Данное изобретение раскрывает новые и полезные соединения фенилоксазолидинона, отличающиеся тем, что они имеют бициклический тиазиновый, либо оксазиновый заместитель. Соединения являются полезными противомикробными агентами, эффективными против ряда патогенов человека и патогенов животных, включая грамположительные аэробные бактерии, такие как множественно-резистентные стафилококки, стрептококки и энтерококки, а также анаэробные организмы, такие как Bacteroides spp. и Clostridia spp., такие как Mycobakterium tuberculosis, Mycobacterium avium и Mycobacterium spp.
Информационные данные
Данные соединения родственны благодаря своей структуре фенилоксазолидинонового кольца, соединениям, раскрытым в публикациях ниже, за исключением того, что соединения изобретения имеют либо бициклический тиазиновый, либо оксазин фениловый заместитель. Рассматриваемые соединения имеют полезную антибактериальную активность.
PCT/US 94/08904 заявка раскрывает оксазолидиноновые антибактериальные соединения, имеющие либо морфолиновый, либо тиоморфолиновый заместитель.
PCT/US 93/03570 заявка раскрывает оксазолидиноны, содержащие замещенную диазиновую часть и их использование в качестве антимикробных средств.
PCT/US 92/08267 заявка раскрывает замещенные арил- и гетероарил-фенил-оксазолидиноны в качестве антибактериальных агентов.
PCT/US 89/03548 заявка раскрывает 5'индолил -5β- амидометилоксазолидиноны, 3-(конденсированное-кольцо замещенный)фенил -5β- амидометилоксазолидиноны, и 3-(азотзамещенный)фенил -5β- амидометилоксазолидиноны, которые используют в качестве антибактериальных агентов.
К другим ссылкам, раскрывающим различные оксазолидиноны относятся Пат. США 4 801 600, 4 921 869, Gregory W.A., et al., J. Med. Chem., 32, 1673-81 (1989); Gregory W.A., et al., J. Med. Chem., 33, 2569-78 (1990); Wang С., et al. , Tetrahedron, 45, 1323-26 (1989); и Brittelli, et al., J. Med. Chem., 35, 1156 (1992).
Евр. Пат. публикация 352 781 раскрывает фенил и пиридилзамещенные фенил оксазолидиноны.
Евр. Пат. публикация 316 594 раскрывает 3-замещенные стирил оксазолидиноны.
Евр. Пат. публикация 312 000 раскрывает фенилметил и пиридинилметилзамещенные фенил оксазолидиноны.
Сущность изобретения
В одном аспекте предметом изобретения является соединение формулы I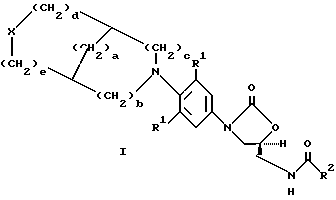
Более предпочтительные соединения, подкласс соединений, описываемых структурной формулой I, представлены структурной формулой II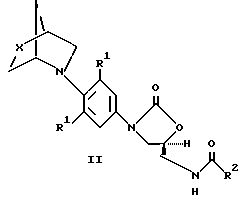
или их фармацевтически приемлемые соли,
где X представляет (a) O, (b) S, (c) SO, (d) SO2;
R1 представляет независимо H, F, Cl или OMe;
R2 представляет (a) водород, (b) C1-C8-алкил, возможно замещенный одним, или более, заместителем, выбранным из: F, Cl, гидрокси C1-C8-алкокси, C1-C8-ацилокси, (c) C3-C6- циклоалкил, (d) амино, (е) C1-C8-алкиламино, (f) C1-C8-диалкиламино, (g) C1-C8-алкокси,
a = 0-3;
b = 0-2;,
c = 0-2, (при условии, что b и c не могут быть оба 0);
d = 0-2; и
e = 0-2, (при условии, что d u e не могут быть оба 0).
В другом аспекте предметом изобретения является способ лечения микробных инфекций у людей или других теплокровных животных путем введения пациенту, нуждающемуся в таком лечении, эффективного количества описанного выше соединения формулы I или II. Соединение можно вводить в фармацевтической композиции либо перорально, парентерально, либо локально. Предпочтительно, соединение вводят в количестве от около 0,1 до около 100 мг/кг веса тела/день, более предпочтительно, от около 3,0 до около 50 мг/кг веса тела/день.
Детальное описание изобретения
Данное изобретение раскрывает новые замещенные бициклические оксазинил- или тиазинилфенилоксазолидиноны вышеописанной структурной формулы I и II. Соединения полезны в качестве антимикробных агентов, эффективных против ряда патогенов человека и патогенов животных, в частности, аэробных грамположительных бактерий, включая множественно-резистентные стафилококки и стрептококки, а также анаэробные организмы, такие как бактероиды и Clostridia виды, и кислотоустойчивые организмы, такие как Mycobacterium tuberculosis и другие микобактериальные виды.
"Алкил" означает цепи углеродных атомов, имеющие указанное число атомов углерода, которые могут быть либо неразветвленными, либо разветвленными.
"Алкокси" означает группу с указанным числом атомов углерода, соединенных с кислородом, такую группу, как метокси (-OCH3), этокси, бутокси и т.д. и ее изомерные формы.
"Ацилокси" означает группу с указанным числом атомов углерода, которые образуют кислоту, где удалена OH группа, такую как ацетил, CH3CO-; бензоил, C6H5CO-.
"Циклоалкил" означает группу с указанным числом атомов углерода, образующих циклопропил, циклобутил, циклопентил, циклогексил и т.д. и ее изомерные формы.
"Амино" означает NH, "алкиламино" означает группу, где одна из позиций водорода замещается алкилом, и "диалкиламино" означает группу, где оба водорода замещаются алкильной группой.
Термин "Фармацевтически приемлемые соли" означает соли присоединения кислоты, которые можно получить любым из известных в данной области способом. К типичным солям присоединения кислоты относятся гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат, ацетат, пропионат, лактат, малат, сукцинат, тартрат, циклогексансульфаматы, метансульфонаты, этансульфонаты, бензолсульфонаты, толуолсульфонаты, фумараты и другие фармацевтически приемлемые противоионы для аминов.
Предпочтительно, X является S.
R1 заместителями являются, предпочтительно, оба фтором и, более предпочтительно, фтором и водородом.
R2 заместителем является, предпочтительно, водород, метил, дихлорметил, гидроксиметил или метокси. Более предпочтительно, R2 является водородом, метокси или метилом. Наиболее предпочтительно, R2 является метилом.
Предпочтительной абсолютной конфигурацией при C-5 оксазолидинонового кольца заявленных соединений данного изобретения является конфигурация, представленная в структурах формул I и II. Эту абсолютную конфигурацию называют (S) по Cahn-Ingold-Prelog системе номенклатуры. Это такой (S)-энантиомер, который фармакологически активен. Рацемическую смесь используют тем же самым путем и для той же самой цели, как чистый (S)-энантиомер; различие состоит в том, что для получения того же самого бактерицидного действия, необходимо применить в два раза больше рацемического вещества. Специалисту в данной области должно быть очевидно, что когда дополнительный хиральный центр (ы) присутствует в бициклическом оксазиновом или тиазиновом фрагменте соединений структурной формулы I и II, то возможны диастереоизомеры. Эти диастереоизомеры в рацемической и энантиомерной формах, также находятся в пределах объема соединений формулы I и II изобретения.
Предпочтительными соединениями формулы I являются
(S)-H-[[3-[3-фтор-4-((1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан- 5-ил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид (Пример 1);
(S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1] гептан-5- ил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид (Пример 2);
(S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-2,2-диоксо-5-азабицикло[2.2.1] гептан-5-ил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид (Пример 3);
(S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)-ил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид (Пример 4);
(S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)-ил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамиl, S-оксид (Пример 5) ;
(S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)-ил) фенил] -2-оксо-5-оксазолидинил]метил]ацетамид, S,S-диоксид (Пример 6);
цис-(S)-N-[[3-[3-фтор-4-[3-окса-7-азабицикло[3.3.0] октан-7-ил] - фенил] -2-оксо-5-оксазолидинил]метил]ацетамид (Пример 7);
(S)-N-{ [3-[3-фтор-4-[(1R, 4R)-2-тиа-5-азабицикло[2.2.1] гептан-5- ил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(2-тиа-6-азабицикло[3.2.0] гептан-6-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-тиа-6-азабицикло[3.2.0] гептан-6-ил)фeнил] -2- oкco-5-oкcaзoлидинил]мeтил]aцeтaмид;
(S)-N-[[3-[3-фтор-4-(3-тиа-7-азабицикло[3.3.1] нонан-7-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-( 3-тиа-9-азабицикло[3.3.1]нонан-9-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-[[3-[3-фтор-4-(2-тиа-6-азабицикло[3.2.1] октан-6-ил)фенил] -2-оксо-5 -оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(2-тиа-6-азабицикло[3.3.1] нонан-6-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-[[3-[3-фтор-4-(7-тиа-3-азабицикло[4.2.1] нонан-3-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(9-тиа-3-азабицикло[3.3.1] нонан-3-иленил]-2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-окса-6-азабицикло[3.2.0]гептан-6-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(6-окса-3-азабицикло[3.1.1]гептан-3-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-окса-7-азабицикло[3.3.1] нонан-7-ил)фенил] -2-оксо-5 -оксазолидинил]метил]ацетамид
(S)-N-[(3-[3-фтор-4-(3-окса-9-азабицикло[3.3.1] нонан-9-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(9-окса-3-азабицикло[3.3.1] нонан-3-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(2-окса-5-азабицикло[2.2.2] октан-5-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(2-окса-6-азабицикло[3.2.1] октан-6-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-окса-7-азабицикло[4.2.0] октан-7-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-окса-8-азабицикло[3.2.1] октан-8-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(6-окса-2-азабицикло[3.2.1] октан-2-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(8-окса-3-азабицикло[3.2.1] октан-3-ил)фенил]-2-оксо -5-оксазолидинил]метил]ацетамид; и
(S)-N-[[3-[3-фтор-4-[(1R, 4R)-2-окса-5-азабицикло[2.2.1] гептан-5-ил] фeнил]-2-oкco-5-oкcaзoлидинил]мeтил]aцeтaмид.
Наиболее предпочтительным соединением является (S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1] гептан-5-ил] фенил] -2- оксо-5-оксазолидинил] метил]ацетамид (Пример 2).
(S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)- ил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид (Пример 4).
(S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)-ил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид, S,S-диоксид (Пример 6).
Фармацевтические композиции данного изобретения можно получить смешиванием (объединением) соединений формулы I или II данного изобретения с твердым или жидким фармацевтически приемлемым носителем, и, произвольно, с фармацевтически приемлемыми адъювантами и эксипиентами, применяя стандартную и общепринятую технологию. К композициям в твердой форме относятся порошки, таблетки, диспергируемые гранулы, капсулы, саше и суппозитории. Твердым носителем может быть, по крайней мере, одно вещество, которое может также функционировать как разбавитель, ароматизирующее средство, солюбилизатор, смазка, суспендирующее средство, связующее, таблетку- дезинтегрирующее средство и инкапсулирующее средство. Инертные твердые носители включают карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, целлюлозные вещества, низкоплавкий воск, масло какао и т.п. Композиции в жидкой форме включают растворы, суспензии и эмульсии. Например, можно разработать растворы соединений данного изобретения, растворенные в воде и системах вода-пропиленгликоль и вода-полиэтиленгликоль, произвольно содержащие подходящие обычные окрашивающие средства, отдушки, стабилизаторы и загущающие средства.
Предпочтительно, фармацевтическую композицию раэрабатывают, применяя стандартную технологию для единичной дозированной лекарственной формы, содержащей эффективные или соответствующие количества активного компонента, т. е. соединение формулы I согласно данному изобретению.
Количество активного компонента, который является соединением формулы I или II согласно данному изобретению, в фармацевтической композиции и ее дозированной единичной лекарственной форме можно варьировать или регулировать в широком диапазоне в зависимости от конкретного применения, эффективности конкретного соединения, требуемой концентрации. В общем, количество активного компонента обычно находится в диапазоне от 0,5 до 90% от веса композиции.
При терапевтическом использовании для лечения бактериальных инфекций или борьбы с бактериальными инфекциями у теплокровных животных соединения или их фармацевтические композиции обычно вводят перорально и/или парентерально при дозе, необходимой для получения или поддержания концентрации, т.е. количества или уровня в крови активного компонента у животного, подвергаемого лечению, которая обычно антибактериально эффективна. В общем, такое антибактериально эффективное количество дозы активного компонента обычно находится в диапазоне от около 0,1 до 100, более предпочтительно, от около 3,0 до около 50 мг/кг веса тела/день. Следует иметь в виду, что дозы могут зависеть от потребности пациента, тяжести бактериальной инфекции, подлежащей лечению, и конкретного подлежащего использованию соединения. Кроме того, следует иметь в виду, что первоначальная вводимая доза может быть увеличена сверх вышеупомянутого верхнего уровня для того, чтобы быстро достичь требуемого уровня в крови, или первоначальная доза может быть меньше, чем оптимум, и дневную дозу можно прогрессивно увеличивать во время хода лечения в зависимости от конкретной ситуации. При необходимости суточную дозу можно также разделить на несколько доз для введения, например, от двух до четырех раз в день.
Соединения формулы I или II согласно данному изобретению вводят парентерально, т.е. при помощи инъекции, например, внутривенной инъекции, или при помощи других парентеральных путей введения. Фармацевтические композиции для парентерального введения обычно содержат фармацевтически приемлемое количество соединения формулы I или II в виде растворимой соли (соли присоединения кислоты или соли основания), растворенной в фармацевтически приемлемом жидком носителе, таком как, например, вода для инъекции или буфер, чтобы обеспечить подходящий забуференный изотонический раствор, например, имеющий pH около 3-7. К подходящим буферным веществам, например, относятся тринатрий ортофосфат, натрий бикарбонат, натрий цитрат, N-метилглюкамин, L(+)-лизин и (L)+-аргинин, не называя других представительных примеров буферных веществ. Соединение формулы I обычно растворяют в носителе в количестве, достаточном для обеспечения фармацевтически приемлемой инъецируемой концентрации в диапазоне от около 1 мг/мл до 400 мг/мл раствора. Полученную жидкую фармацевтическую композицию обычно вводят таким образом, чтобы обеспечить дозу, соответствующую вышеупомянутому противобактериальному эффективному количеству.
Предпочтительный способ получения оксазолидинонов формулы I или II в энантиомерно чистой форме описан в Схемах I-IV.
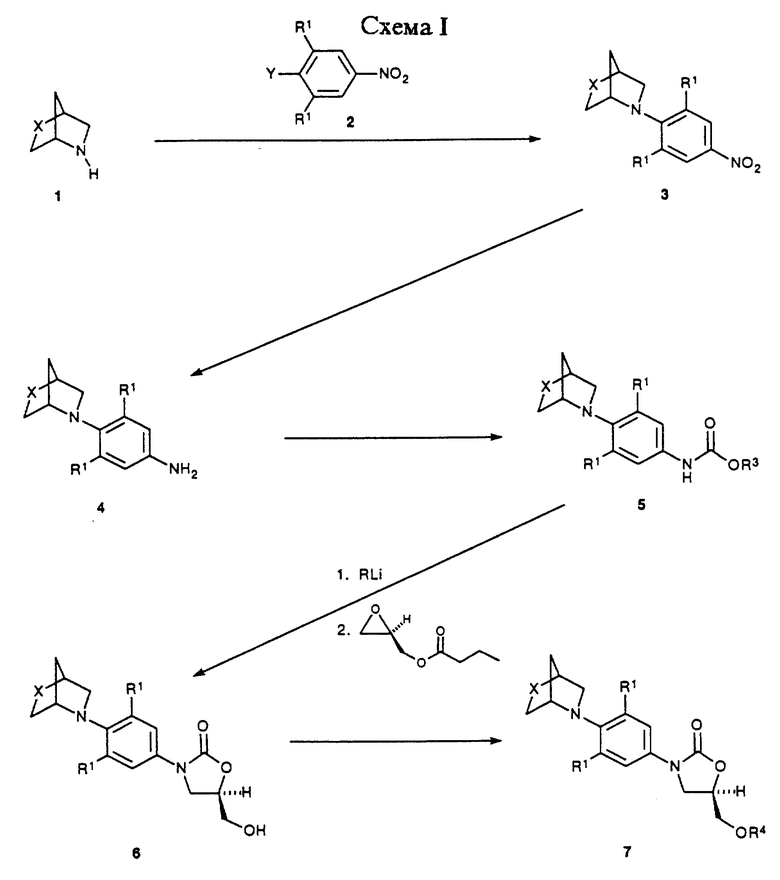
Как показано в Схеме 1, бициклические оксазины и триазины (коммерчески доступные или известные в литературе), такие как (1S,4S)-2-окса-5-азабицикло[2.2.1] гептан (X= 0) и (1S,4S)-2-тиа-5-азабицикло [2.2.1]гептан (X=S) структуры 1, подвергают воздействию с функционализированным нитробензолом 2 (Y= галоген или трифторметансульфонат) в присутствии подходящего основания, такого, как N,N-диизопропилэтиламин, и в подходящем растворителе, таком как ацетонитрил, тетрагидрофуран (ТГФ, THF) или этилацетат, при температуре окружающей среды до температуры кипения флегмы, с получением аддуктов 3. Когда X=0, затем нитрогруппу 3 восстанавливают каталитическим гидрированием в присутствии подходящего катализатора, такого как 10% палладий на угле или W-2 никель Ренея, и в подходящем растворителе, таком как этилацетат, тетрагидрофуран, водный тетрагидрофуран, метанол и их смеси, получая анилины 4. В случае, когда X=S, нитрогруппу 3 можно восстановить действием гидросульфита натрия в водном тетрагидрофуране при температуре окружающей среды до 55oC, получая анилины 4. Альтернативно, восстановление нитрогруппы 3 (X=S) можно осуществить каталитическим гидрированием в присутствии подходящего катализатора, такого как платина на сульфиде углерода или W-2 никель Ренея, и в соответствующей системе-растворителе, например, водный тетрагидрофуран. Последние условия, в частности, используют в том случае, когда реакционную смесь просто фильтруют через Celite® или т.п. для удаления катализатора, и фильтрат, содержащий анилин 4, непосредственно используют в следующей стадии. Наконец, анилины 4 превращают в их бензил (R3CH2Ph) или метил (R3=CH3) карбаматные производные 5, используя стандартные Schotten-Baumann условия или их вариации, известные специалистам в данной области. Затем уретаны 5 депротонируют подходящим основанием, таким как н-бутиллитий, литий диизопропиламид, или литий бис(триметилсилил)амид, в подходящем растворителе, таком как тетрагидрофуран или N,N-диметилформамид, и при подходящей температуре, такой как от -78 до -60oC, получая литированный промежуточный продукт, который затем обрабатывают коммерчески доступным (-)-(R)-глицидил бутиратом. Затем нагревание до температуры окружающей среды непосредственно дает 5-(гидроксиметил)оксазолидиноны 6 в энантиомерно обогащенной форме. Затем соединение 6 превращают в соответствующий мезилат 7 (R4=метансульфонил) или арил сульфонат 7 (R4= ArSO2, например, п-толуолсульфонил) путем действия, например, метансульфонилхлорид/пиридин или метансульфонилхлорид/триэтиламин/ дихлорметан или п-толуолсульфонилхлорид/пиридин.
Как показано в Схеме II, затем полученное сульфонатное производное 7 подвергают взаимодействию с азидным источником, таким как азид натрия или калия, в апротонном растворителе, таком как N,N-диметилформамид (ДМФ, DMF) или 1-метил-2-пирролидинон, возможно в присутствии катализатора, такого как 18-краун-6, при температуре 50-90oC, с получением азида 8. Затем азид восстанавливают гидрированием при помощи палладиевого катализатора на угле или платинового катализатора в соответствующем растворителе, таком как этилацетат или метанол, получая соответствующий амин 9. Альтернативно, и предпочтительно, в случае, когда X=S, азид может быть восстановлен путем обработки соединением трехвалентного фосфора, таким как трифенилфосфин, в соответствующем растворителе, таком как тетрагидрофуран с последующим добавлением воды. Альтернативно, мезилатную или арилсульфонатную группу соединений 7 можно заменить фталимидом калия в ацетонитриле при температуре кипения флегмы с получением промежуточного фталимида 10. Затем фталимид 10 депротонируют путем обработки водным метиламином в кипящем этаноле, получая амин 9. В еще одном альтернативном варианте мезилат 7 подвергают взаимодействию с гидроксидом аммония в горячем изопропаноле или изопропанол/тетрагидрофуране, предпочтительно, в герметизированном реакционном сосуде, получая амин 9. Затем амин 9 ацилируют при помощи реакций, известных специалистам в данной области, получая оксазолидиноны структуры 11. Например, амин может быть подвергнут взаимодействию с хлорангидридом или ангидридом в основном растворителе, таком как пиридин, при температуре в диапазоне от -30oC до 30oC, с получением ацилированного соединения 11 (R2= произвольно замещенный алкил). Для специалистов в данной области очевидно, что другие ацильные группы, не выходящие за рамки объема данного изобретения, могут быть легко введены в амин 9 при помощи стандартных техник ацилирования, например, тех, которые освещены March, J."Advanced Organic Chemistry", 4th ed., John Wiley & Sons: New York, 1992; p 417-425, с получением дополнительных примеров 11. Соединения структуры 11 представляют примеры противобактериальных агентов-бициклических оксазин- и триазинзамещенных оксазолидинонов формулы II, которые являются предметом данного изобретения. Как показано в Схеме III, оксазолидиноны 11, сами по себе, примеры противобактериальных агентов формулы II, могут быть далее преобразованы в дополнительные соединения формулы II. В частности, 11 (X=S) может быть окислен в соответствующий сульфоксид(ы) 12 (X=SO) с помощью метапериодата натрия в смеси воды и метанола. Специалистам в данной области очевидно, что возможны как эндо-, так и экзосульфоксиды, и что обе изомерные формы, а также смеси их, входят в объем данного изобретения. Кроме того, соединения 11 или 12 могут быть окислены в соответствующие сульфоны 13 (X=SO2) путем обработки 4-метилморфолин N-оксидом и каталитическими количествами тетроксида осмия в водном ацетоне. Для специалистов в данной области очевидно, что известны альтернативные условия для окисления 11 (X=S) в 12 или 13, например, те, которые освещены March, J. "Advanced Organic Chemistry", 4th ed.; John Wiley & Sons: New York, 1992; p. 1201-1202.
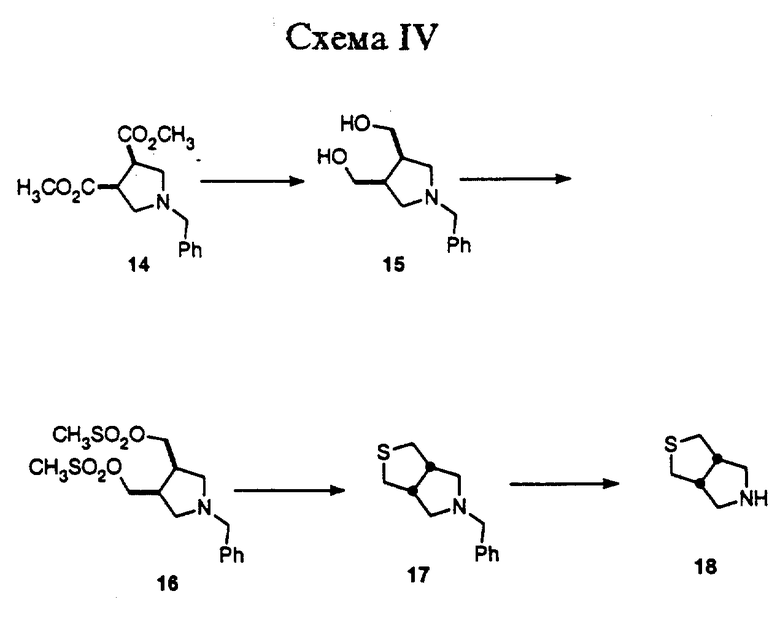
Как показано в Схеме IV, синтез соединений, которые включают тиенопирролидин, начинается с восстановления диэфира 14 в диол 15, используя литийалюминийгидрид в качестве восстанавливающего агента. Затем соединение 15 превращают в бис-мезилат 16 путем реакции с метансульфонилхлоридом и триалкиламиновым основанием. Циклизацию 16 в тиенопирролидин 17 проводят путем взаимодействия с сульфидом натрия, и соединение 17 дебензилируют в тиенопиррол 18 реакцией с водородом в присутствии подходящего катализатора, такого как палладий на угле. Затем получают соединение примера 4 из 18, следуя способам, описанным в общих чертах в Схемах I и II (кроме замещения 1 на 18). Соединения примеров 5 и 6 получают путем окисления соединения примера 4, используя те же методики, которые описаны в Схеме III.
Противомикробную активность испытывают in vivo, используя метод анализа на мышах (Murine Assay). Группы мышей женской особи инъецируют внутрибрюшинно бактериями, которые размораживают непосредственно перед использованием и суспендируют в brain heart вытяжке с 4% пивными дрожжами UC9213 (Staphylococcus aureus) или в brain heart вытяжке (Streptococcus species). Обработку антибиотиком при шести уровнях доз на лекарственное средство проводят спустя час и пять часов после заражения, либо орально, либо подкожно. За выживанием наблюдают ежедневно в течение шести дней. ED50 значения, в расчете на смертность, определяют, используя про бит анализ. Соединения данного изобретения сравнивают с хорошо известным противомикробным средством (Ванкомицин, Vancomicin) в качестве контроля. Результаты представлены в таблице (см. в конце описания).
Для специалистов в данной области очевидно, что описанные синтетические способы являются только иллюстративными и что использование альтернативных бициклических оксазинов и триазинов, известных из патентной литературы и открытой публикации, допускает получение дополнительных примеров структурной формулы 1.
Пример 1: (S)-N-[[3-[4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил] -3- фторфенил] -2-оксо-5-оксазолидинил] метил] ацетамид Стадия 1: 4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-3-фторнитробензол
Смесь коммерчески доступного (1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан гидрохлорида (0,200 г, 1,47 ммоль), кислого дикалий фосфата (1,030 г, 5,90 ммоль) и 3,4-дифторнитробензола (0,195 мл, 1,77 ммоль) в диметилсульфоксиде (6 мл) перемешивают при температуре окружающей среды в атмосфере N2. ТСХ анализ (5% MeOH/CHCl3) после 3 часов обнаруживает, что исходный нитробензол поглощен. Реакционную смесь разбавляют H2O и (60 мл) и экстрагируют CHCl3. Объединенные органические экстракты промывают H2O и рассолом, сушат над Na2SO4 фильтруют и концентрируют при пониженном давлении, получая желтое твердое вещество. Хроматография на силикагеле (60 г), элюируя градиентом 0-2% MeOH/CHCl3, дает, после концентрирования соответствующих фракций, 0,314 г (90%) названного соединения в виде желтого твердого вещества с т. пл. 106,5-108oC и МС (ЭИ) 238 (М+).
Стадия 2: N-(карбобензилокси)-4-[(1S, 4S)-2-окса-5-азабицикло- [2.2.1] гептан-5-ил]-3-фторанилин
Раствор 4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-3-фторнитробензола (0,160 г, 0,672 ммоль) в смеси 3:1 ТРФ/НО (4 мл) обрабатывают уксусной кислотой (0,115 мл) и затем 10% палладий/уголь (0,020 г) в токе N2. Атмосферу замещают H2 (баллон), повторяя вакуумирование и наполнение, и смесь перемешивают при температуре окружающей среды. Через 2 часа ТСХ анализ (6% CH3CN/CHCl3) показывал, что восстановление завершилось. Реакционную смесь фильтруют через Celite® и фильтрат немедленно помещают в атмосферу N2 обрабатывают K2CO3 (0,464 г, 3,36 ммоль), а затем бензил хлорформиатом (0,117 мл, 0,864 ммоль). ТСХ анализ (6% CH3CN/CHCl3) через 0,5 часа показал, что реакция завершилась. Реакционную смесь концентрируют при пониженном давлении и хроматографируют на силикагеле (20 г), элюируя градиентом 1-5% CH3CN/CHCl3. Концентрирование соответствующих фракций дает 0,226 г (98%) названного соединения в виде белого твердого вещества с т. пл. 120-121oC и МС (ЭИ) 342 (М+).
Стадия 3: (R)-[3-[4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5- ил]-3-фторфенил]-2-оксо-5-оксазолидинил]метанол
Раствор N-(карбобензилокси)-4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1] гептан-5-ил] -3-фторанилина (0,169 г, 0,494 ммоль) в сухом ТГФ (2 мл) охлаждают до -78oC в N2 атмосфере и затем подвергают взаимодействию с н-бутиллитием (0,312 мл 1,6 М раствора в гексане, 0,499 ммоль). После перемешивания в течение 10 минут при -78oC, реакционную смесь обрабатывают (R)-глицидил бутиратом (0,070 мл, 0,499 ммоль). По завершении добавления охлаждающую баню удаляют и смесь оставляют перемешиваться при температуре окружающей среды в течение ночи, и в течение этого времени появляется беловатый осадок. ТХС анализ (5% MeOH/CHCl3) показал, что реакция завершилась. Реакционную смесь обрабатывают прибл. 5 каплями насыщенного водного NH4Cl, что приводит к образованию гомогенного раствора. Реакционную смесь концентрируют при пониженном давлении, получая беловатое твердое вещество. Хроматография на силикагеле, элюируя градиентом 1-5% MeOH/CHCl3, дает, после концентрирования соответствующих фракций, 0,116 г (84%) названного соединения в виде белого твердого вещества с т. пл. 138-140oC и МС (ЭИ) 308 (М+). Дополнительно, 0,018 г (10%) второго компонента, идентифицированного как: сложный эфир бутирата названного соединения с помощью 1H ЯМР анализа, получают в виде масла цвета янтаря.
Стадия 4: (R)-[[3-{4-(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5- ил]-3-фторфенил-2-оксо-5-оксазолидинил]метил]метансульфонат
Раствор (R-[3-4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил]-фторфенил] -2-оксо-5- оксазолидинил]метанола (0,765 г, 2,48 ммоль) в сухом CH2Cl2 (30 мл) охлаждают до 0oC в N2 атмосфере и обрабатывают Et3N (0,518 мл, 3,73 ммоль), а затем метансульфонил хлоридом (0,202 мл, 2,61 ммоль). ТХС анализ (5% MeOH/CHCl3) через 0,5 часа показал, что реакция завершилась. Реакционную смесь промывают H2O и рассолом сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая 0,992 г (приблизительно, 100%) названного соединения в виде желтовато-коричневого твердого вещества. Аналитическую пробу получают путем перекристаллизации из 5% CH2Cl2/U-PrOH. Эта проба имела т.пл. 124,5-126oC и МС (ЭИ) 386 (М+).
Стадия 5: (R)-[[3-[4-[(1S, 4S)-2-окса-5-азабицикло [2.2.1]гептан-5-ил] -3-фторфенил]-2-оксо-5-оксазолидинил]метил]азид
Раствор (R)-[[3-[4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил] -3-фторфенил] -2-оксо-5-оксазолидинил] метил] метансульфоната (0,869 г, 2,25 ммоль) в сухом ДМФ (DMF) (10 мл) обрабатывают твердым NaN3 (0,732 г, 11,3 ммоль) при температуре окружающей среды в атмосфере N2. Затем смесь нагревают до 65oC и ход реакции контролируют при помощи ТСХ. После 7,5 ч при этой температуре, ТСХ анализ (5% MeOH/CHCl3) показывает завершение реакции. Реакционную смесь разбавляют EtOAc (100 мл), промывают H2O (3 x 15 мл) и рассолом, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении, получая 0,692 г (92%) названного соединения в виде желтовато-коричневого твердого вещества. Аналитическую пробу получают путем перекристаллизации из 1: 1 EtOAc/гексан в виде беловатого твердого вещества с т.пл. 101-102,5oC и МС (ЭИ) 333 (М+).
Стадия 6: (S)-N-[[3-[4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил] -3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (R)-[[3-[4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил] -3-фторфенил]-2-оксо-5-оксазолидинил]метил]азида (0,652 г, 1,96 ммоль) в MeOH (20 мл) и CH2Cl2 (10 мл) обрабатывают 10% палладий/уголь (0,095 г) в потоке N2. Затем атмосферу заменяют на H2 (баллон), повторяя вакуумирование и наполнение, и смесь перемешивают при температуре окружающей среды под H2. Через 3 ч ТСХ анализ (5% MeOH/CHCl3) показывает завершение реакции. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют при пониженном давлении. Неочищенный 5-(аминометил)оксазолидинон растворяют в CH2Cl2 (20 мл) и обрабатывают пиридином (0,190 мл, 2,35 ммоль) и затем уксусным ангидридом (0,222 мл, 2,35 ммоль). Через 0,5 ч ТСХ анализ (5% MeOH/CHCl3) показывает завершение реакции ацетилирования. Реакционную смесь промывают H2O и рассолом, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая беловатое твердое вещество. Хроматография на силикагеле (70 г), элюируя градиентом 1-3% MeOH/CHCl3, дает, после концентрирования соответствующих фракций, 0,517 г (76%) указанного в заголовке оксазолидинона-антибактериального агента в виде белого твердого вещества с т.пл. 60-65oC и МС (ЭИ) 349 (М+).
Пример 2: (S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1] гептан-5-ил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: 4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1]гептан-5-ил]-3- фторнитробензол
Смесь коммерчески доступного (15,43)-2-тиа-5-азабицикло[2.2.1] гептана (0,500 г, 3,30 ммоль), диизопропилэтиламина (1,434 мл, 8,24 ммоль) и 3,4-дифторнитробензола (0,437 мл, 3,96 ммоль) в сухом ацетонитриле (15 мл) нагревают при температуре кипения флегмы в N2 атмосфере в течение 1 ч и затем охлаждают до температуры окружающей среды на протяжении ночи. Реакционную смесь концентрируют при пониженном давлении, получая желтый сироп. Хроматография на силикагеле (50 г), при элюировании хлороформом, дает, после концентрирования соответствующих фракций, 0,700 г (84%) названного соединения в виде желтого твердого вещества с т.пл. 97-98oC и МС (ЭИ) 254 (М+).
Стадия 2: N-(карбобензилокси)-4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1] гептан-5-ил]-3-фторанилин
Раствор 4-[(1S, 4S)-2-тиа-5- азабицикло[2.2.1]гептан-5-ил]-3-фторнитробензола (1,64 г, 6,46 ммоль) в смеси 20% H2O/ТГФ (50 мл) обрабатывают платиной на сульфиде углерода (0,200 г) в N2 токе. Атмосферу заменяют H2 (баллон), повторяя вакуумирование и наполнение. Через 12 ч, ТСХ анализ показывает, что значительное количество исходного вещества все еще остается. Реакционную смесь переносят в аппарат Парра и трясут при 3,164 кг/см (45 psi) H2. Через 2 ч, ТСХ анализ показывает, что еще остается некоторое количество исходного вещества. Реакционную смесь фильтруют через Celite® и фильтрат, содержащий смесь требуемого анилинового промежуточного и исходного производного нитробензола, охлаждают до 0oC и обрабатывают NaHCO3 (2,170 г, 25,8 ммоль) и бензил хлороформиатом (1,02 мл, 7,10 ммоль). Через 0,5 ч, реакционную смесь концентрируют под пониженным давлением до желтого/зеленого сиропа. Это вещество растворяют в CHCl3, промывают H2O и рассолом, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Фильтрация через набивку силикагеля, при элюировании 20-30% EtOAc/гексан, дает, после концентрирования соответствующих фракций, смесь исходного производного нитробензола и названного соединения. Вещество поглощают в 20% H2O/ТГФ (50 мл) и обрабатывают W-2 никелем Ренея (прибл. 0,400 г). Реакционную смесь встряхивают в аппарате Парра при 3,164 кг/см2 (45 psi) H2. Через 3 ч, реакционную смесь фильтруют через Celite® и фильтрат охлаждают до 0o и обрабатывают NaHCO3 (2,00 г, 23,8 ммоль), а затем бензил хлороформиатом (0,600 мл, 4,19 ммоль). Через 0,5 ч, реакционную смесь концентрируют при пониженном давлении и остаток хроматографируют на силикагеле (125 г), элюируя 10-20% EtOAc/гексан, получая, после концентрирования соответствующих фракций, 2,20 г (95%) названного соединения в виде желтого твердого вещества с т.пл. 91-93oC и МС (ЭИ) 358 (М+).
Стадия 3: (R)-[3-[4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1]гептан-5-ил]-3- фторфенил]-2-оксо-5-оксазолидинил]метанол
Раствор N-(карбобензилокси)-4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1]гептан -5-ил]-3-фторанилина (0,359 г, 1,00 ммоль) в сухом ТГФ (4 мл) в N2 охлаждают до -78oC и затем обрабатывают н-бутиллитием (0,633 мл, 1,6 М раствора в гексане, 1,01 ммоль). Реакционную смесь перемешивают при - 78oC в течение 15 минут и затем обрабатывают (R)-глицидил бутиратом (0,151 мл, 1,00 ммоль). По завершении добавления, охлаждающую баню удаляют, и реакционный смеси дают нагреться до температуры окружающей среды в течение ночи. ТХС анализ (5% MeOH/CHCl3) показал, что реакция завершилась, но присутствует небольшое количество сложного эфира бутирата названного соединения. Добавление 5 капель 25 вес% раствора NaOMe/MeOH, с последующим перемешиванием в течение 20 мин при комнатной температуре, эффективно для превращения этого промежуточного в названное соединение. Реакционную смесь обрабатывают насыщенным водным NH4CI (10 капель) и затем концентрируют при пониженном давлении до масла. Это вещество растворяют в CH2Cl2 и промывают H2O и рассолом, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая неочищенный продукт. Хроматография на силикагеле (50 г), при элюировании градиентом 1-3% MeOH/CHCl3, дает, после концентрирования соответствующих фракций, 0,132 г (41%) названного соединения в виде масла. Растиранием с EtOAc получают осадок, который выделяют и сушат в вакууме, получая беловатое твердое вещество с т.пл. 156-157oC и МС (ЭИ) 324 (М+).
Стадия 4 : (R)-[[3-[4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1]гептан-5-ил-] -3-фторфенил]-2-оксо-5-оксазолидинил]метил]метансульфонат
Раствор (R)-[3-[4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1]гептан-5-ил]-3-фторфенил]-2-оксо-5-оксазолидинил]метанола (1, 68 г, 5,19 ммоль) в сухом CH2Cl2 (100 мл) в N2 охлаждают до 0oC и обрабатывают Et3N (0,793 мл, 5,70 ммоль), а затем метансульфонил хлоридом (0,442 мл, 5,70 ммоль). Через 0,5 ч, при этой температуре, ТХС анализ (5% MeOH/CHCl3) показал, что реакция завершилась. Смесь промывают H2O, насыщают водным NaHCO3 и рассолом, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая 1,65 г (79%) названного соединения в виде белого твердого вещества с т.пл. 139-142oC и МС (ЭИ) 402 (М+).
Стадия 5: (R)-[[3-[4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1]гептан-5-ил] -фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Раствор (R)-[[3-[4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1]гептан-5-ил] -3-фторфенил]-2-оксо-5-оксазолидинил]метил]метансульфоната (1,56 г,3,88 ммоль), 1: 1 ТГФ/u-PrOH (4 мл) и 30% NH4OH (4 мл) нагревают до 95oC в герметизированной трубке в течение 14 часов и затем охлаждают до температуры окружающей среды. ТСХ анализ (5% MeOH/CHCl3) показывает завершение реакции. Смесь разбавляют CH2Cl2 (75 мл), промывают насыщенным NaHCO3 (15 мл) и рассолом (15 мл), сушат над Na2SO4 фильтруют и концентрируют при пониженном давлении, получая сироп. Неочищенный промежуточный 5-(аминометил)оксазолидинон растворяют в CH2Cl2 (75 мл) и обрабатывают пиридином (0,345 мл, 4,27 ммоль) и уксусным ангидридом (0,403 мл, 4,27 ммоль) при температуре окружающей среды. Через 1 час, ТСХ анализ (5% MeOH/CHCl3) показывает завершение реакции ацилирования. Реакционную смесь промывают H2O и рассолом, сушат над Na2SO4 фильтруют и концентрируют при пониженном давлении до твердого вещества янтарного цвета. Хроматографией на силикагеле (125 г), при элюировании 1-3% MeOH/CHCl3 получают, после концентрирования соответствующих фракций, 1,23 г (87%) названного оксазолидинона-антибактериального агента в виде твердого вещества с т.пл. 90-95oC и МС (ЭИ) 365 (М+).
Пример 3: (S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-2,2-диоксо-5-азабицикло [2.2.1]гептан-5-ил]фенил]-2-оксо-5-оксазолидинил]-метил]ацетамид
Раствор (S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-5-азабицикло [2.2.1] гептан-5-ил] фенил] -2-оксо-оксазолидинил] метил]ацетамида (0,300г, 0,82 ммоль) в смеси 25% H2O/ацетон (16 мл) обрабатывают при температуре окружающей среды 4-метилморфолин-N-оксидом (0,288 г, 2,47 ммоль), а затем тетроксидом осмия (0,102 мл, 2,5 вес% раствора в трет-бутаноле, 0,008 ммоль). Через 18 ч ТСХ анализ (10% MeOH/CHCl3) показывает завершение реакции окисления. Реакционную смесь обрабатывают насыщенным водным NaHSO3 затем экстрагируют CHCl3. Объединенные органические экстракты промывают рассолом, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток хроматографируют на силикагеле (10 г), элюируя 1-3% MeOH/CHCl3, получая после концентрирования соответствующих фракций, 0,321 г (98%) названного оксазолидинона-бактерицидного средства в виде белого твердого вещества с т.пл. 95-105oC.
Пример 4: (S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-c] пиррол -5(3H)-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Стадия 1: цис-1-(Фенилметил)-3,4-пирролидиндиметанол
Сложный диметиловый эфир (цис)-1-(фенилметил)-3,4-пирролидиндикарбоновой кислоты получают согласно процедуре Y.Terao, et al. (Chem. Pharm. Bull., 1985, 33, 2762-66). К перемешиваемому раствору этого сложного диэфира (12,14 г, 43,8 ммоль) в сухом ТГФ (175 мл) в атмосфере N2, охлажденному до 0oC, добавляют по каплям раствор литийалюминийгидрида (1 М в ТГФ, 87 мл, 87 ммоль) на протяжении 15 мин. Реакционную смесь перемешивают при 0oC в течение 1 ч, затем при RT в течение 18 ч. Реакционную смесь охлаждают до 0oC и гасят последовательным добавлением H2O (3,2 мл), 5 N NaOH (3,2 мл) и H2O (11,7 мл). Реакционная смесь становится очень густой и перемешивается с трудом. Реакционную смесь разбавляют эфиром (500 мл) и фильтруют через небольшой слой целита. Осадок на фильтре промывают эфиром (250 мл). Фильтрат промывают H2O (1 x 300 мл), и органические фильтраты сушат (MgSO4), фильтруют и концентрируют, получая 9,3 г (41,8 ммоль, 96%) требуемого диола и густого желтого масла. Используют без дополнительной очистки. ВРМС (HRMC) (ББА. FAB) рассчитано для C13H19NO2+H 222, 1494, найдено 222, 1490.
Стадия 2: цис-1-(Фенилметил)-3,4-ди(метилсульфонилокси)-метилпирролидин
К перемешиваемому раствору цис-1-(фенилметил)-3,4-пирролидиндиметанола (9,2 г, 41,6 ммоль) в CH2Cl2 (240 мл), охлажденному до 0oC, добавляют триэтиламин (29 мл, 208,1 ммоль), а затем метансульфонил хлорид (8,1 мл, 104,0 ммоль). Реакционную смесь перемешивают при 0oC в течение 15 мин, затем при комнатной температуре (КТ, RT) в течение 1,5 ч. Реакционную смесь выливают в H2O (240 мл) и фазы разделяют. Водную фазу экстрагируют CH2Cl2 (1 x 100 мл). Объединенные органические фазы сушат (MgSO4), фильтруют и концентрируют. Остаток очищают флеш-хроматографией, используя этилацетат в качестве элюента, и получая 14,2 г (37,5 ммоль, 90%) требуемого бис-мезилата в виде густого желтого масла. ВРМС (HRMC) (ЭИ) рассчитано для C15H23NO6S2 377, 0967, найдено 377, 0958.
Стадия 3: Гексагидро-5-(фенилметил)-1Н-тиено[3,4-c]пиррол
К перемешиваемому раствору цис-1-(фенилметил)-3,4-ди (метилсульфонилокси) метилпирролидина (9,2 г, ммоль) в сухом ДМСО (DMSO) (48 мл) добавляют безводный сульфид натрия (5,7 г, 73,3 ммоль). Темную реакционную смесь нагревают при 120oC в течение 18 ч. Охлажденную реакционную смесь выливают в ледяную H2O (150 мл). Образующуюся смесь экстрагируют эфиром (3 x 200 мл). Объединенные органические фазы сушат (MgSO4), фильтруют и концентрируют. Образующийся остаток очищают флеш-хроматографией, используя этилацетат в качестве элюента и получая 4,2 г (19,1 ммоль, 78%) требуемого продукта в виде густого желтого масла. ВРМС (HRMC) (ЭИ) рассчитано для C13H17NS 219, 1082, найдено 219, 1080. Анал. Рассчитано для C17H17NS: C, 71,19; H, 7,81; N, 6,39. Найдено: С, 70,82; H, 7,83; N, 6,35.
Стадия 4: Гексагидро-1H-тиено[3,4-c]пиррол, гидрохлорид
К перемешиваемому раствору гексагидро-5-(фенилметил)-1Н-тиено [3,4-c] пиррола (1,2 г, 5,3 ммоль) в CH2CI2 (21 мл), охлажденному до 0oC, добавляют по каплям шприцом 1-хлорэтилхлорформиат (1,15 мл, 10,7 ммоль). Реакционную смесь перемешивают при 0oC в течение 20 минут, затем при КТ в течение 90 минут. Реакционную смесь концентрируют. Образующийся остаток очищают флеш-хроматографией, используя 25% этилацетат в гексане в качестве элюента, получая 611,3 мг (2,6 ммоль, 49%) 1-хлорэтилкарбамата. Затем колонку промывают 20% метанольным аммиаком в CHCl3, получая 160,5 мг (1,24 ммоль, 23%) требуемого амина в виде свободного основания. 1-хлорэтилкарбамат (611,3 мг, 2,6 ммоль) растворяют в метаноле (15 мл) и кипятят с обратным холодильником в течение 90 минут. Охлажденную реакционную смесь концентрируют, получая 408,0 (2,5 моль, 47%) требуемого амина в виде HCl соли в расчете на хлоркарбамат). Т. пл. 149-151oC; ВРМС (HRMC) (ЭИ) рассчитано для C6H11NS 129, 0612, найдено 129, 0614. Анал. Рассчитано для C6H12ClNS: С, 43,50; H, 7,30; С, 21,39; N, 8,45; S, 19,35. Найдено: С, 43,39; H, 7,23; N, 8,24; Cl, 21,08; S, 19,12.
Стадия 5: 5-(2-фтор-4-нитрофенил)-гексагидро-1H-тиено[3,4-с] пиррол
К перемешиваемому раствору 5-гексагидро-1H-тиено[3,4-с]пиррол, гидрохлорида (147,3 мг, 0,89 ммоль) в ацетонитриле (5 мл) добавляют 3,4-фторнитробензол (0,11 мл, 0,98 ммоль), затем дииэопропилэтил амин (0,36 мл, 2,05 ммоль). Гомогенную реакционную смесь кипятят с обратным холодильником в течение 18 ч. Охлажденную реакционную смесь концентрируют. Образующийся остаток разбавляют EtOAc (50 мл) и промывают насыщенным водным H4Cl (1 x 25 мл). Водный слой экстрагируют EtOAc (1 x 30 мл). Объединенные органические фазы промывают насыщенным NaHCO3 (1 x 40 мл), рассолом (1 x 40 мл), сушат (MgSO4), фильтруют и концентрируют. Остаток очищают флеш-хроматографией, используя 20% EtOAc в гексане в качестве элюента и получая 202,5 г (0,75 ммоль, 89%) требуемого нитросоединения в виде светлого желтого твердого вещества. Т. пл. 107-109oC. Анал. Рассчитано для C12H13FN2O2S: С 53,72; H 4,88; N, 10,44; S, 11,95. Найдено: С, 53,38; H, 5,03; N, 10,34; S, 11,89.
Стадия 6: 3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-c]пиррол-5(3H)-ил) фенилкарбаминовая кислота, фенилметиловый сложный эфир
К перемешиваемому раствору 5-(2-фтор-4-нитрофенил)гексагидро-1H-тиено [3,4-c] пиррола (1,44 г, 5,4 ммоль) в этаноле (70 мл) добавляют 2 М CuSO4 (2,9 мл). Эту смесь охлаждают до 0oC и порциями добавляют боргидрид натрия (1,10 г, 26,8 ммоль). (Внимание: реакция сильно экзотермична). Затем темную реакционную смесь кипятят с обратным холодильником в течение 2 ч, охлажденная реакционная смесь распределяется между EtOAc и H2O. Фазы разделяют. Водную фазу экстрагируют EtOAc (3 x 100 мл). Объединенные органические фазы сушат (MgSO4), фильтруют и концентрируют. Образовавшийся твердый остаток растворяют в смеси ацетон/H2O (2:1, 60 мл). Этот перемешиваемый раствор охлаждают до 0oC и добавляют твердый NaHCO3 (1,35 г, 16,1 ммоль). Реакционную смесь перемешивают при 0oC в течение 15 мин, затем при КТ в течение 2 ч. Реакционную смесь гасят осторожным добавлением 10% водного NaHSO4 (30 мл). Реакционную смесь выливают в EtOAc (250 мл) и фазы разделяют. Водный слой экстрагируют EtOAc (1 x 100 мл). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют. Остаток очищают флеш-хроматографией, используя 20% EtOAc в гексане, получая 1,6 г (4,3 ммоль, 81%) требуемого карбамата: т. пл. 101-102oC. Анал. Рассчитано для C20H21FN2O2S: С, 64,50; H, 5,68; N, 7,52; S, 8,61. Найдено: С, 64,33; H, 5,56; N, 7,53; S, 8,61.
Стадия 7: (5R)-3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5 (3H)-ил)фенил]-5-(гидроксиметил)-2-оксазолидинон
К перемешиваемому раствору фенилметилового эфира 3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)-ил) фенилкарбаминовой кислоты (1,36 г, 3,6 ммоль) в сухом ТГФ (14 мл) в атмосфере N2, охлажденному до -78oC, добавляют н-бутиллитий (1,6М в гексане, 2,4 мл, 3,8 ммоль). Реакционную смесь перемешивают при -78oC в течение 35 мин и затем добавляют R-(-)-глицидилбутират (0,54 мл, 3,8 ммоль). Реакционную смесь перемешивают при -78oC в течение 30 мин, затем при КТ на протяжении ночи. Образуется густой осадок. Реакционную смесь гасят насыщенным водным NH4CI (14 мл) и выливают в EtOAc (50 мл). Фазы разделяют. Органический слой промывают насыщенным водным NaHCO3 (1 x 30 мл), рассолом (1 x 30 мл), сушат (MgSO4), фильтруют и концентрируют. Остаток очищают флеш-хроматографией, используя EtOAc в качестве элюента, получая 801,6 мг (2,4 ммоль, 65%) требуемого продукта: т.пл. 165-167oC. Анал. Рассчитано для C16H19FN2O3S: С, 56,79; H, 5,66; N, 8,28; S, 9,48. Найдено: С, 56,88; H, 5,74; N,8,21; S, 9,33.
Стадия 8: (5R)-3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5 (3H)-ил)фенил]-5-[[(метилсульфонил)окси]метил]-2-оксазолидинон
К перемешиваемому раствору (5R)-3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-c] пиррол-5(3H)-ил)фенил-5- (гидроксиметил)-2-оксазолидинона (656,5 мг, 1,9 ммоль) в CH2Cl2 (20 мл), охлажденному до 0oC, добавляют триэтиламин (0,41 мл, 2,9 ммоль), а затем метансульфонилхлорид (0,18 мл, 2,3 ммоль). Реакционную смесь перемешивают при 0oC в течение 15 мин, затем при КТ в течение 18 ч. Реакционную смесь выливают в H2O (20 мл). Фазы разделяют. Водный слой экстрагируют CH2Cl2 (1 x 50 мл). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют. Остаток растирают со смесью эфир/гексан, и твердое вещество отделяют фильтрацией и сушат, получая 773,9 мг (1,9 ммоль, 96%) требуемого мезилата. Т. пл. 148-150oC. Анал. Рассчитано для C17H21FN2O5S2: С, 49,03; H, 5,08; N, 6,73; S, 15,40. Найдено: C, 48,56; H, 5,12; N, 6,48; S, 15,41. Найдено: С, 48,46; H, 5,25; N, 6,38.
Стадия 9: (S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3.4-с] пиррол- 5(3H)-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Перемешиваемую суспензию (5R)-3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)-ил)фенил] -5- [метилсульфонил) окси] -метил]-2-оксазолидинона (208,5 мг, 0,5 ммоль) в ТГФ (3 мл) и метанольном аммиаке (3 мл) нагревают в герметизированной трубке при 100oC в течение 48 ч. (Реакционная смесь становится гомогенной при около 80oC). Охлажденную реакционную смесь концентрируют, и образовавшийся остаток растворяют в CH2Cl2 (5 мл) и охлаждают до 0oC. К этой перемешиваемой суспензии добавляют пиридин (0,12 мл, 1,5 ммоль), а затем уксусный ангидрид (60 мкл, 0,6 ммоль). Гомогенную реакционную смесь перемешивают при 0oC в течение 15 мин, затем при КТ в течение 1 ч, затем концентрируют. Остаток очищают с помощью флеш-хроматографии, используя 7% метанол в EtOAc в качестве элюента, получая 148,2 мг (0,4 ммоль, 78%) требуемого ацетамида. Т. пл. 143-144oC; KF-H2O: 0,52%. Анал. Рассчитано для C18H22FN3O3S плюс 0,52% H2O: С, 56,68; H, 5,87; N, 11,01; S, 8,40. Найдено: С, 56,31; H, 5,90; N, 10,74; S, 8,30.
Пример 5: (S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-с] пиррол-5(3H)-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид, S-оксид
К перемешиваемому раствору (S)-N-[[3-[3-фтор-4-(тетра-гидро-1H-тиено [3,4-c] пиррол-5(3H)-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида (216,8 мг, 0,57 ммоль) в CH3OH (4 мл) и H2O (4 мл), охлажденному до 0oC, добавляют метапериодат натрия (134,4 мг, 0,63 ммоль). Реакционную смесь перемешивают при 0oC в течение 1 ч, затем при КТ в течение 18 ч. Твердый осадок удаляют фильтрацией. Твердое вещество промывают CHCl3 (50 мл). Фильтрат промывают H2O (1 x 30 мл). Водный слой экстрагируют CHCl2 (2 x 25 мл). Объединенные органические слои сушат (MgSO4), фильтруют и концентрируют. Остаток очищают с помощью флеш-хроматографии, используя 7% метанол в CH2Cl2 в качестве элюента, получая 195,7 мг (0,5 ммоль, 87%) требуемого сульфоксида. т.пл. 162-164oC; ВРМС (HRMC) (ЭИ) рассчитано для C18H22FN3O4S 395, 1315, найдено 395, 1309. KF-H2O: 2,87%. Анал. Рассчитано для C18H22FN3O4S плюс 2,87% H2O: С, 53,09; H, 5,76; N, 10,32; S, 7,87. Найдено: С, 53,07; H, 6,01; N, 10,20; S, 7,87.
Пример 6: (S)-N-[[3-[3-фтор-4-(тетрагидро-1H-тиено[3,4-c] пиррол-5(3H)-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид, S,S-диоксид
К перемешиваемому раствору (S)-N-[[3-[З-фтор-4-(тетрагидро-1H-тиено[3,4-c] пиррол-5(3H)-ил) фенил] -2-оксо-5-оксазолидинил] метил] ацетамида (213,9 мг, 0,56 ммоль) в смеси 25% ацетон/H2O (8 мл) добавляют N-метилморфолин-N-оксид (198,1 мг, 1,7 ммоль), а затем тетроксид осмия в трет-бутаноле (2,5% по весу) (30 мкл, 0,08 ммоль). Реакционную смесь перемешивают при КТ в течение 18 ч. Реакционную смесь гасят осторожным добавлением насыщенного бисульфита натрия (8 мл). Смесь выливают в CH2Cl2 (50 мл) фазы разделяют. Водную фазу экстрагируют CH2Cl2 (2 x 25 мл). Объединенные органические слои промывают рассолом (1 x 30 мл), сушат (MgSO4), фильтруют и концентрируют. Остаток очищают с помощью флеш-хроматографии, используя 7% метанол в CHCl3 качестве элюента, получая 194,3 мг (0,47 ммоль, 84%) требуемого сульфона. Т. пл. 135-137oC; ВРМС (HRMC) (ЭИ) рассчитано для C18H22FN3O5S 411, 1264, найдено 411, 1263. KF-H2O: 1,10%. Анал. Рассчитано для C18H22FN3O5S плюс 1,10% H2O: С, 51,96; H, 5,45; N, 10,10; S, 7,71. Найдено: С, 51,73; H, 5,62; N, 9,96; S, 7,75.
Пример 7: цис-(S)-N-[[3-[3-фтор-4-[3-окса-7-азабицикло[3.3.0] октан-7-ил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид
Следуя общей методике примера 2 и производя несущественные изменения, но замещая (1S, 4S)-2-тиа-5-азабицикло[2.2.1] -гептан на гексагидро-1H-фтор-(3,4-c)пиррол (Miller, A.D. U.S. Patent 3 975 532 1976) (2,33 г, 20,66 ммоль), получают названное соединение, т.пл. 124-126oC.



Раскрыты соединения формулы I и формулы II, где Х представляет О, S, SO2, SO; R1 представляет независимо Н, F; R2 представляет водород, С1-С8-алкил, возможно замещенный одним или более заместителем, выбранным из группы, включающей F, Сl, гидрокси, С1-С8-алкокси; а = 0-3; b = 0-2; с = 0-2 ( при условии, что в b и c могут быть оба 0); d = 0-2 и е = 0-2 ( при условии, что d и е не могут быть оба 0), или их фармацевтически приемлемые соли, а также способы лечения микробных инфекций на их основе. Изобретение может быть использовано в медицине, полезно против ряда патогенов человека и животных, включая грамположительные аэробные бактерии, а также анаэробные организмы. 4 c. и 11 з.п.ф-лы, 1 табл. 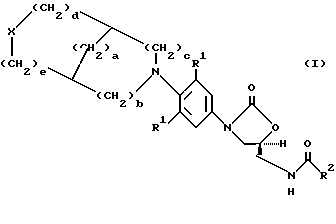

и их фармацевтически приемлемые соли,
где X представляет (a) O, (b) S, (c) SO, (d) SO2;
R1 представляет независимо H, F;
R2 представляет (a) водород, (b) C1-C8-алкил, возможно замещенный одним или более заместителем, выбранным из группы, включающей F, Cl, гидрокси, (g) C1-C8-алкокси;
a = 0 - 3;
b = 0 - 2;
c = 0 - 2 (при условии, что b и c не могут быть оба 0);
d = 0 - 2;
e = 0 - 2 (при условии, что d и e не могут быть оба 0).
а) (S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-окса-5-азабицикло[2.2.1]гептан-5-ил] фенил]-2-оксо-5-оксазолидинил]метил]-ацетамид,
b) (S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-5-азабицикло[2.2.1] гептан-5-ил] фенил]-2-оксо-5-оксазолидинил]метил]-ацетамид, или
c) (S)-N-[[3-[3-фтор-4-[(1S, 4S)-2-тиа-2,2-диоксо-5-азабицикло[2.2.1] гептан-5-ил]фенил]-2-оксо-5-оксазолидинил]метил]-ацетамид.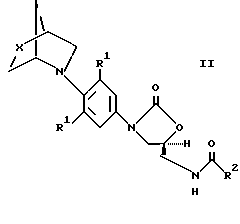
или их фармацевтически приемлемые соли,
где X представляет (a) O, (b) S, (c) SO, (d) SO2;
R1 представляет независимо H, F;
R2 представляет (a) водород, (b) C1-C8-алкил, возможно замещенный одним или более заместителем, выбранным из F, Cl, гидрокси, (g) C1-C8-алкокси.
| RU 94046011 A1, 10.10.96 | |||
| RU 95114378 A1, 10.06.97 | |||
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| ДАТЧИК-ШВОУЛОВИТЕЛЬ ДЛЯ ОБНАРУЖЕНИЯ ШВА ПРИ ОБРАБОТКЕ ДВИЖУЩЕЙСЯ ТКАНИ | 0 |
|
SU312000A1 |
| УСТРОЙСТВО для ПРОДОЛЬНОЙ РЕЗКИ полосового ПОЛИМЕРНОГО МАТЕРИАЛАi:.-K;iu;^:j-M-.xH?!it>&.iiMj| | 0 |
|
SU352781A1 |

