В настоящем изобретении раскрыты новые и полезные соединения фенилоксазолидинона, замещенные азотсодержащим гетероароматическим кольцом, присоединенным по одному из атомов азота. Более конкретно, это 5-членное азотсодержащее ароматическое кольцо, содержащее от 1 до 4 атомов азота, один из которых связан с фенилоксазолидиноном. Эти соединения являются полезными антимикробными агентами, эффективными против ряда патогенов человека и животных, включая грамположительные аэробные бактерии, такие как множественно-устойчивые стафилококки, стрептококки и энтерококки, а также такие анаэробные организмы, как Bacteroides spp. и Clostridia spp. виды и acid-fast организмы, такие как Mycobacterium tuberculosis, Mycobacterium avium и Mycobacterium spp.
Предпосылки изобретения
Соединения настоящего изобретения сходны по структуре фенилоксазолидинонового кольца с соединениями, раскрытыми в приводимых далее публикациях за исключением того, что рассматриваемые соединения имеют замещение гетероароматическим кольцом, содержащим азот, которое присоединено к фенилоксазолидинону через один из атомов азота. Такая точка присоединения облегчает отличающийся, но более легкий синтез, нежели соответствующая углерод-углерод связанная структура, и поэтому предоставляет преимущество по сравнению с соединениями с углерод-углеродной связанной структурой. Соединения настоящего изобретения являются уникальными и обладают полезной антибактериальной активностью.
В заявке PCT/US93/03570 раскрыты оксазолидиноны, содержащие фрагмент замещенного диазина, и их применение в качестве антимикробных агентов.
В PCT/US92/08267 заявке раскрыты замещенные арил и гетероарилфенил-оксазолидиноны, пригодные в качестве антибактериальных агентов. В одном аспекте раскрыты 5-членные азотсодержащие гетероароматические кольца, присоединенные к фенилоксазолидинону, хотя там и не раскрыт способ их синтеза, который позволил бы осуществить присоединение за счет атома азота (См. схему C (V, W, ee и ff)).
В PCT/US89/03548 заявке раскрыты 5'-индолинил-5 β -амидометилоксазолидиноны, 3-(замещенный конденсированным кольцом)фенил-5 β -амидометилоксазолидиноны, и 3-(азотом замещенный)фенил-5 β -амидометилоксазолидиноны, которые можно использовать в качестве антибактериальных агентов.
Другие ссылки, в которых раскрыты различные оксазолидиноны, включают патент США 4801600, 4921869, Gregory W.A., et. al., J. Med. Chem., 32, 1673-81 (1989); Gregory W. A., et. al., J. Med. Chem., 33, 2569-78 (1990); Wang C. et. al., Tetrahedron, 45, 1323-26 (1989); и Brittelli et. al., J. Med. Chem. , 35, 1156 (1992).
В Европейской патентной публикации 352781 раскрыты фенил и пиридил замещенные фенилоксазолидиноны.
В Европейской патентной публикации 316594 раскрыты 3-замещенные стирилоксазолидиноны.
В Европейской патентной публикации 312000 раскрыты фенилметил и пиридинилметилзамещенные фенилоксазолидиноны.
В патенте США 5254577 раскрыты азотсодержащие гетероароматические кольца, присоединенные к фенилоксазолидинону, но не через гетероароматический азот (см. колонку 19 и 43).
Краткое содержание изобретения
В одном аспекте изобретения предложено соединение структурной формулы 1: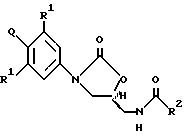
или его фармацевтически приемлемые соли, где Q представляет гетероароматическое 5-членное кольцо, связанное с ароматическим кольцом 1 через азот, следующей структуры: i, ii, iii, iv, v, vi, vii, viii или ix: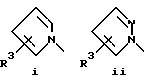
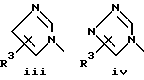
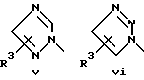
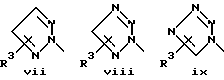
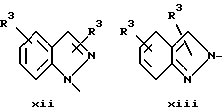
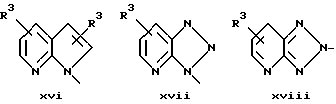
или в другом варианте Q может быть бензоконденсированным гетероароматическим 5 членным кольцом, связанным с ароматическим кольцом 1 через азот, следующей структуры: x, xi, xii, xiii, xiv, xv, xvi, xvii, или xviii: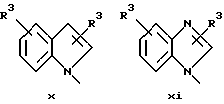

R1 независимо представляет O, OCH3, F или Cl;
R2 независимо представляет: (a) водород,
(b) C1-C8алкил, необязательно замещенный одним или более заместителем, выбранным из F, Cl, гидрокси, C1-C8алкокси, C1-C8ацилокси;
(c) C3-C6циклоалкил;
(d) амино;
(e) C1-C8алкиламино;
(f) C1-C8диалкиламино;
(g) C1-C8алкокси;
R3 каждый независимо выбирают из:
(a) H,
(b) F, Cl, Br,
(c) OR4,
(d) SR4,
(e) S(O)nR4 (n = 1 или 2),
(f) CN
(g) O2CR4,
(h) NHCOR4,
(i) NHCO2R4,
(j) NHSO2R4,
(k) CO2R4,
(l) CON(R4)2,
(m) COR4,
(n) C1-C8 разветвленный или неразветвленный алкил, или C3-C8циклоалкил, необязательно замещенный одним или более заместителем, выбранным из (a) - (m), или
(o) фенил, необязательно замещенный одним или более заместителем из предшествующих групп (a) - (n); и
R4 представляет:
(a) H,
(b) C1-C6 разветвленный или неразветвленный алкил, или C3-C8циклоалкил, необязательно замещенный одним или более заместителем, выбранным из фтора, хлора, гидрокси, C1-C4алкокси, C1-C4ацила, C1-C4ацилокси или O2CCH2N(CH3)2 или
(c) фенил, необязательно замещенный одним или более заместителем, выбранным из фтора, хлора, C1-C4 разветвленного или неразветвленного алкила, гидрокси, C1-C4алкокси, C1-C4ацила, C1-C4ацилокси или O2CCH2N(CH3)2. Подробное описание изобретения
В настоящем изобретении раскрыты новые замещенные 5-членным азотсодержащим гетероциклом фенилоксазолидиноны в соответствии со структурной формулой 1, приведенной выше.
"Алкил" означает цепочки атомов углерода, содержащие указанное число атомов углерода, которые могут быть разветвленными или неразветвленными.
"Алкокси" означает указанное число атомов углерода, присоединенное к кислороду с образованием таких групп, как метокси (-OCH3), этокси, бутокси и т.д., и их изомерные формы.
"Ацилокси" означает указанное число атомов углерода, образующих органическую кислоту, где была исключена группа OH, например ацетил: CH3CO-; бензоил: C6H5CO-.
"Циклоалкил" означает указанное число атомов углерода, образующих циклопропил, циклобутил, циклопентил, циклогексил, и т.д., и их изомерные формы.
"Амино" означает NH2, "алкиламино" соответствует структуре, в которой один атом водорода замещен алкилом, а "диалкиламино" соответствует случаю, когда оба водорода замещены алкильными группами.
"Фармацевтически приемлемые соли" представляют собой соли присоединения кислот, которые можно получить известными специалистам способами. Обычно соли присоединения кислот включают гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат ацетат, пропионат, лактат, малеат, сукцинат, тартрат, циклогексансульфаматы, метансульфонаты, этансульфонаты, бензолсульфонаты, толуолсульфонаты, фумараты, и другие фармацевтически приемлемые противоионы аминов.
Эти соединения пригодны в качестве антимикробных агентов, эффективных против ряда патогенов человека и животных, особенно против аэробных грамположительных бактерий, включая устойчивые ко многим антибиотикам стафилококки и стрептококки, а также таких анаэробных организмов, как бактероиды и виды клостридий, а также таких acid-fast организмов, как Mycobacterium tuberculosis и других видов Mycobacterium.
Фармацевтические композиции настоящего изобретения можно получить, объединяя соединения формулы 1 настоящего изобретения с твердым или жидким фармацевтически приемлемым носителем, и, необязательно, с фармацевтически приемлемыми адьювантами и эксципиентами, используя стандартные и удобные способы. Твердые формы композиций включают порошки, таблетки, диспергируемые гранулы, капсулы, облатки и суппозитории. Твердым носителем может быть, по крайней мере, одно вещество, которое может также действовать как разбавитель, вкусовой агент, солюбилизатор, скользящее, суспендирующий агент, связующее, агент, придающий таблеткам рассыпчатость, и инкапсулирующий агент. Инертные твердые носители включают карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, целлюлозные материалы, низкоплавкие воски, масло какао и т.п. Жидкие формы композиций включают растворы, суспензии и эмульсии. Так, например, можно приготовить растворы соединений настоящего изобретения в воде и смеси воды и пропиленгликоля, воды и полиэтиленгликоля, необязательно содержащие подходящие обычные подкрашивающие агенты, вкусовые агенты, стабилизаторы и загустители.
Предпочтительно приготовлять фармацевтические композиции, используя обычные способы, в единичных дозовых формах, содержащих эффективное или соответствующее количество активного компонента, т.е. соединения формулы 1 настоящего изобретения.
Количество активного компонента, т.е. соединения формулы 1 настоящего изобретения, в фармацевтической композиции и единичной дозовой форме ее может варьироваться или подбираться в широком интервале в зависимости от конкретного применения, эффективности конкретного соединения, желательной концентрации. Обычно количество активного компонента меняется в интервале от 0,5 до 90% от веса композиции.
При терапевтическом использовании для лечения или для борьбы с бактериальными инфекциями теплокровных животных пациенту, страдающему такой антимикробной инфекцией, соединения или фармацевтические композиции настоящего изобретения вводят перорально и/или парэнтерально в дозах для достижения и поддержания концентрации, то есть количества или уровня в крови активного соединения у подлежащего лечению животного, в количестве, которое будет антибактериально эффективным. Обычно такое антибактериально эффективное количество или доза активного компонента находится в интервале от около 0,1 до 100, более предпочтительно от около 3,0 до около 50 мг/кг веса/день. Следует учитывать, что дозировка может меняться в зависимости от потребности пациента, тяжести бактериальной инфекции, подлежащей лечению, и конкретного предполагаемого для использования соединения. Кроме того, следует учитывать, что начально вводимую дозу можно повысить выше верхнего предела, чтобы быстро достичь нужного уровня в крови, или начальная доза может быть меньше оптимальной, и дневную дозу можно прогрессивно увеличивать в ходе лечения в зависимости от конкретной ситуации. При желании дневную дозу можно также разделить на множество доз для приема, например, 2-4 раза в день.
Соединения формулы 1 настоящего изобретения вводят парэнтерально, то есть за счет инъекций, например внутривенных инъекций, или за счет других парэнтеральных способов введения. Фармацевтические композиции для парэнтерального введения обычно содержат фармацевтически приемлемое количество соединения формулы 1 в виде растворимой соли (соли присоединения кислоты или основания), в фармацевтически приемлемом жидком носителе, таком как, например, вода для инъекций, или буфер для получения соответствующим образом буферированного изотонического раствора, например, с pH около 3,5-6. Подходящие буферирующие растворы включают, например, тринатрийортофосфат, бикарбонат натрия, цитрат натрия, N-метилглюкамин, L(+)-лизин и L(+)-аргинин, в качестве нескольких представителей буферирующих агентов. Соединения формулы 1 обычно растворяют в носителе в количестве достаточном для обеспечения фармацевтически приемлемой инъектируемой концентрации в интервале от около 1 до около 400 мг/мл раствора. Полученные жидкие фармацевтические композиции следует вводить таким образом, чтобы обеспечить вышеуказанные антибактериально эффективные дозы. Соединения формулы 1 настоящего изобретения выгодно вводить перорально в твердых или жидких дозовых формах.
Наиболее предпочтительные в этой серии соединения желательно получать в виде оптически чистых энантиомеров с (S)-конфигурацией в соответствии с системой обозначений Cahn-Ingold-Prelog C5 оксазолидинонового кольца. Оптически чистый материал можно получить за счет одного или множества способов асимметрического синтеза или за счет выделения из рацемической смеси путем селективной кристаллизации соли из, например, рацемической модификации промежуточного амина 10 (схема II) соответствующей оптически активной кислотой, такой как дибензоилтартрат или 10-камфорсульфоновая кислота, с последующей обработкой основанием до получения оптически активного амина. Хотя (S)-энантиомер этой серии соединений предпочтителен, так как он фармакологически активен в качестве антибактериального агента, рацемическую модификацию также можно использовать аналогичным образом как и чистый (S)-энантиомер: разница будет в том, что для достижения такого же антибактериального эффекта понадобится вдвое больше рацемического материала. Кроме того, специалистам должно быть ясно, что если хиральный центр существует в любом из заместителей, связанных с Q, тогда возможны диастереоизомеры. Такие диастереоизомеры, как в рацемической, так и в конфигурационно обогащенных формах, входят в объем соединений структурной формулы 1 настоящего изобретения.
Предпочтительный способ получения соединений структурной формулы 1 в виде существенно энантиомерно обогащенной формы представлен на схемах I-VIII.
Как видно на схеме I, 5-членные гетероциклы общей структуры 1 (где n = числу атомов в кольце), которые либо доступны коммерчески, либо их получение описано в литературе (см., например, Katritzky et. al., в "Comprehensive Heterocyclic Chemistry"), обрабатывают замещенным производным нитробензола 2 (Y представляет галоид или трифторметансульфонилокси) в подходящем основании в сочетании растворителей, например карбонат калия в ДМСО, при подходящей температуре, обычно от комнатной до 90oC, до получения аддукта 3. Этот способ хорошо работает для таких 5-членных гетероциклов, как имидазол, пиразол и т.п. Для 5-членных гетероциклов 1, таких как пиррол или индол и т.п., гитероцикл добавляют к суспензии такого сильного основания, как гидрид натрия, трет.-бутоксид калия или т.п. в ТГФ или ДМФ, или аналогичном растворителе до получения соли щелочного металла 4, которую затем обрабатывают нитроароматическим соединением 2 при температуре в интервале от 0oC до температуры кипения с обратным холодильником растворителя до получения аддукта 3. Затем нитропроизводное 3 восстанавливают за счет каталитического гидрирования в присутствии такого подходящего катализатора, как палладий на угле, W-2-никель Рэнея или платина на сероуглероде, в подходящем растворителе, таком как этилацетат, ТГФ, метанол, или их комбинация, до получения производного анилина 5. Если в качестве растворителя для этого восстановления используют ТГФ, нет необходимости отфильтровывать растворитель или выделять производное анилина 5, но можно просто продуть реактор таким инертным газом, как азот, и добавить насыщенный раствор бикарбоната натрия, и обработать полученную реакционную смесь, либо бензилом, либо метилхлорформатом до получения соответствующего бензил (R представляет CH2Ph), или метилкарбамата (R представляет CH3) производных 6.
Любое из производных карбамата 6 можно депротонировать за счет такого литиевого основания, как н-бутиллитий, литийдиизопропиламид /LDA/ или литийбис/триметилсилил/амид/LHMDS/ в подходящем растворителе, таком как ТГФ, N, N-диметилформамид (ДМФ), или их смеси, при соответствующей температуре, обычно от -78oC до -40oC, до получения литированного промежуточного соединения, которое непосредственно обрабатывают (R)-(-)-глицидилбутиратом. Нагревание этой реакционной смеси до комнатной температуры или выше приводит к получению /гидроксиметил/оксазолидинона 7 в существенно энантиомерно обогащенной форме.
На схеме II представлена конверсия /гидроксиметил/оксазолидинона 7 в оксазолидиноновые антибактериальные агенты структурной формулы 1. Как видно, соединение 7 можно превратить в соответствующий мезилат (R = CH3) или тозилат (R = n-CH3C6H4) за счет обработки метансульфонилхлоридом в присутствии триэтиламина или пиридина, или паратолуолсульфонилхлоридом в присутствии пиридина соответственно. Полученный сульфонат 8 можно обработать азидом щелочного металла, например азидом натрия или калия в соответствующем апротонном диполярном растворителе, таком как ДМФ или N,N-метилпирролидон /NMP/ с необязательным катализатором, таким как 18-краун-6 при температуре в интервале 50-90oC до получения азида 9. Этот азид 9 можно восстановить до соответствующего амина 10 за счет гидрирования в присутствии палладиевого, платинового или никелевого катализатора, в таком соответствующем растворителе как этилацетат, ТГФ или метанол. В другом варианте азид 9 можно восстановить до амина 10 за счет обработки трифенилфосфином или другими соединениями трехвалентного фосфора в таком растворителе, как ТГФ, с последующим добавлением воды. Амин 10 можно также получить за счет обработки сульфоната 8 фталимидатом калия в ДМФ при 40-60oC или в кипящем с обратным холодильником ацетонитриле до получения фталимида II, у которого удаляют защиту, обрабатывая, например, водным метиламином в кипящем с обратным холодильником этаноле. Более прямой путь до амина 10 состоит в обработке сульфоната 8 водным раствором аммиака в смеси изопропиловый спирт-ТГФ в запаянной ампуле, нагретой до 75-105oC в масляной бане. Затем амин 10 ацилируют в реакциях хорошо известных специалистам до получения (ациламинометил) оксазолидинонов структурной формулы 12. Так, например, амин 10 можно обработать хлорангидридом или ангидридом кислоты в присутствии такого основания, как пиридин или триэтиламин при температуре в интервале -40 - 40oC до получения ацильного производного 12. Очевидно, что такие другие ацильные производные, как карбаматы, можно получить в аналогичных условиях реакций. Легко видеть, что препарат 12 представляет пример соединения структурной формулы 1. Видно также, что в некоторых случаях 12 можно превратить в другие соединения структурной формулы 1 за счет обработки 40% водным формальдегидом в диметилсульфоксиде при температурах в интервале 90-150oC, необязательно в присутствии кислотного катализатора, до получения соответствующего гидроксиметильного производного 13.
В другом варианте соединение 13 можно получить более сложным способом, при котором возможно получение других региоизомеров, которые нелегко получить за счет гидроксиметилирования. Как видно на схеме III, алкоксикарбонильные производные 5-членных кольцевых гетероциклов 14 (где m = числу метиленов) либо доступны коммерчески, либо их можно получить в соответствии с литературными данными, можно обработать производным нитробензола 2 в таком подходящем растворителе, как ДМСО, ДМФ или тому подобных, в присутствии такого подходящего основания, как карбонат калия, при соответствующей температуре в интервале от комнатной до 90oC до получения биарильного производного 15. Затем полученное нитропроизводное восстанавливают за счет каталитического гидрирования в присутствии такого подходящего катализатора, как палладий на угле, W-2 никель Рэнея, или платина на сероуглероде, в таком подходящем растворителе, как этилацетат, ТГФ, метанол или в их сочетании, до получения производного анилина 16. Если для такого восстановления используют ТГФ в качестве растворителя, нет необходимости отфильтровывать катализатор или выделять производное анилина 16, но можно просто продуть реактор таким инертным газом, как азот, и добавить насыщенный раствор бикарбоната натрия и обработать соответствующую смесь либо бензил, либо метилхлорформатом до получения соответствующего бензил (R = CH2Ph), либо метил (R = CH3) карбаматных производных 17. Сложноэфирную группу любого из карбаматных производных 17 можно восстановить либо литийборгидридом, либо возможно комплексом боран-ТГФ до получения соответствующего спирта 18. Специалисту должно быть ясно, что последующая трансформация 18 потребует защиты гидроксильной группы. Это можно осуществить, например, получая трет.-бутилдиметилсилильный (TBS) эфир 19 (R = Si(CH3)2т. -Bu) за счет обработки 18 трет.-бутилдиметилхлорсиланом в присутствии такого основания, как имидазол или диизопропиламин, необязательно в присутствии 4-диметиламинопиридина в качестве катализатора, в таком подходящем растворителе, как ДМФ, ТГФ или дихлорметан. Любое из карбаматных производных 19 можно подвергнуть депротонированию за счет такого литиевого основания, как литийдиизопропиламид (LDA) или литийбис(триметилсилил)амид (LHMDS) в таком подходящем растворителе, как ТГФ, ДМФ или их смеси, при подходящей температуре, обычно от -78oC до -40oC, до получения литированного производного, которое непосредственно обрабатывают (R)-(-)-глицидилбутиратом. Затем нагревание этой реакционной смеси до комнатной температуры или более высоких температур, приводит к получению (гидроксиметил)оксазолидинона 20 в высоко энантиомерно обогащенной форме.
На схеме IV представлено превращение (гидроксиметил)оксазолидинона 20 в оксазолидиноновые антибактериальные агенты структурной формулы 1. Как видно, соединение 20 можно превратить в соответствующий (ациламинометил)оксазолидинон 21 (R = разветвленной или неразветвленной алкильной цепочкой) в результате последовательности реакций, идентичных той, которую используют для превращения 7 в 12 на схеме II. Затем одним из многочисленных способов можно удалить защиту у (ациламинометил)оксазолидинона 21, например, за счет обработки метанольным раствором HCl при кипении с обратным холодильником, или за счет обработки смесями уксусной кислоты в ТГФ-воде при температуре 40-90oC до получения спирта 22. Спирт 22 представляет пример оксазолидинонового антибактериального агента структурной формулы 1.
Дальнейшую трансформацию, представленную на схеме IV, можно использовать для получения других соединений структурной формулы 1 из соединения 22. Как видно, спирт 22 можно окислить за счет обработки такими реагентами, как пиридинийдихромат в ДМФ, или за счет обработки газообразным кислородом в присутствии катализатора - благородного металла в водном растворе, или другими известными специалистам способами, до получения карбоновой кислоты 23. Карбоновую кислоту 23 можно превратить в сложный эфир за счет обработки соответствующим алкилгалидом в присутствии такого основания, как карбонат калия, в соответствующем растворителе, таком как ацетон, до получения сложного эфира 24 (R = разветвленная или неразветвленная алкильная цепочка). В другом варианте сложный эфир 24 можно получить, добавляя карбоновую кислоту 23 к раствору газообразной HCl, растворенной в соответствующем спирте, или другими известными специалистам способами.
На схеме V представлены способы превращения спирта 22 в другие соединения структурной формулы 1. Как видно, 22 можно превратить в соответствующий мезилат (R = CH3) или тозилат (R = p-CH3C6H4) за счет обработки метансульфонилхлоридом в присутствии триэтиламина или пиридина, или р-толуолсульфонилхлоридом в присутствии пиридина. Полученное производное сульфоната 25 можно обработать солью щелочного металла или меркаптаном (R = разветвленная или неразветвленная алкильная цепочка или арил) в апротонном диполярном растворителе, таком как ДМФ, ДМСО или ацетонитрил в присутствии такого необязательного катализатора, как 18-краун-6, при температуре в интервале от комнатной до 90oC до получения сульфида 26. Затем сульфид 26 можно превратить либо в сульфоксид 27, либо сульфон 28, за счет окисления соответствующими стехиометрическими количествами подходящего окисляющего агента, такого как м-хлорпероксибензойная кислота (MCPBA), в таком подходящем растворителе, как дихлорметан, диэтиловый спирт или т.п. В другом варианте сульфон 28 можно получить непосредственно за счет обработки тетроксидом осмия или тетроксидом рутения в присутствии периодата натрия, или за счет обработки кислым персульфатом калия. Соединения 26, 27 и 28 составляют примеры соединений структурной формулы 1.
Другие возможные превращения спирта 22 представлены на схеме V. Как видно, производное сульфоната 25 можно обработать азидом щелочного металла в таком диполярном апротонном растворителе, как ДМФ, ДМСО, ацетонитрил или т. п., необязательно в присутствии такого катализатора, как 18-краун-6, при температуре в интервале от 50-90oC до получения азида 29. Затем азид 29 можно восстановить до соответствующего амина 30 за счет гидрирования в присутствии платинового, палладиевого или никелевого катализатора, в таком подходящем растворителе, как этилацетат, ТГФ или метанол. В другом варианте азид 29 можно восстановить за счет обработки трифенилфосфином или другими соединениями трехвалентного фосфора, в таком растворителе, как ТГФ, с последующим добавлением воды. Затем амин 30 можно использовать для получения соответствующего производного сульфонамида 31 за счет обработки соответствующим сульфонилхлоридом (R = разветвленная или неразветвленная алкильная цепочка или арил) в таком подходящем растворителе, как пиридин. Легко видеть, что производное сульфонамида 31 представляет пример соединений структурной формулы 1.
Из-за недостатка коммерчески доступных 3-замещенных гетероциклов ряда пиррола, пиразола или триазола, некоторые из возможных соединений структурной формулы 1 невозможно легко получить способами, описанными ранее на схемах. В результате, способы получения других гетероциклических производных оксазолидинона из производного анилина 37 представлены на схемах VI, VII, VIII, IX.
Получение производного анилина 37 представлено на схеме VI. Как видно, нитроароматическое соединение 2 обрабатывают бензиламином в присутствии такого подходящего основания, как триэтиламин или N,N-диизопропилэтиламин, в таком растворителе, как ацетонитрил, до получения продукта замещения 32. Затем нитросоединение 32 можно превратить в оптически активное производное оксазолидинона 36 способами, представленными на схемах I и II для превращения нитропроизводного 3 в оксазолидинон 12. На этой стадии синтеза гидрогенолиз 36 с использованием в качестве катализатора палладия на угле и метанола в качестве растворителя служит как для удаления CbZ защитной группы, так и бензил защищающей группы, с получением анилина 37.
Получение 3-замещенных производных пиррола можно осуществить как представлено на схеме VI. Анилин 37 обрабатывают коммерчески доступным 3,5-диметокси-3-тетрагидрофуранкарбоксальдегидом в условиях кислотного катализа, например, при кипячении с обратным холодильником в уксусной кислоте, что приводит к получению 3-пирролкарбоксальдегида 38. Затем альдегид 38 можно превратить в другие аналоги пиррола, такие как гидроксиметилпиррол 39, сложный эфир 40 или производное оксима 41 известными специалистам способами. Видно, что пирролы 30, 40 и 41 составляют примеры соединений структурной формулы 1.
Превращение анилина 37 в производные замещенного триазола представлено на схеме VII. Как видно, диазотизация анилина 37 за счет обработки азотистой кислотой с последующим добавлением азида натрия приводит к образованию азида 42, который является полезным предшественником для получения триазолов за счет 1,3-диполярного циклоприсоединения замещенного ацетилена 43. Как видно, обработка азида 42 замещенным производным ацетилена 43 в таком растворителе, как бензол, толуол и т. п. , при температуре в интервале от комнатной до 120oC, необязательно в присутствии кислоты Льюиса в качестве катализатора, такой как алюминийхлорид, эфират трехфтористого бора, тетрахлорид титана или т. п. , приводит к получению замещенного производного триазола 44. В другом варианте в том случае, если одна из R3 групп в 43 представляет ацильную группу, например R4CO в ацетилене 45, циклоприсоединение в указанных выше условиях приводит к получению преимущественно 4-ацилтриазольного производного 46 при меньшем количестве соответствующего 5-ацилтриазольного производного 47. В другом варианте допустимые функциональности в R3 44 можно использовать для превращения 44 в ацильное производное 46 и/или 47. 4-ацилтриазолоксазолидинон 46 можно превратить в производное оксима 48 или гидроксисоединение 49 известными специалистам способами. Можно видеть, что 46, 48 и 49 составляют примеры соединений структурной формулы 1.
Превращение анилина 37 в оба возможные региоизомера замещенных производных пиразола представлено на схеме VIII. Как видно, диазотизация анилина 37 с последующим восстановлением диазониевой соли либо хлоридом олова [II], либо ин ситу получаемым сульфитом натрия, дает производное гидразина 50. Видно, что гидрозин 50 можно подвергнуть взаимодействию с триформилметаном 51 (Synthesis, 1989, 858) до получения пиразол-4-карбоксальдегида 52. В другом варианте обработка гидразина 50 4-метокси-2-оксо-бут-3-еновой кислоты этиловым сложным эфиром (Helv. Chim. Acta, 1967, 50, 128) приводит к образованию региоизомерного 3-карбоэтоксипиразола 54. Видно, что альдегид 52 и сложный эфир 54 можно превратить в другую серию аналогов в результате превращений, представленных на схемах IV, V и VI. Видно, что 52 и 54 составляют примеры соединений структурной формулы 1.
Примеры гетероарил-фенилоксазолидинонов, которые можно получить как часть настоящего изобретения, следующие:
(S)-N-[[3-[3-фтор-4-(1H-пиррол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-карбометокси-1H-пиррол-1-ил)фенил]-2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-пиррол-1-ил-3-карбоксальдегид)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[[3-фтор-4-(3-(гидроксииминометил)-1H-пиррол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-(метоксииминометил)-1H-пиррол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-(гидроксиметил)-1H-пиррол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-(2-карбоэтоксивинил)-1H-пиррол-1-ил)фенил]-2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-(2-карбоэтоксиэтил)-1H-пиррол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-(3-гидроксипропил)-1H-пиррол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(3-(3-метансульфониламинопропил)-1H-пиррол- 1-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-имидазол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-метил-1H-имидазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-этил-1H-имидазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[4-(карбометокси)-1H-имидазол-1-ил] фенил]-2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[4-(гидроксиметил)-1H-имидазол-1-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[4-(2-гидроксиэтил)-1H-имидазол-1-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[1H-имидазол-1-ил-4-карбоксальдегид] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-(гидроксииминометил)-1H-имидазол-1- ил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-(метоксииминометил)-1H-имидазол-1- ил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-пиразол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-метил-1H-пиразол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-(гидроксиметил)-1H-пиразол-1-ил] фенил]-2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-(карбометокси)-1H-пиразол-1-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-пиразол-1-ил-3-карбоксальдегид)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-(гидроксииминометил)-1H-пиразол-1- ил]фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-(метоксииминометил)-1H-пиразол-1- ил] фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[4-(гидроксиметил)-1H-пиразол-1-ил] фенил]-2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[4-(карбометокси)-1H-пиразол-1-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[1H-пиразол-1-ил-4-карбоксальдегид] фенил]-2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-гидроксииминометил)-1H-пиразол-1-ил)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-метоксииминометил)-1H-пиразол-1-ил)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-1,2,4-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-меркапто-4H-1,2,4-триазол-4-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[3-метилтио-4H-1,2,4-триазол-4-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-[4H-1,2,4-триазол-1-ил] фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-1,2,3-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-карбометокси-1H-1,2,3-триазол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-(гидроксиметил)-1H-1,2,3-триазол-1-ил)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-(1-гидроксиэтил)-1H-1,2,3-триазол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-1,2,4-триазол-1-ил-4-карбоксальдегид] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-(гидроксииминометил)-1H-1,2,3-триазол-1- ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида оксим;
(S)-N-[[3-[3-фтор-4-(4-метоксииминометил)-1H-1,2,3-триазол-1- ил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-ацетил-1H-1,2,3-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-(1-гидроксииминоэтил)-1H-1,2,3-триазол-1- ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(4-(1-метоксииминоэтил)-1H-1,2,3-триазол-1- ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(2H-1,2,3-триазол-2-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-1,2,3,4-тетразол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(2H-1,2,3,4-тетразол-2-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-индол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил] ацетамид;
(S)-N-[[3-[3-фтор-4-(7-аза-1H-индол-1-ил)фенил]-2-оксо-5- оксазолидинил] метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-бензимидазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-индазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид;
(S)-N-[[3-[3-фтор-4-(1H-бензотриазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид; и
(S)-N-[[3-[3-фтор-4-(1H-1,2,3-триазоло[4,5b] -пиридин-1-ил)фенил]-2- оксо-5-оксазолидинил]метил]ацетамид.
Антибактериальная активность
Оксазолидиноновые антибактериальные агенты настоящего изобретения обладают полезной активностью против различных организмов. Ин витро активность соединения настоящего изобретения можно оценить с помощью стандартных тестовых процедур, таких как определение минимальной ингибирующей концентрации (MIC = МИК) за счет агарного разбавления по способу "Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria^ that Grow Aerobically" (MFT), опубликованному в январе 1983 г National Committee for Chimical Laboratory Standarts 771 East Lancaster Avenue, Villanov Pensilvania 19084, USA. Активность выбранных соединений настоящего изобретения против Staphylococcus aureus и Streptococcus preumoniae представлена в таблице 1.
Антимикробную активность тестируют ин виво, используя процедуру анализа на мышах. Группе из мышей самок (шесть мышей по 18-20 г каждая) вводят внутрибрюшинно бактерии, которые оттаивают непосредственно перед использованием, и суспендируют в мозговой жидкости с 4% пивными дрожжами UC9213 (Staphylococcus aureus) или в мозговой жидкости (Staphylococcus species). Обработку антибиотиком при 6 уровнях доз лекарства вводят через час и через пять часов после инфицирования либо за счет перорального интубирования, либо подкожно ("Subcut. "). Выживание оценивают ежедневно в течение 6 дней. Рассматриваемые соединения сравнивают с хорошо известным антимикробным агентом (Vancomycin) в качестве контроля. Значения ЭД50 рассчитывают на основании данных о смертности, используя probit анализ. Полученные данные представлены в таблице 2.
Пример 1
(S)-N-[[3-[3-фтор-4-(1H-пиррол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
3-фтор-1-нитро-(1H-пиррол-1-ил)бензол:
Суспензию 403 мг (630 мг, 60% в масле, 15,75 ммолей) гидрида натрия в 85 мл ТГФ обрабатывают 1,01 г (1,04 мл, 15 ммолей) пиррола с последующим нагреванием при 50oC в течение 15 минут. Затем этот раствор обрабатывают 2,51 г (1,74 мл, 15,75 ммоля) 3,4-дифторнитробензола с последующим нагреванием при кипении с обратным холодильником в течение 18 часов. Полученную смесь охлаждают и обрабатывают 10 мл насыщенного раствора аммонийхлорида. Полученную смесь разбавляют этилацетатом и экстрагируют водой (2 х). После сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество темно-коричневого цвета, которое обрабатывают хроматографически на 75 г силикагеля (230-400 меш), элюируя 30% /объем/объем/ смесью дихлорметана в гексане. В результате получают 2,05 г (66%) указанного в заглавии производного пиррола в виде твердого вещества светло-желтого цвета.
1H ЯМР /CDCl3/ δ : 14, 7,57, 7,16 и 6,44
3-фтор-1-(фенилметоксикарбониламино)-4-(1H-пиррол-1-ил)бензол:
Раствор 500 мг (2,43 ммоля) полученного ранее производного пиррола в 50 мл смеси 4: 1 ТГФ:вода обрабатывают 75 г 5% платины на сероуглероде с последующим гидрированием при давлении одна атмосфера в течение 18 часов. Затем полученную смесь обрабатывают 10 мл насыщенного раствора бикарбоната натрия и охлаждают до -20oC. Полученную смесь обрабатывают 477 мг (0,42 мл, 2,79 ммоля) бензилхлорформата с последующим нагреванием до комнатной температуры в течение 48 часов. Полученную смесь фильтруют через целит, промывают фильтровальную лепешку метанолом. Полученную смесь концентрируют в вакууме до примерно половины исходного объема, смесь разбавляют этилацетатом и экстрагируют водой (4 х) и насыщенным раствором NaCl. После сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество коричневого цвета, которое обрабатывают хроматографически на 35 г силикагеля (230-400 меш), элюируя смесью 1:3:32 метанол:дихлорметан:гексан, а затем смесью 1: 3: 16 метанол: дихлорметан: гексан. В результате получают 581 мг указанного в заглавии CBZ производного в виде коричневого твердого вещества, пригодного для использования на следующей стадии
1H ЯМР /CDCl3/ δ : 7,50, 7,40, 7,30, 7,09, 6,98, 6,35, 5,23.
(S)-N-[3-[3-фтор-4-(1H-пиррол-1-ил)фенил] -2-оксо-5- оксазолидинил] метанол:
Раствор 300 мг (0,97 ммоля) CBZ производного 2 в 5 мл ТГФ при -78oC обрабатывают 1,06 мл (1,0 М, 1,06 ммоля) литийбис(триметилсилил)амида, а затем перемешивают при -78oC в течение 30 минут. Полученный раствор обрабатывают 153 мг (0,15 ммолей) (R)-(-)глицидилбутирата с последующим нагреванием до 0oC, а затем постепенным нагреванием при комнатной температуре в течение 48 часов. Полученную смесь разбавляют этилацетатом и экстрагируют водой и насыщенным солевым раствором. В результате сушки над сульфатом натрия и концентрирования в вакууме получают красно-коричневое масло, которое обрабатывают с помощью радиальной хроматографии на 2 мм хроматотроновой пластинке, элюируя 2% /объем/объем/ метанолом в дихлорметане, а затем 5% /объем/объем/ метанолом в дихлорметане. В результате этих процедур получают 157 мг (59%) указанного в заглавии оксазалидинона в виде светло-золотисто-коричневого твердого вещества. Масс-спектр высокого разрешения: Рассчитано для C14H13FN2O3: 276,0910. Найдено: 276,0918.
(S)-[3-[3-фтор-4-(1H-пиррол-1-ил)фенил] -2-оксо-5- оксазолидинил] метилазид:
Раствор 1,94 г (7,0 ммоля) полученного ранее спирта и 1,24 г (1,71 мл, 12,25 ммоля) триэтиламина в 60 мл дихлорметана при 0oC обрабатывают 1,0 г (0,68 мл, 8,78 ммоля) метансульфонилхлорида с последующим перемешиванием при 0oC в течение 30 минут. Полученный раствор нагревают до комнатной температуры и разбавляют дихлорметаном с последующей экстракцией водой (3 х) и насыщенным солевым раствором. В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло янтарного цвета, которое растворяют в 120 мл ДМФ, обрабатывают 4,5 г (70 ммолей) азида натрия и нагревают при 60oC в течение 18 часов. Полученную смесь охлаждают и разбавляют 75 мл смеси 1:1 эфир: этилацетат, и экстрагируют водой (6 х 40 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают 2,13 г (100%) указанного в заглавии азида в виде желто-коричневого твердого вещества, достаточно чистого для использования на следующей стадии. Масс-спектр высокого разрешения: Рассчитано для C14H12FN5O2: 302,1053. Найдено: 302,1056.
(S)-N-[3-[3-фтор-4-(1H-пиррол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил] ацетамид:
Раствор 1,0 г (3,32 ммоля) полученного ранее азида в 30 мл метанола и 15 мл ТГФ обрабатывают 200 мг платины на сероуглероде с последующим гидрированием при давлении одна атм. в течение 24 часов. Полученный раствор фильтруют через целит, промывают фильтровальную лепешку ТГФ. Полученный фильтрат концентрируют в вакууме, а остаток растворяют в пиридине и обрабатывают уксусным ангидридом с последующим перемешиванием в течение 48 часов при комнатной температуре. Полученный раствор концентрируют в вакууме, а полученное коричневое твердое вещество растворяют в этилацетате и дважды экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают коричневое твердое вещество, которое обрабатывают хроматографически на 50 г силикагеля (230-400 меш), элюируя 1% /объем/объем/ метанолом в дихлорметане, а затем 2% /объем/объем/ метанолом в дихлорметане. В результате этих процедур получают 582 мг (56%) указанного в заглавии в виде грязно-белого твердого вещества с т. пл. 198-199,5oC. Масс-спектр высокого разрешения: Рассчитано для CHFNO: 317,1176. Найдено: 317,1182.
Пример 2
(S)-N-[3-[3-фтор-4-(1H-пиразол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
3-фтор-1-нитро-4-(1H-пиразол-1-ил)бензол:
Раствор 2,0 г (29,37 ммоля) пиразола и 8,12 г (58,75 ммоля) карбоната калия в 70 мл ДМСО обрабатывают 4,67 г (3,25 мл, 29,37 ммоля) 3,4-дифторнитробензола с последующим нагреванием при 90oC в течение 18 часов. Полученный раствор охлаждают и разбавляют 100 мл воды и экстрагируют этилацетатом (2 х 100 мл). Органические слои экстрагируют водой (5 х 100 мл) и насыщенным солевым раствором (75 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают грязно-белое твердое вещество, которое перекристаллизовывают из горячих гексанов до получения 5,3 г (87%) указанного в заглавии производного пиразола в виде грязно-белого твердого вещества. Т. пл. 128-129oC.
3-фтор-1-(фенилметоксикарбониламино)-4-(1H-пиразол-1-ил)бензол:
Раствор полученного ранее пиразола в 120 мл ТГФ обрабатывают 1,5 г W-2 никеле Рэнея с последующим гидрированием при 40 пси (2,812 кг/см2) водорода в течение 16 часов. Полученный раствор фильтруют через целит, промывают фильтровальную лепешку ацетоном и концентрируют фильтрат в вакууме. Полученный остаток желтого цвета растворяют в 100 мл ТГФ и обрабатывают 75 мл насыщенного раствора бикарбоната натрия с последующим охлаждением до 0oC. Затем полученный раствор обрабатывают 7,30 г (42,72 ммоля) бензилхлорформата с последующим перемешиванием при 0oC в течение 30 минут, а затем нагревая до комнатной температуры в течение 1 часа. Полученную смесь разбавляют 125 мл этилацетата и 100 мл воды. Слои разделяют и водный слой экстрагируют 100 мл этилацетата. Объединенные органические слои экстрагируют насыщенным раствором бикарбоната натрия (2 х 100 мл) и насыщенным раствором NaCl. В результате сушки над сульфатом натрия и концентрирования в вакууме получают светло-бежевый твердый продукт, который перекристаллизовывают из смеси хлороформ-гексан до получения 5,92 г (79%) указанного в заглавии соединения в виде блестящих белых пластинок, т. пл. 82-83oC.
(S)-[3-[3-фтор-4-(1H-пиразол-1-ил)фенил] -2-оксо-5- оксазолидинил]метанол:
Раствор 3,0 г (9,63 ммоля) CBZ производного 2 в 130 мл ТГФ при -78oC обрабатывают 10,6 мл (1,0 М, 10,60 ммоля) литийбис(триметилсилил)амида в ТГФ с последующим перемешиванием при -78oC в течение 30 минут. Затем полученный раствор обрабатывают 1,53 г (1,50 мл, 10,60 ммоля) чистого (R)-(-)глицидилбутирата с последующим перемешиванием при -78oC в течение 30 минут, а затем нагревая до комнатной температуры в течение 18 часов. Полученную смесь обрабатывают 2 мл насыщенного раствора аммонийхлорида с последующим разбавлением 400 мл этилацетата. Полученную смесь экстрагируют водой (2 х 100 мл), насыщенным солевым раствором (200 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество желтого цвета, которое перекристаллизовывают из горячего метанола до получения 2,28 г (85%) соединения 3 в виде белых иголок. Т. пл. 180-181oC.
(S)-[3-[3-фтор-4-(1H-пиразол-1-ил)фенил] -2-оксо-5- оксазолидинил] метилметансульфонат:
Суспензию 403 мг (1,45 ммолей) полученного ранее соединения и 882 мг (1,22 мл, 8,71 ммоля) триэтиламина в 15 мл дихлорметана при 0oC обрабатывают 500 мг (0,34 мл, 4,36 ммоля) метансульфонилхлорида по каплям. Полученный раствор перемешивают при 0oC в течение 30 минут с последующим нагреванием до комнатной температуры. Полученную смесь разбавляют 20 мл дихлорметана и экстрагируют дважды водой (по 25 мл) и насыщенным раствором бикарбоната натрия. В результате сушки над сульфатом натрия и концентрирования в вакууме получают 520 мг (примерно 100%) указанного в заглавии мезилата, достаточно чистого для дальнейшего использования.
1H ЯМР /ДМСО-d6/ δ : 8,16, 7,80, 7,48, 6,55, 5,05, 4,53, 4,23, 3,89, 3,25.
(S)-[3-[3-фтор-4-(1H-пиразол-1-ил)фенил] -2-оксо-5-оксазолидинил] - метилазид:
Раствор 518 мг (1,45 ммоля) полученного ранее мезилата в 15 мл N,N-диметилформамида обрабатывают 1,89 г (29,07 ммоля) азида натрия с последующим нагреванием при 70oC в течение 48 часов. Полученную смесь разбавляют 100 мл этилацетата с последующим экстрагированием водой (6 х 75 мл) и насыщенным раствором NaCl (75 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество бежевого цвета, которое обрабатывают хроматографически на 50 г силикагеля (230-400 меш), элюируя 3% /объем/объем/ ацетоном в дихлорметане, а затем 3,5% /объем/объем/ метанолом в дихлорметане. В результате получают 355 мг (81%) указанного в заглавии азида в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,99, 7,92, 7,76, 7,24, 6,49, 4,83, 4,12, 3,99, 3,74, 3,62
(S)-N-[[3-[3-фтор-4-(1H-пиразол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
Раствор 355 мг полученного ранее азида в 30 мл этилацетата обрабатывают 150 мг палладия на карбонате кальция с последующим гидрированием при давлении 1 атм. в течение 18 часов. Полученный раствор обрабатывают затем 15 мл пиридина и 10 мл уксусного ангидрида, а затем перемешивают при комнатной температуре в течение 18 часов. Полученную смесь разбавляют 100 мл этилацетата и фильтруют через целит, промывают фильтровальную лепешку этилацетатом. Полученную смесь концентрируют в вакууме, а оставшийся пиридин и уксусный ангидрид удаляют за счет молекулярной перегонки в высоком вакууме (0,2 мм рт. ст. /горячая водяная баня). Полученный твердый остаток перекристаллизовывают из горячей смеси ацетон-гексан до получения 192 мг (51%) указанного в заглавии соединения в виде твердого вещества белого цвета. Т. пл. 183-184oC.
Пример 3
(S)-N-[[3-[3-фтор-4-(1H-имидазол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид:
3-фтор-4-(1H-имидазол-1-ил)нитробензол:
Раствор 2,14 г (31,4 ммоля) имидазола и 10,9 г (62,8 ммоля) вторичного кислого фосфата калия в 190 мл ДМСО обрабатывают 5,25 г (3,7 мл, 32,9 ммоля) 3,4-дифторнитробензола с последующим нагреванием при 90oC в течение 18 часов. Полученный раствор разбавляют этилацетатом и экстрагируют водой. Водный слой подвергают обратной экстракции этилацетатом, и объединенные органические слои затем экстрагируют водой дважды. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества коричневого цвета. Этот материал обрабатывают хроматографически на 250 г силикагеля (230-400 меш), элюируя дихлорметаном, а затем 1% /объем/объем/ метанолом в дихлорметане и, наконец, 2% /объем/объем/ метанолом в дихлорметане. В результате получают 6,0 г (92%) указанного в заглавии соединения в виде твердого вещества бронзового цвета.
1H ЯМР /CDCl3/ δ : 8,17, 7,96, 7,63, 7,35, 7,26
3-фтор-4-(1H-имидазол-1-ил)-1-(фенилметоксикарбониламино)бензол:
Раствор 500 мг (2,41 ммоля) полученного ранее нитросоединения в 50 мл ТГФ обрабатывают 100 мг 10% палладия на угле с последующим гидрированием при атмосферном давлении в течение 20 часов. Полученную смесь обрабатывают 60 мг дополнительно 10% палладия на угле и гидрируют при атмосферном давлении в течение 5 часов. Затем полученную смесь фильтруют через целит, промывают фильтровальную лепешку метанолом. Полученный фильтрат концентрируют в вакууме до получения масла коричневого цвета, которое растворяют в 50 мл ТГФ с последующей обработкой 405 мг (4,82 ммоля) твердого бикарбоната натрия. Полученный раствор охлаждают до -20oC с последующим добавлением 493 мг (0,43 мл, 2,89 ммоля) бензилхлорформата по каплям. Полученную смесь нагревают до комнатной температуры в течение 18 часов, затем разбавляют этилацетатом. Полученный раствор экстрагируют водой (трижды) и насыщенным раствором NaCl. В результате сушки над сульфатом натрия и концентрирования в вакууме получают светло-коричневое твердое вещество, которое перекристаллизовывают из горячей смеси хлороформгексан до получения 692 мг (92%) указанного в заглавии соединения в виде светло-коричневых кристаллов. Масс-спектр высокого разрешения: Рассчитано для C17H14FN3O2: 311,1070. Найдено: 311,1092.
(S)-[3-[3-фтор-4-(1H-имидазол-1-ил)фенил]-2-оксо-5- оксазолидинил]метанол:
Раствор 500 мг (1,61 ммоля) полученного ранее производного CBZ в 22 мл ТГФ и 4 мл ДМФ при - 78oC обрабатывают 1,69 мл (1,69 ммоля) литийбис(триметилсилил)амида при перемешивании при -78oC в течение 20 минут. Полученный раствор обрабатывают 243 мг (0,24 мл, 1,69 ммоля) (R)-(-)глицидилбутирата с последующим нагреванием до 0oC, а затем до комнатной температуры. После перемешивания в течение 3 часов при комнатной температуре по данным ТСХ оказывается, что реакция больше не идет. Полученный раствор тогда нагревают при 40oC в течение 2 часов с последующим охлаждением и перемешиванием при комнатной температуре в течение 18 часов. Затем полученный раствор разбавляют этилацетатом и экстрагируют водой (трижды). После высушивания над сульфатом натрия и концентрирования в вакууме получают твердое вещество коричневого цвета, которое обрабатывают хроматографически на 40 г силикагеля (230-400 меш), элюируя 2% /объем/объем/ метанолом в дихлорметане, а затем 5% /объем/объем/ метанолом в дихлорметане. В результате получают 214 мг (48%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета. Масс-спектр высокого разрешения: Рассчитано для C13H12FN3O3: 277,0863. Найдено: 277,0876.
(S)-[3-[3-фтор-4-(1H-имидазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метилазид:
Раствор 150 мг (0,54 ммоля) полученного ранее оксазалидинона и 96 мг (0,13 мл, 0,95 ммоля) триэтиламина в 5 мл дихлорметана при 0oC обрабатывают 77 мг (52 мкл, 0,68 ммоля) метансульфонилхлорида с последующим перемешиванием при 0oC в течение 30 минут. Полученный раствор нагревают до комнатной температуры с последующим разбавлением дихлорметаном и трижды экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают пену светло-коричневого цвета. Этот материал растворяют в 10 мл ДМФ и обрабатывают 525 мг (8,1 ммоля) азида натрия, затем нагревают при 60oC в течение 48 часов. Этот раствор охлаждают и разбавляют этилацетатом, а затем трижды экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают 163 мг (100%) указанного в заглавии азида в виде масла желтого цвета.
1H ЯМР /CDCl3/ δ : 8,15, 7,74, 7,46, 7,35, 7,29, 4,85, 4,12, 3,90, 3,76, 3,59
(S)-N-[[3-[3-фтор-4-(1H-имидазол-1-ил)фенил] -2-оксо-5- оксазолидинил] метил]ацетамид:
Раствор 163 мг (0,54 ммоля) азида в 8 мл пиридина обрабатывают 85 мг палладия на карбонате кальция и 110 мг (0,10 мл, 1,08 ммоля) уксусного ангидрида. Полученную смесь гидрируют при давлении в одну атмосферу в течение 20 часов. Затем полученную смесь обрабатывают 80 мг палладия на карбонате кальция, а затем гидрируют при давлении в одну атмосферу в течение 4 часов. Полученный раствор фильтруют через целит, промывают фильтровальную лепешку этилацетатом. Полученный фильтрат концентрируют в высоком вакууме, а остаток обрабатывают с помощью радиальной хроматографии на 2 мм хроматронной пластинке, элюируя 2% /объем/объем/ метанолом в дихлорметане, а затем 5% /объем/объем/ метанолом в дихлорметане. В результате получают 84 мг (49%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения: Рассчитано для C15H15FN4O3: 318,1128. Найдено: 318,1140.
Пример 4
(S)-N-[[3-[3-фтор-4-(1H-1,2,4-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
3-фтор-1-нитро-4-(1H-1,2,4-триазол-1-ил)бензол:
Раствор 5,0 г (72,4 ммоля) 1H-1,2,4-триазола и 25,22 г (144,8 ммоля) вторичного кислого фосфата калия в 150 мл ДМСО обрабатывают 11,52 г (72,4 ммоля) 3,4-дифторнитробензола, а затем нагревают при 90oC в течение 3 часов. Полученный раствор охлаждают и добавляют до 500 мл воды, полученное твердое вещество белого цвета отфильтровывают и промывают водой. Полученное белое твердое вещество сушат в вакууме (60oC/10 мм рт. ст.) до получения 10,79 г (72%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения: Рассчитано для C8H5FN4O2: 208,0396. Найдено: 208,0408
3-фтор-4-(1H-1,2,4-триазол-1-ил)анилин:
Раствор 8,7 (41,83 ммоля) полученного ранее нитросоединения в 350 мл смеси 1:1 метанол-ТГФ обрабатывают 3,5 г 10% палладия на угле, а затем гидрируют при давлении 45 пси (3,164 кг) водорода в течение 18 часов. Полученный раствор фильтруют через целит, промывают фильтровальную лепешку метанолом. Полученный фильтрат концентрируют в вакууме, а полученный остаток обрабатывают хроматографически на 200 г силикагеля (230-400 меш) элюируя 1% /объем/объем/ метанолом в дихлорметане, а затем 3% /объем/объем/ метанолом в дихлорметане. В результате получают 4,0 г (54%) производного анилина в виде грязно-белого твердого вещества. Масс-спектр высокого разрешения: Рассчитано для C8H7FN4: 178,0655. Найдено: 178,0673.
3-фтор-1-(фенилметоксикарбониламино)-4-(1H-1,2,4-триазол-1-ил)бензол:
Раствор 4,0 г (22,5 ммоля) полученного ранее производного анилина в 50 мл ацетона обрабатывают 50 мл насыщенного раствора бикарбоната натрия, затем охлаждают до 0oC. Полученный раствор обрабатывают 8,0 г (6,78 мл, 47,26 ммоля) бензинхлорформата. Полученный раствор нагревают до комнатной температуры в течение 1 часа, а затем добавляют 300 мл воды и экстрагируют этилацетатом (2 х 300 мл). Органический слой экстрагируют 300 мл воды и сушат над сульфатом натрия. В результате концентрирования в вакууме получают твердое вещество бежевого цвета, которое перекристаллизовывают из смеси хлороформ-гексан до получения 2,6 г (37%) указанного в заглавии производного CBZ. Масс-спектр высокого разрешения: Рассчитано для C16H13FN4O2: 312,1022. Найдено: 312,1033.
(S)-[3-[3-фтор-4-(1H-1,2,4-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил] метанол:
Раствор 2,5 г (8,0 ммоля) полученного ранее производного CBZ в 115 мл ТГФ при -78oC обрабатывают 8,8 мл (1,0 М, 8,8 ммоля) литийбис(триметилсилил)амида, а затем перемешивают при -78oC в течение 30 минут. Полученный раствор обрабатывают 1,27 г (1,25 мл, 8,8 ммоля) (R)-(-)глицидилбутирата, а затем перемешивают с последующим нагреванием до 0oC, а затем при -78oC в течение 15 минут. Полученный раствор затем нагревают до комнатной температуры в течение 18 часов. Полученную смесь обрабатывают 2 мл насыщенного раствора аммонийхлорида, затем разбавляют 100 мл воды и экстрагируют этилацетатом (2 х 100 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество, которое тщательно растирают с хлороформом до получения 1,4 г (57%) указанного в заглавии оксазолидинона. Масс-спектр высокого разрешения: Рассчитано для C12H11FN4O3: 278,0815. Найдено: 278,0836.
(S)-[3-[3-фтор-4-(1H-1,2,4-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил] метилазид:
Суспензию 1,0 г (3,6 ммоля) полученного ранее оксазалидинона и 548 мг (0,76 мл, 5,42 ммоля) триэтиламина в 20 мл дихлорметана при 0oC обрабатывают 495 мг (0,33 мл, 4,32 ммоля) метансульфонилхлорида. Затем полученный раствор нагревают до комнатной температуры в течение 2 часов. Полученный раствор обрабатывают 20 мл воды, и слои разделяют. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения 1,0 г (75%) промежуточного мезилата в виде твердого вещества. Раствор 1,34 г (3,6 ммоля) этого материала и 2,34 г (36 ммоля) азида натрия в 36 мл ДМФ нагревают при 70oC в течение 18 часов. Полученный раствор охлаждают и разбавляют 100 мл этилацетата, а затем экстрагируют водой (5 х 75 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают 1 г (88%) указанного в заглавии азида в виде грязно-белого твердого продукта.
1H ЯМР /CDCl3/ δ : 8,6, 8,13, 7,91, 7,83, 7,32, 4,85, 4,14, 3,92, 3,77, 3,63.
(S)-N-[[3-[3-фтор-4-(1H-1,2,4-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
Раствор 100 мг (0,36 ммоля) азида в 3 мл пиридина обрабатывают 50 мг палладия на карбонате кальция, а затем гидрируют при давлении водорода в одну атмосферу в течение 18 часов. Полученный раствор обрабатывают 0,2 мл уксусного ангидрида, а затем перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют в вакууме, а остаток растворяют в этилацетате и экстрагируют водой. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества светло-бежевого цвета. Этот материал обрабатывают с помощью радиальной хроматографии на 2 мм пластинке, элюируя 2% /объем/объем/ метанолом в дихлорметане, а затем 4% /объем/объем/ метанолом в дихлорметане. В результате получают 62 мг (54%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения: Рассчитано для C14H14FN5O3: 319,1018. Найдено: 319,1087.
Пример 5
(S)-N-[[3-[3-фтор-4-(1H-индол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил] ацетамид:
3-фтор-4-(1H-индол-1-ил)нитробензол:
Суспензию 334 мг (374 мг 60% в масле, 9,35 ммоля) гидрида натрия в 5 мл ТГФ обрабатывают раствором 1,0 г (8,5 ммоля) индола в 5 мл ТГФ, а затем перемешивают при комнатной температуре в течение 15 минут. Полученный раствор обрабатывают 1,35 г (8,5 ммоля) 3,4-дифторнитробензола. Полученный раствор перемешивают при комнатной температуре в течение 48 часов, затем концентрируют в вакууме до получения масла коричневого цвета, которое растворяют в этилацетате и промывают водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло, которое обрабатывают хроматографически на 100 г силикагеля (230-400 меш), элюируя 20% /объем/объем/ этилацетатом в гексане. В результате получают 1,1 г (51%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
1H ЯМР /CDCl3/ δ : 8,23, 7,74, 7,40, 7,35, 7,30 6,80.
3-фтор-4-(1H-индол-1-ил)-1-(фенилметоксикарбониламино)бензол:
Раствор 1,0 г (3,9 ммоля) полученного ранее нитросоединения в 5 мл ТГФ обрабатывают 200 мг W-2 никеля Рэнея, а затем гидрируют при давлении водорода в одну атмосферу в течение 18 часов. Полученный раствор фильтруют через целит, фильтровальную лепешку промывают ТГФ. Полученный фильтрат концентрируют в вакууме, а остаток растворяют в 10 мл ацетона и обрабатывают 8,2 мл насыщенного раствора бикарбоната натрия, затем охлаждают до 0oC и добавляют 699 мг (4,1 ммоля) бензилхлорформата. Полученный раствор нагревают до комнатной температуры в течение 18 часов, разбавляют этилацетатом и экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло, которое обрабатывают хроматографически на 75 г силикагеля (230-400 меш), элюируя 50% /объем/объем/ этилацетатом в гексане. В результате получают 1,03 г (71%) указанного в заглавии соединения в виде твердого вещества желтого цвета.
1H ЯМР /CDCl3/ δ : 7,69, 7,60, 7,40, 7,25, 7,15, 6,70.
(S)-N-[3-[3-фтор-4-(1H-индол-1-ил)фенил] -2-оксо-5- оксазолидинил]метанол:
Раствор 1,0 г (2,77 ммоля) полученного ранее производного CBZ в 5 мл ТГФ при -78oC обрабатывают 3,05 мл (1,0 М, 3,05 ммоля) литийбис(триметилсилил)амида в ТГФ, а затем перемешивают при -78oC в течение 45 минут. Полученный раствор обрабатывают 440 мг (0,43 мл, 3,05 ммоля) (R)-(-)глицидилбутирата, а затем перемешивают при -78oC в течение 15 минут, нагревают до комнатной температуры в течение 18 часов. Полученную смесь разбавляют этилацетатом, и экстрагируют водой. В результате сушки и концентрирования в вакууме получают полу-твердое вещество, которое обрабатывают хроматографически на 60 г силикагеля (230-400 меш), элюируя 2% /объем/объем/ метанолом в дихлорметане. В результате получают 630 мг (70%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,69; 7,50; 7,38; 7,25; 6,71; 4,80; 4,09; 3,79.
(S)-[3-[3-фтор-4-(1H-индол-1-ил)фенил] -2-оксо-5- оксазолидинил]метилазид:
Раствор 484 мг (1,2 ммоля) полученного ранее мезилата в 10 мл ДМФ обрабатывают 390 мг (6 ммолей) азида натрия, затем нагревают при 90oC в течение 18 часов. Полученный раствор охлаждают и разбавляют этилацетатом, затем экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают 336 мг (80%) указанного в заглавии азида в виде смолы, достаточно чистой для дальнейшего использования.
1H ЯМР /CDCl3/ δ : 7,72, 7,68, 7,51, 7,37, 7,20, 6,71, 4,85, 4,13, 3,91, 3,77, 3,62.
(S)-N-[[3-[3-фтор-4-(1H-индол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил] ацетамид:
Раствор 336 мг (0,96 ммоля) полученного ранее азида в 5 мл этилацетата обрабатывают 150 мг 10% палладия на угле, а затем гидрируют при давлении водорода в 1 атмосферу в течение 18 часов. Полученный раствор фильтруют через целит, фильтровальную лепешку промывают этилацетатом. Полученный фильтрат концентрируют в вакууме до получения масла светло-коричневого цвета, которое растворяют в 2 мл пиридина, и обрабатывают 0,2 мл уксусного ангидрида, а затем перемешивают при комнатной температуре в течение 60 часов. Полученный раствор концентрируют в высоком вакууме, а остаток растворяют в хлороформе и экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают смолу, которую обрабатывают с помощью радиальной хроматографии на 4 мм хроматотронной пластинке, элюируя 1,5% /объем/объем/ метанолом в дихлорметане, а затем 2,5% /объем/объем/ метанолом в дихлорметане. В результате получают 277 мг (81%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,70, 7,51, 7,33, 7,20, 6,71, 6,11, 4,84, 4,12, 3,87, 3,70, 2,06
Пример 6
(S)-N-[[3-[3-фтор-4-(1H-1,2,3-триазол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид:
3-фтор-1-нитро-4-(1H-1,2,3-триазол-1-ил)бензол:
Суспензию вторичного кислого фосфата калия, 38,0 г (0,218 моля) и 1H-1,2,3-триазол, 7,53 г (6,3 мл, 0,109 моля) в 325 мл диметилсульфоксида обрабатывают, прикапывая 3,4-дифторнитробензол, 17,3 г (12,1 мл, 0,109 моля) при нагревании до 90oC в течение 18 часов. Полученную смесь охлаждают до комнатной температуры, разбавляют 500 мл воды и экстрагируют этилацетатом (4 х 50 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество бледно-желтого цвета. Этот материал обрабатывают хроматографически на 600 г (230-400 меш) силикагеля, элюируя метиленхлоридом, 1% /объем/объем/, 2% и 5% /объем/объем/ метанолметиленхлоридом до получения 11,38 г (50%) 3-фтор-1-нитро-4-(1H-1,2,3-триазол-1-ил)бензола в виде твердого вещества светло-желтого цвета. Т. пл. 123-124,5oC, наряду с 9,66 г (43%) региоизомера 3-фтор-1-нитро-4-(2H-1,2,3-триазол-1-ил)бензола в виде твердого вещества светло-желтого цвета. Т. пл. 137-139oC.
3-фтор-1-(фенилметоксикарбониламино)-4-(1H-1,2,3-триазол-1-ил)бензол:
Раствор 5,0 г (24,0 ммоля) полученного ранее соединения в 400 мл смеси 1: 1 метанол-ТГФ обрабатывают 3 г W-2 никеля Рэнея. Полученную смесь гидрируют в аппарате Парра при давлении водорода 45 пси (3,164 кг/см2) в течение 18 часов, а затем отфильтровывают и фильтровальную лепешку промывают метанолом. В результате концентрирования в вакууме получают твердое вещество белого цвета, которое растворяют в 500 мл безводного ТГФ, охлаждают до -20oC и обрабатывают бикарбонатом натрия, 4,0 г (48,0 ммоля) и бензилхлорформатом, 4,9 г (4,3 мл, 28,8 ммоля). После перемешивания в течение 72 часов при постепенном нагревании до комнатной температуры растворитель удаляют при пониженном давлении, а оставшееся масло растворяют в 200 мл этилацетата. Полученную смесь экстрагируют водой (3 х 50 мл), сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества желтого цвета. Полученный материал обрабатывают хроматографически на 400 г силикагеля (230-400 меш), элюируя метиленхлоридом, 1% /объем/объем/ и 2% /объем/объем/ метанолметиленхлоридом до получения 6,18 г (82%) указанного в заглавии соединения в виде твердого вещества белого цвета. Т. пл. 121-122oC.
(R)-[3-[3-фтор-4-(1H-1,2,3-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил] метанол:
Раствор 2,0 г (6,4 ммоля) полученного ранее соединения в 100 мл безводного ТГФ при -78oC обрабатывают 6,7 мл (1,0 М, 6,7 ммоля) литийбис(триметилсилил)амида в ТГФ с последующим перемешиванием при -78oC в течение 30 минут. Полученную смесь обрабатывают 960 мг (6,7 ммоля) (R)-(-)-глицидилбутирата, а затем нагревают до 0oC и постепенно нагревают до комнатной температуры. Спустя 18 часов полученную смесь гасят насыщенным водным раствором аммонийхлорида (30 мл) и экстрагируют этилацетатом (3 х 30 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое маслянистое вещество желтого цвета. Сырой материал перекристаллизовывают из кипящей смеси метанол:этилацетат 2:1 до получения 1,10 г (62%) указанного в заглавии соединения в виде твердого белого вещества. Т. пл. 179-180,5oC.
(R)-[3-[3-фтор-4-(1H-1,2,3-триазол-1-ил)фенил] -2-оксо-5- оксазолидинил] метилазид:
Суспензию 1,35 г (4,85 ммоля) полученного ранее соединения и 859 мг (1,18 мл, 8,49 ммоля) триэтиламина в 60 мл дихлорметана при 0oC обрабатывают 695 мг (0,47 мл, 6,07 ммоля) метансульфонилхлорида с последующим нагреванием до комнатной температуры в течение 1 часа. Полученную смесь охлаждают до 0oC, обрабатывают триэталамином, 859 мг (8,49 ммоля) и метансульфонилхлоридом, 695 мг (6,07 ммоля). Спустя 5 минут полученную смесь экстрагируют водой (3 х 20 мл), насыщенным водным раствором NHCO3 (2 х 20 мл), насыщенным водным раствором NaCl (1 х 20 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают 1,89 г соответствующего мезилата в виде твердого вещества светло-желтого цвета, которое растворяют в 90 мл ДМФ и обрабатывают 4,73 г (72,8 ммоля) азида натрия при 60oC в течение 18 часов. Полученную смесь охлаждают до 0oC и разбавляют 200 мл воды. Полученную смесь экстрагируют смесью 1:1 этиловый спирт-этилацетат (5 х 50 мл). Объединенный органические слои экстрагируют затем водой (4 х 50 мл), сушат над сульфатом натрия и концентрируют в вакууме до получения 1,48 г (примерно 100%) указанного в заглавии азида в виде грязно-белого твердого вещества, достаточно чистого для дальнейшего использования.
1H ЯМР /CDCl3/ δ : 8,09, 7,98, 7,87, 7,29, 4,86, 4,16, 3,94, 3,70.
(S)-N-[[3-[3-фтор-4-(1H-1,2,3-триазол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид:
Раствор 300 мг (0,99 ммоля) указанного ранее азида в 12 мл ТГФ обрабатывают 750 мг W-2 никеля Рэнея. Полученную смесь гидрируют при атмосферном давлении в течение 18 часов. Полученную смесь обрабатывают 783 мг (0,80 мл, 9,9 ммоля) пиридина и 505 мг (0,47 мл, 4,9 ммоля) уксусного ангидрида с последующим перемешиванием в течение 2 часов. Полученную смесь фильтруют через целит, промывают фильтровальную лепешку этилацетатом. Полученный фильтрат экстрагируют водой (3 х 30 мл) и насыщенным солевым раствором (1 х 20 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают грязно-белое твердое вещество. Сырой материал перекристаллизовывают из кипящей смеси 10:1 этилацетат:гексан до получения 210 мг (66%) указанного в заглавии соединения в виде твердого белого вещества. Т. пл. 193-194oC.
Пример 7
(S)-N-[[3-[3-фтор-4-(1H-1,2,3-триазол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил]-2,2-дихлорацетамид:
Раствор 275 мг (0,91 ммоля) (R)-[[3-[3-фтор-4-(1H-1,2,3-триазол-1- ил)фенил]-2-оксо-5-оксазолидинил]метилазида (из примера 6) в 15 мл ТГФ обрабатывают 1,5 г W-2 никеля Рэнея и гидрируют при атмосферном давлении. Спустя 18 часов полученную смесь фильтруют через целит, промывают фильтровальную лепешку метанолом. Полученный фильтрат концентрируют в вакууме, а полученное масло перемешивают в 15 мл метиленхлорида, охлаждают до 0oC и обрабатывают 138 мг (0,19 мл, 1,36 ммоля) триэтиламина и 161 мг (0,11 мл, 1,09 ммоля) дихлорацетилхлорида. После перемешивания при комнатной темепературе в течение 24 часов полученную смесь разбавляют метиленхлоридом и экстрагируют водой (3 х 20 мл) и насыщенным водным хлоридом натрия (1 х 20 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают грязно-белое твердое вещество. Неочищенный материал обрабатывают с помощью радиальной хроматографии на 4 мм хроматотронной пластине, элюируя дихлорметаном и 2% /объем/объем/ метанолдихлорметаном. В результате получают 182 мг (52%) указанного в заглавии соединения в виде твердого белого вещества. Т. пл. 195-198oC (с разложением).
Пример 8
(S)-N-[[3-[3-фтор-4-(2H-1,2,3-триазол-2-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
3-фтор-1-(фенилметоксикарбониламино)-4-(2H-1,2,3-триазол-2-ил)бензол:
Раствор 6,0 г (28,82 ммоля) 3-фтор-1-нитро-4-(2H-1,2,3-триазол-2- ил)бензола в 150 мл этилацетата и 25 мл метанола обрабатывают 750 мг W-2 никеля Рэнея. Полученную смесь гидрируют в аппарате Парра при давлении водорода 45 пси (3,164 кг/см2) в течение 24 часов. Полученную смесь фильтруют через целит, и полученный фильтрат концентрируют в вакууме. Остаток растворяют в 150 мл ацетона, обрабатывают 72 мл насыщенного раствора бикарбоната натрия, затем охлаждают до 0oC. Полученную смесь обрабатывают 6,15 г (5,14 ммоля) бензилхлорформата, перемешивают при 0oC в течение 1 часа, а затем нагревают до комнатной температуры в течение 3 часов. Полученную смесь разбавляют 500 мл воды и 400 мл этилацетата. Водную фазу промывают 250 мл этилацетата, и объединенные органические слои экстрагируют 300 мл насыщенного солевого раствора. В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло янтарного цвета, которое перекристаллизовывают из горячей смеси хлороформ:гексан до получения 7,52 г (84%) указанного в заглавии соединения в виде твердого вещества белого цвета. Т. пл. 99,5-101oC.
(R)-[3-[3-фтор-4-(2H-1,2,3-триазол-2-ил)фенил] -2-оксо-5- оксазолидинил] метанол:
Раствор 1,0 г (3,20 ммоля) полученного ранее соединения в 45 мл ТГФ при -78oC обрабатывают 3,52 мл (1,0 М, 3,52 ммоля) литийбис(триметилсилил)амида в ТГФ, а затем перемешивают при -78oC в течение 30 минут. Полученный раствор обрабатывают 508 мг (0,50 мл, 3,52 ммоля) (R)-(-)-глицидилбутирата с последующим перемешиванием при 0oC в течение 30 минут, а затем при комнатной температуре в течение 18 часов. Полученную смесь разбавляют 75 мл этилацетата и экстрагируют водой (2 х 75 мл) и насыщенным солевым раствором (75 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло янтарного цвета, которое растворяют в 75 мл метанола и обрабатывают 1,0 г K2CO3, а затем перемешивают в течение 2 часов. Полученную смесь фильтруют и концентрируют в вакууме до получения масла янтарного цвета, которое обрабатывают хроматографически на 50 г силикагеля (230-400 меш), элюируя 3% /объем/объем/ метанолом в дихлорметане. В результате получают 446 мг (50%) указанного в заглавии соединения в виде твердого белого вещества. Т. пл. 159-151oC.
(R)-[3-[3-фтор-4-(2H-1,2,3-триазол-2-ил)фенил] -2-оксо-5- оксазолидинил] метилазид:
Раствор 350 мг (1,26 ммоля) полученного ранее спирта и 2 мл триэтиламина в 8 мл дихлорметана при 0oC обрабатывают 362 мг (1,64 ммоля) нозилхлорида, а затем перемешивают при 0oC в течение 30 минут и нагревают до комнатной температуры в течение 1 часа. Полученную смесь разбавляют 50 мл дихлорметана и экстрагируют водой (215,8 г х 50 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают 540 мг (92%) соответствующего нозилата, который растворяют в 12 мл N,N-диметилформамида и обрабатывают 2 г азида натрия, а затем перемешивают при комнатной температуре в течение 48 часов. Полученный раствор разбавляют 125 мл этилацетата и экстрагируют водой (5 х 50 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердый продукт желтого цвета, который обрабатывают хроматографически на 50 г силикагеля (230-400 меш), элюируя дихлорметаном, а затем 5% /объем/объем/ ацетоном в дихлорметане. Таким способом получают 333 мг (94%) указанного в заглавии азида в виде твердого вещества бледно-желтого цвета.
1H ЯМР /CDCl3/ δ : 7,87, 7,70, 7,38, 4,84, 4,13, 3,91, 3,75, 3,63.
(S)-N-[[3-[3-фтор-4-(2H-1,2,3-триазол-2-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
Раствор полученного ранее азида в 50 мл обрабатывают 500 мг W-2 никеля Рэнея, а затем гидрируют в аппарате Парра при давлении 30 пси (2,109 кг/см2) водорода в течение 18 часов. Полученную смесь фильтруют через целит, и полученный фильтрат концентрируют в вакууме. Остаток растворяют в 8 мл пиридина и обрабатывают 4 мл уксусного ангидрида с последующим перемешиванием при комнатной температуре в течение 24 часов. Растворитель удаляют в высоком вакууме (0,2 мм рт. ст.), а остаток растворяют в 75 мл этилацетата и экстрагируют водой (2 х 50 мл) и насыщенным солевым раствором (50 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло янтарного цвета, которое обрабатывают с помощью радиальной хроматографии на 4 мм хроматотроновой пластине, элюируя 4% /объем/объем/ метанолом в дихлорметане. Полученный материал перекристаллизовывают из смеси хлороформ-гексан до получения 164 мг (47%) указанного в заглавии соединения.
1H ЯМР /CDCl3/ δ : 7,87, 7,82, 7,70, 7,32, 6,21, 4,83, 4,10, 3,84, 3,68, 2,03.
Пример 9
(S)-N-[[3-[3-фтор-4-(3-меркапто-4H-1,2,4-триазол-4-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
4-(N-бензиламино)-3-фторнитробензол:
Раствор 23,9 г (16,9 мл, 0,15 моля) 3,4-дифторнитробензола и 29,1 г (39,2 мл, 0,23 моля) N,N-диизопропилэтиламина в 285 мл ацетонитрила обрабатывают 19,3 г (19,7 мл, 0,18 моля) бензиламина, а затем нагревают при кипении с обратным холодильником в течение 5 часов. Полученную смесь концентрируют в вакууме, а остаток растворяют в 200 мл этилацетата с последующим экстрагированием водой (3 х 50 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество желтого цвета, которое перекристаллизовывают из горячей смеси этилацетат-гексан до получения 32,25 г (87%) указанного в заглавии соединения в виде иголок желто-оранжевого цвета. Т. пл. 97,5-99oC.
4-(N-бензилоксикарбониламино)-1-(бензилоксикарбониламино)-3-фторбензол:
Раствор 14,25 г (0,058 моля) полученного ранее соединения в 800 мл ТГФ обрабатывают 1,0 г 5% платины на угле, а затем гидрируют в аппарате Парра при давлении 30 пси (2,109 кг/см2) водорода в течение 18 часов. Полученную смесь фильтруют через целит, промывают фильтровальную лепешку этилацетатом. Полученный фильтрат концентрируют в вакууме до получения маслянистого остатка, который растворяют в 1,0 л ТГФ и обрабатывают 15,8 г (16,5 мл, 0,13 моля) N,N-диметилформамида. Полученный раствор охлаждают до 0oC, и прикапывают 21,72 г (18,18 мл, 0,127 моля) бензилхлорформата в течение 30 минут. Раствор перемешивают при 0oC в течение 30 минут, а затем оставляют постепенно нагреваться до комнатной температуры с последующим перемешиванием в течение 23 часов. Полученный раствор концентрируют в вакууме, а остаток растворяют в 1 л этилацетата. Полученную смесь промывают 80 мл холодного 1Н раствора HCl, 80 мл воды и 80 мл насыщенного раствора бикарбоната натрия. Полученный раствор сушат над сульфатом магния и концентрируют в вакууме до получения масла, которое обрабатывают хроматографически на 500 г силикагеля (230-400 меш), элюируя 10-15% /объем/объем/ этилацетата в гексане. В результате получают 12,42 г (44%) указанного в заглавии соединения в виде твердого вещества белого цвета. Т. пл. 101-102,5oC.
(R)-[3-[3-фтор-4-(N-бензилбензилоксикарбониламино)фенил]-2-оксо-5- оксазолидинил]метанол:
Раствор 12,42 г (0,026 моля) указанного ранее соединения в 375 мл ТГФ при -78oC обрабатывают 16,7 мл (1,6 М, 0,027 моля) н-бутиллития в гексане по каплям в течение 6 минут, а затем перемешивают при -78oC в течение 30 минут. Полученный раствор обрабатывают затем 4,6 г (4,5 мл, 0,032 моля) (R)-(-)-глицидилбутирата, а затем перемешивают при -78oC в течение 1 часа, затем нагревают до комнатной температуры в течение 20 часов. Полученный раствор обрабатывают 25 мл насыщенного раствора аммонийхлорида, добавляя затем 25 мл воды и 300 мл этилацетата. Водную фазу экстрагируют этилацетатом, и объединенные органические слои сушат над сульфатом магния и концентрируют в вакууме. Полученное таким образом масло обрабатывают хроматографически на 400 г (230-400 меш) силикагеля, элюируя 1% /объем/объем/ метанолом в дихлорметане. В результате получают 7,56 г (66%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,48, 7,25, 7,07, 6,95, 5,15, 4,18, 4,67, 3,91, 3,69.
(S)-N-[3-[3-фтор-4-(N-бензилбензилоксикарбониламино)фенил] -2- оксо-5-оксазолидинил]метилацетамид:
Раствор 8,23 г (18,3 ммоля) полученного ранее соединения и 3,24 г (4,5 мл, 32,0 ммоля) триэтиламина в 107 мл дихлорметана при 0oC обрабатывают 5,47 г нозилхлорида, а затем перемешивают при 0oC в течение 4 часов. Полученную смесь разбавляют дихлорметаном и экстрагируют водой (2 х). В результате сушки над сульфатом натрия и концентрирования в вакууме получают пену желтого цвета, которую растворяют в 58 мл изопропилового спирта, 93 мл ацетонитрила и 116 мл гидроксида аммония, а затем нагревают при 40oC в течение 18 часов. Полученную смесь охлаждают, разбавляют этилацетатом и экстрагируют водой (3 х) и насыщенным солевым раствором. В результате сушки над сульфатом натрия и концентрирования в вакууме получают пену желтого цвета, которую растворяют в 200 мл этилацетата и обрабатывают 4,67 г (4,3 мл, 45,75 ммоля) пиридина и 7,24 г (7,4 мл 91,5 ммоля) уксусного ангидрида. Полученный раствор перемешивают при комнатной температуре в течение 18 часов, затем экстрагируют трижды водой и насыщенным солевым раствором (1 х). В результате сушки над сульфатом натрия и концентрирования в вакууме получают пену янтарного цвета, которую обрабатывают хроматографически на 450 г силикагеля (230-400 меш), элюируя 1% /объем/объем/ метанолом в дихлорметане. В результате получают 7,04 г (78%) указанного в заглавии соединения в виде пены светло-желтого цвета. Масс-спектр высокого разрешения (с электронным ударом): Рассчитано для C27H26FN3O5: 491,1856. Найдено: 491,1860.
(S)-N-[3-[3-фтор-4-аминофенил]-2-оксо-5-оксазолидинил]метилацетамид:
Раствор 5,6 г (11,4 ммоля) полученного ранее соединения в 200 мл этанола обрабатывают 200 мг 10% палладия на угле с последующим гидрированием в аппарате Парра при давлении водорода 45 пси (3,16 кг/см2) в течение 24 часов. Полученную смесь фильтруют через целит, промывают фильтровальную лепешку метанолом. Полученный фильтрат концентрируют в вакууме до получения грязно-белого твердого вещества, которое перекристаллизовывают из горячей смеси этилацетат-гексан до
получения 2,81 г (92%) указанного в заглавии соединения в виде грязно-белого вещества.
1H ЯМР /CDCl3/ δ : 8,20, 7,29, 6,95, 6,76, 5,00, 4,65, 4,00, 3,63, 3,37, 1,83.
(S)-N-[3-[3-фтор-4-изотиоцианофенил]-2-оксо-5- оксазолидинил]метил]ацетамид:
Раствор 960 мг (4,12 ммолей) 1,1'-тиокарбонилди-2(1H)-пиридона в 30 мл дихлорметана при 0oC обрабатывают 1,0 г (3,74 ммоля) полученного ранее соединения сразу. Полученную смесь перемешивают при 0oC в течение 2,5 часа, а затем нагревают до комнатной температуры в течение 2 часов. Полученную смесь концентрируют в вакууме и полученное твердое вещество тщательно растирают с 15 мл воды. Продукт отфильтровывают и промывают небольшим количеством воды с последующей сушкой в течение ночи в вакуумном шкафу. В результате получают 1,09 г (94%) указанного в заглавии соединения в виде белого твердого вещества. Т. пл. 172,5-176oC.
(S)-N-[3-[3-фтор-4-[3-меркапто-4H-1,2,4-триазол-4-ил] фенил]-2- оксо-5-оксазолидинил]метилацетамид:
Раствор 11,1 г (3,59 ммоля) полученного ранее соединения в 26 мл ТГФ обрабатывают 230 мг (3,77 ммоля) муравьинокислого гидразида с последующим нагреванием при 70oC в течение 3,5 часа. Полученную смесь охлаждают, а остаток отфильтровывают и промывают этилацетатом. Твердую часть сушат в вакуумном шкафу при 60oC до получения 1,2 г твердого продукта. Из этого материала 100 г (0,27 ммоля) суспендируют в 2 мл воды и обрабатывают 0,28 мл (0,97 М, 0,27 ммоля) раствора KOH с последующим перемешиванием в течение 45 минут. Затем раствор обрабатывают 0,28 мл (1,0 н, 0,28 ммоля) раствора HCl. Осадок отфильтровывают и промывают небольшим количеством 1н. раствора HCl. Твердую часть сушат в вакуумном шкафу при комнатной температуре. В результате получают 83 мг указанного в заглавии соединения в виде твердого продукта белого цвета. Т. пл. 269-271oC.
Пример 10
(S)-N-[[3-[3-фтор-4-[3-метилтио-4H-1,2,4-триазол-4-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
Суспензию 200 мг (0,57 ммоля) соединения примера 9 и 121 мг (53 мкл, 0,85 ммоля) метилиодида в 5 мл метанола обрабатывают 53 мг (0,63 ммоля) твердого бикарбоната натрия. Полученную смесь перемешивают при комнатной температуре в течение 23 часов, твердый осадок отфильтровывают и фильтровальную лепешку промывают метанолом. Полученный фильтрат концентрируют в вакууме до получения смолы, которую обрабатывают хроматографически на 25 г силикагеля (230-400 меш), элюируя 5% /объем/объем/ метанолом и 0,5% /объем/объем/ гидроксидом аммония в дихлорметане. В результате получают твердое вещество белого цвета, которое перекристаллизовывают из метанола до получения 128 мг (61%) указанного в заглавии соединения в виде твердого белого вещества. Т. пл. 188,5-190,5oC.
Пример 11
(S)-N-[[3-[3-фтор-4-(4H-1,2,4-триазол-4-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид:
330 мг (0,938 ммоля) меркаптана примера 9 добавляют к 0,6 мл 20% раствора азотной кислоты с последующим нагреванием на паровой бане примерно в течение 1 минуты. После этого добавляют дополнительно 0,6 мл 20% азотной кислоты с последующим нагреванием на паровой бане в течение 6-7 минуты. Причем в этот момент вся твердая часть уходит в раствор. Полученную смесь охлаждают до 0oC и pH устанавливают 9, добавляя раствор гидроксида аммония. Полученную смесь концентрируют в вакууме до получения смолистого масла коричневого цвета, которое обрабатывают хроматографически на 25 г силикагеля (230-400 меш), элюируя 5% /объем/объем/ метанолом и 0,5% /объем/объем/ гидроксидом аммония в дихлорметане до получения твердого вещества, которое перекристаллизовывают из ацетонитрила и получают указанное в заглавии соединение. Т. пл. 204,5-206oC.
Пример 12
(S)-N-[[3-[3-фтор-4-(1H-пиррол-1-ил-3-карбоксальдегид)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
Раствор 2,0 г (7,5 ммоля) производного анилина примера 9 и 1,68 г (10,5 ммоля) 2,5-диметокси-3-тетрагидрофуранкарбоксальдегида в 55 мл ледяной уксусной кислоты кипятят с обратным холодильником в течение 2 часов. Полученный раствор охлаждают и растворитель удаляют в вакууме, азеотропно перегоняя остаток с толуолом для удаления последних следов уксусной кислоты. Остаток обрабатывают хроматографически на силикагеле 300 г (230-400 меш), элюируя дихлорэтаном, а затем 1-3% /объем/объем/ метанолом в дихлорметане. В результате получают 2,21 г указанного в заглавии соединения в виде аморфного твердого вещества светло-желтого цвета. Масс-спектр высокого разрешения: Рассчитано для C17H16FN3O4: 345,1125. Найдено: 345,1129.
Пример 13
(S)-N-[[3-[3-фтор-4-(3-гидроксиметил-1H-пиррол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
Раствор 125 мг (0,36 ммоля) полученного ранее соединения в 4 мл метанола и 2 мл дихлорметана при 0oC обрабатывают 7 мг (0,18 ммоля) боргидрида натрия. Полученный раствор оставляют нагреваться до комнатной температуры в течение 4 часов, затем разбавляют 30 мл дихлорметана и экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество белого цвета, которое перекристаллизовывают из смеси этилацетат-метанол-гексан. В результате получают 106 мг (84%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения (электронный удар): Рассчитано для C17H18FN3O4: 347,1281. Найдено: 347,1274.
Пример 14
(S)-N-[[3-[3-фтор-4-(3-карбометокси-1H-пиррол-1-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид:
Раствор 220 мг (0,64 ммоля) соединения примера 12 в 10 мл смеси 1:1 ацетонитрил-метанол обрабатывают 164 мг (0,64 ммоля) цианида натрия, 1,15 г (13,2 ммоля) активированной двуокиси марганца и 60 мг (58 мкл, 1,0 ммоля) ледяной уксусной кислоты. Полученную смесь перемешивают при комнатной температуре в течение 36 часов и в это время добавляют 550 мг (6,32 ммоля) активированной двуокиси марганца и 60 мг (58 мкл, 1,0 ммоля) ледяной уксусной кислоты. Полученный раствор перемешивают еще 24 часа и фильтруют через целит, промывают фильтровальную лепешку метанолом. Полученный фильтрат концентрируют в вакууме, разбавляют этилацетатом и экстрагируют трижды водой. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества розового цвета. Этот материал обрабатывают с помощью радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя дихлорметаном, а затем 1-2% /объем/объем/ метанолом в дихлорметане. В результате получают 170 мг (71%) указанного в заглавии соединения в виде твердого вещества белого цвета. Элементный анализ: Рассчитано для C18H18FN3O5: C 57,60; H 4,83; N 11,20. Найдено: C 57,29; H 5,19; N 11,07.
Пример 15
(S)-N-[[3-[3-фтор-4-(1H-пиррол-1-ил-карбоксальдегид)фенил] -2- оксо-5-оксазолидинил]метил]ацетамида метоксим:
Раствор 150 мг (0,43 ммоля) соединения примера 12 в 5 мл смеси 1:1 метанол:дихлорметан обрабатывают 44 мг (0,52 ммоля) метоксиламингидрохлорида и 36 мг (0,26 ммоля) карбоната калия с последующим перемешиванием при комнатной температуре в течение 18 часов. Полученную смесь разбавляют дихлорметаном и экстрагируют трижды водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество красно-коричневого цвета. Этот материал обрабатывают хроматографически на 2 мм хроматотроновой пластине, элюируя дихлорметаном, а затем 1-2% /объем/объем/ метанолом в дихлорметане. В результате получают 101 мг (63%) указанного в заглавии соединения в виде твердого вещества белого цвета, которое представляет смесь E- и Z-стереоизомеров по двойной связи. Масс-спектр высокого разрешения (электронный удар): рассчитано для C18H19FN4O4: 347,1390. Найдено: 347,1390.
Пример 16
(S)-N-[[3-[3-фтор-4-(3-(гидроксииминометил)-1H-пиррол-1- ил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамид:
Раствор 150 мг (0,43 ммоля) альдегида примера 12 и 36 мг (0,26 ммоля) карбоната калия в 10 мл смеси 1:1 метанол:дихлорметан обрабатывают 36 мг (0,52 ммоля) гидроксиламингидрохлорида с последующим перемешиванием при комнатной температуре в течение 48 часов. Полученную смесь разбавляют этилацетатом, экстрагируют трижды водой, сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества. Этот материал обрабатывают с помощью радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя 2-4% /объем/объем/ метанолом в дихлорметане. В результате получают 90 мг (58%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /ДМСО/ δ : 11,07, 10,05, 8,24, 8,01, 7,85, 7,70, 7,62, 7,40, 7,30, 7,14, 6,63, 6,48, 4,75, 4,16, 3,77, 3,42, 1,83.
Пример 17
(S)-N-[[3-[3-фтор-4-(4-ацетил-1H-1,2,3-триазол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
(S)-N-[[3-[4-азидо-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид:
Раствор 4,0 г (14,96 ммоля) (S)-N-[3-[3-фтор-4-аминофенил]-2- оксо-5-оксазолидинил]метилацетамида примера 9 в 40 мл 6 н. раствора HCl при 0oC обрабатывают порциями 4,12 г (59,84 ммоля) нитрита натрия с последующим перемешиванием при 0oC в течение 2 часов. Затем полученную смесь обрабатывают несколькими порциями раствором 1,94 г (29,92 ммоля) азида натрия и 24,52 г (0,30 моля) ацетата натрия в 50 мл воды. После завершения добавления раствор экстрагируют этилацетатом, органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения 4,1 г (93%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета, достаточно чистого для дальнейшего использования.
1H ЯМР /CDCl3/ δ : 8,22, 7,59, 7,33, 4,72, 4,10, 3,72, 3,40, 1,90.
(S)-N-[[3-[3-фтор-4-(4-ацетил-1H-1,2,3-триазол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
Раствор 500 мг (1,7 ммоля) полученного ранее азида в 25 мл бензола обрабатывают 347 мг (5,1 ммоля) 3-бутин-2-она с последующим нагреванием при кипении с обратным холодильником в течение 18 часов. Полученную смесь охлаждают и выделяют фильтрованием 407 мг (66%) указанного в заглавии соединения. Масс-спектр высокого разрешения (электронный удар): рассчитано для C16H16FN5O4: 361,1186. Найдено: 361,1183.
Пример 18
(S)-N-[[3-[3-фтор-4-(4-(1-гидроксиэтил)-1H-1,2,3-триазол-1- ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид:
Раствор 150 мг (0,40 ммоля) соединения примера 17 в 10 мл смеси 1:1 ТГФ: метанол обрабатывают 15 мг (0,4 ммоля) боргидрида натрия с последующим перемешиванием при комнатной температуре в течение 18 часов. Полученную смесь концентрируют в вакууме, растворяют в смеси 1:1 метанол:дихлорметан и фильтруют. Полученный фильтрат концентрируют в вакууме, а остаток обрабатывают с помощью радиальной хроматографии на 2 мм хроматотроновой пластине. В результате получают 120 мг (80%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр (электронный удар): m/z: 363, 291, 276, 232, 217, 207, 191, 179, 163, 148, 135, 123, 85, 56.
Пример 19
(S)-N-[[3-[3-фтор-4-(4-(1-гидроксииминометил)-1H-1,2,3-триазол-1- ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид:
Раствор 150 мг (0,40 ммоля) соединения примера 17 в 10 мл смеси 1:1 ТГФ: метанол обрабатывают 66 мг (0,96 ммоля) гидроксиламин гидрохлорида и 66 мг (0,48 ммоля) карбоната калия с последующим перемешиванием при комнатной температуре в течение 8 часов, а затем нагревают при кипении с обратным холодильником в течение 2 часов. Полученную смесь концентрируют в вакууме, а остаток обрабатывают с помощью радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя 5% /объем/объем/ метанолом в дихлорметане. В результате получают 30 мг (20%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр (электронный удар): m/z: 376, 332, 318, 287, 274, 245, 220, 202, 187, 175, 163, 149, 139, 82, 56.
Пример 20
(S)-N-[[3-[3-фтор-4-(4-карбометокси-1H-1,2,3-триазол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
Раствор 1,0 г (3,4 ммоля) азида примера 17 в 50 мл бензола обрабатывают 573 мг (6,8 ммоля) метилпропилата с последующим нагреванием при кипении с обратным холодильником в течение 24 часов. В результате получают 880 мг указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения (электронный удар): Рассчитано для C16H16FN5O5: 377,1135. Найдено: 377,1131.
Пример 21
(S)-N-[[3-[3-фтор-4-(1H-1,2,3-триазол-1-ил-4-карбоксальдегид)фенил] - 2-оксо-5-оксазолидинил]метил]ацетамид:
Раствор 1,70 г (5,8 ммоля) азида примера 17 в 50 мл бензола обрабатывают 4,46 г (34,8 ммоля) 1,1-диэтокси-2-пропина с последующим нагреванием при кипении с обратным холодильником в течение 18 часов. Полученный раствор обрабатывают 1,0 г (7,80 ммоля) 1,1-диэтокси-2-пропина с последующим кипячением с обратным холодильником в течение дополнительно 3 часов. Полученный раствор охлаждают и концентрируют в вакууме до получения масла, которое обрабатывают хроматографически на 200 г силикагеля (230-400 меш), элюируя 2-3% /объем/объем/ метанолом в дихлорметане до получения 2,0 г (80%) (S)-N-[[3-[3-фтор-4-(4-(диэтоксиметил)-1H-1,2,3-триазол-1- ил)фенил] -2-оксо-5-оксазолидинил]метил]ацетамида и
(S)-N-[[3-[3-фтор-4-(5-(диэтоксиметил)-1H-1,2,3-триазол-1- ил)фенил] -2-оксо-5-оксазолидинил] метил] ацетамида в виде неразделимой смеси изомеров. 410 мг (0,973 ммоля) из этого материала растворяют в ТГФ и обрабатывают 1н. раствором HCl с последующим перемешиванием при комнатной температуре в течение 48 часов, а затем нагревают при кипении с обратным холодильником в течение 1 часа. Полученную смесь растворяют в этилацетате и экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло, которое обрабатывают с помощью радиальной хроматографии на 2 мм хроматотроновой пластине, элюируя 7% /объем/объем/ метанолом в дихлорметане. Менее полярная фракция 110 мг (33%) по данным хроматографии является указанным в заглавии соединением и представляет твердое вещество белого цвета.
1H ЯМР /CDCl3/ δ : 8,60, 7,99, 7,85, 7,36, 6,1, 4,86, 4,12, 3,89, 3,72, 2,04.
Пример 22
(S)-N-[[3-[3-фтор-4-(4-гидроксиметил-1H-пиразол-1-ил)фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
10 мл метанола при 0oC обрабатывают, прикапывая 1 мл ацетилхлорида с последующим добавлением 2,0 г (17,84 ммоля) 4-пиразолкарбоновой кислоты с последующим нагреванием смеси при кипении с обратным холодильником в течение 18 часов. Полученный раствор охлаждают и концентрируют в вакууме до получения 2,6 г (90%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CD3OD/ δ : 8,25, 3,86.
4-(4-карбометокси-1H-пиразол-1-ил)-3-фтор-1-нитробензол:
Раствор 2,9 г (17,84 ммоля) полученного ранее соединения и 4,93 г (35,68 ммоля) карбоната калия в 85 мл ДМСО обрабатывают 2,83 г (17,84 ммоля) 3,4-дифторнитробензола. Полученный раствор нагревают при 90oC в течение 16 часов с последующим охлаждением, разбавляют хлороформом и экстрагируют водой (5 х). В результате сушки над сульфатом натрия и концентрирования в вакууме получают 4,17 г (88%) указанного в заглавии соединения в виде грязно-белого твердого вещества.
1H ЯМР /CDCl3/ δ : 8,65, 8,28, 3,91.
4-(4-карбометокси-1H-пиразол-1-ил)-3-фтор-1- (фенилметоксикарбониламино)бензол:
Раствор 4,17 г (15,7 ммоля) полученного ранее соединения в 200 мл ТГФ обрабатывают 1 г W-2 никеля Рэнея с последующим гидрированием в аппарате Парра при давлении водорода 45 пси (3,164 кг/см2) в течение 18 часов. Полученную смесь фильтруют через целит, а полученный фильтрат концентрируют в вакууме до получения грязно-белого твердого вещества, которое растворяют в 30 мл ТГФ и 5 мл ацетона и обрабатывают 34 мл насыщенного раствора бикарбоната натрия, а затем охлаждают до 0oC и добавляют 3,21 г (2,69 мл, 18,8 ммоля) бензилхлорформата. Полученный раствор перемешивают при 0oC в течение 1 часа, а затем оставляют нагреваться до комнатной температуры в течение 18 часов. Полученную смесь концентрируют в вакууме, образующийся водный слой экстрагируют этилацетатом (3 х). Органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения 3,1 г (53%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 8,41, 8,10, 7,79, 7,64, 7,40, 7,10, 6,88, 5,23, 3,88.
4-(4-гидроксиметил-1H-пиразол-1-ил)-3-фтор-1- (фенилметоксикарбониламино)бензол:
Раствор 3,0 г (8,13 ммоля) полученного ранее соединения в 20 мл безводного ТГФ обрабатывают 1,42 г (65,04 ммоля) боргидрида лития с последующим перемешиванием при комнатной температуре в течение 18 часов. Полученный раствор обрабатывает 5 мл насыщенного водного раствора бикарбоната натрия с последующим концентрированием в вакууме. Полученную смесь разбавляют этилацетатом и экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают остаток, который обрабатывают хроматографически на 150 г силикагеля (230-400 меш), элюируя 2% /объем/объем/ метанолом в дихлорметане. В результате получают 1,63 г (59%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения (электронный удар): Рассчитано для C18H16FN3O3: 341,1176. Найдено: 341,1180.
4-(4-[(тетрагидропиран-2-ил)оксиметил] -1H-пиразол-1-ил)-3-фтор-1- (фенилметоксикарбониламино)бензол:
Раствор 1,6 г (4,77 ммоля) полученного ранее соединения в 30 мл дихлорметана обрабатывают 400 мг (4,75 ммоля) дигидропирана и 10 мг р-толуолсульфоновой кислоты. Полученный раствор перемешивают при комнатной температуре в течение 18 часов с последующим экстрагированием 30 мл насыщенного раствора бикарбоната натрия и водой (30 мл). В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество, которое хроматографически обрабатывают на 85 г силикагеля (230-400 меш), элюируя 20% /объем/объем/ этилацетатом в гексане. В результате получают 2,0 г (99%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения (электронный удар): Рассчитано для C23H24FN3O4: 425,1751. Найдено: 425,1747.
(R)-[3-[3-фтор-4-(4-[(тетрагидропиран-2-ил)оксиметил] -1H-пиразол-1- ил)фенил]-2-оксо-5-оксазолидинил]метанол:
Раствор 1,4 г (3,36 ммоля) полученного ранее соединения в 10 мл ТГФ при -78oC обрабатывают 3,7 мл (1,0 М, 3,7 ммоля) литийбис(триметилсилил)амида в ТГФ с последующим перемешиванием при -78oC в течение 30 минут. Полученный раствор обрабатывают 533 мг (0,52 мл, 3,7 ммоля) (R)-(-)-глицидилбутирата с последующим перемешиванием при -78oC в течение 30 минут, нагреванием до 0oC в течение 30 минут и постепенным нагреванием до комнатной температуры в течение 18 часов. Полученный раствор обрабатывают 1 мл насыщенного раствора аммонийхлорида и разбавляют этилацетатом. Полученную смесь экстрагируют водой, сушат над сульфатом натрия и концентрируют в вакууме. Остаток обрабатывают хроматографически на 70 г силикагеля (230-400 меш), элюируя 2% /объем/объем/ метанолом в дихлорметане. В результате получают 910 мг (70%) указанного в заглавии соединения в виде твердого белого вещества. Масс-спектр высокого разрешения (электронный удар): Рассчитано для C19H22FN3O5: 391,1543. Найдено: 391,1542.
(R)-[3-[3-фтор-4-(4-[(тетрагидропиран-2-ил)оксиметил] -1H-пиразол-1- ил)фенил]-2-оксо-5-оксазолидинил]метансульфонилоксиметан:
Раствор 0,820 мг (2,1 ммоля) полученного ранее соединения и 318 мг (3,15 ммоля) триэтиламина в 25 мл дихлорметана при 0oC обрабатывают 360 мг (2,62 ммоля) метансульфонилхлорида. Полученный раствор перемешивают при 0oC в течение 1 часа. Этот раствор разбавляют дихлорметаном и экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают 986 мг (99%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,95, 7,92, 7,74, 7,25, 4,97, 4,75, 4,50, 4,20, 4,01, 3,93, 3,59, 3,12, 1,85, 1,74, 1,56.
(R)-[3-[3-фтор-4-(4-[(тетрагидропиран-2-ил)оксиметил] -1H-пиразол-1- ил)фенил]-2-оксо-5-оксазолидинил]метилазид:
Раствор 986 мг (2,1 ммоля) полученного ранее соединения и 683 мг (10,5 ммоля) азида натрия в 20 мл ДМФ нагревают при 60oC в течение 18 часов с последующим разбавлением этилацетатом и экстрагированием водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают 874 мг (990) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,98, 7,87, 7,74, 7,23, 4,82, 4,72, 4,50, 4,11, 3,90, 3,73, 3,61, 3,55, 1,81, 1,73, 1,58.
(S)-N-[[3-[3-фтор-4-(4-[(тетрагидропиран-2-ил)оксиметил] -1H- пиразол-1-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетатамид:
Раствор 874 мг (2,1 ммоля) полученного ранее соединения в 15 мл этилацетата обрабатывают 200 мг 10% палладий на угле с последующим гидрированием при давлении водорода 1 атм. в течение 1,5 часа. Полученный раствор фильтруют через целит, а фильтрат концентрируют в вакууме до получения твердого вещества коричневого цвета, которое растворяют в 3 мл пиридина и обрабатывают 428 г (4,2 ммоля) уксусного ангидрида с последующим перемешиванием при комнатной температуре в течение 18 часов. Раствор разбавляют этилацетатом и экстрагируют водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердый продукт, который обрабатывают хроматографически на 50 г силикагеля (230-400 меш), элюируя 1-2% /объем/объем/ метанолом в дихлорметане. В результате получают 626 мг (69%) указанного в заглавии соединения в виде твердого белого вещества. Масс-спектр высокого разрешения (электронный удар): Рассчитано для C21H25FN4O5: 432,1809. Найдено: 432,1811.
(S)-N-[[3-[3-фтор-4-(4-гидроксиметил-1H-пиразол-1-ил)фенил]-2- оксо-5-оксазолидинил]метил]ацетамид:
Раствор 300 мг (0,693 ммоля) полученного ранее соединения в 15 мл метанола обрабатывают 40 мг р-толуолсульфоновой кислоты с последующим перемешиванием при комнатной температуре в течение 18 часов. Полученную смесь концентрируют в вакууме, а остаток растворяют в этилацетате и экстрагируют насыщенным раствором бикарбоната натрия и водой. В результате сушки над сульфатом натрия и концентрированием в вакууме получают 140 мг (58%) указанного в заглавии соединения в виде твердого вещества белого цвета. Масс-спектр высокого разрешения (электронный удар): Рассчитано для C16H17FN4O4: 349,1312. Найдено: 349,1321.
Пример 23
(S)-N-[[3-[3-фтор-4-[3-(2-карбоэтоксивинил)-1H-пиррол-1-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетамид:
Раствор 805 мг (3,19 ммоля) диизопропилэтоксикарбонилметилфосфоната в 7 мл ТГФ при 0oC обрабатывают 3,19 мл (1М, 3,19 ммоля) раствора трет.-бутоксида калия в ТГФ с последующим нагреванием до комнатной температуры в течение 1 часа. Полученный раствор охлаждают до -78oC и обрабатывают через канюлю раствором 500 мг (1,45 ммоля) соединения примера 12 в 4 мл ТГФ. Полученный раствор перемешивают при -78oC в течение 30 минут, затем нагревают до комнатной температуры в течение 2 часов. Полученный раствор гасят, добавляя 1 мл насыщенного раствора аммонийхлорида с последующим разбавлением этилацетатом и экстрагированием водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество желтого цвета, которое обрабатывают хроматографически на 100 г силикагеля (230-400 меш), элюируя 1-2% /объем/объем/ метанолом в дихлорметане. В результате получают 497 мг (83%) указанного в заглавии соединения в виде твердого вещества светло-желтого цвета.
1H ЯМР /CDCl3/ δ : 7,65, 7,37, 7,27, 7,20, 6,97, 6,56, 6,17, 6,02, 4,82, 4,24, 4,08, 3,83, 3,70, 2,04, 1,33.
Пример 24
(S)-N-[[3-[3-фтор-4-[3-(2-карбоэтоксиэтил)-1H-пиррод-1-ил] фенил] -2- оксо-5-оксазолидинил]метил]ацетатамид:
Раствор 150 мг (0,36 ммоля) соединения примера 23 и 54 мг (0,54 ммоля) хлорида меди [1] в 15 мл смеси 1:1 метанол:ТГФ при 0oC обрабатывают 136 мг (3,6 ммоля) боргидрида натрия. Полученный раствор перемешивают при 0oC в течение 30 минут, а затем нагревают до комнатной температуры в течение 1 часа. Этот раствор обрабатывают 2 мл насыщенного раствора аммонийхлорида с последующим фильтрованием через целит. Полученный фильтрат концентрируют в вакууме, а остаток разбавляют этилацетатом и экстрагируют трижды водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают 150 мг (99%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,60, 7,34, 7,33, 6,91, 6,81, 6,21, 5,97, 4,81, 4,15, 4,08, 3,82, 3,72, 3,65, 2,87, 2,62, 2,04, 1,26.
Пример 25
(S)-N-[[3-[3-фтор-4-[3-(3-гидроксипропил)-1H-пиррол-1-ил]фенил]-2- оксо-5-оксазолидинил]метил]ацетатамид:
Раствор 275 мг (0,66 ммоля) соединения примера 24 в ТГФ (15 мл) обрабатывают 287 мг (13,2 ммоля) боргидрида лития с последующим перемешиванием при комнатной температуре в течение 18 часов. Полученный раствор обрабатывают 1 мл насыщенного раствора аммонийхлорида, разбавляют этилацетатом и экстрагируют водой (3 х). В результате сушки над сульфатом натрия и концентрирования в вакууме получают масло, которое обрабатывают хроматографически на 20 г силикагеля (230-400 меш), элюируя 2-4% /объем/объем/ метанолом в дихлорметане. В результате получают 96 мг (39%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,74, 7,66, 7,38, 7,27, 7,18, 6,95, 6,61, 5,98, 4,82, 4,37, 4,27, 4,09, 3,83, 3,68, 2,88, 2,82, 2,62, 2,04, 1,79.
Пример 26
(S)-N-[[3-[3-фтор-4-[3-(3-метансульфониламинопропил)-1H-пиррол-1- ил] фенил]-2-оксо-5-оксазолидинил]метил]ацетатамид:
Раствор 100 мг (0,266 ммоля) соединения примера 25 (предварительно высушенного при 60oC и давлении 0,1 мм рт. ст. в течение 18 часов до использования) и 88 мг (120 мкл, 0,88 ммоля) триэтиламина в 2 мл дихлорметана при 0oC обрабатывают 85 мг (58 мкл, 0,73 ммоля) метансульфонилхлорида с последующим перемешиванием при 0oC в течение 30 минут. Полученный раствор разбавляют дихлорметаном и экстрагируют водой дважды, и насыщенным раствором бикарбоната натрия. В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердое вещество, которое растворяют в 2 мл ДМФ и обрабатывают через канюлю при 0oC раствором 38 мг (0,4 ммоля) метансульфонамида в 2 мл ДМФ, который был предварительно обработан 10 мг (17 мг 60% в масле, 0,43 ммоля) гидрида натрия. Затем полученный раствор нагревают при 60oC в течение 18 часов. Полученный раствор охлаждают, и ДМФ удаляют в вакууме, а остаток растворяют в этилацетате и экстрагируют дважды водой. В результате сушки над сульфатом натрия и концентрирования в вакууме получают твердый продукт, который обрабатывают с помощью радиальной хроматографии на 2 мм хроматотронной пластине, элюируя 1-4% /объем/объем/ метанолом в дихлорметане. В результате получают 31 мг (26%) указанного в заглавии соединения в виде твердого вещества белого цвета.
1H ЯМР /CDCl3/ δ : 7,61, 7,56, 7,35, 7,23, 6,92, 6,81, 6,19, 6,06, 4,81, 4,29, 4,07, 3,82, 3,22, 3,67, 2,95, 2,62, 2,04, 1,90.
Примеры получения соединений формулы I, где Q представляет viii и ix.
Пример 27
(S)-N-[[3-[3-Фтор-4-(2H-1,2,3,4-тетразол-2-ил)фенил]-2-оксо-5- оксазолидинил]метил]ацетамид.
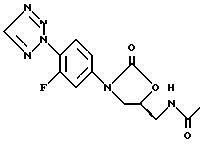
Стадия 1. Раствор 15,83 г 1H-тетразола, 7,19 г 3,4-дифтор-нитробензола и 22,85 г триэтиламина в 90 мл ацетонитрила нагревают при кипении с обратным холодильником в течение 18 часов с последующим охлаждением и концентрированием в вакууме. Остаток растворяют в 250 мл этилацетата и экстрагируют водой (2х100 мл) и насыщенным раствором NaCl (100 мл). В результате сушки (Na2SO4) и концентрирования в вакууме получают 7,1 г черного масла. Его обрабатывают хроматографически на 360 г силикагеля (230-400 меш), элюируя 3% (об/об) смесью ацетона в дихлорметане. В результате получают 0,93 г менее полярного 2H-региоизомерного аддукта в виде желтого твердого вещества и 0,38 г более полярного 1H-региоизомерного аддукта в виде желто-коричневого твердого вещества.
2H-региоизомер:
1H ЯМР (ДМСО) δ : 9,71 (с, 1H), 8,57 (дд, J = 9,6, 1,6 Гц, 1H), 8,32 (om, 2H);
1H-региоизомер:
1H ЯМР (ДМСО) δ : 10,04 (с, 1H), 8,57 (дд, J = 10,4, 2,0 Гц, 1H), 8,34 (д, J = 8,9 Гц, 1H), 8,23 (т, J = 8,1 Гц, 1H).
Стадия 2. Раствор 500 мг 2H-региоизомера, полученного на стадии 1, в 50 мл 1:1 смеси метанола-этилацетата обрабатывают 500 мг никеля Рэнея W-2 с последующей гидрогенизацией при давлении водорода 45 пси (3,164 кг/см2) в течение 18 часов в аппарате Парра. Смесь фильтруют через целит, промывают фильтровальную лепешку этилацетатом. Фильтрат концентрируют в вакууме с получением белого твердого вещества, которое растворяют в 20 мл ацетона, обрабатывают 5 мл насыщенного NaHCO3 с последующим охлаждением до 0oC. Смесь обрабатывают 448 мг (0,38 мл, 2,63 ммол) бензилхлорформата, затем перемешивают при 0oC в течение 1 часа и нагревают до температуры окружающей среды в течение 18 часов. Смесь разбавляют 100 мл этилацетата, экстрагируют водой (2х50 мл) и насыщенным раствором NaCl (50 мл). В результате сушки (Na2SO4) и концентрирования в вакууме получают рыжевато-коричневое твердое вещество, которое перекристаллизовывают из горячей смеси хлороформ-гексан с получением 633 мг (85%) карбамата в виде светло-бежевых игл, т.пл. 120-122oC.
1H ЯМР (CDCl3) δ : 8,70 (с, 1H), 7,75 (т, J = 8,7 Гц, 1H), 7,70 (дд, J = 13,7, 1H), 7,40 (om, 5H), 7,21 (дк, J = 8,8 Гц, 1H), 6,94 (шир.с, 1H), 5,24 (с, 2H).
Масс-спектр высокого разрешения (FAB): рассчитано для C15H12FN5O2+H1: 314,1053. Найдено: 314,1060.
Стадия 3. Раствор 554 мг карбамата, полученного на стадии 2, в 25 мл ТГФ при -78oC обрабатывают 1,94 мл (1,0 М, 1,94 ммол) бис(триметилсилил)амида лития в ТГФ, затем перемешивают при -78oC 20 минут. Раствор затем обрабатывают 280 мг (275 мл, 1,94 ммол) (R)-(-)-глицидилбутирата с последующим нагреванием до 0oC и постепенным нагреванием до температуры окружающей среды в течение 18 часов. Смесь затем обрабатывают 1 мл насыщенного раствора хлорида аммония и разбавляют 100 мл дихлорметана. Смесь экстрагируют водой (2х50 мл) и насыщенным раствором хлорида аммония (50 мл). В результате сушки (Na2SO4) и концентрирования в вакууме получают белое твердое вещество, которое фильтруют через sep-pak® и обрабатывают с помощью радиальной хроматографии на 4 мм хроматотроновой пластине, элюируя 5% (об/об) смесью метанола в дихлорметане с получением 275 мг гидроксиметилоксазолидинона в виде не совсем белого твердого вещества. Т. пл. 145,5-147,5oC.
1H ЯМР (DMCO) 9,66 (с, 1H), 7,96 (т, J = 8,7 Гц, 1H), 7,88 (дд, J = 13,5, 2,4 Гц, 1H), 7,64 (дд, J = 8,9, 1,7 Гц), 5,25 (т, J = 5,6, 1H), 5,78 (м, 1H), 4,17 (т, J = 9,1 Гц, 1H), 3,92 (дд, J = 8,9, 6,0 Гц, 1H), 3,71 (ддд, J = 12,4, 5,5, 3,3 Гц, 1H), 3,59 (ддд, J = 12,3, 5,6, 4,0 Гц, 1H);
Анализ: Рассчитано для C11H10FN5O3: C 47,32; H 3,61; N 25,08. Найдено: C 47,48; H 3,70; N 24,73.
Стадия 4. Суспензию 180 мг гидроксиметилоксазолидинона, полученного на стадии 3, в 4 мл дихлорметана и 0,5 мл триэтиламина при 0oC обрабатывают 171 мг нозилхлорида с последующим перемешиванием при 0oC в течение 3 часов и нагреванием до температуры окружающей среды. Смесь разбавляют 30 мл дихлорметана и экстрагируют водой (2х20 мл) и 0,5 N раствором HCl. В результате сушки (Na2SO4) и концентрирования получают 230 мг соответствующего нозилата. Этот материал растворяют в 6 мл ДМФА и обрабатывают 2 г азида натрия с последующим перемешиванием при комнатной температуре в течение 18 часов. Смесь разбавляют 75 мл этилацетата, затем экстрагируют водой (5х50 мл) и насыщенным раствором NaCl (50 мл). В результате сушки (Na2SO4) и концентрирования в вакууме получают 187 мг азида в виде не совсем белого твердого вещества, достаточно чистого для дальнейшего использования.
1H ЯМР (CDCl3) 8,71 (с, 1H), 7,85 (т, J = 8,8 Гц, 1H), 7,79 (дд, J = 12,6, 2,4 Гц, 1H), 7,43 (дт, J = 8,8, 1,1 Гц, 1H), 4,88 (м, 1H), 4,16 (т, J = 8,8 Гц, 1H), 3,94 (дд, J = 8,9, 6,2 Гц, 1H), 3,79 (дд, J = 13,3, 4,2 Гц, 1H), 3,64 (дд, J = 13,4, 4,2 Гц, 1H);
Стадия 5. Раствор 187 мг азида, полученного на стадии 4, в 60 мл 2:1 раствора этилацетат-метанол обрабатывают 300 мг никеля Рэнея W-2, затем подвергают гидрогенизации при давлении водорода 45 пси (3,164 кг/см2) в аппарате Парра в течение 18 часов. Смесь фильтруют через целит, фильтровальную лепешку промывают метанолом. Фильтрат концентрируют в вакууме и остаток растворяют в 15 мл пиридина, обрабатывают 6 мл уксусного ангидрида с последующим перемешиванием при температуре окружающей среды в течение 24 часов. Раствор концентрируют при высоком вакууме (0,2 мм рт.ст.) и остаток растворяют в 50 мл дихлорметана, и экстрагируют водой (2х25 мл). В результате сушки (Na2SO4) и концентрирования в вакууме получают твердое вещество, которое фильтруют через Sep-Pak® и подвергают радиальной хроматографии на 4-мм хроматотроновой пластине, элюируя 3% (об/об) раствором метанола в дихлорметане. В результате получают 109 мг указанного в заголовке соединения в виде не совсем белого твердого вещества, т. пл. 185-186oC (с разл.).
1H ЯМР (DMCO) 9,65 (с, 1H), 8,25 (м, 1H), 7,96 (т, J = 8,6 Гц, 1H), 7,85 (д, J = 13,4 Гц, 1H), 7,60 (д, J = 8,5 Гц, 1H), 4,79 (м, 1H), 4,20 (т, J = 8,9, 1H), 3,82 (дд, J = 8,8, 6,4 Гц, 1H), 3,44 (м, 1H), 1,83 (с, 3H);
Анализ: Рассчитано для C13H13FN6O3 1/4H2O: C 48,08; H 4,19; N 25,88; Найдено: C 48,22; H 3,94; N 25,75;
Пример 28
(S)-N-[[3-[3-Фтор-(1H-1,2,3,4-тетразол-1-ил)фенил] -2-оксо-5- оксазолидинил]метил]ацетамид.
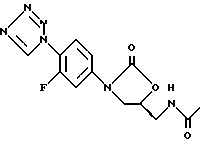
Этот оксазолидинон может быть получен из 1H-региоизомера, полученного на стадии 1 (выше) с последующими процедурами, описанными в стадиях 2-5. Однако, в действительности соединение было получено альтернативным методом (см. табл. 3 в конце описания).
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИМИКРОБНЫЕ ФЕНИЛОКСАЗОЛИДИНОНЫ | 1995 |
|
RU2134692C1 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ИМЕЮЩИЕ С-С-СВЯЗЬ С 4-8-ЧЛЕННЫМИ ГЕТЕРОЦИКЛИЧЕСКИМИ КОЛЬЦАМИ | 1996 |
|
RU2175324C2 |
| БИЦИКЛИЧЕСКИЕ ОКСАЗИН- ИЛИ ТИАЗИН-ОКСАЗОЛИДИНОНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ЛЕЧЕНИЯ МИКРОБНЫХ ИНФЕКЦИЙ | 1995 |
|
RU2128660C1 |
| ОКСАЗОЛИДИНОНОВЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1998 |
|
RU2215740C2 |
| 2-АМИНОИНДАНЫ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ЛИГАНДОВ ДОПАМИНА Д3 | 1996 |
|
RU2161601C2 |
| 14-ЗАМЕЩЕННЫЕ МАРКФОРТИНЫ | 1994 |
|
RU2131877C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ДЕГЕНЕРАЦИЕЙ СОЕДИНИТЕЛЬНОЙ ТКАНИ | 1997 |
|
RU2168497C2 |
| ТРОПОНЗАМЕЩЕННЫЙ ФЕНИЛОКСАЗОЛИДИНОН ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ ГИДРАТЫ | 1993 |
|
RU2119484C1 |
| ПРОТИВОПАРАЗИТАРНЫЕ МАРКФОРТИНЫ И ПАРАГЕРКВАМИДЫ | 1996 |
|
RU2162090C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-4-ФЕНИЛ-4-ОКСОМАСЛЯНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2139850C1 |
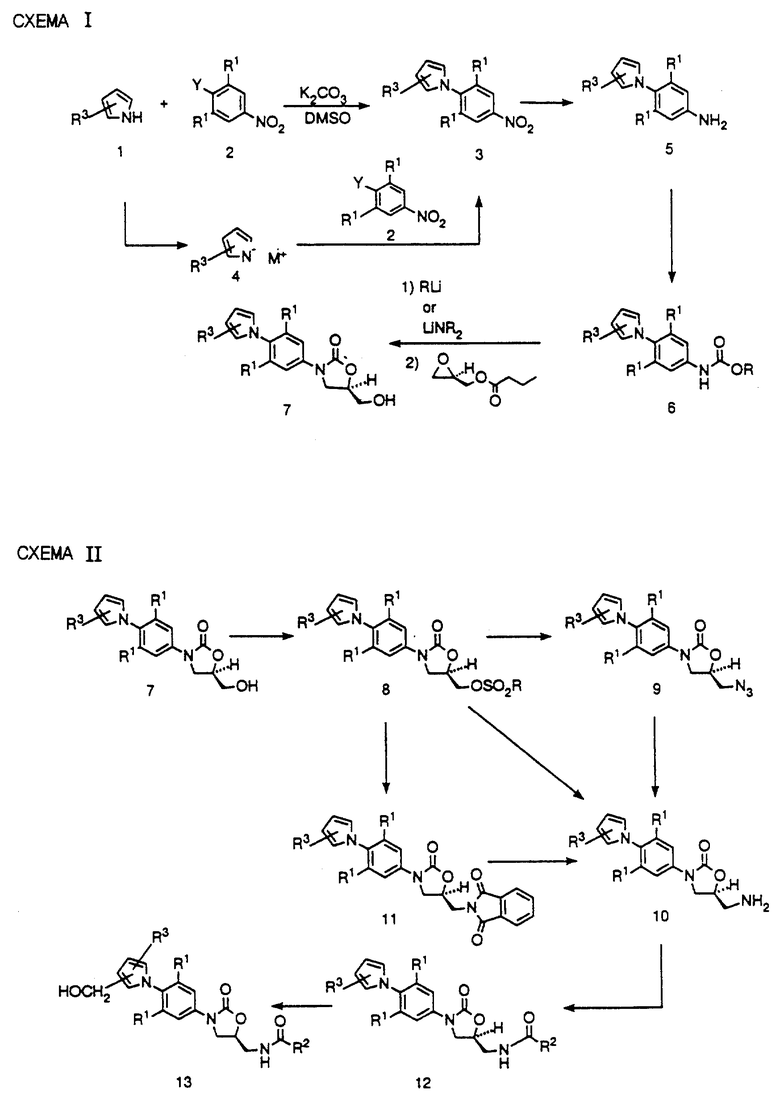
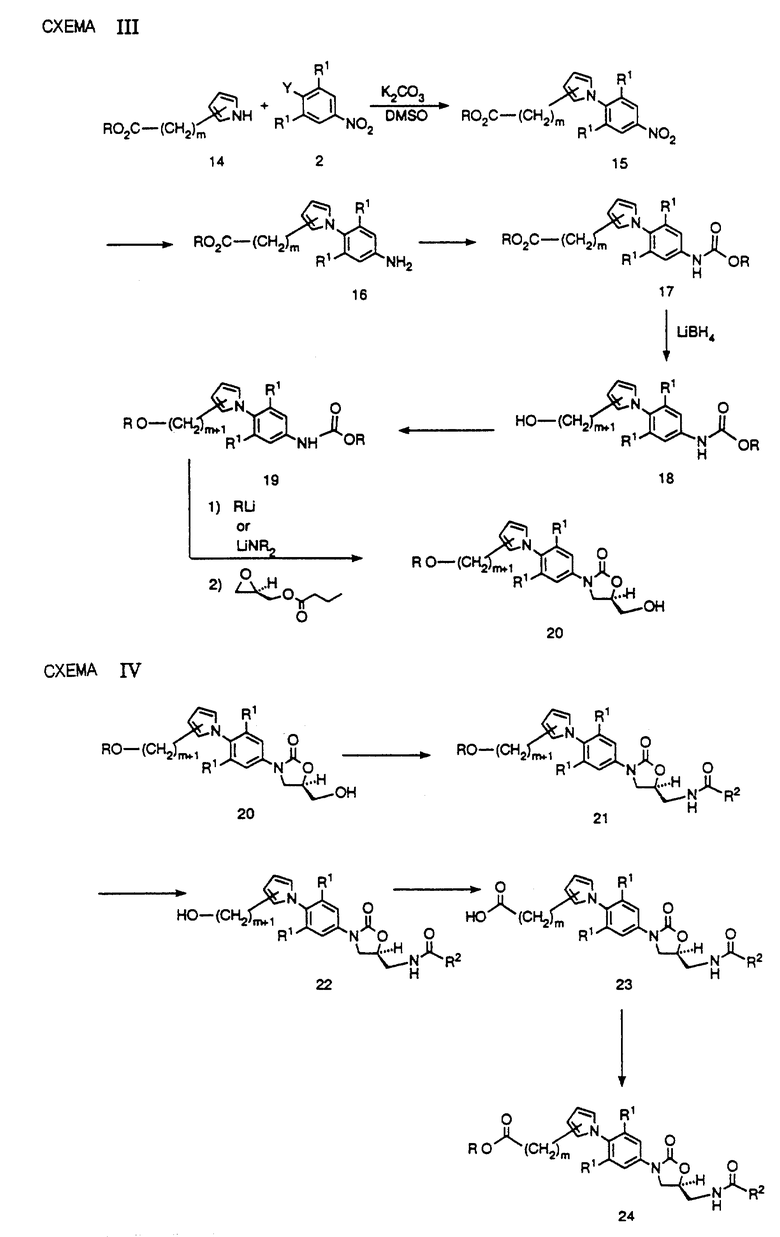
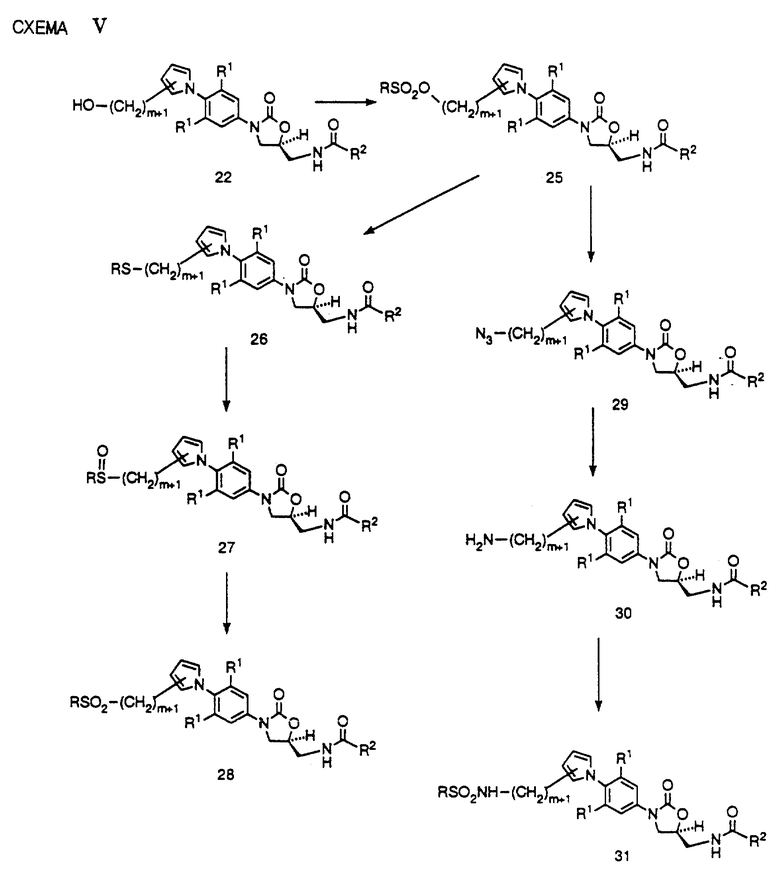
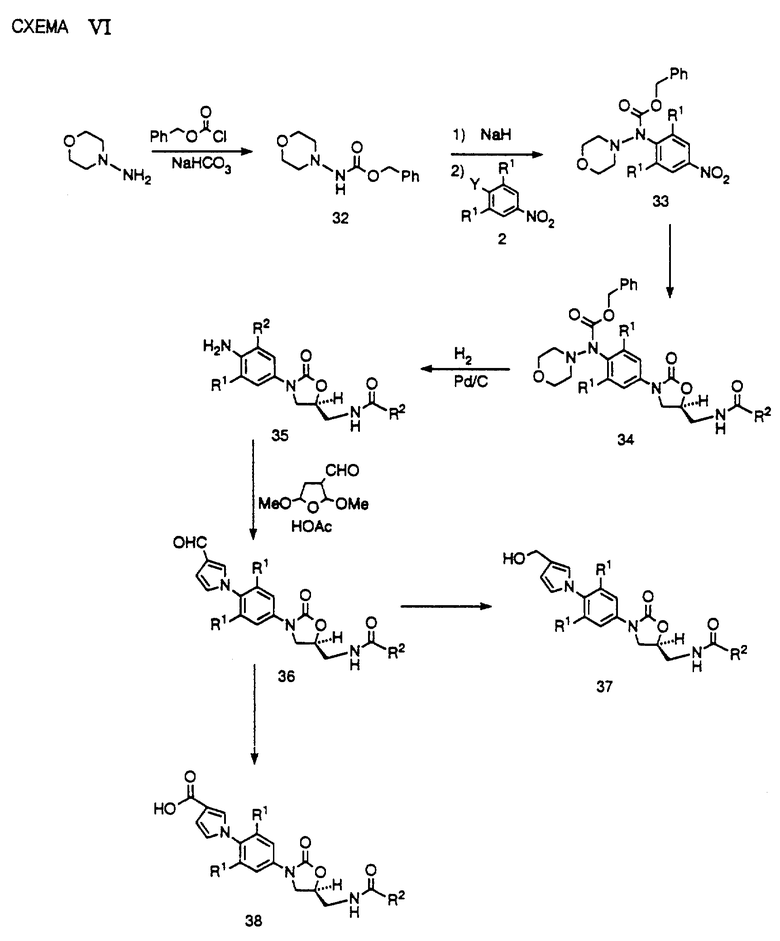
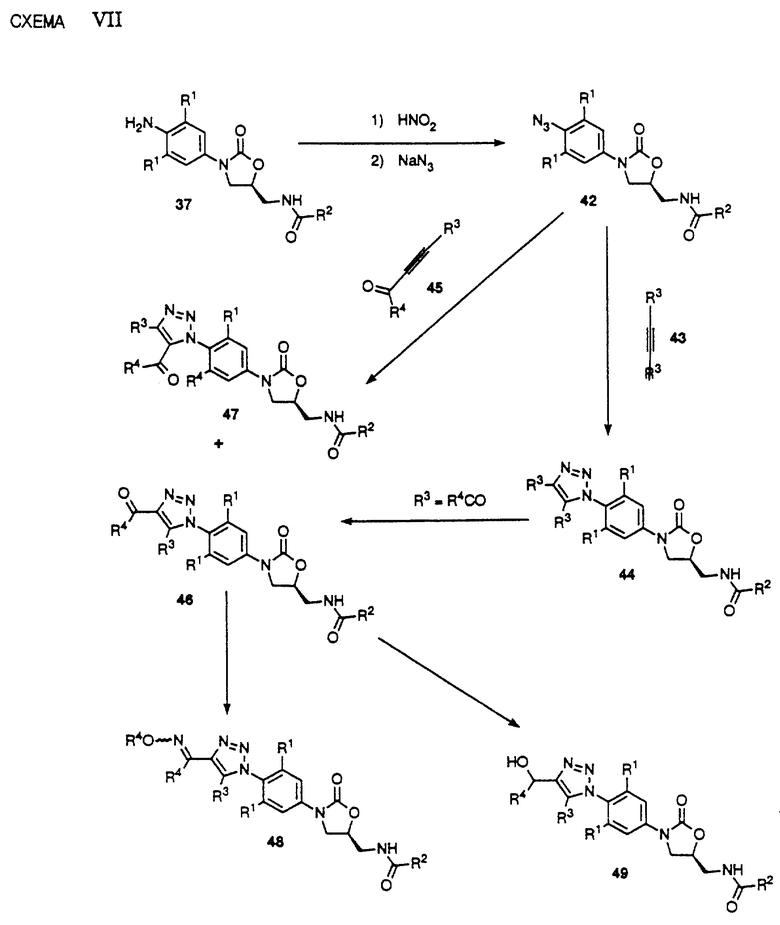
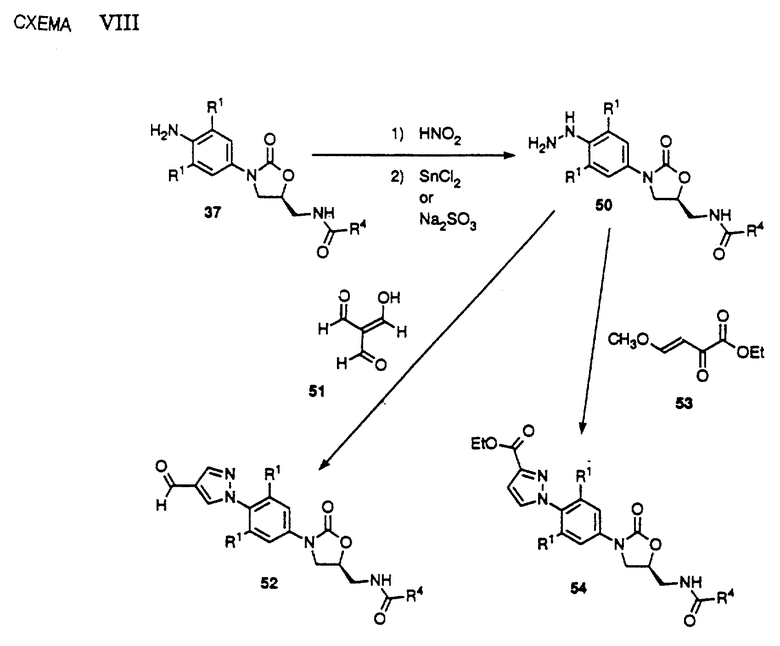
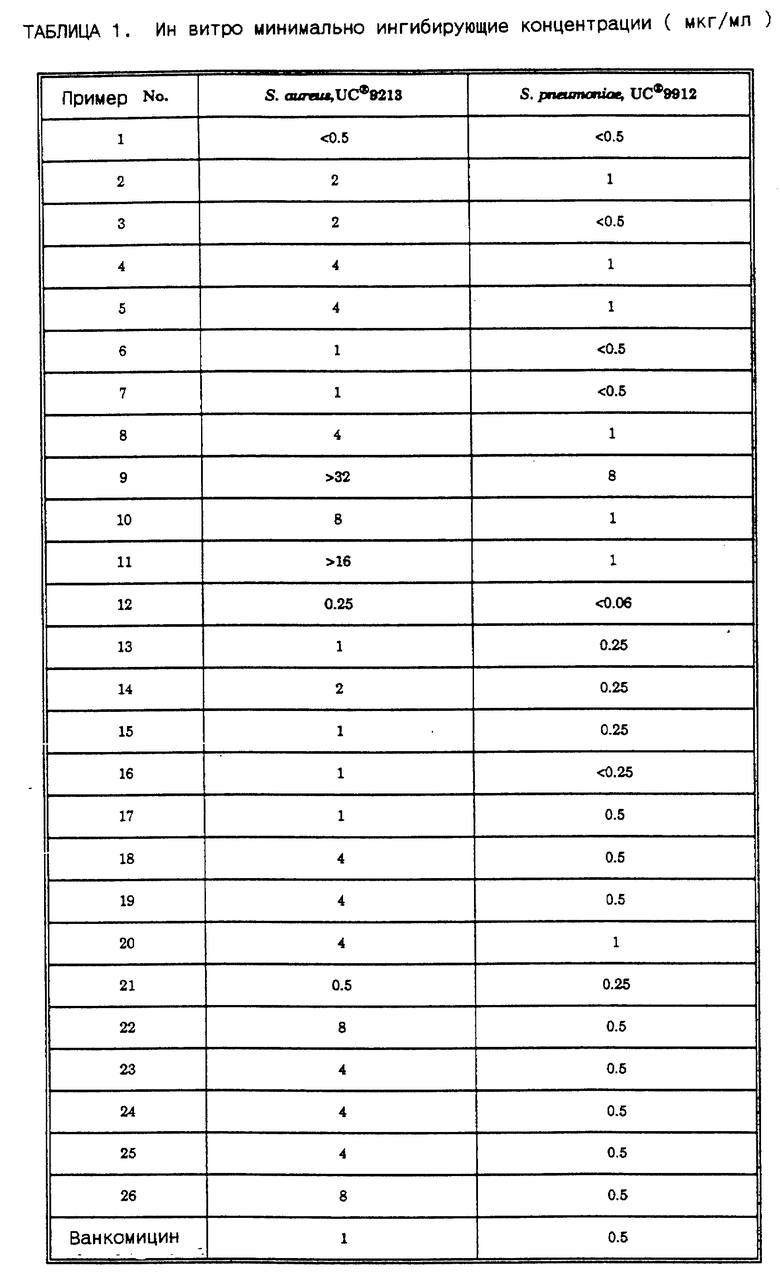


Описываются новые соединения - фенилоксазолидиноны, замещенные в кольце гетероароматическими кольцами формулы (Ia), или его фармацевтические приемлемые соли, где Q представляет гетероароматическое 5-членное кольцо, связанное с ароматическим кольцом 1 по атому азота, структурной формулы: i, ii, iii, iv, v, vi, vii, viii или ix, или в другом варианте Q может представлять конденсированное с бензолом гетероароматическое 5-членное кольцо, связанное с ароматическим кольцом 1 по атому азота, структурной формулы X, R1 независимо представляет Н, F или Cl; R2 независимо представляет C1-C8-алкил, необязательно замещенный одним или более атомами F или Cl; R3 каждый независимо выбирают из: (a) H, (b) OR4, (c) SR4, (d) NHSO2R4, (e) CO2R4, (f) COR4, (g) насыщенного или ненасыщенного C1-C8-алкила, разветвленного или неразветвленного, необязательно замещенного одним или более заместителем, выбранным из (e), (j) -C(R5)=NR6, где R5 представляет C1-C6-алкил, а R6 представляет OH или OC1-C6-алкил; R4 представляет: (a) H, (b) C1-C6 разветвленную или неразветвленную алкильную цепочку. Эти соединения являются полезными антимикробными агентами, эффективными против ряда патогенов человека и животных, включая грамположительные аэробные бактерии, такие как множественно-устойчивые стафилококки, стрептококки и энтерококки, а также такие анаэробные организмы, как Bacteroides spp. и Clostridia spp. виды и acid-fast организмы, такие как Mycobacterium tuberculosis, Mycobacterium avium и Mycobacterium spp. 10 з.п.ф-лы, 8 ил., 3 табл.
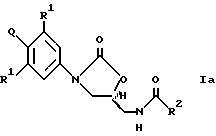
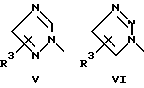

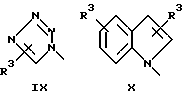
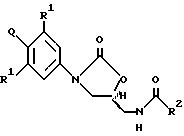
или его фармацевтические приемлемые соли,
где Q представляет гетероароматическое 5-членное кольцо, связанное с ароматическим кольцом 1 по атому азота, структурной формулы: i, ii, iii, iv, v, vi, vii, viii, или ix: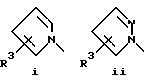
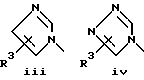
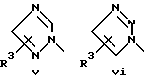
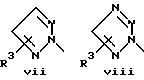
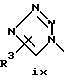
или в другом варианте Q может представлять конденсированное с бензолом гетероароматическое 5-членное кольцо, связанное с ароматическим кольцом 1 по атому азота, структурной формулы х: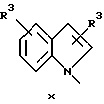
R1 независимо представляет H, F или Cl;
R2 независимо представляет С1 - С8-алкил, необязательно замещенный одним или более атомами F или Cl;
R3 каждый независимо выбирают из: (а) Н, (b) ОR4, (с) SR4, (d) NНSO2R4, (е) СО2R4, (f) СОR4, (g) насыщенного или ненасыщенного С1 - С8-алкила, разветвленного или неразветвленного, необязательно замещенного одним или более заместителем, выбранным из (е), (j) -С(R5)=NR6, где R5 представляет С1-С6-алкил, а R6 представляет ОН или ОС1-С6-алкил;
R4 представляет: (а) Н, (b) С1-С6 разветвленную или неразветвленную алкильную цепочку.
| УСТРОЙСТВО для ПРОДОЛЬНОЙ РЕЗКИ полосового ПОЛИМЕРНОГО МАТЕРИАЛАi:.-K;iu;^:j-M-.xH?!it>&.iiMj| | 0 |
|
SU352781A1 |
| WO 9309103 А, 13.05.1993 | |||
| -Бензимидазолил-этиленовые производные 2,5-дифенилоксазола,как люминофоры | 1977 |
|
SU597680A1 |

