Настоящая заявка является частичным продолжением заявки США рег. N 08/006423, поданной 19 января 1993 г., которая полностью вводится в настоящее описание посредством ссылки и которая, в свою очередь, является продолжением заявки рег. N 07/907261, поданной 1 июля 1992 г. Настоящая заявка является также частичным продолжением заявок США рег. N 07/996455, поданной 24 декабря 1992 г., и N 08/029819, поданной 11 марта 1993 г., обе из которых полностью вводятся в настоящее описание посредством ссылки.
Настоящее изобретение относится к соединениям, обладающим противоопухолевой активностью. Кроме того, настоящее изобретение относится к промежуточным соединениям, используемым для получения соединений, обладающих противоопухолевой активностью.
Впервые, таксол был выделен из коры ствола тихоокеанского тиса, Taxus brevifolianutt (Tаxaceae), и его структура может быть представлена следующим образом (с указанием 2-, 7-, 10- и 13-положений):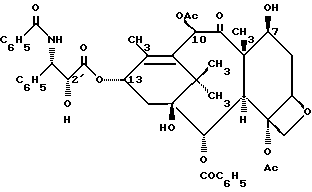
Клинические испытания таксола, финансируемые Национальным онкологическим институтом (NCI), дали многообещающие результаты при лечении запущенных случаев рака яичника, молочной железы и других видов рака. Совсем недавно таксол был апробирован для лечения метастатической карциномы яичника.
По сравнению с другими антимитотическими препаратами таксол является уникальным средством, поскольку он стимулирует сборку стабильных микротрубочек из тубулина даже при прочих неблагоприятных условиях. Связываясь с микротрубочками, таксол стабилизирует их от деполимеризации, нарушая тем самым равновесие "тубулин-микротрубочка" и способствуя ингибированию митоза. Механизм действия, токсикология, клиническая эффективность и другие характеристики таксола рассматриваются в ряде статей, например в статье Rowinsky и др. Taxol: A novel Investigational Antimicrotubule Agent, J. Natl. Cancer Inst. 82, стр. 1247 (1990).
Поскольку было установлено, что это лекарственное средство обладает значительной эффективностью при лечении раковых заболеваний, многие лаборатории приступили к разработке программ по исследованию аналогов таксола в целях поиска наилучших фармакологических профилей. При осуществлении одной из таких программ был получен тасотер формулы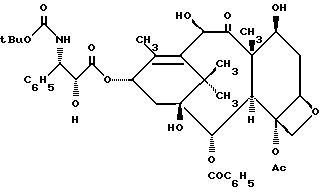
который, как указывалось, обладал такой же, как таксол, эффективностью при стимулировании сборки микротрубочек, а его цитотоксичность приблизительно в два раза выше цитотоксичности таксола. См., например, Biologically Active Taxol Analogues with Deleted A-Ring Side Chain Substitutents and Wariable C-2' Configuration, J. Med. Chem., 34, стр. 1176 (1991); Relationshipso between the Structure of Taxol Analogues and Their Antimitotic Activity, J. Med. Chem., 34, стр. 992 (1991).
За последние годы было установлено, что введение фтора в фармакологически активные соединения приводит к значительным и неожиданным результатам. Для полного ознакомления с успехами в области получения биологически активных фторорганических соединений, см., Advances in the Preparation of Biologically Active Organofluorine Compounds, Tetrahedror, 43, N 14, стр. 3123 (1987). Поэтому целью настоящего изобретения является получение фторированных таксолов и их производных.
Настоящее изобретение относится к фторированному производному таксола формулы I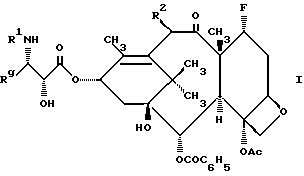
где R1 представляет собой -CORz, в котором Rz является RO- или R;
Rg представляет собой C1-6 - алкил, C2-6 - алкенил, C2-6 - алкинил, C3-6 - циклоалкил, или радикал формулы - W-Rx, в котором W является связью, C2-6 - алкендиилом, или -(CH2)t-, где t = 1-6, а Rx - нафтилом, фурилом, тиенилом или фенилом и, кроме того, Rx может быть необязательно замещенным одной, двумя или тремя одинаковыми или различными C1-6 - алкильной группой, C1-6 - алкоксигруппой, галогеном, или -CF3 - группой;
R2 представляет собой -OCOR, H, PH, -OR, -OSO2R, -OCONRoR, -OCONHR, -OCOO(CH2)tR или -OCOOR, где
R и Ro независимо - C1-6 - алкил, C2-6 - алкенил, C3-6 - циклоалкил, C2-6 - алкинил или фенил, необязательно замещенный 1-3 одинаковыми или различными C1-6 - алкильной группой, C1-6 - алкоксигруппой, галогеном, или -CF3 - группой.
Кроме того, настоящее изобретение относится к фармацевтическим композициям и промежуточным соединениям, используемым для получения фторированных таксолов формулы I. Настоящее изобретение также включает в себя способ лечения опухолевых заболеваний у млекопитающих, который предусматривает использование соединений формулы I.
Настоящее изобретение относится к фторированному производному таксола формулы I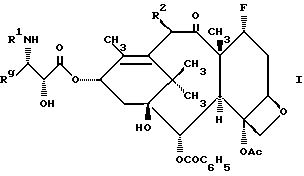
где R1 представляет собой -CORz, в котором Rz - RO- или R;
Rg - C1-6 - алкил, C2-6 - алкенил, C2-6 - алкинил, C3-6 - циклоалкил, или радикал формулы -W-Rx, в котором W является связью, C2-6 - алкенидиилом или -(CH2)t-, где t = 1-6, а Rx - нафтилом, фурилом, тиенилом или фенилом и, кроме того, Rx может быть, но необязательно, замещенным 1-3 одинаковыми или различными C1-6 - алкильной, C1-6 - алкокси-, галогеном или -CF3 - группами;
R2 представляет собой -OCOR, H, OH, -OR, -OSO2R, -OCONRoR, -OCONHR, -OCOO(CH2)tR или -OCOOR, где R и Ro независимо - C1-6 - алкил, C2-6 - алкенил, C3-6 - циклоалкил, C2-6 - алкинил или фенил, необязательно замещенный 1-3 одинаковыми или различными C1-6 - алкильной, C1-6 - алкокси, галогеном или -CF3 - группами.
Синтез фторированного производного формулы I может быть осуществлен различными способами. Приведенные ниже описания синтеза и конкретные примеры его осуществления ставят перед собой лишь иллюстративные цели и не должны рассматриваться как некое ограничение настоящего изобретения какими-либо отдельными способами.
В одном из вариантов осуществления настоящего изобретения соединение формулы I может быть получено способом, проиллюстрированным на схеме 1. Как показано на схеме 1, 2'-гидроксизащищенный таксол формулы II подвергают реакции с трифторидом диэтиламиносеры (DAST), в результате чего получают смесь, состоящую из производного 7 -α- фторотаксола формулы III и производного 8-десметил-7,8-циклопропатаксола (или просто 7,8-циклопропатаксол) формулы XXV (стадия a). Реакция стадии (a) может быть проведена в ряде растворителей, например, таких, как тетрагидрофуран, метиленхлорид, диэтиловый эфир, толуол, 1,1-диметоксиэтан (ДМЭ) и т.п., или в любой их комбинации/смеси. При этом следует отметить, что в том случае, если стадию (a) проводят в смеси ТГФ и диэтилового эфира или в смеси от около 10:1 до 8:1 толуола и тетрагидрофурана, то может быть получено более высокое отношение 7 -α- фторотаксола III к 7,8-циклопропатаксола XXV. В используемом способе R3 обычно является гидроксизащитной группой. Соединение формулы III может быть отделено от соединения XXV, либо эта смесь может быть использована в стадии (b) без разделения, а продукт I может быть отделен от соединения XXVI после стадии (b). Разделение соединений может быть осуществлено любым стандартным способом очистки, обычно используемым в таких случаях специалистами. Примерами способов разделения могут служить хроматография, фракционированная кристаллизация и т. п. Особенно предпочтительным способом разделения является ЖХВД (жидкостная хроматография высокого давления).
Используемые в настоящем изобретении стандартные гидроксизащитные группы (или просто гидроксизащитные группы) представляют собой такие части молекулы, которые могут быть использованы для блокирования или защиты гидроксильных функциональных групп и которые являются хорошо известными каждому специалисту. Предпочтительно использовать такие группы, которые могут быть удалены способами, не оказывающими заметного неблагоприятного воздействия на оставшуюся часть молекулы. Примерами таких легко удаляемых гидроксизащитных групп являются хлороацетил, метоксиметил, 2,2,2-трихлороэтилоксиметил, 2,2,2-трихлороэтилоксикарбонил (или просто трихлороэтилоксикарбонил), тетрагидропиранил, тетрагидрофуранил, дифенилметил, триC1-6-алкилсилил, трифенилсилил и т.п. Другие подходящие защитные группы, которые могут быть использованы в данном случае, описаны в главе 2 "Protecting Groups in Organic Synthesis", Secondi Ed., Theodora W. Greene and Peter G.M. Wuts (1991, John Wiley & Sons). Для соединений формулы II особенно предпочтительный защитной группой является бензилоксикарбонил, которую обычно удаляют путем каталитического гидрирования, или триC1-6алкилсилил, которая может быть удалена с помощью иона фторида.
В другом варианте осуществления настоящего изобретения соединение формулы I2 может быть получено способом, проиллюстрированным в схеме II. В этой схеме, из соединения формулы I1 удаляют (C)-13-боковую цепь путем реакции восстановления с использованием восстановительного агента, такого как борогидрид тетрабутиламмония, в результате чего получают 7 -α- фторо-баккатин III формулы IV (стадия (a)). Затем азетидинон XV подвергают реакции с соединением формулы IV (стадия (b)). Основной класс азетидинонов формулы XV является хорошо известным. Синтез этих соединений или их предшественников описан в заявке на европатент (Holton) 0400971 A2, опубликованной 5 дек. 1990, в заявках на европатент (Holton) 0534709 A1; 0534708 A1 и 0534707 A1, которые все опубликованы 31 марта 1993; а также Ojima и др. в Tetrahedron, 48, N 34, стр. 6985-7012 (1992); Journal of Organic Chemistry, 56, стр., 1681-1683 (1991); и Tetrahedron letters, 33, N 39, стр. 5737-5740 (1992); Brieva и др. , в J. Org. Chem., 58, стр. 1068-1075, и Palomo и др., в Tetrahedron letters, 31, N 44, стр. 6429-6432 (1990); причем все девять указанных работ вводятся в настоящее описание посредством ссылки. Каждому специалисту ясно, что эти способы могут быть соответствующим образом адаптированы для получения других азетидинонов, которые могут быть представлены формулой XV, но которые конкретно не раскрываются ни в данном описании, ни в вышеуказанных девяти работах, ни в каких-либо других работах.
В заявках на европатент 0400971 A2, 0534709 A1, 0534708 A1 и 0534707 A1 и в Tetrahedron, 48, N 34, стр. 6985-7012 (1992) также описываются способы, в которых азетидиноны формулы XV подвергают реакции с (C)13-гидроксигруппой производных баккатина III или его алкоксид металла, в результате чего получают аналоги таксола с различными (C)13-боковыми цепями. В стадии (b) схемы II, превращение гидроксигруппы на (C)13-углероде (помеченном звездочкой) в алкоксид металла предпочтительно осуществлять перед реакцией сочетания. Катион металла указанного алкоксида металла предпочтительно выбирать из металлов группы Ia или IIa. Нужный алкоксид металла может быть получен с помощью реакции соединения формулы IV с сильным металлическим основанием, таким как диизопропиламид лития, C1-6-алкиллитий, бис(триметилсили)амида лития, фениллития, гидрид лития, гидрид натрия, гидрид калия, гидрид лития или т.п. Например, если необходимо получить алкоксид лития, то соединение формулы IV может быть подвергнуто реакции с н-бутиллитием в инертном растворителе, таком как тетрагидрофуран. После удаления R3 из соединения формулы V (в стадии (c)) получают соединение формулы I3. Если R3 является триC1-6алкилсилильной группой, такой как триэтилсилильная группа, то она может быть удалена с использованием иона фторида или минеральной кислоты в спирте или ацетонитриле. Удаление с использованием фторидного иона осуществляют в инертном растворителе, таком как тетрагидрофуран, метиленхлорид, 1,4-диоксан, ДМФ, хлороформ или в аналогичном растворителе; при этом предпочтительно, чтобы реакционная среда была забуферена слабой кислотой, например уксусной кислотой. Присутствие 7,8-циклопропа-производного, если оно имеет место, не оказывает существенного влияния на какую-либо стадию схемы II, при условии, если будет использоваться соответствующее количество реагентов с учетом их потребления, обусловленного присутствием указанного производного. Как правило, бывает предпочтительным отделить 7,8-циклопропа-производное после стадии (a), но перед стадией (b).
В схеме III проиллюстрирован способ получения соединений формулы I5, где R является -OCOR, -OSO2R, -OCONRoR, -OCONHR, -OCOO(CH2)tR или -OCOOR. Исходные соединения формулы XXX хорошо описаны в литературе, или они могут быть получены способами, хорошо известными специалистам. Например, как показано в схеме IIIa, соединение формулы XXXVIII может быть подвергнуто реакции с RC(= O)L, R(CH2)tOC(=O)I, POC(=O)I, LO2R, LCONRoR, LCONHR, O=C=N-R или с их ангидридным производным, где I является обычной уходящей группой, такой как хлоро-, бромо-, мезил, трифторметансульфонил или тозил; и в результате этой реакции образуется соединение формулы XXXIX. Обычно в стадии (a) необходимо использовать основание в целях предварительного удаления протона из C-10-гидроксильной группы. Особенно предпочтительным основанием для стадии (a) является сильное основание, такое как C1-6-алкиллитий, бис(триметилсилил)амид лития, или аналогичное основание, используемое в количестве около 1,1 эквивалента. Депротонирование с помощью основания предпочтительно проводить в аротонном растворителе, таком как тетрагидрофуран, и при низких температурах, обычно в пределах от -40o до 0oC. В стадии (b) соединение формулы XXXIX может быть подвергнуто реакции с азетидиноном формулы XV, в основном таким же образом, как и в стадии (b) схемы II, с получением соединения формулы XI, из которого могут быть удалены группы R3, в результате чего образуется соединение формулы XXX.
Соединение формулы I3, которое принадлежит к ряду соединений формулы I, может быть получено способом, проиллюстрированным на схеме IV. В стадии (a), при обработке соединения VI от 1 до 2 эквивалентами стандартного гидроксизащитного реагента, предпочтительно трихлороэтилхлороформата, может быть получена смесь, одновременно включающая в себя 2'- и 7-гидроксизащитное (соединение формулы XIII) и 2'- и 10-гидроксизащищенное (соединение формулы VII) производные таксола.
Соединение формулы XIII затем подвергают реакции с 1,1,2-трифторо-2-хлоротриэтиламином (стадия (b)) и получают диенеон формулы VIII. В стадии (c) защитные группы R3 удаляют. (Удаление трихлороэтилоксикарбонильной группы может быть осуществлено с использованием цинкового порошка в уксусной кислоте.) В стадии (d) диен соединения формулы IX подвергают каталитическому гидрированию, в результате чего получают соединение формулы X. После этого в стадии (e) 2'-гидроксигруппу еще раз блокируют, на этот раз предпочтительно бензилоксикарбонилом, и получают в результате соединение формулы XI. После обработки этого соединения формулы XI соединением DAST получают фторосоединение формулы XII. Затем защитные группы R3 (стадия (g)) удаляют и получают в результате соединение формулы I3.
В схеме V проиллюстрирован способ получения соединения формулы I4, которое, кроме того, принадлежит к соединениям формулы I. В cтадии (a) соединение формулы VII подвергают реакции cDAST, в результате чего получают соединение формулы XIV. После удаления защитных групп R3 получают соединение I4.
Соединение формулы VI является либо уже известным, либо оно может быть легко получено способом, показанным на cхеме VI. В этой cхеме cтадия (a) в основном идентична cтадии (b) cхемы II. Производное баккатина III формулы XXXIII с защитными группами на 7- и 10-гидроксигруппах также является либо уже известным соединением, либо оно может быть легко получено из 10-диацетилбаккатина III. См., например, заявки на Европатент 0 253 738 A1 и 0 522 958 A1, опубликованные 20 января 1988 г. и 13 января 1993 г. соответственно. После удаления гидроксизащитных групп в стадии (b) получают соединение формулы VI.
В настоящей заявке, числовые индексы, следующие после символа "C", указывают на число атомов углерода, которое может содержать данная группа. Например, понятие "C1-6-алкил" включает в себя алкильные группы с прямой или разветвленной цепью, имеющие от 1 до 6 атомов углерода и представляющие собой метил, этил, н-пропил, изопропил, н-бутил, т-бутил, н-пентил, н-гексил, 3-метилпентил или аналогичные алкильные группы; понятие "C2-6-алкенил" включает в себя прямые или разветвленные алкенильные группы, такие как винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, металлил, 1,1-диметилаллил, 1-гексенил, 2-гексенил или аналогичные группы; понятие "C3-6-циклоалкил" включает в себя цилкопропил, циклобутил, циклопентил или циклогексил; понятие "C2-6-алкинил" включает в себя прямые и разветвленные алкинильные группы, такие как этинил, пропаргил (2-пропинил), 1-пропинил, 2-бутинил, 3-бутинил, 1-гексинил, 4-метил-2-пентинил и аналогичные группы; понятие "C2-6-алкениил" включает в себя такие группы, как этилен-1,2-диил(винилен), 2-метил-2-бутен-1,4-диил, 2-гексен-1,6-диил и аналогичные группы; понятие "C1-6-алкилокси(алкокси) включает в себя прямые или разветвленные алкилоксигруппы, такие, как метокси, этокси, н-пропокси, i-пропокси, н-бутокси, т-бутокси (т-бутилокси), н-пентилокси, н-гексилокси или 3-метилпентилокси и т. п.; а понятие "галоген" включает в себя фтор, хлор, бром или иод. Термин "азетидинон" относится к азетидин-2-ону. В настоящем описании все однажды определенные символы сохраняют свое значение до тех пор, пока им не будет дано новое определение.
Ниже представлены конкретные примеры, которые иллюстрируют синтез характерных соединений настоящего изобретения, но при этом не ограничивают объема настоящего изобретения или области, к которой оно относится. Описанные в примерах методы могут быть адаптированы для получения соединений, входящих в объем настоящего изобретения, но конкретно не раскрываемых в данных примерах. Кроме того, каждому специалисту ясно, что те же самые соединения могут быть получены с использованием некоторых модификаций описанных способов.
Все указанные в примерах температуры даны в градусах Цельсия, если это не оговорено особо. Специальные характеристики, полученные методом ядерного магнитного резонанса (ЯМР), относятся к химическим сдвигам (δ), выраженным в миллионных долях (м.д.) и измеренным по отношению к сигналу тетраметилсилана (ТМС), который использовали в качестве эталона. Относительные области, указанные для различных сдвигов в спектральных данных протонного ЯМР, соответствуют числу атомов водорода определенного функционального типа в данной молекуле. Природа сдвигов в отношении их мультиплетности описывается как: широкий синглет (ш.с), широкий дублет (ш.д), широкий триплет (ш.т.), широкий мультиплет (ш.м.), широкий квартет (ш.кв.), синглет (с), мультиплет (м), дублет (д), квартет (кв.), триплет (т), двойной дублет (дд), двойной триплет (дт) и двойной квартет (д.кв). Для снятия ЯМР-спектров в качестве растворителей использовали ДМСО-d6 (пердейтеродиметилсульфоксид), D2O (тяжелая вода), CDCl3 (дейтерохлороформ) и другие стандартные дейтерированные растворители. Сокращение "Обмен" означает обмениваемый на CD3OD (например, "д + обм. " означает "дублет + обмениваемый сигнал. Полный сигнал коллапсирует именно в дублет, после того как был обменен протон). "Включ." означает "включая".
Описание инфракрасных (ИК) спектров включает в себя лишь абсорбционные волновые числа (см-1), являющиеся показателем идентификации функциональных групп.
Целит представляет собой зарегистрированную торговую марку (Корпорации: Johns - Manville Products Corporation) диатомовой земли.
Используемые в настоящем описании аббревиатуры являются стандартными сокращениями, обычно используемыми в специальной литературе. Ниже приведен список некоторых из таких аббревиатур:
Ac - ацетил;
Ar - арил;
Bz - бензоил;
Cbz - бензилоксикарбонил;
DCl - десорбционная химическая ионизация;
DMF - диметилформамид (ДМФ);
DMSO - диметилсульфоксид (ДМСО);
FAB - бомбардировка быстрыми атомами;
h - часы, ч;
HPM - масс-спектроскопия высокого разрешения (МСВР);
i-PrOH - изопропиловый спирт;
min - минуты (мин);
MS - масс-спектрометрия;
NOBA - м-нитробензиловый спирт;
Ph - фенил;
rt - комнатная температура;
tBu - третичный бутил (т-бутил);
TES - триэтилсилил;
THF - тетрагидрофуран (ТГФ);
tIc - тонкослойная хроматография (ТСХ);
v/v - объем/объем;
V - выход.
В Таблицах 1 и 2 перечислены некоторые соединения, синтез которых описан в представленных ниже Примерах.
Пример 1. 2'-O-(Бензилоксикарбонил)таксол (IIa).
К размешанному раствору таксола (150 мг, 0,176 мМ) и N,N-диизопропилэтиламина (93 мкл, 0,534 мМ, 3 экв.) в безводном CH2Cl2 (4 мл) при комнатной температуре добавляли бензилхлороформат (75 мкл, 0,525 мМ, 3 экв.) при комнатной температуре. Реакционную смесь размещали при комнатной температуре в течение 3 часов. После этого смесь концентрировали до получения объема 2 мл, а остаток очищали на колонке с силикагелем, используя смесь (1:1) EtOAc/гексана в качестве элюента, в результате чего получали 150 мг (0,152 мМ, выход 86%) целевого соединения IIa, в виде белого порошка: т.пл. 140-150oC (разлож. ) [α]
МСВР (масс-спектроскопия высокого разрешения) для C55H58NO16 (MH+: вычислено 988,3756; найдено 988,3766
Элементный анализ для C55H57NO16 • H2O: вычислено: C 65,67, H 5,92; N 1,40; найдено: C 65,99; H 5,64; N 1,33.
Пример 2. 2'-O-Бензилоксикарбонил-7- -α- фторотаксол IIIa.
DAST (18,7 мкл, 0,141 мМ) растворяли в сухом дихлорметане (0,5 мл), и полученный раствор охлаждали до 0oC. Затем добавляли раствор соединения IIa (71 мг, 0,072 мМ) в дихлорметане (1 мл), и полученный раствор выдерживали 30 минут при 0oC и 4 часа при комнатной температуре. После этого реакцию гасили путем добавления к реакционной смеси воды (0,15 мл) и смесь концентрировали. Остаток хроматографировали на колонке с силикагелем (элюируя 40% этилацетатом в гексане), в результате чего получали 61 мг смеси соединения IIIa и 2-O-бензилоксикарбонил-8-десметил-7,8-циклопропатаксола XXVa; 1H-ЯМР (смеси IIIa и XXVa, CDCl3): δ 8,08 (д, J = 8,7 Гц, 2H); 7,65 - 7,17 (м, 18H): 6,85 (обмен. д, J = 9,4 Гц, 1H); 6,49 (с, 1H, H-10); 6,25 - 6,14 (м, 1H, H-13); 5,92 (дд, J = 9,4 Гц, J = 2,4 Гц); 1H, H-3; 5,68 (д, J = 7,2 Гц, 1H, H-2); 5,38 (м, H, H-2); 5,06 (м, 2H); 4,96 (ш.д, 1H, H-5); 4,80 - 4,35 (М, 1H, H-7); 4,31 - 4,20 (м, 2H, H-20); 3,94 (д, J = 7,2 Гц; 1H, H-3); 2,47 - 1,64 (м, 17H, включ. с. при 2,38, 3H, при 2,11, 3H, при 1,78, 3H, 1,65, 3H); 1,10 (c, 3H); 1,07 (с, 3H).
Пример 3. 7 -α- Фторотаксол (1a).
Смесь (1: 1) соединения IIIa и 2'-O-бензилоксикарбонил-8-десметил-7,8-циклопропатаксола (89 мг) растворяли в этилацетате (3 мл), и получаемую смесь слегка размешивали при давлении водорода в 1 атм и в присутствии палладированного угля (10% Pd, 29 мг, 0,027 мМ). Через 12 часов растворитель удаляли, а остаток очищали с помощью хроматографии на сигикагеле, элюируя смесью 40% этилацетата в гексане, в результате чего получали 67,7 мг целевого соединения вместе с 8-десметил-7,8-циклопропатаксолом (XXXIa), в виде белого твердого продукта;
1H-ЯМР (смеси XXVa и Ia, CDCl3) δ 8,11 (д, J = 8,7 Гц, 2H); 7,72 - 7,07 (м, 14H); 6,50 (с, 1H, H-10); 6,14 (ш.т, H-13); 5,80 (дд, J = 9,0 Гц, J = 2,4 Гц, 1H, H-3); 5,74 (д, J = 7,2, 1H, H-2) 4,98 (д, J = 8,1 Гц, 1H, H-5); 4,77 (м, 1H, H-2); 4,70 - 4,40 (м, 1H, H-7); 4,40 - 4,21 (м, 2H, H-20); 4,02 (д, J = 7,2 Гц, 1H, H-3); 2,60 - 1,55 (м, 17H, включ. с при 2,37, 3H, 2,20, 2H, 1,77, 3H, 1,74, 3H); 1,14 (с, 4H); 1,12 (с, 3H).
Для разделения 7 -α- фторотаксола и 8-десметил-7,8-циклопропатаксола были использованы следующие методы ЖХВД:
Метод I:
Оснащение:
Насос: PE Серии 4
Колонка: Shandon Hypercarb (графитированный углерод), 7 мкм, 100 • 4,6 мм, 59864750 (информация относительно размеров препаративной колонки может быть получена из Keyst one Scientific Bellefonte, PA)
Инжектор: PEISS - 100
Детектор: HP-1040M
Условия:
Подвижная фаза: метиленхлорид: гексан 85 : 15. Разделение не дает потерь при смеси метиленхлорид : гексан : изопропиловый спирт 80:19:1
Скорость потока: 2,5 мл/мин
Детектор: 254 нм
Разбавитель: образец, растворенный в метиленхлориде
Метод 2.
При использовании колонки (30 см x 2,5 см) для препаративной ЖХВД FYNMAX-60A (Si 83.121-C) с этилацетатом и гексаном (1:1) в качестве элюента и скоростью потока 10 мл/мин время удерживания для 7 -α- фторотаксола составляло 15,59 мин, тогда как время удерживания для 8-десметил-7,8-циклопропатаксола составляло 16,65 мин.
Пример 4. -α- Фторотаксол (Ia)
Соединение IIa (258 мг, 0,26 мМ) растворяли в ТГФ (1,7 мл) и диэтиловом эфире (3,4 мл), полученный раствор охлаждали до -78oC. К этому раствору добавляли DAST (69 мкл, 0,52 мМ), смесь размешивали 30 минут при -78oC, а затем в течение ночи при комнатной температуре. Реакцию гасили путем добавления воды (0,3 мл), а реакционную смесь концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесь 30% этилацетата в гексане, и получали 87 мг (выход 33,7%) 2'-O-бензилоксикарбонил -7α- фторотаксола (IIIa) в виде аморфного твердого вещества. 1H-ЯМР-спектр был в основном идентичен спектру, указанному в Примере 2; 19F-ЯМР (CDCl3) ⊘ (VS, CF3COOH) 90 (ддд, JF,H7 = 49,6 Гц, JF,H6 = 40,1 Гц, JF,H6 = 21,6 Гц).
После удаления 2'-O-бензилоксикарбонильной группы, как описано в примере 3, получали целевое соединение с выходом 87%, 1H-ЯМР подтвердили предполагаемую структуру. MCBP для MH+: вычислено 856,3344; найдено: 856,3367.
Пример 5. N-Дебензоил-N-бутоксикарбонил-2'-O-триэтилсилил-7- -α- фторотаксол (Va).
Смесь 7 -α- фторотаксола и 2'-O-бензилоксикарбонил-8-десметил- 7,8-циклопропатаксола (572 мг, 3: 2 - смесь) обрабатывали борогидридом тетрабутиламмония (286 мг, 1,111 мМ) в сухом дихлорметане (7 мл) при комнатной температуре. Избыточный борогидрид нейтрализовали уксусной кислотой (0,4 мл); а растворитель выпаривали, в результате чего получали неочищенный продукт. Этот продукт очищали на колонке с силикагелем, элюируя 50% этилацетатом в гексане, и получали 271 мг смеси 7 -α- фторобаккатина III (IV) и 8-десметил-7,8-циклопропабаккатина III (XXVII) в виде белого пенистого продукта. ЯМР - спектр соответствовал нужной структуре.
Раствор смеси соединения IV и 8-десметил-7,8-циклопропабаккатина III (130 мг) в сухом ТГФ (1 мл) охлаждали до -40oC, и по капле в присутствии аргона добавляли н-бутиллитий (1,63 М в гексане, 0,164 мл, 0,260 мМ). Через 15 минут добавляли раствор 1-т-бутоксикарбонил-(3R,4S)-цис-3-триэтилсилилокси-4-фенилазетидинона (XVa) (203 мг, 0,530 мМ) в сухом ТГФ (0,5 мл), и полученную смесь нагревали до 0oC. Затем реакцию оставляли на 90 минут при 0oC, после чего ее гасили насыщенным водным хлоридом аммония. Реакционную смесь экстрагировали этилацетатом. Этилацетатный слой осушали, фильтровали и концентрировали в вакууме, в результате чего получали неочищенный маслянистый продукт. Этот продукт очищали с помощью хроматографии на силикагеле, элюируя 40% этилацетатом в гексане, и получали 143 мг смеси целевого соединения и N-дебензоил- N-т-бутоксикарбонил-2'-O-триэтилсилил-8-десметил-7,8-циклопропатаксола (XXVIIIa) в виде белого пенистого продукта; 1H-ЯМР (смеси XXVIIIa и Va, 300 МГц, CDCl3) δ 8,14 (д, 2H); 7,45-7,17 (м, 8H); 6,56 (с, 0,6H, H-10); 6,32 (с, 0,4H, H-10, H-10); 6,28 (м, 1H, H-13); 5,72 (д, 0,6H, H-2); 5,44 (м, 1H, H-3); 5,28 (обм.м. 1H, N-H); 5,00 (д, 1H, H-5); 4,70-4,45 (м, 1H, H-7); 4,50 (ш.с., 1H, H-2); 4,40-4,35 (м, 2H, H-20) 4,05 (д, 1H, H-3); 2,63-1,15 (м, 32H); 0,73 (м, 9H); 0,34 (м, 6H).
Пример 6. N-Дебензол-N-т-бутоксикарбонил-7 -α- фторотаксол (Ib).
К раствору, содержащему смесь соединения Va и N-дебензоил-N-т- бутоксикарбонил-2'-O-триэтилсилил-8-десметил-7,8-циклопропатаксола (100 мг) в ацетонитриле (1 мл) при -5oC добавляли водную HCl (0,0192 мл, 0,30 мМ, 36% - раствор). Реакционную смесь размешивали в течение 10 минут, а затем разбавляли этилацетатом (1,5 мл). Органическую фазу промывали водой, осушали, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя 40% этилацетата в гексане, в результате чего получали 73 мг смеси целевого продукта и N-дебензоил-N-т-бутоксикарбонил-8-десметил-7,8-циклопропатаксола (XXIXa), в виде пенистого вещества;
1H-ЯМР (смеси Ib и XXIXa, 300 МГц, CDCl3) δ 8,11 (м, 2H); 7,60-7,22 (м, 8H); 6,50 (с, 0,6H, H-10); 6,30 (с, 0,4H, H-10); 6,22 (м, 1H, H-13); 5,72 (д, 0,6H, H-2); 5,61 (д, 0,4H, H-2); 5,50-5,42 (м, 1H, H-3'); 5,28 (обм.ш.д, 1H, N-H); 5,00 (д, 1H, H-5); 4,70-4,40 (м, 1H, H-7); 4,60 (ш.с, 1H, H-2'); 4,40-4,23 (м, 1H, H-20); 4,20 (д, 1H, H-3); 3,40 (обм. ш.с., 1H, O-H); 2,65-1,10 (м, 32H). МСВД для МH+: вычислено 852,3607; найдено 852,3604.
Пример 7. 7 -α- Фторобаккатин III (IV).
В сухую колбу в инертной атмосфере добавляли 2'-O-(бензилоксикарбонил)таксол (IIa) (4 г, 4 мМ) и сухой толуол (80 мл). Полученную суспензию размешивали при температуре окружающей среды, добавляя при этом по капле сухой тетрагидрофуран (16 мл) до тех пор, пока не получали бесцветный раствор. Этот раствор охлаждали до -78oC в бане из сухого льда и ацетона, а затем обрабатывали трифторидом диэтиламиносеры (DAST, 1,2 мл, 2,5 экв. ). Реакционную смесь размешивали в течение 16 часов, и в процессе этого перемешивания смесь постепенно нагревалась до температуры окружающей среды. Полученную суспензию фильтровали, а фильтрат (разведенный этилацетатом (30 мл)) промывали насыщенным водным дикарбонатом натрия, а затем солевым раствором. Органическую фракцию осушали (MgSO4) и концентрировали, в результате чего получали неочищенный продукт в виде белого пенистого вещества. Этот продукт частично очищали с помощью колоночной хроматографии на силикагеле, элюируя 10% CH3CN в CH2Cl2, и получали 1,45 г смеси 7 -α- фторо-производного IIIa и 7,8-циклопропа-аддукта XXVa (смесь 82:18, как было установлено с помощью 1H-ЯМР).
Вышеуказанную смесь (1,45 г) растворяли в этилацетате (60 мл) и обрабатывали палладированным углем (300 мг). После встряхивания в течение 4 часов при давлении водорода 50 фунт/кв.дюйм (344,7 кПа) реакционную смесь вентилировали (т. е. , удаляли газы) и фильтровали через небольшой слой из силикагеля, а затем концентрировали. В результате этой процедуры получали целевой продукт в виде белой пенистой смеси (1,24 г, выход 99%, согласно 1H-ЯМР, смесь имела состав 90:10). После этого смесь 7 -α- фторо- и 7,8-циклопропатаксола растворяли в метиленхлориде (30 мл) и обрабатывали борогидридом тетрабутиламмония (745 мг, 2,9 мМ, 2 экв.) и размешивали в течение 6 часов. Затем реакцию гасили уксусной кислотой (1 мл), разводили дополнительным количеством метиленхлорида (30 мл) и промывали насыщенным водным раствором бикарбоната натрия. Органическую фракцию осушали сульфатом магния и концентрировали. Неочищенный остаток (маточную смесь замещенного таксана) частично очищали с помощью колоночной хроматографии на силикагеле, элюируя 10% CH3CN в CH2Cl2, и получали целевой баккатин III (50 мг, 60%) в виде 90:10 - смеси (как было определено с помощью 1H-ЯМР) 7 -α- фторо- и 7,8-циклопропа-аналогов виде белого пенистого продукта. Этот продукт кристаллизовали из горячего изопропанола, и получали 7 -α- фторобаккатин III (IV) в виде небольших белых игольчатых кристаллов (выход: 410 мг); т.пл. 234-236oC (разлож.); 1H-ЯМР (300 МГц, CDCl3): δ 8,14 (д, 2H, J = 6 Гц); 7,65-7,52 (м, 1H); 7,52-7,49 (м, 2H), 6,57 (с, 1H); 5,72 (д, 1H, J = 9 Гц); 5,03 (д, 1H, J = 9 Гц); 4,86-4,79 (м, 1H); 4,55 (дд, C-7 протон 1H, J = 3,9, JH-F=47,1 Гц); 4,36 (A ABкв., 1H, J = 7,8 Гц); 4,27 (B ABкв., 1H, J=7,8 Гц); 4,12 (д, 1H, J = 6,9 Гц); 2,60-2,48 (м, 2H); 2,30-1,07 (м, 22H, включая синглеты при 2,30; 2,21; 2,08; 1,77; 1,58; 1,13; 1,07).
Пример 8. Получение 1-т-бутоксикарбонил-(3R,4S)-цис-3- триэтилсилилокси-4-фенилазетидинона (XVa) (Схема VII).
Гидрохлорид (L) - треонинметилового сложного эфира (1,26 г, 7,44 мМ) в безводном дихлорметане (15 мл) размешивали с имидазолом (1,01 г, 14,89 мМ) и т-бутоксидифенилсилилхлоридом (2,274 г, 7,816 мМ) при комнатной температуре в течение 16 часов. Реакционную смесь распределяли между водой и дихлорметаном. Органическую фазу промывали 5% водным бикарбонатом натрия и водой, осушали, концентрировали и получали 2,88 г неочищенного маслянистого продукта, который непосредственно использовали в следующей стадии; 1H-ЯМР (CDCl3) δ 7,70-7,25 (м, 10H); 4,40 (м, 1H); 3,62 (с, 3H); 3,31 (д, J = 3 Гц, 1H); 2,12 (ш.с. 2H); 1,3-1,15 (м, 12H).
Полученный маслянистый продукт (548 мг, 1,414 мМ) в безводном дихлорметане (10 мл) обрабатывали бензальдегидом (0,158 мл, 1,55 мМ) при комнатной температуре и в присутствии 4A-молекулярного сита, и получали в результате соединение формулы XVIIa in situ. После охлаждения раствора, содержащего соединение XVIIa, до -40oC добавляли триэтиламин (0,20 мл, 1,698 мМ), а затем добавляли ацетоксиацетилхлорид (XVIa) (0,182 мл, 1,698 мМ) в течение 10 минут. Смесь оставляли на 4 часа для нагревания до комнатной температуры, после чего продукт распределяли между дихлорметаном и водой. Затем органическую фазу промывали водой и солевым раствором, осушали и концентрировали. После хроматографии на силикагеле (элюирование смесью EtOAc/гексана = 1:4) получали 411 мг соединения XVIIIa в виде прибл. 10:1 - смеси 3R, 4S:3S,4R - диастереомеров.
Полученную смесь диастереомеров (245,51 мг, 0,414 мМ) в сухом ТГФ (2 мл) обрабатывали уксусной кислотой (0,15 мл) и фторидом тетрабутиламмония (ФТБФ, 1М в ГТФ, 1,20 мл). Раствор размешивали 14 часов при комнатной температуре, а затем распределяли между этилацетатом и 5% водным бикарбонатом натрия. Органическую фазу осушали и концентрировали. После флеш-хроматографии на силикагеле (с использованием в качестве элюента смесь (1:1) этилацетата и гексана) получали 66 мг (выход 50%) соединения XIXa (один диастереомер) в виде пенистого вещества);
1H-ЯМР (CDCl3) δ: 7,42 - 7,25 (м, 5H); 5,90 (д, J=4,8 Гц, 1H); 5,09 (д, J= 4,8 Гц, 1H); 4,28 (м, 1H); 4,01 (д, J=4,8 Гц, 1H); 3,70 (с, 3H); 1,73 (с, 3H); 1,19 (д, J=6,6 Гц, 3H).
Соединение формулы XIXa (9,8 г, 0,0305 М) в сухом дихлорметане (100 мл) обрабатывали при -78oC триэтиламином (9,40 мл 0,0671 М) и метансульфонилхлоридом (MSCl, 3,50 мл, 0,0457 М). Раствор оставляли на ночь для нагревания до комнатной температуры. После этого реакционную смесь распределяли между собой и дихлорметаном. Органический слой промывали 5% водным бикарбонатом натрия, разбавляли водной HCl, водой и солевым раствором, и концентрировали, в результате чего получали соединение XXa в виде неочищенного маслянистого остатка. Этот остаток (10,0 г) растворяли в дихлорметане (250 мл) и озонировали при -78oC до тех пор, пока окраска раствора не станет синей. Затем добавляли метилсульфид (11 мл), реакционную смесь концентрировали и получали соединение формулы XXIa (неочищенное).
Соединение формулы XXIa растворяли в ТГФ (150 мл) и при -78oC обрабатывали гидратом гидразина (10 мл). Через 2 часа смесь выливали в разбавленную водную HCl и этилацетат и две фазы разделяли. Органическую фазу промывали другой кислотой, водой и солевым раствором, а затем концентрировали и получали неочищенный продукт, который очищали с помощью хроматографии на силикагеле, используя в качестве элюента 1 - 5% метанол в метиленхлориде, и в результате этой процедуры получали 4,40 (выход 71%) соединения формулы XXIIa;
1H-ЯМР (CDCl3) δ 7,38 - 7,24 (м, 5H); 6,31 (ш.с. 1H); 5,87 (ш.м. 1H); 5,04 (д, J=4,8 Гц, 1H); 1,67 (с. 3H).
К охлажденной (-5oC) смеси 1М водного КОН (140 мл) и ацетонитрила (100 мл) по капле добавляли раствор соединения XXIIa (2,39 г, 11,22 мМ) в ацетонитриле (130 мл). Полученную смесь размешивали 1 час при 0oC и разбавляли этилацетатом (300 мл), водой (50 мл) и насыщенным водным бикарбонатом натрия (50 мл). Органическую фазу отделяли, а водный слой экстрагировали этилацетатом (3х200 мл). Органические фазы объединяли, осушали, фильтровали и концентрировали, в результате чего получали соединение XXIIIa (неочищенное), которое перекристаллизовывали из гексана/ацетона; т.пл. 184 - 6oC, выход 1,53 г, 82%.
К азетидиону XXIIIa (580 мг, 3,55 мМ) в сухом ТГФ (5,0 мл) добавляли имидазол (265,5 мг, 3,90 мМ). Полученную смесь размешивали в течение 1 часа. Затем добавляли этилацетат и органический слой промывали солевым раствором, 10% водным раствором HCl и осушали. После хроматографии на силикагеле (с использованием в качестве элюента 25% этилацетата в гексане) получали 670 мг (выход 68%) соединения XXIVa в виде пенистого продукта.
К размешанному раствору соединения XXIVa (2,20 г, 7,92 мМ) в сухом ТГФ (25 мл) добавляли диизопропилэтиламин (1,65 мл, 9,51 мМ) при 0oC и в атмосфере аргона. Полученный раствор размешивали 5 минут, а затем добавляли ди-т-бутилкарбонат (Boc2O, 2,08 г, 9,51 мМ) и 4-диметиламинопиридин (193,6 мг, 1,58 мМ). Реакционную смесь размешивали 60 минут при 0oC. После этого реакционную смесь разводили этилацетатом (25 мл), промывали солевым раствором, 10% водным бикарбонатом натрия, концентрировали и получали в результате маслянистый остаток. После флеш-хроматографии (с использованием в качестве элюента 15% этилацетата в гексане) получали 2,40 г (выход 83%) соединения XVa в виде белого твердого продукта; 1H-ЯМР (CDCl3) δ 7,28 (м, 5H); 5,03 (м, 2H); 1,38 (с, 9H); 0,76 (т, J=7,56, 9H); 0,43 (м, 6H).
Пример 9. 7 -α- Фтор-2'-O-триэтилсилил-3'-дифенил-3'-(2-фурил)- N-дебензоил-N-т-бутоксикарбонилтаксол (V b)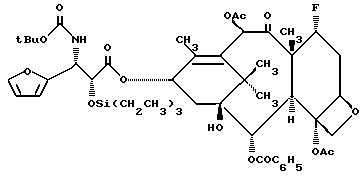
Раствор 7 -α- фтор-баккатина III (IV) (59,3 мг, 0,1 мМ) в сухом тетрагидрофуране (5 мл) продували инертным газом и охлаждали до -55oC в бане из сухого льда и ацетона. Затем к этому раствору с помощью шприца по капле добавляли гексаметилдисилазан лития (0,5 М-раствор в ТГФ, 0,24 мл, 1,2 экв.). Полученный светло-желтый раствор размешивали 5 минут. Затем в течение 5 минут добавляли раствор (в 2 мл тетрагидрофуране) рацемического 1-т-бутоксикарбонил-цис-3-триэтилсилилокси-4-(2-фурил)азетидинона (XV b) (178,4 мг, 6 экв). После этого охлаждающую баню заменяли баней из льда и солевого раствора и полученный раствор размешивали при 0oC в течение 1 часа. Реакцию гасили путем добавления насыщенного раствора NH4Cl (2 мл), а затем разводили этилацетатом (25 мл) и промывали водой (2х10 мл). Органическую фракцию осушали сульфатом магния и концентрировали, в результате чего получали целевой продукт в виде неочищенного бесцветного маслянистого продукта. Этот продукт очищали на силикагеле, элюируя смесью (7:3) гексанов и этилацетата. В результате описанной процедуры получали целевой продукт в виде бесцветного стеклообразного вещества (80,5 мг, выход 84%); 1H-ЯМР (300 МГц, CDCl3): δ 8,13 (д, 2H, J=9,0 Гц); 7,62 - 7,56 (м, 1H); 7,51 - 7,46 (м, 2H); 7,38 (с, 1H); 6,59 (c, 1H); 6,45 (дд, 1H, J=1,8, 3,2 Гц); 6,21 (д, 1H, J=3,2 Гц); 5,76 (д, 1H, J= 7,2 Гц); 5,33 (ш.т. 2H); 5,03 (д, 1H, J=7,5 Гц); 4,75 (с, 1H); 4,57 (дд, C-7 притон 1H, J=4,3, JH-F=46,9 Гц); 4,37 (A из AB кв., 1H, J=8,4 Гц); 4,27 (B из AB кв., 1H, J=8,4 Гц); 4,05 (д, 1H, J=7,2 Гц); 2,49 - 1,16 (м, 11H, включая синглеты при 2,47 (3H), 2,20 (3H), 1,88 (3H); 1,72 (3H); 1,38 (9H); 0,83 (т, 9H, J=5 Гц); 0,55 - 0,37 (м, 6H); 13C-ЯМР (75,6 МГц, CDCl3): δ 206,0; 171,1; 169,4; 167,2; 155,2; 152,1; 141,8; 141,4; 133,6; 131,8; 130,1; 129,2; 128,7; 110,6; 107,1; 96,2; 93,9; 81,9; 80,7; 80,0; 78,7; 77,9; 77,8; 75,0; 72,3; 70,8; 56,9; 56,7; 52,7; 42,6; 40,0; 35,5; 33,9; 33,6; 28,1; 28,0; 25,5; 22,5; 21,2; 20,7; 14,6; 14,5; 14,3; 14,2; 6,4; 4,2;
Пример 10. 7 -α- Фторо-2-O-триэтилсилил-3'-дефенил-3'-(2- тиенил)-N-дебензоил-N-бутоксикарбонилтаксол (Vc)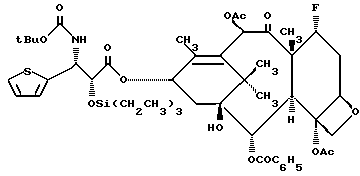
В соответствии с процедурой, описанной в Примере 9, получали целевой продукт в виде белого пенистого вещества (выход 78% по отношению к восстановленному исходному материалу);
1H-ЯМР (300 МГц, CDCl3): δ 8,14 (д, 2Н, J = 9,0 Гц); 7,63 - 7,58 (м, 1H); 7,51 - 7,48 (м, 2H); 7,24 (дд, 2H, J = 2,4, 3,6 Гц); 7,00 - 6,93 (м, 2H); 6,58 (с, 1H); 6,23 (т, 1H, J = 9 Гц); 5,77 (д, 1H, J = 6 Гц); 5,51 - 5,42 (м, 2H); 5,03 (д, 1H, J = 9 Гц); 4,57 (д, 1H, J = 3 Гц); 4,59 (дд, C-7 протон 1H, JH-F = 48 Гц); 4,38 (A из ABКв., J = 6 Гц); 4,27 (B из ABкв., 1H, J = 6 Гц); 4,05 (д, 1H, J = 7 Гц); 2,57 - 1,15 (м, 11H, включая синглеты при 2,44 (3H), 2,20 (3H), 1,88 (3H), 1,70 (3H), 1,32 (9H); 0,86 (т, 9H); 0,86 (т. 9Н; J = 5 Гц); 0,56 - 0,41 (м, 6H); 13C-ЯМР (75,6 МГц, CDCl3) δ 20,6; 171,0; 169,4; 168,8; 167,2; 161,4; 142,9; 141,3; 133,6; 131,8; 130,2; 129,2; 128,7; 126,9; 124,6; 124,5; 96,3; 93,9; 81,9; 80,8; 78,8; 77,9; 77,8; 77,2; 76,5; 75,2; 75,0; 71,0; 65,4; 56,9; 53,7; 42,7; 40,3; 35,6; 33,6; 28,1; 22,7; 21,3; 20,8; 18,8; 14,5; 14,3; 10,4; 6,3; 4,5.
Пример 11. 7 -α- Фторо-3'-дефенил-3'-(2-фурил)-N-дебензоил-N-т-бутоксикарбонилтаксол (Ie).
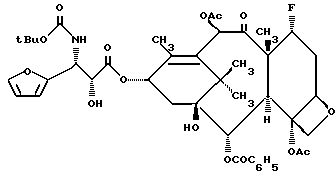
Раствор соединения Vb (80 мг, 0,08 мМ) в ацетонитриле (2 мл) охлаждали до 0oC в бане из льда и солевого раствора. К этому раствору добавляли 1 н. HCl (0,5 мл, 6 экв.), и реакционную смесь размешивали при комнатной температуре в течение 30 минут. Затем растворитель выпаривали в вакууме, а остаток распределяли между этилацетатом (25 мл) и водой (10 мл). Органическую фракцию осушали сульфатом магния и концентрировали, в результате чего получали белое пенистое вещество. Этот неочищенный продукт очищали на силикагеле, элюируя 10% CH3CN в CN2Cl2. Целевой продукт выделяли в виде белого пенистого вещества (45,6 мг, выход 77% по отношению к восстановленному исходному материалу); [α]D = -26,2o (c. 0,8 мг/мл, CH2Cl2); 1H-ЯМР (300 МГц, CDCl3): δ 8,12 (д, 2H, J = 6 Гц); 7,63 - 7,58 (м, 1H); 7,50 (т, 2H, J = 6 Гц); 7,41 (с, 1H); 6,57 (с, 1H); 6,37 - 6,36 (м, 1H); 6,33 - 6,31 (м, 1H); 6,20 (т, 1H, J = 6 Гц); 5,76 (д, 1H, J = 6 Гц); 5,37 - 5,23 (м, 2H); 5,02 (т, 1H, J = 9 Гц); 4,71 (ш. с, 1H); 4,57 (дд, C-7 протон 1H J = 4,2, JH-F = 46,8 Гц); 4,36 (A из ABкв., 1Н, J = 8,7 Гц); 4,27 (B из ABкв., 1H, J = 8,1 Гц); 4,04 (д, 1H, J = 7,2 Гц); 3,28 (ш.с, 1H); 2,59 - 2,20 (м, 5H, включая синглеты при: 2,41 (3H), 2,21 (3H); 1,85 (с, 3H); 1,43 - 1,17 (м, 18H); 13C-ЯМР (75,5 МГц, CDCl3): δ 205,7; 169,2; 169,0; 167,1; 142,3; 140,6; 133,5; 132,1; 130,0; 129,1; 128,6; 110,5; 107,2; 95,9; 93,6; 81,8; 80,6; 78,5; 77,8; 77,7; 74,7; 72,1; 71,6; 56,9; 5,8; 51,5; 42,5; 39,9; 35,4; 33,8; 33,5; 28,0; 27,9; 25,6; 22,2; 20,9; 20,7; 14,5; 14,1; 14,0;
МСВР для MH+ (C43H53NO15F):
вычислено: 842, 3399, найдено 842, 3389.
Пример 12. 7 -α- Фторо-3'-дефенил-3'-(2-тиенил)-N-дебензоил-N- т-бутокситаксол (If).
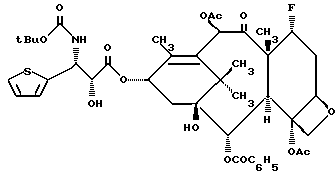
В соответствии с процедурой, описанной в Примере 11, получали целевой продукт в виде белого пенистого вещества 22,5 мг, выход 61%; 1H-ЯМР (300 МГц, CDCl3): δ 8,12 (д, 2H, J = 9 Гц); 7,64 - 7,59 (м, 1H); 7,50 (т, 2H, J = 9 Гц); 7,28 - 7,26 (м, 2H); 7,09 - 7,07 (M, 1H); 7,01 - 6,98 (м, 1H); 6,56 (с, 1H); 6,19 (т, 1H, J = 9 Гц); 5,76 (д, 1H, J = 6 Гц); 5,53 (ш.д, 1H, J = 12 Гц); 5,35 (д, 1H, J = 9 Гц); 5,00 (д, 1H, J = 9 Гц); 4,65 - 4,63 (м, 1,5H, C-7 скрытый протон), 4,48 (д, 0,5 H, J = 9 Гц), 4,36 (A из АВкв., 1H, J = 8,7 Гц); 4,27 (B из АВкв., 1H, J = 8,1 Гц); 4,04 (д, 1H, J = 7,2 Гц); 3,28 (ш. с. 1H); 2,59 - 2,20 (м, 5H, включая синглеты при: 2,39 (3H); 2,18 (3H); 1,72 (с, 3H); 1,43 - 1,17 (м, 18H); 13C-ЯМР (75,6 МГц, CDCl3): δ 205,6; 172,1; 169,3; 169,0; 167,0; 141,4; 140,6; 133,6; 132,1; 130,1; 129,3; 129,0; 128,6; 126,9; 125,2; 95,9; 93,5; 81,8; 80,8; 80,2; 78,5; 77,8; 77,7; 77,3; 76,8; 74,7; 73,3; 72,2; 56,9; 52,5; 42,5; 39,9; 35,5; 33,8; 33,5; 28,0; 27,9; 25,6; 22,3; 20,9; 20,7; 14,6; 14,1; 14,0; [α]D = -156 (с, 0,25 мг/мл, CH2Cl2).
Пример 13. Получение гидробензамида. PhCH(-N = CHPh)2.
В 3-литровую трехгорлую колбу, снабженную механической мешалкой и термометром, добавляли 1 л концентрированного NH4OH (ок.30%, 14,8 М). Затем раствор бензальдегида (265 г, 2,50 М) в 500 мл 2-пропанола добавляли сразу, одной порцией. Полученную смесь энергично размешивали в течение 43 часов примерно при 22oC Полученную суспензию фильтровали, а осадок на фильтре промывали водой (1 л). После сушили в вакууме, получали 242,2 г гидробензамина в виде белого твердого продукта (т.пл.100-102oC с выходом 97,4%).
В соответствии с описанной выше процедурой могут быть получены бис-имины формулы RgCH(-N= CHRg)2: гидрофурамид (Rg = 2-фурил) и гидротиенамид (Rg = 2-тиенил).
Пример 14. (±-цис-3-Ацетокси-1-[(фенил)(бензилиденимидино)метил]-4- фенилазетидин-2-он(XXXVa).
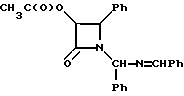
В 1-литровую, трехгорлую, круглодонную колбу, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли гидробензамид (30,00 г, 100,5 мМ) и этилацетат (150 мл). Размешивали в защитной атмосфере азота, реакционную смесь охлаждали до 5oC и добавляли триэтиламин (16,8 мл, 121 мМ). Затем по капле в течение 90 мин добавляли раствор ацетоксиацетилхлорида (12,4 мл, 116 мМ) в этилацетате (200 мл). После выдерживания при этой температуре в течение 16 часов реакционную смесь нагревали до 20oC (1,5 часа) и переносили в разделительную воронку. Органический слой промывали последовательно водным насыщенным NH4Cl (150 мл, 100 мл), водным насыщенным NaHCO3 (120 мл) и солевым раствором (120 мл). На этой стадии целевое соединение в целях его характеристики может быть выделено путем осушки органической фазы сульфатом магния с последующей фильтрацией и удалением растворителя в вакууме. В результате этого получали целевой продукт с количественным выходом в виде неочищенного красного стеклообразного вещества.
Чистота (по ЖХВД, площадь): 87,9% (1:1 - смесь диастереомеров); 1H-ЯМР (CDCl3, 200 МГц): δ 8,45 (с, 1H, N = CH); 7,80 - 7,85 (м, 1H, Ph); 7,60 - 7,65 (м, 1H, Ph); 7,26 - 7,50 (м, 9H, Ph); 7,00 - 7,10 (м, 4H, Ph); 6,28 (с, 0,5 H, NCHN); 6,23 (с, 0,5 H, NCHN); 5,81 (д, J = 4,8 Гц, 0,5 H, H-3); 5,76 (д, J = 4,8 Гц, 0,5H, H-3); 5,30 (д, J = 4,8 Гц, 0,5 H, H-4); 4,75 (д, J = 4,8 Гц, 0,5H, H-4); 1,63 (с, 3H, CH3CO); ИК (KBr): ν (см-1 = 1763 (С=0), 1641 (C = N); УФ (метанол): λмакс (нм) = 261,252.
Пример 15. (±-цис-3-Ацетилокси-4-фенилацетидин-2-он(XXXVIa)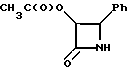
Раствор соединения, полученного в Примере 14, в этилацетате (500 мл) осторожно, в потоке аргона, переносили в 2,0-литровую колбу Парра, содержащую 10% палладий на активированном угле (6,00 г). Полученную смесь в течение 20 часов обрабатывали водородом (4 атм.), после чего катализатор удаляли путем фильтрации через прокладку из целита (Celite-диатомовая земля, Johns Manville). Остаток на фильтре суспендировали в этилацетате (200 мл), размешивали (10 минут) и фильтровали. После этого осадок на фильтре промывали этилацетатом (100 мл), и фильтраты объединяли. Органический слой промывали 10% HCl (300 мл), а затем оба слоя фильтровали через воронку из спеченного стекла, в результате чего выделяли белый осадок (дибензиламин•HCl), который промывали этилацетатом (100 мл). Затем фазы разделяли, и органический слой промывали другой порцией 10% HCl (200 мл). Объединенные 10% HCl - промывки снова экстрагировали этилацетатом (200 мл), а объединенные органические слои промывали водным NaHCO3 (насыщенным) (300 мл) и солевым раствором (250 мл). Органический слой осушали сульфатом магния, фильтровали и концентрировали в вакууме до получения конечного объема 75 мл. Затем смесь охлаждали до 4oC, а осажденный продукт выделяли путем фильтрации. Осадок на фильтре промывали гексаном (200 мл), и получали в результате 16,12 г (78,1% - полный выход исходя из гидробензамида) целевого соединения в виде белых игольчатых кристаллов.
Т. пл = 150 - 151oC; ЖХВД - чистота (площадь): 99,8%; 1H-ЯМР (CDCl3, 200 МГц); δ = 7,30 - 7,38 (м, 5H, Ph); 6,54 (ш.с. обмен., 1H, NH); 5,87 (дд, J = 2,7, 4,7 Гц, 1H, H-3); 5,04 (д, J = 4,7 Гц; 1H, H-4); 1,67 (с, 3H, CH3CO); ИК (KBr): ν (см-1) = 3210 (N-H); 1755, 1720 (C=0); КГ: 0,17%;
Анализ для C11H11NO3:
Вычислено: C 64,38; H 5,40; N 6,83
Найдено: C 64,07; H 5,34; N 6,77
Пример 16. (±)-цис-3-Ацетилокси-1[(2-фурил)(2-фурилметиленимино) метил] -4- (2-фурил)азетидин-2-он (XXXV b)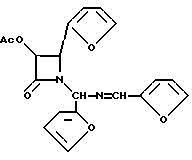
Целевое соединение получали в соответствии с процедурой, описанной в примере 14, за исключением того, что вместо гидробензамида использовали гидрофурамид, а реакцию осуществляли по шкале 18,6 мМ (вместо 100 мМ). Таким образом, используя гидрофурамид (5,00 г, 18,6 мМ), триэтиламин (3,11 мл, 22,3 мМ) и ацетоксиацетилхлорид (2,30 мл, 21,4 мМ), получали 6,192 г (90,4%) целевого соединения в виде бледно-красного сиропа.
Это соединение было получено в виде 1:1-смеси диастереомеров;
1H-ЯМР (CDCl3; 200 МГц): δ 8,211 (с, 0,5H, J = CH); 8,208 (с, 0,5H, N = CH); 7,14-7,59 (м, 3H, фурил); 6,90 (д, J = 3,5 Гц, 0,5H, фурил); 6,83 (д, J = 3,5 Гц, 0,5H, фурил); 6,10-6,53 (м, 6H, фурил, NCHN); 5,90 (д, J = 4,9 Гц, 0,5H, H-3); 5,86 (д, J = 4,8 Гц, 0,5H, H3); 5,35 (д, J = 4,8 Гц, 0,5H, H-4); 4,90 (д, J = 4,9 Гц, 0,5H, H-4); 1,91 (с, 1,5H, CH3CO); 1,88 (с, 1,5H, CH3CO);
ИК (пленка): ν (см-1)=1778, 1753 (C=O), 1642 (C=N); УФ (метанол): λмакс (нм) = 220, 278.
Пример 17. (±)-цис-3-(Ацетилокси)-4-(2-фурил)азетидин-2-он (XXXVIb)
Целевое соединение получали в соответствии с процедурой, описанной в примере 15, за исключением того, что продукт выделяли с помощью препаративной ТСХ, а реакцию осуществляли в молярном масштабе 2,7 мМ на основании исходного количества гидрофурамида. Так, например, неочищенный продукт примера 16 (1,00 г) снова растворяли в этилацетате (50 мл) и добавляли к 10% палладированному активированному углю (150 мг). После очистки неочищенного твердого продукта с помощью препаративной ТСХ (2 мм силикагеля, 1:1-смесь этилацетата и гексана) получали 386 мг (полный выход, скорректированный исходя из гидрофурамида - 65,8%) целевого соединения в виде желтого твердого вещества. Полученный продукт перекристаллизовывали из этилацетата/гексана.
Т.пл. 118-119oC; ЖХВД-чистота (площадь): 99,4%; 1H-ЯМР (CDCl3, 200 МГц); δ 7,44 (т, J = 1,3 Гц, 2H, фурил); 6,39 (д, J = 1,3 Гц, 1H, фурил); 6,21 (ш. с. обмен. , 1H, NH); 5,88 (дд, J = 2,2, 4,6 Гц, 1H, H-3); 5,05 (д, J = 4,6 Гц; 1H, H-4); 1,92 (с, 3H, CH3CO);
ИК (KBr): ν (см-1) = 3203 (N=H), 1756, 1726 (C=O); УФ (метанол): λмакс (нм) = 222.
Пример 18. (±)-цис-3-Ацетилокси-1-[(2-тиенил)(2- тиенилметиленимино)метил]-4-(2-тиенил)азетидин-2-он (XXXVc)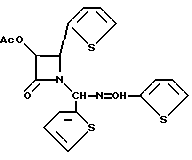
Целевое соединение получали в соответствии с процедурой, описанной в пример 14, за исключением того, что вместо гидробензамида использовали гидротиенамид. Таким образом, используя гидротиенамид (30 г, 94,7 мМ), триэтиламин (15,84 мл, 114 мМ), и ацетоксиацетилхлорид (11,6 мл, 108 мМ), получали целевое соединение в виде вязкого маслянистого вещества. Полученный продукт представлял собой смесь диастереомеров. 1H-ЯМР (CDCl3): δ 8,52 (с, 1H); 8,502 (с, 1H); 7,51 (д, J = 4,9 Гц, 1H); 7,45 (д, J = 4,4 Гц, 1H); 7,41 (д, J = 3,1 Гц, 1H); 7,37 (д, 1H); 7,30 (м, 3H); 7,16 (м, 1H); 7,16 (м, 3H); 7,09 (м, 2H); 6,94 (м, 1H); 6,89 (м, 1H); 6,81-6,74 (м, 4H); 6,48 (с, 1H); 6,43 (с, 1H); 5,85 (м, 2H); 5,59 (д, J = 4,8 Гц, 1H); 5,17 (д, J = 4,8 Гц, 1H); 1,87 (с, 3H); 1,86 (с, 3H).
Пример 19. (±)-цис-3-(Ацетилокси)-4-(2-тиенил)азетидин-2-он (XXXVIc)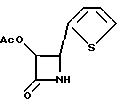
70% водный раствор уксусной кислоты (0,35 мл ледяной уксусной кислоты и 0,15 мл воды) целиком, одной порцией, добавляли к размешанному раствору соединения XXXVc (0,431 г, 1,03 мМ) в дихлорметане (2,93 мл) при 25oC. Затем смесь доводили до температуры перегонки и размешивали в течение 2,5 часов. После этого смесь разбавляли 50 мл дихлорметана, затем промывали двумя 75 мл-порциями насыщенного водного бикарбоната натрия, а затем одной 50 мл-порцией насыщенного солевого раствора. Органический экстракт концентрировали в вакууме с получением коричневого маслянистого остатка, который растворяли в минимальном количестве дихлорметана, а затем помещали в колонку с силикагелем (размером 4'' • 0,5''). В качестве элюента использовали градиент 10-60% EtOAc в гексане и получали в результате менее полярные побочные продукты, а затем целевое соединение (0,154 г, выход 75%) в виде белого твердого продукта. 1H-ЯМР (CDCl3): δ 7,32 (дд, J = 4,7, 1,5 Гц, 1H); 7,03 (м, 2H); 6,75 (ш. с. 1H); 5,86 (дд, J = 4,6, 2,7 Гц, 1H); 5,27 (д, J = 5,3 Гц, 1H); 1,83 (с, 3H); 13C-ЯМР (CDCl3): δ 169,3; 165,5; 138,4; 127,1; 127,07; 126,2; 78,3; 54,0; 20,0.
Пример 20. 7 -α- Фторо-10-дезацетилокситаксол (Ic)
10-Дезацетилтаксол VIa (140 мг, 0,173 мМ) в сухом дихлорметане (3,5 мл) обрабатывали при 0oC пиридином (0,028 мл, 0,346 мМ) и трихлороэтилформатом (0,0724 мл, 0,260 мМ). После выдерживания при этой температуре в течение 1 часа охлаждающую баню удаляли, и смесь размешивали при комнатной температуре. Затем растворитель выпаривали, а остаток хроматографировали на колонке с силикагелем (элюируя 30-50% этилацетатом в гексане), в результате чего получали 92,3 мг (выход 46%) соединения XIIIa в виде пенистого вещества. Продолжая элюирование, получали также соединение VIIa (выход 16%) в виде пенистого вещества.
Соединение XIIIa (92,3 мг, 0,079 мМ) в сухом дихлорметане (2 мл) обрабатывали 1,1,2-трифторо-2-хлоротриэтиламином (0,0384 мл, 0,238 мМ). Раствор размешивали в течение ночи, растворитель выпаривали, а остаток очищали с помощью хроматографии на силикагеле, элюируя 25% этилацетатом в гексане, в результате чего получали 42,8 мг (выход 47%) соединения VIIIa в виде белого твердого вещества.
Диенон VIIIa (39 мг, 0,034 мМ) растворяли в метаноле (0,5 мл) и уксусной кислоте (0,5 мл). Затем добавляли цинковый порошок (66,4 мг, 1,02 мМ), и полученную смесь выдерживали в течение 1 часа при температуре 40oC. Нерастворившийся материал удаляли путем фильтрации. Фильтрат концентрировали, а остаток хроматографировали на силикагеле, элюируя 60% этилацетатом в гексане, в результате чего получали 22 мг (выход 81,5%) соединения IXa в виде пенистого вещества.
Диенон IXa (22 мг, 0,028 мМ) в этилацетате (0,7 мл) подвергали гидрогенизации в течение 5,5 часов в присутствии 10% палладированного угля (14,7) при комнатной температуре и при давлении, слегка превышающем 1 атм. После удаления катализатора путем фильтрации и очистки продукта с помощью хроматографии на силикагеле (с использованием в качестве элюента смеси этилацетата и гексана (1:1)), получали 15 мг (выход 68%) соединения Xa в виде пенистого вещества.
Соединение Xa (27 мг, 0,034 мМ) в дихлорметане (1 мл) обрабатывали бензилхлороформом (0,0146 мг, 0,102 мМ), а затем диизопропилэтиламином (0,0177 мл, 0,102 мМ). Реакционную смесь размешивали 45 минут при 0oC и 12 часов при комнатной температуре. После выпаривания растворителя и хроматографии на силикагеле (с использованием в качестве элюента 40% этилацетата в гексане) получали 25,5 мг (выход 81%) соединения XIa в виде пенистого вещества.
Соединение XIa (25,5 мг, 0,28 мМ) в дихлорометане (0,8 мл) при 0oC обрабатывали DAST (0,0071 мл, 0,055 мМ). После выдерживания реакционной смеси в течение 45 минут при 0oC ее оставляли на 5 часов при комнатной температуре. После выпаривания растворителя и хроматографии получали XIIa в виде неочищенного пенистого продукта. Это соединение растворяли в этилацетате (1 мл) и размешивали в течение 12 часов в присутствии 10% палладированного угля (8,9 мг) при комнатной температуре и при давлении, слегка превышающем 1 атм. Катализатор удаляли путем фильтрации, и после хроматографии продукта на силикагеле получали 10 мг (выход 40% за две стадии) соединения 1c в виде пенистого вещества; 1H-ЯМР (CDCl3) δ 8,08 (д, 2H); 7,70 (д, 2H); 7,68 - 7,28 (м, 11H); 7,04 (д, 1H); 6,04 (ш.т., 1H); 5,75 (дд, 1H); 5,69 (д, 1H); 4,92 (д, 1H); 4,72 (дд, 1H); 4,55 (дд, JH-F 47 Гц); 4,30 - 4,21 (м, 3H); 3,81 (дд, 1H); 3,47 (д, обмен. 1H); 3,37 (ш.д., 1H); 2,48 - 1,30 (м, 13H, включая синглеты при 2,30; 1,72; 1,61); 1,07 (с, 3H); 1,02 (с, 3H); МСВР для MH+: вычислено: 798, 3290, найдено: 798, 3264.
Пример 21. 7 -α- Фторо-10-дезацетилтаксол (1d).
Раствор соединения VIIa (полученного в соответствии с приведенным выше описанием, 120 мг, 0,103 мМ) в дихлорметане (2 мл) охлаждали при 0oC и обрабатывали DAST (0,0266 мл, 0,207 мМ). Полученный раствор размешивали 30 минут при 0oC, а затем 4 часа при комнатной температуре. После этого реакцию гасили путем добавления воды (0,05 мл). Затем реакционную смесь концентрировали, а остаток очищали с помощью хроматографии на силикагеле, элюируя 30% этилацетатом в гексане, в результате чего получали 81 мг (выход 68%) соединения XIVa в виде пенистого вещества. Это соединение (63 мг, 0,054 мМ) растворяли в метаноле (0,5 мл) и уксусной кислоте (0,5 мл) и обрабатывали цинковым порошком (104 мг, 1,62 мМ) при 45oC в течение 90 мин. Затем реакционную смесь фильтровали, а фильтрат концентрировали. Остаток хроматографировали на силикагеле, элюируя 40% гекскном в 60% этилацетате, и получали 38 мг (выход 86%) соединения 1c в виде белого твердого вещества; 1H-ЯМР (CDCl3) δ 8,17 (д, 2H); 7,78 (д, 2H); 7,66 - 7,26 (м, 11H); 7,15 (д, 1H); 6,20 (дд, 1H); 5,83 (дд, 1Н); 5,76 (д, 1H); 5,22 ((с, 1H); 5,01 (д, 1H); 4,80 (м, 1H); 4,56 (дд, JH-F = 47 Гц); 4,40 (м, 3H); 4,10 (д + обм.с, 2H); 3,55 (д, обм. 1H); 2,66 - 1,70 (м, 13H, включая синглеты при 2,41; 1,82, 1,76); 1,12 (с, 3H); 1,03 (с, 3H);
МСВР для MH+: вычислено: 814, 3239; найдено: 814, 3214.
Пример 22. (±)-цис-3-Триэтилсилилокси-4-(2-фурил)- азетидин-2-он (XXXVIIa)
Ацетоксилактам XXXIb (3,78 г, 19,4 мМ) в 60 мл метанола размешивали с K2CO3 (20 мг, 0,14 мМ) в течение 90 минут, и полученный раствор нейтрализовали с помощью Dowex 50W-X8, а затем фильтровали. Фильтрат концентрировали, а остаток растворяли в 80 мл безводного ТГФ и размешивали 30 минут при 0oC с имидазолом (1,44 г, 21,2 мМ) и TESCl (3,4 мл, 20,2 мМ). Полученный раствор разбавляли этилацетатом и промывали солевым раствором, после чего осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле, элюируя смесью (3:1) гексана и этилацетата, и получали 4,47 г (выход 86%) целевого соединения в виде бесцветного маслянистого продукта; ИК (пленка) 3276 (шир.), 1768, 1184, 732 см-1; 1H-ЯМР (CDCl3, 300 МГц) δ 7,38 (с, 1Н); 6,39 (ш.с., 1Н); 6,35 (с, 2Н); 5,05 (дд, J = 4,6, 2,3 Гц, 1Н); 4,78 (д, J = 4,6 Гц, 1H); 0,82 (т, J = 8,5 Гц, 6H); 0,50 (д кв., J = 8,5, 1,8 Гц, 9H); 13C-ЯМР (CDCl3, 75,5 Гц) δ 169,6; 150,4; 142,6; 110,5; 109,1; 79,6; 53,2; 6,4; 4,4.
Пример 23. (±)-цис-3-Триэтилсилилокси-4-(2-фурил)-N-т- бутоксикарбонилазетидин-2-он (XVb)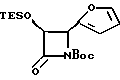
TES-лактам (XXXVIIa) (2,05 г, 7,7 мМ) в 30 мл дихлорметана размешивали при 0oC с диизопропилэтиламином (1,5 мл, 8,6 мМ) и ди-т-бутилкарбонатом (2,0 г, 9,2 мМ), а также с каталитическим количеством диметиламинопиридина (ДМАП). Полученный раствор разбавляли дихлорметаном и промывали солевым раствором, после чего осушали сульфатом магния и концентрировали. Остаток хроматографировали на силикагеле, элюируя смесью 8:1 гексана/этилацетата, и получали 2,0 г (выход 70%) целевого соединения в виде воскообразного твердого продукта.
Пример 24. (±)-цис-3-Триэтилсилилокси-4-(2-тиенил)- азетидин-2-он (XXXVIIb)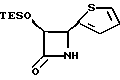
Раствор 3-ацетоксилактама (XXXVIc) (2,5 г, 11,8 мМ) растворяли в метаноле (10 мл) и обрабатывали насыщенным водным бикарбонатом натрия (10 мл), а полученную суспензию размешивали в течение 3 часов. Затем реакционную смесь разбавляли этилацетатом (20 мл) и промывали водой (15 мл). Водную фракцию подвергали обратной экстракции этилацетатом несколько раз, а объединенные органические фракции осушали сульфатом магния и концентрировали, в результате чего получали желтый твердый продукт (выход 1,7 г). Неочищенный продукт растворяли в сухом тетрагидрофуране (20 мл), и полученный раствор охлаждали до 5oC в бане со льдом и водой. После 5-ти минутного перемешивания по капле добавляли триэтилхлоросилан (1,85 мл, 1,1 экв.). Полученную суспензию размешивали 3 часа при указанной температуре, после чего твердые вещества удаляли путем фильтрации. Органическую фракцию промывали водой (2 х 20 мл), затем осушали сульфатом магниям и концентрировали. Полученный продукт очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь (7: 3) гексана и этилацетата, в результате чего получали целевой продукт в виде бесцветного твердого вещества (1,5 г, выход 45%); т.пл. 70-71oC; 1H-ЯМР (300 МГц, CDCl3): δ 7,32 - 7,30 (м, 1H); 7,05 - 6,68 (м, 2H); 5,06 - 5,05 (м, 2H); 0,82 (т, J = 8 Гц); 0,55 - 0,46 (м, 6H); 13C-ЯМР (75,6 МГц, CDCl3): δ 169,1; 139,7; 126,5; 125,8; 79,4; 55,1; 6,3; 4,4.
Пример 25. (±)-цис-3-Триэтилсилилокси-4-(2-тиенил)-N- т-бутоксикарбонил-азетидин-2-он (XVc)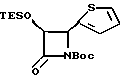
Раствор силилазетидинона XXXVIIb (425,7 мг, 1,48 мМ) растворяли в дихлорметане (10 мл) и охлаждали до 5oC в бане с ледяной водой. Реакционную смесь обрабатывали каталитическим количеством DMAP, диизопропилэтиламином (0,25 мл, 1,0 экв), а затем ди-т-бутилкарбонатом (388,4 мг, 1,2 экв.). После размешивания в течение 2 часов при вышеуказанной температуре реакцию гасили насыщенным водным бикарбонатом натрия (5 мл), а органическую фракцию промывали водой (5 мл), а затем осушали сульфатом магния, пропускали через небольшой слой силикагеля и концентрировали, в результате чего получали целевой продукт в виде бесцветного маслянистого вещества (525,3 мг, выход 93%); 1H-ЯМР (300 МГц, CDCl3): δ 7,31-7,29 (м, 1H); 7,08-7,07 (м, 1H); 7,00-6,58 (м, 1H); 5,31 (д, 1H, J = 6 Гц); 5,03 (д, 1H, J = 6 Гц); 1,40 (с, 9H); 0,83 (т, 9H, J = 8 Гц); 0,56-0,47 (м, 6H); 13C-ЯМР (75,6 МГц, CDCl3): δ 165,5; 147,5; 136,4; 127,6; 126,2; 126,1; 83,3; 77,3; 57,9; 27,7; 6,2; 4,3.
Пример 26.
В соответствии с процедурами, описанными в приведенных выше Примерах, могут быть синтезированы следующие конкретные производные таксола формулы I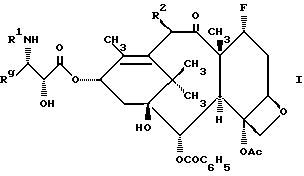
7 -α- Фторо-3'-дефенил-3-(2-тиенил)таксол (Pg = 2-тиенил, R1 = бензоил, R2 = OAc);
7 -α- Фторо-3'-дефенил-3-(2-фурил)таксол (Pg = 2-фурил, R1 = бензоил, R2 = OAc);
7 -α- Фторо-10-десацетил-10-бензоил-3-дефенил-3'-(2-фурил)- таксол (Rg = 2-фурил, R1 = бензоил, R2 = -OCOC6H5)
7 -α- Фторо-10-десацетил-10-бензоил-3'-дефенил-3'-(2-тиенил) таксол (Rg = 2-тиенил, R1 = бензоил, R2 = -OCOC6H5)
7 -α- Фторо-10-десацетил-10-бензоил-3'-дефенил-3'-(2-тиенил) таксол (Rg = 2-тиенил, R2 = OCH3)
7 -α- Фторо-10-десацетил-10-фенилметилкарбонил-3'-дефенил-3'-(2-фурил)- таксол (Rg = 2-фурил, R1 = бензоил, R2 = -OC(=O)OCH2С6H5)
7 -α- Фторо-10-десацетил-10-н-бутилкарбонил-3'-дефенил-3'-(2-тиенил) таксол (Rg = 2-тиенил, R2 = OCOCH2CH2CH2 CH2)
7 -α- Фторо-10-десацетил-10-метилсульфонил-3'-дефенил-3'-(2-фурил)таксол (Rg = 2-фурил, R1 = бензоил, R2 = -OSO2CH3).
Пример 27. Характерный пример селективной дериватизации C-10-положения 10-десацетилбаккатина. 10-Бензоил-10-десацетил-7- триэтилсилилбаккатин (XXXIXa).
В атмосфере аргона производное баккатина формулы XXXVIII, где R3 = SiEt3, (43,5 мг, 0,066 мМ) растворяли в сухом тетрагидрофуране (1,0 мл). Полученный раствор охлаждали до -40oC и медленно добавляли н-BuLi (0,050 мл, 0,82 мМ, 1,6 М - раствор). После 5-минутного размешивания добавляли бензоилхлорид (0,030 мл, 0,26 мМ), и реакционную смесь нагревали до 0oC. Затем реакционную смесь размешивали в течение 1,5 часов, и реакцию гасили в насыщенном растворе хлорида аммония (2 мл). Водную среду экстрагировали этилацетатом (2 х 5 мл), осушали сульфатом магния и выпаривали, в результате чего получали маслянистое вещество. После флеш-хроматографии на силикагеле (с использованием в качестве элюента 50% этилацетата в гексане) получали целевое соединение (30 мг, выход 60%, соединение формулы XXXIX, где R3 = Si(Et)3, Rm = OCOC6H5) в виде пенистого продукта;
1H-ЯМР (CDCl3): δ 8,17-8,05 (м, 4H); 7,64-7,42 (м, 6H); 6,67 (с, 1H); 5,67 (д, 1H); 4,95 (д, 1H); 4,81 (м, 1H); 4,56 (дд, 1H); 4,30 (д, 1H); 4,14 (д, 1H); 3,92 (д, 1H); 2,50 (м, 1H); 2,30-2,0 (м, 18H); 1,92-1,80 (м, 1H); 1,72-1,62 (ш. с., 4H); 1,30 (с, 3H); 1,00 (с, 3H); 0,89 (т, 3H); 0,56 (кв., 6H); MCBP (FAB/NOBA): для C42H54O11Si(MH+): вычислено: 762, 3435, найдено: 762, 3427.
В соответствии с описанной методологией могут быть получены C-10-карбонаты, сульфонаты, карбаматы, сложные эфиры и т.п. При этом лучшие выходы могут быть получены с использованием гексаметилдисилазана лития.
Биологические данные.
Данные in vitro - цитотоксичности
Производные 7-фторотаксола настоящего изобретения показали in vitro цитотоксическую активность против клеток карциномы толстой кишки человека HCT-116 и HCT-116/М46 представляют собой клетки, предварительно отобранные на устойчивость против teniposide и экспрессирующие фенотип множественной лекарственной устойчивости, включая устойчивость к таксолу. Цитотоксичность оценивали в клетках HCN-116 карциномы толстой кишки человека путем анализа, проводимого с использованием XTT (2,3-бис(2-метокси-4-нитро-5-сульфофенил)-5-[ (фениламино)карбонил] 2H-тетразолия гидроксида) и описанного D.A. Scudiero и др., в "Evaluation of Soluble tetrazolum/ formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines", Cancer Res. 48: 4827-4833, 1988. Клетки высевали в 96-луночные планшеты для микротитрирования с плотностью 4000 клеток на лунку, а через 24 часа в эти лунки добавляли лекарственные средства и проводили серийное разведение. Клетки инкубировали 72 часа при 37oC, и в течение этого времени добавляли тетразолиевый краситель (XTT). Дегидрогеназа (фермент), присутствующая в живых клетках, способствует восстановления XTT с образованием соединения, поглощающего свет при 450 нм, которое может быть количественно проанализировано с помощью спектрофотометрии. Чем больше оптическая плотность, тем больше число живых клеток. Полученные результаты выражали в величинах IC50, которые представляют собой концентрации лекарственного средства, необходимые для ингибирования пролиферации клеток (т.е. оптическая плотность при 450 нм), до 50% от пролиферации необработанных контрольных клеток. IC50 - величины для соединений, испытанных в данном анализе, приводятся в Таблице 3.
Мышиная модель M 109.
Гибридным мышам Balb/c х DBA/2 FI имплантировали внутрибрюшинно в соответствии с описанием Wiliam Rose в "Evaluation of Madison 109 Lung Carcinoma as a Model for Soreening Antitumor Drugs, Cancer Treatment Reports, 65, N 3-4 (1981) 0,5 мл 2% (масс./об.) раствора Брея карциномы легких M109.
Затем мышей обрабатывали испытуемым соединением путем внутрибрюшинных инъекций в различных дозах либо на 1,5 и 9 день после имплантации, либо на 5 и 8 день после имплантации опухоли. Этих мышей ежедневно контролировали на выживание в период примерно до 75 - 90 дней после имплантации опухоли. В данном эксперименте одна группа мышей, которая оставалась необработанной, служила в качестве контрольной группы.
Среднее время выживания для мышей, обработанных соединением, (T) сравнивали со средним временем выживания для контрольных мышей. Отношение этих двух величин для каждой обработанной группы мышей умножали на 100, в результате чего получали процентное соотношение (т. е. , % Т/С), которое представлено в Таблице 4 для характерного соединения настоящего изобретения.
Соединения формулы I настоящего изобретения обладают противоопухолевой ингибирующей активностью у млекопитающих. Таким образом, в другом своем варианте настоящее изобретение относится к способу ингибирования опухолей у млекопитающих, восприимчивых к соединению формулы I. Настоящее изобретение также относится к промежуточным соединениям, которые могут быть использованы для получения производных 7-фторотаксола формулы I.
Соединения формулы I могут быть также использованы для получения водорастворимых пролекарств. Некоторые из водорастворимых предшественников таксола были описаны в литературе. См., например, патент США N 5059699, выданный 22 октября 1991 г. Kingston; патент США N 4942184, выданный 17 июля 1990 г. Haugwitz и др. : патент США N 4960790, выданный 2 октября 1990 г. Stella и др. ; при этом все три патента вводятся в настоящее описание посредством ссылки. Водосолюбилизирующие части молекул, описанные в вышеуказанных трех патентах США, могут быть также присоединены к 2 - и/или 10-гидроксигруппе соединения формулы I в целях придания ему большей водорастворимости. Таким образом, настоящее изобретение относится к противоопухолевым соединениям, которые могут быть использованы для получения их предшественников.
Настоящее изобретение также относится к фармацевтическим композициям (препаратам), содержащим соединения формулы I в сочетании с одним или несколькими фармацевтически приемлемыми, инертными или физиологически активными носителями, наполнителями, разбавителями или адъювантами. Примеры получения препаратов, содержащих таксол, или его близкородственных производных (включая возможную дозировку этих соединения), широко описаны в литературе, например в патентах США N 4960790 и 4814470; и в соответствии с этими примерами могут быть получены композиции, содержащие соединения настоящего изобретения. Например, новые соединения могут быть введены в виде таблеток, драже, порошковых смесей, капсул, инъецируемых растворов, суппозиториев, эмульсий, дисперсий, пищевых добавок и в виде других подходящих форм. Обычно фармацевтический препарат выключает в себя активное соединения, соответствующим образом смешанное с нетоксичным фармацевтическим органическим носителем или с нетоксичным фармацевтическим неорганическим носителем, и составляющие приблизительно 0,01 - 2500 мг, или в более высокой дозе, а предпочтительно 50 - 500 мг на одну стандартную лекарственную форму. В качестве фармацевтически приемлемых носителей могут быть использованы такие традиционные носители, как маннит, мочевина, декстраны, лактозы, картофельный или кукурузный крахмал, стеарат магния, тальк, растительные масла, полиалкиленгликоли, этилцеллюлоза, поли(винилпирролидон), карбонат кальция, этилолеат, изопропилмиристат, бензилбензоат, карбонат натрия, желатин, карбонат калия, кремниевая кислота, и другие обычно используемые фармацевтически приемлемые носители. Фармацевтический препарат может также содержать нетоксичные добавки, такие как эмульгирующие агенты, консерванты, смачивающие агенты и др. , например сорбитанмонолаурат, триэтаноламинолеат, полиоксиэтиленмоностеарат, трипальмитат глицерина, сульфосукцинат диоксилнатрия, и т.п.
Соединения настоящего изобретения могут быть также лиофилизованы и, если это необходимо, использованы в сочетании с другими фармацевтически приемлемыми наполнителями при составлении препаратов, пригодных для парентерального введения путем инъекции. Для такого способа введения лиофилизованный препарат может быть восстановлен путем разведения в воде (обычно в физиологическим растворе) или в смеси воды и органического растворителя, такого как пропиленгликоль, этанол и т.п.
Соединения настоящего изобретения могут быть использованы, в основном, тем же способом, каким используется таксол при лечении опухолей у млекопитающих. Способ, доза и схема введения таксола при лечении людей с раковыми заболеваниями широко исследовалась специалистами. См., например, Ann.Int. Med. , III, стр. 273 - 279 (1989). Доза соединений настоящего изобретения независимо от того, является ли она разовой, разделенной или суточной дозой, будет варьироваться от активности данного соединения, способа его введения, веса тела пациента и природы его заболевания. Вводимая доза не ограничена какими-либо конкретными границами, но обычно она представляет собой эффективное количество или молярное эквивалентное количество фармакологически активной свободной формы соединения, продуцируемой из введенного препарата в результате метаболического высвобождения активного лекарственного средства с достижением желаемого фармакологического и физиологического действия. Вводимые дозы обычно варьируются в пределах 0,8 - 8 мг/кг веса тела либо около 50-275 мг на м2 поверхности тела пациента. Обращаясь к проводимым ранее исследованиям по применению таксола и его производных, специалисты-онкологи без излишнего экспериментирования могут легко установить приемлемые схемы для эффективного введения соединений настоящего изобретения.
Дополнительные экспериментальные данные
1. Данные по биологической активности заявленных соединений (см. в конце описания, таблица 5).
Лекарственные средства, используемые для лечения, вводят на 5 и 8 дни после имплантации опухоли, в/бр.
2. Получение и физико-химические характеристики аналогов 2-таксола.
В представленных ниже примерах использована следующая аббревиатура:
1H NMR - 1H ЯМР;
13C NMR - 13C ЯМР;
Hz - Гц;
s - синглет;
d - дублет;
dd - двойной дублет;
t - триплет;
m - мультиплет;
broad - широкий;
proton - протон;
complex - комплексный
HRMS - масс-спектр высокого разрешения.
Пример 28. 7- -α- Фтор баккатин III.
В сухую колбу добавляют 2'-CBZ таксол (4 г, 4 ммоль) и сухой толуол (80 мл) в атмосфере инертного газа. Полученную суспензию перемешивают при температуре окружающей среды, одновременно добавляя по каплям сухой тетрагидрофуран (16 мл) до получения окрашенного раствора. Раствор охлаждают до -78oC на бане сухой лед/ацетон и затем обрабатывают трихлоридом диметиламиносеры (DAST, 1,2 мл, 2,5 экв.). Реакционную массу перемешивают в течение 16 часов, таким образом постепенно нагревая ее до температуры окружающей среды. Полученную суспензию фильтруют и фильтрат (разбавленный этилацетатом (30 мл)) промывают насыщенным водным раствором бикарбоната натрия с последующей промывкой насыщенным солевым раствором. Органическую фракцию сушат (MgSO4) и концентрируют, получая неочищенный продукт в виде белой пены. Неочищенный материал по частям очищают колоночной хроматографией (силикагель, 10% CH3CN в CH2Cl2). Получают 0,75 г 6,7-дегидроаддукта и 1,45 г смеси 7 -α- фтор- и циклопропил-аддуктов (смесь 82 : 18 по данным 1H ЯМР).
Вышеуказанную смесь (1,45 г) помещают в этилацетат (60 мл) и обрабатывают палладием на угле (33 мг). После встряхивания в течение 4 часов при давлении водорода ≈ кг/см2 реактор продувают и реакционную массу фильтруют через небольшой слой силикагеля и концентрируют. Получают смесь желаемых продуктов в виде белой пены (1,24 г 99%, смесь 90 : 10 по данным 1H ЯМР). Смесь 7 -α- фтор и циклопропил таксола помещают в сухой метиленхлорид (30 мл) и обрабатывают борогидридом тетраметиламмония (745 мг, 2,9 моль, 2 экв.) и перемешивают в течение 6 часов. Затем реакцию останавливают добавлением уксусной кислоты (1 мл), разбавляют дополнительным количеством метиленхлорида (30 мл) и промывают насыщенным водным раствором бикарбоната натрия. Органическую фракцию сушат (MgSO4) и концентрируют. Неочищенную смесь замещенных таксановых структур по частям очищают колоночной хроматографией (силикагель, 10% CH3CH в CH2Cl2) с получением желаемого баккатина III (510 мг, 60%) в виде смеси 90:10 (1H ЯМР) 7 -α- фтор- и циклопропил-аналогов в виде белой пены. Полученную пену кристаллизуют из горячего изопропанола с получением 7 -α- фтор баккатина III в виде маленьких игольчатых кристаллов (410 мг). Т.пл. = 234-236oC (разл.).
1H NMR (300 MHz, CDCl3): δ 8.14 (d, 2H, J = 6 Hz); 7,65-7.52 (m, 1H); 7.52-7.49 (m, 2H); 6.57 (s, 1H); 5.72 (d, 1H, J = 9 Hz); 5.03 (d, 1H, J = 9 Hz); 4.86-4.79 (m, 1H); 4.55 (dd, C-7 proton 1H, J = 3.9, JH-F = 47.1 Hz); 4.36 (A of ABq, 1H, J = 7.8 Hz); 4.27 (B of ABq, 1H, J = 7.8 Hz); 4.12 (d, 1H, J = 6.9 Hz); 2.60-2.48 (m, 2H); 2.30-1.07 (m, 22H) включая синглеты на 2.30; 2.21; 2.08; 1.77; 1.58; 1.13; 1.07.
Пример 29. Аналог 7 -α- фтор-2'-O-триэтилсилил-3'-(2-фурил)-3'-N-BOC таксола.
Раствор 7 -α- баккатина III (59.3 мг, 0.1 ммоль) в сухом тетрагидрофуране (5 мл) в атмосфере инертного газа охлаждают до -55oC на бане сухой лед/ацетон. К раствору с помощью шприца по каплям добавляют литий гексаметилдисилазан (0.5 М раствор в ТГФ, 0.24 мл, 1.2 экв.). Полученный бледно-желтый раствор перемешивают в течение 5 минут и затем добавляют раствор рацемического фурилазетидинона (178.4 мг, 6 экв.) в тетрагидрофуране (2 мл) в течение 5 минут. Затем охлаждающую баню заменяют на баню лед/солевой раствор и перемешивают полученный раствор при 0oC в течение 1 часа. Реакцию останавливают добавлением насыщенного раствора NH4Cl (2 мл) и затем разбавляют этилацетатом (25 мл) и промывают водой (2х10 мл). Органическую фракцию сушат (MgSO4) концентрируют, получая желаемый продукт в виде бесцветного масла. Неочищенный продукт очищают на силикагеле, используя смесь гексаны/этилацетат (7: 3) в качестве элюента. Получают целевой продукт в виде бесцветной стеклообразной массы (80.5 мг, 84%).
1H NMR (300 MHz, CDCl3): δ 8.13 (d, 2H, J = 9.0 Hz); 7.62-7.56 (m, 1H); 7.51-7.46 (m, 2H); 7.38 (s, 1H); 6.59 (s, 1H); 6,45 (dd, 1H, J = 1.8, 3.2 Hz); 6.21 (d, 2H, J = 3.2 Hz); 5.76 (d, 1H, J = 7.2 Hz); 5.33 (broad, t, 2H); 5.03 (d, 1H, J = 7.5 Hz); 4.75 (s, 1H); 4.57 (dd, C-7 proton 1H, J = 4.3, JH-F = 46.9 Hz); 4.37 (A of ABq, 1H, J = 8.4 Hz); 4.27 (B of ABq, 1H, J = 8.4 Hz); 4.05 (d, 1H, J = 7.2 Hz); 2.49-1.16 (m, 11H, включая синглеты на 2.47 (3H), 2.20 (3H), 1.88 (3H), 1.72 (3H), 1.38 (9H); 0.83 (t, 9H, J = 5 Hz); 0.55-0.37 (m, 6H).
13C NMR (75.6 MHz, CDCl3): δ 206.0; 171.1; 169.4; 169.1; 167.2; 155.2; 152.1; 141.8; 141.4; 133.6; 131.8; 130.1; 129.2; 128.7; 110.6; 107.1; 96.2; 93.9; 81.9; 80.7; 80.0; 78.7; 77.9; 77.8; 75.0; 72.3; 70.8; 56.9; 56.7; 52.7; 42.6; 40.0; 35.5; 33.9; 33.6; 28.1; 28.0; 25.5; 22.5; 21.2; 20.7; 14.6; 14.5; 14.3; 14.2; 6.4; 4.2.
Пример 30. Аналог 7 -α- фтор-2'-O-триэтилсилил-3'-(2-тиенил)-3'-N-BOC таксола.
Соединение получают по методике примера 29 в виде белой пены. Выход 78% в расчете на исходный материал.
1H NMR (300 MHz, CDCl3): δ 8.14 (d, 2H, J = 9.0 Hz); 7.63-7.58 (m, 1H); 7.51-7.48 (m, 2H); 7.24 (dd, 2H, J = 2.4, 3.6 Hz); 7.00-6.93 (m, 2H); 6.58 (s, 1H); 6.23 (t, 1H, J = 9 Hz); 5,77 (d, 1H, J = 6 Hz); 5.51-5.42 (m, 2H); 5.03 (d, 1H, J = 9 Hz); 4.57 (d, 1H, H = 3 Hz); 4.59 (dd, C-7 proton 1H, J = 6, JH-F = 48 Hz); 4.38 (A of ABq, 1H, J = 6 Hz); 4.27 (B of ABq, 1H, J = 6 Hz); 4.05 (d, 1H, J = 7 Hz); 2.57-1.15 (m, 11H, включая синглеты на 2.44 (3H), 2.20 (3H), 1.88 (3H), 1.70 (3H), 1.32 (9H); 0.86 (t, 9H, J = 5 Hz); 0.56-0.41 (m, 6H).
13C NMR (75.6 MHz, CDCl3): δ 296.0; 171.0; 169.4; 168.8; 167.2; 161.4; 142.9; 141.3; 133.6; 131.8; 130.2; 129.2; 128.7; 126.9; 124.6; 124.5; 96.3; 93.9; 81.9; 80.8; 80.0; 78.8; 77.9; 77.8; 77.2; 76.5; 75.2; 75.0; 71.0; 65.4; 56.9; 53.7; 42.7; 40.3; 35.6; 33.6; 28.1; 22.7; 21.3; 20.8; 18.8; 14.5; 14.3; 10.4; 6.3; 4.5.
Пример 31. Аналог 7 -α- фтор-3'-(2-фурил)-3'-N-BOC таксола (соединение 2).
Раствор 2'-O-силил защищенный субстрат (80 мг, 0.08 ммоль) в ацетонитриле (2 мл) охлаждают до 0oC на бане лед/солевой раствор. К раствору добавляют 1 н. HCl (0.5 мл, 6 экв.) и реакционную массу перемешивают в течение 30 мин при этой температуре. Затем растворитель выпаривают в вакууме и остаток распределяют между этилацетатом (25 мл) и водой (10 мл). Органическую фракцию сушат (MgSO4) и концентрируют с получением белой пены. Неочищенный продукт очищают на силикагеле, используя 10% CH3CN в CH2Cl2 в качестве элюента. Целевой продукт выделяют в виде белой пены (45,6 мг, 77% в расчете на исходный материал). [α]D = -26/2o (конц. 0,8 мг/мл, CH2Cl2).
1HNMR (300 MHz, CDCl3): δ 8,12 (d, 2H, J = 6 Hz); 7,63 - 7,58 (m; 1H); 7,50 (t, 2H, J = 6 Hz); 7,41 (s, 1H); 6,57 (s, 1Н); 6,37 - 6,36 (m, 1H); 6,33 - 6,31 (m, 1H); 6,20 (t, 1H, J = 6 Hz); 5,76 (d, 1H, J = 6 Hz); 5,37 - 5,23 (m, 2H); 5,02 (d, 1H, J = 9 Hz); 4,71 (broad s, 1H); 4,57 (dd, C-7 ptoton 1H, J = 4,2, JH-F = 46,8 Hz); 4,36 (A of ABq, 1H, J = 8,7 Hz); 4,27 (B of ABq, 1H, J = 8,1 Hz); 4,04 (d, 1H, J = 7,2 Hz); 3,28 (broad s, 1H); 2,59 - 2,20 (m, 5H, включая синглеты н 2,41 (3H); 2,21 (3H); 1,85 (s, 3H); 1,43 - 1,17 (m, 18H).
13CNMR (75,6 MHz, CDCl3): δ 205,7; 169,2; 169,0; 167,1; 142,3; 140,6; 133,5; 132,1; 130,0; 129,1; 128,6; 110,5; 107,2; 95,9; 93,6; 81,8; 80,6; 78,5; 77,8; 77,7; 74,7; 72,1; 71,6; 56,6; 55,8; 51,5; 42,5; 39,9; 35,4; 33,8; 33,5; 28,0; 27,9; 25,6; 22,2; 20,9; 20,7; 14,5; 14,1; 14,0.
HRMS вычисл. для MH+ (C43H53NO15F): 842, 3399; найдено: 842, 3389
Пример 32. Аналог 7 -α- фтор-3'-2-(тиенил)-3'-N-BOC таксола (Соединение 3).
Целевое соединение получали по методике примера 31, используя лактам на основе тиофена. Желаемый продукт выделяли в виде белой пены (22,5 мг, 61%).
1HNMR (300 MHz, CDCl3): δ 8,12 (d, 2H, J = 9 Hz); 7,64 - 7,59 (m; 1H); 7,50 (t, 2H, J = 9 Hz); 7,28 - 7,26 (m, 2H); 7,09 - 7,07 (m, 1H); 7,01 - 6,98 (m, 1H); 6,56 (s, 1H); 6,19 (t, 1H, J = 9 Hz); 5,76 (d, 1H, J = 6 Hz); 5,53 (broad d, 1H, J = 12 Hz); 5,35 (d, 1H, J = 9 Hz); 5,00 (d, 1H, J = 9 Hz); 4,65 - 4,63 (m, 15H, C-7 proton hidden); 4,48 (d, 0,5H, J = 9 Hz); 4,36 (A of ABq, 1H, J = 8,7 Hz); 4,27 (B of ABq, 1H, J = 8,1 Hz); 4,04 (d, 1H, J = 7,2 Hz); 3,28 (broad s, 1H); 2,59 - 2,20 (m, 5H, включая синглеты на 2,39 (3H); 2,18 (3H); 1,72 (s, 3H); 1,43 - 1,17 (m, 18H).
13CNMR (75,6 MHz, CDCl3): δ 205,6; 172,1; 169,3; 169,0; 167,0; 141,5; 140,6; 133,6; 132,1; 130,1; 129,3; 129,0; 128,6; 126,9; 125,2; 125,1; 95,9; 93,5; 81,8; 80,8; 80,2; 78,5; 77,8; 77,7; 77,3; 76,8; 74,7; 73,3; 72,2; 56,9; 52,5; 42,5; 39,9; 35,5; 33,8; 33,5; 28,0; 27,9; 25,6; 22,3; 20,9; 20,7; 14,6; 14,1; 14,0.
Пример 33. 7 -α- Фтор-3'-N-бензоил-3'-(3-фурил)паклитаксел (Соединение 5).
1H NMR (300 MHz, CDCl3): δ 8,17 - 8,14 (m, 2H); 7,73 - 7,36 (complex m, 10H); 6,82 (d, 1H, J=9,3 Hz); 6,54 (s, 1H); 6,51 (s, 1H); 6,18 (t, 1H, J=8,6 Hz); 5,76 (d, 2H, J=7,4 Hz); 5,00 (d, 1H, J=8,4 Hz); 4,69 - 4,64 (m, 1H); 4,56 (dd, 1H, J=4,8 Hz, JHF=46,8 Hz); 4,33 (ABq, 2H, J=8,3, 24,6 Hz); 4,06 (d, 1H, J= 7,2 Hz); 3,64 (d, 1H, J=4,8 Hz); 2,55 - 1,11 (complex m, 23H, включая синглеты на 2,39, 2,20, 1,80, 1,68, 1,18, 1,16).
13C NMR (76,5 MHz, CDCl3): δ 20,5; 172,1; 169,2; 166,9; 166,6; 143,5; 140,2; 133,6; 132,4; 131,8; 130,1; 129,1; 128,6; 128,5; 126,9; 122,9; 109,5; 95,9; 93,5; 81,8; 80,9; 78,4; 77,8; 77,7; 76,4; 74,6; 72,6; 71,9; 47,8; 42,4; 39,8; 35,6; 33,9; 33,6; 25,8; 22,2; 20,8; 20,7; 147,7; 14,6; 14,1; 14,0.
Пример 34. 7 -α- Фтор-10-дезацетил-3'-N-BOC-3'-(2-фурил)паклитаксел (Соединение 6).
1H NMR (300 MHz, CDCl3): δ 8,14 - 8,11 (m, 2H); 7,63 - 7,39 (complex m, 4H); 6,37 - 6,30 (m, 2Н); 6,21 (t, 1H, J=9,0 Hz); 5,74 (d, 1H, J=7,1 Hz); 5,33 - 5,24 (m, 9H); 5,00 (d, 1H, J=7,0 Hz); 4,69 (br, d, 1H, J=2,7 Hz); 4,55 (dd, 1H, J=5,0 Hz, JHF=47,0 Hz); 4,34 (ABq, 2H, J=8,3, 46,6 Hz); 4,06 (d, 1H, J= 7,3 Hz); 3,34 (d, 1H, J=4,8 Hz); 2,56 - 1,08 (complex m, 30H, включая синглеты на 2,40, 1,87, 1,34, 1,21, 1,11).
13C NMR (76,5 MHz, CDCl3): δ 205,7; 172,3; 169,2; 167,1; 151,3; 142,3; 138,8; 134,5; 133,6; 130,1; 129,2; 128,7; 110,5; 107,3; 96,1; 93,7; 81,7; 80,7; 80,3; 78,1; 77,9; 77,8; 77,5; 74,9; 72,3; 71,7; 56,7; 56,5; 51,5; 42,5; 39,9; 35,7; 33,9; 33,6; 28,3; 28,1; 25,6; 22,3; 20,3; 19,6; 14,7; 14,6; 14,3; 13,9.
Пример 35. 7 -α- Фтор-3'-N-BOC-3'-(3-метил-2-бутенил)паклитаксел (Соединение 7).
1H NМR (300 MHz, CDCl3): δ 8,10 (d, 2H, J=8,5 Hz); 7,60 (t, 1H, J=6,9 Hz); 7,45 (t, 2H, J=7,5 Hz); 6,59 (s, 1H); 6,11 (t, 1H, J=9,0 Hz); 5,73 (d, 1H, J=7,2 Hz); 5,27 (d, 1H, J=7,2 Hz); 5,00 (d, 1H, J=8,1 Hz); 4,89 (d, 1H, J=8,6 Hz); 4,73 (br t, 1H); 4,55 (dd, 1H, J=2,1 Hz, JHF=47,1 Hz); 4,30 (ABq, 2H, J= 8,4, 26,1 Hz); 4,22 - 4,19 (m, 1H); 4,02 (d, 1H, J=7,0 Hz); 3,43 (d, 1H, J=6,3 Hz); 2,54 - 1,15 (complex m, 37H, включая синглеты на 2,28, 2,19, 1,85, 1,74, 1,70, 1,34, 1,23, 1,15).
13C NМR (76,5 МHz, CDCl3): δ 205,8; 172,7; 169,3; 168,9; 166,9; 155,3; 141,0; 137,7; 133,6; 132,0' 130,1; 129,9; 129,3; 128,6; 120,8; 96,0; 93,6; 81,8; 80,7; 79,7; 78,6; 77,9; 76,5; 74,8; 73,7; 71,9; 56,9; 56,7; 51,4; 42,5; 39,9; 35,7; 33,8; 33,5; 28,1; 27,9; 25,6; 22,1; 21,0; 20,7; 18,5; 14,7; 14,6; 14,2; 14,1.
Пример 36. 7 -α- Фтор-3'-N-BOC-3'-(3-фурил) паклитаксел (Соединение 8).
1H NМR (300 MHz, CDCl3): δ 8,136 (d, 2H, J=7,2 Hz); 7,62 - 7,41 (m, 5H); 6,56 (s, 1Н); 6,44 (d, 1H, J=0,78 Hz); 6,18 (t, 1H, J=8,3 Hz); 5,76 (d, 1H, J= 7,0 Hz); 5,22 - 5,11 (m, 2H); 5,01 (d, 1Н, J=8,1 Hz); 4,56 (dd, 1H, J=4,2 Hz, JHF= 46,8 Hz); 4,53 (d, 1H, J=4,2 Hz); 4,31 (ABq, 2H, J=8,4, 30,3 Hz); 4,04 (d, 1H, J=7,0 Hz); 3,42 (d, 1H, J=5,4 Hz); 2,55 - 1,17 (complex m, 32H, включая синглеты на 2,41, 2,20, 1,85, 1,72 1,66, 1,34, 1,25, 1,77).
13C NMR (76,5 MHz, CDCl3): δ 205,7; 172,5; 169,4; 169,0; 167,1; 155,2; 143,5; 140,7; 140,0; 133,7; 132,2; 130,2; 130,0; 129,2; 128,7; 123,5; 109,4; 96,0; 93,6; 93,3; 84,7; 81,9; 80,9; 80,1; 80,0; 78,6; 77,9; 77,8; 75,7; 74,8; 72,8; 72,1; 56,8; 53,1; 47,1; 42,2; 35,6; 33,6; 33,3; 28,1; 22,3; 20,9; 18,8; 15,2; 14,7; 14,2; 10,2; 8,7;
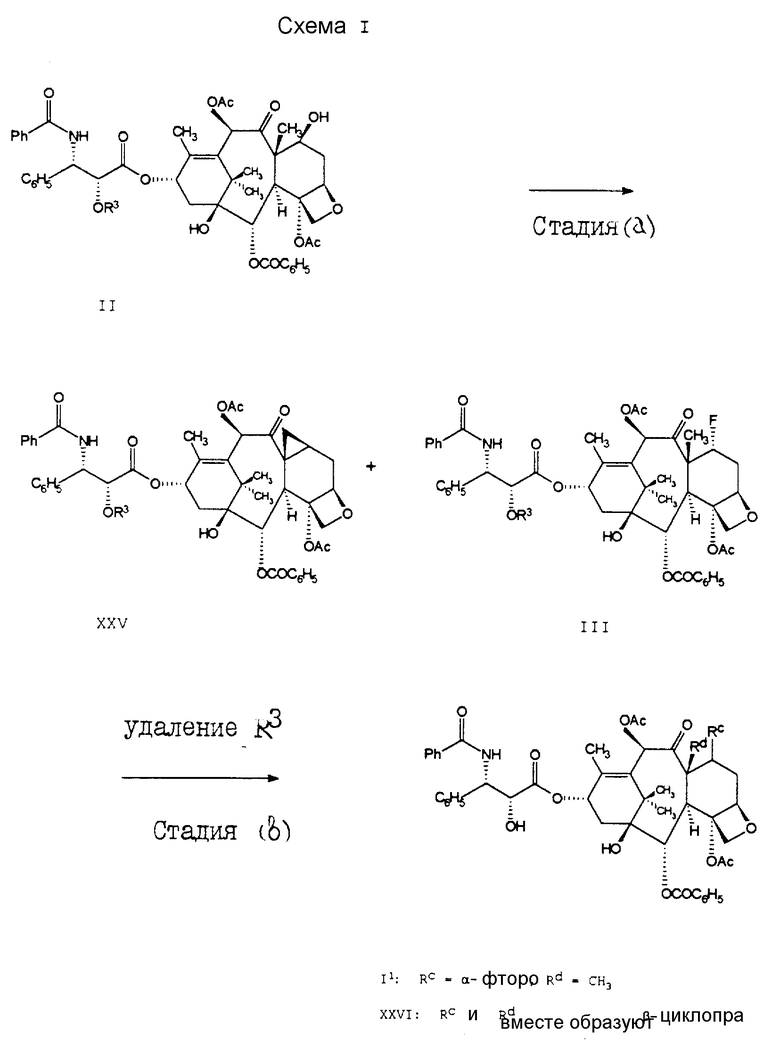
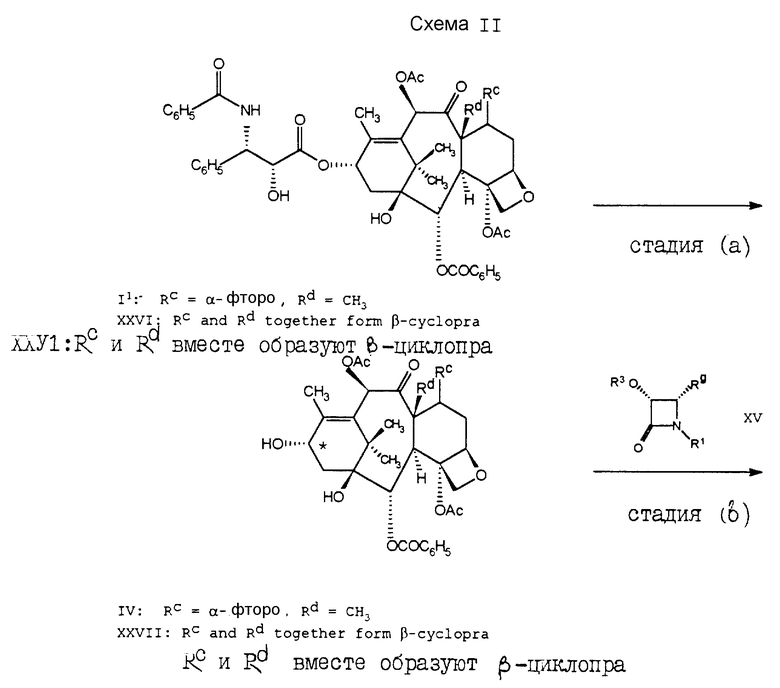
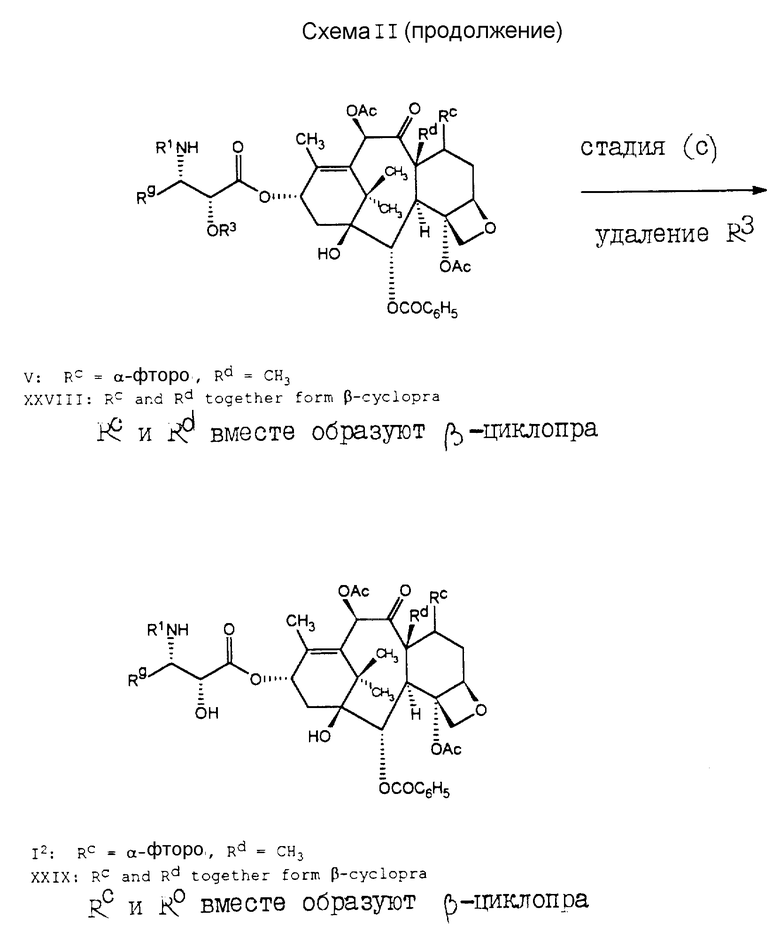
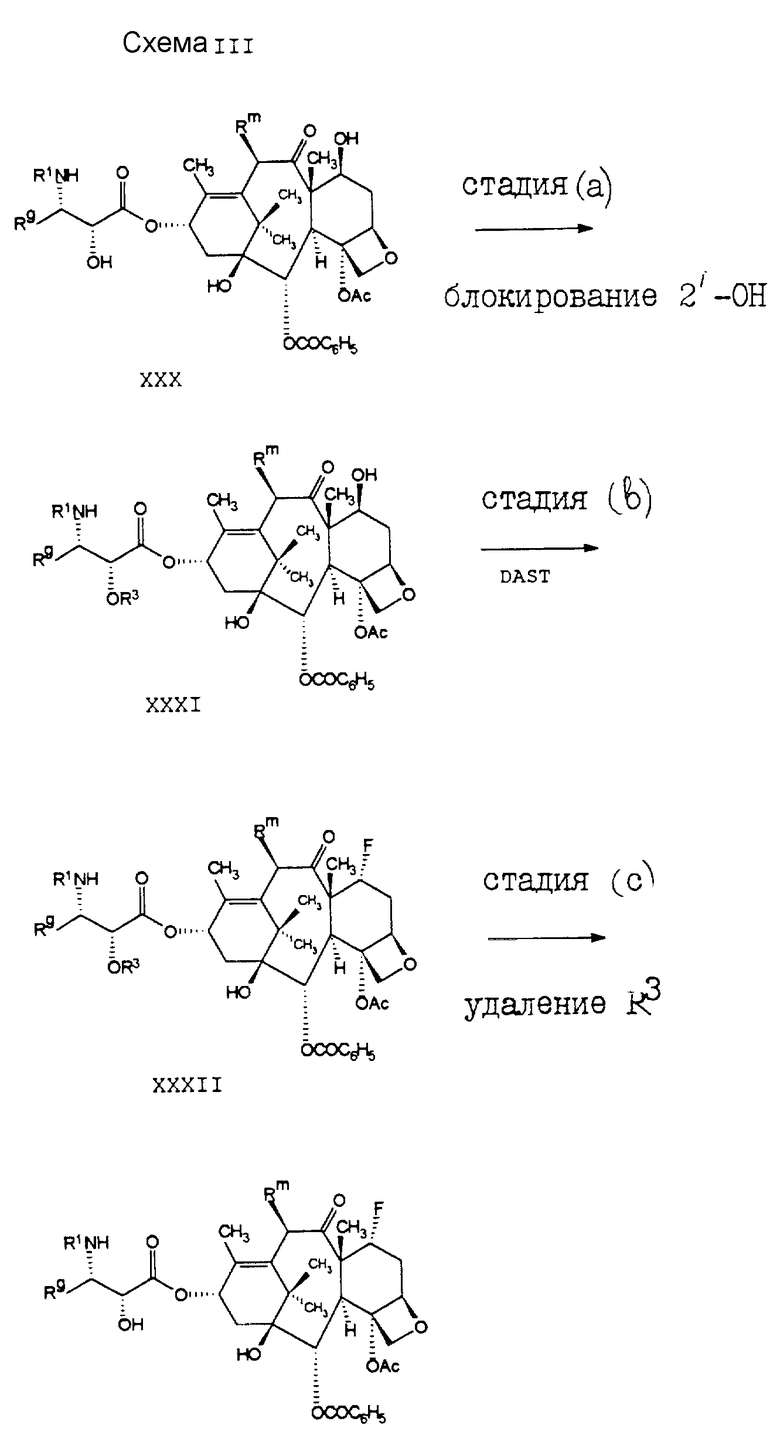
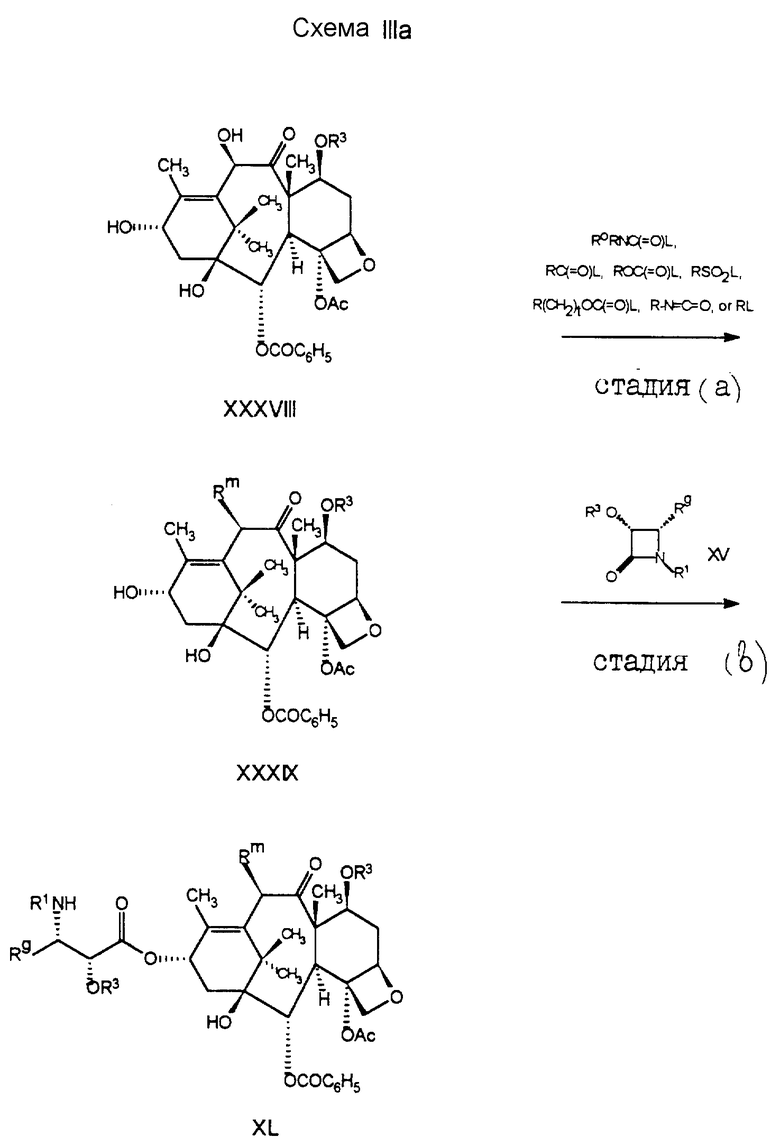
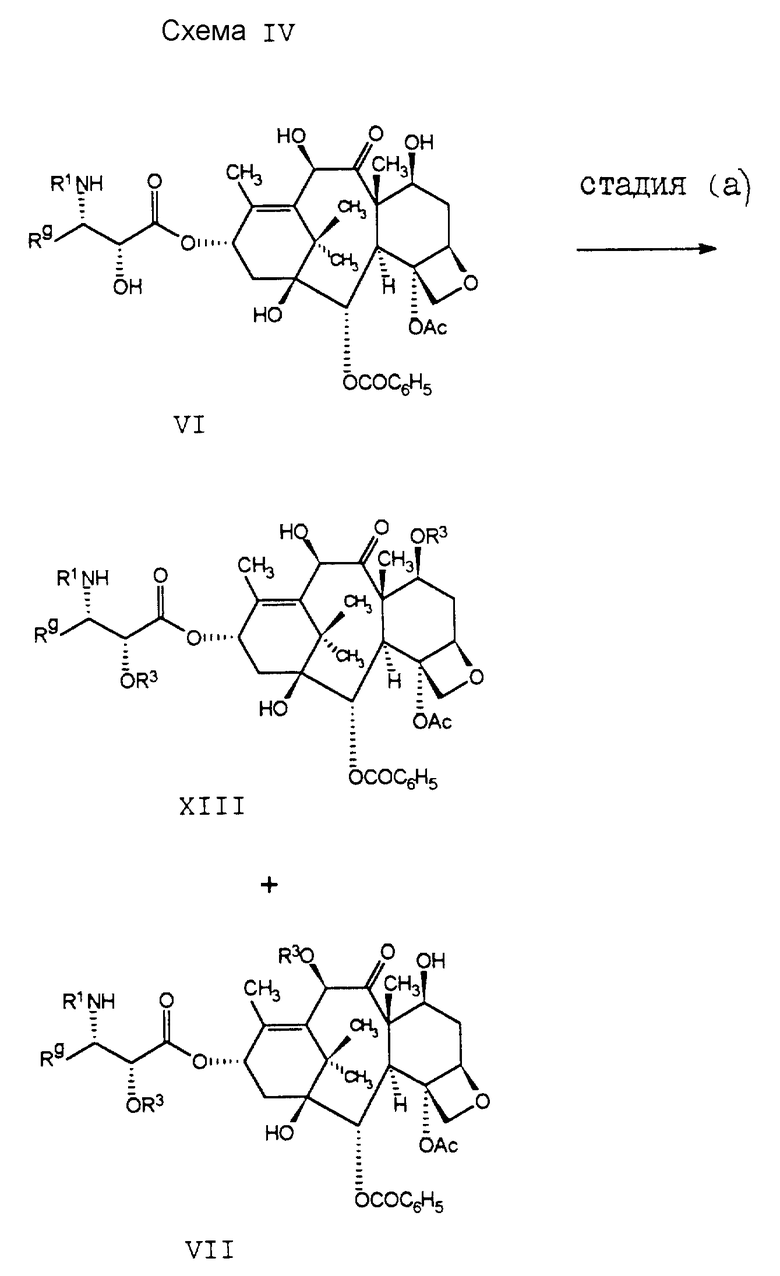
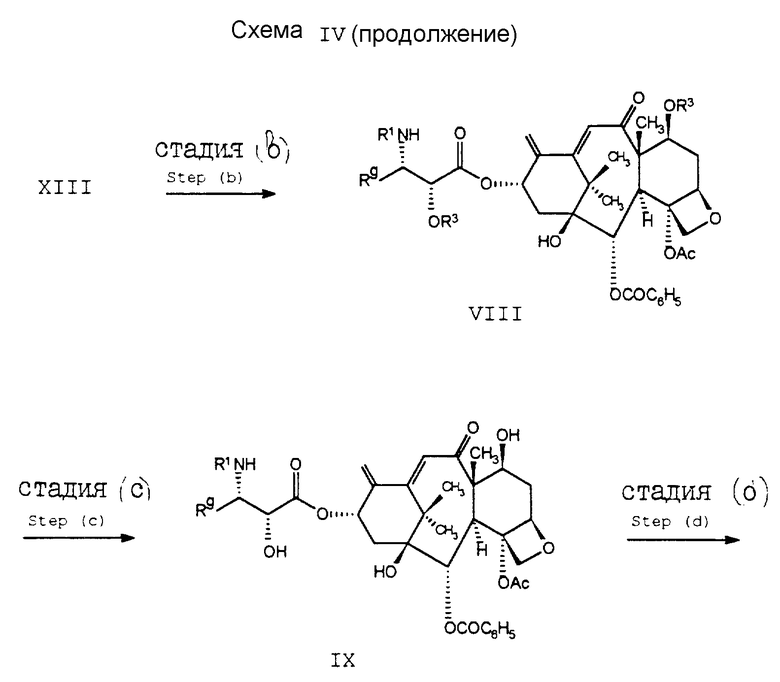
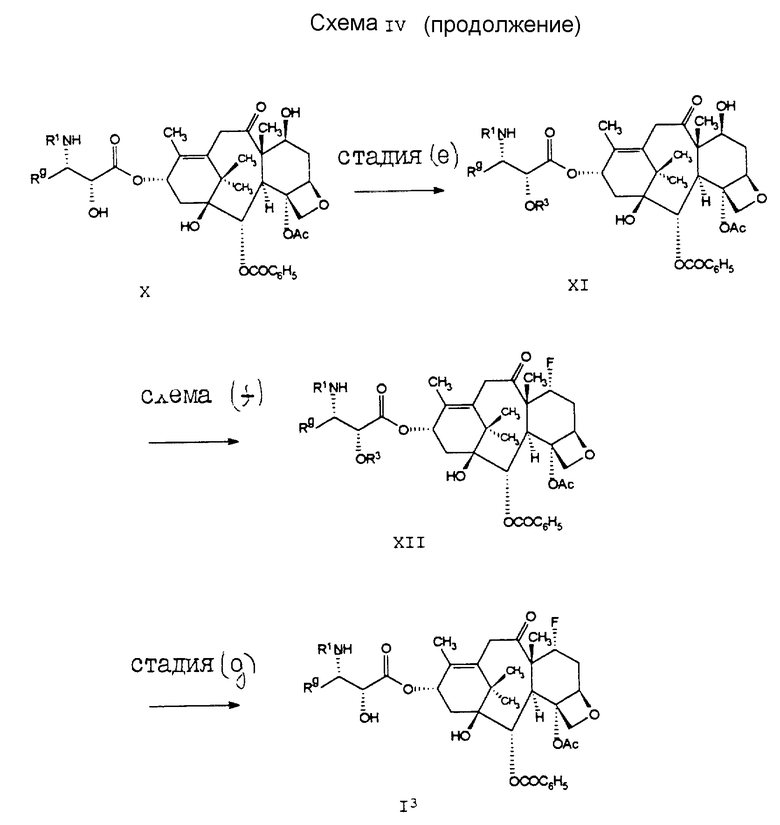
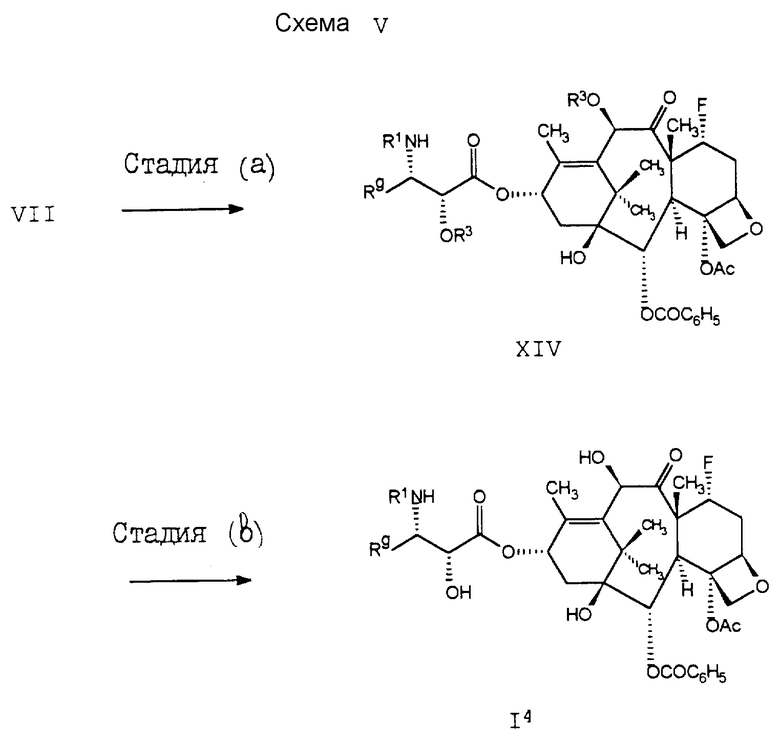
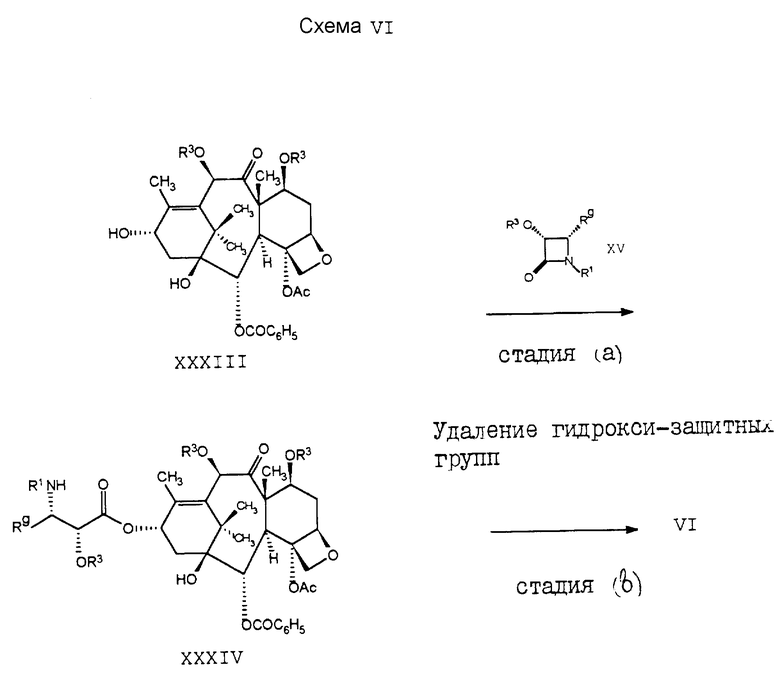
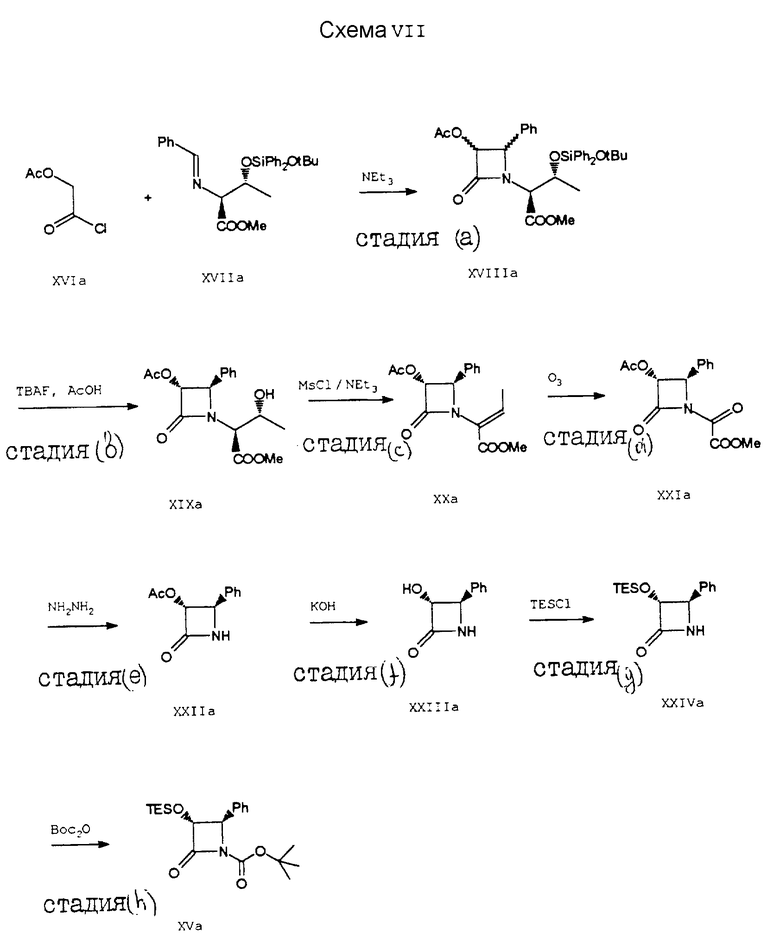
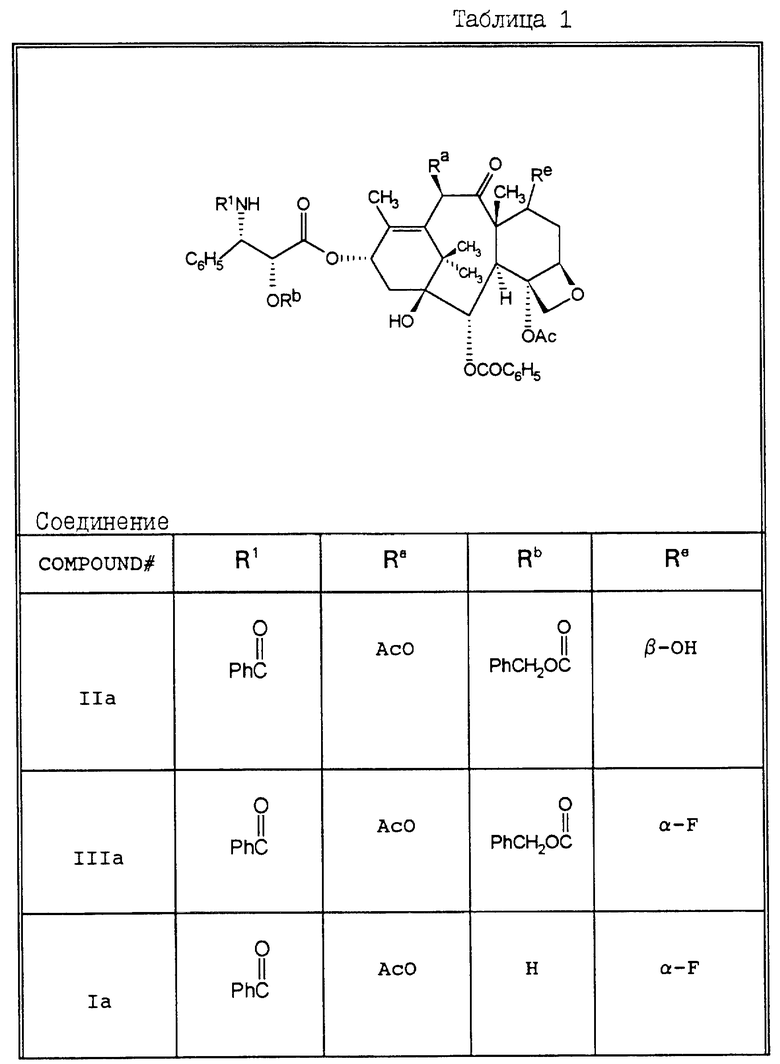
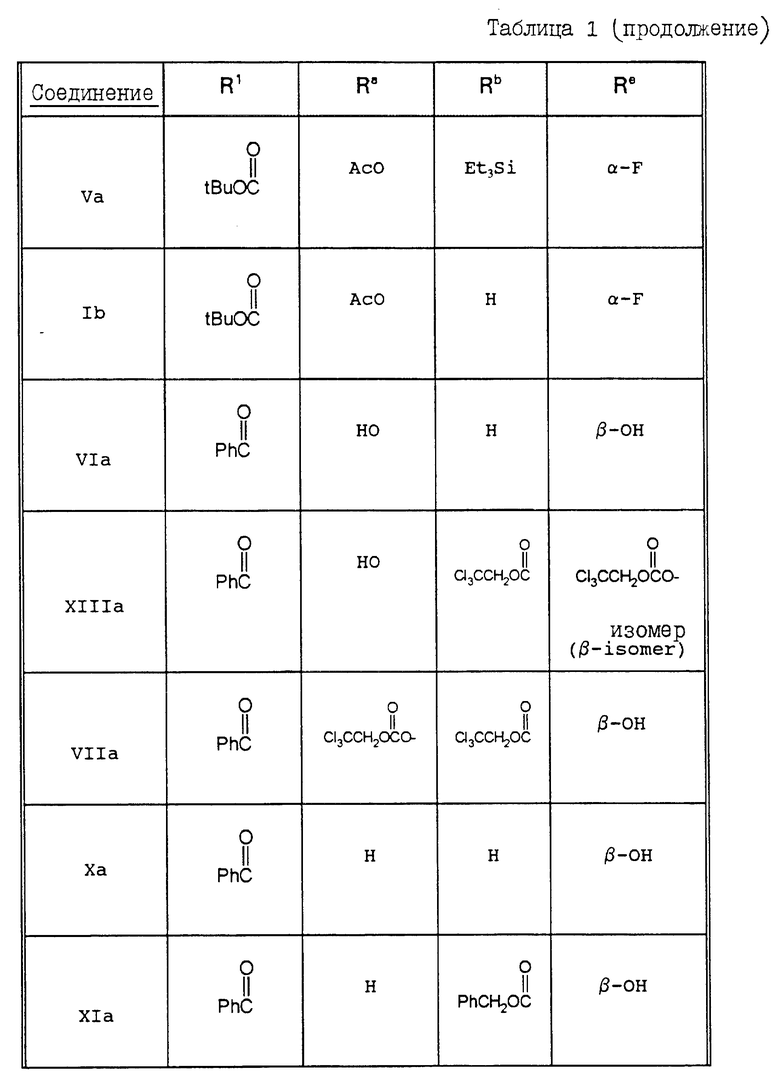
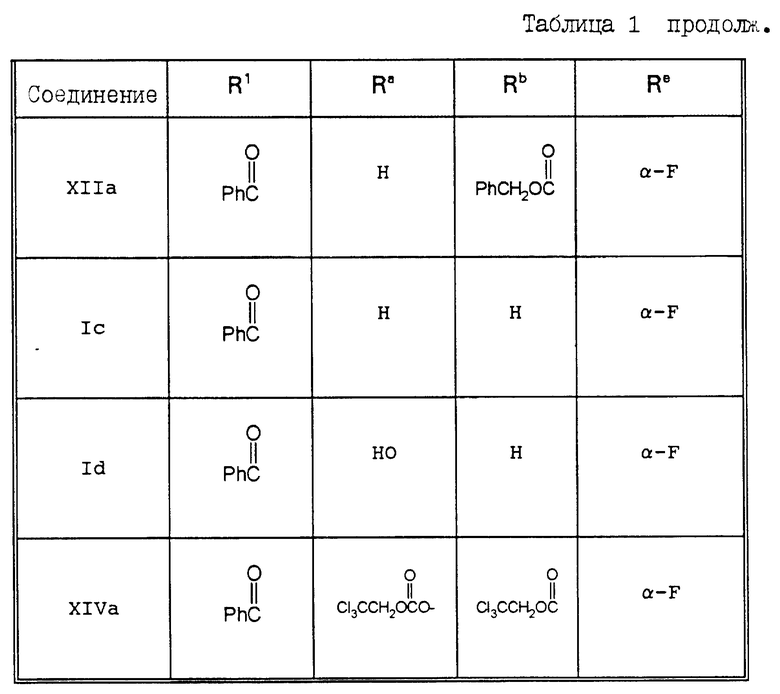
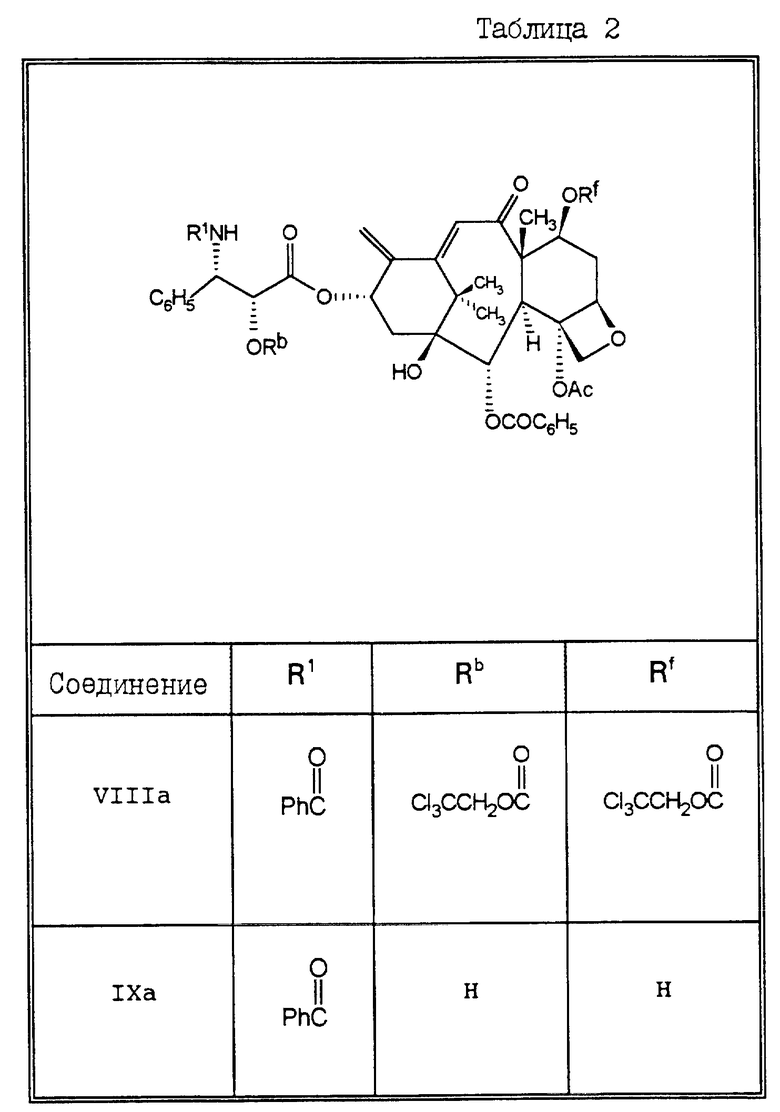
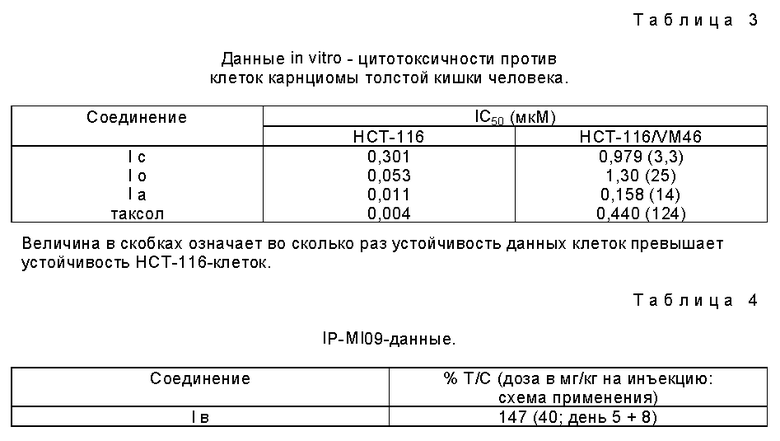
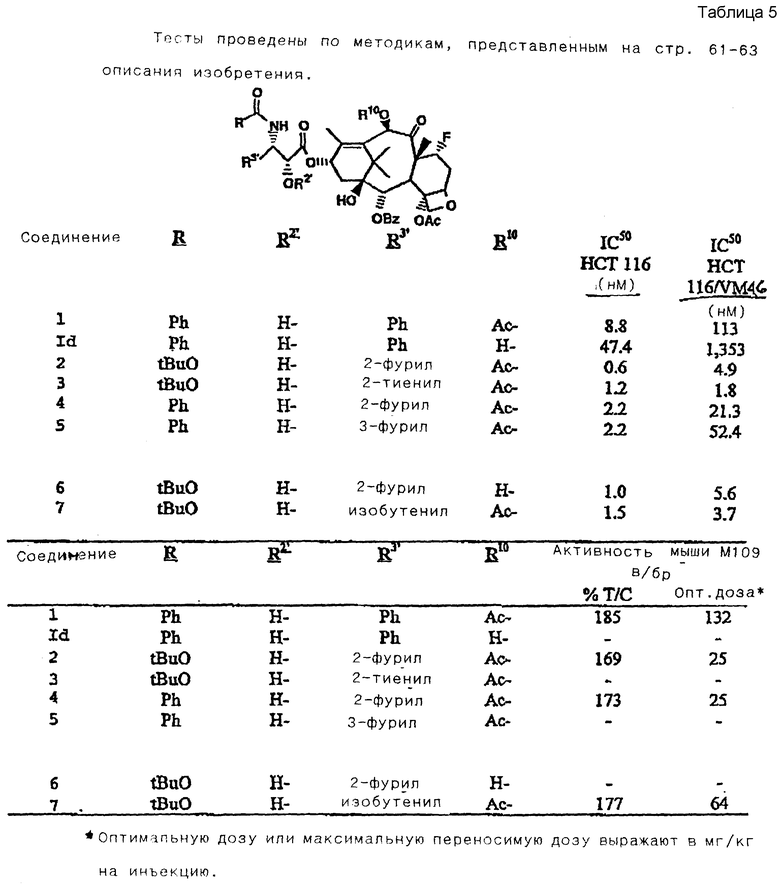
Описываются новые фторотаксолы формулы (I), где R1 представляет собой - CORz, в котором Rz является RO- или R, Rg представляет собой C2-6-алкенил или радикал формулы W-Rx, в котором W является связью, а Rx - фурилом, тиенилом или фенилом; R2 представляет собой -ОСО, Н, ОН, -OR, -OSO2R1-ОСОО(СН2)tR или -ОСООR где t = 1-6, целое число, R независимо представляет собой C1-6 -алкил или фенил. Описываются промежуточные соединения, фармацевтическая композиция, обладающая противоопухолевой активностью на основе соединения формулы (I), а также способ лечения опухолей у млекопитающих с использованием вышеуказанных соединений. 4 с. и 9 з.п.ф-лы, 7 ил., 2 табл.
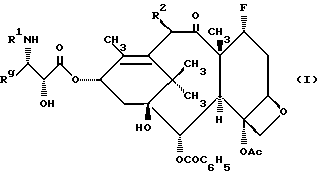
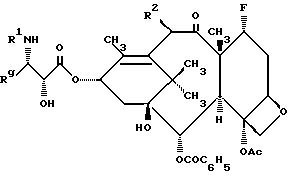
где R1 представляет собой -CORz, в котором Rz является RO- или R;
Rg - C2-6-алкенил или радикал формулы W-Rx, в котором W является связью, а Rx - фурилом, тиенилом или фенилом;
R2 представляет собой -OCOR, H, OH, -OR, -OSO2R, -OCOO(CH2)tR или -OCOOR, где t = 1 - 6, целое число; R независимо - C1-6-алкил или фенил.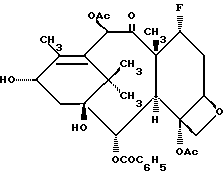
где Ac - ацетил.
Приоритет по пунктам и признакам:
01.07.92 по п. 1: R1 - бензоил (т.е. R1 - -CORz, где Rz - R, где R - фенил) или трет-бутоксикарбонил (т.е. -CORz, где Rz-RO, где R - трет-бутил или C1-6-алкил); R2 - H, OH или ацетокси (т.е. OCOR, где R - метил или C1-6-алкил); R9 - фенил; по п.2: все группы, за исключением R9 - 2-тиенил или 2-фурил; и по п.3 - 6 и 13.
| 0 |
|
SU400971A1 | |
| Узел крепления электрического аппарата | 1971 |
|
SU473326A1 |
| Реле времени | 1974 |
|
SU505047A1 |
| WO 9209589, 11.06.92 | |||
| Противоопухолевая химиотерапия | |||
| Справочник/Под ред | |||
| Н.И.Переводчиковой.-М.: Медицина, 1993, с.6-39 | |||
| Машковский М.Д | |||
| Лекарственные средства.-М.: Медицина, 1993, т.2, с.493-553. | |||

