Это изобретение касается серии бициклических тетрагидропиразолопиридинов, которые являются селективными ингибиторами фосфодиэстеразы (ФДЭ) типа IV или продукции фактора некроза опухолей (здесь далее ФНО), и как таковые применимы для лечения астмы, артрита, бронхита, хронического обструктивного заболевания дыхательных путей, псориаза, аллергического ринита, дерматита и других воспалительных заболеваний; и СПИДа, септического шока и других заболеваний, связанных с выработкой ФНО.
Это изобретение также относится к способу применения таких соединений для лечения вышеназванных заболеваний у млекопитающих, особенно у людей, и к фармацевтическим композициям, пригодным для этого.
С тех пор, как стало известно, что циклический АМФ является внутриклеточным вторичным месседжером (E.W. Sutherland and T.W. Rall, Pharmacol. Rev. , 1960, 12, 265) подавление фосфодиэстераз стало мишенью для модуляции и соответственно терапевтического вмешательства в ряда болезнетворных процессов. Совсем недавно были выявлены отдельные классы ФДЭ (J. A. Beavo and D.H. Reifnyder, TiPS, 1990, 11, 150) и их селективное подавление привело к улучшению лекарственной терапии (C.D. Nicholson, R.A. Challiss and M. Shahid, TiPS, 1991, 12, 19). Более конкретно было установлено, что подавление ФДЭ типа IV может приводить к подавлению выделения медиаторов воспаления (M.W. Verghese et al. , J. Mol. Cell Cardiol., 1989, 12 (Suppl. 11), S 61) и расслабление гладкой мускулатуры дыхательных путей (T.J. Torphy in Directions for New Anti-Asthma Drugs, eds S.R. O'Donnell and C.G.A. Persson, 1988, 37, Birkhauser-Verlag). Таким образом, соединения, которые подавляют ФДЭ типа IV, но которые обладают слабой активностью против других типов ФДЭ, должны угнетать выделение медиаторов воспаления и расслабить гладкую мускулатуру дыхательных путей без оказания воздействия на сердечно-сосудистую систему или тромбоциты.
Установлено, что ФНО участвует в процессах развития многих инфекционных и аутоиммунных заболеваний (W. Friers, FEBS Letters, 1991, 285, 199). Кроме того, было показано, что ФНО является основным медиатором воспалительной реакции, наблюдаемой при сепсисе и септическом шоке (C.E. Spooner et al., Clinical Immunology and Immunophathology, 1992, 62, S 11).
Краткое изложение изобретения
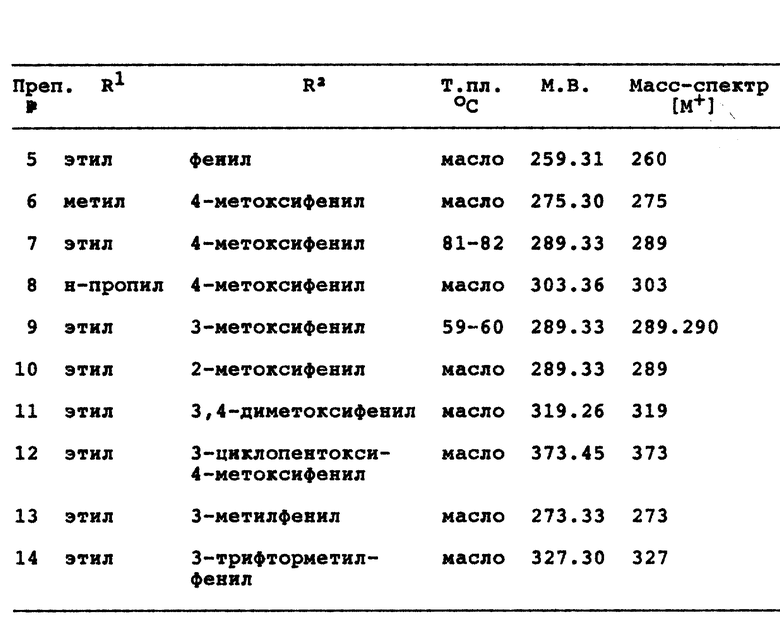
Данное изобретение касается соединений формулы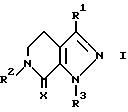
и их фармацевтически приемлемых солей; где
R1 является водородом, (C1-C7)алкилом, (C2-C3)алкенилом, (C3-C5)циклоалкилом или метилен(C3-C5)циклоалкилом, в котором каждый алкил или алкенильная группа могут быть произвольно замещенными вплоть до двух (C1-C2)алкильными или трифторметильными группами или, вплоть до трех, галогенами;
X является кислородом или двумя атомами водорода;
R2 и R3 каждый независимо выбираются из группы, состоящей из водорода, (C1-C14)алкила, (C1-C14)алкоксила, (C2-C7)алкенила, (C4-C7)гетероциклической группы, содержащей кислород, серу, SO2 или NR5, где R5 является водородом или (C1-C4)алкилом или группой формулы
где
a является целым числом от 1 до 5;
b и c является 0 или 1;
R4 является водородом, гидроксилом, (C1-C5)алкилом, (C2-C5)алкенилом, (C1-C5)алкоксилом, (C3-C6)циклоалкоксилом, галогеном, трифторметилом, CO2R6, CONR6R7, NR6R7, NO2 или SO2NR6R7, где R6 и R7 каждый независимо являются водородом или (C1-C4)алкилом;
где Z является кислородом, серой, SO2 или BR8, где R8 является водородом или (C1-C4)алкилом; и
Y является (C1-C5)алкиленом или (C2-C6)алкенилом, произвольно замещенным, числом до 2, (C1-C7)алкильными или (C3-C7)циклоалкильными группами: или группой формулы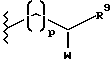
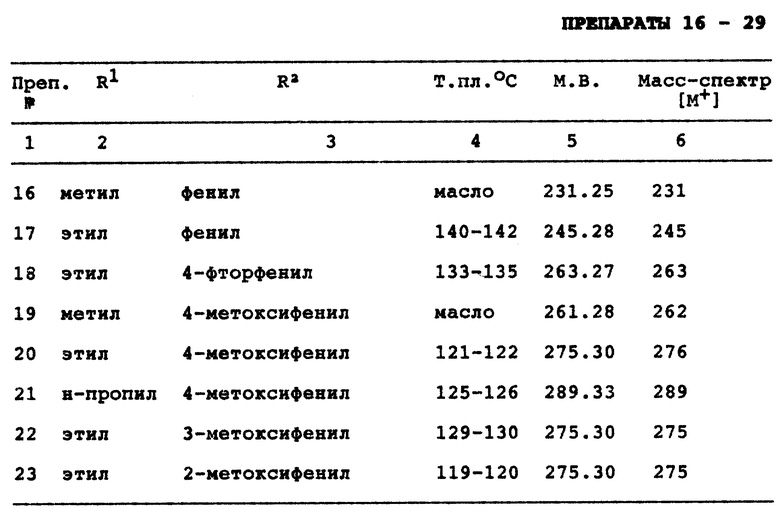
где
p является целым числом от 1 до 3,
W является оксогруппой или гидроксилом,
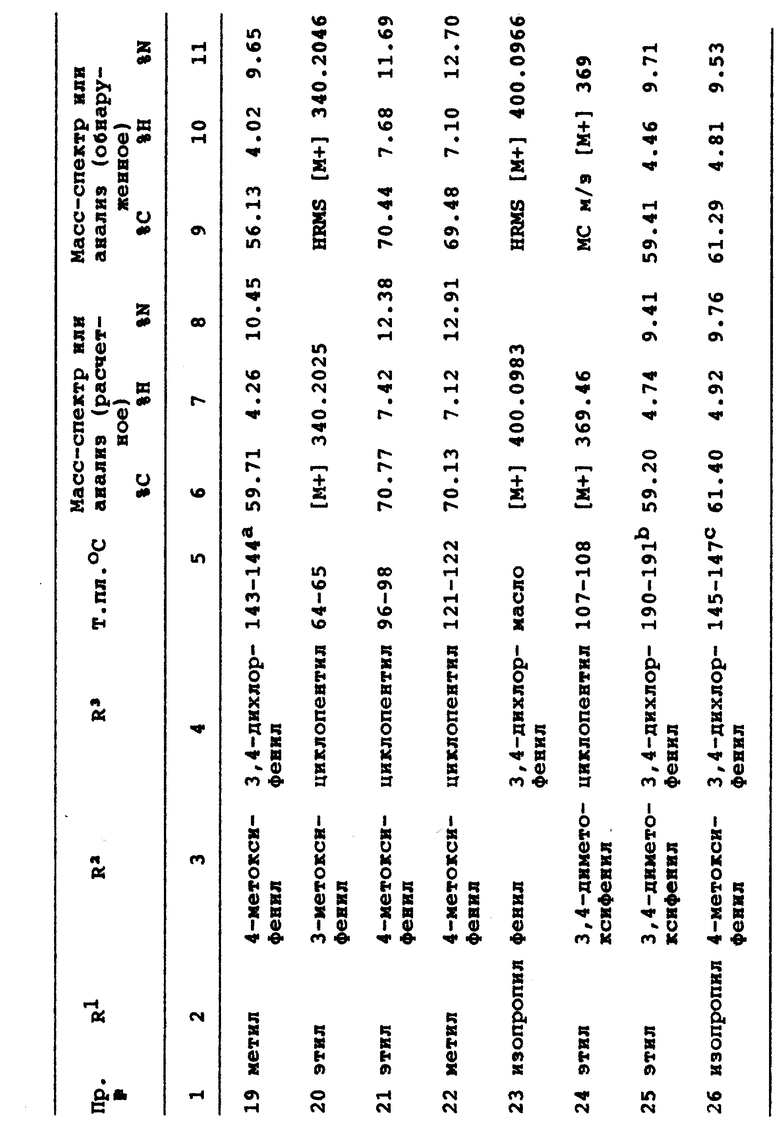
R9 является (C1-C3)алкилом;
где каждый указанный алкил, алкенил, циклоалкил, алкоксиалкил или гетероциклическая группа могут быть произвольно замещенными от одной до четырнадцати, предпочтительно пятью группами из группы, состоящей из (C1-C2)алкила, трифторметила или галогена при условии, что когда R1 является этилом и R2 является 4-метилфенилом, R3 не может быть водородом, метилом, фенилом, 4-фторфенилом или 2-пиридилом, и при условии, что когда R2 является 4-метилфенилом и R3 является 4-фторфенилом, R1 не может быть фенилом, метилом или н-пропилом, и при условии, что когда R1 является этилом, а R2 является фенилом, R3 не может быть 4-хлорфенилом, 4-фторфенилом или 4-метилфенилом, и при условии, что когда R1 является этилом и R2 является 4-метоксифенилом, R3 не может быть 4-фторфенилом.
По одному из осуществлений изобретение относится к соединению с формулой I, где R1 является (C1-C3)алкилом и R2 и R3 каждый независимо выбираются из группы, состоящей из (C3-C7)циклоалкила, (C4-C7)гетероциклической группы, содержащей SO2 или группу с формулой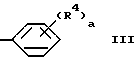
где
a является целым числом от 1 до 5 и
R4 является водородом, гидроксилом, (C1-C5)алкилом, (C1-C5)алкоксилом или галогеном.
В другом осуществлении изобретение относится к соединению с формулой I, где R1 является этилом или изопропилом; R2 является фенилом, 2-метилфенилом, 3-метилфенилом, 2-метоксифенилом, 3-метоксифенилом или 3-трифторметилфенилом и R3 является циклобутилом, циклопентилом, циклогексилом, 3-сульфоланилом, 4-фторфенилом или 3,4-дихлорфенилом.
Данное изобретение, кроме того, относится к фармацевтической композиции для подавления фосфодиэстеразы (ФДЭ) типа IV и продукции фактора некроза опухолей (ФНО), содержащей фармацевтически эффективное количество соединения, соответствующего формуле I, и его фармацевтически приемлемых солей и фармацевтически приемлемый носитель.
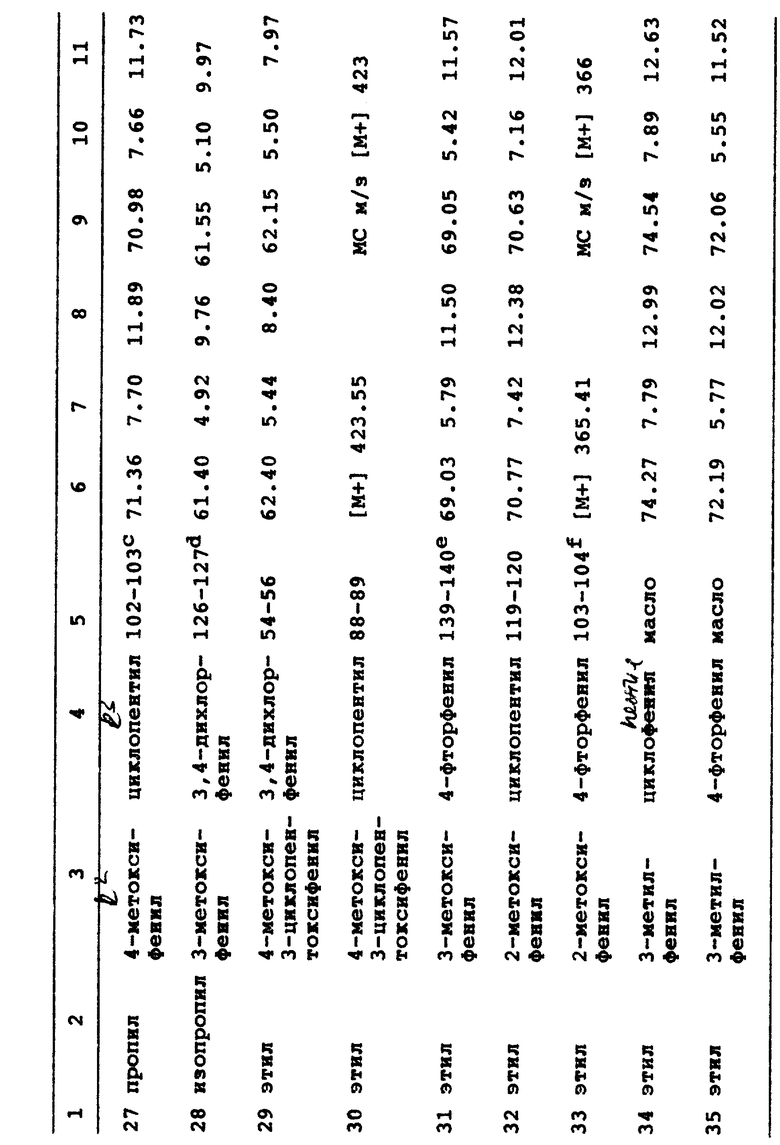
Данное изобретение также относится к способу подавления фосфодиэстеразы (ФДЭ) типа IV и продукции фактора некроза опухолей (ФНО), включающему введение больному эффективного количества соединения по формуле I и его фармацевтически приемлемых солей.
Данное изобретение, кроме того, относится к способу лечения состояния воспаления у млекопитающих, который включает введение указанному млекопитающему противовоспалительного количества соединения с формулой I и его фармацевтически приемлемых солей.
Данное изобретение дополнительно касается фармацевтической композиции для лечения астмы, артрита, бронхита, хронического обструктивного заболевания дыхательных путей, псориаза, аллергического ринита, дерматита и других воспалительных заболеваний, СПИДа, септического шока и других заболеваний, связанных с продукцией ФНО, включающей фармацевтически эффективное количество соединения по формуле I и его фармацевтически приемлемых солей вместе с фармацевтически приемлемым носителем.
Это изобретение, кроме того, относится к способу лечения и предупреждения состояния, выбираемого из группы, состоящей из астмы, артрита, бронхита, хронического обструктивного заболевания дыхательных путей, псориаза, аллергического ринита, дерматита и других воспалительных заболеваний, СПИДа, септического шока и других заболеваний, связанных с выработкой ФНО, включающему введение больному эффективного количества соединения по формуле I и его фармацевтически приемлемых солей.
Конкретными предпочтительными соединениями этого изобретения являются:
3-этил-1-(4-метоксифенил)-6-фенил-7-оксо-4,5,6,7-тетрагидро-1H -пиразоло-[3,4-c]пиридин;
3-этил-1-циклопентил-6-фенил-7-оксо-4,5,6,7-тетрагидро-1H -пиразоло-[3,4-c]пиридин;
3-этил-1(3,4-дихлорфенил)-6-(3-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-циклопентил-6-(3-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло-[3,4-c]пиридин;
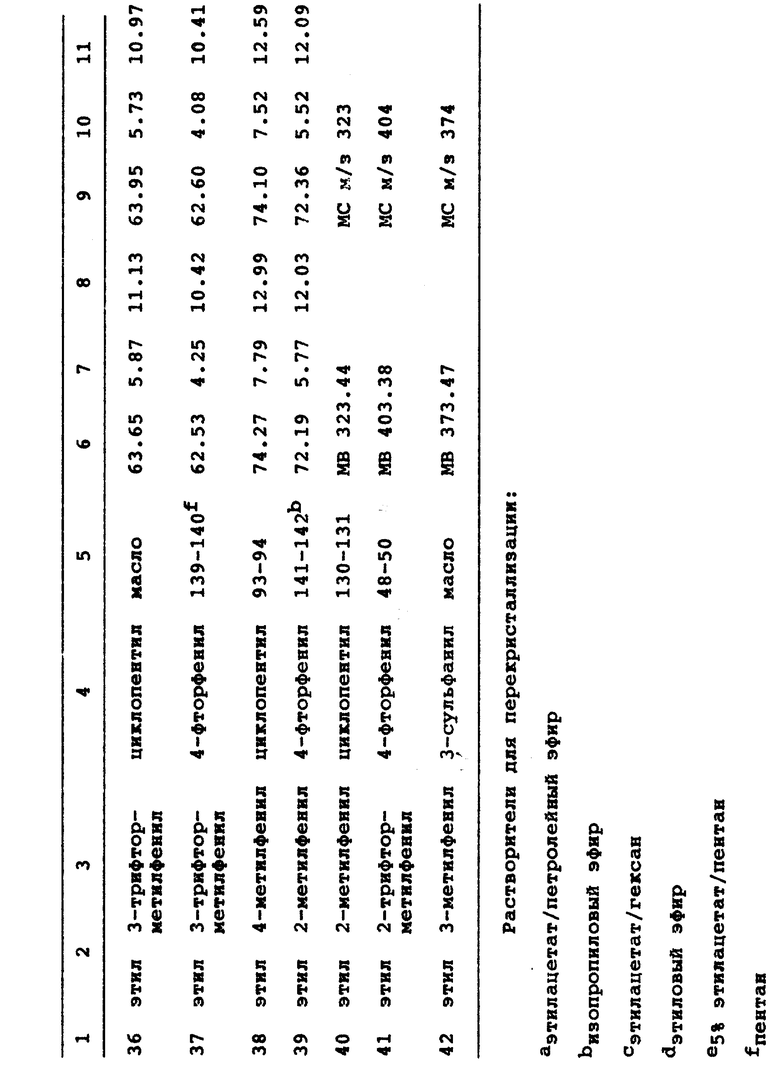
3-этил-1-(4-фторфенил)-6-(2-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло-[3,4-c]пиридин;
3-этил-1-циклопентил-6-(3-метилфенил)-7-оксо-4,5,6,7-тетрагидро -1H-пиразоло-[3,4-c]пиридин;
3-этил-1-циклопентил-6-(3-трифторметилфенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло-[3,4-c]пиридин;
3-этил-1-циклогексил-6-(3-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-изопропил-1-циклопентил-6-(3-метоксифенил)-7-оксо- 4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-циклобутил-6-(3-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-циклопентил-6-фенил-4,5,6,7-тетрагидро-1H-пиразоло [3,4-c] пиридин;
3-этил-1-циклопентил-6-(2-метилфенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
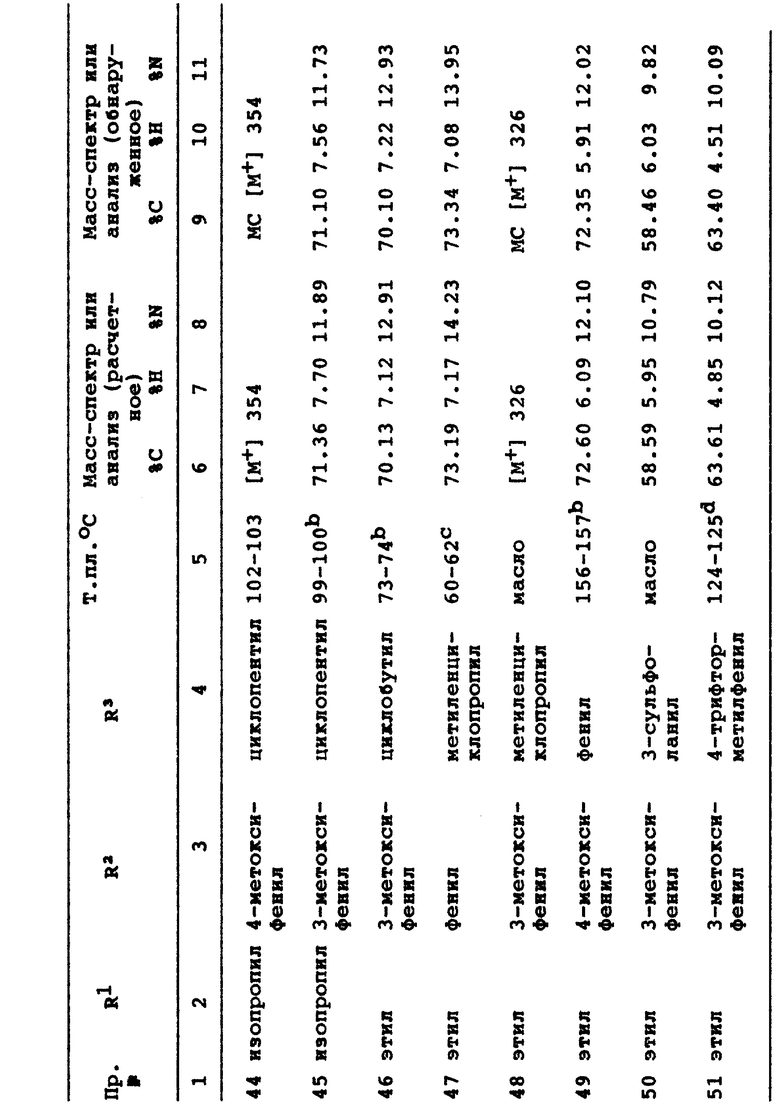
3-этил-1-(3-сульфоланил)-6-(3-метилфенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-(3-сульфоланил)-6-(3-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-циклобутил-6-(3-метилфенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-(3-сульфоланил)-6-(3-трифторметилфенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-циклобутил-6-(3-трифторметилфенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин;
3-этил-1-циклобутил-6-(2-метилфенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин.
Детальное описание изобретения
Термин "галоген", который использован здесь, если не указано иначе, включает хлор, бром и фтор.
Если не указано иначе, алкильные, алкоксильные и алкенильные группы, упоминаемые здесь, могут быть с прямой цепью, или если содержат три или более атомов углерода, могут быть с прямой цепью с разветвленной цепью, циклическими или комбинацией циклической и разветвленной или с прямой цепью частей.
"Воспалительные заболевания", которые можно лечить по этому изобретению, включают, но не ограничиваются ими, астму, хроническое обструктивное заболевание легких, бронхит и артрит.
R1, R2 и R3, как они использованы здесь, если не указано иначе, представляют собой то, чему дано определение выше, со ссылками на формулу I.
Следующие схемы реакции иллюстрируют, но не являются ограничивающими, получение соединений данного изобретения.
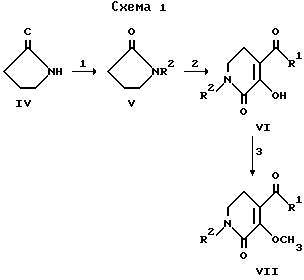
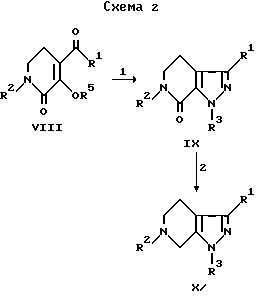
В реакции 1 со схемы 1 2-пирролидиноновое соединение формулы IV превращается в соответствующее N-(арил)-2-пирролидиновое соединение V, где "арил" является группой формулы II, путем взаимодействия IV с чистым арилгалогенидом в присутствии медного катализатора и карбоната калия. Подходящие арилгалогениды включают 1-йод- или 1-бром-4-метоксибензол, 3-метоксибензол, 2-метоксибензол, 3-метилбензол, 4-метилбензол, 2-метилбензол, 3-трифторметилбензол, 2-трифторметилбензол, 3,4-диметоксибензол или 3-циклопентокси-4-метоксибензол. Температура реакции обычно должна быть в интервале от примерно 110oC до примерно 170oC, предпочтительно примерно 150oC, в течение временного периода, равного от примерно 14 часов до примерно 22 часов, предпочтительно около 18 часов, при условии реакции в инертной среде.
В реакции 2 со схемы 1 R1 галогенид добавляется к суспензии магния в безводном апротонном растворителе. Реакционная смесь нагревается в колбе с обратным холодильником до тех пор, пока не израсходуется магний, и затем охлаждается до температуры в интервале от примерно -15oC до примерно 15oC, предпочтительно до примерно 0oC. Затем добавляется N-(арил)-2- пирролидоновое соединение с формулой V, и реакционную смесь нагревают до комнатной температуры при перемешивании в течение периода времени от примерно 1.5 часов до примерно 2.5 часов, предпочтительно примерно в течение 2 часов. Подходящие алкилгалогениды включают бромметан, бромэтан или бромпропан. Подходящим безводным апротонным растворителем является безводный эфир. После завершения реакции желаемое промежуточное соединение может быть выделено обычным путем, например сначала промыванием смешанных органических веществ водой и солевым раствором, затем высушиванием сульфатом натрия, фильтрованием и концентрированием при пониженном давлении с получением легко выделяемого осадка в виде белого твердого вещества.
Вышеуказанный осадок превращается в соответствующее 2,5,6-тетрагидропиридиновое соединение формулы VI путем диспергирования осадка в смеси неполярного апротонного растворителя и основания. После энергичного перемешивания добавляется этилоксалилхлорид, и реакционная смесь нагревается в колбе с обратным холодильником в течение периода времени от примерно 1.5 часов до примерно 4.5 часов, предпочтительно в течение примерно 3.0 часов. Предпочтительно неполярным апротонным растворителем является бензол, а предпочтительным основанием является гидроксид натрия. Растворители удаляются, а полученный в результате осадок обрабатывается раствором алкоксида натрия в этаноле. После нагревания в колбе с обратным холодильником в течение периода времени от примерно 1 часа до примерно 3 часов, предпочтительно примерно в течение 1.5 часов, смесь концентрируют под пониженным давлением и подкисляют до pH 3 хлористоводородной кислотой.
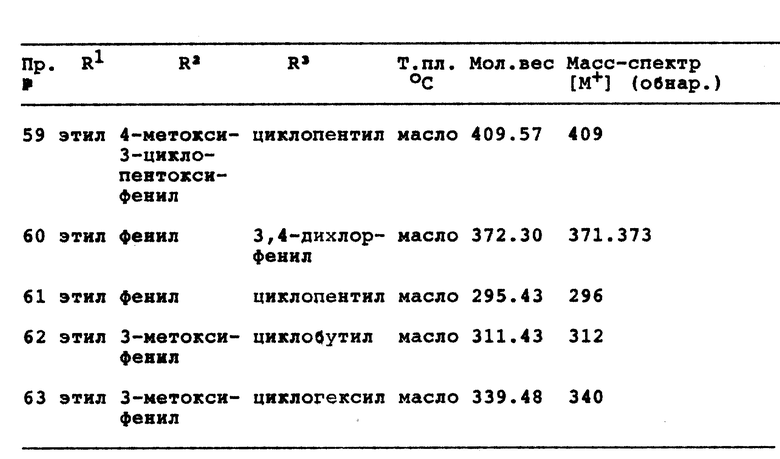
В реакции 3 со схемы 1 соединение формулы VI превращается в соответствующее 3-метокси-1,2,5,6-тетрагидропиридиновое соединение VII путем нагревания в колбе с обратным холодильником реакционной смеси из VI и 3-метил-1-п-толилтриазена в апротонном растворителе. Предпочтительным апротонным растворителем является 1,2-дихлорэтан. Временной период для реакции находится в интервале от примерно 30 минут до примерно 120 минут, предпочтительно составляет примерно 45 минут.
В реакции 1 со схемы 2 1,2,5,6-тетрагидропиридиновое соединение формулы VIII, где R5 является водородом или метилом, превращается в соответствующее 4,5,6,7-тетрагидро-7-оксо-1H-пиразоло [3,4-с]пиридиновое соединение IX путем реакции VIII с гидразином с формулой R3HNNH2. В качестве исходных материалов могут использоваться оба производных соединения формулы VIII, 3-гидрокси- и 3-метокси-, в одном из трех различных наборов условий реакции.
По одному из сочетаний условий реакции 1,2,5,6- тетрагидропиридиновое соединение формулы VIII превращается в соответствующее соединение формулы IX путем реакции VIII с гидрохлоридом гидразина и алкоксидом натрия в безводном полярном протонном растворителе. Предпочтительным алкоксидом натрия является метоксид натрия, а предпочтительным безводным полярным протонным растворителем является безводный этанол. Реакционную смесь нагревают в колбе с обратным холодильником в течение периода времени, равного от примерно 9 часов до примерно 15 часов, предпочтительно примерно 12 часам.
При втором сочетании условий реакции 1,2,5,6-тетрагидропиридиновое соединение превращается в соответствующее соединение формулы IX путем реакции VIII с гидразинбензойной кислотой в безводном полярном протонном растворителе, предпочтительно этаноле. Реакционную смесь нагревают в колбе с обратным холодильником в течение периода времени от примерно 16 часов до примерно 24 часов, предпочтительно в течение 20 часов. Соединение IX, полученное таким образом, может далее реагировать с образованием соответствующего 1-(4-бензамид)-7-оксо-4,5,6,7-тетрагидро-1H-пиразоло[3,4-с]пиридинового соединения путем реакции IX с метоксидом натрия в полярном протонном растворителе, предпочтительно метаноле, в течение периода времени от примерно 15 минут до примерно 45 минут, предпочтительно в течение 30 минут. Полярный протонный растворитель удаляется при пониженном давлении, твердый остаток суспендируют в неполярном апротонном растворителе, предпочтительно бензоле, а затем полярный растворитель удаляется под пониженным давлением. Полученное в результате сухое твердое вещество суспендируется в холодном эфире и обрабатывается оксалилхлоридом и N,N-диметилформамидом и оставляется при перемешивании на срок от примерно 30 минут до примерно 90 минут, предпочтительно на 60 минут. Затем растворитель удаляют и неочищенный остаток растворяют в сухом тетрагидрофуране. Полученный в результате раствор по каплям добавляют к перемешиваемому гидроксиду аммония при температуре от примерно -10oC до примерно 10oC, предпочтительно при 0oC.
При третьем сочетании условий реакции 1,2,5,6-тетрагидропиридиновое соединение формулы VIII превращается в соответствующее соединение формулы IX путем реакции VIII с гидрохлоридом гидразина в полярном протонном растворителе, предпочтительно метаноле. Реакционную смесь нагревают до температуры от примерно 70oC до примерно 110oC, предпочтительно до примерно 90oC при мягком пропускании азота до тех пор, пока весь растворитель не удалится. Неразбавленную смесь затем нагревают до температуры от примерно 120oC до примерно 180oC, предпочтительно до примерно 150oC, в течение периода времени от примерно 30 минут до примерно 90 минут, предпочтительно в течение примерно 60 минут.
Образованные таким образом соединения с формулой IX можно превращать в соответствующее 6-R2-4,5,6,7-тетрагидро-7-оксо- 1H-пиразоло[3,4-с]пиридиновое соединение, где R2 является другой группой, а не II, путем реакции IX в растворе в полярном апротонном растворителе, предпочтительно ацетонитриле, с раствором аммонийцерия (IV) нитрата в воде при температуре от примерно -15oC до примерно 15oC, предпочтительно при примерно 0oC, в течение периода времени от примерно 20 минут до примерно 50 минут, предпочтительно в течение примерно 35 минут. После завершения реакции смесь разбавляют водой и экстрагируют этилацетатом. Смешанные органические вещества затем промывают насыщенным раствором бикарбоната натрия, а затем сульфидом натрия. Полученное таким образом соединение в полярном апротонном растворителе, предпочтительно тетрагидрофуране, обрабатывают гидридом натрия, нагревают в колбе с обратным холодильником при перемешивании в течение периода времени от примерно 30 минут до примерно 60 минут, предпочтительно в течение 45 минут. Реакционную смесь охлаждают до температуры от примерно 20oC до примерно 30oC, предпочтительно до примерно 25oC, и добавляют алкилгалогенид с формулой R2, где R2 является тем, чему дано определение в пояснениях к формуле I, а не группой из формулы II. Реакционную смесь перемешивают и нагревают в колбе с обратным холодильником в течение периода времени от примерно 12 часов до примерно 20 часов, предпочтительно в течение 16 часов.
В реакции 2 со схемы 2 2-оксо-4,5,6,7-тетрагидро-1H-пиразоло[3,4-с] пиридиновое соединение IX превращается в соответствующее соединение формулы X путем реакции IX с восстанавливающим средством, предпочтительно с литийалюминия гидридом, в неполярном апротонном растворителе, предпочтительно эфире. Реакционную смесь перемешивают в течение периода времени от примерно 12 часов до примерно 20 часов, предпочтительно 16 часов. Затем добавляется вода и основание, предпочтительно гидроксид натрия, и реакционную смесь перемешивают в течение периода времени от примерно 1.5 часов до примерно 2.5 часов, предпочтительно в течение 2 часов, и фильтруют. Фильтрат концентрируют до получения твердого белого вещества.
Способность этих соединений или их фармацевтически приемлемых солей подавлять фосфодиэстеразу IV (ФДЭ4) и, следовательно, проявлять эффективность при лечении воспалительных заболеваний, показана с помощью следующих исследований in vitro.
БИОЛОГИЧЕСКОЕ ИСПЫТАНИЕ
(Человеческая легочная ФДЭIV)
Тридцать - сорок грамм ткани человеческого легкого помещают в 50 мл трис/фенилметилсульфонилфторид (ФМСФ) /сахарозного буфера с pH 7.4 и гомогенизируют, используя Tekmar TissumizerR(Tekmar Co., 7143 Kemper Road, Cincinnati, Ohio 45249) при полной скорости в течение 30 секунд. Гомогенат центрифугируют при 48000xg в течение 70 минут при 4oC. Супернатант фильтруют дважды через 0.22 мкм фильтр и наносят на Моно-Q колонку для жидкостной экспресс-хроматографии белков (Pharmacia LKB Biotechnology, 800 Centennial Avenue, Piscataway, New Jersey 08854) предварительно уравновешенную трис/ФМСФ буфером pH 7.4. Для нанесения образца на колонку используют скорость потока 1 мл/мин с последующей скоростью потока 2 мл/мин для последующих промывания и элюирования. Образец элюируют, используя повышающийся поэтапно градиент NaCl в буфере трис/ФМСФ pH 7.4. Собирают фракции по 8 мл. Фракции испытывают на специфическую ФДЭIV активность, определяемую по [3H]цАМФ гидролизу и на способность известного ингибитора ФДЭIV (например, ролипрама) подавлять гидролиз. Соответствующие фракции объединяют, разбавляют этиленгликолем (2 мл этиленгликоля/5 мл препарата фермента) и хранят при -20oC до использования.
Соединение растворяют в ДМСО до концентрации 10 мМ и разбавляют водой 1: 25 (400 мкМ соединения, 4% ДМСО). Дополнительные серийные разведения производят 4% ДМСО до достижения желаемых концентраций. Конечная концентрация ДМСО в исследуемой пробирке составляет 1%. В стеклянную пробирку 12 x 75 мм в двух повторностях добавляли следующее по порядку (все концентрации даны как конечные концентрации в исследуемой пробирке):
I) 25 мкл соединения или ДМСО (1%, для контроля или пустой пробы);
II) 25 мкл трис-буфера pH 7.5;
III) [3H]цАМФ (1 мкМ);
IV) 25 мкл фермента ФДЭIV (для пустой пробы фермент предварительно инкубируется в кипящей воде в течение 5 минут).
Реакционные пробирки встряхивают и помещают на водяную баню (37oC) на 20 минут, по истечении этого времени реакцию останавливают, помещая пробирки в баню с кипящей водой на 4 минуты. В каждую пробирку на ледяной бане добавляется промывающий буфер (0.5 мл, 0.1 М 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (ГЭПЭС)/0.1 М NaCl, pH 8.5). Содержимое каждой пробирки наносят на колонку Affi-Gel 601 (Biorad Laboratories P.O. Box 1229, 85A Marcus Drive, Melville, New York 11747) (подобие (средство) боронатному гелю, объем слоя 1 мл), предварительно уравновешенную отмывающим буфером. [3H] цАМФ промывают 2 x 6 мл промывным буфером и затем [3H]-5'АМФ элюируют 4 мл 0.25 М уксусной кислоты. После перемешивания вращением 1 мл элюата добавляют к 3 мл жидкости для сцинтилляции в подходящем сосуде, перемешивают вращением и просчитывают на [3H].
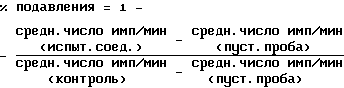
IC50 определяют как концентрацию соединения, которое подавляет 50% специфического гидролиза [3H]цАМФ до [3H]5'АМФ.
(ФНО)
Способность этих соединений или их фармацевтически приемлемых солей подавлять продукцию ФНО, следовательно, проявлять эффективность при лечении заболеваний, связанных с продукцией ФНО, показана в следующих испытаниях in vitro.
Периферическую кровь (100 mls) собирали от людей-добровольцев в этилендиаминтетрауксусную кислоту (ЭДТУ). Моноциты выделяют с помощью Ficoll/Hypaque и три раза промывали в неполном сбалансированном солевом растворе Хэнкса (ССРХ). Клетки повторно суспендируют до конечной концентрации 1 • 106 клеток на мл в предварительно подогретой RPMI (содержащей 5% фетальной телячьей сыворотки, глютамин, пен/степ и нистатин). Моноциты высевали в виде суспензии 1 • 106 клеток в 1.0 мл в 24-ячеечные платы. Клетки инкубируют при 37oC (5% двуокиси углерода) и дают прикрепиться к платам в течение 2 часов, по истечении этого времени неприкрепившиеся клетки удаляют путем осторожного отмывания. Затем добавляют испытуемые вещества в 3 - 4 концентрациях каждое к клеткам и инкубируют в течение 1 часа. В соответствующие ячейки добавляют ЛПС (10 мкл). Платы инкубируют в течение ночи (18 часов) при 37oC. В конце срока инкубации анализировали на ФНО с помощью сэндвичного ELISA (набор R&D Quantikine). Определения IC50 производили для каждого соединения на основе линейного регрессионного анализа.
Фармацевтически приемлемые соли этого изобретения включают, но не ограничиваются ими, те, которые образованы с HCl, HBr, HNO3, H2SO4, H2PO4, CH3SO3H, п-CH3C6H4SO3H, CH3CO2H, глюконовой кислотой, винной кислотой, малеиновой кислотой и янтарной кислотой. Фармацевтически приемлемые катионные соли соединений этого изобретения с формулой I, где R4 является CO2R6 и R6 является водородом, включают, но не ограничиваются ими, те, которые образуются натрием, калием, кальцием, магнием, аммонием, N,N,-дибензилэтилендиамином, N-метилглюкамином (меглумином), этаноламином и диэтаноламином.
Для введения людям при лечении и профилактике воспалительных заболеваний дозы соединений с формулой 1 и их фармацевтически приемлемых солей (здесь далее также называемых активными соединениями данного изобретения) для перорального приема в основном находятся в интервале от 0.1 до 100 мг в сутки для среднего взрослого пациента (70 кг). Таким образом, для типичного взрослого пациента отдельные таблетки или капсулы содержат от 0.1 до 50 мг активного соединения в подходящем фармацевтически приемлемом носителе или наполнителе. Дозы для внутривенного введения обычно находятся в интервале от 0.1 до 10 мг в разовой дозе, когда необходимо. Для интраназального или ингаляционного введения дозированная лекарственная форма обычно создается в виде 0.1-1% раствора (вес/объем). На практике врач будет определять фактическую дозировку, которая будет наиболее подходящей для конкретного пациента и будет изменяться в зависимости от возраста, веса и реакции конкретного больного. Указанные выше дозировки являются примерными для среднестатистического случая, но могут, конечно, существовать примеры, когда требуются более высокие или более низкие уровни дозировки, и подобные дозы охватываются объемом этого изобретения.
При применении у людей для подавления ФНО может использоваться ряд обычных путей введения, включая пероральный, парентеральный и местное применение. В основном активное соединение будет вводиться перорально или парентерально в дозах от примерно 0.1 до 25 мг/кг веса тела субъекта, нуждающегося в лечении, в сутки, предпочтительно от 0.3 до 5 мг/кг. Однако будет встречаться некоторое изменение дозировки по необходимости в зависимости от состоянии субъекта, нуждающегося в лечении. Лицо, ответственное за применение, в любом случае будет определять соответствующую дозу для конкретного субъекта.
При использовании для людей активные соединения данного изобретения могут вводиться в чистом виде, но в основном будут применяться в смеси с фармацевтическим растворителем или носителем в зависимости от намеченного пути введения и стандартов фармацевтической практики. Например, они могут вводиться перорально в форме таблеток, содержащих такие наполнители, как крахмал или лактоза, или в капсулах или пилюлях, или в чистом виде, или в смеси с наполнителями, или в форме эликсиров или суспензий, содержащих улучшающие вкус или подкрашивающие средства. Они могут вводиться в виде инъекций парентерально; например, внутривенно, внутримышечно или подкожно. Для парентерального введения лучше всего их использовать в виде стерильного водного раствора, который может содержать другие вещества: например, достаточное количество солей и глюкозы, для того, чтобы сделать раствор изотоническим.
Таким образом, в дополнительном аспекте это изобретение представляет фармацевтические композиции, содержащие соединение с формулой I и их фармацевтически приемлемые соли вместе с фармацевтически приемлемым растворителем и носителем.
Данное изобретение иллюстрируется следующими примерами, но не ограничивается их деталями.
Пример 1
3-Этил-1-(4-метоксифенил)-6-фенил-7-оксо-4,5,6,7-тетрагидро- 1H-пиразоло[3,4-c]пиридин
Смесь 3-гидрокси-2-оксо-1-фенил-4-пропионил-1,2,5,6- тетрагидропиридина (1.0 г, 4.1 ммоля), 4-метоксифенилгидразина гидрохлорида (0.8 г, 4.6 ммоля) и метоксида натрия (0.11 г, 2 ммоля) в 35 мл безводного этанола (перегнанного с Mg) нагревали в колбе с обратным холодильником. Через 12 часов растворитель удаляли с помощью отгонки на роторном испарителе под пониженным давлением и неочищенный остаток хроматографировали на колонке двуокиси кремния 4 x 20 см, используя эфир/гексан 1:1 в качестве элюента с получением 345 мг вещества, названного в заголовке, в виде красного масла, которое кристаллизовалось при стоянии при комнатной температуре. Желаемый 1-(4-метоксифенил)-региоизомер менее полярен, чем 2-(4-метоксифенил)- побочный продукт.
Т.пл. 43-45oC.
ИК (хлороформ) лактам C=O, 1665 см-1;
1H ЯМР (300 МГц, CDCl3) d 1.32 (t, J = 7.6 Гц, 3H), 2.74 (q, J = 7.6 Гц, 2H), 2.96 (t, J = 6.6 Гц, 2H), 3.79 (s, 3H), 4.10 (t, J = 6.6 Гц, 2H), 6.89 (d, J = 9.0 Гц, 2H), 7.22-7.39 (m, 5H), 7.45 (d, J = 9.0 Гц, 2H);
Расчетный анализ для C21H21N3O2:
C, 72.60; H 6.09; N, 12.09 .
Обнаружено: C, 72.48; H, 6.08; N, 11.66;
MC м/з [M+] 347.
Примеры 2-15 см. в конце описания.
Пример 16
3-Этил-1-(4-фенилкарбоновая кислота)-6-фенил-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин
Смесь 3-гидрокси-2-оксо-1-фенил-4-пропионил-1,2,5,6-тетрагидропиридина (1.0 г, 4.08 ммоля), 4-гидразинбензойной кислоты (0.68 г, 4.49 ммоля) и 30 мл безводного этанола нагревали в колбе с обратным холодильником. Через 20 часов смесь концентрировали в роторном испарителе под пониженным давлением и твердый остаток суспендировали в смеси этилацетата (500 мл) и буфера pH (200 мл). Органический слой отделяли (оставляя большую часть 2-(4-фенилкарбоновой кислоты) - побочного продукта), промывали солевым раствором, сушили сульфатом натрия, фильтровали и концентрировали под пониженным давлением. Перекристаллизация из метанола дает 0.64 грамма соединения, указанного в заголовке, в виде оранжевого твердого вещества.
Т.пл. 261-263oC.
1H ЯМР (300 МГц, ДМСО-d6) 1.23 (t, J = 7.6 Гц, 3H), 2.68 (q, J = 7.6 Гц, 2H), 2.94 (t, J = 6.5 Гц, 2H), 4.05 (t, J = 6.5 Гц, 2H), 7.20-7.41 (m, 5H), 7.65 (d, J = 8.6 Гц, 2H), 7.96 (d, J = 8.6 Гц, 2H), 13.05 (s, 1H);
MC м/з [M+] 362.
Пример 17
1-(4-Бензамид)-3-этил-6-(4-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1H-пиразоло[3,4-c]пиридин
К перемешиваемому раствору метокси натрия в метаноле (полученному из 6.6 мг Na) добавляют 3-этил-6-(4-метоксифенил)-1- (4-фенилкарбоновая кислота)-7-оксо-4,5,6,7-тетрагидро-1H- пиразоло[3,4-c] пиридин (96 мг, 0.25 ммоля). Через 30 минут метанол удаляли под пониженным давлением, твердый остаток суспендировали в бензоле, и бензол удаляли под пониженным давлением. Полученное в результате сухое твердое вещество суспендировали в холодном эфире (ледяная баня) и обрабатывали оксалилхлоридом (31 мкл, 0.35 ммоля) и безводным N, N-диметилформамидом (1 капля). После перемешивания в течение 1 часа летучие вещества удаляли под пониженным давлением, и неочищенный остаток растворяли в безводном тетрагидрофуране. Полученный в результате раствор добавляли по каплям к интенсивно перемешиваемому гидроксиду аммония при 0oC. После нагревания до комнатной температуры в течение 2 часов, реакционную смесь концентрировали под пониженным давлением до тех пор, пока не начинало осаждаться желтое твердое вещество. В это время смесь разбавляли водой до примерно 100 мл и фильтровали, осадок промывали водой с получением 81 мг вещества, названного в заголовке.
Температура разложения 243-245oC.
1H ЯМР (ДМСО-d6) 1.24 (t, J = 7.6 Гц, 3H), 2.68 (q, J = 7.6 Гц, 2H), 2.93 (t, J = 6.5 Гц, 2H), 3.75 (s, 3H), 3.99 (t, J = 6.5 Гц, 2H), 6.94 (d, J = 9.1 Гц, 2H), 7.27 (d, J = 9.0 Гц, 2H), 7.43 (s, 1H), 7.59 (d, J = 8.5 Гц, 2H), 7.90 (d, J = 8.6 Гц, 2H), 8.04 (s, 1H);
Анализ, расчетное C22H22N4O3:
C, 67.68; H, 5.68; N, 14.35.
Обнаруженное: C, 67.19; H, 5.31; N, 13.55.
HRMS расчетный для C22H22N4O3 [M+] 391.1770.
Обнаруженное: 391.1781.
Исходный 3-этил-6-(4-метоксифенил)-1-(4-фенилкарбоновая кислота)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c] пиридин получали, используя соответствующие реагенты, по процедуре из примера 16.
Пример 18
1-(3,4-дихлорфенил)-3-этил-6-(3-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1Н-пиразоло[3,4-c]пиридин
Перемешиваемую смесь 3-метокси-1-(3-метоксифенил)-2-оксо-4- пропионил-1,2,5,6-тетрагидропиридина (0.49 г, 1.7 ммоля), 3,4-дихлорфенилгидразина гидрохлорида (0.40 г, 1.87 ммоля) и метоксида натрия (46 мг, 0.85 ммоля) в безводном этаноле нагревали в колбе с обратным холодильником. Через 16 часов смесь концентрировали под пониженным давлением и хроматографировали на силикагелевой колонке, используя этилацетат/гексан 1:4 в качестве элюента с получением белого твердого вещества. Перекристаллизация из эфира давала 0.46 грамма белых игольчатых кристаллов.
Т.пл. 97-99oC.
1H ЯМР (250 МГц, CDCl3) 1.31 (t, J = 7.5 Гц, 3H), 2.73 (q, J = 7.6 Гц, 2H), 2.96 (t, J = 6.6 Гц, 2H), 3.79 (s, 3H), 4.09 (t, J = 6.6 Гц, 2H), 6.78-6.91 (m, 3H), 7.29-7.49 (m, 3H), 7.73 (d, J = 1.8 Гц, 1H);
МС м/з [M+] 416.
Примеры 19 - 42
Реакция соответствующего гидразингидрохлорида с нужным 4-алканоил-3-метокси-2-оксо-1,2,5,6-тетрагидропиридином, аналогично процедуре из примера 18, дает следующие соединения. (см. в конце описания).
Пример 43
1-Циклогексил-3-этил-6-(3-метоксифенил)-7-оксо-4,5,6,7- тетрагидро-1Н-пиразоло[3,4-c]пиридин
Раствор 3-метокси-1-(3-метоксифенил)-2-оксо-4-пропионил- 1,2,5,6-пиридина (0.80 г, 2.8 ммоля) и циклогексилгидразингидрохлорида (0.54 г, 3.6 ммоля) в метаноле (15 мл) нагревали до 90oC при мягком пропускании азота до тех пор, пока растворитель не удалится. Неразбавленную смесь затем нагревали до примерно 150oC в атмосфере азота в течение 1 часа. После охлаждения до комнатной температуры смесь растворяли в эфире и промывали 1N хлористоводородной кислотой с последующим промыванием солевым раствором, сушили сульфатом натрия, фильтровали и концентрировали под пониженным давлением. Хроматография на силикагеле с использованием этилацетат/гексана 1:1 в качестве элюента дает 0.47 грамма соединения, указанного в заголовке, в виде желтого масла.
1H ЯМР (250 МГц, CDCl3) 1.20-1.52 (m, 6H, включая t при 1.23, J = 7.6 Гц, 3H), 1.64-1.74 (m, 1H), 1.80-2.06 (m, 6H), 2.67 (q, J = 7.6 Гц, 2H), 2.87 (t, J = 6.7 Гц, 2H), 3.82 (s, 3H), 3.97 (t, J = 6.7 Гц, 2H), 5.13 (tt, J = 4.3 и 11.3 Гц, 1H), 6.79-6.93 (m, 3H), 7.31 (t, J = 8.1 Гц, 1H);
HRMS рассчитанный для C21H27N3O2 [M+]: 353.2103.
Обнаруженное: 353.2094.
Примеры 44 - 57
Реакция соответствующего гидразингидрохлорида с нужным 4-алканоил-3-метокси-2-оксо-1,2,5,6-тетрагидропиридином, аналогичная процедуре из примера 43, дает следующие соединения (см. в конце описания).
Пример 58
3-Этил-6-(4-фторфенил)-1-(4-метоксифенил)-4,5,6,7-тетрагидро- 1Н-пиразоло[3,4-c]пиридин
К перемешиваемому раствору 3-этил-6-(4-фторфенил)-1-(4-метоксифенил)- 7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c] пиридина (0.3 г, 0.82 ммоля) в 50 мл эфира добавляли лития-алюминия гидрид (33 мг, 0.86 ммоля). После перемешивания в течение 16 часов добавляли воду (0.5 мл), а затем N гидроксид натрия (1 мл). После перемешивания в течение 2 часов белый осадок отфильтровывали через броунмиллерит и фильтрат концентрировали под пониженным давлением. Хроматография на силикагелевой колонке с использованием этилацетата/гексана 1:3 дает 0.12 грамма соединения, указанного в заголовке, в виде бледно-желтой пасты.
1H ЯМР (250 МГц, CDCl3) 1.28 (t, J = 7.6 Гц, 3H), 2.66 (q, J = 7.6 Гц, 2H), 2.71 (t, J = 5.7 Гц, 2H), 3.49 (t, J = 5.7 Гц, 2H), 3.84 (s, 3H), 4.23 (s, 2H), 6.84-6.99 (m, 6H), 7.36 (d, J = 9.0 Гц, 2H);
МС м/з [M+] 352.
Примеры 59 - 63
Реакция соответствующего 7-оксо-2,5,6,7-тетрагидро-1Н- пиразоло[3,4-c] пиридина с лития-алюминия гидридом, аналогичная процедуре из примера 58, дает следующие соединения (см. в конце описания).
Пример 64
1-Циклопентил-3-этил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридин
Перемешиваемый раствор 1-циклопентил-3-этил-6-(4-метоксифенил)- 7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c] пиридина (2.58 г, 7.60 ммоля) в ацетонитриле (90 мл) при 0oC обрабатывается раствором церий-аммония нитрата (12.5 г, 22.8 ммоля) в воде (110 мл). После перемешивания в течение 35 минут смесь разбавляли водой (550 мл) и экстрагировали этилацетатом (100 х 4). Объединенные органические экстракты промывали 50% насыщенным раствором бикарбоната натрия (250 мл) и затем 10% раствором сульфита натрия до тех пор, пока промывной водный слой не становится бледно-желтым. Затем органический слой дополнительно промывали насыщенным раствором бикарбоната натрия и соляным раствором и обрабатывали убирающим окрашивание углем. После перемешивания в течение 30 минут смесь высушивали сульфатом натрия, фильтровали через броунмиллерит и концентрировали под пониженным давлением. Коричневый осадок перекристаллизовывали из эфира с получением 0.814 грамма желтовато-коричневого твердого вещества.
Т.пл. 143-145oC;
МС (м/з) 234;
1H ЯМР (250 МГц, CDCl3)) 1.21 (t, J = 7.6 Гц, 3H), 1.62-2.13 (m, 8H), 2.62 (q, J = 7.6 Гц, 2H), 2.73 (t, J = 6.8 Гц, 2H), 3.51 (dt, J = 2.7 и 6.8 Гц, 2H), 5.47 (s, 1H), 5.61 (пентет, J = 7.7 Гц, 1H).
Пример 65
1-циклопентил-3-этил-6-циклопропилметил-7-оксо-4,5,6,7- тетрагидро-1Н-пиразоло[4,4-c]пиридин
Раствор 1-циклопентил-3-этил-7-оксо-4,5,6,7-тетрагидро-1Н- пиразоло[3,4-c]пиридина (0.21 г, 0.92 ммоля) в ТГФ (5 мл) обрабатывается 60% раствором гидрида натрия в минеральном масле (40 мг, 1.01 ммоля). После перемешивания при нагревании в колбе с обратным холодильником в течение 45 минут реакционную смесь охлаждали до 25oC и добавляли (бромметил)циклопропан (0.31 г, 2.29 ммоля). Смесь перемешивают при нагревании в колбе с обратным холодильником в течение 16 часов и затем охлаждают до 25oC перед концентрированием при пониженном давлении. Хроматография на силикагеле с элюированием этилацетатом/гексаном дает 0.19 г соединения, названного в заголовке, в виде бесцветного масла.
MC м/з [M+] 288;
1H ЯМР (300 МГц, CDCl3) 0.26-0.31 (m, 2H), 0.50-0.56 (m, 2H), 0.85-1.06 (m, 1H), 1.20 (t, J = 7.6 Гц, 3H), 1.62-2.08 (m, 8H), 2.61 (g, J = 7.6 Гц, 2H), 2.74 (t, J = 6.8 Гц, 2H), 3.39 (d, J = 6.9 Гц, 2H), 3.63 (t, J = 6.8 Гц, 2H), 5.67 (пентет, J = 7.8 Гц, 1H).
Препарат 1
4-Изобутирил-3-метокси-1-фенил-2-оксо-1,2,5,6-тетрагидропиридин
Перемешиваемый раствор свежеперегнанного диизопропиламина (0.16 мл, 2.21 ммоля) в безводном тетрагидрофуране (4 мл) охлаждали до 0oC и обрабатывали 2.5 М н-бутиллитием (0.85 мл 2.11 ммоля). Через 15 минут смесь охлаждали до -78oC и по каплям через канюлю добавляли предварительно охлажденный раствор 4-пропионил-3-метокси-1-фенил-2- оксо-1,2,5,6-тетрагидропиридина (0.52 г, 2.0 ммоля) в тетрагидрофуране (4 мл). Через примерно 20 минут добавляли метилйодид (0.20 мл, 3.0 ммоля) к яркому оранжево-красному раствору, и смеси давали принять комнатную температуру в течение 2.5 часов. Реакционную смесь выливали в насыщенный водный раствор хлорида аммония, и органический слой промывали солевым растворами, сушили сульфатом натрия, фильтровали и концентрировали под пониженным давлением. Хроматография на силикагелевой колонке с использованием этилацетата/гексана 1:4 в качестве элюента дает 0.12 грамма соединения, указанного в заголовке, в виде желтого масла и 0.1 грамма регенерируемого исходного материала.
1H ЯМР (250 МГц, CDCl3) 1.15 (d, 6H), 2.72 (t, 2H), 3.47 (гептет, 1H), 3.82 (t, 2H), 3.97 (s, 3H), 7.21-7.45 (m, 5H);
MC м/з [M+] 274.
Препараты 2 - 3
Реакция соответствующего 3-метокси-2-оксо-4-пропионил-1,2,5,6- тетрагидропиридина с литийдиизопропиламином и метилйодидом, аналогичная процедуре из препарата 1, дает следующие соединения с формулой VII (см. в конце описания)
Препарат 4
3-Метокси-1-(4-метилфенил)-2-оксо-4-пропионил- 1,2,5,6-тетрагидропиридин
Раствор 3-гидрокси-1-(4-метилфенил)-2-оксо-4-пропионил- 1,2,5,6-тетрагидропиридина (5.9 г, 23 ммоля и 3-метил-1-п- толилтриазина (5.1 г, 34 ммоля) в 1,2-дихлорэтане нагревали в колбе с обратным холодильником в течение 45 минут. Смеси давали охладиться до комнатной температуры и выливали в воду и подкисляли 6N хлористоводородной кислотой. Водный слой экстрагировали 3 раза метиленхлоридом и объединенные органические вытяжки промывали 1N хлористоводородной кислотой, затем водой и солевым раствором, сушили сульфатом магния, фильтровали и концентрировали под пониженным давлением. Полученное в результате поддающееся количественному определению коричневое масло было чистым по тонкослойной хроматографии и 1H ЯМР и использовалось без очистки.
1H ЯМР (300 МГц, CDCl3) 1.12 (t, J = 7.2 Гц, 3H), 2.34 (s, 3H), 2.71 (t, J = 6.7 Гц, 2H), 2.93 (g, J = 7.2 Гц, 2H), 3.77 (t, J = 6.8 Гц, 2H), 3.94 (s, 3H), 7.20 (s, 4H);
MC [M+] 273.
Препараты 5 - 14
Реакция соответствующего 3-гидрокси-1-арил-2-оксо-4-алканоил- 1,2,5,6-тетрагидропиридина с 3-метил-1-п-толилтриазином, аналогичная процедуре для препарата 4, дает следующие соединения с формулой VI (см. в конце описания).
Препарат 15
3-Гидрокси-1-(3-метилфенил)-2-оксо-4-пропионил- 1,2,5,6-тетрагидропиридин
К перемешиваемой суспензии магниевых стружек (1.9 г, 79 ммоля) в 30 мл безводного эфира по каплям добавляли бромэтан (5.9 мл, 79 ммоля). Слабое кипение в колбе с обратным холодильником начиналось после добавления примерно 1 мл. После того, как весь магний был израсходован, реакционную смесь охлаждали до 0oC и сразу добавляли N-(3-метилфенил-2-пирролидон (8.7 г, 50 ммолей). После нагревания до комнатной температуры и перемешивания в течение 2 часов реакционную смесь выливали на лед и экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и солевым раствором, сушили сульфатом натрия, фильтровали и концентрировали под пониженным давлением с получением 8.8 грамма белого твердого вещества.
Вышеупомянутое твердое вещество диспергировали в смеси 40 мл бензола и 86 мл 1N гидроксида натрия и при энергичном механическом перемешивании добавляли этилоксалилхлорид (7.2 мл, 64 ммоля). После перемешивания при нагревании в колбе с обратным холодильником в течение 1.5 часов слои разделяются, и водный слой экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и солевым раствором, сушили сульфатом магния, фильтровали и концентрировали под пониженным давлением с получением янтарного масла.
ГХМС [M+] 305.
Вышеуказанное промежуточное соединение растворяли в 20 мл безводного этанола и обрабатывали раствором метоксида натрия в метаноле (приготовленном путем осторожного добавления натрия (1.0 г) к 10 мл безводного метанола). После перемешивания при нагревании в колбе с обратным холодильником в течение 1.5 смесь концентрировали под пониженным давлением и добавляли 100 мл воды. Смесь подкисляли до pH 6 6N хлористоводородной кислотой, и тускло-желтый осадок отфильтровывали и промывали водой. Перекристаллизация из 75 мл изопропилового эфира давала 6.8 г бледно-желтых кристаллов.
Т.пл. 115-116oC;
1H ЯМР (300 МГц, CDCl3) 1.6 (t, J = 7.2 Гц, 3H), 2.37 (s, 3H), 2.74-2.82 (m, 4H), 3.85 (t, J = 6.8 Гц, 2H), 7.08-7.14 (m, 3H), 7.30 (t, J - 7.7 Гц, 1H);
MC м/з [M+] 259.
Препараты 16 - 29
Реакция соответствующего 2-пирролидинона с нужным алкилмагнийбромидом с последующей обработкой этилоксисалилхлоридом и основанием аналогично тем, о которых сообщено в разделе Препарат 15, дает следующие соединения с формулой VI (см. в конце описания).
Препарат 30
N-(2-Метоксифенил)-2-пирролидон
Смесь 2-пирролидона (15.0 г, 176 ммолей), 2-йоданизола (7.6 мл, 59 ммолей), порошка меди (7.5 г, 117 ммолей) и карбоната калия (8.1 г, 59 ммолей) перемешивают в атмосфере азота при 150oC. Через 18 часов реакционную смесь отфильтровывали через подушку силикагеля, элюируя этилацетатом/гексаном с получением бледно-желтого масла. Непрореагировавшие реагенты удалялись с помощью вакуумной дистилляции (0.6 мм, 80-100oC), оставляя 9.2 грамма соединения, указанного в заголовке, в виде медоподобного масла.
1H ЯМР (300 МГц, CDCl3) 2.20 (пентет, 2H), 2.55 (t, 2H), 3.75 (t, 2H), 3.82 (s, 3H), 6.93-7.02 (m, 2H), 7.25-7.30 (m, 2H);
MC м/з [M+] 191.
Препараты 31 - 39
Реакции соответствующего йод- или бромбензола с 2-пирролидиноном, аналогичная той, о которой сообщено в разделе Препарат 30, дает следующие соединения с формулой V (см. в конце описания).
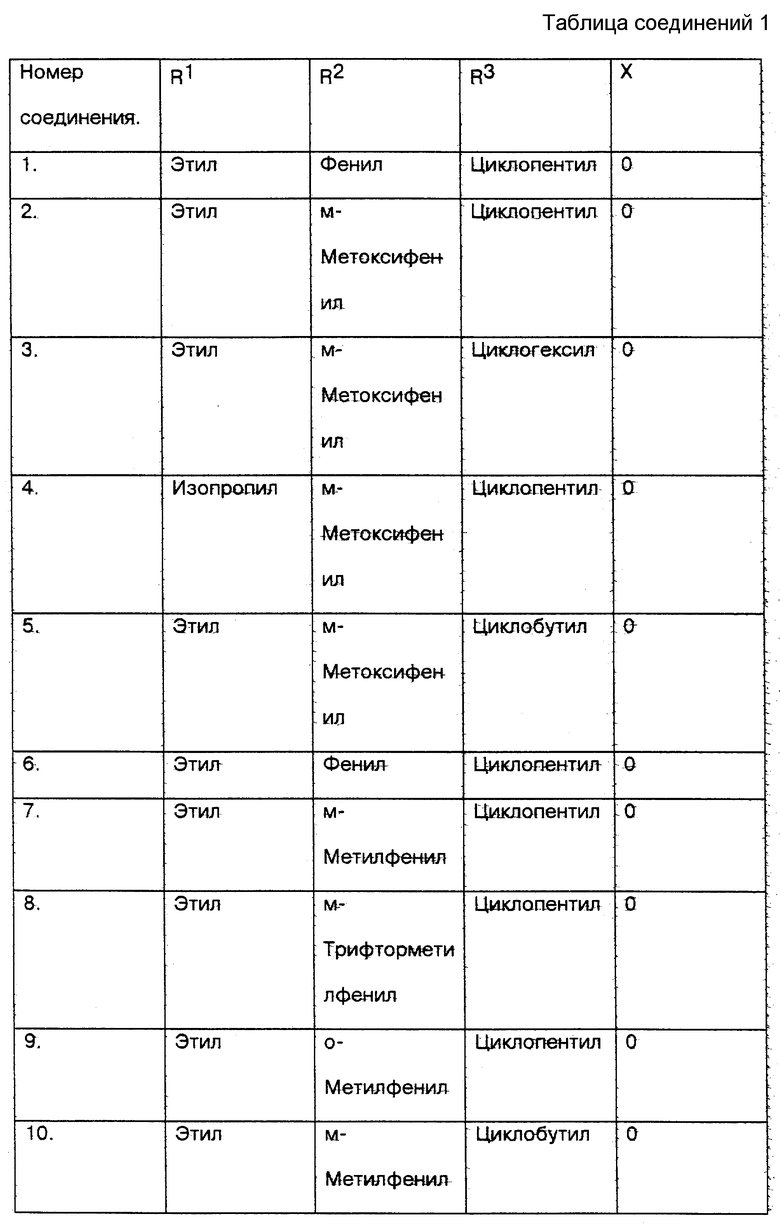
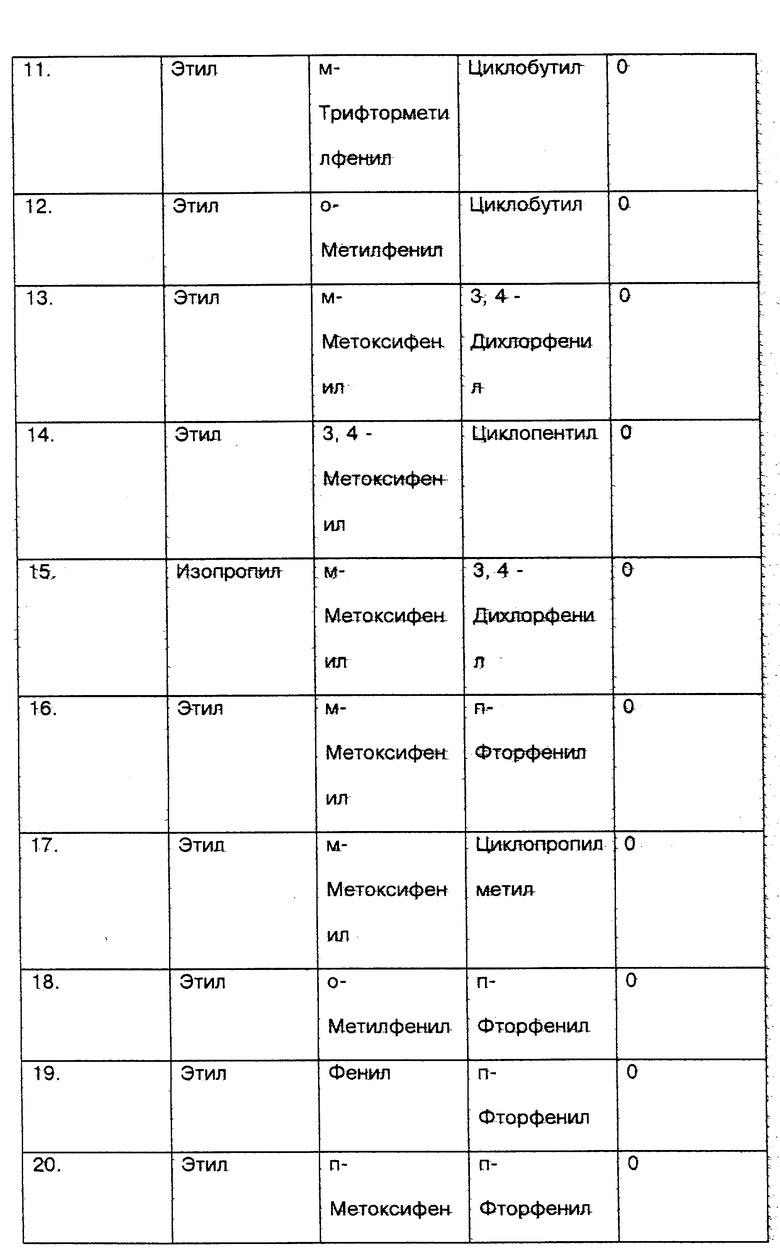
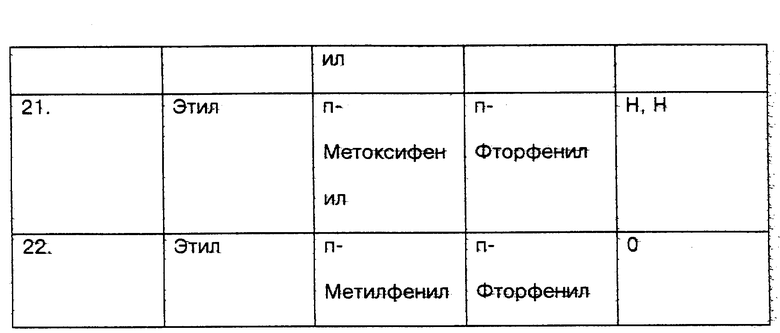
Со стороны заявителя соединения настоящего изобретения (соединения N 1 - 18) и сравниваемые соединения (соединения N 19 - 22) вышеприведенных ссылок были протестированы in vitro с использованием метода анализа PDE4 в легком человека, который признается на современном уровне техники в качестве стандарта для определения того, ингибируют ли указанные соединения фосфодиэстеразу IV (PDE4), и, следовательно, для демонстрации их эффективности для лечения воспалительных заболеваний (см. таблицу соед. 1-22 в конце описания).
Метод анализа PDE4 в легком человека, как он был упомянут в спецификации на странице 10, заключается в следующем.
От тридцати до сорока граммов легочной ткани человека размещалось в 50 мл буфера Трис /фенилметилсульфонилфторид (PMSF)/ сукроза с pH 7,4 и гомогенизировалось с использованием Tekmar TissumizerR (Tekmar Co., 7143 Kemper Road, Cincinnati, Ohio 45249) на полной скорости в течение 30 секунд. Гомогенат центрифугировался при 48000 • g в течение 70 минут при 4oC. Супернатант фильтровался два раза при помощи фильтра 0,22 мкм и вносился в колонку Mono - Q FPLC (Pharmacia LKB Biotechnology, 800 Centennial Avenue, Piscataway, New Jersey 08854), предварительно приведенную в равновесие при помощи буфера Трис/PMSF с pH 7,4. Для внесения образца в колонку использовался расход 1 мл/минута, после чего устанавливался расход 2 мл/минута для последующего промывания и элюирования. Образец элюировался при помощи увеличивающегося ступенчато градиента NaCl в буфере Трис/PMSF с pH 7,4. Были собраны фракции по восемь мл. Фракции анализировались на специфическую активность PDE4, определенную по гидролизу [3H] cAMP и по способности известного ингибитора PDE4 (например, ролипром) ингибировать данный гидролиз. Соответствующие фракции объединялись, разбавлялись этиленгликолем (2 мл этиленгликоля / 5 мл препарата энзима) и хранились при -20oC до момента использования.
Соединения растворяли в диметилсульфоксиде (ДМСО) с концентрацией 10 мМ и разбавляли 1:25 в воде (400 мкМ соединения, 4% ДМСО). Дальнейшие последовательные разбавления проводились в 4% диметилсульфоксиде до достижения требуемых концентраций. Конечная концентрация диметилсульфоксида в пробирке для анализа составляла 1%. В дубликате следующие реактивы, по порядку, были добавлены в стеклянную пробирку 12 х 75 мм (все концентрации приводятся для конечных концентраций в пробирке для анализа).
i) 25 мкл соединения или диметилсульфоксида (1% для контрольного и пустого образца).
ii) 25 мкл Трис буфера с pH 7,5.
iii) [3H] cAMP (1 мкМ).
iv) 25 мл энзима PDE4 (для пустого образца энзим предварительно выдерживался в кипящей воде в течение 5 минут).
Пробирки для реакции встряхивались для перемешивания и помещались в водяную баню (37oC) на 20 минут, после этого времени реакция останавливалась при помещении пробирок в кипящую водяную баню на 4 минуту. В ледяной бане в каждую пробирку добавлялся промывающий буфер (0,5 мл, 0,1 М 4-(2-гидроксиэтил)-1-пиперазин-этансульфоновая кислота (HEPES)/0,1 М NaCl, pH 8,5). Содержимое каждой пробирки вносилось в колонку Affi-Gel 601 (Biorad Laboratories, P. O. Box 1229, 85A Marcus Drive, Melville, New York 11747) (гель со сродством к боронату, объем подложки 1 мл), предварительно приведенную в равновесие промывающим буфером. [3H] cAMP промывался промывающим буфером 2 х 6 мл, и после этого [3H] 5'AMP элюировался 4 мл 0,25-молярной уксусной кислоты. После интенсивного перемешивания 1 мл элюата добавлялся к 3 мл сцинтилляционной жидкости в соответствующей пробирке, проводились интенсивное перемешивание и подсчет [3H].
% ингибирования = 1 - (среднее cpm (cpm. в испытываемом образце) - среднее cpm (в пустом образце))/(среднее cpm (в контрольном образце) - среднее cpm (в пустом образце))
IC50 определяется как такая концентрация соединения, которая ингибирует 50% специфического гидролиза [3H] cAMP с образованием [3H] 5'AMP.
5. Результаты приведенного выше теста следующие:
Номер соединения - HUPDE*4 IC50 мкМ
1. - 0.0299
2. - 0.0119
3. - 0.0062
4. - 0.0136
5. - 0.0036
6. - 0.0170
7. - 0.0123
8. - 0.0306
9. - 0.0015
10. - 0.0006
11. - 0.031
12. - 0.0019
13. - 0.13
14. - 0.22
15. - 0.17
16. - 0.21
17. - 0.15
18. - 0.26
19. - 0.44
20. - 3.84
21. - 9.30
22. - 1.66
* Метод анализа PDE4 в легком человека.
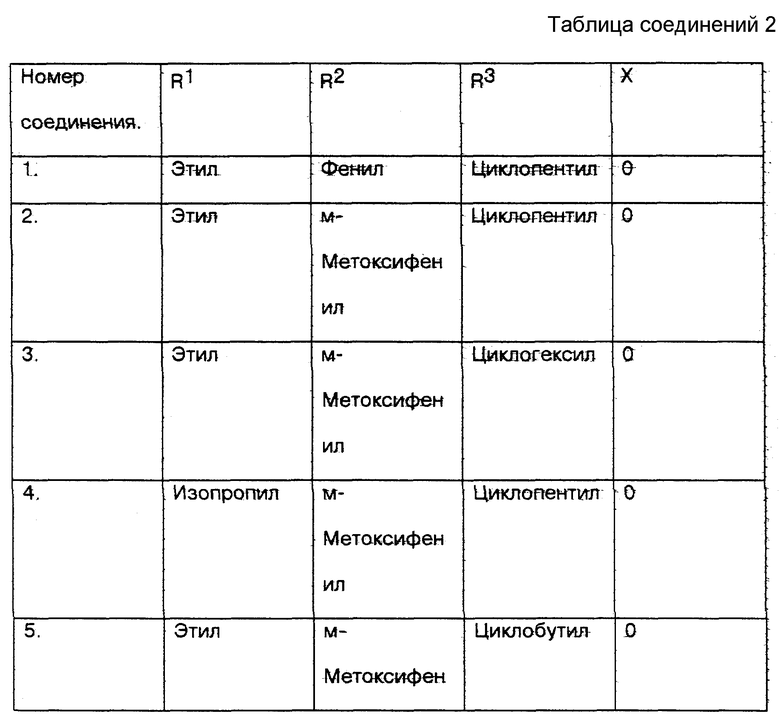
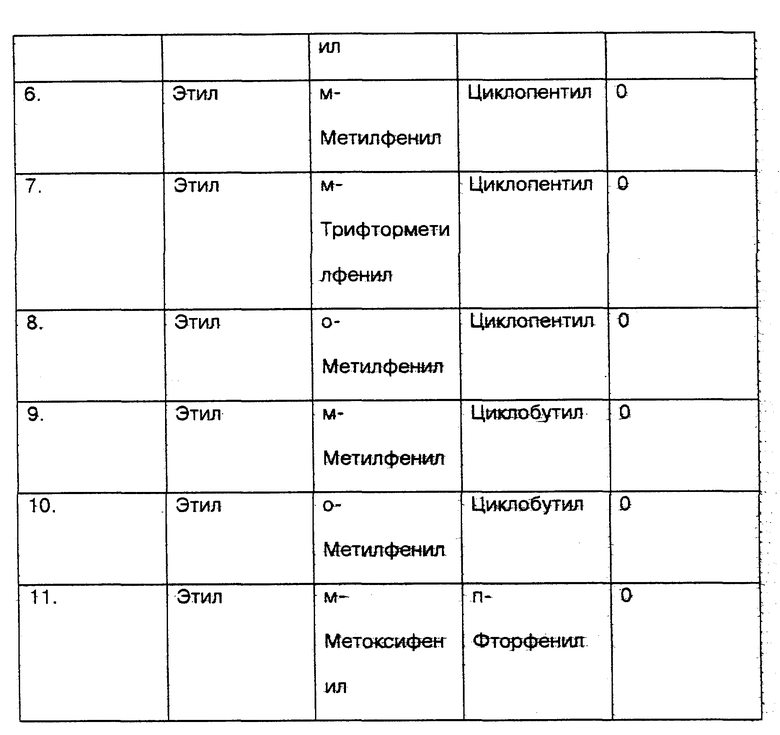
6. С моей стороны соединения настоящего изобретения (соединения N 1 N 10 и сравниваемое соединение N 11) из U.S. 3365459 были протестированы in vitro с использованием метода анализа фактора некроза опухоли (далее называемое TNF), который признан на современном уровне техники в качестве стандарта для определения того, ингибируют ли указанные соединения получение TNF, и, следовательно, для демонстрации их эффективности при лечении заболеваний, при которых образуется TNF (см. таблицу соединений 2 в конце описания).
Метод анализа THF заключается в следующем.
Периферийная кровь (100 мл) от добровольцев была собрана в этилендиаминтетрауксусной кислоте (EДTA). Моноядерные клетки изолировались при помощи Ficoll/Hipaque и промывались три раза в неполном HBSS. Клетки суспендировались еще раз с конечной концентрацией, равной 1 • 104 клеток на миллилитр в предварительно нагретом RPMI (содержащем 5% FCS, глутамин, pen/step и нистатин). Моноциты наносились в виде 1 • 106 клеток в 1,0 мл на пластины с 24 ячейками. Клетки выдерживались при 37oC (5% углекислый газ) и оставлялись прилипать к пластинам в течение 2 часов, после этого времени неприлипшие клетки удалялись при помощи осторожного промывания. После этого добавлялись тестируемые соединения (10 мкл) к клеткам при 3 - 4 концентрациях каждое, и производилось выдерживание в течение 1 часа. LPS (10 мкл) добавлялся в соответствующую ячейку. Пластины выдерживались в течение ночи (18 часов) при 37oC. В конце периода выдерживания TNF был проанализирован при помощи сэндвичевой ELISA (R&D Quantikine Kit). Определения IC50 были проведены для каждого соединения на основе линейного регрессионного анализа.
7. Результаты приведенного выше теста следующие:
Номер соединения - TNF - HM* - IC50 ** мкМ
1. - 0.025
2. - 0.008
3. - 0.043
4. - 0.035
5. - 0.024
6. - 0.011
7. - 0.087
8. - 0.060
9. - 0.014
10. - 0.033
11. - 0.41
* Метод анализа моноцитов человека TNF.
** IC50 на основе линейного регрессионного анализа.
8. Вышеприведенные данные демонстрируют, что соединения заявленного изобретения в вышеуказанном применении обладают неожиданными свойствами, например, лучшим ингибированием фосфодиэстеразы IV и получением фактора некроза опухоли при сравнении с соединениями предшествующего уровня техники.
Датировано: 7 марта 1994 года. Allen J. Duplantierн
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ 5,6-ДИГИДРО-9H-ПИРАЗОЛО[3,4-С]-1,2,4-ТРИАЗОЛО [4,3-А] ПИРИДИНЫ, СПОСОБ ИНГИБИРОВАНИЯ ФОСФОДИЭСТЕРАЗЫ ТИПА IУ И СПОСОБ ИНГИБИРОВАНИЯ ПРОДУЦИРОВАНИЯ ФАКТОРА ОПУХОЛЕСПЕЦИФИЧЕСКОГО НЕКРОЗА | 1996 |
|
RU2161158C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРИМЕНЕНИЕ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2317988C2 |
| ПИРАЗОЛОПИРИМИДИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1993 |
|
RU2124016C1 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ 5-ЛИПОКСИГЕНАЗЫ | 1995 |
|
RU2136666C1 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| ПРОИЗВОДНЫЕ АРИЛСУЛЬФОНИЛАМИНОГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ МАТРИЧНЫХ МЕТАЛЛОПРОТЕИНАЗ И СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2145597C1 |
| АМИНОЗАМЕЩЕННЫЕ ПИРАЗОЛЫ И ПРОМЕЖУТОЧНЫЕ АМИНОЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 1993 |
|
RU2142455C1 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ | 2001 |
|
RU2286343C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1993 |
|
RU2130453C1 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИЙ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2290179C2 |
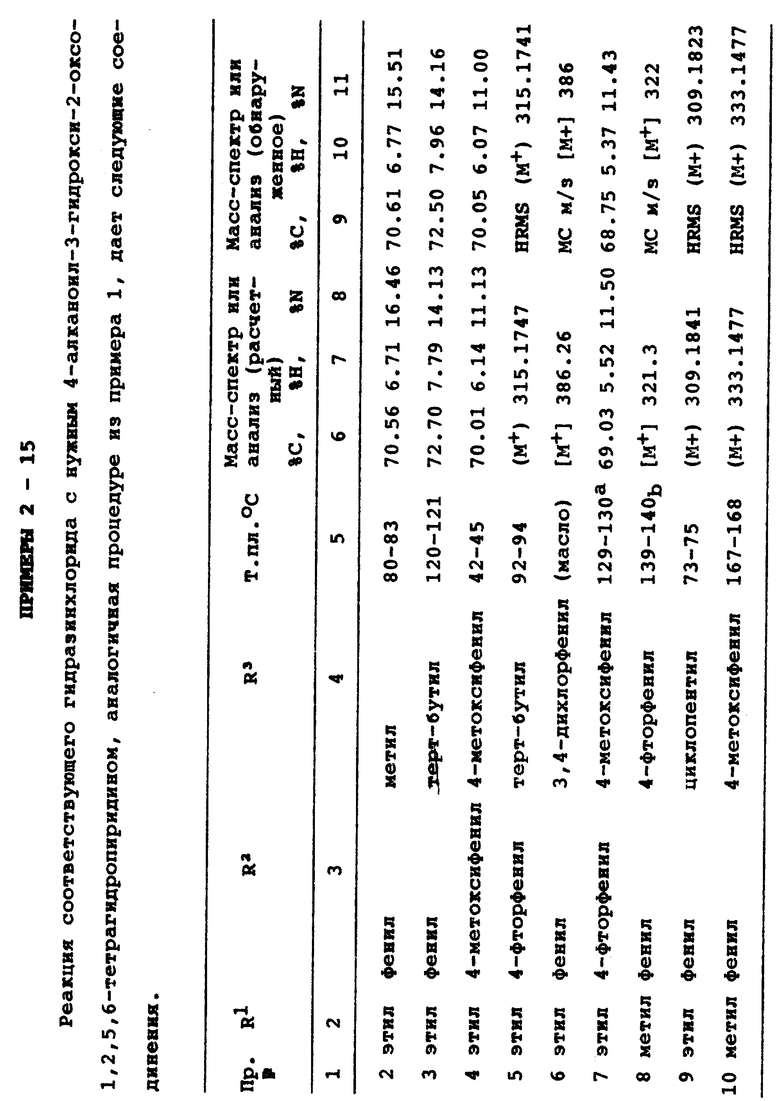





Бициклические тетрагидропиразолпиридины формулы I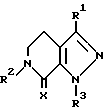
где R1 - С1-3алкил; X - кислород или два атома водорода; R2 C1-4алкил, индалил, С3-7циклоалкил или группа формулы II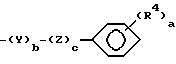
а = 1 или 2; b и с = 0 или 1 ; R4-Н, C1-5алкил, алкоксил, С3-6циклоалкоксил, галоген, СF3, СО2R6, CONR6R7, NR6R7, R6, R7 - Н или C1-4алкил; Z - SO; Y - С1-5алкилен; R3 имеет значения, определенные для R2 и метиленциклопропил, при условии, что R2 и R3 не могут одновременно означать метил, или их фармацевтически приемлемые соли и фармацевтическая композиция на их основе ингибируют фосфодиэстеразу типа IV и продуцирование фактора некроза опухолей. 4 c. и 4 з.п. ф-лы, 2 табл.
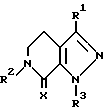
или их фармацевтически приемлемые соли,
где R1 является (С1-С3) алкилом;
Х является кислородом или двумя атомами водорода;
R2 выбирают из группы, состоящей из (С1-С4) алкила; индалила, (С3-С7) циклоалкила или группы формулы II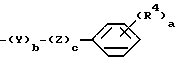
где а является целым числом от 1 до 2;
b и с является 0 или 1;
R4 является водородом, (С1-С5) алкилом, (С1-С5) алкоксилом, (С1-С6) циклоалкоксилом, галогеном, трифторметилом, СО2R6, СОNR6R7, NR6R7, где R6 и R7 являются водородом или (С1-С4) алкилом;
где Z является SO2;
Y является (С1-С5) алкиленом;
R3 имеет значения, определенные для R2 и метиленциклопропил, при условии, что R2 и R3 не могут одновременно означать метил.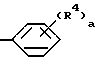
где а является целым числом от 1 до 2;
R4 является водородом, гидроксилом, (С1-С5) алкилом, (С1-С5) алкоксилом и галогеном.
3-этил-1-(4-метоксифенил)-6-фенил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклопентил-6-фенил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-(3,4-дихлорфенил)-6-(3-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклопентил-6-(3-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-(4-фторфенил)-6-(2-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклопентил-6-(3-метилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклопентил-6-(3-трифторметилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклогексил-6-(3-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-изопропил-1-циклопентил-6-(3-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклобутил-6-(3-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклопентил-6-фенил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклопентил-6-(2-метилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-(3-сульфоланил)-6-(3-метилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-(3-сульфоланил)-6-(3-метоксифенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклобутил-6-(3-метилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-(3-сульфоланил)-6-(3-трифторметилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклобутил-6-(3-трифторметилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина;
3-этил-1-циклобутил-6-(2-метилфенил)-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-c]пиридина.
| Способ получения производных 1,3-дигидро-2Н-имидазо/4,5-в/хинолин-2-онов или их фармацевтически приемлемых солей | 1986 |
|
SU1450746A3 |
| Акустический объемный элемент | 1987 |
|
SU1463883A1 |
| Устройство для бескольцевого прядения | 1973 |
|
SU531901A1 |
| US 4450164 A, 1984 | |||
| US 4492695 A, 1985 | |||
| US 4419359 A, 1983 | |||
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| Chemical Abstracts, 1977, vol | |||
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
