей. Колонку 1,7 мл уравновешивали 50 мМ Трис-HCl (pH 8,3) и загружали лиофилизованный материал, полученный после DEAE-FPLC (восстановленный в буфере). Затем колонку подвергали трехкратному промыванию в исходном буфере, а после этого в том же самом буфере, содержащем 3,0 М NaCl, и снова в исходном буфере (каждая промывка составляла трехкратный объем колонки). Расход составлял 0,3 мл/мин. Связанный материал элюировали 10 мл 0,1 М 4-аминобензамидина в 25 мМ HCl. Активные фракции собирали, объединяли, с буферным ионообменом в 50 мМ Трис-HCl pH 8,3 на колонке PD-10 (Pharmacia).
Несвязанный материал элюировали из колонки путем вымывания в исходном буфере, и материал, еще обладающий антитромбинной активностью, снова загружали в колонку до тех пор, пока весь этот материал не будет связан и хроматографирован.
(*) Lundblad, R.L., 1971, Biochemistry, 10: 2501-2506
(d) ОФ-ВЭЖХ
Материал, полученный после аффинной хроматографии, очищали с помощью обращенно-фазовой высокоразрешающей хроматографии (ОФ-ВЭЖХ) на колонке (C4-Vidak 4,6 х 250 мм, 5 мкм) с использованием 20 мМ фосфата натрия (pH 7,5) в качестве первого элюента, и 50% ацетонитрила в воде, в качестве другого элюента. Антитромбинный полипептид элюировали линейным градиентом от 5% до 55% элюента B в течение 45 минут, при комнатной температуре со скоростью 1,0 мл/мин. Полученная в результате хроматограмма показана на фиг. 1.
Пики белка (обнаруживаемые при 220 нм) собирали вручную, концентрировали в вакууме и снова хроматографировали при тех же условиях.
Очищенные после ВЭЖХ C4 антитромбинные полипептиды анализировали на содержание белка, аминокислотный состав, N-концевую последовательность, C-конец, и их активность, которую определяли с помощью in vitro-анализа (ATU/NIH-тест и тест на определение "тромбинового времени"). В результате этих анализов было обнаружено, что каждый из трех пиков белка обладал значительной антитромбинной активностью.
Полные аминокислотные последовательности полипептидов, помеченные P1 и P2 на фиг. 1, определяли посредством N-концевого секвенирования пептидов, полученных из триптического и V8-протеазного переваров. Эти последовательности соответствуют вышеуказанным последовательностям (i) и (ii). Частичная аминокислотная последовательность другого полипептида (P3) соответствует последовательности, обозначенной выше (iii).
Пример 2: Триптическое переваривание и пептидное картирование пиридилэтилированных (ПЭ) P1 и P2
(a) Восстановление/алкилирование - Активные фракции, очищенные с помощью аффинной хроматографии на Тромбин-сефарозе (пример 1c) объединяли и подвергали буферному ионообмену в 10 мМ Трис-HCl (pH 8,3) на PD-10-колонке. Активный пул (около 50 мкг) концентрировали в центрифуге Speed-Vac (Savant) и обрабатывали 100 мкл 1% b-меркаптоэтанола в 6 М гуанидин-HCl/ 50 мМ Трис-HCl (pH 8,5) в присутствии азота, в темном помещении, в течение 2 часов при комнатной температуре. Затем добавляли 4 мкл 4-винилпиридина (чистого) и смесь инкубировали 2 часа, как указано выше4.
Пиридилэтилированные полипептиды были сначала выделены из реакционной смеси посредством ОФ-ВЭЖХ на C4-колонке (Vydac) (4,6 х 250 мм, 5 мкм), элюированной в течение 90 минут линейным градиентом 5-65% ацетонитрила в 0,1% TFA, со скоростью потока 1,0 мл/мин. При таких условиях смесь антитромбинных полипептидов плохо разделяется, поэтому проводили повторную хроматографию на той же колонке с использованием в качестве элюента смеси фосфата натрия и ацетонитрила в условиях, уже описанных в примере 1d.
(b) Трипсиновое переваривание и пептидное картирование ПЭ-P1 и ПЭ-P2
Очищенные ПЭ-P1 и ПЭ-P2 (10 и 20 мкг соответственно) переваривали TPCK-обработанным трипсином (Sigma) в 200 мкл 1% бикарбоната аммония (pH 8,0) в присутствии 0,2 М фосфата натрия. Трипсин добавляли в отношении фермента к субстрату 1:20 (масс./масс.) и инкубировали в течение 4 часов при 37oC. Переваривание прекращали посредством лиофильной осушки в Savant.
Пептиды, полученные посредством триптического переваривания, отделяли на C18-колонке (мк Bondapak, 3,9 х 300 мм, 10 мк Waters) или на C4-Vydac-колонке (4,6 х 250 мм, 5 мкм), элюируя в течение 60 минут линейным градиентом 5 - 65% ацетонитрила в 0,1% TFA, при скорости потока 1,0 мл/мин (фиг. 2). Элюированные пики собирали вручную, концентрировали в Savant, а затем подвергали анализу на аминокислотную последовательность и N-концевую последовательность на импульсном жидкофазном секвенаторе сер. 477A (Applied Biosystems).
Результаты указанного пептидного картирования трипсинпереваренных ПЭ-P1 (A) и ПЭ-P2 (B) показаны в конце описания.
Пример 3: Химический синтез P2-гена.
Нуклеотидкодирующую последовательность конструировали на основе предпочтительных кодонов5 E.coli. Кроме того, BalI-рестрикционный сайт конструировали в непосредственной близости к 5'-концу синтетического гена для возможности вставки указанной последовательности в различные векторы экспрессии. Фактически, тот же самый синтетический ген использовали для экспрессии рекомбинантного белка P2 в клетках бактерий или насекомого. В случае использования клеток насекомого были разработаны методы, которые позволяли получать белок P2 в виде секретированного или цитоплазматического продукта.
Все манипуляции с плазмидными ДНК осуществляли в соответствии со описанием Маниатиса и др.6
Шесть синтетических комплементарных олигонуклеотидов получали с использованием автоматического синтезатора ДНК (Applied Biosystems) (полученные последовательности показаны на фиг. 3). После ферментного фосфорилирования эти 6 олигонуклеотидов собирали с использованием ДНК-лигазы и полученную двухнитевую последовательность вставляли в M13-фаговый вектор mp18, получая тем самым плазмиду M13-P2, которая показана на фиг. 4. Для того, чтобы иметь возможность вставить P2-ген в M13-вектор, к указанным синтетическим олигонуклеотидам также добавляли Hind III и PstI-сайты. Правильность нуклеотидной последовательности проверяли методом Сэнгера, проведенным на однонитевой фаговой ДНК7.
Рекомбинантную плазмиду M13-P2 использовали в качестве источника P2-гена для всех векторов экспрессии, используемых в примерах.
Пример 4: Экспрессия и секреция P2 из клеток Е.coli.
Для получения секреции в периплазму рекомбинантного продукта необходимо молекулу P2 синтезировать в виде белка-предшественника. В частности, аминокислотная последовательность, называемая "лидерным пептидом", и ответственная за эффективную секрецию, должна присутствовать в NH2-конце P28,9. Эта дополнительная последовательность затем отщеплялась in vivo, в течение секреции, посредством специфической лидерной пептидазы Е. coli, в результате чего получали правильную зрелую последовательность10.
В литературе11,12 описано много примеров секреторных систем. Из них были выбраны системы на основе сигнала, стимулирующего секрецию внешнемембранного белка Е.coli (OmpA) (пред. опубл.13). Для этого были построены два дополнительных комплементарных олигонуклеотида, кодирующие лидерный пептид OmpA, которому предшествовала последовательность Shine - Dalgarno OmpA, известная, как последовательность, ответственная за эффективную трансляцию матричной РНК14.
Эта последовательность, показанная на фиг. 5, также включает в себя начало P2-гена, кодирующего первые 10 аминокислот. Присутствие BalI-сайта позволяет связывать эти два синтетических фрагмента с оставшейся P2-кодирующей последовательностью, тогда как наличие расположенного выше (в направлении 3′ __→ 5′) Hind III-сайта позволяет связывать их с M13-вектором. Так, например, синтетический Hind III-BalI-фрагмент лигировали с BalI-BamHI-отрезком из M13-P2 и вставляли в M13 mp18, в результате чего получали новую плазмиду, обозначенную OMP-P2. Указанная новая плазмида схематически изображена на фиг. 5.
Из OMP-P2 может быть вырезан ген P2 в виде Hind III-BamHI фрагмента, который кодирует OmpA-Shine -Dalgarno и лидерный пептид, за которыми следует P2-кодирующая последовательность. Этот рестрикционный фрагмент уже легко может быть вставлен в соответствующий вектор экспрессии. Теоретически, для получения высоких уровней продуцирования гетерологических белков в бактерии может быть использовано несколько экспрессирующих систем. Ранее14, в лаборатории авторов настоящей заявки была успешно использована система на основе промотора Ptrp. К тому же даже в случае выбранного промотора уровни экспрессии данного полипептида являются практически непредсказуемыми.
Плазмида pFC33, показанная на фиг. 6, уже была описана в литературе14. Эта плазмида несет резистентность к ампициллину и содержит бактериальный промотор Ptrp, который управляет экспрессией проаполипопротеина Al. После переваривания pFC33 посредством Hind III и BamHI большой Hind III-BamHI-фрагмент, несущий ген устойчивости к антибиотикам и промотор, выделяли и лигировали с Hind III-BamHI-фрагментом, полученным из OMP-P2, кодирующего P2-ген. Подробности этого конструирования показаны на фиг. 6. Выделенная новая плазмида, обозначенная pFC-P2, представляла собой конечную плазмиду для продуцирования P2 в Е. coli.
Предметом настоящего изобретения является использование штаммов Е. coli типа B для экспрессии и секреции в периплазму P2 и других антитромбинных полипептидов настоящего изобретения. Действительно, было обнаружено, что инсерция плазмиды pFC-P2 в штаммы бактерии Е. coli типа B позволяет получить высокий уровень продуцирования P2. В связи с этим интересно отметить, что различные типы штаммов Е. coli не работают одинаково эффективно, как это может показаться, поэтому выбор этого штамма хозяина является очень важным для продуцирования буфрудина.
Для продуцирования P2 могут быть использованы несколько штаммов типа B Е. coli. Предпочтительными штаммами являются ATCC 12407, ATCC 11303, NCTC 10537. Ниже приводится пример трансформации штамма NCTC 10537 с использованием плазмиды рFС-P2, и последующего культивирования полученного трансформанта.
Компетентные клетки штамма NCTC 10537 получали посредством процедуры с использованием хлорида кальция, описанной Mandel и Higa 15. Приблизительно 200 мкл препарата указанных клеток (при концентрации 1 • 109 клеток на один миллилитр) трансформировали с использованием 2 мкл плазмидной ДНК (прибл. концентрация 5 мкг/мл). Трансформанты отбирали на чашках с L-агаром, содержащим 100 мкг/мл ампициллина. Две небольшие колонии наносили полосами кончиками деревянной палочки, при этом каждую колонию наносили в виде трех полос длиной 1 см на L-агар, содержащий тот же антибиотик. После 12-часового инкубирования при 37oC части полос анализировали на продуцирование P2-белка путем инокуляции в 10 мл LB-среды (содержащей ампициллин в концентрации 150 мкг/мл) и инкубировали в течение ночи при 37oC. На следующий день культуры разводили 1:100 в среде M9, содержащей аналогичную концентрацию ампициллина, и инкубировали в течение 6 часов при 37oC.
20 мл указанной культуры центрифугировали при 12 000 хг в течение 10 минут при 4oC. Бактериальный осадок ресуспендировали в 2 мл 33 мМ HCl-Трис (pH 8), затем добавляли эквивалентный объем второго объема 33 мМ ЭДТК, 40% сахарозы, и полученную смесь инкубировали в условиях мягкого встряхивания в течение 10 минут при 37oC. После центрифугирования клетки с искусственно увеличенной проницаемостью мембраны ресуспендировали в 2 мл холодной воды и выдерживали на льду в течение 10 минут. Полученный супернатант выделяли путем центрифугирования и получали в результате периплазматическую фракцию бактериальной клетки.
Используя хромогенный анализ, который основан на ингибировании способности тромбина к гидролизу синтетического субстрата S-223816, обнаруживали наличие антитромбиновой активности в периплазматической фракции P2-продуцирующих клеток, тогда как в контрольных периплазматических фракциях эта активность отсутствовала.
Аналогичным образом конструировали новую плазмиду экспрессии/секреции для P2, где вместо промотора Ptrp использовали промотор Plpp/lac 17. Эта плазмида, обозначенная pOMP-P2, показана на фиг. 7. После вставки этой плазмиды в штаммы Е. coli типа B также получали высокие уровни активного P2. В качестве исходной плазмиды для конструирования pOMP-P2 использовали плазмиду pIN-III-OmpA3, описанную Ghrayb и др17. Условия культивирования и индуцирования экспрессии с использованием изопропил-β-D-тиогалактопиранозида (IPTG) также были аналогичными условиям, описанным ранее17.
Пример 5: Экспрессия и секреция белка P2 из клеток насекомого
Для секретирования белка P2 из рекомбинантных клеток насекомого P2-кодирующую последовательность соединяли с лидерным пептидом, который является хорошо распознаваемым этими клетками. Для этой цели использовали лидерный пептид G-белка вируса везикулярного стоматита (VSV)18. Аналогичным образом (см. выше) была получена синтетическая ДНК-последовательность, кодирующая лидерный пептид G-белка VSV, которая располагалась за началом P2-гена; полученная в результате нуклеотидная последовательность представлена на фиг. 8. В этом случае также были сконструированы соответствующие рестрикционные сайты (Hind III, BamHI и BalI), которые позволяют лигировать эту последовательность к оставшейся части P2-гена и вектору экспрессии.
Синтетический фрагмент Hind III-BalI лигировали с очищенным BalI-BamHI-фрагментом из M13-P2, несущего P2-ген, и вставляли в M13mp18, предварительно разрезанную Hind III и BamHI. В результате этого конструирования получали новую плазмиду, обозначенную VSV-P2, которая схематически показана на фиг. 9. Из VSV-P2 вырезали ДНК-фрагмент BamHI-BamHI, несущий P2-ген, лигированный с лидерным пептидом VSV, который затем вставляли в вектор pAcYMl19, как показано на фиг. 10. Полученную плазмиду обозначали pAc-P2.
Для осуществления экспрессии в клетках насекомого необходимо, чтобы P2-кодирующая последовательность (VSV) была перенесена в бакуловирусный геном под транскрипционным контролем промотора полиэдрина. Для этих целей клетки насекомого совместно трансфецировали ДНК бакуловируса дикого типа и вектором-переносчиком pAc-P2. В качестве хозяйских клеток использовали клетки Spodoptera frugiperda. Более подробно эксперимент проводили следующим образом.
Клетки S. frugiperda трансфецировали смесью ДНК инфекционного AcNPV и плазмидной ДНК, которые представляли собой отдельные рекомбинантные векторы-переносчики, с использованием модификации процедуры, описанной Summer и др. 20 Один микрограмм вирусной ДНК смешивали с 25-100 мкг плазмидной ДНК и осаждали 0,125 М (конечная концентрация) хлорида кальция в присутствии 20 мМ HEPES-буфера (pH 7,5), 1 мМ динатрийортофосфата, 5 мМ хлорида калия, 140 мМ хлорида натрия и 10 мМ глюкозы (общий объем 1 мл).
ДНК-суспензию инокулировали в монослой 106 S. frugiperda клеток в чашку с тканевой культурой (35 мм) и выдерживали в течение 1 часа при комнатной температуре для ее адсорбции в клетки, а затем заменяли 1 мл среды. После инкубации в течение 3 дней при 28oC надосадочную жидкость собирали и использовали для продуцирования бляшек в монослое клеток S. frugiperda. Бляшки, содержащие рекомбинантный вирус, идентифицировали на отсутствие в них полиэдроза с помощью оптического микроскопа. Затем из этих бляшек удаляли вирус и после очистки эти бляшки использовали для получения полиэдрин-негативного вирусного продукта.
Описанную выше процедуру использовали для выделения рекомбинантного бакуловируса, геном которого содержал P2-ген под контролем промотора полиэдрина и лидерный пептид белка CVSV. Этот вирус был использован для инфицирования клеток S. frugiperda в соответствии с хорошо известной процедурой20 (с множественностью инфекции 10). Затем инфицированные клетки культивировали в культуру с постоянным перемешиванием (спин-культуру) или в монослоях в присутствии 10% околоплодной сыворотке теленка в соответствии со стандартными способами20. В обоих условиях, S-2238-хромогенный анализ показал наличие антитромбиновой активности в супернатантах культур инфицированных клеток.
Пример 6: Экспрессия белка P2 в цитоплазме клеток насекомого
Белок P2 может быть также продуцирован и аккумулирован в цитоплазме клеток S. frugiperda. Этот способ дает обычно более высокий выход гетерологичных белков, поскольку в нем используются сигналы экспрессии полиэдрина, который является несекретируемым вирусным белком.
Способ настоящего изобретения для получения больших количеств рекомбинантного белка P2 основан на экспрессии гибридного полипептида, где первые 18 аминокислот полиэдрина соединены с сохранением рамки считывания с 64 аминокислотами P2. Присутствие NH2-концевой последовательности полиэдрина позволяет получить высокий уровень экспрессии21. Кроме того, между участком полиэдрина и P2-последовательностью находится метиониновый остаток, который способствует выделению P2-части при обработке гибридного белка CNBr.
Аналогично предыдущим способом сначала получали синтетический ДНК-фрагмент, который позволяет соединять BalI-BamHI-фрагмент из M13-P2 с соответствующим вектором экспрессии. Новый синтетический фрагмент, показанный на фиг. 11, включает в себя также BamHI и BalI-сайты, необходимые для последующих манипуляций.
Затем был получен другой вектор-переносчик, pAcFTl, несущий нуклеотидную последовательность, кодирующую первые 18 аминокислот полиэдрина (фиг. 12). Короче говоря, EcoRV-BamHI-фрагмент pAcYMl19 заменяли синтетическим олигонуклеотидом, содержащим последовательность гена полиэдрина от нуклеотида -92 до нуклеотида +55. Соответствующий BamHI-сайт находится после этой последовательности и использовался для инсерции полной P2-кодирующей последовательности в соответствии со схемой, проиллюстрированной на фиг. 13. С помощью этой конструкции была получена новая плазмида, обозначенная pAcFTl-P2, которая была использована для переноса гибридного гена в бакуловирусный геном.
Рекомбинантный бакуловирус получали в соответствии с описанием в примере 5. Инфицирование клеток S.frugiperda проводили согласно стандартным процедурам20. Культивирование инфицированных клеток насекомого приводило к цитоплазматическому аккумулированию гибридного белка. Этот гибридный белок использовался в качестве источника рекомбинантного белка P2. Для расщепления гибрида посредством CNBr22,23 могут быть использованы несколько способов, описанных в литературе. Применение метода Olson и др. 23 позволяет получить правильную полипептидную последовательность P2. Эта молекула показала антитромбиновую активность.
Пример 7
Для получения Tyr61-варианта P2-белка нуклеотиды 5 и 6, описанные выше в gримере 3 и показанные на фиг. 3, заменяли следующими олигонуклеотидами:
Олиго 5-Tyr
5'-CGAAATCTCAGACTGAAGGTGACTTCGAAGAAATTCCGGACGAATACATCCTG AACTAGTAACTGCA 3'
Олиго 6-Tyr
5'-GTTACTAGTTCAGGATGTATTCGTCCGGAATTTCTTCGAAGTCACCTTCA 3'
В Олиго-Tyr-5, триплет TAC (который подчеркнут) кодирует тирозиновый остаток и заменяет первоначально присутствующий GAC-триплет, кодирующий аспарагиновую кислоту. Олиго-Tyr-6 соответствующим образом корректировали для получения полной комплементарности между двумя нитями. Последующие, стадии, приводящие к экспрессии и/или секреции указанного варианта в клетках насекомого или Е. coli, были уже описаны выше, в примерах 4-6.
Пример 8
Для получения глицин-удлиненного производного P2-белка, описанные выше олигонуклеотиды 5 и 6 (см. пример 3) и показанные на фиг. 3, заменяли следующими олигонуклеотидами:
Олиго 5-Gly
5'-CGAAATCTCAGACTGAAGGTGACTTCGAAGAAATTCCGGACGAAGACATCCTGAAC- GGTTAGTAACTGCA-3'
Олиго 6-Gly
5'-GTTACTAACCGTTCAGGATGTCTTCGTCCGGAATTTCTTCGAAGTCACCTTCA-3'
В олиго-5-Gly, триплет GGT (подчеркнут), который кодирует глицин, вставляли перед стоп-кодоном. Олиго 6-Gly был соответственно корректирован для достижения полной комплементарности двух нитей. Последующие стадии, приводящие к экспрессии и/или секреции Gly-удлиненного производного в клетках насекомого или Е. coli, были уже описаны выше, в примерах 4-6.
Пример 9: Клонирование кДНК P1 и P2
(a) Полную РНК из голов Hirudinaria manillensis получали в соответствии со способом Cathala и др.24
(b) Реакцию обратной транскрипции осуществляли следующим образом:
10 мкг полной РНК, полученной из голов пиявок
1 мкг dT17-адапторного праймера
8 мкл 5 мМ смеси dNTPs
8 мкл AMV-буфера 5X
H2O до 40 мкл
объединяли и держали на льду, затем смешивали и смесь нагревали 2 минуты при 65oC с последующим охлаждением льдом. После этого добавляли 10 ед. Араназина (Promega) и 20 ед. AMV - обратной транскриптазы (Boehringer Mannheim) и инкубировали в течение 2 часов при 42oC. Затем реакционную смесь экстрагировали хлороформом и осаждали изопропанолом.
(c) Затем осуществляли полимеразно-цепную реакцию (PCR). Ниже показана основная схема этой реакции:
PCR-смесь:
5 мкл обратно-транскрибированной РНК
10 мкл 10 х PCR-буфера (Cetus /Perkin - Elmer)
16 мкл dNTPs смеси (1,25 мМ каждого dNTP)
2 мкл MgCl2 0,1 М
25-500 мМ каждого праймера
H2O до 100 мкл
Реакционную смесь денатурировали при 95oC в течение 5 минут, затем добавляли 2,5 ед. Taq-полимеразы (Cetus /Perkin - Elmer) и покрывали сверху 80 мкл минерального масла. Реакцию проводили в термоячейке для реакции ДНК (Cetus /Perkin - Elmer).
Ниже приводятся циклические профили:
3 мин 94oC
2 мин 60oC
2 мин 30 сек 72oC 1 цикл
1 мин 94oC
2 мин 60oC
3 мин 30 сек 72oC 30 циклов
1 мин 94oC
2 мин 60oC
5 мин 72oC 1 цикл
7 мин 72oC
оставляли при 25oC
Остаточную Taq-полимеразу инактивировали осаждением фенолхлороформом и этанолом и образцы хранили при -20oC. Для получения полных последовательностей P1 и P2-кДНК проводили три раунда PCR-амплификации. Последовательности каждого праймера показаны ниже. Положения, в которые была введена в олигонуклеотидную последовательность дегенерация, указаны приведенными ниже праймерной последовательности альтернативными нуклеотидами (N означает, что использовались все четыре нуклеотида). Рестрикционные сайты, добавленные для облегчения клонирования продуктов амплификации, подчеркнуты.
dT17-адапторный праймер:
5' GAC TCG AGT CGA CAT CGA TTT TTT TTT TTT TTT TT 3'
XhoI SalI
Адапторный праймер:
5' GAC TCG AGT CGA CAT CG 3'
XhoI SalI
Праймер 3-8
5' ATC GAA GCT TTA TAG CGA TTG TAG NGA 3'
Hind III C A C C
Праймер 52-56
5' CTA AGG ATC CTT CTT CGA AGT CNCC 3'
BamHI C A A
Праймер 32-37
5'ATC GGA ATT CAG TTC TGG AAA TCA GTG CGT 3'
EcoRI
Праймер 5'I
5' CTA AGA ATT CTT CGC AAC TTA TAT GCG TT 3'
EcoRI
Праймер 5' II
5' ATC GGA ATT CTT AAT TCA ATA TAT CTT CAT 3'
EcoRI
Первый раунд амплификации
500 пМ полностью дегенерированных праймеров, имеющих длину от 3 до 8 остатка и от 56 до 52 остатка аминокислотной последовательности P2, использовали в качестве оппозиционных праймеров в PCR-реакции.
Амплификация 3'-концов кДНК (RACE-протокол), Frohman и др.25
Генный специфический праймер, имеющий длину от 32 до 37 остатка, конструировали на основе нуклеотидной последовательности P2, определенной в первом раунде амплификации. Этот праймер использовали вместе с dT-адапторным праймером для амплификации кДНК (фиг. 14).
Амплификация 5'-концов кДНК (RACE-протокол), Frohman и др.25
10 мкг полной РНК, полученной из голов пиявок, подвергали обратной транскрипции в соответствии с описанной выше процедурой, за исключением того, что (dT17-адапторный праймер заменяли 1 мкг генного специфического праймера (5'I) (фиг. 15). Затем реакционную смесь осаждали изопропанолом и продукты однонитевой кДНК подвергали полиаденилированию у ее 5'-концов с использованием терминальной дезоксинуклеотидилтрансферазы (TdT) следующим образом:
22 мкл кДНК
1 мкл 6 мМ dATP
6 мкл 5X TdT-буфера (BRL)
1,1 мкл TdT (BRL)
Эти образцы инкубировали в течение 10 минут при 37oC и нагревали в течение 16 минут при 65oC. Затем реакционную смесь разводили до 500 мкл дистиллированной воды.
10 мкл полиаденилированного продукта амплифицировали с использованием 10 пМ dT17-адапторного праймера, 25 пМ адапторного праймера и 25 пМ второго ген-специфического праймера, расположенного за (в направлении 3′ __→ 5′) первым специфическим праймером, используемым для транскрипции (5' II, см. фиг. 15).
(d) Анализ PCR-продуктов
Амплифицированные продукты расщепляли в рестрикционных сайтах, присутствующих в каждом праймере. Переваренный продукт подвергали гель-очистке и субклонировали в вектор pUC13, предварительно переваренный теми же рестриктирующими ферментами. Плазмиды, несущие нужную инсерцию, идентифицировали с помощью рестрикционного анализа. Плазмидную ДНК секвенировали с использованием Секвеназы (USB), следуя рекомендациям поставщика. В конце описания представлены полученные таким образом кДНК-последовательности P1 и P2 и выведенные аминокислотные последовательности (лидерные последовательности подчеркнуты).
Источники информации
1) Markwardt, F. 1970, Methods in Enzymology, 19, p. 924.
2) Markwardt, F. 1985, Biomed. Biochim. Acta. 44, p. 1007.
3) Markwardt, F. Hauptmann, J., Nowak, G., Klessen, C., and Walsmann, P. 1982. Thromb. Haemostasis 47, p. 226.
4) Dupont D., Keim P., Chui A., Bello R. and Wilson K., Derivatizer-Analyser User Bulletin No. 1, Applied Biosystems Inc., 1987.
5) Grosjeans H. and Fiers W. 1982. Gene, 18, p. 199.
6) Maniatis T., Fritsch E.F. and Sambrook J. 1982. Cold Spring Harbor, NY.
7) Sanger, F. , Nicklen, S., and Coulson, A.R. 1977, Proc. Natl. Acad. Sci. USA 74, p. 5463.
8) Blobel G. and Dobberstain B. 1975. J. Cell Biology, 67, p. 83.
9) Pages J.M. 1983, Biochimie, 65, p. 531.
10) Wolfe P.B. 1983. J. Biol. Chem. 258, p. 12073.
11) Talmadge K. , Stahl S. and Gilbert W. 1980. Proc. Natl. Acad. Sci. USA, 77, p. 3369.
12) Oka T., Sakamoto S., Miyoshi K., Fuwa T., Yoda K., Yamasaki M., Tamura G. and Miyake K. 1985. Proc. Natl. Acad. Sci. USA, 82, p. 7212.
13) Henning V., Royer H.D., Teather R.M., Hindennach I. and Hollenberg C.P. 1979. Proc. Natl. Acad. Sci. USA, 76, p. 4360.
14) Isacchi A., Sarmientos P., Lorenzetti R. and Soria M. 1989, Gene 81, p. 129.
15) Mandel M. and Higa A.J. 1970. J. Mol. Biology, 53, p. 154.
16) Krstenansky, J.K., and Mao, S.J.T. 1987. FEBS Lett. 211, p. 10.
17) Ghrayeb J., Kimura H., Takahara M., Hsiung H., Masui Y. and Inouye M. 1984. EMBO Journal 3, p. 2437.
18) Bailey, M.J., McLeod, D.A., Kang, C., and Bishop, D.H.L. 1989. Virology 169, p. 323.
19) Matsuura, Y., Possee, R.D., Overton, H.A. and Bishop. D.H.L. 1987. J. Gen. Virol. 68, p. 1233.
20) Summers, M.D., and Smith, G.E. 1987, Texas Agricultural Experiment Station Bulletin No. 1555.
21) Luckow, V.A. and Summers, M.D. 1988, Virology, 167, p.56.
22) Gross E. 1967. Methods in Enzymology, 11, p. 238.
23) Olson H., Lind P., Pohl G., Henrichson C., Mutt V., Jornvall H., Josephson S., Uhlen M. and Lake M. 1987, Peptides, 9, p. 301.
24) Cathala, G., Savouret, J.-F., Mendez, B., West, B.L., Karin, M., Martial, J.A. and Baxter, J.D. (1983) DNA, 2,4:329-335.
25) Frohman, M.A., Dush, M.K. and Martin, G.R. (1988) Proc. Natl. Acad. Sci. USA 85: 8998-9002.9




Изобретение относится к генной инженерии, а именно к генно-инженерным способам получения антитромбиновых полипептидов, используемых для лечения венозных тромбозов. Получают последовательность ДНК, кодирующую предшественник целевого полипептида, связанный с лидерным пептидом. Последовательность ДНК или встраивают в подходящий вектор экспрессии, содержащий ген устойчивости к антибиотику и бактериальный промотор. Вектором трансформируют Е.соli типа В. Культивируют трансформированный штамм в условиях, необходимых для экспрессии. Выделяют целевой продукт. Или встраивают в вектор переноса и осуществляют контраксфекцию клеток насекомых этим вектором и ДНК бакуловируса дикого типа. Из трансформированных клеток выделяют рекомбинантную вирусную ДНК, содержащую кодирующую предшественник целевого полипептида последовательность ДНК под контролем полиэндринового промотора, которую используют для инфицирования культуры клеток насекомых. Фрагмент ДНК, кодирующий полипептид, обладающий антитромбиновой активностью, имеет установленную нуклеотидную последовательность. Рекомбинантные векторы экспрессии и переноса включают эту нуклеотидную последовательность. Фармацевтическая композиция для лечения сердечно-сосудистых болезней содержит в качестве активного начала эффективную концентрацию рекомбинантного полипептида с антитромбиновой активностью и фармацевтически приемлемый носитель или разбавитель. Использование изобретения позволяет повысить эффективность лечения венозных тромбозов. 9 с.п. ф-лы, 15 ил.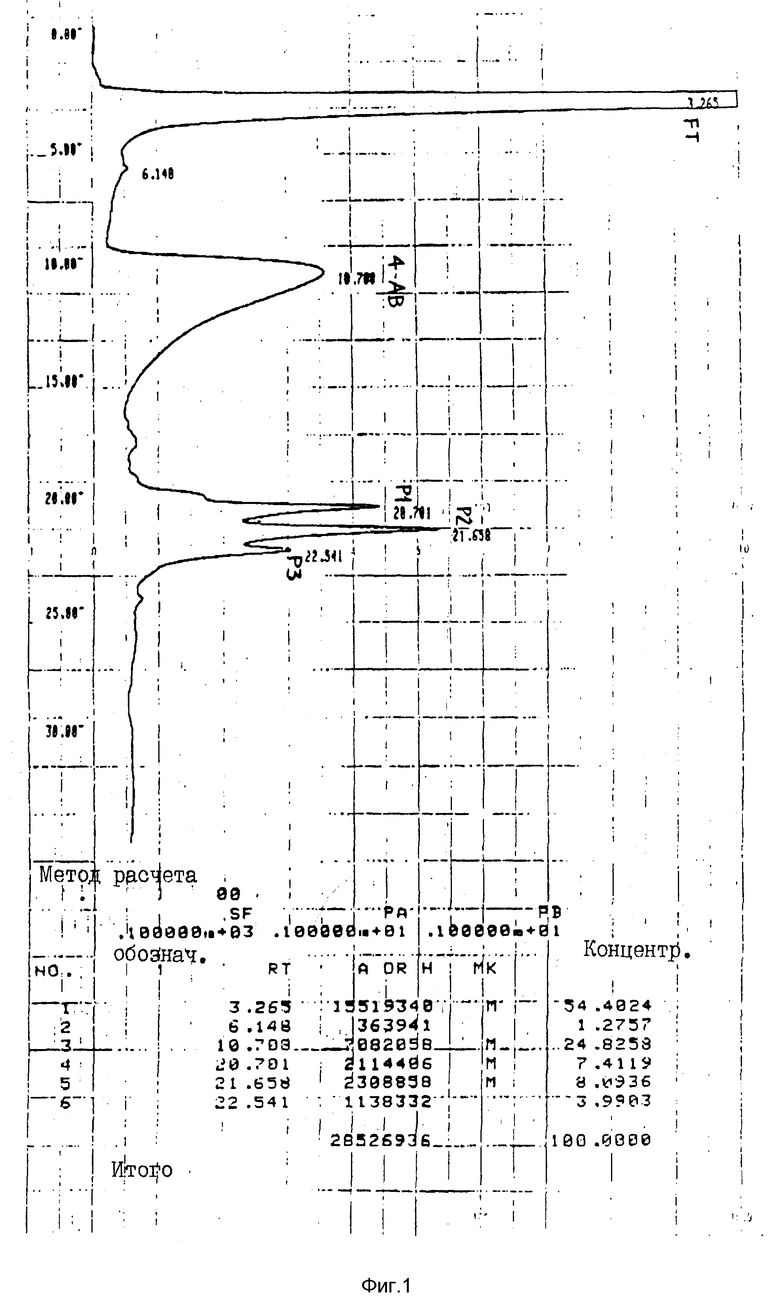
GTTTCTTACACCGACTGCACCGAATCTGGCCAGAACTACTGCCTGTGCGTTGGTTCTAACGTTTGCGGTGAAGGTAAAAACTGCCAGCTGTCTTCTTCTGGTAACCAGTGCGTTCACGGTGAAGGTACCCCGAAACCGAAATCTCAGACTGAAGGTGACTTCGAAGAAATTCCGGACGAAGACATCCTGAACTAG.
| 0 |
|
SU158564A1 | |
| ФОРМИРОВАТЕЛЬ СТУПЕНЧАТОГО СИГНАЛА | 0 |
|
SU352387A1 |
| Способ получения вектора,имеющего нуклеотидную последовательность,кодирующую протеин | 1979 |
|
SU1205777A3 |
