Данная заявка является частичным продолжением одновременно рассматриваемой заявки 08/108015 данных изобретателей, поданной 17 августа 1993 г., которая в свою очередь является частичным продолжением заявки в США N 07/996455, поданной 24 декабря 1992 г. /теперь аннулированной/. Заявка США N 08/108015 включена в данное описание в целом путем ссылки.
Настоящее изобретение относится к противоопухолевым соединениям. В частности, в соответствии с настоящим изобретением предлагаются новые таксановые производные, их фармацевтические композиции и их применение в качестве противоопухолевых веществ.
Таксол (паклитаксел) является природным продуктом, экстрагированным из коры тиса тихоокеанского, Taxus blevifolia. Как оказалось, он обладает превосходной антиопухолевой активностью в моделях животного происхождения (ин виво), а недавние исследования объяснили его уникальный характер действия, включающий анормальную полимеризацию турбулина и нарушение митоза. В настоящее время он проходит клинические испытания против рака яичника, молочной железы и других типов в Соединенных Штатах и Франции, и предварительные результаты подтвердили, что он является наиболее обещающим химиотерапевтическим средством. Результаты клинических исследований паклитаксела рассмотрены у Rowinsky and Donehower, "The Clinical Pharmacology and Use Antimicrotubule Agents in Cancer Chemotherapeutics", Pharmac. Ther., 52 : 35-84, 1991.
Недавно был также обнаружен полусинтетический аналог паклитаксела под названием Таксотер (торговая марка), который показал хорошую противоопухолевую активность в животных моделях. В настоящее время Таксотер тоже проходит клинические испытания в Европе и Соединенных Штатах Америки. Ниже показано строение паклитаксела и Таксотера с традиционной для молекулы паклитаксела системой нумерации.
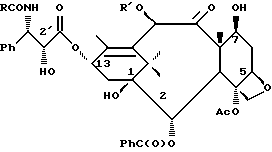
Таксол: R = Ph; R' = ацетил
Таксотер: R = трет-бутокси; R' = водород.
Одним из недостатков паклитаксела является его весьма органическая растворимость в воде, что требует приготовления лекарственных форм в неводных фармацевтических носителях. Одним из обычно используемых носителей является Кремофор EL, который сам оказывает на человека нежелательные побочные эффекты. Поэтому рядом исследовательских групп были получены водорастворимые производные паклитаксела, которые раскрыты в следующих ссылочных материалах:
(a) Хоугвиц и др., патент США N 4942184;
(b) Кингстон и др., патент США N 5059699;
(c) Стелла и др., патент США N 4960790;
(d) Заявка 0558959 A1 на европейский патент, опубликованная 8 сентября 1993 г.;
(e) Vyas ey al. , Bioorganic & Medicinal Chemictry Letters, 1993, 3: 1357-1360; и
(f) Nicolaou et al., Nature, 1993, 364:464-466.
Соединениями в соответствии с настоящим изобретением являются фосфонооксиметиловые эфиры таксоновых производных и их фармацевтически приемлемые соли. Растворимость солей в воде облегчает приготовление фармацевтических композиций.
Настоящее изобретение касается таксановых производных, имеющих формулу (A)
T -- [OCH2(OCH2)mOP(O)(OH)2]n, (А)
где T - таксановый фрагмент, несущий на C13 атоме углерода замещенную 3-амино-2-гидроксипропаноилоксигруппу; n=1, 2 или 3; m=0 или целому числу от 1 до 6 включительно; или их фармацевтически приемлемый солей.
В соответствии с настоящим изобретение предлагаются также таксановые производные, имеющие формулу (B):
T' -- [OCH2(OCH2)mSCH3]n, (В)
где T' - T, в котором не вступающие в реакцию гидроксигруппы защищены, m и n такие, как те, что определены для формулы (A).
Далее, в соответствии с настоящим изобретением предлагаются интермедиаты, имеющие формулу (C):
T' -- [OCH2(OCH2)mOP(O)(ORy)2]n, (С)
где T', m и n такие, как те, что определены для формулы (A), и Ry - фосфонозащитная группа.
Кроме того, в соответствии с настоящим изобретением предлагаются соединения формулы (D):
13-OH-txn -- [OCH2(OCH2)mSCH3]n, (D)
где m и n такие, как те, что определены выше; и txn - таксановый фрагмент; или их C13 алкоксид металла.
Еще в соответствии с настоящим изобретением предлагается способ задерживания роста опухоли в млекопитающем хозяине, включающий в себя введение в организм упомянутого млекопитающего хозяина эффективного против опухоли количества соединения формулы (A).
И еще в соответствии с настоящим изобретением предлагается фармацевтическая композиция, которая содержит эффективное против опухоли количество соединения формулы (A) и фармацевтически приемлемый носитель.
Если в описании конкретно или в контексте не указано иное, следует иметь в виду следующие определения. "Алкил" означает неразветвленную или разветвленную насыщенную углеродную цепь, имеющую от одного до шести атомов углерода; примеры включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изо-бутил, трет-бутил, н-пентил, изопентил и н-гексил. "Алкенил" означает неразветвленную или разветвленную углеродную цепь, имеющую, по крайней мере, одну двойную углерод-углеродную связь и имеющую от двух до шести атомов углерода; примеры включают этенил, пропенил, изопропенил, бутенил, изобутенил, пентенил и гексенил. "Алкинил" означает неразветвленную или разветвленную углеродную цепь, имеющую, по крайней мере, одну тройную углерод-углеродную связь и от двух до шести атомов углерода; примеры включают этинил, пропинил, бутинил и гексинил.
"Арил" означает ароматический углеводород, имеющий от шести до десяти атомов углерода; примеры включают фенил и нафтил. "Замещенный арил" означает арил, замещенный по крайней мере одной группой, выбранной из C1-6 алканоилокси, гидрокси, галогена, C1-6 алкила, трифторметила, C1-6 алкокси, арила, C2-6 алкенила, C1-6 алканоила, нитро, амино и амидо. "Галоген" означает фтор, хлор, бром и иод.
"Фосфоно-" означает группу -P(O)(OH)2, а "фосфонооксиметокси" или "фосфонооксиметиловый эфир" означает тривиально группу -OCH2(OCH2)mOP(O)(OH)2. "(Метилтио)тиокарбонил" означает группу -C(S)SCH3. "Метилтиометил" (представленный также как сокращение МТМ) является тривильным названием группы -CH2SCH3.
"Таксановый фрагмент" (представляемый также как сокращение txn) означает фрагменты, содержащие таксановый скелет из двадцати атомов углерода, представленный структурной формулой, показанной ниже в абсолютной конфигурации.
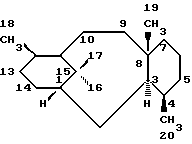
Показанная выше система нумерации, использованная в данном описании, является традиционной для номенклатуры таксанов, например, обозначение C1 относится к углеродному атому, отмеченному цифрой "1"; C5-C20 оксетан относится к оксетановому кольцу, снабженному y углеродных атомов 4, 5 и 20 атомом кислорода; и C9 окси к атому кислорода, присоединенному к углеродному атому, отмеченному цифрой "9", причем упомянутый атом кислорода может быть представлен оксогруппой, α- или β- гидрокси или α- или β- ацилокси.
"Замещенный 3-амино-2-гидроксипропаноилокси" означает остаток, представленный формулой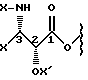
(X - неводородная группа и X' - водород или неводородная группа). Стереохимия этого остатка такая же, как у боковой цепи паклитаксела. Эта группа иногда названа в данном описании как "боковая цепь у C13".
"Таксановое производное" (сокращенно T) относится к соединению, имеющему таксановый фрагмент, несущий боковую цепь у C13.
"Гетероарил" означает пяти- или шестичленное ароматическое кольцо, содержащее от, по крайней мере, одного до четырех неуглеродных атомов, выбранных из кислорода, серы и азота. Примеры гетероарила включают тиенил, фурил, пирролил, имидазолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, тетразолил, тиатриазолил, оксатриазолил, пиридил, пиримидил, пиразинил, пиридазенил, триазинил, тетразинил и подобные кольца.
"Фосфонозащитные группы" означают фрагменты, которые могут быть использованы для блокировки (защиты) фосфоновой функциональной группы; предпочтительно такими защитными группами являются те, что могут быть удалены способами, которые не оказывают заметного влияния на остальную часть молекулы. Подходящими фосфонооксизащитными группами являются хорошо известные специалистам в данной области техники группы, включающие, например, бензильную и аллильную группы.
"Гидроксизащитные группы" включают (но не ограничены этим) простые эфиры, такие как метиловый, трет-бутиловый, бензиловый, пара-метоксибензиловый, пара-нитробензиловый, аллиловый, тритиловый, метоксиметиловый, метоксиэтоксиметиловый, этоксиэтиловый, тетрагидропираниловый, тетрагидротиопираниловый и триалкилсилиловые эфиры, такие как триметилсилиловый и трет-бутилдиметилсилиловый эфиры; сложные эфиры, такие как бензоиловый, ацетиловый, феноацетиловый, формиловый, моно-, ди- и тригалогенацетиловые, такие как хлорацетиловый, дихлорацетиловый, трихлорацетиловый, трифторацетиловый; и карбонаты, такие как метил-, этил-, 2,2,2-трихлорэтил-, аллил-, бензил- и паранитрофенилкарбонаты.
Дополнительные примеры гидрокси- и фосфонозащитных групп можно найти в стандартных ссылочных материалах, таких как Greenе and wuts, Protective Groups in Organic Synthesis, 2-nd Ed., 1991, John Wiley and Sons, and McOmie, Protective Groups in Organic Chemistry, 1975, Plenum Press.
В этих учебниках можно также найти методы введения и снятия защитных групп.
"Фармацевтически приемлемая соль" означает металлическую или аминовую соль кислотной фосфоногруппы, в которой катион не способствует сколь-нибудь существенно токсичности или биологической активности активного соединения. Подходящие соли металлов включают литиевую, натриевую, калиевую, кальциевую, бариевую, магниевую, цинковую и алюминиевую соли. Предпочтительными солями металлов являются соли натрия и калия. Подходящими солями амина являются, например, соли аммиака, трометамина (TRIS), триэтиламина, прокаина, бензатина, дибензиламина, хлорпрокаина, холина, диэтаноламина, триэтаноламина, этилендиамина, глюкамина, N-метилглюкамина, лизина, аргинина, этаноламина и многие другие. Предпочтительными аминовыми солями являются лизиновая, аргининовая и N-метилглюкаминовая соли.
В описании и пунктах формулы изобретения -OCH2(OCH2)mОP(O)(OH)2 охватывает и свободную кислоту, и ее фармацевтически приемлемые соли, если смысл конкретно не указывает, что подразумевается свободная кислота.
В соответствии с настоящим изобретением предлагаются таксановые производные формулы (A)
T --- [OCH2(OCH2)mОP(O)(OH)2]n, (А)
где T - таксановый фрагмент, несущий на C13 углеродном атоме замещенную 3-амино-2-гидроксипропаноилоксигруппу; n = 1, 2 или 3; m = 0 или целому числу от 1 до 6 включительно,
или их фармацевтически приемлемые соли.
В одном из вариантов таксановый фрагмент содержит, по крайней мере, следующие функциональные группы: C1 - гидрокси, C2 - бензилокси, C4 - ацетилокси, C5 - C20 оксетан, C9 - окси и C11 - C12 двойную связь.
В предпочтительном варианте таксановый фрагмент произведен от остатка, имеющего формулу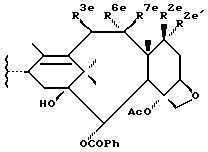
где R2e' - водород и R2e - водород, гидрокси, -OC(O)Rx или -OC(O)ORx; или R2e - водород и R2e' - фтор; R3e - водород, гидрокси, -OC(O)Rx, C1-6 алкилокси или -OC(O)ORx; один из R6e и R7e - водород, а другой - гидрокси или -OC(O)ORx; R6e и R7e вместе образуют оксогруппу; Rx - такой, как тот, что определен ниже.
В другом варианте боковая цепь у C13 произведена от остатка, имеющего формулу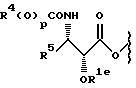
где R1e - водород или -C(O)Rx, -C(O)ORx; R4 и R5 - независимо C1-6 алкил, C2-6 алкенил, C2-6 алкинил или -Z-R6;
Z - прямая связь, C1-6 алкил или C2-6 алкенил; R6 - арил, замещенный арил, C3-6 циклоалкил или гетероарил; и Rx - C1-6 алкил, необязательно замещенный на один - шесть одинаковых или разных атомов галогена, C3-6 циклоалкил, C2-6 алкенил или радикал формулы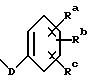
где D - связь или C1-6 алкил; и Ra, Rb и Rc - независимо водород, амино, C1-6 алкиламино, ди-C1-6 алкиламино, галоген, C1-6 алкил или C1-6 алкокси; p = 0 или 1.
В предпочтительном варианте R4 - C1-6 алкил и p = 1 или R4 - -Z-R6 и p = 0. Более предпочтительно R4(O)p - трет-бутокси, фенил, изопропилокси, н-пропилокси или н-бутокси.
В другом предпочтительном варианте R5 - C2-6 алкенил или -Z-R6 и Z и R6 - такие, как те, что определены выше. Более предпочтительно R5 - фенил, 2-фурил, 2-тиенил, изобутенил, 2-пропенил или C3-6 циклоалкил.
В другом варианте соединение формулы (A) может быть более конкретно представлено формулой (I)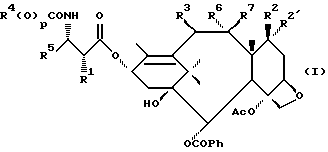
где R1 - гидрокси, -OCH2(OCH2)mОP(O)(OH)2, - OC(O)Rx или - OC(O)ORx; R2' - водород и R2 - водород, гидрокси, -ОСH2(OCH2)mOP(O)(OH)2 или -OC(O)ORx; или R2' - фтор, и R2 - водород; R3 - водород, гидрокси, ацетокси, -OCH2(OCH2)mOP(O)(OH)2 или -OC(O)ORx; один из R6 или R7 - водород, а другой - гидрокси, C1-6 алканоилокси или -OCH2(OCH2)mOP(O)(OH)2; или R6 и R7 вместе образуют оксогруппу; при условии, что, по крайней мере, R1, R2, R3, R6 или R7 - -OCH2(OCH2)mOP(O)(OH)2, R4, R5, Rx, m и p - такие, как те, что представлены выше; или его фармацевтически приемлемыми солями.
В соединениях формулы (I) примеры Rx включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, хлорметил, 2,2,2-трихлорэтил, циклопропил, циклобутил, циклопентил, циклогексил, этенил, 2-пропенил, фенил, бензил, бромфенил, 4-аминофенил, 4-метиламинофенил, 4-метилфенил, 4-метоксифенил и т. п. Примеры R4 и R5 включают 2-пропенил, изобутенил, 3-фуранил (3-фурил), 3-тиенил, фенил, нафтил, 4-гидроксифенил, 4-метоксифенил, 4-фторфенил, 4-трифторметилфенил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, этенил, 2-пропенил, 2-пропинил, бензил, фенетил, фенилэтенил, 3,4-диметоксифенил, 2-фуранил (2-фурил), 2-тиенил, 2-(2-фуранил)этенил, 2-метилпропил, циклопропил, циклобутил, циклопентил, циклогексил, циклогексилметил, циклогексилэтил и т.п.
В одном из предпочтительных вариантов в соответствии с настоящим изобретением предлагаются соединения формулы (I), в которой R5 - C2-6 алкенил или -Z-R6, а Z и R6 - предпочтительно такие, как определенные выше. Более предпочтительно R5 - фенил, 3-фурил, 3-тиенил, 2-пропенил, изобутенил, 2-фурил, 2-тиенил или C3-6 циклоалкил.
В другом предпочтительном варианте в соединениях формулы (I) R4 - C1-6 алкил, при этом p = 1; или R4 - -Z-R6 и Z и R6 - такие, как определенные выше, при этом p = 0. Более предпочтительно R4(O)p - трет-бутокси, фенил, изопропилокси, н-пропилокси, н-бутокси.
В другом предпочтительном варианте в соответствии с настоящим изобретением предлагаются соединения формулы (I), в которой R1 - -OCH2(OCH2)mОP(O)(OH)2. В более предпочтительном варианте R2 - гидрокси, -OCH2(OCH2)mОP(O)(OH)2 или -OC(O)Rx, и Rx - предпочтительно C1-6 алкил. В другом предпочтительном варианте R3 - гидрокси или ацетокси.
В другом предпочтительном варианте в соответствии с настоящим изобретением предлагается соединение формулы (I), где R2 - -OCH2(OCH2)mОP(O)(OH)2; R1 - гидрокси или -OC(O)Rx; и R3 - водород, гидрокси, ацетокси, -OCH2(OCH2)mОP(O)(OH)2 или -OC(O)ORx; и Rx - такой, как определенный выше. В более предпочтительном варианте R1 - гидрокси или -OC(O)ОRx и Rx - предпочтительно C1-6 алкил; и R3 - гидрокси или ацетокси.
В другом предпочтительном варианте в соответствии с настоящим изобретением предлагается соединение формулы (I), в которой R3 - -OCH2(OCH2)mОP(O)(OH)2; R1 - гидрокси или -OC(O)ORx; R2' - водород, а R2 - водород, гидрокси или -OC(O)ORx; или R2' - фтор, а R2 - водород; и Rx - такой, как определенный выше. В более предпочтительном варианте R1 - гидрокси или -OC(O)ORx и Rx - предпочтительно C1-6алкил. В другом более предпочтительном варианте R2 - гидрокси.
В другом предпочтительном варианте m = 0 или 1, когда фосфонооксиметоксигруппа присутствует на C7 таксанового фрагмента.
Предпочтительными фармацевтическими приемлемыми солями соединения формулы (A) являются соли щелочных металлов, включающие литиевые, натриевые и калиевые соли, и соли аминов, включающие соли триэтиламина, триэтаноламина, этаноламина, аргинина, лизина и N-метилглюкамина. Еще более предпочтительными солями являются соли аргинина, лизина и N-метилглюкамина.
Наиболее предпочтительные варианты таксановых производных формулы (A) включают следующие соединения: (1) 7-O-фосфонооксиметилпаклитаксел; (2) 2'-O-(этилоксикарбонил)-7-O-фосфонооксиметилпаклитаксел; (3) 2'-O-фосфонооксиметилпаклитаксел; (4) 2',7-бис-O-(фосфонооксиметил)паклитаксел; (5) 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)- 3'-(2-фурил)-2'-O-этилоксикарбонил-7-O-фосфонооксиметилпаклитаксел; (6) 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)- 3'-(2-тиенил)-2'-O-этилоксикарбонил-7-O-фосфонооксиметилпаклитаксел; (7) 10-дезацетил-3'-N-дезбензоил-3'-N-(трет-бутилоксикарбонил)- 10-O-(фосфонооксиметил)паклитаксел; (8) 2'-O-фосфонооксиметоксиметилпаклитаксел и их соответственные фармацевтически приемлемые соли, в частности соли натрия, калия, аргинина, лизина, N-метилглюкамина, этаноламина, триэтиламина и триэтаноламина.
Соединения формулы (A) могут быть получены из исходного (сырьевого) материала - таксановых производных T - [OH]n, где T и n - такие, как определенный выше. Природу T-[OH]n особенно не ограничивают, пока имеется, по крайней мере, одна реакционноспособная гидроксигруппа, присутствующая на таксановом фрагменте или боковой цепи у C13, чтобы обеспечить возможность образования простой эфирной связи (фосфонооксиметилового эфира). Понятно, что реакционноспособная гидроксигруппа может быть непосредственно присоединена к главной цепи пропаноилокси у C13 (например, 2'-гидроксигруппа паклитаксела) или к таксановому скелету (например, 7-гидроксигруппа паклитаксела), или она может присутствовать на заместителе на боковой цепи у C13 или на заместителе на таксановом скелете. Для получения соединений формулы (A) может быть использована последовательность реакций, показанная на схеме I.
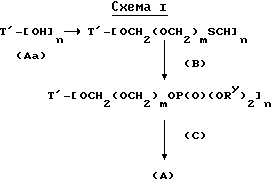
В схеме I T' - таксановое производное, в котором не вступающие в реакцию гидроксигруппы уже заблокированы (защищены); Ry - фосфонозащитная группа; n и m - такие, как определенные выше. Следовательно, соответственно защищенное T', имеющее одну или несколько реакционноспособных гидроксигрупп, сначала превращают в соответствующий метилтиометиловый эфир формулы (B). При использовании в качестве примера паклитаксела T' может представлять собой сам паклитаксел (для осуществления 2', 7-бисметилтиометилирования), 7-O-триэтилсилилпаклитаксел или 2'-O-этоксикарбонилпаклитаксел. Соединение формулы (B), где m = 0, может быть получено путем обработки T'-[OH]n диметилсульфоксидом с уксусным ангидридом или диметилсульфидом и органическим пероксидом. Указанные реакции описаны более полно ниже.
МТМ эфир, имеющий одно посредническое (промежуточное) метиленокси-звено (т.е. соединения формулы (B), где m = 1) может быть получено несколькими возможными способами. В оном способе соединение формулы (B), где m = 0, вводят в химическое взаимодействие с N-иодсукцинимидом (NIS) и метилтиометанолом, чтобы продолжить цепь на одно метиленокси-звено.
T' -- [OCH2SCH3]n + nCH3SCH2--OH 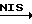 T' -- [OCH2OCH2SCH3]n.
T' -- [OCH2OCH2SCH3]n.
О соединении метилтиометанол и его получении сообщено в Syn. Comm., 1986, 16 (13): 1607 - 1610.
В альтернативном способе T-алкоксид (Ad), полученный путем обработки соединения формулы (Aa) основанием, таким как н-бутиллитий, диизопропиламид лития или гексаметилдисилазид лития, вводят во взаимодействие с хлорметилметилтиометиловым эфиром, чтобы получить соединение формулы (B), где m = 1.
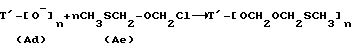
Соединение (Ae) получают введением метилтиометоксида (полученного из метилтиометанола путем обработки основанием, таким как н-бутиллитий, диизопропиламид лития или гексаметилдисилазид лития) во взаимодействие с хлориодметаном. Соединение (Ae) может быть также получено путем обработки 1,1'-дихлордиметилэфира (ClCH2OCH2Cl) стехиометрическим количеством или менее (например, около 0,8 эквивалента) иодида натрия, а затем тиометоксида натрия. О 1,1'-дихлордиметиловом эфире сообщено в Ind. J. Chem., 1989, 28B, стр. 454 - 456.
В еще одном способе соединение формулы (Ae) вводят во взаимодействие с бис(МТМ)эфиром, CH3SCH2OCH2SCH3, и NIS и в результате получают соединение формулы (B), где m = 1.
T' -- [OH]n + nCH3SCH2OCH2SCH3 ---> T'--[OCH2OCH2SCH3]n,
Бис(МТМ)эфир получают путем введения 1,1'-дихлордиметилового эфира во взаимодействие с иодидом натрия, а затем с тиометоксидом натрия.
Описанная выше процедура с использованием метилтиометанола и NIS может быть применена к любому реагенту, имеющему МТМ группу, для продолжения цепи на одно метиленокси-звено за один раз. Например, соединение формулы (B), где m = 1, может быть введено во взаимодействие с NIS для получения соединения формулы (B), в которой m = 2. Процесс может быть повторен для получения соединений формулы (B), в которой m = 3, 4, 5 или 6.
На второй стадии, показанной в схеме 1, метилтиометиловый эфир превращают в соответствующий защищенный фосфонооксиметиловый эфир. Это осуществляют путем обработки МТМ эфира NIS и защищенным фосфатом HOP(O)(ORy)2. На третьей стадии удаляют фосфонозащитную группу и гидроксизащитную(ые) группу(ы) и получают соединение (A). Например, подходящей фосфонозащитной группой является бензил, который может быть снят путем каталитического гидрогенолиза; гидроксизащитные группы, такие как триалкилсилил могут быть сняты посредством фторид-иона, трихлорэтоксикарбонил может быть снят цинком. Снятие защитных групп описано в учебниках, таких как Green and Wuts, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991; and McOmic, Protective in Organic Chemistry, Plenum Press, 1973. Обе стадии описаны более подробно в дальнейшей части описания.
Вариант последовательности реакций, показанной в схеме I, представлен в схеме II.
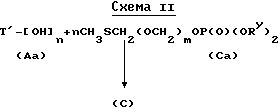
В схеме II соединение формулы (Aa) вводят в химическое взаимодействие с соединением (Ca) и NIS и получают соединение формулы (C), у которого затем снимают защиту и получают в результате соединение формулы (A). Соединения формулы (Ca), в которой m = 0, могут быть получены сначала обработкой метилтиометанола основанием, таким как гексаметилдисилазид натрия, лития или калия, чтобы получить метилтиометоксид; метоксид затем вводят во взаимодействие с защищенным хлорфосфатом, таким как дибензилхлорфосфат, и получают требуемое соединение. Соединения формулы (Ca), в которой m = 1, могут быть получены обработкой CH3SCH2OCH2Cl двузащищенным фосфатом (солью), например, дибензилфосфатами натрия, калия, тетра(н-бутил)аммония; или CH3SCH2OCH2Cl может быть сначала превращено в соответствующее иодистое соединение с использованием иодида натрия до введения во взаимодействие с фосфатом. В соответствии с другим вариантом соединения формулы (Ca), в которой m = 1, могут быть получены путем обработки ClCH2OCH2Cl иодидом натрия с последующей обработкой тиометоксидом натрия, в результате чего получают CH3SCH2OCH2SCH3; это соединение затем обрабатывают NIS-ом и двузащищенным фосфатом, таким как дибензилфосфат, и получают требуемый продукт. Любой из упомянутых выше реагентов, имеющий МТМ группу, может быть продолжен на одно метиленокси-звено за раз путем введения упомянутого реагента во взаимодействие с метилтиометанолом и NIS.
В соответствии с другим способом получения соединения формулы (A) T-алкоксид (Ad) вводят во взаимодействие с иод-фосфатом, как показано в схеме III.
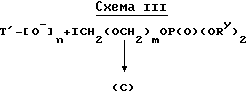
В схеме III иодфосфатное соединение получают путем введения во взаимодействие ClCH2(OCH2)mCl с двузащищенным фосфатом, что дает ClCH2(OCH2)mOP(O)(ORy)2, которое затем обрабатывают иодидом натрия, получая в результате целевой продукт.
Еще один способ, пригодный для получения подгруппы соединений формулы (A), в которых, по крайней мере, одна из фосфонооксиметоксигрупп связана с таксановым фрагментом, показан в схеме IV.
Схема IV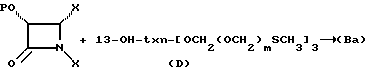
В схему IV m и n - такие, как определенные выше, X - неводородная группа, P - гидроксизащитная группа, txn - таксановый фрагмент. Соединения формулы (D) являются таксанами, имеющими 13 α- гидроксигруппу и один или несколько метилтиометиловых эфиров, связанных непосредственно или косвенно с таксановым скелетом; включены также C13-алкоксиды металлов формулы (D). Примером соединения формулы (D) является 7-O-метилтиометилбаккатин III: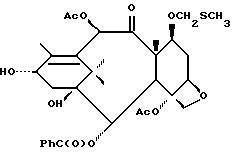
Сочетание таксана (D) с азетидиноном аналогично тому, что показано ниже в схеме VI, и описанная процедура получения соединения формулы (Id) применима также к получению соединения формулы (Ba) [т.е. соединения формулы (B), в котором, по крайней мере, одна МТМ группа связана непосредственно или косвенно с таксановым фрагментом], если вместо соединения формулы (II) в схеме VI использовать соединение формулы (D). Таксан (D) предпочтительно сначала превращают в C13-алкоксид металла, такой как алкоксид натрия, калия или лития (предпочтительным является алкоксид лития). Азетидинон служит предшественником боковой цепи у C13. После реакции сочетания с таксаном удаляют гидроксизащитную группу P и, если требуется, свободная гидроксигруппа на боковой цепи может быть превращена в МТМ эфир или произведена в сложный эфир или карбонат, как здесь описано.
Азетидинон может быть получен описанными ниже способами, которые в общем известны в данной области техники. Соединения формулы (D) могут быть получены обычным образом, описанным выше в связи с получением соединений формулы (B), с использованием подходящим образом защищенного таксана. Однако является более целесообразным получать их из соединения формулы (Ba) путем расщепления боковой цепи у C13 с использованием борогидрида, такого как борогидрид натрия или тетрабутиламмония; например, 7-O-МТМ паклитаксела обрабатывают с борогидридом тетрабутиламмония и получают 7-O-МТМ баккатин III.
Общий процесс схемы I для получения соединения формулы (A) представлен в качестве примера более конкретно в схеме V, которая иллюстрирует получение соединения формулы (I') [т. е. соединения формулы (I), в которой m = 0]. Процедура, используемая в этой схеме синтеза, в общем применима к другим таксановым производным, конкретно не охваченным формулой (I). Кроме того, процедура в схеме V может быть модифицирована в соответствии с содержащимися в данном описании идеями специалистами в данной области техники для получения таксановых производных формулы (A), в которой m = 1 или 2.
Следует понимать, что в схеме V и во всех других местах описания термин "гидроксизащитная группа" может охватывать карбонаты (-OC(O)ORx); следовательно, когда в качестве гидроксизащитной группы используют карбонат, предполагают его удаление на более поздней стадии для образования свободной гидроксигруппы, иначе карбонатный фрагмент останется частью конечного продукта.
В схеме V (см. в конце описания) R1a - гидрокси, защищенный гидрокси, -OC(O)Rx или -OC(O)ORx; R2' - водород и R2a - водород, гидрокси, защищенный гидрокси или -OC(O)ORx; или R2' - фтор и R2a - водород; R3a - водород, гидрокси, защищенный гидрокси, ацетокси или -OC(O)ORx; один из R6a или R7a - водород, а другой - гидрокси, защищенный гидрокси или C1-6 алканоилокси; R6a и R7a вместе образуют оксогруппу; при условии, что, по крайней мере, один из R1a, R2a или R3a, R6a или R7a - гидрокси, R1b - гидрокси, защищенный гидрокси, -OCH2SCH3, -OC(O)Rx или -OC(O)ORx; R2' - водород и R2b - водород, гидрокси, защищенный гидрокси, -OCH2SCH3 или -OC(O)Rx; или R2' - фтор и R2b - водород; R3b - водород, гидрокси, защищенный гидрокси, ацетокси, -OCH2SCH3 или -OC(O)ORx; один из R6b или R7b - водород, а другой - гидрокси, защищенный гидрокси, C1-6 алканоилокси или -OCH2SCH3; или R6b и R7b вместе образуют оксогруппу; при условии, что, один из радикалов R1b, R2b, R3b, R6b или R7b - -OCH2SCH3, R1c - гидрокси, защищенный гидрокси, -OCH2OP(O)(ORy)2, -OC(O)Rx или -OC(O)ORx; R2' - водород, и R2c - водород, гидрокси, защищенный гидрокси, -OCH2OP(O)(ORy)2 или -OC(O)ORx; или R2' - фтор и R2c - водород; R3c - водород, гидрокси, защищенный гидрокси, ацетокси, -OCH2OP(O)(ORy)2 или -OC(O)ORx; один из R6c или R7c - водород, а другой - гидрокси, защищенный гидрокси, C1-6 алканоилокси или -OCH2OP(O)(ORy)2; при условии, что, по крайней мере, один из R1c, R2c, R3c, R6c или R7c - -OCH2OP(O)(ORy)2. R1' - гидрокси, -OCH2OP(O)(OH)2' -OC(O)Rx или -OC(O)ORx; R2''' - водород и R2'' - водород, гидрокси, -OCH2OP(O)(OH)2 или -OC(O)ORx; или R2''' - фтор и R2'' - водород; R3 - водород, гидрокси, ацетокси, -OCH2OP(O)(OH)2 или -OC(O)ORx; один из R6' или R7' - водород, а другой - гидрокси, C1-6 алканоилокси или -OCH2P(O)(OH)2; при условии, что, по крайней мере, один из R1', R2', R3', R6' или R7' - -OCH2OP(O)(OH)2. R4, R5 и Rx - такие, как определенные выше, и Ry - фосфонозащитная группа.
На первой стадии свободную гидроксигруппу соединения формулы (Ia) превращают в соответствующую группу - метилтиометиловый эфир (-OCH2SCH3). Это превращение может быть осуществлено одной из двух процедур (Ia - диметилсульфидный метод) и (Ib - диметилсульфоксидный метод). О диметилсульфидном методе превращения спиртов в метилтиометиловые эфиры сообщено в Medina et al., Tet. Lett., 1988, стр. 3773 - 3776 (относящиеся к данному вопросу части этого материала включены в данное описание путем ссылки). Диметилсульфоксидный метод является хорошо известной реакцией, обычно называемой реакцией Паммерера.
Следует отметить, что реакционная способность гидроксигруппы отличается в зависимости от ее местоположения на исходном материале - таксановом производном формулы (Ia). Хотя в общем 2'- гидроксигруппа более реакционноспособна в реакциях ацилирования, чем 7-гидроксигруппа, которая в свою очередь более реакционноспособна, чем 10-гидроксигруппа, неожиданно было обнаружено, что 7-гидрокси более легко превращается в метилтиометиловый эфир, чем 2'-гидроксигруппа, Третичная гидроксигруппа при C-1 обычно наименее реакционноспособна. Разница в реакционной способности гидроксигруппы может быть использована для управления местоположением и степенью метилтиометилирования.
Таким образом, в случае соединения формулы (Ia), где R1a и R2a - оба гидрокси, преобладающим продуктом метилтиометилирования является соответствующий 7-O-метилтиометиловый эфир. Чтобы получить соединение формулы (Ib), где R1b - метилтиометокси, причем без превращения 7-гидроксигруппы (если она есть) в метилтиометиловый эфир, 7-гидроксигруппу блокируют традиционной гидроксизащитной группой, такой как триэтилсилил. Аналогичным образом, 10-метилтиометиловый эфир может быть получен тоже без превращения 7- и/или 2'-гидроксигрупп (если они присутствуют), когда их блокируют одинаковыми или разными гидроксизащитными группам. Даже если предпочтительной для метилтиометилирования группой является 7-гидрокси, все же желательно защищать 2'-гидроксигруппу, если целевым продуктом является 7-монометилтиометиловый эфир.
Кроме того, можно регулировать условия реакции с тем, чтобы способствовать образованию бис- или трис-метилтиометилэфирных таксановых производных. Например, в случае паклитаксела увеличение времени реакции или использование большего избытка метилтиометилирующих реагентов может привести к более высокому содержанию 2', 7-бис(метилтиометил)эфирного паклитаксела в смеси продуктов.
При процедуре (Ia) в схеме V соединение формулы (Ia) обрабатывают диметилсульфидом и органическим пероксидом, таким как бензоилпероксид. Реакцию проводят в инертном органическом растворителе, таком как ацетонитрил, метиленхлорид и т.п., при температуре, благоприятной для образования продукта; обычно реакцию осуществляют в температурном интервале примерно от -40oC до температуры окружающей среды. Диметилсульфид и бензоилпероксид используют в избытке по отношению к сырьевому таксановому производному (Ia), и диметилсульфид используют в избытке по отношению к бензоилпероксиду.
Относительные количества используемых сырьевых материалов зависят от степени метилтиометилирования, которая должна быть достигнута. Так, когда одна свободная гидроксигруппа сырьевого таксанового производного (Ia) должна быть превращена в метилтиометиловый эфир, диметилсульфид и бензоилпероксид могут быть использованы в до 10-кратном избытке по отношению к таксановому производному (Ia), а избыток диметилсульфида по отношению к бензоилпероксиду предпочтительно равен примерно двух-трехкратному. В случае, когда сырьевой материал (Ia) имеет и 2'- и 7-гидроксигруппы, количество полученного 2',7-бис(метилтиометил)-эфира увеличивается с увеличением относительных количеств диметилсульфида и бензоилпероксида. Когда целевым продуктом является 2',7-бис(метилтиометил)эфир, диметилсульфид, предпочтительно используют примерно в 15 - 20-кратном избытке по отношению к сырьевому таксановому производному, а избыток бензоилпероксида относительно сырьевого таксанового производного равен примерно 5 - 10-кратному.
В соответствии с другим вариантом соединение формулы (Ib) может быть получено путем обеспечения химического взаимодействия соединения формулы (Ia) с диметилсульфоксидом и уксусным ангидридом (процедура Ib). Эта процедура пригодна для превращения не-2'-гидроксигруппы в ее метилтиометиловый эфир. При процедуре (Ib) соединение формулы (Ia) растворяют в диметилсульфоксиде, и к раствору добавляют уксусный ангидрид. Реакцию обычно проводят при комнатной температуре в течение 18 - 24 часов, и в результате получают монометилтиометиловый эфир.
На второй стадии последовательности реакций метилтиометиловый эфир превращают в соответствующий защищенный фосфонооксиметиловый эфир. Превращение метилтиометилового эфира в защищенный фосфонооксиметиловый эфир может быть осуществлено общим способом, описанным в Veeneman et al., Tetrahedron, 1991, v 47, стр. 1547 - 1562 (этот материал включен в данное описание путем ссылки). В этом случае соединение формулы (Ib) с, по крайней мере, одной метилтиометилэфирной группой обрабатывают N-иодсукцинимидом и защищенной фосфорной кислотой, такой как дибензилфосфат. Реакцию осуществляют в инертном органическом растворителе, таком как тетрагидрофуран или галогенированный углеводород, такой как 1,2-дихлорэтан или метиленхлорид, и, хотя и не обязательно, в присутствии дегидратирующего агента, такого как молекулярные сита (цеолиты). Для ускорения реакции может быть также добавлен катализатор, такой как трифторметансульфонат серебра. Реакцию проводят при температуре в интервале от примерно 0oC до примерно комнатной температуры, предпочтительно при комнатной температуре. N-иодсукцинимид и защищенную фосфорную кислоту используют примерно в таком же молярном эквиваленте, что и метилтиометилэфир (Ib), но предпочтительно их используют с небольшим избытком, например, приблизительно 1,3 - 1,5 эквивалента, по отношению к соединению формулы (Ib).
На третьей стадии последовательности реакций удаляют фосфонозащитную группу и гидроксизащитную группу (при наличии). Снятие защитных групп осуществляют традиционными методами, хорошо известными в данной области техники, таким как гидролиз, катализируемый кислотой или основанием, гидрогенолиз, восстановление и т.п. Например, каталитический гидрогенолиз может быть использован для снятия бензильной фосфонозащитной группы, а также бензилоксикарбонильной гидроксизащитной группы . Методы снятия защиты можно найти в обычных учебниках, например, упомянутых выше учебниках Грина и Вутца или Мак-Оми.
Основные соли соединения формулы (I) могут быть образованы традиционные методики, включающими контактирование свободной кислоты соединения формулы (I) с металлическим основанием или с амином. Подходящие металлические основания включают гидроксиды, карбонаты и бикарбонаты натрия, калия, лития, кальция, бария, магния, цинка и алюминия, а подходящие амины включают триэтиламин, аммиак, лизин, аргинин, N-метилглюкамин, этаноламин, прокаин, бензатин, дибензиламин, трометамин (TRIS), хлорпрокаин, холин, диэтаноламин, триэтаноламин и т.п. Основные соли могут быть затем очищены путем хроматографирования с последующей лиофилизацией или кристаллизацией.
Исходные производные таксана
Описанные выше процессы могут быть применены к любым таксановым производным формулы T-[OH]n для образования соединений формулы (A). Многие примеры T-[OH] n указаны в литературе и некоторые из них перечислены ниже: (a) паклитаксел; (b) Таксотер (торговая марка); (c) 10-дезацетилпаклитаксел; (d) таксановые производные, раскрытые в PCT заявке 93/06079 (опубликована 1 апреля 1993 г.), имеющие формулу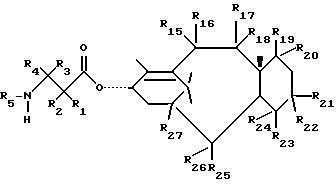
где R1 - -OR6, -SR7 или -NR8R9; R2 - водород, алкил, алкенил, алкинил, арил или гетероарил; R3 и R4 - независимо водород, алкил, алкенил, алкинил, арил, гетероарил или ацил при условии, однако, что R3 и R4 не являются оба ацилом; R5 - -COR10, -COOR10, -COSR10, -CONR8R10, -SO2R11 или -POR12R13, R6 - водород, алкил, алкенил, алкинил, арил, гетероарил, гидроксизащитная группа или функциональная группа, которая повышает растворимость в воде таксанового производного; R7 - алкил, алкенил, алкинил, арил, гетероарил или сульфгидрилзащитная группа; R8 - водород, алкил, алкенил, алкинил, арил, гетероарил; R9 - аминозащитная группа; R10 - алкил, алкенил, алкинил, арил, гетероарил; R11 - алкил, алкенил, алкинил, арил, гетероарил, -OR10 или -NR8R14; R12 и R13 - независимо друг от друга алкил, алкенил, алкинил, арил, гетероарил, -OR10 или -NR8R14; R14 - водород, алкил, алкенил, алкинил, арил, гетероарил; R15 и R16 - независимо друг от друга водород, гидрокси, низший алканоилокси, алкеноилокси, алкиноилокси, арилоилокси или R15 и R16 вместе образуют оксогруппу; R17 и R18 - независимо друг от друга водород, гидрокси, низший алканоилокси, алкеноилокси, алкиноилокси, арилоилокси или R17 и R18 вместе образуют оксогруппу; R19 и R20 - независимо водород или гидрокси или низший алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; R21 и R22 - независимо водород или низший алканоилокси, алкеноилокси, алкиноилокси или арилоилокси или R21 и R22 вместе образуют оксогруппу; R24 - водород или гидрокси или низший алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; или R23 и R24 вместе образуют оксо или метилен или R23 и R24 вместе с атомом углерода, к которому они присоединены, образуют оксирановое кольцо или R23 и R22 вместе с углеродным атомом, к которому они присоединены, образуют оксетановое кольцо; R25 - водород, гидрокси или низший алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; или R26 - водород, гидрокси, или низший алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; или R26 и R25 вместе образуют оксогруппу; и R27 - водород, гидрокси или низший алкокси, алканоилокси, алкеноилокси, алкиноилокси или арилоилокси; (e) таксановые производные, раскрытые в патенте США N 5227400: 3'-дезфенил-3'-(2-фурил)- или 3'-(2-тиенил)производные паклитаксела, Таксотера; (f) таксановые производные, раскрытые в EP 534709, опубликованном 31 марта 1993 г. (производные паклитаксела, в которых фенильные группы боковой цепи независимо заменены на нафтил, стирил или замещенный фенил); см. также PCT 92/09589, опубликованную 11 июня 1992 г.; (g) таксановые производные, раскрытые в EP 534707, опубликованном 31 марта 1993 г. (производные паклитаксела, в которых 3'-N-бензоильная группа замещена на этоксикарбонил или метоксикарбонил); (h) PCT заявка 93/06093, опубликованная 1 апреля 1993 г. (10-дезацетоксипроизводные паклитаксела и Таксотера); (i) EP 524093, опубликованный 20 января 1993 г. (10-, 7- или 7,10-бис-O-(N-замещенные карбамоилпроизводные таксана); (j) 9- α -гидроксианалог паклитаксела раскрыт в Klein, "Synthesis of 9-Dihydrotaxol: A New Bioactive Taxane", Tetrahedron Letters, 1993, 34 (13): 20-47-2050; (k) 14- β -гидроксианалог паклитаксела и Таксотера, полученный из 14 β -гидрокси-10-дезацетилбаккатина III, раскрыт на 205-ом национальном съезде Американского химического общества в Колорадо, 1993 г. (Med. Chem. Division, Abstract N 28); и (l) другие таксаны, такие как C7-фтортаксаны и различные C10-замещенные таксаны, раскрытые в одновременно рассматриваемой заявке 08/062687 на патент США, поданной данными заявителями 20 мая 1993 г., которая полностью включена в данное описание путем ссылки.
Свободную(ые) гидроксигруппу(ы) таксановых производных можно превращать традиционными способами в соответствующие сложные эфиры или карбонаты; например, в соединениях формулы (Ia) один из R1a, R2a или R3a - -OC(O)Rx или -OC(O)ORx и Rx - такой, как определенный выше. Следовательно, таксановое производное T-OH может быть введено во взаимодействие с соединением формулы L-C(O)ORx (L - уходящая группа), таким как хлорформиат, в присутствии основания, такого как третичный амин, чтобы получить соответствующий карбонат; например, реакция паклитаксела с этилхлорформиатом в присутствии диизопропилэтиламина дает 2'-O-этилоксикарбонилпаклитаксел. T-OH может быть также введено во взаимодействие с карбоновой кислотой RxCO2H или ее ацилирующим эквивалентом (например, ангидридом, активным сложным эфиром или ацилгалогенидом), что даст соответствующий сложный эфир.
Кроме того, таксановые производные T-[OH]n могут быть получены путем ацилирования таксанового фрагмента, имеющего C13-гидроксигруппу, подходящим образом замещенной 3-амино-2-гидроксипропановой кислотой, ее ацилирующим эквивалентом или ее предшественником. Подходящими предшественниками замещенной 3-амино-2-гидроксипропановой кислоты являются, например, азетидиноны формулы (III). Примером этой реакции ацилирования является сочетание гидроксизащищенного баккатина III или гидроксизащищенного 10-дезацетилбаккатина III и производного фенилизосерина, что дает производные паклитаксела, как раскрыто, например, в выданных Денису и др. патентах США N 4924011 и N 4924012; или сочетание защищенного баккетина III и азетидинона, дающее паклитаксел и его производные, как раскрыто в заявке 400971 на европейский патент, опубликованной 5 декабря 1990 г. (теперь патент США N 5175315) и патенте США N 5229526.
Такой процесс, как тот, что раскрыт в EP 400971 (процесс Холтона) включает в себя осуществление взаимодействия 1-бензоил-3-(1-этокси)этокси-4-фенил-2-азетидинона с 7-O-триэтилсилилбаккатином III в присутствии N,N-диметиламинопиридина и пиридина при 25oC в течение 12 часов, в результате чего после удаления различных гидроксизащитных групп получают паклитаксел. У Ojima et al. , "New and Efficient Approaches to the Semisynthesis of Taxol and its C-13 Side Chain Analogs by Means of β -Lactam Synthon Method" Tetrahedron, 1992, 48 (34): 6985 - 7012 описано усовершенствование процесса Холтона. Процесс Оджима включает сначала образование натриевой соли 7-триэтилсилилбаккатина III посредством гидрида натрия, а затем введение этой соли во взаимодействие с хиральным 1-бензоил-3-(1-этокси)этокси-4-фенил-2-азетидиноном, в результате чего после снятия гидроксизащитных групп получают паклитаксел. В патенте США N 5229526 Холтон раскрывает сочетание металла алкоксида баккатина III или его производного с 2-азетидиноном, в результате чего получают таксаны с боковой цепью у C13. Этот процесс, как сказано, является высоко диастереоселективным, поэтому могут быть использованы рацемические смеси предшественника 2-азетидинона с боковой цепью. Как недавно сообщено Ojima et al., "A Highly Efficient Route to Taxotere by the β -Lactam Synthon Method", Tetrahedron Letters, 1993, 34(26): 4149 - 4152, сочетание металлалкоксидов 7,10-бис-O-(трихлорэтоксикарбонил)-10-дезацетилбаккатина III с хиральным 1-(трет-бутоксикарбонил)-4-фенил-3-(защищенный гидрокси)-2-азетидиноном дает после снятия защиты Таксотер. Имеющие отношение к описываемому в данной заявке части всех приведенных выше ссылок включен в данное описание путем ссылки.
Процесс взаимодействия баккатина с азетидиноном, распространенный на получение соединений формулы (Ia) показан в схеме VI (см. в конце описания). Этим способом также могут быть получены при использовании соответствующих сырьевых материалов другие таксановые производные, конкретно не охватываемые формулой (Ia).
В схеме VI R2' - водород, а R2d - водород, защищенный гидрокси или -OC(O)ORx; или R2' - фтор, а R2d - водород; R3d - водород, ацетокси, защищенный гидрокси или -OC(O)ORx; один из R6d или R7d - водород, а другой - гидрокси, защищенный гидрокси или C1-6 алканоилокси; или R6d и R7d вместе образуют оксогруппу; P - гидроксизащищенная группа; M - водород или металл группы 1A, такой как литий, натрий или калий; и p, R4, R5 и Rx - такие, как те, что определены выше. Реакцию можно проводить в соответствии с процедурой, раскрытой в EP 400971, при которой производное баккатина III формулы (II), где M - водород, вводят во взаимодействие с азетидиноном формулы (III) в присутствии органического основания, такого как N,N-диметиламинопиридин. Однако является предпочтительным сначала превращать производное баккатина III в 13-алкоксид путем обработки его сильным основанием, таким как гидриды, алкиламиды и бис(триалкилсилил)амиды металлов группы 1A, как раскрыто выше в патенте США N 5229526 и ссылочных материалах автора Оджимы. Является более предпочтительным, когда 13-алкоксидом является алкоксид лития. Образование литиевой соли может быть обеспечено путем осуществления взаимодействия соединения формулы (II), где M - водород, с сильным основанием металла, таким как диизопропиламид лития, C1-6 алкиллитий, бис(триметилсилил)амид лития, фениллитий, гидрид лития или подобное основание.
Реакцию сочетания между таксаном формулы (II) и азетидиноном формулы (III) проводят в инертном органическом растворителе, таком как тетрагидрофуран, при пониженной температуре в интервале примерно от 0oC до -78oC. Азетидиноны формулы (III) могут быть использованы в виде рацемической смеси, сочетаемой с металлалкоксидами таксана формулы (II), в которой M - металл группы IA, причем в этом случае азетидиноновый реагент предпочтительно используют в количестве, по крайней мере, 2 эквивалентов по отношению к таксановому реагенту, а более предпочтительно от примерно 3 до примерно 6 эквивалентов. Могут быть также использованы хиральные азетидиноны, и в этом случае может быть достаточным один эквивалент азетидинона относительно таксана, но предпочтительно азетидинон используется в небольшом избытке, например до 1,5 эквивалентов.
Гидрозащитные группы могут быть одинаковыми или они могут быть выбраны так, чтобы обеспечить возможность избирательного удаления одной или нескольких защитных групп без существенного влияния на другие группы; например, в соединении формулы (Id) R2d и PO оба могут быть триэтилсилилокси, а R3d может быть бензилоксикарбонилом; каталитический гидрогенолиз в присутствии палладия на угле обеспечивает удаление бензилоксикарбонилзащитной группы без удаления триэтилсилильной группы. Таким образом, гидроксизащитные группы соединения формулы (Id) могут быть избирательно сняты, что даст соединение формулы (Ia).
Соединения формулы (II) либо известны из литературы, например баккатин III, 10-дезацетилбаккатин III и их гидроксизащищенные производные, либо могут быть получены из известных соединений традиционными способами, например, превращением гидроксигруппы в карбонат. Дополнительные соединения формулы (II) могут быть получены в соответствии с процедурами, описанными ниже в разделе "Получение сырьевых материалов".
Соединения формулы (III) могут быть получены из соединения формулы (IIIa) в соответствии с общим способом, описанным в EP 400971 и Ojima et al. , Tetrahedron 48:6985-7012, 1992.
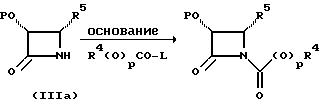
В этом случае соединение формулы (IIIa) сначала обрабатывают основанием, такимкакн-бутиллитийилитриэтиламин,азатемсоединениемформулыR4(O)pCO-L, где L -уходящая группа, и в результате получают соединение формулы (III).
Соединения формулы (IIIa) могут быть получены в соответствии с общим способом, раскрытым в EP 400971, через посредство промежуточного соединения -3-ацетокси-4-замещенного-2-азетидинона (IIIb), или способом, раскрытым в патенте США N 5229526, через посредство промежуточного соединения -3-триэтилсилилокси-4-замещенного-2-азетидинона. В усовершенствованном способе соединение (IIIb) может быть получено путем конденсирования ацетоксиацетилхлорида с бис-имином с последующим гидрогенолизом или кислотным расщеплением для удаления N-иминовой группы; этот способ показан в приведенной ниже схеме, в которой R5' - необязательно замещенный арил или гетероарильная группа, такая как фурил или тиенил. Этот способ раскрыт в одновременно рассматриваемой заявке в США N 08/052434, поданной 23 апреля 1993 г., которая включена в данное описание путем ссылки.
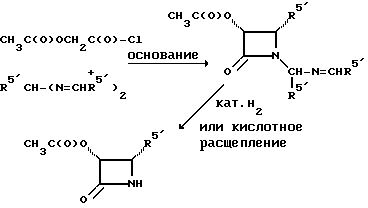
Продуктом (IIIb), получаемым от этих реакций циклоприсоединения, обычно является рацемическая смесь двух цисазетидинонов. Рацемическая смесь может быть разделена традиционными способами, такими как превращение в диастереомеры, дифференциальная адсорбция на колонке, наполненной хиральными адсорбентами, или ферментативно. Например, рацемическая смесь соединений формулы (IIIb) может быть введена в контакт с ферментом, который катализирует гидролиз сложного эфира, например, эстераза или липаза, обеспечивая селективное расщепление 3-ацильной группы одного энантиомера без влияния на другой. (См. Brieva et al., J. Org. Chem., 1993, 58:1068-1075, а также одновременно рассматриваемую заявку N 092170 на патент США, поданную 14 июля 1993 г., заявку N 552041 на европейский патент, опубликованную 29 июля 1993 г.). В соответствии с другим вариантом рацемическая смесь может быть сначала подвергнута катализируемому основанием гидролизу для удаления 3-ацильной группы и образования рацемической смеси соответствующего 3-гидрокси- β- лактама, после чего рацемическую смесь 3- гидрокси β- лактата вводят в контакт с ферментом, способным к катализирующему ацилированию гидроксигруппы, чтобы избирательно ацилировать гидроксигруппу одного энантиомера без влияния на другой. Или же рацемическая смесь 3-гидрокси- β- лактама может быть ацилирована хиральной карбоновой кислотой, и полученная диастереомерная смесь может быть затем разделена известными в данной области техники методами с последующим удалением хирального вспомогательного реагента и получением в результате требуемого энантиомера.
В материалах Ojima et al., J. Org. Chem., 56:1681-1683, 1991; Tet. Lett. , 33: 5737-5740, 1992, и Tetrahedron, 48:6985-7012, 1992 описан синтез ряда хиральных азетидинонов формулы (IIIa) и/или соответствующего N-(p-метоксифенил)-льного родственника, где P - гидроксизащитная группа триизопропилсилил; и R5-4-метоксифенил, 3,4-диметилоксифенил, фенил, 4-фторфенил, 4-трифторметилфенил, 2-фурил, 2-фенилэтенил, 2-(2-фурил)этенил, 2-метилпропил, циклогексилметил, изопропил, фенетил, 2-циклогексилэтил или н-пропил. Имеющие отношение к данной заявке части этих ссылочных материалов включены в данное описание путем ссылки. Другие азетидиноны в пределах определения формулы (III), но конкретно не раскрытые в указанных ссылочных материалах, могут быть получены специалистами в данной области техники другими методами, широко известными в этой области.
Биологическая оценка
Соединения в соответствии с настоящим изобретением являются новыми противоопухолевыми веществами; представленные соединения формулы (A) были оценены при испытаниях на цитотоксичность in vitro и на животных моделях опухоли in vivo.
Данные о цитотоксичности in vitro
Соединения в соответствии с настоящим изобретением проявили цитотоксическую активность in vitro против клеток HCT-116 и HCT-116/VM46 ракового новообразования в толстой кишке человека. Клетки HCT-116/VM46 - это клетки, которые были ранее выбраны за устойчивость к тенипозиду и выражают фенотип, обладающий устойчивостью к многим лекарственным средствам, включая устойчивость к паклитакселу. Цитотоксичность была оценена в клетках HCT-116 рака толстой кишки человека путем испытания XTT (2,3-бис(2-метокси-4-нитро-5-сульфенил)-5-[(фениламино)карбонил] - 2H-тетразолийгидроксид), как описано у D. A. Scudiero et al., "Evaluation of soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines", Cancer Res. , 48: 4827-4833, 1988. Клетки высевали по 4000 клеток/ячейку на пластинах микротитратора с 96 ячейками, и через 24 часа добавляли лекарственные средства и последовательно разводили. Инкубировали клетки при 37oC в течение 72 часов, причем в этот период добавляли тетразолиевый краситель (XTT). Фермент дегидрогеназа в живых клетках уменьшает XTT до формы, которая поглощает свет при 450 нм, что можно количественно определить спектрофотометрическим путем. Чем больше поглощение, тем больше число живых клеток. Результаты выражали через IC50 /концентрация лекарства, необходимая для сдерживания быстрого разрастания клеток (т.е. поглощение при 450 нм) до 50% от разрастания необработанных контрольных клеток. Значения IC50 для оцениваемых соединений при этом испытании даны в таблице 1 в конце описания.
Было также проверено при испытании на цитотоксичность соединение 7-O-метилтиометилпаклитаксел (Пример 1(a)), которое показало IC50 = 0,003 мкМ против HCT-116 и 0,025 мкМ против HCT-116/VM46.
Противоопухолевая активность in vivo
Гибридным мышам Balb /c x DBA2 F1 (CDF1) имплантировали подкожно (sc) 0,1 мл 2%-ного (масса/объем) M109 рака легких (как описано у W.Rose "Evaluation of Madison 109 Lung Carcinoma as a Model for Screening Antitumor Drugs", Cancer Treatment Reports, 65, N 3-4 стр. 299-312, 1981). Испытываемые соединения и стандартное лекарство (пакситаксел) вводили внутривенно группам мышей, причем каждая группа получала соединение с разным уровнем дозы, в результате чего были оценены три-четыре разные дозы на соединение. Для оценки выживания мышей за ними следили ежедневно до их смерти или примерно 75 дней после имплантации опухоли, если они не умирали. Одну группу мышей на эксперимент оставляли нелеченной для контроля. Опухоли измеряли один или два раза в неделю, и размер в миллиметрах использовали для оценки массы опухоли в соответствии с опубликованной процедурой (там же).
Среднее время выживания леченных соединениями (T) мышей сравнивали со средним временем выживания параллельных, контрольных (C) мышей. Отношение двух величин для каждой леченной соединениями группой мышей умножали на 100 и выражали как процент (т.е. % T/C) в таблице II (см. в конце описания) для представительных соединений. Кроме того, в таблице II показана также разница между средним временем для групп, подвергнутых лечению, и средним временем для контрольной группы при росте опухоли до 1 г (выражена как величина T-C в днях). Чем больше величина T-C, тем больше задержка роста первичной опухоли. Соединения, показывающие % T/C ≥ 125% и/или T-C ≥ 4,0 дней, считаются активными в модели (образце) M109 SC.
Соединение примера 3 (триэтаноламиновая соль) было затем оценено в мышиной и человеческой моделях ксенотрансплантированных опухолей (M109, A2780/ с DDP - рак яичника человека, устойчивый к цисплатину, и HCT-116 - рак толстой кишки человека) в сравнении с паклитакселом как положительным контрольным опытом. Модель A2780 /с DDP описана у Rose and Basler, In vivo, 1989, 3: 249-254. М109 пересевали (пассажировали) подкожно раз в две недели в мышей Balb /C и имплантировали подкожно в мышей CDF1 для оценки противоопухолевой активности. A2780 /с DDP и HCT-116 выращивали в лишенных зобной железы мышах и для пересева (каждый две-три недели), и для терапевтических (лечебных) экспериментов. Соединение примера 3 вводили внутривенно в воде или орально в воде с несколькими каплями твина 80, а паклитаксел либо суспендировали в воде с твином 80, либо растворяли в кремофор/этаноле (50%/50%) и разбавляли физиологическим раствором. Режим при испытаниях с М109 (имплантация подкожно) проводили раз в день в течение 5 последовательных дней, начиная с 4 дня после имплантации опухоли. При испытаниях с ксенотрансплантацией опухоли человека соединения вводили 5 раз - один раз в день через день, начиная с момента, когда опухоли находились на стадии 50 - 100 мг по массе.
В одном эксперименте с M109 соединение примера 3, введенное внутривенно, обеспечило макс. % T/C = 155 (T-C = 19 дням) при 36 мг/кг/инъек. (для сравнения, паклитаксел обеспечивал макс. % T/C = 132 (T-C = 13 дням) при 36 или 18 мг/кг/инъек. ). При том же самом эксперименте соединение примера 3, введенное орально, обеспечило макс. % T/C = 158 (T-C = 22,2 дня) при дозе 160 мг/кг/введ., в то время как паклитаксел при той же самой дозе (наивысшей из испытанных), суспендированной в воде и твине 80 не показал активности. При другом эксперименте с M109 введенное внутривенно соединение примера 3 дало макс. % T/C = 170 (T-C = 17 дням) при 48 мг/кг/инъек. (для сравнения, паклитаксел для макс. % T/C = 167 (T-C = 14 дням) при 48 или 36 мг/кг/инъек. ). В том же эксперименте орально введенное соединение 3 дало макс. %T/C = 172 (T-C = 17 дням) при дозе 200 мг/кг/введ., а паклитаксел, растворенный в кремофор/этанол/физиологическом растворе, не показал активности при 60 мг/кг/инъек. В этом эксперименте паклитаксел, растворенный в кремофор/этанол/физиологическом растворе, при дозе более 60 мг/кг/инъек. не вводили из-за ограничений, обусловленных растворимостью и токсичностью.
При эксперименте с A2780/c DDP введенные внутривенно соединения примера 3 показали макс. значение T-C = 29,8 дня при 36 мг/кг/инъек. (ср., паклитаксел дал макс. T-C = 26,3 дня при 36 мг/кг/инъек.). Орально введенное соединение примера 3 дало макс. T-C = 20 дням при дозе 160 мг/кг/введ. При эксперименте с HCT-116 лечение политакселом при введенной внутривенно дозе 24 или 36 мг/кг/инъек. дало 6 излечений из 7 или 6 из 8 (соответственно) леченных мышей, а оральное введение соединения примера 3 при 160 или 240 мг/кг/введ. излечивало соответственно 6 или 7 из 8 леченных мышей. Излечение означает отсутствие опухоли на 80 день после имплантации.
Соединения в соответствии с настоящим изобретением являются фосфонооксиметиловыми эфирами таксановых производных. Формы фармацевтически приемлемых солей проявляют улучшенную растворимость в воде по сравнению с паклитакселом, что обеспечивает возможность удобного фармацевтического изготовления лекарственных форм. Без связи с теорией, думается, фосфонооксометиловые эфиры в соответствии с настоящим изобретением являются лекарственными предшественниками (предшествующим лекарственным сырьем) паклитаксела или его производного, причем фосфонооксиметильный фрагмент расщепляется при контакте с фосфатазой in vivo с образованием затем родственного соединения. Как показано выше, соединения в соответствии с настоящим изобретением являются эффективными агентами, задерживающими рост опухоли. Поэтому настоящее изобретение касается также способа подавления опухолей в организме млекопитающего, который (способ) включает в себя введение в организм несущего опухоль хозяина эффективного против опухоли количества соединения формулы (A).
Соединения формулы (A) в соответствии с настоящим изобретением могут быть использованы таким же образом, как и паклитаксел, поэтому онколог - специалист в области лечения рака, может без нужного экспериментирования назначить соответствующий лечебный курс введения соединения в соответствии с настоящим изобретением. Дозировка, способ и график введения соединений в соответствии с настоящим изобретением особо не ограничиваются и могут меняться с каждым конкретным используемым соединением. Так, соединение в соответствии с настоящим изобретением может быть введено любым подходящим способом, предпочтительно парентерально; доза может, например, находиться в интервале примерно 1-100 мг/кг массы тела или примерно 20 - 500 мг/м2. Соединения формулы (A) могут быть также введены орально, при этом оральная доза может находиться в интервале примерно 5-500 мг/кг массы тела. Конкретная используемая доза будет изменяться в зависимости от конкретно составленной композиции, способа введения и конкретного местонахождения, хозяина и типа подлежащей лечению опухоли. При определении дозы учитывают многие факторы, влияющие на действие лекарства, такие как возраст, масса, пол, диета и физическое состояние больного.
В соответствии с настоящим изобретением предлагаются также фармацевтические композиции, содержащие эффективное против опухоли количество соединения формулы (A) в комбинации с одним или несколькими фармацевтически приемлемыми носителями, эксципиентами, разбавителями или вспомогательными веществами. Примеры изготовления форм паклитаксела или его производных можно найти, например, в патентах США NN 4960790 и 4814470. Например, соединения в соответствии с настоящим изобретением могут быть приготовлены в форме таблеток, пилюль, порошковых смесей, капсул, инъекционных форм, растворов, суппозиториев, эмульсий, дисперсий, пищевых премиксов и в других подходящих формах. Они могут быть также изготовлены в форме стерильных твердых композиций, например, высушенных сублимационно и, если нужно, объединенных с другими фармацевтически приемлемыми эксципиентами. Такие твердые композиции могут быть восстановлены стерилизованной водой, физиологическим солевым раствором или смесью воды и органического растворителя, такого как пропиленгликоль, этанол и т.п., или какой-либо другой стерильной инъекционной средой непосредственно перед использованием для парентерального введения.
Типичными фармацевтически приемлемыми носителями являются, например, манитол, мочевина, декстраны, лактоза, картофельный и маисовый крахмалы, стеарат магния, тальк, растительные масла, полиалкиленгликоли, этилцеллюлоза, поли/винилпирролидон/, карбонат кальция, этилолеат, изопропилмиристат, бензилбензоат, карбонат натрия, желатин, карбонат калия, кремневая кислота. Фармацевтический препарат может также содержать нетоксичные вспомогательные вещества, такие как эмульгирующие, консервирующие, смачивающие средства и т. п. , как, например, сорбитанмонолаурат, триэтаноламинолеат, полиоксиэтиленмоностеарат, глицерилтрипальмитат, диоктилсульфосукцинат натрия и т.п.
В следующих ниже экспериментальных процедурах все температуры, когда нет конкретного указания, представлены в oC. Спектральные характеристики ядерного магнитного резонанса (ЯМР) относятся к химическим сдвигам (δ), выраженным в миллионных долях (м. д.), в сопоставлении с тетраметилсиланом (ТМС) как этанолом для сравнения. Упоминаемая относительная площадь для различных сдвигов в спектральных данных протонного ЯСР соответствует числу водородных атомов конкретного функционального типа в молекуле. Характер сдвигов по мультиплетности указан как широкий синглет (шс), широкий дублет (шд), широкий триплет (шт), широкий квартет (шкв), синглет (с), мультиплет (м), дублет (д), квартет (кв), триплет (т), дублет дублетов (дд), дублет триплетов (дт), и дублет квартетов (дкв). Растворителями, используемыми для получения ЯМР-спектра, являются ацетон-d6 (дейтерированный ацетон), ДМСО-d6 (пердейтеродиметилсульфоксид), D2O (дейтерированная вода), CDCl3 (дейтерохлороформ) и другие традиционные дейтерированные растворители. Описание спектра инфракрасного излучения (ИК-спектра) включает только волновые числа (см-1) поглощения, имеющие идентификационные значения функциональных групп.
Целит - зарегистрированная торговая марка ф. "Джонс-Манвилл продактс корпорейшн" для диатомовой земли.
Использованные в данном описании сокращения являются традиционными сокращениями, широко используемыми в данной области техники. Некоторые из них: МС (масс-спектрометрия); МСВР (масс-спектрометрия высокого разрешения); Ac (ацетил); Ph (фенил); v/v (объем/объем, о/о); FAB (бомбардировка быстрыми атомами); NOBA (мета-нитробензиловый спирт); мин (минута/ы); ч (час/ы); NIS (N-иодсукцинимид); BOC (трет-бутоксикарбонил); CBZ (бензилоксикарбонил); Bn (бензил); Bz (бензоил); TES (триэтилсилил); ДМСО (диметилсульфоксид); ТГФ (тетрагидрофуран); ГМДС (гексаметилдисилазан).
Получение сырьевых (исходных) материалов
Ниже даны примеры получения нескольких конкретных сырьевых материалов, пригодных для получения соединений формулы (А).
Получение 1. 10-Дезацетоксипаклитаксел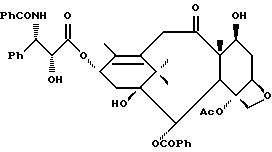
(а) 2'-7-О-бис(2,2,2-трихлорэтоксикарбонил)-10-дезацетилпаклитаксел
10-Дезацетилпаклитаксел (140 мг, 0,173 ммоль) в сухом дихлорметане (3,5 мл) обрабатывали при 0oC пиридином (0,028 мл, 0,346 ммоль) и трихлорэтилхлорформиатом (0,0724 мл, 0,260 ммоль). Через 1 час при этой температуре убирали холодную ванну, и смесь перемешивали при комнатной температуре всю ночь. Выпаривали растворитель, и остаток хроматографировали на силикагеле (30-50% этилацетата в гексане), получив в результате указанное в заголовке соединение в виде пены (92,3 мг, 46%). Дальнейшее элюирование дало непрореагировавший исходный материал (35 мг, 25%) и 2',10-О-бис/2,2,2-трихлорэтоксикарбонил)-10-дезацетилпаклитаксел с выходом 16%.
(b) 2',7-О-бис(2,2,2-трихлорэтоксикарбонил)-10-дезацетокси-11,12- дигидропаклитаксел-10,12(18)-диен
Полученный на стадии (а) продукт (92,3 мг, 0,079 ммоль) в сухом дихлорметане (2 мл) обрабатывали при комнатной температуре 1,1,2-трифтор-2-хлортриэтиламином (0,0384 мл, 0,238 ммоль). Раствор перемешивали всю ночь. Выпаривали растворитель, и остаток очищали путем колоночной хроматографии (25% этилацетата в гексане), получив в результате указанное в заголовке соединение в виде белого порошка (42,8 мг, 47,3%).
(c) 10-Дезацетокси-11,12-дигидропаклитаксел-10,12(18)-диен
Полученный на стадии (b) продукт (39 мг, 0,034 ммоль) растворяли в метаноле (0.5 мл) и уксусной кислоте (0,5 мл) и обрабатывали промытой кислотой цинковой пылью (66,4 мг, 1,020 ммоль). Суспензию нагревали при 40oC в течение 1 часа, фильтровали, и фильтрат упаривали. Хроматография остатка смесью 60% этилацетат/гексан дала указанное в заголовке соединение в виде пены (22 мг, 81%).
(d) 10-Дезацетоксипаклитаксел
Полученный на стадии (с) продукт (22 мг, 0,028 ммоль) в этилацетате (0,7 мл) гидрогенизировали при атмосферном давлении в присутствии палладия на древесном угле (10%, 14,7 мг, 0,014 ммоль Pd). Через 5,5 ч при комнатной температуре фильтрация (промывка этилацетатом), упаривание и хроматография (60% этилацетата в гексане) дали указанный в заголовке продукт (15,0 мг, 68%) в виде белой пены.
Получение 2. 7-Дезокси -7α- фторпаклитаксел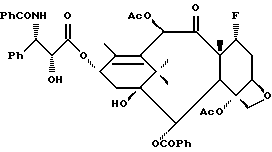
(a) 2'-O-Бензилоксикарбонил-7-дезокси-7 α- фторпаклитаксел
Диэтиламиносеры трифторид (ДАСТ, 18,7 мкл, 0,141 ммоль) растворяли в сухом дихлорметане (0.5 мл), и этот раствор охлаждали до 0oC. Добавляли раствор 2'-O-(бензилоксикарбонил)паклитаксела (71 мг, 0,072 ммоль) в дихлорметане (1 мл), и полученный раствор держали при 0oC 30 мин и при комнатной температуре 4 ч. Затем к реакционной смеси добавляли воду (0,15 мл), чтобы быстро прекратить реакцию, и полученную смесь концентрировали до получения остатка. Остаток хроматографировали на колонке с силикагелем (с элюированием 40% этилацетата в гексане) и получили 61 мг (выход 85,7%) смеси (1: 1) указанного в заголовке соединения и 2'-O-бензилоксикарбонил-8-дезметил-7,8-циклопропапаклитаксела.
(b) 7-Дезокси -7α- фторпаклитаксел
Полученную на стадии (а) смесь продуктов (89 мг) растворяли в этилацетате (3 мл) и смесь перемешивали в среде водорода под давлением немного выше одной атмосферы в присутствии палладия на древесном угле (10% Pd, 29 мг, 0,027 ммоль). Через 12 ч удаляли растворитель, и остаток очищали путем силикагель-хроматографии (с элюированием 40% этилацетата в гексане), получив в результате 67,7 мг указанного в заголовке соединения вместе с 8-дезметил-7,8-циклопропаклитакселом.
Затем использовали метод HPLC (жидкостной хроматографии высокого давления) для разделения 7-дезокси-7α-фторпаклитаксела и 8-дезметил-7,8-циклопропапаклитаксела.
Оборудование
Насос: PE серия 4
Колонка: Шандон гиперкарб (графитизированный углерод), 7 мк, 100 х 4,6 мм, N 59864750 (информация о препаративных колонках может быть получена от Keystone Scientific, Bellefonte, PA)
Инжектор: PE ISS-100
Детектор: HP-104OM
Условия
Подвижная фаза: 85:15 метиленхлорид:гексан
Разделение не исчезало при 80:19:1 метиленхлорид:гексан:изопропиловый спирт
Расход: 2,5 мл/мин
Детектор: 254 нм
Разбавитель: Образец растворяли в метиленхлориде
Получения 3. 7-Дезокси -7α- фторбаккатин III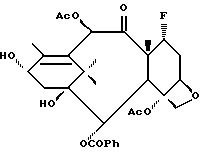
В сухую колбу в инертной атмосфере добавляли 2'-O-(бензилоксикарбонил)паклитаксел (4 г, 4 ммоль) и сухой толуол (80 мл). Полученную суспензию перемешивали при температуре окружающей среды с одновременным добавлением по каплям сухого тетрагидрофурана, пока не получали в результате бесцветный раствор. Этот раствор охлаждали до -78oC в ванне с сухим льдом и ацетоном и затем обрабатывали диэтиламиносеры трифторидом (ДАСТ, 1,2 мл, 2,5 эквив). Реакционной смеси позволяли перемешиваться в течение 16 ч, когда она постепенно нагревалась до температуры окружающей среды. Полученную суспензию фильтровали и фильтрат (разбавленный 30 мл этилацетата) промывали насыщенным водным раствором бикарбоната натрия и затем рассолом. Органическую фракцию высушивали (MgSO4) и концентрировали, получив в результате неочищенный (сырой) продукт в виде белой пены. Неочищенный материал частичного очищали путем хроматографии на колонке с силикагелем (с элюированием 10% CH3CN в CH2Cl2) и получали 1,45 г смеси 2'-O-(бензилоксикарбонил)-7-дезокси -7α- фторпаклитаксела и 2'-O-(бензилоксикарбонил)-8-дезметил-7,8-циклопропапаклитаксела (смесь 82:18 по 1H-ЯМР).
Вышеупомянутую смесь (1,45 г) растворяли в этилацетате (60 мл) и обрабатывали палладием на угле (300 мг). После встряхивания в течение 4 ч в среде водорода под давлением 50 фунтов на квадратный дюйм (3,5 кг/см2) реакционную смесь сообщали с атмосферой и фильтровали через короткий столб силикагеля и концентрировали. Это дало смесь требуемых продуктов - 7-дезокси -7α- фторпаклитаксела и 8-дезметил-7,8-циклопропапаклитаксела - в виде белой пены (1,24 г, выход 99%, смесь 90:10 по 1H-ЯМР). Эту смесь растворяли в сухом метиленхлориде (30 мл) и обрабатывали тетрабутиламмонийборогидридом (745 мг, 2,9 ммоль, 2 эквив.) и позволяли перемешиваться в течение 6 ч. Затем реакционную смесь быстро охлаждали уксусной кислотой (1 мл), разбавляли дополнительным метиленхлоридом (30 мл) и промывали насыщенным водным раствором бикарбоната натрия. Органическую фракцию высушивали (MgSO4) и концентрировали. Неочищенную смесь (замещенные таксоновые ядра) частично очищали путем хроматографии на колонке с силикагелем) с элюированием 10% CH3CN в CH2Cl2) и в результате получили смесь с отношением 90:10 (при определении методом 1H-ЯМР) 7-дезокси-7 α- -фторбаккатина III и 8-дезметил-7,8-циклопропабаккатина III (510 мг, 60%) в виде белой пены. Полученную пену кристаллизовали из горячего изопропанола, получив в результате 7-дезокси -7α- фторбаккатин III (в виде небольших белых игл с выходом 410 мг); т.пл. 234 - 236oC (разложение).
Получение 4. 10-Дезацетокси-7-дезокси -7α- фторпаклитаксел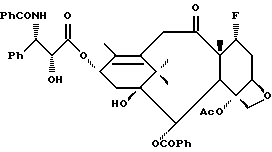
(a) 2'-O-Бензилоксикарбонил-10-дезацетоксипаклитаксел
10-Безацетоксипаклитаксел (27 мг, 0,034 ммоль) в дихлорметане (1 мл) обрабатывали бензилхлорформиатом (0,0146 мл, 0,102 ммоль) и затем диизопропилэтиламином (0,0177 мл, 0,102 ммоль). Реакционную смесь перемешивали при 0oC 45 мин и при комнатной температуре в течение 12 ч. Выпаривание растворителя и хроматографии на колонке с силикагелем (с элюированием 40% этилацетата в гексане) дали 25,5 мг (выход 81%) указанного в заголовке соединения в виде пены.
(b) 10-Дезацетокси-7-дезокси -7α- фторпаклитаксел
Полученный на стадии (а) продукт (25,5 мг, 0,028 ммоль) в дихлорметане (0,8 мл) обрабатывали ДАСТ-ом (0,0071 мл, 0,055 ммоль). Через 45 мин при 0oC реакции позволяли продолжаться в течение 5 ч при комнатной температуре. Выпаривание растворителя и хроматография дали 2'-O-бензилоксикарбонил-7-дезокси -7α- фторпаклитаксел в виде неочищенной пены. Это соединение растворяли в этилацетате (1 мл) и перемешивали в среде водорода при давлении немного выше одной атмосферы в присутствии палладия на древесном угле (10%, 8,9 мг) в течение 12 ч при комнатной температуре. Удаляли путем фильтрации катализатор, и продукт хроматографировали на колонке с силикагелем, то дало 10 мг (выход 40% за две стадии) указанного в заголовке продукта в виде пены.
Получение 5. 10-Дезацетил-7-дезокси -7α- фторпаклитаксел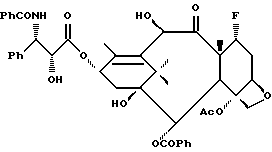
Раствор 2', 10-O-бис(2,2,2-трихлорэтоксикарбонил)-10-дезацетилпаклитаксела (120 мг, 0,103 ммоль) в дихлорметане (2 мл) охлаждали при 0oC и обрабатывали диэтиламиносеры трифторидом (0,0266 мл, 0,207 ммоль). Раствор перемешивали при 0oC 30 мин и при комнатной температуре 4 ч. Реакцию резко прекращали путем добавления воды (0,05 мл). Реакционную смесь концентрировали, и остаток очищали путем хроматографии на колонке с силикагелем (с элюированием 30% этилацетата в гексане), получив в результате 81 мг (выход 68%) 2',10-O-бис(2,2,2-трихлорэтоксикарбонил)-7-дезокси 7α- фторпаклитаксела в виде пены. Это соединение (63 мг, 0,054 ммоль) растворяли в метаноле (0,5 мл) и уксусной кислоте (0,5 мл) и обрабатывали цинковой пылью (104 мг, 1,62 ммоль) в течение 90 мин при 45oC. Реакционную смесь фильтровали, и фильтрат концентрировали. Хроматография на силикагеле (с элюированием 40% гексана в 60% этилацетата) упомянутого остатка дала 38 мг (выход 86%) указанного в заголовке соединения в виде белого твердого вещества.
Получение 6. 7-Дезоксибаккатин III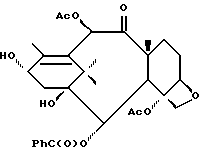
(a) 7-O-[Метилтио)тиокарбонил]баккатин III
Баккатин III (750 мг, 1,278 ммоль) растворяли в сухом тетрагидрофуране (20 мл) и в раствор добавляли одной порцией имидазол (8,7 мг, 0,128 ммоль). Добавляли при комнатной температуре натрия гидрид (50% в минеральном масле, 77 мг, 1,597 ммоль). После прекращения выделения газов (10 мин) добавляли за один раз дисульфид углерода (4,6 мл). Через 3 ч при комнатной температуре желтый раствор обрабатывали метилиодидом (0,238 мл, 3,835 ммоль) и перемешивали всю ночь. Обработка этилацетатом и водой дала указанное в заголовке соединение в виде неочищенного (сырого) масла.
Альтернативная процедура.
Баккатин III (394 мг, 0,672 ммоль) растворяли в тетрагидрофуране (5 мл) и дисульфиде углерода (1 мл). К этому раствору добавляли гидрид натрия (40,3 мг, 60%, 1,009 ммоль). Добавляли также каталитическое количество имидазола. Реакционную смесь перемешивали при комнатной температуре 1,5 ч, после чего добавляли метилиодид (122,8 мкл, 2,016 ммоль). Через 40 минут удаляли растворитель в вакууме, и остаток хроматографировали на силикагеле (с элюированием 20%-50%-60% этилацетата в гексанах), получив в результате указанное в заголовке соединение (260 мг, выход 57,2%) вместе с 7-эпибаккатином (98,5 мг, 25%),
(b) 7-O-[(Метилтио)тиокарбонил]-13-O-триэтилсилилбаккатин III
Продукт стадии (а) в виде неочищенного масла растворяли в сухом диметилформамиде (5 мл) и обрабатывали имидазолом (870 мг, 12,78 ммоль) и триэтилсилилхлоридом (2,10 мл, 12,78 ммоль) при комнатной температуре в течение 15 ч. Добавляли воду с последующей экстракцией в этилацетат. Органический слой долго промывали водой и затем высушивали. Флешхроматография на силикагеле (с элюированием 20% этилацетата в гексане) дала указанное в заголовке соединение в виде стекловидного твердого вещества (выход 209 мг, 209-й выход за две стадии).
Альтернативная процедура
Полученный на стадии (а) продукт (193,4 мг, 0,286 ммоль) растворяли в сухом диметиформамиде (2,86 мл). К этому раствору добавляли имидазол (77,9 мг, 1,14 ммоль), а затем триэтилсилилхлорид (192 мкл, 1,14 ммоль). Реакционную смесь перемешивали всю ночь при комнатной температуре. Через 12 ч реакционную смесь разбавляли этилацетатом (150 мл). Органический слой промывали водой (3 х 10 мл) и рассолом (1 х 10 мл), высушивали и концентрировали в вакууме. Остаток хроматографировали на силикагеле (с элюированием 20% этилацетата в гексанах) и получили в результате указанный в заголовке продукт (163 мг, выход 72,0%).
(c) 7-Дезокси-13-O-триэтилсилилбаккатин III
Полученный на стадии (b) продукт (182 мг, 0,230 ммоль) в сухом бензоле (5 мл) нагревали до 80oC в присутствии гидрида трибутилолова (0,310 мл, 1,150 ммоль) и 2,2'-азобисизобутиронитрила (АИБН, 10 мг). Через 3 ч раствору позволяли охладиться, и растворитель выпаривали в вакууме. Хроматография на силикагеле (с элюированием 20% этилацетата в гексане) полученного остатка дала указанное в заголовке соединения в виде масла.
(d) 7-Дезоксибаккатин III
Полученный на стадии (с) продукт растворяли в тетрагидрофуране (5 мл) и обрабатывали тетрабутиламмонийфторидом (1М в тетрагидрофуране, 0,50 мл, 0,50 ммоль) в течение 2 ч при комнатной температуре. После разбавления этилацетатом и промывки водой и рассолом с последующей хроматографией на силикагеле (с элюированием смесью (1: 1) этилацетата и гексана) получили указанное в заголовке соединение в виде белого стекловидного твердого вещества (63 мг, выход 58% за две стадии).
Получение 7. 10-Дезацетоксибаккатин III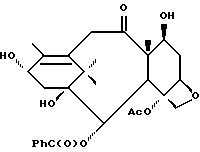
(a) 10-Дезацетил-10-O-(пентафторфенокси)тиокарбонил-7-O- триэтилсилилбаккатин III
7-O-Триэтилсилил-10-дезацетилбаккатин III (см. Green et. al., J. Am. Chem. Soc 110, стр. 5917, 1988) (319 мг, 0,485 ммоль) растворяли в сухом тетрагидрофуране (5 мл), охлаждали до -40oC и обрабатывали н-бутиллитием (1,58 М в гексанах, 0,384 мл, 0,606 ммоль). Через 40 минут при этой температуре добавляли с помощью шприца неразбавленный пентафторфенилхлортиоформиат (0,086 мл, 0,536 ммоль). Реакционную смесь перемешивали при -20oC в течение 90 мин, быстро охлаждали насыщенным раствором аммонийхлорида и экстрагировали этилацетатом. Слой этилацетата высушивали и концентрировали. Остаток очищали путем хроматографии на силикагеле (с элюированием 40% этилацетата в гексане) и получили в результате указанное в заголовке соединение в виде пены (320 мг, выход 74%).
(b) 10-Дезацетокси-7-O-триэтилсилилбаккатин III
Полученный на стадии (a) продукт (119 мг, 0,135 ммоль) растворяли в сухом толуоле (3 мл) и обрабатывали АИБН-ом (2 мг). Раствор дегазировали сухим азотом и затем добавляли трибутилолова гидрид (0,055 мл, 0,202 ммоль). После этого раствор нагревали при 90oC в течение 1 часа. Затем выпаривали растворитель и хроматографировали на силикагеле остаток (с элюированием 40% этилацетата в гексане), получив в результате указанного в заголовке соединения (87 мг, выход 99%) в виде бесцветной пены.
(c) 10-Дезацетоксибаккатин III
Полученный на стадии (b) продукт (120 мг, 0,186 ммоль) растворяли в ацетонитриле (3,5 мл), и раствор охлаждали до -10oC. Добавляли концентрированную HCl (36%, 0,060 мл), и раствор перемешивали 30 мин. Смесь разбавляли этилацетатом (75 мл) и промывали насыщенным водным раствором бикарбоната натрия и рассолом, после чего высушивали и концентрировали. Остаток очищали путем флеш-хроматографии на силикагеле (с элюированием 70% этилацетата в гексане) и получили 10-дезацетилоксибаккатин III в виде пены (75 мг, выход 76%).
Получение 8. 10-Дезацетокси-7-дезоксибаккатин III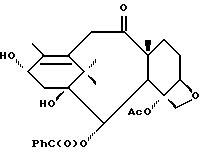
(a) 7-O[(Метилтио)тиокарбонил]-10-дезацетоксибаккатин III
10-дезацетоксибаккатин III (75 мг, 0,142 ммоль) растворили в сухом тетрагидрофуране (2 мл) и дисульфиде углерода (0,5 мл). Затем добавляли гидрид натрия (60% в минеральном масле, 8,5 мг, 0,213 моль), и смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли иодметан (0,026 мл, 0,426 ммоль), и позволяли реакции продолжаться всю ночь. Затем удаляли растворитель, и остаток очищали путем хроматографии на силикагеле (с элюированием 50 - 70% этилацетата), получив в результате указанное в заголовке соединение в виде пены (46,6 мг, выход 53%).
(b) 10-Дезацетокси-7-дезоксибаккатин III
Полученный на стадии (a) продукт (36 мг, 0,058 ммоль) в бензоле (1 мл) нагревали с обратным холодильником в присутствии АИБН (2 мг) и гидрида трибутилолова (0,079 мл, 0,290 ммоль) в среде аргона в течение 3 ч. Концентрация реакционной смеси и флеш-хроматография на силикагеле остатка (с элюированием 40% этилацетата в гексанах) с последующим отделением методом HPLC (жидкостная хроматография высокого давления) от других компонентов дали указанное в заголовке соединение в виде пены (16,8 мг, выход 56%).
Альтернативная процедура
К раствору 7-O-[(метилтио)тиокарбонил] -13-O-триэтилсилилбаккатина III (продукт получения 6, стадия (b), 416,3 мг, 0,527 ммоль) в сухом толуоле (10,5 мл) добавляли каталитическое количество АИБН, и полученный раствор дегазировали сухим N2 в течение 5 мин. Добавляли гидрид трибутилолова (708,7 мкл, 2,63 ммоль), и реакционную смесь нагревали при 100oC в течение 2 ч, после чего добавляли другую часть гидрида трибутилолова (425,3 мкл, 1,581 ммоль). Реакционную смесь нагревали 5,5 ч при 100oC, после чего позволяли охладиться до комнатной температуры. Хроматография на силикагеле (с элюированием 20% этилацетата в гексанах) дала 7-дезокси-10-дезацетокси-13-O-(триэтилсилил)баккатин III (320 мг, выход 97%).
К раствору полученного на вышеописанной стадии продукта (160 мг, 0,255 ммоль) в сухом тетрагидрофуране (2 мл) при комнатной температуре добавляли тетрабутиламмонийфторид (766 мкл, 1 M, 0,766 ммоль). Реакционную смесь перемешивали 1 ч при комнатной температуре. Удаляли растворитель, и остаток хроматографировали на силикагеле (с элюированием 50-70% этилацетата в гексанах), получив в результате указанный в заголовке целевой продукт (115 мг, выход 87,9%).
Получение 9. (3R,4S)-1-трет-Бутоксикарбонил-4-фенил-3-триэтилсилилокси-2-азетидинон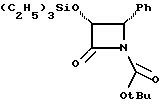
К перемешанному раствору (3R,4S)-4-фенил-3-триэтилсилилокси-2-азетидинона (2,200 г, 7,92 ммоль) в сухом тетрагидрофуране (25 мл) N,N-диизопропилэтиламин (1,65 мл, 9,510 ммоль, 1,2 эквив.) при 0oC в среде аргона. Раствор перемешивали 5 мин, после чего добавляли ди-трет-бутилдикарбонат (2,080 г, 9,510 ммоль, 1,2 эквив.) и 4-диметиламинопиридин (193,6 мг, 1,581 ммоль, 0,20 эквив. ). Реакционную смесь перемешивали при 0oC 60 мин и затем разбавляли этилацетатом (25 мл). Полученный раствор промывали рассолом, 10%-ным NaHCO3, 10%-ным раствором HCl, высушивали (MgSO4) и концентрировали, получив в результате неочищенное соединение (масло). Это соединение затем очищали путем флеш-хроматографии на силикагеле (с элюированием 15% этилацетата в гексанах) и получили указанное в заголовке соединение в виде белого твердого вещества (2,4 г, выход 83%).
Получение 10. (±)-цис-3-Ацетокси-4-фенилазетидин-2-он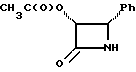
(a) В трехгорлую круглодонную колбу емкостью 1 л, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли гидробензамид (30,00 г, 100,5 ммоль) и этилацетат (150 мл). Перемешивая под защитным слоем аргона, реакционную смесь охлаждали до 5oC и добавляли к ней триэтиламин (16,8 мл, 121 ммоль). Затем добавляли по каплям в течение 90 мин раствор ацетоксиацетилхлорида (12,4 мл, 116 ммоль) в этилацетате (300 мл). Через 16 часов при этой температуре реакционной смеси давали возможность нагреваться до 20oC (1,5 ч) и переносили ее в делительную воронку. Органический слой промывали последовательно насыщенным водным раствором NH4Cl /150 мл, 100 мл/, насыщенным водным раствором NaHCO3 (120 мл) и рассолом (120 мл). Для целей идентификации указанное в заголовке соединение может быть выделено на этой стадии путем высушивания органической фазы над MgSO4, фильтрования и удаления растворителя в вакууме. Это дало (±)-цис-3-ацетилокси-1-[(фенил) (бензилиденимино)метил]-4-фенилазетидин-2-он с количественным выходом сырого продукта в виде красного стекла.
(b) Раствор полученного в части (a) соединения в этилацетате (500 мл) осторожно переносили под струей аргона в 2-литровую колбу Парра, содержащую 10% палладия на активированном угле (6,00 г). Эту смесь обрабатывали водородом (4 атм.) в течение 20 ч, после чего катализатор удаляли путем фильтрации через слой целита. Остаток от фильтрования суспендировали в этилацетата (200 мл), перемешивали (10 мин) и фильтровали. Остаток от фильтрации промывали этилацетатом (100 мл), и фильтраты объединяли. Органический слой промывали 10%-ной HCl (300 мл), и оба слоя фильтровали через воронку со спеченным стеклом для удаления белого осадка (дибензиламин • HCl), который промывали этилацетатом (100 мл). Фазы разделяли, и органический слой промывали другой частью 10%-ной HCl (200 мл). Объединенные промывные воды с 10%-ной HCl вновь экстрагировали этилацетатом (200 мл), и объединенные органические слои промывали насыщенным водным раствором NaHCO3 (300 мл) и рассолом (250 мл). Органический слой высушивали над MgSO4, фильтровали и концентрировали в вакууме до конечного объема 75 мл. Эту смесь охлаждали до 4oC, и осажденный продукт отделяли путем фильтрования. Остаток от фильтрования промывали гексаном (200 мл), получив в результате 16,12 г (78,1% общий выход от гидробензамида) указанного в заголовке соединения в виде белых игл; т.пл. = 150 - 151oC.
Получение 11. (±)-цис-3-Триэтилсилилокси-4-(2-фурил)-N-трет- бутоксикарбонилазетидин-2-он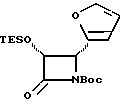
(a) Следовали процедуре, описанной в Получении 10, часть (a), за исключением того, что вместо гидробензамида, использовали гидрофурамид [т.е. 2-фурил-CH-(N= CH-2-фурил)2] , и реакцию проводили в расчете на 18,6 ммоль (вместо 100 ммоль). В результате гидрофурамид (5,00 г, 18,6 ммоль), триэтиламин (3,11 мл, 22,3 ммоль) и ацетоксиацетилхлорид (2,30 мл, 21,4 ммоль) дали 6,192 г (выход 90,4%) соединения (±)-цис-3-ацетилокси-1-[(2-фурил)(2-фурилметиленимино)-метил] -4- (2-фурил)азетидин-2-он в виде бледно-красного сиропа.
(b) Следовали процедуре, описанной в Получении 10, часть (b), за исключением того, что продукт выделяли путем препаративной тонкослойной хроматографии, и реакцию проводили в расчете на 2,7 ммоль, вместо первоначального количества гидрофурамида. Так, полученный в описанной выше части (a) неочищенный продукт вновь растворяли в этилацетате (50 мл) и добавляли к 10% палладия на активированном угле (150 мг). Очистка сырого твердого вещества путем препаративной тонкослойной хроматографии (2 мм силикагеля, элюированного смесью этилацетат-гексан с отношением 1:1) дала 386 мг (откорректированный общий выход от гидрофурамида 65,8%) (±)-цис-3-(ацетилокси)-4-(2-фурил)азетидин-2-она в виде желтого твердого вещества. Это соединение перекристаллизовывали из смеси этилацетат-гексан; т.пл. = 118 - 119oC.
(c) Полученное в описанной выше части (b) соединение (3,78 г, 19,4 ммоль) в 60 мл метанола перемешивали с K2CO3 (20 мг, 0,14 ммоль) в течение 90 мин, и раствор нейтрализовали посредством Dowex 50w-X8 и фильтровали. Фильтрат концентрировали, и остаток растворяли в 80 мл безводного ТГФ (тетрагидрофурана) и перемешивали при 0oC с имидазолом (1,44 г, 21,2 ммоль) и TESCl (3,4 мл, 20,2 ммоль) в течение 30 мин. Раствор разбавляли этилацетатом и промывали рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (с элюированием смесь гексан-этилацетат с отношением 3:1), что дало 4,47 г (выход 86%) (±)-цис-3-триэтилсилилокси-4-(2-фурил)азетидин-2-она в виде бесцветного масла.
(d) Полученный в части (c) продукт (2,05 г, 7,7 ммоль) в 30 мл дихлорметана перемешивали при 0oC с диизопропилэтиламином (1,5 мл, 8,6 ммоль) и ди-трет-бутилдикарбонатом (2,0 г, 9,2 ммоль), а также каталитическим количеством диметиламинопиридина (ДМАП). Раствор разбавляли дихлорметаном и промывали рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (с элюированием смесью гексан-этилацетат с отношением 8:1) и получали в результате 2,0 г (выход 70%) указанного в заголовке соединения в виде воскообразного твердого вещества.
Рацемическая смесь, полученная в части (b), может быть использована в качестве субстрата для ферментативного гидролиза с использованием липазы, такой как PS-30 из Pseudomonas sp. (Амано интернешнл Ко.), чтобы получить (3R, 4R)-3-гидрокси-4-(2-фурил)-азетидин-2-он. Метод ферментативного разделения с использованием липазы PD-30 и других ферментов раскрыт в одновременно рассматриваемой заявке в США N 092170, поданной данными заявителями 14 июля 1993 г., которая путем ссылки включена в данное описание полностью.
Следуя процедурам в частях (c) и (d) с использованием (3R,4R)-3-гидрокси-4-(2-фурил)-азетидин-2-она, получают (3R,4R)-N-(трет-бутоксикарбонил)-3-триэтилсилилокси-4-(2-фурил) азетидин-2-он.
Получение 12. (±)-цис-3-Триэтилсилилокси-4-(2-тиенил)-N-трет- бутоксикарбонилазетидин-2-он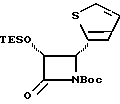
(a) Следовали процедуре, описанной в Получении 10, стадия (a), за исключением того, что вместо гидробензамида, использовали гидротиенамид [т.е. 2-тиенил-CH-(N = CH-2-тиенил)2]. При этом гидротиенамид (30 г, 94,7 ммоль), триэтиламин (15,84 мл, 114 ммоль) и ацетоксиацетилхлорид (11,6 мл, 108 ммоль) дали (±)-цис-3-ацетилокси-1-[(2-тиенил) (2-тиенилметиленимино)метил]-4-(2-тиенил)азетидин-2-он в виде вязкого масла.
(b) К перемешиваемому раствору полученного в части (a) продукта (0,431 г, 1,03 ммоль) в дихлорметане (2,93 мл) при 25oC добавляли 70%-ный водный раствор уксусной кислоты (0,35 мл ледяной уксусной кислоты и 0,15 мл воды). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 2,5 ч. Реакционную смесь разбавляли 50 мл дихлорметана и затем промывали двумя 75 мл порциями насыщенного водного раствора бикарбоната, а затем одной 50 мл порцией насыщенного рассола. Органический экстракт концентрировали в вакууме до получения коричневого масла, растворяли в минимальном количестве дихлорметана и затем помещали на колонку с силикагелем с размерами 4'' x 0,5'' (10,16 см х 1,27 см). Элюирование с использованием градиента 10 - 60% этилацетата в гексане дало менее полярные побочные продукты и затем (±)-цис-3-ацетилокси-4-(2-тиенил)-азетидин-2-он (0,154 г, выход 75%) в виде белого твердого вещества.
(c) раствор полученного в части (b) продукта (2,5 г, 11,8 ммоль) растворяли в метаноле (10 мл) и обрабатывали насыщенным водным раствором бикарбоната натрия (10 мл), и полученной суспензии позволяли перемешиваться при температуре окружающей среды в течение 3 ч. Затем реакционную смесь разбавляли этилацетатом (20 мл) и промывали водой (15 мл). Водную фракцию экстрагирования обратно несколько раз этилацетатом, и объединенные органические фракции высушивали ( MgSO4) и концентрировали, получив в результате желтое твердое вещество (выход 1,7 г). Неочищенное вещество растворяли в сухом тетрагидрофуране (20 мл), и охлаждали раствор до 5oC в водяной ванне со льдом. После перемешивания в течение 5 мин добавляли по каплям триэтилхлорсилан (1,85 мл, 1,1 эквив.). Полученной суспензии позволяли перемешиваться в течение 3 ч при указанной температуре, после чего отделяли путем фильтрования твердые частицы. Органическую фракцию промывали водой (2 х 20 мл), а затем высушивали (MgSO4) и концентрировали. Неочищенный продукт очищали путем хроматографии на колонке с силикагелем (с элюированием смесью гексаны-этилацетат с отношением 7:3) и получили (±)-цис-3-триэтилсилилокси-4-(2-тиенил)азетидин-2-он в виде бесцветного твердого вещества (1,5 г, выход 45%); т.п. 70 - 71oC.
Альтернативная процедура
Полученный в часть (b) продукт (2,0 г, 9,37 ммоль) в 40 мл метанола перемешивали с K2CO3 (60 мг, 0,43 ммоль) 30 мин, и раствор нейтрализовали посредством Dowex 50W-X8 и фильтровали. Фильтрат концентрировали, и остаток растворяли в 50 мл безводного ТГФ и перемешивали при 0oC с имидазолом (0,85 г, 11,3 ммоль) и TESCl (1,9 мл, 12,5 ммоль) в течение 30 мин. Раствор разбавляли этилацетатом и промывали рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (с элюированием смесью гексан-этилацетат с отношением 3:1), получив в результате 2,13 г (выход 86%) указанного в заголовке соединения в виде бесцветного масла.
(d) Раствор полученного в части (c) продукта (425,7 мг, 1,48 ммоль) растворяли в дихлорметане (10 мл) и охлаждали до 5oC в водяной ванне со льдом. Реакционную смесь обрабатывали каталитическим количеством ДМАП, а после чего диизопропилэтиламино (TESCl, 0,25 мл, 1,0 эквив.) и затем ди-трет-бутилдикарбонатом (388,4 мг, 1,2 эквив.). После перемешивания в течение 2 ч при указанной температуре реакцию быстро прекращали насыщенным водным раствором бикарбоната натрия (45 мл), и органическую фракцию промывали водой (5 мл), после чего высушивали (MgSO4), пропускали через короткую пробку из силикагеля и концентрировали, получив в результате целевой продукт в виде бесцветного масла (525,3 мг, выход 93%).
Процедура, описанная выше в Получениях 9, 11 (d) и 12 (d), может быть приспособлена для получения других N - замещенных азетидинонов, пригодных для получения соединений в соответствии с настоящим изобретением. Примеры таких азетидинов перечислены в таблице III; P - гидроксизащитная группа, такая как триэтилсилил, триизопропилсилил и этоксиэтил.
Получение 13. 10-Дезокситаксотер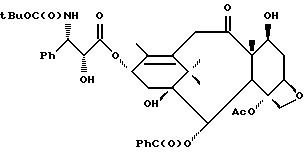
10-Дезацетокси-7-O-триэтилсилилбаккатин III (100 мг, 0,156 ммоль) помещали в колбу под аргоном и растворяли в сухом тетрагидрофуране (1,5 мл). После охлаждения до -40oC добавляли по каплям н-бутиллитий (1,45 М в гексанах, 0,119 мл, 0,170 ммоль), а затем (3R, 4S)-1-трет-бутоксикарбонил-4-фенил-3-триэтилсилилокси-2-азетидинон (94,2 мг, 0,25 ммоль) в тетрагидрофуране (0,5 мл) в течение 2 мин. Смесь сразу же нагревали до 0oC и перемешивали 45 мин, перед тем как резко охладить насыщенным аммонийхлоридом (3 мл). Смесь экстрагировали этилацетатом, высушивали и концентрировали. Хроматография на силикагеле (с элюированием 30% этилацетата в гексане) дала 10-дезокси-2', 7-бис-O-(триэтилсилил)таксотер в виде пены (125 мг, выход 76%). Это соединение (100 мг, 0,098 ммоль) немедленно растворяли в ацетонитриле (2 мл) при -5oC и обрабатывали хлористоводородной кислотой (0,037 мл, 36%, 12 М). Смесь перемешивали 2 ч при -5oC, после чего резко охлаждали водным бикарбонатом, экстрагировали этилацетатом и высушивали. После выпаривания растворителя остаток хроматографировали на силикагеле (с элюированием 75% этилацетата в гексане), что дало указанное в заголовке соединение в виде пены (80,5 мг, выход 80%).
Общая процедура, проведенная в Получении 13, может быть приспособлена для получения других соединений формулы (Ia) с исходными компонентами баккатином III и азетидиноном; примеры других соединений формулы (Ia) перечислены в таблице IV. Понятно, что хотя перечисленные ниже соединения показаны со свободными гидроксигруппами, при надлежащем выборе различных гидроксизащитных групп любая из защитных групп в положении 2', 7 или 10 может быть избирательно снята без влияния на другие имеющиеся защитные группы.
Получение 14 Вис(метилтиометил)эфир
SH3SCH2OCH2SCH3
К раствору 1,1'-дихлордиметилового эфира (3,0 г, 26,3 ммоль) в ацетоне (100 мл) при 0oC добавляли иодид натрия (8,23 г, 55,23 ммоль), и смесь перемешивали при этой температуре 20 мин. Затем добавляли четырьмя порциями тиометоксид натрия (1,84 г, 5,23 ммоль), и полученный раствор перемешивали еще 1 ч. После этого гетерогенный раствор фильтровали через слой целита, и фильтрат концентрировали в вакууме. Остаточное масло распределили между этилацетатом и насыщенным водным раствором бикарбоната натрия. Водный слой удаляли и затем экстрагировали этилацетатом, затем объединенную органику обрабатывали смесью насыщенного водного раствора бикарбоната натрия и 5%-ного водного раствора тиосульфата натрия с отношением 1:1 (объем/объем). Затем органику промывали рассолом, высушивали над сульфатом натрия и концентрировали в вакууме. Остаточное масло очищали путем флеш-хроматографии (30: 1, гексаны: этилацетат) и получили 1,9 г желтого масла, которое затем перегоняли с использованием кугельроровского аппарата (120 - 130oC, 20 мм рт. ст.), что дало 1,5 г (45%) указанного в заголовке соединения в виде бесцветного масла:
1H ЯМР (300 МГц, CDCl3) : δ 4,73 (4H, с), 2,15 (6H, с),
Получение 15. Дибензилметилтиометилфосфат
CH3SCH2OP(O)(OBu)2
К раствору бис(метилтиометил)эфира (30 мг, 2,34 ммоль) и молекулярных сит (300 мг) в тетрагидрофуране (ТГФ) (100 мл) при комнатной температуре добавляли дибензилфосфат (2,74 г, 9,85 ммоль) и затем N-иодсукцинимид (608 мг, 2,71 ммоль), и раствор перемешивали 4 ч. Реакционную смесь затем разбавляли этилацетатом и фильтровали через слой целита. Фильтрат обрабатывали смесью насыщенного водного раствора бикарбоната натрия и 5%-ного водного раствора тиосульфата натрия с отношением 1:1 (объем/объем). Затем бесцветный органический экстракт промывали рассолом, высушивали над сульфатом натрия и концентрировали в вакууме, получив в результате 600 мг (69%) указанного в заголовке соединения:
1H ЯМР (300 МГц, CDCl3) : δ 7,35 (10H, с), 5,29 (2H, д, J = 12,2 Гц), 5,08 (4H, дд, J = 8,0 и 1,0 Гц), 4,68 (2H, с), 2,10 (3H, с).
Примеры
Следующие ниже примеры приведены для иллюстрации синтеза представленных соединений в соответствии с настоящим изобретением и не должны рассматриваться как как-то ограничивающие объем изобретения. Специалисты в данной области техники могут приспособить эти способы (без ненужных экспериментов) к синтезу соединений, хотя конкретно и не раскрытых, но входящих в объем настоящего изобретения.
Пример 1. 7-O-Фосфонооксиметилпаклитаксел и его мононатриевая соль
(a) Получение 7-O-метилтиометилпаклитаксела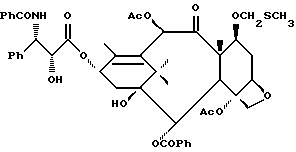
К тщательно перемешанной смеси паклитаксела (0,85 г, 1 ммоль) и диметилсульфида (0,72 мл, 8 ммоль) в сухом ацетонитриле (10 мл) при 0oC добавляли бензоилпероксид (0,98 г, 4 ммоль). Перемешивание продолжали в течение 2,5 часов при 0oC. Ход реакции контролировали путем тонкослойной хроматографии на силикагеле в системе растворителей толуол:кетон (2:1, объем/объем) (Rfтакс.= 0,38, Rfпрод.= 0,64), и когда видели образование продуктов более высокой полярности, то реакцию быстро прекращали путем выпаривания растворителей с использованием роторного испарителя Ротавапор при 30oC. Анализ путем тонкослойной хроматографии показал наличие некоторых количеств непрореагировавших паклитаксела и 2',7-O-бис(метилтиометил) паклитаксела. Отделение указанного в заголовке соединения от реакционной смеси производили путем колоночной флеш-хроматографии на силикагеле 60 (40 - 63 мкм). EM Sciense (100 мл), диаметр колонки 2 дюйма (50,6 мм) с использованием системы растворителей этилацетат-гексан (1: 1, объем/объем) (Rfпрод.=0,34). Продукт (552 мг, выход 60%) извлекали из фракций 12 - 18 (каждая фракция приблизительно 20 мл).
Масс-спектр: (FAB/матрица NOBA, NaI, KI): [M+H]+, m/z 914; [M+Na]+, m/z 936; [M+K]+, m/z 952
Элементный анализ: C - 64,28 (вычисл. 64,39), H - 5,85 (вычисл. 6,07), N - 1,46 (вычисл. 1,53).
УФ-спектр (МеОН): λmax = 226 нм, Е (1%/1 см) = 150, А = 0,2653.
ИК-спектр (KBr): 3432, 3066, 2940, 1726, 1668, 1602, 1582, 1514, 1484, 1452, 1372, 1242, 1178, 1142, 1108, 1068, 1026, 990, 916, 884, 852, 802, 774, 710, 608, 570, 538, 482 см-1.
1H-ЯМР (CDCl3), δ : 1,15 (3H, c), 1,19 (3H, с), 1,73 (3H, с), 1,79 (H, с), 1,90 (3H, д), 2,09 (3H, с), 2,16 (3H, с), 2,29 (2H, д), 2,35 (3H, с), 2,77 (H, м), 3,70 (H, д), 3,83 (H, д), 4,17 (H, д), 4,26 (H, д, перекрывается с H, д), 4,63 (2H, т), 4,77 (Н, дд), 4,91 (H, д), 5,65 (H, д), 5,77 (H, дд), 6,16 (H, дд), 6,48 (H, с), 7,07 (H, д), 7,29 - 7,50 (10H, м), 7,57 (H, м), 7,73 (2H, д), 8,08 (2H, д).
(b) Получение 7-O-дибензилфосфонооксиметилпаклитаксела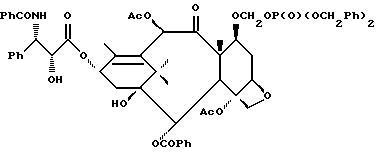
К смеси 7-O-метилтиометилпаклитаксела (119 мг, 0,13 мМ) и порошкообразных молекулярных сит 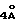 (приблизительно 120 мг) в сухом 1,2-дихлорметане (5 мл) добавляли раствор N-иодсукцинимида (45 мг, 0,2 мМ) и дибензилфосфата (55 мг, 0,2 мМ) в сухом тетрагидрофуране (4 мл). Реакционную смесь перемешивали при комнатной температуре 16 часов. Ход реакции контролировали тонкослойной хроматографией в системе толуол: ацетон (2: 1, объем/объем) (Rfпрод.= 0,48). Молекулярные сита удаляли путем фильтрования через Целит 545, и фильтрат экстрагировали метиленхлоридом (100 мл). Органический слой промывали 1%-ным раствором тиосульфата натрия (примерно 100 мл) и 0,5 М бикарбоната натрия (100 мл) и рассолом. Экстракт фильтровали через сепаратор Ватмана для разделения фаз, и выпаривали растворители. Очистка на флеш-колонке с силикагелем 60 в смеси метиленхлорид:этилацетат (2:1, объем/объем) дала 7-O-дибензилфосфонооксиметилпаклитаксел (41,5 мг).
(приблизительно 120 мг) в сухом 1,2-дихлорметане (5 мл) добавляли раствор N-иодсукцинимида (45 мг, 0,2 мМ) и дибензилфосфата (55 мг, 0,2 мМ) в сухом тетрагидрофуране (4 мл). Реакционную смесь перемешивали при комнатной температуре 16 часов. Ход реакции контролировали тонкослойной хроматографией в системе толуол: ацетон (2: 1, объем/объем) (Rfпрод.= 0,48). Молекулярные сита удаляли путем фильтрования через Целит 545, и фильтрат экстрагировали метиленхлоридом (100 мл). Органический слой промывали 1%-ным раствором тиосульфата натрия (примерно 100 мл) и 0,5 М бикарбоната натрия (100 мл) и рассолом. Экстракт фильтровали через сепаратор Ватмана для разделения фаз, и выпаривали растворители. Очистка на флеш-колонке с силикагелем 60 в смеси метиленхлорид:этилацетат (2:1, объем/объем) дала 7-O-дибензилфосфонооксиметилпаклитаксел (41,5 мг).
(c) Получение 7-O-фосфонооксиметилпаклитаксела и его мононатриевой соли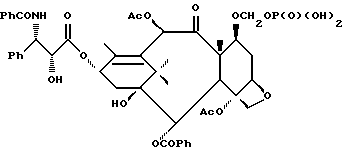
7-O-Дибензилфосфонооксиметилпаклитаксел (41,5 мг) растворяли в этилацетате (5 мл) и добавляли 10%-ный палладий на древесном угле (20 мг). Осуществляли гидрогенизацию при 40 фунтах на кв. дюйм (275 кПа) при комнатной температуре в течение 1 часа. Ход реакции контролировали тонкослойной хроматографией в системе хлороформ:метанол:вода (120:45:8, объем/объем). Очистка путем препаративной тонкослойной хроматографии (несущая силикагель пластика 20х20х0,05 см в аналитической системе) дала 7-O-фосфонооксиметилпаклитаксел (26 мг, выход 75%).
Поскольку при очистке на силикагеле наблюдали разложение 7-O- дибензилфосфонооксиметилпаклитаксела, то процедуру гидрогенизации видоизменили. Так, неочищенный экстракт 7-O- дибензилфосфонооксиметилпаклитаксела гидрогенизировали без всякой очистки. Гидрогенизацию неочищенного экстракта 7-O-дибензилфосфонооксиметилпаклитаксела осуществляли при 60 фунтах на кв. дюйм (400 кПа) в течение 24 часов.
7-O-фосфонооксиметилпаклитаксел (70 мг) растворяли в 5 мл раствора ацетон-вода (1:1) и разбавляли водой до 50 мл. Добавляли сухой бикарбонат натрия (18 мг, 1,2 эквив.). Ацетон выпаривали при комнатной температуре с помощью роторного испарителя и оставшийся водный раствор лиофилизировали. Неочищенную мононатриевую соль 7-O- фосфонооксиметилпаклитаксела очищают путем хроматографии на колонке C18 с обратной фазой в системе вода:ацетонитрил (70: 30, объем/объем). Элюат контролировали путем аналитической HPLC/15 см, колонка C18 Джонса, 1 мл/мин, l=230/270 нм /в буфере ацетонитрил: 0,05 M аммоний ацетат (45:55, объем/объем), pH=7, Rt=2,09 мин. Фракции, содержавшие целевой продукт объединяли, выпаривали ацетонитрил, и оставшийся водный раствор лиофилизировали, получив в результате мононатриевую соль (112 мг) 7-O-фосфонооксиметилпаклитаксела.
Масс-спектр (FAB): [M+H]+, m/z 986; [M+Na]+, m/z 1008.
УФ-спектр (MeOH): λмакс = 230 нм, E (1%/1 см)=248.
ИК-спектр (KBr): 3430, 3066, 2948, 1724, 1652, 1602, 1580, 1518, 1486, 1452, 1372, 1316, 1246, 1178, 1154, 1108, 1070, 1000, 982, 946, 856, 802, 776, 710, 628, 538 см-1.
1H-ЯМР (ацетон-d6/D2O), δ: 8,05 (2H, д), 7,92 (2H, д), 7,65 (1H, дд), 7,58 - 7,35 (9H, м, перекрытие), 7,23 (1H, дд), 6,38 (1H, с), 6,08 (1H, т), 5,65 (1H, д), 5,60 (1H, д), 5,10 (1H, шс), 4,99 (1H, д), 4,97 (1H, шс), 4,80 (1H, д), 4,28 (1H, дд), 4,11 (2H, с), 3,79 (1H, д), 2,94 (1H, м), 2,35 (3H, с), 2,35 - 2,10 (1H, м), 2,13 (3H, с), 1,95 (3H, с), 1,84 (1H, м), 1,67 (3H, с), 1,13 (6H, с, перекрытие).
Пример 2. Альтернативный способ получения 7-O- фосфонооксиметилпаклитаксела
(a) Получение 2'-O-(бензилоксикарбонил)паклитаксела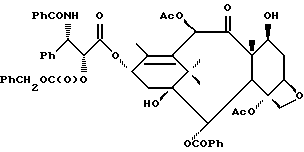
К перемешанному раствору паклитаксела (150 мг, 0,176 ммоль) и N,N-диизопропилэтиламина (93 мкл, 0,534 ммоль, 3 эквив.) в безводном метиленхлориде (4 мл) при комнатной температуре добавляли бензилхлорформиат (75 мкл, 0,525 ммоль, 3 эквив.). Реакционную смесь перемешивали при комнатной температуре 3 часа, концентрировали до 32 мл и очищали на колонке с силикагелем с использованием в качестве элюента смеси (1:1) этилацетат:гексаны, в результате чего получали указанное соединение в виде белого порошка (150 мг, выход 86%); т.пл. 140 - 150oC (разложение).
(b) Получение 2'-O-(бензилоксикарбонил)-7-O- метилтиометилпаклитаксела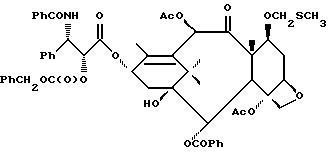
К охлажденному (сухой лед-CCl4, температура ванны -30oC) раствору 2'-O-(бензилоксикарбонил)паклитаксела (4,395 г, 5,0 ммоль) в сухом ацетонитриле (80 мл) добавляли последовательно диметилсульфид (3,6 мл, 40 ммоль) и бензоилпероксид (4,9 г, 20,247 ммоль). Через 10 минут, при -30oC, убирали охлажденную ванну и тщательно перемешивали реакционную смесь в течение 2 ч при комнатной температуре. Затем реакционную смесь разбавляли этилацетатом до объема 200 мл и промывали водой и рассолом. Органический слой высушивали (MgSO4), после чего выпаривали растворитель, получив в результате остаток, который держали под вакуумом 18 ч для удаления диметилсульфоксида, который присутствовал как побочный продукт реакции. Остаток очищали на колонке с силикагелем с использованием в качестве элюента сначала смеси (1:2) этилацетат-гексан для удаления менее полярных примесей, а затем смеси (1:1) этилацетат-гексан, что дало в результате ожидаемое указанное в заголовке соединение в виде пены. Тщательно его растирали с сухим диэтиловым эфиром и фильтровали, получив указанное в заголовке соединение в виде рыхлого твердого вещества (5,0 г, 95%); т.пл. 120 - 122oC.
Масс-спектр (FAB): [MH]+, m/z 1048; [M + Na]+, m/z 1070; [M + K]+, m/z 108.
ИК (KBr): 3440, 3066, 1750, 1722, 1664, 1602, 1583, 1538 см-1.
ЯМР (CDCl3), δ: 1,177 (3H, с), 1,236 (3H, с), 1,745 (3H, с), 2,023 (3H, с), 2,121 (3H, с), 2,162 (3H, с), 2,436 (3H, с), 3,887 (H, д), 4,134 (H, д), 4,197 (H, д), 4,295 (H, м), 4,964 (H, д), 5,161 (2H, д), 5,450 (H, д), 5,703 (H, д), 5,981 (H, дд), 6,257 (H, т), 6,541 (H, с), 6,920 (H, д, NH), 7,322 - 8,22 (15H, м).
Указанное в заголовке соединение было также получено следующим альтернативным способом.
К раствору 2'-O-(бензилоксикарбонил)паклитаксела (2,0 г, 2,0263 ммоль) в сухом диметилсульфоксиде (10 мл) добавляли по каплям уксусный ангидрид (10 мл). Полученную смесь перемешивали при комнатной температуре 18 ч под N2, разбавляли этилацетатом (100 мл) и тщательно промывали холодным 6%-ным раствором бикарбоната натрия (6 х 30 мл), хлоридной водой (6 х 30 мл) и рассолом. Высушивали (MgSO4) органический слой, а растворитель выпаривали, получив остаток. Его очищали на колонке с силикагелем и элюировали метиленхлоридом, смесью метиленхлорид - 5 % ацетонитрила и смесью метиленхлорид - 10% ацетонитрила, получив в результате ожидаемое указанное в заголовке соединение (1,86 г, 87,7%). Это соединение идентично тому, что получили ранее описанным способом с использованием диметилсульфида и бензоилпероксида.
(c) Получение 2'-O-(бнзилоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксела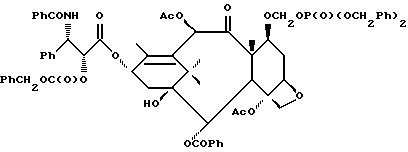
К раствору 2'-O-(бензилоксикарбонил)-7-O- метилтиометилпаклитаксела (5,0 г, 5,5396 ммоль) в сухом 1,2-дихлорэтане (120 мл) добавляли активированные порошкообразные молекулярные сита 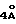 (5,0 г). К этой смеси добавляли по каплям при комнатной температуре раствор смеси N-иодсукцинимида (1,61 г, 7,1632 ммоль) и дибензилфосфата (1,97 г, 7,1632 ммоль) в сухом тетрагидрофуране (90 мл). После интенсивного перемешивания при комнатной температуре в течение 30 мин реакционную смесь фильтровали через целит, и фильтрат упаривали досуха, получив красный остаток. Остаток растворяли в этилацетате (100 мл), промывали холодным 6%-ным раствором NaHSO3 (2 х 50 мл), холодным 6%-ным раствором NaHCO3 (2 х 50 мл) и рассолом (1 х 50 мл). Органический слой высушивали (MgSO4), и выпаривали растворитель, получив в результате твердую массу, которую растирали в порошок с сухим диэтиловым эфиром и фильтровали, получив указанное в заголовке соединение в виде цвета слоновой кости твердого вещества (5,9 г, 97%); т.пл. 124 - 127oC.
(5,0 г). К этой смеси добавляли по каплям при комнатной температуре раствор смеси N-иодсукцинимида (1,61 г, 7,1632 ммоль) и дибензилфосфата (1,97 г, 7,1632 ммоль) в сухом тетрагидрофуране (90 мл). После интенсивного перемешивания при комнатной температуре в течение 30 мин реакционную смесь фильтровали через целит, и фильтрат упаривали досуха, получив красный остаток. Остаток растворяли в этилацетате (100 мл), промывали холодным 6%-ным раствором NaHSO3 (2 х 50 мл), холодным 6%-ным раствором NaHCO3 (2 х 50 мл) и рассолом (1 х 50 мл). Органический слой высушивали (MgSO4), и выпаривали растворитель, получив в результате твердую массу, которую растирали в порошок с сухим диэтиловым эфиром и фильтровали, получив указанное в заголовке соединение в виде цвета слоновой кости твердого вещества (5,9 г, 97%); т.пл. 124 - 127oC.
Масс-спектр (FAB): [MH]+, m/z 1278; [M + Na]+, m/z 1301; [M + K]+, m/z 1316.
ИК-спектр (KBr): 3430, 3066, 3032, 1750, 1726, 1664, 1582, 1532 см-1.
ЯМР (CDCl3), δ: 1,160 (3H, с), 1,703 (3H, с), 1,985 (3H, с), 2,164 (3H, с), 2,420 (3H, с), 3,854 (H, д), 4,151 (H, д), 4,216 (H, м), 4,298 (H, д), 4,873 (H, д), 5,043 (6H, м), 5,140 (2H, д), 5,417 (H, д), 5,670 (H, д), 5,971 (H, дд), 6,241 (H, т), 6,317 (H, с), 6,912 (H, д, NH), 7,280 - 8,115 (25H, м).
(d) Получение 7-O-фосфонооксиметилпаклитаксела
К раствору 2'-O-(бензилоксикарбонил)-7-O- дибензилфосфонооксиметилпаклитаксела (6,0 г, 4,7095 ммоль) в этилацетате (120 мл) добавляли 10% Pd/C (6,0 г), и смесь гидрогенизировали при 60 фунтах на кв. дюйм (400 кПа) в течение 24 часов. Реакционную смесь фильтровали через целит и, выпарив растворитель, получили 4,07 г неочищенного остатка. Его очищали на короткой колонке с силикагелем путем последовательного элюирования смесью хлороформа с 10%, 20% и 40% метанола и получили указанное в заголовке соединение в виде белого твердого вещества (3,2 г, 71%); т. пл. 155 - 158oC.
Этот продукт имел такой же Rf (тонколойная хроматография) и такое же время удерживания (HPLC - жидкостная хроматография высокого давления), что и аутентичная проба.
Масс-спектр (FAB): [MH] +, m/z 964; [M + Na]+, m/z 986; [M + K]+, m/z 1002; [M + K+ + Na+ - H]+, m/z 1024; [M + 2K - H]+, m/z 1040.
УФ-спектр (MeOH): λмакс = 230 нм, E (1%/1 см) = 252,5.
ИК-спектр (KBr): 3432, 3066, 2992, 1722, 1648, 1602, 1580, 1522, 1488, 1452, 1372, 1316, 1246, 1178, 1154, 1110, 1070, 1000, 980, 946, 854, 802, 776, 710, 628, 538 см-1.
1ЯМР (ацетон-d6/D2O), δ: 1,08 (3H, с), 1,10 (3H, с), 1,63 (3H, с), 1,88 (3H, с), 1,96 (H, м), 2,13 (3H, с), 2,32 (3H, с), 2,89 (H, м), 3,76 (H, д), 4,19 (H, м), 4,89 (H, дд), 5,09 (H, дд), 5,55 - 5,60 (2H, перекрывающиеся дублеты), 6,04 (H, т), 6,32 (H, с), 7,20 (H, т), 7,34 - 7,67 (10H, перекрывающиеся мультиплеты), 7,87 (2H, дд), 8,02 (2H, дд).
Пример 3. 2'-O-(Этоксикарбонил)-7-O-фосфонооксиметилпаклитаксел
(a) Получение 2'-O-(этоксикарбонил)паклитаксела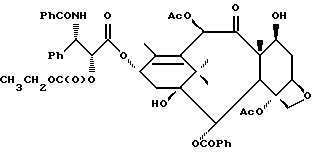
К раствору паклитаксела (4,35 г, 5,1 ммоль) в сухом метиленхлориде (51 мл) добавляли N,N-диизопропилэтиламин (2,67 мл, 15,3 ммоль) и затем этилхлорформиат (1,46 мл, 15,3 ммоль). Реакционную смесь перемешивали при 0oC 2 часа, а затем при комнатной температуре еще 1 час. Реакционную смесь разбавляли этилацетатом (400 мл), органическую фазу промывали насыщенным раствором NaHCO3 (2 х 30 мл) и рассолом (30 мл). Полученную органическую фазу высушивали над MgSO4 и получили неочищенное указанное в заголовке соединение (93%), которое использовали на следующей стадии без дополнительной очистки.
Масс-спектр (FAB/NOBA, NaI, KI): [M + H]+, m/z 926; [M + Na]+, m/z 948; [M + K]+, m/z 964.
Масс-спектр высокого разрешения (FAB/NOBA, внешний эталон CSI/Гли): [M + H] +, m/z 926,3588 наблюдаемое, C50H56NO16, вычисленное значение: 926,3599 (отклонение Δ = 1,2 м.д.).
1H-ЯМР (CDCl3), δ: 1,13 (3H, с), 1,23 (3H, с), 1,30 (3H, т), 1,67 (3H, с), 1,92 (3H, с), 2,21 (3H, с), 2,37 (H, д), 2,45 (3H, с), 2,54 (H, м), 3,80 (H, д), 4,15 - 4,32 (4H, перекрывающиеся мультиплеты), 4,43 (H, дд), 4,96 (H, д), 5,42 (H, д), 5,68 (H, д), 5,98 (H, дд), 6,28 (2H, перекрывающиеся мультиплеты), 7,00 (H, д), 7,34 - 7,59 (11H, перекрывающиеся мультиплеты), 7,74 (2H, д), 8,12 (2H, д).
Альтернативная процедура
Паклитаксел (5,40 г, 6,324 ммоль) в сухом дихлорметане (63 мл) охлаждали до 0oC и обрабатывали неразбавленным N,N-диизопропилэтиламином (3,30 мл, 3 эквив. ) и затем неразбавленным этилхлорформиатом (1,81 мл, 3 эквив.) по каплям в течение 5 минут. Реакцию контролировали посредством тонкослойной хроматографии (50% этилацетата в гексане). Через 2 ч при 0oC и 16 ч при комнатной температуре реакцию завершали, и желто-оранжевый раствор разбавляли этилацетатом (300 мл) и промывали насыщенным бикарбонатом натрия (3 х 75 мл) и рассолом (75 мл). Высушивание (MgSO4) и упаривание дали неочищенное указанное в заголовке соединение, которое очищали путем осаждения: добавляли дихлорметан (около 100 мл), а затем охлаждали и добавляли гексан (примерно 60 мл) до точки помутнения. После охлаждения во льду в течение нескольких часов улавливали путем фильтрования твердое вещество (5,17 г, выход 88%).
Альтернативная процедура
В высушенной над пламенем одногорлой колбе емкостью 3 л растворяли паклитаксел (99,0 г, 115,9 ммоль) в 1350 мл сухого метиленхлорида в среде аргона. Раствор охлаждали до -10oC. Медленно (примерно 3 мин) добавляли N,N-диизопропилэтиламин (52,4 г, 405,7 ммоль), а затем добавляли ClCO2Et (31,45 г, 289,8 ммоль; время добавления примерно 15 минут). Полученную смесь перемешивали всю ночь (16 часов) при -4oC. Тонкослойная хроматография показала, что реакция не была завершена. Поэтому добавляли другую навеску N,N-диизопропилэтиламина (2,62 г, 20,28 ммоль) и затем ClCO2Et (2,20 г, 20,28 ммоль) и продолжали перемешивание 3 часа при -4oC. Тонкослойная хроматография показала отсутствие исходного материала. Холодную смесь разбавляли этилацетатом (1,5 л) и переносили в делительную воронку. Затем ее промывали 5%-ным раствором KHSO4 (2 х 500 мл), водой (1 х 500 мл), 5%-ным раствором KHSO4 (1 х 500 мл), водой (1 х 500 мл), насыщенным раствором NaHCO3 (2 х 500 мл) и рассолом (2 х 500 мл), высушивали (MgSO4) и, удалив растворитель в вакууме, получили 147 г неочищенного продукта. Остаток растворяли в горячем метиленхлориде (800 мл, температура ванны 42oC), и по каплям добавляли гексаны (530 мл) с перемешиванием и поддержанием при этом указанной температуры. Кристаллизующуюся смесь отставили в сторону на 3 часа при комнатной температуре, а затем поставили в холодную комнату (0oC) на всю ночь. Тяжелые белые кристаллы улавливали фильтрованием и промывали смесью гексаны - CH2Cl2 (1: 1, объем/объем) (2 х 200 мл). После высушивания на вакуумном фильтре в течение 1 часа их высушивали в вакууме (примерно 1,0 мм рт. ст.) всю ночь и получили в результате 95,7 г (выход 89%) указанного в заголовке соединения (индекс однородности при измерении путем HPLC = 98,5%).
(b) Получение 2'-O-(этоксикарбонил)-7-O-метилтиометилпаклитаксела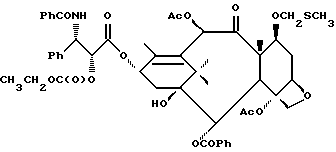
К раствору 2'-O-(этоксикарбонил)паклитаксела (4,38 г, 4,7 ммоль) в сухом диметилсульфоксиде (12,5 мл) добавляли уксусный ангидрид (12,5 мл). Реакционную смесь перемешивали 24 часа при комнатной температуре и затем разбавляли этилацетатом (500 мл), промывали насыщенным раствором NaHCO3 (3 х 40 мл) и водой (2 х 40 мл). Полученный органический слой высушивали над MgSO4, и выпаривали растворители в вакууме досуха. Остаток очищали путем хроматографии на силикагеле (40% этилацетата в гексанах) и получили в результате требуемое указанное в заголовке соединение (4,39 г, 94%).
Масс-спектр (FAB/NOBA, NaI, KI): [M + H]+, m/z 986; [M + Na]+, m/z 1008; [M + K]+, m/z 1024.
MCBP (FAB/NOBA, внешний эталон CSI/Гли): [M + H] +, m/z 986,3646 (вычисленное значение: 986,3633, отклонение Δ = 1,3 м.д.).
1H ЯМР (CDCl3),: δ: 1,18 (3H, с), 1,20 (3H, с), 1,30 (3H, с), 1,75 (3H, с), 1,84 (H, м), 2,09 (3H, с), 2,11 (3H, с), 2,16 (3H, с), 2,24 (H, д), 2,37 (H, д), 2,45 (3H, с), 2,80 (H, м), 3,68 (H, д), 4,08 - 4,33 (5H, м, перекрытие), 4,65 (2H, с), 4,96 (H, д), 5,43 (H, д), 5,69 (H, д), 5,98 (H, дд), 6,26 (H, т), 6,55 (H, с), 7,00 (H, д), 7,32 - 7,61 (11H, м, перекрытие), 7,73 (2H, дд), 8,11 (2H, дд).
Альтернативная процедура
2'-O-(Этоксикарбонил(паклитаксела (2,260 г, 2,4406 ммоль) растворяли в безводном диметилсульфоксиде (6 мл), и добавляли одной порцией при комнатной температуре уксусный ангидрид (6 мл). Реакцию контролировали путем HPLC (аналитическая колонка C18; 60% ацетонитрила - 40% 10 мМ аммонийфосфатного буфера, pH 6). Через 30 ч раствор разбавляли этилацетатом (250 мл) и промывали насыщенным водным бикарбонатом (3 раза), а затем водой и рассолом. После высушивания над сульфатом магния и фильтрования неочищенный продукт хроматографировали на силикагеле (40% этилацетата в гексане) и получили указанное в заголовке соединение в виде белой пены (2,030 г, 91%) с чистотой 90% при определении путем HPLC (жидкостная хроматография высокого давления). Часть продукта дополнительно очищали на второй колонке (5% ацетонитрила в дихлорметане), получив в результате материал, имеющий чистоту приблизительно 97% при определении путем HPLC.
Альтернативный способ получения 2'-O-(этоксикарбонил)-7-O- метилтиометилпаклитаксела
2'-O-(Этоксикарбонил)паклитаксел (4,170 г, 4,503 ммоль) растворяли в безводном ацетонитриле (68 мл) при -40oC, и добавляли диметилсульфид (3,2 мл, 44,10 ммоль), а затем бензоилпероксид (4,400 г, 18,24 ммоль). Смесь помещали в ванну со льдом и перемешивали при 0oC, а ход реакции контролировали путем тонкослойной хроматографии (40% этилацетата в гексане). Через 3 ч, не обнаружив исходного материала, раствор обрабатывали путем добавления этилацетата (250 мл) и насыщенного водного раствора бикарбоната натрия (100 мл). Затем органическую фазу промывали бикарбонатом, водой и рассолом, после чего высушивали над сульфатом магния и фильтровали. Остаток очищали путем флеш-хроматографии на силикагеле (4% ацетонитрила в дихлорметане) и получили в результате указанное в заголовке соединение в виде белой пены (2,571 г, выход 58%). Чистоту этого образца оценивали путем HPLC и она была более 97%. ЯМР - спектр был идентичен тому, что представлен выше.
Альтернативная процедура получения 2'-O-(этоксикарбонил)-7-O-метилтиометилпаклитаксела
2'-O-(Этоксикарбонил)паклитаксел (49,3 г, 53,2 ммоль) помещали в высушенную над пламенем одногорлую колбу емкостью 1 л и растворяли в сухом ацетонитриле (500 мл) при комнатной температуре. Посредством шприца быстро добавляли метилсульфид (39,1 мл, 0,532 ммоль). Перемешанную реакционную смесь охлаждали до -16oC в ванне с солью и льдом, и к смеси добавляли одной порцией твердый бензоилпероксид (51,6 г. 0,213 ммоль). (Для полного осуществления реакции необходимы полных четыре эквивалента). Продолжали перемешивание в течение 30 минут, в течение которых температура выросла до примерно -10oC. В продолжение всего этого периода реакционная среда оставалась гетерогенной (бензоилпероксид не растворился полностью). Охлаждающую ванну заменяли водой со льдом, в результате чего температура повысилась до 0oC, и оставшийся бензоилпероксид растворялся примерно 5 минут после повышения температуры. После перемешивания в течение еще 2,5 часов при 0oC реакция, как показала тонкослойная хроматография, была завершена. Путем удаления раствора на роторном испарителе уменьшали объем раствора до примерно 200 мл, после чего перенесли раствор в делительную воронку, где его промывали гептаном (5 х 500 мл). Слой ацетонитрила разбавляли этилацетатом (1,5 л) и промывали смесью (3:1) насыщенного NaHCO3 и 5% K2CO3 (объем/объем) (2 х 500 мл), насыщенным NaHCO3 (2 х 500 мл), полунасыщенным рассолом (1 х 500 мл) и рассолом (1 х 500 мл), высушивали (MgSO4) и, удалив растворитель в вакууме, получили 67,0 г неочищенного продукта. Его растворяли в ацетоне (200 мл), нагревали до 40oC в водяной бане, после чего добавляли по каплям с перемешиванием гексаны, до тех пор пока не заметили помутнение (400 мл). Кристаллизующуюся смесь оставляли стоять в течение 3 часов при комнатной температуре, а затем переносили в холодную камеру (0oC), где ее держали всю ночь (16 часов). Образовался толстый слой слежавшегося осадка. Твердое вещество собирали путем фильтрования и промывали смесью (3:1, объем/объем) гексаны-ацетон (2 х 50 мл). Полученные белые кристаллы высушивали на вакуумном фильтре в течение 1 часа и затем в вакууме (примерно 0,5 мм рт. ст.) всю ночь, и в результате получили 47,5 г (выход 91%) указанного в заголовке соединения (индекс однородности при измерении путем HPLC был равен 94,8%).
(c) Получение 2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксела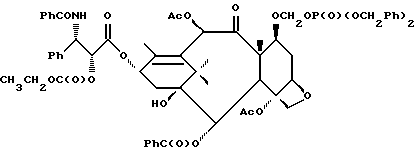
К смеси 2'-O-(этоксикарбонил)-7-O-метилтиометилпаклитаксела (5,677 г, 5,76 ммоль) и молекулярных сит  (5,7 г) в метиленхлориде (100 мл) при комнатной температуре добавляли раствор N-иодсукцинимида (1,953 г, 8,65 ммоль) и дибензилфосфата (2,41 г, 8,65 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали 40 мин при комнатной температуре. После этого периода реакция, как показала тонкослойная хроматография, была завершена. Реакционную смесь фильтровали через целит, и фильтрат концентрировали в вакууме, получив в результате буроватый остаток, который разбавляли этилацетатом (800 мл), органическую фазу промывали 1%-ным Na2SO3 (2 x 80 мл), а затем 5%-ным рассолом (2 x 50 мл). Органическую фазу концентрировали в вакууме и высушивали. Хроматография полученного остатка (50 - 60% этилацетата в гексанах) дала целевое указанное в заголовке соединение (6,23 г, 89%).
(5,7 г) в метиленхлориде (100 мл) при комнатной температуре добавляли раствор N-иодсукцинимида (1,953 г, 8,65 ммоль) и дибензилфосфата (2,41 г, 8,65 ммоль) в тетрагидрофуране. Реакционную смесь перемешивали 40 мин при комнатной температуре. После этого периода реакция, как показала тонкослойная хроматография, была завершена. Реакционную смесь фильтровали через целит, и фильтрат концентрировали в вакууме, получив в результате буроватый остаток, который разбавляли этилацетатом (800 мл), органическую фазу промывали 1%-ным Na2SO3 (2 x 80 мл), а затем 5%-ным рассолом (2 x 50 мл). Органическую фазу концентрировали в вакууме и высушивали. Хроматография полученного остатка (50 - 60% этилацетата в гексанах) дала целевое указанное в заголовке соединение (6,23 г, 89%).
Масс-спектр (FAB/NOBA, NaI, KI): [M + Na]+, m/z 1238; [M + K]+, m/z 1254.
Масс-спектр высокого разрешения (FAB/NOBA, внешний эталон CSI/гли): [M + Na]+, m/z 1216, 4291
(C65H71NO20P, вычисленное значение: 1216, 4307, отклонение Δ = 1,3 м.д. ).
1H ЯМР (CDCl3), δ: 1,18 (3H, с), 1,21 (3H, с), 1,30 (3H, т), 1,67 (6H, с), 1,80 (H, с), 1,93 (H, м), 1,99 (3H, д), 2,18 (3H, с), 2,23 (H, м), 2,38 (H, м), 2,45 (3H, с), 2,80 (H, м), 3,86 (H, д), 4,14 - 4,32 (5H, перекрывающиеся мультиплеты), 4,88 (H, д), 5,00 - 5,07 (4H, перекрывающиеся мультиплеты), 5,42 (H, д), 5,68 (H, д), 5,96 (H, дд), 6,26 (H, т), 6,33 (H, с), 6,95 (H, д), 7,30 - 7,61 (11H, перекрывающиеся мультиплеты), 7,75 (2H, дд), 8,12 (2H, дд).
Альтернативная процедура
К раствору 2'-O-(Этоксикарбонил)-7-O-метилтиометилпаклитаксела (350 мг, 0,355 ммоль) в безводном тетрагидрофуране (8 мл) добавляли раствор N-иодсукцинимида (120 мг, 0,532 ммоль) и дибензилфосфата (148 г, 0,532 ммоль) в тетрагидрофуране (5 мл). Реакцию контролировали путем HPLC (колонка C18; 70% ацетонитрила, 30% 10 мМ аммонийфосфата, pH 6). Через 2 часа реакция была завершена, о чем свидетельствовало менее 5% обнаруженного исходного материала. Раствор разбавляли этилацетатом (75 мл) и промывали 1%-ным водным раствором бисульфита (2 x 50 мл) и рассолом (50 мл). После быстрого высушивания над сульфатом магния и фильтрования выпаривали растворитель. Флеш-хроматография на силикагеле (45% этилацетата в гексане) дала указанное в заголовке соединение в виде белой пены (281 мг, 65%). Анализ путем HPLC показал чистоту приблизительно 95%.
Альтернативная процедура
Раздробленные молекулярные сита  помещали в высушенную над пламенем одногорлую колбу емкостью 1 л, которую затем соединяли с вакуумной линией (примерно 0,5 мм рт.ст.). Сита нагревали с помощью прибора для сушки горячим воздухом в течение примерно 10 мин с встряхиванием вручную. После охлаждения под вакуумом в колбу вводили аргон и добавляли 2'-O-(Этоксикарбонил)-7-O-метилтиометилпаклитаксел (37,5 г, 38,03 ммоль), а затем дибензилфосфат (14,3 г, 53,24 ммоль) и тетрагидрофуран (400 мл). Гетерогенную смесь интенсивно перемешивали 15 минут при комнатной температуре посредством магнитной мешалки. В отдельной высушенной над пламенем колбе растворяли N-иодсукцинимид (10,7 г, 47,54 ммоль) в тетрагидрофуране (50 мл) под аргоном. (Во время приготовления раствора NIS переноса жидкости и хода реакции сосуды покрывали алюминиевой фольгой для защиты от света). Затем к реакционной смеси медленно (10 мин) добавляли посредством шприца приготовленный раствор. Колбу, содержащую NIS, промывали 5 мл ТГФ и переносили в реакционную смесь, которую затем перемешивали 2 часа при комнатной температуре. Анализ путем тонкослойной хроматографии показал отсутствие исходного материала. Раствор темно-красного цвета фильтровали через слой целита непосредственно в интенсивно перемешиваемую двухфазную смесь, содержащую этилацетат (500 мл) 10%-ный водный раствор тиосульфата натрия (300 мл) и насыщенный раствор бикарбоната натрия (200 мл). Через несколько секунд красный цвет исчез и раствор стал бесцветным. Слой целита промывали этилацетатом (примерно 100 мл) и оба слоя жидкости переносили в делительную воронку. Органический слой разбавляли 1 литром этилацетата, разделяли слои, и органический слой промывали смесью насыщенного NaHCO3 и 5%-ного K2CO3, а затем насыщенным NaHCO3 (2 x 500 мл), полунасыщенным рассолом (1 x 500 мл) и рассолом (1 x 500 мл). Экстракт высушивали безводным MgSO4 и фильтровали. Затем его обрабатывали 5,0 г нейтрального норита (древесный уголь) путем перемешивания при комнатной температуре в течение 15 минут. Опять фильтровали его через слой целита, и, удалив растворитель при пониженном давлении, получили 52 г неочищенного продукта. Его растворяли в смеси толуола и метиленхлорида (280 мл/25 мл), и к полученному раствору добавляли по каплям гексаны (20 мл). После 3 часов выдерживания при комнатной температуре кристаллизующуюся смесь всю ночь держали при 0oC. На стенках колбы образовалось твердое вещество бледно-желтого цвета. После декантирования маточного раствора остаток тритурировали с толуолом (50 мл), фильтровали, промывали толуолом и высушивали на вакуумном фильтре в течение 30 минут. Затем переносили его в эксикатор с драйеритом, после чего высушивали в вакууме (примерно 0,5 мм рт.ст.) в течение 4 часов, получив в результате 24,4 г (выход 53%) указанного в заголовке соединения (показатель гомогенности при определении путем HPLC был равен 95,9%). Маточный раствор упаривали досуха, тритурировали с тоуолом (100 мл), фильтровали, промывали толуолом и высушивали на вакуумном фильтре в течение 30 минут. После высушивания в эксикаторе, как описано выше, получили 12,5 г (выход 27%) того же самого продукта (показатель гомогенности при определении путем HPLC был равен 97,1%).
помещали в высушенную над пламенем одногорлую колбу емкостью 1 л, которую затем соединяли с вакуумной линией (примерно 0,5 мм рт.ст.). Сита нагревали с помощью прибора для сушки горячим воздухом в течение примерно 10 мин с встряхиванием вручную. После охлаждения под вакуумом в колбу вводили аргон и добавляли 2'-O-(Этоксикарбонил)-7-O-метилтиометилпаклитаксел (37,5 г, 38,03 ммоль), а затем дибензилфосфат (14,3 г, 53,24 ммоль) и тетрагидрофуран (400 мл). Гетерогенную смесь интенсивно перемешивали 15 минут при комнатной температуре посредством магнитной мешалки. В отдельной высушенной над пламенем колбе растворяли N-иодсукцинимид (10,7 г, 47,54 ммоль) в тетрагидрофуране (50 мл) под аргоном. (Во время приготовления раствора NIS переноса жидкости и хода реакции сосуды покрывали алюминиевой фольгой для защиты от света). Затем к реакционной смеси медленно (10 мин) добавляли посредством шприца приготовленный раствор. Колбу, содержащую NIS, промывали 5 мл ТГФ и переносили в реакционную смесь, которую затем перемешивали 2 часа при комнатной температуре. Анализ путем тонкослойной хроматографии показал отсутствие исходного материала. Раствор темно-красного цвета фильтровали через слой целита непосредственно в интенсивно перемешиваемую двухфазную смесь, содержащую этилацетат (500 мл) 10%-ный водный раствор тиосульфата натрия (300 мл) и насыщенный раствор бикарбоната натрия (200 мл). Через несколько секунд красный цвет исчез и раствор стал бесцветным. Слой целита промывали этилацетатом (примерно 100 мл) и оба слоя жидкости переносили в делительную воронку. Органический слой разбавляли 1 литром этилацетата, разделяли слои, и органический слой промывали смесью насыщенного NaHCO3 и 5%-ного K2CO3, а затем насыщенным NaHCO3 (2 x 500 мл), полунасыщенным рассолом (1 x 500 мл) и рассолом (1 x 500 мл). Экстракт высушивали безводным MgSO4 и фильтровали. Затем его обрабатывали 5,0 г нейтрального норита (древесный уголь) путем перемешивания при комнатной температуре в течение 15 минут. Опять фильтровали его через слой целита, и, удалив растворитель при пониженном давлении, получили 52 г неочищенного продукта. Его растворяли в смеси толуола и метиленхлорида (280 мл/25 мл), и к полученному раствору добавляли по каплям гексаны (20 мл). После 3 часов выдерживания при комнатной температуре кристаллизующуюся смесь всю ночь держали при 0oC. На стенках колбы образовалось твердое вещество бледно-желтого цвета. После декантирования маточного раствора остаток тритурировали с толуолом (50 мл), фильтровали, промывали толуолом и высушивали на вакуумном фильтре в течение 30 минут. Затем переносили его в эксикатор с драйеритом, после чего высушивали в вакууме (примерно 0,5 мм рт.ст.) в течение 4 часов, получив в результате 24,4 г (выход 53%) указанного в заголовке соединения (показатель гомогенности при определении путем HPLC был равен 95,9%). Маточный раствор упаривали досуха, тритурировали с тоуолом (100 мл), фильтровали, промывали толуолом и высушивали на вакуумном фильтре в течение 30 минут. После высушивания в эксикаторе, как описано выше, получили 12,5 г (выход 27%) того же самого продукта (показатель гомогенности при определении путем HPLC был равен 97,1%).
(d) Получение 2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела и его мононатриевой, монокалиевой, триэтиламиновой, аргининовой, лизиновой, этаноламиновой, N-метилглюкаминовой и триэтаноламиновой солей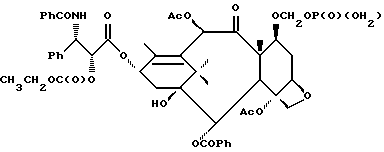
К раствору 2'-O-(этоксикарбонил)-7-O-дибензилфосфоно-оксиметилпаклитаксела (1,23 г, 1,01 ммоль) в сухом этилацетате (40 мл) добавляли 10%-ный палладий на угле (428 мг, 10%, 0,404 ммоль). Реакционную смесь подвергали гидрогенизации при 60 фунтах на кв. дюйм (400 кПа) с непрерывным встряхиванием в течение 24 часов. Твердое вещество отфильтровывали через целит, после чего целит несколько раз промывали этилацетатом. Фильтрат концентрировали и получили свободную кислотную форму указанного в заголовке с соединения (1,01 г, 80%-ная чистота при определении путем HPLC). На следующей стадии удаляли примеси путем препаративной хроматографии на колонке C-18.
Масс-спектр (FAB/NOBA, NaI, KI): [M + Na]+, m/z 1058; [M + K]+, m/z 1074; [M + 2Na - H]+, m/z 1080; [M + Na + K - H]+, m/z 1096; [M + 2K - H]+, m/z 1112.
Масс-спектр высокого разрешения (FAB/NOBA, внешний эталон CSI/Гли): [M + Na]+, m/z 1058, 3163
(C51H58NO20PNa, вычисленное значение: 1058, 3188; отклонение Δ = 2,3 м. д.).
1H ЯМР (ацетон-d6/D2O), δ: 1,13 (3H, с), 1,21 (3H, с), 1,66 (3H, с), 1,87 (H, м), 1,93 (3H, с), 2,14 (3H, с), 2,18 (H, м), 2,44 (3H, с), 2,95 (H, м), 3,81 (H, д), 4,12 (2H, с), 4,15 - 4,27 (3H, перекрывающиеся мультиплеты), 4,92 - 4,99 (2H, перекрывающиеся широкие мультиплеты), 5,15 (H, шс), 5,48 (H, д), 5,61 (H, д), 5,84 (H, дд), 6,07 (H, т), 6,96 (H, с), 7,25 (H, т), 7,28 - 7,69 (10H, перекрывающиеся мультиплеты), 7,89 (2H, дд), 8,08 (2H, дд), 8,86 (H, д).
Альтернативная процедура
2'-O-(Этоксикарбонил)-7-O-(дибензилфосфонооксиметил)- паклитаксел (490 мг, 0,402 ммоль) в этилацетате (20 мл) гидрогенизировали в аппарате Парра для встряхивания при 60 фунтах на кв. дюйм (400 кПа) в присутствии палладия на древесном угле (10%, масса/масса, 150 мг). Контроль осуществляли путем тонкослойной хроматографии и жидкостной хроматографии высокого давления (HPLC). Когда не обнаруживали (26 ч) больше ни исходного материала, ни промежуточного продукта (предпочтительно монобензилфосфата), фильтровали суспензию через целит и упаривали ее досуха. Анализ путем HPLC показал чистоту 88 - 92%.
Альтернативная процедура
Описанную ниже триэтиламиновую соль 2'-O-(этоксикарбонил)- 7-O-фосфонооксиметилпаклитаксела (5,4 г, 4,75 ммоль) интенсивно разделяли между этилацетатом (100 мл) и 5%-ным NaHSO4 (45 мл) с перемешиванием при 0oC в течение 30 мин. Водный слой отделяли и экстрагировали этилацетатом (20 мл). Объединенный слой этилацетата промывали полунасыщенным рассолом (25 мл), рассолом (25 мл х 2), высушивали над NaSO4 и фильтровали, получив в результате раствор кислоты (примерно 4,75 ммоль) в этилацетате (примерно 150 мл). Этот раствор затем концентрировали досуха на роторном испарителе и получили 3,75 г указанного в заголовке соединения в виде свободной кислотной формы с 95%-ным выходом.
Мононатриевую соль получали следующим образом.
Пробу 2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела (1,6 г, 1,55 ммоль) растворяли в ацетонитриле (30 мл), подвергнув ультразвуковой обработке. Это раствор разбавляли водой (30 мл) и добавляли к нему 1,1 М раствор NaHCO3 (2,11 мл, 2,32 ммоль), поочередно встряхивали и обрабатывали ультразвуком, чтобы получить раствор (5 - 20 мин). Раствор несколько молочного цвета наносили на колонку C-18, промывали водой в количестве двух объемов колонки, а затем элюировали мононатриевую соль 25% ацетонитрила в воде. Соответствующие фракции объединяли, выпаривали ацетонитрил и лиофилизировали водную фазу, получив в результате мононатриевую соль указанного в заголовке соединения (850 мг, примерно 50%) с чистотой, равной 97% при определении путем HPLC.
Масс-спектр (FAB/NOBA, NaI, KI); [M + Na]+, m/z 1180.
Масс-спектр высокого разрешения (FAB / NOBA, внешний эталон CSI/Гли): [M + Na] +, m/z 1080, 2968 (C51H57NO20PNa2), вычисленное значение: 1080, 3007; отклонение D=3,6 м.д.).
Элементный анализ: C 52,65 (выч. 56,72), H 5,06 (выч. 5,23), N 1,20 (выч. 1,30), Na 2,74 (выч. 2,12).
ИК-спектр (KBr): 3430, 3066, 2988, 1746, 1722, 1660, 1602, 1582, 1526, 1488, 1452, 1374, 1246, 1178, 1150, 1108, 1070, 1052, 1026, 1002, 966, 912, 834, 792, 776, 710, 628, 538 см-1.
1H ЯМР (ДМСО-d6, D2O-ацетон-d6), δ: 1,10 (6H, c), 1,23 (3H, т), 1,64 (3H, c), 1,70 (H, м) 1,90 (3H, с), 1,99 (H, м), 2,14 (3H, с), 2,37 (3H, с), 2,98 (H, м), 3,74 (H, д), 4,07 (2H, с), 4,13 - 4,26 (3H, м, перекрывающийся), 4,80 (H, шдд), 4,97 (H, д), 5,09 (H, шт), 5,44 (H, д), 5,55 (H, д), 5,99 (H, т), 6,34 (H, с), 7,22 (H, т), 7,43 - 7,69 (10H, м, перекрывающийся), 7,92 (2H, д), 8,06 (2H, дд).
Натриевую соль получали следующим образом.
Неочищенный 2'-O-(этоксикарбонил)-7-O-фосфонооксиметилпаклитаксел (89%; 70 мг, 0,060 ммоль) в этилацетате (2 мл) обрабатывали раствором этилгексаноата натрия (87,5 мМ в этилацетате, 1,0 мл, 0,0875 ммоль) при комнатной температуре с перемешиванием. После перемешивания при комнатной температуре в течение 1 ч добавляли гексан (1,2 мл) до точки помутнения. После выстаивания при -20oC в течение 2 ч, отфильтровывали (с некоторым затруднением, очень медленно) тонкий аморфный порошок через тонкую фильтровальную бумагу и получили 45 мг (70%) натриевой соли. Она имела чистоту (определенную путем HPLC) 95,2% и содержала небольшое количество этилгексановой кислоты (ЯМР).
Триэтаноламиновую соль получали следующим образом.
Полученный в результате гидрогенизации (89% по HPLC 2'-O-(этоксикарбонил)-7-O-фосфонооксиметилапаклитаксел (0,69 г, 0,593 ммоль после поправки на примеси) растворяли в этилацетате (10 мл) и медленно перемешивали, добавляя при этом по каплям раствор триэтаноламина (0,11 М в этилацетате, с использованием 5,1 мл, 0,95 эквив.). Молочного цвета раствор, полученный при этой процедуре, дигерировали при 0oC в течение 2 ч, после чего фильтровали через тонкую фильтровальную бумагу, промывая холодным этилацетатом. Получили 499 мг (80%) аморфного тонкого неэлектростатического порошка, который высушивали всю ночь в вакууме. HPLC показала 96,6%-ную чистоту (C-18, 45% 5 мМ Q12+ 10 мМ аммонийфосфата с pH 6,55% ацетонитрила). ЯМР-спектр (D2O/ацетон/ДМСО) показал следы этилацетата и отсутствие других четкообозначенных примесей. Производили анализ на 2-3-х гидрат.
Затем очищали триэтаноламиновую соль, полученную ранее в результате другого эксперимента, следующим образом. Триэтаноламинову соль (приблизительно 2 г) растворяли в примерно 30% ацетонитрила в воде. Этот раствор элюировали при небольшом давлении азота через колонку C-18 (Бейкербонд) с градиентом 20-40% ацетонитрила в воде. Улавливали фракции, содержавшие требуемую триэтаноламиновую соль, и удаляли ацетонитрил путем выпаривания на роторном испарителе при пониженном давлении. Водные растворы замораживали и лиофилизировали всю ночь, получив в результате 1,4 г триэтаноламиновой соли с чистотою 97,5%.
Триэтаноламиновая соль может быть также получена следующим образом.
2'-O-(Этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела триэтиламиновую соль (3,0 г, 2,64 ммоль) распределяли между этилацетатом (60 мл) и 5%-ным NaHSO4 (30 мл) с интенсивным перемешиванием при 0oC в течение 15 минут. Водный слой отделяли и экстрагировали этилацетатом (10 мл). Объединенный слой этилацетата промывали рассолом (15 мл), высушивали над Na2SO4, фильтровали, и получили раствор кислоты (примерно 2,64 ммоль) в этилацетате (примерно 70 мл). К этому раствору при комнатной температуре добавляли по каплям при интенсивном перемешивании N(CH2CH2OH)3 (0,35 мл, 2,64 ммоль) в течение 5 минут. Полученную суспензию перемешивали еще 1 ч, после чего ее фильтровали, промывали этилацетатом (15 мл х 2), высушивали в вакууме, и получили в результате 2,8 г триэтаноламиновой соли с выходом 89%. Анализ путем HPLC показал, что индекс однородности равен 98,7%; т.пл.: выше 157oC, с разложением.
Элементный анализ: вычислено для C56H73N2O23P•2,0 H2O•0,3 этилацетата: C 55,60; H 6,48; N 2,27; KF (H2O) 2,92. Найдено: C 55,94; H 6,59; N 2,43; KF (H2O) 3,50.
Триэтиламиновую соль получали следующим образом.
К раствору 2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксела (10 г, 8,23 ммоль) в этилацетате (350 мл) при комнатной температуре добавляли 10% палладий на угле (2 г, 20%-ая загрузка). Полученную суспензию дегазировали путем отсасывания воздуха с последующей продувкой аргоном. Этот процесс повторяли еще два раза. Затем аргон заменяли водородом, после чего осуществляли ту же самую процедуру дегазирования. Полученную суспензию перемешивали под давлением водорода из баллона (2-3 фунта на кв. дюйм, т.е. 0,14-0,21 кг/см2) в течение 16 ч при комнатной температуре с интенсивным перемешиванием. Отсасывали водород и заменяли его аргоном три раза с последующим осуществлением процедуры дегазации. Полученную суспензию фильтровали через слой целита. К этому гомогенному фильтрату медленно добавляли Et3N (8,23 ммоль, 1,14 мл) в течение 5 минут при интенсивном перемешивании. Полученную тонкодисперсную белую суспензию перемешивали еще 30 минут. Фильтровали ее через воронку со спеченным пористым фильтром с пористостью E. Остаток от фильтрования высушивали в вакууме (1 мм рт.ст.) в течение 16 ч, и получали в результате 8,22 г указанной в заголовке триэтиламиновой соли с выходом 88%. Анализ путем HРLC дал показатель гомогенности 97,4%; т.пл.: выше 178oC с разложением.
Элементный анализ: вычислено для C57H73N2O20P • 4,5 H2O: C 56,19; H 6,79; N 2,30; KF (H2O) 6,65.
Вычислено: C 56,33; H 6,87; N 2,32; KF (H2O) 7,96.
Альтернативная процедура получения триэтиламиновой соли.
В колбу емкостью 250 мл добавляли 2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (5,67 г, 4,66 ммоль) и растворяли его в этилацетате (150 мл). Колбу снабжали трехходовым клапаном, одно из присоединений которого соединяли с вакуумной линией, а другое - с линией аргона. Путем использования клапана в колбе создавали частичное разрежение, а затем продували ее аргоном. Этот процесс повторяли еще два раза. В колбу добавляли палладий на активированном угле (10% Pd) (0,85 г). Линию аргона, присоединенную к трехходовому клапану, заменяли баллоном, заполненным водородом. Посредством клапана в колбе создавали разрежение, а затем продували ее водородом. Это процесс повторяли еще четыре раза. Полученную смесь перемешивали при комнатной температуре в среде водорода из баллона на протяжении всей ночи. Анализ путем тонкослойной хроматографии через 17 часов после начального воздействия водородом показал отсутствие исходного материала. Баллон с водородом, присоединенный к трехходовому клапану, заменяли линией аргона. Посредством клапана создавали в колбе частичное разрежение, после чего продували ее аргоном. Этот процесс повторяли еще два раза. Содержимое колбы подвергали вакуумному фильтрованию через слой целита. Целит промывали этилацетатом (2 х 10 мл). К перемешиваемому фильтрату добавляли триэтиламин (0,650 мл, 4,66 ммоль). Полученную суспензию перемешивали при комнатной температуре два часа, после чего уменьшали объем до примерно 150 мл путем упаривания на роторном испарителе. Твердое вещество отфильтровывали, промывали этилацетатом (2 х 10 мл) и высушивали под вакуумом, получив в результате 4,76 г (выход 90%) указанной в заголовке триэтиламиновой соли в виде белого порошка (анализ путем HPLC показал, что индекс гомогенности равен 96,6%).
Альтернативная процедура получения триэтиламиновой соли.
В колбу емкостью 250 мл вводили 2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (5,17 г, 4,25 ммоль) и растворяли его в этилацетате (150 мл). Колбу снабжали трехходовым клапаном, одно из присоединений которого соединяли с вакуумной линией, а другое - с линией аргона. Пользуясь клапаном, создавали в колбе частичное разрежение, после чего продували аргоном. Этот процесс повторяли еще два раза. Добавляли в колбу палладий на активированном угле (10% Pd, 0,86 г). Линию аргона, присоединенную к трехходовому клапану заменяли баллоном, заполненным водородом. Посредством клапана создавали в колбе частичное разрежение, после чего продували водородом. Это процесс повторяли еще пять раз. Полученную смесь перемешивали при комнатной температуре в среде водорода из баллона на протяжении всей ночи. Анализ путем тонкослойной хроматографии через 16 часов после начального воздействия водородом показал отсутствие исходного материала. Баллон с водородом, присоединенный к трехходовому клапану, заменяли линией аргона. Посредством клапана создавали в колбе частичное разрежение, после чего продували аргоном. Этот процесс повторяли еще два раза. Содержимое колбы подвергали вакуумному фильтрованию через слой целита. Целит промывали этилацетатом (4 х 10 мл). К перемешиваемому фильтрату добавляли триэтиламин (0,590 мл, 4,25 ммоль). Полученную суспензию перемешивали при комнатной температуре один час, после чего уменьшали объем до примерно 140 мл путем упаривания на роторном испарителе. Твердое вещество отфильтровывали, промывали этилацетатом (10 мл) и высушивали под вакуумом, получив в результате 4,46 г (выход 92%) указанной в заголовке триэтиламиновой соли в виде белого порошка (определенный путем HPLC-анализа показатель гомогенности составлял 96.7%).
Лизиновую соль получали следующим образом.
К суспензии 10% палладия на угле (20%-ная загрузка, 3 г) в этаноле (600 мл, крепость 200) при 0oC добавляли порциями 2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (15,0 г, 12,34 ммоль). Полученную суспензию дегазировали путем отсасывания воздуха и продувки аргоном. Этот процесс повторяли еще два раза. Затем аргон заменяли водородом, после чего выполняли такую же процедуру дегазирования при интенсивном перемешивания при 0oC в течение 2 ч. Удаляли охлаждающую ванну, и реакционную смесь перемешивали при температуре окружающей среды еще 4,5 часа. Реакционную смесь дегазировали путем отсасывания водорода и продувки аргоном три раза, фильтровали ее под аргоном через слой целита. К полученному фильтрату медленно добавляли раствор лизина (1,63 г, 0,94 эквив.) в смеси вода:этанол (1:1, крепость 200, 20 мл) в течение 5 минут при интенсивном перемешивании. К полученной белой суспензии добавляли дистиллированную воду (110 мл), и перемешивали суспензию 30 минут. Нагревали ее примерно до 55oC. Полученный гомогенный раствор держали в установке с масляной ванной при 50oC и медленно охлаждали до комнатной температуры 16 часов и до 4oC в течение 3 часов, фильтровали его и, подвергнув вакуумному высушиванию в течение 16 часов, получили 11,8 г (выход примерно 80%) лизиновой соли, имеющей показатель однородности 99,0% при определении путем HPLC; т.пл. выше 170oC с разложением.
Элементный анализ: вычислено для C57H72N3O22P • 8,0 H2O:
C 51,62; H 6,69; N 3,17; KF (H2O) 10,87.
Найдено: C 51,76; H 6,57; N 3,48; KF (H2O) 11,42.
Этаноламиновую соль получали следующим образом
2'-O-(Этоксикарбонил)-7-O-фосфонооксиметилпаклитаксела триэтиламиновую соль (3,0 г, 2,64 ммоль) распределяли между этилацетатом (60 мл) и 5%-ным NaHSO4 (30 мл) с интенсивным перемешиванием при 0oC в течение 15 минут. Водный слой отделяли и экстрагировали этилацетатом (15 мл). Объединенный слой этилацетата промывали рассолом (15 мл), высушивали над Na2SO4, фильтровали, и в результате получили раствор свободной кислоты (примерно 2,64 ммоль) в этилацетате (примерно 70 мл). К этому раствору при комнатной температуре добавляли по каплям с интенсивным перемешиванием раствор H2NCH2CH2OH (0,15 мл, 2,64 ммоль) в этилацетате (5 мл) в течение 5 минут. Полученную суспензию перемешивали еще 1 час, после чего ее фильтровали, промывали этилацетатом (15 мл х 2) и высушивали в вакууме, в результате чего получили 2,6 г указанной в заголовке этаноламиновой соли с выходом 89%. Показатель однородности при определении путем HPLC-анализа был равен 97,8%; т.пл. выше 130oC с разложением.
Элементный анализ: вычислено для C53H65N2O21P • 2,5 H2O:
C 55,73; H 6,18; N 2,45; KF (H2O) 3,94.
Найдено: C 55,76; H 6,39; N 2,45; KF (H2O) 6,00.
Аргининовую соль получали следующим образом.
К суспензии 10% палладия на угле (загрузка 20%, 6 г) в этаноле (900 мл, крепость 200) при 0oC добавляли по частям 2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (30,0 г, 24,69 ммоль). Полученную суспензию дегазировали путем отсасывания воздуха и продувки аргоном. Этот процесс повторяли еще два раза. Затем аргон заменяли водородом, после чего опять осуществляли описанную выше процедуру дегазирования при интенсивном перемешивании. Полученную смесь перемешивали при 0oC в течение 2 часов. Убирали охлаждающую ванну, и перемешивали реакционный раствор при окружающей температуре еще 24 часа. Реакционную смесь дегазировали путем отсасывания водорода и продувки аргоном, осуществляя указанную выше процедуру три раза. Фильтровали ее под аргоном через слой целита. Фильтрат разделяли на две равные части, и к каждой части добавляли этанол (190 мл, крепость 200). К одной части (примерно 630 мл) медленно добавляли раствор аргинина (2,0 г, 0,94 эквив. ) в смеси вода: этанол (2:1, крепость 200, 20 мл) в течение 5 минут при интенсивном перемешивании. К полученной белой суспензии добавляли дистиллированную воду (100 мл) и перемешивали 30 минут, после чего нагревали до примерно 60oC. Фильтровали ее горячей, и фильтрат держали в установке с масляной ванной при 50oC, позволив ему охладиться до комнатной температуры, и держали при комнатной температуре 2 часа и при 4oC 2 часа. Фильтровали его и промывали холодной смесью 3% воды в этаноле (100 мл) и высушивали отсасыванием в течение 16 часов, в результате чего получили 12,95 г (примерно 86%-ный выход) указанной в заголовке аргининовой соли с показателем однородности, равным 96,7%.
Этот материал (12,95 г) растворяли в смеси 15% воды в этаноле (примерно 700 мл) при 55oC. Раствор охлаждали и держали при 30oC в течение 3,5 часов, при комнатной температуре в течение 16 часов и при 4oC в течение 3 часов. Полученные кристаллы отфильтровывали, промывали холодной смесью 2% воды в этаноле (50 мл х 2), подвергали высушиванию отсасыванием в течение 4 часов и затем высушивали в вакууме (1 мм рт.ст.) 16 часов, в результате чего получили 10,2 г (80%-ный выход) указанной в заголовке аргининовой соли (показатель однородности 98,5%); т.пл.: выше 176oC с разложением.
Элементный анализ: вычислено для C57H72N5O22P • 6,4 H2O:
C 51,65; H 6,45; N 5,28; KF (H2O) 8,7.
Найдено: C 51,86; H 6,65; N 5,53; KF (H2O) 8,72.
N-Метилглюкаминовую соль получали следующим образом.
К суспензии 10% палладия на угле (20%-ная загрузка, 6 г) в метаноле (900 мл, крепость 200) при 0oC добавляли частями 2'-O-(этоксикарбонил)-7-O-дибензилфосфонооксиметилпаклитаксел (30,0 г, 24,69 ммоль). Полученную суспензию дегазировали путем отсасывания воздуха и продувки аргоном. Этот процесс повторяли еще два раза. Затем аргон заменяли водородом, после чего осуществляли вышеописанную процедуру при интенсивном перемешивании. Полученную смесь перемешивали при 0oC 2 часа. Убирали охлаждающую ванну, и реакционный раствор перемешивали при температуре окружающей среды еще 24 часа. Реакционную смесь дегазировали путем отсасывания водорода и продувки аргоном, следуя вышеописанной процедуре три раза. Фильтровали ее под аргоном через слой целита. Фильтрат разделяли на две равные части, и к каждой части добавляли этанол (190 мл, крепость 200). К одной части (примерно 600 мл) медленно добавляли раствор N-метилглюкамина (2,24 г, 0,94 эквив.) в смеси вода:этанол (1: 1, крепость 200, 20 мл) в течение 5 минут при интенсивном перемешивании. К полученной белой суспензии добавляли дистиллированную воду (100 мл), и суспензию перемешивали 30 минут и затем нагревали до примерно 49oC. Прозрачный гомогенный раствор держали в установке с масляной ванной при 50oC, позволив ему охладиться до комнатной температуры, и держали при комнатной температуре 2 часа и при 4oC 1,5 часа. Раствор фильтровали, промывали смесью 3% воды в этаноле, и высушивали отсасыванием при комнатной температуре 16 часов, получив в результате 9,65 г (примерно 64%-ный выход) указанной в заголовке N-метилглюкаминовой соли с показателем однородности 96,4%.
Этот материал (9,65 г) растворяли в смеси 15% воды в этаноле (примерно 450 мл) при 52oC. Затем раствор охлаждали и держали при 28oC 3,5 часа, при комнатной температуре 16 часов и при 4oC 3 часа. Полученные кристаллы отфильтровывали, промывали холодной смесью 2% воды в этаноле (50 мл х 2), высушивали отсасыванием 4 часа и затем высушивали под вакуумом (1 мм рт.ст.), что дало в результате 7,5 г (примерно 80%-ный выход) указанной в заголовке N-метилглюкаминовой соли (показатель однородности при определении путем HPLC составлял 98,6%); т.пл.: выше 154oC с разложением.
Элементный анализ: вычислено для C58H75N2O25Р • 5,0 H2O:
C 52,72; H 6,48; N 2,12; KF (H2O) 6,82.
Найдено: C 53,09; H 6,50; N 2,08; KF (H2O) 7,12.
Пример 4. 2'-O-(фосфонооксиметил)паклитаксел
(a) Получение 2'-O-(метилтиометил)-7-O-(триэтилсилил)-паклитаксела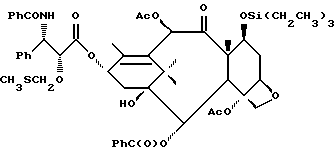
К охлажденному (0 - (-5oC)) раствору 7-O-(триэтилсилил)паклитаксела (2,46 г, 2,5439 ммоль) в сухом ацетонитриле (100 мл) добавляли диметилсульфид (1,348 г, 1,59 мл, 21,6976 ммоль), а затем бензоилпероксид (2,628 г, 10,8488 ммоль). Гетерогенную смесь перемешивали при 0oC 1 час и держали при 5oC 18 часов. Получили желтый раствор. Его упаривали досуха и очищали на колонке с силикагелем (элюирование смесью этилацетат:гексан с отношением 1:4, 1: 3 и 1:2), получив в результате указанное в заголовке соединение (1,0 г, 38%). Оно было использовано как таковое для следующей стадии.
Масс-спектр: [M + H]+, 1028; [M + Na]+, 1050; [M + K]+, 1066.
(b) Получение 2'-O-(метилтиометил)паклитаксела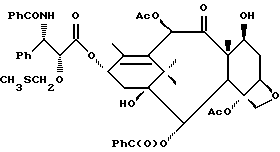
К охлажденному (-15oC) раствору полученного на стадии (a) продукта (1,0 г, 0,9737 ммоль) в сухом ацетонитриле (30 мл) добавляли пол каплям 0,5 н. HCl (3 мл). Полученный раствор перемешивали при -15oC в течение 1 часа и при 5oC в течение 18 часов. Разбавляли его этилацетатом (20 мл) и промывали холодным 6%-ным раствором NaHCPО3 и рассолом. Высушивали (МgSO4) и упаривали досуха. Полученный продукт очищали на покрытой силикагелем пластинке (метиленхлорид 15% ацетонитрила) и получали чистое указанное в заголовке соединение (280 мг, 31,4%).
ИК-спектр: 3446, 3064, 2940, 1726, 1666, 1582, 1516, 1486 см-1.
ЯМР (CDCl3): δ 1,118 (с, 3H), 1,229 (с, 3H), 1,622 (с, 3H), 1,689 (с, 3H), 1,871 (с, 3H), 2,209 (с, 3H), 2,450 (с, 3H), 3,800 (д, H), 4,119 (д, H), 4,305 (д, H), 4,413 (м, H), 4,563 (д, H), 4,703 (д, H), 4,940 (д, H), 4,958 (дд, H), 5,667 (д, H), 5,822 (дд, H), 6,263 (м, 2H), 7,019 (д, NH), 7,293 - 8,127 (м, 15H).
Масс-спектр: [M+H]+, 914, [M+Na]+, 936; [M+K]+, 952.
Масс-спектр высокого разрешения: MH+ - 914,3394 (вычислено 914,3422)
(c) Получение 2'-O-(дибензилфосфонооксиметил)паклитаксела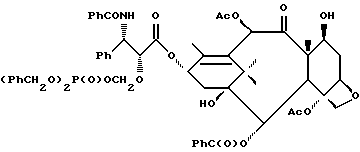
К перемешиваемому раствору полученного на стадии (b) продукта (0,89 г, 0,9748 ммоль) в сухом 1,2-дихлорэтане (12 мл) добавляли порошкообразные 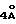 молекулярные сита (1,0 г), а затем добавляли по каплям раствор смеси N-иодсукцинимида (0,33 г, 1,4622 ммоль) и дибензилфосфата (0,41 г, 1,4622 ммоль) в сухом тетрагидрофуране (8 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 часа, после чего фильтровали через целит. Фильтрат упаривали досуха, и красный остаток растворяли в этилацетате (50 мл) и промывали холодным 6%-ным NaHSO4, холодным 6%-ным NaHCO3 рассолом. Раствор высушивали (MgSO4) и упаривали, получив в результате пену, ее очищали посредством покрытой силикагелем пластины (метиленхлорид-20% ацетонитрила) и получали чистый продукт (0,77 г, 69%).
молекулярные сита (1,0 г), а затем добавляли по каплям раствор смеси N-иодсукцинимида (0,33 г, 1,4622 ммоль) и дибензилфосфата (0,41 г, 1,4622 ммоль) в сухом тетрагидрофуране (8 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 часа, после чего фильтровали через целит. Фильтрат упаривали досуха, и красный остаток растворяли в этилацетате (50 мл) и промывали холодным 6%-ным NaHSO4, холодным 6%-ным NaHCO3 рассолом. Раствор высушивали (MgSO4) и упаривали, получив в результате пену, ее очищали посредством покрытой силикагелем пластины (метиленхлорид-20% ацетонитрила) и получали чистый продукт (0,77 г, 69%).
ИК-спектр: 3854, 3744, 3362, 3066, 1960, 1722, 1602, 1580 см-1.
ЯМР (CDCl3): δ 1,075 (с, 3H), 1,167 (с, 3H), 1,651 (с, 3H), 1,799 (с, 3H), 2,209 (с, 3H), 2,296 (с, 3H), 2,464 (м, H), 3,686 (м, H), 4,121 (д, H), 4,240 (д, H), 4,293 (м, H), 4,808-4,957 (м, 6H), 5,006 (м, H), 5,565-5,649 (м, 2H), 6,034 (т, H), 6,194 (3, H), 7,100-8,132 (м, 26H).
Масс-спектр: [M+H]+, 1144; [M+Na]+, 1166; [M+K]+, 1182.
(d) Получение 2'-O-(фосфонооксиметил)паклитаксела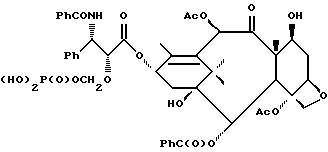
Смесь полученного на стадии (с) продукта (0,9 г, 0,7874 ммоль) и 10% Рd/C (1 г) в этилацетате (20 мл) гидрогенизировали при 60 фунтах на кв. дюйм (400 кПа) в течение 24 часов. Реакционную смесь фильтровали через целит, и фильтрат упаривали досуха. Остаток очищали посредством покрытой силикагелем пластины (метиленхлорид-40% метанола) и получили указанный в заголовке продукт (0,254 г, 33,4%);
т.пл. 202 - 205oC (разложение).
ИК-спектр (KBr): 3438, 3066, 2942, 1722, 1652, 1602 см-1.
ЯМР (ацетон-d6/D2O): δ 1,081 (с, 6H) 1,571 (с, 3H), 1,847 (с, 3H), 2,115 (с, 3H), 2,357 (с, 3H), 3,707 (д, H), 4,08 (м, 2H), 4,275 (м, H), 4,941 - 5,085 (м, 4H), 5,231 (т, H), 5,430 (д, H), 5,544 (д, H), 5,970 (т, H), 6,376 (м, H), 6,961- 8,017 (м, 16H).
Масс-спектр: [M+Na]+, 986, [M+K]+, 1002; [M+2Na-H]+, 1008; [M+Na+K-H]+, 1024; [M+2K-H]+, 1040.
Масс-спектр высокого разрешения: MNa+ - 986,2955 (вычислено 986,2976).
Пример 5. 2',7-O-(Фосфонооксиметил)паклитаксела натриевая соль
(a) Получение 2',7-O-бис(метилтиометил)паклитаксела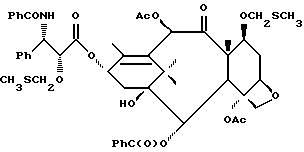
К перемешанному раствору паклитаксела (0,853 г, 1 ммоль) и диметилсульфида (1,465 г, 20 моль) в ацетонитриле (20 мл) при 0oC добавляли твердый бензоилпероксид (1,995 г, 8 ммоль). Реакционную смесь интенсивно перемешивали при 0oC в течение 3 часов. Ход реакции контролировали путем тонкослойной хроматографии в смеси гексан:этилацетат (1:1, объем/объем), Rf паклитаксела = 0,24, Rf продукта = 0,60. После исчезновения исходного материала (приблизительно через 3 часа) реакцию быстро прекращали путем выпаривания растворителей досуха при 25oC с использованием вакуумной линии в помещении. Сухой остаток разделяли посредством колонки с силикагелем (EM Science, 40 - 63 мкм), 100 мл сухого силикагеля, размер колонки : Ф = 3/4 дюйма (19 мм), система растворителей: гексан: этилацетат (3:2, объем/объем), объем каждой фракции: приблизительно 25 мл. Из фракции 15 - 19 было извлечено указанное в заголовке соединение (0,515 г, 53%-ный выход).
Масс-спектр (FAB/матрица NOBA, NaI, KI): [M+H]+, m/z 974; [M+Na]+, m/z 996; [M+K]+, m/z 1012.
УФ-спектр (MeOH): λмакс = 204 нм, E (1%/1 см)= 243,45; λмакс = 228 нм, E (1%/ 1 см) = 313,99.
ИК-спектр (KBr): 3440, 3064, 2926, 1724, 1668, 1602, 1582, 1514, 1484, 1452, 1372, 1314, 1266, 1242, 1178, 1142, 1068, 1026, 990, 915, 886, 848, 800, 774, 710, 646, 606, 570, 540, 480 см-1.
1Н ЯМР (CDCl3), δ: 1,17 (3H, с), 1,20 (3H, с), 1,68 (3H, с), 1,74 (3H, с), 1,84 (H, дд), 2,04 (3H, д), 2,09 (3H, с), 2,15 (3H, с) перекрывается с (H, м), 2,37 (H, дд), 2,51 (3H, с), 2,79 (H, ддд), 3,78 (H, д), 4,18 (H, д), 4,28 (H, м), 4,31 (H, д), 4,53 - 4,74 (4H, два перекрывающихся AB м), 4,93 (H, д), 4,95 (H, д), 5,68 (H, д), 5,82 (H, дд), 6,24 (H, дд), 6,54 (H, с), 7,05 (H, д), 7,28 - 7,59 (10Н, перекрывающийся м), 7,57 (H, м), 7,76 (2H, д), 8,09 (2H, д).
(b) Получение 2',7-O-(дибензилфосфонооксиметил)паклитаксела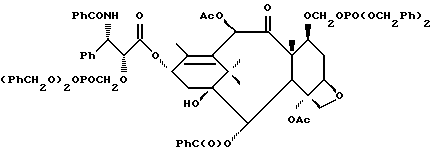
К смеси 2', 7-O-бис(метилтиометил)пакклитаксела (198 мг, 0,2 ммоль) и  молекулярных сит (примерно 200 мг) в метиленхлориде (12 мл) при комнатной температуре добавляли раствор N-иодсукцинимида (135 мг, 0,5 ммоль) и дибензилфосфата (167 мг, 0,5 ммоль) в сухом тетрагидрофуране (8 мл). Реакционную смесь перемешивали 1,5 часа затем молекулярные сита офильтровывали на целите, промывали метиленхлоридом (10 мл), и выпаривали растворители досуха при комнатной температуре с использованием бытовой вакуумной линии. Остаток растворяли в этилацетате (100 мл) и промывали в делительной воронке 1%-ным тиосульфатом натрия (50 мл), 0,5 м бикарбонатом натрия (50 мл) и дважды водой (2 х 50 мл). Органическую фазу высушивали над сульфатом магния, упаривали досуха и вновь растворяли в этилацетате (1 мл). Продукт осаждали 50 мл смеси диэтиловый эфир: гексан (1:1) и дважды промывали одной и той же системой растворителей (2 х 50 мл). Получили сырой продукт (218 мг) с 74%-ным выходом. Очистку этого продукта осуществляли путем загрузки его раствора в метиленхлориде (3 мл) на силикагель (Ф = 3/4 дюйма (19 мм) х L = 1 дюйм (25,4 мм)) и элюирования продукта 50 мл смеси метиленхлорид: этилацетат (3: 1). Получили указанное в заголовке соединение (172,7 мг) с выходом 59,3%.
молекулярных сит (примерно 200 мг) в метиленхлориде (12 мл) при комнатной температуре добавляли раствор N-иодсукцинимида (135 мг, 0,5 ммоль) и дибензилфосфата (167 мг, 0,5 ммоль) в сухом тетрагидрофуране (8 мл). Реакционную смесь перемешивали 1,5 часа затем молекулярные сита офильтровывали на целите, промывали метиленхлоридом (10 мл), и выпаривали растворители досуха при комнатной температуре с использованием бытовой вакуумной линии. Остаток растворяли в этилацетате (100 мл) и промывали в делительной воронке 1%-ным тиосульфатом натрия (50 мл), 0,5 м бикарбонатом натрия (50 мл) и дважды водой (2 х 50 мл). Органическую фазу высушивали над сульфатом магния, упаривали досуха и вновь растворяли в этилацетате (1 мл). Продукт осаждали 50 мл смеси диэтиловый эфир: гексан (1:1) и дважды промывали одной и той же системой растворителей (2 х 50 мл). Получили сырой продукт (218 мг) с 74%-ным выходом. Очистку этого продукта осуществляли путем загрузки его раствора в метиленхлориде (3 мл) на силикагель (Ф = 3/4 дюйма (19 мм) х L = 1 дюйм (25,4 мм)) и элюирования продукта 50 мл смеси метиленхлорид: этилацетат (3: 1). Получили указанное в заголовке соединение (172,7 мг) с выходом 59,3%.
Масс-спектр (FAB, матрица NOBA/NaI, KI): [M + Na]+ , m/z 1456; [M + K]+, m/z 1472.
Уф спектр (MeCN): λмакс = 194 нм, E (1%/1 см) = 1078,36; λмакс = 228 нм, E (1%/1 см) = 311,95.
ИК-спектр (KBr): 3430, 3066, 3032, 2958, 1744, 1726, 1664, 1602, 1582, 1532, 1488, 1456, 1372, 1270, 1244, 1158, 1108, 1068, 1016, 1000, 952, 886, 800, 776, 738, 698, 604, 498 см-1.
1H-ЯМР (CDCl3), δ: 1,12 (3H, с), 1,14 (3H, с), 1,56 (H, м), 1,67 (3H, с), 1,84 (3H, д), 1,90 (H, м), 2,17 (3H, с), 2,29 (3H, с), 2,73 (H, м), 3,73 (H, д), 4,08 (H, д), 4,15 (H, м), 4,20 (H, д), 4,77 (H, м), 4,79 (H, д), 4,91 - 5,04 (10H, перекрывающийся м), 5,25 (H, дд), 5,38 (H, дд), 5,54 - 5,64 (2H, перекрывающийся м), 5,99 (H, шир. дд), 6,25 (H, с), 7,11 - 7,14 (2H, м), 7,24 - 7,64 (28H, перекрывающийся м), 7,94 (2H, дд), 8,04 (2H, дд), 8,30 (H, д).
(c) Получение натриевой соли 2',7-O-бис(фосфонооксиметил)паклитаксела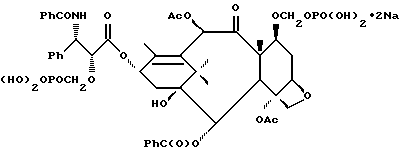
Пробу 2', 7-O-бис(дибензилфосфонооксиметил)паклитаксела (112 мг, 0,078 ммоль) растворяли в этилацетате (7 мл) и гидрогенизировали на 10% палладия на угле (50 мг) при комнатной температуре и 60 фунтах на кв. дюйм (400 кПа) в течение 2 часов. Катализатор удаляли путем фильтрования на целите. Целит промывали этилацетатом (10 мл). Фильтрат обрабатывали твердым бикарбонатом натрия (20 мг, 3 эквив.), после чего выпаривали растворитель досуха. Сухой остаток вновь растворяли в 5 мл смеси вода: ацетон (4:1, объем/объем) и очищали путем хроматографии на колонке C-18 с обратной фазой (55 - 105 мк C-18, Уотерс, 50 мл сухого C-18, Ф = 3/4 дюйма (19 мм), в смеси вода: ацетон (4: 1, объем/объем). Элюент на аналитической колонке C-18 Джонса для НPL C (15 см, 1 мл/мин, λ = 230 нм) в буфере ацетонитрил: фосфат с pH 6 (50/50, объем/объем) с добавлением коктейля Q12 из ионных пар (Regis), Rt = 4,7 минут. Фракции, содержащие указанный в заголовке продукт, объединяли, выпаривали ацетон под бытовым вакуумом при 20oC, и раствор лиофилизировали. Был получен указанный в заголовке продукт (44,2 мг) с выходом 58,8%.
Масс-спектр (FAB, матрица NOBA/NaI, KI): [M + H]+, m/z 1118; [M + Na]+, m/z 1140.
УФ-спектр (MeCN): λмакс = 192 нм, E (1%/1 см) = 129,73; λмакс = 230 нм, E (1%/1 см) = 26,43.
ИК-спектр (KBr): 3430, 3066, 2956, 1724, 1658, 1604, 1582, 1520, 1486, 1452, 1374, 1316, 1256, 1152, 1110, 1070, 1026, 966, 914, 802, 772, 710, 538 см-1.
1H-ЯМР (ацетон-d6/D2O), δ: 0,97 (3H, с), 1,02 (3H, с), 1,47 (H, м), 1,54 (3H, с), 1,70 (H, м), 1,75 (3H, с), 1,85 (H, м), 2,11 (3H, с), 2,30 (3H, с), 2,88 (H, м), 3,64 (H, д), 4,03 (H, м), 4,06 (H, д), 4,16 (H, д), 4,74 (H, м), 4,86 (H, м), 51,11 (H, шт), 5,22 (H, д), 5,42 (H, д), 5,90 (H, шт), 6,21 (H, с), 7,06 (H, шт), 7,32 - 7,69 (10H, перекрывающийся м), 7,80 (2H, д), 7,93 (2H, д).
Пример 6. 7-O-Метилтиолметилбаккатин III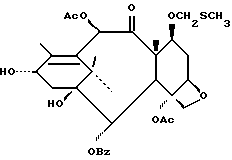
К раствору 2'-O-этилоксикарбонил-7-O-метилтиометилпактилтаксела (соединение примера 3 (b), 27 г, 27,4 ммоль) в 100 мл тетрагидрофурана и 500 мл метанола добавляли свежеизмельченный K2CO3 (2,7 г, 19 ммоль). Раствор перемешивали 30 минут и нейтрализовали смолой IP-120 (Н+), фильтровали и концентрировали. Затем неочищенный фильтрат растворяли в 200 мл дихлорметана и перемешивали 24 часа с тетрабутиламмонийборогидридом (10 г). Раствор разбавляли дихлорметаном и промывали водой, насыщенным раствором бикарбоната и рассолом. После этого органическую фракцию высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (1:1, гексан:этилацетат) и получили 9,4 г указанного в заголовке соединения (53%) с точкой плавления 269oC.
Масс-спектр FAB (NOBA) M + H вычисл. для C33H43SO11: 647.
Найдено: 647.
ИК-спектр (KBr): 3474, 1746, 1724, 1712, 1270, 1240, 1070 см-1.
1H-ЯМР (CDCl3, 300 МГц), δ: 8,08 (д, J = 7,1 Гц, 2H), 7,58 (т, J = 7,5 Гц, 1H), 7,45 (т, J = 7,8 Гц, 2H), 6,55 (с, 1H), 4,94 (д, J = 8,1 Гц, 1H), 4,83 (шир. кв., J = 5,1 Гц, 1H), 4,66 (АВкв, J = 14,7 и 12,3 Гц, 2H), 4,30 (м, 2H), 4,13 (д, J = 8,4 Гц, 1H), 3,91 (д, J = 6,6 Гц, 1H), 2,79 (м, 1H), 2,27 (с, 3H), 2,25 (м, 2H), 2,19 (с, 3H), 2,16 (с, 3H), 2,10 (с, 4H), 1,81 (м, 1H), 1,72 (с, 3H), 1,61 (м, 2H), 1,16 (с, 3H), 1,03 (с, 3H).
13C ЯМР (CDCl3, 75,5 Гц), δ: 202,3, 170,8, 169,3, 167,0, 144,2, 132,6, 132,1, 130,1, 129,4, 128,6, 83,9, 80,9, 78,7, 75,7, 74,5, 73,9, 67,9, 57,6, 47,6, 42,7, 38,3, 26,7, 22,6, 21,0, 20,1, 15,2, 15,0, 10,8.
Пример 7. Триэтаноламиновая соль 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3' -(2-фурил)-2'-O-этилоксикарбонил-7-O-фосфонооксиметилпалитаксела
(a) Получение 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3'-(2-фурил)-7-O- метилтиометилпаклитаксела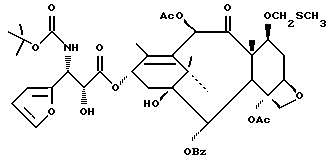
К раствору гексаметилдисилазана (0,40 мл, 1,90 ммоль) в 15 мл тетрагидрофурана добавляли раствор н-бутиллития (0,75 мл, 2,5 М в гексанах, 1,88 ммоль) и перемешивали 5 минут при -55oC. К этому раствору добавляли 7-метилтиометилбаккатин III (соединение примера 6, 1,03 г, 1,59 ммоль) в 10 мл тетрагидрофурана и перемешивали 10 минут до добавления 10 мл раствора (3R, 4R)-1-(трет-бутилоксикарбонил)-4-(2-фурил)- 3-(триэтилсилилокси)-2-азетидинона (883 мг, 2,40 ммоль). Убирали холодную ванну, и заменяли ее ванной при 0oC, после чего реакционную смесь перемешивали 30 минут. Раствор разбавляли этилацетатом и промывали насыщенным раствором NH4Cl, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (2,5:1, гексан:этилацетат) и получили 1,5 г продукта реакции сочетания 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3'- (2-фурил)-7-O-метилтиометил-2'-O-триэтилсилилпаклитаксела (93%).
Масс-спектр FAB /NOBA/ [M+Na] вычисл. для C50H71NSSiO16 :1036, найдено: 1036.
ИК-спектр (пленка): 3446 (с), 1720, 1368, 1242, 1166, 1144, 1124, 1066 см-1.
1H ЯМР (CDCl3, 300 МГц), δ: 8,07 (д, J = 7,2 Гц, 2H), 7,56 (м, 1H), 7,46 (т, J = 7,5 Гц, 2H), 7,36 (м, 1H), 6,56 (с, 1H), 6,33 (м, 1H), 6,20 (м, 2H), 5,67 (д, J = 6,9 Гц, 1H), 5,29 (шс, 2H), 4,94 (д, J = 7,8 Гц, 1H), 4,75 (с, 1H), 4,65 (с, 2H), 4,28 (м, 2H), 4,16 (д, J = 8,1 Гц, 1H), 3,89 (д, J = 6,9 Гц, 1H), 2,80 (м, 1H), 2,46 (с, 3H), 2,37 (м, 1H), 2,22 (м, 1H), 2,16 (с, 3H), 2,10 (с, 3H), 2,04 (с, 3H), 1,84 (м, 1H), 1,74 (с, 3H), 1,65 (м, 1H), 1,33 (с, 9H), 1,20 (с, 3H), 1,19 (с, 3H), 0,81 (т, J = 7,8 Гц, 9H), 0,47 (м, 6H).
13C ЯМР (CDCl3; 75,5 Гц), δ: 202,0, 171,2, 170,3, 169,3, 167,1, 155,3, 152,0, 141,9, 141,0, 133,6, 132,9, 130,2, 129,2, 128,7, 110,7, 107,3, 84,0, 81,1, 80,2, 78,7, 76,1, 75,7, 74,7, 74,1, 72,4, 71,1, 57,4, 52,8, 47,1, 43,3, 35,2, 33,0, 28,1, 26,3, 22,9, 21,2, 21,0, 15,0, 14,5, 10,9, 6,5, 4,3.
К раствору полученного выше 2'-триэтилсилилового эфира (330 мг, 0,32 ммоль) в 7 мл тетрагидрофурана добавляли тетрабутиламмонийфторид (0,35 мл, 1,0 М а ТГФ, 0,35 ммоль) и перемешивали 10 минут. Раствор разбавляли этилацетатом и промывали рассолом, высушивали над MgSO4 и концентрировали, а остаток хроматографировали на силикагеле (2:1, гексан:этилацетат) и получили в результате 301 мг указанного в заголовке соединения (95%).
Масс-спектр FAB (NOBA) M+H вычисл. для C45H58NO16S: 900. Найдено: 900.
ИК-спектр (пленка): 3442, 1720, 1242, 1066, 1026 см-1.
1H ЯМР (CDCl3, 300 МГц), δ: 8,07 (д, J = 7,3 Гц, 2H), 7,57 (т, J = 7,3 Гц, 1H), 7,45 (т, J = 7,8 Гц, 2H), 7,38 (с, 1H), 6,53 (с, 1H), 6,34 (д, J = 3,2 Гц, 1H), 6,29 (д, J = 3,2 Гц, 1H), 6,17 (т, J = 8,1 Гц, 1H), 5,65 (д, J = 6,9 Гц, 1H), 5,29 (м, 2H), 4,92 (д, J = 8,0 Гц, 1H), 4,70 (м, 1H), 4,64 (д, J = 4,6 Гц, 2H), 4,29 (м, 2H), 4,14 (д, J = 8,3 Гц, 1H), 3,86 (д, J = 6,8 Гц, 1H), 3,37 (д, J = 5,8 Гц, 1H), 2,77 (м, 1H), 2,38 (с, 3H), 2,32 (м, 2H), 2,16 (с, 3H), 2,10 (с, 3H), 2,02 (с, 3H), 1,77 (м, 3H) 1,73 (с, 3H), 1,33 (с, 9H), 1,17 (с, 3H), 1,12 (с, 3H).
13C ЯМР (CDCl3, 75,5 Гц), δ 202,0, 172,6, 170,3, 169,2, 167,0, 155,2, 151,3, 142,4, 140,4, 133,7, 133,2, 130,2, 129,1, 128,7, 110,7, 107,4, 83,9, 81,2, 80,5, 78,6, 76,5, 76,1, 75,4, 74,6, 74,0, 72,5, 71,8, 57,4, 51,7, 47,2, 43,2, 35,2, 32,8, 28,1, 26,4, 22,6, 20,9, 15,2, 14,6, 10,9, 8,3.
(b) Получение 3'-N-дебензоил-3'-дезфенил-3'-N-(трет- бутилоксикарбонил)-3'-(2-фурил)-2'-O-этилоксикарбонил-7-O- метилтиометилпаклитаксела
К раствору полученного на стадии (a) продукта (864 мг, 0,96 ммоль) в 50 мл дихлорметана при 0oC добавляли диизопропилэтиламин (2,0 мл, 11,5 ммоль) и этилхлорформиат (0,50 мл, 5,25 ммоль) и перемешивали 4 часа. Раствор разбавляли дихлорметаном и промывали насыщенным раствором бикарбонат, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (1:1, гексан:этилацетат) и получили 884 мг 2'-этилкарбоната указанного в заголовке соединения (95%).
Масс-спектр FAB (NOBA) M+H вычисл. для C48H62NO18S: 972,3688. Найдено: 972,3654.
ИК-спектр (пленка): 1752, 1720, 1370, 1244, 1196, 1176, 1064 см-1.
1H ЯМР (CDCl3, 300 МГц), δ: 8,09 (д, J = 7,8 Гц, 2H), 7,57 (т, J = 7,5 Гц, 1H), 7,46 (т, J = 7,8 Гц, 2H), 7,38 (с, 1H), 6,55 (с, 1H), 6,35 (м, 1H), 6,27 (м, 1H), 6,22 (т, J = 7,8 Гц, 1H), 5,67 (д, J = 7,2 Гц, 1H), 5,51 (д, J = 9,9 Гц, 1H), 5,34 (д, J = 2,4 Гц, 1H), 5,25 (д, J = 10,2 Гц, 1H), 4,95 (д, J = 8,1 Гц, 1H), 4,65 (с, 2H), 4,30 (м, 2H), 4,22 (м, 2H), 3,88 (д, J = 7,2 Гц, 1H), 2,81 (м, 1H), 2,41 (с, 3H), 2,36-2,21 (м, 2H), 2,16 (с, 3H), 2,11 (с, 3H), 2,09 (с, 3H), 1,83 (м, 1H), 1,74 (с, 3H), 1,67 (с, 1H), 1,59 (с, 1H), 1,34 (с, 9H), 1,29 (т, J = 7,2 Гц, 3H), 1,20 (с, 3H), 1,18 (с, 3H).
13C ЯМР (CDCl3, 75,5 Гц), δ: 202,1, 169,9, 169,1, 167,6, 167,0, 154,0, 150,1, 142,6, 141,0, 133,6, 132,9, 130,2, 129,2, 128,7, 110,7, 107,5, 83,9, 81,1, 80,7, 78,7, 76,0, 75,7, 75,1, 74,7, 74,2, 71,8, 65,1, 57,4, 49,7, 47,1, 43,2, 35,0, 33,0, 28,1, 26,3, 22,6, 21,1, 20,9, 15,1, 14,5, 14,1, 10,9.
(c) Получение 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3'-(2-фурил)- 2-'-O-этилоксикарбонил-7-O-дибензилфосфонооксиметилпаклитаксела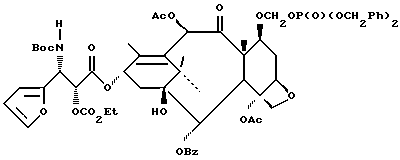
К раствору полученного на стадии (b) продукта (230 мг, 0,236 ммоль) в 10 мл безводного тетрагидрофурана добавляли 300 мг 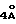 молекулярных сит, дибензилфосфат (270 мг, 0,98 ммоль) и перекристаллизованный NIS (62 мг, 0,28 ммоль). К этому раствору добавляли трифторметансульфонат серебра (45 мг, 0,17 ммоль), и раствор перемешивали 3 часа. Раствор фильтровали через целит и разбавляли этилацетатом, промывали 10%-ным раствором NaS2O8, насыщенным раствором бикарбоната и рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (15% ацетонитрила в хлороформе) и получили 219 мг дибензилфосфата указанного в заголовке соединения (77%).
молекулярных сит, дибензилфосфат (270 мг, 0,98 ммоль) и перекристаллизованный NIS (62 мг, 0,28 ммоль). К этому раствору добавляли трифторметансульфонат серебра (45 мг, 0,17 ммоль), и раствор перемешивали 3 часа. Раствор фильтровали через целит и разбавляли этилацетатом, промывали 10%-ным раствором NaS2O8, насыщенным раствором бикарбоната и рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (15% ацетонитрила в хлороформе) и получили 219 мг дибензилфосфата указанного в заголовке соединения (77%).
Масс-спектр FAB (NOBA) вычисл. для C61H72NPO22Na: 1224. Найдено: 1224.
ИК-спектр (пленка): 3422 (шир. ), 1750, 1722, 1370, 1244, 1160, 1036, 1016, 1000, 976, 944 см-1.
1H ЯМР (CDCl3, 300 МГц), δ: 8,08 (д, J = 6,9 Гц, 2H); 7,58 (т, J = 7,2 Гц, 1H), 7,46 (т, J = 7,8 Гц, 2H), 7,39 (с, 1H), 7,31 (м, 1H), 6,35 (м, 2H), 6,28 (с, 1H), 6,21 (т, J =7,8 Гц, 1H), 5,64 (д, J = 6,9 Гц, 1H), 5,50 (д, J = 10,5 Гц, 1H), 5,39 (д, J = 6,6 Гц, 1H), 5,32 (д, J = 2,4 Гц, 1H), 5,25 (д, J = 9,9 Гц, 1H), 5,01 (дд, J = 8,1 и 6,3 Гц, 5H), 4,86 (д, J = 8,4 Гц, 1H), 4,29 - 4,09 (м, 4H), 3,85 (д, J = 6,9 Гц, 1H), 2,77 (м, 1H), 2,40 (с, 3H), 2,30 (м, 2H), 2,16 (с, 3H), 1,99 (с, 3H), 1,94 (м, 1H), 1,70 (с, 3H), 1,67 (с, 1H), 1,54 (с, 1H), 1,34 (с, 9H), 1,28 (т, J = 7,2 Гц, 3H), 1,20 (с, 3H), 1,17 (с, 3H).
13C ЯМР (CDCl3, 75,5 Гц), δ: 201,8, 169,9, 169,2, 167,7, 167,0, 155,1, 154,0, 150,0, 142,74, 141,1, 133,7, 132,9, 130,2, 129,1, 128,7, 128,5, 128,4, 128,0, 110,7, 107,6, 93,8, 84,1, 81,6, 80,8, 80,7, 78,8, 76,3, 75,1, 74,6, 71,8, 69,3, 69,2, 65,1, 57,0, 49,7, 46,7, 43,2, 35,0, 28,1, 26,4, 22,6, 21,2, 20,8, 14,6, 14,1, 10,5.
(d) Получение триэтаноламиновой соли 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3'- (2-фурил)-2'-O-этилоксикарбонил-7-О-фосфонооксиметилпаклитаксела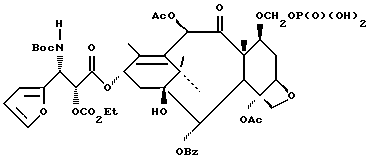
К раствору полученного на стадии (c) продукта (311 мг, 0,259 ммоль) в 25 мл этилацетата добавляли 60 мг палладия на угле (10%), и раствор перемешивали в среде водорода в течение 30 минут. Удаляли катализатор путем фильтрования через целит, и фильтрат концентрировали в вакууме. Остаток растворяли в 3 мл этилацетата, и добавляли триэтаноламин (2,3 мл, 0,1 М в этилацетоне, 0,23 ммоль). Раствор концентрировали и остаток хроматографировали на C18 (40% ацетонитрила в воде) и лиофилизировали, получив в результате 205 мг триэтаноламиновой соли (67%) фосфоновой кислоты.
Масс-спектр FAB (NOBA): M + Na вычисл. для C47H60HPO22Na: 1044. Найдено: 1044.
ИК-спектр (пленка): 3432 (шир. ), 1752, 1722, 1372, 1246, 1158, 1108, 1096, 1070, 1002 см-1.
1H ЯМР (d6 ацетон/D2O, 300 МГц), δ: 8,09 (д, J = 7,2 Гц, 2H), 7,62 (м, 2H), 7,52 (т, J = 7,5 Гц, 2H), 6,48 (д, J = 3,3 Гц, 1H), 6,42 (м, 2H), 6,16 (т, J = 8,7 Гц, 1H), 5,65 (д, J = 6,9 Гц, 1H), 5,46 (д, J = 3,6 Гц, 1H), 5,30 (д, J = 3,6 Гц, 1H), 5,17 (шс, 1H), 5,01 (шд, J = 9,0 Гц, 1H), 4,19 (шс, 1H), 4,18 (м, 5H), 3,95 (м, 6H), 3,87 (д, J = 6,9 Гц, 1H), 3,68 (с, 7H), 3,50 (шт, J = 4,8 Гц, 6H), 2,95 (м, 1H), 2,44 (с, 3H), 2,41 (м, 2H), 2,16 (с, 3H), 1,9 (с, 3H), 1,94 (м, 1H), 1,68 (с, 3H), 1,34 (с, 9H), 1,24 (т, J = 6,9 Гц, 3H), 1,17 (с, 6H).
Пример 8. Триэтаноламиновая соль 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3'- (2-тиенил)-2'-O-этилоксикарбонил-7-O-фосфонооксиметилпаклитаксела
(a) Получение 3'-N-дебензоил-3'-дезфенил-3'-N-(трет- бутилоксикарбонил)-3'-(2-тиенил)-7-O-метилтиометилпаклитаксела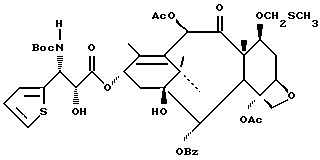
К раствору ГМДС (0,5 мл, 2,4 ммоль) в 18 мл ТГФ при -55oC добавляли н-бутиллитий (0,85 мл, 2,5 М в гексанах, 2,1 ммоль). Через 10 минут добавляли по каплям 7-метилтиометилбаккатин III (1,15 г, 1,78 ммоль) в 18 мл ТГФ и перемешивали на холоду в течение 10 минут. Добавляли (±)цис-1- (трет-бутилоксикарбонил)-4-(2-тиенил)-3-(триэтилсилилокси)- 2-азетидинон (2,80 г, 7,3 ммоль) в 18 мл ТГФ, и позволяли холодной ванне медленно нагреваться до 0oC в течение 30 минут. Раствор разбавляли этилацетатом, промывали насыщенным раствором NH4Cl, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (5 : 1, гексан : этилацетат), получив 1,87 г рекуперированного лактамами (3 : 1, гексан : этилацетат), что дало 1,44 г продукта реакции сочетания 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3'- (2-тиенил)-7-O-метилтиометил-2'-O-триэтилсилилпаклитаксела (78%).
Масс-спектр FAB (NOBA): M + Na вычисл. для C51H71NO15S2SiNa: 1052. Найдено: 1052.
ИК-спектр (пленка): 3442 (шир. ), 1720, 1490, 1368, 1270, 1242, 1162, 1110, 1064, 1024, 984, 754 см-1.
1H ЯМР (CDCl3, 300 МГц), δ: 8,09 (д, J = 7,2 Гц, 2H), 7,57 (т, J = 7,6 Гц, 1H), 7,47 (т, J = 7,8 Гц, 2H), 7,22 (м, 1H), 6,95 (м, 2H), 6,55 (с, 1H), 6,21 (т, J = 9,3 Гц, 1H), 5,68 (д, J = 6,9 Гц, 1H), 5,49 (шд, 1H), 5,39 (шд, J = 9,6 Гц, 1H), 4,94 (д, J = 7,8 Гц, 1H), 4,65 (с, 2H), 4,57 (с, 1H), 4,28 (м, 2H), 4,17 (д, J = 8,4 Гц, 1H), 3,88 (д, J = 6,9 Гц, 1H), 2,80 (м, 1H), 2,46 (с, 3H), 2,37 (м, 1H), 2,20 (м, 1H), 2,17 (с, 3H), 2,10 (с, 3H), 2,03 (с, 3H), 1,84 (м, 1H), 1,74 (с, 3H), 1,68 (с, 1H), 1,62 (с, 1H), 1,31 (с, 9H), 1,20 (с, 6H), 0,84 (т, J = 7,8 Гц, 9H), 0,50 (м, 6H).
13C ЯМР (CDCl3, 75,5 Гц), δ: 201,9, 171,1, 170,7, 170,1, 169,3, 167,0, 155,1 142,8, 140,9, 133,6, 132,9, 130,2, 129,2, 128,7, 126,9, 124,6, 83,9, 81,2, 80,1, 78,8, 77,4, 76,0, 75,7, 75,2, 74,8, 74,1, 71,3, 57,4, 53,8, 47,0, 43,3, 35,3, 33,3, 28,1, 26,3, 23,0, 21,3, 20,9, 14,9, 14,4, 10,9, 6,6, 4,5.
К раствору полученного выше 2'-триэтилсилилового эфира (1,41 г, 1,37 ммоль) в 14 мл тетрагидрофурана добавляли тетрабутиламмонийфторид (1,4 мл, 1,0 М в ТГФ, 1,40 ммоль). Раствор перемешивали 30 минут, разбавляли этилацетатом, промывали рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (гексан : этилацетат, 1 : 1) и получили 1,16 г указанного в заголовке соединения (92%).
Масс-спектр FAB (NOBA): M + Na вычисл. для C45H57NO15S2Na: 938. Найдено: 938.
ИК-спектр (пленка): 3440 (шир.), 1720, 1368, 1242, 1168, 1106, 1066, 710 см-1.
1H ЯМР (CDCl3, 300 МГц), δ: 8,08 (д, J = 7,2 Гц, 2H), 7,59 (м, 1H), 7,47 (т, J = 7,8 Гц, 2H), 7,24 (м, 1H), 7,07 (м, 1H), 6,99 (м, 1H), 6,53 (с, 1H), 6,18 (т, J = 8,1 Гц, 1H), 5,66 (д, J = 6,9 Гц, 1H), 5,49 (д, J = 9,6 Гц, 1H), 5,32 (д, J = 9,6 Гц, 1H), 4,92 (д, J = 7,8 Гц, 1H), 4,63 (м, 3H), 4,28 (м, 2H), 4,15 (д, J = 8,4 Гц, 1H), 3,86 (д, J = 6,9 Гц, 1H), 2,80 (м, 1H), 3,47 (д, J = 5,4 Гц, 1H), 2,78 (м, 1H), 2,36 (с, 3H), 2,34 ( , 2H), 2,17 (с, 3H), 2,10 (с, 3H), 2,00 (с, 3H), 1,83 (м, 1H), 1,74 (с, 3H), 1,72 (с, 1H), 1,61 (с, 1H), 1,33 (с, 9H), 1,21 (с, 3H), 1,18 (с, 3H).
13C ЯМР (CDCl3, 75,5 МГц), δ: 201,9, 172,3, 170,3, 169,2, 167,0, 154,0, 141,5, 140,2, 133,7, 133,3, 130,2, 129,1, 128,7, 127,0, 125,4, 125,4, 83,9, 81,3, 80,4, 78,6, 76,1, 75,4, 74,5, 74,0, 73,4, 72,5, 57,5, 52,8, 47,2, 43,2, 35,3, 32,9, 28,2, 26,4, 22,6, 20,9, 15,1, 14,7, 10,8.
(b) Получение 3'-N-дебензоил-3'-дезфенил-3'-N- (трет-бутилоксикарбонил)-3'-(2-тиенил)-2'-O-этилоксикарбонил- 7-O-метилтиометилпаклитаксела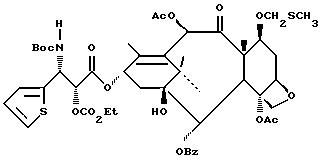
К раствору полученного на стадии (a) продукта (621 мг, 0,677 ммоль) в 35 мл дихлорметана при 0oC добавляли диизопропилэтиламин (1,20 мл, 6,89 ммоль) и этилхлорформиат (0,35 мл, 3,7 ммоль) и перемешивали 1 час. Убирали холодную ванну, и раствор перемешивали 2 часа, разбавляли дихлорметаном, промывали насыщенным раствором бикарбоната, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (гексан : этилацетат = 1 : 1) и получили 528 мг указанного в заголовке соединения (79%).
Масс-спектр FAB (NOBA): M + Na вычисл. для C43H61NO17S2Na: 1010. Найдено: 1010.
ИК-спектр (пленка): 3510, 3440, 1752, 1720, 1370, 1244, 1198, 1170, 1026, 988, 756 см-1.
1H-ЯМР (CDCl3, 300 МГц), δ: 8,09 (д, J=7,2 Гц, 2H), 7,58 (м, 1H), 7,48 (т, J= 7,8 Гц, 2H), 7,26 (м, 1H), 6,99 (, 2H), 6,55 (с, 1H), 6,23 (т, J=9,0 Гц, 1H), 5,68 (д, J=6,9 Гц, 2H), 5,33 (д, J=9,9 Гц, 1H), 5,25 (д, J=2,4 Гц, 1H), 4,94 (д, J=7,8 Гц, 1H), 4,65 (с, 2H), 4,33 - 4,08 (м, 5H), 3,88 (д, J= 6,9 Гц, 1H), 2,80 (м, 1H), 2,40 (с, 3H), 2,40 - 2,20 (м, 2H), 2,16 (с, 3H), 2,11 (с, 3H), 2,07 (с, 3H), 1,83 (м, 1H), 1,74 (с, 3H), 1,69 (с, 1H), 1,60 (с, 1H), 1,33 (с, 9H), 1,31 (т, J=7,2 Гц, 3H), 1,20 (с, 3H), 1,19 (с, 3H).
13C-ЯМР (CDCl3, 75,5 Гц), δ: 202,0, 169,7, 169,1, 167,5, 167,1, 154,0, 140,9, 133,6, 132,9, 130,2, 129,2, 128,7, 127,2, 125,4, 125,3, 83,9, 81,2, 80,6, 78,8, 76,9, 76,0, 75,7, 74,7, 74,2, 72,8, 72,0, 65,2, 57,4, 50,9, 47,1, 43,3, 35,1, 33,0, 28,1, 26,4, 22,7, 21,2, 20,9, 15,1, 14,5, 14,1, 10,9.
(c) Получение 3'-N-(дебензоил-3'-дезфенил-3'-N-(трет- бутилоксикарбонил)-3'-(2-тиенил)-2'-O-этилоксикарбонил-7-O- дибензилфосфонооксиметилпаклитаксела
К раствору полученного на стадии (b) продукта (516 мг, 0,522 ммоль) в 15 мл безводного тетрагидрофурана добавляли 530 мг молекулярных сит 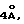 дибензилфосфат (576 мг, 2,09 ммоль) и перекристаллизованный N-иодсукцинимид (136 мг, 0,604 ммоль). К этому раствору добавляли трифторметансульфонат серебра (50 мг, 0,194 ммоль), и раствор перемешивали 1 час. Раствор фильтровали через целит, разбавляли этилацетатом, промывали 10%-ным раствором NaS2O8, насыщенным раствором бикарбоната и рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (15% ацетонитрила в хлороформе) и получили 535 мг указанного в заголовке соединения (84%).
дибензилфосфат (576 мг, 2,09 ммоль) и перекристаллизованный N-иодсукцинимид (136 мг, 0,604 ммоль). К этому раствору добавляли трифторметансульфонат серебра (50 мг, 0,194 ммоль), и раствор перемешивали 1 час. Раствор фильтровали через целит, разбавляли этилацетатом, промывали 10%-ным раствором NaS2O8, насыщенным раствором бикарбоната и рассолом, высушивали над MgSO4 и концентрировали. Остаток хроматографировали на силикагеле (15% ацетонитрила в хлороформе) и получили 535 мг указанного в заголовке соединения (84%).
Масс-спектр FAB (NOBA): M+Na вычисл. для C16H72NO21PSNa: 1240. Найдено: 1240.
ИК-спектр (пленка): 3424 (шир.), 1750, 1722, 1370, 1244, 1016, 1000, 944 см-1.
1H-ЯМР (CDCl3, 300 МГц), δ: 8,08 (д, J=7,0 Гц, 2H), 7,58 (м, 1H), 7,47 (т, J=7,5 Гц, 2H), 7,28 (м, 11H), 6,99 (м, 2H), 6,33 (с, 1H), 6,22 (т, J=7,8 Гц, 1H), 5,66 (м, 2H), 5,39 (т, J=6,6 Гц, 1H), 5,34 (д, J=12 Гц, 1H), 5,22 (д, J= 2,4 Гц, 1H), 5,01 (дд, J=8,1 и 6,0 Гц, 5H), 4,86 (д, J=7,8 Гц, 1H), 4,29 - 4,08 (м, 5H), 3,85 (д, J=6,6 Гц, 1H), 2,76 (м, 1H), 2,39 (с, 3H), 2,35 - 2,18 (м, 2H), 2,16 (с, 3H) 1,97 (с, 4H), 1,69 (с, 4H), 1,33 (с, 9H), 1,30 (т, J=7,2 Гц, 3H), 1,20 (с, 3H), 1,17 (с, 3H).
13C ЯМР (CDCl3, 75,5 Гц), δ: 197,4, 165,4, 164,9, 163,3, 162,7, 150,6, 149,7, 136,7, 136,0, 129,4, 128,6, 125,9, 124,7, 124,3, 124,2, 124,1, 123,6, 122,9, 121,1, 121,0, 89,4, 79,8, 77,3, 76,5, 76,3, 74,4, 72,0, 70,7, 70,3, 67,7, 64,9, 64,9, 60,9, 52,7, 46,5, 42,3, 38,9, 30,7, 23,8, 22,0, 18,3, 17,0, 16,4, 10,3, 9,8, 6,2.
(d) Получение триэтаноламиновой соли 3'-N-дебензоил-3'-дезфенил-3'-N-(трет-бутилоксикарбонил)-3'-(2- тиенил)-2'-O-этилоксикарбонил-7-O-фосфонооксиметилпаклитаксела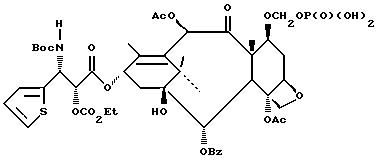
К раствору полученного на стадии (c) продукта (512 мг, 0,42 ммоль) в 30 мл этилацетата добавляли 53 мг палладия на угле (10%), и раствор перемешивали в среде водорода в течение 3 часов. Удаляли катализатор путем фильтрования через целит, и фильтрат концентрировали в вакууме. Остаток растворяли в 2 мл этилацетата, и добавляли триэтаноламин (4,0 мл, 0,1 М этилацетате, 0,40 ммоль). Раствор концентрировали, и остаток хроматографировали на C18 (40% ацетонитрила в воде) и лиофилизировали, получив в результате 280 мг триэтаноламиновой соли (56%). Анализ путем HPLC показал чистоту соли, равную 96%.
Масс-спектр FAB (NOBA): M+Na вычисл. для C47H60NO21PS:1060. Найдено: 1060.
ИК-спектр (KBr): 3422 (шир.), 1750, 1720, 1372, 1246, 1162, 1096, 1068, 1000 см-1.
1H ЯМР (d6 ацетон/D2O, 300 МГц), δ: 8,06 (д, J=7,2 Гц, 2H), 7,63 (т, J= 7,2 Гц, 1H), 7,52 (т, J=7,8 Гц, 2H), 7,38 (д, J=4,2 Гц, 1H), 7,16 (д, J=3,5 Гц, 1H), 7,01 (дд, J=5,1 и 3,6 Гц, 1H) 6,37 (с, 1H), 6,11 (т, J=8,7 Гц, 1H), 5,61 (д, J=6,9 Гц, 1H), 5,60 (с, 1H), 5,26 (д, J=4,5 Гц, 1H), 5,14 (д, J=6,6 Гц, 1H), 5,00 (д, J=8,4 Гц, 1H), 4,86 (дд, J=12,0 и 6,3 Гц, 1H), 4,17 (м, 5H), 4,00 (с, 7H), 3,92 (т, J=4,8 Гц, 6H), 3,84 (д, J=6,9 Гц, 1H), 3,48 (т, J= 5,4 Гц, 6H), 2,94 (м, 1H), 2,42 (с, 3H), 2,36 (м, 1H), 2,27 (м, 1H), 2,15 (с, 3H), 1,95 (с, 4H), 1,66 (с, 3H), 1,30 (с, 9H), 1,23 (т, J=7,2 Гц, 3H) 1,14 (с, 6H).
Пример 9. 10-дезацетил-3'-N-дезбензоил-3'-N-(трет- бутилоксикарбонил)-10-O-(фосфонооксиметил)паклитаксел
(a) Получение 10-дезацетил-10-O-бензилоксикарбонил-7-O- триэтилсилилбаккатина III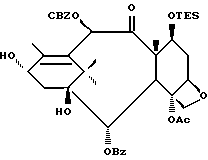
В сухую колбу в среде аргона, содержащую 7-O-триэтилсилил-10- дезацетилбаккатин III (2,093 г, 3,177 ммоль), добавляли сухой тетрагидрофуран (30 мл) и охлаждали до -70oC. Туда же добавляли по каплям 1,6 M н-бутиллитий (2,38 мл, 3,81 ммоль). После перемешивания в течение 15 минут добавляли по каплям бензилхлорформиат (0,91 мл, 6,35 ммоль). Полученную смесь перемешивали 3 часа с постепенным нагреванием до окружающей температуры. Реакцию быстро прекращали посредством 25 мл насыщенного раствора NH4Cl, смесь промывали рассолом и высушивали над MgSO4. Флеш-хроматография (силикагель, 30 - 45% этилацетата в гексане) дала 2,24 г (89%) указанного в заголовке соединения в виде белой пены.
1H ЯМР (CDCl3, 300 МГц), δ: 8,10 (д, J=8,0 Гц, 2H), 7,63 - 7,58 (м, 1H), 7,47 (т, J= 8,0 Гц, 2H), 7,41 - 7,26 (м, 5H), 6,29 (с, 1H), 5,61 (д, J=7,0 Гц, 1H), 5,20 (кв. J=12,2 Гц, 2H), 4,96 (д, J=9,0 Гц, 1H), 4,87 - 4,84 (м, 1H), 4,48 (дд, J=6,7 и 10,4 Гц, 1H), 4,30 (д, J=8,5 Гц, 1H), 4,14 (д, J=8,5 Гц, 1H), 3,84 (д, J=7,0 Гц, 1H), 2,58 - 2,48 (м, 1H), 2,29 (м, 4H), 2,20 (с, 3H), 2,03 (д, J= 5,0 Гц, 1H), 1,92 - 1,83 (м, 1H), 1,68 (с, 3H), 1,17 (с, 3H), 1,04 (с, 3H), 0,91 (т, J=7,5 Гц, 9H), 0,57 (кв. J=7,4 Гц, 6H).
(b) Получение 10-дезацетил-10-O-бензилоксикарбонил-3'- N-дезбензоил-3'-N-(трет-бутилоксикарбонил)-2'-7-бис-O- триэтилсилилпаклитаксела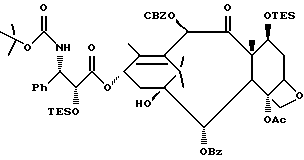
В сухую колбу, содержащую полученный на стадии (a) продукт (3,50 г, 4,42 ммоль), добавляли небольшое количество толуола, после чего раствор концентрировали под вакуумом. Эту колбу помещали в среду аргона, и добавляли в нее 100 мл сухого тетрагидрофурана. Колбу охлаждали до -70oC, и добавляли в нее по каплям 1,0 М гексаметилдисилазид лития (6,19 мл, 6,19 ммоль). После перемешивания в течение 20 минут добавляли по каплям раствор (3R,4S)-1-(трет-бутилоксикарбонил)-4-фенил-3-триэтилсилилокси-2- азетидинона (2,58 г, 7,07 ммоль) в 10 мл сухого ТГФ. Реакционную смесь перемешивали 3,5 часов, постепенно нагревая до температуры окружающей среды. Затем ее быстро охлаждали 70 мл насыщенного раствора NH4Cl, промывали рассолом и высушивали над MgSO4. Флеш-хроматография (силикагель, 5 - 15% этилацетата в гексанах) дала 5,12 г (99%) указанного в заголовке соединения в виде белой пены.
1H ЯМР (CDCl3, 300 МГц), δ: 8,11 (д, J= 8,0 Гц, 2H), 7,60 - 7,58 (м, 1H), 7,48 (т, J = 8,0 Гц, 2H), 7,24 - 7,26 (м, 10H), 6,32 - 6,26, (м, 2H), 5,69 (д, J = 7 Гц, 1H), 5,47 (шд, J = 9,7 Гц, 1H), 5,31 - 5,10 (м, 3H), 4,94 (д, J = 8,5 Гц, 1H), 4,56 (с, 1H), 4,46 (дд, J = 6,9 и 10,6 Гц, 1H), 4,31 (д, J = 8,3 Гц, 1H), 4,17 (д, = 8,3 Гц, 1H), 3,81 (д, J = 7,0 Гц, 1H), 2,53 (с, 3H), 2,48 - 2,33 (м, 1H), 2,22 - 2,17 (м, 1H), 2,09 (с, 3H), 1,95 - 1,86 (м, 1H), 1,70 - (с, 3H), 1,65 (с, 1H), 1,52 (с, 1H), 1,30 (с, 9H), 1,26 - 1,19 (м, 6H), 0,94 - 0,87 (м, 9H), 0,80 - 0,75 (м, 9H), 0,61 - 0,53 (м, 6H), 0,48 - 0,30 (м, 6H).
(c) Получение 10-дезацетил-3'-N-дебензоил-3'-N-(трет-бутилоксикарбонил)-7-O- триэтилсилилпаклитаксела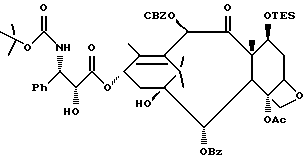
Полученный на стадии (b) продукт (5,12 г, 4,40 ммоль) растворяли в 100 мл этилацетата, переносили в сосуд Парра и помещали под защитный слой аргона. Туда добавляли 10% палладия на угле (2,4 г), и реакционную смесь помещали на гидрогенизационную установку Парра (55 фунтов на кв. дюйм, т.е. 3,87 кг/см2) на 8 часов. Реакционную смесь фильтровали через целит и концентрировали. Флеш-хроматография (силикагель, 15 - 20% этилацетата в гексане) дала 3,24 г (79%) указанного в заголовке соединения в виде белой пены. Гидролиз 2'-триэтилсилильной группы продукта, полученного на стадии Парра.
1H ЯМР (CDCl3, 300 МГц), δ: 8,10 (д, J = 8,0 Гц, 2H), 7,63 - 7,58 (м, 1H), 7,49 (д, J = 8,0 Гц, 2H), 7,39 - 7,26 (м, 5H), 6,27 - 6,17 (м, 1H), 5,64 (д, J = 7,2 Гц), 5,42 (д, J = 9,4 Гц, 1H), 5,28 - 5,25 (м, 1H), 5,12 (с, 1H), 4,92 (д, J = 8,6 Гц, 1H), 4,62 (шс, 1H), 4,38 - 4,28 (м, 3H), 4,17 (д, J = 8,5 Гц, 1H), 3,85 (д, J = 6,7 1H), 3,36 (д, J = 5,3 Гц, 1H), 2,49 - 2,40 (м, 1H), 2,36 (с, 3H), 2,25 (шд, J = 8,7 Гц, 2H), 1,99 - 1,91 (м, 1H), 1,85 (с, 3H), 1,74 (с, 3H), 1,69 (с, 1H), 1,67 (с, 1H), 1,35 (с, 9H), 1,22 (с, 3H), 1,11 (с, 3H), 0,93 (т, J = 7,5 Гц, 9H), 0,61 - 0,49 (м, 6H).
(d) Получение 10-дезацетил-2'-O-бензилоксикарбонил-3'-N-дебензоил-3'-N-(трет- бутилоксикарбонил)-7-O-триэтилсилилпаклитаксела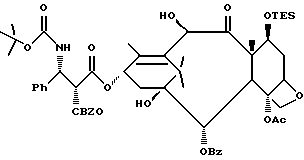
В колбу, содержащую полученный на стадии (с) продукт (3,24 г, 3,51 ммоль), добавляли 30 мл сухого дихлорметана. Колбу помещали под аргон и охлаждали до 0oC. К реакционной смеси добавляли N,N-диизопропилэтиламин (1,22 мл, 7,02 ммоль), а затем добавляли по каплям бензилхлорформиат (1,00 мл, 7,02 ммоль). Через 15 минут охлажденную ванну убирали, и позволяли реакционной смеси перемешиваться при температуре окружающей среды в течение 7 часов. Затем смесь резко охлаждали посредством 30 мл насыщенного раствора NH4Cl, промывали рассолом и высушивали над MgSO4. Флеш-хроматография (силикагель, 7 - 20% этилацетата в гексане) дала 3,24 г (89%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (CDCl3, 300 МГц), δ: 8,10 (д, J = 8,0 Гц, 2H), 7,62 - 7,57 (м, 1H), 7,48 (т, J = 8,0 Гц, 2H), 7,40 - 7,26 (м, 10H), 6,33 - 6,27 (м, 1H), 5,66 (д, J = 7,0 Гц, 1H), 5,49 - 5,42 (м, 2H), 5,31 (с, 1H), 5,22 - 5,13 (м, 3H), 4,93 (д, J = 9,4 Гц, 1H), 4,38 (дд, J = 6,5 и 10,7 Гц, 1H), 4,34 - 4,28 (м, 2H), 4,18 (д, J = 8,3 Гц, 1H), 3,90 (д, J = 6,7 Гц, 1H), 2,52 - 2,30 (м, 4H), 2,24 - 2,20 (м, 1H), 1,97 - 1,87 (м, 3H), 1,74 (с, 3H), 1,59 (с, 3H), 1,32 (с, 9H), 1,26 (с, 3H), 1,11 (с, 3H), 0,96 - 0,88 (м, 9H), 0,61 - 0,48 (м, 6H).
(e) Получение 10-деацетил-2'-O-бензилоксикарбонил-3'-N-дебензоил-3'-N-(трет- бутилоксикарбонил)-10-O-(дибензилфосфонооксиметил)-7-O- триэтилсилилпаклитаксела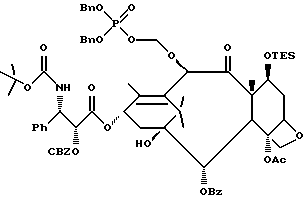
Полученный на стадии (d) продукт растворяли в 13,5 мл (54%) ДМСО, 8,75 мл (35%) уксусного ангидрида и 2,75 мл (11%) ледяной уксусной кислоты и помещали в атмосферу аргона. Реакционную смесь перемешивали 56 часов, после чего ее разбавляли этилацетатом до объема 60 мл. Раствор промывали насыщенным раствором NaHCO3, пока он не стал нейтральным при проверке индикаторной бумаги для определения pH, и затем промывали рассолом. Органическую фракцию высушивали MgSO4 и концентрировали. Флеш-хроматография (15 - 20% этилацетата в гексане) дала 3,12 г неочищенной белой пены с требуемым тиометилацетальным продуктом (т. е. 10-дезацетил-2'-O-бензилоксикарбонил-3'-N-дебензоил- 3'-N-(трет-бутилоксикарбонил)-10-O-(метилтиометил)-7-O- триэтилсилилпаклитакселом), составляющим 70% материала по ЯМР.
Затем упомянутую выше неочищенную смесь (3,12 г) растворяли в 1,2-дихлорэтане (61 мл) и помещали под защитный слой аргона. Добавляли порошкообразные молекулярные сита  (3,12 г), и полученную гетерогенную смесь интенсивно перемешивали. Добавляли к ней посредством канюли раствор перекристаллизованного N-иодсукцинимида (0,830 г, 3,69 ммоль) и дибензилфосфата (1,027 г, 3,69 ммоль) в сухом тетрагидрофуране (46 мл). Полученную смесь перемешивали 5 часов, фильтровали через слой целита и разбавляли до объема 250 мл этилацетата. Промывали ее холодным 2%-ным раствором NaHCO3 (2 х 125 мл), холодным 6%-ным раствором NaHCO3 (2 х 125 мл) и рассолом. Органическую фазу высушивали (MgSO4) и концентрировали. Флеш-хроматография (силикагель 25 - 35% этилацетата в гексане) дала 1,52 г (40%) указанного в заголовке соединения в виде белого твердого вещества.
(3,12 г), и полученную гетерогенную смесь интенсивно перемешивали. Добавляли к ней посредством канюли раствор перекристаллизованного N-иодсукцинимида (0,830 г, 3,69 ммоль) и дибензилфосфата (1,027 г, 3,69 ммоль) в сухом тетрагидрофуране (46 мл). Полученную смесь перемешивали 5 часов, фильтровали через слой целита и разбавляли до объема 250 мл этилацетата. Промывали ее холодным 2%-ным раствором NaHCO3 (2 х 125 мл), холодным 6%-ным раствором NaHCO3 (2 х 125 мл) и рассолом. Органическую фазу высушивали (MgSO4) и концентрировали. Флеш-хроматография (силикагель 25 - 35% этилацетата в гексане) дала 1,52 г (40%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (CDCl3, 300 МГц), δ: 8,08 (д, J= 7,0 Гц, 2H), 7,59 - 7,55 (м, 1H), 7,46 (т, J = 7,2 Гц, 2H), 7,38 - 7,25 (м, 20H), 6,30 (т, J = 8,5 Гц, 1H), 5,65 (д, J = 6,8
Гц, 1H), 5,49 - 5,39 (м, 4H), 5,32 (с, 1H), 5,18 - 4,19 (м, 4H), 4,93 (д, J = 9,2 Гц, 1H), 4,44 (дд, J = 6,6 и 10,2 Гц, 1H), 4,31 (д, J = 8,4 Гц, 1H), 4,16 (д, J = 8,5 Гц, 1H), 3,80 (д, J = 6,9 Гц, 1H), 2,69 - 2,39 (м, 4H), 2,33 - 2,23 (м, 3H), 2,03 (с, 3H), 1,90 (т, J = 12,6 Гц, 1H), 1,68 - 1,63 (м, 6H), 1,28 (с, 9H), 1,16 - 1,10 (м, 6H), 0,93 (т, J = 7,4 Гц, 9H), 0,55 (кв., J = 7,8 Гц, 6H).
13C ЯМР (CDCl3, 75,5 МГц), δ: 204,1, 169,7, 167,9, 167,1, 151,1, 140,7, 135,7, 133,6, 130,2, 129,2, 128,9, 128,8, 128,7, 128,6, 128,5, 128,4, 128,3, 128,2, 128,0, 127,8, 126,4, 90,4, 84,2, 81,1, 80,4, 79,3, 78,8, 74,9, 72,8, 72,0, 70,5, 69,2, 69,1, 69,0, 58,1, 46,8, 43,2, 37,1, 35,0, 28,1, 26,5, 22,8, 21,0, 14,1, 10,0, 6,9, 5,5.
Масс-спектр (FAB): m/z+: 1345.
(f) Получение 10-дезацетил-2'-O-бензилоксикарбонил-3'-N-дебензоил-3'-N-(трет -бутилоксикарбонил)-10-O-(дибензилфосфонооксиметил)паклитаксела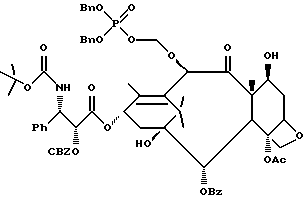
Раствор полученного на стадии (е) продукта (50,8 мг, 0,038 ммоль) в сухом тетрагидрофуране (2,5 мл) под аргоном охлаждали до -40oC. К этому раствору добавляли по каплям тетрабутиламмонийфторид (0,057 мл, 0,057 ммоль) в тетрагидрофуране (1,0 М). Реакционную смесь перемешивали 1,5 часа при постепенном нагреве до -20oC. Смесь резко охлаждали 15 мл насыщенного раствора NH4Cl и разбавляли 30 мл этилацетата. Органическую фазу промывали NaHCO3 (2 х 15 мл) и рассолом. Затем ее высушивали (MgSO4) и концентрировали. Препаративная тонкослойная хроматография (силикагель, 50% этилацетата в гексане) дала 36 мг (77%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР (CDCl3; 300 МГц), δ: 8,10 (д, J = 8,5 Гц, 2H), 7,60 - 7,55 (м, 1H), 7,49 - 7,44 (м, 2H), 7,36 - 7,18 (м, 20H), 6,27 - 6,22 (м, 1H), 5,78 (с, 1H), 5,67 (д, J = 7,0 Гц, 1H), 5,44 - 5,34 (м, 3H), 5,27 (д, J = 2,2 Гц, 1H), 5,24 - 5,05 (м, 4H), 5,01 - 4,91 (м, 4H), 4,39 - 4,28 (м, 2H), 4,17 (д, J = 8,2 Гц, 1H), 3,87 (д, J = 7,0 Гц, 1H), 2,58 - 2,51 (м, 1H), 2,41 (с, 3H), 2,40 - 2,18 (м, 2H), 2,00 - 1,87 (м, 5H), 1,73 - 1,69 (м, 4H), 1,30 (с, 9H), 1,22 - 1,15 (м, 6H).
Масс-спектр (FAB): m/z+: 1231.
(g) Получение 10-дезацетил-3'-N-дезбензоил-3'-N-(трет-бутилоксикарбонил)-10-O -(фосфонооксиметил)паклитаксела триэтаноламиновой соли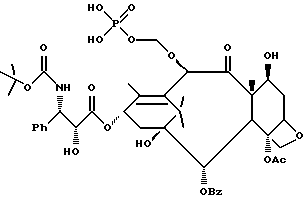
Колбу Парра емкостью 500 мл загружали 10-дезацетил-2'-O- бензилоксикарбонил-3'-N-дезбензоил-3'-N-(тетра-бутилоксикарбонил)-10-O- (дибензилфосфонооксиметил)паклитакселом (264,9 мг, 0,215 ммоль) и этилацетатом (20 мл). Потом колбу обдували струей аргона, и добавляли в нее 10% Pd/C (318 мг). Полученную смесь помещали на установку Парра в среду водорода под давлением 55 фунтов на кв. дюйм (3,86 кг/см2). Реакцию контролировали путем HPLC (70:30 буфер CH3CN/Q8, pH 6,0, 1,00 мл/мин, колонка Зорбакса C-18, 25,0 см, λ = 230 нм), до тех пор, пока не стало видно исходного материала (12,5 часов). Смесь фильтровали через слой целита, который промывали этилацетатом и небольшим количеством дихлорметана. Полученный фильтрат концентрировали, и остаток растворяли в дихлорметане (5 мл). Добавление гексана вызвало образование осадка, из которого было выделено 140,3 мг свободной кислоты (чистота при определении путем HPLC - 80%) в виде белого твердого вещества. Этот материал направляли прямо на следующую стадию.
В колбу, содержащую упомянутую выше свободную кислоту (140 мг, 0,153 ммоль), добавляли дихлорметан (10 мл). Затем полученный раствор обрабатывали 0,100 М раствором триэтаноламина в этилацетате (1,16 мл, 0,116 ммоль), в результате чего раствор стал мутным. Добавляли приблизительно 2 мл гексана и смесь держали при -20oC всю ночь. Полученный осадок отфильтровывали через воронку с пористой (4,0 - 5,5 мкм) стеклянной пластинкой. Твердое вещество собирали и помещали под вакуум на 4 часа, что дало 69,9 мг (42%) указанной в заголовке триэтаноламиновой соли в виде серого порошка, который имел чистоту 95 - 96% при определении путем HPLC-анализа. (TR = 2,05 мин, Буфер 70:30
CH3CN/Q8, pH 6,0, 1,00 мл/мин, Зорбакс C-18, 25,0 см, λ = 230 нм).
1H ЯМР (d6-ацетон/D2O, 300 МГц), δ: 8,03 (д, J = 7,4 Гц, 2H), 7,65 (т, J = 7,3 Гц, 1H), 7,54 (т, J = 7,6 Гц, 2H), 7,42 - 7,33 (м, 5H), 7,21 (т, J = 7,0 Гц, 1H), 6,09 (т, J = 9,0 Гц, 1H), 5,81 (с, 1H), 5,59 (д, J = 7,0 Гц, 1H), 5,12 (шс, 2H), 4,93 (д, J = 8,4 Гц, 2H), 4,56 (д, J = 4,9 Гц, 1H), 4,31 - 4,26 (м, 1H), 4,11 (с, 2H), 3,41 - 3,37 (м, 6H), 2,42 - 2,32 (м, 5H), 2,15 (шс, 1H), 1,97 (с, 3H), 1,77 - 1,64 (м, 2H), 1,58 (с, 3H), 1,13 (с, 9H), 1,15 - 1,07 (м, 6H).
13C ЯМР (d6-ацетон, D2O, 75,5 МГц), δ: 171,6, 166,9, 156,6, 141,8, 135,1, 134,2, 131,0, 130,7, 129,4, 129,3, 128,4, 128,1, 88,3, 85,4, 81,9, 79,7, 78,6, 78,1, 76,8, 76,0, 74,8, 71,9, 71,2, 47,4, 44,0, 37,1, 36,3, 28,5, 27,0, 23,1, 22,0, 14,7, 10,4.
Масс-спектр высокого разрешения: MNa+ - 940,3142,
Вычисленное для C44H56NO18PNa = 940,3133.
Пример 10. 2'-O-фосфонооксиметоксиметилпаклитаксел
(a) Получение 2'-O-(метилтиометоксиметил)-7-O-триэтилсилилпаклитаксела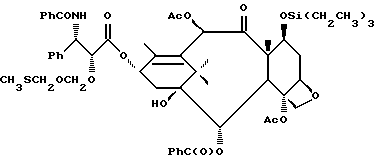
К раствору 7-O-триэтилсилилпаклитаксела (70,0 мг, 72,2 ммоль), бис (метилтиометил)эфира (90 мг, 72,2 ммоль), молекулярных сит (70 мг) и N-иодсукцинимида (160 мг, 72,2 ммоль) в тетрагидрофуране (2,0 мл) при комнатной температуре добавляли трифлат серебра (5,0 мг, 19,5 ммоль), и полученный раствор перемешивали 2 часа. Затем реакционную смесь разбавляли этилацетатом и фильтровали через слой целита. Фильтрат промывали насыщенным водным раствором бикарбоната натрия, а затем смесью (1:1, объем/объем) насыщенного водного раствора бикарбоната натрия и 5%-ного водного раствора тиосульфата натрия и наконец рассолом. Органику затем высушивали над сульфатом натрия и концентрировали в вакууме. Остаточное масло очищали путем флеш-хроматографии (3: 1, гексаны-этилацетат) и получили в результате 22,0 мг (29%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (CDCl3, 300 МГц), δ: 8,12 - 7,20 (15H, м), 7,04 (1H, д, J = 8,9 Гц), 6,41 (1H, с), 6,25 (1H, м), 5,81 (1H, дд, J = 8,9 и 2,4 Гц), 5,68 (1H, д, J = 7,0 Гц), 4,93 (1H, д, J = 8,0 Гц), 4,79 (2H, м), 4,71 (1H, д, J = 2,4 Гц), 4,45 (1H, дд, J = 10,5 и 6,6 Гц), 4,30 (1H, д, J = 8,3 Гц), 4,28 (1H, д, J = 11,7 Гц), 4,17 (1H, д, J = 8,3 Гц), 4,04 (1H, д, J = 11,7 Гц), 3,80 (1H, д, J = 6,9 Гц), 2,48 - 1,13 (25H, м, вкл. синглеты при 2,51, 2,13, 2,05, 2,01, 1,69, 1,19, 1,16), 0,98 - 0,85 (9H, м), 0,65 - 0,50 (6H, м).
(b) Получение 2'-O-(дибензилфосфонооксиметоксиметил)-7- триэтилсилилпаклитаксела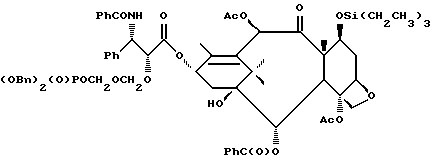
К раствору полученного на стадии (a) продукта (15 мг, 0,0141 ммоль) и молекулярных сит (15 мг) в ТГФ (0,5 мл) при комнатной температуре добавляли дибензилфосфат (20,0 мг, 0,089 ммоль) и затем N-иодсункцинимид (4,2 мг, 0,0187 ммоль), и раствор перемешивали 1 час. В этот момент анализ реакционной смеси путем тонкослойной хроматографии показал присутствие только исходного материала. После этого добавляли тремя частями в течение 2 часов трифлат серебра (5,0 мг, 0,0189 ммоль), и реакционную смесь перемешивали еще 1 час. Затем реакционную смесь разбавляли этилацетатом, и полученный раствор фильтровали через слой целита. Фильтрат обрабатывали смесью (1: 1 объем/объем) насыщенного водного раствора бикарбоната натрия и 5%-ного водного раствора тиосульфата натрия. Органический экстракт затем промывали рассолом, высушивали над сульфатом натрия и концентрировали в вакууме. Остаточное масло очищали путем флеш-хроматографии (1:1, гексан:этилацетат) и получили 5,0 мг (33%) указанного в заголовке соединения.
1H ЯМР (CDCl3, 300 МГц), δ: 8,08-7,16 (25H, м), 7,18 (1H, д, J = 8,8 Гц), 6,41 (1H, с), 6,21 (1H, м), 5,82 (1H, дд, J = 9,0 и 3,1 Гц), 5,66 (1H, д, J = 7 Гц), 5,01-4,65 (10H, м), 4,56 (1H, дд, J = 14,7 и 5,6 Гц), 4,43 (1H, дд, J = 10; 4 и 6,7 Гц), 4,29 (1H, д, J = 8,3 Гц), 4,16 (1H, д, J = 8,3 Гц), 3,78 (1H, д, J = 7,0 Гц), 2,60 - 1,13 (22H, м, вкл. синглеты при 2,49, 2,15, 1,93, 1,66, 1,15, 1,13, 3H каждый), 0,95 - 0,84 (9H, м), 0,63 - 0,45 (6H, м).
(c) Получение 2'-O-фосфонооксиметоксиметилпаклитаксела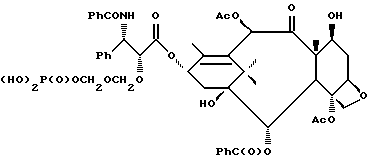
Полученный на стадии (b) продукт обрабатывали тетрабутиламмонийфторидом в соответствии с процедурой, описанной в примере 9 (f), для удаления 7-O-триэтилсилилзащитной группы. Полученное при этом соединение подвергали каталитической гидрогенизации в соответствии с процедурой, описанной в предшествующих примерах, и получили указанное в заголовке соединение.
Пример II. 2'-O-фосфонооксиметоксиметилпаклитаксел (Альтернативный способ)
(a) Получение 2'-O-триэтилсилилпаклитаксела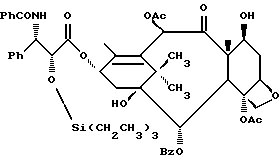
К раствору паклитаксела (20,0 г, 0,0234 моль) и имидазола (3,59 г, 0,052 моль) в 150 мл ДМФ (диметилформамида) при 0oC добавляли порциями по 2 мл в течение 20 мин триэтилсилилхлорид (6,0 мл, 0,053 моль). Затем реакционную смесь перемешивали при 0oC в течение 1 часа. Далее смесь разбавляли этилацетатом и насыщенным водным раствором аммонийхлорида. Органический слой отделяли, промывали рассолом, высушивали над сульфатом натрия и концентрировали в вакууме, получив в результате желтое масло. Очистка неочищенного продукта путем флеш-хроматографии (гексаны:этилацетат, 1:3, а затем 1:1) дала 21,07 г (выход 98%) целевого указанного в заголовке соединения в виде неокрашенного белого твердого вещества.
1H ЯМР (CDCl3, 300 МГц), δ: 8,15 (2H, м), 7,70 (2H, м), 7,65 - 7,30 (11H, м), 7,15 (1H, д, J = 8,9 Гц), 6,30 (1H, с), 6,25 (1H, м), 6,70 - 6,10 (2H, м), 4,94 (1H, д, J = 7,9 Гц), 4,67 (1H, д, J = 2,0 Гц), 4,40 (1H, м), 4,29 (1H, д, J = 8,4 Гц), 4,18 (1H, д, J = 8,4 Гц), 3,81 (1H, д, J = 7,1 Гц), 2,65 - 1,10 (22H, включая синглеты при 2,55, 2,20, 1,88 1,69, 1,22, 1,13, 3H, каждый).
(b) Получение 2'-O-триэтилсилил-7-O-бензилоксикарбонилпаклитаксела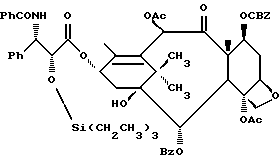
К раствору 2'-O-триэтилсилилпаклитаксела (22,3 г, 24,1 ммоль) в ТГФ (250 мл), охлажденном до -50oC, добавляли по каплям в течение 10 минут бутиллитий (1,6 М в гексанах, 12,9 мл, 8,06 ммоль). Полученный раствор перемешивали 20 мин, поддерживая при этом температуру в пределах между -50oC и -35oC. Затем реакционную смесь охлаждали до -50oC и добавляли в нее по каплям в течение 5 минут бензилхлорформиат (5,0 мл, 29,8 ммоль). Реакционную смесь выдерживали при -40oC в течение 30 минут, после чего приводили в равновесие до 0oC в течение приблизительно 30 минут. Затем смесь разбавляли этилацетатом и насыщенным водным раствором аммоний хлорида, и полученный органический слой промывали рассолом, высушивали над сульфатом натрия и концентрировали в вакууме. 1H ЯМР-анализ неочищенной смеси показал наличие требуемого 2'-O-триэтилсилил-7-O-бензилоксикарбонилпаклитаксела, а также 2'-O-триэтилсилил-7-эпигидроксипаклитаксела (отношение 3: 1 соответственно). Эту смесь продуктов использовали на следующей стадии без дальнейшей очистки и затем разделения изомеров. Аналитическую пробу основного продукта 2'-O-триэтилсилил-7-O-бензилоксикарбонилпаклитаксела очищали путем флеш-хроматографии.
1H ЯМР (CDCl3, 300 МГц), δ: 8,12 (2H, м), 7,72 (1H, м), 7,65 - 7,27 (1H, д, J = 8,8 Гц), 6,41 (1H, м ), 6,20 (1H, м), 5,72 - 5,65 (2H, м), 5,52 (1H, м), 5,24 (1H, д, J = 12,3 Гц), 5,16 (1H, д, J = 12,3 Гц), 4,95 (1H, д, J = 8,7 Гц), 4,69 (1H, с), 4,35 (1H, д, J = 8,3 Гц), 4,25 (1H, д, J = 8,3 Гц), 3,94 (1H, д, J = 6,8 Гц), 2,70 - 1,12 (22H, вкл. синглеты при 2,54, 2,14, 2,01, 1,80, 1,20, 1,15, 3H каждый), 0,81 - 0,73 (9H, м), 0,55 - 0,31 (6H, м).
(c) Получение 7-O-бензилоксикарбонилпаклитаксела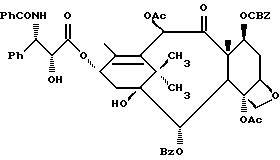
К раствору полученного на стадии (b) продукта (24,0 г, 22,6 ммоль) в ацетонитриле (250 мл), охлажденном до 0oC, добавляли хлористоводородную кислоту (6 н., 1,0 мл, 6,0 ммоль). Через 10 минут анализ путем тонкослойной хроматографии (гексаны: этилацетат, 1: 1) показал, что реакция завершена. Реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия, а затем этилацетатом и органический слой отделяли, промывали рассолом, высушивали сульфатом натрия и концентрировали в вакууме. Остаточное масло очищали путем флеш-хроматографии (гексаны:этилацетат, 1:3, а затем 1:1) и получили в результате 11,4 г (48% за две стадии) указанного в заголовке соединения и 4,8 г (20%) 7-эпигидроксипаклитаксела.
1Н ЯМР (CDCl3, 300 МГц), δ: 8,09 (2H, м), 7,71 (2H, м), 7,65 - 7,27 (16H, м), 7,10 (1H, д, J = 8,9 Гц), 6,39 (1H, с), 6,16 (1H, м), 5,81 (1H, д, J = 8,9 и 2,4 Гц), 5,65 (1H, д, J = 6,9 Гц), 5,49 (1H, дд, J = 10,6 и 7,2 Гц), 5,20 (1H, д, J = 11,9 Гц), 5,12 (1H, д, J = 11,9 Гц), 4,91 (1H, д, J = 8,4 Гц), 4,78 (1H, м), 4,30 (1H, д, J = 8,4 Гц), 4,15 (1H, д, J = 8,4 Гц), 3,91 (1H, д, J = 6,8 Гц), 3,39 (1H, д, J = 4,9 Гц), 2,65 - 1,10 (22H, вкл. синглеты при 2,39, 2,18, 1,81, 1,75, 1,21, 1,15, 3H каждый).
(d) Получение 2'-O-(метилтиометоксиметил)-7-O- бензилоксикарбонилпаклитаксела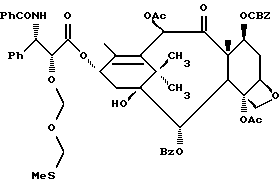
К раствору 7-O-бензилоксикарбонилпаклитаксела (5,53 г, 5,71 ммоль), простого 1,1'-дитиометилдиметилового эфира (7,8 г, 57,1 ммоль), N-иодсукцинимида (6,35 г, 28,3 ммоль) и высушенных в печи порошкообразных молекулярных сит (5,0 г), в ТГФ (110 мл) при комнатной температуре добавляли трифлат серебра (300 мг, 1,17 ммоль). Анализ путем тонкослойной хроматографии (гексаны:этилацетат, 1:1) реакционной смеси через 20 минут показал превращение примерно 40% исходного материала в высшие фракции. Затем добавляли трифлат серебра (150 мг, 0,585 ммоль), и контролировали реакцию путем тонкослойной хроматографии, которая показала, что спустя 30 минут реакция была завершена приблизительно на 65%. Смесь разбавляли этилацетатом (100 мл), фильтровали через слой целита, и фильтрат выливали в делительную воронку, содержавшую 20 мл насыщенного водного раствора бикарбоната натрия и 50 мл 5%-ного водного раствора тиосульфата натрия. Органический слой отделяли, промывали рассолом, высушивали над сульфатом натрия и концентрировали в вакууме. Остаточное масло очищали путем флеш-хроматографии (гексаны:этилацетат, градиентное элюирование, от 4:1 до 3:2) и получили 3,0 г (выход 54%) указанного в заголовке соединения в виде светло-желтого твердого вещества.
1H ЯМР (CDCl3, 300 МГц), δ: 8,10 (2H, м), 7,74 (2H, м), 7,66 - 7,25 (18H, м), 7,05 (1 H, д, J = 8,9 Гц), 6,40 (1H, с), 6,26 (1H, м), 5,77 (1H, дд, J = 8,8 и 2,5 Гц), 5,71 (1H, д, J = 6,9 Гц), 5,51 (1H, дд, J = 10,6 и 7,1 Гц), 5,21 (1H, д, J = 11,9 Гц), 5,14 (1H, д, J = 11,9 Гц), 4,92 (2H, м), 4,68 (1H, д, J = 2,5 Гц), 4,31 (1H, д, J = 11,8 Гц), 4,30 (1H, д, J = 8,5 Гц), 4,16 (1H, д, J = 8,5 Гц), 4,10 (1H, д, J = 11,8 Гц), 3,93 (1H, д, J = 6,9 Гц), 2,65 - 1,10 (25H, включ. синглеты при 2,50, 2,15, 2,05, 1,74, 1,72, 1,20, 1,15, 3H каждый).
(e) Получение 2'-O-дибензилфосфонооксиметоксиметил)-7-O- бензилоксикарбонилпаклитаксела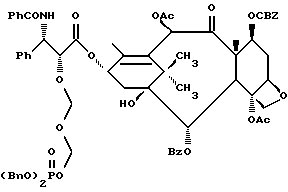
К раствору 2'-O-(метилтиометоксиметил)-7-O-бензилоксикарбонилпаклитаксела (1,06 г, 1,07 ммоль) и высушенных в печи порошкообразных молекулярных сит (1,0 г), в ТГФ (20 мл) при комнатной температуре дибензилфосфат (1,49 г, 5,30 ммоль), а затем сразу же N-иодсукцинимид (2,65 г, 1,18 ммоль). Анализ путем тонкослойной хроматографии (гексаны:этилацетат, 1:1) реакционной смеси через 2,5 часа показал, что реакция завершена приблизительно на 60%. После этого добавляли N-иодсукцинимид (175 мг, 0,78 ммоль), и реакционную смесь перемешивали еще 30 минут, после чего анализ путем тонкослойной хроматографии показал, что реакция завершена. Далее реакционную смесь разбавляли этилацетатом (50 мл) и фильтровали через слой целита. Фильтрат выливали в делительную воронку, содержавшую 100 мл насыщенного раствора бикарбоната натрия и 20 мл 5%-ного водного раствора тиосульфата натрия. Органический слой отделяли, промывали рассолом, высушивали над сульфатом натрия, и концентрировали в вакууме. Остаточное масло очищали путем флеш-хроматографии (гексаны:этилацетат, градиентное элюирование, от 3:1 до 1:1) и получили 750 мг (62%-ный выход целевого указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (CDCl3, 360 МГц), δ: 8,10 (2H, м), 7,79 (2H, м), 7,65 - 7,24 (26H, м), 7,10 (1H, м), 6,41 (1H, с), 6,20 (1H, м), 5,79 (1H, дд, J = 8,8 и 3,6 Гц), 5,65 (1H, д, J = 7,0 Гц), 5,52 (1H, м), 5,20 (1H, д, J = 11,8 Гц), 5,11 (1H, д, J = 11,8 Гц), 5,04 - 4,85 (6H, м), 4,75 - 4,60 (4H, м), 4,30 (1H, д, J = 8,4 Гц), 4,15 (1H, д, J = 8,4 Гц), 3,92 (1H, д, J = 7,0 Гц), 2,65 - 11,0 (22H, включ. синглеты при 2,48, 2,19, 1,95, 1,80, 1,20, 1,10, 3H каждый).
(f) получение триэтаноламиновой соли 2'-O-фосфонооксиметоксиметилпаклитаксела
К раствору 2'-O-(дибензилфосфонооксиметоксиметил)-7-O- бензилоксикарбонилпаклитаксела (500 мг, 0,382 ммоль) в этилацетате (40 мл) в колбе Парра добавляли палладий (10%) на угле. Колбу закрепляли на аппарате Парра, и реакционную смесь подвергали воздействию водорода при 50 фунтах на кв. дюйм (3,5 кг/см2). Реакционную смесь встряхивали 6,5 ч, а затем фильтровали через воронку с пористой стеклянной пластинкой. К этому фильтрату добавляли триэтаноламин (0,1 н. в этилацетате, 4,0 мл), и полученный раствор концентрировали в вакууме. Неочищенное твердое вещество суспендировали в примерно 5,0 мл этилацетата, и декантировали растворитель. Этот процесс повторяли три раза, в результате чего получили указанную в заголовке триэтаноламиновую соль (300 мг) с чистотою 87% при определении путем HPLC-анализа. Дальнейшая очистка этого соединения путем хроматографирования на колонке C18 (вода:ацетонитрил, 3: 1) дала указанное в заголовке целевое соединение (120 мг, 34%) с чистотою 95% при определении путем HPLC.
1H ЯМР (CD СОCD3, D2O, 300 МГц), δ: 9,05 (1H, д, J = 8,7 Гц), 8,15 - 7,12 (21H, м), 6,40 (1H, м), 6,05 (1H, м), 5,69 - 5,55 (2H, м), 5,01 - 4,85 (6H, м), 4,35 (1H, м), 4,14 (2H, м), 3,96 - 3,85 (6H, м), 3,25 (1H, д, J = 7,1 Гц).
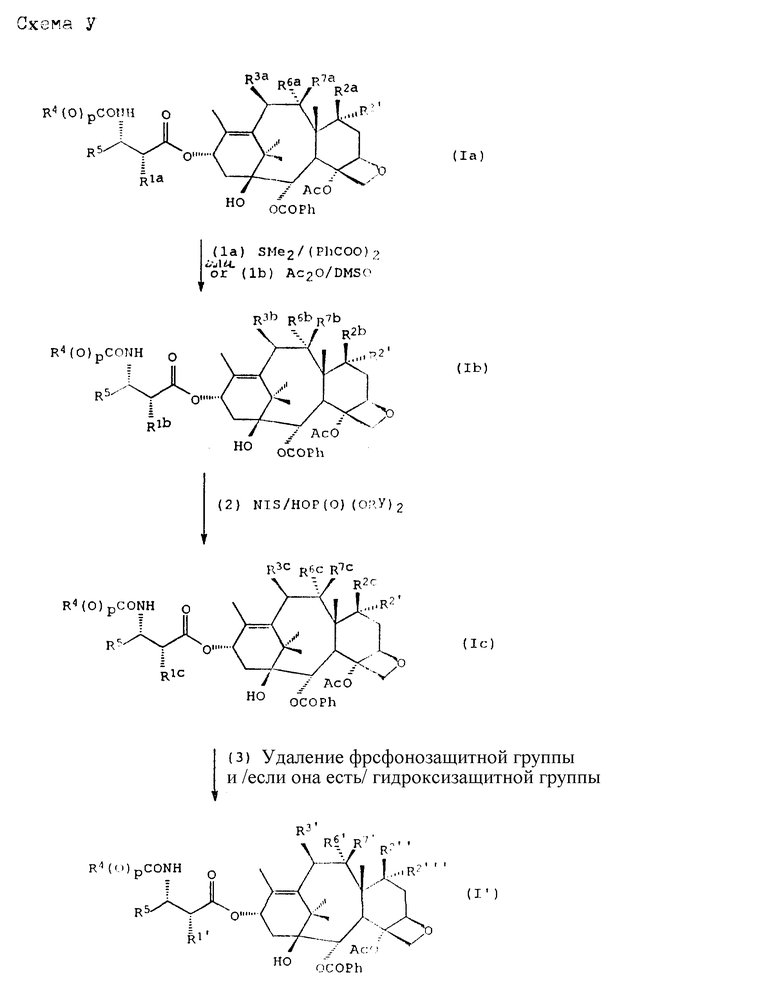
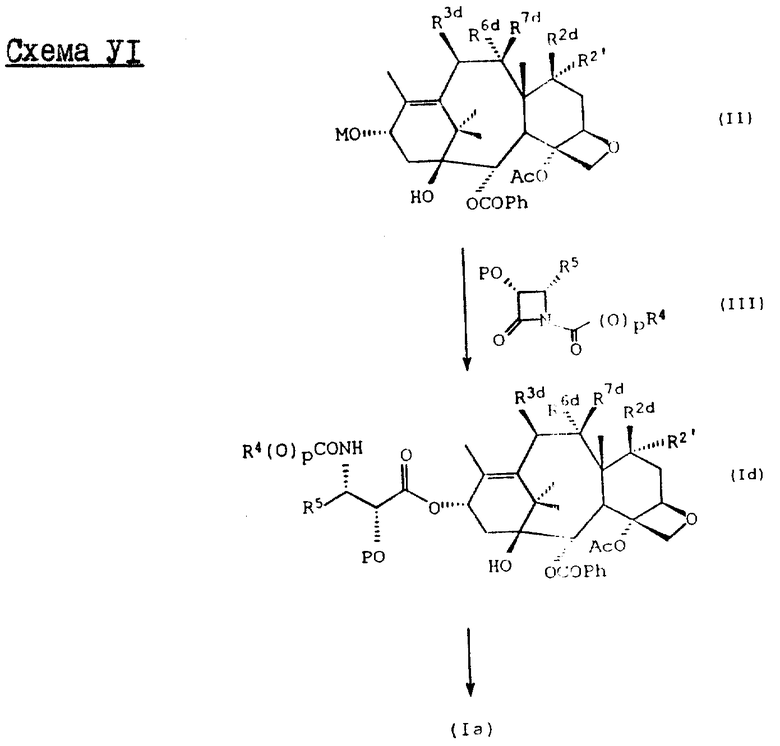
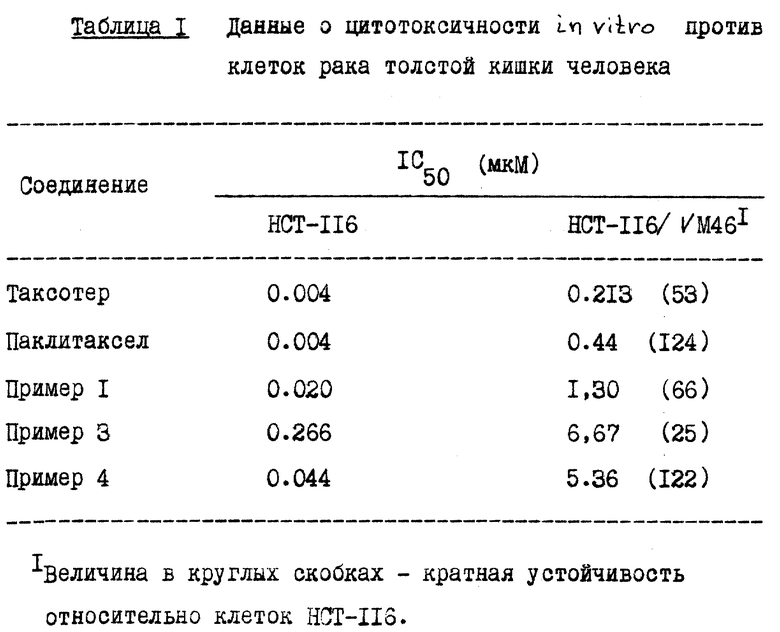

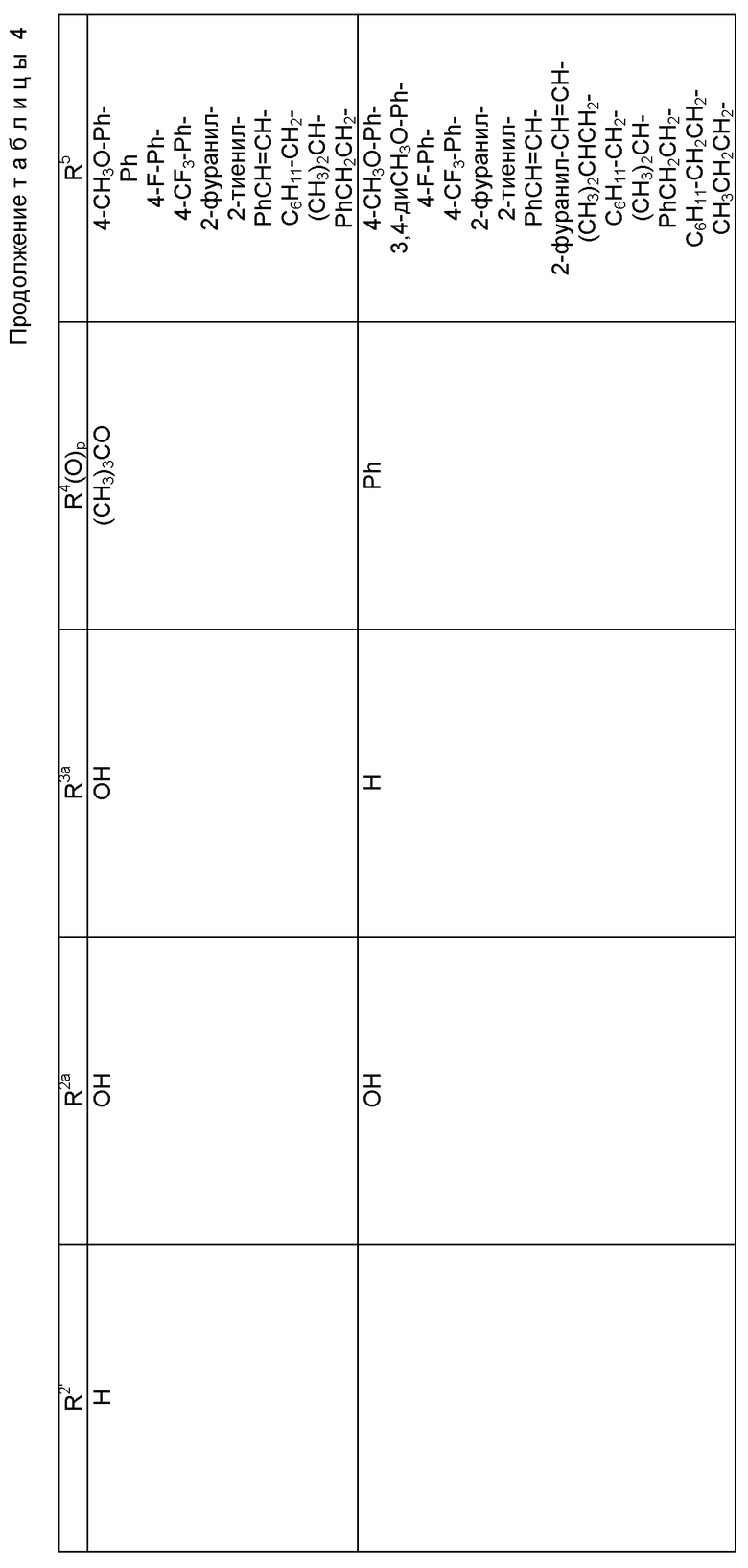
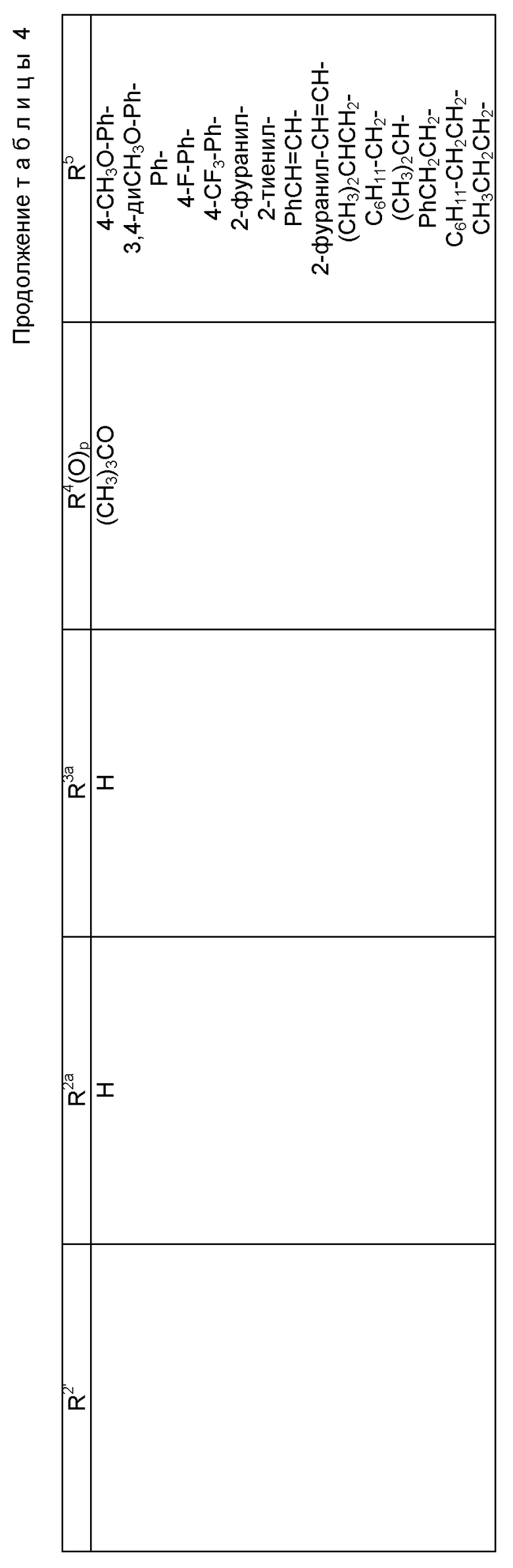
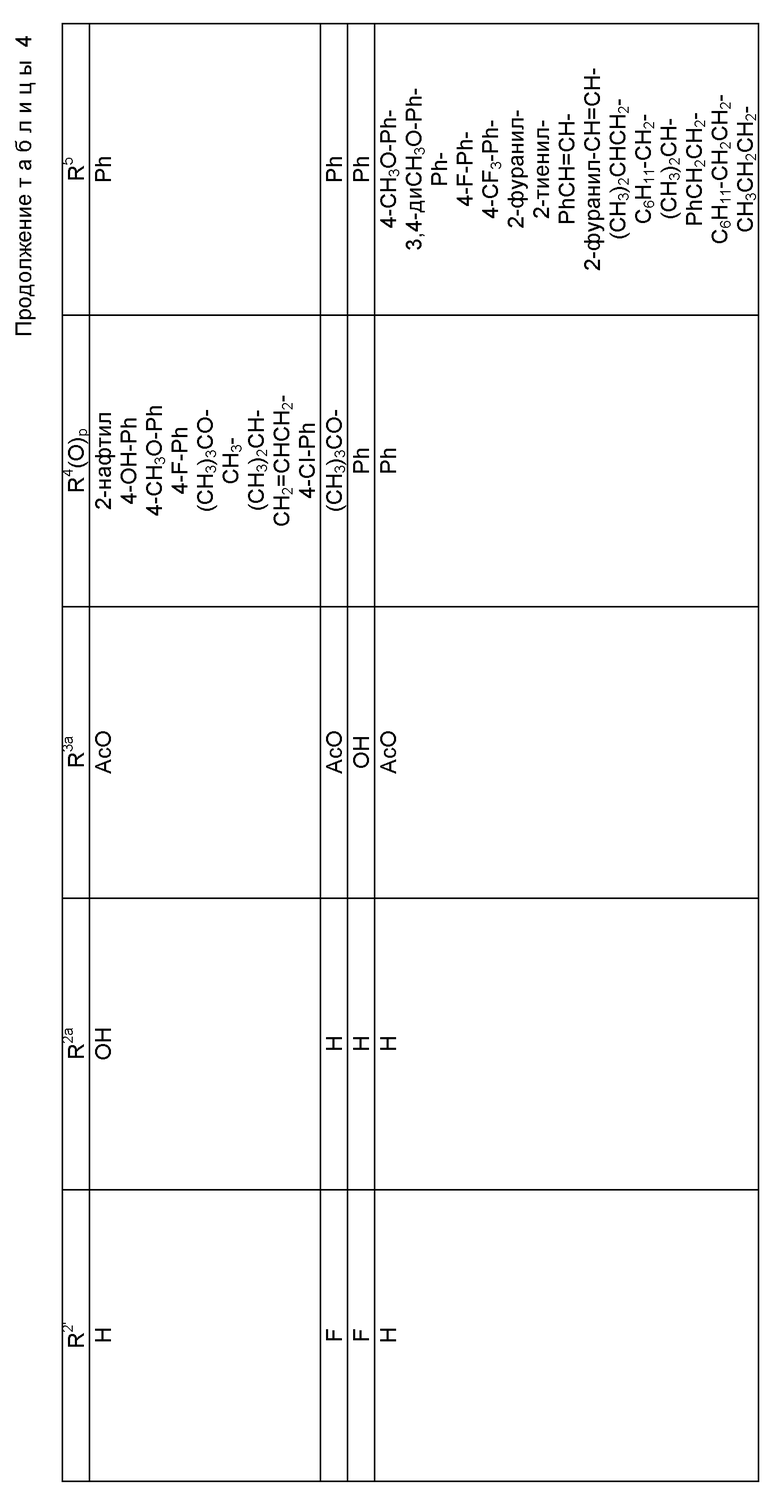
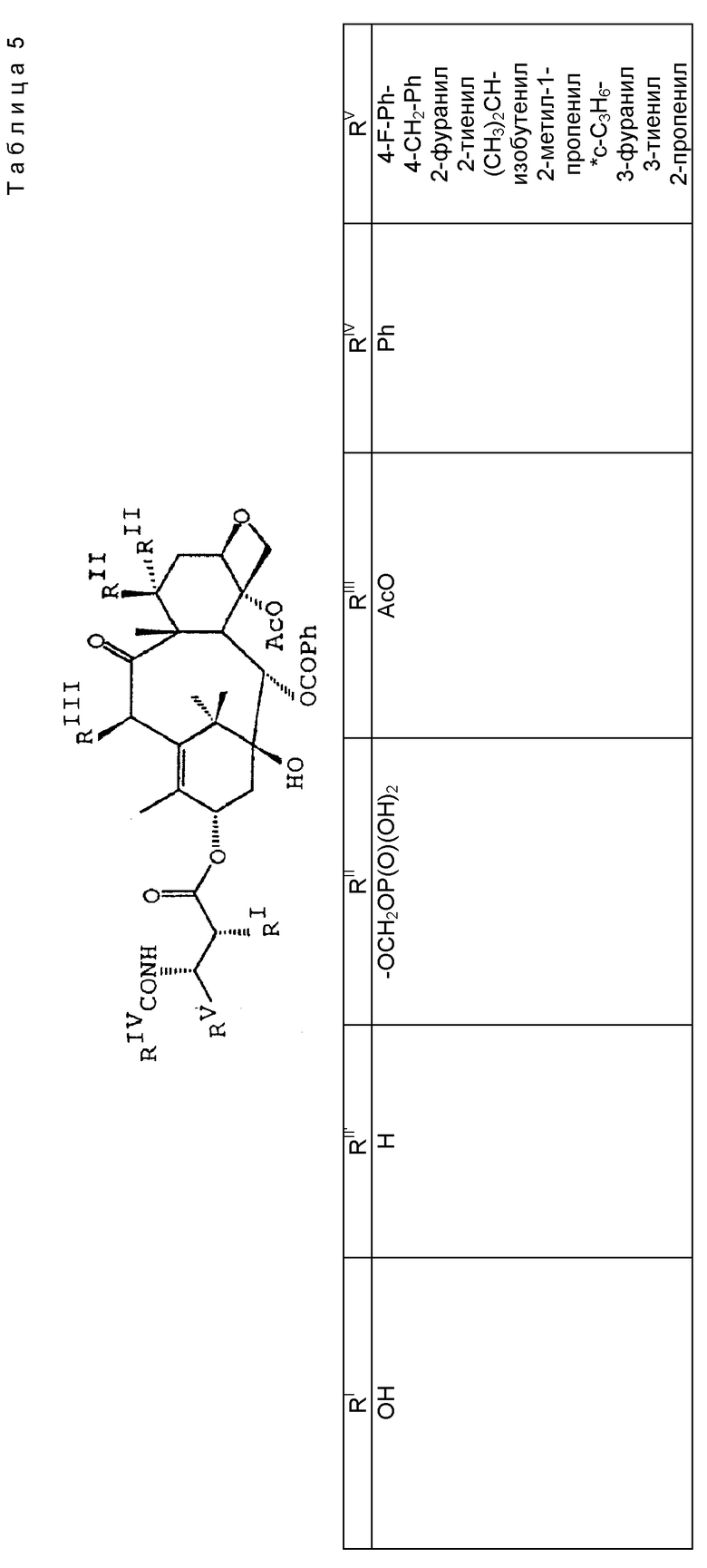
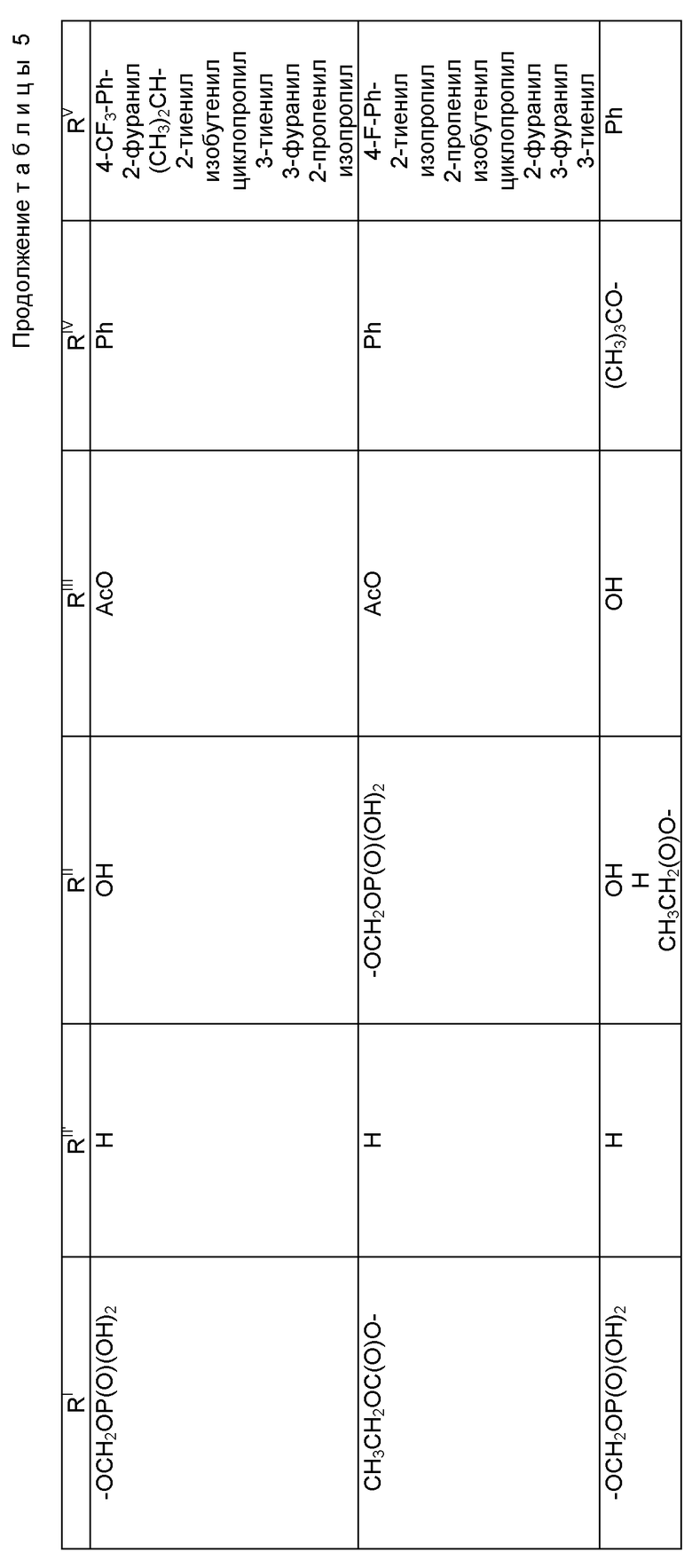
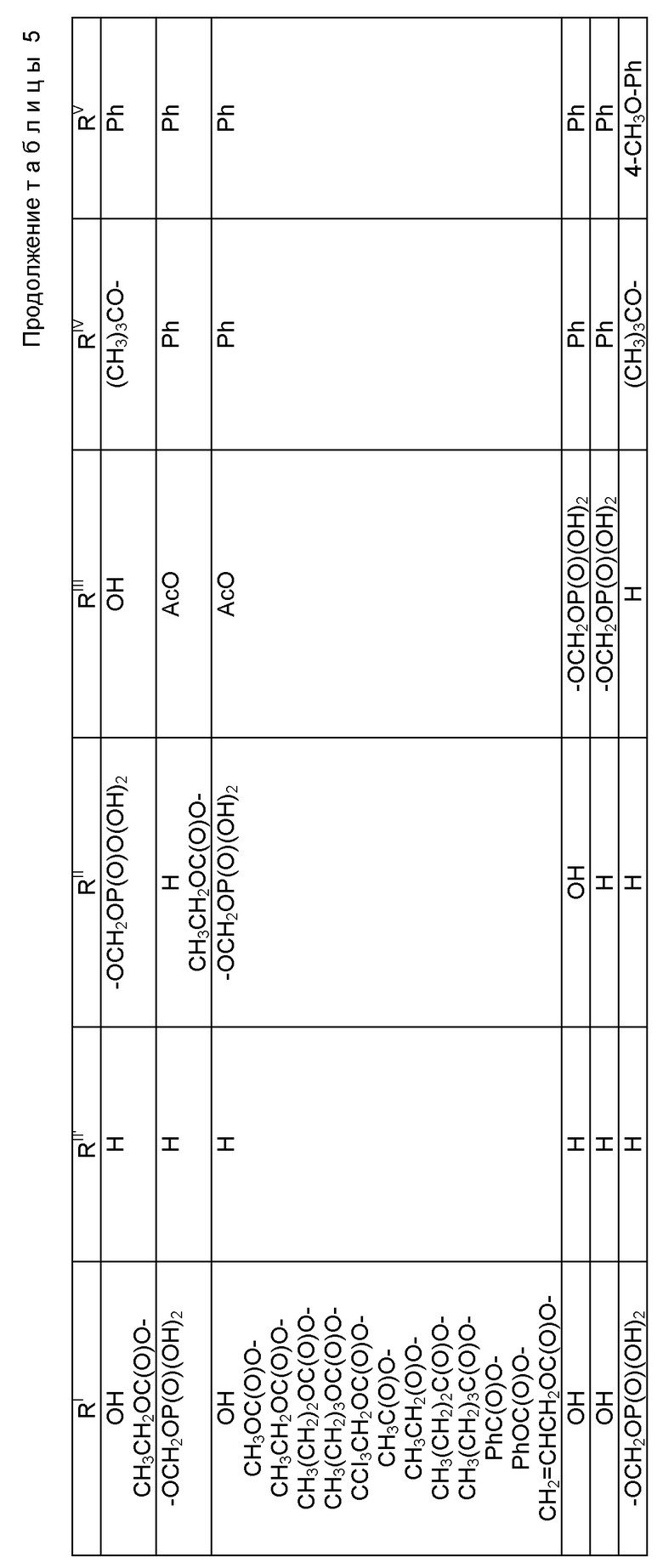
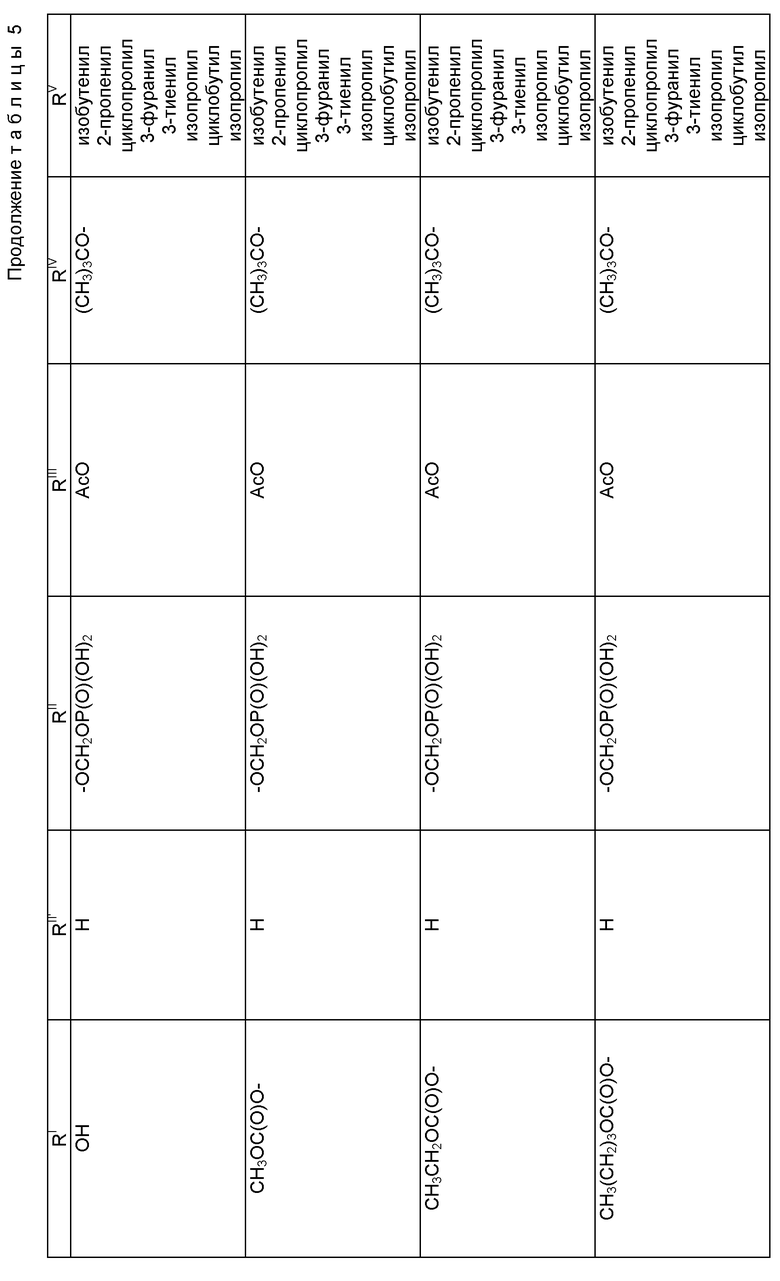
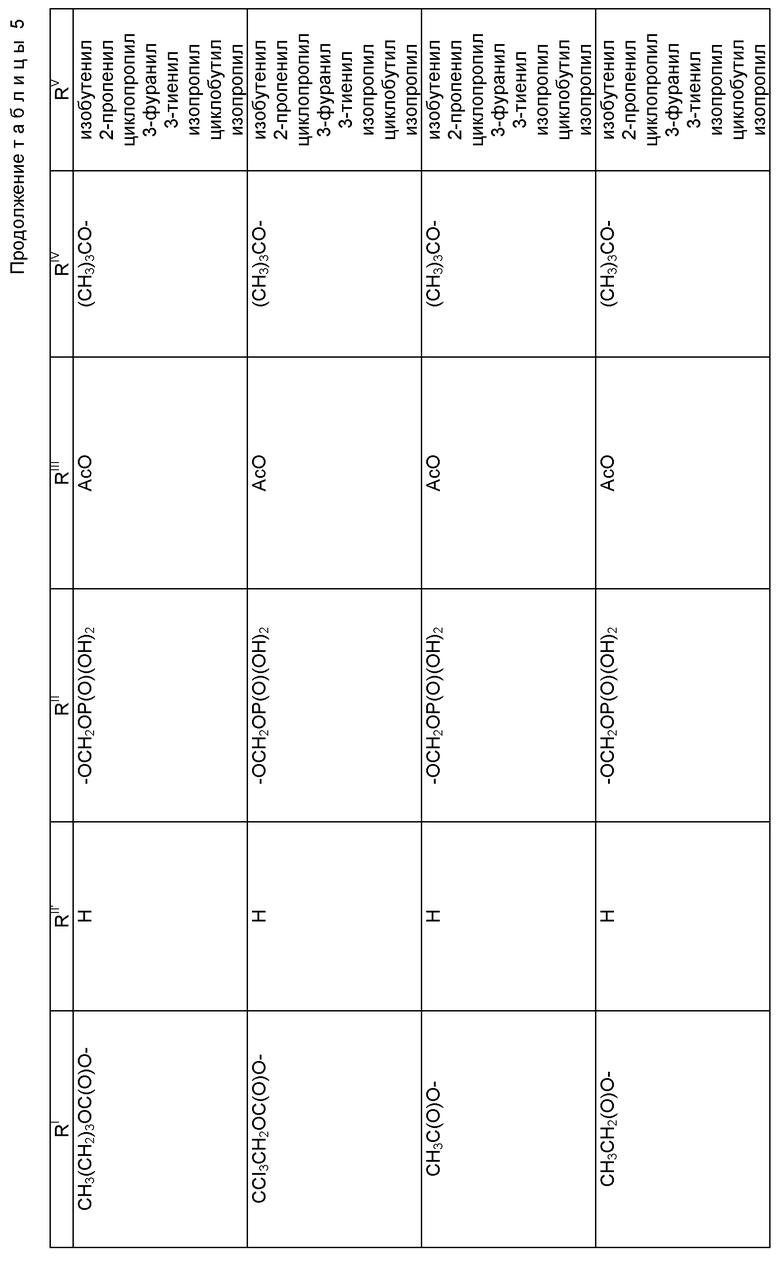
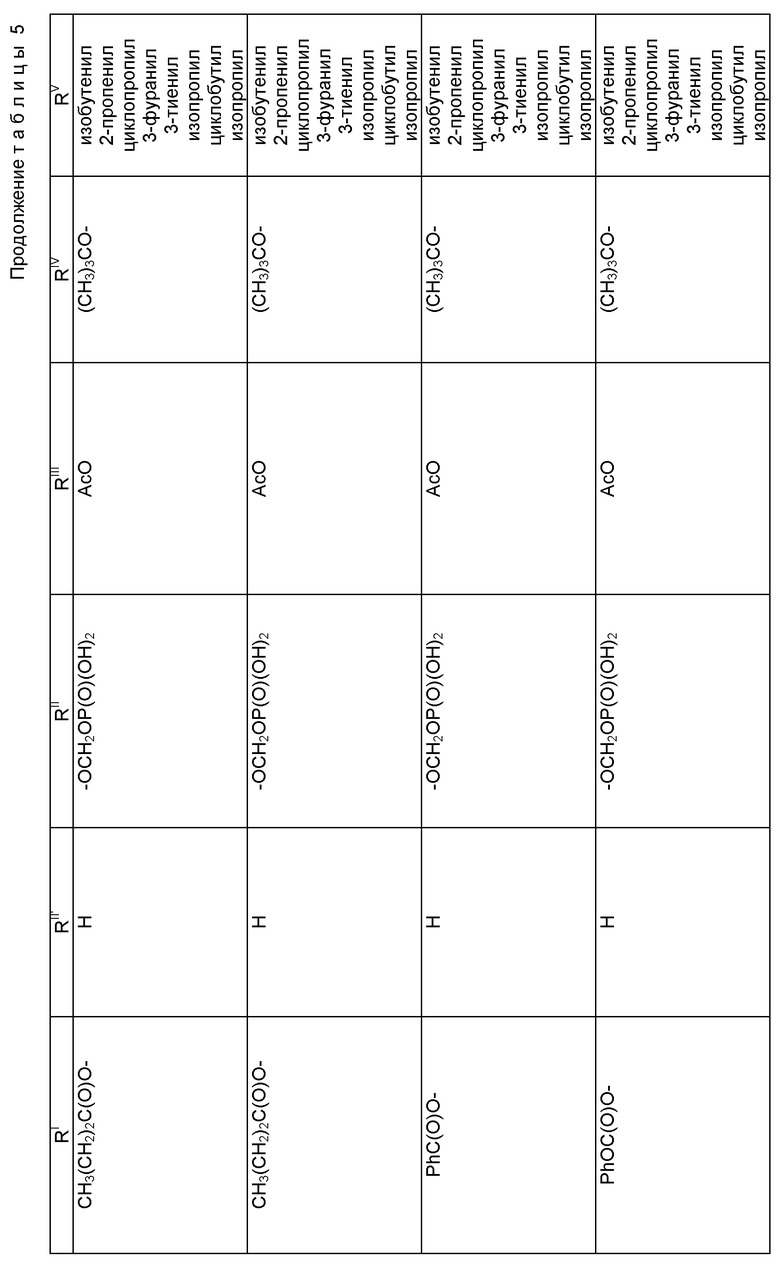
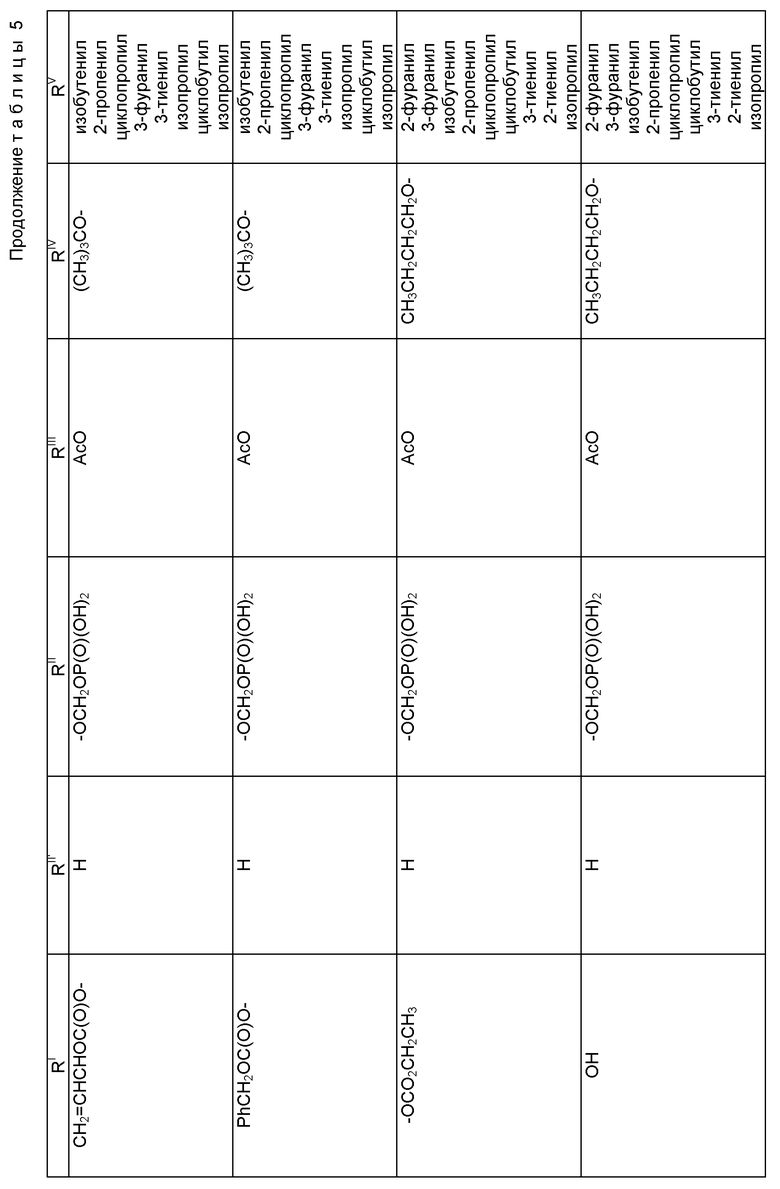
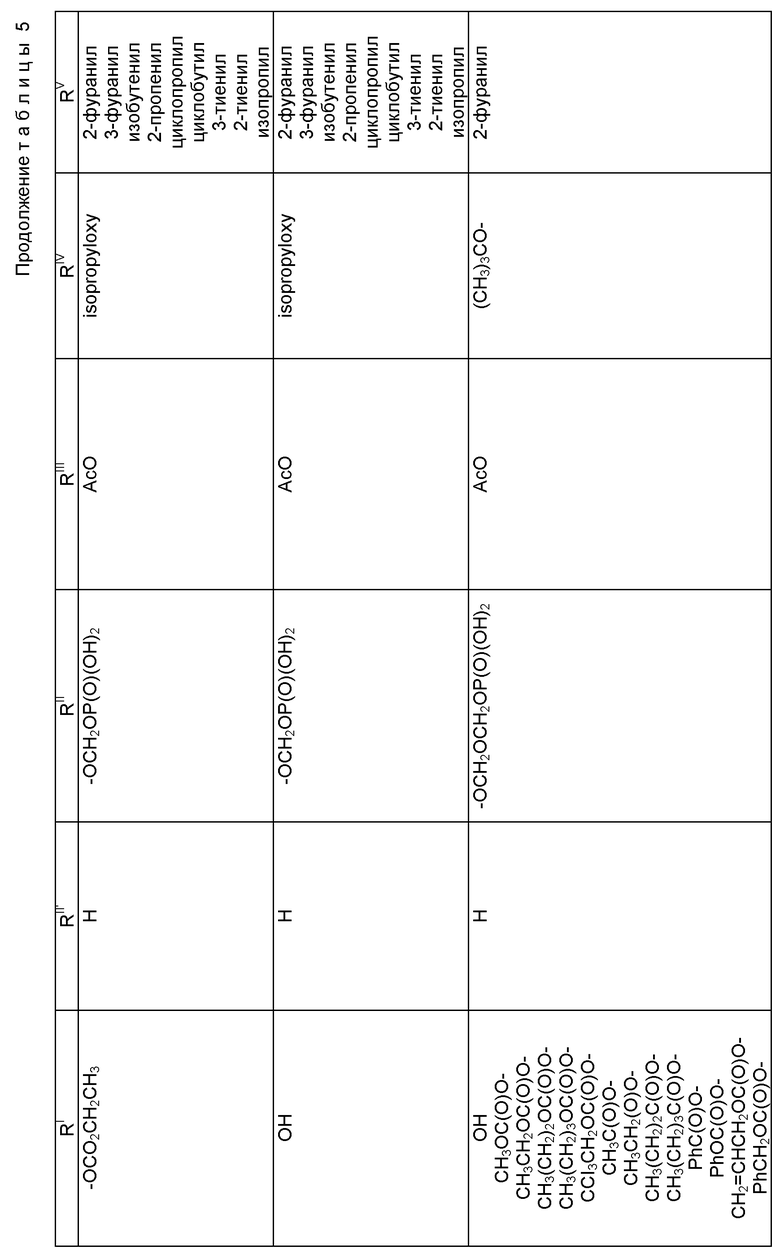
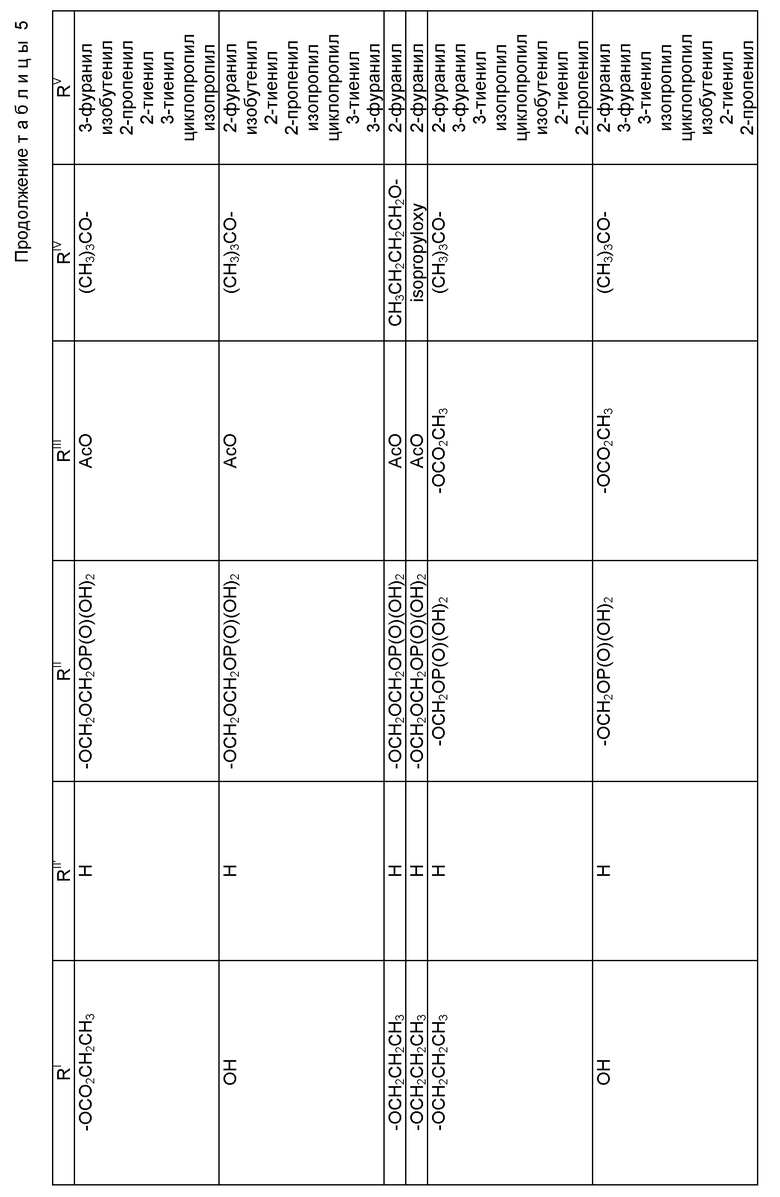
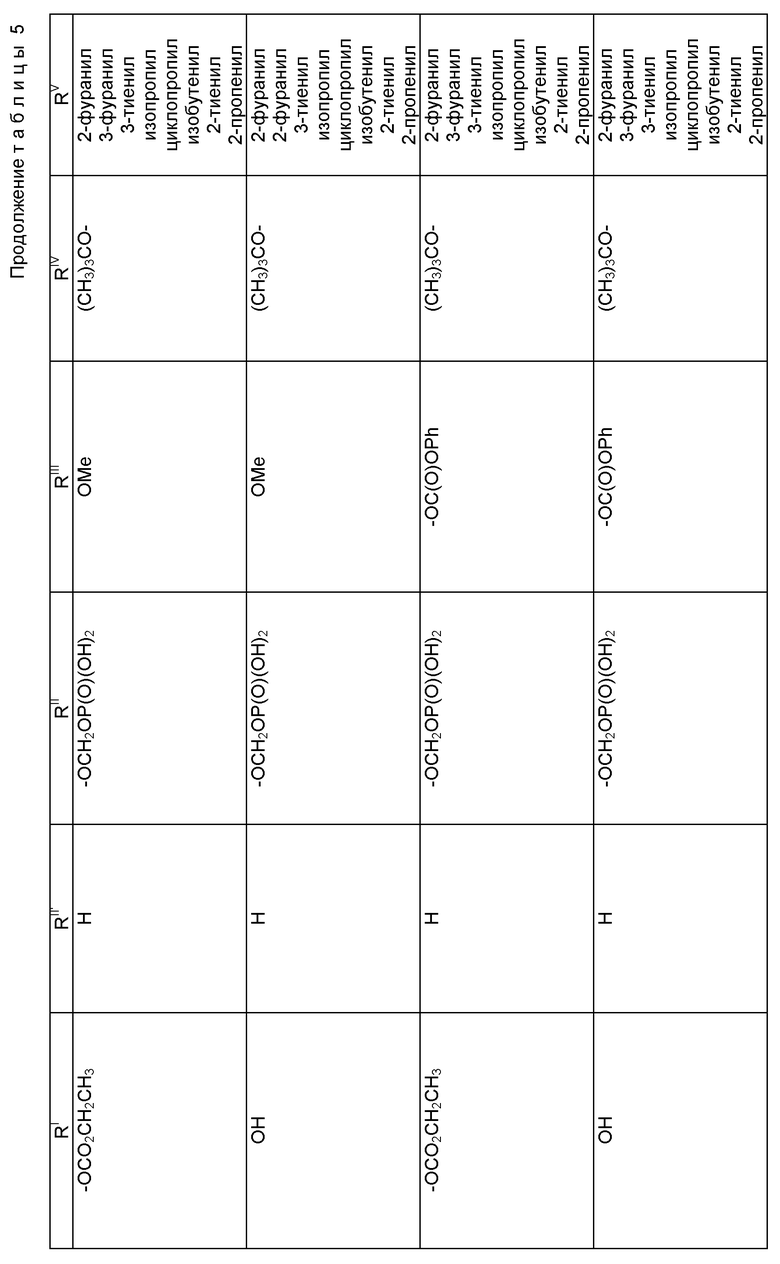
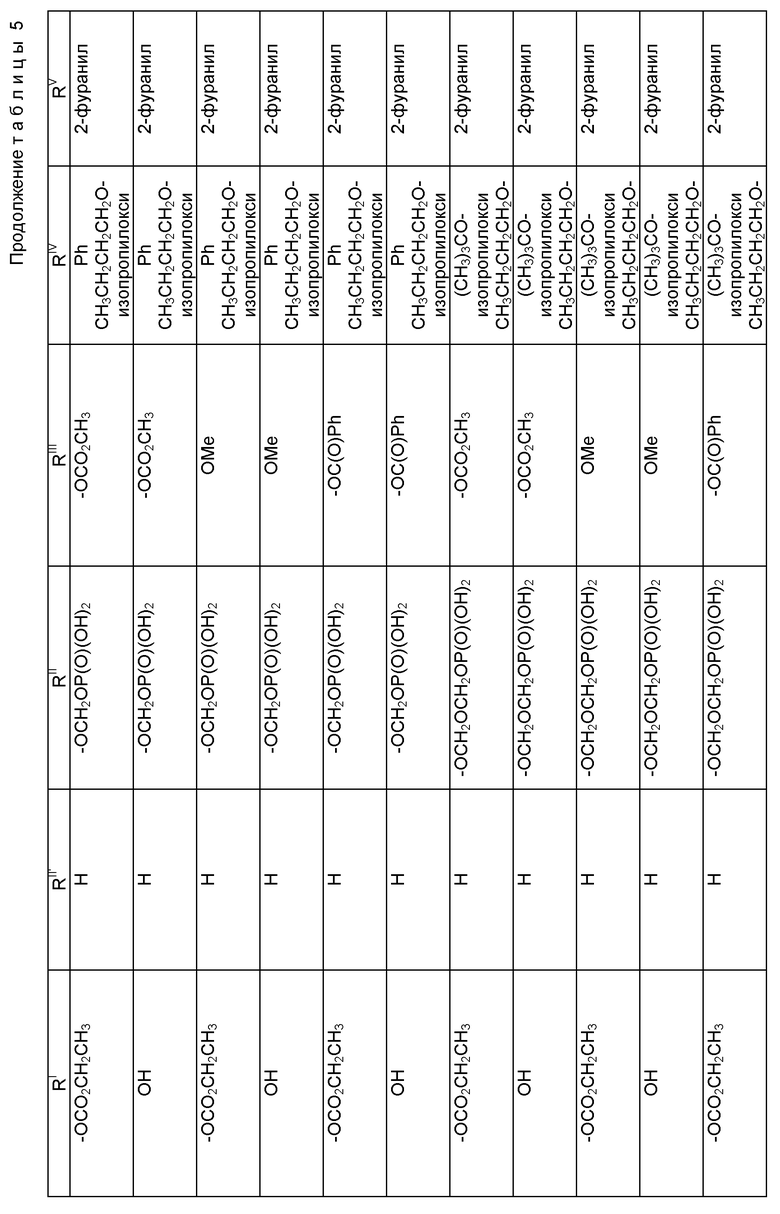
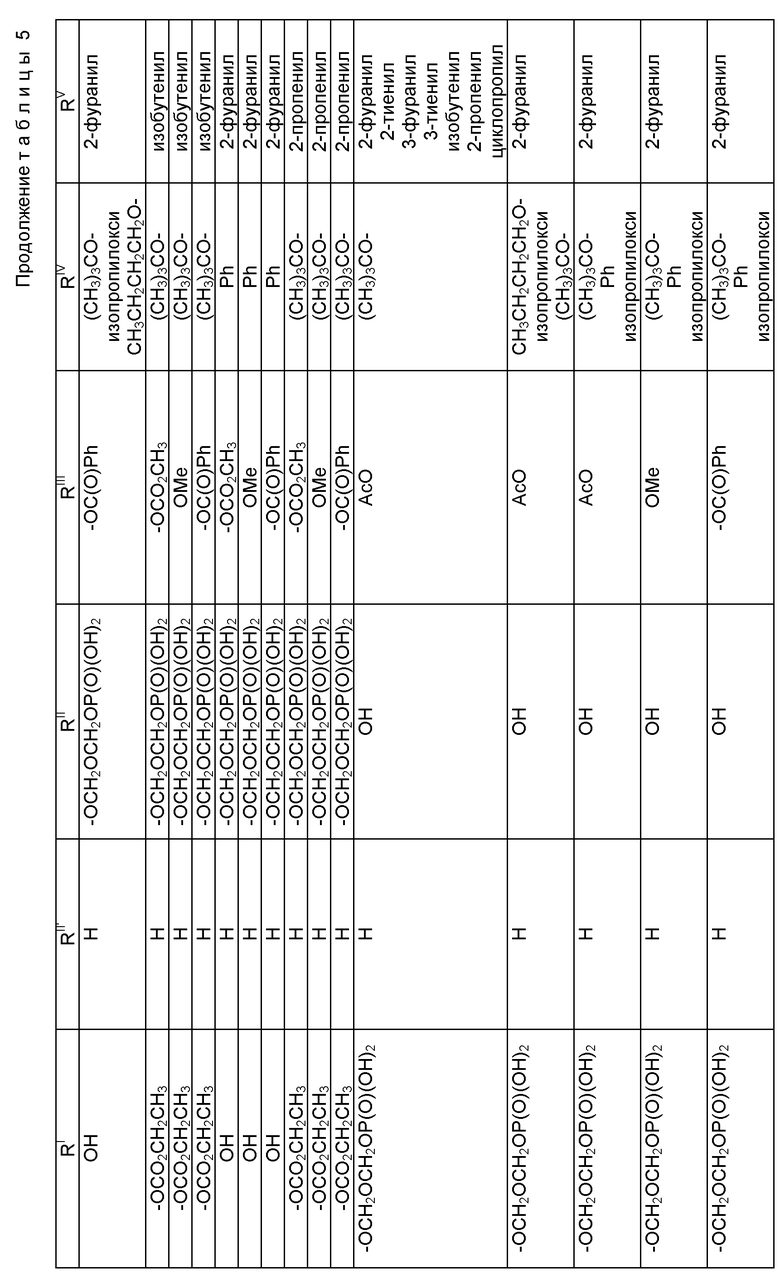
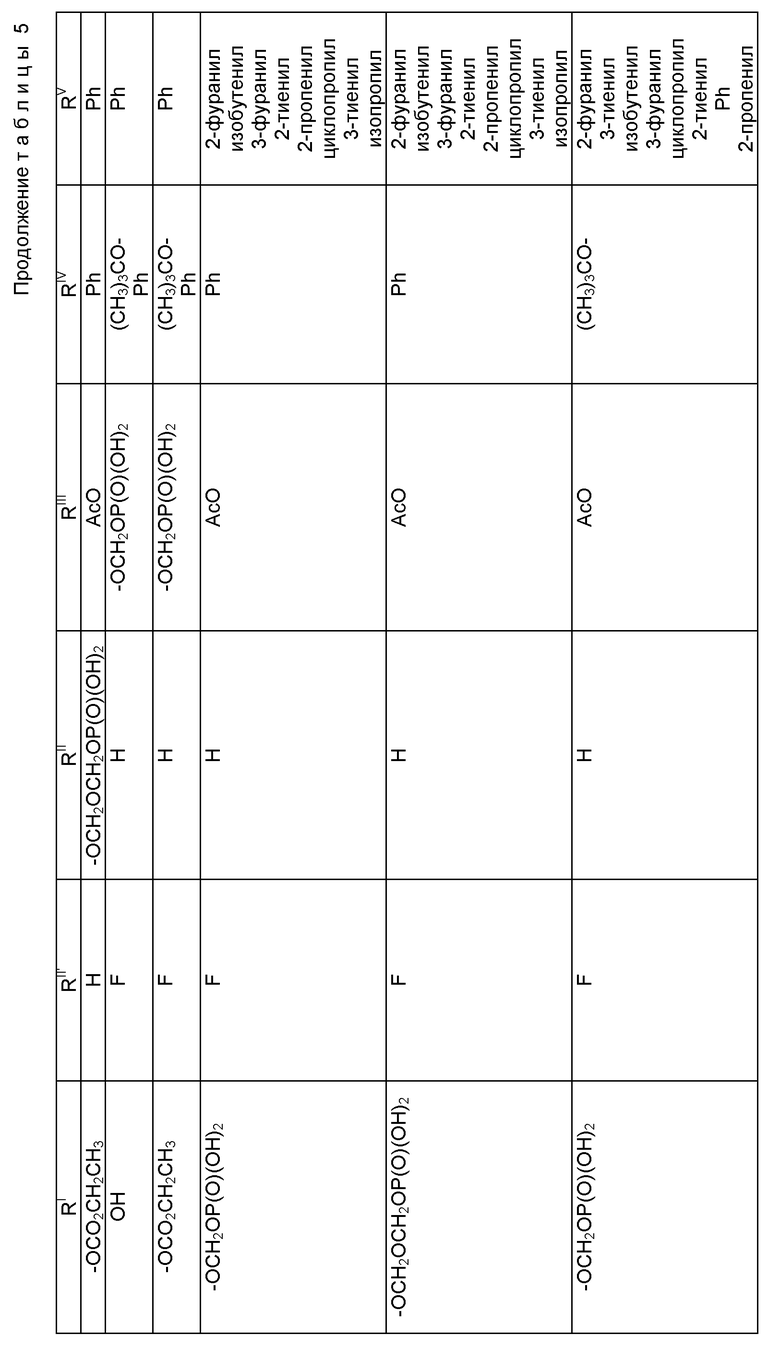
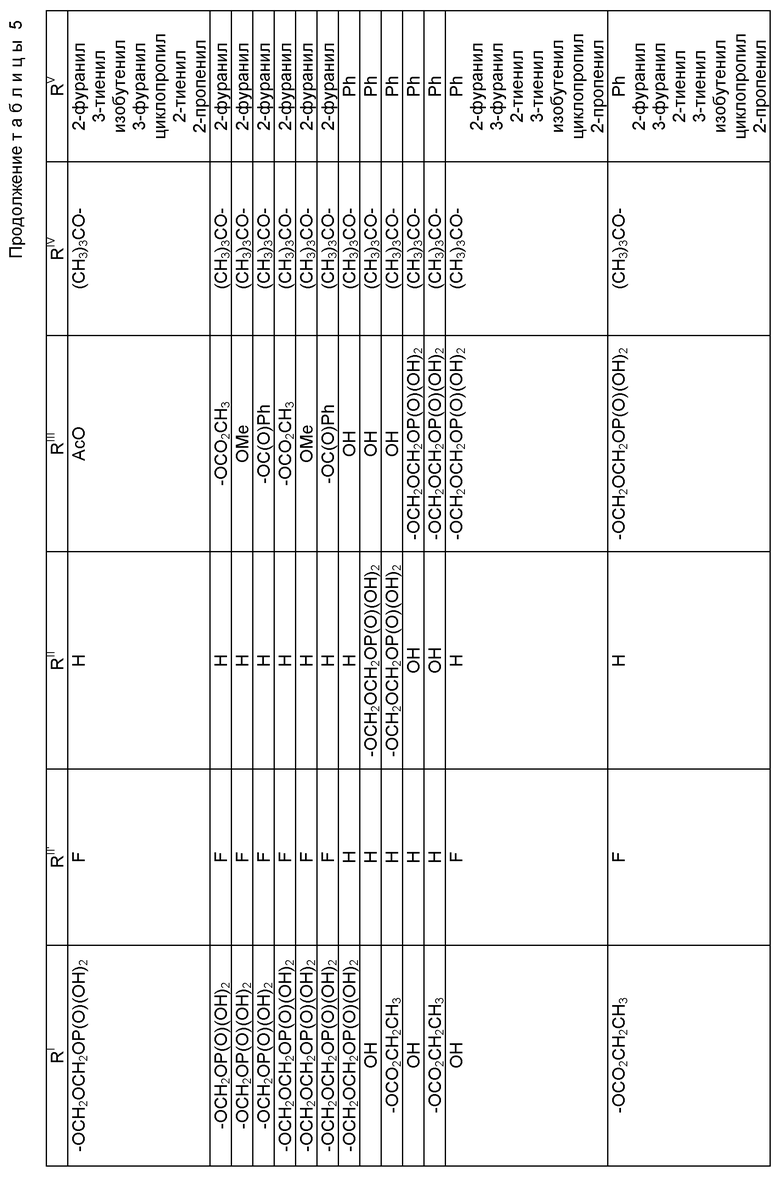
Фосфонооксиметиловые эфиры таксановых производных формулы I, где R1 - гидрокси, -OCH2(OCH2)mOP(O)(OH)2 или -OC(O)ORx; R2' - водород; R2 - гидрокси, -OCH2(OCH2)mOP(O)(OH)2, -OC(O)ORx или R2' - фтор, а R2 - водород; R3 - H, гидрокси, ацетокси; R4, R5 - C1-6алкил или Z-R6; Z - прямая связь; R6' - арил или гетероарил; Rx - C1-6алкил; p = 0 или 1; R6, R7 вместе образуют оксогруппу, при условии, что по крайней мере один из R1, R2 - -OCH2(OCH2)mOP(O)(OH)2, m = 0 или 1, или их фармацевтически приемлемые соли обладают противоопухолевой активностью. 8 с. и 46 з.п.ф-лы, 5 табл.
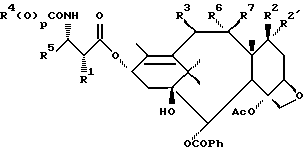
где R1 - гидрокси -OCH2(OCH2)mOP(O) (OH)2 или OC(O)ORx;
R2' - водород и R2 - гидрокси, -OCH2(OCH2)mOP(O) (OH)2 или -OC(O)ORx или R2' - фтор,
R2 - водород;
R3 - водород, гидрокси, ацетокси;
R4, R5 - независимо (C1 - C6)алкил или Z - R6';
Z - прямая связь;
R6' - арил или гетероарил,
Rx - (C1 - C6)алкил;
p = 0 или 1;
R6 и R7 вместе образуют оксогруппу при условии, что по крайней мере один из R1, R2 - OCH2(OCH2)mOP(O) (OH)2;
m = 0 или 1,
или их фармацевтически приемлемые соли.
или его C13алкоксид металла.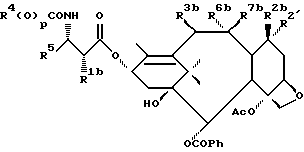
где R1b - гидрокси, OCH2SCH3 или OC(O)ORx;
R2' - водород или R2b - гидрокси, гидрокси, защищенный фосфоновой группой -OCH2 S CH3 или R2' - фтор, а R2b - водород;
R3b - гидрокси, защищенный триалкилсилильной группой ацетокси;
R4, R5 - независимо (C1 - C6)алкил или Z - R6';
R6' - арил или гетероарил;
Rx - (C1 - C6)алкил,
p = 0 или 1;
R6b и R7b вместе образуют оксогруппу при условии, что по крайней мере один из R1b, R2b - OCH2SCH3.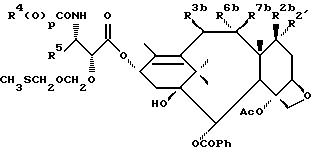
где R2', R2b, R3b, R4, R5, R6b, R7b и p определены выше.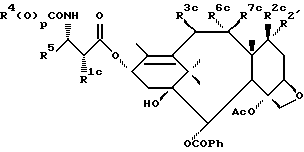
где R1k - гидрокси, -OCH2OP(O)(OCH2Ry)2 -или -OC(O)ORx;
R2' - водород, R2c - гидрокси, гидрокси, защищенный триалкилсилильной группой, OCH2OP(O)(OCH2Ry)2 или R2' - фтор,
R2c - водород; R3c - ацетокси; Rx - фосфонозащитная группа;
R6c и R7c вместе образуют оксогруппу при условии, что по крайней мере один из R1c, R2c - -OCH2OP(O)(OCH2Ry)2;
R4, R5 - независимо (C1 - C6)алкил или Z - R6;
z - прямая связь;
R6' - арил или гетероарил;
Rx - (C1 - C6)алкил,
p = 0 или 1;
Ry - фосфонозащитная группа.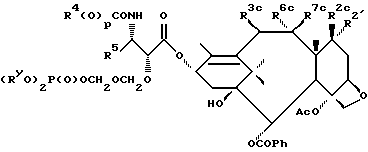
где R2', R2c, R3c, R4, R5, R6c, R7c, Ry и p определены выше.
Приоритет по пунктам:
24.12.92 - по пп.1-17, 22-31, 34, 37-39, 47;
17.08.93 - остальные пункты формулы.
| US 4857653 A, 15.08.89 | |||
| Машковский М.Д | |||
| Лекарственные средства, ч.2, - М., 1988, с.425-435 | |||
| US 4942184 A, 17.07.90 | |||
| US 5059699 A, 10.05.93 | |||
| Лигатура | 1976 |
|
SU558959A1 |

