Предшествующий уровень
Данное изобретение раскрывает новые и полезные соединения фенилоксазолидинона, имеющие либо пирролидинильную, либо азетидинильную часть. Соединения являются полезными антимикробными агентами, эффективными против ряда патогенов человека и патогенов животных, включая грам-положительные аэробные бактерии, такие как множественно-резистентные стафилококки, стрептококки и энтерококки, а также анаэробные организмы, такие как Bacteroides spp. и Clostridia spp. виды, и кислотоустойчивые организмы, такие как Mycobacterium tuberculosis, Mycobacterium avium и Mycobacterium spp.
Информационные данные
Данные соединения родственны, благодаря своей структуре фенилоксазолидинонового кольца, соединениям, раскрываемым в публикациях ниже, за исключением того, что соединения изобретения имеют мультизамещенную пирролидинильную или азетидильную часть. Рассматриваемые соединения уникальны и обладают полезной антибактериальной активностью.
PCT/US93/03570 заявка раскрывает оксазолидиноны, содержащие замещенную диазиновую часть и их использование в качестве антимикробных средств.
PCT/US92/08267 заявка раскрывает замещенные арил- и гетероарил-фенил-оксазолидиноны, используемые в качестве бактерицидных средств.
PCT/US89/03548 заявка раскрывает 5' индолил-5 β -амидометилоксазолидиноны, 3-(конденсированное-кольцо замещенный)фенил-5 β - амидометилоксазолидиноны, и 3-(азот замещенный)фенил-5 β -амидометилоксазолидиноны, которые используют в качестве бактерицидных средств.
К другим ссылкам, раскрывающим различные оксазолидиноны, относятся Пат. США 4 801 600, 4,921 869, Gregory W.A., et al., J. Med. Chem., 32, 1673-81 (1989); Gregory W.A., et al., J,Med.chem., 33, 2569-78 (1990); Wang С., et al. , Tetrahedron, 45, 1323-26 (1989); Brittelli, et al., J.Med. Chem., 35, 1156 (1992).
Евр. Пат. публикация 352 781 раскрывает фенил и пиридил замещенные фенилоксазолидиноны.
Евр. Пат. публикация 316 594 раскрывает 3-замещенные стирил оксазолидиноны.
Евр. Пат. публикация 312 000 раскрывает фенилметил и пиридинилметил замещенные фенил оксазолидиноны.
Сущность изобретения
В одном аспекте предметом изобретения является соединение формулы I: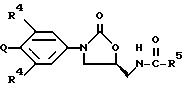
или его фармацевтически приемлемые соли, где
Q выбирают из структур i, ii, iv и v: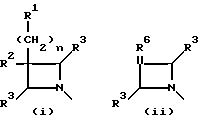
R1 представляет
(a) H,
(b) OR7,
(c) NR8R9,
(d) C1-C8ацил, который может быть замещен гидроксилом, C1-C8алкокси - бензоилом;
каждый R2 выбирают из
(a) H,
(b) OH,
(c) OR, где R представляет собой C1-C6алкилом;
каждый R3 выбирают из
(a) H,
(b) C1-C3алкил;
R4 представляет собой фтор;
R5 представляет собой
(a) C1-C8алкил, необязательно замещенный одним-тремя заместителями, выбранными из F, Cl,
(b) C3-C6циклоалкил;
R6 представляет
(a) O,
(b) NR10, где R10 является OR7, где R7 имеет нижеуказанные значения,
(c) O(CH2)mO;
R7 представляет
(a) H,
(b) фтор,
(c) C1-C8алкил;
R8 и R9 являются C1-C8алкилом, который может быть замещен фтором;
n равен 0 или 1 и m равен 2 или 3.
В одном аспекте предметом изобретения является соединение структурной формулы i: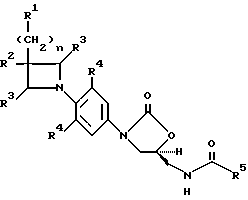
В другом аспекте предметом изобретения является соединение структурной формулы ii:
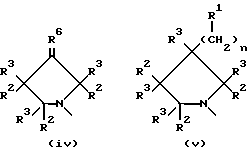
В еще одном аспекте предметом изобретения является соединение структурной формулы iv: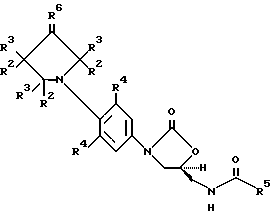
В еще одном аспекте предметом изобретения является соединение структурной формулы v:
Соединения обладают очень низкой токсичностью.
Способ лечения микробных инфекций у людей или других теплокровных животных осуществляется путем введения пациенту, при необходимости, эффективного количества описанного выше соединения формулы I (i-v). Соединение можно вводить в фармацевтической композиции либо перорально, парентерально, либо локально. Предпочтительно соединение вводят в количестве от около 0,1 до около 100 мг/кг веса тела/день, более предпочтительно от около 3,0 до около 50 мг/кг веса тела/день.
Детальное описание изобретения
Данное изобретение раскрывает новые замещенные азетидинил- и пирролидинил-фенилоксазолидиноны вышеописанной структурной формулы I (и структурно представленных в формулах i-v). Соединения применяют в качестве противомикробных средств, эффективных против ряда патогенов человека и патогенов животных, в частности аэробных грам-положительных бактерий, включая множественно-резистентные стафилококки, энтерококки и стрептококки, а также анаэробные организмы, такие как бактероиды и clostridia виды, и кислото-устойчивые организмы, такие как Mycobacterium tuberculosis и другие микобактериальные виды.
R4 заместители предпочтительно оба представляют фтор и более предпочтительно фтор и водород.
R5 заместитель предпочтительно представляет водород, метил, дихлорметил, гидроксиметил или метокси. Более предпочтительно R5 представляет водород, метокси или метил. Наиболее предпочтительно, чтобы R5 представлял метил.
R6 заместитель предпочтительно является O, OCH2CH2O, NOH и NOCH3.
"Алкил" означает цепи углеродных атомов, имеющие указанное число атомов углерода, которые могут быть либо неразветвленными, либо разветвленными.
"Алкокси" означает группу с указанным числом атомов углерода, соединенных с кислородом, такую как метокси (-OCH3), этокси, бутокси и т.д. и ее изомерные формы.
"Ацилокси" означает группу с указанным числом атомов углерода, которые образуют органическую кислоту, где удалена OH группа, такую как ацетил, CH3CO-; бензоил, C6H5CO-.
"Циклоалкил" означает группу с указанным числом атомов углерода, образующих циклопропил, циклобутил, циклопентил, циклогексил и т.д. и их изомерные формы.
"Ph" означает фенил. "Карбонил" означает -C(=O)-часть. "Амино" означает NH2, "алкиламино" означает группу, где одна из позиций водорода замещена алкилом, и "диалкиламино" означает группу, где оба водорода замещены алкильной группой.
Термин "фармацевтически приемлемые соли" означает соли присоединения кислоты, которые можно получить любым из известных в данной области способом. К типичным солям присоединения кислоты относятся гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат, ацетат, пропионат, лактат, малат, сукцинат, тартрат, циклогексансульфаматы, метансульфонаты, этансульфонаты, бензолсульфонаты, толуолсульфонаты, фумараты и другие фармацевтически приемлемые противоионы для аминов.
Азетидинил-фенилоксазолидиноны.
Предпочтительной абсолютной конфигурацией при C-5 оксазолидинонового кольца соединений, заявляемых в данном изобретении, является конфигурация, представленная в структурах формулы i и ii. Эту абсолютную конфигурацию называют (S) по Cahn-Ingold-Prelog номенклатурной системе. Это тот (S)-энантиомер, который фармакологически активен как антибактериальный агент. Рацемическую смесь используют тем же самым путем и для той же самой цели, как и чистый (S)-энантиомер; разница состоит в том, что для получения такого же антибактериального эффекта необходимо использовать в два раза больше рацемического вещества. Для специалиста в данной области очевидно, что когда хиральный центр (R не Н) присутствует в азетидиновом фрагменте соединений структурной формулы i, ii, то возможны диастереоизомеры. Эти диастереоизомеры в рацемической и энантиомерно обогащенной формах, также охватываются объемом соединений формулы i, ii изобретения.
Предпочтительный способ получения оксазолидинонов формулы i, ii в энантиомерно чистой форме представлен Схемами I-VI. Схемы I-VI содержат общие структурные представления получения различных соединений данного изобретения, центром которых является циклическая структура ядра, где Q, является i, ii.
Как показано в Схеме I, 3-гидроксиазетидины структуры 1 могут быть депротонированы подходящим основанием, например гидридом натрия, в подходящем растворителе, например тетрагидрофуран (ТГФ), и затем алкилированы алкил галоидами, например метилиодидом, с получением азетидиновых эфиров 2 (R13 = алкил). При необходимости азетидиновые эфиры 2 (R13 = произвольно замещенный фенил) можно получить из азетидинола 1, используя известные способы (Taylor, C. R. , Jr. , ; Cale, A.D., Jr.; Stauffer, H.F., Jr. U.S. Patent 4 956 359, 1990). Бензгидрильную группу 1 и 2 удаляют гидрогенолизом в присутствии минеральной кислоты, например хлористоводородной кислоты, и в присутствии подходящего катализатора, например гидроксида палладия-на-угле, в подходящем растворителе, таком как метанол, с получением азетидинов 3 (R13 = H, алкил, фенил). Затем соединение структуры 3 подвергают взаимодействию с функционализированным нитробензолом 4 (X = галоген или трифторометансульфонат) в присутствии подходящей комбинации основание/растворитель, например двухосновный фосфат калия в диметилсульфоксиде или N,N-диизопропилэтиламин в ацетонитриле или ТГФ, и при соответствующей температуре, обычно от температуры окружающей среды до 70oC, получая аддукт 5. Если требуется, чтобы R1 соединения структурной формулы i или преобразованного промежуточного был гидрокси, то подходящую защитную группу, такую как трет-бутилдиметилсилильная часть, вводят путем взаимодействия 5 (R13 = Н) с трет-бутилдиметилсилил хлоридом в присутствии имидазола в соответствующем растворителе, таком как N,N-диметилформамид (ДМФ), при температуре окружающей среды, получая 5 (R13 = трет-бутилдиметилсилил). Для специалиста в данной области очевидно, что эта защитная группа является просто иллюстративной, и что можно применять другие альтернативные защитные группы, такие как группы, описанные Greene, T.W.; Wuts, P.G.M. "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, New York, 1991. Нитрогруппу 5 затем восстанавливают путем каталитического гидрирования в присутствии соответствующего катализатора, такого как 10% палладий/углерод или W-2 никель Ренея, и в соответствующем растворителе, например ТГФ/H2O. Когда используют эту последнюю систему растворителя, реакционную смесь сначала фильтруют, чтобы удалить катализатор, и фильтрат, содержащий промежуточный анилин, затем обрабатывают, например, бикарбонатом натрия и бензил или метил хлорформиатом, получая соответствующие бензил (R14 = CH2Ph) или метил (R14 = CH3) уретан производные 6. Уретаны 6 затем депротонируют подходящим основанием, таким как н-бутиллитий (н-BuLi), литий диизопропиламид (ЛДА, LDA) или литий бис(триметилсилил)амид, в соответствующем растворителе, таком как тетрагидрофуран (ТГФ), и при соответствующей температуре, такой как от -78oC до -40oC, получая литиевый интермедиат, который затем обрабатывают коммерчески доступным (-)-(R)-глицидил бутиратом. Нагревание до температуры окружающей среды затем непосредственно дает 5-(гидроксиметил)оксазолидиноны 7 в энантиомерно обогащенной форме.
Как показано в Схеме II, затем соединение 7 превращают в соответствующий мезилат 8 (R15 = метил) или арилсульфонат 8 (R15 = ArSO2, например п-толуолсульфонил) путем действия, например, метансульфонил хлорид/пиридина или метансульфонил хлорид/триэтиламин/дихлорметана или п-толуолсульфонил хлорид/пиридина. Полученное сульфонатное производное 8 затем подвергают взаимодействию с источником азида, таким как азид натрия или калия, в апротонном растворителе, таком как N,N-диметилформамид (ДМФ) или 1-метил-2- пирролидинон, произвольно в присутствии катализатора, такого как 18-краун-6, при температуре 50-90oC, получая азид 9. Затем азид восстанавливают гидрированием на катализаторе палладий-на-угле или платиновом катализаторе в соответствующем растворителе, таком как этилацетат или метанол, получая соответствующий амин 10. Альтернативно, азид 9 может быть восстановлен обработкой соединением трехвалентного фосфора, таким как трифенилфосфин, в соответствующем растворителе, таком как тетрагидрофуран, с последующим добавлением воды. Интермедиатный амин 10 можно также получить обработкой фталимидного производного 11 (полученным путем взаимодействия сульфоната 8 с фталимидом калия в подходящем растворителе, например ацетонитриле, при температуре кипения с обратным холодильником) метиламином в этанол/Н2O при температуре кипения с обратным холодильником. Альтернативно, амин 10 можно получить непосредственно из мезилата 8 путем аммонолиза водным гидроксидом аммония в системе растворителя, состоящей из H2О/изопропанол/ТГФ, в герметизированном реакционном сосуде, помещенном в 70-95oC масляную баню. Затем амин 10 ацилируют при помощи реакций, известных специалистам в данной области, получая оксазолидиноны структуры 12. Например, амин может быть подвергнут взаимодействию с хлорангидридом или ангидридом в основном растворителе, таком как пиридин, при температуре в диапазоне от -30 до 30oC, с получением ацилированного соединения 12 (R5 = произвольно замещенный алкил). Специалистам в данной области очевидно, что другие карбонильные группы, находящиеся внутри объема данного изобретения, могут быть легко введены в амин 10 при помощи стандартных техник ацилирования, например изложенными March, J. "Advanced Organic Chemestry", 3rd ed.; John Wiley & Sons: New York, 1985; p. 370-375, с получением дополнительных примеров 12. Если требуется, чтобы R13 был H, то введенную трет-бутилдиметилсилильную защитную группу выбранных примеров 12 удаляют, используя соответствующие условия, такие как отмечены Greene, T.W.; Wuts, P.G.M. "Protective Groups in Organic Synthesis" 2nd ed.; John Wiley & Sons, New York, 1991, в частности водную хлористоводородную кислоту в ацетонитриле при температуре окружающей среды, получая соответствующий спирт. Соединения структуры 12 (R13 = H, алкил, произвольно замещенный фенил) представляют примеры бактерицидных средств-азетидинзамещенных оксазолидинонов формулы I, которые являются предметом данного изобретения.
Азетидин-содержащие оксазолидиноны 12, сами по себе, примеры антибактериальных агентов структурной формулы I, кроме того, могут быть преобразованы в дополнительные соединения формулы i и также примеры структурной формулы ii, как показано в Схеме III. Например, 12 (R13 = Н) можно окислить в соответствующий азетидинон 13 путем взаимодействия его с каталитическим количеством тетра-н-пропиламмоний перрутената в присутствии N-метилморфолин N-оксида и молекулярных сит в ацетонитрил/дихлорметане. Соединение 13 является примером антибактериальных агентов структурной формулы ii. Кетоновая часть 13 поддается дальнейшей модификации. Реакция 13 с Lawesson's реагентом или альтернативными реагентами, описанными March, J. "Advanced Organic Chemistry", 4rd ed.; John Wiley & Sons: New York, 1992; p. 893-894, дает соответствующий тиокетон 14 (R6 = s).
Оксимы 14 (например, R6 = NOH и NOCH3) легко получают взаимодействием 13 с, например, гидрохлоридом гидроксиламина или гидрохлоридом метоксиламина, в присутствии соответствующего основания, такого как пиридин, и в соответствующем растворителе, например метаноле, при температуре окружающей среды. Различные производные гидразона (R6 = NNHR7 ) можно получить путем взаимодействия 13 с гидразинами, как описано Greene, T.W.; Wuts, P.G.M. "Protective Groups in Organic Synthesis", 2nd ed.; John Wiley & Sons, New York, 1991, p. 212-213 и March, J. "Advanced Organic Chemistry, 4rd ed.; John Wiley & Sons: New York, 1992; p. 904-905. Имины 14 (R6 = N-алкил или N-арил) синтезируют путем обработки 13 первичными аминами, как описано March, J. "Advanced Organic Chemestry", 4rd ed.; John Wiley & Sons: New York, 1992; p. 896-897. Олефиновые производные (14: R6 = CR11R12) получают взаимодействием 13 с различными олефинизирующими реагентами, такими как улиды фосфора и т.п., которые известны специалистам в данной области техники. Иллюстративные примеры описаны March, J. "Advanced Organic Chemistry", 4rd ed.; John Wiley & Sons: New York, 1992; p. 956-963. Для случая, когда R6 является CF2, следует включить обработку кетона 13 с (NaO2CCF2Cl) натрий дифторхлорацетатом и трифенилфосфатом, как описано в Tetrahedron Letters 1964, р. 1461. Полученную олефиновую связь можно восстановить каталитическим гидрированием или другими способами, известными специалистам в данной области, чтобы получить примеры структурной формулы i. Другие соединения структуры 14, например, циклические и ациклические кетали и дитио кетали, можно получить взаимодействием соединения 13 с диолами, дитиолами, спиртами или тиолами при условиях, например, описанных Greene, T.W.; Wuts, P.G.M. "Protective Groups in Organic Synthesis", 2nd.; John Wiley & Sons, New York, 1991, p. 177-201. Соединение 12 (R13 = H) можно также превратить в различные производные 15 (R16 = произвольно замещенный ацил, алкоксикарбонил, карбоксамид и т.д.) путем обработки 12 различными карбонильными производными, такими как ангидриды, алкил хлорформиаты, изоцианаты и т.п., в присутствии соответствующих оснований, и в подходящих растворителях, известных специалистам в данной области. Соединения 14 и 15 представляют примеры оксазолидинонов - бактерицидных средств структурной формулы i и ii.
Схемы IV-VI описывают в общих чертах техники получения примеров структурной формулы i, в которых R1 является группой другой, чем OR7. Как показано в Схеме IV, мезилат 16, полученный взаимодействием азетидинола - исходного вещества 1 с метансульфонил хлоридом в присутствии триэтиламина и дихлорметана, подвергают взаимодействию с рядом нуклеофилов. Например, обработка 16 аммиаком, первичными аминами или вторичными аминами, приводит к 3-аминоазетидинам структуры 17. Аналогично, обработка 16 тиолатами или цианидом дает аддукты 18 и 19, соответственно. Соединение 19 можно восстановить литийалюминийгидридом и т.п. в соответствующий 3- (аминометил)азетидин 20. Соединение 19, кроме того, можно превратить в карбоксилатное производное 21. Специалисты в данной области могут, кроме того, трансформировать карбоксилатную часть 21 в соответствующий карбоксамид или различные ацильные группы, которые после дополнительного преобразования составляют дополнительные примеры антибактериальных агентов структурной формулы i. Соединения 17 и 20 можно дополнительно N-алкилировать способами, известными специалистам в данной области, включая обработку 17 и 20 алкилгалогенидами или тозилатами в присутствии подходящего основания. Альтернативно, выбранные алкильные группы можно ввести по азоту 17 и 20 путем восстановительного алкилирования, как описано March, j. "Advanced Organic Chemistry", 4rd ed; John Wiley & Sons: New York, 1992; p. 898-900. В случаях амино интермедиатов 17 и 20, когда присутствует свободная NH, необходимо защитить эти части, чтобы сделать возможным протекание обычным путем некоторых последовательных реакций. Аминогруппа может быть превращена в соответствующий т-бутил карбамат (BOC), бензил карбамат (Cbz), трифторацетамид, или производные фталимида и т.п., в том виде, как требуется, используя способы, описанные Greene, T.W.; Wuts, P.G.M. "Protective Groups in Organic Synthesis, 2nd ed.; John Wiley & Sons, New York, 1991 p. 309-403. Бензгидрильную защитную группу 17-21 затем удаляют путем гидрогенолиза в присутствии подходящего катализатора, например гидроксида палладия-на-угле, и в присутствии минеральной кислоты, например хлористоводородной кислоты, с получением промежуточных азетидинов 22 (R1 не OR7).
Схема V описывает в общих чертах преобразование промежуточных азетидинов 22 в примеры структурной формулы i (R1 не OR7). Химия, в основном, идентична описанной в Схеме I. Соединение 22 сначала превращают в производное нитробензола 23. Восстановление и превращение в карбоматы структуры 24 осуществляют, как описано ранее. Как описано в Схеме I, промежуточное соединение 24 преобразуют в оптически активный 5-(гидроксиметил)оксазолидинон 25. Применение способов, описанных в общих чертах в Схеме II, затем позволяет превратить 25 в соединение структуры 26. Удаление любых введенных защитных групп, например BOC групп, на соединениях, где R1 есть амино, достигают способами, известными специалистам в данной области, например, описанными Greene, T.W., Wuts, P.G.M. "Protective Groups in Organic Synthesis", 2nd ed.; John Wiley & Sons, New York, 1991. Полученная свободная аминогруппа затем может быть N-алкилирована или N-ацилирована, при необходимости, с получением дополнительных соединений структуры 26, которые являются примерами оксазолидинонов антибактериальных агентов структурной формулы i.
Схема VI изображает вариацию химии, описанной в общих чертах в Схеме IV, где промежуточный 12-гидроксиазетидинилфенилоксазолидинон (R13 = ОН) превращают в соответствующий мезилат 27 путем действия метансульфонил хлорида в присутствии соответствующего основания, такого как триэтиламин, и в соответствующем растворителе, например дихлорметане. Те же самые нуклеофильные замещения, описанные в Схеме IV для мезилата 16, могут быть проведены на более функционизированном мезилате 27 с получением соединений 28-30. Применяя способы, аналогичные способам, описанным в Схеме IV, 30 можно превратить в соединения 31 и 32. Соединения 28-32 представляют примеры оксазолидинонов - антибактериальных агентов структурной формулы i, которые являются предметом данного изобретения. Для специалистов в данной области очевидно, что соединения 28-32 являются просто иллюстративными примерами, и эти соединения, сами по себе, поддаются дальнейшей химической модификации с получением дополнительных примеров структурной формулы i. 2-азетидиноны структуры 33 обрабатывают соответствующим основанием, например гидридом натрия, в соответствующем растворителе, например ТГФ, получая депротонированное промежуточное соединение, которое затем обрабатывают функционизированным производным нитробензола 4 (X = галоген), получая аддукт 34. Специалисту в данной области очевидно, что соответствующие защитные группы, например, описанные Greene, T.W.; Wuts, P.G.M. "Protective Groups in Organic Synthesis", 2nd ed. ; John Willey & Sons, New York, 1991, требуются для выбранных R18 и R19 заместителей соединения 33. Интермедиат 34 может быть превращен при помощи стадий, описанных в общих чертах в Схемах I и II в соединения структуры 35, сами по себе примеры структурной формулы iii, которые являются предметом данного изобретения. Альтернативно, азетидинил-замещенное промежуточное 36, полученное по технологии, описанной в Схемах I и II, окисляют в соответствующий азетидинон 35 (R17 = O) при помощи, например, каталитического количества тетроксида рутения в присутствии подходящего окислителя, такого как метапериодат натрия, и в подходящей системе растворителя, например этилацетат/вода. Чтобы получить примеры соединения 35, где R17 есть S, выбранные азетидиноновые интермедиаты подвергают взаимодействию с Lawesson's реагентом или т.п.
Примерами азетидинил-фенилоксазолидинонов, которые могут быть получены как часть данного изобретения, являются следующие:
1. (S)-N-[[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид
2. (S)-N-[[3-[3-фтор-4-(3-гидрокси-1-азетидинил)фенил] -2-оксо- 5-оксазолидинил]метил]ацетамид
3. (S)-N-[[3-[3-фтор-4-[3-[N-(2-фторэтил)-N-метиламино] -1-азетидинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
4. (S)-N-[[3-[3-фтор-4-(3-оксо-1-азетидинил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид
5. (S)-N-[[3-[3-фтор-4-[3-(метоксиимино)-1-азетидинил]-фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
6. (S)-N-[[3-[3-фтор-4-(3-метокси-3-метил-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
7. (S)-N-[[3-[3-фтор-4-(3-гидрокси-2-метил-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
8. (S)-N-[[3-[3-фтор-4-(2-оксо-1-азетидинил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид
9. (S)-N-[[3-[3-фтор-4-(3-гидрокси-2-оксо-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
10. (S)-N-[[3-[3-фтор-4-(3-метокси-2-оксо-1-азетидинил)фенил]-2- оксо-5-оксазолидинил]метил]ацетамид
11. N-[[3-[3-фтор-4-(3-метил-2-оксо-1-азетидинил)фенил]-2-оксо-(5S)- оксазолидинил]метил]ацетамид
12. (S)-N-[[3-[3-фтор-4-(3,3-диметил-2-оксо-1-азетидинил)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид
13. (S)-N-[[3-[3-фтор-4-[3-(гидроксиимино)-1-азетидинил] фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид
14. N-[[3-[3-фтор-4-[(2R)-метил-3-оксо-1-азетидинил] фенил] - 2-оксо-(5S)-оксазолидинил]метил]ацетамид
15. N-[[3-[3-фтор-4-[(2S)-метил-3-оксо-1-азетидинил] фенил] - 2-оксо-(5S)-оксазолидинил]метил]ацетамид
16. N-[[3-[3-фтор-4-[3-(метоксиимино)-(2R)-метил-1-азетидинил] фенил]-2-оксо-(5S)-оксазолидинил]метил]ацетамид
17. N-[[3-[3-фтор-4-[3-(метоксиимино)-(2S)-метил-1-азетидинил] фенил] - 2-оксо-(5S)-оксазолидинил]метил]ацетамид
18. (S)-N-[[3-[3-фтор-4-[3-(дифторметилен)-1-азетидинил] фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид
19. (S)-N-[[3-[3-фтор-4-[3-(метоксиметилен)-1-азетидинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
20. (S)-N-[[3-[3-фтор-4-[3-(гидроксиацетил)-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
21. (S)-N-[[3-[3-фтор-4-[3-(метоксиамино)-1-азетидинил] фенил] 2-оксо-5-оксазолидинил]метил]ацетамид
22. N-[[3-[3-фтор-4-[2,4-диметил-3-оксо-1-азетидинил] фенил]-2-оксо-(5S)-оксазолидинил]метил]ацетамид
23. N-[[3-[3-фтор-4-[2,4-диметил-3-(метоксиимино)-1-азетидинил] фенил] -2-оксо-(5S)-оксозолидинил]метил]ацетамид
24. N-[[3-[3-фтор-4-[2,4-диметил-3-метокси-1-азетидинил] фенил] -2-оксо-(5S)-оксазолидинил]метил]ацетамид
25. (S)-N-[[3-[3-фтор-4-[(3S)-метокси-(2R)-метил-1-азетидинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
26. (S)-N-[[3-[3-фтор-4-[(3R)-метокси-(2S)-метил-1-азетидинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
27. (S)-N-[[3-[3-фтор-4-[(3S)-гидрокси-(2R)-метил-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
28. (S)-N-[[3-[3-фтор-4-[(3R)-гидрокси-(2S)-метил-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
29. (S)-N-[[3-[3-фтор-4-(3-фтор-1-азетидинил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид
30. (S)-N-[[3-[3-фтор-4-[3-фтор-(2R)-метил-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
31. (S)-N-[[3-[3-фтор-4-[3-фтор-(2S)-метил-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
32. (S)-N-[[3-[3-фтор-4-[3-[(гидроксиацетил)амино]-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
33. (S)-N-[[3-[3-фтор-4-[3-[(метансульфонил)амино]-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
34. (S)-N-[[3-фтор-4-[3-[(формил)амино]-1-азетидинил]фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
35. (S)-N-[[3-[3-фтор-4-[3-[(ацетил)амино] -1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
36. (S)-N-[[3-[3-фтор-4-[3-[(метоксикарбонил)амино]-1-азетидинил] фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
37. (S)-N-[[3-[3-фтор-4-[3-[(бензилоксикарбонил)амино] -1-азетидинил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Пирролидинил-фенилоксазолидиноны.
Данное изобретение раскрывает новые замещенные пирролидинил фенилоксазолидиноны согласно вышеупомянутым формулам iv и v. Соединения представляют полезные антимикробные средства, эффективные против ряда патогенов человека и патогенов животных, в частности аэробных грам-положительных бактерий, включая устойчивые к многим антибиотикам стафилококки и стрептококки, а также анаэробные организмы, такие как бактероиды и clostridia виды, и кислотостойкие организмы, такие как Mycobacterium tuberculosis и другие микобактериальные виды.
Наиболее предпочтительные соединения в этой серии желательно получать в виде оптически чистых энантиомеров, имеющих (S)-конфигурацию, согласно Cahn-Ingold-Prelog системе обозначения, при C5 кольца оксазолидинона. Оптически чистое вещество обычно получают путем одного из ряда асимметрических синтезов или путем выделения из рацемической модификации селективной кристаллизацией соли из, например, рацемической модификации интермедиатного амина 11 (описанного в Схеме IX ) с соответствующей оптически активной кислотой, такой как дибензоил тартрат или 10-камфорсульфокислота, с последующей обработкой основанием, получая оптически активный амин. Хотя (S)-энантиомер этой серии соединений предпочтителен, поскольку он фармакологически активен как бактерицидное средство, рацемическую модификацию также используют таким же образом, как и чистый (S)-энантиомер; причем различие состоит в том, что рацемического вещества требуется в два раза больше для достижения того же самого бактерицидного действия. Кроме того, специалисту в данной области очевидно, что когда присутствует хиральный центр на пирролидиновой части соединений структурной формулы iv и v возможны диастереоизомеры. Эти диастереизомеры, либо в рацемической, либо в конфигурационно обогащенной формах, также входят в объем соединений формул iv и v данного изобретения.
Предпочтительный способ получения пирролидинил фенилоксазолидинонов формулы iv и v в энантиомерно чистой форме представлен Схемами VIII-XVI. Схемы VIII-XVI содержат общие структурные представления для получения различных соединений изобретения, центром которых является циклическая структура ядра, где Q есть iv или v.
Как показано в Схеме VIII, производные структуры 1, либо коммерчески доступные, либо полученные модификацией опубликованных способов (US 4 753 953), могут быть защищены диолом, таким как этиленгликоль, в условиях кислотного катализа с азеотропным удалением воды, с получением ацеталя 2. N-бензильную группу 2 затем удаляют гидрогенолизом в присутствии катализатора благородного металла, такого как палладий-на-угле или гидроксид платины-на-угле, в подходящем растворителе, получая пирролидиновое производное 3. Пирролидиновое производное 3 может быть обработано производным нитробензола 4 (Y = галоген или трифторметансульфонат) в подходящей комбинации основания и растворителя, например двуосновный фосфат калия в ДМСО (DMSO), и соответствующей температуре, обычно от температуры окружающей среды до 90oC, с получением аддукта 5. Нитрогруппу 5 затем восстанавливают каталитическим гидрированием в присутствии катализаторов, таких как палладий-на-угле или W-2 никель Ренея, в соответствующем растворителе, таком как этилацетат или тетрагидрофуран (ТГФ), получая производное анилина 6. Когда используют ТГФ в качестве растворителя для этого восстановления, то нет необходимости удалять катализатор путем фильтрации или выделять анилиновое производное 6, а можно просто очистить реакционный сосуд инертным газом, таким как азот, добавить насыщенный водный раствор бикарбоната натрия и обработать полученную охлажденную смесь либо бензил, либо метил хлорформиатом, получая соответствующие бензил (R14 = CH2Ph) или метил карбаматные (R14 = CH3) производные 7. Каждый из карбаматных производных 7 может быть депротонирован литиевым основанием, таким как н-бутиллитий, литий диизопропил амид (ЛДА, LDA ), или литий бис (триметилсилил)амид (LHMDS ), в соответствующем растворителе, таком как ТГФ, N,N-диметилформамид (ДМФ), или их смеси, при соответствующей температуре, такой как от -78oC до -40oC, получая литиевое промежуточное соединение, которое непосредственно обрабатывают коммерчески доступным (R)-(-)-глицидил бутиратом. Нагревание этой реакционной смеси до температуры окружающей среды затем дает (гидроксиметил)оксазолидиноны 8 в высоко энантиомерно обогащенной форме.
Как показано в Схеме IX, соединение 8 можно превратить в соответствующий мезилат (R15 = CH3) или тозилат (R15 = п-CH3C6H4) путем обработки метансульфонил хлоридом в присутствии триэтиламина или пиридина или толуолсульфонил хлоридом в присутствии пиридина.
Полученное сульфонатное производное 9 может быть затем обработано азидом щелочного металла, таким как азид натрия или калия, в апротонном диполярном растворителе, таком как ДМФ или N-метилпирролидинон (NMП, NMP), с произвольным катализатором, таким как 18-краун-6, при температуре в диапазоне 50-90oC, с получением азида 10. Азид 10 может быть восстановлен в соответствующий амин 11 гидрированием в присутствии палладиевого, платинового или никелевого катализатора, в соответствующем растворителе, таком как этилацетат, ТГФ или метанол. Альтернативно, азид 10 может быть восстановлен обработкой трифенилфосфином или другими соединениями трехвалентного фосфора в растворителе, таком как ТГФ, с последующим добавлением воды. Амин 11 можно получить обработкой сульфоната 9 фталимидом калия в ДМФ при 40-90oC или кипящем ацетонитриле с получением фталимида 12, который затем депротонируют обработкой, например, водным метиламином, в кипящем этаноле, получая 11. Более прямой путь получения амина 11 заключается в обработке сульфоната 9 водным раствором аммиака в системе растворителя изопропиловый спирт-ТГФ в герметизированной трубке, нагретой при 75-105oC на масляной бане. Амин 11 затем ацилируют при помощи реакций, известных специалистам в данной области, получая (ациламинометил)оксазолидиноны структуры 13. Например, амин 11 может быть обработан хлорангидридом кислоты или ангидридом в присутствии основания, такого как пиридин или триэтиламин, при температуре в диапазоне -40 - 40oC, с получением ацильного производного 13 (R5 = произвольно замещенный алкил). Можно видеть, что в аналогичных условиях реакции легко могут быть получены другие ацильные производные, такие как карбаматы. Наконец, обработкой 13 водной кислотой, такой как п-толуолсульфокислота в водном ацетоне, можно гидролизовать ацетальную функциональность, получая соответствующее карбонильное производное 14, которое представляет пример пирролидинон-замещенных бактерицидных средств формулы iv. Для специалистов в данной области очевидно, что из 14 можно получить другие варианты воплощения соединения формулы iv. Реакция 14 с Lawesson's реагентом или другими альтернативными реагентами, такими как сульфид водорода, обычно дает производное тиокетона 15 (R6 = S). Оксимы (R6 = NHOH и NHOCH3) можно получить из 14 обработкой либо гидрохлоридом гидроксиламина, либо гидрохлоридом метоксиамина в присутствии основания, такого как пиридин или ацетат натрия, в растворителе, таком как метанол. Производные гидразона (R6 = NNHR12) можно получить обработкой 14 производными гидразина. Аналогично, имины (R6 = NR12) можно получить обработкой 14 первичным амином. Олефиновые производные (R6 = CR11R12) можно получить обработкой 14 различными реагентами олефинирования, такими как улиды фосфора (Witting реагенты), фосфонатные сложные эфиры (Horner-Emmons Реагенты) или другими реагентами, известными специалистам в данной области. Легко видеть, что восстановление олефиновой связи каталитическим гидрированием или другими способами может привести к получению примеров структурной формулы V.
Схемы X-XII описывают в общих чертах получение примеров структурной формулы v, где R1 = OR7, R3 = H и n = 0. Как показано в Схеме X, промежуточное 1, описанное в Схеме VIII, восстанавливают в спирт 16 путем обработки любым из ряда стандартных гидридных восстанавливающих агентов, таких как борогидрид натрия, литийалюминийгидрид или т.п. Бензильную защитную группу 16 затем в дальнейшем можно удалить гидрогенолизом, используя катализатор, такой как гидроксид палладия-на-угле, или 10% палладий-на-угле, получая аминоспирт 17. Здесь следует отметить, что некоторые примеры 17 могут быть коммерчески доступными, но следует предусмотреть возможность de novo синтеза 17 для того, чтобы включить более сложные примеры, представляющие большой интерес с точки зрения их антибактериального действия. Аминоспирт 17 затем может быть подвергнут взаимодействию с производным нитробензола, таким как 4 (Y = галоген или трифторметансульфонат), в присутствии соответствующего основания, такого как двуосновный фосфат калия, в подходящем растворителе, таком как диметилсульфоксид, при температурах в диапазоне 40-90oC, чтобы получить 18. Для специалистов в данной области очевидно, что могут потребоваться последовательные трансформации 18 для того, чтобы защитить гидроксильную группу. Это можно осуществить, например, путем получения трет-бутилдиметилсилилового эфира 19 (R = Si(CH3)2t-Bu) обработкой 18 трет-бутилдиметилхлорсиланом в присутствии основания, такого как имидазол или диизопропилэтиламин, произвольно в присутствии 4-диметиламинопиридина в качестве катализатора, в подходящем растворителе, таком как ДМФ, ТГФ или дихлорметан. Нитрогруппу 19 можно восстановить гидрированием в присутствии катализатора, такого как 10% палладий-на-угле или W-2 никель Ренея, в растворителе, таком как ТГФ или этилацетат, получая анилиновое производное 20. Анилиновое производное 20 не выделяют рутинным способом, а непосредственно подвергают обработке насыщенным бикарбонатом натрия и алкил хлорформиатным производным, таким как бензил хлорформиат или метил хлорформиат, чтобы получить соответствующее бензил (R14 = CH2Ph) или метил (R14 = CH3) карбаматное производное 21. Карбаматное производное 21 затем можно депротонировать при помощи основания, такого как н-бутиллитий или литий диизопропиламид (LDA) или литий бис(триметилсилил) амид (LHMDS), в растворителе, таком как ТГФ или ДМФ или их смеси, при температуре в диапазоне от -78 до -40oC, получая литиевое производное, которое обрабатывают коммерчески доступным (R)-(-)-глицидил бутиратом. Полученную смесь затем нагревают до температуры окружающей среды, непосредственно получая (гидроксиметил)оксазолидиноны 22.
Специалистам в данной области понятно, что промежуточный оксазолидинон 22 может быть использован для ряда способов получения различных вариантов воплощения соединений структурной формулы iv. Как показано в Схеме XI, оксазолидинон 22 может быть превращен в (ациламинометил)оксазолидинон 23 при помощи последовательности реакций, идентичной той, которую используют для превращения соединения 8 в соединение 13 согласно Схеме IX. Защитную группу 23 (R = Si(CH3)2т-Bu) можно удалить стандартными способами, известными специалистам в данной области, таким как обработка тетрабутиламмоний фторидом в ТГФ с получением спирта 24, который является примером соединения структурной формулы v. Соединение 24 может быть превращено в ряд производных 23 (R = R7 = произвольно замещенный ацил, алкоксикарбонил, карбоксамид, и т.д.) путем обработки различными карбонильными производными, такими как ангидриды, хлорангидриды уксусной кислоты, алкил и арил хлорформиаты, изоцианаты, и т.п., используя соответствующие основания и катализаторы в подходящих растворителях, известных специалисту в данной области. Так, соединения 23 и 24 представляют примеры оксазолидинонов - антибактериальных агентов структурной формулы v.
Другое использование интермедиатного оксазолидинона 22 иллюстрируется Схемой XII. Как показано, 22 (R = Si(CH3)2т-Bu) можно защитить, например, обработкой третбутилдифенилхлорсиланом и соответствующим основанием, таким как диизопропилэтиламин, и катализатором, таким как 4-диметиламинопиридин, в соответствующем растворителе, таком как дихлорметан, ТГФ, или т.п., получая бис-защищенное производное 25 (R = Si(CH3)2т-Bu, R1 = SiPh2т-Bu). Бис-защищенное производное 25 селективно депротонируют, чтобы удалить трет-бутилдиметилсилил эфирную защитную группу, путем обработки уксусной кислотой, используя сорастворитель воды и ТГФ в целом ряде соотношений при температурах в диапазоне 50-100oC, получая спирт 26. Спирт 26 можно проалкилировать по незащищенной гидроксильной группе обработкой подходящим основанием, таким как гидрид натрия, в диполярном апротонном растворителе, таком как ТГФ, ДМФ или т. п. , в присутствии алкил галогенида, такого как метил иодид, или его замещенных производных, чтобы получить силиловый эфир 25 (R = R7 алкил с неразветвленной или разветвленной цепью, R1 = SiPh2т-Bu). Содержащую кремний защитную группу 25 затем можно удалить обработкой фторидом тетрабутиламмония в растворителе, таком как ТГФ, получая спирт 27 (R = алкил с неразветвленной или разветвленной цепью). Спирт 27 затем может быть превращен в (ациламинометил)-оксазолидиноновое производное 28 (R7 = алкил с неразветвленной или разветвленной цепью) при помощи последовательности реакций, идентичной той, которую используют для превращения 8 в 13 по Схеме IX. (Ациламинометил)оксазолидиноновое производное 27 является примером оксазолидинона - антибактериального агента структурной формулы v.
Схемы XIII-XVI описывают в общих чертах получение примеров структурной формулы v, где R1 является замещенной амино частью. Схема XVI описывает в общих чертах случай, где R1 = NHR и n = 0. Как показано, аминопирролидин 29 либо доступный коммерчески, либо полученный способами, известными в данной области, произвольно защищенный как либо трифторацетамид (R = COCF3) или трет-бутоксикарбонильное производное (R=CO2 т-Bu), гидрируют в присутствии подходящего катализатора, такого как гидроксид палладия-на-угле, в соответствующем растворителе, таком как метанол, чтобы удалить бензильную защитную группу, получая пирролидин 30. Пирролидин 30 может быть подвергнут взаимодействию с нитроароматическим соединением, таким как 4, в присутствии подходящего основания, такого как двуосновный фосфат калия, в соответствующем растворителе, таком как диметил сульфоксид (ДМСО) или ДМФ, чтобы получить продукт замещения 31. Нитро группу 31 восстанавливают гидрированием в присутствии 10% палладия-на-угле или W-2 никеля Ренея в подходящем растворителе, таком как ТГФ или этилацетат, получая производное анилина 32.
Анилиновое производное 32 не выделяют рутинным способом, а обычно обрабатывают непосредственно насыщенным водным раствором бикарбоната натрия и алкил хлорформиатным производным, таким как бензил хлорформиат или метил хлорформиат, получая соответствующий бензил (R14 = CH2Ph) или метил (R14 = CH3) карбаматное производное 33. В случае, когда 33 защищен ацил производным, необходимо удалить защитную группу соответствующим способом, известным специалистам в данной области, чтобы получить производное первичного амина 34. Амин 34 затем обрабатывают производным диалкил мочевины, таким как N, N-диметил мочевина или N,N-дибензил мочевина, в присутствии формальдегида, в условиях, открытых Knapp, et al.2, чтобы получить либо диметил (R = CH3), либо дибензил (R = CH2Ph) триазон производное 35. Триазон 35 затем можно депротонировать с помощью основания, такого как н-бутиллитий, литий диизопропиламин (LDA) или литий бис(триметилсилил)амид в растворителе, таком как ДМФ или смеси ДМФ и ТГФ, при температурах в диапазоне от -78oC до -40oC, чтобы получить литиевое производное, которое непосредственно обрабатывают коммерчески доступным (R)-(-)-глицидил бутиратом. Полученную смесь затем нагревают до температуры окружающей среды, получая (гидроксиметил)оксазолидинон 36.
Схема XIV описывает в общих чертах превращение триазон производного 36 в соединения формулы v. Как показано, триазон производное 36 может быть превращено в (ациламинометил)оксазолидинон 37 способом, идентичным тем, которые используют для превращения соединения 8 в 13 в Схеме IX. Триазон группу 37 можно удалить гидролизом водной хлористо-водородной кислотой или обработкой насыщенным раствором хлорида аммония, получая 38. Соединение 38 может быть превращено в ряд карбонильных производных 39 (R = произвольно замещенный алкил, алкокси, алкиламино, и т. д.) обработкой различными карбонильными производными, такими как хлорангидриды карбоновой кислоты, ангидриды, алкил и арил хлорформиаты, изоцианаты и т.п., используя соответствующие основания и катализаторы, в соответствующих растворителях, известных специалисту в данной области. Кроме того, соединение 38 можно превратить в ряд алкил производных 40 (R = произвольно замещенный неразветвленный или разветвленный цепной алкил) путем обработки соответствующим альдегидом или кетоном в присутствии газа-водорода и катализатора, такого как 10% палладий-на-угле, или реагента, такого как цианоборгидрид натрия, в протонном растворителе, таком как метанол. Альтернативно, 38 обычно обрабатывают произвольно замещенным алкил галогенидом в присутствии основания, такого как карбонат натрия, в подходящем растворителе, таком как ТГФ или ацетонитрил, произвольно в присутствии воды. Таким образом, полученные соединения 39 и 40 представляют примеры соединений формулы v, где n = 0.
Схема XV описывает получение примеров соединений структурной формулы v, где R3 = H, R1 = NR8R9, а n = 1. Как показано, пиррролидин производное 41, доступное известными способами 1a, 3 произвольно защищенное как либо трифторацетамид (R = COF3) или трет-бутоксикарбонил производное (R = CO2 т-Bu), дебензилируют путем гидрогенолиза в присутствии катализатора, такого как гидроксид палладия-на-угле или 10% палладий-на-угле, в подходящем растворителе, таком как метанол, получая 42. Пирролидин 42 затем может быть подвергнут взаимодействию с нитробензольным производным, таким как 4, в присутствии основания, такого как двуосновный фосфат калия, в растворителе, таком как диметилсульфоксид (ДМСО), чтобы получить продукт 43 с ароматическим замещением. Арил пирролидин 43 может быть восстановлен гидрированием в присутствии катализатора, такого как 10% палладий-на-угле или W-2 никель Ренея, в растворителе, таком как ТГФ или этилацетат, с получением анилин производного 44. Анилин производное 44 не выделяют рутинным способом, а обычно обрабатывают непосредственно насыщенным раствором бикарбоната натрия и алкил хлорформиатом, таким как бензил или метил хлорформиатом, чтобы получить соответствующий бензил (R14 = CH2Ph) или метил (R14 = CH3) карбамат производное 45. В том случае, когда 45 защищают как ацил производное, необходимо удалить защитную группу путем соответствующего способа, известного специалистам в данной области, чтобы получить производное 46 первичного амина. Амин 46 затем обрабатывают производным диалкил мочевины, таким как N,N'-диметил мочевина или N,N'-бензил мочевина, в присутствии формальдегида в условиях, открытых Knapp et al.2, получая либо диметил (R = CH3), либо дибензил (R = CH2Ph) триазон производное 47. Триазон 47 может быть депротонирован при помощи основания, такого как н-бутиллитий, литий диизопропиламид (LDA), или литий бис(триметилсилил)амид, в растворителе, таком как ДМФ или смеси ДМФ и ТГФ, при температурах в диапазоне от -78oC до -40oC, чтобы получить литиевое производное, которое непосредственно обрабатывают коммерчески доступным (R)-(-)глицидил бутиратом. Полученную смесь затем нагревают до температуры окружающей среды, получая (гидроксиметил) оксазолидинон 48.
Схема XVI описывает превращение триазон производного 48 в соединения структурной формулы v. Как показано, триазон производное 48 может быть превращено в (ациламинометил)оксазолидинон 49 способом, идентичным тем, которые используют для превращения соединения 8 в 13 в Схеме IX. Триазон группу 49 можно удалить гидролизом водным раствором хлористоводородной кислоты или обработкой горячим насыщенным раствором хлорида аммония, получая амин 50. Амин 50 затем может быть превращен в ряд карбонильных производных 51 (R = произвольно замещенный алкил, алкокси, алкиламино и т.д.) путем обработки различными карбонильными производными, такими как хлорангидриды уксусной кислоты, ангидриды, алкил и арил хлорформиаты, изоцианаты и т.п., используя соответствующие основания и катализаторы, в подходящих растворителях, известных специалисту в данной области. Кроме того, амин 50 может быть превращен в ряд алкил производных 52 (R = произвольно замещенный неразветвленный или разветвленный цепной алкил) путем обработки 50 соответствующим альдегидом или кетоном в присутствии газа-водорода и катализатора, такого как 10% палладий-на-угле, или реагента, такого как цианоборогидрид натрия, в протонном растворителе, таком как метанол. Альтернативно, 50 обычно обрабатывают произвольно замещенным алкил галогенидом в присутствии основания, такого как карбонат натрия, в подходящем растворителе, таком как ТГФ или ацетонитрил, произвольно в присутствии воды, получая 52. Таким образом, полученные соединения 51 и 52 представляют примеры оксазолидинонов-бактерицидных средств структурной формулы v, где n = 1.
Для специалистов в данной области очевидно, что описанные способы синтеза являются просто иллюстративными по природе, и что известны альтернативные способы синтеза, например, некоторые из тех, на которые здесь ссылаются.
Примерами пирролидинил-фенилоксазолидинонов, которые могут быть получены как часть данного изобретения, являются следующие:
1. (S)-N-[[3-[3-фтор-4-(3-оксопирролидинил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид
2. (S)-N-[[3-[3-фтор-4-(3-оксо-4-метилпирролидинил)-фенил]-2- оксо-5-оксазолидинил]метил]ацетамид
3. (S)-N-[[3-[3-фтор-4-(2,4-диметил-3-оксопирролидинил)фенил]-2- оксо-5-оксазолидинил]метил]ацетамид
4. (S)-N-[[3-[3-фтор-4-(2,2-диметил-3-оксопирролидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
5. (S)-N-[[3-[3-фтор-4-(4,4-диметил-3-оксопирролидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
6. (S)-N-[[3-[3-фтор-4-(2,4-диметил-3-оксопирролидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
7. (S)-N-[[3-[3-фтор-4-(3-изонитрозопирролидинил)фенил] -2-оксо- 5-оксазолидинил]метил]ацетамид
8. (S)-N-[[3-[3-фтор-4-(О-метил-3-изонитрозопирролидинил)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид
9. (S)-N-[[3-[3-фтор-4-(3-гидроксипирролидинил)-фенил] -2-оксо-5- оксазолидинил]метил]ацетамид
10. (S)-N-[(3-[3-фтор-4-(цис-3-гидрокси-4-метилпирролидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
11. (S)-N-[[3-[3-фтор-4-(транс-3-гидрокси-4-метилпирролидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
12. (S)-N-[[3-[3-фтор-4-(3-метоксипирролидинил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид
13. (S)-N-[[3-[3-фтор-4-(цис-3,4-дигидроксипирролидинил)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид
14. (S)-N-[[3-[3-фтор-4-(транс-3,4-дигидроксипирролидинил)фенил]- 2-оксо-5-оксазолидинил]метил]ацетамид
15. (S)-N-[[3-[3-фтор-4-(3-(гидроксиацетиламино)пирролидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
16. (S)-N-[[3-[3-фтор-4-[3-(фенилметоксиацетиламино) пирролидинил]фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
17. (S)-N-[[3-[3-фтор-4-(3-(метоксиацетиламино)пирролидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
18. (S)-N-[[3-[3-фтор-4-(3-метоксикарбониламино)пирролидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
19. (S)-N-[[3-[3-фтор-4-(3-этоксикарбониламино)-пирролидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
20. (S)-N-[[3-[3-фтор-4-(цис-3-(фенилметоксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
21. (S)-N-[[3 [3-фтор-4-(цис-3-(гидроксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
22. (S)-N-[[3-[3-фтор-4-(цис-3-(метоксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
23. (S)-N-[[3-[3-фтор-4-(цис-3-(метоксикарбониламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
24. (S)-N-[[3-[3-фтор-4-(цис-3-(этоксикарбониламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
25. (S)-N-[[3-[3-фтор-4-(транс-3-(фенилметоксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
26. (S)-N-[[3-[3-фтор-4-(транс-3-(гидроксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
27. (S)-N-[[3-[3-фтор-4-(транс-3-(метоксиацетиламино)-4- метилпирролидинил)-фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
28. (S)-N-[[3-[3-фтор-4-(транс-3-(метоксикарбониламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
29. (S)-N-[[3-[3-фтор-4-(транс-3-(этоксикарбониламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
30. (S)-N-[[3-[3-фтор-4-(3-(фенилметоксиацетиламино)- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
31. (S)-N-[[3-[3-фтор-4-(3-(гидроксиацетиламино) метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
32. (S)-N-[[3-[3-фтор-4-(3-(метоксиацетиламино) метилпирролидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
33. (S)-N-[[3-[3-фтор-4-(3-(метоксикарбониламино)метил- пирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
34. (S)-N-[[3-[3-фтор-4-(3-(этоксикарбониламино) метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
35. (S)-N-[[3-[3-фтор-4-(цис-3-(фенилметоксиацетиламино) метил-4-метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил] ацетамид
36. (S)-N-[[3-[3-фтор-4-(цис-3-(гидроксиацетиламино) метил-4-метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил] ацетамид
37. (S)-N-[[3-[3-фтор-4-(цис-3-(метоксиацетиламино)метил-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
38. (S)-N-[[3-[3-фтор-4-(цис-3-(метоксикарбониламино)метил 4-метилпирролидинил)фенил]-2-oкco-5-oкcaзoлидинил]метил]ацетамид
39. (S)-N-[[3-[3-фтор-4-(цис-3-(этоксикарбониламино)метил-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
40. (S)-N-[[3-[3-фтор-4-(транс-3-(фенилметоксиацетиламино) метил-4-метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил] ацетамид
41. (S)-N-[[3-[3-фтор-4-(транс-3-(гидроксиацетиламино)метил-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
42. (S)-N-[[3-[3-фтор-4-(транс-3-(метоксиацетиламино)метил-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетиламид
43. (S)-N-[[3-[3-фтор-4-(транс-3-(метоксикарбониламино)метил-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
44. (S)-N-[[3-[3-фтор-4-(транс-3-(этоксикарбониламино)метил-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
45. (S)-N-[[3-[3-фтор-4-[транс-3-(фенилметокси)ацетиламино-4- гидроксипирролидинил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
46. (S)-N-[[3-[3-фтор-4-(транс-3-(метоксиацетиламино)-4- гидроксипирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
47. (S)-N-[[3-[3-фтор-4-(транс-3-(метоксикарбониламино)-4- гидроксипирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
48. (S)-N-[[3-[3-фтор-4-(транс-3-(этоксикарбониламино)-4- гидроксипирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Антибактериальная активность
Оксазолидиноновые антибактериальные агенты данного изобретения имеют полезную активность против ряда организмов. In vitro активность соединений данного изобретения можно оценить при помощи стандартных методик испытания, таких как определение минимальной ингибирующей концентрации (МИК, МIС) путем разбавления агаром, описанного в "Methods for Dilution Antimicrobial Susceptibility Test for Bacteria That Grow Aerobically" (MFT) published Jan. 1983 by the National Committee for Clinical Laboratory Standards, 771 East Lancaster Avenue, Villanova, Pennsylvania 19084, USA. Активность выбранных соединений данного изобретения против Staphylococcus aureus и streptococcus pneumoniae представлены в таблице.
Азетидинил-фенилоксазолидиноны
Пример 1: (S)-N-[[3-[3-фтор-4-(3-метокси-1-азетидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: 1-(дифенилметил)-3-метоксиазетидин
Суспензию гидрида натрия (0,440 г 60% дисперсии в масле, 11,0 ммоль) в сухом тетрагидрофуране (125 мл) в атмосфере азота охлаждают с помощью бани со льдом до 0oC и обрабатывают твердым 1-(дифенилметил)-3-азетидинолом (2,393 г, 10,0 ммоль) на протяжении 5 мин. После перемешивания в течение 30 мин при 0oC добавляют иодометан (1,490 г, 0,654 мл, 10,5 ммоль). По завершении добавления охлаждающую баню удаляют и реакционной смеси дают возможность нагреться в течение ночи до комнатной температуры. Реакционную смесь выливают в pH 7 фосфатный буфер и экстрагируют этилацетатом. Объединенные органические экстракты промывают рассолом, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт хроматографируют на силикагеле (300 мл), заполненным дихлорметаном, элюируя градиентом смеси 1-10% этилацетат/дихлорметан. Концентрирование соответствующих фракций дает 2,081 г (82%) названного соединения в виде бледно-желтого сиропа с МС (ЭИ) 253 (М+).
Стадия 2: 3-фтор-4-(3-метокси-1-азетидинил) нитробензол
Раствор 1-(дифенилметил)-3-метоксиазетидина (2,00 г, 7,91 ммоль) в смеси 25% тетрагидрофуран/этанол (100 мл) обрабатывают 5 HCl (5,0 мл) и гидроксидом палладия-на-угле (Pearlman's катализатор, 0,500 г). Смесь встряхивают на аппарате Парра при 3,164 кг/см2 (45 psi) H2. Через 16 ч некоторое количество исходного вещества все еще остается, как показано с помощью ТСХ (TLC) анализа (силикагель, 6% ацетонитрил/хлороформ). Дополнительные 0,500 г Pearlman's катализатора добавляют и гидрогенолиз продолжают еще 16 ч, и за это время реакция, по-видимому, завершается. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют при пониженном давлении до масла янтарного цвета. Это вещество растворяют в диметилсульфоксиде (30 мл) и обрабатывают кислым динатрий фосфатом (6,88 г, 39,6 ммоль) и 3,4-дифторнитробензолом (1,05 мл, 9,49 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 ч, и при этом времени ТСХ анализ (силикагель, 6% ацетонитрил/хлороформ) показывает, что реакция завершена. Реакционную смесь разбавляют водой (150 мл) и экстрагируют хлороформом (3 x 40 мл). Объединенные органические экстракты промывают водой (3 x 25 мл), рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая неочищенный продукт. Очистку выполняют с помощью хроматографирования на силикагеле (100 г), элюируя градиентом смеси 0-1% ацетонитрил/хлороформ. Концентрирование соответствующих фракций дает 1,50 г (84%) названного соединения в виде светло-желтого твердого вещества с т.пл. 57,5-58oC и МС (ЭИ) 226 (М+).
Стадия 3: N-(карбобензилокси)-3-фтор-4-(3-метокси-1-азетидинил) анилин
Раствор 3-фтор-4-(3-метокси-1-азетидинил)-нитробензола (1,50 г, 6,64 ммоль) в смеси 1:1 метанол/тетрагидрофуран (35 мл) обрабатывают 10% палладий-на-угле и затем аммоний формиатом (1,26 г, 19,9 ммоль) при комнатной температуре. Через 20 мин окраска реакционной смеси изменяется с желтой на бесцветную. Реакционную смесь фильтруют через Celite® фильтрат концентрируют при пониженном давлении. Выделенное масло немедленно растворяют в смеси 3:1 ацетон/вода (25 мл) и обрабатывают карбонатом натрия (2,75 г, 19,9 ммоль) и бензил хлорформиатом (1,31 г, 8,30 ммоль). Через 30 мин ТСХ анализ (6% ацетонитрил/хлороформ) показал, что реакция завершена. Реакционную смесь экстрагируют хлороформом (3 х 20 мл). Объединенные органические экстракты промывают рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до масла пурпурного цвета. Это вещество хроматографируют на силикагеле (100 мл), элюируя градиентом смеси 1-3% ацетонитрил/хлороформ, получая после концентрирования соответствующих фракций 1,24 г (56%) названного соединения в виде беловатого твердого вещества с т.пл. 95-96,5oC и МС (ЭИ) 330 (М+).
Стадия 4: (R)-[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метанол
Раствор N-(карбобензилокси)-3-фтор-4-(3-метокси-1-азетидинил) анилина (0,865 г, 2,62 ммоль) в сухом тетрагидрофуране (10 мл) охлаждают до -78oC в атмосфере азота и обрабатывают н-бутиллитием (1,65 мл 1,6 М раствора в гексанах, 2,65 ммоль). После перемешивания при -78oC в течение 15 мин реакционную смесь обрабатывают (R)-глицидил бутиратом (0,374 мл, 2,65 ммоль). Затем охлаждающую ванну удаляют и реакционной смеси дают возможность нагреться до температуры окружающей среды. Через 1 ч по данным ТСХ анализа считают, что реакция завершилась. Реакционную смесь гасят путем добавления насыщенного водного хлорида аммония (0,5 мл) и концентрируют при пониженном давлении до желтого твердого вещества. Хроматографированием на силикагеле (10 г) при элюировании градиентом смеси 1-2% метанол/хлороформ получают после концентрирования соответствующих фракций 0,530 г (68%) названного соединения в виде беловатого твердого вещества с т.пл. 131-132oC и МС (ЭИ) 296 (М+).
Стадия 5: (R)-[[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил]-2- оксо-5-оксазолидинил]метил]метансульфонат
Раствор (R)-[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил] -2-оксо-5- оксазолидинил] метанола (0,492 г, 1,66 ммоль) в сухом дихлорметане (25 мл) в атмосфере азота охлаждают с помощью бани со льдом до 0oC, обрабатывают триэтиламином (0,254 мл, 1,83 ммоль) и затем метансульфонил хлоридом (0,141 мл, 1,83 ммоль). Через 1 ч обработки при 0oC ТСХ анализ (10% метанол/хлороформ) показал, что реакция завершилась. Реакционную смесь промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до беловатого твердого вещества. Хроматографированием на силикагеле (100 г) при элюировании градиентом смеси 1-3% метанол/хлороформ получают после концентрирования соответствующих фракций 0,601 г (97%) названного соединения в виде белого твердого вещества с т.пл. 122,5-123,5oC и МС (ЭИ) 374 (М+).
Стадия 6: (R)-[[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метил]азид
(R)-[[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил] -2-oкco-5- oкcaзoлидинил] метил] метaнcульфoнaт (0,508 г, 1,36 ммоль) объединяют с азидом натрия (0,106 г, 1,63 ммоль) в N,N-диметилформамиде (4 мл). Реакционную смесь нагревают до 65oC в атмосфере азота. Через 2 ч небольшое количество исходного мезилата еще остается, как показано с помощью ТСХ анализа (5% метанол/хлороформ). Добавляют дополнительно 0,044 г азида натрия и реакционную смесь нагревают еще 1,5 ч, и при этом времени ТСХ анализ показал, что реакция завершилась. Реакционную смесь фильтруют и концентрируют в вакууме. Остаток хроматографируют на силикагеле (25 г), элюируя градиентом смеси 1-3% метанол/хлороформ. Соответствующие фракции объединяют и концентрируют в вакууме, получая 0,426 г (98%) названного соединения в виде беловатого твердого вещества. Аналитический образец получают путем перекристаллизации этого вещества из смеси 3:1 этилацетат/гексан, получая белое твердое вещество с т. пл. 111-112,5oC и МС (ЭИ) 321 (М+).
Стадия 7: (R)-N-[[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (R)-[[3-[3-фтор-4-(3-метокси-1-азетидинил)фенил] -2-оксо- 5-оксазолидинил]метил]азида (0,393 г, 1,22 ммоль) в смеси 5:1 метанол/дихлорметан (20 мл) обрабатывают 10% палладием-на-угле (0,030 г) в токе азота. Затем атмосферу замещают водородом (баллон). После перемешивания в течение 3 часов в атмосфере водорода о завершении восстановления судят по данным ТСХ анализа (5% метанол/хлороформ). Реакционную смесь фильтруют через Celite® и концентрируют при пониженном давлении. Неочищенный 5-(аминометил)оксазолидинон растворяют в дихлорметане (15 мл) и обрабатывают пиридином (0,118 мл, 1,46 ммоль) и уксусным ангидридом (0,138 мл, 1,46 ммоль) в атмосфере азота при температуре окружающей среды. Через 2 ч о том, что реакция завершилась, судят по данным ТСХ. Реакционную смесь промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Хроматографированием на силикагеле (50 г) при элюировании градиентом смеси 1-3% метанол/хлороформ получают после концентрирования соответствующих фракций 0,281 г (68%) названного антибактериального средства в виде белого твердого вещества. Аналитическая проба, полученная перекристаллизацией из смеси 2:1 этилацетат/гексан, представляла белое твердое вещество с т.пл. 160-161,5oC и МС (ЭИ) 337 (М+).
Пример 2: (S)-N-[[3-[3-фтор-4-(3-гидрокси-1-азетидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: 3-фтор-4-(3-гидрокси-1-азетидинил)нитробензол
1-(Дифенилметил)-3-азетидинол гидрохлорид (2,00 г, 7,29 ммоль) растворяют в метаноле (75 мл) и обрабатывают 6N HCl (1,20 мл, 7,29 ммоль). Затем добавляют в потоке азота гидроксид палладия-на-угле (0,200 г). Затем реакционную смесь встряхивают на аппарате Парра при 2,81 кг/см2 (40 psi) H2. Через 16 ч ТСХ анализ (5% метанол/хлороформ) показал, что исходное вещество израсходовано. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют при пониженном давлении до масла янтарного цвета. Это вещество растворяют в диметилсульфоксиде (29 мл) и атмосферу замещают азотом. Добавляют кислый динатрий фосфат (5,07 г, 29,2 ммоль) и затем 3,4-дифторонитробензол (0,966 мл, 8,75 ммоль). Смесь перемешивают при температуре окружающей среды в атмосфере азота. Через 3 ч ТСХ анализ (5% метанол/хлороформ) показал, что реакция завершена. Реакционную смесь разбавляют водой (250 мл) и экстрагируют хлороформом (4 x 50 мл). Объединенные органические экстракты промывают водой (2 x 50 мл) и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая желтое твердое вещество. Хроматографированием на силикагеле (100 г) при элюировании градиентом смеси 3-7% ацетонитрил/хлороформ получают после концентрирования соответствующих фракций 1,00 г (65%) названного соединения в виде оранжевого твердого вещества с т.пл. 130,5-132oC и МС (ЭИ) 212 (М+).
Стадия 2: 4-[3-[(трет-бутилдиметилсилил)окси-1-азетидинил]-3- фторнитробензол
Раствор 3-фтор-4-(3-гидрокси-1-азетидинил)нитробензола (5,51 г, 26,0 ммоль) в N,N-диметилформамиде (104 мл) в атмосфере азота охлаждают до 0oC на бане со льдом и обрабатывают имидазолом (1,86 г, 27,3 ммоль) и затем трет-бутилдиметилсилил хлоридом (4,12 г, 27,3 ммоль). Реакционную смесь перемешивают при 0oC в течение 30 мин и затем при комнатной температуре в течение ночи. ТСХ анализ (5% метанол/хлороформ) показал, что небольшое количество исходного вещества все еще остается к этому времени. Добавляют дополнительное количество трет-бутилдиметилсилил хлорида (0,392 г). После перемешивания в течение ночи ТСХ анализ показал, что реакция завершилась. Реакционную смесь разбавляют водой (500 мл) и экстрагируют дихлорметаном (4 х 70 мл). Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении до желтого твердого вещества. Хроматографированием на силикагеле (200 г) при элюировании смесями 5 и 10% этилацетат/гексан получают после концентрирования соответствующих фракций 6,30 г (74%) названного соединения в виде желтого твердого вещества с т.пл. 98,5-99,5oC и МС (ЭИ) 326 (М+).
Стадия 3: N-(карбобензилокси)-4-[3-[(трет-бутилдиметилсилил)окси]-1-азетидинил] -3-фторанилин
4-[3-[(трет-Бутилдиметилсилил)окси] -1-азетидинил] -3-фторнитробензол (1,00 г, 3,07 ммоль) соединяют с 10% палладием-на-угле (0,100 г) в смеси 3:1 тетрагидрофуран/вода (25 мл) в атмосфере азота. Атмосферу заменяют водородом (баллон), повторяя вакуумирование и наполнение. Через 2 ч первоначальная желтая окраска реакционного раствора исчезает и ТСХ анализ (15% этилацетат/гексан) показал, что восстановление завершено. Реакционную смесь c фильтруют через Celite® и фильтрат немедленно помещают в атмосферу азота и обрабатывают бикарбонатом натрия (1,41 г, 16,8 ммоль) и бензил хлорформиатом (0,528 мл, 3,69 ммоль). Через 30 мин при температуре окружающей среды по данным ТСХ анализа (15% этилацетат/гексан) реакция завершается. Реакционную смесь концентрируют при пониженном давлении до беловатого твердого вещества. Хроматографированием на силикагеле (125 г) при элюировании смесью 5-30% этилацетат/гексан получают после концентрирования соответствующих фракций 0,565 г (43%) названного соединения в виде белого твердого вещества с т.пл. 91-93oC и МС (ЭИ) 430 (М+).
Стадия 4: (R)-[3-[4-[3-[(трет-бутилдиметилсилил)окси] -1-азетидинил]-3-фторфенил]-2-оксо-5-оксазолидинил]метанол
Раствор N-(карбобензилокси)-4-[3-[(трет-бутилдиметилсилил) окси]-1-азетидинил]-3-фторанилина (6,30 г, 14,7 ммоль) в сухом тетрагидрофуране (100 мл) в атмосфере азота охлаждают до -78oC и обрабатывают н-бутиллитием (9,16 мл, 1,6 М раствора в гексанах, 14,7 ммоль). После окончания добавления реакционную смесь перемешивают при -78oC в течение 15 мин и затем обрабатывают (R)-глицидил бутиратом (2,21 мл, 14,7 ммоль). По окончании добавления охлаждающую баню удаляют и реакционной смеси позволяют перемешиваться при температуре окружающей среды в течение 1,5 ч. После этого добавляют насыщенный водный хлорид аммония (20 мл). Через 3 мин к реакционной смеси добавляют насыщенный водный раствор бикарбоната натрия (10 мл) и органический растворитель удаляют с помощью роторного испарения при пониженном давлении. Добавляют дихлорметан (100 мл) и смесь промывают водой и рассолом. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Хроматографированием на силикагеле (200 г) при элюировании градиентом смеси 1-3% метанол/хлороформ получают после концентрирования соответствующих фракций 4,39 г (75%) названного соединения в виде беловатого твердого вещества с т.пл. 183-186oC и МС (ЭИ) 396 (М+).
Стадия 5: (R)-[[3-[4-[3-[(трет-бутилдиметилсилил)окси]-1-азетидинил]-3- фторфенил]-2-оксо-5-оксазолидинил]метил]метансульфонат
Неочищенный (R)-[3-[4-[3-[(трет-бутилдиметилсилил)окси]-1-азетидинил]-3- фторфенил] -2-оксо-5-оксазолидинил]метанол (5,38 г, 13,6 ммоль) растворяют в сухом дихлорметане (70 мл), охлажденном до 0oC на бане со льдом, и обрабатывают триэтиламином (2,08 мл, 14,9 ммоль) и затем метансульфонил хлоридом (1,15 мл, 14,9 ммоль). Через 30 мин при 0oC по данным ТСХ анализа (5% метанол/хлороформ) реакция завершается. Реакционную смесь промывают насыщенным водным раствором бикарбоната натрия, водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении до беловатого твердого вещества. Аналитическая проба, полученная растиранием неочищенного продукта с изопропанолом и диэтиловым эфиром, с последующей фильтрацией и высушиванием в вакууме, дает беловатое твердое вещество с т.пл. 142-145oC и МС (ЭИ) 474 (М+).
Стадия 6: (R)-[[3-[4-[3-[(трет-бутилдиметилсилил)окси]-1- азетидинил]-3-фторфенил]-2-оксо-5-оксазолидинил]метил]азид
Неочищенный (R)-[[3-[4-(3-[трет-бутилдиметилсилил)окси] -1-азетидинил] -3-фторфенил] -2-оксо-5-оксазолидинил]метансульфонат (9,42 ммоль) растворяют в сухом N,N-диметилформамиде (50 мл) и обрабатывают азидом натрия (4,42 г, 68,0 ммоль) при температуре окружающей среды. Реакционную смесь нагревают до 65oC в атмосфере азота в течение 4 ч. ТСХ анализ (5% метанол/хлороформ) к этому времени показывает, что реакция завершилась. Реакционную смесь разбавляют этилацетатом (100 мл) и промывают водой (3 x 25 мл) и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до твердого вещества янтарного цвета. Хроматографированием на силикагеле (125 г) при элюировании градиентом смеси 10-20% этилацетат/гексан получают после концентрирования соответствующих фракций 2,21 г (56% для 3 стадий) названного азида в виде белого твердого вещества с т.пл. 121-122,5oC и МС (ЭИ) 421 (М+).
Стадия 7: (S)-N-[[3-[4-[3-[(трет-бутилдиметилсилил)окси]-1- азетидинил] -3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (R)-[[3-[4-[3-[(трет-бутилдиметилсилил)окси]-1- азетидинил]-3-фторфенил] -2-оксо-5-оксазолидинил] метил]азида (2,04 г, 4,85 ммоль) в смеси 1:1 этилацетат/метанол (200 мл) обрабатывают 10% палладием-на-угле (0,300 г) в потоке азота. Затем атмосферу заменяют H2 (баллон), повторяя вакуумирование и наполнение. Через 3 ч по данным ТСХ анализа (5% метанол/хлороформ), реакция завершается. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют при пониженном давлении до белого твердого вещества. Неочищенный 5-(аминометил)оксазолидинон растворяют в дихлорметане (100 мл), охлажденном до 0oC в атмосфере и обрабатывают пиридином (0,431 мл, 5,33 ммоль) и уксусным ангидридом (0,503 мл, 5,33 ммоль). Через 30 мин при 0oC по данным ТСХ анализа (5% метанол/хлороформ) ацетилирование завершается. Реакционную смесь промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая неочищенный продукт желтовато-коричневого цвета. Хроматографированием на силикагеле (125 г) при элюировании градиентом 1-3% метанол/хлороформ получают после концентрирования соответствующих фракций 1,77 г (84%) названного соединения в виде белого твердого вещества с т.пл. 183,5-184oC и МС (ЭИ) 437 (М+).
Стадия 8: (S)-N-[[3-[3-фтор-4-(3-гидрокси-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
Ацетонитрильный (55 мл) раствор (S)-N-[[3-[4-[3- [(трет-бутилдиметилсилил)окси] -1-азетидинил] -3-фторфенил]- -2-оксо-5-оксазолидинил]метил]ацетамида (1,23 г, 2,81 ммоль) в полипропиленовой колбе обрабатывают 38% водной фтористоводородной кислотой (15 мл) при температуре окружающей среды. По данным ТСХ анализа снятие защиты завершается через 3 ч. Реакционную смесь разбавляют водой (15 мл) и нейтрализуют твердым бикарбонатом натрия. Добавляют еще воды (50 мл) и смесь экстрагируют хлороформом. Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая названное оксазолидиноновое антибактериальное средство в виде беловатого твердого вещества с т.пл. 174-177oC и МС (ЭИ) 323 (М+)
Пример 3: (S)-N-[[3-[3-фтор-4-[3-[N-(2-фторэтил)-N-метиламино] -1-азетидинил]фенил]-2-oкco-5-oкcaзoлидинил]метил]ацетамид
Стадия 1: 1-(Дифенилметил)-3-[N-(2-фторэтил)-N-метиламино] азетидин
2-(Дифенилметил)-3-(метиламино)азетидин (5,00 г, 19,8 ммоль) соединяют с карбонатом натрия (16,4 г, 119 ммоль) и 2-фторэтил тозилатом (6,50 г, 29,8 ммоль) в смеси 6% вода-ацетонитрил (200 мл). Смесь кипятят с обратным холодильником в атмосфере азота. Через 3 ч кипячения с обратным холодильником по данным ТСХ анализа (5% метанол/хлороформ) остается небольшое количество исходного вещества. Добавляют еще 1,8 г 2-фторэтил тозилата и реакционную смесь кипятят с обратным холодильником еще 2 ч. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют при пониженном давлении. Хроматографированием на силикагеле (200 г) при элюировании градиентом 1-5% ацетонитрил/хлороформ получают после концентрирования соответствующих фракций 4,07 г (69%) названного соединения в виде сиропа янтарного цвета с ИК 1598, 1452, 1362, 1029 см-1.
Стадия 2: 3-фтор-4-[3-[N-(2-фторэтил)-N-метиламино]-1-азетидинил] нитробензол
Сосуд Парра загружают 1-(дифенилметил)-3-[N-(2-фторэтил)-N- метиламино] азетидин (3,88 г, 13,0 ммоль) и смесь 25% тетрагидрофуран/этанол (130 мл). Затем в потоке азота добавляют гидроксид палладия-на-угле (1,9 г). Затем смесь встряхивают в аппарате Парра при 3,16 кг/см2 (45 psi) H2. Через 20 ч по данным ТСХ анализа (5% метанол/хлороформ) гидрогенолиз завершается. Реакционную смесь фильтруют через Celite® и концентрируют при пониженном давлении до бесцветного масла. Это вещество растворяют в диметилсульфоксиде (50 мл) и обрабатывают динатрий кислым фосфатом (13,6 г, 78,0 ммоль), с последующей обработкой 3,4-дифторнитробензолом (1,72 мл, 15,6 ммоль). Затем реакционную смесь перемешивают при температуре окружающей среды и за развитием реакции следят с помощью ТСХ анализа (5% метанол/хлороформ). Через 4 ч реакционную смесь выливают в H2O (500 мл) и экстрагируют дихлорметаном (4 x 75 мл). Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до желтого сиропа. Хроматографированием на силикагеле (200 г) при элюировании градиентом 0-2,5% ацетонитрил/дихлорметан получают после концентрирования соответствующих фракций 2,87 г (81%) названного соединения в виде желтого твердого вещества с т.пл. 46,5-48oC и МС (ББА, FAB) 272 (М+H)+.
Стадия 3: N-(Карбобензилокси)-3-фтор-4-[3-[N-(2-фтopэтил)-N- метиламино] -1-азетидинил]анилин
Раствор 3-фтор-4-[3-[N-(2-фторэтил)-N-метиламино] -1-азетидинил]нитробензола (2,66 г, 9,82 ммоль) в смеси 2:1 тетрагидрофуран/вода (50 мл) и уксусной кислоты (2,0 мл) обрабатывают 10% палладий/углерод в потоке азота.
Атмосферу заменяют H2 (баллон) путем повторения вакуумирования и наполнения, и реакционную смесь перемешивают в атмосфере H2 в течение ночи. По данным ТСХ анализа (6% ацетонитрил/хлороформ) к этому времени восстановление завершается. Реакционную смесь фильтруют через Celite®. Экспозицию на воздухе сводят к минимуму, так как при этих условиях быстро развивается пурпурное окрашивание. Фильтрат охлаждают на бане со льдом до около 0oC и обрабатывают карбонатом натрия (6,8 г, 49 ммоль) и бензилхлороформиатом (1,63 мл, 10,3 ммоль). Реакционную смесь перемешивают при около 0oC в течение 1 ч и затем нагревают до комнатной температуры на протяжении 30 мин. По данным ТСХ анализа (6% ацетонитрил/хлороформ) к этому времени реакция завершается. Реакционную смесь разбавляют водой (200 мл) и экстрагируют хлороформом (3 x 75 мл). Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до сиропа темного янтарного цвета. Хроматографированием на силикагеле (200 г) при элюировании градиентом 1-3% метанол/хлороформ получают после концентрирования соответствующих фракций 3,71 г (100%) названного соединения в виде сиропа янтарного цвета. Аналитическую пробу готовят с помощью дополнительной хроматографической очистки, получая твердое вещество светло-янтарного цвета с т. пл. 57-59oC и МС (ЭИ) 375 (М+).
Стадия 4: (R)-N-[3-[3-фтор-4-[3-N-(2-фторэтил)-N-метиламино] -1-азетидинил]фенил]-2-оксо-5-оксазолидинил]метанол
Раствор N-(карбобензилокси)-3-фтор-4-[3-[N-(2-фтopэтил)-N-метилaминo]-1- aзетидинил] анилина (3,38 г, 9,01 ммоль) в сухом тетрагидрофуране (40 мл) в атмосфере азота охлаждают до -78oC и обрабатывают н-бутиллитием (5,69 мл 1,6 М раствора в гексане, 9,10 ммоль). Реакционную смесь нагревают до -40oC и затем повторно охлаждают до -78oC и обрабатывают (R)-глицидил бутиратом (1,29 мл, 9,10 ммоль). После завершения добавления реакционной смеси дают возможность нагреться до температуры окружающей среды. Через 1 ч по данным ТСХ анализа (5% метанол/хлороформ) считают, что реакция завершилась. Реакционную смесь гасят насыщенным водным раствором хлорида аммония (1 мл), разбавляют водой (100 мл) и экстрагируют хлороформом (3 x 50 мл). Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме до сиропа янтарного цвета. Хроматографированием на силикагеле (200 г) при элюировании градиентом смеси 1-4% метанол/хлороформ получают после концентрирования соответствующих фракций 1,69 г (55%) названного соединения в виде беловатого твердого вещества с т. пл. 124-125oC и МС (ЭИ) 341 (М+).
Стадия 5: (R)-[[3-[4-[3-[N-(2-фторэтил)-N-метиламино] 1-азетидинил]-3-фторфенил]-2-оксо-оксазолидинил]метил]метансульфонат
Раствор (R)-N-[3-[3-фтор-4-[3-[N-(2-фторэтил)-N-метиламино] -1-азетидинил] фенил] -2-оксо-5-оксазолидинил] метанола (1,40 г, 4,11 ммоль) в сухом дихлорметане (16 мл) обрабатывают триэтиламином (0,628 мл, 4,52 ммоль) и затем охлаждают до 0oC в атмосфере азота. Затем добавляют метансульфонил хлорид (0,348 мл, 4,52 ммоль) и смесь перемешивают при 0oC. Через 2 ч обработки ТСХ анализ (5% метанол/хлороформ) показал, что еще остается небольшое количество исходного вещества. Добавляют дополнительную порцию метансульфонил хлорида (0,100 мл, 1,30 ммоль) и реакцию продолжают еще 1 ч при 0oC. Реакционную смесь промывают насыщенным водным раствором бикарбоната натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая 1,88 г (100%) названного соединения в виде светло оранжевого твердого вещества. 1H ЯМР анализ показал, что это вещество имеет высокое качество. Аналитический образец получают путем хроматографирования 200 мг неочищенного продукта на силикагеле (10 г), элюируя градиентом 1-4% метанол/ хлороформ, после концентрирования соответствующих фракций 99 мг названного соединения в виде беловатого твердого вещества с т.пл. 96,5-98oC и МС (ЭИ) 419 (М+).
Стадия 6: (R)-[[3-[4-[3-[N-(2-фторэтил)-N-метиламино]-1-азетидинил]-3- фторфенил]-2-оксо-5-оксазолидинил]метил]азид
Раствор (R)-[[3-[4-[3-[N-(2-фторэтил)-N-метиламино] -1-азетидинил] -3- фторфенил]-2-оксо-5-оксазолидинил]метил]метансульфоната (1,65 г, 3,94 ммоль) в сухом в N,N-диметилформамиде (15 мл) обрабатывают твердым азидом натрия (0,768 г, 11,8 ммоль). Реакционную смесь нагревают до 65oC в атмосфере азота. Через 3 ч небольшое количество исходного мезилата еще остается, как показано с помощью ТСХ анализа (5% метанол/хлороформ). Добавляют дополнительную порцию азида натрия (0,256 г, 3,94 ммоль) и реакционную смесь нагревают при 65oC в течение 1 ч. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют и концентрируют при пониженном давлении. Остаток хроматографируют на силикагеле (100 г), элюируя градиентом смеси 1-4% метанол/хлороформ, получая после концентрирования соответствующих фракций 1,24 г (86%) названного соединения в виде беловатого твердого вещества с т.пл. 60-63oC и МС (ЭИ) 366 (М+).
Стадия 7: (S)-N-[[3-[3-фтор-4-[3-[N-(2-фторэтил)-N-метиламино]-1 -азетидинил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (R)-[[3-[4-[3-[N-(2-фторэтил)-N-метиламино] -1-азетидинил]-3-фторфенил] -2-оксо-5-оксазолидинил] метил]азида (1,14 г, 3,11 ммоль) в метаноле (20 мл) обрабатывают 10% палладий/углерод (0,114 г) в потоке азота. Атмосферу замещают H2 (баллон), повторяя вакуумирование и наполнение. После 2,5 ч согласно данным ТСХ анализа (5% метанол/хлороформ) считали, что восстановление было полным. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют в вакууме до беловатого твердого вещества. Это вещество растворяют в дихлорметане (20 мл) и обрабатывают пиридином (0,264 мл, 3,27 ммоль) и уксусным ангидридом (0,309 мл, 3,27 ммоль) в атмосфере азота. Через 30 мин ТСХ анализ (5% метанол/хлороформ) показал, что ацетилирование завершилось. Реакционную смесь промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая масло янтарного цвета. Хроматографированием на силикагеле (125 г) при элюировании градиентом смеси 1-4% метанол/хлороформ получают после концентрирования соответствующих фракций 0,892 г (75%) названного оксазолидинона - антибактериального агента в виде беловатого твердого вещества с т.пл. 125,5-127oC и МС (ЭИ) 382 (М+).
Пример 4: (S)-N-[[3-[3-фтор-4-(3-оксо-1-азетидинил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (S)-N-[[3-[3-фтор-4-(3-гидрокси-1-азетидинил)фенил] -2-оксо-5-оксазолидинил] метил] ацетамида (Пример 2, 0,440 г, 1,36 ммоль) в смеси 10% ацетонитрил/дихлорметан (100 мл) обрабатывают порошкообразными молекулярными ситами с размером пор 4 ангстрема (0,682 г), 4-метилморфолин N-оксидом (0,319 г, 2,72 ммоль) и затем тетра-п-пропиламмоний перрутенатом (0,024 г, 0,068 ммоль) при температуре окружающей среды. После перемешивания в течение 1,5 часов при температуре окружающей среды ТСХ анализ (10% метанол/хлороформ) показал, что еще остается небольшое количество исходного спирта. Добавление дополнительного количества 4-метилморфолин N-оксида не расходует исходное вещество. Реакционную смесь концентрируют при пониженном давлении до темного пурпурного твердого вещества, которое затем хроматографируют на силикагеле (20 г), элюируя градиентом смеси 1-3,5% метанол/хлороформ, получая после концентрирования соответствующих фракций 0,204 г (47%) названного оксазолидинона-антибактериального средства в виде белого твердого вещества с т.пл. 192-193oC и МС (ЭИ) 321 (М+).
Пример 5: (S)-N-[[3-[3-фтор-4-[3-(метоксиимино)-1-азетидинил] фенил] - 2-oкco-5-oкcaзoлидинил]метил]ацетамид
Раствор (S)-N-[[3-[3-фтор-4-(3-оксо-1-азетидинил)фенил] -2-оксо- 5-оксазолидинил] метил] ацетамида (Пример 4, 0,200 г, 0,623 ммоль) в смеси 5% метанол/дихлорметан (10 мл) обрабатывают пиридином (0,201 мл, 2,49 ммоль) и затем гидрохлоридом метоксиламина (0,052 г, 0,623 ммоль) при температуре окружающей среды. Через 1,5 ч ТСХ анализ (10% метанол/хлороформ) показал, что реакция завершилась. Реакционную смесь концентрируют при пониженном давлении до белого твердого вещества. Хроматографированием на силикагеле (10 г), элюируя градиентом смеси 1-3% метанол/хлороформ, получают после концентрирования соответствующих фракций 0,204 г (94%) названного оксазолидинона-антибактериального агента в виде беловатого твердого вещества. В другом опыте по синтезу получили вещество с т.пл. 189- 192oC и МС (ЭИ) 350 (М+).
Пример 6: (S)-N-[[3-[3-фтор-4-(3-метокси-3-метил-1-азетидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: 1-(дифенилметил)-3-метокси-3-метилазетидин
Суспензию 1-(дифенилметил)-3-метил-3-азетидинол гидрохлорида (5,00 г, 17,2 ммоль) в сухом тетрагидрофуране (200 мл) в атмосфере азота охлаждают с помощью бани со льдом до 0oC и обрабатывают гидридом натрия (2,10 г 60% дисперсии в масле, 51,8 ммоль). Охлаждающую баню удаляют и смесь кипятят с обратным холодильником в течение 15 минут. После охлаждения до комнатной температуры добавляют иодометан (7,40 г, 3,22 мл, 51,8 ммоль). По завершении добавления реакционную смесь кипятят с обратным холодильником в течение 15 ч. ТСХ анализ (9:1 гексан/этилацетат) показал, что метилирование завершилось. Реакционную смесь гасят насыщенным водным аммоний хлорида. Смесь переносят в делительную воронку с этилацетатом и водой и затем экстрагируют этилацетатом. Объединенные органические экстракты промывают рассолом, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт хроматографируют на силикагеле, элюируя смесью 9:1 этилацетат/гексан. Концентрирование соответствующих фракций дает 2,30 г (50%) названного соединения в виде бесцветного масла с МС (ЭИ) 267 (М+).
Стадия 2: 3-фтор-4-(3-метокси-3-метил-1-азетидинил)нитробензол
Раствор 1-(дифенилметил)-3-метокси-3-метилазетидина (2,20 г, 8,2 ммоль) в смеси 25% тетрагидрофуран/этанол обрабатывают ледяной уксусной кислотой (1,60 г, 1,50 мл, 24,7 ммоль) и затем гидроксидом палладия-на-угле (0,220 г) в потоке азота. Реакционную смесь встряхивают на аппарате Парра при 2,46 кг/см2 (35 psi) H2. К этому времени ТСХ (TLC) анализ (9:1 гексан/этилацетат) показал, что реакция завершилась. Смесь фильтруют через Celite® (промывают этилацетатом) и фильтрат концентрируют в вакууме, получая масло. Это вещество растворяют в диметилсульфоксиде (15 мл) и раствор обрабатывают кислым динатрий фосфатом (8,60 г, 49,2 ммоль) и 3,4-дифторнитробензолом (1,30 г, 0,886 мл, 8,0 ммоль) при температуре окружающей среды. Желтую реакционную смесь перемешивают при температуре окружающей среды в течение 18 ч. Реакционную смесь разбавляют водой и экстрагируют дихлорметаном. Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая желтое твердое вещество. Хроматографированием на силикагеле, элюируя смесью 8:1 гексан/этилацетат, получают после концентрирования соответствующих фракций 1,80 г (90%) названного соединения в виде желтого твердого вещества с т.пл. 110-111oC и МС (ЭИ) 240 (М+).
Стадия 3: N-(карбобензилокси)-3-фтор-4-(3-метокси-3-метил-1- азетидинил)анилин
Раствор 3-фтор-4-(3-метокси-3-метил-1-азетидинил)нитробензола (1,60 г, 6,7 ммоль) в смеси 25% тетрагидрофуран/метанол (35 мл) дегазируют, повторяя вакуумирование и заполнение N2 и затем обрабатывают 10% палладий/углерод (0,160 г). Затем добавляют аммоний формиат (2,10 г, 33,3 ммоль) и смесь дегазируют. Реакционную смесь перемешивают при температуре окружающей среды в течение 20 мин, и в течение этого времени окраска реакционной смеси изменяется с желтой на бесцветную. ТСХ анализ (2:1 гексан/этилацетат) показывает, что реакция завершена. Реакционную смесь фильтруют через Celite® (промывают дихлорметаном и метанолом) и фильтрат концентрируют в вакууме, получая пурпурную смолу, которую немедленно растворяют в смеси 1:1 ацетон/вода (30 мл) в атмосфере азота. Смесь охлаждают на бане со льдом до 0oC и обрабатывают бикарбонатом натрия (1,10 г, 13,4 ммоль) и бензил хлорформиатом (1,20 г, 1,00 мл, 7,37 ммоль). Охлаждающей бане дают возможность постепенно нагреваться в течение 3 ч. После перемешивания в течение дополнительных 2 ч при температуре окружающей среды реакционную смесь переносят в делительную воронку с дихлорметаном (50 мл). Органическую фазу отделяют, промывают водой, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая пурпурное масло. Хроматографированием на силикагеле, элюируя смесью 6:1 гексан/этилацетат, получают после концентрирования соответствующих фракций 2,04 г (89%) названного соединения в виде вязкого масла, которое отверждается при стоянии с образованием воскообразного вещества с т.пл. 73-75oC и МС (ЭИ) 344 (М+).
Стадия 4: (R)-[3-[3-фтор-4-(3-метокси-3-метил-1-азетидинил) фенил]-2-оксо-5-оксазолидинил]метанол
Раствор N-(карбобензилокси)-3-фтор-4-(3-метокси-3-метил-1- азетидинил)анилина (1,85 г, 5,4 ммоль) в сухом тетрагидрофуране (20 мл) охлаждают до -78oC в ванне со смесью сухой лед/ацетон и обрабатывают н-бутиллитием (3,50 мл 1,6 М раствора в гексане, 5,7 ммоль) с помощью шприца. Через 5 мин при этой температуре с помощью шприца добавляют (R)-глицидил бутират (0,823 г, 0,807 мл, 5,7 ммоль) и реакционную смесь оставляют на ночь с постепенным нагреванием охлаждающей ванны до температуры окружающей среды. Согласно данным ТСХ анализа (5% метанол/хлороформ) исходное Cbz производное израсходовано. Реакционную смесь концентрируют при пониженном давлении, получая воскообразное желтое твердое вещество. Остаток растворяют в смеси 20% метанол/дихлорметан, промывают последовательно насыщенным раствором хлорида аммония, насыщенным раствором бикарбоната натрия и рассолом. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая неочищенный продукт. Радиальная хроматография на силикагеле при элюировании 2, 3 и затем 5% метанол/хлороформ дает после концентрирования соответствующих фракций 0,811 г (48%) названного соединения в виде бесцветной смолы, которая является очень чистой по данным 1H ЯМР анализа. Аналитическую пробу готовят путем перекристаллизации из смеси дихлорметан/диэтиловый эфир, получая белое твердое вещество с т.пл. 113-114oC и МС (ЭИ) 310 (М+).
Стадия 5: (R)-[[3-[3-фтор-4-(3-метокси-3-метил-1-азетидинил) фенил]-2-оксо-5-оксазолидинил]метил]метансульфонат
Раствор (R)-[3-[3-фтор-4-(3-метокси-3-метил-1-азетидинил)фенил] -2-оксо-5- оксазолидинил]метанола (0,700 г, 2,25 ммоль) в сухом дихлорметане (10 мл) охлаждают с помощью бани со льдом до 0oC. Затем добавляют триэтиламин (0,250 г, 0,345 мл, 2,48 ммоль) и метансульфонил хлорид (0,271 г, 0,183 мл, 2,37 ммоль) и реакционную смесь перемешивают в атмосфере азота до тех пор, пока охлаждающая ванна не нагреется. Через 3 ч ТСХ анализ (5% метанол/хлороформ) показал, что мезилирование в основном завершилось. Реакционную смесь разбавляют дополнительным количеством дихлорметана и промывают 0,5 N хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и рассолом. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Радиальная хроматография на силикагеле при элюировании смесью 3% метанол/хлороформ, дает после концентрирования соответствующих фракций 0,022 г непрореагировавшего исходного спирта и 0,700 г (80%) названного соединения в виде белого твердого вещества с МС (ЭИ) 388 (М+).
Стадия 6: (S)-N-[[3-[3-фтор-4-(3-метокси-3-метил-1-азетидинил) фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Смесь (R)-[[3-[3-фтор-4-(3-метокси-3-метил-1-азетидинил)фенил] -2 -оксо-5-оксазолидинил] метил] метансульфоната (0,680 г, 1,75 ммоль) в смеси 2:1 аммоний гидроксид/изопропанол (30 мл) кипятят с обратным холодильником (холодильник, охлаждаемый сухим льдом). Через 7 ч ТСХ анализ (5% метанол/хлороформ) показал присутствие большого количества исходного вещества в смеси. Реакцию продолжают еще 46 ч. После охлаждения до температуры окружающей среды ТСХ анализ показал, что исходный мезилат в основном израсходован. Реакционную смесь переносят в делительную воронку вместе с дихлорметаном (100 мл) и промывают водой. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Неочищенный 5-(аминометил)оксазолидинон растворяют в сухом дихлорметане (10 мл) охлажденном до 0oC с помощью бани со льдом, и обрабатывают пиридином (0,698 г, 0,708 мл, 8,75 ммоль) и уксусным ангидридом (0,357 г, 0,330 мл, 3,50 ммоль). Затем охлаждающую баню удаляют и реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь разбавляют дополнительным дихлорметаном (60 мл) и промывают 5% водным раствором хлористоводородной кислоты, насыщенным водным раствором карбоната натрия, водой и рассолом. Затем органический слой сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая неочищенную смолу. Радиальная хроматография на силикагеле при элюировании смесью 1, 2 и 4% метанол/хлороформ дает после концентрирования соответствующих фракций 0,481 г (78%) названного оксазолидинона-антибактериального средства в виде белого вспененного твердого вещества. Лиофилизированный образец имеет т.пл. 132-134oC и МС (ЭИ) 351 (М+).
Пример 7: (S)-N-[[3-фтор-4-(3-гидрокси-3-метил-1-азетидинил)фенил -2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: 3-фтор-4- (3-гидрокси-3-метил-1-азетидинил) нитробензол
Раствор 1-(дифенилметил)-3-метил-3-азетидинил гидрохлорида (3,00 г, 10,4 ммоль) в 2: 1 метанол/тетрагидрофуран (60 мл) обрабатывают триэтиламином (1,00 г, 1,40 мл, 10,4 ммоль) и затем катализатором гидроксид палладия/углерод (0,300 г) в потоке N2. Смесь встряхивают на аппарате Парра при давлении 2,81 кг/см2 (40 psi) H2. По данным ТСХ анализа (2:1 гексан/этилацетат) спустя 2,5 ч гидрогенолиз полностью завершается. Реакционную смесь фильтруют через Celite® (промывают дихлорметаном) и фильтрат концентрируют при пониженном давлении, получая масло, которое немедленно помещают в сухой диметилсульфоксид (15 мл) и обрабатывают дикалий кислым фосфатом (3,40 г, 19,8 ммоль) и 3,4-дифторнитробензолом (1,60 г, 1,10 мл, 9,9 ммоль) при температуре окружающей среды в атмосфере N2. После перемешивания 16 ч при этой температуре по данным ТСХ анализа (2:1 гексан:этилацетат) реакция в основном завершается. Реакционную смесь переносят в делительную воронку с H2O (150 мл) и дихлорметаном (100 мл). После встряхивания органическую фазу отделяют. Водную фазу вновь экстрагируют дихлорметаном и объединенные органические экстракты промывают водой, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая масло. Хроматография на силикагеле при элюировании смесью 3:1 гексан/этилацетат дает после концентрирования соответствующих фракций 2,15 г (98%) названного соединения в виде оранжевого твердого вещества с т.пл. 109-110oC и МС (ЭИ) 226 (М+).
Стадия 2: 4-[3-[(трет-бутилдиметилсилил)окси]-3-метил-1- азетидинил]-3-фторнитробензол
Раствор 3-фтор-4-(3-гидрокси-3-метил-1-азетидинил)нитробензола (1,80 г, 8,0 ммоль) в сухом N,N-диметилформамиде (15 мл) охлаждают до 0oC с помощью бани со льдом и затем обрабатывают имидазолом (0,572 г, 8,4 ммоль) и трет-бутилдиметилсилил хлоридом (1,30 г, 8,4 ммоль) в атмосфере N2. Реакционную смесь перемешивают на протяжении ночи с постепенным нагреванием охлаждающей бани окружающим воздухом. Затем реакционную смесь переносят в делительную воронку вместе с водой и смесью 1:1 гексан/диэтиловый эфир. После встряхивания слои разделяют и водную фазу вновь экстрагируют дополнительной смесью 1: 1 гексан/диэтиловый эфир. Объединенные органические экстракты промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая масло. Хроматография на силикагеле при элюировании смесью 3% диэтиловый эфир/гексан, и затем смесью 1:1 гексан/этилацетат, дает после концентрирования соответствующих фракций 0,536 г исходного спирта и 1,90 г (70%) названного соединения в виде светло-желтого твердого вещества с т.пл. 98-99,5oC и МС (ЭИ) 340 (М+).
Стадия 3: N-(карбобензилокси)-4-[3-[(трет-бутил-диметилсилил)окси]-3-метил-1- азетидинил]-3-фторанилин
Раствор 4-[3-[(трет-бутилдиметилсилил)окси] -3-метил-1- азетидинил]-3-фторнитробензола (1,70 г, 5,0 ммоль) в смеси 2:1 метанол/тетрагидрофуран (30 мл) дегазируют, повторяя вакуумирование и заполнение N2, и затем обрабатывают 10% палладий/углерод (0,170 г) в атмосфере азота. Добавляют твердый аммоний формиат, реакционную смесь дегазируют некоторое время и смесь охлаждают с помощью бани со льдом из-за ее нагревания. Цвет реакционной смеси изменяется от желтой до бесцветной. По данным ТСХ анализа (2:1 гексан/этилацетат) восстановление завершается. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют в вакууме, получая промежуточный анилин в виде твердого вещества, которое немедленно растворяют в смеси 1:1 ацетон/вода, охлаждают до 0oC с помощью бани со льдом и обрабатывают бикарбонатом натрия (0,840 г, 10,0 ммоль) и затем бензил хлорформиатом (0,938 г, 0,785 мл, 5,5 ммоль). Реакционную смесь оставляют перемешиваться на протяжении ночи в атмосфере N2 с постепенным нагреванием охлаждающей ванны окружающим воздухом. В сумме, через 16 часов, реакционную смесь переносят в делительную воронку вместе с дихлорметаном (100 мл). После встряхивания органическую фазу отделяют, промывают водой, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая неочищенное масло. Хроматографированием на силикагеле при элюировании смесью 9:1 гексан/этилацетат получают после концентрирования соответствующих фракций 1,68 г (76%) названного соединения в виде смолы с МС (ЭИ) 444 (М+).
Стадия 4: (R)-[3-[-4-[3-[(трет-бутилдиметилсилил)окси]-3-метил- 1-азетидинил]-3-фторфенил]-2-оксо-5-оксазолидинил]метанол
Раствор N-(карбобензилокси)-4-[3-[(трет-бутилдиметилсилил)окси] -3- метил-1-азетидинил]-3-фторанилина (1,60 г, 3,8 ммоль) в сухом тетрагидрофуране (20 мл) в атмосфере N2 охлаждают до -78oC с помощью бани со смесью сухой лед/ацетон и обрабатывают н-бутиллитием (2,40 мл, 1,55 М раствора в гексане, 3,8 ммоль) с помощью шприца. Через 5 мин при этой температуре добавляют (R)-глицидил бутират (0,548 г, 0,538 мл, 3,8 ммоль). По окончании добавления охлаждающую ванну удаляют и реакционной смеси дают нагреться до температуры окружающей среды в атмосфере N2. Через 2 ч при этой температуре ТСХ анализ (2:1 гексан/этилацетат) показал, что исходное Cbz производное израсходовано. Реакционную смесь концентрируют в вакууме и остаток растворяют в этилацетате, промывают водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая неочищенный продукт. Хроматографированием на силикагеле при элюировании 30% этилацетат/дихлорметан получают после концентрирования соответствующих фракций 1,10 г (71%) названного 5-(гидроксиметил)оксазолидинона в виде белого твердого вещества с т.пл. 143,5-144,5oC и МС (ЭИ) 410 (М+).
Стадия 5: (R)-[[3-[4-[3-[(трет-бутилдиметилсилил) окси]-3-метил-1-азетидинил]-3-фторфенил]-2-оксо-5-оксазолидинил] метил]метансульфонат
Раствор (R)-[3-[4-[3-[(трет-бутилдиметилсилил)окси] -3-метил- 1-азетидинил] -3-фторфенил] -2-оксо-5-оксазолидинил]метанола (1,00 г, 2,43 ммоль) в сухом дихлорметане (15 мл), охлаждают до 0oC на бане со льдом, и обрабатывают триэтиламином (0,295 г, 0,406 мл, 2,92 ммоль) и затем метансульфонил хлоридом (0,321 г, 0,217 мл, 2,80 ммоль) в атмосфере N2. Через 2 ч при 0oC по данным ТСХ анализа (5% метанол/хлороформ) реакция завершается. Реакционную смесь переносят в делительную воронку вместе с дополнительным количеством дихлорметана (75 мл). Органический слой промывают 0,5 N хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия, водой и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая 1,03 г (94%) названного соединения в виде белого твердого вещества с т.пл. 113-115oC и МС (ЭИ) 488 (М+).
Стадия 6: (R)-[[3-[4-[3-[(трет-бутилдиметилсилил)окси] -3-метил-1-aзетидинил]-3-фторфенил]-2-оксо-5-оксазолидинил]метил]азид
Раствор (R)-[[3-[4-[3-[(трет-бутилдиметилсилил)окси-3-метил-1-азетидинил] -3-фтopфенил] -2-оксо-5-оксазолидинил] метил]метансульфоната (0,960 г, 2,10 ммоль) в сухом N,N-диметилформамиде (20 мл) в атмосфере N2 обрабатывают азидом натрия (0,273 г, 4,2 ммоль) при температуре окружающей среды. Реакционную смесь нагревают до 70oC в течение 3 ч и затем охлаждают до комнатной температуры. ТСХ анализ (5% метанол/хлороформ) к этому времени показывает, что реакция завершилась. Растворитель удаляют в вакууме и остаток переносят в дихлорметан (100 мл) и воду. Органическую фазу отделяют, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая 0,820 г (90%) названного соединения в виде белого твердого вещества с т.пл. 127-128oC и МС (ЭИ) 435 (М+).
Стадия 7: (S)-N-[[3-[4-[3-[(трет-бутилдиметилсилил)окси]-3- метил-1-азетидинил]-3-фторфенил-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (R)-[[3-[4-[3-[(трет-бутилдиметилсилил)окси] - 3-метил-1-азетидинил]-3-фторфенил]-2-оксо-5-оксазолидинил]метил]азида (0,750 г, 1,72 ммоль) в этилацетате (20 мл) дегазируют, повторяя вакуумирование и заполнение N2. Затем раствор обрабатывают 10% палладий/углерод (0,075 г) в потоке азота. Затем атмосферу заменяют H2 (баллон), повторяя вакуумирование и наполнение, и реакционную смесь перемешивают при температуре окружающей среды. Через 6 ч по данным ТСХ анализа (5% метанол/хлороформ) реакция завершается. Реакционную смесь фильтруют через Celite® фильтрат концентрируют при пониженном давлении. Остаток растворяют в сухом дихлорметане (10 мл), охлаждают с помощью бани со льдом и обрабатывают пиридином (0,680 г, 0,696 мл, 8,60 ммоль) и уксусным ангидридом (0,263 г, 0,243 мл, 2,58 ммоль). Охлаждающую ванну удаляют и реакционную смесь перемешивают при температуре окружающей среды в атмосфере N2. Через 3 ч реакционную смесь переносят в делительную воронку вместе с дихлорметаном (100 мл). Смесь промывают 10% хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и рассолом, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Радиальная хроматография на силикагеле при элюировании 3% и затем 5% метанол/хлороформом, дает после концентрирования соответствующих фракций 0,643 г (83%) названного соединения в виде белого твердого вещества с т.пл. 137-138oC и МС (ЭИ) 451 (М+).
Стадия 8: (S)-N-[(3-[3-фтор-4-(3-гидрокси-3-метил-1-азетидинил) фенил] -2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (S)-N-[[3-[4-[3-[(трет-бутилдиметилсилил) окси]-3-метил-1-азетидинил] -3-фторфенил] -2-оксо-5-оксазолидинил]метил] ацетамида (0,550 г, 1,22 ммоль) в ацетонитриле 20 мл) в полипропиленовой колбе обрабатывают 40% водной фтористоводородной кислотой (5 мл) при температуре окружающей среды. По данным ТСХ анализа (5% метанол/хлороформ) снятие защиты завершается, в основном, через 16 ч. Реакционную смесь переносят в склянку Эрленмейера вместе с некоторым количеством дихлорметана (150 мл). Перемешиваемую смесь осторожно обрабатывают насыщенным водным раствором бикарбоната натрия (300 мл, выделение газа). После прекращения выделения газа смесь переносят в делительную воронку и фазы разделяют. Водную фазу нейтрализуют до pH 7 путем добавления 6 N хлористоводородной кислоты и экстрагируют дихлорметаном. Объединенные органические экстракты сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Радиальная хроматография на силикагеле при элюировании 5% метанол/хлороформ дает после концентрирования соответствующих фракций 0,374 г (91%) названного оксазолидинона-антибактериального агента в виде белого твердого вещества с т.пл. 152-153oC и МС (ЭИ) 337 (М+).
Примеры пирролидинон-фенилоксазолидинонов
Пример 8: (S)-N-[[3-[4-(1-Аза-5,5-диметил-4,6-диоксабицикло [3.3.0]октан-1-ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид
3-Фтор-1-нитро-4-(2,5-дигидропиррил)бензол
Раствор 2,30 г (14,47 ммоль) 2,4-динитрофторбензола и 5,04 (28,93 ммоль) двуосновного фосфата калия в 30 мл ДМСО обрабатывают 1,0 г (14,47 ммоль) 3-пирролина, а затем нагревают при 60oC в течение 24 ч. Раствор охлаждают и разбавляют 100 мл хлороформа, а затем экстрагируют водой (5 x 75 мл). Сушка (Na2SO4) и концентрирование в вакууме дает желтое твердое вещество, которое перекристаллизовывают из горячей смеси этилацетат-гексан, получая 2,58 г (86%) названного соединения в виде желтых призм, т.пл. 132-134oC.
3-Фтор-1-нитро-4-(цис-3,4-дигидроксипирролидинил)бензол
Раствор 1,25 г (6,02 ммоль) предыдущего соединения и 881 мг (7,52 ммоль) N-метилформалин N-оксида в 60 мл ацетона и 13 мл воды обрабатывают 4 мл 2,5% раствора тетроксида фосфора в трет-бутиловом спирте с последующим перемешиванием при температуре окружающей среды в течение 24 ч. Раствор 500 мг бисульфита натрия, 2 г магнезола (magnesol) и 10 мл воды добавляют при перемешивании в течение 20 мин. Смесь фильтруют через целит, промывают осадок на фильтре ацетоном. Фильтрат концентрируют в вакууме и разбавляют 100 мл этилацетата и 150 мл воды. Водный слой экстрагируют этилацетатом (3 x 100 мл) и объединенные органические слои сушат (Na2SO4) и концентрируют в вакууме, получая желтое твердое вещество. Это вещество перекристаллизовывают из горячей смеси ацетон-гексан, получая 1,33 г (91%) названного соединения в виде желтого твердого вещества, т.пл. 151-153oC.
1-(1-Азa-5,5-диметил-4,6-диоксабицикло[3.3.0]октан-1-ил)-2-фтор- 4-нитробензол (3)
Раствор 1,27 г (5,24 ммоль) предыдущего диола в 10 мл диметилформамида обрабатывают 1,09 г (1,3 мл, 10,49 ммоль) 2,2-диметоксипропаном и несколькими кристаллами п-толуолсульфокислоты, а затем перемешивают при температуре окружающей среды в течение 24 ч. Раствор разбавляют 50 мл хлороформа и экстрагируют водой (5 х 30 мл). Сушка (Na2SO4) и концентрирование в вакууме дает 1,45 г (98%) названного соединения в виде желтого твердого вещества, достаточно чистого для дальнейшего использования. Аналитический образец получают перекристаллизацией из горячей смеси ацетон-гексан, т.пл. 113-114oC.
1-(1-Аза-5,5-диметил-4,6-диоксабицикло[3.3.0]октан-1-ил)-2- (фенилметоксикарбонил)аминобензол
Раствор 1,45 г (5,14 ммоль) предыдущего соединения в 50 мл тетрагидрофурана обрабатывают 260 мг 10% палладия-на-угле, а затем гидрируют при одной атмосфере в течение 3 ч. Реакционный сосуд продувают азотом и обрабатывают 20 мл насыщенного раствора бикарбоната натрия с последующим охлаждением до 0oC и добавлением 1,75 г (1,47 мл, 10,27 ммоль) бензил хлорформиата. Раствор перемешивают при 0oC в течение 30 мин с последующим нагреванием до температуры окружающей среды в течение 18 ч. Смесь разбавляют 50 мл этилацетата и 50 мл воды и фильтруют через целит, промывая осадок на фильтре этилацетатом. Фильтраты отделяют и органическую фазу экстрагируют 50 мл воды и 50 мл насыщенного раствора хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме дает твердое вещество светло-пурпурного цвета, которое перекристаллизовывают из горячей смеси этилацетат-гексан, получая 1,56 г (79%) названного соединения в виде тонких белых кристаллов. 1H ЯМР (CDCl3) δ 7,39, 7,25, 6,93, 6,67, 5,19, 4,80, 3,73, 2,97, 1,53, 1,37.
(R)-[3-[4-(1-Аза-5,5-диметил-4,6-диоксабицикло[3.3.0]октан-1- ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метанол (5)
Раствор 1,36 г (3,52 ммоль) Cbz производного предыдущего соединения в 48 мл тетрагидрофурана при -78oC обрабатывают, добавляя по каплям, 2,41 мл (1,6 М, 3,87 ммоль) н-бутиллития в гексане с последующим перемешиванием при -78oC в течение 30 мин. Затем раствор обрабатывают 558 мг (0,55 мл, 3,87 ммоль) чистого (R)-(+)-глицидил бутирата, а затем нагревают до 0oC в течение 30 мин и затем при температуре окружающей среды в течение 18 ч. Затем раствор охлаждают до 0oC и обрабатывают 4 мл насыщенного водного раствора аммоний хлорида, затем разбавляют 200 мл дихлорметана и экстрагируют водой (2 x 50 мл) и насыщенным раствором аммоний хлорида (50 мл). Сушка (Na2SO4) и концентрирование в вакууме дает коричневое твердое вещество, которое хроматографируют на 80 г 230-400 меш силикагеле, элюируя 2% (об./об.) метанола в дихлорметане. Эти операции дают 840 мг названного соединения в виде беловатого твердого вещества. 1H ЯМР (CDCl3) δ : 7,34, 7,06, 6,71, 4,80, 4,75, 4,01, 3,73, 3,67, 3,56, 2,95, 1,50, 1,36.
(S)-[[3-[4-(1-Аза-5,5-диметил-4,6-диоксабицикло[3.3.0] октан-1- ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]метансульфонат
Раствор 791 мг (2,25 ммоль) предыдущего спирта и 283 мг (0,39 мл, 2,81 ммоль) триэтиламина в 11 мл дихлорметана при 0oC обрабатывают 296 мг (0,20 мл, 2,58 ммоль) метансульфонил хлорида, а затем перемешивают при 0oC в течение 30 мин. Смесь нагревают до температуры окружающей среды и разбавляют 75 мл дихлорметана с последующей экстракцией водой (2 х 30 мл) и насыщенным раствором бикарбоната натрия (30 мл). Сушка (Na2SO4) и концентрирование в вакууме дает 810 мг (84%) названного мезилата в виде беловатого твердого вещества, достаточно чистого для дальнейшего использования. 1H ЯМР (CDCl3) δ : 7,36, 7,07, 6,73, 4,90, 4,82, 4,48, 4,41, 4,10, 3,89, 3,77, 3,10, 3,02, 1,51, 1,37.
(S)-N-[[3-[4-(1-Аза-5,5-диметил-4,6-диоксабицикло[3.3.0] октан-1- ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор 810 мг (1,88 ммоль) предыдущего мезилата в 8 мл смеси 1:1 тетрагидрофуран-изопропиловый спирт в повторно-герметизируемой толстостенной ампуле обрабатывают 8 мл гидрохлорида аммония. Ампулу запаивают и нагревают при 100oC в течение 22 ч. Ампулу охлаждают, и реакционная смесь распределяется между хлороформом и водой. Органический слой сушат (Na2SO4) и концентрируют в вакууме, получая 520 мг желтовато-коричневого твердого вещества, которое используют растворенным в 2 мл пиридина и обрабатывают 357 мг (0,33 мл, 3,50 ммоль) уксусного ангидрида с последующим перемешиванием при температуре окружающей среды в течение 18 ч. Раствор разбавляют хлороформом (20 мл) и экстрагируют водой (4 х 20 мл). Органический слой сушат (Na2SO4) и концентрируют в вакууме, и полученное твердое вещество перекристаллизовывают из смеси хлороформ/гексан, получая 350 мг (59%) названного ацетамида в виде беловатых игл, т.пл. 195-7oC.
Пример 9: (S)-N-[[3-[4-(3,4-цис-дигидроксипирролидинил)-3- фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид (9)
Раствор 249 мг (0,63 ммоль) соединения, полученного в Примере 8, в 2 мл тетрагидрофурана обрабатывают 2 мл раствора 2 N хлористоводородной кислоты, затем перемешивают при температуре окружающей среды в течение 18 ч. Смесь концентрируют в вакууме и остаток обрабатывают 30 мл насыщенного раствора бикарбоната натрия. Смесь экстрагируют этилацетатом (2 x 50 мл), сушат (Na2SO4) и концентрируют в вакууме, получая 110 мг беловатого твердого вещества.
Это вещество подвергают радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя 7% (об./об.) метанола в дихлорметане. Эти операции дают 60 мг (28%) названного диола в виде белого твердого вещества, т.пл. 183-189oC (разлагается).
Пример 10: (S)-N-[[3-[4-(3-гидроксипирролидинил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид
3-Фтор-4-(3-гидроксипирролидинил) нитробензол
К раствору 3-пирролидинола (1,82 г) и 3,4-дифторнитробензола (3,0 г) в диметилформамиде (30 мл) добавляют карбонат натрия (3,94 г). Смесь перемешивают при комнатной температуре в течение ночи и затем фильтруют с последующей промывкой дихлорметаном. Объединенный фильтрат концентрируют при пониженном давлении. Полученный остаток хроматографируют на колонке с силикагелем смесью этилацетат/гексан с увеличивающимся содержанием этилацетата (от 50 до 60%). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (4,83 г); 1H ЯМР (CD3OD, 270 МГц), δ : 0,85 (2H, м), 2,25-2,7 (4H, м), 3,26 (1H, м), 5,47 (1H, т, J = 8,6 Гц), 6,62 (1H, дд, J= 2,4, 14,0 Гц), 6,69 (1H, дд, J = 2,4, 8,6 Гц).
3-Фтор-4-[3-(трет-бутилдиметилсилилокси)пирролидинил]нитробензол
Смесь предыдущего соединения (4,83 г), триэтиламина (9,5 мл) и диметиламинопиридина (2,79 г) в тетрагидрофуране (100 мл) обрабатывают трет-бутилдиметилсилил хлоридом (5,15 г) при 0oC и перемешивают при комнатной температуре в течение 2 дней. Смесь фильтруют и нерастворимое вещество промывают дихлорметаном. Объединенный фильтрат промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный остаток хроматографируют на колонке с силикагелем, элюируя смесью этилацетат/гексан (70/30). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (11) (6,45 г); 1H ЯМР (CDCl3) δ : 0,09 (6H, с), 0,88 (9H, с), 2,02 (2H, м), 3,42-3,78 (4H, м), 4,52 (1H, м), 6,53 (1H, т, J= 8,9 Гц), 7,87 (1H, дд, J = 2,4, 14,0 Гц), 7,94 (1H, дд, J = 2,4, 8,9 Гц).
3-Фтор-4-[3-(трет-бутилдиметилсилилокси)пирролидинил]анилин
Палладий-на-угле (10%, 500 мг) добавляют к смеси предыдущего соединения (5,0 г) в смешанном растворителе дихлорметана (20 мл) и метанола (100 мл). Смесь перемешивают при 1 атмосфере водорода в течение 6 ч, затем фильтруют и палладий-на-угле промывают метанолом и дихлорметаном. Объединенные фильтраты концентрируют при пониженном давлении, получая названное соединение (4,52 г); 1H ЯМР (CDCl3) δ : 0,08 (6H, с), 0,88 (9H, с), 1,87 (1H, м), 2,11 (1H, м), 3,05 (1H, м), 3,29 (2H, м), 3,56 (1H, м), 4,48 (1H, м), 6,38 (1H, дд, J = 2,4, 8,9 Гц), 6,44 (1H, дд, J = 2,4, 14,0 Гц), 6,61 (1H, т, J = 8,9 Гц).
1-Бензилоксикарбониламино-4-[3-(трет-бутилдиметилсилилокси) пирролидинил]-3-фторбензол
Бензил хлорформиат (3,0 мл) добавляют к смеси предыдущего соединения (4,52 г) и бикарбоната натрия (1,76 г) в тетрагидрофуране (50 мл) при 0oC. Смесь перемешивают при комнатной температуре в течение ночи и затем фильтруют с последующей промывкой тетрагидрофураном. Объединенные фильтраты сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный остаток подвергают обработке на колонке с силикагелем, элюируя гексан/этилацетат (90/10). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (4,40 г); 1H ЯМР (CDCl3) δ/ : 0,08 (6H, с), 0,88 (9H, с), 1,87 (1H, м), 2,11 (1H, м), 3,05 (1H, м), 3,29 (2H, м), 3,56 (1H, м), 4,48 (1H, м), 5,18 (2H, с), 6,59 (1H, т, J = 8,9 Гц), 6,88 (1H, дд, J = 2,4, 8,9 Гц), 7,10 (1H, дд, J = 2,4, 14,0 Гц), 7,36 (5H, м).
(S)-[[3-[4-[3-(трет-бутилдиметилсилилокси)пирролидинил] -3- фторфенил]] -2-оксо-5-оксазолидинил]метанол
Раствор предыдущего соединения (4,40 г) в сухом тетрагидрофуране (50 мл) охлаждают до -78oC в азоте и обрабатывают 7,3 мл (1,6 М в гексане) н-BuLi при перемешивании в течение 5 мин. Смесь обрабатывают 1,65 мл (R)-(+)-глицидил бутирата и дают ей медленно нагреться до комнатной температуры в течение ночи. Реакцию гасят путем добавления насыщенного раствора сульфата аммония (10 мл) и смесь экстрагируют дихлорметаном (3 x 50 мл). Объединенный органический слой промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный остаток хроматографируют на колонке с силикагелем, элюируя смесью гексан/этилацетат с возрастающим содержанием этилацетата (50 до 70%). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (1,44 г); 1H ЯМР (CDCl3) δ : 0,08 (6H, с), 0,88 (9H, с), 1,91-2,10 (2H, м), 3,18 (1H, м), 3,39-3,48 (2H, м), 3,63 (1H, м), 3,78 (1H, м), 3,87-4,01 (3H, м), 4,48 (1H, м), 4,66 (1H, м), 6,63 (1H, т, J=0 9,5 Гц), 7,04 (1H, дд, J=2,4, 9,5 Гц), 7,32 (1H, дд, J=2,4, 14,6 Гц).
(S)-[[3-[4-[3-(трет-бутилдиметилсилилокси)пирролидинил] -3-фторфенил]-2-оксо-5-оксазолидинил]метил]метансульфонат
К раствору предыдущего соединения (1,44 г) в пиридине (10 мл) добавляют при перемешивании п-толуолсульфонил хлорид (0,88 г) при 0oC. Смесь перемешивают при комнатной температуре в течение ночи. Медленно к смеси при перемешивании добавляют воду (40 мл) и смесь дополнительно перемешивают в течение 1 ч, получая кристаллы. Кристаллы собирают фильтрацией, промывают водой и сушат при 40oC при пониженном давлении в течение ночи, получая названное соединение (1,96 г); 1H ЯМР (CDCl3) δ : 0,09 (6H, с), 0,88 (9H, с), 1,90-2,08 (2H, м), 2,46 (3H, с), 3,21 (1H, м), 3,40-3,49 (2H, м), 3,65 (1H, м), 3,81 (1H, дд, J = 5,9, 8,9 Гц), 4,03 (1H, т, J=8,9 Гц), 4,24 (2H, м), 4,49 (1H, м), 4,80 (1H, м), 6,63 (1H, т, J=9,5 Гц), 6,96 (1H, дд, J =2,4, 9,5 Гц), 7,23 (1H, дд, J=2,4, 14,6 Гц), 7,36 (2H, д, J=7,8 Гц), 7,79 (2H, д, J= 7,8 Гц).
(S)-N-[[3-[4-[3-(трет-бутилдиметилсилилокси)пирролидинил] -3- фторфенил] -2-оксо-5-оксазолидинил]метил]ацетамид
Смесь предыдущего соединения (1,96 г) в диметилформамиде (20 мл) обрабатывают азидом натрия (455 мг) и нагревают при 60oC в течение ночи. После охлаждения до комнатной температуры смесь фильтруют и осадок промывают дихлорметаном. Объединенные фильтраты концентрируют при пониженном давлении. Полученный остаток хроматографируют на колонке с силикагелем, элюируя смесью гексан/ацетон (80/20). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая соединение (1,40 г). Раствор этого соединения в сухом тетрагидрофуране (20 мл) обрабатывают трифенилфосфином (868 мг) и перемешивают при комнатной температуре в течение 2 ч. Смесь обрабатывают водой (0,5 мл) и перемешивают при 40oC в течение 4 ч, затем при комнатной температуре в течение ночи. Смесь концентрируют при пониженном давлении и сушат путем азеотропной перегонки с толуолом. Полученный остаток суспендируют в дихлорметане (20 мл) и обрабатывают пиридином (0,5 мл) и уксусным ангидридом (0,6 мл) при 0oC. Смесь перемешивают при комнатной температуре в течение ночи и концентрируют при пониженном давлении. Остаток подвергают обработке на колонке с силикагелем, элюируя смесью метанол/дихлорметан с возрастающим содержанием метанола (1 до 3%). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (1,502 г); 1H ЯМР (CDCl3) δ : 0,08 (6H, с), 0,88 (9H, с), 1,89-2,09 (2H, м), 2,02 (3H, с), 3,18 (1H, м), 3,36-3,67 (6H, м) 3,71 (1H, дд, J= 6,8, 8,9 Гц), 3,99 (1H, т, J= 8,9 Гц), 4,48 (1H, м), 4,76 (1H, м), 6,20 (1H, широкий, NH), 6,61 (1H, т, J = 9,2 Гц), 6,98 (1H, дд, J =2,4, 9,2 Гц), 7,30 (1H, дд, J = 2,4, 15,1 Гц).
(S)-N-[[3-[4-[3-(гидроксипирролидинил)-3-фторфенил] -2-оксо-5- оксазолидинил]метил]ацетамид
Смесь предыдущего соединения (1,50 г) в сухом тетрагидрофуране (8 мл) обрабатывают тетрабутиламмоний фторидом (6,7 мл; 1,0 М в тетрагидрофуране) при 0oC и дают возможность нагреться до комнатной температуры при перемешивании в течение 4 ч. Смесь обрабатывают водой (50 мл) и экстрагируют дихлорметаном (30 мл x 4). Объединенный органический слой промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток подвергают обработке на колонке с силикагелем, элюируя смесью ацетон/гексан (80/20). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (0,77 г); 1H ЯМР (DMSO-d6) δ : 1,78-2,00 (2H, м), 1,80 (3H, с), 3,12 (1H, м), 3,26-3,54 (5H, м), 3,67 (1H, дд, J = 6,8, 8,9 Гц), 4,04 (1H, т, J = 8,9 Гц), 4,34 (1H, м), 4,68 (1H, м), 4,91 (1H, д, J = 4,1, OH), 6,72 (1H, т, J= 9,7 Гц), 7,07 (1H, дд, J = 2,4, 9,7 Гц), 7,38 (1H, дд, J = 2,4, 16,2 Гц), 8,23 (1H, т, J = 5,7 Гц, NH).
Пример 11: (S)-N-((3-(3-фтор-4-(3-оксопирролидинил)фенил)-2- оксо-5-оксазолидинил)метил)ацетамид
7-Бензил-7-аза-1,4-диоксаспиро(4,5)нонан
К суспензии 1-N-бензил-3-пирролидинона (1,92 г) и этиленгликоля (6,81 г) в бензоле (70 мл) добавляют моногидрат п-толуолсульфокислоты (1,93 г). Смесь кипятят с азеотропным удалением воды с помощью ловушки Дин-Старка (Dean-Stark) в течение 6 ч. После охлаждения до комнатной температуры смесь промывают насыщенным водным раствором бикарбоната натрия (50 мл х 3). Затем водный слой экстрагируют дихлорметаном (30 мл х 3). Органические слои объединяют, промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении, получая названное соединение (1,57 г), 1H ЯМР (CDCl3) δ : 2,06 (2H, т, J = 7,3 Гц), 2,64 (2H, с), 2,66 (2H, т, J = 7,3 Гц), 3,61 (2H, с), 3,89 (4H, м), 7,28 (5H, м).
7-Аза-1,4-диоксаспиро(4,5)нонан
Гидроксид палладия-на-угле (20%, 200 мг) добавляют к раствору предыдущего соединения (1,57 г) в смешанном растворителе дихлорметана (10 мл) и метанола (20 мл). Смесь перемешивают под давлением 3 кгс/см2 водорода в течение 2 дней, затем фильтруют и гидроксид палладия-на-угле промывают метанолом и дихлорметаном. Объединенный фильтрат концентрируют при пониженном давлении, получая названное соединение (1,38 г); 1H ЯМР (CDCl3) δ : 2,17 (2H, т, J=7,3 Гц), 3,35 (2H, с), 3,52 (2H, т, J=7,3 Гц), 3,99 (4H, с).
4-(7-Аза-1,4-диоксоспиро(4,5)нонан-7-ил)-3-фтор-нитробензол
Следуя общей методике Примера 9 и внося несущественные изменения, за исключением того, что используют в качестве исходного вещества предыдущее соединение (1,38 г), получают названное соединение (1,50 г), 1H ЯМР (CDCl3) δ : 2,18 (2H, т, J = 7,3 Гц), 3,67 (2H, с), 3,72 (2H, т, J=7,3 Гц), 4,02 (4H, с), 6,53 (1H, т, J=7,3 Гц), 7,88 (1H, дд, J=2,4, 14,0 Гц), 7,94 (1H, дд, J= 2,4, 8,9 Гц).
4-(7-Аза-1,4-диоксаспиро(4,5)нонан-7-ил)-3-фтор-1- (фенилметоксикарбониламино)-бензол
Следуя общей методике Примера 9 и внося несущественные изменения, за исключением того, что используют в качестве исходного вещества предыдущее соединение (1,49 г), получают названное соединение (2,44 г), 1H ЯМР (CDCl3) δ : 2,16 (2H, т, J= 6,8 Гц), 3,41 (2H, т, J = 6,8 Гц), 3,46 (2H, с), 3,98 (4H, с), 5,18 (2H, с), 6,60 (1H, т, J=9,2 Гц), 6,91 (1H, дд, J =2,4 9,2 Гц), 7,22 (1H, дд, J =2,4, 14,0 Гц), 7,37 (5H, м).
(R)-(3-(3-фтор-4-(7-аза-1,4-диоксаспиро(4,5)нонан-7-ил)фенил) -2-оксо-5-оксазолидинил)метанол
Раствор предыдущего соединения (2,44 г) в сухом диметилформамиде (25 мл) охлаждают до -78oC в азоте и обрабатывают 6,6 мл (1,0 М в тетрагидрофуране) литий бис(триметилсилил)амида при перемешивании в течение 5 мин. Смесь обрабатывают 0,93 мл (R)-(-)-глицидил бутирата и медленно нагревают до комнатной температуры на протяжении ночи. Hебольшой избыток литий бис(триметилсилил)амида гасят путем добавления воды (5 мл). Смесь концентрируют при пониженном давлении и сушат с помощью азеотропной перегонки с толуолом. Остаток суспендируют в дихлорметане (100 мл) и нерастворимое вещество удаляют фильтрованием. Фильтрат концентрируют при пониженном давлении и полученный остаток подвергают обработке на колонке с силикагелем, элюируя смесью гексан/ацетон (50/50). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (1,79 г); 1H ЯМР (DMSO-d6) δ : 2,07 (2H, т, J = 7,3 Гц), 3,35 (4H, перекрываемый сигналами, обусловленными растворителем), 3,50-3,69 (2H, м), 3,78 (1H, дд, J=7,3, 9,2 Гц), 3,92 (4H, с), 4,02 (1H, т, J=9,2 Гц), 4,67 (1H, м), 5,20 (1H, т, J= 5,9 Гц, OH), 6,77 (1H, т, J= 10,3 Гц), 7,13 (1H, дд, J=2,4, 10,3 Гц), 7,46 (1H, дд, J=2,4, 16,2 Гц).
(R)-N-((3-(3-фтор-4-(7-аза-1,4-диоксаспиро(4,5)нонан-7-ил)фенил) -2-оксо-5-оксазолидинил)метил)азид
Следуя общей методике Примера 9 и внося несущественные изменения, за исключением того, что используют в качестве исходного вещества предыдущее соединение (1,78 г), получают названное соединение (1,76 г), 1H ЯМР (CDCl3) δ : 2,14 (2H, т, J= 7,3 Гц), 2,44 (3H, с), 3,45 (2H, т, J= 7,3 Гц), 3,49 (2H, с), 3,73- 4,13 (2H, м), 3,98 (4H, с), 4,24 (2H, м), 4,80 (1H, м), 6,60 (1H, т, J= 9,5 Гц), 6,96 (1H, дд, J=2,4, 9,5 Гц), 7,23 (1H, дд, J=2,4, 14,6 Гц), 7,36 (2H, д, J=7,8 Гц), 7,78 (2H, д, J=7,8 Гц).
(R)-N-((3-(3-фтор-4-(7-аза-1,4-диоксаспиро(4,5)нонан-7-ил)фенил) -2-оксо-5-оксазолидинил)метил)ацетамид
Следуя общей методике Примера 9 и внося несущественные изменения, за исключением того, что Lindlar катализатор (150 мг) затем добавляют к смеси предыдущего соединения (17) (1,45 г) в смешанном растворителе дихлорметана (10 мл) и метанола (40 мл). Смесь перемешивают при атмосферном давлении водорода в течение 2 дней, затем фильтруют и катализатор промывают метанолом и дихлорметаном. Объединенный фильтрат концентрируют при пониженном давлении. Полученный остаток суспендируют в дихлорметане (20 мл) и обрабатывают пиридином (0,3 мл) и уксусным ангидридом (0,7 мл) при 0oC. Смесь перемешивают при комнатной температуре в течение ночи и концентрируют при пониженном давлении. Остаток подвергают обработке на колонке с силикагелем, элюируя смесью метанол/дихлорметан с возрастающим содержанием метанола (2 до 5%). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (0,72 г); 1H ЯМР (CDCl3) δ : 2,01 (3H, с), 2,16 (2H, т, J=7,3 Гц), 3,43 (2H, т, J=7,3 Гц), 3,47 (2H, с), 3,53-3,67 (2H, м), 3,71 (1H, дд, J=6,8, 9,2 Гц), 3,98 (5H, м), 4,75 (1H, м), 6,27 (1H, широкий), 6,61 (1H, т, J=9,2 Гц), 6,99 (1H, дд, J=2,4, 9,2 Гц), 7,33 (1H, дд, J= 2,4, 15,1 Гц).
(S)-N-((3-(3-фтор-4-оксопирролидинил)фенил)-2-оксо-5-оксазолидинил) метил)ацетамид
Смесь предыдущего соединения (610 мг) в ацетоне (12 мл) и воды (3,7 мл) обрабатывают моногидратом п-толуолсульфокислоты (609 мг) и кипятят с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры смесь концентрируют при пониженном давлении. Остаток обрабатывают 5 мл триэтиламина и экстрагируют дихлорметаном (30 мл х 5). Органические слои объединяют, промывают рассолом, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток подвергают обработке на колонке с силикагелем, элюируя смесью гексан/ацетон (50/50). Соответствующие фракции объединяют и концентрируют при пониженном давлении, получая названное соединение (390 мг); 1H ЯМР (CDCl3) δ : 2,03 (3H, с), 2,65 (2H, т, J=6,8 Гц), 3,47-3,78 (7H, м), 4,02 (1H, т, J = 8,9 Гц), 4,76 (1H, м), 6,18 (1H, т, J=5,9 Гц, NH), 6,77 (1H, т, J=9,2 Гц), 7,09 (1H, дд, J= 2,4, 9,2 Гц), 7,43 (1H, дд, J=2,4, 15,1 Гц).
Пример 12: (S)-N-((3-(3-Фтор-4-(3-(фенилметоксиацетиламино) пирролидинил)фенил)-2-оксо-5-оксазолидинил)метил)ацетамид
Стадия 1: 3-Фтор-1-нитро-4-(3-трифторацетиламино)- пирролидинилбензол
Раствор 4,49 г (20,54 ммоль) 3-(трифторацетиламино)-пирролидин гидрохлорида и 2,97 г (18,68 ммоль) 3,4-дифторнитробензола и 7,16 г (41,10 ммоль) двуосновного фосфата калия в 87 мл ДМСО нагревают при 90oC в течение 18 ч. Смесь охлаждают и разбавляют 300 мл этилацетата. Раствор экстрагируют водой (5 x 100 мл) и сушат (Na2SO4). Концентрация в вакууме дает желтое твердое вещество, которое перекристаллизовывают из горячей смеси этилацетат-гексан, получая 5,0 г (84%) названного продукта в виде желтого кристаллического твердого вещества, т.пл. 165-167oC.
Стадия 2: 3-Фтор-1-(фенилметоксикарбонил)амино-4- (3-трифторацетиламино)пирролидинилбензол
Раствор 4,10 г (12,76 ммоль) нитросоединения в 100 мл этилацетата обрабатывают 500 мг 10% палладия-на-угле с последующим гидрированием при атмосферном давлении в течение 4 часов. Смесь фильтруют через целит, промывая осадок на фильтре этилацетатом. Фильтрат концентрируют в вакууме и остаток растворяют в 50 мл ацетона и обрабатывают 75 мл насыщенного раствора NaHCO3 с последующим охлаждением до 0oC и обработкой 4,36 г (3,64 мл, 25,53 ммоль) бензил хлорформиата. Затем раствор нагревают до температуры окружающей среды в течение 18 ч. Смесь разбавляют 200 мл этилацетата и экстрагируют водой (2 x 50 мл) с последующей сушкой (Na2SO4). Концентрация в вакууме дает бежевое твердое вещество, которое перекристаллизовывают из горячей смеси этилацетат-гексан, получая 4,41 г (81%) названного Cbz производного в виде бледно-бежевых игл, т.пл. 143-145oC.
Стадия 3: 3-Фтор-1-(фенилметоксикарбонил)амино-4-(3-(1,3- диметилгексагидро-2-оксо-1,3,5-триазин-5-ил)пирролидинил)бензол
Раствор 1,86 г (4,37 ммоль) Cbz производного в 36 мл ТГФ обрабатывают 50 мл 2 N NaOH с последующим кипячением с обратным холодильником в течение 36 ч. Раствор охлаждают и разбавляют 50 мл воды с последующей экстракцией этилацетатом (4 x 75 мл). Объединенные органические слои экстрагируют насыщенным раствором NaCl с последующей сушкой (Na2SO4) и концентрированием в вакууме. Полученное масло янтарного цвета растворяют в 20 мл диоксана и 40 мл толуола и образующийся раствор обрабатывают 385 мг (4,37 ммоль) N,N'-диметилмочевины и 5 мл 37% водного раствора формальдегида. Смесь нагревают при температуре кипения флегмы до тех пор, пока прибл. 5 мл воды не соберется в ловушке Дин-Старка. Раствор охлаждают и концентрируют в вакууме. Остаток хроматографируют на 180 г 230-400 меш силикагеле, элюируя 2% (об./об.) метанолом в дихлорметане. Эти операции дают 1,40 г (73%) названного продукта в виде тонких белых кристаллов, т.пл. 192-194oC.
Стадия 4: (R)-(3-(3-Фтор-4-(3-(1, З-диметилгексагидро-2-оксо-1,3,5- триазин-5-ил)пирролидинил)фенил)-2-оксо-5-оксазолидинил)метанол
Раствор 737 мг (1,67 ммоль) триазона в 10 мл ДМФ при -50oC обрабатывают 1,84 мл (1,84 ммоль) литий гексаметилдисилазида в ТГФ. Раствор перемешивают при -50oC в течение 5 мин, затем добавляют 265 мг (0,26 мл, 1,84 ммоль) (R)-(+)-глицидил бутирата. Затем раствор нагревают до температуры окружающей среды в течение 18 ч. Смесь концентрируют в вакууме (около 0,3 мм Hg) и остаток хроматографируют на 36 г 230-400 меш силикагеле, элюируя 3% (об./об.) метанолом в дихлорметане. Эти операции дают 400 мг (59%) названного продукта в виде белого твердого вещества. 1H ЯМР (CDCl3) δ : 7,36, 7,05, 6,65, 4,72, 4,27, 4,18, 3,95, 3,77, 3,46, 2,88, 2,17, 1,94.
Стадия 5: (R)-(3-(3-Фтор-4-(3-(1,3-диметилгексагидро-2-оксо-1,3,5 -триазин-5-ил)пирролидинил)фенил)-2-оксо-5-оксазолидинил) метансульфонат
Раствор 395 мг (0,97 ммоль) оксазолидинона и 146 мг (0,20 мл, 1,45 ммоль) триэтиламина в 5 мл дихлорметана при 0oC обрабатывают 471 мг (0,32 мл) метансульфонил хлорида. Раствор перемешивают при 0oC в течение 30 мин с последующим нагреванием до температуры окружающей среды. Раствор обрабатывают водой и водный слой экстрагируют дихлорметаном. Объединенные органические слои сушат (Na2SO4) и концентрируют в вакууме, получая 460 мг (98%) названного продукта в виде беловатого твердого вещества, используемого на следующей стадии. 1H ЯМР (CDCl3) δ : 7,33, 7,03, 6,63, 4,89, 4,45, 4,27, 4,18, 4,12, 3,89, 3,76, 3,55, 3,47, 3,38, 3,10, 2,88, 2,16, 2,08, 1,92.
Стадия 6: (S)-N-((3-(3-Фтор-4-(3-(фенилметоксиацетиламино)- пирролидинил)фенил)-2-оксо-5-оксазолидинил)метил)ацетамид
Раствор 460 мг (0,97 ммоль) мезилата в 15 мл изопропилового спирта и 15 мл гидроксида аммония в герметизированной трубке нагревают при 100oC в течение 18 ч. Смесь охлаждают и разбавляют водой с последующей экстракцией хлороформом. Органический слой сушат (Na2SO4) и концентрируют в вакууме. Остаток растворяют в 4 мл пиридина и обрабатывают 0,5 мл уксусного ангидрида с последующим перемешиванием при температуре окружающей среды в течение 18 ч. Смесь концентрируют в вакууме, получая масло, которое растворяют в хлороформе и экстрагируют водой. Органический слой сушат (Na2SO4) и концентрируют в вакууме, получая 472 мг производного ацетамида, которое непосредственно растворяют в 4 мл 1N HCl раствора с последующим перемешиванием при температуре окружающей среды в течение 18 часов. Смесь концентрируют в вакууме и растворяют в 4 мл ацетона и обрабатывают 2 мл насыщенного раствора NaHCO3 с последующей обработкой 217 мг (0,14 мл, 1,18 ммоль) бензилоксиацетил хлорида, за которым следует перемешивание при температуре окружающей среды в течение 1 ч. Смесь концентрируют в вакууме и остаток хроматографируют на 25 г 230-400 меш силикагеле, элюируя смесью 2% (об./об.) метанола в дихлорметане и затем 3% (об./об.) метанола в дихлорметане. Эти операции дают 189 мг (40%) продукта - производного бензилоксиацетамида в виде белого твердого вещества. 1H ЯМР (CDCl3) δ : 7,33, 7,02, 6,81, 6,68, 6,08, 4,75, 4,60, 4,01, 3,71, 3,61, 3,34, 2,32, 2,02, 1,91.
Пример 13: (S)-N-((3-(3-Фтор-4-(3-(гидроксиацетиламино) пирролидинил)фенил)-2-оксо-5-оксазолидинил)метил)ацетамид
Продолжая процедуру Примера 12, стадию 6, раствор 189 г (0,39 ммоль) бензилоксиацетамида в 5 мл метанола и 5 мл ТГФ обрабатывают 200 мг 10% палладия-на-угле с последующим гидрированием при атмосферном давлении в течение 1 ч. Раствор разбавляют метанолом и фильтруют через целит, промывая осадок на фильтре метанолом. Фильтрат концентрируют в вакууме, получая масло, которое хроматографируют на 10 г 230-400 меш силикагеле, элюируя смесью 7% (об. /об.) метанола в дихлорметане. Эти операции дают 95 мг (62%) продукта-производного гидроксиацетамида в виде розового твердого вещества. 1H ЯМР (CDCl3) δ : 7,41, 7,10, 6,78, 4,77, 4,53, 4,10, 4,00, 3,77, 3,57, 3,32, 2,30, 1,99, 1,98.
Пример 14: (S)-N-[[3-Фтор-4-(цис-3-(метоксикарбониламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: цис-3-(1,1-диметилэтоксикарбониламино)-4- метилпирролидин
Раствор 1,5 г (5,2 ммоль) цис-1-фенилметил-3- (1,1-диметилэтоксикарбониламино)-4-метилпирролидина (ср. Pat US 4 753 953) в 50 мл метанола обрабатывают 200 мг Pd(OH)2/C с последующим гидрированием при одной атмосфере H2 в течение 3,5 ч. Смесь фильтруют через целит, промывая осадок на фильтре метанолом. Фильтрат концентрируют в вакууме, получая бесцветное масло, которое используют непосредственно в следующей стадии. 1H ЯМР (CD3OD) δ : 4,85, 4,03, 3,14, 3,05, 2,66, 2,45, 2,23, 1,46, 0,95.
Стадия 2: 3-Фтор-1-нитро-4-[цис-3-(1,1- диметилэтоксикарбониламино)-4-метилпирролидинил]бензол
Раствор ранее полученного соединения и 1,8 г (10,3 ммоль) двуосновного фосфата в 40 мл ДМСО нагревают при 90oC в течение 18 ч. Смесь охлаждают и разбавляют 125 мл этилацетата, затем экстрагируют водой (4 х 75 мл). Органический слой сушат (Na2SO4) и концентрируют в вакууме, получая 1,75 г желтого твердого вещества, достаточно чистого для дальнейшего использования. МС (ЭИ) м/з 339 (М+), 339, 282, 266, 223, 222, 208, 207, 154, 70, 57.
Стадия 3: 3-Фтор-1-нитро-[цис-3-(1,3-диметилгексагидро-2-оксо- 1,3,5-триазин-5-ил)-4-метилпиppoлидинил]бензол
Раствор 1,75 г (5,17 ммоль) предыдущего соединения в 50 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 18 ч. Раствор концентрируют в вакууме и полученное красно-коричневое масло растворяют в этилацетате и осторожно экстрагируют насыщенным раствором NaHCO3 (4х). Объединенные водные слои экстрагируют этилацетатом, и объединенные органические слои сушат (Na2SO4) и концентрируют в вакууме. Остаток растворяют в 90 мл толуола и 45 мл диоксана и обрабатывают 1,12 г (12,7 ммоль) N, N'-диметилмочевины и 35 мл водного формальдегида, а затем кипятят с обратным холодильником в течение 2,5 часа. Смесь охлаждают и концентрируют в вакууме. Остаток хроматографируют на 200 г 230-400 меш силикагеле смесью 1-2% (об. /об.) метанола в хлороформе. Эти операции дают 3,65 г (86%) требуемого продукта в виде маслянисто-желтоватого вещества. МС (ЭИ) м/з 351 (М+), 223, 222, 221, 208, 207, 182, 169, 154, 130, 113.
Стадия 4: 3-Фтор-1-(фенилметоксикарбонил)амино-[цис-3-(1,3- диметилгексагидро-2-оксо-1,3,5-триазин-5-ил)-4-метилпирролидинил] бензол
Раствор 1,82 г (5,17 моль) в 200 мл ТГФ обрабатывают 750 мг 10% палладия-на-угле, а затем гидрируют при давлении водорода 3,16 кг/см2 (45 psi) в течение 3 ч. Смесь охлаждают до -20oC с последующим добавлением 30 мл насыщенного раствора NaHCO3. Затем смесь обрабатывают 1,10 г (0,92 мл, 6,46 ммоль) бензил хлорформиата, с последующим нагреванием до температуры окружающей среды в течение 18 ч. Смесь фильтруют через целит, промывая осадок на фильтре метанолом и дихлорметаном. Фильтрат концентрируют в вакууме и остаток растворяют в дихлорметане и экстрагируют водой (3 x 30 мл). Сушка (Na2SO4) и концентрирование в вакууме дает пурпурную пену, которую хроматографируют на 125 г 230-300 меш силикагеля, элюируя смесью 1-5% метанола в дихлорметане. Эти операции дают 1,74 г (74%) названного соединения в виде стойкой пены. 1H ЯМР (CDCl3) δ : 7,34, 6,92, 6,53, 5,18, 4,19, 4,13, 3,69, 3,58, 3,52, 3,41, 3,15, 2,86, 2,37, 1,05.
Стадия 5: (R)-(3-(3-Фтор-4-(цис-3-(1,3-диметилгексагидро-2-оксо- 1,3,5-триазин-5-ил)-4-метилпирролидинил)фенил)- 2-оксо-5-оксазолидинил)метанол
Раствор 860 мг (1,89 ммоль) предыдущего соединения в 25 мл ТГФ при -78oC обрабатывают 1,3 мл (2,08 ммоль) н-бутиллития в гексане, с последующим нагреванием до -50oC. После перемешивания при -50oC в течение 20 мин добавляют 286 мг (0,28 мл, 1,98 ммоль) (R)-(-)-глицидил бутирата. Смесь нагревают до 0oC в течение 30 мин и затем до температуры окружающей среды в течение 18 ч. Смесь обрабатывают насыщенным раствором NH4Cl и разбавляют этилацетатом и затем экстрагируют водой (3 х). Водную фазу экстрагируют этилацетатом и объединенные органические слои сушат (Na2SO4) и концентрируют в вакууме, получая коричневое масло. Это вещество хроматографируют на 40 г 230-400 меш силикагеля, элюируя метанолом в дихлорметане. Эти операции дают 512 мг (64%) названного соединения в виде белой стойкой пены. 1H ЯМР (CDCl3) δ : 7,35, 7,03, 6,60,4,72, 4,22, 4,14, 3,94, 3,75, 3,70, 3,60, 3,54, 3,43, 3,18, 2,87, 2,39, 1,05.
Стадия 6: (R)-(3-(3-Фтор-4-(цис-3-(1,3-диметилгексагидро-2-оксо- 1,3,5-триазин-5-ил)-4-метилпирролидинил)фенил)-2-оксо-5-оксазолидинил) метансульфонилоксиметан
Раствор 1 г (2,37 ммоль) предыдущего соединения и 480 мг (0,66 мл, 4,74 ммоль) триэтиламина в 15 мл дихлорметана при 0oC обрабатывают 407 мг (0,28 мл, 3,56 ммоль) метансульфонил хлорида. Раствор перемешивают при 0oC в течение 1,5 ч с последующим нагреванием, разбавлением дихлорметаном и экстракцией водой (3 х). Органический слой сушат (Na2SO4) и концентрируют в вакууме, получая названное соединение. 1H ЯМР (CDCl3 δ : 7,45, 7,02, 6,92, 4,92, 4,50 (1H), 4,43, 4,23, 4,17, 3,91, 3,83, 3,65, 3,50, 3,29, 3,11, 2,89, 2,48, 1,12.
Стадия 7: (S)-(N-((3-Фтор-4-(цис-3-(1,3-диметил-гексагидро-2- оксо-1,3,5-триазин-5-ил)-4-метилпирролидинил)фенил)-2-оксо- 5-оксазолидинил)метил)ацетамид
Раствор предыдущего мезилата в 6 мл ТГФ и 6 мл изопропилового спирта обрабатывают 6 мл концентрированного гидроксида аммония. Смесь нагревают до 100oC в герметизированной трубке в течение 48 ч. Смесь охлаждают и разбавляют этилацетатом и водой. Смесь экстрагируют этилацетатом (2 х). Смесь обрабатывают насыщенным раствором NaCl и экстрагируют хлороформом (3 х). Объединенные органические слои сушат (Na2SO4) и концентрируют в вакууме. Остаток растворяют в 15 мл пиридина и охлаждают до 0oC. Раствор обрабатывают 0,5 мл уксусного ангидрида с последующим нагреванием до температуры окружающей среды в течение 20 ч. Смесь концентрируют в вакууме и остаток хроматографируют на 75 г 230-400 меш силикагеля, элюируя метанолом в дихлорметане. Эти операции дают 934 мг (85%) названного соединения в виде белой стойкой пены. МС (ББА, FAB) м/z 463 (М+Н), 464, 463, 462, 461, 364, 363, 361, 333, 101, 44.
Стадия 8: (S)-(N)-((3-Фтор-4-(цис-3-амино-4-метил-пирролидинил) фенил)-2-оксо-5-оксазолидинил)метил)ацетамид
Раствор 875 мг (1,89 ммоль) предыдущего соединения в 5 мл раствора 2N HCl перемешивают при температуре окружающей среды в течение 20 ч. Раствор концентрируют в вакууме и остаток сушат путем азеотропной перегонки с толуолом. Эти операции дают 662 мг (прибл. 100%) названного соединения в виде стойкой пены янтарного цвета. МС (ББА) м/z, 351 (М+Н), 427, 352, 351, 350, 349, 255, 123, 101, 89, 44. ВРМС(БАА) рассч. для C17H23FN4O3+H1 351, 1832, найдено 351, 1840.
Стадия 9: (S)-N-[[3-Фтор-4-(цис-3-(метоксикарбониламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор 150 мг (0,43 ммоль) предыдущего соединения в 6 мл ацетона и 3 мл воды обрабатывают 108 мг (1,29 ммоль) NaHCO3 и охлаждают до 0oC. Раствор затем обрабатывают 89 мг (73 мкл, 0,95 ммоль) метил хлорформиата. Раствор затем разбавляют этилацетатом и экстрагируют водой (3 х). Сушка (Na2SO4) и концентрирование в вакууме дает белое твердое вещество, которое подвергают радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя метанолом в дихлорметане. Эти операции дают 155 мг (53%) названного соединения в виде белого твердого вещества. МС (ЭИ) м/z 408 (М+), 409, 408, 364, 334, 333, 318, 289, 276, 215, 56.
Пример 15: (S)-N-[[3-Фтор-4-(цис-3-(гидроксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: (S)-N-[[3-Фтор-4-(цис-3-(фенилметоксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор 300 мг (0,90 ммоль) амина (Пример 14, Стадия 8) в 14 мл ацетона и 7 мл воды обрабатывают 302 мг (3,60 ммоль) NaHCO3 и охлаждают до 0oC. Раствор затем обрабатывают 414 мг (0,35 мл, 2,25 ммоль) бензилоксиацетил хлорида, с последующим нагреванием до температуры окружающей среды в течение 72 ч. Смесь разбавляют этилацетатом и водой и органический слой экстрагируют водой (3 х). Органический слой сушат (Na2SO4) и концентрируют в вакууме, получая коричневое масло. Это вещество хроматографируют на 30 г 230-400 меш силикагеля, элюируя метанолом в дихлорметане. Эти операции дают 132 мг (30%) названного соединения в виде белого твердого вещества. 1H ЯМР (CDCl3) δ : 7,57, 7,36, 7,03, 5,98, 4,77, 4,67, 4,63, 4,04, 3,74, 3,70, 3,63, 3,55, 3,45, 3,38, 2,71, 2,03, 1,07.
Стадия 2: (S)-N-[[3-Фтор-4-(цис-3-(гидроксиацетиламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор 107 мг (0,21 ммоль) предыдущего соединения в 25 мл метанола обрабатывают 100 мг 10% палладия-на-угле с последующим гидрированием при одной атмосфере в течение 72 ч. Смесь фильтруют через целит, и фильтрат концентрируют в вакууме. Остаток подвергают радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя метанолом в дихлорметане. Эти операции дают 24 мг (8%) названного соединения в виде светло-бежевого твердого вещества. МС (ЭИ) м/z 408 (М+), 408, 334, 333, 318, 289, 215, 70, 57, 56, 55.
Пример 16: (S)-N-[[3-Фтор-4-(цис-3-(метансульфониламино)-4- метилпирролидинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Суспензию 200 мг (0,57 ммоль) амина (пример 14, Стадия 8) в 10 мл дихлорметана обрабатывают 144 мг (0,20 мл, 1,43 ммоль) триэтиламина с последующим охлаждением полученного раствора до 0oC. Смесь обрабатывают 83 мг (56 мкл, 0,73 ммоль) метансульфонил хлорида с последующим нагреванием при температуре окружающей среды в течение 18 ч. Смесь разбавляют 50 мл этилацетата с последующей экстракцией водой (3 х 30 мл). Органический слой сушат (Na2SO4) и концентрируют в вакууме, получая беловатое твердое вещество, которое подвергают радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя метанолом в дихлорметане. Эти операции дают 11 мг (45%) названного соединения в виде белого твердого вещества. МС (ЭИ) м/z 428 (М+), 429, 428, 384, 300, 280, 238, 236, 177, 70, 56.
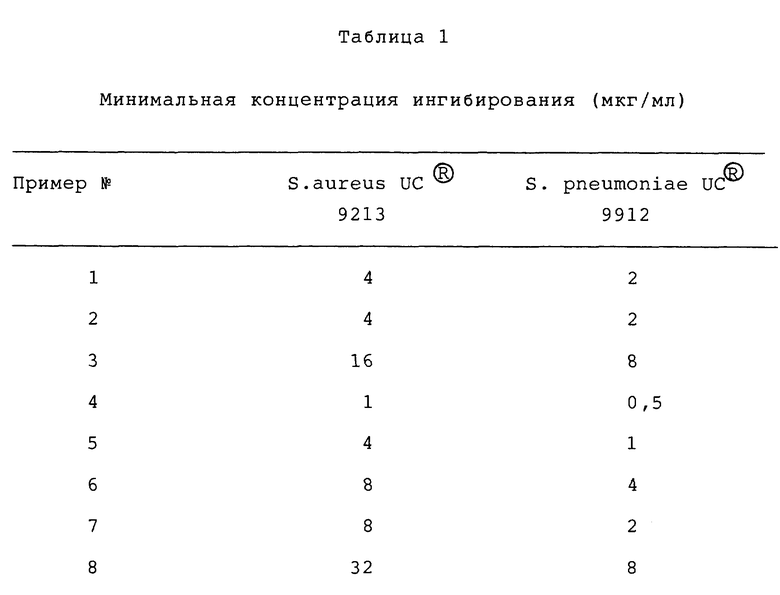

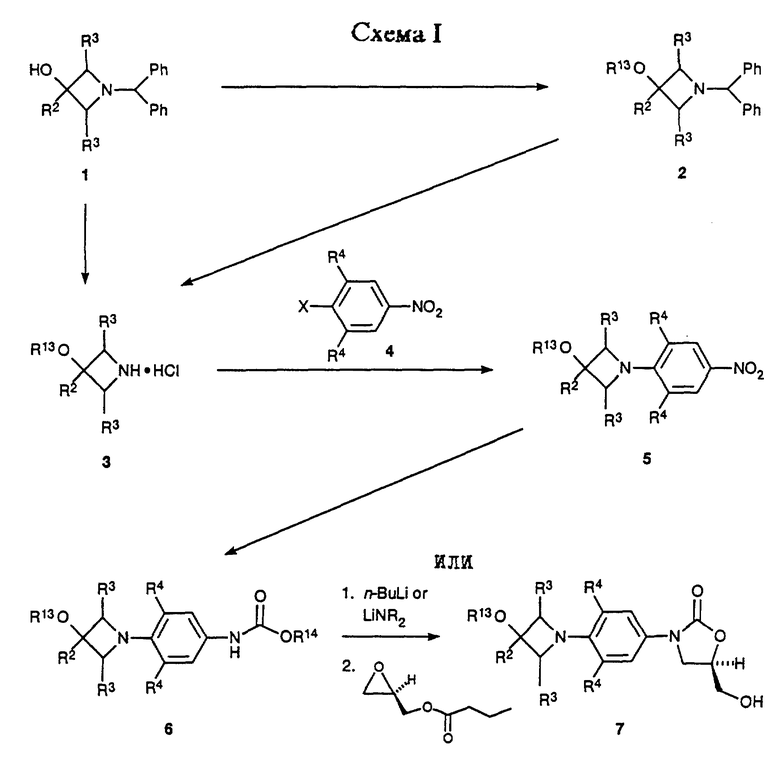
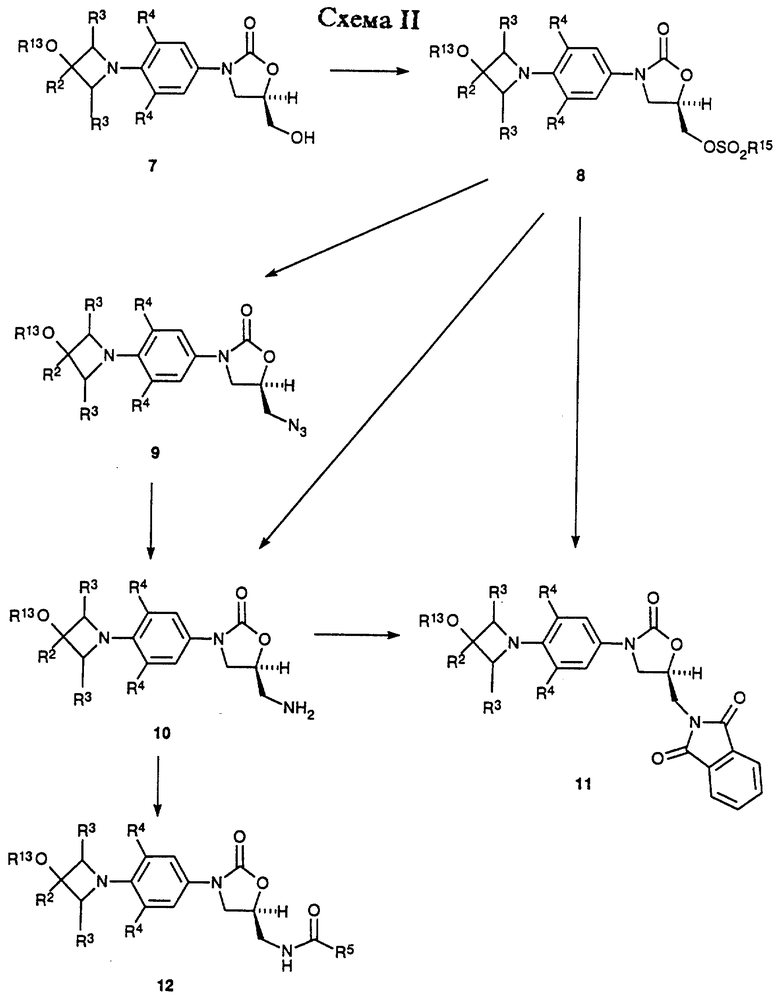
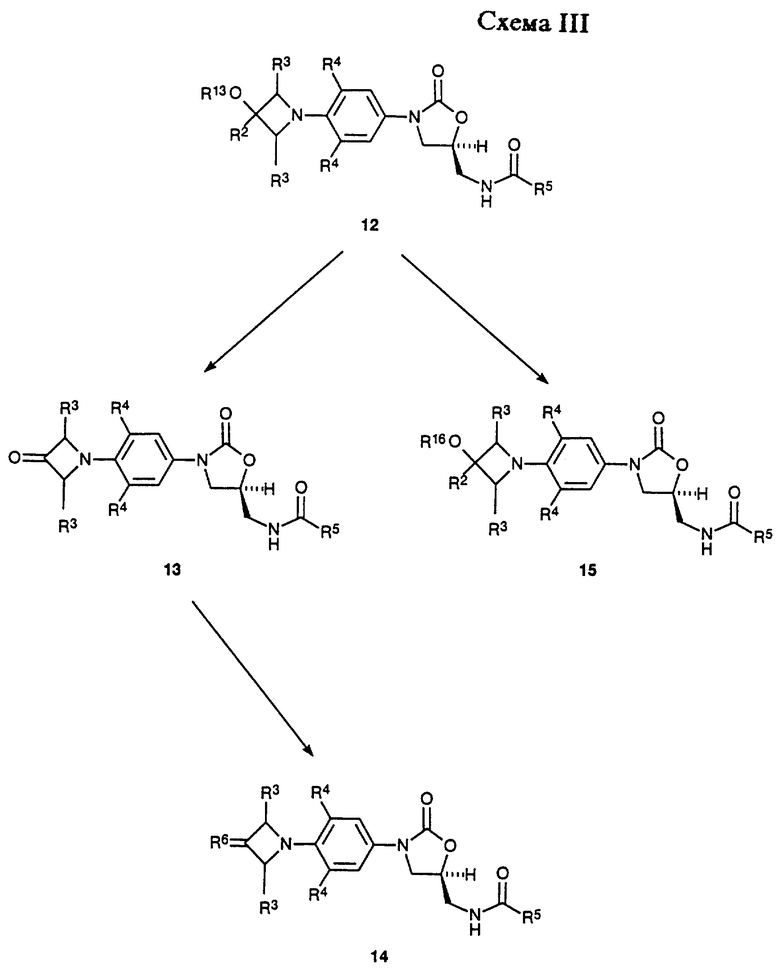


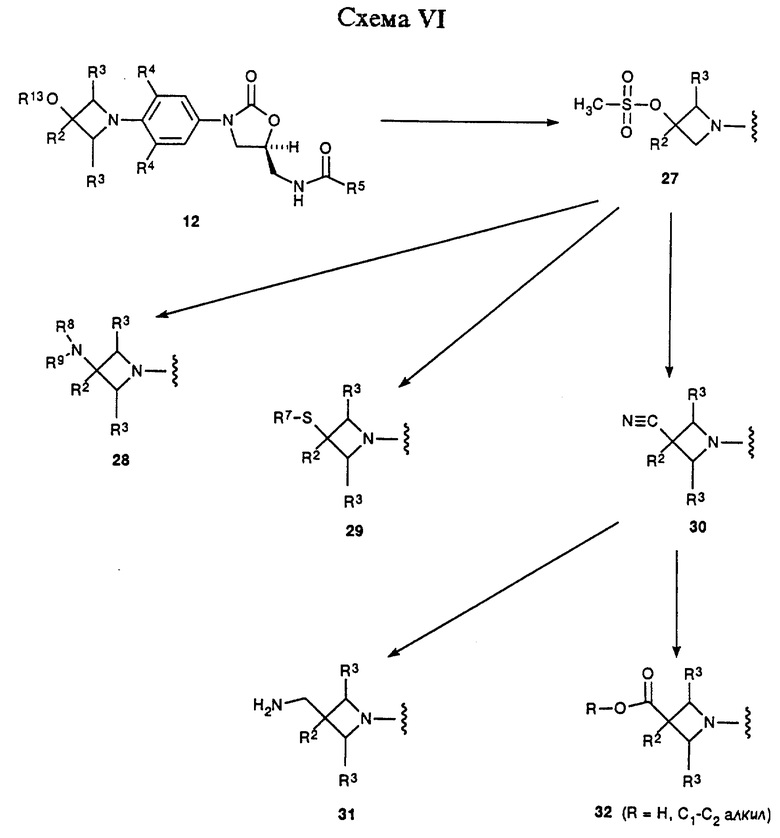
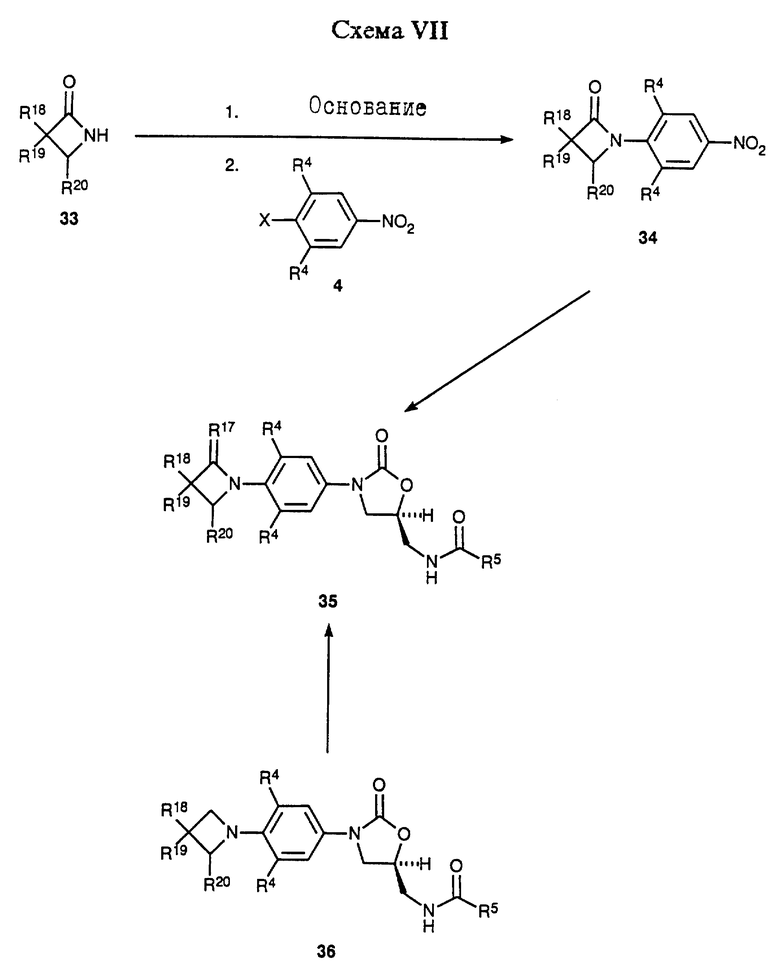
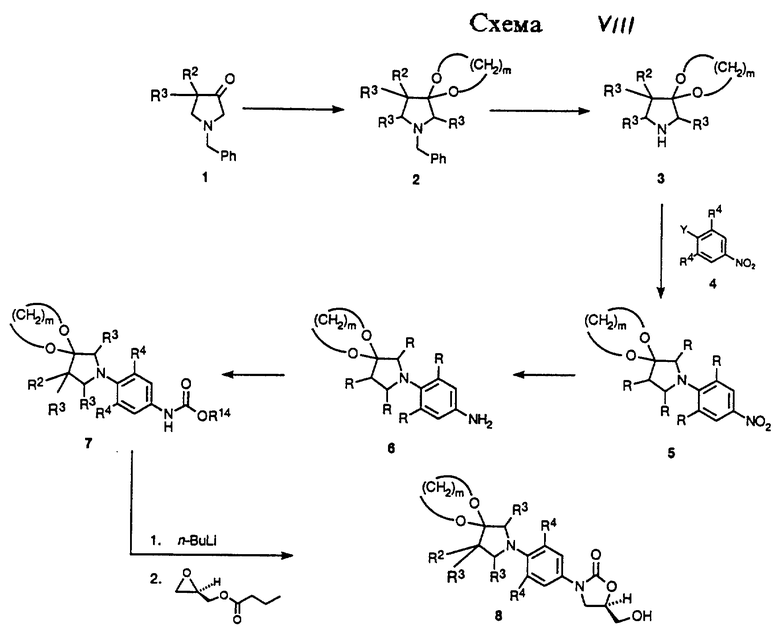
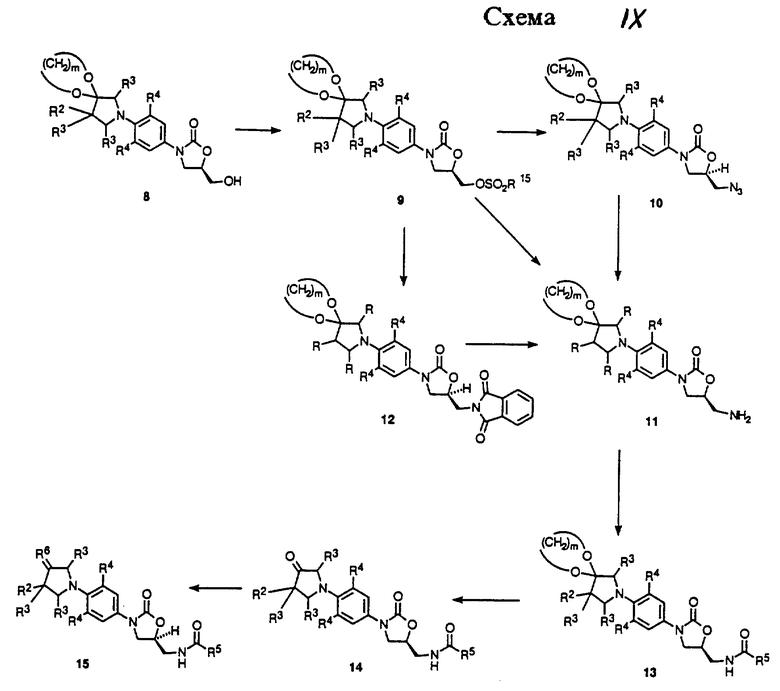
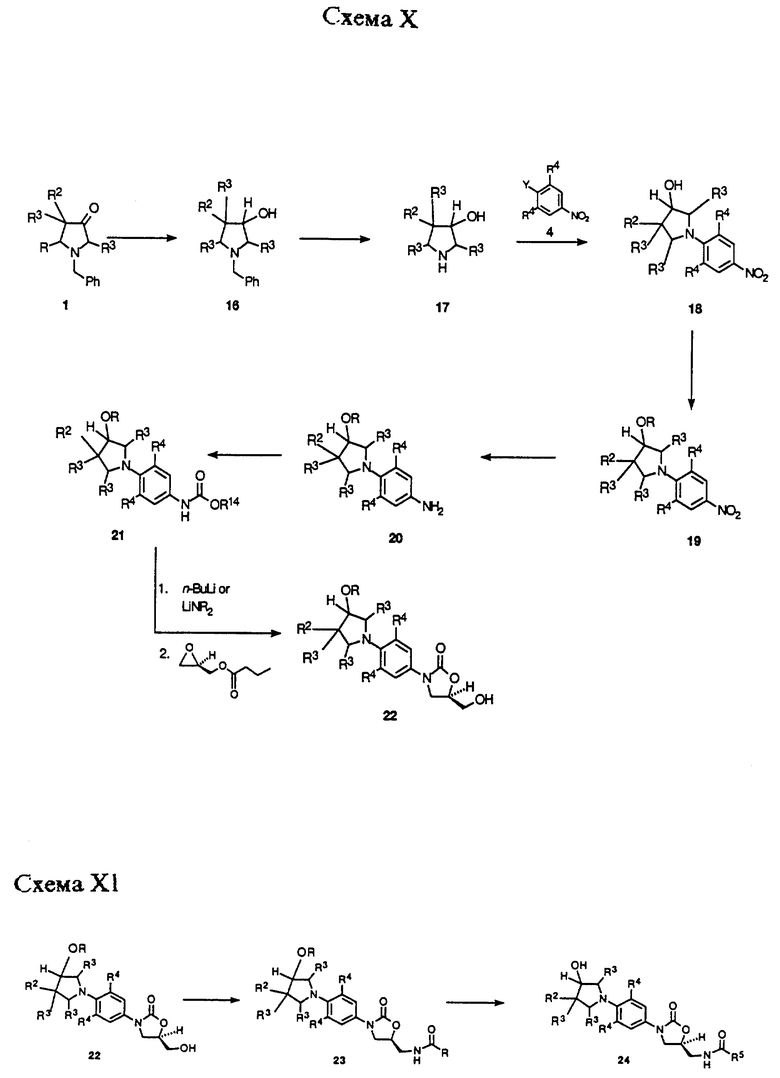
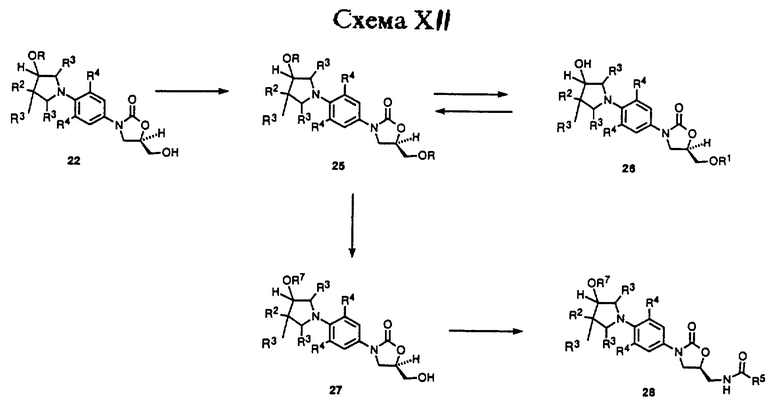
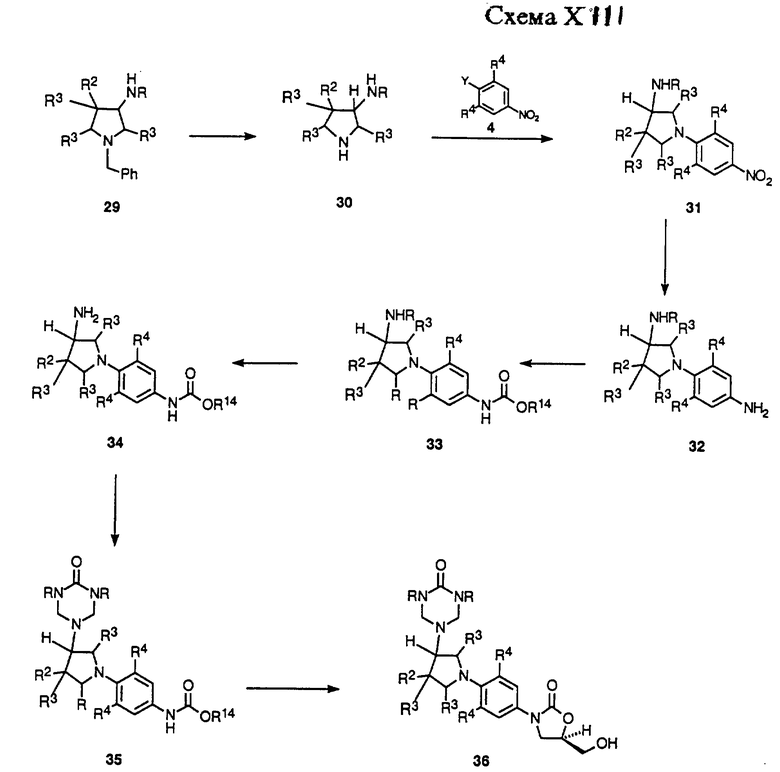
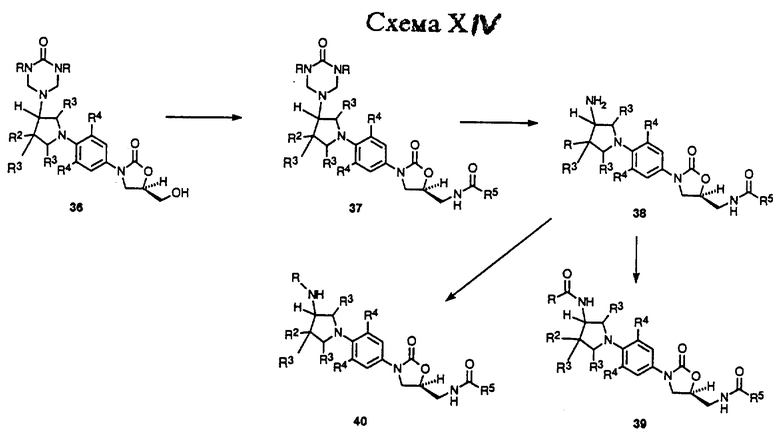

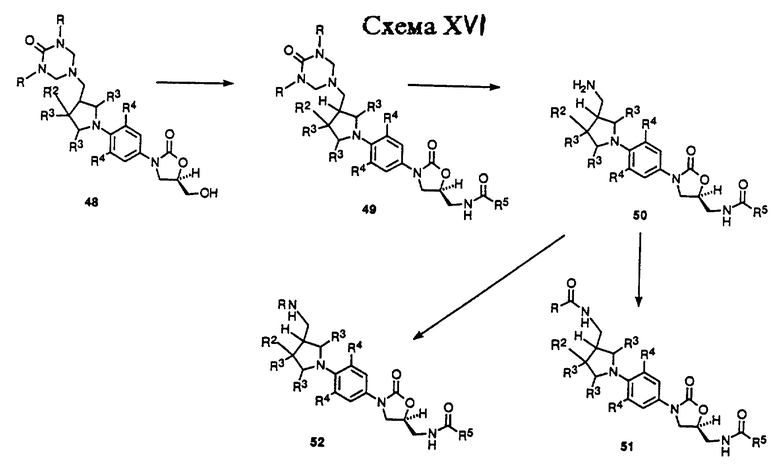
Изобретение относится к новым производным фенилоксазолидинона общей формулы (I), в которой Q выбирают из формулы  V, R1 представляет водород, OR7, где R7 является водородом, фтором, C1-C8-алкилом, NR8R4, где R8 и R4 является C1-C8-алкилом, который может быть замещен фтором, C1-C8-ацилом, который может быть замещен гидрооксилом, C1-C8-алкокси, бензоилом, R2 представляет водород, гидроксигруппу, OR, где R является C1-C6-алкилом, R3 представляет водород, C1-C3-алкил, R4 представляет C1-C8-алкил, который может быть замещен одним-тремя заместителями, выбранными из фтора, хлора, C3-C6-циклоалкила, R6 представляет кислород, NR10, где R10 является OR7, где R7 имеет вышеуказанные значения, O(CH2)mO, n равен 0 или 1 и m равен 2 или 3, и их фармацевтически приемлемые соли. 8 з.п.ф-лы, 1 табл., 15 схем.
V, R1 представляет водород, OR7, где R7 является водородом, фтором, C1-C8-алкилом, NR8R4, где R8 и R4 является C1-C8-алкилом, который может быть замещен фтором, C1-C8-ацилом, который может быть замещен гидрооксилом, C1-C8-алкокси, бензоилом, R2 представляет водород, гидроксигруппу, OR, где R является C1-C6-алкилом, R3 представляет водород, C1-C3-алкил, R4 представляет C1-C8-алкил, который может быть замещен одним-тремя заместителями, выбранными из фтора, хлора, C3-C6-циклоалкила, R6 представляет кислород, NR10, где R10 является OR7, где R7 имеет вышеуказанные значения, O(CH2)mO, n равен 0 или 1 и m равен 2 или 3, и их фармацевтически приемлемые соли. 8 з.п.ф-лы, 1 табл., 15 схем.

где Q выбирают из формул 
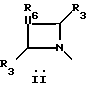
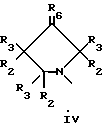
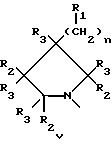
R1 представляет H, OR7, где R7 является H, F, C1 - C8-алкилом, NR8R9, где R8 и R9 являются C1 - C8-алкилом, который может быть замещен F; C1 - C8-ацил, который может быть замещен гидроксилом, C1 - C8-алкокси, бензоилом;
R2 представляет H, OH, OR, где R является C1 - C6-алкилом;
R3 представляет H, C1 - C3-алкил;
R4 представляет F;
R5 представляет C1 - C8-алкил, который может быть замещен одним-тремя заместителями, выбранными из F, Cl; C3 - C6-циклоалкил;
R6 представляет O, NR10, где R10 является OR7, где R7 имеет вышеуказанные значения, O(CH2)mO;
n равен 0 или 1;
m равен 2 или 3,
или их фармацевтически приемлемые соли.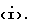
3. Соединение по п.1, в котором Q представляет структуру 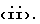
4. Соединение по п.1, в котором Q представляет структуру 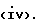
5. Соединение по п.1, в котором Q представляет структуру (V).
| УСТРОЙСТВО для АВТОМАТИЧЕСКОГО ОТКРЫВАНИЯ ЗАДНЕГО БОРТА СА.МОСВАЛЬНОГО КУЗОВА | 0 |
|
SU316594A1 |
| УСТРОЙСТВО для ПРОДОЛЬНОЙ РЕЗКИ полосового ПОЛИМЕРНОГО МАТЕРИАЛАi:.-K;iu;^:j-M-.xH?!it>&.iiMj| | 0 |
|
SU352781A1 |
| М.Д.Машковский | |||
| Лекарственные средства | |||
| - М.: Медицина, т | |||
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Букса для железнодорожного подвижного состава | 1922 |
|
SU329A1 |

