Маркфортины являются известными соединениями и описаны Polonsky et al. в [Journal Chemical Society Chemical Communication, 1980, 601-602 (Маркфортин А) и Tetrahedron Letters, 1981, 22, 1977-1980 (Маркфортины В и С)]. Эти соединения являются грибковыми метаболитами Penicillium roqueforti. Маркфортины близки по структуре парагерквамидам, которые также являются известными соединениями. Парагерквамиды описаны Yamazaki et al. (Tetrahedron Lett. 1981 22 135-136) и Blanchflower et al. (Journal of Antibiotics, 1991, 44, 492-497). Применение маркфортинов А, В и С описано в Патентах США 4866060 и 4923867 и некоторые их производные применяются для лечения и профилактики паразитарных заболеваний у животных.
WO 92/22555 (опубл. 23 декабря 1992) в основном описано производное маркфортина или парагерквамида (т.е., конкретной формулы (III), замещенной в положении 14 метилом или метилом и гидроксилом), однако нет описания того, как получить такие производные 14-метил-14гидроксимаркфортина.
Парагерквамид имеет следующую структуру: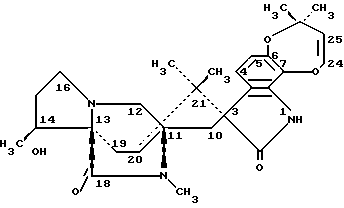
Маркфортин А имеет следующую структуру: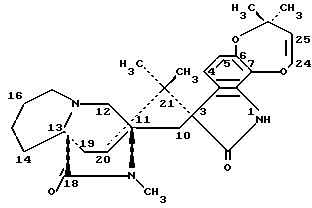
Маркфортин B имеет следующую структуру: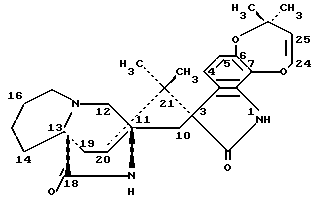
Маркфортин C имеет следующую структуру: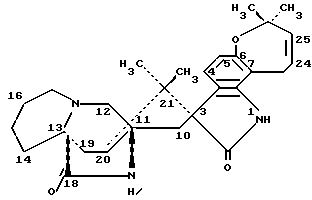
Маркфортин D имеет следующую структуру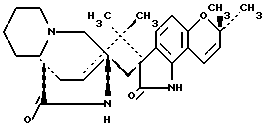
WO 91/09961 (опубл. 11 июля 1991) описаны различные производные маркфортина и парагерквамида и их 12а-N-оксидов также, как получение VM 29919 (парагерквамида) и VM 55596 (12а-N-оксида парагерквамида) inter alia из Penicillium Sp. IMI 332995.
В патенте США 4873247 описаны производные парагерквамида и штамм Penicillium charlessi MF 5123 (ATCC 20841) для получения парагерквамида. В патенте США 4873247 (а также как и ЕР 390532-А, ЕР 301742-А) описаны различные синтетические производные парагерквамида, а также получение парагерквамида из Penicillium charlessi MF 5123 (АТСС 20841).
SmithKIine Beecham в международной заявке WO 92/22555 (опубл. 23 декабря 1992) в основном описывает 14α-гидрокси-маркфортиновые соединения и способ, который использует 14-гидрокси-14-метилмаркфортиновые соединения для получения противопаразитических лекарств. Однако, никакого описания каких-либо способов получения производных 14α- гидрокси-маркфортина или 14α- гидрокси -14β метилмаркфортина не приводится.
Сущность изобретения.
Данное изобретение касается синтезов 14-замещенных маркфортинов А, В, С и D и их производных и применения этих соединений в качестве противопаразитических агентов. Таким образом, целью данного изобретение описание этих производных маркфортина. Дальнейшей целью данного изобретения является описание способов получения этих соединений. Еще одной целью является описание применения настоящих соединений в качестве противопаразитических агентов для лечения и профилактики паразитических заболеваний. Еще одной целью изобретения является описание композиций для лечения паразитических заболеваний, которые содержат новые соединения по данному изобретению в качестве активного ингредиента. Дальнейшие цели станут очевидны из ознакомления с нижеследующим описанием.
Описание изобретения
Соединения по настоящему изобретению представлены формулой (I):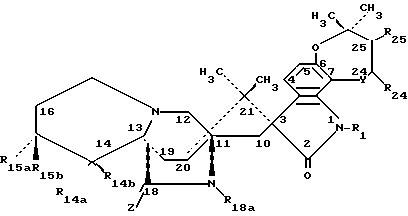
или их фармацевтически приемлемые соли,
где Z обозначает О,
Y обозначает атом кислорода (-O-),
R1 обозначает атом водорода, C2-C7 алканоил (-C(O)C2-C7 алкил) или -C(O)NR4R5 группу-, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют морфолиновое кольцо,
R14a - атом водорода, гидроксил или C1-C7 алкил,
R14b - атом водорода или гидроксил,
R18a - C1-C7 алкил,
R24 - атом водорода,
R25 - атом водорода,
R15a и R15b оба обозначают атом водорода,
при условии, что, когда один из R14a или R14b обозначает гидроксил, а другой обозначает атом водорода или метил, то R15a и R15b могут быть атомом водорода или метилом;
пунктирная линия между атомами углерода 24 и 25 представляет одинарную линию или двойную связь; при общем условии, что R14a и R14b оба не являются атомами водорода.
Более конкретно, изобретение относится к соединению, где пунктирная линия обозначает двойную связь между атомами углерода 24 и 25.
Предпочтительными являются соединения, выбранные из группы, включающей:
14α- гидроксимаркфортин А;
14α- гидрокси -14β- метилмаркфортин А;
14β- гидроксимаркфортин А;
N-оксид 14α- гидроксимаркфортин А;
14α- гидрокси -14β- этилмаркфортин А;
14α- гидрокси-N(1)-морфолинокарбонилмаркфортин А;
14α- гидрокси-N(1)-ацетилмаркфортин А;
N-оксид 14α- гидрокси -14β- метилмаркфортин А;
14α- гидрокси -14β- метил-N(1)-ацетилмаркфортин А;
14α- гидрокси -15α- метилмаркфортин А; или
14α- гидрокси -14β- метил -15α- метилмаркфортин А.
Еще более предпочтительными являются соединения, выбранные из группы, включающей:
14β- метилмаркфортин А;
14β- этилмаркфортин А;
14α- гидрокси -14β- этилмаркфортин А;
14α- гидрокси -14β- метилмаркфортин А; или
14α- гидрокси -14β- метил -15β- метилмаркфортин А.
Наиболее предпочтительными являются
14α- гидроксимаркфортин А, 14α- гидрокси -15β- маркфортин А и 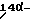 гидрокси -14β- метилмаркфортин А.
гидрокси -14β- метилмаркфортин А.
Другой аспект данного изобретения касается производных 14- замещенного маркфортина А и В формулы IA: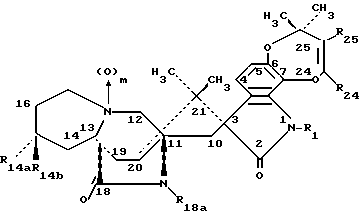
Другой аспект данного изобретения касается производных 14- замещенного тиомаркфортина А и В формулы IB: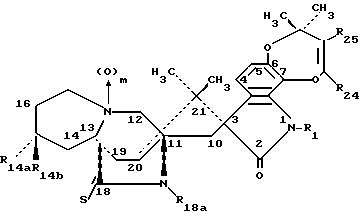
Другой аспект данного изобретения касается производных 14- замещенного маркфортина С и D формулы II: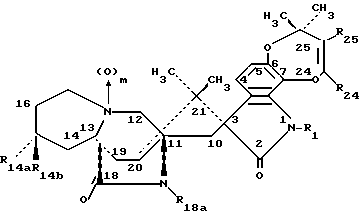
Другой аспект данного изобретения касается производных 14-замещенного тиомаркфортина С и D формулы III: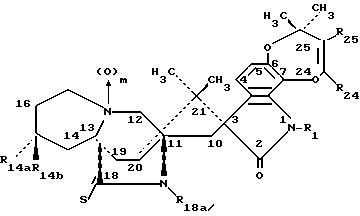
Содержание углерода в различных углеводородсодержащих радикалах указывается приставкой, обозначающей минимальное и максимальное число атомов углерода в радикале, например, приставка "Сi-Сj" указывает содержание атомов углерода от целого "i" до целою "j" включительно. Так, C1-С3 алкил относится к алкилу с 1-3 атомами углерода включительно, или к метилу, этилу, пропилу и изопропилу.
В связи с вышесказанным, "C1-C7 алкил" включает алкильные группы с от 1 до 7 атомов углерода либо прямой, либо разветвленной цепи. Примеры таких низших алкильных групп являются метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, гексил, гептил и тому подобное.
Цикло(С3-С8)алкил включает алкильные 3-8 членные кольца. Примерами цикло(С3-С8)алкильных групп являются циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, бицикло[2.2.1]гептил и тому подобное.
C1-C8 алкокси включает алкоксильные группы с от 1 до 8 атомов углерода либо прямой, либо разветвленной цепи. Примеры таких C1-C8 алкоксильных групп являются метокси, этокси, пропокси, изопропокси, бутокси, втор-бутокси, пентокси, гексилокси, гептилокси и тому подобное.
C2-С7 алканоил включает алканоильные группы с от 2 до 7 атомов углерода прямой или разветвленной цепи. Примеры таких С2-С7 алканоильных групп включают ацетил, пропионил, изопропионил, бутирил, пентаноил, гексаноил и тому подобное.
С10-С24 алканоил (-C(O)C9-C23 алкил) включает алканоильные группы с от 9 до 23 атомов углерода либо прямой, либо разветвленной цепи. Примеры таких C10-C14 алканоильных групп включают деканоил [- С(O)(CH2)9CH3], лаурил [-C(O)(CH2)10CH3] , тридеканоил [-С(O)(CH2)11CH3], миристоил [-С(O)(CH2)12CH3] , пентадеканоил [-С(O)(CH2)13CH3] , пальмитоил [-С(O)(CH2)14CH3], магароил [-С(O)(CH2)15CH3] , стеароил [-C(O)(CH2)16CH3], арахидоил [-С(O)(CH2)18CH3] , генеикоизаноил [-С(O)(CH2)19CH3], бегеноил [-С(O)(CH2)20CH3], трикозаноил [-С(O)(CH2)21CH3], тетракозаноил [-C(O)-(CH2)22CH3] и тому подобное.
C10-C24 алкеноил (-C(O)C9-C23 алкенил) включает ненасыщенные группы с от 10 до 24 атомов углерода либо прямой, либо разветвленной цепи. Примеры таких C10-C24 алкеноильных групп включают ундециленоил [-C(O)(CH2)7CH:CHCH3], олеоил [-С(O)(CH2)7CH: CH(CH2)7CH3] , линолил [-С(O)(CH2)7CH: CHCH2CH: CH(CH2)4CH3], и тому подобное.
Термин "C2-C8 алкоксиалкил" включает алкоксизамещенные низшие алкильные группы, содержащие от 2 до 8 атомов углерода и от 1 до 3 атомов кислорода либо прямой, либо разветвленной цепи. Примеры таких C2-C8 алкоксиалкильных групп включают метоксиметил, метоксиэтоксиметил, этоксиэтил и тому подобное. Примеры C1-C8 алкоксиметилов являются метоксиметил, этоксиметил, пропоксиметил, бутоксиметил, пентоксиметил, гексоксиметил и гептоксиметил и их изомерные формы.
Термин "алканоилоксиметилен" включает алканоилокси-замещенные метилены, содержащие от 2 до 8 атомов углерода в прямой или разветвленной цепи. Примеры таких C2-C8 алканоилоксиметиленовых групп включают ацетоксиметил, трет-бутоксиметил, н-пропоксиметил, валероксиметил и тому подобное.
Термин "замещенный бензоилоксиметилен" включает такие бензоилоксиметильные группы, в которых бензольное кольцо имеет от 0 до 3 заместителей, выбранных из низшего алкила, трифторметила, C1-C7 алкокси, нитро или цианогрупп и атомов галогена.
Термин "замещенный бензолсульфонил" включает такие бензолсульфонилгруппы, в которых бензольное кольцо замещено от 0 до 3 заместителями, выбранными из низшего алкила, трифторметила, C1-C7 алкокси, нитро или цианогрупп и атомов галогена.
Термин "галоген" включает атомы галогенов: фтора, хлора, брома и иода.
Термин "галоген C1-C7 алкил" включает галогензамещенные C1-C7 алкильные группы, содержащие от 1 до 7 атомов углерода в прямой или разветвленной цепи и от 1 до 3 атомов галогена. Примеры галоген C1-C7 алкилов включают фторметил, 2-бромэтил, 3-хлорпропил, 5-иодпентил, трифторметил и тому подобное.
Термин "C2-C8 алкенил" включает низшие алкильные группы, содержащие от 2 до 8 атомов углерода либо прямой, либо или разветвленной цепи, которые содержат от 1 до 2 двойных углерод-углеродных связей. Примеры таких C2-C8 алкенильных групп включают аллил, 3-бутенил, 2,4-пентадиенил, гексенил и тому подобное.
Термин "C2-C8 алкинил" включает алкинильные группы, содержащие от 1 до 8 атомов углерода либо прямой, либо разветвленной цепи, которые содержат от 1 до 2 тройных углерод-углеродных связей. Примеры таких C2-C8 алкинильных групп включают пропаргил, 2-бутинил, 2,4-пентадиинил, 5-гексинил и им подобные.
Примеры "алкоксикарбонилов" (-C(= O)O-(CH2)p-C1-C7 алкокси) включают этоксикарбонил, изопропоксикарбонил, метоксикарбонил, бутоксикарбонил, гексоксикарбонил и тому подобное. С1-C7 алканоил включает алкильные группы с от 1 до 7 углеродных атомов либо прямой, либо разветвленной цепи. Примеры C1-С7 алканоилов включают ацетил, пропионил, изобутирил, валерил, 5-метилгексаноил и тому подобное.
Примеры аминокарбонилов (-C(= O)-NR4R5) включают диметиламинокарбонил, пропилметиламинокарбонил, дибутиламинокарбонил, изо-пропиламинокарбонил, гексиламинокарбонил и тому подобное.
Примеры аминотиокарбонилов (-C(=S)-NR4R5) включают диметиламинотиокарбонил, пропилметиламинотиокарбонил, дибутиламинотиокарбонил, изопентиламинотиокарбонил, гексиламинотиокарбонил и тому подобное.
Примеры группы -P(=X)(R2)(R3) включают диэтилтиофосфорил, фенил-метоксифосфонил, 2-тиоксо-1,2,3-диоксафосфоринанил, N,N-диметилметоксифосфорамидил, дифенилфосфинил и тому подобное.
Примеры -SR6 включают 2,4-динитробензолсульфенил, диметиламиносульфенил, этоксикарбонилсульфенил, трихлорметилсульфенил, 4-морфолиносульфенил, этоксикарбонилсульфенил и тому подобное.
Примеры -SO2NR4R5 включают диметилсульфамоил, фенилметилсульфамоил, 4-морфолиносульфамоил, пиперидинилсульфамоил и тому подобное.
Термин "P-содержащее гетероциклическое кольцо" включает 1,3-диокса-2-фосфоринан, 1-аза-3-окса-2-фосфолан, 1,3-диаза-2-фосфолан, 1-тиа-3-окса-2-фосфолан и тому подобное.
Примеры гетероциклических аминовых колец, соответствующих, согласно -NR4R5, -NR'4R'5 и -NR7R8, являются:
4-морфолин,
4-фенил-1-пиперазин,
4-(2-пиридинил)-1-пиперазин,
2,6-диметил-4-морфолин,
1-пирролидин,
4-метил-1-пиперазин,
1-пиперидин,
4-фенил-1-пиперидин,
тиазолидин,
4-фенил-1,2,3,6-тетрагидропиридин,
4-фенилпиперидин,
этилпролинат,
тетрагидрофуриламин,
3-пирролин,
тиазолидин-4-карбоновая кислота,
тиоморфолин,
нипекотамид,
2-метилпиперидин,
3-метилпиперидин,
4-метилпиперидин,
N-метилпиперазин,
1-метилгомопиперазин,
1-ацетилпиперазин,
N-карбоэтоксипиперазин.
Фармацевтически приемлемые соли обозначают соли, используемые для введения соединений по данному изобретению и включают: мезилат, гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат, ацетат, пропионат, лактат, малеат, малат, сукцинат, тартрат и тому подобное. Эти соли могут быть в гидратированной форме.
Предпочтительными соединениями по данному изобретению являются соединения формулы IA и IB, где R14a обозначает гидроксил и водород, R14b обозначает водород, метил и этил; R24 и R25 обозначают водород; R18a обозначает водород, C1-C4 алкил, C2-C4 алкоксиалкил, С4-С4 алкенил или бензил; и пунктирная линия между атомами углерода 24 и 25 обозначает двойную связь.
Примеры предпочтительных соединений по данному изобретению приведены в конце описания.
Общие способы получения гетероатомных N-оксидов можно найти в гл. 2 "Chemistry of the Heterocyclic N-oxides", A.R. Katritzky and J.M. Lagowsky, опублик. 1971 Academic Press (Vol. 19 of ORGANIC CHEMISTRY - A Series of Monographs). Обычно, N-оксид образуется при взаимодействии с пероксикарбоновой кислотой в подходящем растворителе. Более всего для этого подходит ароматическая надкислота в неполярном растворителе, тогда как реакцию можно обычно проводить при комнатной температуре. Подходящими ароматическими надкислотами являются надбензойная кислота, хлорнадбензойная кислота и надфталевая кислота.
Получение исходного продукта
N-18a-замещенные маркфортины В и С и С24-С25-модифицированные маркфортины легко получаются способами, описанными в патенте США 4 923 867, описание которого включено здесь в качестве ссылки.
Маркфортины А, В и С выделяются, вместе с ранее описанными рокфортинами, в качестве грибковых метаболитов Penicillium roqueforti, с использованием стандартных методов ферментации и выделения. Выделение, а также аналитические и структурные характеристики маркфортина А подробно описаны Polonsky et al. (Journal of the Chemical Society Chemical Communications 1980, 601-602). Выделение, а также аналитические и структурные характеристики маркфортинов В и С детально описаны Polonsky et al. (Tetrahedron Lettyкы , 1981, 22, 1977-1980).
Альтернативно и более предпочтительно, маркфортины А, С и D могут быть выделены из Penicillium sp. UC 7780 (номер штамма в Upjohn Culture Collection, UC 7780, The Upjohn Company, Kalamazoo, MI). Этот штамм выделен из грязного образца, хранящегося в Иллинойсе, депонированном в собрании патентных культур Министерства сельского хозяйства США (U.S. Department of Agriculture, patent cultire collection in Peoria, IL) и ей присвоен номер NRRL 18887. Для последующей характеристики грибка было проведено таксономическое исследование согласно способам и материалам, описанных в I. John Pitt, The Genus Penicillium, Academic Press, London, (1979). Споры и покровные поверхности были изучены методом сканирующей электронной микроскопии по методу, (A. Dietz, J. Matthews, J. Appl. Microbiology 18:694-696 (1969). Интактные конидиофоры визуализировались методом световой микроскопии [A.H.S. Onions et al. , "Smith's Introduction to Industrial Mycology, John. Willey and Sons, New York, pp. 301-302, (1979)] после приготовления предметного стекла с культурой(ами): стеклянную чашку Петри, содержащую предметное стекло, образец и покровное стекло, стерилизуют. На предметное стекло помешают небольшой прямоугольный кусок агара картофельной декстрозы и инокулируют с четырех сторон культурой плесени. Покровное стекло помещают на инокулированный прямоугольный кусок агара и добавляют стерильной воды для поддержания влажности. Камеру подвергают инкубации в течение шести дней при 24oC. Микроскопические препарат(ы) готовят, снимая покровное стекло и помещая его на каплю лактофенольного хлопкового голубого красителя.
Характеристики Penicillium sp. UC 7780 (NRRL 18887) следующие:
Морфология - бивертициллатный пенициллус (два разветвления между конидиумом и ножкой). Эти разветвления (метулы) поддерживают фиалиды, или структуры, несущие конидии. Конидиофоры (примерно 35 мкм) ограничены мутовками из 2-5 (10-14 мкм) митул. Фиалиды имеют форму амфор (т.е. похожи на древнегреческие сосуды для вина) в мутовках 2-5 (7 мкм). Конидии - гладкие и сфероидальные (2 мкм), обычно образующие длинные колонны. Стенки ножки гладкие.
Культуру инокулируют на трех чашках Петри диаметром 6 см, одна - с дрожжевым агаром Чапека (Czapek yeast agar, CYA), две другие - с агаром из солодового экстракта (malt extract agar, МЕА) и 25%-ым агаром нитроглицерина (G25N). Инсоляция проводится из полужидкой суспензии (0,5 мл 0,2%-го агара с 0,05%-ым Tween 80). Инокулируемую порцию конидии добавляют в пробирку и перемешивают. Порцию суспензии инокулируют на образец в трех местах чашки. Для точечной инокуляции на 6 см-ую чашку используют иглу. Режим инкубации: одна чашка CYA, чашки МЕА и G25N при 24oC, одна чашка CYA при 37oC и 6 см чашка CYA при 5oC. Через 7 дней записывают диаметры колоний, цвета и другие характеристики (приведены в Таблице 1). На агаре картофельной декстрозы (PDA, Difco) образуется глубокое красное окрашивание на дне или обратной стороне колонии.
Никакой половой стадии отмечено не было. Результаты по культуре (NRRL 18887) классифицировали по определителю Penicillium для нахождения подвида. Внутри определителя самого вида Penicillium определяется подвид, к которому относятся образцы. Эти образцы имеют некоторые характеристики, отличающие их от подвида Biverticillium, даже если пенициллиус является бивертицилатным. Образцы главным образом образуют колонии с диаметром более 10 мм в течение 7 дней в G25N. Метулы являются более длинными, чем фиалиды и находятся на мутовках 25. Эти характеристики относят данный образец Penicillium (NRRL 18887) к подвиду Furcatum.
Приведенное описание иллюстрирует вытяжку из образца Penicillium UC 7780 (NRRL 18887), которая может использоваться для получения маркфортина и его производных. Однако, настоящее изобретение охватывает также и мутантов описанного выше вида микроорганизмов. Например, такие мутанты получены естественным отбором или под воздействием мутагенов, включая ионизирующие излучения, такие как УФ-излучение, или химические мутагены. такие как нитрозогуанидин, или им подобных воздействий. Эти мутанты также входят в предмет данного изобретения.
Настоящее описание включает интер- и интраспецифические рекомбинанты, полученные генетическими методами, хорошо известными специалистам, такими как, например, конъюгация, транзульция и генная инженерия.
Penicillium. sp. UC 7780 (NRRL 18887) может быть выращен в аэробных условиях способом, аналогичным обычно используемому для выращивания известных культур рода Penicillium.
В качестве компонентов сред могут использоваться любые из хорошо известных питательных для Penicillium материалов. Например, в качестве источников усваиваемого углерода могут использоваться глюкоза, глицерин, мальтоза, декстрин, крахмал, лактоза, сахароза, меласса, соевое масло, масло семян хлопка и т.п., предпочтительно - глюкоза и глицерин. Источником усваиваемого азота могут служить: соевая мука, мука земляных орехов, мука семян хлопка, рыбная мука, кукурузная патока, пептон, рис, отруби, мясной экстракт, дрожжи, дрожжевой экстракт, нитрат натрия, нитрат аммония, сульфат аммония и т.п. Такие неорганические соли, как хлорид натрия, фосфаты, карбонат кальция и т.п. могут добавляться в культивационную среду. Незначительное количество соли металла добавляется, если это необходимо. Кроме того, незначительное количество тяжелого металла может добавляться при необходимости.
Для культивирования образцов Penicillium (NRRL 18887), особенно в анаэробных условиях, могут с успехом использоваться обычные аэробные методы выращивания, такие как твердая культура, культура при аэрации и взбалтывании, перемешиваемая культура и т.п.
При культивировании с аэрацией и взбалтыванием могут должным образом использоваться пеногасители, например, силиконовое масло, растительные масла, ПАВ и т.п.
Значение pH среды обычно находится в пределах от 3 до 9, предпочтительно, вблизи нейтральной реакции, а температура обычно поддерживается порядка 20-З0oC, в частности предпочтительна температура 21oC.
Культивирование продолжается до тех пор, пока маркфортин А накапливается в среде, обычно в течение от 20 до 240 часов, предпочтительно - от 48 до 168 часов; после культивирования маркфортин А может быть извлечен и выделен из культивационного бульона подходящим сочетанием различных методов. Например, это может быть экстракция органическим растворителем, например, эфиром, этилацетатом или хлороформом, растворение в более полярном растворителе, например, ацетоне или спирте, удаление примесей менее полярным растворителем, например, петролейным эфиром или гексаном, адсорбционная хроматография на активированном угле или силикагеле, гель-фильтрация через колонку Sephadex (получаемую от Pharmacia Co., Ltd, U.S.A.) и т.д.
Указанные соединения по настоящему изобретению являются неожиданно сильными парагерквамидапаразитными агентами против эндо- и эктопаразитов, в частности паразитических червей и членистоногих, вызывающих многочисленные паразитические заболевания у людей, животных и растений.
Паразитические заболевания могут вызываться как эндопаразитами, так и эктопаразитами. Эндопаразиты - это паразиты, которые живут внутри тела хозяина, как внутри органа (такого как желудок, легкое, сердце, кишка и т.п.), так и просто под кожей. Эктопаразиты - это паразиты, которые живут на внешней поверхности хозяина, но поглощают питательные вещества из него.
Эндопаразитические заболевания, обычно называемые гельмитозом, возникают вследствие заражения хозяина паразитическими червями, известными как гельмиты. Гельмитоз - это преобладающая и серьезная мировая экономическая проблема, поскольку инфицированию подвергаются домашние животные, такие как свиньи, овцы, лошади, крупный рогатый скот, козы, собаки, кошки и домашняя птица. Многие из этих инфекций вызваны группой червей, описанных как нематоды, которые вызывают заболевания у многих видов животных во всем мире. Эти заболевания часто являются серьезными и могут привести к смерти инфицированного животного. Наиболее распространенные подвиды нематод, заражающих животных, о которых говорилось выше, это Haemonchus, Trichostrongylus, Ostertagia, Nematodirus, Cooperia, Ascaris, Bunostomum, Oesophagostomum, Chabertia, Trichuris, Strongylus Trichonema, Dictyocaulus, Capillaria, Heterakis, Toxocara, Ascaridia, Oxyuris, Ancylastoma, Uncinaria, Toxascaris и Parascaris. Многие паразиты-видоспецифичны (инфицируют только одного хозяина) и многие также имеют предпочтительное место, поражаемое внутри животного. Так, Haemonchus и Ostertagia первично инфицируют желудок, тогда как Nematodirus и Cooperia атакуют главным образом кишечник. Другие паразиты предпочтительно поселяются в сердце, глазах, легких, кровеносных сосудах и т.п., в то время, как остальные являются подкожными паразитами. Гельмитоз приводит к слабости, потере веса, анемии, повреждению кишечника, потере аппетита и поражению других органов. В отсутствие лечения, эти заболевания могут привести к смерти животного.
Инфекции эктопаразитными членистоногими, такими как клещи, чесоточные клещи, вши, слепни, шершни, мясные мухи, блохи и им подобные, являются серьезной проблемой. Инфекции этими паразитами приводят к потере крови, поражениям кожи и могут вмешиваться в нормальные привычки в еде, вызывая таким образом потерю веса. Эти инфекции могут также привести к переносу серьезных заболеваний, таких как энцефалит, анаплазмоз, сыпь у свиней и им подобных, которые могут быть смертельными.
Животные могут быть одновременно инфицированы несколькими видами паразитов, поскольку заражение одним паразитом может ослабить животное и сделать его восприимчивым к заражению следующим видом паразита. Таким образом, соединение с широким спектром действия имеет особые преимущества при лечении таких заболеваний. Соединения по данному изобретению имеют неожиданно высокую активность против паразитов и, кроме того, активны против Dirofilaria у собак, Nematorspiroides Syphacia у грызунов, сосущих насекомых и мигрирующих диптероидных личинок нематод, таких как виды Hypoderma у крупного рогатого скота и Gastrophilus у лошадей.
Указанные соединения также используются против эндо- и эктопаразитов, которые вызывают паразитические заболевания у человека. Примеры таких эндопаразитов, инфицирующих человека, являются желудочно-кишечные паразиты вида Ancylostma, Necator, Ascaris, Strongyloides, Ttichinella, Capillaria, Trichuris, Enterobius и тому подобное. Другие эндопаразиты, которые инфицируют человека, обнаруживаются в крови или других органах. Примерами таких паразитов являются ленточные черви Wucheria, Brugia, Onchocerca и им подобные, равно как и черви Strongylides и Trichinella на внекишечных стадиях. Эктопаразиты, паразитирующие на человеке, включают клещей, блох, чесоточных клещей, вшей и им подобных, и, как и в случае домашних животных, инфекции этими паразитами могут приводить к переносу серьезных и даже смертельных заболеваний. Настоящие соединения активны против этих эндо- и эктопаразитов, и, кроме того, эффективны против сосущих насекомых и других диптероидных паразитов, раздражающих человека. Настоящие соединения при введении перорально или парентерально вводятся дозами от 0,05 до 20 мг/кг веса тела животного.
Настоящие соединения также применяются против обычных паразитов жилищ, таких как Btatella sp. (тараканы), Tineola sp. (платяная моль), Attagenus sp. (клопы), Musca domestica (домовые мухи) и против Solenopsis Invicta (неаборигенные рыжие муравьи).
Кроме этого, соединения используются против сельскохозяйственных вредителей, таких как тля Acyrthiosiphon sp, (тли), саранча и долгоносики, а также против вредных насекомых, которые паразитируют на хранящемся зерне, таких как Tribolium sp., и против насекомых на незрелых стадиях, живущих в тканях растений. Соединения также используются как наматоциды для борьбы с почвенными нематодами, что важно для сельского хозяйства.
При использовании в качестве парагерквамидапаразитных агентов для животных, настоящие соединения могут вводиться внутрь как перорально или путем инъекций, так и в виде жидких обтираний или шампуней.
При пероральном введении соединения могут вводиться в виде капсул, таблеток, в форме пропитанных шариков, или, альтернативно, могут смешиваться с пищей животных. Капсулы, таблетки и пропитанные шарики включают активный ингредиент в сочетании с подходящим носителем, таким как крахмал, тальк, стеарат магния или дикальций фосфат. Эти дозированные формы готовятся путем тонкого смешивания активного ингредиента с подходящими хорошо измельченными инертными ингредиентами, включающими разбавители, наполнители, диспергаторы, суспендирующие агенты и/или связующие так, чтобы образовывались однородная смесь, раствор или суспензия. Инертным является ингредиент, который не реагирует с настоящими соединениями и нетоксичен для животного, подвергаемого лечению. Подходящие инертные ингредиенты включают крахмал, лактозу, тальк, стеарат магния, растительные смолы и масла и тому подобное. Такие составы могут содержать большое многообразие количеств активных и неактивных ингредиентов, зависящее от многочисленных факторов, таких как размер и тип животною вида, подвергаемого лечению, типа и разновидности инфекции. Активный ингредиент также может вводиться в качестве пищевой добавки путем простого смешивания с пищей или нанесения на ее поверхность. Альтернативно, активный ингредиент может смешиваться с инертным носителем, и порченная композиция может смешиваться с пищей или непосредственно скармливаться животному. Подходящими инертными носителями являются кукурузная мука, цитрусовая мука, ферментационные осадки, соя грубого помола, высушенное зерно и тому подобное. Активные ингредиенты тонко смешиваются с этими инертными носителями путем размола, перемешивания и толчения, так, чтобы конечная композиция содержала от 0,001 до 5,0% (масс.) активного ингредиента.
Соединения альтернативно могут вводиться парентерально путем инъекции состава, состоящего из активного ингредиента, растворенного в инертном жидком носителе. Инъекция может быть внутримышечной, внутриягодичной или подкожной. Вводимый состав состоит из активного ингредиента, смешанного и подходящим инертным жидким носителем. Приемлемые жидкие носители включают растительные масла, такие как арахисовое масло, масло семян хлопка, кунжутное масло и тому подобное, также, как и органические растворители, такие как монокеталь, формилглицерин и тому подобное. Как альтернатива могут использоваться водные парентеральные составы. Растительные масла являются предпочтительными жидкими носителями. Составы готовят растворением или суспендированием активного ингредиента в жидком носителе так, чтобы конечный состав содержал от 0,005 до 20% (масс.) активного ингредиента.
Наружное применение настоящих соединений является возможным при использовании жидких обтираний или шампуней, содержащих настоящие соединения в виде водных растворов или суспензий. Эти составы, в общем случае, содержат суспендирующий агент, такой как бентонит и, как правило, также содержат пеногаситеяь. Допускаются составы, содержащие от 0,5 до 5% (масс.) настоящих соединений.
Настоящие соединения в первую очередь используются как противопаразитические агенты для лечения и/или профилактики гельминтоза у домашних животных, таких как крупный рогатый скот, овцы, лошади, собаки, кошки, козы, свиньи и у домашней птицы. Они также применяются для профилактики и лечения у этих животных паразитных инфекций эктопаразитами, такими как клещи, чесоточные клещи, вши, блохи и им подобные. Соединения также эффективны при лечении паразитических инфекций у человека. При лечении таких инфекций соединения по данному изобретению могут использоваться индивидуально или в комбинации друг с другом или с другими неродственными противопаразитическими агентами. Дозировка настоящих соединений, требуемая для достижения наилучших результатов, зависит от различных факторов, таких как вид и размер животного, тип и разновидность инфекции, метода введения и используемого соединения. Пероральное введение настоящих соединений в дозах на уровне от 0,005 до 50 мг/кг веса тела животного в виде разовой дозы или нескольких доз в течение нескольких дней обычно дает хорошие результаты. Разовая доза настоящих соединений обычно дает превосходный результат, однако многократные дозы назначаются для борьбы с повторным заражением или для борьбы с необычно устойчивыми паразитами. Методики введения этих соединений животным хорошо известны специалистам в области ветеринарии.
Соединения по данному изобретению также могут использоваться для борьбы с вредителями сельского хозяйства, поражающими урожай как на поле, так и при хранении. В этих целях соединения применяются в виде аэрозолей, порошков, эмульсий и им подобных как к произрастающим растениям, так и к убранному урожаю. Методики применения этих соединений в данном случае хорошо известны специалистам по сельскому хозяйству.
Следующие примеры приводятся для лучшего понимания данного изобретения; они не являются ограничениями изобретения.
Процесс N 1: Получение и выделение маркфортина А
Ферментативный процесс:
Высеваемые ферментаты инокулируют затравками агара индивидуального образца Penicillium UC 7780 (NRRL 18887), хранившегося в жидком азоте. Три затравки размораживают и используют как инокуляты.
GS-7 состоит из глюкозы и муки семян хлопка (продается Traders Protein, Procter & Gamble Oilseed Products Co., Memphis, TN, U.S.A. под торговой маркой "Pharmamedia"). Вода, стоявшая в незакрытой таре, используется для гидратации компонентов среды; pH среды доводится NH4OH до 7,2. Среду разливают порциями по 300 мл в неперегороженные колбы закрытой системы вместимостью 1000 мл и стерилизуют 30 минут в автоклаве при 121oC. Каждую колбу закрытой системы, содержащую 300 мл среды GS-7, инокулируют тремя затравками агара Penicillium ps. UC 7780 (NRRL 18887) и перемешивают на роторном смесителе при 250 об/мин в течение 36 часов при 22oC.
Вторичный ферментативный процесс:
Выращенные культуры используют в качестве инокулята для вторичной среды при посевном соотношении 0,3%. Вторичная среда состоит из 25 г моногидрата глюкозы (продаваемого С.Р.С. International под торговой маркой Cerelosc), 25 г муки семян хлопка (продается под торговой маркой "Pharmamedia"), 329,8 мг MgCl2 • 6H2), 11,4 мг MnSO4 • H2O, 3,2 мг FeSO4 • 7H2O, 1,8 мг Na2MoO4 • 2H2), 367,6 мг CaCl2 • 2H2O, 84,2 мг NaCl, 5,8 мг KCl, 0,1 мг ZnSO4 • 7H2O, 0,1 мг CoCl2 • 6H2O, 3,1 мг CuSO4 • 5H2O и 0,5 мл силиконового пеногасителя (продается под торговой маркой SAG-471 Antifoam) на литр воды квалификации "прошедшая обратный осмос". Компоненты среды, в количестве для приготовления 200 л вторичной среды, гидратируют водой квалификации "прошедшая обратный осмос", до объема 190 л в 250-литровом ферментере. После составления среды pH доводят до 7,2 и затем среду стерилизуют 30 минут при 121oC. Две колбы закрытой системы созревшей первичной культуры используют в качестве инокулята при посевном соотношении 0,3%. Вторичную культуру инкубируют при 22oC и аэрации 125 slm и давлении 5 psig при 250 об/мин в течение 36 часов.
Производственный ферментативный процесс:
Производственная среда состоит из 50 г свекольной мелиссы, 16 г рыбной муки (продается под торговой маркой Menhaden Select Fish Meal), 10 г дрожжевого экстракта (продается под торговой маркой Fidco), 329,8 мг MgCl2 • 6H2), 11,4 мг MnSO4 • H2O, 3,2 мг FeSO4 • 7H2O, 1,8 мг Na2MoO4 • 2H2O, 367,6 мг CaCl2 • 2H2O, 84,2 мг NaCl, 5,8 мг KCl, 0,1 мг ZnSO4 • 7H2O, 0,1 мг CoCl2 • 6H2O, 3,1 мг CuSO4 • 5H2O и 0,5 мл силиконового пеногасителя (продается под торговой маркой SAG-471 Antifoam) на литр воды (квалификации "прошедшая обратный осмос").
Компоненты среды, в количестве для приготовления 5000 л среды, гидратируют водой, квалификации "прошедшая обратный осмос" до объема 4700 л в 5000-литровом ферментере. После составления среды pH доводят КОН до 7,0 и затем среду стерилизуют 30 минут при 123oC. Созревшую вторичную культуру используют в качестве инокулята при посевном соотношении 1,0%. Вторичную культуру инкубируют при 22oC и аэрации 2500 slm и давлении 5 psig при 250 об/мин в течение 96 часов.
Выделение маркфортина А:
Ферментационный объем 4900 л получают путем пропускания через миксер с большим сдвиговым усилием в приемный сосуд. После переливания добавляют 4% (масс. ) диатомовой земли и 1/2 объема метиленхлорида. Раствор после ферментации затем фильтруют на фильтр-прессе. Осадок с фильтра дважды промывают метиленхлоридом в количестве 10% от объема.
Полученный фильтрат декантируют для удаления воды (водной фазы). Оставшегося фракцию метиленхлорида, обогащенную продуктом, затем концентрируют до объема 44 л. Концентрат затем разбавляют 20%-ым количеством метиленхлорида (9 л) и пропускают через диатомовую землю на фильтре.
53 л обработанного концентрата далее очищают для отделения маркфортина А от других соединений хроматографией на силикагеле и кристаллизацией.
Перед хроматографированием обработанный концентрат делят примерно на равные аликвоты. Каждую аликвоту хроматографируют на свеженабитой колонке диаметром 9'' (23 см), загруженной 25 кг сухого силикагеля (объем 59 л). Загруженные колонки элюируют 120 л 10%-го раствора ацетона в метиленхлориде, 120 л 20%-го раствора ацетона в метиленхлориде, 120 л 30%-го раствора ацетона в метиленхлориде, 160 л 40%-го раствора ацетона в метиленхлориде и 130 л ацетона, собирая 30 и 40%-ые элюаты в виде фракций по 20 л. Элюаты контролируют методом ТСХ с использованием, например, элюента, состоящего из 6% изо-пропанола и 0,3% гидроксида аммония в метиленхлориде на силикагельных пластинах Whatman LK6DF. Фракции маркфортина А, содержащие небольшие количества маркфортина D, который совместно хроматографируется, кристаллизуют из ацетона. Соответствующие фракции (40-100 л) концентрируют под пониженным давлением до объема примерно 5 л. Раствор (иногда, слегка мутный) переносят затем в роторный испаритель и продолжают концентрировать при пониженном давлении. В ходе концентрирования добавляют несколько порций ацетона объемом 1 л до полного вытеснения метиленхлорида. Полученную суспензию в ацетоне (объемом примерно в 1 л) вымораживают в течение суток, собирают кристаллы маркфортина А, промывают несколькими небольшими порциями холодного ацетона и высушивают в вакууме. Образовавшиеся кристаллы могут быть загрязнены несколькими процентами маркфортина D. Повторная перекристаллизация из смеси метиленхлорид/ ацетон (с вытеснением метиленхлорида, как это описано выше) дает чистый маркфортин А.
Выделение маркфортина D:
Ферментационный объем 4900 л получают путем пропускания через миксер с большим сдвиговым усилием в приемный сосуд. После переливания добавляют 4% (масс. ) диатомовой земли и 1/2 объема метиленхлорида. Раствор после фермeнтaции затем фильтруют на фильтр-прессе. Осадок с фильтра дважды промывают метиленхлоридом в количестве 10% от объема.
Полученный фильтрат декантируют для удаления воды (водной фазы). Оставшуюся фракцию метиленхлорида, обогащенную продуктом, затем концентрируют до объема 44 л. Концентрат затем разбавляют 20%-ым количеством метиленхлорида (9 л) и пропускают через диатомовую землю на фильтре.
53 л обработанного концентрата далее очищают для отделения маркфортина А от других соединений хроматографией на силикагеле и кристаллизацией.
Перед хроматографированием обработанный концентрат делят примерно на равные аликвоты. Каждую аликвоту хроматографируют на свеженабитой колонке диаметром 9'' (23 см), загруженной 25 кг сухого силикагеля (объем 59 л). Загруженные колонки элюируют 120 л 10%-го раствора ацетона в метиленхлориде, 120 л 20%-го раствора ацетона в метиленхлориде, 120 л 30%-го раствора ацетона в метиленхлориде, 160 л 40%-го раствора ацетона в метиленхлориде и 130 л ацетона, собирая 30 и 40%-ые элюаты в виде фракций по 20 л. Элюаты контролируют методом ТСХ с использованием, например, элюента, состоящего из 6% изопропанола и 0,3% гидроксида аммония в метиленхлориде на силикагельных пластинах Whatman LK6DF. Фракции маркфортина А, содержащие маркфортин D, концентрируют. Один грамм концентрата растворяют в 20 мл 93%-ой муравьиной кислоты и выдерживают при комнатной температуре в течение 16 часов. После удаления летучих компонентов при пониженном давлении хроматографируют на силикагеле (элюент- MeOH: CH2Cl2 1:20), полчая 100 мг маркфортина D в виде белого твердого вещества. Структура вещества может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] : вычислено для C28H35N3O3 462.2756, найдено 462.2739.
Процесс 1А Получение и выделение маркфортинов А и С.
Ферментативный процесс:
Высеваемые ферментаты инокулируют затравками агара индивидуального образца Penicillium UC 7780 (NRRL 18887), хранившегося в жидком азоте. Три затравки размораживают и используют как инокуляты для 100 мл посевной середы GS-7. GS-7 состоит из глюкозы и муки семян хлопка (продается Traders Protein, Procter & Gamble Oilseed Products Co., Memphis, TN, U.S.A. под торговой маркой "Pharmamedia"), к каждой порции добавляется вода, стоявшая в незакрытой таре до концентрации 25 г/л. После составления среды pH GS-7 доводится NH4OH до 7,2. Среду разливают порциями по 100 мл в неперегороженные колбы вместимостью 500 мл и стерилизуют 30 минут в автоклаве. Стерильные колбы с GS-7 инокулируют, как это описано выше, и перемешивают при 250 об/мин в течение 35-58 часов при 23oC.
Вторичный ферментативный процесс:
Выращенные культуры используют в качестве инокулята для вторичной среды при посевном соотношении 1%. Вторичная среда состоит из 45 г глюкозы, 25 г энзимно-расщепленного казеина (продаваемого Sheffield Products, Norwich, N. Y. , U.S.A. под торговой маркой Peptonized Milk Nutrient), 2,5 г дрожжевого экстракта (продается Difco Laboratories, Detroit, MI под торговой маркой ВАСТО Yeast Extract, код: 0127) на литр воды, хранившейся в незакрытой посуде. После составления pH производственной среды доводят до 7,0 с использованием гидроксида калия. Среду затем автоклавируют в течение 30 минут порциями по 100 мл в 500 мл закрытых ферментационных колбах. Стерильную производственную среду инокулируют, как описано выше, и перемешивают в течение 7-14 дней при 250 об/мин при 21oC.
Производственный ферментационный процесс (танки Labraferm):
Выращенные посевные культуры используют в качестве инокулята для стерильной производственной среды при посевном отношении 0,5%. Производственная среда описана выше. После установления pH 7,0 с использованием КОН 10 л среды автоклавируют 90 минут в 12-литровых танках (New Brunswick Scientific Co. , Inc.). Танки инокулируют при посевном отношении 0,5% и перемешивают массу при 500 об/мин и 20oC в течение 5-9 дней. Расход воздуха поддерживают около 10-15 л/мин.
Выделение маркфортинов А и С:
Весь ферментационный бульон (35 л) мацерируют при малой скорости в большом мерном смесителе и смешивают с равным количеством метиленхлорида. Смесь оставляют на сутки при охлаждении, а затем подвергают центрифугированию для расслоения эмульсии. Образующийся чистый слой метиленхлорида отделяют и выпаривают при пониженном давлении. Концентрированный раствор остатка (37,4 г) в метиленхлориде переносят на колонку, набитую силикагелем (1 кг) в метиленхлориде. Колонку элюируют смесью метиленхлорида с увеличивающимся содержанием ацетона (10%, 20%, 30%, 40% и 50% ацетона). Фракции контролируют методом ТСХ и соответствующие фракции выпаривают и кристаллизуют из ацетона. Получают маркфортин А и маркфортин С.
Процесс 1В Производство и выделение маркфортинов А и С.
Ферментативным процесс:
Высеваемые ферментаты инокулируют затравками агара индивидуального образца Penicillium ps. UC 7780 (NRRL 18887), хранившегося в жидком азоте. Три затравки размораживают и используют как инокуляты для 100 мл посевной среды GS-7. GS-7 состоит из глюкозы и муки семян хлопка (продается Traders Protein, Procter & Gamble Oilseed Products Co., Memphis, TN, U.S.A. под торговой маркой "Pharmamedia"), к каждой порции добавляется вода, стоявшая в незакрытой таре до концентрации 25 г/л. После составления среды pH GS-7 доводится NH4OH до 7,2. Среду разливают порциями по 100 мл в неперегороженные колбы вместимостью 500 мл и стерилизуют 30 минут в автоклаве. Стерильные колбы с GS-7 инокулируют, как это описано выше, и перемешивают при 250 об/мин в течение 35-58 часов при 23oC.
Производственный ферментативный процесс (Шейкерная колба)
Выращенные посевные культуры используют в качестве инокулята для стерильной производственной среды при посевном отношении 1%. Производственная среда состоит из 20 г глюкозы, 15 мл глицерина, 20 г муки семян хлопка (продается Traders Protein, Procter & Gamble Oilseed Products Co., Memphis, TN, U.S.A. под торговой маркой "Pharmamedia"), 10 г муки соевых бобов и 3 г K2HPO4 на литр воды, стоявшей в незакрытой таре. После составления среды устанавливают pH 6,8 с использованием КОН. Среду автоклавируют 30 минут порциями по 100 мл в перегороженных ферментационных колбах вместимостью 500 мл. Стерильную производственную среду инокулируют как это описано выше и перемешивают массу при 250 об/мин и 21oC в течение 7-14 дней.
Производственный ферментативный процесс (танки Labraferm):
Выращенные посевные культуры используют в качестве инокулята для стерильной производственной среды при посевном отношении 0,5%. Производственная среда описана выше. После установления pH 7,0 с использованием КОН 10 л среды автоклавируют 90 минут в 12-литровых танках Labraferm (New Brunswick Scientific Co. , Inc.). Танки инокулируют при посевном отношении 0,5% и перемешивают массу при 500 об/мин и 20oC в течение 5-9 дней. Расход воздуха поддерживают около 10-15 л/мин.
Выделение маркфортинов А и С:
Весь ферментационный бульон (35 л) мацерируют при малой скорости в большом мерном смесителе и смешивают с равным количеством метиленхлорида. Смесь оставляют на сутки при охлаждении, а затем подвергают центрифугированию для расслоения эмульсии. Образующийся чистый слой метиленхлорида отделяют и выпаривают при пониженном давлении. Концентрированный раствор остатка (37.4 г) в метиленхлориде переносят на колонку, набитую силикагелем (1 кг) в метиленхлориде. Колонку элюируют смесью метиленхлорида-с увеличивающимся содержанием ацетона (10%, 20%, 30%, 40% и 50% ацетона). Фракции контролируют методом ТСХ и соответствующие фракции выпаривают и кристаллизуют из ацетона. Получают маркфортин А и маркфортин С.
Синтез 14-замещенных маркфортинов.
Обработка маркфортина А (формула Ia, пункт А) циануриодидом дает смесь (формула 5) 16α- иодо 17β- цианомаркфортина А и 16β- иодо -17α- цианомаркфортина А, которые могут быть разделены хроматографией на силикагеле. Дегидроиодирование этой смеси гидроксидом калия в метаноле приводит к 16,17-дегидро-17- цианомаркфортину А (формула 6), который окисляется диоксидом селена до 17-кетомаркфортина А (формула 7). Введение двойной связи между положениями С15 и С16 сопровождается селенированием по положению 16 (фенилселенилхлорид и LDA) с последующим окислением селенового промежуточного продукта перекисью водорода. Последующее отщепление фенилселеновой кислоты дает 15,16-дегидро-17-кетомаркфортин А (формула 8). Это соединение является ключевым промежуточным продуктом в синтезе 14α- гидроксимаркфортина А (формула 10), в который он может превращен любым из двух различных синтетических путей.
По первому пути, аллильное окисление этого материала по положению 14 с использованием бис(триметилсилил)амида калия и 2-фенилсульфонил-3-фенилоксазеридина сопровождается окислением по положению 16, приводя к смеси требуемого 14α- гидрокси-15,16- дегидро-17-кетомаркфортина А (формула 9а) и 15,16-дегидро-16- гидрокси-17-кетомаркфортина А (формула 9b). Эти два продукта разделяют хроматографией на силикагеле. Соединение формулы 9а восстанавливают алюмогидридом лития в ТГФ до 14α- гидроксимаркфортина А (формула 10), соединения, указанного в заголовке описания изобретения. Альтернативно, соединение формулы 8 (схема В) окисляют диоксидом селена в диоксане, получая смесь 2: 1 14α- гидрокси-15,16-дегидро-17-кетомаркфортина А (формула 9а) и 15,16-дегидро-14,17-дикетомаркфортина А (формула 11). Их разделяют хроматографией на силикагеле. Каждое из этих соединений независимо превращается в 14α- гидрокси-17-кетомаркфортин А (формула 12а): соединение формулы 9а путем восстановления двойной связи между положениями 15 и 16 с помощью триэтилборгидрида лития; соединение формулы 11-путем восстановления боргидридом лития карбонила по положению 14. В последнем случае, также образуется равное количество 14β- гидрокси-17-кетомаркфортина А (формула 12b), который удаляется хроматографией. Соединение формулы 12а восстанавливают комплексом бора с ТГФ с образованием 14α- гидроксимаркфортина А (формула 10).
14α- гидрокси-15,16-дегидро-17-кетомаркфортин А (формула 9а, схема С) восстанавливают триэтилборгидридом лития до 14α- гидрокси- 17-кетомаркфортина А (формула 12а). Его далее превращают в 14,17-дикетомаркфортин А (формула 13) путем окисления по Сверну (Swern) с использованием оксалилхлорида и ДМСО. Обработка метилмагнийбромидом по реакции Гриньяра (Grignard) дает смесь 14α- гидрокси -14β- метил-7- кетомаркфортина А (формула 14а) и 14β- гидрокси -14α- метил-7-кетомаркфортина А (формула 14b), которую разделяют хроматографией на силикагеле. Соотношение продуктов зависит от используемого растворителя: в метиленхлориде соотношение 6:1, тогда как в ТГФ оно > 50:1. Восстановление алюмогидридом лития соединения формулы 13а дает 14α- гидрокси -14β- метил-7-метилмаркфортин А (формула 15).
Окисление по Сверну 14α- гидроксимаркфортина А (формула 10, схема D) дает 14-кетомаркфортин А (формула 16), который восстанавливают до 14β- гидроксимаркфортина А (формула 17). Обработкой 14-кетомаркфортина А (формула 16) этилмагнийбромидом по реакции Гриньяра получают гидрокси 14α- 14этилмаркфортин А (формула 19). Обработка 14α- гидроксимаркфортина А (формула 10) м-хлорпероксибензойной кислотой дает N-оксид 14α- гидроксимаркфортина А (формула 18). 14β- метилмаркфортин А может быть получен из 14α- гидрокси -14β- метилмаркфортина А путем дегидроксилирования. Так, 14α- гидрокси -14β- метилмаркфортин А обрабатывают фенилхлортионоформиатом в присутствии основания. Это тиоформиатное производное 14α- гидрокси -14β- метилмаркфортина А восстанавливают гидридом три-н-бутилолова с образованием 14β- метилмаркфортина А.
Альтернативно, 14α- гидроксимаркфортин А может быть синтезирован из маркфортина А (схема Е). Обработка маркфортина А бикарбонатом натрия и иодом в водном ТГФ дает 17-кетомаркфортин А (формула 7), который может быть дисульфенилирован с использованием LDA и фенилдисульфида с образованием 16-дитиофенил-17- кетомаркфортина А (формула 20, схема Е) из маркфортина А с выходом 60%. Окисление м-хлорпероксибензойной кислотой приводит к 16- тиофенил-16-сульфоксифенил-17-кетомаркфортину А (формула 21), который отщепляет при кипячении толуол с образованием 15,16-дегидро-16-тиофенил-17-кетомаркфортина А (формула 22). Последующая обработка м-хлорпероксибензойной кислотой дает 15,16-дегидро-16- сульфоксифенил-17-кетомаркфортин А (формула 23), который подвергают перегруппировке с использованием диэтиламина в метаноле с образованием 15,16-дегидро -14α- гидрокси-17-кетомаркфортинаА (формула 9 а).
14α- гидрокси -15α- метилмаркфортин А (формула 35, схема О) может быть синтезирован из 15,16-дегидро -14α- гидрокси-17- кетомаркфортинаА (формула 9а, схема G). Так, 15,16-дегидро 14α- гидрокси-17-кетомаркфортина А (формула 9а) обрабатывают либо метилмагиийбромидом, либо литийдиметилмедью с образованием 15α- метил -14α- гидрокси-17-кетомаркфортина А (формула 34), который восстанавливают комплексом бора с диметилсульфидом с получением 15α- метил -14α- гидроксимаркфортина А (формула 35). 15α- метил -14α- гидрокси-17-кетомаркфортин А (формула 34) превращают в 15α- метил-14,17-дикетомаркфортин А (формула 36) путем окисления по Сверну с использованием оксалилхлорида в ДМСО. Обработка метилмагнийбромидом по реакции Гриньяра дает 15α- метил -14α- гидрокси -14β- метил-17-кетомаркфортин А (формула 37), который восстанавливают комплексом бора с диметилсульфидом с образованием 15α метил 14α- гидрокси 14β- метилмаркфортина А (формула 38).
Эти предварительно описанные процессы могут использоваться для получения производных 14-замещенных маркфортинов В, С и D.
Получение 16-иод-17-цианомаркфортина А в виде смеси диастереомеров (формула 5)
Твердый циануриодид (11,7 г, 76,5 ммоль) добавляют к раствору маркфортина А (10,5 г, 22 ммоль) в CHCl3 (150 мл) и реакционную смесь кипятят с обратным холодильником до полного растворения всего маркфортина А (около 5 часов). Попорченный черный раствор охлаждают до комнатной температуры, разбавляют 100 мл CH2Cl2, промывают сначала насыщенным раствором NaHCO3, а затем - раствором Na2SO3. Отделяют органическую фазу, высушивают над MgSO4 и упаривают досуха. Образующееся неочищенное твердое вещество хроматографируют на силикагеле (элюент-этилацетат: гексан 3:2) с последующим выделением 16-иодо-17-цианомаркфортина А (12,5 г, 90%) в виде белого твердого порошка. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Получение 16,17-дегидро-17-цианомаркфортина А (формула 6)
16-иод-17-цианомаркфортин А (9,5 г, 15 ммоль) растворяют в 150 мл метанола и добавляют 3 мл 45%-го водного раствора КОН. Реакционную смесь перемешивают 2 часа при комнатной температуре. Добавляют воды и отфильтровывают образующийся белый осадок; осадок промывают водой и высушивают в вакууме в течение суток. Получают 16,17-дегидро-17-цианомаркфортин А (6,6 г, 75%) в виде белого порошка. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Масс-спектр: M/Z [М+H] : 501.
Получение 17-кетомаркфортина А (формула 7)
К раствору 16,17-дегидро-17-цианомаркфортинаА (6,0 г, 10 ммоль) в 100 мл 95%-го этанола добавляют 2,9 г (26 ммоль) диоксида селена и 2 часа перемешивают реакционную смесь при комнатной температуре. Реакцию прекращают добавлением 100 мл насыщенного раствора NaHCO3. Образующуюся смесь экстрагируют CH2Cl2 (2 • 200 мл). Экстракты объединяют, осушают (MgSO4) и концентрируют; получают 7 г неочищенного продукта. Продукт очищают хроматографией на силикагеле (элюент-этанол); получают 17-кетомаркфортин А (3,6 г, 75%) в виде белого твердого вещества. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Масс-спектр: M/Z [M+H] вычислено для C28H33N3O5 + H: 492,2498, измерено: 492,2.478.
Альтернативно и более предпочтительно, указанное в заглавии соединение может быть синтезировано с использованием п-толуолсульфокислоты. Так, моногидрат п-толуолсульфокислоты (1 г) добавляют к раствору 10 г 16,17-дегидро-17-цианомаркфортина А в 50 мл 95%-го метанола и 1 час перемешивают реакционную смесь при комнатной температуре. К смеси добавляют 2 мл триэтиламина и отгоняют растворитель. Осадок обрабатывают 10%-ым водным раствором карбоната натрия (100 мл) и отфильтровывают и высушивают; получают указанное в заглавии соединение в виде твердого вещества (выход 90%). Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Получение 15,16-дсгидро-17-кетомаркфортинаА (формула 8)
Раствор диизопропиламида лития готовят из 1,6 М раствора н-бутиллития (9,9 мл, 15,4 ммоль) в гексане и 2,2 мл (15,7 ммоль) диизопропиламина. Этот раствор разбавляют 20 мл безводного ТГФ и замораживают до -78oC. Раствор 2,0 г (4,1 ммоль) 17-кетомаркфортина А в 20 мл безводного ТГФ по каплям прибавляют в реакционную смесь, повышая температуру до -40oC в течение 1 часа. Смесь снова охлаждают до -78oC и прибавляют по каплям 19 мг (5,2 ммоль) хлорида фенилселения в 10 мг ТГФ. Через 5 минут реакцию прекращают добавлением насыщенного NaHCO3, смесь экстрагируют CH2Cl2, осушают (MgSO4) и концентрируют; получают желтое твердое вещество, которое далее используют без очистки. Это вещество растворяют в 150 мл ТГФ и обрабатывают 1,5 мл 30%-го H2O при 0oC. Убирают охлаждающую баню и реакционную смесь перемешивают 30 минут при комнатной температуре. Реакцию прекращают добавлением 100 мл 1 н. NaOH. Экстракты объединяют, осушают (MgSO4) и концентрируют; получают неочищенный продукт. Этот продукт очищают хроматографией на силикагеле (элюент-этилацетат); получают 1,3 г (65%) 15,16-дегидро-17-кетомаркфортина А в виде белого твердого вещества. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Масс-спектр: M/Z [М+H] вычислено для C28H31N3O5 + H: 490,2342, измерено: 490,2345.
Получение 14α- гидрокси-15,16-дегидро-17-кетомаркфортина А (формула 9а) с использованием реакции окисления
1 мл 0,5 М раствора бис(триметилсилил)амида калия (0,5 ммоль) по каплям прибавляют к раствору 66 мг (0,14 ммоль) 15,16-дегидро- 17-кетомаркфортинаА в 2 мл ТГФ при -78oC. Получаемый мутный бледно-желтый раствор нагревают за 1 час до -40oC. Реакционную смесь охлаждают до -78oC и по каплям прибавляют раствор 42 мг (0,16 ммоль) 2-фенилсульфонил-3-фенилоксазеридина в 2 мл ТГФ. Смесь перемешивают 5 минут, после чего реакцию прекращают добавлением NaHCO3. Смесь экстрагируют CH2Cl2 (2 • 25 мл). Экстракты объединяют, осушают (MgSO4) и концентрируют; получают неочищенный продукт. Продукт очищают препаративной тонкослойной хроматографией (ТСХ) на силикагеле (элюент-этилацетат). Получают α- гидрокси- 15,16-дегидро-17-кетомаркфортин А (8 мг, 12%) в виде белого твердого вещества. Структура может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [M+H] вычислено для C28H31N3O6 + H: 506,2291, измерено: 506,2280. 15,16-дегидро-16- гидрокси17-кетомаркфортин А (14 мг, 20%) также может быть получен из слоя. Его структура может быть подтверждена спектроскопией ЯМР.
Получение α- гидрокси-15,16-дегидро-17-кетомаркфортина А (формула 9 а). 15.16-дегидро-14,17-дикетомаркфортина А (формула 11). 14,15-дегидро-16,17-дикетомаркфортина А (формула 24) с использованием диоксида селена
1,29 г (2,6 ммоль) 15,16-дегидро-17-кетомаркфоритна А растворяют в 30 мл п-диоксана и обрабатывают 390 мг диоксида селена. Смесь кипятят 1 час с обратным холодильником и отгоняют растворитель в вакууме. Осадок обрабатывают 30 мл метиленхлорида и отфильтровывают. Фильтрат концентрируют, осадок хроматографируют на силикагеле (метанол: этилацетат 1:20). Получают 14α- гидрокси-15,16-дегидро-17-кетомаркфортин А (430 мг, 32%) в виде твердого вещества. 15,16-дегидро-14,17- дикетомаркфортинА (формула 11, 212 мг, 16%) также выделяют в ходе хроматографирования. 14,15-дегидро-16,17-дикетомаркфортин А (формула 24, 106 мг, 8%) также выделяют в ходе хроматографирования. Структура этих продуктов может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Превращение 14α- гидрокси-15,16-дегидро-17-кетомаркфортина А (формула 9а) в 15,16-дегидро-14,17-дикетомаркфортина А (формула 11)
14α- гидрокси-15,16-дегидро-17-кетомаркфортин А (60 мг, формула 9а) растворяют в 10 мл метиленхлорида и обрабатывают 60 мг диоксида марганца. Смесь перемешивают 1 час при комнатной температуре и концентрируют. С помощью препаративной ТСХ на силикагеле (элюент-50% метиленхлорид в этилацетате) получают 15,16-дегидро-14,17-дикетомаркфортин А (35 мг, 60%, формула 11). Структура соединения может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Пример 1. Получение α- гидроксимаркфортина А (формула 10)
20 мг 14α- гидрокси-15,16-дегидро-17-кетомаркфортина А (0,040 ммоль) растворяют в 5 мл ТГФ и обрабатывают 1 М раствором алюмогидрида лития (0,11 мл, 0,11 ммоль) в ТГФ при 0oC. Смесь перемешивают 1/2 часа при 0oC, после чего добавляют 10%-ый раствор NaHCO3. Смесь экстрагируют CH2Cl2 (2 • 10 мл). Экстракты объединяют, осушают (MgSO4) и упаривают растворитель. Препаративной ТСХ на силикагеле осадка (элюент - 10% метанол в этилацетате) получают 14α- гидроксимаркфортин А (3 мг, 15%) в виде белого твердого вещества. Структура может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. MCBP M/Z [M+H] вычислено для C28H35N3O5 + H: 494,2655, измерено: 494.2653.
Получение 14α- гидрокси-17-кетомаркфортина А (формула 12) из 14α- гидрокси-15,16-дегидро-17-кетомаркфортина A (формула 9a)
50 мг 14α- гидрокси-15,16-дегидро-17-кетомаркфортина А (0,1 ммоль) растворяют в 5 мл ТГФ и обрабатывают 1 М раствором алюмогидрида лития (0,7 мл, 0,7 ммоль) в ТГФ при -78oC. Смесь перемешивают 1/2 часа при -78oC. Реакцию прекращают добавлением 1 мл метанола, и смесь концентрируют. Образующееся твердое вещество подвергают хроматографии на силикагеле (метанол:метиленхлорид 1:20); получают 14α- гидрокси-17-кетомаркфортин А (43 мг, 86%) в виде белого твердого вещества. Структура может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. MCBP M/Z [М+H] вычислено для C28H33N3O6 + H: 508,2447, измерено: 508,2437.
Получение 14α- гидрокси-17-кетомаркфортина А (формула 12) из 15,16-дегидро-14,17-дикетомаркфортина А (формула 11)
470 мг 15,16-дегидро-14,17-дикетомаркфортина А (0,93 ммоль) растворяют в ТГФ и обрабатывают 1 М раствором боргидрида лития (2 мл) в ТГФ при комнатной температуре. Смесь перемешивают 2 часа, после чего добавляют 10%-ый раствор NaHCO3. Смесь экстрагируют CH2Cl2 (2 • 20 мл). Экстракты объединяют, осушают (MgSO4) и упаривают растворитель. Осадок содержит смесь двух эпимеров, которые легко разделяются хроматографией на силикагеле (метанол:этилацетат 1: 20) 14α- гидрокси-17-кетомаркфортин А (90 мг, 19%) и β- гидрокси-17-кетомаркфортин А (94 мг, 20%). Структура этих соединений может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Получение 14α- гидроксимаркфортина А (формула 10) из 14α- гидрокси-17-кетомаркфортина А (формула 12а)
413 мг 14α- гидрокси-17-кетомаркфортина (0,81 ммоль) растворяют в 20 мл ТГФ и обрабатывают 1 М раствором комплекса бора с ТГФ (2,43 мл) в ТГФ при 0oC. Смесь перемешивают 2 1/4 часа. После этого перемешивают еще 1/2 часа, затем прибавляют 3 мл метанола. После отгонки растворителя осадок хроматографируют на силикагеле (метанол: этилацетат 1:16). Получают 14α- гидроксимаркфортин А (250 мг, 92% от прореагировавшего исходного вещества) и 14α- гидрокси-17-кетомаркфортин А (исходное вещество, 140 мг, 34%).
Получение 14,17-дикетомаркфортина А (формула 13)
Раствор оксалилхлорида (40 мкл) в безводном CH2Cl2 (5 мл) обрабатывают ДМСО (45 мкл) при -78oC. Смесь перемешивают 1 час при -78oC. Добавляют по каплям раствор 27 мг 14α- гидрокси-17-кетомаркфортина А в 2 мл CH2Cl2. Реакционную смесь перемешивают 20 мин при -78oC. В реакционную смесь добавляют 0,3 мл триэтиламина, нагревают в течение 20 минут до комнатной температуры. Добавляют 10 мл 10%-го раствора Na2CO3 и 10 мл CH2Cl2. Органический слой осушают (MgSO4) и концентрируют. Осадок хроматографируют на силикагеле (метанол: этилацетат 1:20). Получают 14,17-дикетомаркфортин А (22 мг, 80%) в виде белого твердого вещества. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C28H31N3O6 + H: 506,2291, измерено: 506,2290.
Получение 14α- гидрокси -14β- метил-17-кетомаркфортина А (формула 14a)
Раствор 16 мг (0,032 ммоль) 14,17-дикетомаркфортина А в 5 мл CH2Cl2 при -78oC обрабатывают 3 М раствором метилмагнийбромида (0,16 мл, 0,48 ммоль) в диэтиловом эфире при -78oC. Реакцию прекращают добавлением нескольких капель 10%-го Na2CO3. Смесь разбавляют 10 мл CH2Cl2 осушают (MgSO4) и концентрируют. Осадок хроматографируют на силикагеле (метанол: этилацетат 1:20). Получают 14α- гидрокси -14β- метил-17-кетомаркфортин А (8 мг, 50%, Rf=0,4) в виде белого твердого вещества. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C29H35N3O6 + H: 522.2604, измерено: 522,2620. Также получают из слоя 14β- гидрокси -14α- метил-17-кетомаркфортин А (1,2 мг, 7%, Rf=0,4). Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C29H35N3O6 + H: 522,2604, измерено: 522,2620. Соотношение полученных таким образом продуктов, равное 6:1, увеличивается более, чем до 50:1 при замене используемого в качестве растворителя CH2Cl2 на ТГФ.
Пример 2: Получение 14α- гидрокси -14β- метилмаркфортина А (формула 15)
Раствор 5 мг (0,01 ммоль) 14α- гидрокси -14β- метил-17- кетомаркфортина А в 5 мл ТГФ обрабатывают 1 М раствором алюмогидрида лития (0,03 мл, 0,03 ммоль) в ТГФ при 0oC. Смесь перемешивают 1/2 часа при 0oC, после чего добавляют 10%-ый раствор NaHCO3. Смесь экстрагируют CH2Cl2 (2 • 5 мл). Экстракты объединяют, осушают (MgSO4) и отгоняют растворитель. Препаративной ТСХ осадка на силикагеле (метанол: этилацетат 1:20) получают 14α- гидрокси 14β- метилмаркфортин А (2 мг, 40%). Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C29H37N3O5 + H: 508,2811, измерено: 508,2816.
Получение 14-кетомаркфортина A (формула 16)
Раствор оксалилхлорида (150 мкд) в безводном CH2Cl2 (20 мл) обрабатывают ДМСО (170 мкл) при -78oC. Смесь перемешивают 1 час при -78oC. Добавляют по каплям раствор 110 мг 14α- гидроксимаркфортина А в 5 мл CH2Cl2.
Реакционную смесь перемешивают 20 мин при -78oC. В реакционную смесь добавляют 1 мл триэтиламина, нагревают в течение 20 минут до комнатной температуры. Добавляют 20 мл 10%-го раствора Na2CO3 и 20 мл CH2Cl2. Органический слой осушают (MgSO4) и концентрируют. Осадок хроматографируют на силикагеле (метанол: метиленхлорид 1:25). Получают 14-кетомаркфортин А (82 мг, 75%) в виде белого твердого вещества. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C28H33N3O5 + H: 492,2498, измерено: 492,2510.
Пример 3: Получение 14β- гидроксимаркфортина А (формула 17)
Раствор 10 мг (0,01 ммоль) 14α- кетомаркфортина А в 2 мл метанола обрабатывают 5 мг боргидрида натрия при 0oC. Смесь перемешивают 1/2 часа при 0oC, после чего добавляют 10%-ый раствор NaHCO3. Смесь экстрагируют CH2Cl2 (2 • 10 мл). Экстракты объединяют, осушают (MgSO4) и отгоняют растворитель. Препаративной ТСХ осадка на силикагеле (метанол: этилацетат 1:16) получают 14β- гидроксимаркфортин А (5 мг, 50%). Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C28H35N3O5 + H: 494,2655, измерено: 494,2653.
Пример 4: Получение N-оксида 14α- гидроксимаркфортина А (формула 18)
Раствор 15 мг 14α- гидроксимаркфортина А в 3 мл CH2Cl2 обрабатывают 15 мг м-хлорпероксибензойной кислоты при 0oC. После перемешивания в течение 1/2 часа при 0oC смесь обрабатывают 30 мкл триэтиламина и концентрируют. Препаративной ТСХ осадка на силикагеле (метанол: метиленхлорид 1:8) получают N-оксид 14α- гидроксимаркфортина А (12 мг, 80%). Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C28H35N3O6 + H: 510,2604, измерено: 510,2615.
Пример 5: Получение 14α- гидрокси -14β- этилмаркфортина А (формула 19)
Раствор 25 мг (0,05 ммоль) 14-кетомаркфортина А в 5 мл ТГФ обрабатывают 3 М раствором этилмагнийбромида (0,15 мл, 0,45 ммоль) в диэтиловом эфире при -78oC. Смесь перемешивают 1/2 часа при -78oC. Реакцию прекращают добавлением нескольких капель 10%-го Na2CO3. Смесь разбавляют 10 мл CH2Cl2, осушают (MgSO4) и концентрируют. Осадок хроматографируют на силикагеле (метанол: метиленхлорид 1:20). Получают 14α- гидрокси -14β- метилмаркфортин А (10 мг, 45%). Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией. МСВР M/Z [М+H] вычислено для C30H39N3O5 + H: 522,2968, измерено: 522,2983.
Получение 14β- метилмаркфортина А из 14α- гидрокси -14β- метилмаркфортина А
1 мл 0,5 М раствора бис(триметилсилил)амида калия (0,5 ммоль) по каплям прибавляют к раствору 66 мг (0,14 ммоль), 14α- гидрокси -14β- метилмаркфортина А в 2 мл ТГФ при -78oC. Получаемый мутный бледно-желтый раствор нагревают за 1 час до -40oC. Реакционную смесь охлаждают до -78oC, перемешивают 15 минут, а затем по каплям прибавляют раствор фенилхлортионоформиата (0,094 мл, 0,7 ммоль) в 2 мл ТГФ. Через 10 минут баню с сухим льдом убирают. Взаимодействие продолжается еще 3 часа, после чего реакцию прекращают добавлением NaHCO3. Смесь экстрагируют CH2Cl2 (2 • 25 мл). Экстракты объединяют, осушают (MgSO4) и концентрируют; получают неочищенный продукт. Продукт очищают препаративной тонкослойной хроматографией (ТСХ) на силикагеле (элюент-этилацетат). Получают 14α- O-фенокситиокарбонил -14β- метилмаркфортин А.
К раствору 14α- O-фенокситиокарбонил -14β- метилмаркфортина А (64 мг, 0,1 ммоль) в толуоле прибавляют AIBN (3,3 мг) с последующим добавлением гидрида трибутилолова (54 мкл, 0,2 ммоль). Смесь кипятят 3 часа с обратным холодильником. После отгонки растворителя осадок очищают препаративной ТСХ (силикагель, этилацетат). Получают 14β- метилмаркфортин А. Структура может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
Альтернативный синтез 17-кетомаркфортина А (формула 7, пункт E)
К 65 г (0,136 моль) маркфортина А и 137 г (1,63 моль) бикарбоната натрия в 2 л ТГФ и 1, 25 л воды по каплям при кипячении с обратным холодильником прибавляют в течение 1 часа 206 г (0,81 моль) иода в 1,25 л ТГФ. Альтернативно, смесь можно перемешивать 16 часов при комнатной температуре. После медленного охлаждения до комнатной температуры (2,5 часа) реакцию прекращают добавлением 1,5 л насыщенного раствора тиосульфата натрия (Na2S2O3) и экстрагируют этилацетатом (2 • 1 л). Объединяют обе фракции органического слоя и промывают насыщенным раствором тиосульфата натрия (1 л), осушают (MgSO4), фильтруют и высушивают в течение суток в вакуумной печи (65oC). Получают 62. г неочищенного 17-кетомаркфортина (формула 7) в виде желтого твердого вещества. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 7,68 (с, 1Н), 6,80 (д, 1Н), 6,70 (д, 1Н), 6,32 (д, 1Н), 4,90 (д,1Н), 3,75 (кв, 2Н), 3,23 (т, 1Н), 3,09 (с, 3H), 2,80 (д, 1Н), 2,65 (д, 1Н), 2,49-2,21 (м, 2Н), 2.08 (д, 1Н), 1,98-1,45 (м, 5Н), 1,46 (с, 3H), 1,44 (с, 3H), 1,09 (с, 3H), 0,90 (с, 3H).
Альтернативно, вместо иода можно использовать ICl.
Синтез 16-дитиофенил-17-кетомаркфортина А (формула 20)
5 г (10,2 ммоль) неочищенного 17-кетомаркфортина А в 150 мл ТГФ при -78oC шприцем добавляют в раствор диизо-пропиламида лития, приготовленный прибавлением по каплям 1,6 М раствора н-бутиллития (24,8 мл, 0,04 ммоль) к 5,7 мл (0,041 ммоль) диизо-пропиламина в 100 мл ТГФ при 0oC. Реакционную смесь медленно нагревают до -50oC в течение примерно часа. Образующуюся мутную красно-коричневую смесь затем обрабатывают 4,4 г (0,02 ммоль) фенилдисульфида. Реакцию немедленно прекращают добавлением 100 мл насыщенного раствора бикарбоната натрия; реакционную смесь экстрагируют метиленхлоридом (300 мл). Органическую фазу осушают (MgSO4), концентрируют (8 г) и хроматографируют на силикагеле (120 г, элюент-60%-ый этилацетат в гексане). Получают указанное в заголовке соединение (4,4 г, 61%, считая на маркфортин А) в виде белого твердого вещества. МСВР [М++H]: 708; 1H ЯМР (300 Мгц, CDCl3): δ, м. д. 7,74 (с, 1Н), 7,71 (д, 2Н), 7,64 (д, 2Н), 7,45-7,30 (м, 6Н), 6,81 (д, 1Н), 6,72 (д, 1Н), 6,32 (д, 1Н), 4,91 (д, 1Н), 3,70 (кв, 2Н), 3,16 (т, 1Н), 3,01 (с, 3H), 2.75 (д, 1Н), 2,53 (дт, 1Н), 2,35 (дт, 1Н), 2,15-1,50 (м, 5Н), 1,47 (с, 3H), 1,45 (с, 3H), 1.06 (с, 3H), 0,82 (с, 3H).
Синтез 16-тиофенил-16-сульфоксифенил-17-кетомаркфортина А (формула 21)
К 10 г (14 ммоль) 16-дитиофенил-17-кетомаркфортина А в 250 мл CH2Cl2 при -78oC в атмосфере азота 15 минут по каплям добавляют 4,2 г (15,5 ммоль) 64%-ой м-хлорпероксибензойной кислоты (м-ХПБК) в 200 мл CH2Cl2. Реакцию немедленно прекращают насыщенным раствором тиосульфата натрия (200 мл), разбавленным насыщенным раствором NaHCO3 (200 мл); массу экстрагируют 200 мл CH2Cl2, осушают (MgSO4), после чего концентрируют при пониженном давлении. Получают 11 г неочищенного 16-тиофенил-16-сульфоксифенил-17-кетомаркфортина А (формула 21).
1H ЯМР (300 Мгц, CDCl3) δ, м.д. 8,0-7,29 (м, 11Н), 6,80 (д, 1Н), 6,70 (д, 1Н), 6,31 (д, 1Н), 4,90 (д, 1Н), 3,68 (д, 1Н), 3,41 (д, 1Н), 3,14 (т, 1Н), 3,07 (с, 3H), 2,82 (дт, 1Н), 2,80 - 2,65 (м, 2Н), 2,16 (дт, 1Н), 2,05 - 1,1 (м, 4Н), 1,47 (с, 3H), 0,96 (с, 3H), 0,83 (с, 3H).
Синтез 16-тиофенил-15,16-дегидро-17-кетомаркфортина А (формула 22)
11 г неочищенного 16-тиофенил-16-сульфоксифенил-17- кетомаркфортина А (формула 21) 45 минут кипятят с обратным холодильником в 250 мл толуола, охлаждают до комнатной температуры, разбавляют насыщенным раствором бикарбоната натрия (300 мл) и экстрагируют 300 мл этилацетата. Органический слой осушают (MgSO4) и концентрируют. Получают 10,6 г неочищенного 16-тиофенил-15,16- дегидро-17-кетомаркфортина А (формула 22). МСВР M/Z (М++H, C34H35N3O5S + H1): вычислено 598,2376, найдено 598,2387. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 8,18 (с, 1Н), 7,55 - 7,45 (м, 2Н), 7,29 - 7,45 (м, 3H), 6,83 (д, 1Н), 6,70 (д, 1Н), 6,34 (д, 1Н), 5,92 (дт, 1Н), 4,91 (д, 1Н), 3,87 (кв, 1Н), 3,30 (дд, 1Н), 3,21 (т, 1Н), 3,08 (с, 3H), 2,80 (д, 1Н), 2,35 (дд, 1Н), 2,10 (д, 1Н), 2,03 (дд, 1Н), 1,78 (дд, 1Н), 1,46 (с, 3H), 1,44 (с, 3H), 1,11 (с, 3H), 0,88 (с, 3H).
Синтез 16-сульфоксифенил-15,16-дегидро-17-кетомаркфортина А (формула 23)
К 10,6 г неочищенного 16-тиофенил-15,16-дегидро-17- кетомаркфортина А (формула 22) в 300 мл метиленхлорида при -78oC по каплям добавляют 2,8 г 64%-ой м-ХПБК в 125 мл CH2Cl2. Реакцию прекращают добавлением 300 мл насыщенного раствора тиосульфата натрия и 300 мл насыщенного раствора бикарбоната натрия. Затем массу экстрагируют 300 мл метиленхлорида. Органический слой осушают (MgSO4), фильтруют и концентрируют. Получают 13 г неочищенного 16-сульфоксифенил-15,16-дегидро-17-кетомаркфортина А (формула 23). 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 7,75 - 7,3 (м, 5Н), 6,81 (с, 1Н), 6,75 - 6,6 (м, 2H), 6,31 (д, 1H), 4,90 (д, 1Н), 3,78 - 3,58 (м, 2Н), 3,22 (т, 1Н), 2,98 (с, 3H), 2,88 - 2,45 (м, 2H), 2,12 - 1,55 (м, 5Н), 2,03 (дд, 1H), 1,46 (с, 3H), 1,44 (с, 3H), 1,12 (с, 3H), 0,88 (с, 3H).
Синтез 14α- гидpoкcи-15,16-дeгидpo-17-кeтoмapкфopтин А (формула 9а) из 16-сульфоксифенил-15,16-дегидро-17-кетомаркфортина А (формула 23)
К 13 г неочищенного 16-сульфоксифенил-15,16-дегидро-17- кетомаркфортина А (формула 23) в водном метаноле (10/1, 300 мл) добавляют 15 мл диэтиламина. После кипячения с обратным холодильником в течение 1/2 часа реакционную смесь охлаждают до комнатной температуры, разбавляют 450 мл воды и экстрагируют 300 мл метиленхлорида. Органический слой осушают (MgSO4), после чего концентрируют и хроматографируют на силикагеле (130 мл, элюент - 30% ацетон в метиленхлориде). Получают 3,6 г (выход 50%, считая на 16-дитиофенил-17-кетомаркфортин А) 14α- гидрокси-15,16-дегидро- 17-кетомаркфортина А (формула 9а) в виде белого твердого вещества.
Пример 6. Синтез 14α- гидрокси-N(1)- морфолинокарбонилмаркфортина А (формула 25)
К раствору 30 мг 14α- гидроксимаркфортина А в 3 мл сухого ТГФ прибавляют гидрид калия (70 мг, 50%-ая дисперсия в масле). Раствор перемешивают 1 час при комнатной температуре, затем добавляют 30 нл морфолинокарбонилхлорида в 3 мл сухого ТГФ. Смесь перемешивают 1 час при комнатной температуре, после чего добавляют 3 мл 5%-го водного раствора бикарбоната натрия и 3 мл метиленхлорида. Слои разделяют; водный слой экстрагируют метиленхлоридом (3 мл). Объединенные экстракты осушают (MgSO4), фильтруют и упаривают в вакууме. Препаративной хроматографией осадка на пластинах с силикагелем (элюент-5% метанол в метиленхлориде) получают 15 мг 14α- гидрокси-N(1)- морфолинокарбонилмаркфортина А. МСВР M/Z [М+H] : вычислено для C33H42N4O7 + H: 607,3132; найдено: 607,3153.
Пример 7. Синтез α- ацетоксимаркфортина А (формула 26)
50 мг 14α- гидроксимаркфортина А растворяют в смеси ацетонитрил/ метиленхлорид (4 мл/ 2 мл). Раствор обрабатывают 50 мкл уксусного ангидрида. Смесь кипятят с обратным холодильником 1 час и охлаждают до комнатной температуры: перемешивают при комнатной температуре 16 часов, после чего добавляют 3 мл 5%-го водного раствора бикарбоната натрия и 3 мл метиленхлорида. Слои разделяют; водный слой экстрагируют метиленхлоридом (3 мл). Объединенные экстракты осушают (MgSO4), фильтруют и упаривают в вакууме. Препаративной хроматографией осадка на пластинах с силикагелем (элюент-2,5% метанол в метиленхлориде) получают 30 мг 14α- ацетоксимаркфортина А. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 8,70 (с, NH), 6,80 (д, J=8,1 Гц, С4-Н), 6,68 (д, J=8,l Гц, C5-Н). 6,37 (д, J=7,7 Гц, C24-Н), 5,44 (т, J=2,5 Гц, C14-Н), 4,92 (д, J= 7,7 Гц, C25-Н), 3,75 (д, J=11,8 Гц, C12-Н), 3,12 (с, 3H, N-Me), 3,04 (т, J= 10,2 Гц, C20-Н), 2,6 - 2,8 (м, 2Н), 2,5 - 2,1 (м, 3Н), 2,16 (с, 3H, O-ацетил), 2,0- 1,5 (м, 4Н), 1,44 (с, 6Н, C27-Н & C28-H), 1,12 (с, 3H), 0,88 (с, 3H).
Пример 8. Синтез 14α- O-этоксикарбонил(аминокарбонил)маркфортина А (формула 27)
33 мг 14α- гидроксимаркфортина А растворяют в смеси ацетонитрил/метиленхлорид (2 мл/ 1 мл). Раствор обрабатывают 30 мкл этоксикарбонил-изоцианата. Смесь перемешивают 1 час при комнатной температуре, после чего концентрируют. Препаративной хроматографией осадка на пластинах с силикагелем (элюент-5% метанол в метиленхлориде) получают 25 мг α- O-этоксикарбонил(аминокарбонил)маркфортина А. 1H ЯМР(300 Мгц, CDCl3): δ, м.д. 8,80 (ушир. , 1H, NH), 7,50 (с, NH), 6,80 (д, J=8,1 Гц, C4-Н), 6,68 (д, J=8,1 Гц, C5-Н), 6,37 (д, J=7,7 Гц, C24-Н), 5,42 (т, J=2,5 Гц, C14-Н), 4,92 (д, J=7,7 Гц, C25-Н), 4,24 (кв, 2H, J=7,1 Гц, O-CH2), 3,72 (д, J=11,8 Гц, C12-Н), 3,13 (с, 3H, N-Me), 3,08 (т, J=10,2 Гц, C20-Н), 2,6 - 2,8 (м, 2Н), 2,5 - 2,1 (м, 3Н), 2,0 - 1,5 (м, 4Н), 1,44 (с, 6Н, C27-H & C28-H), 1,28 (т, 3H, J=7,1 Гц, OCH2-CH3), 1,12 (c, 3H), 0,88 (c, 3H).
Пример 9. Синтез α- гидрокси-N(1)-ацетилмаркфортина А (формула 28)
К раствору 40 мг 14α- гидроксимаркфортина А в 3 мл сухого ТГФ прибавляют гидрид калия (70 мг, 50%-ая дисперсия в масле). Раствор перемешивают 1 час при комнатной температуре, затем добавляют 50 мкл уксусного ангидрида. Смесь перемешивают 1 час при комнатной температуре, после чего добавляют 3 мл 5%-го водного раствора бикарбоната натрия и 3 мл метиленхлорида. Слои разделяют; водный слой экстрагируют метиленхлоридом (3 мл). Объединенные экстракты осушают (MgSO4), фильтруют и упаривают в вакууме. Препаративной хроматографией осадка на пластинах с силикагелем (элюент-5% метанол в метиленхлориде) получают 10 мг 14α- гидрокси- N(1)-ацетилмаркфортина А. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 6.88 (кв, J=8,1 Гц, С4-H & С5-Н), 6,37 (д, J=7,7 Гц, C24-Н), 4,09 (ушир., C14-Н), 4,86 (д, 3,75, J=7,7 Гц, C25-Н), 3,23 (д, J= 11,3 Гц, C12-Н), 3,11 (с, 3H, N-Me), 3,05 - 1,5 (м, C12-Н), 2,61 (с, 3H, N-ацетил), 1,44 & 1,46 (с, 6Н, C27-H & C28-H), 1,08 (с, 3H), 0,82 (с, 3H).
Пример 10. Синтез 14α- O-(10-ундеценоил)маркфортина А (формула 29)
33 мг 14α- гидроксимаркфортина А растворяют в смеси ацетонитрил/метиленхлорид (4 мл/2 мл). Раствор обрабатывают 40 мкл 10-ундеценоилхлорида, 40 мкл триэтиламина и 18 мг 4-диметиламинопиридина. Смесь кипятят с обратным холодильником 16 часов и охлаждают до комнатной температуры, после чего добавляют 3 мл 5%-го водного раствора бикарбоната натрия и 3 мл метиленхлорида. Слои разделяют; водный слой экстрагируют метиленхлоридом (3 мл). Объединенные экстракты осушают (MgSO4), фильтруют и упаривают в вакууме. Препаративной хроматографией осадка на пластинах с силикагелем (элюент-2,5% метанол в метиленхлориде) получают 7 мг α- O-910-ундеценоил)маркфортина А. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 8,42 (с, NH), 6,80 (д, J=8,1 Гц, С4-Н), 6,68 (д, J= 8,1 Гц, C5-Н), 6,35 (д, J=7,7 Гц, C24-Н), 5,78 (м, 1Н), 5,44 (ушир., C14-Н), 4,89 - 5,0 (м, 3H), 3,74 (д, J=11,8 Гц, C12-Н), 3,12 (с, 3H, N-Me), 3,05 (т, J=10,2 Гц, C20-Н), 2,6 - 2,8 (м, 2Н), 2,9 - 1,2 (м, 28Н), 1,44 (с, 6Н, С27-H & С28-Н), 1,11 (с, 3H), 0,82 (с, 3H).
Пример 11. Получение 14α- гидрокси β- винилмаркфортина А (формула 30)
Раствор 200 мг (0,4 ммоль) 17-кетомаркфортина А в 5 мл ТГФ обрабатывают при -78oC 1М раствором винилмагнийбромида (4,0 мл, 4 ммоль) в ТГФ при -78oC. Полученную смесь перемешивают 2 часа при -78oC и нагревают до комнатной температуры. При комнатной температуре перемешивают еще 2 часа. Реакцию прекращают добавлением 10%-го Na2CO3 смесь разбавляют 30 мл CH2Cl2, промывают насыщенным раствором хлорида аммония, осушают (MgS04) и концентрируют. Осадок очищают хроматографией на силикагеле (элюент-гексан:ацетон 6:4); получают 120 мг (60%, Rf=0,45) α- гидрокси β- винилмаркфортина А в виде белого твердого вещества.
1H ЯМР (300 Мгц, CDCl3): δ, м.д. 7,86 (с, NH), 6,78 & 6,67 (д, J=8,1 Гц, C4-H & C5-Н), 6,32 (д, J=7,7 Гц, C24-Н), 6,58 (дд, J=17,4, 10,9 Гц, 1Н, винил), 5,43 (д, J=17,4 Гц, 1Н, винил), 5,18 (д, J= 10,9 Гц, 1Н, винил), 4,89 (д, J= 7,7 Гц, C25-Н), 3,7 (ушир., 1Н), 3,11 (с, 3H, N-Me), 2,95 (т, 1Н, C20-Н), 2,8 - 1,5 (м, 12H), 1,44 (c, 6H, C27-H & C28-H), 1,08 (с, 3H), 0,82 (с, 3H).
MC M/Z [М+H]: 520.
Пример 12. Синтез 14α- O-морфолинокарбонил-N(1)- морфолинокарбонил-маркфортина А (формула 31)
К раствору 25 мг 14α- гидроксимаркфортина А в 3 мл сухого ТГФ прибавляют гидрид калия (50 мг, 50%-ая дисперсия в масле). Раствор перемешивают 1 час при комнатной температуре, затем добавляют 30 мкл уксусного ангидрида. Смесь перемешивают 3 час при комнатной температуре, после чего добавляют 3 мл 5%-го водного раствора бикарбоната натрия и 3 мл метиленхлорида. Слои разделяют; водный слой экстрагируют метиленхлоридом (3 мл). Объединенные экстракты осушают (MgSO4), фильтруют и упаривают в вакууме. Препаративной хроматографией осадка на пластинах с силикагелем (элюент-5 /о метанол в метиленхлориде) получают 17 мг α- O-морфолинокарбонил-N(1)- морфолинокарбонилмаркфортина А. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 6,84 (кв, J=8,1 Гц, C4-H & С5-Н), 6,41 (д, J=7,7 Гц, C24-Н), 3,8 - 3,2 (м, 17Н), 2,98 (с, 3H, N-Ме), 3,0 - 1,5 (м, 13Н), 1,45 (с, 6Н, C27-H & C28-H), 1,41 (с, 3H, C14-Me), 1,18 (с, 3H), 0,83 (с, 3H).
Пример 13. Получение N-оксида 14α- гидрокси -14β- метилмаркфортина А (формула 32)
Раствор 30 мг 14α- гидроксимаркфортина А в 3 мл CH2Cl2 обрабатывают 20 мг м-хлорпероксибензойной кислоты при 0oC. После перемешивания в течение 1/2 часа при 0oC к смеси добавляют 10 мл 5%-го водного раствора бикарбоната натрия и 20 мл метиленхлорида. Слои разделяют; водный слой экстрагируют метиленхлоридом (10 мл). Объединенные экстракты осушают (MgSO4), фильтруют и упаривают в вакууме при 0oC, обрабатывают 30 мкл триэтиламина и концентрируют. Получают 20 мг указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (300 Мгц, CD3OD): δ, м.д. 6,91 & 6,70 (д, J=8,1 Гц, C4-H & С5-Н), 6,36 (д, J=7,7 Гц, C24-Н), 4,91 (д, J=7,7 Гц, C25-Н), 4,08 & 3,76 (AB, кв, J=12,9 Гц, 2H, C12-Н), 3,5-3,1 (M, 4H), 3,12 (c, 3H, N-Me), 2,8 - 1,6 (м, 7H), 1,46 & 1,44 (2с, 6Н, С27-Н & C28-H), 1,50 (с, 3H, C14-Me), 1,20 (с, 3H), 0,93 (с, 3H).
Пример 14. Синтез 14α- гидрокси -14β- метил-N(1)- ацетилмаркфортина А (формула 33)
К раствору 25 мг 14α- гидроксимаркфортина А в 3 мл сухого ТГФ прибавляют гидрид калия (50 мг, 50%-ая дисперсия в масле). Раствор перемешивают 1 час при комнатной температуре, затем добавляют 50 мкл уксусного ангидрида. Смесь перемешивают 1 час при комнатной температуре, после чего добавляют 3 мл 5%-го водного раствора бикарбоната натрия и 3 мл метиленхдорида. Слои разделяют; водный слой экстрагируют метиленхлоридом (3 мл). Объединенные экстракты осушают (MgSO4), фильтруют и упаривают в вакууме. Препаративной хроматографией осадка на пластинах с силикагелем (элюент-5% метанол в метиленхлориде) получают 20 мг 14α- гидрокси -14β- метил-N(1)- ацетилмаркфортина А. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 6,88 (кв, J=8,l Гц, С4-H & С5-Н), 6,37 (д, J=7,7 Гц, C24-Н), 4,86 (д, 3,75, J=7,7 Гц, C25-Н), 3,68 (д, C12-Н), 3,11 (c, 3H, N-Me), 2,9 - 2,7 (м, 2Н), 2,61 (с, 3H N-ацетил),), 2,5 - 1,5 (м, 10Н), 1,47 & 1,46 (с, 6Н, C27-H & C28-Н), 1,50 (с, 3H, C12-Me), 1,08 (с, 3H), 0,82 (с, 3H).
Получение 14α- гидрокси -15α- мeтил-17-кетомаркфортина А (формула 35)
Раствор 300 мг (формула 9а) 14α- гидрокси-15,16-дегидро-17- кетомаркфортина А в 12 мл ТГФ обрабатывают 3 М раствором метилмагнийбромида (1,0 мл, 5 эквивалентов) при комнатной температуре. Реакцию прекращают добавлением 3 мл насыщенного раствора хлорида аммония. Смесь разбавляют 30 мл CH2Cl2, промывают насыщенным раствором хлорида аммония, осушают (MgSO4) и концентрируют. Осадок хроматографируют на силикагеле (метанол: этилацетат 1:20). Получают 14α- гидрокси -15α- метилмаркфортин А (90 мг, 30%) в виде белого твердого вещества. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 7,90 (с, NH), 6,8 & 6,70 (д, J=8,1 Гц, C4-H & С5-Н), 6,32 (д, J=7,7 Гц, C24-Н), 4,89 (д, 3,75, J=7,7 Гц, C25-Н), 4,36 (ушир., 1H, C14-Н), 3,74 (ушир., 2Н, C12-Н), 3,20 (т, 1H, C20-H), 3,06 (с, 3H, N-Ме), 2,78 & 2,10 (д, 2Н, J=15,8 Гц, C10-Н), 2,5 - 1,8 (м, 5Н), 1,46 & 1,44 (2с, 6Н, С27-H & С28-Н), 1,13 (д, 3H, C15-Me), 1,12 (с, 3H), 0,88 (с, 3H).
Альтернативно, указанное в заголовке соединение может быть получено с использованием диметилкупрата лития. К 0,4 г (0,002 моль) иодида меди в ТГФ по каплям при 0oC добавляют 1,4 М раствор метиллития (9 мл, 0,013 моль). Полученную бледно-желтую смесь перемешивают 15 минут, после чего обрабатывают раствором 0,5 г (0,001 моль) 14α- гидрокси-15,16-дегидро-17-кетомаркфортина А в 12 мл ТГФ при 0oC. После 15 минут перемешивания мутной оранжевой смеси реакцию прекращают добавлением 25 мл насыщенного раствора хлорида аммония, экстрагируют 30 мл этилацетата. Органический слой осушают (MgSO4), фильтруют и концентрируют. Получают неочищенный продукт, который очищают на хроматотроне (пластина 4 мм, 4%-ый метанол в метиленхлориде).
Выход: 0,23 г (44%) белого твердого вещества.
Пример 15. Получение 14α- гидрокси -15α- метилмаркфортина А (формула 35)
90 мг (0,18 ммоль); 14α- гидрокси -15α- метил-17-кетомаркфортина А растворяют в 10 мл ТГФ и обрабатывают 0,18 мл 12 М раствора комплекса бора с диметилсульфидом при 0oC. Смесь перемешивают 2 часа при 0oC, затем добавляют 0,4 мл метанола и перемешивают еще 1 час. После отгонки растворителя осадок хроматографируют на силикагеле (элюент-ацетон: метиленхлорид 30:70). Получают 14α- гидрокси -15α- метилмаркфортин А (20 мг) в виде твердого вещества. 1H ЯМР (300 Мгц, CDCl3): δ, м.д. 8,39 (с, NH), 6,79 & 6,70 (д, J=8,1 Гц, C4-Н & C5-H), 6,36 (д, J=7,7 Гц, C24-Н), 4,91 (д, J=7,7 Гц, C25-Н), 3,81 (ушир. , 1Н, C14-H), 3,67 (д, J=11,7 Гц, C12-Н), 3,03 (т, 1Н, C20-H), 3,11 (с, 3H, N-Me), 2,68 & 1,86 (д, 2Н, J=15,7 Гц, C10-Н), 2,7 - 1,2 (м, 8Н), 1,02 (д, 3H, J=6,8 Гц, С15-Me), 1,11 (с, 3H), 0,85 (с, 3H).
MCBP M/Z [М+H] : вычислено для C29H37N3O5 + H: 508,2811; найдено: 508,2840.
Получение 14,17-дикето -15α- метилмаркфортина А (формула 36)
Раствор оксалилхлорида (40 мкл) в безводном CH2Cl2 (5 мл) обрабатывают ДМСО (45 мкл) при -78oC. Смесь перемешивают 1 час при -78oC. Добавляют по каплям раствор 27 мг α- гидрокси-17- кетомаркфортина А в 2 мл CH2Cl2. Реакционную смесь перемешивают 20 мин при -78oC. В реакционную смесь добавляют 0,3 мл триэтиламина, нагревают в течение 20 минут до комнатной температуры. Добавляют 10 мл 10%-го раствора Na2CO3 и 10 мл CH2Cl2. Органический слой осушают (MgSO4) и концентрируют. Осадок хроматографируют на силикагеле (метанол: этилацетат 1:20). Получают 14,17-дикетомаркфортин А (22 мг, 80%) в виде белого твердого вещества. Структура продукта может быть подтверждена спектроскопией ЯМР и масс-спектрометрией.
MCBP M/Z [М+H] вычислено для C28H31N3O6 + H: 506,2291, измерено: 506,2280.
Получение 14α- гидрокси -14β- метил -15α- метил-17-кетомаркфортина А (формула 37)
Раствор 25 мг (0,05 ммоль) 14,17-дикето α- метилмаркфортина А в 5 мл CH2Cl2 при -78oC обрабатывают 3 М раствором метилмагнийбромида (0,2 мл, 0,6 ммоль) в диэтиловом эфире при -78oC. Образующуюся смесь перемешивают 1/2 часа при -78oC. Реакцию прекращают добавлением нескольких капель 10%-го Na2CO3. Смесь разбавляют 10 мл CH2Cl2, осушают (MgSO4) и концентрируют. Осадок хроматографируют на силикагеле (метанол: метиленхлорид 1:25). Получают 14α- гидрокси -14β- метил -15α- мeтил-17- кeтoмapкфopтин A (16 мг, 62%) в виде белого твердого вещества.
1H ЯМР(300 Мгц, CDCl3) δ, м.д. 8,13 (с, 1Н), 6,78 (д, 1Н), 6,70 (д, 1Н), 6,33 (д, 1Н), 4,91 (д, 1Н), 3,75 (кв, 2Н), 3,16 (т, 1Н), 3,05 (с, 3H), 2,78 (д, 1Н), 2,86 - 2,57 (м, 1Н), 2,42 - 2,0 (м, 6Н), 1,64 (с, 3H), 1,45 (с, 3H), 1,44 (с, 3H), 1,11 (с, 3H), 1,04 (д, 3H), 0,92 (д, 3H).
Пример 16. Получение 14α- гидрокси -14β- метил -15α- метилмаркфортина А (формула 38)
15 мг (0,028 ммоль) 14α- гидрокси -14β- метил -15α- метил-17- кетомаркфортина A растворяют в 10 мл ТГФ и обрабатывают 10 М раствором комплекса бора с диметилсульфидом (0,02 мл) при 0oC. Смесь перемешивают 2 часа при 0oC, добавляют 0,4 мл метанола и перемешивают еще 1 час. После отгонки растворителя осадок хроматографируют на силикагеле (элюент-ацетон: метиленхлорид 30: 70). Получают 14α- гидрокси -14β- метил -15α- метилмаркфортин А (4 мг, 29%) в виде твердого вещества.
1H ЯМР (300 Мгц, CDCl3): δ, м.д. 7,82 (с, 1Н), 6,78 (д, 1Н), 6,79 (д, 1Н), 6,67 (д, 1Н), 6,33 (д, 1Н), 4,90 (д, 1Н), 3,65 (д, 1Н), 3,09 (с, 3H), 2,98 (т, 1Н), 2,69 (д, 1Н), 2,60 - 2,22 (м, 7Н), 2,06 (дд, 1Н), 1,87 (д, 1Н), 1,85 - 1,75 (м, 1Н), 1,44 (с, 6Н), 1,43 (с, 3H), 1,10 (с, 3H), 0,94 (д, 3H), 0,86 (д, 3H).
Подходящим образом С-24, С-25, N-1 и N-18a замещенные производные маркфортинов А, В, С и D в качестве исходных материалов в приведенных выше последовательностях реакций могут быть легко получены способами, данными в Патенте США 4 923 867. Взаимодействие осуществляют в подходящем инертном растворителе, таком как пиридин, коллидин, толуол (предпочтительно), ксилол, диоксан, тетрагидрофуран и им подобных, при температуре от 10oC до 18oC, предпочтительно от 80oC до 140oC.
Альтернативно, С-24, С-25 и N-1 производные могут быть получены из маркфортинов. Например, большие серии аналогов маркфортина могут быть получены алкилированием положения N-1 в N-18a-замещенных маркфортинах. Эти производные могут быть легко получены последующей обработкой раствора маркфортина А, N-18а-замещенных N-18a-замещенного маркфортина С в апротонном органическом растворителе, таком как тетрагидрофуран, эфир, бензол и им подобных, избытком сильного основания, такого как гидрид калия (предпочтительно), гидрид натрия, бутиллитий, трет-бутоксид калия и им подобными, с последующей обработкой подходящим алкилирующим агентом при температуре от 0oC до 50oC в течение от 1/4 часа до 48 часов. Подходящие алкилирующие агенты включают алкилбромиды, алкилиодиды, алкилсульфонаты, алкенилиодиды, алкинилбромиды, алкоксилхлориды и тому подобное.
Дополнительные серии производных могут быть получены модификацией маркфортинов А, В и С с двойной связью между С-24 и С-25. Их 24,25-дигидроаналоги легко получаются при перемешивании раствора подходящего маркфортина в спиртовом растворителе, таком как метанол, этанол, пропанол и им подобных, с катализатором, таким как палладий, платина, трис-(трифенилфосфин)хлорродий и им подобными, в присутствии газообразного водорода. Проект, являющийся аналогом 24,25-дигидромаркфортина, может быть выделен и очищен с использованием методов, известных специалистам в данной области. Примечательно, что реакции для модификации других видов маркфортиновых структур, описанные выше, могут также применяться к аналогам 24,25-дигидромаркфортина для получения соответствующих 24,25-дигидроаналогов. Кроме того, маркфортиновые аналоги с модифицированным кольцом "G" могут быть получены через 24,25-дибромиды, которые легко получают обработкой раствора маркфортина в галогенированном растворителе, таком как дихлорметан, хлороформ, четыреххлористый углерод и им подобных, 1 молем эквивалентов брома при температуре от -20oC до 25oC в течение от 1/4 часа до 8 часов. Реакция дает соответствующее 24,25-дибромопроизводное 24,25-дигидромаркфортина, которое может быть выделено и очищено с использованием методов, известных специалистам в данной области. Примечательно, что 24,25-дихлораналог может быть получен в ходе замещения брома на хлор в описанном выше процессе. Аналоги 24,25-дибром-24,25-дигидромаркфортина, описанные выше, используются для получения аддитивных производных. Так, обработка раствора дибромида в спиртовом растворителе, таком как метанол, этанол, пропанол и им подобных сильным основанием, таким как 1,8-диазабицикло- [5.4.0] ундецена-7 (ДБУ) при температуре от 0oC до 30oC в течение от 1/4 часа до 24 часов дает аналоги 24-алкокси-25-бром-24,25- дигидромаркфортина, которые могут быть выделены и очищены с использованием методов, известных специалистам в данной области. Эти аналоги 24-алкокси-25-бромпроизводные могут быть дебромированы путем обработки раствора соединения в апротонном органическом растворителе, таком как бензол, толуол, гексан и им подобных, гидридом соединения олова в качестве восстанавливающего агента, таим как гидрид трибутилолова, гидрид трифенилолова и им подобными с добавкой радикального инициатора, такого как азобис-изобутиронитрил (АИБН) (или без нее) при температуре от 25oC до 120oC в течение от 1/2 часа до 48 часов. Этот процесс даст соответствующие 24-алкоксипроизводные маркфортина (R24= низший алкоксил в общей структуре), которые могут быть выделены и очищены с использованием методов, известных специалистам в данной области.
Haemonchus contortus/ Trichostrongylus colubriformis/ Jird Assay:
В этом опыте in vivo используют песчанок, инфицированных двумя важными для исследования паразитами жвачных H. contortus/ и Т, colubriformis (могут использоваться антигельминтно-чувствительные или - устойчивые черви). Первоначально, была достигнута активность только против Н. contortus, как это описано в GA. Conder et al.. J. Parasitol. 76, 168-170 (1990), тогда как в последующих опытах изучалась активность против обоих видов паразитов с использованием методик, приведенных в GA. Conder et al., J. Parasitol. 77, 621-623 (1991). Активность 14α- гидpoкcимapкфopтинa A, 14α- гидрокси -14β- метилмаркфортина А, 14β- гидроксимаркфортина A, N-оксида 14α- гидроксимаркфортина А и маркфортина D приведена в Таблице 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВОПАРАЗИТАРНЫЕ МАРКФОРТИНЫ И ПАРАГЕРКВАМИДЫ | 1996 |
|
RU2162090C2 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ЗАМЕЩЕННЫЕ В КОЛЬЦЕ ГЕТЕРОАРОМАТИЧЕСКИМИ КОЛЬЦАМИ, В КАЧЕСТВЕ АНТИМИКРОБНЫХ АГЕНТОВ | 1996 |
|
RU2154645C2 |
| НОВЫЕ ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ КАМПТОТЕЦИНА (СРТ-11) И РОДСТВЕННЫХ СОЕДИНЕНИЙ | 1996 |
|
RU2164917C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ДЕГЕНЕРАЦИЕЙ СОЕДИНИТЕЛЬНОЙ ТКАНИ | 1997 |
|
RU2168497C2 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ИМЕЮЩИЕ С-С-СВЯЗЬ С 4-8-ЧЛЕННЫМИ ГЕТЕРОЦИКЛИЧЕСКИМИ КОЛЬЦАМИ | 1996 |
|
RU2175324C2 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНТИОАЛКИЛЬНЫЕ ИЛИ АЛКИЛЭФИРНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИНГИБИРОВАНИЯ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИРУСОВ | 1996 |
|
RU2167155C2 |
| БИЦИКЛИЧЕСКИЕ ОКСАЗИН- ИЛИ ТИАЗИН-ОКСАЗОЛИДИНОНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ЛЕЧЕНИЯ МИКРОБНЫХ ИНФЕКЦИЙ | 1995 |
|
RU2128660C1 |
| ОКСАЗОЛИДИНОНОВЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1998 |
|
RU2215740C2 |
| АНТИМИКРОБНЫЕ ФЕНИЛОКСАЗОЛИДИНОНЫ | 1995 |
|
RU2134692C1 |
| АМИНОГУАНИДИНКАРБОКСИЛАТЫ | 1995 |
|
RU2162462C2 |































































































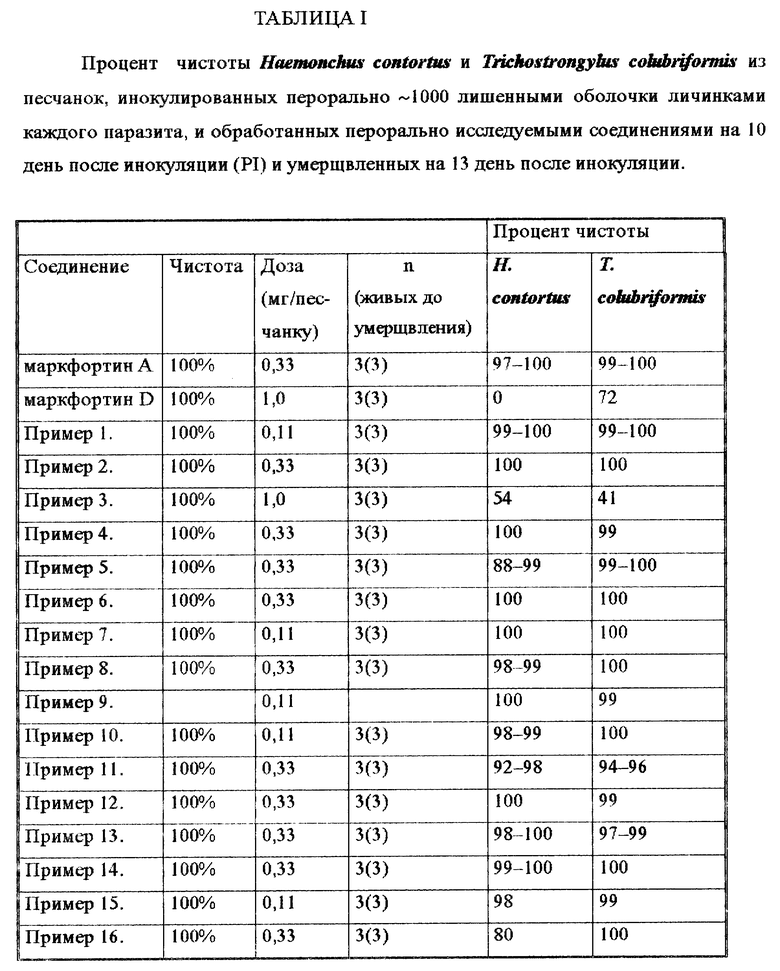
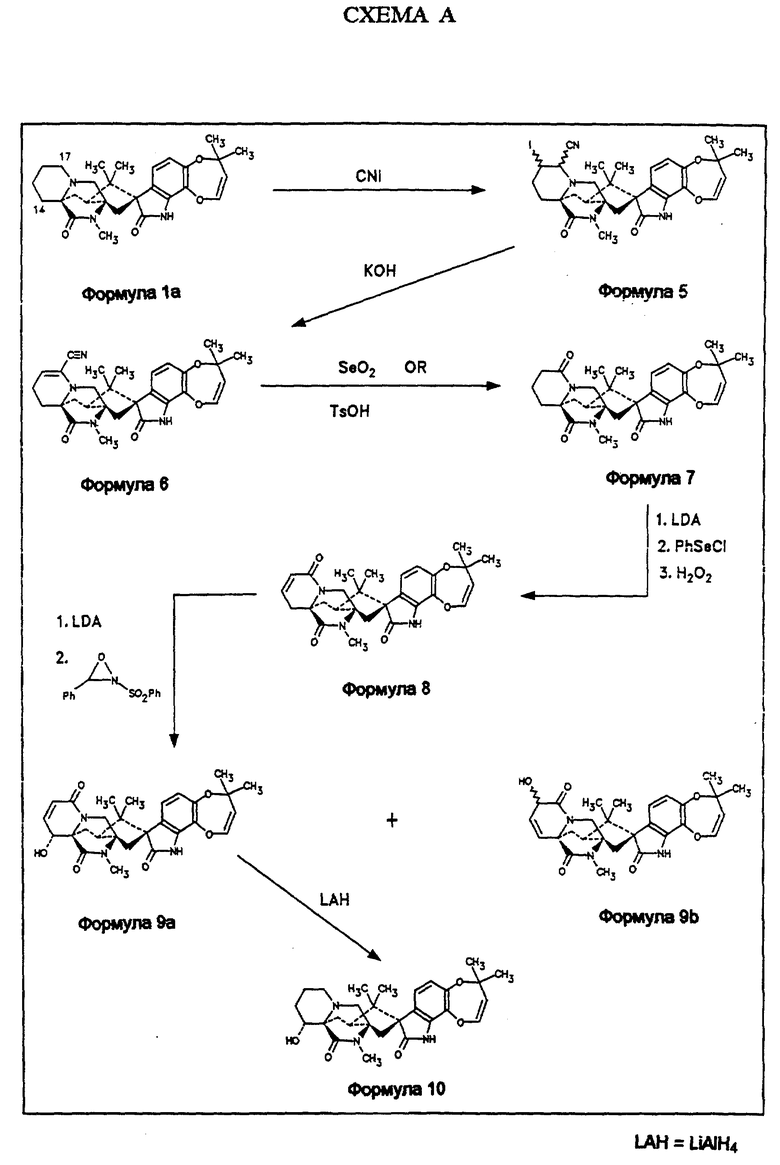
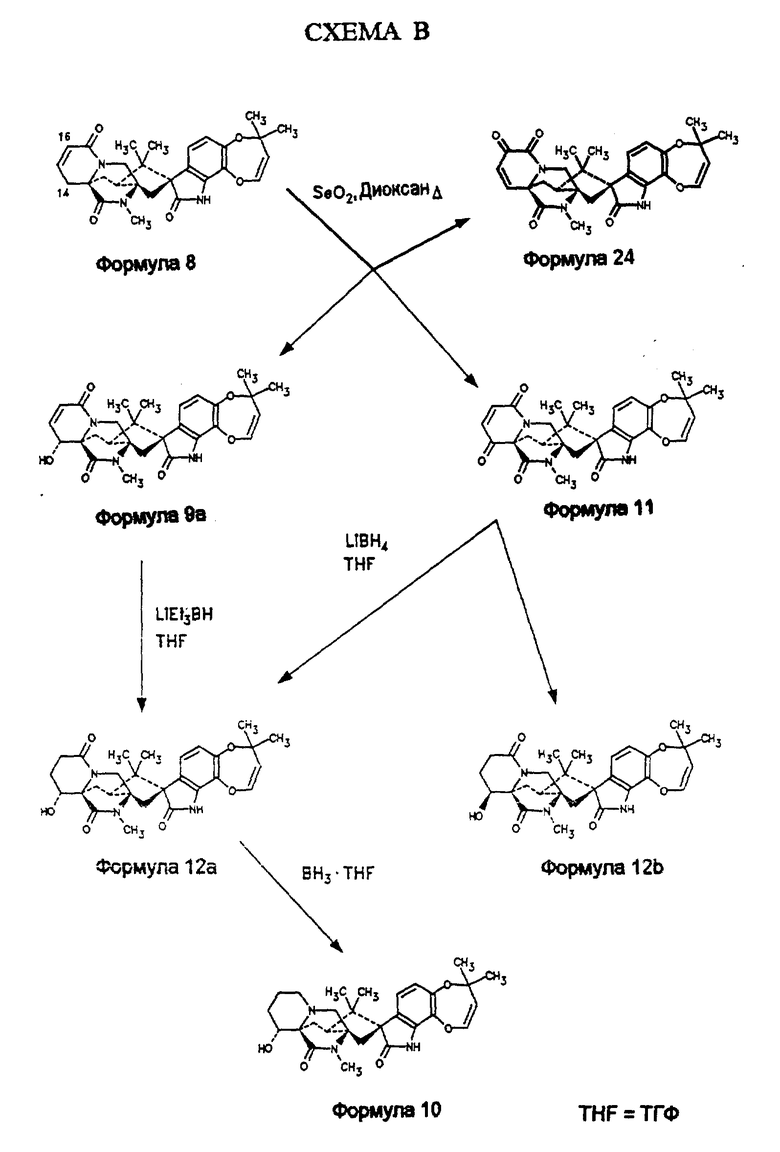
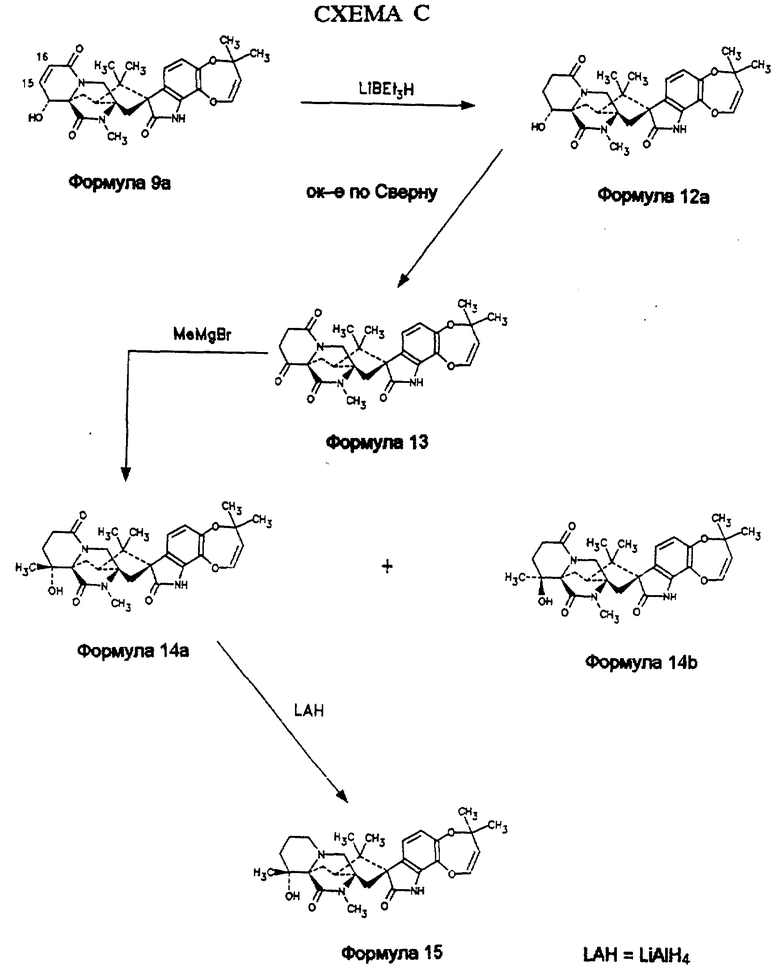
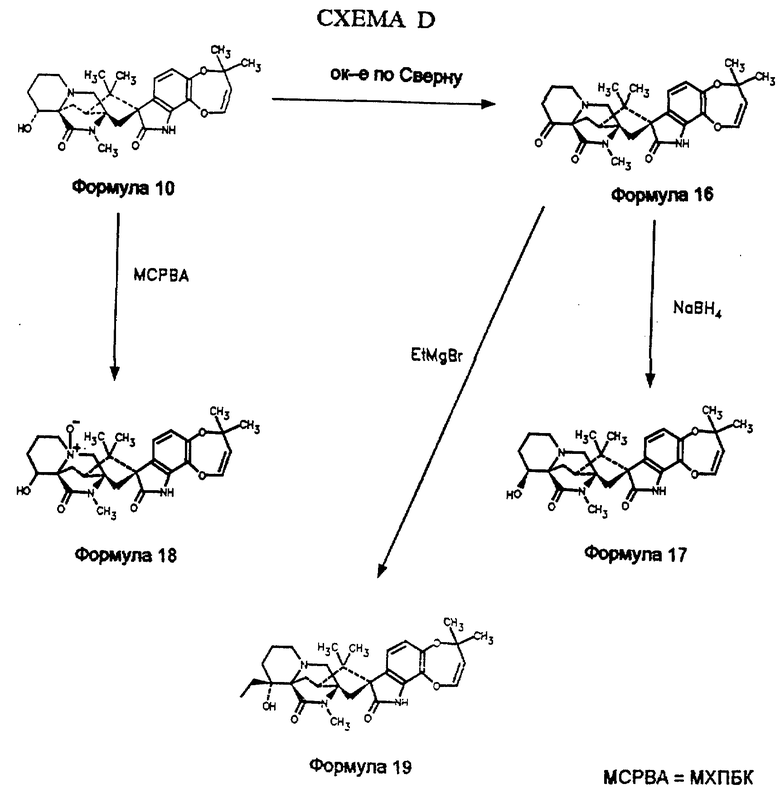
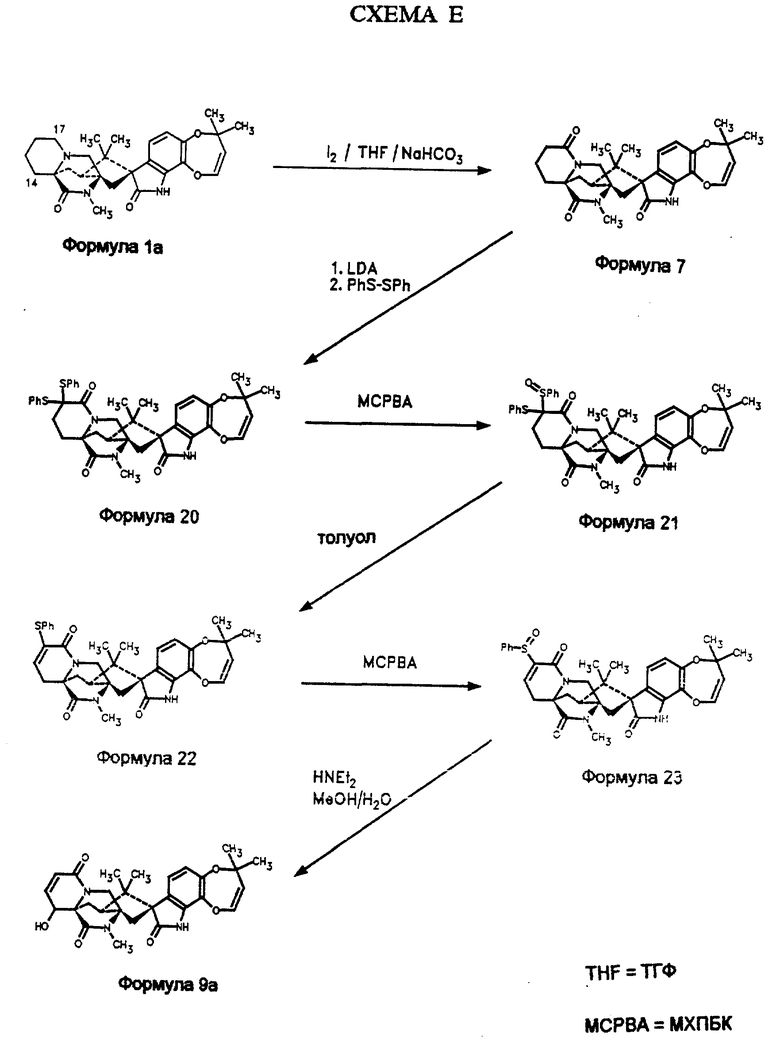
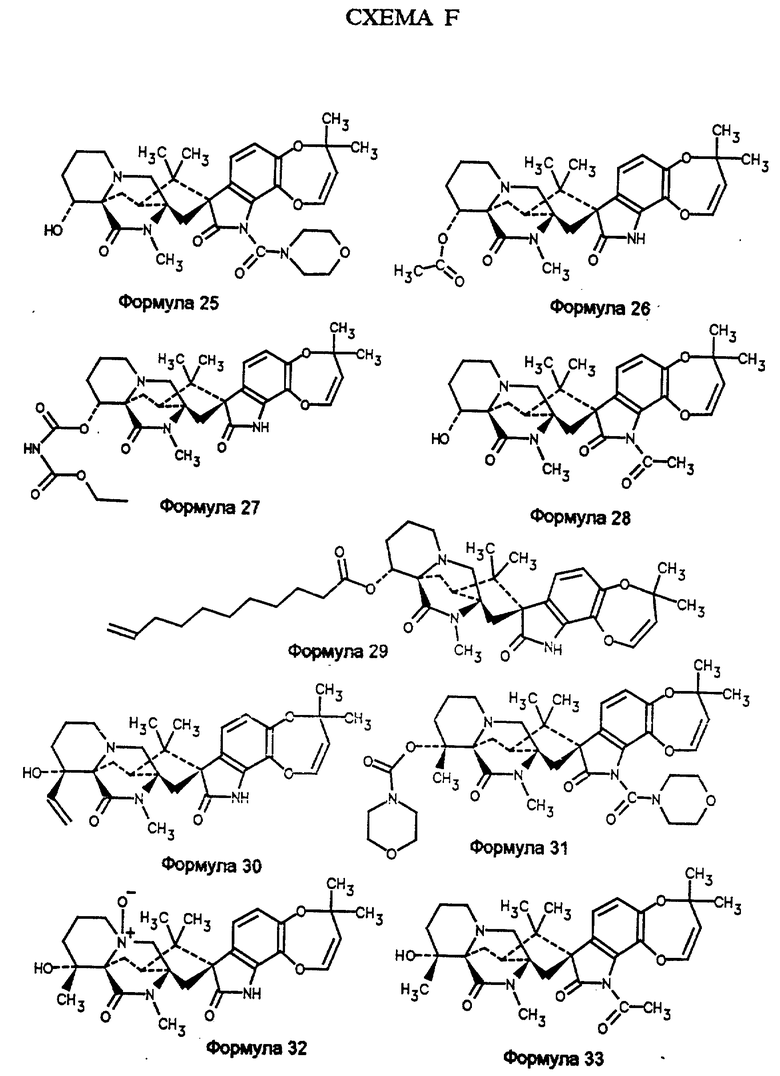
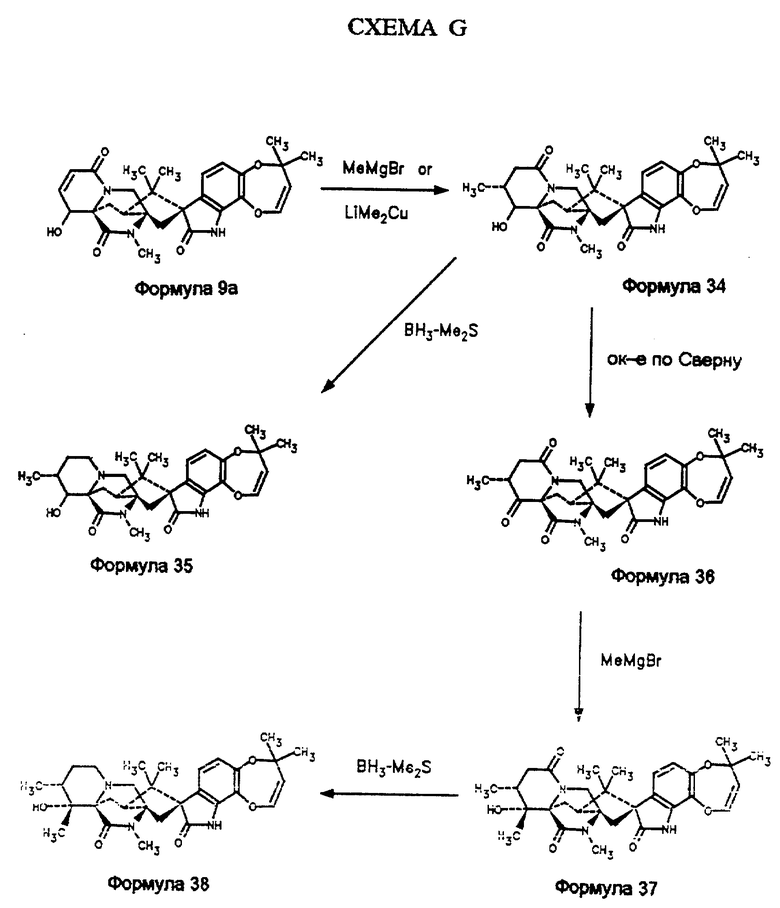
14-Замещенные маркфортины общей формулы I
или их фармацевтически приемлемые соли, или 12-N-оксид,
где m = 0;
Z, Y - кислород, R1 - Н, С2-7 алканоил или -С(O)NR4N5;
R4 и R5 вместе с атомом азота, к которому они присоединены, образуют морфолиновое кольцо;
R14a - атом водорода, гидроксил или С1-7 алкил;
R14b - Н или гидроксил;
R18а - алкил;
R15a, R15b, R24, R25 - атом водорода. Соединения формулы I - сильные парагерквамидапаразитные агенты против эндо- и эктопаразитов, в частности паразитических червей и членистоногих, вызывающих многочисленные паразитические заболевания у людей, животных и растений. 6 з.п. ф-лы, 1 табл., 7 ил.
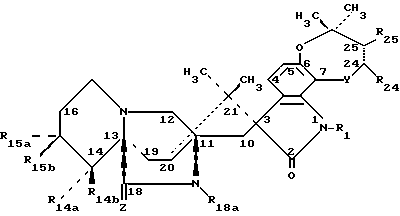
или их фармацевтически приемлемые соли,
где Z обозначает О;
Y - атом кислорода (-О-);
R1 - атом водорода, С2-С7 алканоил (-С(О) С2-С7 алкил) или -С(О)NR4R5 группу, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют морфолиновое кольцо;
R14а - атом водорода, гидроксил или С1-С7 алкил;
R14b - атом водорода или гидроксил;
R18а - С1-С7 алкил;
R24 - атом водорода;
R25 - атом водорода;
R15а и R15b оба обозначают атом водорода; при условии, что, когда один из R14а или R14b обозначает гидроксил, а другой обозначает атом водорода или метил, то R15а и R15b могут быть атомом водорода или метилом;
пунктирная линия между атомами углерода 24 и 25 представляет одинарную линию или двойную связь; при общем условии, что R14а и R14b оба не являются атомами водорода.
| US 4978656 A, 1990 | |||
| US 5164389 A, 1990 | |||
| УСТРОЙСТВО для ВЫЧИСЛЕНИЯ ПОПРАВКИ ДОЗИРОВАНИЯ АВТОМАТИЧЕСКОГО ДОЗАТОРА НЕПРЕРЫВНОГО ДЕЙСТВИЯ | 0 |
|
SU390532A1 |
| WO 9200300 A1, 1992 | |||
| WO 9222555 A1, 1992 | |||
| WO 9107961 A1, 1989 | |||
| КРИОТРОННЫЙ АССОЦИАТИВНЫЙ ЗАПОМИНАЮЩИЙЭЛЕМЕНТ | 0 |
|
SU301742A1 |
| EP 322937 A1, 1989 | |||
| ОГНЕУПОРНОЕ ПОКРЫТИЕ | 0 |
|
SU354615A1 |
| WO 9310120 A1, 1993. | |||

