Изобретение относится к новым оксазолидиноновым соединениям или к их фармацевтически приемлемым солям и к содержащим их в качестве активного ингредиента фармацевтическим агентам для профилактики или лечения инфекционных заболеваний. Эти соединения являются уникальными оксазолидинонами, имеющими гексагидро-1,4-диазепин-5-оновое замещение.
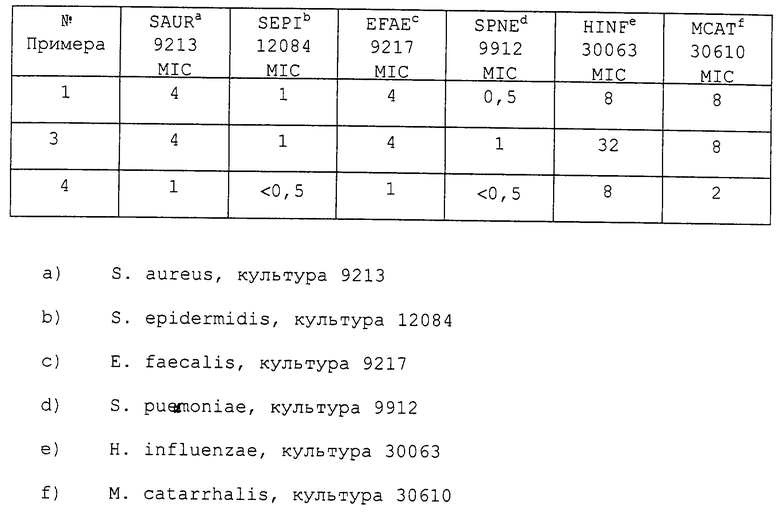
Более конкретно, новые оксазолидиноновые соединения настоящего изобретения относятся к полезным антимикробным агентам, эффективным против различных патогенов, встречающихся у человека и в ветеринарии, включающих грамм-положительные аэробные организмы, такие как staphylococci и streptococci с множественной устойчивостью к лекарственным препаратам, грамм-отрицательные организмы, такие как Н. influenzae и М. catarrhalis, а также анаэробные организмы, такие как разновидности bacteroides и clostridia и кислотоустойчивые организмы, такие как Mycobacterium tuberculosis и Mycobacterium avium.
ИНФОРМАЦИЯ ОБ УРОВНЕ ТЕХНИКИ
Международная публикация 97/27188 раскрывает аналоги пиперазин-3-она, которые являются гомологами соединений данного изобретения.
Международная публикация WО 93/23384 раскрывает оксазолидиноны, содержащие замещенный диазиновый (пиперазиновый) фрагмент и их применение в качестве антимикробных агентов.
Международная публикация WО 93/09103 раскрывает замещенные арил- и гетероарил-фенил-оксазолидиноны, полезные в качестве антимикробных агентов.
Международная публикация WО 90/02744 раскрывает 5'-индолинил-5-амидометилоксазолидиноны, 3-(конденсированный цикл-замещенный)фенил-5-амидометилоксазолидиноны, 3-(азот-замещенный)фенил-5-амидометилоксазолидиноны, полезные в качестве антибактериальных агентов.
Другие ссылки, раскрывающие различные оксазолидиноны, включают патенты США 5547950, 4801600, J. Med. Chem., 32, 1673-81 (1989); J. Med. Chem., 33, 2569-78 (1990); Tetrahedron, 45, 1323-26 (1989); J. Med. Chem., 35, 1156 (1992).
Публикация европейского патента 352781 раскрывает фенил- и пиридил-замещенные фенилоксазолидиноны.
Публикация европейского патента 316594 раскрывает 3-замещенные стирилоксазолидиноны.
Публикация европейского патента 312000 раскрывает фенилметил- и пиридилметил-замещенные фенилоксазолидиноны.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение предлагает оксазолидиноновое производное, представленное общей структурной формулой I, или его фармацевтически приемлемую соль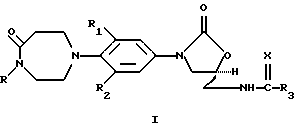
или его фармацевтически приемлемую соль,
где R обозначает Н, С2-6алкенил, С2-7алкинил, C1-6алкил или C1-6алкил, замещенный одним или двумя заместителями, выбранными из следующих групп:
a) F, b) С1, с) СF3, d) -ОН, e) С1-4алкокси, f) -СН2С(=O) С1-4алкила, g) -OC(=O)N(R4)2, h) С1-4алкилS(O)n, (где n равно 0-2), i) -CN, j) карбокси, k) С1-4алкокси, 1) -C(=O)N(R4)2, m) -N(R4)SO2С1-4алкила, n) -N(R4)C(= O)С1-4алкила, о) -N(R4)C(=O)N(R4)2, p) -N(R4)C(=O)С1-4алкокси, q) арила, или r) Het;
арил является фенилом, необязательно замещенным одним или двумя заместителями, выбранными из:
a) F, b) С1, c) Вr, d) СF3, e) CN, f) C1-3алкокси, или g) C1-3алкилтио;
Het обозначает 5- или 6-членный гетероароматический фрагмент, имеющий 1-3 атома N, О или S, необязательно замещенный следующими заместителями:
a) F, b) С1, c) C1-3алкокси, d) C1-3алкилтио, или e) CN; R1 и R2 независимо представляют собой Н, F или С1; R3 представляет собой
а) C1-6алкил, необязательно замещенный 1-3 атомами F или 1-2 атомами С1, b) C1-6алкокси, c) амино, d) C1-6алкиламино, e) C1-6диалкиламино, f) С3-6циклоалкил, g) С1-6алкилтио или
h) 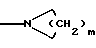
(где m = 0, 1, 2, 3 или 4);
R4 представляет собой a) Н или b) C1-3алкил; и Х представляет собой О или S.
Настоящее изобретение также предлагает антимикробный агент или фармацевтическую композицию, которая содержит в качестве активного ингредиента оксазолидиноновое соединение или его фармацевтически приемлемую соль. Антимикробный агент, содержащий активный ингредиент настоящего изобретения, может использоваться для лечения или профилактики инфекционных заболеваний.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения формулы I, структура которых раскрыта выше, являются полезными антимикробными агентами. Обычно, как дополнительно поясняется ниже, эти соединения могут быть введены в качестве антибактериальных агентов в интервале доз приблизительно от 0,1 до 100 мг/кг, или, предпочтительно, приблизительно от 3,0 до 50 мг/кг веса тела в день.
В структурной формуле, приведенной выше, содержание углерода в различных углеводород-содержащих фрагментах определяется префиксом, обозначающим минимальное и максимальное число атомов углерода во фрагменте, т.е. префикс Ci-Cj определяет число атомов углерода, находящееся в интервале от целого числа "i" до целого числа "j" включительно.
Термин "Ci-6алкил", использующийся в данном документе, относится к алкильной группе, имеющей от одного до шести атомов углерода, такой как, например, метил, этил, пропил, бутил, пентил, гексил и их изомерные формы; предпочтительно, метил, этил, пропил и их изомерные формы.
Термин "С2-6алкенил" относится к алкенильной группе, имеющей по меньшей мере одну двойную связь и от двух до шести атомов углерода, такой как, например, винил, 1-пропенил, 2-пропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил, 1-пентенил, 2-пентенил, 1-гексенил и их изомерные формы, предпочтительно, алкенильная группа имеет от 2 до 6 атомов углерода, и, более предпочтительно, алкенильная группа имеет от 2 до 4 атомов углерода.
Термин "С2-7алкинил" относится к алкинильной группе, имеющей по меньшей мере одну тройную связь и от двух до семи атомов углерода, такой как, например, этинил, пропинил, бутинил, пентинил, гексинил, гептинил и их изомерные формы.
Термин "C1-6алкиламино" относится к алкильной группе, имеющей от одного до шести атомов углерода, соединенной с амино-фрагментом.
Термин "C1-6диалкиламино" относится к двум алкильным группам, имеющим от одного до шести атомов углерода, соединенным с амино-фрагментом.
Термин "С1-4алкокси" относится к алкильной группе, имеющей от одного до четырех атомов углерода, соединенной с атомом кислорода гидроксильной группы, такой как, например, метоксильная, этоксильная, пропоксильная, бутоксильная группы и их изомерные формы, предпочтительно, алкоксильная группа имеет от 1 до 2 атомов углерода.
Термин "C1-6алкилтио" относится к алкильной группе, имеющей от одного до шести атомов углерода, соединенной с тиольным фрагментом, такой как, например, метилтиольная, этилтиольная, пропилтиольная и их изомерные формы, предпочтительно, алкилтиольная группа имеет от 1 до 2 атомов углерода.
Термин "С3-6циклоалкил" относится к 3-6 атомам углерода, образующим циклопропил, циклобутил, циклопентил, циклогексил и их изомерные формы.
Термин "арил" относится к фенильному фрагменту, необязательно замещенному одной или двумя из групп: F, C1, Вr, -СF3, -CN, -C1-3алкокси или -C1-3алкилтио.
Термин "Het" относится к 5- или 6-членному гетероароматическому фрагменту, имеющему от одного до трех атомов, выбранных из группы, состоящей из атомов N, О или S, такому как, например, фуран, тиофен, пиррол, пиразол, триазолы, оксазол, тиазол, изотиазол, оксадиазолы, оксатиазол, пиридин, пиридазин, пиримидин, пиразин, пиперазин и триазины, каждый из которых может быть необязательно замещен одним заместителем, выбранным из группы, состоящей из F, С1, C1-3алкокси, C1-3алкилтио, CN.
Соединения настоящего изобретения могут быть превращены в соли в соответствии с традиционными способами.
Фармацевтически приемлемые соли обозначают соли присоединения кислоты, использующиеся для введения соединений данного изобретения, и включают гидрохлорид, гидробромид, сульфат, фосфат, ацетат, пропионат, лактат, мезилат, малеат, сукцинат, тартрат, цитрат, 2-гидроксиэтилсульфонат, фумарат и т.п., если присутствует основная группа. Эти соли могут находиться в гидратированной форме. Некоторые из соединений данного изобретения могут образовывать соли металлов, такие как соли натрия, калия, кальция и магния, которые охватываются термином "фармацевтически приемлемые соли".
Благодаря конфигурации при С-5 оксазолидинонового цикла соединений, как представлено в структуре формулы I, соединения данного изобретения могут существовать в виде геометрических, оптических и других изомерных форм, и данное изобретение охватывает любой из этих изомеров. Полагают, что рацемическая смесь и энантиомеры являются полезными в качестве антибактериальных агентов. Однако, предпочтительная абсолютная конфигурация при С-5 оксазолидинонового цикла соединений является такой, как представлена в структуре формулы I. По номенклатурной системе Канна-Ингольда-Прелога (Cahn-Ingold-Prelog) эта абсолютная конфигурация называется (S). Считается, что определяющая часть фармакологической активности связана с (S)-энантиомером, обеспечивая антибактериальный эффект.
Соединения формулы I могут быть получены так, как показано на схеме I, где Р представляет собой спиртовую защитную группу, такую как бензил или трет-бутилдиметилсилил. Структуру 2 этой схемы получают в соответствии со способами, описанными в примере 1, стадии 1 и 2. На схеме I спирты 2 защищают, получая бензиловые эфиры. В процедуре, подходящей для этой реакции, раствор спирта 2 в таком растворителе, как Et2O или THF, подвергают взаимодействию вначале с гидридом натрия при 0-25oС и затем с бензилбромидом и иодидом тетрабутиламмония при 0-25oС, получая структуру 3. Этиленкеталь 3 затем может быть удален с помощью кислого катализатора, такого как п-толуолсульфоновая кислота в ацетоне (как описано в примере 1, стадия 2), с получением структуры 5, где Р является бензилом. Альтернативно, может быть удален кеталь 2, полученную структуру 4 подвергают взаимодействию с трет-бутилдиметилсилилхлоридом и имидазолом в DMF или трет-бутилдиметилсилилхлоридом и триэтиламином в метиленхлориде, получая структуру 5, где спиртовая защитная группа (Р) является трет-бутилдиметилсилилом. Затем кетон 5 подвергают взаимодействию с гидрохлоридом гидроксиламина и ацетатом натрия в метаноле-метиленхлориде, получая оксим 6 (см. пример 1, стадия 3). Перегруппировку по Бекману (Beckmann) структуры 6 проводят с п-толуолсульфонилхлоридом и карбонатом натрия в водном ацетоне при 20-40oС, получая структуру 7. Для соединений формулы I, в которых R не является водородом, соединения 7 могут быть алкилированы с помощью R'Y, где Y представляет собой Вr, I, СН3SО3 или п-СН3РhSО3, a R' является соответствующим алкильным заместителем. В одном способе такого алкилирования соединения структуры 7 подвергают взаимодействию с гидридом натрия и R'Y в таком растворителе, как DMF, при 0-25oС, получая соединение 8. Альтернативно, структура 7 может взаимодействовать с R'Y, гидроксидом калия и бромидом тетрабутиламмония в THF или ацетонитриле при 20-50oС, получая соединение 8. Удаление защитных групп со спиртов 7 или 8 дает структуру 9. Если Р является бензиловым эфиром, этот процесс проводят путем гидрогенолиза с использованием водорода и палладиевого катализатора в этаноле или с использованием формиата аммония и палладиевого катализатора в метаноле при 10-30oС. Защитная группа трет-бутилдиметилсилил может быть удалена в кислых условиях или с использованием иона фтора. Удаление защитной группы может быть проведено, например, с использованием трифторуксусной кислоты в метиленхлориде при 25oС, или с использованием фторида тетрабутиламмония в THF при 25oС, с получением спирта 9. Превращение спирта 9 в амин 11 может быть проведено, как описано в примере 1, стадии 1. Альтернативно, взаимодействие соединения 9 с м-нитробензолсульфонилхлоридом и триэтиламином в метиленхлориде при 5-25oС дает м-нитробензолсульфонат 10, который взаимодействует с гидроксидом аммония в THF или ацетонитриле-изопропаноле при 30-60oС, с получением амина 11. Взаимодействие соединения 11 с галогенангидридами или ангидридами кислот, изоцианатами, изотиоцианатами или дитиоэфирами приводит к получению соединений формулы I.
Соединения формулы I, в которых R является водородом, а Х является кислородом, обычно получают путем взаимодействия соединений 12 с п-толуолсульфонилхлоридом и карбонатом натрия в водном ацетоне при 20-40oС (см. пример 1, стадия 4).
Соединения данного изобретения используются для лечения микробных инфекций у людей и у теплокровных животных путем либо парентерального, перорального, либо местного введения.
Термин "лечение" в данном описании означает частичное или полное уменьшение симптомов заболевания, от которого страдает пациент; термин "профилактика" в данном описании означает частичное или полное предотвращение симптомов заболевания у пациента, у которого в соответствии с диагнозом врача может возникнуть такое заболевание или связанное с ним состояние, если не принять превентивных мер.
Фармацевтические композиции данного изобретения могут быть получены путем объединения соединений формулы I данного изобретения с твердым или жидким фармацевтически приемлемым носителем и, необязательно, с фармацевтически приемлемыми адъювантами и эксципиентами, с помощью стандартных и традиционных методик. Твердые формы композиций включают порошки, таблетки, диспергируемые гранулы, капсулы и суппозитории. Твердый носитель может представлять собой по меньшей мере одно вещество, которое может также функционировать как разбавитель, вкусовая добавка, солюбилизатор, смазочный агент, суспендирующий агент, связующий агент, средство для дезинтеграции таблеток и капсулирующий агент. Инертные твердые носители включают карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, целлюлозные вещества, воск с низкой температурой плавления, кокосовое масло и т. п. Жидкие формы композиции включают растворы, суспензии и эмульсии. Например, они могут представлять растворы соединения данного изобретения, растворенных в воде и системах вода-пропиленгликоль и вода-полиэтиленгликоль, необязательно содержащие традиционные окрашивающие агенты, вкусовые добавки, стабилизаторы и загустители.
Предпочтительно, фармацевтическую композицию получают, используя традиционные методики, в стандартной дозировочной форме, содержащей эффективные количества активного компонента, то есть соединения формулы I в соответствии с данным изобретением.
Количество активного компонента, то есть соединения формулы I, в фармацевтической композиции и ее стандартной форме могут широко варьировать или приспосабливаться в зависимости от конкретного способа применения, активности конкретного соединения и желательной концентрации. Обычно количество активного компонента варьирует от 0,5 до 90% от массы композиции.
При терапевтическом лечении или подавлении бактериальных инфекций у людей и у животных, у которых были обнаружены бактериальные инфекции, соединения или их фармацевтические композиции, вводят перорально, парентерально и/или местно в такой дозе, чтобы получить и поддерживать такую концентрацию, то есть количество, или уровень активного компонента в крови у животного, которого подвергают лечению, которая является антибактериально-эффективной. Обычно такое антибактериально-эффективное количество дозы активного соединения находится в интервале приблизительно от 0,1 до 100 мг/кг, более предпочтительно, приблизительно от 3,0 до 50 мг/кг веса тела/день. Следует понимать, что дозы могут варьировать в зависимости от потребностей пациента, тяжести бактериальной инфекции, подлежащей лечению и конкретно используемого соединения. Кроме того, надо понимать, что начальная вводимая доза может быть увеличена свыше указанного ранее верхнего уровня, для того чтобы быстро достигнуть желательного уровня в крови, или начальная доза может быть меньше оптимальной, и суточная доза может быть прогрессивно увеличена в процессе курса лечения, в зависимости от конкретной ситуации. При желании суточная доза может быть также разделена на несколько доз для введения, например, от 2 до 4 раз в день.
Соединения формулы I вводят парентерально, то есть путем инъекции, например, путем внутривенной инъекции или путем других парентеральных способов введения. Фармацевтические композиции для парентерального введения обычно содержат фармацевтически приемлемое количество соединения, соответствующего формуле I, в виде растворимой соли (соли присоединения кислоты или основной соли), растворенной в фармацевтически приемлемом жидком носителе, таком как, например, вода для инъекции и подходящий буферный изотонический раствор, например, имеющий рН, приблизительно составляющий 3,5-6. Подходящие буферные агенты включают, например, ортофосфат натрия, бикарбонат натрия, N-метилглюкамин, L(+)-лизин и L(+)-аргинин (здесь указаны некоторые из таких агентов). Соединения в соответствии с формулой I обычно растворяют в носителе в количестве, достаточном для того, чтобы обеспечить фармацевтически приемлемую концентрацию для инъекции в интервале приблизительно от 1 до 400 мг/мл. Полученную жидкую фармацевтическую композицию вводят так, чтобы получить вышеупомянутое антибактериально-эффективное количество (дозу). Соединения формулы I в соответствии с данным изобретением преимущественно вводят перорально в твердых и жидких дозированных формах.
Для местного применения эффективное количество соединения формулы I вводят в фармацевтически приемлемую гелевую или кремовую основу для лекарства, которое может наноситься на кожу пациента в области, подлежащей лечению. Способы получения таких кремов и гелей хорошо известны в данной области и могут включать агенты, способствующие увеличению проницаемости.
Соединения данного изобретения являются полезными антимикробными агентами, эффективными против различных патогенов человека и животных, включающих грамм-положительные аэробные организмы, такие как staphylococci и streptococci с множественной устойчивостью к лекарственным препаратам, грамм-отрицательные организмы, такие как Н. influenzae и М. catarrhalis, а также анаэробные организмы, такие как разновидности bacteroides и clostridia и кислотоустойчивые организмы, такие как Mycobacterium tuberculosis и Mycobacterium avium.
Для того чтобы более полно иллюстрировать содержание изобретения и способ его применения, приводятся следующие экспериментальные примеры, но они не должны приниматься как ограничивающие объем изобретения.
ПРИМЕР 1
Получение (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксo-l,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил]метил] ацетамида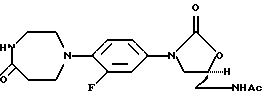
Стадия 1: Получение (S)-N-[3-(3-фтор-4-пиперидин-1-илфенил) -2-оксо-оксазолидин-5-илметил)-ацетамида
Диизопропилэтиламин (15,7 мл) и 3,4-дифторнитробензол (5,0 мл) последовательно добавляют к этилацетатному раствору (70 мл) пиперидина (5,77 г) и смесь перемешивают при комнатной температуре в течение двух дней. К реакционному раствору добавляют воду, отделенный этилацетатный слой промывают водой и насыщенным солевым раствором и сушат над безводным сульфатом натрия. Растворитель упаривают, получая нитросоединение (10,1 г) с выходом 100%. Палладий на угле (10%, 1,0 г) добавляют к этилацетатному раствору (101 мл) нитросоединения (10,1 г) и смесь перемешивают при комнатной температуре в течение 4 часов в атмосфере водорода. Палладий на угле отфильтровывают и фильтрат концентрируют в вакууме, получая амин (8,75 г, 100%). Гидрокарбонат натрия (5,0 г) и бензилоксикарбонилхлорид (8,4 мл) добавляют последовательно к тетрагидрофурановому (THF) раствору (100 мл) амина (8,75 г) и смесь перемешивают при комнатной температуре в течение 14 часов. К реакционному раствору добавляют воду, отделенный THF слой промывают водой и насыщенным солевым раствором и сушат над безводным сульфатом натрия. Растворитель упаривают и остаток очищают с помощью колоночной хроматографии на силикагеле (растворитель: этилацетат/гексан/хлороформ= 1/6/4), получая бензилкарбамат (14,5 г) с выходом 98%. Бутил лития (1,6 М раствор в гексане: 5,2 мл) добавляют к THF раствору (24 мл) бензилкарбамата (2,75 г) при -78oС и смесь перемешивают в течение пяти минут. При той же температуре (R)-(-)-глицидилбутират (1,25 мл) добавляют к перемешиваемому раствору и смесь перемешивают в течение 14 часов пока температура медленно поднимается до комнатной температуры. К реакционному раствору добавляют воду, отделенный THF слой промывают водой и насыщенным солевым раствором и сушат над безводным сульфатом натрия. Растворитель упаривают и остаток очищают с помощью колоночной хроматографии на силикагеле (растворитель: этилацетат/гексан=3/1), получая спирт (2,20 г) с выходом 89%. Тозилхлорид (2,85 г) добавляют к пиридиновому раствору (8 мл) спирта (2,20 г) и смесь перемешивают при комнатной температуре в течение 6 часов. К реакционному раствору добавляют воду (32 мл) и смесь перемешивают в течение 1 часа. Полученный осадок собирают путем фильтрации и промывают водой, после чего сушат под вакуумом при комнатной температуре, получая тозилат (3,28 г) с выходом 98%. Азид натрия (3,80 г) добавляют к диметилформамидному (DMF) раствору (23 мл) тозилата (3,28 г) при комнатной температуре и смесь перемешивают при 65oС в течение 5,5 часа. После охлаждения реакционной смеси до комнатной температуры добавляют воду и смесь экстрагируют этилацетатом; органический слой концентрируют под вакуумом. Полученный остаток растворяют в этилацетате, промывают водой и насыщенным солевым раствором и сушат над безводным сульфатом натрия. Растворитель упаривают и остаток очищают с помощью колоночной хроматографии на силикагеле (растворитель: этилацетат/гексан=1/1), получая азид (2,20 г) с выходом 94%. Уксусный ангидрид (0,65 мл) и пиридин (1,0 мл) добавляют к этилацетатному раствору (19 мл) азида (2,20 г) при комнатной температуре; после добавления палладия на угле (10%, 0,22 г) смесь перемешивают при комнатной температуре в течение 6 часов в атмосфере водорода при давлении 1 атм. Палладий на угле отфильтровывают и фильтрат промывают водой и насыщенным солевым раствором и сушат над безводным сульфатом натрия. Растворитель упаривают и остаток очищают с помощью колоночной хроматографии на силикагеле (растворитель: ацетон/гексан=1/1), получая указанное в заголовке соединение.
Стадия 2: Получение (S)-N-{3-[3-фтор-4-(4-оксо-пиперидин-1-ил)-фенил]-2-оксо-оксазолидин-5-илметил}-ацетамида
Используя коммерчески доступный 1,4-диоксо-8-аза-спиро [4.5] декан, (S)-N-{ 3-[4-(1,4-диокса-8-аза-спиро[4.5] дец-8-ил)-3-фтор-фенил]-2-оксо-оксазолидин-5-илметил}-ацетамид синтезируют с помощью такого же способа, как и в стадии 1. К ацетоновому раствору (70 мл) этого соединения (3,79 г) последовательно добавляют воду (20 мл) и моногидрат п-толуолсульфоновой кислоты (3,66 г) и смесь нагревают с обратным холодильником в течение 3 часов. После охлаждения реакционной смеси до комнатной температуры ацетон отгоняют и водный слой нейтрализуют триэтиламином. Раствор экстрагируют метиленхлоридом, органический слой промывают насыщенным солевым раствором и сушат над безводным сульфатом натрия. Растворитель упаривают и остаток очищают с помощью колоночной хроматографии на силикагеле (растворитель: хлороформ/метанол= 50/1-25/1), получая указанное в заголовке соединение.
Стадия 3: Получение (S)-N-{3-[3-фтор-4-(4-гидроксиимино-пиперидин-1-ил)-фенил]-2-оксо-оксазолидин-5-илметил}-ацетамида
Ацетат натрия (517 мг) и гидрохлорид гидроксиламина (219 мг) последовательно добавляют к раствору 1,00 г продукта стадии 2 в метанол-метиленхлоридной смеси (10-10 мл) и смесь перемешивают при комнатной температуре в течение 2 дней. Растворитель упаривают и остаток растворяют в метаноле, затем добавляют силикагель (8 г). Метанол упаривают и остаток очищают с помощью колоночной хроматографии на силикагеле (растворитель: хлороформ/метанол= 50/1-25/1), получая указанное в заголовке соединение.
Стадия 4: Получение (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксо-1,4-диазепин-1-ил)-фенил]-2-оксо-5-оксазолидинил]метил]-ацетамида
Смесь соединения продукта стадии 3 (0,200 г, 0,549 ммоль) в ацетоне (5,3 мл) в атмосфере азота при перемешивании обрабатывают вначале 5% водным карбонатом натрия (5,3 мл) и затем по каплям в течение 3 минут раствором п-толуолсульфонилхлорида (0,16 г, 0,82 ммоль) в ацетоне (2,7 мл). Вначале эта смесь представляет собой двухфазный раствор; однако, приблизительно через 25 минут начинает образовываться осадок. Его выдерживают при комнатной температуре (23oС) в течение 4 часов и фильтруют. Фильтрат концентрируют при пониженном давлении для удаления ацетона и водный остаток экстрагируют CH2Cl2. Экстракт сушат (МgSO4) и концентрируют, получая небольшое количество неочищенного продукта. Основная часть продукта находится в водном слое, который концентрируют в вакууме. Остаток объединяют с неочищенным продуктом из СН2Сl2 экстракта и подвергают хроматографии на силикагеле, элюируя смесями MeOH-NH4OH-CH2Cl2, затем 3-5% МеОН и 0,3-0,5% NH4OH. Продукт кристаллизуют из MeOH-EtOAc, получая указанное в заголовке соединение. Т. пл. 140-146oС;
МС m/z (относительная интенсивность) 364 (М+, 96,1), 320 (100), 306 (6,7), 294 (10,9), 236 (41,8);
МСВР рассч. для C17H21FN4O4; 364,1547 (М+); обнаружено 364,1545;
1H ЯМР [300 МГц, (СD3)2S0] δ 1,81 (с, 3Н), 2,57 (м, 2Н), 3,07 (м, 4Н), 3,24 (м, 2Н), 3,38 (т, 2Н), 3,67 (д, д, 1Н), 4,06 (т, 1Н), 4,68 (м, 1Н), 7,08 (т, 1Н), 7,13 (д, д, 1Н), 7,45 (д, д, 1Н), 7,65 (т, 1Н), 8,21 (т, 1Н).
ПРИМЕР 2
Получение (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксо-1,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил]метил]тиоацетамида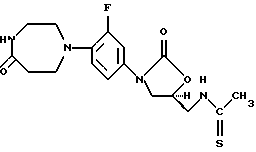
Стадия 1: Получение трет-бутилдиметилсилилового эфира (S)-[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксо-1,4-диазепин-1-ил)-фенил] -2-оксо-5-оксазолидинил]метила
Перемешиваемый раствор 10,6 г (0,03 моль) (S)-[3-[4-(1,4-диоксо-8-азаспиро[4,5] дец-8-ил)-3-фторфенил] -2-оксо-5-оксазолидинил]метанола, промежуточного соединения формулы 2 (схема 1) для получения (S)-N-{3-[3-фтор-4-(4-оксопиперидин-1-ил)фенил] -2-оксооксазолидин-5-илметил}-ацетамида (пример 1, стадия 2), в ацетоне (230 мл) обрабатывают водой (65 мл) и моногидратом п-толуолсульфоновой кислоты (11,4 г, 0,06 моль), нагревают с обратным холодильником в атмосфере азота в течение 5 часов и выдерживают при температуре окружающей среды (24oС) 18 часов. Затем его концентрируют в вакууме для удаления ацетона. Водный остаток нейтрализуют бикарбонатом натрия и экстрагируют этилацетатом; экстракт промывают насыщенным бикарбонатом натрия, водой и разбавленным хлоридом натрия, сушат (Nа2SO4) и концентрируют, получая кетон, соединение формулы 4 (схема 1). Раствор кетона и триэтиламина (12,5 мл, 0,009 моль) в метиленхлориде (100 мл) при перемешивании обрабатывают трет-бутилдиметилсилилхлоридом (6,03, г, 0,04 моль) и выдерживают в атмосфере азота при температуре окружающей среды в течение 23 часов. Добавляют дополнительное количество трет-бутилдиметилсилилхлорида (3,0 г) и смесь выдерживают при температуре окружающей среды еще 20 часов. Снова добавляют дополнительное количество триэтиламина (3,0 мл) и трет-бутилдиметилсилилхлорида (3,0 г) и смесь выдерживают при температуре окружающей среды 4 дня, разбавляют метиленхлоридом, промывают водой и разбавленным хлоридом натрия, сушат (Na2SO4) и концентрируют. Хроматография остатка на силикагеле с использованием в качестве элюента смесей ацетон-гептан, которые содержат 20-30% ацетона, дает 7,72 г трет-бутилдиметилсилилового (TBDMS) эфира, соединения формулы 5 (схема 1), где Р представляет собой TBDMS. Раствор TBDMS эфира (7,27 г, 17,2 ммоль) в метаноле (150 мл) при перемешивании обрабатывают по каплям раствором гидрохлорида гидроксиламина (1,44 г, 0,021 моль) и ацетата натрия (1,72 г, 0,021 моль) в воде (15 мл) и выдерживают при температуре окружающей среды в течение 20 часов. Смесь концентрируют при пониженном давлении. Раствор остатка, белого твердого вещества, в метиленхлориде промывают водой и разбавленным хлоридом натрия, сушат (Na2SO4) и концентрируют, получая 7,25 г оксима, соединения формулы 6 (схема 1). Раствор оксима в ацетоне (165 мл) в атмосфере азота, при перемешивании, обрабатывают 5% водным карбонатом натрия (165 мл) и затем по каплям, в течение 20 минут, раствором п-толуолсульфонилхлорида (4,92 г, 0,0258 моль) в ацетоне (80 мл). Смесь выдерживают при температуре окружающей среды в течение 18 часов и концентрируют при пониженном давлении. Раствор остатка в метиленхлориде промывают водой и разбавленным хлоридом натрия, сушат (Na2SO4) и концентрируют. Хроматография остатка на силикагеле с использованием в качестве элюента смеси 3% метанол-0,3% гидроксид аммония-метиленхлорид дает 5,98 г указанного в заголовке соединения.
Стадия 2: Получение (S)-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксо-1,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил]метил]амина
Охлажденную во льду смесь продукта примера 2, стадии 1 (0,22 г, 0,50 ммоль), в тетрагидрофуране (THF; 15 мл) при перемешивании, в атмосфере азота, обрабатывают по каплям, в течение 2 минут, 1М раствором фторида тетрабутиламмония в THF (1,5 мл). Смесь выдерживают на ледяной бане в течение 10 минут и при температуре окружающей среды (24oС) в течение 1 часа 25 минут, разбавляют этилацетатом, промывают водой и насыщенным солевым раствором, сушат (Na2SO4) и концентрируют. Хроматография остатка на силикагеле с использованием в качестве элюента смесей метанол-метиленхлорид, содержащих 3-6% метанола, дает 0,15 г спирта, соединения формулы 9 (схема 1), где R является водородом: MC(ES) m/z 324 (М+Н+). Перемешиваемую суспензию спирта (0,15 г, 0,46 ммоль) в метиленхлориде (15 мл) и THF (8 мл) в атмосфере азота обрабатывают триэтиламином (0,5 мл, 1,4 ммоль) и затем порциями, в течение 1 минуты, при температуре окружающей среды, 0,14 г (0,56 ммоль) м-нитробензолсульфонилхлорида. Смесь перемешивают 90 минут, смешивают с дополнительным количеством метиленхлорида (10 мл), получая раствор, и выдерживают при температуре окружающей среды 1 час. Затем его выдерживают несколько дней при -11oС, разбавляют метиленхлоридом, промывают насыщенным бикарбонатом натрия, водой и насыщенным солевым раствором, сушат (Na2SO4) и концентрируют, получая 0,21 г м-нитробензолсульфоната, соединения формулы 10 (схема 1). Перемешиваемая смесь м-нитробензолсульфоната (0,21 г, 0,44 ммоль), ацетонитрила (10 мл), 2-пропанола (10 мл) и 29% гидроксида аммония (10 мл) нагревают при 45-50oС с конденсатором, содержащим смесь сухого льда с ацетоном, в течение 4,5 часов и выдерживают при температуре окружающей среды 18 часов. Добавляют дополнительное количество гидроксида аммония (5 мл) и смесь нагревают при 45-50oС в течение 4,5 часов, выдерживают при температуре окружающей среды 1 час, обрабатывают 5 мл гидроксида аммония и выдерживают при температуре окружающей среды 18 часов. Затем его концентрируют, получая желтое твердое вещество, которое подвергают хроматографии на сили-кагеле, элюируя смесями метанол-метиленхлорид, содержащими 5-7,5% метанола, затем смесью 8% метанол-0,2% гидроксид аммония-метиленхлорид, получая указанный в заголовке продукт.
Стадия 3: Получение (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксо-1,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил]метил]тиоацетамида
Перемешиваемый раствор 0,12 г продукта примера 2, стадии 2, и 0,40 мл триэтиламина в смеси метиленхлорида (10 мл) и метанола (10 мл) в атмосфере азота обрабатывают этилдитиоацетатом (0,05 мл) и выдерживают при температуре окружающей среды в течение 145 часов. Дополнительные порции по 0,05 мл этилдитиоацетата добавляют через 24, 31 и 49 часов; через 49 часов добавляют также дополнительное количество триэтиламина (1,0 мл). Смесь концентрируют до небольшого объема, разбавляют этилацетатом, промывают водой и насыщенным солевым раствором, сушат (Na2SO4) и концентрируют. Хроматография остатка на силикагеле с использованием в качестве элюента смеси 3,5% метанол-метиленхлорид, дает 0,061 г указанного в заголовке продукта.
1H ЯМР [300 МГц, (СD3)2SO] δ 2,42 (с, 3Н), 2,56 (м, 2Н), 3,07 (м, 4Н), 3,24 (м, 2Н), 3,76 (дд, 1Н), 3,87 (м, 2Н), 4,11 (т, 1Н), 4,91 (м, 1Н), 7,12 (м, 2Н), 7,46 (дд, 1Н), 7,67 (шир. с, 1Н), 10,35 (шир. с, 1Н).
ПРИМЕР 3
Получение (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-4-метил-5-оксо-1,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида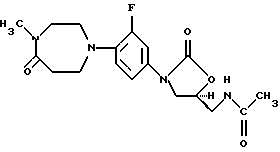
Стадия 1: Получение (S)-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-4-метил-5-оксо-1,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил]метил]амина
Смесь 0,63 г (1,4 ммоль) продукта примера 2, стадии 2, метилиодида (0,093 мл) и THF (40 мл) добавляют по каплям в течение 12 минут в атмосфере азота к перемешиваемой смеси порошкообразного гидроксида калия (0,12 г) и бромида тетрабутиламмония (0,096 г) в THF (10 мл) и выдерживают при температуре окружающей среды 20 часов. Затем его разбавляют этилацетатом, промывают водой и насыщенным солевым раствором, сушат (МgSO4) и концентрируют. Хроматография остатка на силикагеле с использованием в качестве элюента смесей ацетона-метиленхлорида, которые содержат 10-40% ацетона, дает 0,46 г (71%) метилированного продукта, соединения формулы 8 (схема 1), где R' является метилом. При охлаждении во льду и перемешивании смесь этого продукта (0,17 г, 0,38 ммоль) и THF (12 мл) в атмосфере азота обрабатывают по каплям 1М раствором фторида тетрабутиламмония в THF (1,2 мл). Выдерживают в бане со льдом 15 минут и при температуре окружающей среды 3 часа смешивают с водой со льдом и экстрагируют этилацетатом. Экстракт промывают водой и насыщенным солевым раствором, сушат (МgSO4) и концентрируют, получая 0,15 г спирта, соединения формулы 9 (схема 1). Перемешиваемый, охлажденный во льду раствор спирта (0,52 г, 1,5 ммоль) и триэтиламина (0,60 мл) в метиленхлориде (45 мл) обрабатывают в течение 5 минут порциями м-нитробензолсульфонилхлорида (0,42 г). Смесь выдерживают в бане со льдом 15 минут и при температуре окружающей среды 3 часа разбавляют метиленхлоридом, промывают насыщенным бикарбонатом натрия, водой и насыщенным солевым раствором, сушат (MgSO4) и концентрируют, получая м-нитробензолсульфонат, соединение формулы 10 (схема 1). Смесь этого продукта, ацетонитрила (35 мл), 2-пропанола (35 мл) и концентрированного гидроксида аммония (35 мл) выдерживают при перемешивании при 45-50oС с холодильником с сухим льдом-ацетоном в течение 4,5 часов и при температуре окружающей среды в течение 20 часов. Добавляют дополнительное количество гидроксида (6 мл) и смесь выдерживают при 45-50oС в течение 5,5 часов и 18 часов при температуре окружающей среды. Затем смесь концентрируют при пониженном давлении для удаления органических растворителей и водный остаток экстрагируют вначале этилацетатом и затем метиленхлоридом. Экстракты промывают водой и насыщенным солевым раствором, сушат (MgSO4) и концентрируют. Хроматография остатка на силикагеле с использованием смесей метанол-метиленхлорид, содержащих 7,5-10% метанола, дает указанное в заголовке соединение.
Стадия 2: Получение (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-4-метил-5-оксо-1,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида
При перемешивании и охлаждении во льду смесь 0,10 г (0,30 ммоль) продукта примера 3, стадии 1, и пиридина (1,74 мл) в атмосфере азота обрабатывают по каплям уксусным ангидридом (0,57 мл, 6,04 ммоль) и выдерживают в бане со льдом 15 минут и при температуре окружающей среды 3,5 часа. Затем смесь концентрируют в вакууме; остаток смешивают с водой со льдом и насыщенным бикарбонатом натрия и экстрагируют этилацетатом. Экстракт промывают водой и насыщенным солевым раствором, сушат (MgSO4) и концентрируют. Кристаллизация остатка из смеси этилацетат-метанол дает 0,053 г указанного в заголовке соединения.
T. пл. 203-204oС.
MC(ES) m/z 379 (М+Н+, 401 (M+Na+).
Аналитич. рассчит. для C18H23FN4O4: С, 57,13; Н, 6,13; N, 14,81. Обнаружено: С, 57,05; Н, 6,23; N, 14,85.
ПРИМЕР 4
Получение (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-4-метил-5-оксо-1,4-диазепин-1-ил)фенил]-2-оксо-5-оксазолидинил] метил] тиоацетамида
Раствор 0,18 г (0,535 ммоль) продукта примера 3, стадии 1 и триэтиламина (0,21 мл) в THF (8 мл) и метиленхлориде (10 мл), охлаждая во льду и перемешивая, обрабатывают раствором этилдитиоацетата (0,074 мл, 0,64 ммоль) в THF (2 мл). Смесь выдерживают при температуре окружающей среды в течение 20 часов, обрабатывают дополнительно одной каплей этилдитиоацетата и выдерживают при температуре окружающей среды 7 часов. Затем ее концентрируют в токе азота. Остаток смешивают с метиленхлоридом, промывают насыщенным бикарбонатом натрия, водой и насыщенным солевым раствором, сушат (Nа2SO4) и концентрируют. Хроматография остатка на силикагеле с использованием в качестве элюента смесей метанол-метиленхлорид, содержащих 2-4% метанола, и кристаллизация продукта из этилацетата дают 0,13 г указанного в заголовке соединения, т. пл. 157-158oС.
Аналитич. рассчит. для С18Н23FN4О3S: С, 54,81; Н, 5,88; N, 14,20. Обнаружено: С, 54,83; Н, 5,93; N, 14,11.
ПРИМЕР 5
Способ тестирования MIC
MIC анализы in vitro тестируемых соединений определяются стандартным методом агарного разведения. Базовый (исходный) раствор каждого аналога соединения готовят в предпочтительном растворителе, обычно DMSO:H2O (1:3). Серийные 2-кратные разведения каждого образца делают, используя аликвоты объемом 1,0 мл стерильной дистиллированной воды. К каждой аликвоте лекарственного препарата объемом 1,0 мл добавляют 9 мл расплавленной агарной среды Mueller Hinton. Агар с лекарственным препаратом смешивают, выливают в 15х100 мм чашки Петри и оставляют для отверждения и высыхания перед инокуляцией.
Пробирки с каждым из тестируемых организмов поддерживают в замороженном состоянии в паровой фазе жидкого азота замораживающего агента. Тестируемые культуры выращивают в течение ночи при 35oС на среде, соответствующей данному организму. Колонии собирают с помощью стерильного тампона и клеточные суспензии готовят в питательной среде Trypticase Soy (TSB) так, чтобы мутность составляла 0,5 стандарта McFarland. Разведение каждой суспензии 1:20 делают в TSB. Плашки, содержащие агар с добавленным лекарственным препаратом, заражают 0,001 мл каплей суспензии клеток, используя репликатор Steers, получая приблизительно от 104 до 105 клеток на пятно. Плашки инкубируют в течение ночи при 35oС.
После инкубации минимальную концентрацию ингибирования (MIC мкг/мл), самую низкую концентрацию лекарственного препарата, которая ингибирует видимый рост организма, просчитывают и регистрируют. Данные показаны в таблице.

Изобретение относится к новым оксазолидиноновым производным формулы (I), их энантиомерам или их фармацевтически приемлемым солям, где R - водород или С1-6алкил, необязательно замещенный группой, выбранной из циано, фтора, хлора гидрокси или карбомоила; R1 и R2 независимо - фтор или водород; R3 - C1-6алкил; Х - О и S. Фармацевтическая композиция, обладающая антимикробным действием, включающая в качестве фармацевтически активного компонента соединение формулы I и фармацевтически приемлемый носитель. Технический результат - получение нового соединения, обладающего антимикробным действием. 2 с. и 6 з.п. ф-лы, 1 табл.
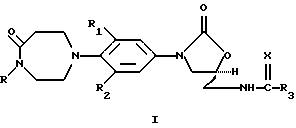

его энантиомер или его фармацевтически приемлемая соль,
где R - водород или C1-6алкил, необязательно замещенный группой, выбранной из циано, фтора, хлора, гидрокси или карбамоила;
R1 и R2 независимо - фтор или водород;
R3 - C1-6алкил;
Х - О или S.
(a) (S)-N[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксо-1,4-диазепин-1-ил)фенил] -2-оксо-5-оксазолидинил] метил] ацетамид,
(b) (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-5-оксо-1,4-диазепин-1 -ил)фенил] -2-оксо-5-оксазолидинил] метил] тиоацетамид,
(c) (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-4-метил-5-оксо-1,4-диазепин-1-ил)фенил] -2-оксо-5-оксазолидинил] метил] ацетамид или
(d) (S)-N-[[3-[3-фтор-4-(1,2,3,4,6,7-гексагидро-4-метил-5-оксо-1,4-диазепин-1-ил)фенил] -2-оксо-5-оксазолидинил] метил] тиоацетамид.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| RU 94026079 A1, 27.05.1996. | |||

