Данная заявка является частичным продолжением одновременно рассматриваемой заявки 07/995443 данных заявителей, поданной 23 декабря 1992 г., которая включена в данное описание путем ссылки.
Настоящее изобретение относится к способам получения таксанов с боковой цепью и их интермедиатов и к новым соединениям, полученным этими способами.
Таксаны являются дитерпеновыми соединениями, которые нашли применение в области изготовления лекарственных форм. Например, было обнаружено, что эффективным противоопухолевым средством является таксол - таксан, имеющий структуру: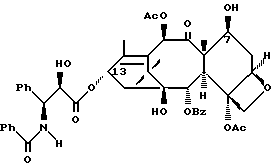
где
Ph - фенил, Ac - ацетил и BZ - бензол. Природные таксаны, такие, как таксол, можно найти в растительных материалах и выделить их оттуда. Но в растительных материалах такие таксаны могут присутствовать в относительно небольших количествах, и потому в случае, например, таксола может потребоваться большое количество деревьев медленно растущего тиса, являющегося источником получения этого соединения. Поэтому продолжались поиски путей синтетического, включая полусинтетическое, получения таксанов, таких, как таксол и его аналоги, а также путей получения промежуточных продуктов (интермедиатов), используемых в получении этих соединений.
В соответствии с настоящим изобретением предлагается новый общий способ получения новых таксанов с боковой цепью, включающий в себя следующие стадии (a)-(e):
(a) получение оксазолинового соединения следующей формулы I или его соли: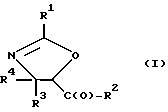
где
R1> - R5, R7-O-, R7 - или (R5)(R6)N-;
R2 - R7-O-, R7- или (R5)(R6)N-;
R3 и R4 - независимо R5, R5-O-C(O)- или (R5)(R6)N-C(O)-;
R5 и R6 - независимо водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло; и
R7 - алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло;
(b) превращение оксазолина формулы I или его соли в оксазолин формулы II или его соль: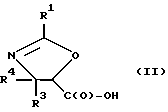
где
R1, R3 и R4 - такие, как указаны выше;
(c) сочетание оксазолина формулы II или его соли с таксаном, имеющим гидроксильную группу, непосредственно связанную с его атомом C-13, или его солью для образования несущего оксазолиновую боковую цепь таксана следующей формулы III или его соли: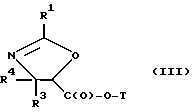
где
R1, R3 и R4 - такие, как указаны выше,
T - таксановый фрагмент, предпочтительно соединение формулы IX, связанное непосредственно через C-13 упомянутого фрагмента;
(d) контактирование несущего оксазолиновую боковую цепь таксана формулы III или его соли с водной кислотой, способной разрывать оксазолиновое кольцо соединения формулы III или его соли, для образования несущего боковую цепь таксана следующей формулы X или его соли: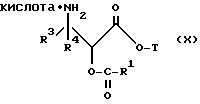
где
R1, R3, R4 и T - такие, как указаны выше, и кислая соль при аминогруппе в упомянутой формуле X образуется в результате контакта с упомянутой разрывающей кольцо кислотой; и
(e) контактирование несущего боковую цепь таксана формулы X или его соли с основанием для образования несущего боковую цепь таксана следующей ниже формулы IV или его соли: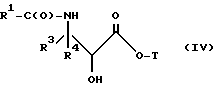
где
R1, R3, R4 и T - такие, как указаны выше.
Кроме того, в соответствии с настоящим изобретением предлагаются индивидуальные способы каждой из стадий (a)-(e), являющиеся новыми способами, и новые соединения формул I, II, III, IV, IX и X и их соли и гидраты, такие, как описаны ниже. Предлагаются также новые лекарственные предшественники этих соединений.
Термины "алкил" или "алк", используемые в данном описании отдельно или как часть другой группы, означают необязательно замещенные неразветвленные и разветвленные насыщенные углеводородные группы, предпочтительно имеющие 1-10 атомов углерода в нормальной цепи, наиболее предпочтительно низшие алкильные группы. Примерами незамещенных таких групп являются метил, этил, пропил, изопропил, н-бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, октил, 2,2,4 - триметилпентил, нонил, децил, ундецил, додецил и т.п. Примерными заместителями могут быть одна или несколько следующих групп: галоген, алкокси, алкилтио, алкенил, алкинил, арил (напр., для образования бензильной группы), циклоалкил, циклоалкенил, гидрокси или защищенный гидрокси, карбоксил (-COOH), алкилоксикарбонил, алкилкарбонилокси, алкилкарбонил, карбамоил (NH2-CO-), замещенный карбамоил (R5)(R6)N-CO-, где R5 или R6 - такие, как указаны выше, за исключением, когда, по крайней мере, один из радикалов R5 или R6 не является водородом), амино (-NH2), гетероцикло, моно- или диалкиламино или тиол (-SH).
Термины "низший алкил" или "низший алк" при использовании в данном описании означают такие необязательно замещенные группы, как те, что описаны выше для алкила, имеющего 1-4 атомов углерода в нормальной цепи.
Термины "алкокси" или "алкилтио" означают такую, как описаны выше, алкильную группу, присоединенную через кислородный (-O)- или серный (-S-) мостик соответственно. Термин "алкилоксикарбонил" при использовании в данном описании означает алкоксигруппу, присоединенную через карбонильную группу. Термин "алкилкарбонил" при использовании в данном описании означает алкильную группу, присоединенную через карбонильную группу. Термин "алкилкарбонил" при использовании в данном описании означает алкильную группу, присоединенную через карбонильную группу, которая, в свою очередь, присоединена через кислородный мостик. Термины "моноалкиламино" или "диалкиламино" означают аминогруппу, замещенную на одну или две описанные выше алкильные группы соответственно.
Термин "алкенил" при использовании в данном описании отдельно или как части группы означает необязательно замещенные углеводородные группы с неразветвленными и разветвленными цепями, содержащей, по крайней мере, одну углерод-углеродную двойную связи в цепи и предпочтительно имеющие 2-10 атомов углерода в нормальной цепи. Примеры незамещенных таких групп включают этенил, пропенил, изобутенил, бутенил, пентенил, гексенил, гептенил, октенил, ноненил, деценил, и т. п. Примерами заместителей могут быть одна или несколько следующих групп: галоген, алкокси, алкилтио, алкил, алкинил, арил, циклоалкил, циклоалкенил, гидрокси или защищенный гидрокси, карбоксил (-COOH), алкилоксикарбонил, алкилкарбонилокси, алкилкарбонил, карбамоил (NH2-CO-), замещенный карбамоил ((R5)(R6)N-CO-, где R5 или R6 - такие, как указаны выше, если, по крайней мере, один из радикалов R5 или R6 не являются водородом), амино (-NH2), гетероцикло, моно- или алкиламино или тиол (-SH).
Термин "алкинил" при использовании в данном описании отдельно или как части другой группы означает необязательно замещенные неразветвленные или разветвленные углеводородные группы, содержащие, по крайней мере, одну углерод-углеродную тройную связь в цепи и предпочтительно имеющие 2-10 атомов углерода в нормальной цепи. Примеры незамещенных таких групп включают этинил, пропинил, бутинил, пентинил, гексинил, гептинил, октинил, нонинил, децинил и т.п. Примеры заместителей могут выключать одну или несколько следующих групп: галоген, алкокси, алкилтио, алкил, алкенил, арил, циклоалкил, циклоалкенил, гидрокси или замещенный гидрокси, карбоксил (-COOH), алкилоксикарбонил, алкилкарбонилокси, алкилкарбонил, карбамоил (NH2-CO-), замещенный карбамоил ((R5)(R6)N-CO-, где R5 или R6 - такие, как указаны выше, если, по крайней мере, один из R5 или R6 не является водоводом амино (-NH2), гетероцикло, моно- или диалкиламино или тиол (-SH).
Термин "циклоалкил" при использовании в данном описании отдельно или как части другой группы означает необязательно замещенные насыщенные циклические углеводородные кольцевые системы, предпочтительно содержащие 1-3 кольца и 3-7 атомов углерода на кольцо. Примерами незамещенных таких групп являются циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил, циклододецил и адамантил. Примеры заместителей включают одну или несколько таких, как описаны выше, алкильных групп или одну или несколько групп, описанных выше в качестве алкильных заместителей.
Термин "циклоалкенил" при использовании в данном описании отдельно или как части другой группы означает такие необязательно замещенные группы, как те, что описаны выше для циклоалкила, но дополнительно содержащие, по крайней мере, одну углерод-углеродную двойную связь, образующую частично ненасыщенное кольцо.
Термины "ар" или "арил" при использовании в данном описании отдельно или как части другой группы означает необязательно замещенные гомоциклические ароматические группы, предпочтительно содержащие 1 или 2 кольца 6-12 атомов углерода в кольце. Примерами незамещенных таких групп являются фенил, бифенил и нафтил. Примеры заместителей включают одну или более, предпочтительно три или меньше нитрогрупп, алкильных групп, таких, как описаны выше или группы, описанных выше в качестве алкильных заместителей.
Термины "гетероцикло" или "гетероциклический" при использовании в данном описании отдельно или как части другой группы означают необязательно замещенные полностью ненасыщенные или ненасыщенные ароматические или неароматические циклические группы, имеющие, по крайней мере, один гетероатом в, по крайней мере, одном кольце, предпочтительно, моноциклические или бициклические группы, имеющие 5 или 6 атомов в каждом кольце. Гетероциклическая группа может, например, иметь 1 или 2 атома кислорода, 1 или 2 атома и/или 1-4 атома азота в кольце. Каждая гетероциклическая группа может быть присоединена через любой атом углерода или гетероатом кольцевой системы. Примерными гетероциклическими группами являются следующие: тиенил, фурил, пирролил, пиридил, имидазолил, пирролидинил, пиперидинил, азепинил, индолил, азоиндолил, хинолинил, изохинолинил, бензотиазолил, бензоксазолил, бензимидазолил, бензоксадиазолил и бензофуразонил. Примерами заместителей являются одна или несколько таких, как описаны выше, алкильных групп или одна или более групп, описанных выше в качестве алкильных заместителей. К таким примерам относятся также меньшие гетероциклы, такие, как эпоксиды и азиридины.
Термины "галоген", "гало" или "гал" при использовании в данном описании отдельно или как части другой группы означает хлор, бром, фтор и иод.
Термин "таксановый фрагмент" при использовании в данном описании отдельно или как части другой группы означает фрагменты, содержащие структуру ядра: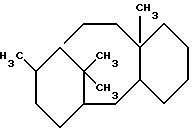
которая может быть замещенной и может содержать этиленовую ненасыщенную в кольцевой системе.
Термин "таксан" при использовании в данном описании означает соединения, содержащие описанный выше таксановый фрагмент.
Термин "гидрокси(или гидроксил)защитная группа" при использовании в данном описании означает любую группу, способную защищать свободную гидроксильную группу и могущую быть снятой после осуществления реакции без разрушения остальной части молекулы. Сведения о таких группах их синтезе можно найти в "Protective Groups in Organic Synthesis" oy T.W. Greene John Wiley and Sons, 1991 г., или Fieser and Fieser. Примеры гидроксизамещенных групп включают метоксиметил, 1-этоксиэтил, 1-метокси-1-метилэтил, бензилоксиметил, (β- триметилсилилэтокси)метил, тетрагидропиранил, 2,2,2-трихлорэтоксикарбонил, трет-бутил (дифенил)силил, триалкилсилил, трихлорметоксикарбонил и 2,2,2-трихлорэтоксиметил.
Термин "соль" охватывает кислые и/или основные соли, образованные неорганическими и/или органическими кислотами и основаниями. Примеры кислых солей включают соли, образованные минеральными кислотами, такими как HCl, H2SO4 или HNO3, или карбоновыми кислотами, такими, как трифторуксусная кислота или уксусная кислота. Примеры основных солей включают соли, образованные аминами, такими, как триэтиламин, диизопропилэтиламин или пиридин, или аминокислотами, такими, как аргинин или гуанидин. "Солями" в данном описании считают и соли гидроксильных групп, такие, как алкоксиды металлов (например, щелочного или щелочноземельного металла). Алкоксиды металлов могут быть, например, образованы путем контактирования гидроксильной группы с металлирующим агентом.
Ссылка на соединение, используемое в соответствии с настоящим изобретением или полученное этими способами, охватывает и его соли и гидраты, если не указано иное.
Получение оксазолиновых соединений формулы I и их солей
В соответствии с настоящим изобретением предлагаются новые способы получения оксазолиновых соединений формулы I и их солей, в частности, способы дегидратации, замещения и обмена, описанные ниже.
В соответствии с настоящим изобретением предлагаются также новые оксазолиновые соединения формулы I и их соли, включающие все их стереоизомеры или либо, по существу, свободные от других стероизомеров, либо в смеси с другими выбранными или всеми другими стероизомерами, при условии, что когда R1 - фенил и один из радикалов R3 или R4 - водород, то либо R2 - не метокси, когда другой из R3 или R4 - пентадецил, бензил или метоксикарбонил, либо R2 - не этокси, когда другой из радикалов R3 или R4 - этоксикарбонил; когда R1 - метил и один из радикалов R3 или R4 - водород, то R2 - не 8-фенилментилокси, когда другой из радикалов R3 или R4 - метилпропил; и когда R1 - ацетилметил и R3 и R4 - водород, то R2 - не этокси или NH2.
Предпочтительными являются оксазолины формулы Ia и их соли, описанные ниже, особенно соединения формулы Ia, имеющие те заместители, которые указаны ниже в разделе под названием "Предпочтительные соединения".
Способ дегидратации
Оксазолиновые соединения формулы I или их соли могут быть получены способом дегидратации, включающим в себя стадию контактирования соединения следующей ниже формулы V или его соли: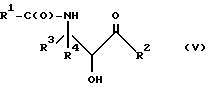
где
R1, R2, R3 и R4 - такие, как указаны выше, с кислотой, способной осуществлять дегидратацию соединения формулы V или его соли, для образования соединения формулы I или его соли.
Исходные соединения формулы V и их соли могут быть получены так, как описано в заявке N 07/975453 на патент США, поданной 12 ноября 1992 г. от имени Патела и др.; Ojima et al., J. Org. Chem., 56, 1681-1683 (1991); Georg et al., Tetrahedron Lett., 32, 3151-3154 (1991); Denis et al., J. Org. Chem. , 51, 46-50 (1986); Corey et al., Tetrahedron Lett., 32, 2857-1860 (1991); Deng et al., J. Org. Chem., 57, 4320-4323 (1992); Ojima et al., Tetrahedron, 48 6985-7012 (1992); Commercon et al., Tett. Lett., 33, 5185-5188 (1992); Denis et al., J.Org. Chem., 56(24), 6939-6942 (1991) (например, с последующими этерификацией и обработкой кислотой); и Denis et al., J. Org. Chem., 55, 1957-1959 (1990), причем все эти материалы включены в данное описание путем ссылки.
В способе дегидратации в соответствии с настоящим изобретением может быть использована любая кислота, способная осуществлять дегидратацию. Примерными кислотами являются сульфокислоты, такие, как пиридиновая паратолуолсульфокислота, паратолуолсульфокислота, камфарсульфокислота и метансульфокислота, карбоновые кислоты, такие, как трифторуксусуная или уксусная кислота, или минеральные кислоты, такие как HCl, H2 SO4 или HNО3. Молярное отношение кислота:соединение формулы V предпочтительно составляет от примерно 1: 100 до примерно 1:1.
Реакцию предпочтительно проводят при температуре от примерно 0o до примерно 200oC и при давлении от примерно 1 атмосферы до примерно 5 атмосфер. Реакцию предпочтительно проводят в среде инертного газа, такого как аргон.
В качестве растворителей предпочтительно используют инертные органические растворители, такие как толуол, тетрагидрофуран, ацетонитрил, бензол или ксилол. Количество используемого растворителя предпочтительно обеспечивает содержание исходного соединения формулы V около 2,5% по массе от общей массы растворителя и соединения формулы V.
Оксазолиновое кольцо соединений формулы I пронумеровано следующим образом: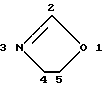
В отношении атомов углерода в 4 и 5 положениях оксазолиновые соединения формулы I могут существовать в виде четырех стереоизомеров Ia, Ib, Ic и Id, представленных ниже:
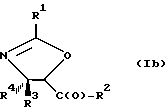
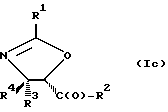
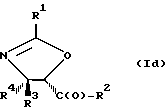
Соединения формулы V также могут существовать в виде четырех стереоизомеров по отношению к углеродным атомам в соответствующих положениях. Этими стереоизомерами являются следующие соединения Va, Vb, Vc и Vd: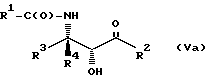
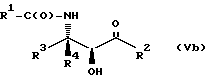
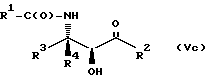
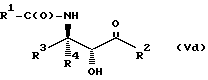
Требуемый стереоизомер соединения формулы I может, например, быть получен предлагаемым способом дегидратации с использованием подходящего стереоизомера исходного соединения формулы V. Так, использование соединения Va даст соединение Ia, использование соединения Vb даст соединение Id, использование соединения Vc даст соединение Ic и использование соединения Id даст соединение Ib. Является предпочтительным использовать в предлагаемом способе дегидратации один отдельный стереоизомер исходного соединения V, хотя могут быть использованы и смеси стереоизомеров. Особенно предпочтительным является использование соединения Va для получения соединения Ia, в частности для получения соединения Ia, имеющего те заместители, которые указаны ниже в разделе "Предпочтительные соединения".
Способ замещения
Оксазолиновые соединения формулы I или их соли могут быть также получены способом замещения, включающим в себя стадию контактирования соединения формулы V или его соли (в присутствии основания) с активирующим веществом, способным активировать гидроксильную группу соединения формулы V или его соли для обеспечения возможности внутримолекулярного замещения и образования соединения формулы I или его соли, при условии, что когда R1 - фенил и один из радикалов R3 или R4 - водород, то или R2 - не этокси, когда другой из радикалов R3 или R4 - этоксикарбонил, или R2 - не метокси, когда другой из радикалов R3 или R4 - бензил.
В качестве активирующего вещества в способе замещения в соответствии с настоящим изобретением может быть использовано любое соединение, способное активировать гидроксильную группу соединения формулы V и осуществлять внутримолекулярное замещение. Примерами активаторов являются сульфогалогениды, такие, как алкилсульфогалогениды (напр., метилсульфохлорид), или арилсульфогалогениды (напр., бензолсульфохлорид или паратолуолсульфохлорид), фосфороксихлорид (POCl3), фосфорпентахлорид (PCl5) или тионилхлорид (SOCl2). Молярное отношение активатор: соединение формулы V предпочтительно составляет от примерно 1:1 до примерно 2:1.
Активирование гидроксильной группы соединения формулы V или его соли может дать новое промежуточное соединение формулы VI или его соль: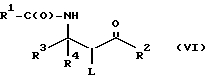
где
R1, R2, R3 и R4 - такие, как указаны выше, и L - уходящая группа такая, как алкилсульфонилокси (напр., метилсульфонилокси), арилсульфонилокси (напр. , бензолсульфонилокси или паратолуолсульфонилокси), хлор или фосфороксигруппа (PO2- или PO-). Настоящее изобретение дает вышеупомянутые новые соединения формулы VI и их соли, включающие все их стереоизомеры, либо, по существу, свободные от других стереоизомеров, либо в связи с другими выбранными или всеми другими стереоизомерами, при условии, что, когда R1 - фенил, R2 - метокси и один из радикалов R3 или R4 - водород, а другой - бензил, то L - не хлор.
Основания, которые могут быть использованы, включают в себя органические основания, такие, как амины (напр., пиридин, триэтиламин, диизопропилэтиламин, лутидин или 1,8-диазобицикло- [5.4.0]ундец-7-ен) или гексаметилдисилазид лития, или неорганические основания, такие, как карбонаты щелочных металлов (например, карбонат калия). Молярное отношение основание: соединение формулы V предпочтительно больше, чем примерно 2:1.
Реакцию предпочтительно проводят при температуре от примерно -20oC до примерно 100oC, в частности 0oC, и при давлении около 1 атмосферы. Реакцию предпочтительно проводят в среде инертного газа, такого, как аргон.
В качестве растворителей предпочтительно используют инертные органические растворители, такие, как хлороформ, метиленхлорид, толуол, тетрагидрофуран, ацетонитрил, или, что наиболее предпочтительно, основные органические растворители, способные действовать в качестве как растворителя, так и основания для данного способа, такие, как пиридин, триэтиламин или лутидин. Количество используемого растворителя предпочтительно обеспечивает содержание исходного вещества формулы V около 10% по массе от общей массы растворителя и соединения формулы V.
Требуемый стереоизомер соединения формулы I может, например, быть получен предлагаемым способом замещения с использованием подходящего стереоизомера исходного соединения формулы V. При этом использование соединения Va даст соединение Ic, использование соединения Vb даст соединение Ib, использование соединения Vc даст соединение Ia и использование соединения Vd даст соединение Id. Является предпочтительным использовать в предлагаемом способе замещения один отдельный стереоизомер исходного соединения формулы V, хотя могут быть использованы и смеси стереоизомеров. Особенно предпочтительным является использование соединения Vc для образования соединения Ia, в частности для получения соединения Ia, имеющего те заместители, которые указаны ниже в разделе под названием "Предпочтительные соединения".
Способ обмена
Оксазолиновые соединения формулы I, где R1 - R1', такой, как указан ниже, или их соли могут быть также получены способом обмена, включающим стадию контактирования соединения следующей ниже формулы VII или его соли: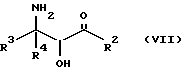
где
R2, R3 и R4 такие, как указаны выше, с соединением следующей ниже формулы VIII или его солью: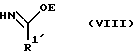
где
R1' и E - независимо алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло; при условии, что когда E - этил, один из R3 или R4 - водород и R1' - фенил, то R2 - не метокси, когда другой из радикалов R3 или R4 - метоксикарбонил, и R2 - не этокси, когда другой из R3 или R4 - этоксикарбонил, а когда R1' - метил, то R2 - не 8-фенилментилокси, когда другой из R3 или R4 - 2-метилпропил.
При использовании одновременно обоих исходных соединений VII и VIII в качестве кислых солей при группах NH2 и HN соответственно, для образования свободной группы NH2 и/или HN (соответственно) может быть использовано аминовое основание, такое, как аммиак или органическое аминовое основание, чтобы обеспечить возможность эффективного продолжения реакции. При этом может быть использовано любое аминовое основание, способное образовывать свободную(ые) групп(ы) NH2 и/или HN. Предпочтительными являются третичные аминовые основания, такие, как триэтиламин, диизопропилэтиламин, лутидин, пиридин или 1,8-диазобицикло [5.4.0]унден-7-ен. Молярное отношение аминового основания к соединению формулы VII предпочтительно составляет от примерно 1:1 до примерно 10:1.
Исходные соединения формулы VII и их соли могут быть получены такими способами, как те, что описаны в заявке N 07/975453 на патент США, поданной от имени Патела и др. 12 ноября 1992 г; Commercon et al., Tetrahedron Lett., 33 (36), 5185-5188 (1992); Corey et al., Tetrahedron Lett., 32, 2857-1860 (1991); Ojima et al. , Tetrahedron, 48, 6985-7012 (1992); и Ojima et al., Tetrahedron Lett., 33, 5737-5740 (1992); все эти материалы включены в данное описание путем ссылки. Исходные соединения формулы VIII и их соли могут быть получены такими способами, как те, что описаны в Kimball et al., Org. Synth. Coll., т. 11, стр. 284 (1943). Использование кислых солей соединений формулы VIII, например, солей, образованных карбоновыми, сульфо- или минеральными кислотами, в качестве исходных материалов является предпочтительным, так как такие соединения относительно устойчивы и легки в обращении. Вышеупомянутые соли могут быть нейтрализованы при контакте с используемым основанием, таким, как описаны выше. Молярное отношение соединения формулы VIII к соединению формулы VII предпочтительно составляет от примерно 1:1 до примерно 2: 1.
Реакцию предпочтительно проводят при температуре от примерно 0oC до примерно 100oC и при давлении примерно 1 атмосфера. Реакцию предпочтительно осуществляют в инертной среде, например, в среде аргона или азота.
Используемыми растворителями предпочтительно являются инертные органические растворители, такие, как толуол, тетрагидрофуран, дихлорметан, 1,2-дихлорэтан или хлороформ. Количество используемого растворителя предпочтительно обеспечивает содержание исходного соединения формулы VII около 6% по массе от общей массы растворителя и соединения формулы VII.
Соединения формулы VII могут, как и соединения формулы V, существовать в виде четырех стереоизомеров по отношению к углеродным атомам в соответствующих положениях. Эти стереоизомеры представляют собой следующие соединения VIIa, VIIb, VIIc и VIId: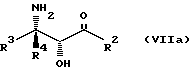
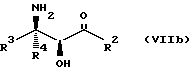
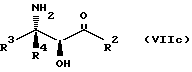
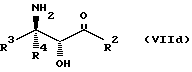
Требуемый стереоизомер соединения формулы I может, например, быть получен предлагаемым способом обмена с использованием подходящего стереоизомера исходного соединения формулы VII. Так, использование соединения VIIa даст соединение Ia, использование соединения VIIb даст соединение Id, использование соединения VIIc даст соединение Ic и использование соединения VIId даст соединение Ib. Является предпочтительным использовать один отдельный стереоизомер исходного соединения VII в предлагаемом способе обмена, хотя могут быть использованы и смеси стереоизомеров. Особенно предпочтительным является использование соединения VIIa для получения соединения Ia, в частности для получения соединения Ia, имеющего заместителя, указанные ниже в разделе под названием "Предпочтительные соединения".
Получение оксазолиновых соединений формулы II и их солей
Оксозолиновые соединения формулы II и их соли могут быть получены из оксазолиновых соединений формулы I и их солей путем превращения группы -C(O)-R2 в группу -C(O)-OH.
Может быть использовано любое вещество, способное к вышеупомянутому превращению. Например, когда R2- алкокси, такое, как метокси или этокси, соединение формулы I или его соль можно деалкилировать для образования соединения формулы II путем использования подходящего нуклеофильного вещества, такого, как метантиоловые соли щелочных и щелочноземельных металлов. В соответствии с другим вариантом можно использовать гидрирование, например, для превращения групп, таких, как бензилоксикарбонил в карбоксил, путем использования гидрирующего вещества, например, водорода и катализатора гидрирования, такого, как палладий.
Превращение группы -C(O)-R2 в карбоксильную группу предпочтительно проводят путем гидролиза. При этом в качестве гидролизующего агента проводят путем гидролиза. При этом в качестве гидролизующего агента можно использовать любое соединение, способное к осуществлению гидролиза. Примеры гидролизующих агентов включают водные основания, такие как гидроксиды (например, гидроксиды металлов, такие, как гидроксид бария, или предпочтительно гидроксиды щелочных металлов, такие, как гидроксид лития, натрия или калия). Молярное отношение основания к соединению формулы I предпочтительно составляет от примерно 1:1 до примерно 3:1. Молярное отношение воды к соединению формулы I предпочтительно составляет от примерно 1:1 до примерно 100:1.
Реакцию предпочтительно проводят при температуре от примерно -20oC до примерно 100oC и при давлении примерно 1 атм. Гидроксидное омыление соединений формулы I или их солей, где R2 - -N(R5)(R6), предпочтительно проводят при более высоких температурах вышеуказанного диапазона температур или при температурах, близких к или равных температуре нагревания с обратным холодильником, используемой жидкой средой. Реакцию предпочтительно осуществляют в среде азота, аргона или воздуха.
Растворители могут быть выбраны из неорганических или органических жидкостей, таких, как вода, спирты, толуол, тетрагидрофуран, диоксан, ацетонитрил или диметилформамид или их смеси. Является предпочтительным использовать в качестве растворителя смесь воды с органической жидкостью, такой, как тетрагидрофуран. Количество используемого растворителя предпочтительно обеспечивает содержание исходного соединения формулы I около 7% по массе от общей массы растворителя и соединения формулы I.
В соответствии с настоящим изобретением предлагаются также новые соединения формулы II и их соли, включающие все их стереоизомеры, либо, по существу, свободные от других стереоизомеров, либо в смеси с другими выбранными или всеми другими стереоизомерами, при условии, что когда R1 - фенил и один из радикалов R3 или R4 - водород, то другой из радикалов R3 или R4 - не COOH. Как и оксазолины формулы I, оксазолины формулы II могут существовать в виде четырех стереоизомеров по отношению к углеродным атомам в 4 и 5 положениях. Эти стереоизомеры представляют собой следующие соединения IIa, IIb, IIc и IId: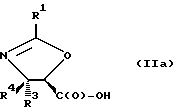
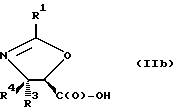
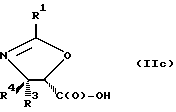
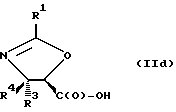
Предпочтительными являются оксазолины формулы IIa и их соли, в частности, соединения формулы IIa имеющие те заместители, которые указаны ниже в разделе под названием "Предпочтительные соединения".
Стереоконфигурация исходного соединения формулы I или его соли может быть в предлагаемом способе сохранена и/или перевернута. Так, например, гидролиз соединения формулы I, имеющего заместители, находящиеся в цис-положении относительно друг друга в положениях 4 и 5, может дать соединение формулы II, имеющее соответствующую цис-конфигурацию, соединение формулы II, имеющее соответствующую транс-конфигурацию, где карбоксильный заместитель в положении 5 перевернут относительно исходного соединения, или смесь вышеуказанных цис- и транс-соединений. Основания, которые при использовании для гидролиза лишают протона углеродный атом, посредством которого присоединена группа -C(O)-R2, и которые затем возвращают протон вышеупомянутому атому углерода с противоположной стороны кольцевой системы, обеспечивают инверсию (обращение) стереоконфигурации. Примерами таких оснований являются основания, описанные выше, или карбонаты щелочных металлов, такие, как карбонат калия, аминовые основания или алкоксиды металлов, таких как щелочные или щелочноземельные металлы, которые (алкоксиды) могут быть образованы до их присоединения или in situ (например, путем присоединения металлирующего агента, такого, как н-бутиллитий, вместе с алканолом, таким, как этанол).
При обращении стереоконфигурации, как описано выше, во время осуществления данного способа, может иметь место образование в качестве промежуточного продукта соединения формулы I, имеющего перевернутую (обращенную) стереоконфигурацию относительно исходного соединения формулы I (т.е. эпимеризации). Так, например, когда исходное соединение формулы I имеет заместители в положениях 4 и 5, находящиеся в цис-положении относительно друг друга, соответствующее транс-соединение формулы I, где заместитель -C(O)-R2 в положении 5 перевернут относительно исходного соединения, может быть образовано в качестве промежуточного соединения в процессе реакции гидролиза. Вышеописанный способ обращения (инверсии) также входит в пределы объема настоящего изобретения.
Сочетание для получения несущих оксазолиновую боковую цепь формулы III и их солей
Несущий боковую цепь таксан формулы III или его соль можно получить способом, включающим стадию контактирования оксазолинового соединения формулы II или его соли с таксаном, имеющим гидроксильную группу, непосредственно присоединенную к его углеродному атому C-13, или его солью в присутствии сочетающего агента. Является предпочтительным использовать в данном способе оксазолины формулы IIa или их соли, в частности соединения формулы IIa, имеющие те заместители, которые указаны ниже в разделе "Предпочтительные соединения".
Таксаны являются соединениями, имеющими структуру ядра
которая может быть замещенной и может иметь, как описано выше, этиленовую ненасыщенность в кольцевой системе. В данном способе может быть использован любой таксан, содержащий гидроксильную группу, непосредственно присоединенную к углеродному атому C-13, или его соль (такую, как алкоксид металла по гидроксильной группе у C-13). Таксановым исходным материалом, используемым в способе по настоящему изобретению, может быть соединение, такое как те, что описаны в публикации N 400971 Европейского патента, включенной в данное описание путем ссылки, и соединение, содержащее таксановый фрагмент, описанный (и полученный способами, описанными или аналогичными им) в заявке N 07/907261 на патент США, поданной 1 июля 1992 г. от имени Чена и др., или в заявке N 07/981151 на патент США, поданной 24 ноября 1992 г. от имени Уеда и др. , которые включены в данное описание путем ссылки. Примерами таких таксанов являются таксаны, имеющие следующую формулу IX: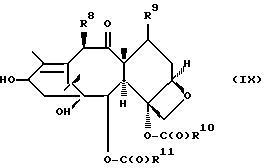
где
R8 - водород, гидроксил, R14-O-, R15-C(O)-O- или R15-O-C(O)-O-;
R9 - водород, гидрокси, фтор, R14-O-, R15-C(O)-O- или R15-O-C(O)-O-;
R10 и R11 - независимо водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, R16-O-, арил или гетероцикло;
R14 - гидроксизащитная группа;
R15 - водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло; и
R16 - алкил;
или их соли.
Предлагается использовать в способе сочетания по настоящему изобретению все стереоконфигурации нерегламентируемых хиральных центров соединения формулы IX. Предпочтительным является использование одного отдельного стереоизомера, но могут быть использованы и смеси стереоизомеров. Используемыми в качестве исходного материала соединения формулы IX являются предпочтительно 7-триалкилсилилбаккатины III, а наиболее предпочтительно 7-триметилсилилбаккатин III или 7-триэтилсилилбаккатин III.
Другой группой соединений формулы IX, служащих в качестве предпочтительных исходных материалов, являются соединения, в которых R8 - OC(O)CH3, R9 - гидроксил или гидроксилзащитная группа, например O-триметилсилил или O-триэтилсилил, R10 - такой, как указан выше, кроме метила, и R11 - арил, например, бензил. Последние соединения считаются новыми, как и способы их получения, которые описаны ниже. Особенно предпочтительными из вышеуказанных соединений являются те, в которых R10 - циклоалкил или OR16.
Вышеупомянутые соединения получают по следующей общей схеме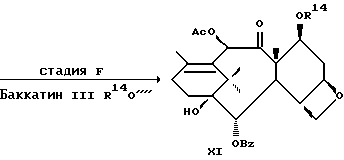
R14 - такой, как указан выше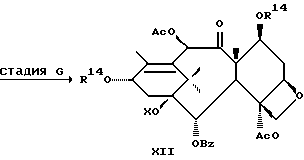
где
R14 - такой, как указан выше, и X - триметилсилан или диметилсилан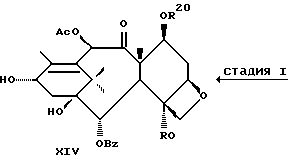
где
R20 - водород или R14 и R - такой как указано выше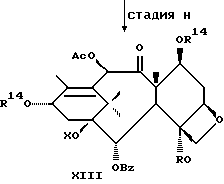
где
R - C(O)R10 и R14 и X - такие, как указаны выше
Стадия F
Осуществляют защиту баккатина III у C-7 и C-13 посредством реакции с подходящим агентом, таким, как галотриалкилсилан, например, триметил или триэтил, 2,2,2-трихлорэтилхлорформиат или карбобензилокси. Может быть использован любой инертный органический растворитель, в котором растворим баккатин III, например, ТГФ (тетрагидрофуран), ДМФ (диметилформамид), MeCl2 и диоксан. Реакцию осуществляют в присутствии третичного аминового основания такого, как пиридин или имидазол. Температуру реакции можно изменять от -30oC до комнатной температуры, причем замещение у C-7 происходит предпочтительно при температуре от -30oC до 0oC, у C-13 - при температуре от 0oC до комнатной температуры. Концентрацию реагента с защитной группой предпочтительно берут в молярном избытке (1-10) для осуществления замещения и у C-7, и у C-13.
Стадия C
Затем защищают интермедиат XI по гидрокси у C-1 посредством реакции с триметилсиланом или предпочтительно диметилсиланом, например, хлортриметилсиланом или предпочтительно хлордиметилсиланом в, например, ДМФ, ТГФ, диоксане и различных простых эфирах. Как и на стадии F, реакцию предпочтительно осуществляют в присутствии третичного аминового основания такого, как имидазол или пиридин. Температура может находиться в пределах от -30oC до комнатной температуры, а предпочтительно равна 0oC.
Стадия H
(A) Затем восстанавливают интермедиат XII у C-4 до гидрокси посредством реакции с подходящим восстановителем таким, как Red-AI или алюмогидрид лития. Восстановитель обычно присутствует в молярном избытке (1-5 эквивалентов). Растворителем могут быть ТГФ, диоксан или различные подходящие простые эфиры, а температура реакции может находиться в пределах от -30oC до 0oC, а предпочтительно равна 0oC.
(B) Интермедиат XIII(A), где группа у C-4 - гидрокси, превращают до соответствующего заместителя у C-4 посредством реакции с подходящим ацилхлоридом, ангидридом кислоты или смешанным ангидридом, например акрилсилхлоридом, бензоилхлоридом, циклоалкилкарбонилхлоридом, алкилхлорформиатом, в присутствии аниона щелочного металла (Li, Na или K) вторичного аминового основания. Растворители для реакции включают ТГФ, диоксан и т.д. Температура реакции находится в пределах от -30oC до комнатной температуры, а предпочтительно равна примерно 0oC.
Стадия I
(A) Затем снимают защиту у интермедиата XIII стадии H(B) путем проведения реакции с пиридинийфторидом (водный раствор фторводорода в пиридине) в ацетонитриле, а затем с тетрабутиламмонийфторидом в ТГФ или фторидом цезия в ТГФ. Далее смесь разводят в спирте, промывают органической или неорганической кислотой и отделяют.
(B) Затем в соединение XIV может быть, как и на стадии F, введена защитная группа для гидрокси у C-7 при параметрах реакции, благоприятствующих описанному выше замещению у C-7.
Далее может быть введена подходящая боковая цепь при C-13 в соответствии с новым способом, раскрытым в данном описании или по методике Нолтона, раскрытой в патентах США N 5227400, N 5175315 и N 5229526, описания к которым включены в данное описание путем ссылки.
Новыми конечными продуктами в соответствии с настоящим изобретением являются, таким образом, соединения формулы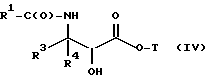
где
R1 - R5, R7-O- или (R5)(R6)-;
R3 и R4 - независимо R5, R5-O-C(O)- или (R5)(R6)-C(O)-;
R5 и R6 - независимо водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло; и
R7 - алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло;
T представляет собой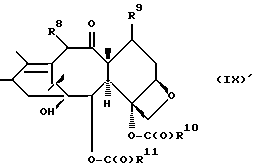
где
R8 - водород, гидроксил, R14-O-, R15-C-(O)- или R15-O-C(O)-O-;
R9 - водород, гидроксил, фтор, R14-O-, R15-C(O)-O- или R15-O-C(O)-O-;
R10 и R11 - независимо водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, R16-O-, арил или гетероцикло;
R14 - гидроксилзащитная группа; и
R15 - водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло; а
R16 - алкил, при условии, что R10 - не метил;
или их соли или гидраты.
Предпочтительные соединения
Особенно предпочтительными среди новых соединений формулы IV являются те соединения, в которых R10 - циклоалкил или OR16. Наиболее предпочтительными из новых соединений формулы IV являются соединения, в которых R10 - циклоалкил; R1 - арил, предпочтительно фенил, или алкокси, предпочтительно трет-бутилокси; R3 - арил, предпочтительно фенил, гетероцикло, предпочтительно 2- или 3-фуранил или тиенил, изобутенил, 2-пропенил, изопропил или (CH3)2CH-; R4 - водород; R8 - предпочтительно гидроксил или алкилкарбонилокси, например, ацетилокси; R9 - гидрокси; и R11 - арил, предпочтительно фенил.
В качестве сочетающего агента в данном способе может быть использовано любое соединение, способное осуществлять этерификацию гидроксильной группы у C-13 (или ее соли) исходного таксана через карбоксильную группу оксазолина формулы II или его соли. Примеры сочетающих агентов включают те соединения, которые образуют активированный сложный оксазолиновый эфир (например, 1-гидроксибензотриазол или N-гидроксисукцинимид) или ангидрид (например, хлорангидрид, такой, как пивалоилхлорид или бис(2-оксо-3-оксазолидинил)фосфинохлорид) при контактировании с оксазолином формулы II, в частности сочетающие агенты, содержащие соединение, такое, как карбодиимид (например, дициклогексилкарбодиимид (DCC), 1,3-диизопропилкарбодиимид (DIC) или 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид) бис(2-оксо-3-оксазолидинил)фосфинохлорид), карбонилдиимидазол (CDI), пивалоилхлорид или 2,4,6-трихлорбензоилхлорид, причем вышеупомянутые соединения предпочтительно используют вместе с таким соединением, как 1-гидроксибензотриазол (HOBt) или N-гидроксисукцинимид (HO-S4), или амином, таким как триэтиламин, пиридин или пиридин, замещенный в положении 4 на -N(R16)(R17), где R16 и R17 - независимо выбраны из алкила, алкенила, алкинила, циклоалкила, циклоалкенила или гетероцикло (для образования соединения, такого как 4-диметиламинопиридин (DMAP)), или где R16 и R17 вместе с атомом азота, к которому они присоединены, образуют гетероциклогруппу (для образования соединения, такого как 4-морфолинопиридин или 4-пирролидинопиридин). Молярное отношение сочетающего агента к исходному таксану предпочтительно составляет от примерно 1:1 до примерно 2: 1. Молярное отношение оксазолина формулы 11 к исходному таксану предпочтительно составляет от примерно 1:1 до примерно 2:1.
Реакцию предпочтительно проводят при температуре от примерно 0oC до примерно 140oC и при давлении примерно 1 атм. Предпочтительно реакцию осуществляют в среде инертного газа, такого как аргон.
Предпочтительно используемыми растворителями являются инертные органические жидкости, такие как толуол, ацетонитрил, 1,2-дихлорэтан, хлороформ, тетрагидрофуран, пиридин, метиленхлорид или диметилформамид. Количество используемого растворителя предпочтительно обеспечивает содержание исходного таксана около 20% по массе от общей массы растворителя и таксанового соединения.
Стереоконфигурация заместителей в положениях 4 и 5 исходного оксазолина в продукте сочетания формулы III может быть сохранена и/или инвертирована, например, предполагается, что может иметь место эпимеризация от цис- к транс, когда заместитель в положении 5 инвертирован относительно исходного материала.
В соответствии с настоящим изобретением предлагаются также новые несущие оксазолиновую боковую цепь таксаны формулы III и их соли, включающие все их стереоизомеры, либо, по существу, свободные от других стереоизомеров, либо в смеси с другими выбранными или всеми другими стереоизомерами.
Разрыв кольца для образования таксанов формулы X и их солей
Несущий боковую цепь таксан формулы X или его соль можно получить из несущего оксазолиновую боковую цепь таксана формулы III или его соли способом, включающим стадию контактирования таксана формулы III или его соли с водной кислотой, способной разрывать (раскрывать) кольцо оксазолиновой группы, присоединенной через C-13 таксанового фрагмента упомянутого таксанового соединения, для образования упомянутого соединения формулы X или его соли.
В способе по настоящему изобретению может быть использована любая водная кислота, способная осуществлять вышеупомянутый разрыв кольца. Примеры разрывающих кольцо кислот включают карбоновые кислоты, такие как уксусная или трифторуксусная кислота, или предпочтительно, минеральные кислоты, такие, как хлористоводородная, фтористоводородная или серная кислота, в воде. Молярное отношение разрывающей кольцо кислоты к соединению формулы III предпочтительно составляет от примерно 1:1 до примерно 10:1. Молярное отношение воды к соединению формулы III предпочтительно составляет от примерно 1:1 до примерно 100:1.
Реакцию раскрытия кольца предпочтительно проводят при температуре от примерно -20oC до примерно 40oC и при давлении примерно 1 атмосфера. Реакцию предпочтительно осуществляют в среде азота, аргона или воздуха.
Предпочтительно используемыми растворителями являются инертные органические жидкости в отдельности или в смеси с водой, такие как тетрагидрофуран, спирты (предпочтительно низшие спирты, такие, как метанол), диоксан, толуол, ацетонитрил или их смеси. Количество используемого растворителя предпочтительно обеспечивает содержание исходного соединения формулы III около 5% по массе от общей массы растворителя и соединения формулы III.
Предпочтительный вариант осуществления настоящего изобретения дополнительно включает стадию снятия защиты с одной или нескольких групп, в частности для освобождения гидроксильных групп, на таксановом фрагменте для получения таксанов формулы X. Снятие защиты может, например, быть осуществлено до или после (или одновременно с ним) вышеупомянутого разрыва кольца путем использования снимающего защиту агента. В качестве снимающего защиту агента может быть использовано любое соединение, способное удалять защитную группу. Например, для удаления силильных защитных групп можно использовать кислоты, такие, как фтористоводородная кислота или водные протиевые кислоты, или тетраталкиламмонийфториды, такие, как тетра-н-бутиламмонийфторид; бензильные защитные группы могут быть удалены путем гидрирования; трихлорэтоксикарбонильные защитные группы могут быть удалены путем контактирования с цинком; и ацетальные или кетальные защитные группы могут быть удалены путем использования протиевых кислот или кислот Льюиса.
Предпочтительный вариант осуществления настоящего изобретения включает одновременно осуществление разрыва кольца и снятия защиты с одной или нескольких гидроксильных групп на таксановой кольцевой структуре, в частности при C-7. Особо предпочтительный вариант включает стадию одновременного осуществления разрыва кольца и снятия защиты путем использования кислоты (например, минеральной кислоты, такой, как хлористоводородная), способной осуществлять обе реакции. Так, например, использование кислоты при условиях реакции, описанных выше для разрыва кольца, может обеспечить одновременное осуществление разрыва кольца и удаления отщепляемых кислотой гидроксилзащитных групп при C-7, таких как триалкилсилил (например, триметилсилил или триэтилсилил).
В соответствии с настоящим изобретением предлагаются также новые промежуточные соединения (интермедиаты) формулы X и их соли, образованные при разрыве кольца и, необязательно, снятии защиты, которые включают все их стереоизомеры либо, по существу, свободные от других стереоизомеров, либо в смеси с другими выбранными или всеми другими стереоизомерами.
Контактирование с основанием для образования таксанов формулы IV и их солей
Обработка соединения формулы X или его соли основанием дает соединение формулы IV или его соль. В способе по настоящему изобретению может быть использовано любое основание, позволяющее перемещение ацильной группы -C(O)-R1 к аминогруппе -NH2, в результате чего образуется соединение формулы IV или его соль. Примерами таких оснований являются бикарбонаты щелочного металла такие, как бикарбонат натрия или бикарбонат калия. Молярное отношение основания к соединению формулы X предпочтительно составляет от примерно 1:1 до примерно 5:1.
Реакцию предпочтительно проводят при температуре от примерно -20oC до примерно 80oC и при давлении 1 атмосфера. Предпочтительно реакцию осуществляют в среде аргона, азота или воздуха.
Предпочтительно используемыми растворителями являются инертные органические жидкости по отдельности или в смеси с водой, такие как тетрагидрофуран, спирты (предпочтительно низшие спирты, такие как этанол), толуол, ацетонитрил, диоксан или их смеси. Количество используемого растворителя предпочтительно обеспечивает содержание соединения формулы X от примерно 1% до примерно 5% по массе от общей массы растворителя и соединения формулы X.
Снятие защиты с защищенных групп может быть осуществлено одновременно с использованием основания или после, но предпочтительно снятие защиты осуществляют, как описано выше, до контактирования с основанием, в частности, одновременно с раскрытием кольца.
Разделение
Продукты, полученные способами в соответствии с настоящим изобретением, могут быть выделены и очищены такими способами, как экстракция, перегонка, кристаллизация и колоночное хроматографирование.
Продукты из таксанов с боковой цепью
Несущие боковую цепь таксаны формулы IV и их соли, полученные способами в соответствии с настоящим изобретением сами фармакологически активны или являются соединениями, которые могут быть превращены в фармакологически активные продукты. Фармакологически активные таксаны, такие, как таксол, могут быть использованы в качестве противоопухолевых средств для лечения больных раком, таким, как рак молочной железы, яичника, толстой кишки или легкого, меланомой или лейкемией. Полезность таких таксанов с боковой цепью описана, например, в публикации N 400971 Европейского патента, патентах США N 4876399, и N 4857653, N 4814470, N 4924012, N 4924011, заявке N 07/907261 на патент США, поданной 1 июля 1992 г. от имени Чена и др., и заявке N 07/981151 на патент США, поданной 24 ноября 1992 г. от имени Уеда и др.; все эти материалы включены в данное описание путем ссылки.
Получаемыми в конечном счете предпочтительными несущими боковую цепь таксанами формулы IV являются особо предпочтительный таксол, имеющий показанную выше структуру, и таксотер, имеющий структуру, показанную ниже: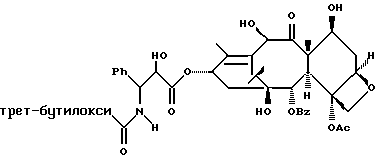
Когда нужно, могут быть использованы или получены любым способом в соответствии с настоящим изобретением сольваты (такие, как гидраты) реагентов или продуктов.
В объем настоящего изобретения входят также водорастворимые лекарственные предшественники соединений формулы IV. Такие лекарственные предшественники соединений формулы IV получают путем введения в C-7 или C-10 и/или в положение 2' боковой цепи фосфонтоксигруппы общей формулы
-OCH2(OCH2)mOP(O)(OH)2,
где
m - O или целое число от 1 до 6 включительно.
Новые лекарственные предшественники имеют формулу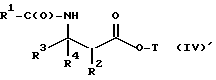
где
R1 - - R5, R7 -O- или (R5)(R6)N-;
R3 и R4 - независимо R5, R5-O-C(O)- или (R5)(R6) N-C(O)-;
R5 и R6 - независимо водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло; и
R7 - алкил, алкенил, алкинил, циклоалкил, циклоалкенил, арил или гетероцикло;
T представляет собой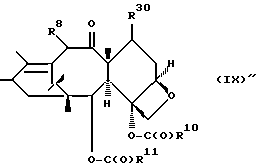
где
R8 - водород, гидроксил, R14 -O-, R15 -C(O)-O-, R15-O-C(O)-O- или -OCH2(OCH2)mOR(O)(OH)2;
R10 и R11 - независимо водород, алкил, алкенил, алкинил, циклоалкил, циклоалкенил, R16 -O-, арил или гетероцикло;
R20 - водород, -OCH2(OCH2)mOR(O)(OH)2, -OC(O)R21 или -OC(O)R21, где R21 - C1-C6 - алкил, необязательно замещенный на один-шесть атомов галогена, C3-C6 циклоалкил, C2-C6 алкенил или радикал формулы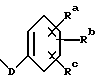
где
D - связь или C1-C6 алкил и Ra, Rb и Rc - независимо водород, амино, C1-C6 моно- или диалкиламино, галоген, C1-C6 алкил или C1-C6 алкокси;
R14 - гидроксизащитная группа;
R16 - алкил;
R30 - водород, гидрокси, фтор, -OCH2(OCH2)mOR(O)(OH)2 или -OC(O)OR21, где R21 - такой, как указан выше;
R15 - водород, алкил, алкенил, алкинил, циклоалкенил, циклоалкил, арил или гетероцикло;
m - O или целое число от 1 до 6 включительно, при условии, что, по крайней мере, один из радикалов R8, R20 и R30 -OCH2(OCH2)mOR(O)(OH)2 и R10 - не метил;
и их основные соли по фосфоноксигруппе.
Предпочтительные соединения формулы IV' включают те, в которых R10 - циклоалкил или OME или OEt; R1 - арил, предпочтительно, фенил или алкокси, предпочтительно трет-бутилокси;
R3 - арил, предпочтительно фенил, или гетероцикло, предпочтительно фурил, или тиенил, или алкенил, предпочтительно пропенил или изобутенил; R4 - водород; R3 - гидрокси или алкилкарбонилокси, предпочтительно ацетилокси; R11 - арил, предпочтительно фенил;
R20 - -OCH2(OCH2)m OR(O)(OH)2 или -OC(O)OR21, где R21 - этил или N -пропил; R30 - -OCH2(OCH2)m OR(O)(OH)2и m = 0 или 1.
Фосфоноксигруппу вводят путем синтеза конечных продуктов формулы IV способами, описанными в заявке N 08/108015 на патент США, поданной 17 августа 1993 г., которая включена в данное описание путем ссылки.
В ходе получения упомянутых выше новых лекарственных предшественников образуются различные новые промежуточные продукты при условиях реакции, описанных в общем в упомянутой заявке N 08/108015. Соединения формулы IV используют в качестве исходных веществ, в которых ненужные гидроксигруппы защищены. Подходящим образом защищенное соединение формулы IV, в котором реакционно-способные гидроксигруппы находятся либо в положении 2', либо в положениях 7 или 10, либо во многих положениях, сначала соединяют с соответствующим метилтиометиловым эфиром -OCH2(OCH2)mSCH3. Затем, в зависимости от значения m, эфир может быть присоединен к защищенному фосфоноксиметиловому эфиру в соответствии с различными стадиями, описанными в вышеупомянутой заявке. Фосфонозащитная(ые) группа(ы) и гидроксизащитные группы могут быть затем удалены традиционными способами.
Затем свободная кислота может быть превращена в требуемую основную соль традиционными способами, включающими контактирование свободной кислоты с металлическим основанием или с амином. Подходящими металлическими основаниями являются гидроксиды, карбонаты и бикарбонаты натрия, калия, лития, кальция, бария, магния, цинка и алюминия, а подходящими аминами являются триэтиламин, аммиак, лизин, аргинин, N-метилглюкамин, этаноламин, прокаин, бензатин, дибензиламин, трометамин (TRIS), хлорпрокаин, холин, диэтаноламин, триэтаноламин и т. п. Основные соли могут быть дополнительно очищены путем хроматографии с последующей лиофилизацией или кристаллизацией.
Лекарственные предшественники могут быть введены орально или парентерально в соответствии с тем, как описано в упомянутой выше заявке N 08/108015. Соединения формул IV и IV' являются новыми противоопухолевыми веществами, проявляющими in vitro цитотоксическую активность против клеток рака толстой кишки человека линий HCT-116 и HCT-116/VM46 и линии М109 клеток рака легких.
Ниже настоящее изобретение описано на примерах, которые следует рассматривать лишь как иллюстративные и ни в какой мере не ограничивающие объем настоящего изобретения, определенный прилагаемой формулой изобретения.
Пример 1
Получение этилового эфира (4S-транс)-4,5-дигидро-2,4-дифенил-5- оксазолкарбоновой кислоты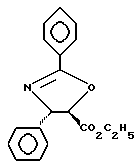
В высушенную в печи колбу емкостью 10 мл, продутую аргоном, добавляли (2R, 3S)-N-бензоил-3-фенилизосеринэтиловый эфир (0,104 г, 0,332 ммоль) и суспендировали в толуоле (5,0 мл). Добавляли пиридиниевую паратолуолсульфокислоту (42 мг, 0,167 ммоль). После перемешивания при комнатной температуре в течение примерно 1 часа смесь нагревали с обратным холодильником. При нагреве получали светлый гомогенный раствор. Примерно после 1 часа нагрева реакционная смесь мутнела. ТСХ (тонкостенная хроматография) после 16,5 часов нагревания показала, что реакция закончена (этилацетат /EtOAc/:гексан 1:1, ФМК (фосфорно-молибденовая кислота)/этанол, ультрафиолет (УФ)).
Реакционную смесь разбавляли 10 мл хлороформа, промывали 5 мл насыщенного водного раствора NaHCO3, высушивали над N2SO4, фильтровали и концентрировали, получив в результате 97,8 мг желтоватого масла (выход = 100%). 1H ЯМР показал, что был получен указанный в заголовке транс-оксазолиновый продукт с незначительным (< 5%) примесями, ни одна из которых не была соответствующим цис-оксазолином.
Пример 2
Получение этилового эфира (4S-транс)-4,5-дигидро-2,4-дифенил-5- оксазолкарбоновой кислоты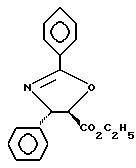
В высушенную над пламенем колбу емкостью 5 мл, продутую аргоном, добавляли (2S, 3S)-N-бензоил-3-фенилизосеринэтиловый эфир (0,100 г, 0,319 ммоль), растворенный в пиридине (1,0 мл), и охлаждали до 0oC. Добавляли по каплям метилсульфонилхлорид (38 мг, 0,335 ммоль и желтоватый раствор перемешивали при 0oC в течение 1 3/4 часа, после чего нагревали до комнатной температуры. Через 1 1/2 часа при комнатной температуре тонкослойная хроматография (ТСХ) показала, что реакция закончена (этилацетат: гексан = 1:1, ФМК/этанол, УФ).
Гетерогенную смесь разбавляли 5 мл этилацетата и промывали 1/3 насыщенным водным раствором CuSO4 (10 мл). Водную фракцию экстрагировали этилацетатом (2х5 мл). Объединенные органические фракции промывали 5 мл насыщенного водного раствора NaCl высушили над Na2SO4, фильтровали и концентрировали, получив в результате 0,12 г желтоватого масла.
Указанный в заголовке продукт очищали путем хроматографии на силикагеле (колонка диаметром 20 мм и длиною 50 мм) этилацетат:гексан = 1:1, и в результате получили 92,6 мг желтоватого масла (выход = 98,3%) 1H ЯМР и масс-спектрография показали, что был получен указанный в заголовке транс-оксазолиновый продукт. Удельные вращения (c = 0,1, CHCl3: [α]D= +15,6°, [α]578= +16,3°, [α]546= +18,7°, [α]436= +33,1°.
Исходное соединение (2S,3S)-N-бензоил-3-фенилизосеринэтиловый эфир получали отдельно следующим образом:
в колбу емкостью 500 мл, содержащую раствор (4S-цис)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты этилового эфира (0,79 г, 2,67 ммоль) в метаноле (MeOH) (57 мл) при 0oC, добавляли 1 н. HCl (57 мл) с перемешиванием в течение 10 минут. Во время добавления HCl имело образование осадка, который растворялся при добавлении тетрагидрофурана (ТГФ). Затем добавляли ТГФ (57 мл) для осветления раствора, и полученную смесь перемешивали при 0oC в течение 2 часов 15 минут. Насыщенным NaHCO3 (120 мл) регулировали pH раствора до 9,0, после чего позволяли смеси перемешиваться при комнатной температуре в течение 18 часов. (Реакцию контролировали путем ТСХ (силикагель) с использованием в качестве элюента смеси EtOAc:гексан = 4:6, причем Rf для исходного материала = 0,71, Rf для продукта = 0,42, УФ проявление).
Реакционную смесь разбавляли этилацетатом (200 мл) и, отделив водный слой, экстрагировали его этилацетатом (100 мл х 1). Затем объединенный раствор этилацетата промывали рассолом (150 мл х 1), высушивали над Na2SO4, фильтровали и концентрировали, получив а результате неочищенный (2S,3S)-N-бензоил-3-фенилизосеринэтиловый эфир в виде твердого вещества (0,810 г). Растворяли его в горячем MeOH (15 мл) и оставляли стоять при комнатной температуре в течение 30 минут и затем при 4oC в течение 1 часа. Отфильтровывали твердое вещество, промывали его холодным MeOH (2 мл) и высушивали в вакууме, получив в результате 0,43 г (2S,3S)-N- бензоил-3-фенилизосеринэтилового эфира в качестве первой порции. Вторую порцию (0,24 г) получали так же, как описано выше, что давало в общем 0,67 г (80%) (2S,3S)-N-бензоил-3-фенилизосеринэтилового эфира (белое вещество: т.пл. = 160-161oC, [α]D= -40,3° (c = 1, CHCl3)).
Элементный анализ: C18H19NO4 • 0,03H2O
Вычислено: C - 68,86; H - 6,12; N - 4,46; H2O - 0,20
Найдено: C - 68,99; H - 6,7; N - 4,60; H2O - 20.
Пример 3
Получение этиловых эфиров (4-транс)- и (4-цис)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты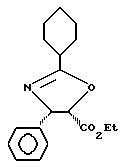
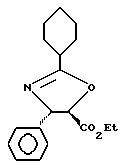
В высушенную в печи колбу емкостью 10 мл, продутую аргоном, добавляли (2S, 3S)-N-бензоил-3-финилизосеринэтиловый эфир (66,88 мг, 0,213 ммоль) и суспендировали его в толуоле (4 мл). Добавляли пиридиновую паратолуолсульфокислоту (49 мг, 0,195 ммоль). Колбу оборудовали ловушкой Дина-старка (заполненной молекулярными ситами с порами 4 ангстрема). Реакционную смесь нагревали с обратным холодильником (большая часть твердых частиц растворялась при нагревании). Пятичасовая ТСХ показала, что реакция была близка к завершению (EtOAc: гексаны = 1:1, ФМК/EtOH, УФ).
Нагревание с обратным холодильником продолжали всю ночь. После 22 часов нагревания реакционную смесь охлаждали до комнатной температуры. Из раствора выпадало маслянистое вещество. При дальнейшем охлаждении до комнатной температуры это масло затвердевало. При добавлении примерно 5 мл EtOAc твердое вещество заметно не растворялось. Чтобы растворить все твердое вещество добавляли примерно 3 мл CHCl3. Тонкослойная хроматография показала отсутствие исходного материала.
Затем раствор промывали 5 мл насыщенного водного раствора NaHCO3, высушивали над Na2SO4, фильтровали и концентрировали, получив в результате 64,3 мг частично кристаллизованного желтого масла. 1H и 13C ЯМР показали, что отношение указанной в заголовке цис-оксазолиновый продукт: указанный в заголовке транс-оксазолиновый продукт: примесь равно примерно 5: следы: 1. Указанный в заголовке транс-оксазолиновый продукт был отнесен за счет следового количества (2R,3S)-N-бензоил-3-фенилизосеринэтилового эфира, присутствовавшего в исходном материале. Продукт хроматографировали на силикагеле при отношениях EtOAc/гексан = 1:1 и 2:1 (Rf = 0,57 (EtOAc : гексаны = 1:1) и получили в результате 49,3 мг маслянистого желтоватого твердого вещества (выход = 78,4; ); 1H ЯМР показал, что отношение (цис:транс) указанных в заголовке цис- и транс-оксазолиновых продуктов составляет приблизительно 10:1.
Пример 4
Получение метилового эфира (4S-транс)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты
Гидрохлорид этилового эфира бензолкарбоксимидной кислоты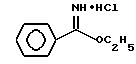
В высушенную над пламенем и продутую аргоном колбу емкостью 100 мл добавляли бензонитрил (30,3 г, 294 ммоль) и этанол (14,2 г, 308 ммоль) и охлаждали до 0oC. HCl барботировали через перемешивающийся раствор в течение 20-минутного периода, в конце которого взвешивание тары показало, что было добавлено 17,5 г HCl. Добавление HCl прекращали, и раствор перемешивали при 0oC. Через примерно 1 час начал образовываться осадок.
После перемешивания при 0oC в течение примерно 2 1/2 часов гетерогенную смесь переносили в холодное (4oC) помещение. После 3 1/2 дней нахождения при 4oC твердую массу измельчали и растирали со 150 мл холодного (4oC) диэтилового эфира. Смеси позволяли стоять при 4oC в течение 6 часов. Смесь фильтровали в вакуум-фильтре и быстро промывали холодным диэтиловым эфиром (2 х 100 мл) и высушивали под высоким вакуумом (0,5 мм рт.ст., 17 часов), получив в результате 51,6 г (95,5%) белого сыпучего порошка указанного в заголовке соединения.
(b)
Метиловый эфир (4S-транс)4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты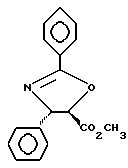
Растворяли (2R,3S)-3-фенилизосеринметилового эфира гидрохлорид (5,76 г, 24,9 ммоль) в 1,2-дихлорэтане (75 мл). Добавляли триэтиламин (2,77 г, 27,3 ммоль) и полученную смесь перемешивали 15 минут, прежде чем добавить одной порцией полученный на описанной выше стадии (a) бензимидат (4,62 г, 24,9 ммоль). Смесь перемешивали 10 минут, после чего нагревали с обратным холодильником. После 4 1/2 часов нагревания ТСХ показала, что реакция закончена (этилацетат:гексан = 1:1, ФМК/этанол, УФ).
Реакционную смесь разбавляли 150 мл дихлорметана и 150 мл 10%-ного K2CO3 и взбалтывали. Разделяли слои и водную фракцию экстрагировали 3 х 50 мл CH2Cl2. Объединенные органические фракции промывали 50 мл насыщенного раствора NaCl, высушивали над Na2SO4, фильтровали и концентрировали, получив в результате желтое масло, которое очищали на колонке с силикагелем (сухой объем примерно 750 мл; заполненная колонка: диаметр 100 мл и длина 110 мм) при отношении этилацетат: гексан = 1:2, в результате чего получили 6,05 г указанного в заголовке соединения в виде очень мало окрашенного масла, которое затвердевало при выдерживании при комнатной температуре. Выход = 86,4%
Пример 5
Получение этилового эфира (4S-цис)-4,5-дигидро-2,4-дифенил-5- оксазолкарбоновой кислоты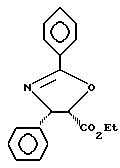
В 100 мл колбу, содержащую раствор (2R,3S)-N-бензоил-3-фенилизосеринэтилового эфира (2,00 г, 6,38 ммоль) в пиридине (20 мл) при 0oC, добавляли по каплям метансульфонилхлорид (0,52 мл, 6,70 ммоль) в течение 2 минут. Раствор перемешивали при 0-4oC в течение 90 минут и затем при 65-70oC в течение 18 часов (реакцию контролировали посредством ТСХ с использованием в качестве элюента смеси этилацетата с толуолом в отношении 1:2, Rf для исходного материала = 0,48 и Rf для указанного в заголовке цис-продукта = 0,78, УФ проявление).
Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (80 мл) и 1/3 насыщенным раствором CuSO4 (80 мл) (1/3 насыщенный раствор CuSO4 получали путем разбавления насыщенного раствора CuSO4 до 1/3 его первоначальной концентрации). Водный слой отделяли и экстрагировали этилацетатом (40 мл х 1). Затем объединенный раствор этилацетата промывали рассолом (80 мл х 1), высушивали над Na2SO4, фильтровали, концентрировали и азеотропировали гептаном (20 мл х 2), получив в результате неочищенный указанный в заголовке цис-оксазолиновый продукт в виде твердого вещества (1,88 г). Растворяли его в горячем этилацетата (8 мл) и затем добавляли гексан (4 мл). Кристаллизующуюся смесь выдерживали при комнатной температуре 20 минут и затем при 4oC в течение 30 минут. Твердое вещество отфильтровывали, промывали холодным этилацетатом (10%) в гексане и высушивали на воздухе, получив в результате 1,34 г (71,3%) указанного в заголовке цис-оксазолинового продукта, имеющего температуру плавления 135oC. [α]D= -9,25° (c = 1,0,CHCl3).
Пример 6
Получение (4S-транс)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты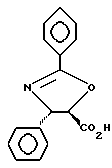
Этиловый эфир (92 мг, 0,311 ммоль) (4S-транс)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты переносили в пузырек на 1 драхму (3,888 г) и растворяли в тетрагидрофуране (ТГФ) (0,8 мл). Добавляли по каплям LiOH (вод. , 1 н., 0,343 ммоль) и полученную двухфазную смесь интенсивно перемешивали при комнатной температуре. За 5 минут был получен гомогенный раствор. Спустя 45 минут тонкослойная хроматография показала отсутствие исходного материала (этилацетат:гексан = 1:1, ФМК/этанол (EtOH), УФ).
Раствор охлаждали до 0oC и затем разбавляли 2,0 мл ТГФ. Резко прекращали реакцию 0,34 мл 1 н. HCl (1,1 экв). После нагревания до комнатной температуры раствор разбавляли 5 мл EtOAc и 5 мл H2O и взбалтывали. Разделяли слои. Водную фракцию экстрагировали 3х5 мл EtOAc. (После экстракции pH водной фракции составлял примерно 6). Объединенные органические фракции высушивали над Na2SO4, фильтровали и концентрировали, получив в результате 72,1 мг белого твердого вещества. Выход = 87%. 1H и 13C ЯМР и МС показали наличие указанного в заголовке продукта, имеющего температуру плавления 201-203oC. [α]D= +25,6°, [α]578= +26,9°, [α]546= +30,7°, [α]436= +53,8 (c = 1,0, CHCl3 : CH3OH = 1 : 1).
Пример 7
Получение (4-транс)-4,5дигидро-2,4дифенил-5-оксазолкарбоновой кислоты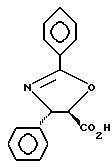
В 10 мл колбу добавляли (4S-транс)-4,5-дигидро-2,2-дифенил-5-оксазолкарбоновой кислоты метиловый эфир (0,509 г, 1,81 ммоль) и растворяли в тетрагидрофуране (4,7 мл). Добавляли по каплям гидроксид лития (1 н. в H2O, 2,0 мл, 1,99 ммоль). Двухфазную смесь интенсивно перемешивали. В пределах двух минут после завершения добавления гидроксида лития был получен светлый раствор. Через 15 минут ТСХ показала, что реакция закончена (этилацетат : гексан = 1:1, ФМК/этанол).
Затем реакционную смесь разбавляли 10 мл ТГФ, и полученный мутный раствор охлаждали до 0oC. Реакцию резко прекращали путем добавления по каплям 2,0 мл 1 н. водной CHl. Далее раствор разбавляли 20 мл этилацетата и 15 мл воды и взбалтывали. Разделяли слои, и водную фракцию экстрагировали 3 х 10 мл этилацетата (pH водного слоя экстракции был равен приблизительно 6). Объединенные органические фракции высушивали над Na2SO4, фильтровали и концентрировали. Полученный концентрат был растворим в смеси бензола и метанола и менее растворим в метаноле, CHCl3, этилацетате или их смеси. Концентрат высушивали под высоким вакуумом всю ночь и получали в результате 0,448 г указанного в заголовке продукта в виде белого твердого вещества. (Выход = 93%). Т. пл. = 201 - 203oC. [α]D= +25,6°, [α]578= +26,9°, [α]546= +30,7°, [α]436= +53,8 (c = 1,0, CHCl3 : CH3OH = 1:1)
Пример 8
Получение (4-транс)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты
Этанол (0,1 мл) смешивали с тетрагидрофураном (1,0 мл), и смесь охлаждали до -78oC. Добавляли по каплям н-бутиллитий (н-BuLi) (2,12 М, 0,050 мл) и смесь нагревали до 0oC. Добавляли твердый этиловый эфир (4S-цис)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновой кислоты, имеющий структуру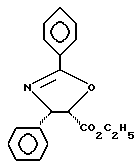
(20 мг, 0,0678 ммоль) и реакционную смесь перемешивали 1 час (в присутствии небольшого количества воды). Смесь исходного цис-оксазолинэтилового эфира и соответствующего транс-оксазолинэтилового эфира (обращение в положение 5) была обнаружена путем тонкослойной хроматографии (очень незначительный гидролиз был отмечен в этот момент). Реакционную смесь перемешивали еще час, после чего оставляли в ледяной бане на всю ночь (от 0oC до комнатной температуры). Через 18 часов ТСХ показала наличие в основном указанного в заголовке продукта - транс-кислоты и следов исходного вещества - цис-эфира (системы растворителей: гексан EtOAc = 2:1 (следы цис-эфира) и EtOAc : ацетон : H2O : MeOH = 7:1:1:1 (указанный в заголовке продукт).
Реакцию резко прекращали фосфатным (pH 4,3) буфером, и реакционную смесь экстрагировали этилацетатом (5 х 10 мл). Высушив органический слой, удаляли растворитель, получив в результате примерно 17 мг (93%) указанного в заголовке продукта. (ЯМР показал указанный в заголовке продукт - транс-кислоту). Т. пл. = 135oC, [α]D= -92,5° (c = 1,0, CHCl3).
Пример 9
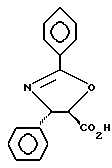
Получение (4S-транс)- и (4S-цис)-4,5, -дигидро-2,4,дифенил-5-оксазолкарбоновых кислот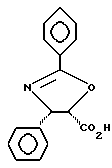
Этиловый эфир (202 мг, 0,6890 ммоль) (4S-цис)-4,5,дигидро-2,4,-дифенил-5-оксазолкарбоновой кислоты растворяли в тетрагидрофуране (1,5 мл), и в полученный раствор добавляли по каплям гидроксид лития (1 н. вод., 0,718 мл). Получали гетерогенный раствор. Реакционную смесь перемешивали всю ночь при комнатной температуре, после чего раствор стал прозрачным. (Тонкослойная хроматография /этилацетат : гексан = 1 : 1/ показала небольшое количество исходного материала. Тонкослойная хроматография (этилацетат : гексан : метанол : вода : ацетон = 7:1:1:1) показала указанные в заголовке цис- и транс-оксазолиновые продукты).
Добавляли 1 н. HCl (0,718 мл) с последующим добавлением насыщенного NaCl (приблизительно 10 мл) и этилацетата (приблизительно 10 мл). Водный слой промывали этилацетатом 5 раз (приблизительно 10 мл), после чего водный слой, имевший pH приблизительно 5,5, подкисляли до pH 3,4 и экстрагировали примерно 10 мл этилацетата. Объединенные органические слои высушивали над MgSO4 и фильтровали. Этилацетат выпаривали при пониженном давлении, получив в результате 183 мг (100%) смеси указанных в заголовке цис- и транс-продуктов (в соответствии с 1H ЯМР отношение цис:транс = 3:1).
Пример 10
Получение 7-триэтилсилил-13-[[(4S-транс)-4,5,-дигидро-2,4,- дифенил-5-оксазолил]-карбонил]баккатина III
(a) 7-Триэтилсилилбаккатин III
(I) 12b - ацетилокси-2a, 3,4,4a,5,6,9,10,11,12,12a,12b- додекагидро-6,9,11, -тригидрокси-4a-8,13,13-тетраметил- 5-оксо-4-[(триэтилсилил)окси]-7,11-метано-1H-цикло-[3,4]бенз[1,2-b]оксет-12-иловый эфир 2aR(2aα,-4β,4aβ,6β,9α,11β,12α,12aα,12α)] -бензойной кислоты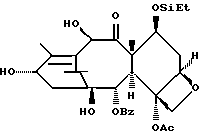
В высушенную над пламенем и продутую аргоном 3-горлую колбу емкостью 1 л (снабженную механической мешалкой и цифровым термометром) добавляли 10-дезацетилбаккатин III (27,4 г, 50,3 ммоль, содержащий H2O - 1,57%, CH3OH - 1,6%, этилацетат - 0,09% и гексан - 0,03%) и 4-диметиламинопиридин (2,62 г, 21,4 ммоль, мас.% H2O (Карл Фишер, или "К.Ф.") = 0,09 и растворяли в сухом диметилформамиде (122 мл, мас.% H2O (К.Ф.) = < 0,01). Добавляли метиленхлорид (256 мл, мас.% H2O (К.Ф.) = < 0,01) (температура реакционного раствора росла во время добавления метиленхлорида от 23oC до 25oC), и полученный гомогенный раствор охлаждали до -50oC.
Добавляли по каплям в течение 3 минут триэтиламин (16 мл, 120 ммоль, мас.% H2O (К.Ф.) = 0,08) и полученный раствор перемешивали при -50oC в течение 5 минут, после чего по каплям добавляли неразбавленный (чистый) триэтилсилилхлорид (18,6 мл, III ммоль). Добавление триэтилсилилхлорида проводили 10 минут, в течение которых температура реакционной смеси не повышалась выше -50oC. Во время добавления триэтилсилилхлорида реакционный раствор становился очень мутным.
Полученную смесь перемешивали при примерно -50oC в течение 1 часа, а затем позволяли ей стоять (без перемешивания) в холодильнике при -48oC в течение 22 часов. (Отдельный эксперимент показал, что перемешивание реакционной смеси при -45oC в течение 8 часов обеспечивает примерно 60%-ное превращение). Затем смесь извлекали из холодильника и нагревали до примерно -10oC. ТСХ анализ смеси (растворитель: этилацетат, проявитель: фосфорномолибденовая кислота/этанол) показал отсутствие исходного материала и одно единственное пятно продукта ((Rf = 0,60). Холодную смесь объединяли с этилацетатом (1 л) и промывали водой (890 мл).
Полученный водный слой отделяли и экстрагировали этилацетатом (250 мл). Объединенные органические слои промывали 5,7%-ным водным раствором NaH2PO4 (2 х 250 мл, измеренный pH 5,7-ного водного раствора NaH2PO4 был равен 4,30±0,05, измеренный pH объединенных промывных вод NaH2PO4 был равен 5,75±0,05), полунасыщенным водным раствором NaCl (250 мл), насыщенным водным раствором NaCl (250 мл), высушивали над Na2SO4, фильтровали и концентрировали на роторном испарителе. (Все операции концентрирования на роторном испарителе в данном примере проводили при температуре водяной бани 35oC).
Полученное полутвердое вещество дополнительно высушивали под высоким вакуумом (примерно 1 мм рт. ст. в течение 20 минут) и получили в результате 41,5 г белого твердого вещества. Неочищенный продукт затем растворяли в CH2Cl2 (400 мл) (для растворения твердого вещества использовали нагревание в водяной бане при 35oC), и объем полученного раствора уменьшали до примерно 150 мл на роторном испарителе. Сразу же началась кристаллизация, после чего смеси позволили стоять при комнатной температуре в течение 1 часа. Добавляли гексаны (100 мл) и подвергали смесь плавному кручению с образованием водорода. Давали смеси возможность постоять в холодном помещении при 4oC в течение 16,5 часов. Твердое вещество отфильтровывали, промывали (3 х 250 мл) смесью CH2Cl2 : гексаны = 1:9 на вакуум-фильтре и высушивали под высоким вакуумом (примерно 0,2 мм рт. ст. в течение 42 часов), получив в результате 26,1 г (79%) указанного в заголовке продукта в виде белого порошка. Маточный раствор концентрировали на роторном испарителе и остатком кристаллизовали из CH2Cl2, получив в результате 4,5 г (14%) указанного в заголовке продукта в виде белых кристаллов. Перекристаллизацию проводили так же, как и при получении первой порции продукта: твердое вещество растворяли в CH2Cl2 (100 мл) без нагрева, и объем полученного раствора уменьшали на роторном испарителе до примерно 7 мл. Кристаллизация начиналась в пределах 5 минут. Смеси давали возможность постоять при комнатной температуре в течение 1 часа, а затем в холодном помещении при 4oC в течение 42 часов. Кристаллы отфильтровывали, промывали смесью CH2Cl2 : гексаны = 1:9 (3 х 50 мл) на вакуум-фильтре и высушивали под высоким вакуумом (примерно 0,2 мм рт. ст. в течение 18 часов). 1H ЯМР - спектр этой порции был идентичен 1H ЯМР - спектру первой порции продукта. Общий выход двух порций составлял 93% (без поправок) (см. таблицу 1).
Т. пл.:239-242oC (разложение)
[α]
TCX: Rf = 0,60 (силикагель, Е ОАс). Проявитель - фосфорномолибденовая кислота/этанол.
(II) [2аR - (2aα,4β,4aβ,6β,9α,11β,12α,12aα,12bα)] - 6,12b-о-Бис(ацетилокси)-12-(бензоилокси-2a, - 3,4,4a, 5,6,9,10,11,12,12a,12b - додекагидро-9,11-дигидро-4a, 8,13,13-тетраметил-4-[(триэтилсилил)окси]-7,11-метано-1H-циклодека[3,4]бенз[1,2-b]оксет-5-он
(7-триэтилсилилбаккатин III)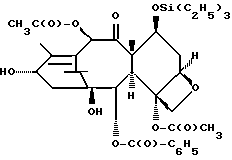
Полученный на описанной выше стадии (I) указанный в заголовке продукт (21,4 г, 32,4 ммоль) добавляли в высушенную над пламенем и продутую аргоном 3-горлую колбу емкостью 1 л (снабженную механической мешалкой и цифровым термометром) и растворяли в ТГФ (360 мл, только что отогнанный из натрий/бензофенона). Полученный раствор охлаждали до -70oC. Добавляли по каплям в течение 23 минут раствор н-бутиллития (н-BuLi) (14,6 мл 2,56 М-го раствора в гексанах, 37,3 ммоль, титрованного трижды с дифенилуксусной кислотой в ТГФ при 0oC). Во время добавления температура реакционной смеси не повышалась выше -68oC. При добавлении н-бутиллития образовывались твердые частицы, которые не растворялись при -70oC. Полученную смесь перемешивали при -70oC в течение 20 минут и затем нагревали до -48oC. При нагревании до -48oC был получен прозрачный гомогенный раствор.
После перемешивания при -48oC в течение 1/2 часа добавляли по каплям в течение 7 минут уксусный ангидрид (4,6 мл, 40 ммоль, отогнанный (137-138oC, 1 атм) в среде аргона перед использованием). Температура реакционной смеси во время добавления не повышалась выше -45oC. Полученный раствор перемешивали при -48oC в течение 20 минут, а затем при 0oC в течение 1 часа. Раствор разбавляли этилацетатом (350 мл), промывали насыщенным водным раствором NH4Cl (250 мл) и разделяли слои. Водный слой экстрагировали этилацетатом (200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl, высушивали над Na2SO4, фильтровали и концентрировали на роторном испарителе. (Все операции концентрировали на роторном испарителе в данном примере проводили при температуре водяной бани 35oC). Воздействие на полутвердое вещество высоким вакуумом (примерно 1,5 мм рт. ст. в течение 1/2 часа) дало 24,7 г белого твердого вещества.
Неочищенный продукт растворяли в CH2Cl2 (300 мл) и объем полученного раствора уменьшали на роторном испарителе до примерно 70 мл. В пределах одной минуты начиналась кристаллизация. Смесь оставляли стоять при комнатной температуре в течение 45 минут, а затем в холодном помещении при 4oC в течение 18 часов. Кристаллы отфильтровывали, промывали смесью CH2Cl2 : гексаны = 1: 9 (3 х 100 мл) на вакуум-фильтре и высушивали под высоким вакуумом (примерно 0,2 мм рт. ст. в течение 19 часов), получив в результате 20,9 г (92,0%) указанного в заголовке продукта в виде мелких белых игольчатых кристаллов. Маточный раствор концентрировали на роторном испарителе и остаток кристаллизовали из смеси CH2Cl2 и гексанов, получив в результате 0,82 г (3,6%) указанного в заголовке продукта в виде небольших белых кристаллов.
Кристаллизацию маточного раствора проводили следующим образом. Остаток растворяли в CH2Cl2 (10 мл) и объем полученного раствора уменьшали на роторном испарителе до примерно 5 мл. Выдержка при комнатной температуре в течение 1/2 часа не привела к образованию кристаллов. Добавляли гексаны (5 мл) порциями по 1 мл и раствор взбалтывали. При этом выпало немного кристаллов. Смесь оставили стоять при комнатной температуре в течение 1/2 часа (образовалось больше кристаллов), а затем в холодном месте при 4oC в течение 18 часов. Кристаллы отфильтровывали, промывали смесью CH2Cl2 и гексанов с отношением 1:9 на вакуум-фильтре и высушивали под высоким вакуумом (примерно 0,15 мм рт. ст. в течение 21 часа). Общий выход двух полученных порций составлял 95,5%. Т. пл. = 218-219oC (разложение); [α]
(b) 7-Триэтилсилил-13-[[(4S-транс)-4,5-дигидро-2,4-дифенил-5- оксазолил] карбонил]баккатин III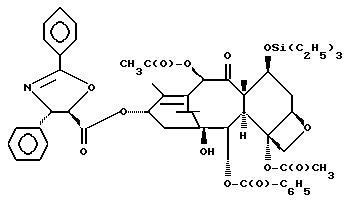
7-Триэтилсилилбаккатин III, полученный на описанной выше стадии (a), (0,209 г, 0,298 ммоль), оксазолин, полученный как указанный в заголовке продукт примера 6 (80,2 мг, 0,300 ммоль), дициклогексилкарбодиимид (84 мг, 0,407 ммоль) и 4-диметиламинопиридин (25 мг, 0,205 ммоль) добавляли в склянку на 1 драхму (3,888 г), высушенную в печи и продутую аргоном, и суспендировали в толуоле (1,0 мл). После перемешивания в течение 1 часа при комнатной температуре некоторая часть твердого вещества была растворена, причем смесь имела желтоватый цвет. Гетерогенную смесь нагревали до 85oC. ТСХ через 2 1/2 часа показала наличие исходного материала (этилацетат:гексан = 1: 1, ФМК/этанол, УФ). Продолжали нагревание при 85oC. Через 5 часов ТСХ показала, по существу, то же самое. Реакционной смеси дали возможность перемешиваться при комнатной температуре всю ночь. Через 14 часов выдержки при комнатной температуре результаты ТСХ были теми же самыми. Гетерогенную смесь разбавляли 1 мл этилацетата (был замечен некоторый осадок) и фильтровали через слой целита. Целит промывали 3х1 мл этилацетата и фильтрат концентрировали, получив в результате 0,349 г желтоватого твердого вещества. 1H ЯМР показал, что указанный в заголовке продукт и 7-триэтилсилилбаккатин III были представлены в отношении приблизительно 8:1 соответственно; были также отмечены некоторое количество 1,3-дициклогексилмочевины и следы либо исходного оксазолина, либо примесей.
Смесь частично разделяли на силикагеле (колонка: диаметр 20 мм, длина 90 мм) при отношении этилацетата к гексану от 1:2 до 1:1. ТСХ анализ показал наличие небольшого пятна с несколько более низким Rf, чем у указанного в заголовке продукта. Смешанные фракции этой примеси и продукта были объединены. Первое пятно: указанный в заголовке продукт (0,267 г небелого твердого вещества, выход = 94%). (1H ЯМР показал отношение целевого продукта и вышеупомянутой примеси, равное приблизительно 18:1); первое пятно и вышеупомянутые смешанные фракции: 11,5 мг масла 1H ЯМР показал отношение целевого продукта и отличающейся примеси, разное приблизительно 2:1). Т. пл. = 139 - 142oC, [α]D= -49,5°, [α]578= -52,6°, [α]546= -63,5°, [α]436= -157,0° (с - 1,0, CHCl3).
Пример 11
Получение 7-триэтилсилил-13-[[(4S-транс)-4,5, -дигидро-2,4- дифенил-5-оксазолин]-карбонил]баккатина III
В склянку на 1 драхму (3,888 г), высушенную над пламенем и продутую аргоном, добавляли (4S-транс)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновую кислоту (96,0 мг, 0,359 ммоль), 7-триэтилсилилбаккатин III (0,252 г, 0,359 ммоль) и 4-диметиламинопиридин (30 мг, 0,246 ммоль) и суспендировали в толуоле (1,2 мл). Сразу же добавляли диизопропилкарбодиимид (63 г, 0,503 ммоль) и слегка желтоватую гетерогенную смесь перемешивали при комнатной температуре. По прошествии некоторого времени был получен очень мутный желтый раствор. В этот момент склянку герметически закрывали и погружали в масляную баню при температуре 80oC. После 3 часов выдерживания при примерно 80oC получали более темный оранжевый раствор. Реакционную смесь непосредственно концентрировали. 1H ЯМР показал отношение требуемого продукта сочетания к 7-триэтилсилилбаккатину III, равное приблизительно 6:1. Продукт частично очищали путем хроматографии на силикагеле с этилацетат : гексан = 1:3, и в результате получили 0,300 г небелого твердого вещества. ТСХ показала выделенный продукт и побочный продукт диизопропилмочевину.
1H ЯМР показал только требуемый продукт сочетания и примесь в отношении примерно 25: 1 и побочный продукт диизопропилмочевину. Отношение целевого продукта сочетания к побочному продукту диизопропилмочевине составляло примерно 12:1.
В соответствии с этими результатами выход целевого продукта составлял примерно 85%. Т. пл. = 139 - 142oC. [α]D= -49,5°, [α]578= -52,6°, [α]546= -63,5°, [α]436= -157,0°, (с = 1,0, CHCl3).
Пример 12
Получение 7-триэтилсилил-13-[[(4S-транс)-4,5, -дигидро-2,5- дифенил-5-оксазолил]-карбонил]баккатина III
7-Триэтилсилилбаккатин III (обозначенный в данном примере как "A" и (4S-транс)-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновую кислоту (обозначенную в данном примере как "B") вводили в контакт друг с другом при условиях, указанных в приведенной таблице 2, и получили указанное в заголовке соединение.
Т. пл. = 139-142oC. [α]D= -49,5°, [α]578= -52,6°, [α]546= -63,5°, [α]436= -157,0°, (с = 1,0, CHCl3).
Ключ к таблице 2
R2POCl =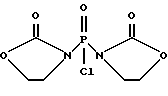
= бис(2-оксо-3-оксазолидиниил)фосфинхлорид
DCC = дициклогексилкарбодиимид
DMAP = 4-диметиламинопиридин
DIC = диизопропилкарбодиимид
ArCOCl = 2,4,6-трихлорбензоилхлорид =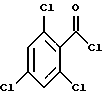
CDI = карбонилдиимидазол
t - BuCOCl = пивалоилхлорид
1,2 - DCE = 1,2-дихлорэтан
NEt3 = триэтиламин
THF = тетрагидрофуран
PhCH3 = толуол
Пример 12a
Все реагенты смешивали вместе перед добавлением растворителя. Путем хроматографии было выделено 108% (90 г) указанного в заголовке продукта (содержащего около 10% примеси). ЯМР не показал наличия исходного соединения A. (Концентрация соединения B в этом примере составляла 0,29 М. Отдельный эксперимент, в котором были использованы DCC (2 экв.), DMAP (3 экв.), DMAP•HCl (2 экв. ) в CHCl3 и 1 экв. соединения B, показал, что молярная концентрация B, равная 0,07 М, не позволяла продолжать реакцию достаточно быстро, чтобы наблюдать образование указанного в заголовке продукта посредством ЯМР в течение 27 часов).
Пример 12b
Все реагенты смешивали вместе перед добавлением растворителя. Указанный в заголовке продукт был получен в отношении к исходному соединению A, равном примерно 9: 1 (ЯМР). Путем хроматографии было выделено 87% (0,63 г) указанного в заголовке продукта.
Пример 12c
Все реагенты смешивали вместе перед добавлением растворителя. Указанный в заголовке продукт был получен в отношении к исходному соединению A, равном примерно 1:1 (ЯМР). Через 1 час дальнейшего хода реакции не наблюдалось/
Пример 12d
В течение 1 часа позволяли образование активированного оксазолина B (путем добавления R2POCl к исходному соединению B) перед добавлением исходного соединения A. Указанный в заголовке продукт был получен в отношении к исходному соединению A, равном примерно 1:6 (ЯМР). После 5 часов дальнейший ход реакции был незначительным.
Пример 12e
В течение 1 часа осуществляли контактирование CDI и исходного соединения B перед добавлением исходного соединения A. Через 4 часа добавляли DMAP. Было получено указанное в заголовке соединение в отношении к исходному соединению A и примеси, равном примерно 1:1:1 (ЯМР). Было отмечено, что до добавления DMAP не было никакой реакции и что избыточный нагрев вызывая некоторое разложение.
Пример 12f
Последним добавляли ArCOCl. Получили отношение указанного в заголовке продукта к исходному соединению A, равное примерно 1:1 (ЯМР). Через 1,5 часа дальнейшего хода реакции не наблюдалось.
Пример 12g
Последним добавляли ArCOCl. Получили отношение указанного в заголовке продукта к исходному соединению A, равное примерно 1:1 (ЯМР). Через 1 час дальнейшего хода реакции не наблюдалось.
Пример 12h
Последним добавляли ArCOCl. Получили отношение указанного в заголовке продукта к исходному соединению A, равное примерно 1:1 (ЯМР). Никакого дальнейшего хода реакции не наблюдалось через 3,5 часа.
Пример 12i
Перед добавлением исходного соединения A обеспечивали предварительно образование смешанного ангидрида в течение 1 часа (путем добавления t - BuCOCl к исходному соединению B). Получили (ЯМР) отношение указанного в заголовке продукта к исходному соединению A, равное примерно 1:2. Никакого дальнейшего хода реакции не наблюдалось через 2 часа.
Пример 12j
Перед добавлением исходного соединения A обеспечивали предварительное образование смешанного ангидрида в течение 1 часа (путем добавления t - BuCOCl к исходному соединению B). Получили (ЯМР) отношение указанного в заголовке продукта к исходному соединению A, равное примерно 3:1. Никакого дальнейшего хода не наблюдалось через 1 час.
Пример 12k
Перед добавлением исходного соединения A обеспечивали предварительное образование смешанного ангидрида в течение 1 часа (путем добавления t - BuCOCl к исходному соединению B). Спустя 1 час после добавления исходного соединения A добавляли DMAP. Получили (ЯМР) отношение указанного в заголовке продукта к исходному соединению A, равное примерно 1:4. Никакой реакции без DMAP не наблюдалось; через 2 часа после добавления DMAP дальнейшего хода реакции не наблюдалось.
Пример 12l
Перед добавлением исходного соединения A обеспечивали предварительное образование смешанного ангидрида в течение 1 часа (путем добавления t - BuCOCl к исходному соединению B). Спустя 16 часов добавляли DMAP при 55oC. Получили (ЯМР) отношение указанного в заголовке продукта к исходному соединению A, равное примерно 1:6. До добавления DMAP не наблюдалось совсем или наблюдалось очень незначительная реакция.
Пример 13
Получение 7-триэтилсилил-13-[[(4S-транс-4,5-дигидро-2,4- дифенил-5-оксазолил]-карбонил]баккатина III
В высушенную над пламенем и продутую аргоном склянку на 1 драхму (3,888 г) добавляли (4S-транс-4,5-дигидро-2,4-дифенил-5-оксазолкарбоновую кислоту (65,0 мг, 0,243 ммоль), 7-триэтилсилилбаккатин III (0,142 г, 0,203 ммоль), DCC (75 мг, 256 ммоль) и пирролидинопиридин (38 мг, 256 ммоль) и частично растворяли в толуоле (1,0 мл). Полученную желтоватую гетерогенную смесь перемешивали при комнатной температуре. ТСХ через 3 часа показала присутствие указанного в заголовке продукта (EtOAc:гексан = 1:1, ФМК/EtOH, УФ). (ТСХ через 7 и 23 часа после перемешивания при комнатной температуре показала отсутствие каких-либо изменений).
Реакционную смесь разбавляли этилацетатом (1 мл), фильтровали через слой целита и концентрировали, получив в результате 0,275 г маслянистого желтоватого твердого вещества. 1H ЯМР показал требуемый указанный в заголовке продукт и 7-триэтилсилилбаккатин III в отношении примерно 8:1. Присутствовал также содержащий N-ацилмочевину побочный продукт сочетающего агента в примерно таком же количестве, что и 7-триэтилсилилбаккатин III.
Твердое вещество хроматографировали на силикагеле с EtOAc:гексан = 1:2, получив в результате 0,176 небелого твердого вещества. Выход примерно 92%. 1H ЯМР показал только целевой продукт сочетания и N-ацилмочевину в отношении приблизительно 11:1.
Пример 14
Получение 7-триэтилсилил-13-[[(4S-транс)-4,5-дигидро-2,4- дифинил-5-оксазолил]-карбонил]баккатина III
В высушенную над пламенем и продутую аргоном склянку на 1 драхму добавляли (4S-транс)-4,5, -дигидро-2,4, -дифинил-5-оксазолкарбоновую кислоту (66,7 мг, 0,250 ммоль), 7-триэтилсилилбаккатин III (0,146 г, 0,208 ммоль), дициклогексилкарбодиимид (79 мг, 0,383 ммоль) и 4-морфолинопиридин (41 мг, 0,250 ммоль) и частично растворяли в толуоле (1 мл). Полученную желтую гетерогенную смесь перемешивали при комнатной температуре. ТСХ через 3 часа показала присутствие указанного в заголовке продукта (этилацетат:гексан = 1: 1, ФМК/EtOH, УФ). (ТСХ через 7 и 23 часа после перемешивания при комнатной температуре показала, что никаких дальнейших изменений не произошло).
Реакционную смесь разбавляли этилацетатом (1 мл), фильтровали через слой целита и концентрировали, получив в результате 0,280 г желтоватого твердого вещества. 1H ЯМР показал наличие целевого указанного в заголовке продукта сочетания и отсутствие 7-триэтилбаккатина III (хотя следы его обнаружены посредством ТСХ). Содержащий N-ацилмочевину побочный продукт от сочетающего агента присутствовал в отношении к указанному в заголовке продукту, равном примерно 1:9.
Твердое вещество подвергали хроматографии на силикагеле с EtOAc:гексаны = 1:2 и получили 0,196 г белого твердого вещества. 1H ЯМР показал только указанный в заголовке продукт сочетания и N-ацилмочевину в отношении примерно 15: 1. Выход превышал 90%. Т. пл. = 139-142oC. [α]D= -49,5°, [α]578= -52,6°, [α]546= -63,5°, [α]436= -157,°0 (с = 1,0, CHCl3).
Пример 15
Получение 7-триэтилсилил-13-[[(4S-транс)-4,5-дигидро-2,4- дифенил-5-оксазолил] -карбонил] баккатина III и 7-триэтилсилил- 13-[[(4S-цис)-4,5-дигидро-2,4-дифенил-5-оксазолил]-карбонил]- баккатина III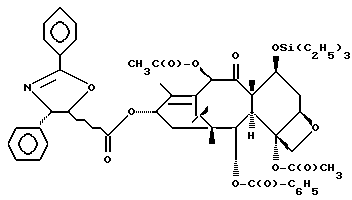
Приготавливали смесь цис- и транс-оксазолиновых продуктов, указанных в заголовке примера 9, в отношении цис:транс = 3:1 (100 мг), 7-трисилилбаккатина III (219 мг, 0,3121 ммоль), DCC (97 мг) и DMAP (23 мг) и толуола (0,9 мл). После 1 часа нагрева при 80oC значительное количество 7-триэтилсилилбаккатина III оказалось непрореагировавшим. Добавляли другую навеску DMAP (97 мг) и DCC (23 мг) и смесь нагревали при 80oC всю ночь. Тонкослойная хроматография (ТСХ) (гексан:этилацетат = 2:1) показала наличие незначительных количеств исходного материала.
Полученную реакционную смесь разбавляли метиленхлоридом (20 мл), добавляли насыщенный раствор бикарбоната натрия (10 мл, водн.) и водяной слой экстрагировали метиленхлоридом /2 х 10 мл/, причем объединенные органические слои высушивали над безводным MgSO4. После концентрирования в вакууме продукт очищали путем ВЭЖХ (гексан:этилацетат = 4:1) и получили смесь указанных в заголовке продуктов, содержавшую дициклогексилмочевину. После ресуспендирования в этилацетате и удаления некоторой части дициклогексилмочевины путем фильтрации продукт весил 260 мг. Другая очистка путем ВЭЖХ дала 117 мг чистого указанного в заголовке транс-продукта (45%) и 45 мг (15%) смеси указанных в заголовке продуктов (транс:цис = ≈2:1). Смесь очищали посредством препаративной ТСХ (гексаны:этилацетат = 1:1) и получили 11 г цис-продукта, указанного в заголовке.
Пример 16
Получение таксола
Полученный в примере 10 указанный в заголовке продукт сочетания (0,102 г, 0,107 ммоль) загружали в колбу емкостью 10 мл и растворяли в тетрагидрофуране (1,2 мл). Затем добавляли метанол (1,2 мл), и гомогенный раствор охлаждали до 0oC. Добавляли по каплям HCl (вод., 1 н., 0,59 мл, 0,59 ммоль) и светлый гомогенный раствор перемешивали при 0oC. Спустя 3 часа нахождения при 0oC ТСХ (этилацетат:гексан = 1:1, ФМК/этанол, УФ) показала, что исходный материал остался, и светлый гомогенный раствор был перенесен в холодное место при 4oC. Через 18 часов выдержки при 4oC ТСХ-анализ показал, что реакция, по существу, закончена (этилацетат : гексан = 1:1, ФМК/этанол, УФ). Получили следующее соединение: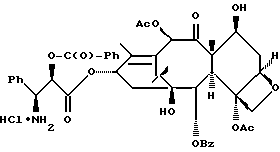
Светлый гомогенный раствор нагревали до комнатной температуры. Добавляли 3,5 мл насыщенного водного раствора NaHCO3 (наблюдалось выделение пузырьков), в результате чего получили гетерогенную смесь. Добавление 5 мл тетрагидрофурана и 2 мл воды существенно не увеличило растворимость. Гетерогенную смесь энергично перемешивали при комнатной температуре. После перемешивания при комнатной температуре в течение 1 часа смесь все еще была гетерогенной. Далее смесь разбавляли 7 мл воды и 4 мл тетрагидрофурана. Полученный светлый гомогенный раствор перемешивали при комнатной температуре.
ТСХ через 2 1/2 часа после добавления NaHCO3 показала только присутствие таксола (этилацетат:гексан = 2:1, ФМК/этанол, УФ). Реакционную смесь разбавляли 25 мл этилацетата и 25 мл воды и встряхивали. Разделяли слои, и водную часть экстрагировали этилацетатом (3 х 25 мл). Объединенные органические фракции высушивали над Na2SO4, фильтровали и концентрировали, получив в результате 104 мг не совсем белого стекловидного твердого вещества. 1H ЯМР показал, что это таксол. Полученное твердое вещество очищали путем хроматографии на силикагеле (колонка: диаметр 20 мм и длина 70 мм) при отношении этилацетата к гексану от 2:1 до 4:1 и получили 79,0 мг указанного в заголовке соединения в виде белого твердого вещества. Выход 86,4%.
Пример 17
Получение 7,13-бис TES баккатина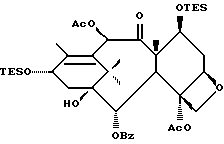
Баккатин III (3,102 г, 5,290 ммоль) растворяли в сухом диметилформамиде (ДМФ) (21 мл). К этому раствору при 0oC добавляли имидазол (1,80 г, 26,5 ммоль), а затем хлортриэтилсилан (TESCl) (4,45 мл, 26,5 ммоль). Реакционную смесь перемешивали при комнатной температуре всю ночь, разбавляли этилацетатом (350 мл) и промывали водой (4х20 мл) и рассолом. Органический слой высушивали и концентрировали в вакууме и остаток хроматографировали (20% этилацетата в гексанах), что дало 4,00 г (89,1%) целевого продукта.
Пример 18
Получение 1-DMS-7,13-TES баккатина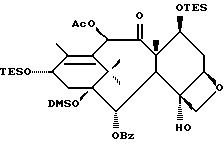
7,13-TES баккатин (2,877 г, 3,534 ммоль) растворяли в сухом ДМФ (17,7 мл). К этому раствору при 0oC добавляли имидазол (720,9 мг, 10,60 ммоль), а затем диметилхлорсилан (91,18 мл, 10,60 ммоль). Реакционную смесь перемешивали при указанной температуре в течение 45 минут и затем разбавляли этилацетатом (300 мл) и промывали водой (4 х 20 мл). Органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (10% этилацетата в гексанах) и получили 2,632 г (85,4%) целевого продукта.
Пример 19.
Получение 4-гидрокси-7,13-бис TES-1-DMS баккатина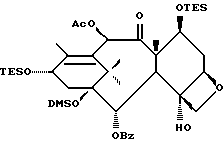
Силилированное производное баккатина из примера 18 (815 мг, 0,935 ммоль) растворяли в ТГФ (15,6 мл). К этому раствору при 0oC добавляли Red-Al (0,910 мл, 60 мас.%, 4,675 ммоль). Через 40 минут реакцию резко прекращали насыщенным раствором тартрата натрия (7 мл). Спустя 5 минут реакционную смесь разбавляли этилацетатом (250 мл). Органическую фазу промывали водой и рассолом и высушивали. Затем органический слой концентрировали в вакууме и остаток хроматографировали (10-20% этилацетата в гексанах), получив в результате 590 мг (76,0%) целевого C4-гидроксильного аналога баккатина.
Пример 20
Получение C-4-циклопропилового эфира 7,13-бис TES-1-DMS баккатина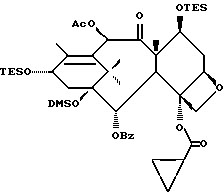
C-4 гидроксильное производное баккатина из примера 19 (196 мг, 0,236 ммоль) растворяли в сухом ТГФ (4,7 мл). Этот раствор при 0oC обрабатывали LHMDS (0,283 мл, IM, 0,283 ммоль) и через 30 минут выдержки при этой температуре добавляли циклопропанкарбонилхлорид (0,032 мл, 0,354 ммоль). Реакционную смесь перемешивали при 0oC в течение 1 часа и затем резко охлаждали насыщенным раствором NH4Cl (3 мл). Реакционную смесь экстрагировали этилацетатом (100 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Полученный остаток хроматографировали (10% этилацетата в гексанах) и в результате получили 137 мг (65%) целевого продукта.
Пример 21.
Получение C4-циклопропанбаккатина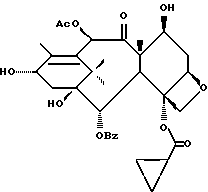
7,13-TES-1-DMS-4-циклопропанбаккатина из примера 20 (673 мг, 0,749 ммоль) растворяли в сухом ацетонитриле (6 мл) и ТГФ (2 мл). К этому раствору при 0oC добавляли пиридин (2,25 мл), а затем 48%-ный раствор HF (6,74 мл). Через 30 минут выдержки при 0oC добавляли тетрабутиламмонийфторид (TBAF) (2,25 мл, 1 М, 2,25 ммоль). До тех пор пока не был израсходован исходный материал (ТСХ - анализ), добавляли дополнительную дозу тетрабутиламмонийфторида. Реакционную смесь концентрировали до сиропа, а затем разбавляли этилацетатом (350 мл) и промывали 1 н. HCl, насыщенным раствором NaHCO3, рассолом и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах), получив в результате 366 мг (80%) целевого продукта.
Пример 22
Получение 7-TES-4-циклопропанбаккатина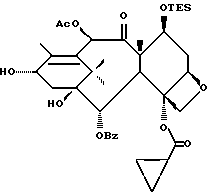
4-циклопропанбаккатин из примера 21 (46,6 мг, 0,076 ммоль) растворяли в сухом диметилформамиде (1 мл). К этому раствору при 0oC добавляли имидазол (20,7 мг, 0,305 ммоль). Реакционную смесь промывали водой и рассолом, высушивали и затем концентрировали в вакууме. Остаток хроматографировали (30-50% этилацетата в гексанах) и в результате получили 36 мг (65,1%) целевого продукта.
Пример 23
Получение 2',7-бис TES-4-циклопропанпаклитаксела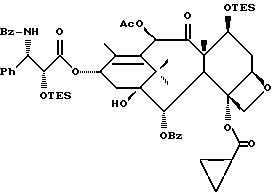
Раствор соединения из примера 22 (30,0 мг, 0,0413 ммоль) в ТГФ (1 мл) охлаждали до -40oC и обрабатывали LHMDS (0,062 мл, 0,062 ммоль). Спустя 5 минут добавляли раствор β - лактама (Способы получения этого β - лактама описаны в патенте США N 5175315) (23,6 мг, 0,062 ммоль) в ТГФ (0,5 мл). Реакционную смесь перемешивали при 0oC в течение 1 часа и затем резко охлаждали насыщенным раствором NH4Cl (1 мл). Далее реакционную смесь экстрагировали этилацетатом (40 мл) и промывали водой и рассолом. Органическую фазу высушивали и концентрировали в вакууме и остаток хроматографировали (20-30-60% этилацетата в гексанах), получив в результате 24,5 мг (53,6%) целевого продукта вместе с 5,1 мг (17%) исходного материала.
Пример 24
Получение 4-циклопропанового эфира паклитаксела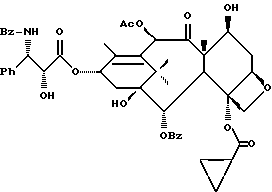
Раствор в ацетонитриле (0,5 мл) продукта из примера 23 (22,0 мг, 0,020 ммоль) обрабатывали при 0oC пиридином (0,60 мл), а затем 48%-ным раствором HF (0,180 мл), реакционную смесь держали при 5oC всю ночь, после чего разбавляли этилацетатом (30 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах), получив в результате 10 мг (57,2%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,10-8,06 (м, 2H), 7,76-7,26 (м, 13H), 7,04 (д, J = 9,1 Гц, 1H), 6,14 (м, 1H), 5,82 (д, J = 9,1 Гц, 1H), 5,65 (д, J = 6,9 Гц, 1H), 4,85 (м, 2H), 4,39 (м, 1H), 4,19 (AB кв., J = 8,4 Гц, 2H), 3,80 (д, = 6,9 Гц, 1H), 3,59 (д, J = 4,8 Гц, 1H), 2,60-1,13 (м, 24H, включ. синглеты при 2,23, 1,77, 1,66, 1,23, 1,14, 3H каждый).
MCBP: вычислено для C49H54NO14 (MH+ - 880,3544, найдено - 880,3523.
Пример 25
Получение 4-циклобутилового эфира 7,13-TES-1-DMS-баккатина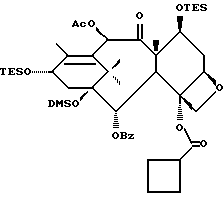
Раствор продукта из примера 19 (113,6 мг, 0,137 ммоль) в ТГФ (2,6 мл) обрабатывали при 0oC LHMDS (0,178 мл, 1 М, 0,178 ммоль). После 30 минут выдержки при 0oC добавляли циклобутилкарбонилхлорид (24,4 мг, 0,206 ммоль). Реакционную смесь перемешивали при 0oC в течение 1 часа и резко охлаждали насыщенным раствором NH4Cl (2 мл). Далее реакционную смесь экстрагировали этилацетатом (75 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (10% этилацетата в гексанах) и получили 80 мг (64,1% целевого продукта).
Пример 26
Получение 4-циклобутилбаккатина
К раствору соединения из примера 25 в ацетонитриле (3 мл) при 0oC добавляли сухой пиридин (0,61 мл), а затем 48%-ную HF (1,83 мл). После 1 часа выдержки при 0oC добавляли тетрабутиламмонийфторид (0,61 мл, 1 М, 0,61 ммоль). До тех пор пока не был израсходован весь исходный материал добавляли дополнительный тетрабутиламмонийфторид. Затем частично удаляли растворитель и остаток разбавляли этилацетатом (150 мл) и промывали 1 н. HCl и насыщенным раствором NaHCO3. Органический слой затем высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получили в результате 95,6 мг (75%) целевого продукта.
Пример 27
Получение 4-циклобутилового эфира 7-TES-баккатина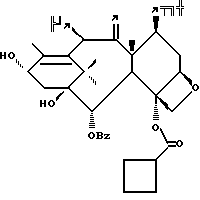
4-циклобутилбаккатин из примера 26 (85 мг, 0,136 ммоль) растворяли в сухом диметилформамиде (1,4 мл). К раствору при 0oC добавляли имидазол (36,9 мг, 0,543 ммоль) и TESCl (91,2 мкл, 0,543 ммоль). Реакционную смесь разбавляли этилацетатом (75 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (40% этилацетата в гексанах) и в результате получили 74 мг (73,6%) целевого продукта.
Пример 28
Получение 2',7-TES-4-циклобутилпаклитаксела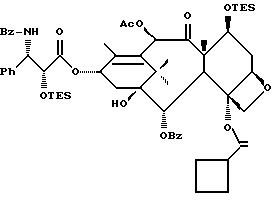
7-TES-4-циклобутилбаккатин из примера 27 (41 мг, 0,055 ммоль) растворяли в ТГФ (1 мл). Этот раствор охлаждали до -40oC и обрабатывали LHMDS (0,083 мл, 1 М, 0,083 ммоль), а затем раствором β -лактама из примера 23 (31,7 мг, 0,083 ммоль) в ТГФ (0,5 мл). Реакционную смесь держали при 0oC в течение 1 часа, после чего резко охлаждали NH4Cl (2 мл). Далее реакционную смесь экстрагировали этилацетатом (50 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме и остаток хроматографировали (20-30% этилацетата в гексанах), получив в результате 56 мг (90,2%) целевого продукта.
Пример 29
Получение 4-циклобутилпаклитаксела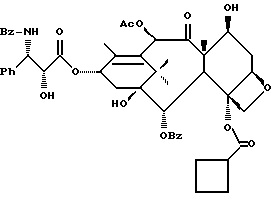
2', 7-TES-4-циклобутилтаксол из примера 28 (47 мг, 0,042 ммоль) растворяли в ацетонитриле (1 мл), к этому раствору при 0oC добавляли пиридин (0,125 мл), а затем 48%-ную HF (0,375 мл). Реакционную смесь держали при 5oC всю ночь. Затем реакционную смесь разбавляли этилацетатом (50 мл) и промывали 1 н. HCl, насыщенным раствором NaHCO3 и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получили в результате 31,8 мг (84,9%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,15-8,12 (м, 2H), 7,73-7,26 (м, 13H), 6,96 (д, J = 9,0 Гц, 1H), 6,26 (с, 1H), 6,17 (м, 1H), 5,80 (д, J = 9,0 Гц, 1H), 5,66 ( д. J = 7,1 Гц, 1H), 4,83 (м, 2H), 4,41 (м, 1H), 4,26 (AB кв, J = 8,4 Гц, 2H), 3,78 (д, J = 7,0 Гц, 1H), 3,57 (д, J = 5,2 Гц, 1H), 3,42 (м, 1H) 2,61-1,14 (м, 25H, включ. синглеты при 2,23, 1,76, 1,68, 1,23, 1,14, 3H каждый).
MCBP: вычислено для C50H56NO14 (MH+) - 894,3701, найдено - 894,3669.
Пример 30
Получение 4-циклопентилового эфира 7,13-TES-1-DMS-баккатина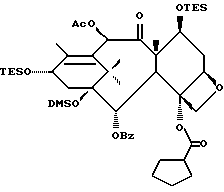
Раствор соединения из примера 19 (147 мг, 0,177 ммоль) в ТГФ (3,5 мл) обрабатывали при 0oC LHMDS (0,230 мл, 1 М, 0,230 ммоль). Спустя 30 минут добавляли циклопентилкарбонилхлорид (32,3 мкл, 0,266 ммоль). Реакционную смесь при указанной температуре перемешивали 1 час, после чего резко охлаждали насыщенным раствором NH4Cl. Далее реакционную смесь экстрагировали, промывали, высушивали и концентрировали в вакууме. Остаток хроматографировали (10% этилацетата в гексанах) и получили в результате 90 мг (55%) целевого продукта.
Пример 31
Получение 4-циклопентилбаккатина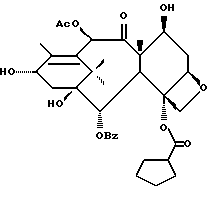
Раствор продукта из примера 30 (75 мг, 0,081 ммоль) в ацетонитриле (1,6 мл) обрабатывали при 0oC пиридином (0,24 мл), а затем 48%-ной HF (0,72 мл). Реакционную смесь перемешивали при 0oC в течение 1 часа, после чего добавляли тетрабутиламмонийфторид (0,405 мл, 1 М, 0,405 ммоль). Через 1 час добавляли еще пять эквивалентов реагента. Реакционную смесь разбавляли этилацетатом (100 мл) и промывали 1 н. HCl и насыщенным раствором NaHCO3 и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (50% этилацетата в гексанах) и получили 44 мг (85%) целевого продукта.
Пример 32.
Получение 4-циклопентилового эфира 7-TES-баккатина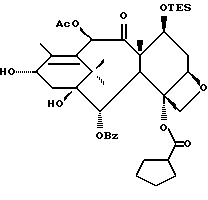
Циклопентилбаккатин (35 мг, 0,055 ммоль) растворяли в сухом диметилформамиде (1 мл). К этому раствору при 0oC добавляли имидазол (14,9 мг, 0,219 ммоль), а затем TESCl (36 мкл, 0,219 ммоль). Реакционную смесь перемешивали при 0oC в течение 30 минут и разбавляли этилацетатом (50 мл). Органический слой промывали и высушивали и концентрировали в вакууме. Остаток хроматографировали (40% этилацетата в гексанах), получив в результате 31 мг (75%) целевого продукта.
Пример 33
Получение 4-циклопентилового эфира 2',7-TES-баккатина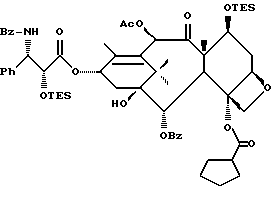
Раствор продукта из примера 32 (24,5 мг, 0,0324 ммоль) в ТГФ (0,6 мл) обрабатывали при -40oC LHMDS (0,049 мл, 1 М, 0,049 ммоль) а затем раствором β - лактама из примера 23 (18,6 мг, 0,049 ммоль) в ТГФ (0,3 мл). Реакционную смесь перемешивали при 0oC в течение 1 часа и затем резко охлаждали насыщенным раствором NH4Cl. Далее реакционную смесь экстрагировали этилацетатом (35 мл) и промывали, а затем высушивали и концентрировали в вакууме. Остаток хроматографировали (20-30-50% этилацетата в гексанах) и получили в результате 15,5 мг (42%) целевого продукта вместе с 7,8 мг (31,8%) непрореагировавшего исходного материала.
Пример 34.
Получение 4-циклопентилового эфира паклитаксела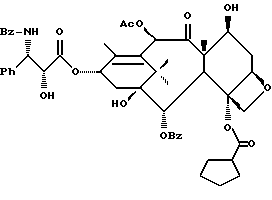
Раствор продукта на примере 33 (13 мг, 0,0115 ммоль) в ацетонитриле 0,3 мл) обрабатывали при 0oC пиридином (0,035 мл), а затем 48%-ной HF (0,103 мл). Реакционную смесь держали при 5oC всю ночь. Затем реакционную смесь разбавляли этилацетатом (30 мл) и промывали 1 н. HCl, NaHCO3 и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (50% этилацетата в гексанах) и получили в результате 7,3 мг (70,3%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,17-8,14 (м, 2H), 7,74-7,26 (м, 13H), 6,90 (д, J = 8,9 Гц, 1H), 6,27 (с, 1H), 6,20 (м, 1H), 5,75 (д, J = 8,9 Гц, 1H), 5,69 (д, J = 7,0 Гц, 1H), 4,79 (м, 2H), 4,44 (м, 1H), 4,24 (AB кв, J = 8,4 Гц, 2H), 3,81 (д, J = 7,0 Гц, 1H), 3,46 (д, J = 4,7 Гц, 1H), 3,06 (м, 1H) 2,56-1,15 (м, 27H, вкл. синглеты при 2,24, 1,82, 1,68, 1,33, 1,15, 3H каждый).
MCBP: вычислено для C51H57NO14Na(MNa+) - 930,3677, найдено 930,3714.
Пример 35.
Получение 2',7-силилированного-4-циклопропантаксана с фуриловой боковой цепью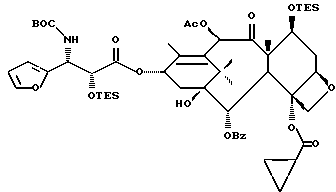
Раствор продукта из примера 22 (75,8 мг, 0,104 ммоль) в ТГФ (2 мл) обрабатывали при -40oC LHMDS (0,136 мл, 1 М, 0,136 ммоль) и β - лактамом (способы получения бета-лактама описаны в патенте США N 5227400) (57,3 мг, 1,156 ммоль). Реакционную смесь перемешивали при 0oC в течение 1 часа и затем резко охлаждали смешением с насыщенным раствором NH4Cl (1 мл). Реакционную смесь затем экстрагировали этилацетатом, промывали и высушивали и концентрировали в вакууме. Остаток хроматографировали (20% этилацетата в гексанах) и получили в результате 113 мг (100%) целевого продукта.
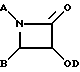
A = Бензоилоксикарбонил
B = 2-Фурил
D = Силильная защитная группа
Пример 36.
Получение 4-циклопропилового эфира таксана с фуриловой боковой цепью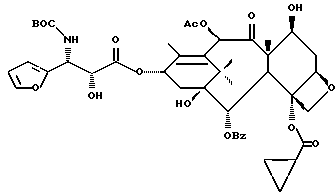
Раствор продукта на примере 35 (2 мл) в ацетонитриле обрабатывали при 0oC пиридином (0,27 мл), а затем 48%-ной HF (0,81 мл). Реакционную смесь держали при 5oC в течение 3 часов, разбавляли этилацетатом (75 мл) и промывали 1 н. HCl, насыщенным раствором NaHCO3, рассолом и высушивали и концентрировали в вакууме. Остаток хроматографировали (50-60% этилацетата в гексанах) и получили 68 мг (88,2%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,09-8,06 (м, 2H), 7,62-7,37 (м, 3H), 7,26 (с, 1H), 6,37-6,30 (м, 3H), 6,19 (м, 1H), 5,65 (д, J = 7,0 Гц, 1H), 5,37 (д, J = 9,9 Гц, 1H), 5,23 (д, J = 9,9 Гц, 1H), 4,82 (д, J = 8,3 Гц, 1H), 4,76 (д, J = 4,1 Гц, 1H), 4,42 (м, 1H), 4,18 (AB кв, J = 8,4 Гц, 2H), 3,85 (д, J = 6,9 Гц, 1H), 3,37 (д, J = 5,4 Гц, 1H), 2,55-1,01 (м, 33H, вкл. синглеты при 2,23, 1,90, 1,66, 1,26, 1,14, 3H каждый, 1,33 9H).
MCBP: вычислено для C45H56NO16 (MH+) - 866,3599, найдено - 886,3569.
Пример 37.
Получение 4-циклобутилового эфира 2',7-силилированного таксана с фуриловой боковой цепью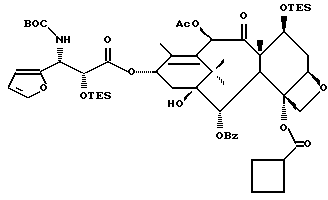
Раствор продукта из примера 22 в ТГФ (0,8 мл) обрабатывали при -40oC LHMDS (0,050 мл, 1 М, 0,050 ммоль). Через 2 минуты добавляли β -лактам из примера 35 (18,2 мг, 0,050 ммоль). Реакционную смесь перемешивали при 0oC в течение 1 часа и охлаждали смешением с насыщенным раствором NH4Cl. Реакционную смесь затем экстрагировали и промывали, а затем высушивали и концентрировали в вакууме. Остаток хроматографировали (20% этилацетата в гексанах) и в результате получили 33,0 мг (89,4%) целевого продукта.
Пример 38
Получение 4-циклобутилового эфира таксана с фуриловой боковой цепью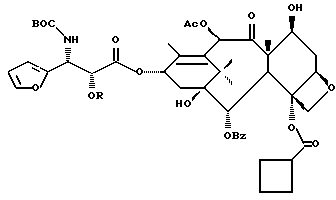
Раствор продукта из примера 37 (30 мг, 0,027 ммоль) в ацетонитриле (1 мл) обрабатывали при 0oC пиридином (0,081 мл), а затем 48%-ной HF (0,243 мл). Реакционную смесь держали при 5oC всю ночь и разбавляли этилацетатом (50 мл), промывали 1 н. HCl, насыщенным раствором NaHCO3 и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получили 22 мг (92,4%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,13-8,10 (м, 2H), 7,62-7,45 (м, 3H), 6,42-6,38 (м, 3H), 6,30 (с, 1H), 6,19 (м, 1H), 5,65 (д, J = 7,1 Гц, 1H), 5,34 (д, J = 9,6 Гц, 1H), 5,18 (д, J = 9,8 Гц, 1H), 4,90 (д, J = 7,7 Гц, 1H), 4,73 (дд, J = 2,0 Гц, J = 5,7 Гц, 1H), 4,45 (м, 1H), 4,25 (AB кв, J = 8,4 Гц, 2H), 3,80 (д, J = 7 Гц, 1H), 3,50 (м, 1H), 3,27 (д, J = 5,8 Гц, 1H), 2,61-1,15 (м, 34H, вкл. синглеты при 2,24, 1,86, 1,68, 1,26, 1,15 3H каждый, 1,33 9H).
Пример 39.
Получение 7,13-TES-1-DMS-4-бутиратбаккатина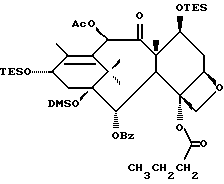
C4-гидроксильное производное баккатина из примера 19 (181 мг, 0,218 ммоль) растворяли в сухом ТГФ (4,4 мл). Этот раствор при 0oC обрабатывали LHMDS (0,262 мл, 1 М, 0,262 ммоль) и спустя 30 минут при этой температуре добавляли бутирилхлорид (0,34 мл, 0,33 ммоль). Реакционную смесь перемешивали при 0oC в течение 1 часа, после чего резко охлаждали смешением с насыщенным NH4Cl (3 мл). Далее реакционную смесь экстрагировали этилацетатом (100 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Полученный остаток хроматографировали (10% этилацетата в гексанах) и в результате получили 138 мг (70,3%) целевого продукта.
Пример 40
Получение C4-бутирилового эфира баккатина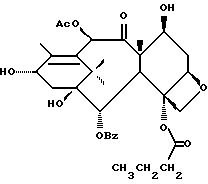
7,13-TES-I-DMS-4-Бутиратбаккатин из примера 39 (527 мг, 0,586 ммоль) растворяли в сухом ацетонитриле (19,5 мл). К этому раствору при 0oC добавляли пиридин (1,95 мл), а затем 48%-ный раствор HF (5,86 мл). После 30 минут выдержки при 0oC реакционную смесь держали при 5oC всю ночь. Затем разбавляли этилацетатом (400 мл) и промывали 1 н. HCl, насыщенным раствором NaHCO3, рассолом и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получали 286 мг (80%) целевого продукта.
Пример 41
Получение 7-TES-4-бутиратбаккатина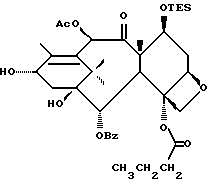
4-Бутиритбаккатин из примера 40 (286 мг, 0,466 ммоль) растворяли в сухом диметилформатиде (2,3 мл). К этому раствору при 0oC добавляли имидазол (127 мг, 1,86 ммоль), а затем TESCl (0,313 мл, 1,86 ммоль). Реакционную смесь перемешивали при 0oC в течение 30 минут и затем разбавляли этилацетатом (100 мл). Далее реакционную смесь промывали водой и рассолом и высушивали, а затем концентрировали в вакууме. Остаток хроматографировали (30-50% этилацетата в гексанах) и получили в результате 283,3 мг (83,5%) целевого продукта.
Пример 42
Получение 2',7-бис TES-C-4-бутиратпаклитаксела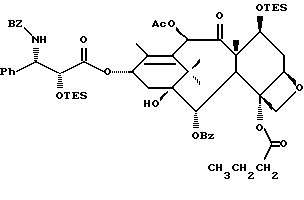
Раствор продукта из примера 41 (300,6 мг, 0,413 ммоль) в ТГФ (8,3 мл) охлаждали до - 40oC и обрабатывали LHMDS (0,619 мл, 0,619 ммоль). Через 5 минут добавляли раствор β -лактама из примера 23 (236 мг, 0,619 ммоль) в ТГФ (4,1 мл). Реакционную смесь перемешивали при 0oC в течение 1 минуты и затем охлаждали смешением с насыщенным раствором NH4Cl (3 мл). Далее реакционную смесь экстрагировали этилацетатом (150 мл) и промывали водой и рассолом. Органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (20-30-60% этилацетата в гексанах) и получили 377 мг (82,3%) целевого продукта.
Пример 43
Получение C4-бутиратпаклитаксела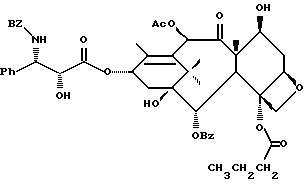
Раствор продукта из примера 42 (366 мг, 0,334 ммоль) в ацетонитриле (15,3 мл) обрабатывали при 0oC пиридином (0,926 мл), а затем 48%-ным раствором HF (2,78 мл). Реакционную смесь держали при 5oC всю ночь и разбавляли этилацетатом (200 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получили в результате 274 мг (94,5%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,12-8,09 (м, 2H), 7,71-7,32 (м, 13H), 7,00 (д, J = 8,9 Гц, 1H), 6,25 (с, 1H), 6,16 (м, 1H), 5,73 (д, J = 8,8 Гц, 1H), 5,64 (д, J = 7,0 Гц, 1H), 4,85 (д, J = 9,4 Гц, 1H), 4,76 (м, 1H), 4,38 (м, 1H), 4,20 (AB кв, J = 8,4 Гц, 2H), 3,77 (д, J = 6,9 Гц, 1H), 3,70 (д, J = 4,3 Гц, 1H), 2,66-0,85 (м, 26H, вкл. синглеты при 2,20, 1,76, 1,65, 1,21, 1,11, 3H каждый, триплет при 0,88 3H).
Пример 44
Получение 7,13-TES-1-DMS-этилкарбонатбаккатина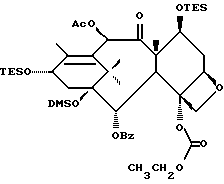
Раствор продукта из примера 19 (205 мг, 0,247 ммоль) в ТГФ (5 мл) обрабатывали при 0oC LHMDS (0,296 мл, 1 М, 0,296 ммоль). После 30 минут выдержки при 0oC добавляли этилхлорформиат (0,0354 мл, 0,370 ммоль). Реакционную смесь перемешивали при 0oC в течение 1 часа и охлаждали смешением с насыщенным раствором NH4Cl (3 мл). Далее реакционную смесь экстрагировали этилацетатом (100 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (10% этилацетата в гексанах) и получили 155 мг (69,6%) целевого продукта.
Пример 45
Получение C4-этилкарбонатбаккатина
К раствору продукта из примера 44 (152 мг, 0,169 ммоль) в ацетонитриле (5,6 мл) при 0oC добавляли сухой пиридин (0,56 мл), а затем 48%-ный раствор HF (1,69 мл). После 30 минут выдержки при 0oC реакционную смесь выдерживали всю ночь при 5oC. Затем остаток разбавляли этилацетатом (150 мл) и промывали 1 н. HCl и насыщенным раствором NaHCO3. Органический слой высушивали и концентрировали в вакууме. Полученный остаток хроматографировали (60% этилацетата в гексанах) и получили в результате 99 мг (95,4% целевого продукта).
Пример 46
Получение 7-TES-C4-этилкарбонатбаккатина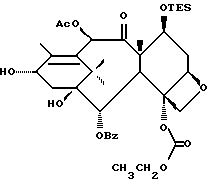
4-Этилкарбонатбаккатин из примера 45 (95 мг, 0,154 ммоль) растворили в сухом диметилформамиде (0,771 мл). К этому раствору при 0oC добавляли имидазол (42 мг, 0,617 ммоль) и TESCl (104 мкл, 0,617 ммоль). Реакционную смесь разбавляли этилацетатом (100 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (40% этилацетата в гексанах) и получили в результате 95 мг (84,4%) целевого продукта.
Пример 47
Получение 2',7-TES-C4-этилкарбонатпаклитаксела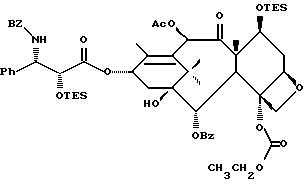
7-TES-С4-этилкарбонатбаккатин из примера 46 (93,4 мг, 0,128 ммоль) растворяли в тетрагидрофуране (2,6 мл). Этот раствор охлаждали до -40oC и обрабатывали LHMDS (0,192 мл, 1М, 0,192 ммоль), а затем раствором β -лактама из примера 23 (73,1 мг, 0,192 ммоль), в тетрагидрофуране (1,3 мл). Реакцию проводили при 0oC в течение 1 часа и прекращали смешением с NH4Cl (3 мл). Реакционную смесь экстрагировали этилацетатом (10 мл) и промывали водой и рассолом. Органический слой высушивали и концентрировали в вакууме и остаток хроматографировали (20-30% этилацетата в гексанах), получив в результате 118 мг (83,0%) целевого продукта.
Пример 48
Получение C4-этилкабонатпаклитаксена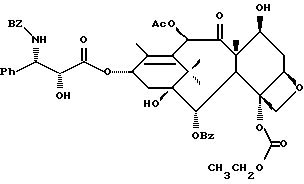
2', 7-TES-4-этилкарбонаттаксол из примера 47 (114 мг, 0,103 ммоль) растворяли в ацетонитриле (5,1 мл) и к этому раствору при 0oC добавляли пиридин (0,285 мл), а затем 48%-ную HF (0,855 мл). Реакцию проводили при 5oC всю ночь. Затем реакционную смесь разбавляли этилацетатом (100 мл), промывали 1 н. HCl, насыщенным раствором NaHCO3 и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получили 75 мг (82,8%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,09-8,06 (м, 2H), 7,75-7,24 (м, 13H), 7,14 (д, J = 9,0 Гц, 1H), 6,24 (с, 1H), 6,10 (м, 1H), 5,79 (д, J = 7,1 Гц, 1H), 5,66 (д, J = 6,9 Гц, 1H), 4,95 (д, J = 8,2 Гц, 1H), 4,75 (м, 1H), 4,41-4,16 (м, 5H), 3,89 (д, J = 4,3 Гц, 1H), 3,81 (д, J = 6,9 Гц, 1H), 2,56-1,11 (м, 23H, вкл. синглеты при 2,21, 1,75, 1,65, 1,18, 1,11, 3H каждый, триплет при 1,22 3H).
Пример 49
Получение 2', 7-силилированного-C4-бутираттаксана с фуриловой боковой цепью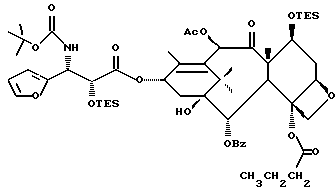
Раствор 7-силил-4-бутиратбаккатина из примера 41 (266 мг, 0,365 ммоль) в ТГФ (7,3 мл) обрабатывали при -40oC LHMDS )0,548 мл, 1 М, 0,548 ммоль). Спустя 2 минуты добавляли раствор β - лактама из примера 35 (201 мг, 0,548 ммоль) в тетрагидрофуране (3,6 мл). Реакционную смесь перемешивали при 0oC в течение 1 часа и охлаждали смешением с насыщенным раствором NH4Cl. Далее реакционную смесь экстрагировали и промывали, высушивали и концентрировали в вакууме. Остаток хроматографировали (20% этилацетата в гексанах), получив в результате 399,0 мг (99%) целевого продукта.
Пример 50
Получение C4-бутираттаксана с фуриловой боковой цепью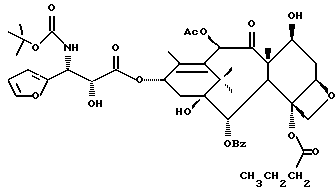
Раствор продукта из примера 49 (399 мг, 0,364 ммоль) в ацетонитриле (18,2 мл) обрабатывали при 0oC пиридином (1,01 мл), а затем 48%-ной HF (3,03 мл). Реакционную смесь держали при 5oC всю ночь, после чего разбавляли этилацетатом (200 мл), промывали 1 н. HCl, насыщенным раствором NaHCO3 и рассолом. Затем органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получили 305 мг (96,5%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,05-8,02 (м, 2H), 7,56-7,35 (м, 4H), 6,33-6,26 (м, 3H), 6,15 (м, 1H), 5,59 (д, J = 7,0 Гц, 1H), 5,40 (д, J = 9,7 Гц, 1H), 5,26 (д, J = 9,7 Гц, 1H), 4,85 (д, J = 9,5 Гц, 1H), 4,66 (м, 1H), 4,39 (м, 1H), 4,17 (AB кв, J = 8,4 Гц, 2H), 3,76 (д, J = 6,9 Гц, 1H), 3,64 (J = 6,0 Гц, 1H), 2,65-0,91 (м, 35H, вкл. синглеты при 2,18, 1,82, 1,62, 1,21, 1,09, 3H каждый, 1,28, 9H, триплет при 0,94 3H).
Пример 51
Получение 7,13-бис TES-1-DMS-C4-метилкарбонатбаккатина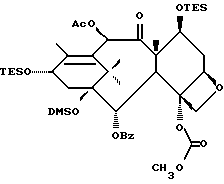
Соединение из примера 19 (118 мг, 0,150 ммоль) растворяли в ТГФ (3 мл). К этому раствору при 0oC добавляли LHMDS (0,180 мл, 1 М, 0,180 ммоль). Через 30 минут добавляли метилхлорформиат (0,174 мл, 0,225 ммоль). Еще через 30 минут реакцию прекращали смешением с NH4Cl. Реакционную смесь экстрагировали этилацетатом (100 мл). Органический слой промывали водой (10 мл х 2) и рассолом (10 мл). Затем органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (5-10% этилацетата в гексанах) и получали в результате 104 мг (82,1% целевого продукта.
Пример 52
Получение C4-метилкарбонатбаккатина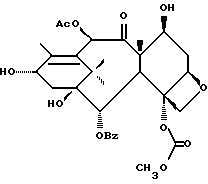
Соединение из примера 51 (89,0 мг, 0,105 ммоль) растворяли в CH3CN (3,5 мл). К этому раствору при 0oC добавляли пиридин (0,30 мл), а затем 48%-ную HF (1,05 мл). Реакционную смесь перемешивали при 0oC в течение 6 часов, а затем разбавляли этилацетатом (100 мл). Далее реакционную смесь промывали 1 н. HCl (10 мл) и насыщенным раствором (NaHCO3 (10 х 3). Органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (50% EtOAc/гексаны), получив 70 мг (100%) целевого продукта.
Пример 53
Получение 7-TES-C4-метилкарбонатбаккатина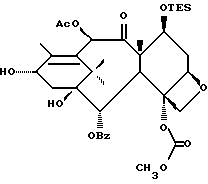
Соединение из примера 52 (115,5 мг, 0,192 ммоль) растворяли в диметилформамиде (0,960 мл). К этому раствору при 0oC добавляли имидазол (52,2 мг, 0,767 ммоль), а затем TESCl (0,128 ил 0,767 ммоль). Через 30 минут реакционную смесь разбавляли этилацетатом (100 мл). Органический слой промывали водой (10 мл х 2) и рассолом (10 мл). Затем органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (40% этилацетата в гексанах) и получили в результате 133 мг (96,8%) целевого продукта.
Пример 54
Получение 2',7-силилированного-C4-метилкарбонаттаксана с фуриловой боковой цепью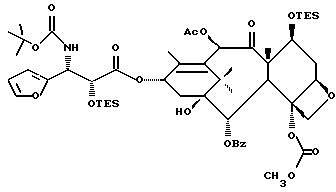
Раствор 7-силил-4-метилкарбонатбаккатина из примера 53 (227,8 мг, 0,318 ммоль) в ТГФ (6,4 мл) обрабатывали при -40oC LHMDS (0,350 мл, 1 М, 0,350 ммоль). Через 2 минуты добавляли раствор β - лактама из примера 35 (140 мг, 0,382 ммоль) в ТГФ (3,6 мл). Реакционную смесь перемешивали при 0oC в течение 1 часа и охлаждали смешением с насыщенным раствором NH4Cl. Далее реакционную смесь экстрагировали и промывали, а затем высушивали и концентрировали в вакууме. Остаток хроматографировали (20% этилацетата в гексанах) и получили в результате 332,0 мг (96,3%) целевого продукта.
Пример 55
Получение C4-метилкарбонаттаксана с фуриловой боковой цепью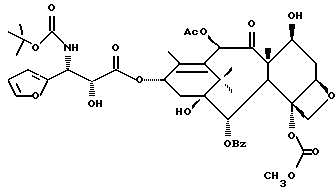
Раствор продукта из примера 54 (332,0 мг, 0,307 ммоль) в ацетонитриле (15,3 мл) обрабатывали при 0oC пиридином Э (1,7 мл), а затем 48%-ной HF (5,1 мл). Реакционную смесь держали при 5oC всю ночь, после чего разбавляли этилацетатом (200 мл) и промывали 1 н. HCl, насыщенным раствором NaHCO3 и рассолом. Затем органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах) и получили 260 мг (99,0%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,05-8,02 (м, 2H), 7,53-7,37 (м, 4H), 6,29-6,15 (м, 4H), 5,62 (д, J = 6,9 Гц, 1H), 5,40 (д, J = 9,6 Гц, 1H), 5,30 (д, J = 9,6 Гц, 1H), 4,91 (д, J = 9,3 Гц, 1H), 4,68 (м, 1H), 4,34 (м, 1H), 4,16 (AB кв, J = 8,5 Гц, 2H), 3,88 (c, 3H), 3,80 (д, J = 8,9 Гц, 1H), 3,69 (д, J = 5,5 Гц, 1H), 2,63-1,08 (м, 28H, вкл. синглеты при 2,18, 1,85, 1,60, 1,20, 1,08, 3H каждый, 1,26, 9H).
Пример 56
Получение 2',7-силилированного-C4-метилкарбонатпаклитаксела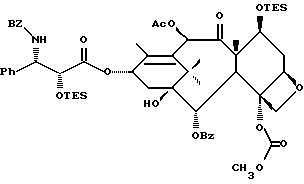
Соединение из примера 53 (113,3 мг, 0,158 ммоль) растворяли в ТГФ (3,16 мл). К этому раствору при -40oC добавляли 1 HM (0,237 мл, 1 М, 0,237 ммоль), а затем β-лактам из примера 23 (90,43 мг, 0,237 ммоль). Следуя той же процедуре, что описана выше, получили 159 мг (91,6%) целевого продукта.
Пример 57
Получение C4-метилкарбонатпаклитаксела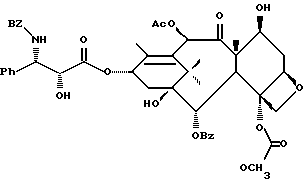
Соединение из примера 56 (149 мг, 0,136 ммоль) растворяли в CH3CN (6,8 мл). К этому раствору при 0oC добавляли пиридин (0,377 мл), а затем 48%-ную HF (1,132 мл). Следуя той же процедуре, что описана выше, получили 103,4 мг (87,6%) целевого продукта.
Пример 58
Получение C4-циклопропилового эфира 7-TES-13-оксаиолбаккатина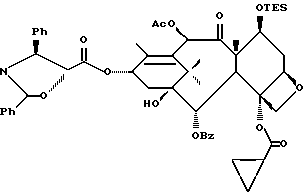
К суспензии продукта из примера 22 (72 мг, 0,099 ммоль) и продукта из примера 6 (29,4 мг, 0,110 ммоль) в толуоле (2 мл) при комнатной температуре добавляли 4-диметиламинопиридин (DMAP) (13,4 мг, 0,110 ммоль). Через 10 минут добавляли дициклогексилкарбодиимид (DCC) (22,6 мг, 0,110 ммоль). Реакционную смесь перемешивали при комнатной температуре 2 часа. Затем реакционную смесь фильтровали через целит и промывали этилацетатом. Органический слой концентрировали в вакууме. Остаток хроматографировали (30% этилацетата в гексанах) и получили целевой продукт (99 мг) с выходом 100%.
1H ЯМР (CDCl3): δ 8,27-8,24 (м, 2H), 8,03-7,26 (м, 13H), 6,42 (с, 1H), 6,08 (м, 1H), 5,67 (д, J = 7,0 Гц, 1H), 5,60 (д, J = 6,0 Гц, 1H), 4,92 (д, J = 6,1 Гц, 1H), 4,87 (д, J = 8,3 Гц, 1H), 4,50 (дд, J = 6,6 Гц J = 10,3 Гц, 1H), 4,16 (AB кв, J = 8,3 Гц, 2H), 3,85 (д, J = 6,9 Гц, 1H), 2,56-0,52 (м, 39H, вкл. синглеты при 2,15, 2,02, 1,68, 1,20, 1,18, 3H каждый триплет при 0,92, 9H).
MCBP: вычислено для C55H66NO13Si (MH+) - 976,4303, найдено - 976,4271.
Пример 59
Получение C-4 циклопропилового эфира паклитаксела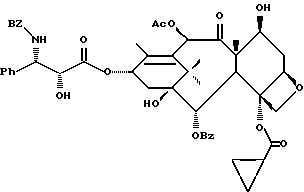
К раствору продукта из примера 58 (83,4 мг, 0,084 ммоль) в ТГФ (0,8 мл) и метаноле (0,8 мл) при 0oC добавляли 1 н. HCl (0,42 мл). Реакционную смесь держали при 4oC в течение 14 часов, а затем нагревали до комнатной температуры и добавляли к ней насыщенный раствор NaHCO3 (2,1 мл). Далее реакционную смесь перемешивали при комнатной температуре 3 часа, после чего вливали в H2O и экстрагировали этилацетатом (4 х 20 мл). Объединенный органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах), получив в результате целевой продукт (45 мг) с выходом 60%.
Пример 60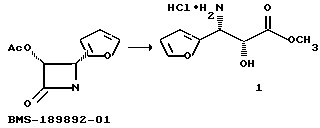
В высушенной печи и продутой аргоном колбе емкостью 25 мл растворяли BMS-189892-01 (485 мг, 3,0 ммоль) (примечание 1) в сухом метаноле (5 мг). В эту колбу добавляли по каплям посредством шприца триметилсилихлорид (326 мг, 3,0 ммоль) при 0oC. Реакционную смесь перемешивали при 0oC в течение 5 минут, после чего удаляли водоледяную баню. Далее реакционную смесь перемешивали 14 часов при комнатной температуре. Затем реакционную смесь концентрировали в вакууме и высушивали под высоким вакуумом, получив в результате соединение 1 (691 мг), 100%) в виде белой пены.
1. Chem Abs.: 34408-064-33.
В колбе емкостью 25 мл растворяли в насыщенном растворе NaHCO3 (10 мл) полученное в примере 60 соединение 1 (691 мг, 3,0 моль). К этому раствору добавляли бензоилхлорформиат (512 мг, 3,0 ммоль) при комнатной температуре. Реакционную смесь перешивали 14 часов при комнатной температуре, и в течение этого периода образовывался белый осадок. Белый осадок отфильтровывали и промывали водой (2 х 5 мл) и гексаном (2 х 5 мл). Твердое вещество высушивали под высоким вакуумом, что дало продукт 2 в виде небелого твердого вещества (745 мг, 86%).
Пример 62
В высушенной в печи и продутой аргоном колбе емкостью 25 мл, оборудованной ловушкой Дина-Старка, растворяли соединение 2 (745 мг, 2,58 ммоль) в толуоле (12 мл) и диметилформамиде (2,5 мл). К этому раствору добавляли пиридиниевую паратолуолсульфокислоту (502 мг, 2,0 ммоль). Реакционную смесь нагревали с обратным холодильником при перемешивании в течение 28 часов. Далее смесь разбавляли этилацетатом (50 мл) и промывали водой (20 мл). Водный слой экстрагировали этилацетатом (50 мл). Объединенные органические фракции высушивали над MgSO4, фильтровали и концентрировали в вакууме, получив в результате неочищенный продукт 3 (630 мг, 77%) в виде темного масла. Неочищенный продукт 3 очищали путем колоночной хроматографии (силикагель, колонка 2 х 12 см, 10% этилацетата в гексане в качестве элюента), получив в результате продукт 3 в виде густого бесцветного масла (540 мг, 66%).
Пример 63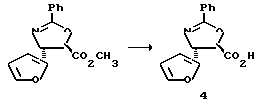
В колбе емкостью 25 мл растворяли продукт 3 (540 мг, 2,1 ммоль) в ТГФ (6 мл) и H2O (3 мл). К этому раствору добавляли одной порцией при комнатной температуре твердый 1 OH (82 мг, 2,0 ммоль). Полученную смесь перемешивали 0,5 часа при комнатной температуре. Реакцию прекращали путем добавления HCl (2,4 мл 1 н. раствора) по каплям при комнатной температуре. Затем смесь вливали в Н2O (10 мл), экстрагировали CH2Cl2 (4 х 15 мл), высушивали над MgSO4, фильтровали и концентрировали в вакууме, получив в результате неочищенный продукт 4 (420 мг, 82%) в виде желтого масла, которое использовали непосредственно на следующей стадии без дальнейшей очистки.
Пример 64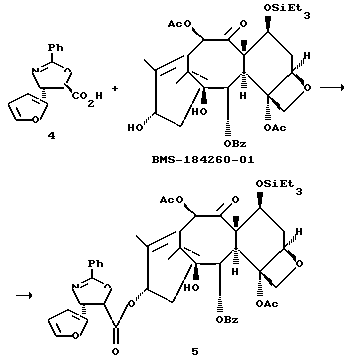
В высушенной в печи и продутой аргоном колбе емкостью 25 мл суспендировали в толуоле (10 мл) при комнатной температуре продукт 4 (140 мг, 0,54 ммоль), ВМ-184260-01 (346 мг, 0,495 ммоль) и диметиламинопиридин (66 мг, 0,54 ммоль). После перемешивания суспензии в течение 20 минут добавляли одной порцией 1,3-дициклогексилкарбодиимид (CC) (111 мг, 0,54 ммоль), и смесь перемешивали при комнатной температуре 2 часа. Затем к реакционной смеси добавляли диметиламинопиридин (66 мг, 0,54 ммоль) и 1,3-дициклогексилкарбодиимид (CC) (111 мг, 0,54 ммоль). Реакционную смесь перемешивали 14 часов. Далее смесь вливали в насыщенный раствор H4Cl (20 мл) и экстрагировали этилацетатом (100 мл). Органический экстракт фильтровали через целит, после чего слой целита промывали этилацетатом (4 х 50 мл). Объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали в вакууме, получив в результате неочищенный продукт 5 (582 мг, 125%). Неочищенный продукт очищали путем колоночной хроматографии (силикагель, 2 х 12 см, 5% этилацетата в гексане как элюента) и получили продукт 5 (413 мг, 89% в виде бесцветного масла.
Пример 65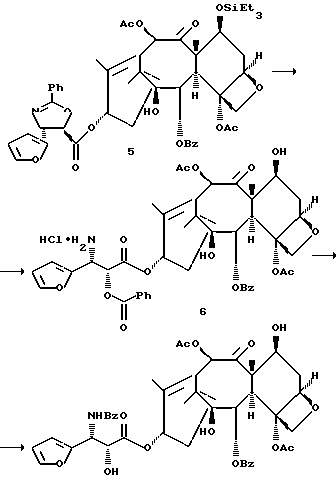
В высушенной в печи и продутой аргоном колбе емкостью 25 мл растворяли в ТГФ (2,0 мл) и метаноле (2,0 мл) продукт 5 (92 мг, 0,094 ммоль). В эту колбу добавляли водную HCl (0,5 мл 2,0 н. раствора) при 0oC. Затем раствор помещали в холодную (6oC) баню на 14 часов. Реакционную смесь нагревали до комнатной температуры и в упомянутую колбу добавляли насыщенный раствор NaHCO3 (5,0 мл). Реакционную смесь перемешивали 3 часа при комнатной температуре. Далее вливали в H2O (10 мл) и экстрагировали CH2Cl2 (4 х 20 мл). Объединенные органические фракции высушивали над MgSO4, фильтровали и концентрировали в вакууме, получив в результате неочищенный продукт (70 мг) в виде белого твердого вещества. К горячему раствору неочищенного продукта, растворенного в CH3OH (3,0 мл), добавляли H2O (примерно 1,0 мл), до тех пор пока раствор не становился мутным. Раствор охлаждали в холодильнике всю ночь. Отфильтровывали белое твердое вещество, использовав средний фильтр с пористой стеклянной пластинкой, и высушивали его под высоким вакуумом, в результате чего получили конечный продукт (51 мг, 64%) в виде белого твердого вещества.
Пример 66
Получение C-4 циклопропилового эфира 7-TES баккатина с C-13 оксаиолилфуриловой боковой цепью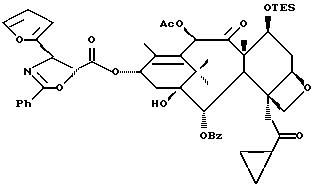
К суспензии в толуоле (2,5 мл) продукта из примера 22 (92,3 мг, 0,127 ммоль) и продукта из примера 63 (36,0 мг, 0,140 ммоль) при комнатной температуре добавляли 4-диметил аминопиридин (17,1 мг, 0,140 ммоль). Через 10 минут добавляли также DCC (28,8 мг, 0,140 ммоль). После 2 часов выдержки при комнатной температуре добавляли вторую порцию реагентов. Реакционную смесь перемешивали всю ночь при комнатной температуре. Затем реакционную смесь фильтровали и промывали этилацетатом. Органический слой концентрировали в вакууме. Остаток хроматографировали (30% этилацетата в гексанах) и получили в результате целевой продукт (125 мг) с выходом 100%.
1H ЯМР (CDCl3): δ 8,20-7,80 (м, 4H), 7,62-7,39 (м, 7H), 6,38 (м, 3H), 6,08 (м, 1H), 5,67 (м, 2H), 5,20 (д, J = 5,9 Гц, 1H), 5,20 (д, J = 5,9 Гц, 1H), 4,88 (д, J = 9,2 Гц, 1H), 4,49 (дд, J = 6,6 Гц, J = 10,2 Гц, 1H), 4,16 (AB кв, J = 8,4 Гц, 2H), 3,86 (д, J = 6,8 Гц, 1H), 2,54-0,52 (м, 39H, вкл. синглеты при 2,14, 2,03, 1,67, 1,21, 1,15, 3H каждый триплет при 0,91, 9H).
MCBP: вычислено для C53H64NO14Si (MH+) - 966,4096, найдено - 966,4134.
Пример 67
Получение C-4 циклопропилового эфира таксана с фуриловой боковой цепью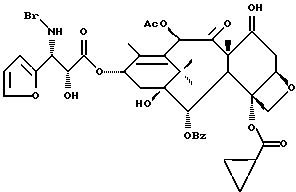
Соединение из примера 66 (69 мг, 0,0715 ммоль) растворяли в ТГФ (1,4 мл) и MeOH (1,4 мл). Затем этот раствор обрабатывали при 0oC 1 н. HCl (0,716 мл). После выдержки в течение 17 часов при 4oC реакционную смесь нагревали до комнатной температуры и обрабатывали насыщенным раствором NaHCO3 (6,5 мл). Через 6 часов выдержки при комнатной температуре реакционную смесь экстрагировали этилацетатом (4 х 20 мл). Объединенный органический слой концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах), что дало целевой продукт (37,4 мг) с выходом 60%.
1H ЯМР (CDCl3): δ 8,12-8,09 (м, 2H), 7,74-7,26 (м, 7H), 6,85 (д, J = 9,3 Гц, 1H), 6,39 (с, 2H), 6,30 (с, 1H), 6,20 (м, 1H), 5,93 (д, J = 9,3 Гц, 1H), 5,67 (д, J = 7,0 Гц, 1H), 4,88 (с, 1H), 4,82 (д, J = 7,7 Гц, 1H), 4,42 (м, 1H), 4,20 (AB кв, J = 8,5 Гц, 2H), 3,85 (д, J = 6,8 Гц, 1H), 2,54-0,88 (м, 24H, вкл. синглеты при 2,23, 1,88, 1,67, 1,24, 1,14, 3H каждый).
MCBP: вычислено для C47H52NO15 (MH+) - 870,3337, найдено - 870,3307.
Пример 68
Получение C-4 циклопропиловый эфир-2'-этилкарбоната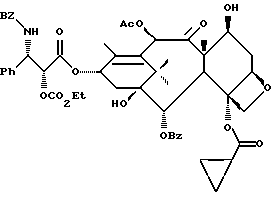
К раствору продукта из примера 24 (1,333 г, 1,52 ммоль) в дихлорметане (22,8 мл) при 0oC добавляли EtPr2N (1,586 мл, 9,10 ммоль), а затем EtOCOCl (0,87 мл, 9,10 ммоль). Реакционную смесь перемешивали при 0oC в течение 6 часов. Затем реакционную смесь разбавляли этилацетатом (200 мл), промывали водой (20 мл х 3) и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (50% этилацетата в гексанах) и получили в результате целевой продукт (1,281 г) с выходом 88,8% вместе с 86 мг исходного материала (6,5%).
1H ЯМР (CDCl3): δ 8,12-8,10 (м, 2H), 7,76-7,26 (м, 13H), 6,90 (д, J = 9,4 Гц, 1H), 6,27 (м, 2H), 6,01 (дд, J = 2,1 Гц, J' = 9,3 Гц, 1H), 5,68 (д, J = 7,0 Гц, 1H), 5,55 (д, J = 2,4 Гц, 1H), 4,83 (д, J = 8,2 Гц, 1H), 4,44 (м, 1H), 4,23 (м, 1H), 3,83 (д, J = 7,0 Гц, 1H), 2,53-0,87 (м, 27H, вкл. синглеты при 2,22, 0,95, 1,87, 1,67, 1,26, 3H каждый триплет при 1,32, 3H).
MCBP: вычислено для C52H58NO16 (MH+) - 952,3756, найдено - 952,3726.
Пример 69
Получение C-4 циклопропан-2'-этилкарбонат-7-замещенного предшественника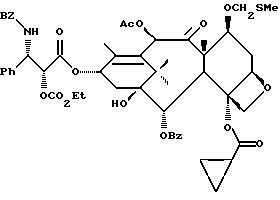
2'-Этилкарбонат из примера 68 (53 мг, 0,056 ммоль) растворяли в диметилсульфоксиде (0,5 мл), после чего добавляли Ac2O (0,5 мл). Реакционную смесь перемешивали при комнатной температуре 14 часов. Затем смесь разбавляли этилацетатом (50 мл), промывали водой (5 мл х 3), насыщенным раствором NaHCO3 и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (40% этилацетата в гексанах) и в результате получили 56,3 мг целевого продукта с выходом 100%.
1H ЯМР (CDCl3): δ 8,10-8,07 (м, 2H), 7,76-7,26 (м, 13H), 6,90 (д, J = 9,4 Гц, 1H), 6,56 (с, 1H), 6,23 (м, 1H) 6,03 (д, J = 9,5 Гц, 1H), 5,70 (д, J = 6,9 Гц, 1H), 5,58 (д, J = 2,1 Гц, 1H), 4,84 (д, J = 8,9 Гц, 1H), 4,66 (c, 2H), 4,21 (м, 5H), 3,91 (д, J = 6,8 Гц, 1H), 2,80-0,87 (м, 30H, вкл. синглеты при 2,17, 2,12, 2,11, 1,75, 1,22, 1,20, 3H каждый триплет при 1,32, 3H).
Пример 70
Получение C-4 циклопропан-2'-этилкарбонат-7-фосфатного предшественника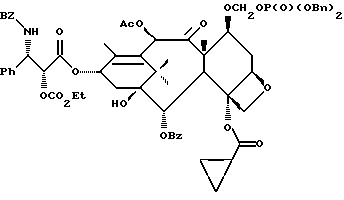
К раствору продукта на примера 69 (1,30 мг, 1,286 ммоль) в дихлорметане (25,7 мл) добавляли 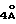 молекулярные сита (1,30 г), а затем раствор NIS (434 мг, 1,929 ммоль) в ТГФ (25,7 мл) и дибензилфосфат (537 мг, 1,929 ммоль). Реакционную смесь перемешивали при комнатной температуре 5 часов. Затем реакционную смесь фильтровали через целит, промывали этилацетатом. Удаляли растворитель и остаток растворяли в этилацетате (200 мл), промывали 1%-ным раствором NaHSO3 и рассолом и высушивали над MgSO4. Органическую фазу концентрировали в вакууме. Остаток хроматографировали (50% этилацетата в гексанах) и получили 1,278 мг продукта с выходом 80,1%.
молекулярные сита (1,30 г), а затем раствор NIS (434 мг, 1,929 ммоль) в ТГФ (25,7 мл) и дибензилфосфат (537 мг, 1,929 ммоль). Реакционную смесь перемешивали при комнатной температуре 5 часов. Затем реакционную смесь фильтровали через целит, промывали этилацетатом. Удаляли растворитель и остаток растворяли в этилацетате (200 мл), промывали 1%-ным раствором NaHSO3 и рассолом и высушивали над MgSO4. Органическую фазу концентрировали в вакууме. Остаток хроматографировали (50% этилацетата в гексанах) и получили 1,278 мг продукта с выходом 80,1%.
1H ЯМР (CDCl3): δ 8,10-8,07 (м, 2H), 7,76-7,26 (м, 23H), 6,90 (д, J = 9,4 Гц, 1H), 6,35 (с, 1H), 6,23 (м, 1H) 6,02 (д, J = 9,5 Гц, 1H), 5,68 (д, J = 6,8 Гц, 1H), 5,56 (с, 1H), 5,40 (м, 1H), 5,04 (м, 4H), 4,75 (д, J = 9,0 Гц, 1H), 4,20 (м, 5H), 3,89 (д, J = 6,8 Гц, 1H), 2,78-0,86 (м, 27H, вкл. синглеты при 2,18, 1,99, 1,67, 1,18, 1,05, 3H каждый триплет при 1,31, 3H).
Пример 71
Получение C-4 циклопропан-2'-этилкарбонат-7-фосфата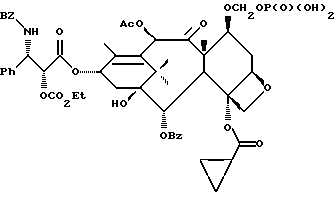
Соединение из примера 70 (1,278 г, 1,03 ммоль) растворяли в сухом ацетонитриле (41,2 мл). К этому раствору добавляли катализатор Pd/C (438 мг, 10%, 0,412 ммоль). Реакционную смесь гидрировали при 50 фунтах на кв. дюйм (3,5 кг/см2) в течение 12 часов. Затем реакционную смесь фильтровали и концентрировали в вакууме, получив в результате 1,08 г неочищенного продукта с выходом 100%.
Неочищенный продукт переносили на следующую стадию без последующей идентификации.
Пример 72
Получение триэтиламиновой соли C-4-циклопропан-2'-этилкарбонат-7-фосфата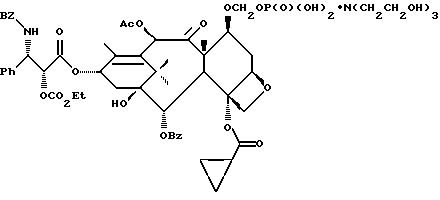
К раствору продукта из примера 71 (1,08 г, 1,02 ммоль) в этилацетате (6,8 мл) добавляли 0,100 м раствор триэтаноламина (6,8 мл, 0,15 М) в этилацетате. Полученную смесь держали при -20oC всю ночь. Затем смесь фильтровали, твердое вещество промывали холодной смесью 10% этилацетата и гексана и высушивали под вакуумом в течение 12 часов, получив в результате целевой лекарственный предшественник (1,00 г) с выходом 81,2%. Чистоту конечного продукта определяли путем ВЭЖХ, что дало приблизительно 97%-ную чистоту.
1H ЯМР (CD3OD): δ 8,10-8,07 (м, 2H), 7,80-7,26 (м, 14H), 6,38 (с, 1H), 6,07 (м, 1H), 5,89 (д, J = 5,2 Гц, 1H), 5,63 (д, J = 7,0 Гц, 1H), 5,55 (д, J = 5,2 Гц, 1H), 5,22 (м, 1H), 4,87 (м,2H), 4,23 (м, 5H), 3,88 (д, J = 7,0 Гц, 1H), 3,80 (м, 6H), 3,30 (м, 1H), 3,18 (м, 6H), 2,97-0,86 (м, 26H, вкл. синглеты при 2,15, 1,94, 1,69, 1,57, 1,13, 3H каждый триплет при 1,30, 3H).
MCBP: вычислено для C53H61NO20P (MH+), М=кислота) - 1062, 3525, найдено - 1062, 3550.
Пример 73
Получение 7-TES-13-TMS баккатина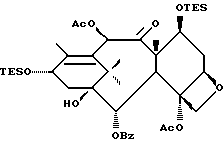
7-TES баккатин из примера 10 (1,895 г, 2,707 ммоль) растворяли в сухом диметилформамиде (10,8 мл). К этому раствору при 0oC добавляли имидазол (736,4 мг, 10,83 ммоль), а затем триметилхлорсилан (TMSCI) (1,37 мл, 10,83 ммоль). Реакционную смесь перемешивали 0oC в течение 1,5 часов. Затем реакционную смесь разбавляли этилацетатом (400 мл) и промывали водой (20 мл х 3) и рассолом (15 мл). Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (20% этилацетата в гексанах) и получили в результате 1,881 г (90%) целевого продукта.
Пример 74
Получение 7-TES-13-TMS-1-DMS баккатина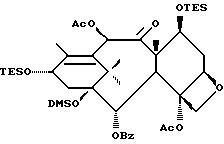
7-TES-13-TMS баккатин из примера 73 (305 мг, 0,430 ммоль) растворяли в сухом диметилформамиде (2 мл). К этому раствору при 0oC добавляли имидазол (87,6 мг, 1,289 ммоль), а затем диметилхлорсилан (122 мг, 1,289 ммоль). Через 1 час реакционную смесь разбавляли этилацетатом (150 мл). Промывали водой (10 мл х 3) и рассолом (10 мл). Полученный органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (10% этилацетата в гексанах), получив в результате 305 мг (92,4%) целевого продукта.
Пример 75
Получение 7-TES-13-TMS-1-DMS-C4-гидроксибаккатина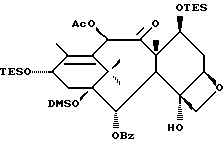
1-DMS-7-TES-13-TMS баккатин из примера 74 растворяли в сухом ТГФ (8 мл). К этому раствору при 0oC добавляли Red-Al (0,314 мл, 60%, 1,61 ммоль). Смесь перемешивали при 0oC в течение 40 минут, после чего реакцию прекращали добавлением насыщенного раствора тартрата натрия (1 мл) в течение 2 минут. Реакционную смесь экстрагировали этилацетатом (150 мл) и промывали водой (15 мл х 2) и рассолом (15 мл). Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (10-20% этилацетата в гексанах) и получили в результате 143,8 мг (45,3%) целевого продукта.
1H ЯМР (300 МГц, CDCl3): δ 8,10-8,06 (м, 2H), 7,55-7,39 (м, 3H), 6,39 (с, 1H), 5,59 (д, J = 5,5 Гц, 1H), 4,68 (дд, J1 = 3,9 Гц, J2 = 9,6 Гц, 1H), 4,61 (м, 1H), 4,53 (м, 1H), 4,21 (AB кв, J = 7,8 Гц, 2H), 4,03 (дд, J1 = 6,1 Гц, J2 = 11,6 Гц, 1H), 3,74 (с, 1H), 3,48 (д, J = 5,7 Гц, 1H), 2,74-0,48 (м, 34H, вкл. синглеты при 2,15, 2,06, 1,54, 1,16, 0,92, 3H каждый), 0,28 (с, 9H), -0,015 и -0,32 (дублеты, 3H каждый).
Пример 76
Получение 7-TES-13-TMS-1-DMS-C4-[OC(O)CH=CH2]баккатина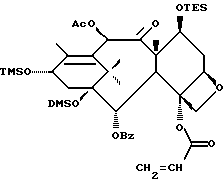
Соединение из примера 75 (99 мг, 0,125 ммоль) растворяли в сухом ТГФ (2,5 мл). К этому раствору при 0oC добавляли LHMDS (0,150 мл, 1 М, 0,150 ммоль). Спустя 30 минут добавляли акрилоилхлорид (0,153 мл, 0,188 ммоль). Еще через 30 минут реакцию прекращали путем добавления насыщенного раствора NH4Cl. Реакционную смесь экстрагировали этилацетатом (100 мл) и промывали водой (10 мл х 2) и рассолом (10 мл). Органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (5-10% этилацетата в гексанах) и получили в результате 57,5 мг (54,6%) целевого продукта.
Пример 77
Получение C4-[OC(O)CH=CH2] баккатина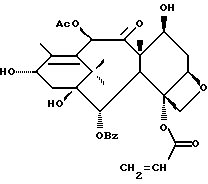
Соединение из примера 76 (105 мг, 0,125 ммоль) растворяли в CH2CN (2,5 мл). К этому раствору при 0oC добавляли пиридин (0,374 мл), а затем 48%-ную HF (1,12 мл). Реакционную смесь держали при 4oC всю ночь. Затем реакционную смесь разбавляли этилацетатом (75 мл). Органический слой промывали 1 н. HCl (5 мл), насыщенным раствором NaHCO3 (5 мл х 3) и рассолом. Органическую фазу затем высушивали и концентрировали в вакууме. Остаток хроматографировали (60% этилацетата в гексанах), получив в результате 60,6 мг (81,3%) целевого продукта.
Пример 78
Получение TES-C4-[OC)O)CH=CH2]баккатина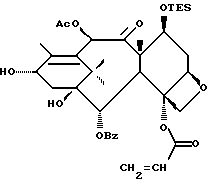
Триол из примера 77 (60,0 мг, 0,100 ммоль) растворяли в сухом диметилформамиде (0,66 мл). К этому раствору при 0oC добавляли имидазол (27,2 мг, 0,400 ммоль), а затем TESCI (0,0672 мл, 0,400 ммоль). Через 30 минут реакционную смесь разбавляли этилацетатом (75 мл) и промывали водой (5 мл х 3) и рассолом. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (40% этилацетата в гексанах), получив в результате 56,0 мг (78,4%) целевого продукта.
Пример 79
Получение 2',7-бис TES-4-[OC(O)CH=CH2]паклитаксела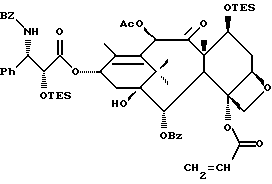
Баккатин из примера 78 (50 мг, 0,0702 ммоль) растворяли в сухом ТГФ (1,4 мл). К этому раствору при -40oC добавлялся LHMDS (0,0843 мл, 1 М, 0,0843 ммоль), а затем сразу же раствор β -лактама из примера 23 (40,1 мг, 0,105 ммоль) в ТГФ (0,7 мл). После 2 минут выдержки при -40oC реакционную смесь перемешивали при 0oC в течение 1 часа. Затем реакцию прекращали путем добавления в реакционную смесь насыщенного раствора NH4Cl. Далее реакционную смесь экстрагировали этилацетатом и промывали водой. Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (20-30% этилацетата в гексанах), получив в результате 66 мг (86%) целевого продукта.
Пример 80
Получение C4-[OC(O)CH=CH2]паклитаксела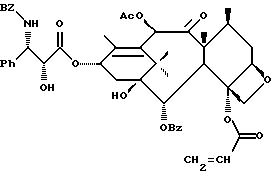
Соединение из примера 79 (46 мг, 0,0421 моль) растворяли в CH2CN (0,85 мл). К этому раствору при 0oC добавляли пиридин (0,125 мл), а затем 48%-ную HF (0,375 мл). Реакцию проводили при 4oC всю ночь. Затем реакционную смесь разбавляли этилацетатом (40 мл) и промывали 1 н. HCl (3 мл) и насыщенным раствором NaHCO3 (3 мл х 3). Органический слой высушивали и концентрировали в вакууме. Остаток хроматографировали (70% этилацетата в гексанах) и получили в результате 28 мг (76,9%) целевого продукта.
Пример 81
Получение 7, 13-бис TES-1-DMS-C4-[C(O)C6H5] баккатина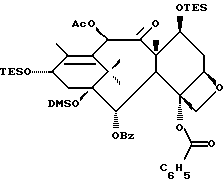
Соединение из примера 19 (279 мл, 0,336 ммоль) растворяли в сухом ТГФ (7 мл). К этому раствору при 0oC добавляли LHMDS (0,403 мл, 1 М, 0,403 ммоль). Через 30 минут добавлялся бензоилхлорид (0,0585 мл, 0,504 ммоль). Спустя еще 30 минут реакцию прекращали путем добавления к реакционной смеси насыщенного раствора NH4Cl. Реакционную смесь экстрагировали этилацетатом (150 мл). Органический слой промывали водой и рассолом и высушивали и концентрировали в вакууме. Остаток хроматографировали (10% этилацетата в гексанах) и получили в результате 215,5 мг (68,6%) целевого продукта.
Пример 82
Получение C4-бензоилбаккатина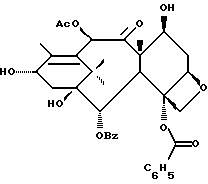
Соединение из примера 81 (161 мг, 0,172 ммоль) растворяли в CH3CN. К этому раствору при 0oC добавляли пиридин (0,57 мл), а затем 48%-ную HF (1,80 мл). После 5 часов выдержки при 4oC добавляли другую порцию реагента. Реакцию проводили при 4oC всю ночь. Затем реакционную смесь разбавляли этилацетатом (100 мл) и промывали 1 н. HCl (5 мл) и раствором NaHCO3 (5 мл х 3). Органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (30-50% этилацетата в гексанах) и получили в результате 48 мг (43,0%) целевого конечного продукта.
Пример 83
Получение 7-TES-C4-бензоилбаккатина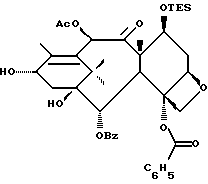
Триол из примера 82 (48,0 мг, 0,074 ммоль) растворяли в диметилформамиде (0,40 мл). К этому раствору при 0oC добавляли имидазол (20,1 мг, 0,296 ммоль), а затем TESC1 (0,0496 мл, 0,296 ммоль). Спустя 30 минут реакционную смесь разбавляли этилацетатом (45 мл) и промывали водой (1 мл х 3) и рассолом. Органическую фазу высушивали и концентрировали в вакууме. Остаток хроматографировали (40% этилацетата в гексанах) и получали в результате 48 мг (85,0%) целевого конечного продукта.
Пример 84
Получение C4-бензоилпаклитаксела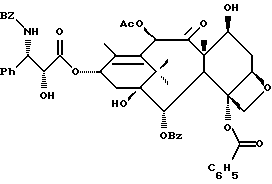
Соединение из примера 83 (364,6 мг, 0,478 ммоль) растворяли в ТГФ (9,6 мл). К этому раствору при -40oC добавляли LHMDS (0,718 мл, 1 М 0,718 ммоль), а затем β лактам из примера 23 (273,5 мг, 0,718 ммоль). Следуя той же самой процедуре, что описана в предыдущих примерах, получили 415 мг (75,9%) соединения. После этого может быть получен аналог пактитаксела со снятой защитой путем растворения вышеупомянутого соединения в CH3CN (16,5 мл) и добавления при 0oC пиридина (0,36 мл), а затем 48%-ной HF (3,0 мл). Следуя стадиям, описанным в примере 80, получили аналог паклитаксела с выходом 315 мг (94,8%).
Пример 85
Получение 4-циклобутантаксана с фуриловой боковой цепью
(А)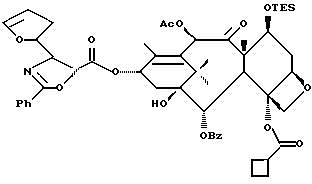
7-TES-4-циклобутилбаккатин из примера 27 (154 мг, 0,208 ммоль) растворяли в сухом толуоле (4 мл). К этому раствору при комнатной температуре добавляли свободную кислоту из примера 63 (64,2 мг, 0,250 ммоль) и 4-диметиламинопиридин (30,5 мг, 0,250 ммоль). Спустя 10 минут добавляли дициклогексилкарбодиимид (51,4 мг, 0,250 ммоль). Реакционную смесь перемешивали 2 часа, и в то же время добавляли другую порцию дициклогексилкарбодиимда и 4-диметиламинопиридина. Реакционную смесь опять перемешивали в течение 12 часов. Затем реакционную смесь фильтровали через целит и отфильтрованный твердый остаток промывали этилацетатом. Объединенный органический слой концентрировали в вакууме и полученный остаток хроматографировали (30-40% этилацетата в гексанах), получив в результате 222 мг (100%) целевого продукта.
(В)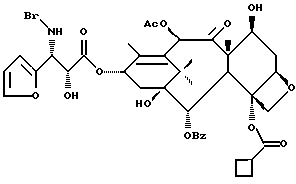
Раствор соединения (А) (182 мг, 0,186 ммоль) в ТГФ (2 мл) и MeOH (2 мл) обрабатывали при 0oC 1 н. HCl (1,86 мл). После 1 часа выдержки 0oC реакционную смесь держали всю ночь при 4oC. Затем реакционную смесь обрабатывали насыщенным раствором NaHCO3 (9,6 мл). После 5 часов выдержки при комнатной температуре реакционную смесь разбавляли этилацетатом (120 мл) и промывали водой (4 х 10 мл). Затем органический слой высушивали MgSO4 и концентрировали в вакууме. Остаток хроматографировали (40-60% этилацетата в гексанах) и получили 77 мг (47% целевого продукта).
1H ЯМР (CDCl3): δ 8,15-8,12 (м, 2H), 7,73-7,35 (м, 9H), 6,87 (д, J = 9,2 Гц, 1H), 6,44 (м, 2H), 6,28 (с, 1H), 6,20 (м, 1H), 5,89 (д, J = 9,2 Гц, 1H), 5,66 (д, H = 7,1 Гц, 1H), 4,90 (д,J = 8,1 Гц, 1H), 4,85 (с, 1H), 4,44 (м, 1Н), 4,27 (AB кв, J 8,4 Гц, 2H), 3,80 (д, J = 7,0 Гц, 1H), 3,56 (м, 1H), 2,61-0,92 (м, 25H, вкл. синглеты при 2,22, 1,83, 1,69, 1,23, 1,13, 3H каждый), 13C ЯМР (CDCl3): 203,6 174,4 172,4 171,2 166,9 166,8 150,9 142,5 142,0 133,5 133,3 132,9 131,9 130,1 130,0 129,7 129,0 128,5 127,0 110,8 108,0 84,6 80,8 78,9 76,4 75,4 75,0 72,0 71,3 58,5 50,1 45,6 43,1 38,8 35,6 35,5 26,7 25,3 25,1 21,9 20,7, 18,2 14,6 9,5. MCBP: вычислено для C48H54NO15 (MH+) - 884,3493, найдено - 884,3472.
Пример 86
Получение паклитаксела
В колбу емкостью 5 мл добавляли соединение из примера 10 (b) и растворяли его в ТГФ. Добавляли метанол и слегка желтоватый гомогенный раствор охлаждали до 0oC. Добавляли HCl и полученный гомогенный раствор перемешивали при 0oC в течение 1/2 часа и затем переносили в холодное помещение с температурой 4oC. Через 19 1/2 часов после добавления HCl тонкослойная хроматография (ТСХ) показала отсутствие исходного материла. Реакционный раствор вливали в колбу, содержащую мл полунасыщенного раствора NaCl. Полученную гетерогенную смесь перемешивали при комнатной температуре в течение минут. Смесь фильтровали и твердое вещество промывали 15 мл воды и подвергали воздушной сушке в воронке с пористой стеклянной пластинкой. Затем белое вещество, растворив в ТГФ, вливали через фритту в другую колбу и концентрировали, получив в результате 0,169 г стекловидного твердого вещества. Материал переносили в пробирку и растворяли в 1,0 мл ТГФ. Добавляли триэтиламин (NEt3) (4 экв., 0,3 моль, 88 мл), в результате чего образовывался осадок. Гетерогенную смесь перемешивали при комнатной температуре. ТСХ показала, что реакция была, по существу, закончена спустя 4,25 часа после добавления триэтиламина. Смесь разбавляли 5 мл этилацетата и 5 мл воды и встряхивали, разделяли слои. Водную часть экстрагировали дважды 5 мл этилацетата. Объединенные органические фракции промывали 5 мл HCl (нераст.), 5 мл насыщенного водного раствора NaCl, высушили над Na2SO4, фильтровали и концентрировали, получив в результате 0,127 г белого твердого вещества (паклитаксела) с выходом 93,9%.
Результаты по изучению активности на модели заболевания М109 у мышей in vivo (данные приведены после формулы изобретения)
Методика. Мышам-гибридам Balb/c x DBA/2 F1 внутрибрюшинно имплантировали 0,5 мл 2% экстракта легочной карциномы М109, как описано в статье "Оценка легочной карциномы как модели для скрининга противоопухолевых лекарственных средств" (Evaluation of Madison 109 Lung Carcinoma as a Model for Screening Abtitumor Drugs), Cancer Treatment Report, 65, 3-4 (1981).
Далее мышам также внутрибрюшинно вводят инъекции исследуемых соединений при различных дозах на 5- и 8-е сутки после имплантации. Мышей отслеживают на выживание примерно в течение 75 суток после имплантации. Одну группу мышей для каждого эксперимента оставляют контрольной.
Среднее время выживания обработанных мышей (T) сравнивают со средним временем выживания для контрольной группы (C). Отношение этих двух значений для каждой группы мышей, обработанных исследуемым соединением, выражают в % (вида % Т/С) в таблице 3.
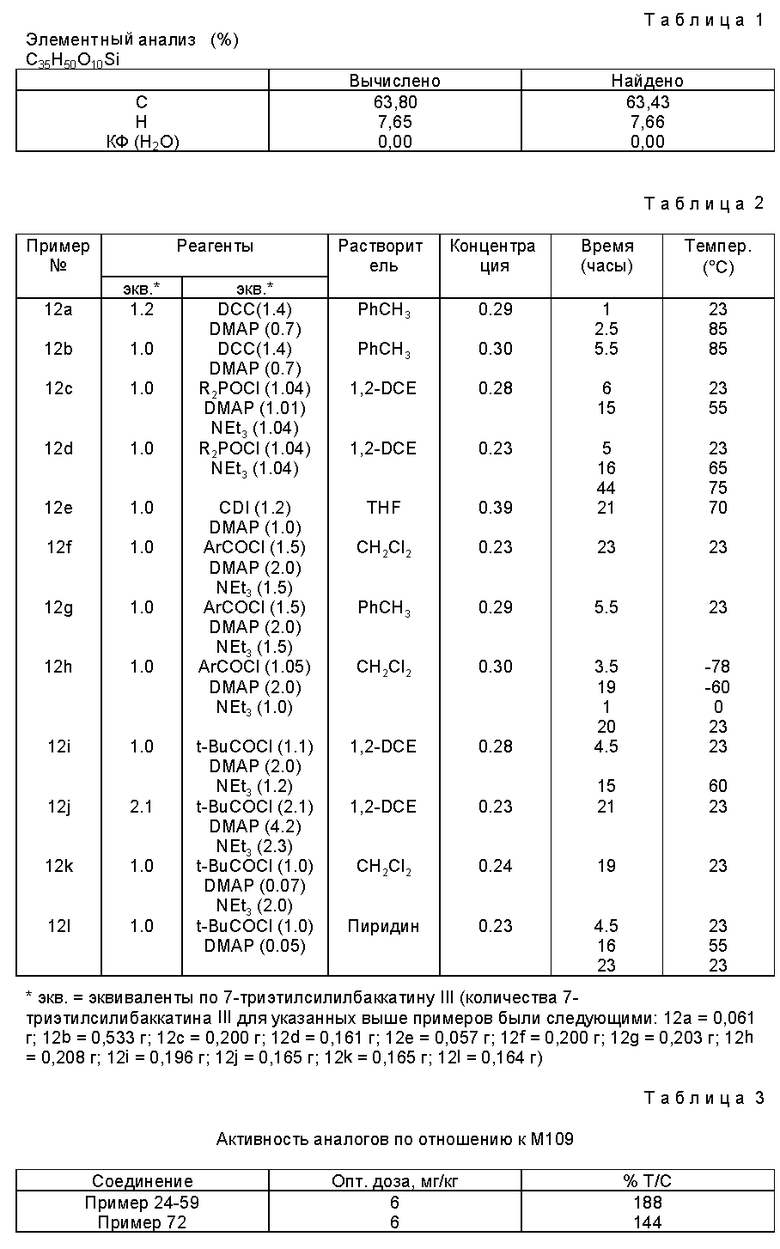
Изобретение относится к способам получения таксанов с боковой цепью и их интермедиатов и к новым соединениям формулы III, полученным этими способами. Таксаны с оксазолиновой боковой цепью формулы III, где R1 - фенил, алкил, алкенил, алкокси, R4 - Н; R3 - фенил, бензил или фурил, R8 и R9 - Н, ОН или R14O - группа, R10 и R11 - независимо - атом Н, алкил, алкенил, C3-7 - циклоалкил, фенил, R14 - гидроксизащитная группа и их соли проявляют противоопухолевую активность. 22 c. и 52 з.п. ф-лы, 3 табл.
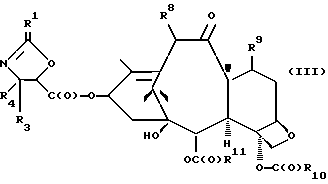
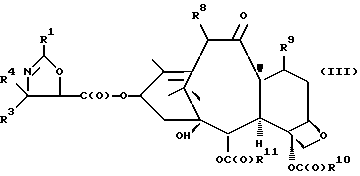
где R1 = R5 или OR7, где R5-фенил, C1 - C10-алкил, C2 - C20-алкенил и R7 - C1 - C10-алкил;
R4 - водород;
R3 - фенил, бензил или фурил;
R8 и R9 - водород, гидроксигруппа или R14O - группа;
R10, R11 - независимо водород, C1 - C10-алкил, C2 - C10-алкенил, C3 - C7-циклоалкил, фенил;
R14 - гидроксизащитная группа,
отличающийся тем, что оксазолин формулы II или его соль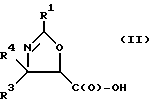
где R1, R3 и R4 имеют вышеуказанные значения,
подвергают взаимодействию с таксаном, формулы XI, имеющим гидроксильную группу, непосредственно присоединенную к его C - 13, или его солью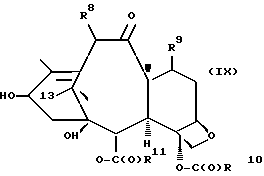
где R8, R9, R10 и R11 имеют вышеуказанные значения,
в присутствии агентов сочетания.
где R1, R3, R4, R8, R9, R10 и R11 имеют указанные в п.1 значения.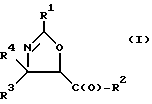
где R1, R3 и R4 имеют указанные в п.1 значения;
R2-R7O - группа, где R7 - C1 - C10-алкил.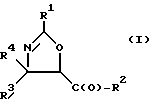
где R1, R2, R3 и R4 имеют указанные в пп.1 и 12 значения,
отличающийся тем, что включает в себя стадию контактирования соединения формулы V или его соли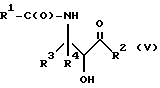
где R1, R2, R3 и R4 имеют указанные в пп.1 и 12 значения,
с кислотой, способной осуществлять дегидратацию соединения формулы V или его соли для образования соединения формулы I или его соли.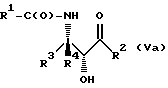
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
для получения в качестве соединения формулы I или его соли следующего соединения Ia или его соли:
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
или в качестве соединения формулы V или его соли используют следующее соединение Vc или его соль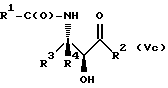
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
для получения в качестве соединения I или его соли следующего соединения Ic или его соли: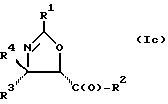
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения.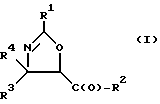
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
отличающийся тем, что включает в себя стадию контактирования соединения следующей формулы V или его соли: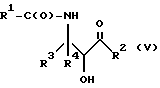
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
в присутствии основания с активирующим агентом, способным активировать гидроксильную группу соединения формулы V или его соли, чтобы осуществить внутримолекулярное замещение и образование соединения формулы I или его соли при условии, что когда R1 - фенил и R4 - водород, то R2 - не этоксигруппа или R2 - не метоксигруппа, когда R3 - бензил.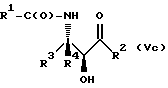
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
для получения в качестве соединения формулы I или его соли следующее соединение Ia или его соль: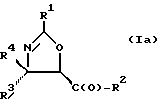
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
или тем, что в качестве соединения формулы V используют соединение или его соль формулы Va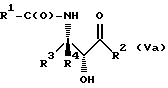
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
для получения в качестве соединения формулы I или его соли соединение или его соль формулы Ic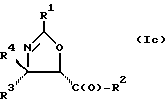
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения.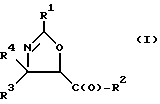
где R1 - C1 - C10 - алкил или фенил; R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
отличающийся тем, что включает в себя стадию контактирования соединения формулы VII или его соли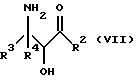
где R2, R3 и R4 имеют указанные в пп.1 и 10 значения
с соединением формулы VIII или его солью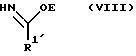
где R1′- имеет вышеуказанные значения;
E - C1 - C10 алкил,
при условии, что когда E - этил, R4 - водород и R1′- фенил, то R2 - не этоксигруппа.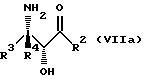
где R1′, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
для получения в качестве соединения формулы I или его соли соединение Ia или его соль: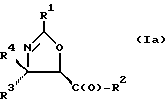
где R2, R3 и R4 имеют вышеуказанные значения,
или тем, что в качестве соединения формулы VII или его соли используют соединение VIIc или его соль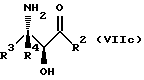
где R2, R3 и R4 имеют вышеуказанные значения, для получения в качестве соединения формулы I или его соли соединение Iс или его соль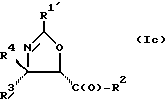
где R1′, R2, R3 и R4 имеют вышеуказанные значения.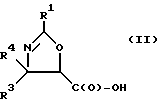
где R1, R3 и R4 имеют указанные в п.1 значения,
отличающийся тем, что проводят гидролиз оксазолина формулы I или его соли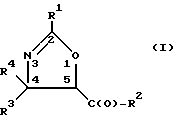
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения.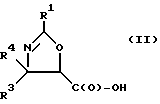
где R1, R3 и R4 имеют указанные в п.1 значения.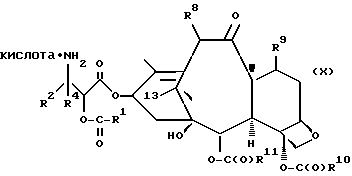
где R1, R3, R4, R8, R9, R10 и R11 имеют указанные в п.1 значения,
отличающийся тем, что включает в себя стадию контактирования таксана с оксазолиновой боковой цепью формулы III или его соли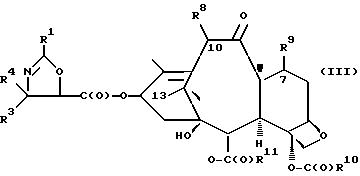
где R1, R3, R4, R8, R9, R10 и R11 имеют указанные в п.1 значения,
с водной кислотой, способной раскрывать оксазолиновое кольцо соединения формулы III или его соли, для образования таксана с боковой цепью формулы X или его соли и кислую соль при аминогруппе в соединении формулы X получают путем контактирования с раскрывающей кольцо кислотой.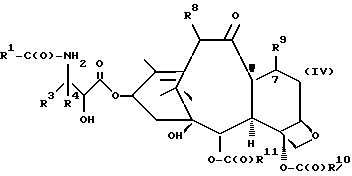
где R1, R3, R4, R8, R9, R10 и R11 имеют указанные в п.1 значения,
отличающийся тем, что включает в себя стадию контактирования таксана с боковой цепью формулы X или его соли
где R1, R3, R4, R8, R9, R10 и R11 имеют указанные в п.1 значения,
с основанием для образования таксана, несущего боковую цепь формулы IV или его соли.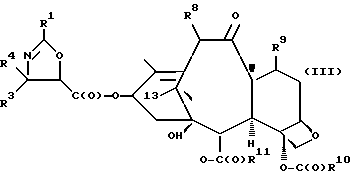
где R1, R3, R4, R8, R9, R10 и R11 имеют указанные в пп.1 и 10 значения,
с водной кислотой, способной раскрывать кольцо оксазолиновой группы таксана формулы III или его соли, с образованием соединения X или его соли.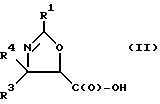
где R1, R3 и R4 имеют указанные в п.1 значения,
с таксаном, имеющим гидроксильную группу, присоединенную непосредственно к его C-13, или его солью формулы IX, где R8, R9, R10 и R11 имеют значения, указанные в п. 1, в присутствии агента сочетания с образованием таксана, несущего боковую цепь, формулы III или его соли.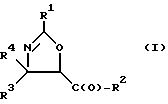
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения.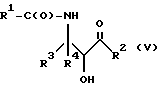
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
с активирующим агентом, способным активировать гидроксильную группу соединения формулы V или его соли для обеспечения возможности внутримолекулярного замещения и образования соединения формулы I или его соли, в присутствии основания.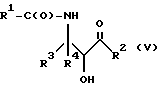
где R1, R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
с кислотой, способной осуществлять дегидратацию соединения формулы V или его соли, с образованием соединения формулы I или его соли.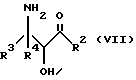
где R2, R3 и R4 имеют указанные в пп.1 и 10 значения,
с соединением формулы VIII или его соли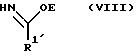
где R1′ имеют вышеуказанные значения;
E - C1 - C10 - алкил.
(a) получение оксазолинового соединения следующей формулы I или его соли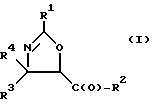
где R1, R2, R3 и R4 имеют значения, указанные в пп.1 и 10;
(b) превращение оксазолина формулы I или его соли в оксазолин формулы II или его соль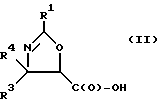
где R1, R3 и R4 имеют указанные в п.1 значения;
(c) сочетание оксазолина формулы II или его соли с таксаном, имеющим гидроксильную группу, присоединенную непосредственно к его C-13, или его солью в присутствии агента сочетания для образования таксана, несущего оксазолиновую боковую цепь следующей формулы III или его соли: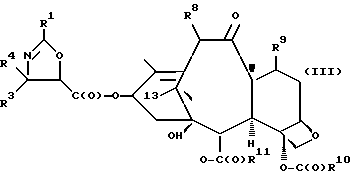
где R1, R3 и R4 имеют указанные в п.1 значения;
(d) контактирование таксана с оксазолиновой боковой цепью формулы III или его соли с водной кислотой, способной раскрывать оксазолиновое кольцо соединения формулы III или его соли, для получения таксана с боковой цепью формулы X или его соли.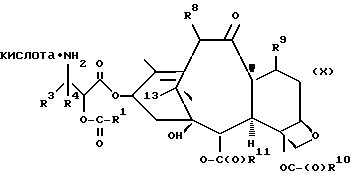
где R1, R3, R4, R8, R9, R10 и R11 имеют указанные в п.1 значения.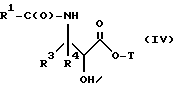
где R1 - R5 или R7 - O - группы;
R3 - фенил, бензил или фурил,
R4 - водород;
R - C1 - C10 - алкил, C2 - C10 - алкенил или фенил;
R7 - C1 - C10 - алкил;
T представляет собой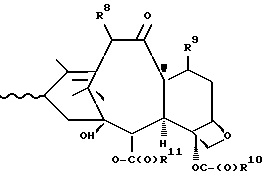
где R8 - водород, гидроксигруппа, R14 - O - группа;
R9 - водород, гидроксигруппа, R14 - O - группа;
R10 и R11 - независимо водород, C1 - C10 - алкил, C2 - C10 - алкенил, C3 - C7 - циклоалкил или фенил;
R14 - гидроксизащитная группа,
при условии, что R10 - не метил.
где R10 выбран из группы, состоящей из циклопропила, циклобутила и циклопентила.
где R10 выбран из группы, состоящей из циклопропила, циклобутила и циклопентила.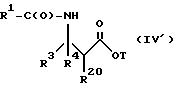
где R1, R3 и R4 имеет указанные в п.1 значения;
T представляет собой фрагмент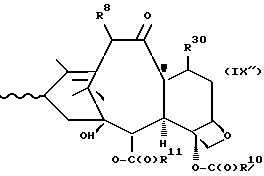
где R8 - водород, гидроксигруппа, или R14 - O -группа, где R14 - гидроксизащитная группа;
R10 и R11 - независимо водород, C1 - C10 - алкил, C2 - C10 - алкенил, C3 - C7 - циклоалкил или фенил;
R20 - водород, -OC/O/OR21 группа, где R21 - C1 - C6 - алкил,
R30 - гидроксигруппа или -OCH2(OCH2)mOP(O)(OH)2 группа, где m = 0 - 6, при условии, что, когда R30 - группа -OCH2(OCH2)mOP(O)(OH)2, то R10 - не метил,
и его основные соли фосфоноксигруппы.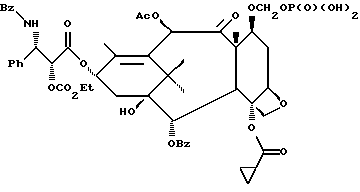
69. Соединение формулы IV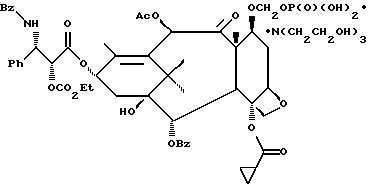
70. Способ получения соединения формулы XIV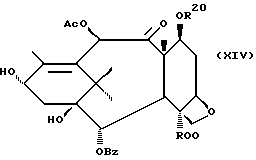
где R20 - гидроксизащитная группа или водород;
R - C/O/R10 группа, где R10 - водород, C1 - C10 - алкил, C2 - C10 алкенил, C3 - C7 циклоалкил;
Ac - ацетил;
Bz - бензил,
отличающийся тем, что включает в себя
A взаимодействие баккатина III формулы A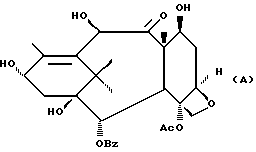
с подходящим агентом в избытке для защиты гидроксигрупп при C-7 и C-13 в инертном органическом растворителе при температуре от -30oC до комнатной;
(B) взаимодействие продукта стадии (A) с триметилсиланом или диметилсиланом в присутствии третичного аминового основания при температуре от -30oC до комнатной;
(C) восстановление продукта стадии (B) реакцией с или алюмогидридом лития при температуре от -30 до 0oC;
(D) взаимодействие продукта стадии (C) с ацилхлоридом, ангидридом кислоты или смешанным ангидридом в присутствии аниона щелочного металла вторичного аминового основания при температуре от -30oC до комнатной;
(E) снятие защиты с продукта стадии (D) реакцией с пиридиний-фторидом в ацетонитриде, а затем с тетрабутиламмонийфторидом или фторидом цезия в тетрагидрофуране (ТГФ), и, если требуется (C-7 - гидроксизащитная группа).
(F) взаимодействие продукта стадии (E) с подходящим агентом для осуществления защиты гидроксигруппы у C-7.
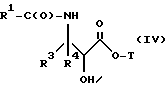
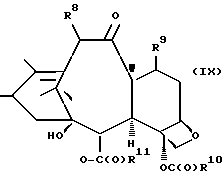
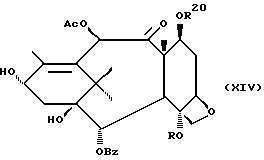
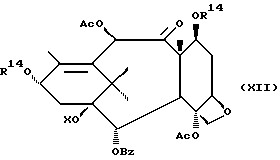
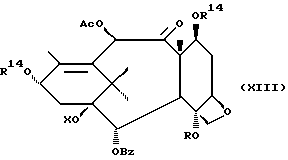
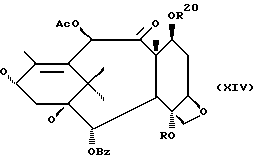
| US 4924011 A, 1990 | |||
| US 5128478 A, 07.06.92 | |||
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| US 4206221 A, 1980 | |||
| US 5059699 A, 1991. | |||

