Изобретение относится к новому классу сульфонамидов, которые являются ингибиторами аспартил-протеазы. В одном варианте настоящее изобретение относится к новому классу ингибиторов HIV аспартил-протеазы, отличающихся специфическими структурными и физиохимическими характеристиками. Настоящее изобретение относится также к фармацевтическим композициям, содержащим эти соединения. Соединения и фармацевтические композиции настоящего изобретения особенно хорошо подходят для ингибирования HIV-1 и HIV-2 протеазной активности, и, соответственно, могут быть с выгодой использованы в качестве вирусных агентов против HIV-1 и HIV-2 вирусов. Настоящее изобретение относится также к способам ингибирования активности HIV аспаратил-протеазы за счет использования соединений настоящего изобретения и способам скринирования соединений по поводу их анти-HIV активности.
Вирус иммунодефицита человека ("HIV") является агентом, вызывающим синдром приобретенного иммунодефицита ("AIDS")-заболевания, характеризующегося разрушением иммунной системы, особенно CD4+ T-клеток, сопровождающегося подверженностью другим инфекциям - и его предшественник AIDS - связанный комплекс ("ARC") - синдром, характеризующийся такими симптомами как персистентная общая лимфаденопатия, лихорадка и потеря веса.
Как и в случае некоторых других ретровирусов, HIV кодирует продуцирование протеазы, которая осуществляет посттрансляционное расщепление предшественников полипептидов в процессе, необходимом для образования инфекционных вирионов (S. Crawford et al., "A Deletion Mutation in 5' Pastof the pc1 Jene of Moloney Murine Leukemia Virus Blocks Proteolytic Processing of the gag and po1 Polyproteins", J. Virol., 53, p.899 (1985)). Эти генные продукты включают po1, который кодирует вирион РНК-зависимую ДНК полимеразу (обратную транскриптазу), эндонуклеазу, HIV протеазу и gag, который кодирует кор-протеины вирионов (H. Toh et al., "Close Structural Resemblance Between Putative Polymerase of Diosophila Transposable Genetic Element 17,6 и pol генный продукт Moloney Murine Leukemia Virus", EMBO J., 4, p.1267 (1985), L. H. Pearl et al., "A Structural Model for the Retroviral Protease", Nature, pp 329-351 (1987), M.D.Power et al., "Nucleotide Seguence of SRV-1, Type D, Simian Acguired Immune Deficiency Syndrome Retrovirus", Science, 231, p.1567 (1986)).
Ряд синтетических антивирусных агентов был сконструирован для того, чтобы отметить различные стадии в цикле репликации HIV. Эти агенты включают соединения, которые блокируют вирусное связывание CD4+ T-лимфоцитов (например, растворимых CD4), и соединений, которые мешают вирусной репликации за счет ингибирования вирусной обратной транскриптазы (например, диданозин и зидовудин (AZT)) и ингибируют интеграцию вирусной ДНК в клеточную ДНК (M. S. Hirsh and P.J.D'Agulia, "Therapy for Human Immundeficiency Virus Infection", N. Eng, J.Med., 328, p. 1686 (1993)). Однако, такие агенты, которые направлены, главным образом, на ранние стадии вирусной репликации, не предотвращают продуцирование инфекционных вирионов в хронически инфицированных клетках. Более того, введение некоторых таких агентов в эффективных количествах приводит к клеточной токсичности и нежелательным побочным эффектам, таким как анемия и подавление костного мозга.
Позднее все внимание по конструированию антивирусных лекарств было сосредоточено на создании соединений, которые ингибируют образование инфекционных вирионов за счет нарушения процессинга вирусных полипротеиновых предшественников. Процессинг этих предшественников протеинов требует действия вирус-кодирующих протеаз, которые существенны для репликации (Kohl, N.b. et al. , "Active HIV Protease is Reguired for Viral Infectivity", Proc. Natl. Acad. Sci. USA., 85, p.4686 (1988). Антивирусный потенциал HIV протеазного ингибирования был продемонстрирован с использованием пептидных ингибиторов. Такие пептидные соединения, однако, обычно являются крупными и сложными молекулами, которые имеют тенденцию демонстрировать слабую биодоступность и обычно не годятся для орального приема. См. также международную патентную заявку WO-A-92 08701 и J.R.Huff, Journal of Medicinal Chemistry, 34/8/ pp 2305-14 (1991). Соответственно, все еще существует необходимость в соединениях, которые могут эффективно ингибировать действие вирусных протеаз, для использования в качестве агентов для предотвращения и лечения хронических и острых вирусных инфекций.
В настоящем изобретении предложен новый класс соединений и их фармацевтически приемлемых производных, которые полезны в качестве ингибиторов аспартил-протеаз, в частности, HIV аспартил-протеаз. Эти соединения можно использовать отдельно или в сочетании с другими терапевтическими или профилактическими агентами, такими как антивирусные агенты, антибиотики, иммуномодуляторы или вакцины, для лечения или профилактики вирусной инфекции.
В соответствии с предпочтительным вариантом соединения настоящего изобретения способны ингибировать HIV вирусную репликацию в CD4+ T-клетках человека. Эти соединения пригодны в качестве терапевтических и профилактических агентов для лечения или предотвращения инфицирования HIV-1 и родственными вирусами, что может привести к асимптоматической инфекции, AIDS - связанным комплексом ("ARC"), синдрому приобретенного иммунодефицита ("AIDS"), или аналогичным заболеваниям иммунной системы.
Основной целью настоящего изобретения является создание нового класса сульфонамидов, которые являются ингибиторами аспартил-протеазы, и особенно ингибиторами HIV аспартил-протеазы. Этот новый класс сульфонамидов представлен формулой I: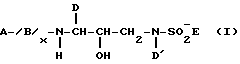
A выбирают из группы, состоящей из H; Het; -R1-Het; -R1-C1-C6 алкила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из гидрокси, C1-C4 алкокси, Het. -O-, Het, -NR2-CO-N/R2//R2/ и -CO-N/R2//R2; и -R1-C2-C6алкенила, который может необязательно быть замещен одной или более из групп, выбранных из группы, состоящей из гидрокси, C1-C2алкокси; Het; -O-Het, -NR2-CO-N/R2//R2/ и -CO-N/R2//R2;
каждый R1 независимо выбирают из группы, состоящей из -C/O/-, -S/O/2, -C/O/-C/O/-, -O-C/O/-, -O-S/O/2- -NR2-S/O/2-, -NR2-C/O/- и NR2/O/-C/O/; каждый Het независимо выбирают из группы, состоящей из C3-C7-циклоалкила, C5-C7циклоалкенила, C6-C10-арила, и 5-7 членного насыщенного или ненасыщенного гетероцикла, содержащего один или более из гетероатомов, выбранных из N, N/R2/, О, S и S/O/n, где указанный гетероцикл может быть необязательно конденсирован с ароматическим кольцом и где любой член указанного Het может быть необязательно замещен одним или более из заместителей, выбранных из группы, состоящей из оксо, -OR2, -R2, N/R2//R2/, -R2-OH, -CN, -CO2R2, -C/O/-N/R2//R2/, -S/O/2-N/R2//R2/, -N/R2/-C/O/-R2, -C/O/-R2, -S/O/n-R2,
-OCF3, -S/O/n-Ar, метилендиокси, -N/R2/-S/O/2/R2/, галоида, -CF3, NO2, Ar и -O-Ar,
каждый R2 независимо выбирают из группы, состоящей из H и C1-C3-алкила, необязательно замещенного Ar,
B, если присутствует, представляет -N/R2/-C/R3//R3/-C/O/-,
x представляет 0 и 1,
каждый R3 независимо выбирают из группы, состоящей из H, Het, C1-C6алкила, C2-C6-алкенила, C3-C6циклоалкила и C5-C6-циклоалкенила, причем любой из членов указанного R3, за исключением H, может быть необязательно замещен одним или более из заместителей, выбранных из группы, состоящей из -OR2, -C/O/-NH-R2, -S/O/n-N/R2//R2/, Het, -CN, -SR2, -CO2R2, NR2-C/O/-R2,
каждый n независимо представляет 1 или 2,
D и D' независимо выбирают из группы, состоящей из Ar, C1-C4-алкила, который может быть необязательно замещен одной или более из групп, выбранных из C3-C6циклоалкила, -OR2, -R3, O-Ar и Ar, C2-C4алкенила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из C3-C6циклоалкила, -OR2, R3, -O-Ar и Ar, C3-C6циклоалкила, который может быть необязательно замещен или конденсирован с Ar, и C5-C6-циклоалкенила, который может быть необязательно замещен или конденсирован с Ar,
каждый Ar независимо выбирают из группы, состоящей из фенила, 3-6 членного карбоциклического кольца и 5-6 членного гетероциклического кольца, содержащего один или более из гетероатомов, выбранных из O, N, S, S/O/n и N/R2/, причем указанное карбоциклическое или гетероциклическое кольцо может быть насыщенным или ненасыщенным, и необязательно замещенным одной или более из групп, выбранных из группы, состоящей из оксо, -OR2, -R2, -N/R2//R2/, -N/R2/-C/O/-R2, -R2-OH, -CN, -CO2R2, -C/O/-N/R2//R2/, галоида и -CF3,
E выбирают из группы, состоящей из Het, O-Het, Het-Het, -O/R3, -NR2R3, C1-C6алкила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het; C2-C6алкенила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het; C3-C6насыщенного карбоцикла, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het, и C5-C6 ненасыщенного карбоцикла, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het, и
каждый R4 независимо выбирают из группы, состоящей из -OR2, -C/O/-NHR2, -S/O/2-NHR2, галоида -NR2-C/O/-R2 и -CN.
Целью настоящего изобретения является также создание фармацевтических композиций, содержащих сульфонамиды формулы I, и способы их использования в качестве ингибиторов HIV аспартил-протеазы.
Еще одной целью настоящего изобретения является создание нового класса HIV аспартил-протеазу ингибирующих соединений, отличающихся новым сочетанием структурных и физико-химических характеристик:
(1) первый и второй акцепторный фрагмент водородной связи, по крайней мере, один из которых более сильно поляризуем, нежели карбонил, причем указанные фрагменты одинаковы или различны, и способны образовывать водородную связь с атомами водорода "щитковой" молекулы воды HIV аспартилпротеазы, если это соединение с ней связано,
(2) существенно гидрофобные фрагменты, которые связаны с P1 и P1' связывающими "карманами" указанной HIV аспартилпротеазы, если это соединение с ней связано,
(3) третий фрагмент, осуществляющий водородную связь, который может быть либо донором, либо акцептором водородной связи, способный одновременно образовывать водородную связь с Asp25 и Asp25' указанной HIV аспартил-протеазы, если это соединение с ней связано,
(4) дополнительно занятый объем пространства, величиной, по крайней мере 100 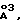 если это соединение связано с активным сайтом указанной HIV аспартил-протеазы, причем указанное пространство перекрывается с объемом пространства, которое должно быть заполнено нативным субстратом указанной HIV аспартилпротеазы или ее негидролизуемой изостерой (isostere);
если это соединение связано с активным сайтом указанной HIV аспартил-протеазы, причем указанное пространство перекрывается с объемом пространства, которое должно быть заполнено нативным субстратом указанной HIV аспартилпротеазы или ее негидролизуемой изостерой (isostere);
(5) энергия деформации связывания соединения с указанной HIV аспартил-протеазой не более 10 ккал/моль, и
(6) нейтральный или благоприятный вклад энтальпии за счет суммы всех электростатических взаимодействий между этим соединением и протеазой, если это соединение связано с указанной HIV аспартил-протеазой.
Целью настоящего изобретения является создание фармацевтических композиций, содержащих соединения, обладающие указанными характеристиками, и способов их использования в качестве ингибиторов HIV аспартил-протеазы.
Еще одной целью настоящего изобретения является создание способа идентификации, конструирования или предсказания ингибиторов HIV аспартил-протеазы, включающий следующие стадии:
(а) отбора соединений-кандидатов с определенной химической структурой, содержащих первый и второй акцепторный фрагмент водородной связи, по крайней мере, один из которых более сильно поляризуется, нежели карбонил, причем указанные фрагменты одинаковы или различны, третий фрагмент, образующий водородную связь, который может быть либо донором, либо акцептором водородной связи, и по крайней мере, два существенно гидрофобных фрагмента,
(b) определения низкоэнергетической конформации для связывания указанного соединения с активным сайтом HIV аспартил-протеазы,
(c) оценки способности указанных первого и второго акцепторных фрагментов водородной связи к образованию водородных связей с "щитовыми" молекулами воды указанной HIV аспартил-протеазы, если указанное соединение связано с ней в указанной конформации,
(d) оценки способности указанных существенно гидрофобных фрагментов к ассоциации с P1 и P1' связывающими карманами указанной HIV аспартил протеазы, если указанное соединение с ней связано в указанной конформации,
(e) оценки способности указанного третьего фрагмента, образующего водородную связь, к образованию водородных связей с Asp25 и Asp25' указанной HIV аспартил-протеазы, если указанное соединение связано с ней в указанной конформации,
(f) оценки перекрывания объема, занимаемого указанным соединением, если указанное соединение связано с указанной HIV аспартил-протеазой в указанной конформации, и объема, занимаемого субстратом HIV аспартил-протеазы или ее негидролизуемым изостером, если указанный полипептид связан с указанной HIV аспартил-протеазой,
(g) оценки энергии деформации связывания указанного соединения с указанной HIV аспартил-протеазой,
(h) оценки вклада энтальпии за счет суммы всех электростатических взаимодействий между указанным соединением и указанной HIV аспартил-протеазой, если указанное соединение с ней связано в указанной конформации, и
(i) принятия или отклонения указанного соединения-кандидата в качестве ингибитора HIV протеазы, на основании определения и оценок, осуществленных на стадиях (b) - (h).
Краткое описание рисунков
Фиг. 1 представляет собой стереоизображение низкоэнергетической конформации соединения 140, предложенного в результате компьютерного моделирования.
На фиг. 2 предложено стереоизображение реальной кристаллической структуры соединения 140 по данным рентгено-кристаллографии.
На фиг. 3 представлено стереоизображение корреляции между предсказанной (тонкая линия) и наблюдаемой (жирная линия) конформацией соединения 140.
Подробное описание изобретения.
Для более полного описания настоящего изобретения предлагается подробное описание. В этом описании использованы следующие сокращения:
Обозначение - Реагент или фрагмент
Ac - ацетил
Me - метил
Et - этил
Bzl - бензил
Trityl - трифенилметил
Asn D или Z-аспарагин
Jle D- или Z-изолейцин
Phe D- или Z-фенилаланин
Val D- или Z-валин
Boc - трет-бутоксикарбонил
Cbz - бензилоксикарбонил/карбобензилокси/
Fmoc - 9-фторенилметиоксикарбонил
DCC - дициклогексилкарбодиимид
DJC - диизопропилкарбодиимид
EDC - 1-/3-диметиламинопропил/-3-этилкарбодиимида гидрохлорид
HoBt - 1-гидроксибензотриазол
HoSu - 1-гидроксисукцинимид
TFA - трифторуксусная кислота
DJEA - диизопропилэтиламин
DBU - 1,8-диазабицикло/5.4.0/ундец-7-ен
EtOAc - этилацетат
Использованы следующие термины.
Если нет других указаний, термины /SO2-'' и "S/O/2-" относятся к сульфону или производному сульфона (то есть, обе группы связаны с атомом серы), но не к сложному эфиру - сульфинату.
Для соединений формулы I и их промежуточных соединений стереохимия, указанная для гидроксила, определяется относительно D на соседнем атоме углерода, если молекула представлена в виде растянутого зигзага (так как это показано для соединений формул XI, XV, XXII и XXXI). Если и OH и D находятся по одно сторону от плоскости, определяемой растянутой основной цепью соединения, стереохимия гидроксила определяется как "син" (syn). Если OH и D расположен по разные стороны от этой плоскости, стереохимия гидроксида указывается как "анти". Термин "гетероцикл" относится к стабильным 5-7 членным моноциклическим или 8-11 членным бициклическим гетероциклам, которые могут быть либо насыщенными, либо ненасыщенными и которые в случае моноциклических гетероциклов могут быть конденсированы с бензольным кольцом. Каждый гетероцикл состоит из гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы. В том смысле, как здесь использован, термин "гетероатомы - азот и сера" включает любые окисленные формы азота и серы, и четвертичные кватернизованные формы любого основного азота. Гетероциклическое кольцо может быть присоединено за счет любого гетероатома цикла, что приводит к образованию стабильной структуры. Предпочтительные гетероциклы, определенные выше, включают, например, бензимидазолил, имидазолил, имидазолиноил, имидазолидинил, хинолил, изохинолил, индолил, пиридил, пирролил, пирролинил, пиразолил, пиразинил, хиноксолил, пиперидинил, морфолинил, тиамофолинил, фурил, тиенил, триазолил, тиазолил, β- карболинил, тетразолил, тиазолидинил, бензофураноил, тиаморфолинилсульфон, бензоксазолил, оксопиперидинил, оксопирролидинил, оксоацепинил, азепинил, изоксазолил, тетрагидропиранил, тетрагидрофуранил, тиадиазолил, бензодиоксолил, тиофенил, тетрагидротиофенил и сульфоланил.
Термины "HIV протеаза" и "HIV аспартил-протеаза" используют взаимозаменяемо, и они относятся к аспартил-протеазе, кодируемой вирусом иммунодефицита человека типа 1 или 2. В предпочтительном варианте изобретения эти термины относятся к аспартил-протеазе вируса иммунодефицита человека типа 1. Термин "гидрофобный" относится к фрагменту, который плохо растворим в воде и часто бывает растворим в жирах. Гидрофобные фрагменты включают (но не ограничиваются ими) такие углеводороды, как алканы, алкены, алкины, циклоалканы, циклоалкены, циклоалкины и ароматические углеводороды, такие как арилы, некоторые насыщенные и ненасыщенные гетероциклы, и фрагменты, которые практически аналогичны боковым цепям гидрофобных природных и синтетических α- аминокислот, включая валин, лейцин, изолейцин, метионин, фенилаланин, α-аминоизобутилмасляную кислоту, аллоизолейцин, тирозин и триптофан.
Термин "существенно гидрофобный" относится к гидрофобному фрагменту, который может необязательно содержать полярные атомы или группы на том участке фрагмента, который экспонирован растворителю, если соединение связано в активном сайте аспартил-протеазы.
Термин "линкерный фрагмент" относится к группе внутри соединения, причем указанная группа состоит из скелета из 1-6 атомов, выбранных из группы, состоящей из C, N, O, S и P, причем указанный скелет замещен или конденсирован c,) или каким-либо другим образом ассоциирован с существенно гидрофобной группой, способной к ассоциации с P1 или P1' связывающим "карманом" HIV аспартил-протеазы, если указанное соединения с ней связано. В альтернативных вариантах настоящего изобретения такие линкерные фрагменты могут быть необязательно замещены группой или группами, которые занимают объем пространства, перекрывающийся с объемом пространства, которое должно быть заполнено нативным субстратом HIV аспартил-протеазы, или ее негидролизуемым изостером.
Термин "более сильно поляризуемый нежели карбонил" относится к фрагменту, обладающему более высокой поляризуемостью (α) нежели у карбонильной группы соответствующего альдегидного, кетонного, сложноэфирного или амидного фрагмента.
Термин "фармацевтически эффективное количество" относится к количеству, которое эффективно при лечении HIV инфекции у пациента. Термин "профилактически эффективное количество" относится к количеству, которое эффективно для предотвращения HIV инфицирования пациента. В том смысле, как здесь использован, термин "пациент" относится к млекопитающим, включая человека.
Термин "фармацевтически приемлемый носитель или адьювант" относится к нетоксичному носителю или адъюванту, который можно вводить пациенту вместе с соединением настоящего изобретения, и который не нарушает его фармакологической активности.
В том смысле, как здесь использовано, соединения настоящего изобретения, включая соединения формулы I, включают и их фармацевтически приемлемые производные. Термин "фармацевтически приемлемое производное" означает любую фармацевтически приемлемую соль, сложный эфир или соль такого эфира соединения настоящего изобретения или любого другого соединения, которое, после его введения реципиенту, способно обеспечить (прямо или косвенно) соединение настоящего изобретения или его анти-вирусноактивный метаболит или остаток.
Фармацевтически приемлемые соли соединений настоящего изобретения включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислот включают соляную, бромистоводородную, серную, азотную, перхлоркислоту, фумаровую, малеиновую, фосфорную, гликолевую, молочную, салициловую, янтарную, толуол-парасульфокислоту, винно-каменную, уксусную, лимонную, метансульфоновую, муравьиную, бензойную, малоновую, нафталин-2-сульфоновую и бензолсульфокислоту. Такие другие кислоты, как щавелевая, можно использовать (хотя они сами и не являются фармацевтически приемлемыми) для получения солей, пригодных в качестве промежуточных соединений для получения соединений настоящего изобретения и их фармацевтически приемлемых солей присоединения кислот.
Соли, полученные из соответствующих оснований, включают соли щелочных металлов (например, натрия), соли щелочноземельных металлов (например, магния), аммония и N-/C1-4-алкил/4 + соли.
Термин "тиокарбаматы" относится к соединениям, содержащим функциональную группу N-SO2-O.
Соединения настоящего изобретения содержат один или более асимметричных атомов углерода и существуют в виде рацематов и рацемических смесей, отдельных энантиомеров, смесей диастереоизомеров и отдельных диастереоизомеров. Все такие изомерные формы этих соединений включены в настоящее изобретение. Каждый стереогенный углерод может иметь как R, так и S конфигурацию. Предпочтительно также, чтобы идентифицированный гидроксил был в положении "син" к D, в растянутой зигзагообразной конформации между азотами, указанными в соединениях формулы I.
Комбинациями заместителей и переменных, предусматриваемыми настоящим изобретением, являются лишь те, которые приводят к образованию стабильных соединений. Термин "стабильный" в том смысле, как здесь использован, относится к соединениям, которые обладают стабильностью, достаточной для того, чтобы обеспечить изготовление и введение млекопитающему в соответствии со способами, известными специалистам. Обычно такие соединения стабильны при температуре 40oC или меньше, в отсутствии влаги или других химически реактивных условий, в течение, по крайней мере, одной недели.
Соединения настоящего изобретения можно использовать в форме солей, полученных из органических или неорганических кислот. Можно указать, среди других таких солей кислот, например, следующие: ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глицерофосфат, полу-сульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат и унлеканоат.
Настоящее изобретение охватывает также кватернизованные формы любых основных азотсодержащих групп соединений, обсуждавшихся ранее. Основной азот может быть кватернизован за счет любых агентов, известных специалистам, включая, например, такие галоиды низших алкилов, как метил-, этил-, пропил- и бутил-хлориды, бромиды и иодиды; такие диалкилсульфаты, как диметилдиэтил-, дибутил- и диамил-сульфаты; такие длинноцепочечные галоиды, как децил-, лаурил-, миристил и стеарил-хлориды, бромиды и иодиды; и аралкилгалоиды включая бензил и фенэтилбромид. В результате такой кватернизации можно получать маслорастворимые или диспергируемые продукты.
Новые сульфонамиды настоящего изобретения представлены следующей формулой I: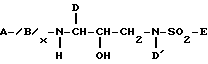
где A выбирают из группы, состоящей из H, Het, -R1-Het, -R1-C1-C6алкила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из гидрокси, C1-C4алкокси, Het, -O-, Het, -NR2-CO-N/R2//R2/ и -CO/N/R2//R2/, и -R1-C2-C6-алкенила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из гидрокси, C1-C4алкокси, Het, -O-Het, -NR2-CO-N/R2//R2/ и -CO-N/R2//R2/,
каждый R1 независимо выбирают из группы, состоящей из -C/O/S/O2/-, -C/O/-C/O/-, -O-C/O/-, -O-S/O/2-, -NR2-C/O/ и -NR2-C/O/-C/O/-,
каждый Het независимо выбирают из группы, состоящей из C3-C7-циклоалкила; C5-C7циклоалкенила, C6-C10арила; и 5-7 членных насыщенных или ненасыщенных гетероциклов, содержащих один или более из гетероатомов, выбранных из N,N/R2/, O, S и S/O/n, причем указанный гетероцикл может необязательно быть сконденсирован с бензольным кольцом, и где любой член из указанных Het может быть необязательно замещен одним или более из заместителей, выбранных из группы, состоящей из оксо, -OR2, -N/R2//R2/, -R2-OH, -CN, -CO2R2, -C/O/-N/R2//R2/, -S/O/2- N/R2//R2/, -N/R2/-C/O/-R2, -C/O/-R2, -S/O/n-R2, -OCF3, -S/O/n-Ar, метилендиокси, -N/R2/-S/O/2/R2/, галоида, -CF3, -NO2, Ar и -O-Ar,
каждый R2 независимо выбирают из группы, состоящей из H и C1-C3алкила, необязательно замещенного Ar, B (если присутствует) представляет -N/R2/-C/R3/R3/-C/O/-, x равен 0 или 1,
каждый R3 независимо выбирают из группы, состоящей из H, Het, C1-C6алкила, C2-C6алкенила, C3-C6циклоалкила и C5-C6циклоалкенила, причем каждый член из указанной группы R3, за исключением H, может быть необязательно замещен одним или более из заместителей, выбранных из группы, состоящей из -OR2, -C/O/-NH-R2, -S/O/n-N/R2//R2/, Het, -CN, -SR2, NR2-C/O/-R2,
каждый n независимо равен 1 или 2.
D и D' независимо выбирают из группы, состоящей из Ar, C1-C4алкила, который может быть необязательно замещен одной или более из групп, выбранных из C3-C6циклоалкила, -OR2, -R3, -O-Ar и Ar; C2-C4алкенила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из C3-C6циклоалкила, -OR2, -R3, -O-Ar и Ar; C3-C6циклоалкила, который может быть необязательно замещен или конденсирован с Ar; и C5-C6циклоалкенила, который может быть необязательно замещен или сконденсирован с Ar; каждый Ar независимо выбирают из группы, состоящей из фенила 3-6 членного карбоциклического кольца и 5-6 членного гетероциклического кольца, содержащего один или более из гетероатомов, выбранных из группы O, N, S, S/O/n и N/R2/, причем указанное карбоциклическое или гетероциклическое кольцо может быть насыщенным или ненасыщенным, и необязательно замещенным одной или более из групп, выбранных из группы, состоящей из оксо, -OR2, -R2, -N/R2//R2/, -N/R2/- C/O/R2, -R2-OH, -CN, -CO2R2, -C/O/-N/R2//R2/, галоида и -CF3,
E выбирают из группы, состоящей из Het, O-Het, Het-Het; -O-R3; -NR2R3; C1-C6алкила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het; C2-C6алкенила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het; C3-C6насыщенного карбоцикла, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het; и C5-C6 ненасыщенного карбоцикла, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из R4 и Het, и
каждый R4 независимо выбирают из группы, состоящей из -OR2, -C/O/-NHR2, -S/O/2-NHR2, галоида, -NR2-C/O/-R2 и -CN.
За исключением тех случаев, когда имеются другие указания, определения переменных A, R1-R4, Het, B, x, n, D, D', Ar и E соответствуют вышеприведенным значениям, указанным для соединений формулы I.
В соответствие с одним вариантом изобретения подклассом соединений являются соединения формулы I и их фармацевтически приемлемые соли, где:
A выбирают из группы, состоящей из H, -R1-Het-; -R1-C1-C6-алкила, который может быть необязательно замещен одной или более из групп, выбранных из группы, состоящей из гидрокси, C1-C4алкокси, Het и -O-Het; и -R1-C2-C6алкенила, который может быть необязательно замещен одной или более из групп, необязательно выбранных из гидрокси, C1-C4алкокси, Het и -O-Het,
каждый R1 независимо выбирают из группы, состоящей из -C/O/-S/O/2, -C/O/-C/O/-, -O-CO-, -O-S/O/2- и NR2-S/O/2-,
каждый Het независимо выбирают из группы, состоящей из C3-C7циклоалкила; C5-C7циклоалкенила; C6-C10арила; и 5-7 членного насыщенного или ненасыщенного гетероцикла, содержащего один или более из гетероатомов, выбранных из N, O и S, который может быть необязательно сконденсирован с бензольным кольцом; причем каждый член указанного Het может быть необязательно замещен одним или более из заместителей, выбранных из группы, состоящей из оксо, -OR2, -R2, -N/R2/2, -R2-OH, -CN, -CO2R2, -C/O/-N/R2/2 и -S/O/2-N/R2/2, каждый R2 независимо выбирают из группы, состоящей из H и C1-C3алкила,
B (если присутствует) представляет -NH-CH/R3/-C/O/-,
x равно 0 или 1,
R3 выбирают из группы, состоящей из Het, C1-C6алкила, C2-C6алкенила, C3-C6циклоалкила и C5-C6циклоалкенила, причем любой член из указанных R3 может быть необязательно замещен одним или более из заместителей, выбранных из группы, состоящей из -OR2, -C/O/-NH-R2, -S/O/n-N/R2/2, Het и -CN,
n равно 1 или 2,
D и D' независимо выбирают из группы, состоящей из Ar; C1-C4алкила, который может быть необязательно замещен C3-C6циклоалкилом или Ar; C2-C4алкенила, который может быть необязательно замещен C3-C6циклоалкилом или Ar; C3-C6циклоалкила, который может быть необязательно замещен или сконденсирован с Ar; и C5-C6циклоалкенила, который может быть необязательно замещен или сконденсирован с Ar; при условии, что если D присоединен к N, D не может быть метилом или C2алкенилом,
Ar выбирают из группы, состоящей из фенила, 3-6 членного карбоциклического кольца и 5-6 членного гетероциклического кольца, содержащего один или более из гетероатомов, выбранных из O, N и S, причем указанное карбоциклическое или гетероциклическое кольцо может быть насыщенным или ненасыщенным и необязательно замещенным одной или более из групп, выбранных из группы, состоящей из оксо, -OR2, -R2, N/R2/2, -N(R2)-C(O)R2, -R2-OH, -CN, -CO2R2, C-(O)-N(R2)2, галоида и -CF3,
E выбирают из группы, состоящей из Het, -O-R3, -NR2R5, C1-C6алкила, который может быть необязательно замещен одним или более из R4 или Het; C2-C6алкенила, который может быть необязательно замещен одним или более из R4 или Het; C3-C6 насыщенного карбоцикла, который может быть необязательно замещен одним или более из R4 или Het; и C5-C6 ненасыщенного карбоцикла, который может быть необязательно замещен одним или более из R4 или Het;
каждый R4 независимо выбирают из группы, состоящей из -OR2, -C(O)-NHR2, -S(O)2-NHR2, галоида и -CN; и
каждый R5 независимо выбирают из группы, состоящей из H и R3, при условии, что по крайней мере, один R5 не представляет H.
Предпочтительным подклассом соединений настоящего изобретения являются соединения формулы I, с молекулярным весом менее чем около 700 г/моль. Более предпочтителен подкласс соединений формулы I с молекулярным весом менее чем около 600 г/моль.
Другими предпочтительными подклассами настоящего изобретения являются соединения формул XXII, XXIII и XXXI: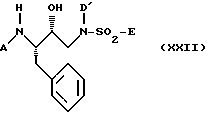
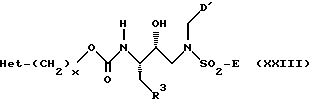
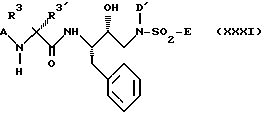
где A, R3, Het, D, D', x и E имеют указанные ранее значения для соединений формулы I. Для удобства ссылок два R3 фрагмента в формуле XXXI были обозначены как R3 и R3'.
Для соединений формулы XXII наиболее предпочтительны соединения, в которых A представляет R1-Het, а D' представляет C1-C3 алкил или C3алкенил, причем указанные алкил или алкенил могут быть необязательно замещены одной или более из групп, выбранных из группы, состоящей из C3-C6-циклоалкила, -OR2, -O-Ar и Ar (причем все остальные переменные имеют значения, указанные ранее для соединений формулы I). Для соединений формулы XXIII наиболее предпочтительными соединениями являются те, в которых R3 представляет C1-C6алкил, C2-C6алкенил, C5-C6циклоалкил, C5-C6циклоалкенил или 5-6 членный насыщенный или ненасыщенный гетероцикл, в котором любой член из указанных R3 может быть необязательно замещен одним или более из заместителей, выбранных из группы, состоящей из -OR2, -C(O)-NHR2, -S(O)nN(R2)(R2), Het, -CN, -SR2, -C(O)2R2 и NR2-C(O)-R2, а D' представляет C1-C3алкил или C3алкенил, причем указанный алкил или алкенил могут быть необязательно замещены одной или более из групп, выбранных из группы, состоящей из C3-C6циклоалкила, -OR2, -O-Ar и Ar (причем все остальные переменные имеют указанные ранее значения для соединений формулы I).
Для соединений формулы XXXI наиболее предпочтительны соединения, в которых A представляет R1-Het, каждый R3 представляет независимо C1-C6алкил, который может быть необязательно замещен заместителем, выбранным из группы, состоящей из -OR2, -C(O)-NH-R2, -S(O)nN(R2)(R2), Het, -CN, SR2, -CO2R2 и -NR2-C(O)-R2, D' представляет C1-C4алкил, который может быть необязательно замещен группой, выбранной из группы, состоящей из C3-C6циклоалкила, -OR2, -O-Ar; а E представляет Het, Het-Het и -NR2R3.
Сульфонамиды настоящего изобретения включают следующие конкретные соединения, представленные в таблицах I-VI. В таблицах I-IV и VI A присоединен за счет правой связи, если нет других указаний. Все остальные заместители в таблицах I-VI (см. в конце описания) присоединены за счет левых связей, если нет других указаний.
Предпочтительные соединения настоящего изобретения (специалистам должно быть ясно, что возможны различные варианты наименований химических соединений, поэтому в таблицах I-VI представлены их структуры):
(S)-N-1-(3-((3-ацетиламино-4-фторбензолсульфонил)-бензиламино)- (1S, 2син)-1-бензил-2-гидроксипропил) -2((хинолин-2-карбонил)-амино)-сукцинамид и (S)-N-1-(3-((4-ацетиламино-3-фторбензолсульфонил)-бензил-амино)- (1S, 2син)-1-бензил-2-гидрокси-пропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 2),
(S)-N-1-(3-((5-ацетиламино-3-метил-тиофен-2-сульфонил)- бензиламино)-(1S, 2син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 5),
(S)-N-1-(1-бензил-3-(бензил)-5-изоксазол-3-ил-тиофен-2-сульфонил) -амино)-(1S, 2син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 6),
(S)-N-1-(3-((бензо[1,2,5] оксадиазол-4-сульфонил)-бензиламино)- (1S, 2син)-1-бензил-2-гидрокси-пропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 9),
N-1-(1-(S)-бензил-3-(бензил-3-сульфамоил-бензолсульфонил)- амино)-2-(син)-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 10),
(S)-N-1-(1-(S)-бензил-2-(син)-гидроксил-3-(изобутил)-5- пиридин-2-ил-тиофен-2-сульфонил)-амино)-пропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 12),
(S)-N-1-(3-((4-бензолсульфонил-тиофен-2-сульфонил)-изобутил- амино)-(1S, 2син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 13),
(S)-N-1-(1-(S)-бензил-3-((4-фторбензолсульфонил)-изобутиламино)- 2-(син)-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 14),
(S)-N-1-(3-((4-ацетиламино-3-фторбензолсульфонил)-изобутил- амино)-(1S,2 син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 15),
(S)-N-1-(3-((3-ацетиламино-4-фторбензолсульфонил)-изобутиламино) -(1S,2 син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 16),
(S)-N-1-(1-(S)-бензил-3-((4-ацетиламино-бензосульфонил)- изобутиламино)-2-(син)-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 17),
(S)-N-1-(3-((5-ацетиламино-3-метил-тиофен-2-сульфонил)- изобутил-амино)-(1S, 2 син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 18),
(S)-N-1-(3-((3-ацетиламино-бензолсульфонил)-изобутиламино)- (1S, 2 син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 19),
(S)-N-1-(3-((бензо[1,2,5] оксадиазол-4-сульфонил)-изобутиламино) -(1S,2 син)-1-бензил-2-гидроксипропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 20),
N-1-((1S, 2 син)-1-бензил-2-гидрокси-3-(1-изобутил-3,3- диметилсульфонилмочевина)-пропил) -2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 21),
N-1-(3-((4-ацетиламино-бензолсульфонил)-изобутиламино)- (1S, 2 син)-1-бензил-2-гидроксипропил) -2-(пиридин-2-ил-метоксикарбонил)-сукцинамид (соединение 22),
(N)-1-(3-((4-ацетиламино-бензолсульфонил)-изобутиламино)- (1S, 2 син)-1-бензил-2-гидроксипропил)-2-пиридин-4-ил-метоксикарбонил)- сукцинамид (соединение 23),
N-1-(3-((4-фтор-бензолсульфонил)-изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-(пиридин-2-ил-метоксикарбонил)- сукцинамид (соединение 26),
4-фтор-N-((2-син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 35),
3,4-дихлор-N-(22син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран -3-илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 37),
N-(4-(((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-ил- метоксикарбониламино)бутил)-изобутил-сульфамоил)-фенил)-ацетамид (соединение 44),
2,4-диметил-тиазол-5-сульфокислоты (1,1-диметил-этоксикарбониламино)-(2- син, 3S)-2-гидрокси-4-фенил-бутил)-изобутиламид (соединение 46),
N-/4-(((2-син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-изобутил-сульфамоил)-фенил/-ацетамид (соединение 48),
4-фтор-N-((2 син.3S)-2-гидрокси-4-фенил-3-((R)-тетрагидрофуран- 3-илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид и 4-фтор-N-((2син,3S)-2-гидрокси-4-фенил-3-((R)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 52),
бензо[1,2,5] оксадиазол-5-сульфокислоты ((2син,3S)-2-гидрокси-4-фенил-3-(пиридин-3-ил)метоксикарбониламино)- бутил)-изобутиламид (соединение 66),
N-(4-(((2син, 3S)-2-гидрокси-4-фенил-3-((R)-тетрагидрофуран- 3-илоксикарбониалмино)-бутил)-изобутил-сульфамоил-фенилацетамид и N-(4-(((2син,3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)бутил-)-изобутил-сульфамоил)-фенил)-ацетамид (соединение 86),
N-(2-фтор-5-(((2-син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран- 3-илоксикарбониалмино)-бутил)-изобутилсульфамоил)-фенил)-ацетамид (соединение 88),
N-(3-(((2син,3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониалмино)-бутил)-изобутилсульфамоил)-фенил)-ацетамид (соединение 91),
4-фтор-N-((2 син,3S)-2-гидрокси-4-фенил-3-((R)-тетрагидрофуран- 3-илоксикарбониламино)-бутил-N-изобутилбензолсульфонамид (соединение 93),
N-(4-(((син)-2-гидрокси-(S)-4-фенил-3-((тетрагидрофуран(R)-3-ил)- оксикарбониламино)-бутил)-изобутил-сульфамоил)-фенил)-ацетамид (соединение 94),
4-фтор-N-(2 син,3S)-2-гидрокси-4-фенил-3-((тетрагидрофуран-(R)- 3-илметоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид и 4-фтор-N-(2 син, 3S)-2-гидрокси-4-фенил-3-((тетрагидрофуран-(S)-3- илметоксикарбониламино)-бутил)-N-изобутилбензосульфонамид (соединение 97),
4-фтор-N-((2 син,3S)-2-гидрокси-4-фенил-3-(пиридин-3-ил- метоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 98),
4-хлор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илкарбониламино)-бутил)-изобутилбензолсульфонамид (соединение 99),
N-((2 син,3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)-бутил-N-изобутил-4-метоксибензолсульфонамид (соединение 100),
4-фтор-N-(2-(син)-гидрокси-3-((2-оксазолидон)-(S)-4-ил)- метоксикарбониламино)-4-(S)-фенил-бутил)-N-изобутил-бензолсульфонамид (соединение 109),
бензол-1,3-дисульфокислоты 1-амид-3-((2 син,3S)-2-гидрокси-4-фенил-3-(3-(S)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-изобутил-амид (соединение 112),
фуран-3-сульфокислоты (2 син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-изобутил-амид (соединение 113),
N-((3-аллилоксикарбониламино)-(2 син,3S)-2-гидрокси-4-фенил- бутил)-N-циклопентилметил-4-фторбензолсульфонамид (соединение 114),
N-циклопентилметил-N-((3-этоксикарбониламино)-(2син, 3S)-2- гидрокси-4-фенил-бутил)-4-фторбензолсульфонамид (соединение 115),
4-хлор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-бензолсульфонамид (соединение 116),
4-хлор-N-циклопентилметил-N-((2 син,3S)-2-гидрокси-4-фенил-3- (пиридин-3-илметоксикарбонил)-бутил)-бензолсульфонамид (соединение 118),
N-(4-(циклопентилметил-((2 син,3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-сульфамоил)-фенил)- ацетамид (соединение 125),
3-хлор-N-((2син, 3S)-2-гидрокси-4-фенил-3-(S)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-N-изобутил-бензолсульфонамид (соединение 138),
4-хлор-N-циклопентилметил-N-(2(син)-гидрокси-3-((2-оксазолидон- 4-(S)-илметил)-оксикарбониламино)-4-фенил-бутил)-бензолсульфонамид (соединение 139),
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-4-метокси- бензолсульфонамид (соединение 140),
N-((3-аллилоксикарбониламино)-(2син, 3S)-2-гидрокси-4-фенил- бутил)-N-циклопентилметил-4-метокси-бензолсульфонамид (соединение 141),
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-(3- пиридин-3-ил-метоксикарбониламино-бутил-4-метоксибензолсульфонамид (соединение 142),
пеиридин-3-сульфокислоты ((2 син,3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-изобутиламид, соль трифторуксусной кислоты (соединение 144),
5-изоксазол-3-ил-тиофен-2-сульфокислоты ((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-изобутил-амид (соединение 145),
N-(4-3-аллилоксикарбониламино)-(2 син, 3S)-2-гидрокси-4- фенилбутил)-циклопентилметилсульфамоил)-фенил)-ацетамид (соединение 146),
N-(4-(циклопентилметил)-((2син, 3S)-2-гидрокси-4-фенил-3- (пиридин-3-илметоксикарбониламино)-бутил)-сульфамоил)-фенил)-ацетамид (соединение 147),
N-циклопентилметил-N-((2син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-бензолсульфонамид (соединение 148),
пиридин-3-сульфокислоты циклопентилметил-((2-син,3S)-2-гидрокси- 4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-амид (соединение 148),
пиперидин-1-сульфокислоты ((2 син,3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-изобутиламид (соединение 150),
N-4-((2-(син)-гидрокси-3-((2-метоксиметил- аллилоксикарбониламино)-4-(S)-фенилбутил)-изобутилсульфамоил)- фенил)-ацетамид (соединение 155),
1-ацетил-2,3-дигидро-1H-индол-6-сульфокислоты ((аллилоксикарбониламино)-(2-син, 3S)-2-гидрокси-4-фенилбутил) циклопентилметил-амид (соединение 156),
1-ацетил-2,3-дигидро-1H-индол-6-сульфокислоты циклопентилметил-((2 син, 3S)-2-гидрокси-4-фенил-3((S)-тетрагидрофуран-3-илоксикарбониламино)- бутил)-амид (соединение 157),
N-циклогексилметил-N-((2 син,3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониалмино)-бутил)-4- метоксибензолсульфонамид (соединение 158),
N-циклогексилметил-4-фтор-N-((2 син,3S)-2-гидрокси-4-фенил-3- ((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-бензолсульфонамид (соединение 159),
N-4-(циклогексилметил)-((2 син,3S)-2-гидрокси-4-фенил-3- ((S)-тетрагидрофуран-3-илоксикарбониалмино)-бутил)-сульфамоилфенил)- ацетамид (соединение 160),
N-((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-4- илметоксикарбониламино)-бутил)-N-изобутил-4-метокси-бензолсульфонамид (соединение 163),
N-(2 син, 3S)-2-гидрокси-4-фенил-3-((син)-тетрагидрофуран-3- илоксикарбониалмино)-бутил)-N-изобутил-4-метилбензолсульфонамид (соединение 165),
N-циклопентилметил-4-гидрокси-N-((2 син, 3S)-2-гидрокси-4- фенил-3-(пиридин-3-ил-метоксикарбониламино)-бутил)-бензосульфонамид (соединение 166),
N-((2 син,3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)-бутил)-N-изобутил-4-нитробензолсульфонамид (соединение 167),
4-амино-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 168),
N-циклопентилметил-4-гидрокси-N-((2 син, 3S)-2-гидрокси-4-фенил-3 -((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-бензолсульфонамид (соединение 169),
N-циклопентилметил-N-((2 син,3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-4- нитробензолсульфонамид (соединение 170),
4-амино-N-циклопентилметил-N-((2 син,3S)-2-гидрокси-4-фенил-3- ((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-бензолсульфонамид (соединение 171),
2,4-диамино-N-циклопентилметил-N-((2-син, 3S)-2-гидрокси- 4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил- бензолсульфонамид (соединение 173).
4-гидрокси-N-(2-син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-N-изобутилзолсульфонамид (соединение 175).
N-циклопентилметил-4-фтор-N-((2 син, 3S)-2-гидрокси-4-фенил-3- ((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)- бензолсульфонамид (соединение 182).
3,4-дихлор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил- 3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)- бензолсульфонамид (соединение 183),
бензилоксикарбонил-(S)-изолейцин-N-(5-((3-амино-(2 син, 3S)-2- гидрокси-4-фенилбутил)-изобутил)-сульфамоил)-2-фтор-фенил)- ацетамид (соединение 187) и
N-((2- син, 3S)-4-циклогексил-2-гидрокси-3-((S)-тетрагидрофуран- 3-илоксикарбониламино)-бутил)-N-циклопентилметил-4-метокси- бунзолсульфонамид (соединение 195).
Наиболее предпочтительные соединения настоящего изобретения:
(S)-N-1-(1-S)-бензил-2-(син)-гидрокси-3-(изобутил- (5-пиридин-2-ил-тиофен-2-сульфонил)-амино)-пропил)-2-((хинолин- 2-карбонил)-амино)сукцинамид (соединение 12),
(S)-N-1-(1-(S)-бензил-3-((4-фторбензолсульфонил)- изобутиламино)-2-(син)-гидроксипропил)-2-((хинолин-2-карбонил)- амино)-сукцинамид (соединение 14),
(S)-N-1-(3-((4-ацетиламино-3-фторбензолсульфонил)- изобутил-амино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-(( хинолин-2-карбонил)-амино)-сукцинамид (соединение 15),
(S)-N-1-(3-((бензо[1,2,5] оксидиазол-4-сульфонил)- изобутиламино)-(1S, 2 син)-1-бензил-гидроксипропил)-2- ((хинолин-2-карбонил)-амино)-сукцинамид (соединение 20),
N-1-((1S, 2 син)-1-бензил-2-гидрокси-3-(1-изобутил- 3,3-диметилсульфонилмочевина)-пропил)-2-((хинолин-2-карбонил)- амино)-сукцинамид (соединение 21),
N-/4-(((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран- 3-илоксикарбониламино)-бутил)-изобутилсульфамоил)-фенил) ацетамид (соединение 48),
N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3- илоксикарбониламино)-бутил-N-изобутил-3-метоксибензолсульфонамид (соединение 100),
4-хлор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4- фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)- бензолсульфонамид (соединение 116),
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил- 3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-4- метокси-бензолсульфонамид (соединение 140),
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил- 3-(3-пиридин-3-ил-метоксикарбониламино)-бутил)-4-метоксибензол сульфонамид (соединение 142),
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил- 3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)- бензолсульфонамид (соединение 148),
N-циклогексилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3- ((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-4- метоксибензолсульфонамид (соединение 158),
N-(4-(циклогексилметил)-((2 син, 3S)-2-гидрокси-4- фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)- сульфамоил-фенил)-ацетамид (соединение 160),
N-циклопентилметил-4-гидрокси-N-((2 син, 3S)-2-гидрокси- 4-фенил-3-(пиридин-3-ил-метоксикарбониламино)-бутил)- бензолсульфонамид (соединение 166),
4-амино-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил-N-изобутил- бензолсулфонамид (соединение 168),
4-амино-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4- фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)- бензолсульфонамид (соединение 171),
2,4-диамино-N-циклопентилметил-N -((2 син, 3S)-2- гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)- бутил)-бензолсульфонамид (соединение 173),
4-гидрокси-N-(2 син, 3S)-2-гидрокси-4-фенил-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-N- изобутилбензолсульфонамид (соединение 175), и
N-((2 син, 3S)-4-циклогексил-2-гидрокси-3-((S)- тетрагидрофуран-3-илоксикарбониламино)-бутил)-N-циклопентилметил -4-метокси-бензолсульфонамид (соединение 195).
Сульфонамиды настоящего изобретения можно получить, используя обычные методики. Удобно синтезировать эти соединения обычными способами, синтезируя их из легко доступных исходных материалов. Соединения настоящего изобретения относятся к наиболее легко синтезируемым HIV протеазным ингибиторам (из уже известных). Описанные ранее ингибиторы HIV протеазы часто содержат четыре или более центра, ряд пептидных связей и/или требуют анти-смысловых реагентов (таких, как металлоорганические комплексы), для осуществления их синтеза. Относительная простота, с которой можно получать соединения настоящего изобретения, представляет ряд преимуществ при крупномасштабном производстве этих соединений.
Обычно, сульфонамиды формулы I удобно получать из производных α-аминокислот общей формулы A-(B)x-NH-CH(D)-COOH, где символы A, B, X и D имеют указанные ранее значения для соединений формулы I. Такие производные α-аминокислот часто являются коммерчески доступными или их можно легко получить из коммерчески доступных производных α-аминокислот известными способами. См., например, J.W.Green and P.G.M. Wuts, "Protective Groups in Organic Synthesis", 2 nd. Ed., John Wiley and Sons (1991). Хотя в объем изобретения входят использование рацемических смесей таких исходных материалов (если x=0), предпочтительны отдельные энантиомеры в S конфигурации.
Используя известные методики, производные α-аминокислот общей формулы A-(B)x-NH-CH(D)-COOD можно легко превратить в производные аминокетона общей формулы
A-(B)x-NH-CH(D)-CO-CH2-X,
где X представляет отщепляемую группу, которая подходящим образом активирует α-углерод (т.е. переводит метилен в состояние, доступное для нуклеофильной атаки). Подходящие отщепляемые группы хорошо известны специалистам и включают галиды и сульфонаты, такие как метансульфонат, трифторметансульфонат или 4-толуолсульфонат. X может также представлять гидрокси, который ин ситу превращается в отщепляемую группу (например, за счет обработки триалкил- или триалилфосфином в присутствии диалкилазодикарбоксилата). Способы получения таких производных аминокетона также хорошо известны специалистам (см., например, Fittkau, J. Prakt. Chem., 315, p. 1037 (1973)). В другом варианте некоторые производные аминокетонов коммерчески доступны (например, от Bachem Bioscience, Inc., Philadelphia, Pennsylvania).
Затем производное аминокетона можно восстановить до соответствующего аминоспирта, представленного формулой:
A-(B)x-NH-CH(D)-CH(OH)-CH2-X. Специалистам известны многие методики восстановления производных аминокетонов, таких как A-(B)x-NH-CH(D)-CO-CH2-X (Larock, R. C. "Comprehensive Organic Transformations", p.527-547, VCH Publichers, Inc. , 1989 и приведенные там ссылки). Предпочтительным восстанавливающим агентом является боргидрид натрия. Реакцию восстановления ведут при температуре от около -40oC до около 40oC (предпочтительно при температуре от около 0oC до около 20oC) в подходящем растворителе или системе растворителей, например в водном или чистом тетрагидрофуране или низшем спирте, таком как этанол или метанол. Хотя настоящее изобретение охватывает как стереоспецифичное, так и нестереоспецифичное восстановление производного аминокетона общей формулы A-(B)x-NH-CH(D)-CO-CH2-X, предпочтительно стереоспецифическое восстановление.
Стереоселективное восстановление можно осуществить за счет использования хиральных реагентов, которые известны специалистам. В настоящем изобретении стереоселективного восстановления достигают, например, в условиях нехелатирующего восстановления, когда хиральное индуцирование вновь образуемых гидроксильных групп задается стереохимией D группы (т.е. присоединение гидрида по Felkin-Ahn). Особенно предпочтительно стереоселективное восстановление, когда образующийся гидроксил находится в син-положении по отношению к D. Было обнаружено, что если гидроксил находится в син-положении по отношению к D, конечный сульфонамид является ингибитором HIV протеазы, обладающим большей эффективностью, нежели антидиастереоизомер.
Гидроксильная группа аминоспирта может быть необязательно защищена любой из известных защищающих кислород групп (таких, как триалкилсилил, бензил или алкилоксиметил) до получения защищенного аминоспирта формулы A-(B)x-NH-CH(D)-C(OR6)-CH2-X, где R6 представляет H или любую подходящую гидроксизащищенную группу. Некоторые подходящие защитные группы описаны у T.W. Greene и P.G.M. Wuts "Protective Groups in Organic Synthesis". 2nd Ed. John Wiley and Sons, 1991).
Затем аминоспирт можно подвергнуть взаимодействию с нуклеофильным соединением амина до получения промежуточного соединения формулы (III)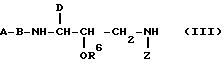
где D и R6 имеют указанные ранее значения, а Z представляет либо D' (как указано для соединений формулы I), либо водород.
В наиболее выгодной схеме синтеза одновременную активацию метилена и защиту спирта можно осуществить за счет образования N-защищенного аминоэпоксида из кислорода и соседнего с ними метилена до получения промежуточного соединения формулы: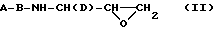
где A, B и D имеют указанные ранее значения для соединений формулы I. Подходящие системы растворителей для получения N-защищенного аминоэпоксида включают этанол, метанол, изопропанол, тетрагидрофуран, диоксан, диметилформамид и т. п. (включая их смеси). Подходящими основаниями для получения эпоксида являются гидроксиды щелочных металлов, трет.-бутоксид калия, DBU и т.д. Предпочтительным основанием является гидроксид калия.
Реакцию N-защищенного аминоэпоксида или другого подходящим образом активированного промежуточного соединения с амином ведут в чистом (то есть без растворителя) виде или в таком полярном растворителе, как низшие спирты, вода, диметилформамид или диметилсульфоксид. Реакцию удобно вести при температуре от 0oC до 120oC и предпочтительно от около 20oC до 100oC. В другом варианте реакцию можно вести в присутствии активирующего агента, такого как активированная окись алюминия, в инертном растворителе, предпочтительно в эфире, таком как диэтиловый эфир, тетрагидрофуран, диоксан или трет.бутилметиловый эфир, обычно при температуре от комнатной до около 110oC, по способу Posner and Rogers, J. Am. Chem. Soc., 99, p.8208 (1977). Другие активирующие реагенты включают такие низшие соединения триалкилалюминия, как триэтилалюминий или диалкилалюминий - галиды, например диэтилалюминийхлорид (Overman and Flippin, Tetrahedron Zetters, p 195 (1981)). Реакции, которые включают эти соединения, обычно ведут в таких инертных растворителях, как дихлорметан, 1,2-дихлорэтан, толуол или ацетонитрил, при температуре от 0oC до около 110oC. Другие способы удаления защитных групп или раскрытия эпоксидов аминами или их эквивалентами, такими как азиды или триметилсилилцианид (Gassman and Guggenheim, J. Am. Chem. Soc., 104, p. 5849 (1982)), известны и очевидны для специалистов.
Соединения формул II и III, а также их функционально-защищенные производные пригодны в качестве промежуточных соединений для получения соединений формулы I. В тех случаях, когда Z представляет D', соединения формулы III можно превратить в соединение формулы I за счет реакции с сульфонил-активированными соединениями до получения сульфонамидов, сульфонилмочевин, тиокарбаматов и т.п. Способы получения таких сульфонил-активированных соединений хорошо известны специалистам. Обычно для получения сульфонамидов используют сульфонилгалоиды. Многие сульфонилгалоиды коммерчески доступны; другие можно легко получить, используя обычные методики синтеза (Gilbert, B.E. "Recent Development in Preparative Sulfonation and Sulfation", Synthesis, 1969: 3 (1969) и в указанных там ссылках: Hoffman, R.V. "M-Trifluoromethylbenzenesulfonyl Chloride", Org. Synth. Coll. Vol. VII, John Wiley and Sons /1990/; Hartman, G. D. et al., "4-Substituted Thiophene - and Furan-2-Sulfonamides as Topical Carbonic Anhydrase Inhibitors", J.Med.Chem., 35, p. 3822 (1992) и в указанных там ссылках. Сульфонилмочевины обычно получают в результате взаимодействия амина с сульфурилхлоридом или с подходящим эквивалентом, таким как сульфурил-бис-имидазол или сульфурил-бис-N-метилимидазол. Тиокарбаматы обычно получают при взаимодействии спирта с сульфурилхлоридом или таким подходящим эквивалентом, как сульфурил бис-имидазол или сульфурил-бис-N-метилимидазол.
В случае соединений формулы III, где Z представляет водород, конверсию полученного первичного амина во вторичный амин можно осуществить известными способами. Такие методики включают реакцию с алкилгалоидом или алкилсульфонатом, или за счет восстановительного алкилирования альдегида или карбоновой кислоты или ее активированного производного с использованием, например каталитического гидрирования, или с помощью цианоборгидрида натрия (Borch et al., J. Am. Chem. Soc., 93 p. 2897 (1971)). В другом варианте первичный амин можно ацилировать, а затем восстановить с помощью борана или другого подходящего восстанавливающего агента, например, по способу Cushman et al., J. Org. Chem. , 56, p. 4161 (1991). Эта методика особенно подходит для соединений формулы III, в которых B отсутствует, а A представляет такую защитную группу, как трет.-бутоксикарбонил (Вос) или бензилоксикарбонил (CbZ).
Если переменная A в конкретном соединении формулы I представляет удаляемую защитную группу, удаление этой защитной группы с последующей реакцией полученного амина с соответствующим активированным реагентом приведет к получению другого соединения формулы I. Так, например, реакция с активированным карбоксилатом, таким как ацилгалоид, (например, фторангидридом, хлорангидридом и бромангидридом), таким активированным сложным эфиром, как нитрофениловый сложный эфир или сложный эфир I-гидроксисукцинимида (HOSU), или таким ангидридом, как симметричный ангидрид или изобутил-ангидрид, или смешанные угольно-фосфорный или угольно-фосфиновый ангидриды, приведет к получению соответствующего амида. Мочевины можно получить при взаимодействии с изоцианатами или аминами в присутствии бис-активированных производных угольной кислоты, таких как фосген или карбонилдиимидазол. Карбаматы можно получить при взаимодействии хлоркарбонатов с карбонатами, этерифицированными такими отщепляемыми группами, как I-гидроксибензотриазол (HOBT) или HOSU, или со спиртами в присутствии производных бисактивированной угольной кислоты, таких как фосген или карбонилдиимидазол. Легко понять, что для облегчения специфических реакций может понадобиться защита одной или более из потенциально реакционноспособных групп, с последующим удалением этих групп. Такие модификации схем реакций, указанных ранее, очевидны для специалистов.
Если переменная B в конкретном соединении формулы I отсутствует, а переменная A представляет удаляемую защитную группу, удаление A с последующей реакцией полученного амина с аминокислотой или с соответствующим образом N-защищенным производным, с последующей реакцией свободного α-амина, если он присутствует, как указано ранее, приводят к получению соединения формулы I. Присоединение аминокислот и их производных осуществляют хорошо известными способами пептидного синтеза. Некоторые из этих способов раскрыты Bonanszky и Bodanszky в "The Practice of Peptide Synthesis" Springer - Verlag, Berlin, Germany /1984/, и в книге "The Peptides" Gross and Meinhofer /Eds/; Academic Press, 1979, Vol. I-III (включены сюда по ссылке).
Обычно для синтеза пептидов в растворе α-амин подлежащей присоединению аминокислоты защищают Boc, CbZ, аллилоксикарбонилом (AIIoc) или 9-фторенилметоксикарбонилом (Fmoc), в то время как свободный карбоксил активируют за счет взаимодействия с таким карбодиимидом, как дициклогексилкарбодиимид (ДОС), 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (EDC), или диизорпропилкарбодиимид (DIC), необязательно в присутствии такого катализатора как HOBT, HOSU или диметиламинопиридин (DMAP). Можно использовать и другие способы, которые включают применение активированных сложных эфиров, хлорангидридов, энзим-активированных аминокислот и ангидридов, включая N-карбокси-ангидриды, симметричные ангидриды, смешанные угольные ангидриды, угольно-фосфоновые и угольно-фосфорные ангидриды. После образования пептида защитные группы можно удалить такими способами, известными из приведенных ранее ссылок, как гидрирование в присутствии палладиевого, платинового или родиевого катализаторов, за счет обработки натрием в жидком аммиаке, соляной, фтористоводородной, бромистоводородной, муравьиной, трифторметансульфоновой или трифторуксусной кислотой, вторичными аминами, ионами фтора, триметилсилилгалоидами, включая бромид и иодид, или щелочью.
Одна из наиболее подходящих схем для получения сульфонамидов формулы XV, представлена далее: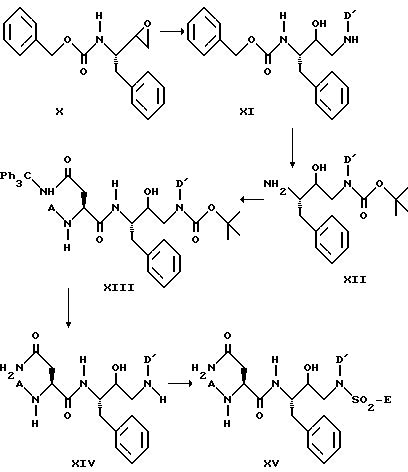
Соединения формулы X можно с успехом получить из легко доступных исходных материалов (см. D.P.Getman, J. Med. Chem., 36, p. 288 (1983)). Каждую стадию вышеприведенной схемы синтеза можно вести, как описано ранее.
Наиболее подходящая схема синтеза для получения предпочтительных сульфонамидов формулы XXII представлена далее: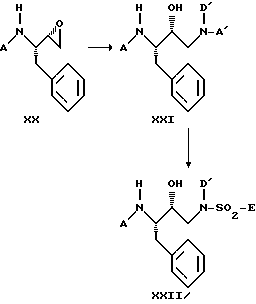
Соединения формулы XX можно с успехом получить из легко доступных исходных материалов (см. B.E.Evans et al., J. Org. Chem., 50, p. 4615 (1985)).
Каждую стадию вышеприведенной схемы синтеза можно осуществлять в соответствии с приведенным описанием.
После превращения соединения формулы XX в соединение формулы XXI, как подробно раскрыто в предшествующей схеме, соединение формулы XXI можно альтернативно подвергнуть взаимодействию с аминокислотой или производным аминокислоты, как раскрыто ранее, до получения предпочтительного соединения формулы XXXI. Наиболее подходящая схема синтеза, в которой используется такой подход, представлена на стр. 83.
Как должно быть очевидно специалистам, вышеуказанные схемы синтеза не должны включать исчерпывающий список всех способов, с помощью которых описанные и заявленные соединения могут быть синтезированы. Специалистам должны быть очевидны и другие способы.
Соединения настоящего изобретения можно модифицировать, вводя соответствующие функциональности для улучшения селективных биологических характеристик. Такие модификации известны специалистам и включают те, которые повышают биологическое проникновение в биологическую систему (то есть, кровь, лимфатическую систему, центральную нервную систему), повышают доступность при пероральном приеме, повышают растворимость для того, чтобы обеспечить возможность введения за счет инъекций, меняют метаболизм и изменяют скорость выведения.
Соединения формулы I характеризуются превосходной способностью ингибировать активность HIY протеазы и вирусную репликацию. Мы считаем, что это связано со специфическими стерическими и электронными взаимодействиями между протеазой и соединениями формулы I. Это предположение основано на анализе структурных оснований для активности соединений формулы I, с учетом известных кристаллических структур HIY протеазы и таких связанных ингибиторов, как структуры, описанные Miller et al., "Structure of Complex of Synthetic HIV-1 Protease with a Substrate - Based Inhibitor at 2,3 A Resolution", Science, Vol. 246, pp 1149-1152 (1989)), (включено сюда по ссылке), а также структур, определенных в наших лабораториях, В соответствии с этими структурами активный сайт HIY аспартилпротеазы определяют с использованием глубоких канавок, содержащих субкарманы для накопления различных боковых цепей протеазного субстрата - называемых P1-Pn и P'1-P'n, в соответствии с обычной номенклатурой для протеазы. В центре канавки расположены два остатка аспарагиновой кислоты (Asp 25 и Asp25', в соответствии с системой нумерации Miller et al.), так как это типично для активных сайтов аспартатов известных аспартиловых протеаз, которые, как считают, являются каталитическими остатками энзима. Канавка покрыта двумя C2-симметрично расположенными "щитками", которые также осуществляют различные непосредственные и косвенные контакты со связанными субстратами.
Мы считаем, что заместители A, D, D' и E соединений формулы I ассоциируются с HIY протеазой за счет гидрофобных сил в связывающих карманах энзима. Мы также считаем, что водород сульфонамидной группы прочно связывается с молекулой воды, удерживаемой за счет водородных связей на "щитках" протеазы ("молекулы воды на щитках"; молекулы воды 511, в соответствии с системой нумерации Meller et al.).
С учетом этого открытия, альтернативный вариант настоящего изобретения относится к новым ингибиторам HIY протеазы, обладающим определенными структурными и физико-химическими особенностями. Мы обнаружили, что соединения, обладающие следующими новыми сочетаниями отличительных черт, удивительно эффективны в качестве ингибиторов HIV протеазы:
(1) первый и второй фрагмент-акцептор водородной связи, по крайней мере, один из которых более сильно поляризуется, нежели карбонил, причем указанные фрагменты одинаковы или различны и способны образовывать водородную связь с атомами водорода молекулы воды "щитка" HIV аспартил протеазы, если соединение с ней связано,
(2) существенно гидрофобные фрагменты, которые связаны с P1 и P1', связывающими карманами указанной аспартил протеазы, если соединение с ней связано,
(3) третий фрагмент, связывающий водород, который может быть донором или акцептором водородной связи, способный к одновременному водородному связыванию с Asp25 и Asp25', указанной аспартил протеазы, если соединение с ней связано,
(4) дополнительно занятый объем пространства, по крайней мере, 100 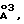 если соединение связано с активным сайтом указанной аспартил протеазы, причем указанное пространство перекрывается с объемом пространства, который должен заполниться нативным субстратом указанной HVI аспартил протеазы или ее негидролизуемым изостером,
если соединение связано с активным сайтом указанной аспартил протеазы, причем указанное пространство перекрывается с объемом пространства, который должен заполниться нативным субстратом указанной HVI аспартил протеазы или ее негидролизуемым изостером,
(5) энергия деформации связи соединения указанной HVI аспартил протеазы с указанным соединением не превышает 10 ккал/моль и
(6) нейтральный или благоприятный вклад энтальпии от суммы всех электростатических взаимодействий между соединением и протеазой, когда соединение связано с указанной HVI аспартил протеазой.
Соединения, обладающие вышеперечисленными особенностями, можно легко идентифицировать или сконструировать известными специалистам способами, используя сочетания химических соображений и расчетных способов. Так, например, специалисты могут легко идентифицировать или выбрать связывающие водород и гидрофобные фрагменты или группы, отвечающие условиям п. (1)-(3), при этом особенности (4)-(6) можно определить, используя хорошо известные расчетные способы определения структур (например, конформацией) и энергетические свойства молекул.
Более того, соединения, характеризующиеся особенностями (1)-(6), перечисленными ранее, можно получить, используя обычные методики, включая химический синтез и выделение природных продуктов. Мы предпочитает схему синтеза, подробно описанную ранее для соединений формулы I.
Мы обнаружили, что если ингибитор HVI протеазы образует водородные связи с молекулой воды "щитка" за счет двух связывающих водород фрагментов, по крайней мере, один из которых более сильно поляризуется нежели карбонил, способность этих соединений ингибировать активность HVI протеазы очень сильно возрастает по сравнению с обычными ингибиторами HVI протеазы.
Без какого-либо желания ограничиваться теорией мы считаем, что сильные водородные связи, которые создаются между молекулой воды "щитка" и двумя связывающими водород фрагментами, по крайней мере, один из которых более сильно поляризуется, нежели карбонил, снижает полную связывающую энергию ингибитора. Большинство ингибиторов HVI протеазы из известных специалистам используют только карбонильные группы для связывания с молекулой воды щитка и поэтому хуже ингибиторов настоящего изобретения. Мы считаем, что возрастание поляризации за счет большего дипольного момента сильно поляризуемого связывающего водород фрагмента (по сравнению с дипольным моментом карбонильного фрагмента) создает более сильную и прочную водородную связь с молекулой воды щитка. Мы предпочитаем использовать четырехвалентную окисленную серу, шестивалентную окисленную серу и пятивалентный окисленный фосфор в качестве сильно поляризуемого связывающего водород фрагмента. Наиболее предпочтительны четырехвалентная окисленная сера и шестивалентная окисленная сера в качестве сильно поляризуемого связывающего водород фрагмента. Шестивалентная окисленная сера (-SO2-) наиболее предпочтительна.
Мы обнаружили, что если сильно поляризуемый связывающий водород фрагмент представляет сульфонамид, полная энергия связывания ингибитора особенно низка. Мы считаем, что такое увеличение стабильности связано с конкретными конформационными характеристиками сульфонамидной S-N связи.
Конкретно, сульфонамидная S-N связь существует только в двух низко-энергетичных ротамерах (см. J.B.Nicholas et al., J.Phys.Chem., 95, p. 9803 (1991) и R.D.Bindas et al., J.Am.Chem.Soc., 112, p. 7861 (1990). Это оказывает эффект фиксации этой части молекулы в благоприятной конформации, где один или оба сильно поляризуемых S=О кислорода могут быть включены во взаимодействия водородных связей со щитковой водой.
Остальные пять структурных и физикохимических особенностей, перечисленных ранее (т. е. особенностей (2)-(6) обычно рассматриваются специалистами, как повышающие способность соединения к конкурирующей активности и нгибированию HIV протеазы. Хотя существуют несколько других отличительных особенностей, которые, как считают, улучшают ингибирующую особенность (как, например, связывание основной цепи ингибитора с энзимом), мы обнаружили, что комбинация только пяти вышеуказанных элементов, вместе с новым элементом (1), типифицирует эффективные HIV протеазные ингибиторы. Вообще, энергия связывания конкретного ингибитора протеазы понижается, если гидрофобные фрагменты ингибитора расположены так, чтобы ассоциировать с гидрофобными связывающими карманами энзима. В случае HIV-I протеазы расположение и природа P1 и P'1 связывающих карманов известны специалистам (см., например, M.Miller et al. ранее). Существенно гидрофобные боковые цепи, которые входят в хорошо определенные P1, P'1 связывающие карманы, также известны специалистам. Предпочтительные боковые цепи расположены внутри 4 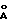 энзима, если связаны с HIV протеазой. Предпочтительные гидрофобные боковые цепи включают те, которые практически аналогичны цепям гидрофобных природных и синтетических α- аминокислот, включая аланин, валин, лейцин, изолейцин, метионин, фенилаланин, α- аминоизобутилмасляную кислоту, аллоизолейцин, тирозин и триптофан. До тех пор, пока часть этой боковой цепи находится в контакте с объемным растворителем, или простирается наружу из энзима, нельзя считать, что она полностью находится в P1 или P'1 и может содержать такую полярную функциональность, как заряженный амин в этом положении.
энзима, если связаны с HIV протеазой. Предпочтительные гидрофобные боковые цепи включают те, которые практически аналогичны цепям гидрофобных природных и синтетических α- аминокислот, включая аланин, валин, лейцин, изолейцин, метионин, фенилаланин, α- аминоизобутилмасляную кислоту, аллоизолейцин, тирозин и триптофан. До тех пор, пока часть этой боковой цепи находится в контакте с объемным растворителем, или простирается наружу из энзима, нельзя считать, что она полностью находится в P1 или P'1 и может содержать такую полярную функциональность, как заряженный амин в этом положении.
Было также установлено специалистами, что наличие гидроксильной группы внутри водородной связи вблизи к двум каталитическим остаткам аспарагиновой кислоты HIV протеазы (Asp25 и Asp25'), является важной особенностью для эффективного ингибитора HIV протеазы (см., например, R.Bone et al., "X-ray Crystal Structure of the HIV Protease Complex with L-700417 an Inhibitor with Pseudo C2 Symmetry", J. Am.Chem.Soc. 113, p. 9382-84 (1991)). Кроме того, следует учитывать, что геометрия Asp-связанного водородной связью фрагмента является особенно важной. Хотя мы и предпочтитаем использовать в этом положении гидроксильную группу, приемным может быть любой фрагмент, образующий водородную связь, который способен образовывать водородные связи с Asp остатаками. Такие образующие водородные связи фрагменты хорошо известны специалистам (например, фосфиновая кислота (D.Grobelhy et.al, Biochem.Biophys.Res. Commun., 169 p. 1111 (1990)).
Следует также учитывать, что связывание конкурирующих ингибиторов с HIV протеазой оптимальным образом осуществляется за счет наличия ингибиторного пересечения объема, перекрывающего объем, занятый нативным полипептидным субстратом, если он связан с активным сайтом энзима. Эффективные ингибиторы HIV протеазы обычно имеют относительно небольшую разницу в энергии между их связью и свободным состоянием (то есть, небольшую энергию деформации связывания). Наиболее предпочтительные ингибиторы HIV протеазы настоящего изобретения имеют энергию деформации связывания не более чем 10 ккал/моль (предпочтительно не более 7 ккал/моль). Однако, следует отметить, что ингибиторы HIV протеазы могут взаимодействовать с HIV протеазой более чем в одной конформации, которая аналогична полной энергии связывания (см. K.H.Murthy, J. Biol. Chem. 267 (1992)). В этих случаях энергия деформации связывания принимается равной разности между энергией свободного соединения и средней энергией конформацией, наблюдаемых в тех случаях, когда ингибитор связан с энзимом.
Кроме того, следует учитывать, что большинство эффективных ингибиторов протеазы не имеет отталкивающих электростатических взаимодействий с целевой протеазой в их связанном состоянии. Такие некомплементарные (например, электростатические) взаимодействия включают отталкивающие заряд-заряд, диполь-диполь и заряд-диполь взаимодействия. Более конкретно, в большинстве предпочтительных ингибиторов HIV протеазы настоящего изобретения сумма всех электростатических взаимодействий между соединением и энзимом, если соединение связано с энзимом, составляет нейтральный или благоприятный вклад в энтальпию связывания.
Предпочтительные соединения, отличающиеся вышеуказанными чертежами (1)-(6), являются соединениями формулы XL:
Z1-Q1-L1-M-L2-Q2-Z2, (XL)
где Q1 и Q2 независимо представляют акцепторный фрагмент водородной связи, способный связываться с атомами водорода молекулы воды щитка HIV аспартил-протеазы, при условии, что, по крайней мере, один из Q1 или Q2 более сильно поляризуется, нежели карбонил.
M является связывающим водород фрагментом, который может быть либо донором, либо акцептором водородной связи, способным к одновременному связыванию с Asp25 и Asp25' указанной HIV аспартил-протеазы,
Z1 и Z2 являются независимо ациклическими или циклическими линкерными фрагментами и
каждый из Z1 и Z2 может присутствовать необязательно, и, если присутствует, его независимо выбирают из групп, которые занимают объем пространства, перекрывающийся с объемом пространства, которое должно быть занято нативным субстратом указанной HIV аспартил протеазы.
Более предпочтительные соединения формулы XL содержат, по крайней мере, одну группу Q1 или Q2, содержащую -SO2-. Наиболее предпочтительные соединения формулы XL содержат, по крайней мере, одну группу Q1 или Q2, содержащую замещенный сульфонамид.
В одном варианте настоящего изобретения соединения формулы XL могут быть дополнительно "затруднены" "конформационными блокировками", такими как макроциклические кольцевые структуры. Такие затруднения хорошо известны специалистам пептидомиметики и могут привести к соединениям с высокой биологической активностью. См., например, Dhanoa, D.S. et.al. "The Synthesis of Potent Macrocyclic Renin Inhibitors" Tetrahedron Lett. 33, 1725 1992) and Flynn, G.A.et al. "An Acyl - Iminium Ion Cyclization Route to a Novel Conformationally Restricted Dipeptide Mimic : Applications to Angiotensin - Converling Enzyme Inhibition" J.Am. Chem. Soc. 109, 7914 (1989)).
Настоящее изобретение включает также новые способы для точной идентификации, конструирования или предсказания HIV ингибиторов, отличающихся структурными и физико-химическими отличительными особенностями (1) - (6). С помощью этих способов специалисты могут легко предсказать и получить особенно эффективные ингибиторы HIV протеазы.
Мы обнаружили, что наиболее эффективны следующие способы идентификации, конструирования или предсказания эффективных ингибиторов HIV протеазы:
(а) выбор соединения-кандидата определенной химической структуры, которое содержит первый и второй акцепторный фрагмент для образования водородной связи, причем, по крайней мере, один из них более поляризуем, нежели карбонил, и указанные фрагменты одинаковы или различны; третий фрагмент, образующий водородную связь, который может быть либо донором, либо акцептором водородной связи; и по крайней мере, два существенно гидрофобных фрагмента,
(b) определение низкоэнергетической конформации для связывания указанного соединения с активным сайтом HIV аспартил-протеазы,
(c) оценка способности указанных первого и второго акцепторных фрагментов, образующих водородную связь, к образованию водородной связи с молекулой воды щитка указанной HIV аспартил-протеазы, если указанное соединение связано с ней в указанной конформации,
(d) оценка способности указанных существенно гидрофобных фрагментов к ассоциации с P1 и P'1 связывающими карманами указанной аспартил-протеазы, если указанное соединение связано с ней в указанной конформации;
(e) оценка способности указанного третьего образующего водородную связь фрагмента образовывать водородные связи с Asp25 и Asp25' указанной HIV аспартил-протеазы, если указанное соединение связано с ней в указанной конформации,
(f) оценка перекрывания объема, занятого указанным соединением, если указанное соединение связано с указанной HIV аспартил-протеазой в указанной конформации, и объема, занятого нативным субстратом HIV аспартил-протеазы или ее негидролизуемым изостером, если указанный полипептид связан с указанной аспартил-протеазой,
(g) оценка энергии деформации связывания указанного соединения с указанной аспартил-протеазой,
(h) оценка вклада в энтальпию суммы всех электростатических взаимодействий указанного соединения и указанной HIV аспартил-протеазы, если указанное соединение связано с ней в указанной конформации, и
(i) принятие или отклонение указанного соединения-кандидата в качестве ингибитора HIV протеазы, на основании определений и оценок, осуществленных в п. (b) - (h).
Используя новые комбинации изложенных ранее стадий в способе скринирования, специалисты могут с успехом избежать потерь времени и дорогостоящих экспериментов для определения энзимативной ингибирующей активности конкретных соединений. Этот способ также пригоден для облегчения рационального конструирования ингибиторов HIV протеазы и анти-HIV-вирусных агентов, включая терапевтические и профилактические агенты против HIV инфекций. Соответственно, настоящее изобретение относится к таким ингибиторам и антивирусным агентам, которые получают в результате описанного способа скринирования.
Для осуществления вышеописанных оценок можно использовать различные обычные методики. Обычно эти способы включают определение положения и близости связывания указанного фрагмента, объем пространства, занимаемый связанным соединением, энергию деформации связывания указанного соединения и энергии электростатического взаимодействия. Примеры обычных методик, подходящих для вышеуказанных оценок, включают методы квантовой механики, молекулярной динамики, способ Монте-Карло, способы систематического поиска и дистанционной геометрии (G.R. Marshall, Ann, Ref. Pharmacol. Toxicol., 27, p. 193 (1987)). Для осуществления этих способов были созданы специальные компьютерные программы. Примеры таких программ включают: Gaussian 92, вариант C, (M.J. Frisch, Gaussian, Inc., Pittsburgh, PA 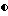 1992); AMBER, version 3.0 (U. C. Singh, University of California at San Franciscо,
1992); AMBER, version 3.0 (U. C. Singh, University of California at San Franciscо, 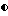 1992); QUANTA/CHARMM (Molecular Simulations, Inc., Burlington, MA
1992); QUANTA/CHARMM (Molecular Simulations, Inc., Burlington, MA  1992), and Insight II/Discover (Biosysm Technologies Inc., San Diego, CA
1992), and Insight II/Discover (Biosysm Technologies Inc., San Diego, CA 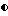 1992). Эти программы можно дополнить, например, используя Silicon Graphies Workstation, IRIS 4D/35 or IBM RISC/6000 workstation model 550.
1992). Эти программы можно дополнить, например, используя Silicon Graphies Workstation, IRIS 4D/35 or IBM RISC/6000 workstation model 550.
Остальные системы обеспечения и пакеты программ известны, и их применение очевидно для специалистов.
Дополнительный анализ реальных детализированных взаимодействий комплекса HIV протеаза-ингибитор можно использовать для более конкретного определения связывающих ассоциаций между энзимом и связанным ингибитором. Такой анализ можно осуществить, например, изучая раствор комплекса с помощью одномерных и многомерных методик ЯМР. Выгодно по возможности обогатить энзим и/или ингибитор такими стабильными изотопами, как 13C, 15N и 2H для более точного определения связывающей конформации и расстояний. Для повышения разрешения при наблюдении взаимодействий можно использовать такие методики, как введение изотопов.
В качестве дополнительных или альтернативных исследований комплекса HIV протеаза-ингибитор можно изучать дифракцию рентгеновских лучей. Процесс определения структур комплексов протеин/ингибитор с использованием рентгеноскопических методик, описанных ранее, хорошо известен и уже использовался для многих различных комплексов (см. N.J. Blundel and L.N. Johnson, Protein Crystallography, Academic Press (1976) and Methods in Entymology, Vol. 114 and 115, H.W. Wyckoff et al., eds., Academic Press (1985)).
Этот способ можно использовать, например, с высокой степенью очистки препаратами HIV протеазы, закомплексованными с представляющим интерес ингибитором в буферированном растворе (обычно при pH от около 4,5 до около 8,0). Комплексы дают возможность закристаллизоваться в присутствии осаждающего агента (например, сульфата аммония) в условиях, которые приводят к образованию отдельных кристаллов комплекса. Конкретные условия для кристаллизации HIV протеазы с различными ингибиторами были хорошо документированы (см., например, G.B. Dreyert et al,, Biochemistry, 31, p. 6646 (1992)). Применение сфокусированного пучка рентгеновских лучей и соответствующим образом подготовленного и установленного кристалла (предпочтительно пучка рентгеновских лучей от вращающегося анодного генератора рентгеновских лучей или синхротрона) обеспечивает получение дифракционной картины от отраженного пучка рентгеновских лучей.
Определение дифракционной картины можно осуществить с помощью визуализации на фотобумаге, экспонируемой рентгеновским лучом, или в другом варианте, используя специальный детектор (такой, как изготовляемый Siemens Analytical X-Ray Instruments, Inc. (Madison, W1) или систему изображений R-axi II от Rigaku Corporation (поставляемую Molecular Structurе Corporation, the Woodlands, TX). Другие системы для создания и обработки данных по дифракции рентгеновских лучей известны специалистам.
Расшифровка данных по дифракции рентгеновских лучей дает трехмерную структуру. Для этой расшифровки создана компьютерная программа (X-PLOR) Yate University, 1992, распространяемая Molecular Simulation, Inc.).
Вообще, используя вышеописанные методики с соответствующим образом подготовленным кристаллическим комплексом, можно определять структуру с точностью около 2 - 3  (при R = 0,25 или менее). Как очевидно специалистам, эти значения достаточны для определения взаимодействий между HIV протеазой и данным соединением, так что становится ясно, выполняются ли условия (1) - (6) и, соответственно, является ли данное соединение ингибитором HIV аспартил-протеазы. Итак, в соответствии с настоящим изобретением могут быть сконструированы и предсказаны исполнительные ингибиторы на основании сочетания кристаллографической структурной информации и расчетных методов.
(при R = 0,25 или менее). Как очевидно специалистам, эти значения достаточны для определения взаимодействий между HIV протеазой и данным соединением, так что становится ясно, выполняются ли условия (1) - (6) и, соответственно, является ли данное соединение ингибитором HIV аспартил-протеазы. Итак, в соответствии с настоящим изобретением могут быть сконструированы и предсказаны исполнительные ингибиторы на основании сочетания кристаллографической структурной информации и расчетных методов.
Так, например, для того, чтобы предсказать связывание предполагаемого ингибитора по способу настоящего изобретения, этот ингибитор исследуют на предмет определения того, содержит ли эта молекула функциональности, которые не достаточно хорошо представлены в существующих моделях силового поля в CHARMM (Molecular Simulation Incorporated Burnington, MA) или AMBER (профессор. P. A. Kollman, UC SF). Если какая-либо функциональность представлена недостаточно хорошо, мы исследуем всю опубликованную информацию по структурам для молекул, содержащих такие функциональности, и в некоторых случаях проводим высокоэффективные ab initio расчеты на простых молекулах, содержащих такие функциональности, для определения их предпочтительных конформаций и разностей энергий между различными конформациями. Более точные параметры, описывающие эти функциональные группы, можно затем получить для силовых полей CHARMM и/или AMBER и использовать их в дальнейших расчетах.
Далее, предполагаемый ингибитор располагают в трехмерном пространстве наряду с другими, родственными ингибиторами, конформации связей для которых были ранее определены с помощью кристаллографических исследований рентгеновских лучей. Для управления процессом сравнения используют как Ван-дер-Вальсовский объем, так и электростатические потенциалы. Такое сравнение обычно осуществляют с помощью таких программ, как Quantal (Molecular Simulations) или Insight 11 (Biosym Technologies, San Diego, CA).
Такое сравнение можно осуществить и вручную, используя эти программы, или за счет автоматизированной процедуры в таких программах (например, методом "наложения" или Quanta или "APEX" модулей Insight 11). Результат такого сравнения является первой информацией о конформации "связей" кандидата в ингибиторы. Затем этот ингибитор помещают в активный сайт HIV протеазы и минимизируют энергию конформации при фиксированных в пространстве атомах энзима. Такую минимизацию обычно осуществляют, используя CHARMM или AMBER силовые поля.
Так как ингибиторы могут иногда связываться в множественные или неожиданные конформации внутри активного сайта, мы часто осуществляем дополнительные поиски конформации связей комплекса энзим-ингибитор. Так, например, можно использовать различные методики поиска Монт-Карло (как, например, в Cоnformational Search Module of Quanta), наряду с высокотемпературной динамикой и симулированным отжигом. Эти методы поиска позволяют определить, существуют ли альтернативные, разумные низкоэнергетические конформации, в которых ингибитор может быть связан с энзимом. Эффект сольвации и десольвации при образовании различных комплексов энзим-ингибитор может быть оценен за счет таких программ, как DEZPH1 (Biosym), Polaris (Molecular Simulations) и AMSOL (профессор C. Cramer, University of Minnesota).
В результате таких поисков получают набор из одной или более связанных конформаций для кандидата в ингибиторы.
Для каждой из таких низкоэнергетических конформаций можно затем добавить молекулы воды к активным сайтам энзима и релаксировать всю систему. И, наконец, симуляции молекулярной динамики можно использовать для изучения подробных движений энзима, ингибитора и связанных молекул воды.
Окончательный набор оставшихся низкоэнергетических конформаций (обычно их остается очень мало) представляет предполагаемые конформации связей кандидата в ингибиторы. Каждая конформация включает нашу оценку динамической гибкости всей системы (ингибитор, энзим и молекулы воды).
Для изучения первых нескольких соединений в серии обычно используют более усовершенствованную методику, если существуют очень большие неопределенности возможных типов связывания в активном сайте энзима. Для остальных соединений в серии низкоэнергетические конформеры, полученные в результате исследований первых соединений, обеспечивают информацию для возможных энергетических конформеров ингибиторных соединений. Кроме того, кристаллографическая информация о конформации связанных комплексов первых соединений в серии часто бывает доступна. Такие предварительные расчеты и структурные исследования значительно облегчают предсказания конформации связей молекул-кандидатов в ингибиторы.
В качестве примера указанного способа скринирования мы осуществили следующую оценку соединения 140 (таблица II), предпочтительного соединения настоящего изобретения, как указано далее.
Предсказание конформации и энергии связывания соединения 140 с HIV протеазой.
Силовое поле для бензолсульфонамидной части соединения 140 получают из ab initio расчетов и включают в AMBER силовое поле. Последние CHARMM параметры силового поля для этого фрагмента, как оказалось, соответствовали результатам, полученным при исследовании минимизации энергии, и были использованы во всех Quanta /CHARMM расчетах.
Низкоэнергетические конформеры, полученные в результате конформационных исследований первых соединений в ряду сульфонамидов (таких, как соединение 16), обеспечили информацию о возможных низкоэнергетических конформерах соединения 140. Эти низкоэнергетические конформеры сравнивают в трехмерном пространстве с другими родственными ингибиторами, для которых конформации связей были ранее определены кристаллографически с использованием рентгеновских лучей. Такой процесс сравнения осуществляют с помощью Quanta и, в некоторых случаях, с помощью "конформационного поиска", необязательного для Quanta. При таком сравнении в качестве сравнительной кристаллической структуры используют комплекс HIV-1 протеазы с соединением 16. Энергию этой структуры ингибитора минимизируют в активном сайте энзима, используя Quanta/CHARMM. В процессе такой минимизации атомы энзима остаются фиксированными. Включают только воду "щитков". Последние симуляции позволяют энзиму релаксировать и используют различные диэлектрические приближения. Получают единственную низкоэнергетическую конформацию, которая согласуется со всеми полученными ранее конформационными симуляциями и кристаллографическими данными (см. фиг. 1). Как было позднее обнаружено, предсказанная конформация связывания находится практически в соответствии с результатами, полученными с помощью кристаллографии с использованием рентгеновских лучей (см. фиг. 2 и 3).
Как обсуждалось ранее, новые соединения настоящего изобретения являются превосходными лигандами для аспартил протеаз, особенно HIV-1 и HIV-2 протеаз. Соответственно, эти соединения способны метить и ингибировать последние стадии HIV репликации, то есть процессинг вирусных полипротеинов HIV кодируемыми протеазами. Такие соединения ингибируют протеолитический процессинг предшественников вирусных полипротеинов за счет ингибирования аспартил-протеазы. Так как аспартил-протеаза существенна для продуцирования зрелых вирионов, ингибирование такого процессинга эффективно блокирует размножение вирусов за счет ингибирования инфекционных вирионов, особенно из хронически инфицированных клеток. Соединения настоящего изобретения с успехом ингибируют способность HIV-1 вирусов инфицировать иммортализированные T клетки человека на протяжении дней, что определено в анализе внеклеточного p24 антигена-специфического маркера вирусной репликации. Остальные антивирусные анализы подтвердили эффективность этих соединений.
Соединения настоящего изобретения можно использовать обычным образом для борьбы с вирусами, такими как HIV и HTV, которые зависят от аспартил-протеаз для обязательных актов в их жизненном цикле. Такие способы лечения, уровни доз и другие требования могут быть подобраны специалистами на основании доступных методик и способов. Так, например, соединение настоящего изобретения можно объединить с фармацевтически приемлемым адъювантом для введения инфицированному вирусом пациенту фармацевтически приемлемым способом и в количестве, эффективном для уменьшения серьезности вирусной инфекции.
В другом варианте соединения настоящего изобретения можно использовать в вакцинах и в способах защиты индивидуумов от вирусной инфекции на протяжении длительного промежутка времени. Эти соединения можно использовать в таких вакцинах либо отдельно, либо вместе с другими соединениями настоящего изобретения способом, соответствующим обычному применению ингибиторов протеазы в вакцинах. Так, например, соединение настоящего изобретения можно объединить с фармацевтически приемлемыми адъювантами, которые обычно используют в профилактически эффективных количествах для защиты индивидуумов на протяжении длительного промежутка времени против HIV инфекции. Как таковые, новые ингибиторы протеазы настоящего изобретения можно вводить как агенты для лечения или предотвращения HIV инфицирования млекопитающих.
Соединения формулы I, особенно те, которые имеют молекулярный вес менее чем около 700 г/моль, можно легко абсорбировать в потоке крови млекопитающих при пероральном приеме. Соединения формулы I с молекулярным весом менее чем около 600 г/моль в большинстве своем, по-видимому, демонстрируют пригодность при пероральном введении. Такая столь удивительная пероральная доступность делает такие соединения превосходными агентами для лечения и профилактики с пероральным введением против HIV инфекции.
Соединения настоящего изобретения можно вводить здоровым или HIV инфицированным пациентам либо в виде единственного агента, либо в сочетании с другими антивирусными агентами, которые нарушают цикл репликации HIV. За счет введения соединений настоящего изобретения с другими антивирусными агентами, которые нацелены на различные акты жизненного цикла вирусов, терапевтический эффект этих соединений повышается. Так, например, совместно вводимый антивирусный агент может быть таким, который нацелен на ранние акты жизненного цикла вирусов, например, на проникновение в клетку, обратную транскрипцию и интеграцию вирусной ДНК в клеточную ДНК. Анти-HIV агенты, которые нацелены на такие ранние акты в жизненных циклах вирусов, включают диданозин (ddI), альцитабин (ddC), d4T, зидовудин (AZT), полисульфатированные полисахариды, ST4 (растворимый CD4), ганикловир, дидеоксицитидин, тринатрийфосфоноформат, элфорнитрин, рибавирин, ацикловир, альфа-интерферон и трименотрексат. Кроме того, не-нуклеозидные ингибиторы обратной транскриптазы, такие как TJBO или невирапин, можно использовать для усиления эффекта соединений настоящего изобретения, а также такие, как ингибиторы утраты вирусной оболочки, ингибиторы транс-активирующих протеинов, такие как tat или rev, или ингибиторы вирусной интегразы.
Объединенные терапии способа настоящего изобретения обладают синергетическим действием при ингибировании HIV репликации, так как каждый компонент-агент комбинации действует на различные сайты HIV репликации. Использование таких комбинаций также выгодно снижает дозы данного обычного антиретровирусного, которые должны были бы понадобиться для целевого терапевтического или профилактического эффекта, при сравнении с тем, если бы этот агент вводили как монотерапевтический. Эти комбинации могут снизить или исключить побочные эффекты обычных отдельных антиретровирусных терапий, и при этом не ухудшить антиретровирусную активность этих агентов. Такие комбинации снижают потенциал устойчивости при терапии за счет одного агента, минимизируя при этом сопутствующую токсичность. Эти комбинации могут также повысить эффективность обычных агентов, не повышая при этом их токсичности. В частности, мы обнаружили, что эти соединения действуют синергетически по предотвращению репликации HIV в T клетках человека. Предпочтительные комбинированные терапии включают введение соединения настоящего изобретения с AZT, dd1, ddC или d4T.
В другом варианте, соединения настоящего изобретения можно вводить совместно с другими ингибиторами HIV протеазы, такими как Ro 31-8959 (Roche), Z-735524 (Merck), XM 323 (Du-Pont Merck) и A-80987 (Abbot) для повышения эффекта лечения или профилактики против различных вирусных мутантов или членов других квази-видов HIV.
Мы предпочитаем введение соединений настоящего изобретения в виде отдельных агентов или в сочетании с ингибиторами обратной транскриптазы ретровирусов, например, производными AZT, или другими ингибиторами HIV аспартил-протеазы. Нам кажется, что совместное введение соединений настоящего изобретения с ингибиторами обратной транскриптазы ретровирусом или ингибиторами HIV аспартил-протеазы может оказывать существенный синергетический эффект, за счет чего наблюдается предотвращение, существенное снижение или полное исключение вирусной инфекционности и связанных с этим симптомов.
Соединения настоящего изобретения можно также вводить в сочетании с иммуномодуляторами (например, бропиримином, антиальфаинтерферон человека антителами, IL-2, GM-CSF, метионинэнкефалином, альфа-интерфероном, диэтилдитиокарбаматом, фактором некроза опухоли, налтрексоном и гЕРО) и антибиотиками (например, пентамидинизэтиоратом) для предотвращения или борьбы с инфекциями и заболеваниями, связанными с HIV инфекциями, такими как AIDS и ARC.
Если соединения настоящего изобретения вводят в комбинированной терапии с другими агентами, их можно вводить последовательно или одновременно. В другом варианте фармацевтические или профилактические композиции настоящего изобретения могут содержать комбинацию ингибитора аспартил-протеазы настоящего изобретения и другого терапевтического или профилактического агента.
Хотя настоящее изобретение сфокусировано на использовании раскрытых здесь соединений для предотвращения и лечения HIV инфекции, соединения настоящего изобретения можно также использовать в качестве ингибиторных агентов для других вирусов, которые зависят от аналогичных аспартил-протеаз для обязательных актов своих жизненных циклов. Эти вирусы включают также как и другие AIDS-подобные заболевания, вызываемые ретровирусами, такими как вирусы иммунодефицита simian (но не ограничиваются ими) HTLV-I и HTLV-II. Кроме того, соединения настоящего изобретения можно также использовать для ингибирования других аспартил-протеаз, и, в частности, других аспартил-протеаз человека, включая ренин, и аспартил-протеазы, которые процессируют предшественники эндотелина.
Фармацевтические композиции настоящего изобретения содержит любое из соединений настоящего изобретения и их фармацевтически приемлемые соли с любыми фармацевтически приемлемыми носителями, адъювантами или разбавителями. Фармацевтически приемлемые носители, адъюванты и разбавители, которые можно использовать в фармацевтических композициях настоящего изобретения, включают (хотя и не ограничиваются ими) ионообменные агенты, окись алюминия, стеарат алюминия, лецитин, протеины сыворотки, такие как альбумин сыворотки человека, такие буферные вещества, как фосфаты, глицин, сорбиновую кислоту, сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамин-сульфат, динатрий кислый фосфат, кислый фосфат калия, хлорид натрия, соли цинка, коллоидную двуокись кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу, полиакрилаты, воски, блок-сополимеры полиэтилен-полиоксиэтилена, полиэтиленгликоль и ланолин.
Фармацевтические композиции настоящего изобретения можно вводить перорально, парэнтерально, за счет ингаляций, поверхностно, ректально, через нос, под язык, вагинально или с помощью имплантаций контейнера. Предпочтительны пероральное введение и инъекции. Фармацевтические композиции настоящего изобретения могут содержать любые обычные нетоксичные, фармацевтически-приемлемые носители, адъюванты или наполнители. Термин парэнтерально, используемый здесь, включает подкожное, внутрикожное, внутривенное, внутримышечное, внутрисуставное, внутрисиновиальное, внутригрудинное, внутрикапсульное, внутриязвенное и внутриопухолевое введение за счет инъекций или инфузий.
Фармацевтические композиции могут быть в форме стерильных препаратов для инъекций, например, в виде стерильных водных или масляных суспензий для инъекций. Эти суспензии могут быть получены известными специалистам способами с применением диспергирующих или смачивающих агентов (таких, как например, Твин 80) и суспендирующих агентов. Стерильные препараты для инъекций могут быть также стерильными растворами или суспензиями для инъекций в нетоксичном, парэнтерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди подходящих носителей и растворителей, которые можно использовать, примерами могут служить маннит, вода, раствор Рингера и изотоничный раствор хлорида натрия. Кроме того, в качестве растворителей или суспендирующей среды обычно используют стерильные фиксированные масла. Для этих целей можно использовать любые смешанные масла, включая синтетические моно- и диглицериды. Жирные кислоты, такие как олеиновая кислота и ее диглицеридные производные, можно использовать для получения препаратов для инъекций, таких как природные фармацевтически приемлемые масла, например оливковое или касторовое масло, особенно в их полиэтоксилированном виде. Такие масляные растворы или суспензии могут также содержать длинноцепочечные спиртовые растворители или дисперсанты, такие как Ph.Helv или аналогичный спирт.
Фармацевтичесикие композиции настоящего изобретения можно вводить перорально в любых приемлемых для перорального приема дозовых формах, включая (но не ограничиваясь ими) капсулы, таблетки и водные суспензии и растворы. В случае таблеток для перорального применения, носители, которые обычно используют, включают лактозу и кукурузный крахмал. Такие скользящие, как стеарат магния, также обычно добавляют. Для перорального введения в форме капсул подходящие разбавители включают лактозу и высушенный кукурузный крахмал. Если перорально принимают водные суспензии, активный ингредиент соединяют с эмульгирующим и суспендирующим агентами. При желании можно добавлять подслащивающие, и/или ароматизирующие, и/или окрашивающие агенты,
Фармацевтические композиции настоящего изобретения можно также вводить в форме суппозиториев для ректального введения. Такие композиции можно приготовить за счет перемешивания соединения настоящего изобретения с подходящим нераздражающим эксципиентом, который остается твердым при комнатной температуре, но становится жидким при ректальной температуре и поэтому плавится в ректуме с высвобождением активных компонентов. Такие материалы включают (хотя и не ограничиваются ими) масло какао, пчелиный воск и полиэтиленгликоли.
Поверхностное нанесение фармацевтических композиций настоящего изобретения особенно подходит, если желательное лечение включает участки или органы, легко доступные при поверхностном нанесении. Для поверхностного нанесения на кожу фармацевтические композиции должны быть сформулированы с подходящими мазями, содержащими активные компоненты, суспензированные или растворенные в носителе. Носители для поверхностного введения соединений настоящего изобретения включают (но не ограничиваются ими), минеральные масла, жидкую нефть, белый петролеум, пропиленгликоль, соединения полиоксиэтилен-полиоксипропилена, эмульгирующий воск и воду. В другом варианте фармацевтические композиции можно получить из подходящих лосьонов или кремов, содержащих активное соединение, суспензированное или растворенное в носителе. Подходящие носители включают (но не ограничиваются ими) минеральные масла, сорбитанмоностеарат, полисорбат 60, воск цетиловых сложных эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Фармацевтические композиции настоящего изобретения можно также наносить поверхностно на нижнюю часть кишечного тракта с помощью композиций ректальных суппозиториев или в виде подходящих клизм. В объем настоящего изобретения включены также поверхностно-трансдермальные повязки.
Фармацевтические композиции настоящего изобретения можно вводить в виде аэрозолей через нос или за счет ингаляций. Такие композиции приготавливают в соответствии с хорошо известными специалистам-фармацевтам способами, и могут быть приготовлены в виде физиологических растворов с использованием бензилового спирта или других подходящих консервантов, ускорителей абсорбции для повышения биодоступности, фторуглеводородов и/или других солюбилизирующих агентов, известных специалистам.
Уровни доз от около 0,01 до около 100 мг/кг веса тела в день, предпочтительно от около 0,5 до около 50 мг/кг веса тела в день активного соединения, представляются подходящими для предотвращения и лечения вирусных инфекций, включая HIV инфекцию. Обычно фармацевтические композиции настоящего изобретения принимают от 1 до 5 раз в день, или, в другом варианте, в виде непрерывных вливаний. Такое лечение можно применять в качестве терапии острых или хронических состояний. Количество активного ингредиента, которое можно объединить с материалами носителя для получения единичной дозовой формы, будет меняться в зависимости от подлежащего лечению хозяина и конкретного способа введения. Обычно препарат содержит от около 5% до около 95% активного соединения (вес/вес). Предпочтительно, чтобы такие препараты содержали от около 20% до около 80% активного соединения.
После улучшения состояния пациента поддерживающую дозу соединения, композиции или комбинации настоящего изобретения можно вводить при желании. Соответственно, доза или частота приема или оба они могут быть уменьшены, в зависимости от симптомов, до уровня, при котором улучшившееся состояние сохраняется, когда симптомы бывают снижены до нужного уровня, лечение следует прекратить. Для пациента, однако, может понадобиться промежуточное лечение на длительной основе после возврата симптомов заболевания.
Как должно быть очевидно специалистам, более низкие или более высокие дозы, нежели те, которые указаны ранее, могут понадобиться. Конкретные дозы и схему лечения для каждого конкретного пациента определяют различные факторы, включающие активность конкретно используемого соединения, возраст, вес тела, общее состояние здоровья, пол, питание, время приема, скорость выведения, комбинацию лекарств, тяжесть и ход заболевания, предрасположенность пациента к инфекции и точку зрения лечащего врача.
Соединения настоящего изобретения могут быть полезны также в качестве коммерческих реагентов, которые эффективно связываются с аспартил-протеазами, особенно HIV аспартил-протеазой. В качестве коммерческих реагентов соединения настоящего изобретения и их производные можно использовать для блокировки протеолиза целевых пептидов или можно получить их производные, которые свяжутся со стабильной смолой в качестве связанного субстрата для применения в афинной хроматографии. Эти и другие применения, которые характеризуют коммерческие ингибиторы аспартил-протеазы, должны быть очевидны специалистам.
Для более полного понимания настоящего изобретения далее приводятся следующие примеры. Эти примеры даны только в иллюстративных целях и никоим образом не ограничивают объем изобретения.
Общие материалы и способы
Все температуры даны в градусах Цельсия. Тонкослойную хроматографию (ТСХ) осуществляют, используя Е.Merck силикагель 60 F254 пластины толщиной 0,25 мм и элюируя указанной системой растворителей. Детектирование соединений осуществляют, обрабатывая пластины соответствующим визуализирующим агентом, таким как 10% раствор фосфомолибденовой кислоты в этаноле, или 0,1% раствором нингидрина в этаноле с последующим нагреванием и/или экспонированием. УФ свету или парам иода. Хроматографию на толстом слое силикагеля также осуществляют на пластинах Merck 60 F254 ("преп пластины") толщиной 0,5, 1,0 или 2,0 мм. После проявления пластин полоски силикагеля, содержащие целевое соединение, выделяют и элюируют соответствующим растворителем. Аналитическую ВЭЖХ осуществляют, используя Water' Delta Pak, 5 мкм двуокись кремния, колонку с обращенной фазой C18, 3,9 мм внутр.диаметр x 15 см длины при скорости потока 1,5 мл/мин, используя следующую таблицу:
Подвижная фаза: A = 0,1% CF3CO2H в H2O
B = 0,1% CF3CO2H в CH3CN
Градиент: T = 0 мин, A (95%), B (5%)
T = 20 мин, A (0%), B (100%)
T = 22,5 мин, A (0%), B (100%)
Препаративную ВЭЖХ осуществляют так же, используя среду с обращенной фазой C18. Времена удерживания для ВЭЖХ фиксируют в минутах. Спектра ЯМР записаны на спектрометре Bruker АМХ 500, снабженном либо обратным, либо QNP зондом, на 500 МГц, и получены в указанном растворителе.
Были определены константы ингибирования для каждого соединения относительно HIV-I протеазы с использованием метода, описанного в основном M.W.Pennington et al., Peptides 1990, Gimet, E. and D, Andrew, Eds., Escom; Leiden, Netherlands (1990).
Соединения формулы I тестируют по их антивирусной эффективности в нескольких вирологических анализах. В первом анализе соединения добавляют в виде раствора в ДМСО к культуре тестовых клеток CCRM-CEM, штамма CD4+ Т-клеток лимфомы человека, предварительно остро инфицированных HIVIIIb, в соответствии со стандартным протоколом (см. MEEk, T.D. et al., "Inhibition of HIV-1 protease in infected T-lymphocytes by synthetic peptide analogues", Nature 343, p.90 (1990). Предпочтительными соединениями являются те, которые способны ингибировать 90% вирусной инфективности при концентрациях 1 мкМ или менее. Более предпочтительными соединениями являются те, которые способны ингибировать 90% вирусной инфекционности при концентрациях 100 нМ или менее.
Действие соединений на ингибирование репликации вирусов измеряют, определяя концентрацию HIV внеклеточного p24 антигена, используя коммерческий энзимный иммуноанализ (от Coulter Corporation, Hialeah, F.Z.).
В зависимости от типа клеток и нужных значений, образование синцитий, активность обратной транскриптазы (RT) или цитопатический эффект в результате способа захвата красителя можно также использовать как величины антивирусной активности. См. H.Mitsuya and S. Broder, "Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphoadenopathy - dideoxynucleosides", Proc. Natl. Acad. Sci. USA. y. 83, pp. 1911-1915 (1986). Действие соединений формулы I на клинические изоляты других HIV-1 штаммов определяют, получая (Iow-passaged) вирус от HIV-инфицированных пациентов и анализируя действие ингибиторов в плане предотвращения инфицирования HIV вирусами в свежеприготовленных моноядерных клетках периферической крови человека (РВМС).
Поскольку соединения формулы I способны ингибировать репликацию HIV вируса в T-клетках человека, и, кроме того, их можно поставлять млекопитающим перорально, очевидно, что они имеют клиническую применимость для лечения HIV инфекции. Эти тесты предсказывают способность соединений ингибировать HIV протеазу ин виво.
Пример 1
A. Соединение XI ((син)-OH, D' = бензил). 184 г нейтральной окиси алюминия (Brockman Suрer 1) суспендируют в достаточном количестве диэтилового эфира, для того, чтобы получить перемешиваемую густую суспензию, и обрабатывают 7,48 мл бензиламина. После перемешивания в течение 5 минут добавляют 7,28 г (1S,2S)-1-(N- бензилоксикарбонил)-амино-2-фенилэтил-оксирана, и полученную смесь перемешивают в течение 15 часов. Полученную смесь обрабатывают 15,28 г ди-трет-бутилпирокарбоната и 4,70 мл диизопропилэтиламина. Полученную смесь перемешивают в течение 3,5 часа, затем обрабатывают 600 мл метанола, оставляют выстаиваться в течение 3,5 часа и фильтруют до получения масла желтого цвета, которое очищают хроматографически на силикагеле, используя градиент от 0,5 до 1,5% метанола в метиленхлориде до получения 3,88 г целевого продукта в виде белого твердого вещества. После дальнейших промывок фильтровальной лепешки метанолом и 3% гидроксидом аммония в метаноле, получают 2,2 г 4-бензиламино-2-N-бензилоксикарбониламино-3-гидрокси- 1-фенилбутана в нескольких порциях. Каждую из этих порций обрабатывают отдельно, в виде раствора в метиленхлориде, 1,1 молярными экивалентами каждого из ди-трет-бутилпирокарбоната и диизопропилэтиламина, с последующей обработкой водой, 10% водным KHSO4 и рассолом, затем сушат над сульфатом магния и концентрируют в вакууме. Объединенные продукты этих реакций очищают хроматографически на силикагеле, используя градиент от 5% до 15% диэтилового эфира в метиленхлориде. Полученные чистые фракции собирают и комбинируют с очищенным ранее продуктом, получая 5,49 г белого твердого вещества. ТСХ: Rf = 0,56 метанол (CH2Cl2), 1H-/ЯМР/CDCl3/ соответствует структуре.
B. Соединение XII ((син)-OH, D' = бензил). Раствор 5,49 г полученного соединения примера 1A в 40 мл этанола гидрируют при слегка избыточном давлении водорода в присутствии 380 мг 10% палладия на угле в течение 16 часов. После фильтрования и концентрирования в вакууме целевой продукт получают в виде 4,03 г белого твердого продукта. ТСХ: Rf = 0,21, 95:5:0,5 (CH2Cl2) (метанол) концентрированный NH4OH.
C. Соединение XIII ((син)-OH, A - бензилоксикарбонил, D' = бензил). Раствор 3,02 г соединения, полученного в примере 1B, в 150 мл метиленхлорида обрабатывают 4,35 г Nα- Cbz- Nδ- тритиласпарагина, 1,16 г гидроксибензотриазолгидрата и 1,64 г 1-(3-диметиламинопропил)-3-этил-карбодиимидгидрохлорида. Полученную смесь перемешивают в течение 16 часов, разбавляют 3 объемами диэтилового эфира и промывают последовательно водой, насыщенным бикарбонатом натрия, 10% раствором KHSO4 и рассолом. После сушки над сульфатом магния и концентрирования в вакууме, получают масло желтого цвета, которое хроматографически очищают на колонке Florisil, используя градиент от 0 до 25% EtOAc в CH2Cl2 в качестве элюента до получения 8,00 г указанного в заглавии соединения в виде белой пены. ТСХ: Rf = 0,51, 5% метанол) (CH2Cl2, )/1H/-ЯМР (CDCl3) соответствует структуре.
C. Соединение XIV ((син)-OH, A = H, D' = бензил)
Раствор 7,90 г соединения примера 1C в 150 мл этанола гидрируют при несколько избыточном давлении водорода в присутствии 550 мг 10% палладия на угле в течение 2,5 часа, затем добавляют примерно 50 мг 10% палладия на угле, полученную смесь фильтруют и концентрируют в вакууме до получения целевого продукта в виде 6,66 г белого твердого вещества, которое используют без дальнейшей очистки, ТСХ: Rf = 0,26, 95:5:0,5 CH2Cl2 (метанол) концентрированный NH4OH.
E. Соединение XIV ((син)-OH, A = зинолин-2-карбонил, D' = бензил). Суспензию 1,51 г хинальдиновой кислоты и 6,17 г соединения, полученного в примере 1D, в 150 мл ацетонитрила, обрабатывают 1,52 мл диизопропилэтиламина и 3,58 г ВОР реагента. Полученную смесь перемешивают в течение 14 часов, затем концентрируют в вакууме. Смолистый остаток разделяют между эфиром и водой, органический слой промывают последовательно рассолом, насыщенным раствором NaHCO3, водой, 10% KHSO4 раствором и рассолом, затем сушат над сульфатом магния и концентрируют в вакууме. Последующая хроматографическая очистка на силикагеле с использованием от 0 до 8,5% растворителя A в метиленхлориде (где растворитель A представляет смесь 90:10:1 метиленхлорид/метанол/концентрированный гидроксид аммония), дает 5,79 г указанного в заглавии соединения в виде белой пены, наряду с 600 мг слегка загрязненных боковых фракций. ТСХ: Rf = 0,41, 5% метанол /CH2Cl2, /1H/-ЯМР/CDCl3/ соответствует структуре.
F. Соединение 1. Порцию в 58 мг соединения, полученного в примере 1E, обрабатывают 1 мл 90% водного TFA и оставляют выстаиваться в течение 17 часов. Полученную смесь концентрируют в вакууме, а остаток помещают в 3 мл CH2Cl2, обрабатывают 100 мкл DJEA и охлаждают до 0oC. К этому раствору добавляют 26 мкл бензолсульфонилхлорида, и полученную смесь перемешивают в течение 18 часов, медленно нагревая до комнатной температуры. После концентрирования этой смеси в вакууме, остаток очищают с помощью тонкослойной хроматографии на силикагеле, используя 5% MeOH/CH2Cl2 в качестве элюента, а затем с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 40 до 100% CH3CN/H2O с 0,1% TFA для элюирования, до получения 8,3 мг указанного в заглавии соединения. ТСХ: Rf = 0,50, 5% MeOH/CH2Cl2. ВЭЖХ: Rт = 17,8 мин.
ЯМР (ДМСО-d6) δ: 2,62 (дд, 1H), 2,76 (дд, 2H), 2,80 (дд, 1H), 3,11 (д, 2H), 3,34 (дд, 1H), 4,59 (шир.с., 1H), 4,68 (шир.с., 1H), 3.97 (м, 1H), 4,20 (д, 1H), 4,35 (д, 1H), 4,68 (дд, 1H), 6,39 (д, 1H), 6,74 (т, 1H), 6,81 (т, 2H), 6,93 (д, 2H), 7,12-7,24 (м, 6H), 7,51 (т, 2H), 7,57 (т, 1H), 7,62 (дд, 1H), 7,77 (т, 2H), 7,97 (д, 1H), 8,09 (д, 1H), 8,16 (д, 1H), 8,31 (д, 1H), 8,53 (д, 1H).
Пример 2
Соединение 2. Порцию в 150 мг соединения, полученного в примере 1E, растворяют в 1 мл 90% водного TFA и перемешивают при комнатной температуре в течение ночи, затем концентрируют в вакууме. Остаток неочищенной соли TFA растворяют в 7 мл сухого метиленхлорида, и pH раствора устанавлвают pH 8 с помощью 1н NaOH. 56 мг смеси 4-фтор-3-ацетамидобензолсульфонилхлорида и 3-фтор-4-ацетамидобензолсульфонилхлорида (примерно 1:1) добавляют, и полученную смесь интенсивно перемешивают в течение 3 часов, после чего добавляют еще 25 мг, и реакции дают протекать еще 12 часов. Затем реакционную смесь разбавляют 50 мл этиленхлорида, и органический слой промывают последовательно водой и рассолом, сушат над сульфатом магния и концентрируют в вакууме. Сырой остаток очищают, используя флеш-хроматографию на колонке с силикагелем, используя градиент от 3 до 5% MeOH в метиленхлориде в качестве элюента до получения 60 мг указанных в заглавии соединений. TCX: Rf = 0,50, 1% MeOH/CH2Cl2, ВЭЖХ: Rт = 13,93 мин.
ЯМР (CDCl3): δ: 0,5 (с, 1H), 8,65 (д, 0,5H), 8,58 (т, 0,5H), 8,20 (дд, 0,5H), 7,85 (д, 1H), 7,74 (м, 0,5H), 7,45-7,63 (м, 1,5H), 7,14-7,25 (м, 6H), 6,78-6,95 (м, 5H), 6,70 (д, 1H), 6,41 (с, 0,5H), 6,25 (с, 0,5H), 6,18 (с, 0,5H), 6,10 (с, 0,5H), 4,88 (а, 0,5H), 4,81 (м, 0,5H), 4,37 (д, 1H), 4,35 (м, 1H), 4,21 (д, 1H), 4,00 (м, 1H), 3,46 (м, 0,5H), 3,35 (м, 0,5H) 3,27 (д, 0,5H), 3,16 (д, 0,5H), 3,14 (д, 1H), 2,45-2,75 (м, 5H), 2,16, 2,20 (2с, 3H все).
Пример 3.
Соединение 3. Порцию в 23 мг соединения, полученного в примере IE, обрабатывают 1 мл 90% водного TFA и оставляют выстраиваться в течение 15 часов. Полученную смесь концентрируют в вакууме, а остаток помещают на 2 мл CH2Cl2, обрабатывают 6 мкл DJEA и охлаждают до 0oC. К этому раствору добавляют 23 мг 3,5-диметилизоксазол-4-сульфонилхлорида, и полученную смесь перемешивают в течение 18 часов, медленно нагревая до комнатной температуры. После концентрирования смеси в вакууме остаток очищают с помощью препаративной хроматографии с обращенной фазой C18, используя линейный градиент от 35% до 100% CH2/CN/H2O с 0,1% TFA для элюирования до получения 1,1 мг указанного в заглавии соединения. TCX: Rf = 0,55, 10% MeOH/CH2Cl2. ВЭЖХ: Rт = 14,5 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 4.
Соединение 4. Порцию в 33 мг соединения, полученного в примере IE, обрабатывают 1 мл 90% водного TFA и оставляют выстаиваться в течение 15 часов. Полученную смесь концентрируют в вакууме, и остаток помещают в 3 мл CH2Cl2, обрабатывают 16 мкл DJEA и охлаждают до 0oC. К этому раствору добавляют 10 мкл 3-трифторметилбензолсульфонилхлорида, и полученную смесь перемешивают в течение 18 часов, медленно нагревают до комнатной температуры. После концентрирования смеси в вакууме остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA для элюирования до получения 1,1 мг указанного в заглавии соединения. ТСХ: Rf = 0,55, 10% MeOH/CH2Cl2. ВЭЖХ: Rт = 14,5 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 5.
Соединение 5. Порцию в 20 мг соединения, полученного в примере IE, обрабатывают 1 мл 90% водного TFA и оставляют выстаиваться в течение 18 часов. Полученную смесь концентрируют в вакууме, а остаток помещают в 1 мл CH2Cl2, обрабатывают 10 мкл DJEA и охлаждают до 0oC. К этому раствору добавляют 13 мг 2-ацетамидо-4-метил-5-тиазолсульфонилхлорида, и полученную смесь перемешивают в течение 17 часов, медленно нагревали до комнатной температуры. После концентрирования смеси в вакууме остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA для элюирования до получения 0,40 мг указанного в заглавии соединения. ТСХ: Rf = 0,5, 10% MeOH/CH2Cl2. ВЭЖХ: Rт = 13,8 мин. /1H-ЯМР (CDCl3) соответствует структуре.
Пример 6.
Соединение 6. Порцию 33 мг соединения, полученного в примере IE, обрабатывают 1 мл 90% водного TFA и оставляют выстаиваться в течение 16 часов. Полученную смесь концентрируют в вакууме, а остаток помещают в 2 мл CH2Cl2, обрабатывают 16 мкл DJEA и охлаждают до 0oC. К этому раствору добавляют 11 мг 5-(изоксазол-3-ил)тиофен-2-сульфонилхлорида, и полученную смесь перемешивают в течение 18 часов, медленно нагревая до комнатной температуры. После концентрирования смеси в вакууме, остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA для элюирования до получения 1,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,7, 10% MeOH/CH2Cl2. ВЭЖХ: Rт = 14,7 мин.
/1H/-ЯМР (CDCl3) соответствует структуре.
Пример 7.
Соединение 7. Порцию в 35,5 мг соединения, полученного в примере I E, обрабатывают 1 мл 90% водного TFA и оставляют выстаиваться в течение 18 часов. Полученную смесь концентрируют в вакууме, а остаток помещают в 3 мл CH2Cl2, обрабатывают 16 мкл DJEA и охлаждают до 0oC. К этому раствору добавляют 10 мг 3-хлорсульфонилбензойной кислоты, и полученную смесь перемешивают в течение 16 часов, медленно нагревая до комнатной температуры. После концентрирования смеси в вакууме, остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA для элюирования до получения 1,6 мг указанного в заглавии соединения. ТСХ: Rf = 0,7 10% MeOH/CH2Cl2. ВЭЖХ: Rт = 13,6 мин.
/1H/-ЯМР (CDCl3) соответствует структуре.
Пример 8.
Соединение 8. 0,04 ммоля соединения примера 10A превращают в свободное основание, разделяя между EtOAc с насыщенным NaHCO3. Обработка полученного соединения избытком 1% HCl/MeOH и концентрирование в вакууме дает гидрохлоридную соль в виде белого твердого вещества. Это соединение суспендируют в CH2Cl2 и обрабатывают достаточным количеством DJEA для того, чтобы довести pH до более 10 (влажная pH индикаторная бумага). Полученный раствор обрабатывают 7 молярными эквивалентами хлорметилсилана и перемешивают в течение 15 часов в атмосфере азота, затем обрабатывают 0,06 ммолями метансульфонилхлорида и перемешивают в течение 1 часа. Полученную смесь концентрируют до небольшого объема, наносят непосредственно на толстую пластину силикагеля и элюируют 7% MeOH/CH2Cl2. Основную поглощающую УФ полосу выделяют и далее очищают с помощью препаративной ВЭЖХ с обращенной фазой до получения указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,65, 10% CH3OH/CH2Cl2, ВЭЖХ: Rт = 12,3 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Примеры 9 и 192
A. Соединение XIV ((син, анти-OH. A = хинолин-2-карбонил, D' = изобутил). Раствор 317 мг (0,425 ммолей) соединения, полученного в примере 17B, диастереоизомера B, и 0,11 мл (0,637 ммолей) диизопропилэтиламина в 7 мл дихлорметана обрабатывают 139,1 мг (0,637 ммоля) ди-трет-бутилдикарбоната. Спустя 24 часа полученную смесь разбавляют дихлорметаном. Полученную смесь промывают водой, 5% NaHCO3, 0,5 н HCl, рассолом, а затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученный остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 20% этилацетат/дихлорметан в качестве элюента до получения 81,2 мг быстро перемещающегося гидроксильного диастереизомера, 65,8 мг медленно перемещающегося гидроксил-диастереизомера и 65,8 мг смеси диастереизомеров, TCX: Rf = 0,67, 40% EtOAc/CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
B. Соединения 9 и 192. Раствор 35,1 мг (0,041 ммоля) полученной смеси диастереизомеров (1:1) примера 9/192A в 0,8 мл дихлорметана обрабатывают 0,8 мл трифторуксусной кислоты. Спустя 4 часа полученную смесь концентрируют в вакууме. TCX: Rf = 0,11, 10% CH3OH/CH2Cl2. К раствору полученной соли трифторуксусной кислоты (весь выход) в 1 мл дихлорметана последовательно добавляют 0,3 мл насыщенного NaHCO3, небольшое количество твердого NaHCO3 и 11,8 мг (0,054 ммоля) бензофуразан-4-сульфонилхлорида. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют один раз дихлорметаном. Объединенный органический слой промывают рассолом, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 2,0 мг соединения 9 в виде белого твердого вещества: TCX:Rf = 0,20, 5% CH3OH/CH2Cl2, ВЭЖЧ, Rт = 14,2 мин. 2,7 мг соединения 192 также получают в виде белого твердого вещества, которое по данным ЯМР и ВЭЖХ содержит примерно 25% соединения 9:TCX:Rf = 0,20, 5% CH3OH/CH2Cl2, ВЭЖХ, Rт = 14,2 мин /1H/-ЯМР соответствует структуре.
Пример 10.
А. Соединение XV ((син)-OH, A = хинолин-2-карбонил, D'=бензил; TFA соль). Раствор при 0oC 1,027 г соединения, полученного в примере IE, в 5 мл CH2Cl2, обрабатывают 5 мл TFA и оставляют выстаиваться в течение 3 часов. Полученную смесь концентрируют в вакууме до получения 0,95 г указанного в заглавии соединения, которое используют без дальнейшей очистки.
В. Соединение 10. Раствор 30,2 мг соединения, полученного в примере 10A, в 3 мл CH2Cl2, обрабатывают 0,33 мл DJEA и 31,1 мг m-бензолсульфонилхлорида. Полученную смесь перемешивают в течение 2 часов, затем обрабатывают 2 мл концентрированного водного гидроксида аммония. Двухфазную смесь перемешивают еще в течение 16 часов, концентрируют в вакууме, а остаток разделяют между этилацетатом и рассолом. Органический слой сушат над сульфатом магния и концентрируют в вакууме, а остаток очищают с помощью препаративной хроматографии на толстом слое силикагеля, используя 3% MeOH/CH2Cl2 в качестве элюента до получения 4,5 мг указанного в заглавии соединения. TCX:Rf = 0,5, 3% MeOH/CH2Cl2. ВЭЖХ:Rт = 13,4 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 11
Соединение 11. Раствор 57,9 мг соединения, полученного в примере 10A, в 5 мл CH2Cl2, обрабатывают 30 мкл DJEA и 9,3 мкл диметилсульфонилхлорида. Полученную смесь перемешивают в течение 12 часов, затем обрабатывают дополнительными 30 мкл DJEA и 9,3 мкл диметилсульфамоилхлорида и реакцию ведут еще 12 часов. Полученную смесь разбавляют затем CH2Cl2 и промывают насыщенным NH4Cl, водный слой промывают CH2Cl2, а объединенные органические экстракты сушат над сульфатом магния. В результате фильтрования и концентрирования получают остаток, который обрабатывают на хроматографической колонне, используя 2,5% MeOH/EtOAc в качестве элюента. Получают продукт с небольшими примесями, его далее очищают с помощью препаративной ВЭЖХ, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA для элюирования. ВЭЖХ:Rт = 13,0 мин. ЯМР (CDCl3): δ 9,15 (д, 1H), 8,34 (д, 1H), 8,22 (д, 1H), 8,18 (д, 1H), 7,90 (д, 1H), 7,80 (т, 1H), 7,65 (т, 1H), 7,16 - 7,38 (м, 5H), 7,05 (д, 1H), 6,95 (т, 1H), 6,87 (т, 1H), 5,85 (шир.с, 1H), 5,62 (шир.с, 1H), 4,87 (м, 1H), 4,46 (с, 2H), 4,08 (м, 1H), 3,66 (м, 1H), 3,30 (м, 2H), 2,59 - 2,94 (м, 4H), 2,81 (с, 6H).
Пример 12.
А. Соединение XIV ((син)-OH, A = хинолин-2-карбонил, D'=бензил, соль трифторуксусной кислоты). К раствору 1,027 г (1,164 ммоля) соединения, полученного в примере IE, в CH2Cl2 (5 мл), при температуре от 0 до 5oC добавляют трифторметансульфоновую кислоту (5 мл). После перемешивания в течение 3 часов реакционную смесь концентрируют в вакууме до получения 0,95 г светло-желтого, смолистого продукта, содержащего один эквивалент трифенилметанола, который используют без последующей очистки.
В. Соединение 12. К раствору 30,2 мг (0,038 ммоля) соединения, полученного в примере 12A, в CH2Cl2 (3 мл), добавляют диизопропиэтиламин (0,33 мл, 0,189 ммолей) и 2-(пирид-2-ил)-тиофен-5-сульфонилхлорид (13 мг, 0,249 ммоля). Спустя 14 часов полученную смесь разбавляют этилацетатом, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной хроматографии с обращенной фазой, используя 5% - 100% градиент H2O/ацетонитрила в качестве элюента, до получения указанного в заглавии продукта.
Пример 13.
Соединение 13. К раствору 30 мг (0,038 ммолей) соединения, полученного в примере 12A, в CH2Cl2 (3 мл) добавляют диизопропилэтиламин (0,33 мл, 0,189 ммолей) и 2-(3-фенил-сульфонил)-тиофенсульфонилхлорид (0,113 ммоля). После перемешивания в течение 2 часов, реакционную смесь разделяют на две фазы, добавляя 30% раствор гидроксида аммония (2 мл). После перемешивания в течение дополнительных 16 часов полученную смесь концентрируют в вакууме, снова растворяют в этилацетате, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и сушат в вакууме. В результате очистки с помощью препаративной хроматографии получают целевое соединение.
Пример 14.
Соединение 14. Соединение, полученное в примере 17B, диастереоизомер B (170 мг) обрабатывают 1 мл 90% водной TFA и оставляют выстаиваться в течение 12 часов. Полученную смесь концентрируют в вакууме, а остаток помещают в 5 мл сухого CH2Cl2. К этому раствору добавляют 3 мл насыщенного водного бикарбоната натрия и 50 мг 4-фторбензолсульфонилхлорида, и полученную смесь перемешивают в течение 3 часов. Полученную смесь разбавляют CH2Cl2 и промывают водой, сушат над сульфатом магния и фильтруют. После концентрирования смеси в вакууме часть остатка очищают с помощью препаративной хроматографии с обращенной фазой C18, используя линейный градиент от 35% до 100 CH3CN/H2O с 0,1% TFA для элюирования до получения 3,0 мг указанного в заглавии соединения. TCX:Rf = 0,25, 5% CH3OH в CH2Cl2. ВЭЖХ:Rт = 14,78 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 15.
Соединение 15. Образец смеси 4-фтор-3-ацетамидобензолсульфонилхлорида и 3-фтор-4 -ацетамидобензолсульфонилхлорида (примерно, 1:1, полученный от Maybridge Chemicals) разделяют на соответствующие региоизомеры с помощью хроматографии на силикагеле, используя 10% изопропиловый спирт/гексан в качестве элюента. Раствор 4-ацетамидо-3-фторбензолсульфонилхлорида (30 мг) и соединения, полученного в примере 17B, диастереизомер B (80 мг) в 10 мл CН2Cl2, подвергают взаимодействию по способу примера 14. После обработки и очистки части продукта с помощью препаративной хроматографии с обращенной фазой C18, используя линейный градиент от 35% до 100 CH3CN/H2O с 0,1% TFA в качестве элюента, получают указанное в заглавии соединение в виде белого твердого вещества. TCX: Rf = 0,25, 5% CH3OH в CH2Cl2. ВЭЖХ:Rт = 12,91 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 16.
Соединение 16. 80 мг соединения, полученного в примере 17B, диастереоизомера B, подвергают взаимодействию с 45 мг 3-ацетамидо-4-фторбензолсульфонилхлорида по способу примера 14. После обработки и очистки части продукта с помощью препаративной хроматографии с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента, получают 1,4 мг указанного в заглавии соединения. TCX:Rf = 0,25, 5% CH3OH в CH2Cl2. ВЭЖХ:Rт = 12,91 мин /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 17.
А. (2S)-2-((1S, 2R, син, анти)-3-(2-метилпропил)-амино-1- бензил-2-гидроксипропил)-N1-((хинолин-2-карбонил)-амино) -N4-тритилсукцинамид. Раствор 683,1 мг (0,96 ммолей) соединения, полученного в примере 191Д, и 1,9 мл (19,2 ммоля) изобутиламина в 10 мл ацетонитрила в запаянной ампуле нагревают при 90 - 100oC в течение 24 часов. После охлаждения до комнатной температуры полученную смесь концентрируют в вакууме. Остаток помещают в дихлорметан и промывают водой, рассолом, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 783,8 мг смеси диастереоизомеров. TCX:Rf = 0,11, 10% CH3OH/CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
В. Соединение XIII (син, анти)-OH, A = хинолин,-2-карбонил, D' = изобутил). Раствор 583,3 мг соединения, полученного в примере 17A, и 0,2 мл диизопропилэтиламина в 10 мл дихлорметана, обрабатывают 256 мг ди-трет-бутилдикарбоната. Спустя 24 часа полученную смесь разбавляют дихлорметаном. Полученную смесь промывают водой, 5% NaHCO3, 0,5 н HCl, рассолом, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают на хроматографической колонке с силикагелем, используя 20% этилацетат/дихлорметан в качестве элюента до получения 154,6 мг быстрого перемещающегося диастереоизомера A, который, как было обнаружено позднее, обладает антиконфигурацией по гидроксильному центру, 98,8 мг более медленно перемещающегося диастереоизомера B, обладающего син-конфигурацией по гидроксильному центру, и 204,6 мг смеси диастереоизомеров A и B. TCX:Rf = 0,60, 0,67, 40% EtOAc/CH2Cl2.
С. Соединение 17. Раствор 64,6 мг соединений, полученных в примере 17B, диастереоизомера B в 1,5 мл дихлорметана обрабатывают 1,5 мл трифторуксусной кислоты. Спустя 4 часа полученную смесь концентрируют в вакууме до получения соли амина и трифторуксусной кислоты. TCX:Rf = 0,11, 10% CH3OH/CH2Cl2. К раствору 17,8 мг полученной соли трифторуксусной кислоты в 1 мл дихлорметана последовательно добавляют 0,3 мл насыщенного NaHCO3, небольшое количество твердого NaHCO3 и 10,7 мг 4-ацетамидобензолсульфолнилхлорида. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют один раз дихлорметаном. Объединенный слой промывают рассолом, а затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 14,4 мг указанного в заглавии соединения в виде твердого вещества белого цвета. TCX:Rf = 0,54, 10% CH3OH/CH2Cl2, ВЭЖХ: Rт = 13,58 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 18
Соединение 18. К раствору 20,8 мг (0,041 ммоля) неочищенной соли трифторуксусной кислоты, полученной, как в примере 17B, диастереоизомера B, в 1 мл дихлорметане последовательно добавляют 0,3 мл насыщенного раствора NaHCO3, небольшое количество твердого NaHCO3 и 13,6 мг (0,054 ммоля) 2-ацетамидо-4-метил-5-тиазолсульфонихлорида. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют один раз дихлорметаном. Объединенный органический слой промывают рассолом, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 4,8 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ:Rf = 0,50, 10% CH3OH/CH2Cl2 ВЭЖХ:Rт = 13,35 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 19.
A. 3-Ацетамидобензолсульфонат натрия. Раствор 118,6 мг (0,55 ммоля) 3-ацетамидобензолсульфоновой кислоты в 0,5 мл воды обрабатывают 0,55 мл (0,55 ммоля) 1,0 н NaOH при 0oC. После перемешивания при комнатной температуре в течение 4 часов полученную смесь концентрируют досуха и используют без дальнейшей очистки.
B. 3-Ацетамидобензолсульфонилхлорид. Неочищенную смесь примера 19A охлаждают до 0oC, и добавляют 0,29 г (1,38 ммоля) пятихлористого фосфора. Полученную смесь твердую перемешивают в течение 3 часов, затем добавляют 5 мл дихлорметана. Спустя 24 часа полученную суспензию фильтруют и концентрируют в вакууме до получения 81,4 мг твердого продукта, который используют без дальнейшей очистки. ТСХ: Rf = 0,50, 40% EtOAc/CH2Cl2.
C. Соединение 19. Раствор 82,7 мг (0,098 ммоля) диастереоизомера B, полученного в примере 17B, в 2 мл дихлорметана обрабатывают 2 мл трифторуксусной кислоты. Спустя 4 часа полученную смесь концентрируют в вакууме до получения соли амина и трифторуксусной кислоты, которую используют без дальнейшей очистки: ТСХ: Rf = 0,11, 10% CH3OH/CH2Cl2. Раствор этой соли (весь выход) в 2 мл дихлорметана обрабатывают последовательно 0,5 мл насыщенного NaHCO3, небольшим количеством твердого NaHCO3 и раствором 81,4 мг (0,046 ммоля) соединения, полученного в примере 19B. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют один раз дихлорметаном. Объединенный органический слой промывают рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 24,7 мг указанного в заглавии соединения в виде белого твердого вещества, ТСХ: Rf = 0,42, 10% CH3OH/CH2Cl2, ВЭЖХ: Rт = 13,8 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 20.
Соединение 20. Раствор 209,0 мг (0,24 ммоля) соединения, полученного в примере 17B, диастереоизомера B, в 5 мл дихлорметана, обрабатывают 5 мл трифторуксусной кислоты. Спустя 4 часа полученную смесь концентрируют в вакууме. ТСХ: Rf = 0,11, 10% CH3OH/CH2Cl2. К раствору этого остатка в 2 мл дихлорметана последовательно добавляют 0,5 мл насыщенного NaHCO3, небольшое количество твердого NaHCO3 и 70,2 мг (0,32 ммоля) бензофуран-4-сульфонилхлорида. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют один раз дихлорметаном. Объединенный органический слой промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 108,0 мг указанного в заглавии соединения в виде белого твердого веществ, ТСХ: Rf = 0,60, 10% CH3OH/CH2Cl2, ВЭЖХ: Rт = 14,95 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 21.
Соединение 21. Соединение, полученное в примере 17B, диастереоизомер B, (228 мг, 0,27 ммоля) растворяют в 1:1 CH2Cl2/TFA (10 мл), и реакционную смесь перемешивают в течение 3,5 часа, затем концентрируют досуха до получения соли трифторуксусной кислоты в виде желтого твердого вещества, которое используют в следующей реакции без очистки. К раствору этого остатка (34,7 мг, 0,05 ммоля) в CH2Cl2 (3 мл) добавляют основание Хеунига (41 мкл, 0,24 ммоля) и диметилсульфамоилхлорида (11 мкл, 0,09 ммоля), и реакционную смесь перемешивают в течение 17 часов при комнатной температуре. Затем реакционную смесь разбавляют CH2Cl2 и промывают насыщенным NH4Cl, а органический слой сушат над сульфатом магния. После фильтрования и концентрирования получают остаток, который обрабатывают на хроматографической колонке с силикагелем, используя 8% CH3OH/CH2Cl2 в качестве элюента, получая целевое соединение, которое затем очищают с помощью препаративной ВЭЖХ. ВЭЖХ: Rт = 13,8 мин. ТСХ: Rf = 0,40, 8% CH3OH/CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 22.
A. Nα- изоциано-L валина метиловый сложный эфир. К соли HCl метилового сложного эфира валина (2,08 г, 12,40 ммоля) в 20 мл толуола добавляют 20% раствор фосгена в толуоле (32 мл, 62,0 ммоля), и полученный раствор нагревают до кипения с обратным холодильником в течение 12 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме до получения бледно-желтой жидкости, которую используют в последующей реакции без дополнительной очистки. ТСХ: Rf = 0,88, 50% гексан/EtOAc, /1H/-ЯМР (CDCl3) соответствует структуре.
B. Nα- /2-пиридилметил/оксикарбонил-L-валина метиловый сложный эфир. Смесь 2-пиридилкарбинола (941 мкл, 9,75 ммоля) и соединения, полученного в примере 22A (1,28 г, 8,12 ммоля), оставляют при перемешивании в CH2Cl2 (7 мл) в течение 12 часов, затем реакционную смесь концентрируют, а остаток обрабатывают хроматографически, элюируя 50% гексан/EtOAc до получения 2,03 г указанного в заглавии соединения в виде бесцветного масла. ТСХ: Rf = 0,26, 50% гексан/EtOAc, /1H/-ЯМР (CDCl3) соответствует структуре.
C. Nα- /2-пиридилметил/оксикарбонил-L-валин. Раствор соединения, полученного в примере 22B (634 мг, 2,38 ммолей) в смеси 1/1 1 н HCl/ТГФ (6 мл), содержащей 12н HCl (0,5 мл), оставляют при перемешивании при комнатной температуре в течение 15 часов, но по данным ТСХ присутствует все еще много исходного материала. Затем добавляют 1 мл HCl (более 12н), и реакционную смесь перемешивают еще 48 часов. Затем реакционную смесь концентрируют досуха и разбавляют CH2Cl2, получая целевую карбоновую кислоту в виде нерастворимой смолы, которую затем промывают дополнительной порцией CH2Cl2, получая соединение 22C, которое содержит небольшие количества 22B. Этот материал используют в последующей реакции без дальнейшей очистки. ТСХ: Rf = 0,11, 8% CH3OH/CH2Cl2/, /1H/-ЯМР (CDCl3) соответствует структуре.
D. Соединение XXX (A = /2-пиридилметил/-оксикарбонил, R3-изопропил, R3' = H, D' = изобутил, A' = трет.-бутоксикарбонил). К соединению, полученному в примере 21B (277 мг, 0,82 ммоля), в CH2Cl2 (5 мл), добавляют 1-/3-диметиламино-пропил/-3-этилкарбодиимидгидрохлорид (210 мг, 1,1 ммоля), кислоту 22C (402 мг, 1,10 ммоля), 1-гидроксибензотриазол гидрат (148 мг, 1,10 ммолей). Реакция протекает в течение 12 часов при комнатной температуре, затем реакционную смесь разбавляют CH2Cl2 и промывают последовательно насыщенным NH4Cl и NaHCO3, а органический слой сушат над сульфатом магния. В результате фильтрования и концентрирования получают остаток, который обрабатывают на хроматографической колонке с силикагелем, используя 17% THF/CH2Cl2 в качестве элюента, в результате чего получают 396 мг продукта. ТСХ: Rf = 0,26, 17% THF/CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
E. Соединение 22. Соединение, полученное в примере 22D (396, мг, 0,69 ммоля), растворяют в 90% водной TFA (11 мл), и реакционную смесь перемешивают в течение 3 часов при комнатной температуре, затем концентрируют досуха. К раствору этого остатка (231 мг, 0,33 ммоля) в CH2Cl2 (5 мл) добавляют избыток твердого NaHCO3 (приблизительно 1 г) и насыщенный водный NaHCO3 (20 мкл), а затем N-ацетилсульфанилхлорид (116 мг, 0,50 ммоля), и реакцию ведут 12 часов при комнатной температуре. Затем реакционную смесь разбавляют CH2Cl2 и промывают насыщенным NaHCO3, и органический слой сушат над сульфатом магния. После фильтрования и концентрирования получают остаток, который обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 8% CH3OH/CH2Cl2. Полученный остаток очищают с помощью препаративной ВЭЖХ (76,1 мг ВЭЖХ: Rт = 12,1 минуты. ТСХ: Rf = 0,46, 8% CH3OH/CH2Cl2,
ЯМР (CDCl3), 8,76 (д, 1H), 8,40 (шир. с., 1H), 8,26 (т, 1H), 7,72 (д, 2H), 7,76 (д, 2H), 7,58 (д, 2H), 7,37 (д, 1H), 7,25 (м, 4H), 7,16 (шир. д, 1H), 6,47 (д, 1H), 5,65 (д, 1H), 5,26 (д, 1H), 4,32 (м, 1H), 3,91 (т, 1H), 3,83 (м, 1H), 3,23 (д, 1H), 3,05 (м, 2H), 2,68-3,10 (м, 3H), 2,22 (м, 3H), 2,0 (м, 1H), 1,82 (м, 1H), 0,85 (д, 3H), 0,80 (д, 3H), 0.71 (д, 3H), 0,65 (д, 3H).
Пример 23.
Соединение 23. Получают по способу примера 22, за исключением того, что 4-пиридилкарбонил используют в реакции с продуктом примера 22A. ВЭЖХ, Rт = 12,0 мин. ТСХ: Rf = 0,50 (8% CH3OH/CH2Cl2). /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 24
Соединение 24. Раствор соединения, полученного после удаления защиты трифторуксусной кислотой у соединения примера 22D (по способу примера 22E, 215 мг, 0,31 ммоля) в CH2Cl2 при комнатной температуре, обрабатывают диизопропилэтиламином (214 мкл, 1,23 ммоля) и диметилсульфамоилхлоридом (40 мкл, 0,37 ммоля) в CH2Cl2 при комнатной температуре в течение 12 часов. Реакционную смесь концентрируют и обрабатывают хроматографически на колонке с силикагелем, используя в качестве элюента 5% CH3OH/CH2CH2. Получают целевое соединение, которое затем очищают с помощью препаративной хроматографии (ВЭЖХ, выход 9,5 мг). ВЭЖХ: Rт = 14,4 мин. ТСХ: Rf = 0,88, 11% CH3OH/CH2CH2, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 25.
Соединение 25. Это соединение получают по способу примера 22, за исключением того, что 3-пиридинкарбонил используют для реакции с соединением примера 22A, а в реакции, соответствующей 22E, материал с удаленной защитой (трифторацетат) подвергают взаимодействию с бензофуран-4-сульфонилхлоридом. ВЭЖХ: Rт = 9,4 мин. ТСХ: Rf = 0,10, 11% CH3OH/CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 26.
Соединение 26. Раствор соединения, полученного после удаления трифторуксусной кислотой защиты у соединения примера 22D (как указано в примере 22E, 27 мг, 0,14 ммолей), в CH2Cl2 обрабатывают избытком твердого NaHCO3 (пример 1 г), и насыщенным NaHCO3 (7 мкл), затем интенсивно перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь дикантируют от твердого вещества, концентрируют, затем остаток очищают непосредственно с помощью препаративной ВЭЖХ (3,0 мг белого твердого вещества - выход). ВЭЖХ: Rт = 14,7 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 27
Соединение 27. Раствор 33 мг соединения, полученного в примере 40A, в CH2Cl2, обрабатывают последовательно при комнатной температуре в атмосфере азота, 20 мг N,N-диизопропилэтиламина и 9,3 мг аллилхлорформата. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над безводным сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя 2:1 смесь (5:10:85 NH4OH/CH3OH/CH2Cl2): диэтиловый эфир до получения 24 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,53, 5:10:85 NH4OH/CH3OH/CH2Cl2. ВЭЖХ: Rт = 14,53 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 28.
Соединение 28. Раствор 47,5 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота 28,7 мг N,N-диизопропилэтиламина и 15,2 мг изобутилхлорформата. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя 2:1 смесь (5:10:85 NH4OH/CH3OH/CH2Cl2): диэтиловый эфир до получения 45 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,60, 5:10:85 NH4OH/CH3OH/CH2Cl2. ВЭЖХ: Rт = 15,58 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 29.
Соединение 29. Раствор 35,6 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 21,5 мг N,N-диизопропилэтиламина и 0,083 нл 1,0 М изопропилхлорформата. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, а затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя 2:1 смесь (5:10:85 NH4OH/CH3OH/CH2Cl2): диэтиловый эфир до получения 33,2 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,56, 5:10:85 NH4OH/CH3OH/CH2Cl2. ВЭЖХ: Rт = 14,81 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 30.
A. (2-Пирролидинонил-гидроксиэтил-N-гидроксисукцинимидилкарбоната). Раствор 572 мг 1-(2-гидроксиэтил)-2-пирролидинона и 1,70 г N,N-дисукцинимидилкарбоната в ацетонитриле обрабатывают при комнатной температуре в атмосфере азота, 1717 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 14 часов и концентрируют в вакууме. Остаток помещают в этилацетат и промывают насыщенным NaHCO3, насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 200 мг белого твердого веществ. ТСХ: Rf = 0,56, 10% изопропанол в CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
B. Соединение 30. Раствор 68 мг соединения, полученного в примере 30A, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота к раствору 32 мг соединения, полученного в примере 40A, и 39 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 4 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток обрабатывают с помощью препаративной тонкослойной хроматографии на силикагеле, используя 2:1 смесь (5: 10: 85 NH4OH/CH3OH/CH2Cl2): диэтиловый эфир до получения 45 мг остатка. Около 20 мг этого остатка очищают с помощью препаративной ВЭЖХ до получения 13,5 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,47, 5:10:85 NH4OH/CH3OH/CH2Cl2. ВЭЖХ: Rт = 12,79 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 31.
Соединение 31. Раствор 39,7 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно, при комнатной температуре в атмосфере азота 24 мг N,N-диизопропилэтиламина и 14,5 мг фенилхлорформата. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя 2:1 смесь (5:10:85 NH4OH/CH3OH/CH2Cl2/: диэтиловый эфир, до получения 39,7 мг указанного в заглавии соединения. ТСХ: Rf = 0,53, 5:10:85 NH4OH/CH3OH/CH2Cl2. ВЭЖХ: Rт = 15,22 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 32.
Соединение 32. Раствор 391 мг соединения, полученного в примере 39A, в 4: 1 CH2Cl2 (насыщенном водном NaHCO3) обрабатывают последовательно при комнатной температуре в атмосфере азота 271 мг 4-фторбензолсульфонилхлорида и 117 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 5% диэтиловый эфир в CH2Cl2 в качестве элюента до получения 420 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,20, 5% диэтиловый эфир в CH2Cl2. ВЭЖХ: Rт = 17,41 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 33.
Соединение 33. Раствор 30 мг соединения, полученного в примере 40A, в CH2Cl2, обрабатывают последовательно при комнатной температуре, в атмосфере азота 18,1 мг N,N-диизопропилэтиламина и 9,3 мг бензилизоцианата. Полученную смесь перемешивают в течение 14 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя смесь 5: 10:85 NH4OH/CH3OH/CH2Cl2, до получения 30,2 мг указанного в заглавии соединения в виде белого 30.2 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,56, 5:10:85 NH4OH/CH3OH/-CH2Cl2. ВЭЖХ: Rт = 14,36 мин. /1/-ЯМР (CDCl3) соответствует структуре.
Пример 34.
Соединение 34. Раствор 55 мг соединения примера 40A в CH2Cl2 обрабатывают последовательно при комнатной температуре, в атмосфере азота 33,3 мг N, N-диизопропилэтиламина и 17,8 мг 2-метоксиэтилхлорформата. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя смесь 2: 1 (5:10:85 NH4OH/CH3OH/CH2Cl2): диэтиловый эфир, до получения 48,1 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,56, 5: 10: 85 NH4OH/CH3OH/CH2Cl2. ВЭЖХ: Rт = 13,43 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 35.
A. Соединение XXI (D'=изобутил, A'=4-фторфенил-гидрохлорид). Раствор 398 мг соединения, полученного в примере 32, в этилацетате обрабатывают при -20oC газообразной HCl. HCl барботируют через смесь в течение 20 минут, причем за это время температура повышается до 20oC. Затем через смесь барботируют азот в течение 15 минут, и растворитель удаляют в вакууме до получения 347 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,82, 5:10:85 NH4OH/CH3OH/CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
B. Соединение 35. Добавляют раствор III мг соединения, полученного в примере 35A, в CH2Cl2, при комнатной температуре и атмосферном давлении азота, к раствору 118 мг соединения, полученного в примере 48A и 133 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток обрабатывают хроматографически с помощью тонкослойной хроматографии на силикагеле, используя 5% CH3OH в CH2Cl2 до получения 98,8 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,48, 5% CH3OH в CH2Cl2. ВЭЖХ: Rт = 15,18 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 36.
Соединение 36. Раствор 48 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота 29,0 мг N, N-диизопропиламина и 15,1 мг 3-бутенилхлорформата. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl, насыщенным NaCl, а затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя смесь 2:1 (5:10:85 NH4OH/CH3OH/CH2Cl2): диэтиловый эфир, до получения 43,8 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,83, 5:10:85 NH4OH/CH3OH/CH2Cl2, Rf = 0,24, 5% диэтиловый эфир в CH2Cl2. ВЭЖХ: Rт = 14,76 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 37.
Соединение 37. Раствор 99 мг соединения, полученного в примере 51D в 4:1 CH2Cl2 (насыщенном водном NaHCO3) обрабатывают последовательно при комнатной температуре в атмосфере азота 83,2 мг 3,4-дихлорбензолсульфонилхлорида и 29 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток обрабатывают с помощью препаративной тонкослойной хроматографии, используя 5% CH3OH в CH2Cl2 до получении 107 мг указанного в заглавии соединения в виде белого твердого продукта. ТСХ: Rf = 0,35 (5% CH3OH в CH2Cl2) ВЭЖХ: Rт = 17,27 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 38.
Соединение 38. К раствору 32 мг соединения, полученного в примере 35A, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота 14 мг бензилхлорформата и 21 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 4 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 10% диэтиловый эфир в CH2Cl2 в качестве элюента, до получения 33 мг продукта. ТСХ: Rf = 0,62, 10% диэтиловый эфир в CH2Cl2. ВЭЖХ: Rт = 17,27 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 39.
A. Соединение XXI (D'=изобутил, A = трет.бутоксикарбонил, A'=H). Раствор 4,1 г эпоксида XX (A = Boc) в 30 мл этанола обрабатывают 22,4 мг изобутиламина и нагревают при кипячении с обратным холодильником в течение 1 часа. Полученную смесь концентрируют до получения указанного в заглавии соединения в виде белого твердого продукта, который используют без последующей очистки. ЯМР (CDCl3): δ 0,91 (д, 3H), 0,93 (д, 3H), 1,37 (с, 9H), 1,68 (шир. с. 2H), 2,40 (д, 2H), 2,68 (д, 2H), 2,87 (дд, 2H), 2,99 (дд, 1H), 3,46 (дд, 1H), 3,75 (шир. с. 1H), 3,80 (шир.с. 1H), 4,69 (д, 1H), 7,19 - 7,32 (м, 4H).
B. Соединение 39. К раствору 514,1 мг соединения, полученного в примере 39A, в дихлорметана (10 мл), добавляют водный бикарбонат натрия (5 мл) и N-ацетилсульфанилилхлорид (428,4 мг). Спустя 14 часов полученную смесь разбавляют этилацетатом, промывают бикарбонатом натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 20% этилацетат в дихлорметане в качестве элюента, до получения 714,4 мг указанного в заглавии продукта. ТСХ: Rf = 0,63, 60% этилацетат/дихлорметан, ВЭЖХ: Rт = 15,3 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 40.
A. Соединение XXII (D'=изобутил, A=H, E=4-ацетамидофенил), гидрохлорид. Через раствор 691,4 мг (1,296 ммоля) соединения, полученного в примере 39B, в 20 мл этилацетата при -20oC барботируют безводный газообразный HCl в течение 10 минут. Ледяную баню удаляют, и спустя еще 15 минут реакционную смесь продувают азотом, затем концентрируют в вакууме до получения 610 мг указанного в заглавии продукта, который используют без дополнительной очистки.
B. Соединение 4. Раствор 41,5 мг неочищенного соединения примера 40A в 5 мл дихлорметана обрабатывают последовательно при комнатной температуре в атмосфере азота 18,1 мг L-дигидрооротовой кислоты, 0,031 мл (0,176 ммоля) диизопропилэтиламина, 15,5 мг (0,115 ммолей) 1-гидроксибензотриазолгидрата, 22 мг (0,115 ммоля) ЕДС. Спустя 1 час суспензию обрабатывают 1 мл диметилформамида. Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают водой, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя (1/2/17 объем/объем/объем) 30% гидроксид аммония/метанол/дихлорметан/ в качестве элюента, до получения 34,2 мг указанного в заглавии продукта. ТСХ: Rf = 0,33, 1/2/17 объем/объем/объем 30% гидроксид аммония/метанол/дихлорметан/. ВЭЖХ: Rт = 11,3 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 41.
Соединение 41. К раствору 42,8 мг соединения, полученного в примере 40A, в 5 мл дихлорметана последовательно добавляют при комнатной температуре в атмосфере азота 17,2 мг N-трет.бутилглиоксалевой кислоты, 0,032 мл диизопропилэтиламина, 16 мг гидрата 1-гидроксибензотриазола, 22,6 мг ЕДС. Полученную смесь перемешивают в течение 16 часов, затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают водой, 0,5 н соляной кислотой, промывают бикарбонатом натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 40% этилацетат/дихлорметан в качестве элюента до получения 14,9 мг указанного в заглавии продукта. ТСХ: Rf = 0,47, 40% этилацетата/дихлорменан, ВЭЖХ: Rт = 15,2 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 42.
Соединение 42. К раствору 43,5 мг неочищенного соединения, полученного в примере 40A, в 5 мл дихлорметана, добавляют последовательно при комнатной температуре в атмосфере азота 13,0 мг сукцинамовой кислоты, 0,024 мл диизопропилэтиламина, 15,0 мг 1-гидроксибензотриазолгидрата и 21,3 мг ЕДС. Полученную смесь перемешивают в течение 16 часов, затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают бикарбонатом натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью тонкослойной препаративной хроматографии, используя /1/2/11 объем/объем/объем 30% гидроксид аммония/метанол/дихлорметан/ в качестве элюента до получения 35,3 мг указанного в заглавии продукта. ТСХ: Rf = 0.25, 1/2/11 объем/объем/объем 30% гидроксидаммония/метанол/дихлорметан, ВЭЖХ: Rт = 11,6 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 43
Соединение 43. К раствору 42,8 мг соединения, полученного в примере 40A, в 5 мл дихлорметана добавляют последовательно при комнатной температуре в атмосфере азота 14,1 мг L-пироглутамовой кислоты, 0,024 мл диизопропилэтиламина, 14,8 мг 1-гидроксибензотриазолгидрата, 20,9 мг ЕДС. Полученную смесь перемешивают в течение 16 часов, затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают водой, 0,5 н соляной кислотой, промывают бикарбонатом натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя /1/2/11 объем/объем/объем 30% гидроксид аммония/метанол/дихлорметан/ в качестве элюента до получения 29,9 мг указанного в заглавии соединения. ТСХ: Rf = 0,33, 1/2/11 объем/объм/объем 30% гидроксид аммония/метанол/дихлорметан, ВЭЖХ: Rт = 11,7 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 44
A. 3-пиридилметил-N-гидроксисукцинимидилкарбонат.
К раствору 181,0 мг 3-пиридинкарбинола в 5 мл ацетонитрила добавляют последовательно при комнатной температуре в атмосфере азота 0,72 мл диизопропилэтиламина и 354,1 мг N,N-дисукцинимидилкарбоната. Спустя 4 часа полученную смесь концентрируют в вакууме до получения твердого вещества желтого цвета, которое используют без дополнительной очистки.
B. Соединение 44. К раствору 59,1 мг неочищенного соединения, полученного в примере 40A, в 3 мл дихлорметана, добавляют последовательно при комнатной температуре в атмосфере азота 0,075 мл диизопропилэтиламина и 46,3 мг соединения, полученного в примере 44A. Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в диэтиловый эфир и экстрагируют 3х25 мл 0,5 н HCl. Объединенные водные экстракты доводят до pH 8 твердым бикарбонатом натрия и экстрагируют 3х25 мл этилацетата. Объединенные органические экстракты промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью тонкослойной препаративной хроматографии, используя /1/2/17/20 об./об. /об. 30% гидроксидаммония/метанол/дихлорметан/диэтиловый эфир/ в качестве элюента, до получения 10,3 мг указанного в заглавии соединения. ТСХ: Rf = 0,4, 1/2/17/20, об./об./об/ 30% гидроксид аммония/метанол/дихлорметан/диэтиловый эфир, ВЭЖХ: Rт = 11,8 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 45.
Соединение 45. К раствору 28,3 мг соединения, полученного в примере 39A, в 4 мл дихлорметана добавляют 1 мл насыщенного водного раствора бикарбоната натрия, 9,2 мг бикарбоната натрия и 0,013 мл бензолсульфонилхлорида. Спустя 14 часов полученную смесь разбавляют этилацетатом, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 10% диэтиловый эфир/дихлорметан в качестве элюента до получения 19.3 мг указанного в заглавии соединения. ТСХ: Rf = 0,84, 25% диэтиловый эфир/дихлорметан, ВЭЖХ: Rт = 17,2 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 46
Соединение 46. К раствору 47,0 мг (0,140 ммолей) соединения, полученного в примере 39A, в 4 мл дихлорметана добавляют 1 мл насыщенного водного раствора бикарбоната натрия, 17,6 мг твердого бикарбоната натрия и 41,1 мг 2,4-диметилтиазол-5-сульфонилхлорида. Спустя 14 часов полученную смесь разбавляют этилацетатом, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 25% этилацетат/дихлорметан в качестве элюента, до получения 34,6 мг указанного в заглавии продукта. ТСХ: Rf = 0,44, 25% диэтиловый эфир/дихлорметан, ВЭЖХ: Rт = 16,4 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Соединение 47. К раствору 50,7 мг соединения, полученного в примере 39A, в 4 мл дихлорметана, добавляют 1 мл насыщенного водного раствора бикарбоната натрия, 15,2 мг бикарбоната натрия и 2-фторбензолсульфонилхлорида 35,2 мг. Спустя 14 часов полученную смесь разбавляют этилацетатом, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 10% диэтиловый эфир/дихлометан в качестве элюента, до получения 40,5 мг указанного в заглавии продукта. ТСХ: Rf = 0,44, 25% диэтиловый эфир/дихлорметан, ВЭЖХ: Rт = 17,2 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 48
A. N-сукцинимидил-/S/-3-тетрагидрофурилкарбонат.
К раствору 12,5 мл 1,93 М фосгена в толуоле при 0-5oC, добавляют 1,3 г /S/ /-/+/-3-гидрокситетрагидрофурана. После перемешивания в течение 2 часов реакционную смесь продувают азотом, а затем концентрируют досуха в вакууме до получения 1,486 г неочищенного хлорформата. Этот материал помещают в 10 мл ацетонитрила и обрабатывают последовательно при комнатной температуре в атмосфере азота 1,17 г N-гидроксисукцинимида и 1,41 мл триэтиламина. После перемешивания в течение 14 часов, реакционную смесь концентрируют в вакууме до получения 3,44 г указанного в заглавии продукта в виде белого твердого вещества.
B. Соединение 48. К раствору 87,2 мг соединения, полученного в примере 40A, в 5 мл дихлорметана, добавляют последовательно при комнатной температуре в атмосфере азота 0,113 мл диизопропилэтиламина пропилэтиламина и 68 мг соединения, полученного в примере 48A. Полученную смесь перемешивают в течение 16 часов, затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают водой, 0,5 н HCl, насыщенным бикарбонатом натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии на силикагеле, используя /3/6/20/65 об./об./об./об. 30% гидроксид аммония/метанол/диэтиловый эфир/дихлорметан/ в качестве элюента, с последующей кристаллизацией из смеси дихлорметана, диэтилового эфира и гексане до получения 58 мг указанного в заглавии продукта. ТСХ: Rf = 0,17, 75% этилацетат/дихлорметан, ВЭЖХ: Rт = 13,1 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 49
Соединение 49. В соответствии со способом примера 83 раствор соединения, полученного в примере 39A, в CH2Cl2, подвергают взаимодействию с 2,4-дифторбензолсульфонилхлоридом в присутствии воды и NaHCO3. После разбавления дополнительным количеством CH2Cl2 и обработки водой полученный продукт сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографически на силикагеле, используя соответствующую систему растворителей, до получения указанного в заглавии продукта.
Пример 50
Соединение 50. Раствор 30 мг соединения, полученного в примера 58, и 9 мкл диметилсульфамоилхлорида в 10 мл CH2Cl2 подвергают взаимодействию по способу примера 14. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O и 0,1% TFA в качестве элюента, получают 6,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,2, 3% CH3OH в CH2Cl2, ВЖЭХ: Rт = 15,96 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 51
A. Соединение XXI /A-трет-бутоксикарбонил, D'=изобутил, A'=бензилоксикарбонил/. К раствору соединения, полученного в примере 39A /2,5 г, 7,43 ммоля/ в CH2Cl2 (50 мл), добавляют триэтиламин (2,1 мл, 14,9 ммоля) с последующим добавлением бензилового эфира хлоругольной кислоты (1,2 мл, 8,1 ммоля). Смесь оставляют при перемешивании при комнатной температуре в течение 6 часов. Раствор разбавляют 1 л CH2Cl2 и промывают водой. Органические слои сушат над безводным MgSO4, концентрируют при пониженном давлении, затем чистят с помощью хроматографии на силикагеле. Система с градиентом растворителя: CH2Cl2, затем 3:97 метанол /CH2Cl2. Указанный в заглавии продукт (2,97 г) получают в виде бесцветного масла. ТСХ: Rf = 0,14, 3:97 метанол/CH2Cl2/, /1H/-ЯМР (CDCl3) соответствует структуре.
B. Соединение XXI /A=H, D'=изобутил, A' = бензилоксикарбонил, гидрохлорид/. В раствор 1,5 г (3,187 ммоля) соединения, полученного в примере 51A, в этилацетате (25 мл) продувают безводный газ HCl в течение 10 мин. Удаляют баню со льдом, и после этого реакционную смесь дополнительно продувают в течение 15 мин азотом, затем концентрируют в вакууме до получения 1,29 г указанного в заглавии продукта в виде белого твердого вещества, которое используют непосредственно для последующей реакции. ТСХ: Rf = 0,14, 10% метанол/CH2Cl2.
C. Соединение XXI /A=/S/-3-тетрагидрофурилоксикарбонил, D'=изобутил, A'= бензилоксикарбонил/. К раствору 1,077 г неочищенного соединения, полученного в примере 51B (2,647 ммоля) в ацетонитриле (10 мл), последовательно добавляют при комнатной температуре в атмосфере азота 1,61 мл (9,263 ммоля) диизопропилэтиламина и 910 мг (3,97 ммоля) соединения, полученного в примере 48A. После перемешивания в течение 3 часов добавляют дополнительно 223 мг (0,973 ммоля) соединения, полученного в примере 48A. Смесь перемешивают в течение 16 часов и затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают водой, 0,5 HCl, насыщенным раствором бикарбоната натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток чистят с помощью хроматографии на колонке с силикагелем при низком давлении, используя градиент от 10% до 25% этилацетата с CH2Cl2 в качестве элюента, с выходом 1.025 г указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0.10, этилацетат /CH2Cl2/, /1H/-ЯМР (CDCl3) соответствует структуре.
D. Соединение XXI /A=(S)-3-тетрагидрофурилоксикарбонил, D'=изобутил, A'= H/. Раствор 872 мг /1,799 ммоля/ соединений, полученных в примере 51C, в (10 мл) этилового спирта добавляют, при комнатной температуре в атмосфере азота, к суспензии 87 мг (10 вес.%) 10% палладия на угле в (5 мл) этилового спирта и гидрируют в течение 16 часов при небольшом положительном давлении водорода. Смесь фильтруют и концентрируют в вакууме до получения 553,2 мг указанного в заглавии продукта в виде бесцветного стекла, которое используют непосредственно в последующей реакции. ТСХ: Rf = 0,46, 10% метанол/CH2Cl2.
E. Соединение 51. К раствору 72,7 мг (0,207 ммоля) соединения, полученного в примере 51D, в CH2Cl2 (4 мл) добавляют водный раствор бикарбоната натрия (1 мл), твердого бикарбоната натрия 22,6 мг (0,27 ммоля) и 2-/пирид-2-ил/-тиофен-5-сульфонилхлорида 64,6 мг (0,249 ммоля). Спустя 14 часов полученную смесь разбавляют этилацетатом, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью тонкослойной препаративной хроматографии, используя 15-30% этилацетат /CH2Cl2 в качестве элюента до получения 53 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,25, 25% этилацетат /CH2Cl2, ВЭЖХ: Rт = 15,3 мин, /1H/-ЯМР соответствует структуре.
Пример 52
A. N-гидроксисукцинимидил-/RS/-3-гидроксил-тетрагидрофурилкарбонат. Указанное в заглавии соединение получают, как описано в примере 48A, исходя из 0,1 г /RS/-3-гидрокси-тетрагидрофурана и получая 2,33 г твердого белого продукта.
B. Соединение 52. К раствору 105 мг соединения, полученного в примере 35A, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота, 112 мг соединения, полученного в примере 52A, и 126 мг N,N-диизопропилэтиламина. Смесь перемешивают в течение 4 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, затем сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 5% CH3OH в CH2Cl2 в качестве элюента, с выходом продукта 101.4 мг. ТСХ: Rf = 0,52, 5% CH3OH в CH2Cl2, ВЭЖХ: Rт = 15,05 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 53
Соединение 53. К раствору 72,3 мг (0,19 ммоля) соединения, полученного в примере 51D, в CH2Cl2 (4 мл) добавляют водный раствор бикарбоната натрия (1 мл), твердый бикарбонат натрия 19,2 мг (0,228 ммоля), и 4-ацетамидо-3-хлорбензолсульфонилхлорид 61,1 мг (0,228 ммоля). Спустя 14 часов полученную смесь разбавляют EtOAc, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 20-45% EtOAc/CH2Cl2 в качестве элюента, до получения 49,1 мг указанного в заглавии продукта. ТСХ: Rf = 0,29, 50% EtOAc/CH2Cl2, ВЭЖХ: Rт = 13,9 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 54
Соединение 54. Раствор 260 мг соединения, полученного в примере 39A, и 45 мг 3-ацетамидо-4-фторбензолсульфонилхлорида в 10 мл CH2Cl2 подвергают взаимодействию по способу примера 14. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O и 0.1% TFA в качестве элюента, получают 1,4 мг указанного в заглавии соединения. ТСХ: Rf=0,25, 5% CH3OH в CH2Cl2. ВЭЖХ: Rт = 15,63 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 55
Соединение 55. 35,0 мг соединения, полученного в примере 54, обрабатывают 1 мл 90% водного раствора TFA и оставляют на 12 часов. Смесь концентрируют в вакууме, и остаток помещают в 10 мл сухого CH2Cl2, обрабатывают 34 мкл DIEA (0,23 ммоля) и 20 мг 1-бензил-3-трет-бутил-1H-пиразол-5-карбонилхлорида. Смесь перемешивают в течение 1,5 часов, затем разбавляют CH2Cl2 и промывают 1H раствором HCl. После высушивания над MgSO4 и концентрирования в вакууме часть смеси очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O и 0,1% TFA для элюирования до получения 1,1 мг указанного в заглавии соединения. ТСХ: Rf = 0,8, 5% CH3OH в CH2Cl2. ВЭЖХ: Rт =18,25 мин, /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 56
A. S/-/-1-фенилэтил-N-гидроксисукцинимидилкарбонат.
Указанное в заглавии соединение получают из 9,5 мкл S/-/-I-фенилэтанола и 30 мг N,N-дисукцинимидилкарбоната по способу примера 44A. Полученное вещество используют без последующей очистки; /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение 56. 45,0 мг соединения, полученного в примере 58, обрабатывают 1 мл 90% водного раствора TFA и оставляют на 12 часов. Смесь концентрируют в вакууме, и остаток помещают в 15 мл сухого CH2Cl2, обрабатывают смешанным ангидридом, приведенным выше, и 65 мкл триэтиламина. Смесь перемешивают в течение 14 часов, затем разбавляют этилацетатом и промывают насыщенным раствором бикарбоната натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Часть смеси очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя гдрадиент от 35% до 100% CH3CN/H2O и 0,1% TFA для элюирования до получения 1,1 мг указанного в заглавии соединения. TCX: Rf = 0,5, 3% CH3OH в CH2Cl2. ВЭЖХ: Rт = 17,44 мин, /1/-ЯМР/CDCl3/ соответствует структуре.
Пример 57
Соединение 57. 30 мг соединения, полученного в примере 58, обрабатывают 1 мл 90% водного TFA и оставляют на 12 часов. Смесь концентрируют в вакууме, и остаток помещают в 25 мл сухого CH2Cl2, промывают насыщенным раствором бикарбоната натрия, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Раствор 14 мг полученного свободного амина в 10 мл CH2Cl2 обрабатывают 6 мкл феноксиацетилхлорида и 12 мкл триэтиламина. Смесь перемешивают в инертной атмосфере в течение 1 часа, затем разбавляют CH2Cl2 и промывают 1н раствором HCl. После высушивания над MgSO4 и концентрирования в вакууме часть смеси очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O и 0,1% TFA в качестве элюента до получения 16,5 мг указанного в заглавии соединения. TCX: Rf = 0,25, 3% MeOH в CH2Cl2. ВЭЖХ: Rт = 16,6 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 58
Соединение 58. Раствор 500 мг соединения, полученного в примере 39A, и 370 мг бензофуразан-4-сульфонилхлорида в 10 мл CH2Cl2 подвергают взаимодействию по способу примера 14. После обработки получают соединение, указанное в заглавии, кристаллизацией из горячего этанола, дальнейшая очистка этого вещества с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O и 0,1% TFA в качестве элюента, дает 2,0 мг указанного в заглавии соединения. TCX: Rf = 0,35, 3% CH3OH в CH2Cl2. ВЭЖХ: Rт = 17,00 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 59
A. R/+/-1-фенилэтил-N-гидроксисукцинимидилкарбонат.
Указанное в заглавии соединение получают из R/+/-1-фенилэтанола по способу примера 56A с получением белого твердого вещества. Полученное вещество используют непосредственно для последующей реакции; /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение 59. порцию в 36 мг соединения, полученного в примере 58, и 0,21 мк ммоля полученного соединения 59A подвергают взаимодействию по способу, описанному в примере 56B. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O и 0,1% TFA в качестве элюента, получают 1,0 мг указанного в заглавии соединения в виде белого твердого вещества. TCX: Rf = 0,45, 3% MeOH в CH2Cl2. ВЭЖХ: Rт = 17,34 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 60
Соединение 60. К раствору 70 мг соединения, полученного в примере 51D, в 10 мл CH2Cl2 добавляют 3 мл насыщенного водного раствора бикарбоната натрия, 50 мг бикарбоната натрия и 53 мг бензофуразан-4-сульфонихлорида. Смесь интенсивно перемешивают в течение 4 часов, затем полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния и фильтруют. После концентрирования смеси в вакууме остаток очищают с помощью хроматографии на толстом слое силикагеля, используя 5% MeOH/CH2Cl2 в качестве элюента до получения 80 мг указанного в заглавии соединения в виде белого твердого вещества. TCX: Rf = 0,80, 5% MeOH в CH2Cl2, ВЭЖХ: Rт = 14,96 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 61
Соединение 61. К раствору 35,5 мг (0,076 ммоля) соединения, полученного в примере 16, в 1 мл дихлорметана, последовательно добавляют 27,6 мкл (0,159 ммоля) диизопропилэтиламина и 12 мкл (0,083 ммоля) бензилхлорформата. Спустя 1 час смесь концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии с 50% смесью этилацетат/дихлорметан в качестве элюента до получения 38,7 мг указанного в заглавии соединения, в виде белого твердого вещества. TCX: Rf = 0,63, 50% этилацетат/дихлорметан, ВЭЖХ: Rт = 15,45 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 62
A. Бензофуран-4-сульфоновая кислота. К раствору 252,0 мг (1,05 ммоля) натриевой соли о-нитроанилин-м-сульфоновой кислоты в 1 мл воды добавляют 0,52 мл 2н раствора HCl. Спустя 1/2 часа добавляют 0,68 мл (1,05 ммоля) гидроокиси тетрабутиламмония (40% в воде). Спустя 2 часа смесь концентрируют в вакууме. Раствор остатка в 7 мл уксусной кислоты подвергают обработке 488,5 мг (1,10 ммоля) тетраацетата свинца. Спустя 24 часа осадок фильтруют и промывают небольшим количеством уксусной кислоты. Твердое вещество далее сушат в вакууме до получения 267,9 мг продукта. TCX: Rf = 0,09, 10% CH3OH/CH2Cl2.
B. Бензофуразан-4-сульфонилхлорид. К раствору 137,0 мг (0,522 ммоля) трифенилфосфина в 0,5 мл дихлорметана медленно добавляют 47 мкл (0,594 ммоля) хлористого сульфурила при 0oC. Удаляют баню со льдом и медленно добавляют неочищенное соединение, полученное в примере 62A, в 0,5 мл дихлорметана. Спустя 3 часа смесь подвергают обработке 30 мл 50% смеси эфир/гексан. Декантируют надосадочную жидкость в сухую склянку и концентрируют в вакууме. Остаток очищают фильтрованием через прессованный силикагель 25% этилацетатом в качестве элюента до получения 23 мг продукта. TCX: Rf = 0,6, 10% CH3OH/CH2Cl2, /1H/-ЯМР/CDCl3/ соответствует структуре.
C. Соединение 62. К раствору 55,7 мг (0,166 ммоля) соединения, полученного в примере 39A, в 1 мл дихлорметана последовательно добавляют 0,5 мл насыщенного NaHCO3, небольшое количество твердого NaHCO3 и соединение, полученное в примере 62B. Спустя 3 часа смесь разбавляют дихлорметаном. Разделяют два слоя, и водный слой экстрагируют один раз дихлорметаном. Объединенный органический слой промывают рассолом, затем сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 5,3 мг указанного в заглавии соединения в виде белого твердого вещества, TCX: Rf = 0,40, 50% этилацетат/дихлорметан, ВЭЖХ Rт = 16,5 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 63
A. Раствор 3,0 мг (0,0058 ммоля) соединения, полученного в примере 62, в 2 мл этилацетата подвергают обработке газом HCl (умеренный поток) в течение 3 минут. Смесь концентрируют в вакууме до получения неочищенной гидрохлоридной соли амина. TCX: Rf = 0,20, 10% CH3OH/CH2Cl2.
B. Соединение 63. К раствору неочищенного соединения, полученного в примере 63A, в 1 мл дихлорметана последовательно добавляют 2,1 мкл (0,0121 ммоля) диизопропилэтиламина и 0,9 мкл (0,0064 ммоля) бензилхлорформата. Спустя 1 час смесь концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии 90% смесью дихлорметан/метанол в качестве элюента, до получение 2,6 мг указанного в заглавии соединения в виде белого твердого вещества. TCX: Rf = 0,34, 50% этилацетат/дихлорметан, ВЭЖХ, Rт = 17,1 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 64
A. 5-/Диметиламино/тиоксометокси/-бензофуразан. К раствору 500 мг (3,67 ммоля) 5-гидроксибензофуразана в 10 ДМФА добавляют 140 мг (4,59 ммоля) NaH небольшими порциями. Полученную смесь перемешивают при комнатной температуре до тех пор, пока не прекратится выделение газа. Затем склянку погружают в холодную водяную баню и добавляют 540 мг (4,41 ммоля) диметилтиокарбамоилхлорида (от Aldrich). Спустя 5 минут водяную баню удаляют, и смесь нагревают до 80oC в течение 1 часа. После охлаждения до комнатной температуры, смесь выливают в 20 мл 0,5н NaOH три раза и три раза в воду. Твердое вещество сушат в вакууме до получения 580 мг продукта, который используют в следующей реакции без дальнейшей очистки, TCX: Rf = 0,20, 20% этилацетат/гексан, /1H/-ЯМР/CDCl3/ соответствует структуре.
B. 5-/Диметиламинокарбонил/тио-бензофуразан.
Неочищенный продукт, 510 мг (2,28 ммоля) из примера 64A, нагревают до 190oC в запаянной трубке. Спустя 5 часов его охлаждают до комнатной температуры, и добавляют этилацетат. Раствор фильтруют через прессованный силикагель и концентрируют в вакууме до получения 360 мг продукта, который вновь используют в следующей реакции без дальнейшей очистки. TCX: Rf = 0,20, 20% этилацетат/гексан.
C. 5-Меркаптобензофуразан. К раствору 357,4 мг (1,60 ммоля) соединения, полученного в примере 64B, в 2 мл метанола добавляют 7 мл 6н NaOH. Смесь нагревают до 90oC в течение 2 часов. Смесь выливают в 100 мл льда и подкисляют концентрированной HCl. Суспензию фильтруют и промывают три раза водой. Остаток сушат в вакууме до получения 145,6 мл продукта. TCX: Rf = 0,70, 20% этилацетат/гексан, /1H/-ЯМР/CDCl3/ соответствует структуре.
D. Бензофуразан-5-сульфонилхлорид. Газообразный хлор пробулькивают через раствор 39,9 мг (0,26 ммоля) соединения, полученного в примере 64C, в смеси 1 мл этилацетата и 0,5 мл воды в течение 3 минут. Затем смесь неоднократно промывают рассолом, до тех пор, пока осадок больше не образуется. Органический слой сушат над MgSO4, фильтруют и концентрируют до получения 30 мг продукта (52%). TCX: Rf = 0,22, 20% Этилацетат/гексан.
E. Соединение 64. Раствор соединений, полученных в примерах 52D и 39A (суммарные выходы) в смеси 1 мл дихлорметана, 0,3 мл насыщенного раствора NaHCO3 и небольшого количества твердого NaHCO3 перемешивают при комнатной температуре в течение 2 часов. Раствор разбавляют 30 мл дихлорметана, и разделяют два слоя. Водный слой один раз экстрагируют дихлорметаном. Объединенный органический слой промывают рассолом, сушат над MgSO4 и концентрируют. Остаток очищают с помощью препаративной тонкослойной хроматографии 90% смесью дихлорметан/эфир в качестве элюента до получения 30 мг указанного в заглавии продукта в виде белого твердого вещества, ТСХ: Rf = 0,46, 10% Et2O/CH2Cl2, ВЭЖХ Rт = 17,6 мин, /1H-ЯМР/CDCl3/: δ 8,45 (с), 1H, 7,96 (д), 1H, 7,65 (д), 1H, 7,25 (м), 5H, 4,65 (д), 1H, 3,85 (м), 1H, 3,78 (м), 1H, 3,30 (д), 2H, 3,10 (м), 2H, 2,90 (м), 2H, 1,90 (м), 1H, 1,40 (с), 9H, 0,90 (д), 6H.
Пример 65
Соединение 65. Раствор 13,1 (0,025 ммоля) соединения, полученного в примере 64E, в 1,5 мл этилацетата обрабатывают газообразным HCl (равномерный поток) при 0oC в течение 3 минут. Растворитель удаляют до получения твердого остатка, который используют в следующей реакции без дальнейшей очистки, ТСХ: Rf = 0,52, 10% CH3OH/CH2Cl2. Раствор этой гидрохлоридной соли (полный выход) в 1 мл дихлорметана последовательно обрабатывают 9,2 мкл (0,053 ммоля) диизопропилэтиламина и 4,0 мкл (0,028 ммоля) бензилхлорформата. Спустя 3 часа смесь концентрируют и очищают с помощью препаративной тонкослойной хроматографии 90% смесью дихлорметан/эфир, в качестве элюента до получения 11,7 мг указанного в заглавии соединения в виде белого твердого вещества, ТСХ: Rf = 0,65, 10% Et2O/CH2Cl2, ВЭЖХ Rт = 17,6 мин, /1H-ЯМР /CDCl3/: δ 8,45 (с), 1H, 7,96 (д), 1H, 7,65(д), 1H, 7,25 (м), 10H, 5,00 (м), 2H, 4,85 (д), 1H, 3,86 (м), 2H, 3,60 (шир.с.) 1H, 3,25 (м), 12H, 3,05 (д), 2H, 2,96 (м), 1H, 2,98 (м), 1H, 1,88 (м), 1H, 0,90 (дд), 6H.
Пример 66
Соединение 66. Раствор 100 мг (0,46 ммоля) соединения, полученного в примере 64D, и 101 мг (0,286 ммоля) соединения, полученного в примере 48A, в смеси 2 мл дихлорметана и 0,5 мл насыщенного раствора NaHCO3 и небольшого количества твердого NaHCO3, перемешивают при комнатной температуре в течение 2 часов. Раствор разбавляют 50 мл дихлорметана, и разделяют два слоя. Водный слой экстрагируют один раз дихлорметаном. Объединенный органический слой промывают рассолом, сушат над MgSO4 и концентрируют. Остаток очищают с помощью препаративной тонкослойной хроматографии 20% смесью этилацетат/гексан в качестве элюента до получения 82 мг указанного в заглавии продукта в виде неочищенного светло-желтого твердого вещества. Вещество далее очищают с помощью препаративной ВЭЖХ с линейным градиентом системы растворителя от 35% до 100% ацетонитрил/воды (0,1% TFA) в течение 80 минут. После удаления растворителей получают 50 мг белого твердого вещества. ТСХ: Rf = 0,46, 10% Et2O/CH2Cl2, ВЭЖХ, Rт = 17,6 мин, /1H-ЯМР/CDCl3/: δ 8,45 (с), 1H, 7,96 (д), 1H, 7,65 (д), 1H, 7,25 (м), 5H, 5,15 (м), 1H, 4,85 (д), 1H, 3,82 (м), 4H, 3,68 (д), 1H, 3,20 (м), 2H, 3,05 (д), 2H, 2,96 (м), 1H, 2,88 (м), 1H, 2,14 (м), 1H, 1,92 (м), 2H, 1,50 (шир.с), 1H, 0,90 (дд), 6H,
Пример 67
Соединение 67. В соответствии со способом примера 40B, раствор соединения, полученного в примере 40A, в CH2Cl2 подвергают обработке бис-//карбоксамидо/-амино/-уксусной кислотой, диизопропилэтиламином, HOBt и EDC в 1: 1: 1:1:1 молярном отношении. Смесь перемешивают в течение 16 часов при комнатной температуре, в то же время защищая от влаги, затем дополнительно разбавляют CH2Cl2 и последовательно промывают H2O, насыщенным раствором NaHCO3 и рассолом, затем сушат над MgSO4 и концентрируют в вакууме. Остаток очищают с помощью хроматографии на силикагеле, используя соответствующий элюент до получения указанного в заглавии продукта.
Пример 68
Соединение 68. Это соединение получают в соответствии со способом, описанным в примере 26, за исключением того, что в качестве амина для взаимодействия используют соединение, полученное в примере 39A (146 мг, 0,43 ммоля), и в качестве ацилирующего агента 4-фторфенилсульфонилхлорид (27 мг, 0,14 ммоля). После хроматографической очистки на колонке с силикагелем с использованием 8% CH3OH/CH2Cl2 в качестве элюента получают 92,8 мг указанного в заглавии соединения. ВЭЖХ: Rт = 15,9 минут. ТСХ: Rf = 0,54 8% MeOH/CH2Cl2, /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 69
A. Соединение, полученное в примере 68 (72,1 мг, 0,167 ммоля), растворяют в 90% TFA (3,3 мл), и реакционную смесь перемешивают в течение 3 часов при комнатной температуре, затем концентрируют досуха. ТСХ: Rf = 0,29, 8% MeOH/CH2Cl2.
B. Соединение 69. К раствору соединения, полученного в примере 69A (41,7 мг, 0,09 ммоля), в CH2Cl2 (2 мл) добавляют диизопропилэтиламин (47 мкл, 0,27 ммоля) и соединение, полученное в примере 48A (33 мг, 0,15 ммоля), и позволяют реакции протекать в течение 14 часов при комнатной температуре. Затем реакционную смесь концентрируют, и остаток очищают хроматографически на колонке с силикагелем, используя 8% ТГФ/CH2Cl2 в качестве элюента, получая желаемое соединение, которое далее подвергают очистке с помощью препаративной ВЭЖХ, получая 7,8 мг белого твердого вещества. ВЭЖХ: Rт = 13,5 минут. ТСХ: Rf = 0,36, 8% TFA/CH2Cl2, /1H-ЯМР/CDCl3/ соответствует структуре.
Пример 70
Соединение 70. Раствор 30 мг соединения, полученного в примере 54, и 17,6 мг 3-ацетамидо-4-фторбензолсульфонилхлорида в 10 мл CH2Cl2 подвергают взаимодействию в соответствии со способом примера 14. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH2Cl2/H2O и 0,10 TFA в качестве элюента, получают 2,0 мг указанного в заглавии соединения. ТСХ: Rf = 0,5, 10% CH3OH/CH2Cl2. ВЭЖХ: Rт = 13,74 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 71
Соединение 71. Порцию в 30 мг соединения, полученного в примере 58, подвергают взаимодействию с трифторуксусной кислотой для снятия защиты, и полученное соединение подвергают взаимодействию с 9 мкл диметилсульфамоилхлорида в 10 мл CH2Cl2 в соответствии со способом примера 14. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100 CH3CN/H2O и 0,1% TFA в качестве элюента, получают 6,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,2, 3% MeOH в CH2Cl2. ВЭЖХ: Rт = 15,96 мин, /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 72
Соединение 72. К раствору соединения, полученного после снятия защиты трифторуксусной кислотой в примере 69A (31 мг, 0,07 ммоля) в CH2Cl2 (2 мл) добавляют диизопропилэтиламин (47 мкл, 0,27 ммоля) и диметилсульфамоилхлорид (22 мкл, 0,20 ммоля), и реакция протекает в течение 16 часов при комнатной температуре. Реакционную смесь концентрируют, и остаток подвергают хроматографированию на пластине с толстым слоем силикагеля (1,00 мм), используя 5% ТГФ/CH2Cl2 в качестве элюента, получая желаемое соединение, которое далее подвергают очистке с помощью препаративной ВЭЖХ до получения 7,8 мг белого твердого вещества. ВЭЖХ: Rт = 14,8 мин, ТСХ: Rf = 0,44, 5% ТГФ/CH2Cl2.
Пример 73
Соединение 73. Порцию в 43 мг соединения, полученного в примере 54, подвергают обработке 1 мл 90% водного раствора TFA и оставляют на 12 часов. Смесь концентрируют в вакууме, и остаток помещают в 5 мл CH2Cl2. К этому раствору добавляют 3 мл насыщенного водного раствора бикарбоната натрия и 25 мг 2,5-диметоксибензолсульфонилхлорида, и смесь перемешивают в течение 12 часов, позволяя медленно нагреваться до комнатной температуры. После концентрирования смеси в вакууме остаток очищают с помощью хроматографии на толстом слое силикагеля, используя 3% MeOH/CH2Cl2 в качестве элюента с последующей очисткой с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% и 0,1% TFA в качестве элюента до получения 5,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,20, 3% MeOH/CH2Cl2. ВЭЖХ: Rт = 15,15 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 74
A. Соединение XXI /A = трет-бутоксикарбонил, D' = циклопропилметил, A' = H/. К раствору соединения XX /A = трет-бутоксикарбонил/ (0,8 г, 2,67 ммоля) в этаноле (30 мл) добавляют раствор KOH (0,18 г, 3,2 ммоля) в этаноле (20 мл), и смесь перемешивают в течение 45 минут при комнатной температуре. В отдельной склянке к раствору циклопропилметиламингидрохлорида (1,44 г, 13,3 ммоля) в этаноле (20 мл) добавляют КОН (0,75 г, 13,3 ммоля). Смесь перемешивают в течение 30 минут при комнатной температуре. Растворы объединяют и нагревают при 85oC в течение 3 часов. Раствор концентрируют при пониженном давлении, и остаток помещают в диэтиловый эфир и фильтруют. Эфирный слой концентрируют до получения 0,32 г белого твердого вещества, /1H-ЯМР /CDCl3/ соответствует структуре.
B. Соединение 74. К раствору соединения, полученного в примере 74A (0,1 г, 0,30 ммоля), в CH2Cl2 (20 мл) добавляют насыщенный раствор бикарбоната натрия, с последующим добавлением твердого бикарбоната натрия (30 мг, 0,36 ммоля), затем 4-фторбензолсульфонилхлорида (0,07 г, 0,36 ммоля). Смесь оставляют перемешиваться при комнатной температуре в течение 4 часов. Органическую часть экстрагируют 250 мл CH2Cl2, сушат над безводным MgSO4, концентрируют при пониженном давлении, затем очищают с помощью жидкостной хроматографии среднего давления, используя градиент CH2Cl2, затем 0,5:99,5 метанол/CH2Cl2, затем 1:99 метанол/CH2Cl2. Получают 35 мг указанного в заглавии соединения в виде бесцветной пены. ВЭЖХ: Rт = 16,8 мин, ТСХ: Rf = 0,32, 3:97 метанол/CH2Cl2, /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 75
A. Соединение XXI /A = трет-бутоксикарбонил, D' = изопропил, A' = H/. Раствор соединения XX /A = трет-бутоксикарбонил/ (1,67 ммоля) в этаноле (10 мл) подвергают обработке изопропиламином (10 мл). Раствор нагревают до 85oC в течение 72 часов. Раствор фильтруют затем концентрируют при пониженном давлении до получения 0,56 г указанного в заглавии соединения, которое используют без последующей очистки. /1H-ЯМР /CDCl3/ соответствует структуре.
B. Соединение 75. К раствору соединения, полученного в пример 75A (0,2 г, 0,65 ммоля) в CH2Cl2 (10 мл) добавляют насыщенный раствор бикарбоната натрия (3 мл), с последующим добавлением твердого бикарбоната натрия (0,11 г, 1,31 ммоля), затем р-фторбензолсульфонилхлорида (0,25 г, 1,28 ммоля). Смесь перемешивают в течение ночи при комнатной температуре. Органическую часть экстрагируют 100 мл CH2Cl2, сушат над безводным MgSO4, концентрируют при пониженном давлении, затем очищают с помощью хроматографии, среднего давления на силикагеле, используя систему с градиентом CH2Cl2, затем 1:99 метанол/CH2Cl2. Получают 200 мг указанного в заглавии соединения в виде бесцветной пены. ТСХ: Rf = 0,22, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 16,48 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 76
A. Соединение XXI /A = трет-бутоксикарбонил, D' = морфолинил, A' = H/. К раствору соединения XX /A = Boc/ в этаноле добавляют 3 молярных эквивалента N-аминоморфолина. Смесь нагревают при кипячении с обратным холодильником в течение 12 часов, охлаждают, и смесь концентрируют в вакууме. Остаток очищают с помощью препаративной хроматографии с обращенной фазой, используя линейный градиент от 5% до 100% ацетонитрил/H2O в качестве элюента, до получения указанного в заглавии соединения.
B. Соединение 76. В соответствии со способом примера 81, раствор соединения, полученного в примере 76A, в CH2Cl2 подвергают взаимодействию с 4-фторбензолсульфонилхлоридом в присутствии воды и NaHCO3. После разбавления дополнительной порцией CH2Cl2 и водной обработки полученный продукт сушат над MgSO4, фильтруют и концентрируют в вакууме. Затем остаток очищают с помощью хроматографии на силикагеле, используя соответствующую систему растворителей, до получения указанного в заглавии продукта.
Пример 77
A. Соединение XXI /A = трет.-бутоксикарбонил, D' = 4-/N,N-диметиламино/-бензил, A' = H/. К раствору соединения XX /A = Boc/ в этаноле добавляют 3 молярных эквивалента 4-аминометил-/N,N-диметил/анилина. Смесь нагревают при кипячении с обратным холодильником в течение 12 часов, охлаждают и смесь концентрируют в вакууме. Остаток очищают с помощью хроматографии на силикагеле, используя соответствующую систему растворителя в качестве элюента, до получения указанного в названии продукта.
B. Соединение 77. В соответствии со способом примера 81, раствор соединения, полученного в примере 77A, в CH2Cl2 подвергают взаимодействию с 4-фторбензолсульфонилхлоридом в присутствии воды и NaHCO3. После разбавления дополнительной порцией CH2Cl2 и водной обработки полученный продукт сушат над MgSO4, фильтруют и концентрируют в вакууме. Затем остаток очищают с помощью хроматографии на силикагеле, используя соответствующую систему растворителей, до получения указанного в заглавии продукта.
Пример 78
A. Соединение XXI /A = трет-бутоксикарбонил, D' = циклопентил, A' = H/.
К раствору соединения XX /A = Boc/ в этаноле добавляют 10 молярных эквивалентов циклопентиламина. Смесь нагревают при кипячении с обратным холодильником в течение 12 часов, и смесь концентрируют в вакууме. Остаток используют без последующей очистки.
B. Соединение 78. В соответствии со способом примера 81, раствор соединения, полученного в примере 78A, в CH2Cl2 подвергают взаимодействию с 4-фторбензолсульфонилхлоридом в присутствии воды и NaHCO3. После разбавления дополнительной порцией CH2Cl2 и водной обработки полученный продукт сушат над MgSO4, фильтруют и концентрируют в вакууме. Затем остаток очищают с помощью хроматографии на силикагеле, используя соответствующую систему растворителей до получения указанного в заглавии продукта.
Пример 79
A. Соединение XXI /A = трет-бутоксикарбонил, D' = 2-/4-пиридил/этил A' = H/. К раствору соединения XX /A = Boc/ в этаноле добавляют 3 молярных эквивалента 4-аминоэтилпиридина. Смесь нагревают при кипячении с обратным холодильником в течение 12 часов, охлаждают и смесь концентрируют в вакууме. Остаток очищают с помощью препаративной хроматографии с обращенной фазой, используя линейный градиент от 5% до 100% ацетонитрил/H2O в качестве элюента, до получения указанного в заглавии продукта.
B. Соединение 79. По способу примера 81 раствор соединения, полученного в примере 79A, в CH2Cl2 подвергают взаимодействию с 4-фторбензолсульфонилхлоридом в присутствии воды и NaHCO3. После разбавления дополнительным количеством CH2Cl2 и обработки водой полученный продукт сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Затем остаток очищают хроматографически на силикагеле, используя соответствующую систему растворителей, до получения указанного в заглавии продукта.
Пример 80
A. 4-цианотетрагидро-4H-пиран. В соответствии со способом Yoneda, R. "Cyanophosphate: Эффективные промежуточные соединения для превращения карбонильных соединений в нитрилы", Tetrahedron Lett, 30, 3681 (1989), раствор тетрагидро-4H/пиранона (9,9 г, 97,8 ммоля) в сухом ТГФ (50 мл) подвергают взаимодействию с цианидом лития (9,7 г, 294 ммоля) и диэтилцианофосфонатом (24 г, 146 ммоля). Смесь перемешивают в течение 24 часов при комнатной температуре. Реакцию гасят добавлением 100 мл H2O. Продукт экстрагируют 1,5 л диэтилового эфира, сушат над безводным MgSO4 и затем концентрируют при пониженном давлении. Остаток растворяют в сухом ТГФ (30 мл) и трет-бутиловом спирте (7,25 г, 97,8 ммоля). Этот раствор медленно добавляют к 75 мл 1 М раствора SmI2. Смесь перемешивают в течение 15 часов при комнатной температуре. Реакцию гасят добавлением 100 мл насыщенного водного раствора NH4Cl. Образующуюся смесь экстрагируют диэтиловым эфиром и органические слои сушат над безводным MgSO4 и концентрируют при пониженном давлении. Очистка с помощью хроматографии на силикагеле дает указанный в заглавии продукт.
B. 4-/аминометил/тетрагидро-4H-пиран.
К раствору соединения примера 80A (10 г, 8+9,9 ммоля) в абсолютированном этаноле (200 мл) добавляют Никель Ренея (2,0 г, 50% суспензия в воде). Смесь перемешивают в течение 24 часов при комнатной температуре при давлении водорода 2,8 кг/см2. Раствор фильтруют через целит, и раствор концентрируют при пониженном давлении. Остаток помещают в эфир (2 л) и промывают рассолом, сушат над безводным MgSO4, затем концентрируют при пониженном давлении до получения указанного в заглавии соединения.
C. /1S, 2R/-N-/1-Бензил-3-/N-/4-/аминометил/тетрагидро-4H- пиран//-2-гидроксипропил/-трет.-бутоксикарбониламин.
К раствору соединения примера 80B (5 г, 48,5 ммоля) в абсолютированном этаноле (20 мл) добавляют соединение XX /A - Boc/ (2,55 г, 9,7 ммоля). Смесь перемешивают в течение 24 часов при комнатной температуре. Раствор концентрируют при пониженном давлении, и сырой продукт очищают на хроматографической колонке до получения указанного в заглавии соединения.
D. Соединение XXII /A = Boc, D' = /4-тетрагидро-4H-пиранил/ метил A' = H/. К раствору соединения XX /A = Boc/ в этаноле добавляют 3 молярных эквивалента соединения, полученного в примере 80C. Смесь нагревают при кипячении с обратным холодильником в течение 12 часов, охлаждают и смесь концентрируют в вакууме. Остаток очищают с помощью препаративной хроматографии с обращенной фазой, используя линейный градиент от 5% до 100% ацетонитрил/H2O в качестве элюента до получения указанного в заглавии соединения.
К раствору соединения XX /A = Boc/ в этаноле добавляют 3 молярных эквивалента N-аминоморфолина. Смесь нагревают при кипячении с обратным холодильником в течение 12 часов, охлаждают и смесь концентрируют в вакууме. Остаток очищают с помощью препаративной хроматографии с обращенной фазой, используя линейный градиент от 5% до 100% ацетонитрил/H2O в качестве элюента, до получения указанного в заглавии соединения.
E. Соединение 80. В соответствии со способом примера 81, раствор соединения, полученного в примере 80D, в CH2Cl2 подвергают взаимодействию с 4-фторбензолсульфонилхлоридом в присутствии воды и NaHCO3. После разбавления дополнительной порцией CH2Cl2 и водной обработки полученный продукт сушат над MgSO4, фильтруют и концентрируют в вакууме. Затем остаток очищают с помощью хроматографии на силикагеле до получения указанного в заглавии соединения.
Пример 81
A. Соединение XXII /A = трет,-бутоксикарбонил. D' = изобутил, E = 3,4-дихлорфенил/. Раствор 316 мг соединения, полученного в примере 39A, в смеси 4: 1 CH2Cl2/ насыщенный водный раствор NaHCO3 последовательно подвергают обработке при комнатной температуре в атмосфере азота, 276 мг. 3,4-дихлорбензолсульфонилхлорида и 95 мг бикарбоната натрия. Смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным раствором NaCl, затем сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 5% диэтиловый эфир /CH2Cl2 в качестве элюента до получения 490 мг продукта. ТСХ: Rf = 0,26, 5% диэтиловый эфир в CH2Cl2. ВЭЖХ: Rт = 18,92 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = изобутил, E = 3,4-дихлорфенил, гидрохлоридная соль/. Раствор 467 мг соединения, полученного в примере 81A, в этилацетате подвергают обработке при -20oC газообразным HCl. HCl пробулькивают через смесь в течение 20 минут, время, в течение которого температуре позволяют подняться до 20oC. Затем через смесь пробулькивают азот в течение 15 минут, и удаляют растворитель в вакууме до получения 412 мг продукта в виде белого твердого вещества, которое используют без последующей очистки.
C. Соединение 81. Раствор 91 мг соединения, полученного в примере 81B, в CH2Cl2 последовательно подвергают обработке при комнатной температуре в атмосфере азота 25 мг аллилхлорформата и 52 мг N,N-диизопропилэтиламина. Смесь перемешивают в течение 4 часов и затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным раствором NaCl, затем сушат над MgSO4, фильтруют и концентрируют в вакууме до получения 89 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,53, 5% диэтиловый эфир в CH2Cl2 ВЭЖХ: Rт = 17,95 мин, /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 82
A. /3-Пиридил/-метил-4-нитрофенил-карбонат. К раствору 3,65 г бис-/нитрофенил/карбоната в 25 мл CH2Cl2 при 0oC последовательно добавляют 0,97 мл 3-пиридилкарбинола и 1,3 мл 4-метилморфина. После перемешивания при комнатной температуре в течение 24 часов полученную смесь разбавляют 100 мл CH2Cl2, промывают насыщенным раствором бикарбоната натрия, водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают фильтрованием через прессованный силикагель, используя 0 - 40% EtOAc/CH2Cl2 в качестве элюента, до получения 1,68 г указанного в заглавии продукта. ТСХ: Rf = 0,19, 50% EtOAc/гексан.
B. Соединение XXII /A = трет-бутоксикарбонил, D' = изобутил, E = 3,4-бензофуразан/. К раствору 498,6 мг соединения, полученного в примере 39A, в 10 мл CH2Cl2 последовательно добавляют 2 мл насыщенного раствора бикарбоната натрия, небольшое количество твердого бикарбоната натрия и 518, 4 мг соединения, полученного в примере 64D. После перемешивания при комнатной температуре в течение 3 часов полученную смесь разбавляют 60 мл CH2Cl2, промывают насыщенным раствором бикарбоната натрия и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии на силикагеле, используя 5% диэтиловый эфир/гексан в качестве элюента, до получения 300 мг белого твердого вещества, ТСХ: Rf = 0,80, 50% EtOAc/гексан.
C. Соединение XXII /A = H, D' = изобутил, E = 3,4-бензофуразан, гидрохлоридная соль/. Раствор 60,3 мг соединения, полученного в примере 82B, в 3 мл EtOAcv при -20oC подвергают обработке безводным газообразным HCl в течение 5 минут. Удаляют баню со льдом, и после дополнительных 10 минут реакционную смесь подвергают барботированию азотом, а затем концентрируют в вакууме, и получающееся белое твердое вещество используют без последующей очистки для последующей реакции.
D. Соединение 82. К раствору соединения, полученного в примере 82C (полный выход), в 2 мл CH2Cl2, последовательно добавляют 45 мкл диизопропилэтиламина и 35,1 мг соединения, полученного в примере 82A. Смесь перемешивают в течение 24 часов и затем концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 60% эфир/CH2Cl2 в качестве элюента с последующей очисткой с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 40% до 100% CH3CN/H2O и 0,1% TFA в качестве элюента. Полученную TFA соль соединения, указанного в заглавии, промывают насыщенным раствором бикарбоната натрия до получения 6,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,15, 20% EtOAc/CH2Cl2, ВЭЖХ: Rт = 13,52 мин /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 83
A. Соединение XXII /A = трет-бутоксикарбонил, D' = изобутил, E = 4-ацетамидо-3-хлорфенил/. Раствор 339 мг соединения, полученного в примере 39A, в 4: 1 CH2Cl2, насыщенный водный раствор NaHCO3 последовательно подвергают обработке при комнатной температуре в атмосфере азота, 324 мг 4-ацетамидо-3-хлорбензолсульфонилхлорида и 102 мг бикарбоната натрия. Смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным раствором NaCl, затем сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 20% диэтиловый эфир в CH2Cl2 в качестве элюента, до получения 498 мг продукта, ТСХ: Rf = 0,27 /20% диэтиловый эфир в CH2Cl2/. ВЭЖХ: Rт = 16,20 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = изобутил, E = 4-ацетамидо-3-хлорфенил, гидрохлоридная соль/. Раствор 474 мг соединения, полученного в примере 83A, в этилацетате подвергают обработке при -20oC газообразным HCl. HCl пробулькивают через смесь в течение 20 минут, время, в течение которого позволяют температуре подняться до 20oC. Затем через смесь пробулькивают азот в течение 15 минут и растворитель удаляют в вакууме до получения 421 мг продукта в виде белого твердого вещества, которое используют без последующей очистки.
C. Соединение 83. Раствор 92 мг соединения, полученного в примере 83B, в CH2Cl2 последовательно подвергают обработке при комнатной температуре в атмосфере азота, 24 мг аллилхлорформата и 52 мг N,N-диизопропилэтиламина. Смесь перемешивают в течение 4 часов и затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным раствором NaCl, затем сушат над MgSO4, фильтруют и концентрируют в вакууме до получения 106 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,38 /20% диэтиловый эфир в CH2Cl2/. ВЭЖХ: Rт = 15,28 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 84
Соединение XXII /A = трет-бутоксикарбонил, D' = изобутил, E - 3,4- дихлорфенил/. К раствору соединения, полученного в примере 51D (220 мг, 0,61 ммоля), в CH2Cl2 (10 мл) добавляют 3,4-дихлорбензолсульфонилхлорид (300 мг, 1,22 ммоля) с последующим добавлением насыщенного раствора бикарбоната натрия (3 мл) и с последующим добавлением 0,1 г твердого бикарбоната натрия. Смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют 100 мл CH2Cl2, органическую часть отделяют, сушат над безводным MgSO4, и органическую часть концентрируют при пониженном давлении до получения 0,17 г сырого продукта. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя CH2Cl2 с последующей обработкой смесью 0,5: 99,5 метанол/CH2Cl2, а затем раствором 1:99 метанол/CH2Cl2 в качестве системы растворителей до получения 103 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,56 /3:97 метанол/CH2Cl2/. ВЭЖХ: Rт = 19,78 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 85
A. /3-тетрагидрофурил/-метил-4-нитрофенилкарбонат. К раствору 1,21 г пара-нитрофенилхлорформата в 20 мл CH2Cl2 при 0oC добавляют последовательно 0,51 г тетрагидро-3-фуранметанола и 0,66 мл 4-метилморфолина. Перемешивают в течение 2 часов и смесь концентрируют в вакууме. Остаток очищают за счет фильтрования через лепешку силикагеля, используя 0 - 50% EtOAc/CH2Cl2 в качестве элюента до получения 1,17 г указанного в заглавии соединения в виде бледно-желтого твердого продукта. ТСХ: Rf = 0,20, 50% EtOAc/гексан.
B. Соединение 85. К раствору 70 мг соединения, полученного в примере 81B, в 1 мл ТГФ добавляют последовательно 56 мкл диизопропилэтиламина и раствор 46,6 мг соединения, полученного в примере 85A в 1 мл ТГФ. Полученную смесь перемешивают в течение 24 часов, а затем концентрируют в вакууме. Остаток разбавляют 60 мл CH2Cl2, промывают 5% бикарбонатом натрия и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 120 мг неочищенного продукта. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 20% EtOAc/CH2Cl2 в качестве элюента до получения 82 мг указанного в заглавии соединения. ТСХ: Rf = 0,4, 20% EtOAc/CH2Cl2. ВЭЖХ: Rт = 17,08 мин. /1H-ЯМР /CDCl3/ соответствует структуре.
Пример 86
Соединение 86. Раствор 42 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 41 мг продукта примера 52A и 46 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя в качестве элюента этилацетат, до получения 43 мг продукта. ТСХ: Rf = 0,44 (20% этилацетата). ВЭЖХ: Rт = 13,14 мин.
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 87
A. Соединение XXII /A = H, D' = изобутил, E = 4-ацетамидо, 3-фторо/. Раствор 25 мг соединения, полученного в примере 54, в EtOAc (10 мл) при 0oC обрабатывают безводным газообразным хлористым водородом в течение 10 минут, и оставляют выстаиваться в течение 12 часов, при этом раствор нагревается до комнатной температуры. Затем полученную смесь концентрируют в вакууме до получения соединения в виде белого твердого продукта, который используют без последующей очистки в последующей реакции.
B. Соединение 87. Порцию в 0,045 ммолей соединения, полученного в примере 87A, помещают в 5 мл CH2Cl2. К этому раствору добавляют 40 мкл диизопропилэтиламина и 6 мкл аллилхлорформата при 0oC, и полученную смесь перемешивают в течение 12 часов, медленно нагревая до комнатной температуры. Полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над безводным сульфатом магния и фильтруют. После концентрирования в вакууме остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента, до получения 11,6 мг указанного в заглавии соединения. ТСХ: Rf = 0,20, 5% MeOH/CH2Cl2 ВЭЖХ: Rт = 14,6 мин;
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 88
Соединение 88. Порцию в 0,033 ммоля соединения, полученного в примере 87A, помещают в 5 мл CH2Cl2. К этому раствору добавляют 26 мкл триэтиламина и 12 мг соединения, полученного в примере 48A, и перемешивают в течение 12 часов. Полученную смесь разбавляют CH2Cl2, промывают насыщенным водным раствором бикарбоната натрия и насыщенным рассолом, сушат над сульфатом магния и фильтруют. После концентрирования смеси в вакууме остаток очищают с помощью хроматографии на толстом слое силикагеля, используя 5% MeOH/CH2Cl2 в качестве элюента, с последующей препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента, до получения 7,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,30, 5% MeOH/CH2Cl2. ВЭЖХ: Rт = 13,38 мин;
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 89
Соединение 89. Раствор 28 мг соединения, полученного в примере 81B, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 8 мг н-пропилхлорформата и 17 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат, и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 31 мг указанного в заглавии продукта в виде белого твердого продукта. ТСХ: Rf = 0,35 /5% диэтиловый эфир в CH2Cl2/ ВЭЖХ: Rт = 18,12 мин,
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 90
Соединение 90. Раствор 28 мг соединения, полученного в примере 83B, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 7 мг н-пропилхлорформата и 15 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 30 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,47 /20% диэтиловвый эфир в CH2Cl2/. ВЭЖХ: Rт = 15,41 мин.
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 91
A. 3-ацетамидобензосульфоновая кислота. Раствор 1,48 г 3-амидобензолсульфоновой кислоты в 1:1 смеси тетрагидрофуран/ вода обрабатывают при 0oC 1,43 г бикарбоната натрия. Спустя 5 минут прикапывают 1,30 г уксусного ангидрида, и реакционной смеси дают нагреться до комнатной температуры в атмосфере азота в течение 14 часов. Реакционную смесь пропускают через колонку ионообменной смолы Amberlyst 15, элюируют водой и концентрируют в вакууме до получения масла, которое после обработки бензолом и азеотропного удаления воды в вакууме получают 1,8 г указанного в заглавии продукта в виде белого кристаллического твердого продукта.
/11H/-ЯМР /CDCl3/ соответствует структуре.
B. 3-ацетамидобензолсульфоновой кислоты, натриевая соль. Соединение, полученное в примере 91A, в воде обрабатывают при 0oC 8,5 мл 1 н гидроксида натрия. Полученную смесь перемешивают в течение 3 часов и концентрируют в вакууме до получения масла, которое после обработки бензолом и азеотропного удаления воды в вакууме дает указанный в заглавии продукт в виде твердого продукта желто-коричневого цвета, который используют непосредственно в следующей реакции.
C. 3-ацетамидобензолсульфонилхлорид. Соединение, полученное в примере 91B, в CH2Cl2 обрабатывают при 0oC 4,5 г пентахлорида фосфора в атмосфере азота. Полученную смесь перемешивают в течение 14 часов, экстрагируют CH2Cl2 и концентрируют в вакууме до получения 1,7 г указанного в заглавии продукта в виде масла коричневого цвета. ТСХ: Rf = 0,21 /1:1 толуол/диэтиловый эфир/. /11H/-ЯМР /CDCl3/ соответствует структуре.
D. Соединение XXII /A = трет.бутоксикарбонил, D' = изобутил, E = 3-ацетамидофенил/. Раствор 280 мг соединения, полученного в примере 39 A в смеси 4 : 1 CH2Cl2/ насыщенный водный NaHCO3, обрабатывают последовательно при комнатной температуре в атмосфере азота, 252 мг соединения, полученного в примере 91C, и 105 мг бикарбоната натрия. Полученную смесь перемешивают в течение 60 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 20% диэтиловый эфир в CH2Cl2 до получения 156 мг указанного в заглавии продукта. ТСХ: Rf = 0,14 /20% диэтиловый эфир в CH2Cl2/. ВЭЖХ: 15,39 мин.
/11H/-ЯМР /CDCl3/ соответствует структуре.
E. Соединение XXII/A=H, D'=изобутил, E = 3-ацетамидофенил гидрохлорид/. Раствор 123 мг соединения, полученного в примере 91D, в этилацетате, обрабатывают при -20oC газообразным HCl. HCl барботируют через смесь в течение 20 минут, и за это время температура смеси повышается до 20oC. Затем через смесь продувают азот в течение 15 минут, растворитель удаляют в вакууме до получения 118 мг указанного в заглавии продукта в виде твердого белого вещества, которое используют непосредственно в последующих реакциях.
F. Соединение 91. Раствор 49 мг соединения, полученного в примере 91E, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота к раствору 48 мг соединения, полученного в примере 48A, и 54 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток обрабатывают с помощью тонкослойной хроматографии на силикагеле, используя 5% CH3OH в CH2Cl2 до получения 42 мг продукта. ТСХ: Rf = 0,32 /5% CH3OH в CH2Cl2/. ВЭЖХ: Rт = 13,27 мин. /11H/-ЯМР /CDCl3/ соответствует структуре.
Пример 92
Соединение 92. К раствору 63,5 мг соединения, полученного в примере 17B, диастереоизомера B, в 1 мл ТГФ добавляют последовательно 52 мкл диизопропилэтиламина, и раствор 43,3 мг соединения, полученного в примере 85 A, в 1 мл ТГФ. Полученную смесь перемешивают в течение 24 часов, а затем концентрируют в вакууме. Остаток разбавляют 60 мл CH2Cl2, промывают 5% бикарбонатом натрия и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 70,7 мг неочищенного продукта. Остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 30% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента, до получения 43,9 мг указанного в заглавии соединения. ТСХ: Rf = 0,29, 100% EtOAc. ВЭЖХ: Rт = 13,24 мин. /11H/-ЯМР /CDCl3/ соответствует структуре.
Пример 93
A. N-гидроксисукцинимидил-/R/-3-гидрокситетрагидрофурил-карбонат. Указанное в заглавии соединение получают по способу примера 48A, исходя из 81 мг /R/-3-гидрокситетрагигидрофурана, до получения 56 мг указанного в заглавии соединения и виде белого твердого вещества.
/1H/ ЯМР /CDCl3/ соответствует структуре.
B. Соединение 94. К раствору 43 мг соединения, полученного в примере 35A, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота 27 мг соединения, полученного в примере 93A, и 39 мг N,N-диизопропиламина. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии в силикагеле, используя 2% CH3OH в CH2Cl2 в качестве элюента, до получения 45 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,52 /5% CH3OH, CH2Cl2/. ВЭЖХ: Rт = 14,94 мин.
/1H/ ЯМР /CDCl3/ соответствует структуре.
Пример 94
Соединение 94. Раствор 47 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно, при комнатной температуре в атмосфере азота 28 мг продукта, полученного в примере 93A, и 39 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографически, используя 5% метанол в CH2Cl2 в качестве элюента до получения 40 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,38 (этилацетат). ВЭЖХ: Rт = 13,09 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 95
Соединение 95. К раствору 72,0 мг (0,189 ммолей) соединения, полученного в примере 51D, в CH2Cl2 (4 мл) добавляют водный раствор бикарбоната натрия (1 мл), бикарбонат натрия (19,1 мг, 0,227 ммоля) и 57,1 мг 2,3-дихлортиофенсульфонилхлорида (0,227 ммоля). Спустя 14 часов полученную смесь разбавляют EtOAc, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя от 5% до 12% EtOAc/CH2Cl2 в качестве элюента, до получения 49,1 мг указанного в заглавии продукта. ТСХ: Rf = 0,62, 25% EtOAc/CH2Cl2/, ВЭЖХ: Rт = 17,3 мин;
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 96
A. /4-ацетамидо/-фенилметил-4-нитрофенилкарбонат. К раствору 242,8 мг р-нитрофенилхлорформата в 5 мл ацетонитрила при 0oC добавляют последовательно 165, 2 мг 4-ацетамидобензилового спирта и 0,13 мл 4-метилморфолина. Полученную смесь перемешивают в течение 24 часов и концентрируют в вакууме. Остаток помещают в CH2Cl2, и промывают 5% бикарбонатом натрия и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 320 мг указанного в заглавии соединения.
ТСХ: Rf = 0,23 50% EtOAc/гексан.
B. Соединение 96. К раствору соединения, полученного в примере 40A, в 1 мл THF добавляют последовательно 56 мкл диизопропилэтиламина и 63 мг соединения, полученного в примере 96A. Полученную смесь перемешивают в течение 24 часов, концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 10% метанол/CH2Cl2 в качестве элюента, с последующей обработкой с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 30% до 100% CH3CN/H2O, с 0,1% TFA в качестве элюента, до получения 50,2 мг указанного в заглавии соединения. ТСХ: Rf = 0,43 10% метанол/CH2Cl2/. ВЭЖХ: Rт = 13,54 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 97
Соединение 97. К раствору 60 мг соединения, полученного в примере 35A, в 1 мл THF добавляют последовательно 54 мкл диизопропилэтиламина и раствор 48,9 мг соединения, полученного в примере 85A, в 1 мл THF. Полученную смесь перемешивают в течение 24 часов, а затем концентрируют в вакууме. Остаток разбавляют 60 мл CH2Cl2, промывают 5% бикарбонатом натрия и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя в качестве элюента 20%EtOAc/CH2Cl2, до получения 46,9 г мг указанного в заглавии соединения. ТСХ: Rf = 0,31, 20% EtOAc/CH2Cl2, ВЭЖХ: Rт = 15,18 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 98
Соединение 98. К раствору 61,0 мг соединения, полученного в примере 35A, в 1 мл THF, добавляют последовательно 49 мкл диизопропилэтиламина и раствор 44 мг соединения, полученного в примере 82A в 1 мл THF. Полученную смесь перемешивают в течение 24 часов, концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 5% метанол/CH2Cl2 в качестве элюента до получения 61,0 мг в виде белого твердого вещества. ТСХ: Rf = 0,19, 5% метанол/CH2Cl2/. ВЭЖХ: Rт = 13,28 мин;
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 99
Соединение 99. Раствор 75 мг соединения, полученного в примере 51D, и 45 мг 4-хлорбензолсульфонилхлорида подвергают взаимодействию по способу примера 60. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, с использованием линейного градиента от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента, получают 24,6 мг указанного в заглавии соединения. ТСХ: Rf = 0,3, 4% MeOH/CH2Cl2/. ВЭЖХ: Rт = 15,87 мин;
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 100
Соединение 100. Раствор 40 мг соединения, полученного в примере 51D, и 45 мг 4-метоксибензолсульфонилхлорида подвергают взаимодействию по способу примера 60. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента, получают 21,4 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,2, 4% MeOH/CH2Cl2. ВЭЖХ: Rт = 14,85 мин;
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 101
Соединение 101. Это соединение получают из соединения, полученного в примере 128, за счет обработки газообразным хлористым водородом и последующего взаимодействия с соединением, полученным в примере 48A по способу примера 132. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1 TFA в качестве элюента на порцию неочищенной смеси, получают 4,2 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,2, 4% MeOH/CH2Cl2. ВЭЖХ: Rт = 11,53 мин.
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 102
Соединение 102. Раствор 36 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно в атмосфере азота 8 мг метилхлорформата и 22 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 30% диэтиловый эфир с CH2Cl2 в качестве элюента, до получения 27 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,10 (30% диэтиловый эфир в CH2Cl2) ВЭЖХ: Rт = 13,49 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 103
Соединение 103. Раствор 103. Раствор 29 мг соединения, полученного в примере 81B, с CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 6 мг метилхлорформата и 17 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 30% диэтиловый эфир в CH2Cl2 в качестве элюента до получения 27 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,10/30% диэтиловый эфир в CH2Cl2/. ВЖЭХ: Rт = 13,49 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Соединение 103
Соединение 103. Раствор 29 мг соединения, полученного в примере 81B, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 6 мг метиленхлорформата и 17 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 30% диэтиловый эфир в CH2Cl2 в качестве элюента до получения 27 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,10 (30% диэтиловый эфир в CH2Cl2). ВЭЖХ: Rт = 13,49 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 103
Соединение 103. Раствор 29 мг соединения, полученного в примере 81B, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота 6 мг метилхлорформата и 17 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают на хроматографической колонке низкого давления на силикагеле, используя 5% диэтиловый эфир/ CH2Cl2 в качестве элюента, до получения 29 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,24 (5% диэтиловый эфир в CH2Cl2).ВЭЖХ: Rт = 17,07 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 104
Соединение 104. Раствор 31 мг соединения, полученного в примере 85A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 8 мг метилхлорформата и 21 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 5% диэтиловый эфир /CH2Cl2 в качестве элюента до получения 24 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,23 (5% диэтиловый эфир в CH2Cl2), ВЭЖХ: Rт = 15,41 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 105
A. N-гидроксисукцинимидилметаллилкарбонат. К раствору 2,9 мл 1,93 М фосфогена в толуоле при -10oC добавляют 857 мг металлилового спирта. Полученную смесь перемешивают в течение 2 часов при -10oC до получения 1,9М раствора указанного в заглавии соединения, которое используют непосредственно в последующих реакциях.
B. Сравнение 105. Раствор 39 мг соединения, полученного в примере 40A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 0,05 мл соединения, полученного в примере 105A, и 24 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью тонкослойной хроматографии, используя этилацетат в качестве элюента, до получения 18 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,67 (этилацетат). ВЭЖХ: Rт = 14,97 мин. /1H/-ЯМР /CDCl3/ соответствует структуре
Пример 106
Соединение 106. Раствор 31 мг соединения, полученного в примере 81B, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота 0,04 мл соединения, полученного в примере 105A, и 18 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 2 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления, используя 5% диэтиловый эфир/CH2Cl2 в качестве элюента до получения 19 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,34 /5% диэтиловый эфир/ CH2Cl2/. ВЭЖХ: Rт = 18,24 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 107
Соединение 107. Раствор 28 мг соединения, полученного в примере 35A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота, 0,05 мл соединения, полученного в примере 105A, и 19 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NаCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 5% диэтиловый эфир в CН2Cl2 в качестве элюента до получения 18 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,25 (5% диэтиловый эфир в CH2Cl2). ВЭЖХ: Rт = 16,68 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 108
Соединение 108. К раствору 62,5 мг 124B в 1 мл THF добавляют последовательно 56 мкл диизопропилэтиламина и раствор 49,6 мг соединения, полученного в примере 82A, в 1 мл TF. Полученную смесь перемешивают в течение 24 часов, а затем концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 50% EtOAc/CH2Cl2 в качестве элюента, с последующей обработкой с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 30% до 100% СH3CN/H2O с 0,1% TFA в качестве элюента на порцию неочищенной смеси, в результате чего получают 4,2 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,16, 10% метанол/CH2Cl2. ВЭЖХ: Rт = 13,67 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 109
A. /S/-4-метоксикарбонил-оксазолидин-2-он. К раствору 4,88 г гидрохлорида серинового сложного эфира в 25 мл воды добавляют 6,94 г карбоната калия. Полученную смесь охлаждают до 0oC, и прикапывают 19,5 мл фосгена. После перемешивания при 0oC в течение 3 часов воду удаляют и получают белое твердое вещество, которое промывают большим количеством CH2Cl2. Затем органический слой сушат над сульфатом магния, фильтруют и концентрируют до получения 3,26 г указанного в заглавии продукта в виде прозрачного масла.
/1H/-ЯМР /D2O/: δ = 3,82 (с, 3H), 4,43 (дд, 1H), 4,53 (дд, 1H), 4,67 (т, 1H), 6,29 (с, 1H).
B. /S/-4-гидроксиметилоксазолидин-2-он. К раствору 3,26 г соединения, полученного в примере 109 A, в 20 мл этанола при 0oC добавляют 0,85 г боргидрида натрия небольшими порциями. Ледяную баню удаляют, и спустя еще 3 часа к полученной смеси добавляют 20 мл 2,0 н хлористого водорода; смесь концентрируют до получения масла. Остаток экстрагируют EtOAc, и органический раствор сушат над сульфатом магния, фильтруют и концентрируют до получения 2,50 г указанного в заглавии соединения.
/1H/-ЯМР /CDCl3/: δ = 2,48 (с, 1H), 3,69 (дд, 1H), 4,08 (м, 1H), 3,31 (т, 1H), 4,57 (т, 1H).
C. 4-нитрофенил-//S/-4-оксазолидин-2-онил/-метилкарбонат. К раствору 1,04 г р-нитрофенилхлорформата в 20 мл CH2Cl2 при 0oC добавляют последовательно 0,5 г соединения, полученного в примере 109B, и 0,6 мл 4-метилморфолина. Полученную смесь перемешивают в течение 2 часов при комнатной температуре, а затем концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 20% EtOAc в CH2Cl2 в качестве элюента, до получения 0,57 г указанного в заглавии соединения, ТСХ: Rf = 0,10, 50% EtOAc/гексан.
D. Соединение 109. К раствору 60 мг соединения, полученного в пример 35A, в 1 мл THF добавляют последовательно 56 мкл диизопропиламина и раствор 51,1 мг соединения, полученного в примере 109C в 1 мл ацетонитрила. Полученную смесь перемешивают в течение 24 часов, а затем концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 5% метанол/CH2Cl2 в качестве элюента до получения 60,4 мг указанного в заглавии соединения. ТСХ: Rf = 0,38 5% метанол/CH2Cl3. ВЭЖХ: Rт = 14,11 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 110
Соединение 110. К раствору 60 мг соединения полученного в примере 40A, 1 мл ацетонитрила добавляют последовательно 51 мкл диизопропилэтиламина и раствор 46,8 мг соединения, полученного в примере 109C в 1 мл ацетонитрила. Полученную смесь перемешивают в течение 48 часов, а затем концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 10% метанол/CH2Cl2 в качестве элюента, с последующей обработкой с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1 TFA в качестве элюента, до получения 16 мг указанного в заглавии соединения, TCX:Rf = 0,28, 50% EtOAc/CH2Cl2. ВЭЖХ: Rт = 12,47 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 111
К раствору 0,067 ммоля соединения, полученного в примере 114Д, в 5 мл тетрагидрофурана добавляют 20 мкл диизопропилэтиламина, а затем прикапывают раствор соединения, полученного в примере 82A, в 5 мл тетрагидрофурана в течение 1 часа. Полученную смесь перемешивают в течение 16 часов, затем концентрируют в вакууме. Неочищенный остаток очищают с помощью хроматографии на толстом слое силикагеля, используя 5% MeOH/CH2Cl2 в качестве элюента до получения 21,8 мг указанного в заглавии соединения. TCX:Rf = 0,45, 5% MeOH/CH2Cl2; /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 112
А. Соединение XXII /A = трет.-бутоксикарбонил, D'= изобутил E = 3-сульфонамидофенил/. К раствору 96,6 мг (0,287 ммоля) соединения, полученного в примере 39A, в CH2Cl2 (4 мл) добавляют водный бикарбонат натрия (1 мл), бикарбонат натрия 36,2 мг (0,431 ммоля) и м-бензолдисульфонилхлорид 86,9 мг (1,08 ммоля). После перемешивания в течение 1 часа добавляют 30% гидроксид аммония (10 мл). Спустя 14 часов полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью флеш-хроматографии, используя градиент от 0% до 100% метанол/CH2Cl2 в качестве элюента, до получения 49,3 мг указанного в заглавии продукта. /1H/-ЯМР /CDCl3/ соответствует структуре.
В. Соединение XXII /A = H,D' = изобутил, E = 3-сульфонамидофенил, гидрохлорид/. Раствор 49,3 мг (0,089 ммоля) соединения, полученного в примере 112A, в EtOAc (10 мл) при -20oC обрабатывают безводным газообразным HCl в течение 10 минут. Ледяную баню убирают, и спустя еще 15 минут реакционную смесь продувают азотом, концентрируют в вакууме до получения 53,1 мг указанного в заглавии продукта в виде соли HCl.
/1H/-ЯМР /CDCl3/ соответствует структуре.
С. Соединение 112. К раствору 53,1 мг соединения, полученного в примере 112B (0,089 ммоля) в CH2Cl2 (3 мл) добавляют последовательно при комнатной температуре в атмосфере азота 0,031 мл (0,177 ммолей) диизопропилэтиламина и 24,3 мг (0,106 ммолей) соединения, полученного в примере 48A. Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в CH2Cl2 и промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученный остаток очищают с помощью хроматографии низкого давления на колонке с силикагалем, используя градиент от 5% до 20% EtOAc в CH2Cl2 в качестве элюента до получения 10,8 мг указанного в заглавии продукта. TCX:Rf = 0,4 25% EtOAc в CH2Cl2. ВЭЖХ:Rт = 13,3 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 113.
А. 3-фурансульфонилхлорид. В высушенной в пламени стеклянной колбе в атмосфере азота к раствору 428 мг (2,909 ммоля) 3-бромфурана в безводном тетрагидрофуране при -78oC добавляют 2,0 мл н-бутиллития (3,2 ммоля при 1,6 молярном растворе в гексане). Спустя 45 минут полученный раствор через канюлю добавляют к 20oC раствору сульфурилхлорида в диэтиловом эфире (5 мл+2 мл смолы). Через 1 час реакцию гасят 0,5 н соляной кислотой, и экстрагируют диэтиловым эфиром. Эфирные экстракты промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 158 мг указанного в заглавии продукта.
1H/-ЯМР /CDCl3/ соответствует структуре.
В. Соединение XXII/A = трет.-бутоксикарбонил, D' = изобутил, E = 3-фурил/. К раствору 289,7 мг (0,861 ммоля) соединения, полученного в примере 39A, в CH2Cl2 (8 мл) добавляют водный раствор бикарбоната натрия (2 мл), твердый бикарбонат натрия (108 мг, 1,292 ммоля) и полученный в примере 113A продукт (157,8 мг, 1,08 ммоля). После перемешивания в течение 1 часа добавляют 30% гидроксид аммония (10 мл). Спустя 14 часов полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью флеш-хроматографии, используя градиент от 1% до 15% EtOAc/CH2Cl2.
С. Соединение XXII/A = H, D'= изобутил, E = 3-фурил, гидрохлорид/. Раствор 217,3 мг (0,581 ммоля) соединения, полученного в примере 113B, в EtOAc (15 мл) при -20oC обрабатывают безводным газообразным HCl в течение 10 минут. Ледяную баню удаляют, и после еще 15 минут реакционную смесь продувают азотом, затем концентрируют в вакууме до получения 228 мг указанного в заглавии продукта в виде HCl соли. TCX:Rf = 0,52 10% метанол/CH2Cl2.
Д. Соединение 113. К раствору 65,3 мг соединения, полученного в примере 113C (0,162 ммоля), в 3 мл CH2Cl2 добавляют последовательно при комнатной температуре в атмосфере азота 0,056 мл (0,324 ммоля) диизипропиламина и 44,6 мг (0,194 ммоля) соединения, полученного в примере 48A.
Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в CH2Cl2 и промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя градиент от 3% до 20% EtOAc в CH2Cl2, до получения 10,8 мг указанного в заглавии продукта. TCX:Rf = 0,6, 25% EtAOc/CH2Cl2. ВЭЖХ:Rт = 13,9 мин;
1Н-ЯМР /CDCl3/ соответственно структуре.
Пример 114
А. Аминометилциклопентан. К раствору LiAlH4 (38 г, 1,0 ммоль) в 2 л диэтилового эфира добавляют циклопентанкарбонитрил (73,2 г, 0,77 ммоля) в виде раствора в 250 мл эфира. Этот раствор перемешивают в течение ночи при комнатной температуре, а затем гасят, добавляя органику к 3 л насыщенного раствора калий, натрий тартрата. Амин экстрагируют 3 эфира, сушат над безводным K2CO3, затем концентрируют перегонкой до примерно 400 мл полного объема. Неочищенный продукт очищают перегонкой, получая 58,2 г указанного в заглавии соединения в виде бесцветного масла.
/1H/-ЯМР /CDCl3/ соответствует структуре.
В. Соединение XXI /A = трет.-бутоксикарбонил, D' = циклопентил метил, A' = H/. К полученному в примере 114A соединению (20 г, 0,2 ммоля) добавляют соединение XX (A - Boc) (5,84 г), и полученную смесь перемешивают в течение 24 часов при комнатной температуре. Полученный раствор концентрируют перегонкой при пониженном давлении. Остаток тщательно растирают с гексаном, и твердую часть собирают фильтрованием с подсосом и промывают гексаном до получения 7,08 г белого твердого продукта, который используют без дальнейшей очистки. TCX:Rf = 0,59 (1:10:90 конц. NH4OH/ метанол CH2Cl2/,
/1H/-ЯМР /CDCl3/ соответствует структуре.
С. Соединение XXII /A = трет.-бутоксикарбонил, D'= циклопентилметил, E = 4-фторфенил/. К раствору соединения, полученного в примере 114B (200 мг, 0,55 ммоля) в CH2Cl2 (10 мл) добавляют 4-фторбензолсульфонилхлорид (210 мл, 1,1 ммоля), а затем добавляют насыщенный раствор бикарбоната натрия (3 мл), и твердый бикарбонат натрия (0,1 г, 1,2 ммоля). Полученную смесь оставляют выстаиваться при комнатной температуре в течение ночи. Раствор разбавляют 100 мл CH2Cl2, органическую часть выделяют, сушат над сульфатом магния и органику концентрируют при пониженном давлении до получения 0,33 неочищенного продукта. Этот материал очищают с помощью жидкостной хроматографии среднего давления, используя CH2Cl2, а затем 0,5:99,5 метанол /CH2Cl2, после чего 1: 99 раствор метанол /CH2Cl2 в качестве системы растворителей до получения 120 мг (выход 42%) указанного в заглавии соединения в виде белого твердого вещества. TCX:Rf = 0,48/ 3:97 метанол /CH2Cl2; ВЭЖХ:Rт = 18,22 минуты.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Д. Соединение XXII /A = H, D'= циклопентилметил, E = 4-фторфенил, гидрохлорид/. Раствор 266 мг соединения, полученного в примере 114C, в этилацетате обрабатывают при -20oC газообразным HCl в течение 20 минут, причем за это время температура повышается до 20oC. Затем через смесь продувают азот в течение 15 минут, и растворитель удаляют в вакууме до получения 224 мг белого твердого вещества, которое используют непосредственно в дальнейшей реакции.
E. Соединение 114. Раствор 31 мг соединения, полученного в примере 114D, В CH2Cl2, обрабатывают последовательно при комнатной температуре в атмосфере азота, 9 мг аллилхлорформата и 19 мг, N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 34 мг указанного в заглавии продукта в виде белого твердого вещества. TCX:Rf = 0,34 (5% диэтиловый эфир в CH2Cl2). ВЭЖХ:Rт = 17,21 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 115
Соединение 115. Раствор 31 мг соединения, полученного в примере 114B, в CH2Cl2, обрабатывают последовательно при комнатной температуре в атмосфере азота 8 мг этилхлорформата и 19 мг N,N-диизопропилэтиламина. Полученную смесь перешивают в течение 3 часов, затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 35 мг указанного в заглавии продукта в виде белого твердого вещества. TCX:Rf = 0,32 (5% диэтиловый эфир). /CH2Cl2/ ВЭЖХ:Rт = 16,86 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 116
A. Соединение XXII/A = трет.-бутоксикарбонил, D' = циклопентилметил, E = 4-хлорфенил/. Соединение, полученное в примере 114B (252 мг), подвергают взаимодействию с 4-хлорбензолсульфонилхлоридом (175 мг) по способу примера 166A. После обработки и чистки с помощью хроматографии на силикагеле с использованием EtOAc/CH2Cl2 в качестве элюента, получают продукт в виде белого твердого вещества, /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII/A = H, D' = циклопентилметил, E = 4-хлорфенил, гидрохлорид/. Раствор 320 мг соединения, полученного в примере 116A, в 20 мл EtOAc обрабатывают безводным газообразным HCl в течение 5 минут. Реакционную смесь продувают азотом, затем концентрируют в вакууме до получения белого твердого вещества, которое используют непосредственно в последующей реакции.
C. Соединение 116. К раствору 63,4 мг соединения, полученного в примере 116B, в 1 мл THF, добавляют последовательно 54 мкл диизопропилэтиламина и раствор 39,9 мг соединения, полученного в примере 48A, в 1 мл THF. Полученную смесь перемешивают в течение 24 часов, а затем концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 20% EtOAc в CH2Cl2 в качестве элюента, до получения 0,62 г указанного в заглавии соединения. TCX: Rf = 0,71, 40% EtOAc/CH2Cl2. ВЭЖХ: Rт = 16,88 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Соединение 117. Раствор 66,1 мг соединения, полученного в примере 116B, в 1 мл THF обрабатывают последовательно 56 мкл диизопропилэтиламина и 19,3 мкл аллилхлорформата. Полученную смесь перемешивают в течение 4 часов, и концентрируют в вакууме. Остаток помещают в 50 мл EtOAc, и промывают 1,0 н HCl, насыщенным бикарбонатом натрия, рассолом, сушат над сульфатом магния, фильтруют и концентрируют. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 20% EtOAc в гексане в качестве элюента, до получения 69,7 мг указанного в заглавии соединения. TCX: Rf = 0,20, 20% EtOAc/ гексан. ВЭЖХ: Rт = 17,83 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 118
Соединение 118. К раствору 65,3 мг соединения, полученного в примере 116B, в 1 мл THF, добавляют последовательно 55 мкл диизопропилэтилена и раствор 49,2 мг соединения, полученного в примере 82A, в 1 мл THF. Полученную смесь перемешивают в течение 24 часов и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 40% EtOAc в CH2Cl2 в качестве элюента, а затем с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 40% до 80% CH3CN/H2O для элюирования, в результате чего получают 70,7 мг указанного в заглавии соединения. TCX: Rf = 0,27, 40% EtOAc/CH2Cl2. ВЭЖХ: Rт = 14,85 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 119
Соединение 119. Раствор 26 мг соединения, полученного в примере 81B, в CH2Cl2 обрабатывают последовательно, при комнатной температуре в атмосфере азота, 6 мг этилхлорформата и 15 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат, и промывают 0,5 н HCl, насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления, на колонке с силикагелем, используя 5% диэтиловый эфир /CH2Cl2 в качестве элюента, до получения 26 мг указанного в заглавии продукта в виде белого твердого вещества. TCX: Rf = 0,19/5% диэтиловый эфир в CH2Cl2/. ВЭЖХ: Rт = 17,50 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 120
Соединение 120. Раствор 30 мг соединения, полученного в примере 40A, в CH2Cl2, обрабатывают последовательно при комнатной температуре в атмосфере азота, 8 мг этилхлорформата и 18 мг N,N-диизопропилэтилена. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат, и промывают 0,5 н HCl и насыщенным NaCl, а затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии на силикагеле, используя в качестве элюента до получения 25 мг указанного в заглавии продукта в виде белого твердого продукта. TCX: Rf = 0,60 (этилацетат). ВЭЖХ: Rт = 13,86 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 121
Соединение 121. Раствор 26 мг соединения, полученного в примере 35A, в CH2Cl2 обрабатывают последовательно, при комнатной температуре в атмосфере азота 7 мг этилхлорформата и 17 мг N,N-диизопропиламина. Полученную смесь перемешивают в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 5% диэтиловый эфир/ CH2Cl2, до получения 22 мг указанного в заглавии продукта в виде белого твердого вещества. TCX: Rf = 0,14 (5% диэтиловый эфир /CH2Cl2). ВЭЖХ: Rт = 15,95 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 122
Соединение 122. Раствор 27 мг соединения, полученного в примере 35A, в CH2Cl2 обрабатывают последовательно, при комнатной температуре в атмосфере азота, 8 мг аллилхлорформата и 18 мг N,N-диизопропиламина. Полученную смесь перемешивают в в течение 3 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают 0,5 н HCl и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 5% диэтиловый эфир в CH2Cl2. ВЭЖХ: Rт = 16,28 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 123
A. Соединение XXII /A = трет.-бутоксикарбонил, D' = изобутил, E = 3,4-диметоксифенил/. К раствору 401 мг (1,192 ммоля) соединения, полученного в примере 39A, в CH2Cl2 (12 мл) добавляют водный бикарбонат натрия (3 мл), твердый бикарбонат натрия (130 мг, 1,549 ммоля) и 3,4-диметоксибензолсульфонилхлорид (33,8 мг, 1,43 ммоля). Спустя 14 часов полученную смесь разбавляют EtOAc, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью флеш-хроматографии, используя в качестве элюента градиент от 5% до 25% EtOAc/CH2Cl2 до получения 440,1, мг указанного в заглавии продукта. TCX: Rf = 0,72, 20% EtOAc/CH2Cl2.
B. Соединение XXII /A = H, D' = изобутил, E = 3,4 - диметоксифенил гидрохлорид/. Раствор 440 мг (0,820 ммоля) соединения, полученного в примере 123A, в EtOAc (15 мл) при -20oC обрабатывают безводным газообразным HCl в течение 10 минут. Ледяную баню удаляют, и после дополнительных 15 минут, реакционную смесь продувают азотом, затем концентрируют в вакууме до получения 610 мг указанного в заглавии продукта в виде соли HCl. TCX: Rf = 0,44, 10% метанол/CH2Cl2.
C. Соединение 123. Раствор 38,9 мг соединения, полученного в примере 123B (0,170 ммоля), в CH2Cl2 (3 мл) обрабатывают последовательно при комнатной температуре, в атмосфере азота, 0,49 мл (0,283 ммоля) диизопропилэтиламина и 66,9 мг (169,6 ммоля) соединения, полученного в примере 48A. Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в CH2Cl2 и промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя градиентное элюирование (10% до 25% диэтиловый эфир в CH2Cl2 до получения 57,6 мг указанного в заглавии продукта. TCX: Rf = 0,39, 25% этиловый эфир/CH2Cl2. ВЭЖХ: Rт = 14,3 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 124
A. Соединение XXII /A - трет.-бутоксикарбонил, D' = изобутил, E = 3,4 дифторфенил/. К раствору 332,7 мг (0,989 ммоля) соединения, полученного в примере 39A, в CH2Cl2 (12 мл), добавляют 3 мл водного бикарбоната натрия, 125 мг твердого бикарбоната натрия (1,483 ммоля) и 3,4-дифторбензолсульфонилхлорид (231 мг, 1,088 ммоля). Спустя 14 часов полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью флеш-хроматографии, используя градиентное элюирование /от 5% до 25% диэтиловый эфир /CH2Cl2/ до получения 313,6 мг указанного в заглавии продукта. /1H/-ЯМР /CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = изобутил, E = 3,4 - дифторфенил, гидрохлорид/. Раствор 312,6 мг (0,610 ммоля) соединения, полученного в примере 124A в EtOAc (15 мл) при -20oC обрабатывают безводным газообразным HCl в течение 10 минут. Ледяную баню удаляют, и спустя еще 15 минут реакционную смесь продувают азотом, затем концентрируют в вакууме до получения 280 мг указанного в заглавии продукта в виде белого твердого вещества. TCX: Rf = 0,46, 10% метанол /CH2Cl2.
C. Соединение 124. К раствору 64,7 мг соединения, полученного в примере 124B (0,144 ммоля) в CH2Cl2 (3 мл), добавляют последовательно при комнатной температуре в атмосфере азота 0,050 мл (0,288 ммоля) диизопропилэтиламина и 39,6 мг (172,9 ммоля) соединения, полученного в примере 48A. Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в CH2Cl2 и промывают насыщенным рассолом, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя градиентное элюирование / от 5% до 20% диэтиловый эфир в CH2Cl2 /до получения 44 мг указанного в заглавии продукта. TCX: Rf = 0,54, 25% диэтиловый эфир /CH2Cl2. ВЭЖХ; Rт = 15,4 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 125
Соединение 125. Это соединение получают из соединения, полученного в примере 146B, по способу примера 88. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, с использованием линейного градиента элюента (от 35% до 100% CH3CN/H2O/ с добавлением 0,1% TFA), получают 10,5 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,4, 4% MeOH/CH2Cl. ВЭЖХ: Rт = 14.06 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 126
A. Соединение XXI /A = трет.бутоксикарбонил, D' = метил, A' = H/. К раствору соединения XX /1,7 ммоля/ в 20 мл этанола добавляют газообразный метиламин при комнатной температуре, в течение 30 минут. Этот раствор перемешивают в течение ночи, концентрируют при пониженном давлении, до получения 0,47 г указанного в заглавии соединения, которое используют без дополнительной очистки. ТСХ: Rf = 0,19, 1:10:90 NH4OH/метанол/ CH2Cl2, /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение 126. К раствору продукта примера 126 A /0,15 г, 0,51 ммоля / в CH2Cl2 (10 мл) добавляют насыщенный раствор бикарбоната натрия /3 мл/, затем добавляют твердый бикарбонат натрия (90 мг, 1,1 ммоля), затем добавляют 3,4-дихлорбензолсульфонилхлорид (0,25 г, 1,0 ммоля). Полученную смесь перемешивают при комнатной температуре в течение ночи. Органическую часть экстрагируют 100 мл CH2Cl2, сушат над безводным сульфатом магния, концентрируют при пониженном давлении, а затем очищают с помощью хроматографии среднего давления на силикагеле, используя градиентную систему элюирования: CH2Cl2, а затем 5 : 95 эфир /CH2Cl2. Указанное в заглавии соединение получают в виде бесцветной пены (210 мг). ТСХ: Rf = 0,42 /3 : 97 метанол/CH2Cl2/, ВЭЖХ: Rт = 17,2 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 127
Соединение 127. К раствору продукта примера 126A /0,15 г 0,51 ммоля/ в CH2Cl2 (10 мл) добавляют насыщенный раствор бикарбоната натрия (3 мл), затем добавляют твердый бикарбонат натрия (100 мг, 1,0 ммоля), затем добавляют 4-фторбензолсульфонилхлорид (0,20 г, 1,0 ммоль). Полученную смесь перемешивают при комнатной температуре в течение ночи. Органику экстрагируют 100 мл CH2Cl, сушат над безводным сульфатом магния, концентрируют при пониженном давлении, затем очищают с помощью хроматографии среднего давления на силикагеле, используя градиентное элюирование: Ch2Cl2, а затем 5:95 эфир / CH2Cl2. Указанное в заглавии соединение получают в виде белого твердого вещества /104 мг/. ТСХ: Rf = 0,36, 3 : 97 метанол / CH2Cl2. ВЭЖХ: Rт = 15,85 мин; /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 128
Соединение 128. К раствору продукта примера 126 A (0,15 г, 0,51 ммоля / в CH2Cl2/ 6 мл) добавляют насыщенный раствор бикарбоната натрия (3 мл), затем добавляют твердый бикарбонат натрия (90 мг, 1,0 ммоля), затем добавляют ацетамидобензолсульфонилхлорид (0,24 г, 1,02 ммоля). Полученную смесь перемешивают при комнатной температуре в течение ночи. Органику экстрагируют 100 мл CH2Cl2, сушат над безводным сульфатом магния, концентрируют при пониженном давлении, очищают с помощью хроматографии среднего давления на силикагеле, используя градиент системы растворитель : CH2Cl2, а затем 5 : 95 EtOAc/CH2Cl2, а затем 10 : 90 EtOAc/CH2Cl2. Указанное в заглавии соединение получают в виде 244 мг белого твердого вещества. ТСХ: Rf = 0,13, 3 : 97 метанол /CH2Cl2, ВЭЖХ; Rт = 13,47 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 129
A. Соединение XXI / A = трет.бутоксикарбонил, D' = /2-тетра-гидрофурил/-метил, A' = H/. К раствору соединения XX /3,3 ммоля/ в 30 мл этанола добавляют тетрагидрофурфуриламин (1,03 мл, 10 ммолей). Полученную смесь нагревают до 85oC и перемешивают в течение ночи. Раствор фильтруют, концентрируют при пониженном давлении до получения 1,29 г указанного в заглавии соединения, которое используют без дополнительной очистки. ТСХ: Rf = 0,52, 1 : 10 : 90 NH4OH/метанол/CH2Cl2.
B. Соединение 129. К раствору соединения, полученного в примере 129 A /200 мг, 0,55 ммоля/ в CH2Cl2 (6 мл) добавляют 4-фторбензолсульфонилхлорид (320 мг, 1,6 ммоля), затем насыщенный раствор бикарбоната натрия (3 мл) и твердый бикарбонат натрия (0,1 г, 1,2 ммоля). Полученную смесь перемешивают при комнатной температуре в течение ночи. Этот раствор разбавляют 100 мл CH2Cl2, органику выделяют, сушат над сульфатом магния, органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиент системы растворителей /CH2Cl2, затем 5 : 95 эфир/ CH2Cl2, затем 10 : 90 эфир/CH2Cl2, до получения 130 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,35, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 16,37 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 130
A. Соединение XXI / A = трет.-бутоксикарбонил, D' = изобутил, A'=H/. К раствору соединения XX / A = трет.-бутоксикарбонил/ (2,5 ммоля) в 30 мл этанола добавляют раствор 2-метилаллиамингидрохлорида (1,34 г, 12,5 ммоля) и KOH (0,70 г, 12,5 ммоля) в этаноле (20 мл). Полученную смесь перемешивают в течение 30 минут при комнатной температуре. Растворы объединяют и нагревают при 85oC в течение 24 часов. Полученный раствор фильтруют и концентрируют при пониженном давлении до получения 0,82 г указанного в заглавии соединения, которое используют без дополнительной очистки. ТСХ: Rf = 0,45, 1 : 10 : 90 конц. NH4OH/метанол/CH2Cl2.
B. Соединение 130. К раствору продукта примера 130A (0,20 г, 0,60 ммоля) в CH2Cl2 (6 мл) добавляют насыщенный раствор бикарбоната натрия (3 мл), затем твердый бикарбонат натрия (0,1 г, 1,2 ммоля), а затем p-фторбензолсульфонилхлорид (0,35 г 1,78 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Органику экстрагируют 100 мл CH2Cl2, сушат над безводным сульфатом магния, концентрируют при пониженном давлении, а затем очищают с помощью хроматографии среднего давления на силикагеле, используя градиентную систему CH2Cl2, затем 1 : 99 метанол/CH2Cl2. Указанное в заглавии соединение получают в виде белого твердого вещества (180 мг) ТСХ: Rf = 0,35, 3 : 97 метанол/CH2Cl2. ВЭЖХ: Rт = 16,82 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 131
Соединение 131. К раствору соединения, полученного в примере 130A (200 мг, 0,60 ммоля), в 6 мл CH2Cl2 добавляют 4-ацетамидобензолсульфонилхлорид (410 мг, 1,76 ммоля), затем насыщенный раствор бикарбоната натрия (3 мл) и твердый бикарбонат натрия (0,1 г, 1,2 ммоля). Полученную смесь перемешивают при комнатной температуре в течение ночи. Полученный раствор разбавляют 100 мл CH2Cl2, органику выделяют, сушат над сульфатом магния, органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 30:70 EtOAc/CH2Cl2, до получения 140 мг указанного в заглавии соединения в виде белого твердого вещества.
ТСХ: Rf = 0,19, 3 : 97 метанол / CH2Cl2, ВЭЖХ: Rт = 15,06 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 132
A. Соединение XXII /A = H, D' = /2-тетрагидрофурил/-метил, E = 4-фторфенил, гидрохлорид/. К раствору соединения, полученного в примере 129B (30 мг, 0,057 ммоля), в 3 мл EtOAc добавляют 30% вес/вес HCl в EtOAc (1 мл). Полученную смесь перемешивают в течение ночи при комнатной температуре. Полученный раствор концентрируют при пониженном давлении до получения 16 мг указанного в заглавии соединения в виде белого твердого вещества, которое используют без последующей очистки.
ТСХ: Rf = 0,60 /1:10:90/ NH4OH/метанол/CH2Cl2/.
B. Соединение 132. К раствору соединения, полученного в примере 132A (16 мг), в CH2Cl2 (5 мл) добавляют триэтиламин (0,1 мл, 0,72 ммоля), затем соединение примера 48A (20 мг, 0,09 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор концентрируют при пониженном давлении, и сырой продукт очищают с помощью хроматографии среднего давления на колонке с силикагелем, используя 20:80 EtOAc/CH2Cl2 в качестве растворителя, до получения 7,4 мг указанного в заглавии соединения. Rf = 0,37 /3:97 метанол/ /CH2Cl2/, ВЭЖХ: Rт = 14,19 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 133
A. Соединение XXII / A = трет.бутоксикарбонил, D' = /2-тетрагидрофурил/-метил, E = 4-ацетамидофенил/. К раствору полученного в примере 129A соединения (200 мг, 0,55 ммоля) в CH2Cl2 (6 мл) добавляют 4-ацетамидобензолсульфонилхлорид (380 мг, 1,6 ммоля), затем насыщенный раствор бикарбоната натрия (3 мл) и твердый бикарбонат натрия (0,1, 1,2 ммоля), полученную смесь перемешивают при комнатной температуре в течение ночи. Полученный раствор разбавляют 100 мл CH2Cl2, органику выделяют, концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 10:90 EtOAc/CH2Cl2, затем 30: 70 EtOAc/CH2Cl2 до получения 120 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,13, 3:97 метанол/CH2Cl2, /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = /2-тетрагидрофурил/-метил, E = 4-ацетамидофенил, гидрохлорид/. К раствору соединения, полученного в примере 133 A (120 мг, 0,22 ммоля) в 5 мл EtOAc, добавляют 30% вес/вес HCl в EtOAc (2 мл). Полученную смесь перемешивают в течение ночи при комнатной температуре. Полученный раствор концентрируют при пониженном давлении до получения указанного в заглавии соединения, которое используют без дополнительной очистки. ТСХ: Rf = 0,50, 1:10:90 NH4OH/метанол/CH2Cl2.
C. Соединение 133. К раствору соединения, полученного в примере 133B, в CH2Cl2 (5 мл) добавляют триэтиламин (0,2 мл, 1 ммоля), затем соединения примера 48A (73 мг, 0,32 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор концентрируют при пониженном давлении, и сырой продукт очищают с помощью хроматографии среднего давления на колонке, используя градиентную систему растворителей: CH2Cl2, затем 1 : 99 метанол/CH2Cl2, затем 3 : 97 метанол/ /CH2Cl2, до получения 87,8 мг указанного в заглавии соединения ТСХ : Rf = 0,09, 3:97 метанол/ CH2Cl2, ВЭЖХ: Rт = 12,53 мин, /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 134.
A. Соединение XXII /A = H, D' - изобутенил, E - 4-ацетамидофенил, гидрохлорид/. К раствору соединения, полученного в примере 131 /40 мг, 0,075 ммоля/ в EtOAc (5 мл) добавляют 30% вес/вес HCl в EtOAc (2 мл). Полученную смесь перемешивают в течение ночи при комнатной температуре. Раствор концентрируют при пониженном давлении до получения указанного в заглавии соединения, которое используют без дополнительной очистки. TCX: Rf=0,38, 1:10:90 NH4OH/метанол/ CH2Cl2.
B. Соединение 134. К раствору соединения, полученного в примере 134 A, в CH2Cl2 (5 мл) добавляют триэтиламин (0,1 мл, 0,72 ммоля), затем соединение примера 48 A (26 мг, 0,11 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор концентрируют при пониженном давлении, и сырой продукт очищают на хроматографической колонке среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 1 : 99 метанол /CH2Cl2, затем 3 : 97 метанол/ CH2Cl2 до получения 10,1 мг указанного в заглавии соединения. Rf = 0,11 /3:97 метанол/CH2Cl2/, ВЭЖХ=12,86 мин,
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 135
A. Соединение XXI /A = H, D' = /изобутенил, E = 4-фторфенил, гидрохлорид/. К раствору соединения, полученного в примере 130B (50 мг, 0,10 ммоля) в EtOAc (5 мл) добавляют 30% вес/вес HCl в EtOAc (1 мл). Полученную смесь перемешивают в течение ночи при комнатной температуре. Полученный раствор концентрируют при пониженном давлении до получения указанного в заглавии соединения, которое используют без дополнительной очистки. ТСХ: Rf = 0,48, 1:10:90 NH4OH /метанол/CH2Cl2.
B. Соединение 135. К раствору соединения, полученного в примере 135 A, в CH2Cl2 /5 мл/ добавляют триэтиламин (0,1 мл, 0,72 ммоля), затем соединение примера 48A (35 мг, 0,15 ммолей). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Раствор концентрируют при пониженном давлении, и сырой продукт очищают с помощью хроматографии среднего давления на колонке, используя градиентную систему растворителей: CH2Cl2, затем 20 : 80 EtOAc/CH2Cl2 в качестве системы растворителей, до получения 12 мг указанного в заглавии продукта. ТСХ: Rf = 0,34, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 14,64 мин.
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 136
A. Соединение XXI /A = трет.-бутоксикарбонил, D'=2-фурфурил, A'=H/. К раствору соединения XX (2,5 ммоля) в этаноле (30 мл) добавляют фурфуриламин (0,67 мл, 7,5 ммоля), полученную смесь нагревают до 85oC в течение 24 часов. Полученный раствор фильтруют и концентрируют при пониженном давлении до получения 0,80 г указанного в заглавии соединения, которое используют без дополнительной очистки. ТСХ: Rf = 0,38, 1:10:90 конц.
NH4OH, /метанол/ CH2Cl2.
B. Соединение XXII /A = трет.-бутоксикарбонил, D'= 2-фурил, E= 4-фторфенил/. К раствору продукта примера 136A (0,20 г, 0,609 ммоля/ в CH2Cl2 (6 мл) добавляют насыщенный раствор бикарбоната натрия (3 мл), затем добавляют твердый бикарбонат натрия (0,1 г 1,2 ммоля), затем p-фторбензолсульфонилхлорид /0,32 г, 1,6 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Органику экстрагируют 100 мл CH2Cl2, сушат над безводным сульфатом магния, концентрируют при пониженном давлении, очищают с помощью хроматографии среднего давления на силикагеле, используя градиентную систему: CH2Cl2, затем 1:99 метанол/CH2Cl2. Указанное в заглавии соединение получают в виде белого твердого вещества (86,1 мг). ТСХ: Rf = 0,17, 3:97 метанол/ CH2Cl2, ВЭЖХ: Rт = 16,5 мин;
/1H/-ЯМР/CDCl3/ соответствует структуре.
C. Соединение XXII /A = H, D' = 2-фурил, E = 4-фторфенил, гидрохлорид/. К раствору соединения, полученного в примере 136B (16 мг, 0,031 ммоля) в EtOAc (3 мл) добавляют 30% вес/вес HCl в EtOAc (1 мл). Полученную смесь перемешивают в течение ночи при комнатной температуре. Полученный раствор концентрируют при пониженном давлении до получения указанного в заглавии соединения, которое используют без дополнительной очистки. ТСХ: Rf = 0,48, 1: 10:90 NН4OH/метанол/CH2Cl2.
D. Соединение 136. К раствору соединения, полученного в примере 136C (5 мл) добавляют триэтиламин (0,1 мл, 0,72 ммоля), затем соединение, полученное в примере 48 A (11 мг, 0,05 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Полученный раствор концентрируют при пониженном давлении, и сырой продукт очищают с помощью хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 20: 80 EtOAc/CH2Cl2, до получения 4,9 мг указанного в заглавии соединения. ТСХ: Rf = 0,28 /3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 14,57 мин.
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 137
A. Соединение XXII /A = трет.бутоксикарбонил, D' = 2-фурил, E = 4-ацетамидофенил/. К раствору соединения, полученного в примере 136 B /200 мг, 0,55 ммоля/ в CH2Cl2 (6 мл) добавляют 4-ацетамидобензолсульфонилхлорид (390 мг, 1,7 ммоля), затем насыщенный раствор бикарбоната натрия (3 мл) и твердый бикарбонат натрия (0,1 г, 1,2 ммоля). Полученную смесь перемешивают при комнатной температуре в течение ночи. Этот раствор разбавляют 100 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, и органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 10: 90 EtOAc /CH2Cl2, затем 30:70 EtOAc/CH2Cl2, до получения 100 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,19, 3:97 метанол/CH2Cl2.
/1H/-ЯМР /CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = 2 фурил, E = 4-ацетамидофенил, гидрохлорид/. К раствору соединения, полученного в примере 137A /30 мг, 0,054 ммоля/ в EtOAc (3 мл) добавляют 30% вес/вес HCl в EtOAc (1 мл). Полученную смесь перемешивают в течение ночи при комнатной температуре. Этот раствор концентрируют при пониженном давлении до получения указанного в заглавии соединения, которое используют без последующей очистки. ТСХ: Rf = 0,37 /1:10:90 NH4OH/метанол/CH2Cl2/.
C. Соединение 137. К раствору соединения, полученного в примере 137A, в CH2Cl2 (5 мл) добавляют триэтиламин (0,1 мл, 0,72 ммоля), затем соединение примера 48 A (19 мг, 0,083 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор концентрирую при пониженном давлении, и сырой продукт очищают с помощью хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 1:99 метанол/CH2Cl2, затем 3:97 метанол/CH2Cl2 до получения 8,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,11 /3:97 метанол/CH2Cl2/, ВЭЖХ: Rт = 12,69 мин;
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 138
Соединение 138. Раствор 75 мг соединения, полученного в примере 51D, и 45 мг 3-хлорбензолсульфонилхлорида подвергают взаимодействию по способу примера 60. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN /H2O с 0,1% TFA в качестве элюента, получая 29,7 мг указанного в заглавии соединения. ТСХ Rf = 0,3, 4% MeOH/CH2Cl2; ВЭЖХ: Rт = 15,83 мин.
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 139
Соединение 139. К раствору 67, 9 мг соединения, полученного в примере 116B, в 1 мл THF добавляют последовательно 57 мкл диизопропилэтиламина и раствор 52,6 мг соединения, полученного в примере 109 C, в 1 мл THF. Полученную смесь перемешивают в течение 24 часов и концентрируют в вакууме. Остаток очищают с помощью препаративной хроматографии в толстом слое силикагеля, используя в качестве элюента 7% метанол в CH2Cl2 до получения 70,0 мг указанного в заглавии соединения. ТСХ: Rf = 0,30, 5% метанол/CH2Cl2.ВЭЖХ: Rт = 15,78 мин.
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 140
A 3/S/-амино-2/син/-гидрокси-4-фенил-1-хлорбутанформат (соль). К суспензии 16,33 г 10% палладия на угле /25 вес.%/ в метаноле и тетрагидрофуране /400 мл, 1:1/ добавляют в атмосфере азота 65,35 г 3/S/-N-/бензилоксикарбонил/-амино-1-хлор-2/син/-гидрокси-4- фенилбутана /195,77 ммоля/ в виде раствора в метаноле и тетрагидрофуране /1,2 л/. К этой суспензии добавляют 540 мл муравьиной кислоты. Спустя 15 часов, реакционную смесь фильтруют через диатомовую землю и концентрируют досуха. Полученное масло суспендируют в толуоле и выпаривают, затем тщательно растирают последовательно в диэтиловом эфире и CH2Cl2 до получения 47,64 г продукта в виде гранул твердого вещества желто-коричневого цвета. ТСХ: Rf = 0,17, 5% уксусная кислота/этилацетат.
B. 3/S/-N-/3/S/-тетрагидрофурилоксикарбонил/-амино-1- хлор-2/син/-гидрокси-4-фенилбутан. К раствору соединения, полученного в примере 140A /1,97 г, 7,95 ммоля/ в CH2Cl2 /20 мл/ добавляют насыщенный раствор бикарбоната натрия (5 мл), затем твердый бикарбонат натрия (1,33 г, 17,9 ммоля) и полученное в примере 48A соединение (2,0 г, 8,7 ммоля). Полученную смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния и концентрируют при пониженном давлении. Остаток перекристаллизовывают из смеси этилацетат/гексан до получения 1,01 г указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,35, 3:97 метанол/ /CH2Cl2/. /1H/-ЯМР /CDCl3/ соответствует структуре.
C. Соединение XX /A = 3/S/-тетрагидрофурилоксикарбонил/. К раствору соединения, полученного в примере 140B /1,0 г, 3,2 ммоля/ в абсолютированном этаноле (15 мл) добавляют твердый КOH (0,21 г, 3,8 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 1,0 часа. Этот раствор фильтруют через слой Целита, затем концентрируют при пониженном давлении. Остаток помещают в эфир (100 мл), промывают рассолом, сушат над сульфатом магния, концентрируют при пониженном давлении до получения 0,88 г указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,49 /3 : 97 метанол/ CH2Cl2/, /11H/-ЯМР /CDCl3/ соответствует структуре.
D. Соединение XXI/A = /S/-3-тетрагидрофурилоксикарбонил, D'=циклопентилметил, A'= H/. Соединение, полученное в примере 140C (0,88 г, 3,2 ммоля), добавляют к соединению, полученному в примере 114A (5,0 г, 50,4 ммоля), и перемешивают в течение 24 часов при комнатной температуре. Полученный раствор концентрируют при пониженном давлении. Остаток тщательно растирают с гексаном, твердую часть собирают фильтрованием с подслоем, и промывают гексаном до получения 0,93 г указанного в заглавии соединения. ТСХ: Rf = 0,44, 1 : 10 : 90 конц. NH4OH/метанол/CH2Cl2/; /11H/-ЯМР /CDCl3/ соответствует структуре.
E. Соединение 140. К раствору соединения, полученного в примере 140D (0,93 г, 2,47 ммоля) в CH2Cl2 (20 мл) добавляют насыщенный раствор бикарботана натрия (5 мл), а затем добавляют твердый бикарбонат натрия (0,42 г, 4,94 ммоля) и 4-метоксибензолсульфонилхлорид (0,61 г, 2,96 ммоля), полученную смесь перемешивают при комнатной температуре в течение 4 часов. Раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, и органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя CH2Cl2, а затем 1 : 99 метанол/CH2Cl2 раствор в качестве системы элюентов, до получения 1,28 г указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,26, 3 : 97 метанол/CH2Cl2/, ВЭЖХ: Rт = 15,66 мин, /11H/-ЯМР /CDCl3/ соответствует структуре.
Пример 141
A. Соединение XXII /A= H, D'= циклопентилметил, E = 4-метокси фенил, гидрохлорид/. Раствор 71,3 мг соединения, полученного в примере 166 A, в 25 мл EtOAc при 0oC обрабатывают безводным HCl в течение 10 минут, оставляют выстаиваться 12 часов при комнатной температуре, концентрируют при пониженном давлении, и полученное белое твердое вещество используют без очистки в последующей реакции.
B. Соединение 141. Соединение, полученное в примере 141A (0,134 ммоля), подвергают взаимодействию с аллилхлорформатом по способу примера 87B. После концентрации смеси в вакууме и обработки остаток очищают с помощью хроматографии в толстом слое силикагеля, используя 5% MeOH/CH2Cl2 в качестве элюента, а затем с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента до получения 21,6 мг указанного в заглавии соединения. ТСХ: Rf = 0,45, 5% MeOH/CH2Cl2/. ВЭЖХ: Rт = 16,96 мин.
Пример 142
Соединение 142. К раствору 0,4 г соединения, полученного в примере 141A, в 45 мл THF добавляют последовательно 1,96 мл диизопропилэтиламина и раствор 2,68 г соединения, полученного в примере 82A, в 45 мл THF. Полученную смесь перемешивают в течение 24 часов и концентрируют в вакууме. Остаток помещают в CH2Cl2, промывают насыщенным бикарбонатом натрия и рассолом сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя от 20% до 40% EtOAc в гексане в качестве элюента, до получения 3,69 г указанного в заглавии соединения. ТСХ: Rf = 0,41, 50% EtOAc/CH2Cl2.
Пример 143
Соединение 143. Раствор 3,69 г соединения, полученного в примере 142, в 100 мл этилового эфира обеспечивают безводным газообразным HCl в течение 10 минут. Через реакционную смесь продувают азот, затем фильтруют. Твердую часть помещают в метанол и концентрируют до получения 3,71 г указанного в заглавии соединения. ТСХ: Rf = 0,62, 90 : 10 : 1 CH2Cl2/MeOH/AcOH. ВЭЖХ: Rт = 13,87 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 145
Соединение XXII /A= трет.-бутоксикарбонил, D'=изобутил, E = 2-/5-изоксазол-3-ил/-тиофен/. К раствору 342,5 мг (1,02 ммоля) соединения, полученного в примере 39A в CH2Cl2 (8 мл), добавляют водный бикарбонат натрия (2 мл), твердый бикарбонат натрия 257 мг (3,1 ммоля) и 5-/изоксазол-3-ил/-тиофенсульфонилхлорида 254,2 мг (1,02 ммоля). Спустя 14 часов полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью флеш-хроматографии, используя 5% до 25% EtOAc/CH2Cl2 в качестве элюента, и перекристаллизовывают из смеси эфир /CH2Cl2 до получения 228,6 мг указанного в заглавии продукта.
/1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D'=изобутил, E = 2-/5-изоксазол-3-ил/-тиофен, гидрохлорид/. Раствор 228,6 мг (0,416 ммоля) соединения, полученного в примере 145 A, в EtOAc (15 мл) при -20oC обрабатывают безводным газообразным HCl в течение 10 минут. Ледяную баню удаляют, и после дополнительных 15 минут реакционную смесь продувают азотом, концентрируют в вакууме до получения 223,6 мг указанного в заглавии соединения в виде соли HCl. ТСХ: Rf = 0,48, 10% метанол/CH2Cl2.
C. Соединение 143. Раствор 78,5 мг соединения, полученного в примере 145 B (0,162 ммоля) в CH2Cl2 (3 мл) обрабатывают последовательно при комнатной температуре в атмосфере азота 0,07 мл (0,408 ммоля) диизопропилэтиламина и 55,6 мг (0,243 ммоля) соединения, полученного в примере 48A. Полученную смесь перемешивают 16 часов, а затем концентрируют в вакууме. Остаток помещают в CH2Cl2 и промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 48,7 мг указанного в заглавии продукта. ТСХ: Rf = 0,36, 25% EtOAc/CH2Cl5. ВЭЖХ: Rт = 15,2 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 146
A. Соединение XXI /A = трет.-бутоксикарбонил, D' = циклопентилметил, E = 4-ацетамидофенил/. К раствору соединения, полученного в примере 114B (300 мг, 0,83 ммоля), в CH2Cl2 (15 мл) добавляют 4-ацетамидобензолсульфонилхлорид (580 мг, 2,48 ммоля), а затем добавляют насыщенный раствор бикарбоната натрия (4 мл) и твердый бикарбонат натрия (0,14 г, 1,67 ммоля). Полученную смесь перемешивают при комнатной температуре в течение ночи. Этот раствор разбавляют 150 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 10 : 90 EtOAc/CH2Cl2, до получения 310 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,10, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 15,96 мин /1H/-ЯМР /CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = циклопентилметил, E = 4-ацетамидогидрохлорид/. К раствору соединения, полученного в примере 146A (210 мг, 0,38 ммоля), добавляют 30% вес/вес HCl в EtOAc (15 мл). Полученную смесь перемешивают в течение 1 часа при комнатной температуре. Раствор концентрируют при пониженном давлении до получения 180 мг указанного в заглавии соединения, которое используют без дополнительнойочистки. ТСХ: Rf = 0,14, 1:10:90 NH4OH/метанол/CH2Cl2.
C. Соединение XXII /A = аллилоксикарбонил, D' = циклопентилметил, E = 4-ацетамидофенил/. К раствору соединения, полученного в примере 146B (100 г, 0,20 ммоля) в CH2Cl2 (10 мл) добавляют триэтиламин (0,1 мл, 0,72 ммоля), затем аллилхлорформат (0,04 мл, 0,3 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор разбавляют 150 мл CH2Cl2, промывают водой, сушат над безводным сульфатом магния, и органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 1 : 99 метанол/CH2Cl2, затем 3 : 97 метанол/CH2Cl2 до получения 103 мг продукта. Rf = 0,22, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 15,29 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 147
Соединение 147. К раствору соединения, полученного в примере 146B (80 мг, 0,16 ммоля) в CH2Cl2 (5 мл) добавляют триэтиламин (0,07 мл, 0,48 ммоля), затем медленно добавляют за 3 часа соединение, полученное в примере 82A (53 мг, 0,19 ммоля) в виде раствора в CH2Cl2 (3 мл). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор разбавляют 100 мл CH2Cl2, промывают водой, сушат над безводным сульфатом магния, органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 1 : 99 метанол/CH2Cl2, затем 2 : 98 метанол/CH2Cl2, до получения 71,7 мг указанного в заглавии соединения. Rf = 0,06; 3 : 97 метанол/CH2Cl2, ВЭЖХ: Rт = 12,61 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 148
A. Соединение XXII /A = трет.-бутоксикарбонил, D' = циклопентилметил, E = фенил/. Раствор 297 мг соединения, полученного в примере 114B, в 4 : 1 CH2Cl2/ насыщенном водном NaHCO3 обрабатывают последовательно, при комнатной температуре в атмосфере азота, 217 мг бензолсульфонилхлорида и 103 мг бикарбоната натрия. Полученную смесь перемешивают в течение 6 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над безводным сульфатом магния, фильтруют и концентрируют в вакууме до получения 426 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,32, 5% диэтиловый эфир/CH2Cl2. /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = циклопентилметил, E = фенил, гидрохлорид/. Раствор 400 мг соединения, полученного в примере 148A, в этилацетате обрабатывают при -20oC газообразным HCl в течение 20 минут, причем за это время температуре дают повыситься до 20oC. Через смесь продувают азот в течение 15 минут, и растворитель удаляют в вакууме до получения 349 мг белого твердого продукта, который используют непосредственно в следующей реакции.
C. Соединение 148. Раствор 40 мг соединения, полученного в примере 148B, в CH2Cl2, добавляют при комнатной температуре в атмосфере азота к раствору 31 мг соединения, полученного в примере 48A, и 35 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, затем сушат на сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 20% диэтиловый эфир/CH2Cl2 в качестве элюента, до получения 45 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,46, 20% диэтиловый эфир/CH2Cl2. ВЭЖХ: Rт = 15,48 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 149
A. Соединение XXII /A = трет-бутоксикарбонил, D' = циклопентил-метил, E = 3-пиридил/. К раствору 153 мг (0,422 ммоля) соединения, полученного в примере 114B, в CH2Cl2 (4 мл) добавляют водный бикарбонат натрия (1 мл), твердый бикарбонат натрия (141,7 мг, 1,69 ммоля), и полученное в примере 144A соединение (156,1 мг, 0,879 ммоля). Спустя 14 часов полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью флеш-хроматографии, используя от 20% до 40% EtOAc/ CH2Cl2 в качестве элюента, до получения 64,7 мг указанного в заглавии продукта. ТСХ: Rf = 0,24, 20% EtOAc/CH2Cl2.
B. Соединение XXII /A = трет.-бутоксикарбонил, D'= циклопентилмел, E = 3-пиридил гидрохлорид/. Раствор 273.1 мг ммоля (0,572 ммоля) соединения, полученного в примере 149 A, в EtOAc (15 мл) при -20oC обрабатывают безводным HCl (газообразным) в течение 10 минут. Ледяную баню удаляют, и спустя еще 15 минут реакционную смесь продувают азотом, затем концентрируют в вакууме. К раствору полученного остатка в CH2Cl2 (3 мл) добавляют последовательно при комнатной температуре в атмосфере азота, 0,076 мл (0,437 ммоля) диизопропилэтиламина и 34,3 мг (0,150 ммоля) соединения, полученного в примере 48A. Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя градиент до 200 до 50% в CH2Cl2 в качестве элюента до получения 11,3 мг указанного в заглавии соединения. ТСХ: Rf = 0,15 40% EtOAc/CH2Cl2. ВЭЖХ: Rт = 13,7 мин;
/1H-ЯМР/CDCl3/ соответствует структуре.
Пример 150
A. 1-пиперидинсульфонилхлорид. Раствор 4 г сульфонилхлорида в ацетонитриле обрабатывают, прикапывая 861 мг пиперадина при комнатной температуре в атмосфере азота. После окончания добавления полученную смесь кипятят с обратным холодильником в течение 18 часов, охлаждают до комнатной температуры и концентрируют в вакууме до получения указанного в заглавии соединения в виде масла красного цвета. ТСХ: Rf = 0,86 CH2Cl2 /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII/A = трет.бутоксикарбонил, D'= изобутил, E = пиперидинил/. Раствор 73 мг соединения, полученного в примере 39A, в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота 121 мг соединения полученного в примере 150A, и 84 мг N,N'-диизопропилэтиламина. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над безводным сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 5% диэтиловый эфир/ CH2Cl2 в качестве элюента, до получения 70 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,21 /5% диэтиловый эфир в CH2Cl2/. ВЭЖХ: Rт = 17,40 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
C. Соединение XXII /A = H,D'= Изобутил, E = пиперидинил, гидрохлорид/. Раствор 70 мг полученного в примере 150B соединения, в этилацетате, обрабатывают при -20oC газообразным HCl в течение 20 мин, причем за это время температуре дают возможность повыситься до комнатной (20oС). Затем через смесь продувают азот в течение 15 минут, растворитель удаляют в вакууме до получения вязкого масла, которое используют непосредственно в следующей реакции.
D. Соединение 150. Раствор соединения, полученного в примере 150C, в CH2Cl2, добавляют при комнатной температуре в атмосфере азота, к раствору 50 мг соединения, полученного в примере 48A, и 56 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 20% диэтиловый эфир/CH2Cl2 в качестве элюента, до получения 16 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,45 /0% диэтиловый эфир/CH2Cl2. ВЭЖХ: Rт = 15,00 мин.
/1H-ЯМР/CDCl3/ соответствует структуре.
Пример 151
A. Соединение XXII /A= трет.-бутоксикарбонил, D'= циклопентилметил, E= 4-трифторметоксифенил/. Раствор 71 мг соединения, полученного в примере 114B, в смеси 4:1 CH2Cl2/ насыщенный водный NaHCO3, обрабатывают последовательно при комнатной температуре в атмосфере азота 76 мг 4-трифторметоксибензолсульфонилхлорида и 25 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления, на силикагеле, используя 5% диэтиловый эфир /CH2Cl2 в качестве элюента, до получения 92 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,34, 5% диэтиловый эфир/ CH2Cl2/. /1H-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII /A=H, D'=циклопентилметил, E = 4-трифторметоксифенил, гидрохлорид/. Раствор 92 мг соединения, полученного в примере 151 A, в этилацетате, обрабатывают при -20oC газообразным HCl в течение 20 минут, причем за это время температура повышается до 20oC. Затем через смесь барботируют азот в течение 15 минут, растворитель удаляют в вакууме до получения 83 мг белого твердого вещества, которое используют непосредственно в последующей реакции.
C. Соединение 151. Раствор 22 мг соединения, полученного в примере 151B, в CH2Cl2, добавляют при комнатной температуре в атмосфере азота, к раствору 15 мг соединения, полученного в примере 48A, и 16 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 60 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 20% диэтиловый эфир/CH2Cl2 в качестве элюента до получения 23 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,44, 20% диэтиловый эфир/ CH2Cl2. ВЭЖХ: Rт = 16,99 мин. /1H/-ЯМР/CDCl3 соответствует структуре.
Пример 152
A. Соединение XXII /A = трет.-бутоксикарбонил, D' = изобутил, E = 4-трифторметоксифенил/. Раствор 97 мг соединения, полученного в примере 39A, в смеси 4:1 CH2Cl2/ насыщенный водный NaHCO3, обрабатывают последовательно при комнатной температуре в атмосфере азота 113 мг 4-трифторметоксибензолсульфонилхлорида и 36 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 5% диэтиловый эфир/CH2Cl2 до получения 120 мг указанного в заглавии продукта в виде белого твердого вещества. ТСХ: Rf = 0,34, 5% диэтиловый эфир/CH2Cl2. ВЭЖХ: Rт = 18,54 мин.
/1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII/A=H, D' = изобутил, E=4-трифторметоксифенил гидрохлорид/. Раствор 100 мг соединения, полученного в примере 152А, в этилацетате, обрабатывают при -20oC газообразным HCl в течение 20 минут, причем за это время температура повышается до 20oC. Азот барботируют через смесь в течение 15 минут, растворитель удаляют в вакууме до получения 89 мг твердого белого вещества, которое используют непосредственно в последующей реакции.
C. Соединение 152. Раствор 41 мг соединения, полученного в примере 152B, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота к раствору 28 мг соединения, полученного в примере 4A, и 32 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 5% диэтиловый эфир/CH2Cl2, до получения 30 мг указанного в заглавии соединения в виде белого твердого продукта. ТСХ: Rf = 0,08 /5% диэтиловый эфир/CH2Cl2/. ВЭЖХ: Rт = 16,52 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 153.
A. Соединение XXII /A = трет.бутоксикарбонил, D' = изобутил, E=4-метоксифенил/. К раствору соединения, полученного в примере 39A /600 мг, 1,77 ммоля/ в CH2Cl2 (10 мл) добавляют 4-метоксибензосульфонилхлорид (0,55 г, 2.66 ммоля), а затем добавляют насыщенный раствор бикарбоната натрия (3 мл) и 0.30 г твердого бикарбоната натрия. Полученную смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют 200 мл CH2Cl2, и органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 5:95 эфир/CH2Cl2, до получения 630 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,48, 3: 97 метанол/CH2Cl2. /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII/A = H, D'= изобутил, E = 4-метоксифенил, гидрохлорид/. К раствору соединения, полученного в примере 153A/ 0,63 г, 1,24 ммоля/, в EtOAc (5 мл) добавляют 30% вес/вес HCl в EtOAc (5 мл). Полученную смесь перемешивают в течение 6 часов при комнатной температуре. Раствор концентрируют при пониженном давлении, получая 0,59 г белого твердого вещества, которое используют непосредственно в последующей реакции. ТСХ: Rf = 0,12, 3: 97 метанол/CH2Cl2.
C Соединение XXII /A=/3-пиридил/метоксикарбонил, D' = изобутил, E = 4-метоксифенил/. К раствору соединения, полученного в примере 153B /100 мг, 0,23 ммоля/, в CH2Cl2 (5 мл) добавляют за 3 часа соединения, полученное в примере 82A (75 мг, 0,27 ммоля) в виде раствора в CH2Cl2 (5 мл). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Органику концентрируют при пониженном давлении, и сырой продукт очищают на хроматографической колонке среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 1:99 метанол/CH2Cl2, затем 3:97 метанол/CH2Cl2 до получения 49,3 мг указанного в заглавии соединения. Rf = 0,33, 3:97 метанол/CH2Cl2. ВЭЖХ: Rт = 13,18 мин.
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 154
Соединение 154. К раствору соединения, полученного в примере 153B (100 мг, 0,20 ммолей), в CH2Cl2 (5 мл) добавляют триэтиламин (0,25 мл, 1,8 ммоля), а затем аллилхлорформат (0,1 мл, 0,94 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Полученный раствор концентрируют при пониженном давлении, и сырой продукт очищают на хроматографической колонке среднего давления, используя градиентную систему растворителей: CH2Cl2; затем 1:99 метанол/CH2Cl2 до получения 94 мг указанного в заглавии соединения. Rf = 0,71, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 16,12 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 155
A. N-гидроксисукцинимидил-1-метоксипропан-3-карбонат. К раствору 355 мг 2-метилен-1,3 пропандиола в ацетонитриле (30 мл) добавляют последовательно при комнатной температуре 65 мг гидрида натрия и 0,25 мл иодометана. Полученную смесь перемешивают в течение 12 часов и концентрируют в вакууме. Остаток помещают в 15 мл ацетонитрила и обрабатывают последовательно при комнатной температуре в атмосфере азота, 1,3 г N,N-дисукцинимидилкарбоната и 1,6 мл триэтиламина. После перемешивания в течение 14 часов реакционную смесь концентрируют в вакууме, а остаток разбавляют CH2Cl2, промывают насыщенным бикарбонатом натрия и насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии на силикагеле, используя в качестве элюента EtOAc, до получения 95 мг указанного в заглавии соединения. /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение 155. Раствор 0,056 ммолей соединения, полученного в примере 40A, подвергают взаимодействию с соединением, полученным в примере 155A, по способу примера 132. После концентрирования смеси в вакууме и обработки остаток очищают с помощью хроматографии в толстом слое силикагеля, используя 7% MeOH/CH2Cl2 в качестве элюента, а затем с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента, до получения 3,7 мг указанного в заглавии соединения. ТСХ: Rf = 0,45, 7% MeOH/CH2Cl2 ВЭЖХ: Rт = 13,78 мин.
Пример 156
A. 1-ацетилиндолин-5-сульфонилхлорид. Порцию в 1,02 г, 1-ацетил индолина обрабатывают 2 мл хлорсульфоновой кислоты при 0oC. Полученную смесь нагревают при 60oC в течение 2 часов, затем обрабатывают измельченным льдом, фильтруют и сушат до получения 1,3 г указанного в заглавии соединения, которое используют непосредственно в последующей реакции. ТСХ: Rf = 0,18, 50% EtOAc /гексан. /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII / A = трет.бутоксикарбонил, D' = циклопентилметил, E = 5-/B-ацетил/-индолин. К раствору 60 мг соединения, полученного в примере 114B, в 15 мл CH2Cl2 добавляют 5 мл насыщенного водного раствора бикарбоната натрия, 50,0 мг бикарбоната натрия и 60 мг соединения, полученного в примере 156A. Спустя 4 часа полученную смесь разбавляют CH2Cl2, промывают насыщенным рассолом, сушат над сульфатом магния и фильтруют. Полученную смесь концентрируют в вакууме до получения целевого продукта, который используют непосредственно в последующей реакции. /1H/-ЯМР/CDCl3/ соответствует структуре.
C. Соединение 156. Раствор 37 мг соединения, полученного в примере 156B, в EtOAc (15 мл) при 0oC обрабатывают безводным газообразным HCl в течение 10 минут и оставляют выстаиваться в течение 12 часов, причем за это время температура повышается до комнатной. Этот сырой материал подвергают взаимодействию с аллилхлорформатом по способу примера 87B. После концентрирования смеси в вакууме и обработки остаток очищают с помощью хроматографии в толстом слое силикагеля, используя в качестве элюента 7% MeOH/CH2Cl2, а затем с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1 TFA в качестве элюента до получения 10,5 мг указанного в заглавии соединения. ТСХ: Rf = 0,75, 10% MeOH/CH2Cl2. ВЭЖХ: Rт = 15,78 мин;
/1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 157
Соединение 157. Раствор 37 мг соединения, полученного в примере 156B, в EtOAc (15 мл) при 0oC обрабатывают безводным газообразным HCl в течение 10 минут и оставляют выстаиваться в течение 12 часов, при этом температура повышается до комнатной. Этот сырой материал затем подвергают взаимодействию с соединением, полученным в примере 48A, по способу примера 88. После концентрирования смеси в вакууме остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент растворителей от 35% до 100% CH3CN/H2O с 0,1% TFA до получения 17,9 мг указанного в заглавии соединения. ТСХ: Rf = 0,6, 10% MeOH/CH2Cl2. ВЭЖХ: Rт = 14,68 мин. /1H/-ЯМР/CDCl3/ соответствуют структуре.
Пример 158
A. Соединение XXII / A = трет.-бутоксикарбонил, D'=циклогексилметил, E= H/. К раствору соединения XX /A=Boc/ (5,0 ммоля) в 20 мл этанола добавляют циклогексилметиламин (3,25 мл, 2,83 ммоля), и полученную смесь перемешивают в течение 3 часов при комнатной температуре. Этот раствор фильтруют и концентрируют при пониженном давлении до получения 1,49 г белого твердого вещества, которое используют непосредственно в последующей реакции. ТСХ: Rf = 0,14, 3:97 метанол/CH2Cl2.
/1H/-ЯМР/CDCl3 соответствует структуре.
B. Соединение XXII/A = трет.-бутоксикарбонил, D'=циклогексилметил, E = 4-метоксифенил/. К раствору соединения, полученного в примере 158A (400 мг, 1,06 ммоля), в CH2Cl2 (10 мл) добавляют 4-метоксибензолсульфонилхлорид (0,66 г, 3,1 ммоля) с последующим добавлением насыщенного раствора бикарбоната натрия (3 мл) и 0,18 г твердого бикарбоната натрия. Полученную смесь перемешивают при комнатной температуре в течение ночи. Этот раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя CH2Cl2, затем 1:90 метанол/CH2Cl2 в качестве системы растворителей, до получения 340 мг указанного в заглавии соединения в виде белого твердого вещества. ТСХ: Rf = 0,39, 3:97 метанол/CH2Cl2.
/1H/-ЯМР/CDCl3/ соответствует структуре.
C. Соединение XXI /A=H, D'-циклогексилметил, E = 4-метоксифенил, гидрохлорид/. К раствору соединения, полученного в примере 158B (0,34 г, 0,62 ммоля) в 10 мл EtOAc, добавляют 5 мл 30% вес/вес HCl в EtOAc. Полученную смесь перемешивают в течение 3 часов при комнатной температуре. Этот раствор концентрируют при пониженном давлении до получения 0,3 г белого твердого вещества, которое используют непосредственно в последующей реакции ТСХ: Rf = 0,12, 3:97 метанол/CH2Cl2.
D. Соединение 158. К раствору соединения, полученного в примере 158C (100 мг, 0,21 ммоля), в CH2Cl2 (8 мл) добавляют триэтиламин (0,2 мл, 1,44 ммоля), затем соединение, полученное в примере 48A (71 мг, 0,31 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 6 часов. Этот раствор разбавляют CH2Cl2 (200 мл), промывают насыщенным раствором бикарбоаната натрия (30 мл), органику выделяют, сушат над безводным сульфатом магния и концентрируют при пониженном давлении, а сырой продукт очищают с помощью хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 10:90 EtOAc/CH2Cl2, до получения 84,9 мг указанного в заглавии соединения. ТСХ: Rf = 0,48, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 16,35 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 159
A. Соединение XXII / A = трет.-бутоксикарбонил, D'=циклогексилметил, E = 4-фторфенил/. К раствору соединения, полученного в примере 158A (400 мг, 1,06 ммоля), в 10 мл CH2Cl2 добавляют 4-фторбензолсульфонилхлорид (0,62 г, 3,2 ммоля), а затем добавляют насыщенный раствор бикарбоната натрия (3 мл) и 0,18 г твердого бикарбоната натрия. Полученную смесь перемешивают при комнатной температуре в течение ночи. Полученный раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя в качестве системы растворителей CH2Cl2, затем 1:99 метанол/CH2Cl2, до получения 280 мг белого твердого вещества. ТСХ: Rf = 0,47, 3:97 метанол/CH2Cl2. /1H/-ЯМР соответствует структуре.
B. Соединение XXII / A = H, D' = циклогексилметил, E = 4-фторфенил, гидрохлорид/. К раствору соединения, полученного в примере 159A, (0,28 г, 0,52 ммоля) добавляют 10 мл 30% вес/вес HCl в EtOAc. Полученную смесь перемешивают в течение 3 часов при комнатной температуре. Раствор концентрируют при пониженном давлении до получения 0,23 г белого твердого вещества, которое используют непосредственно в последующей реакции. ТСХ: Rf = 0,13 /3:97 метанол/CH2Cl2, /1H/-ЯМР/CDCl3/ соответствует структуре.
C. Соединение 159. К раствору соединения, полученного в примере 159C (100 мг, 0,21 ммоля), в CH2Cl2 (8 мл) добавляют триэтиламин (0,2 мл, 1,44 ммоля), затем соединение, полученное в примере 48A (73 мг, 0,32 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 6 часов. Этот раствор разбавляют CH2Cl2, (200 мл), промывают насыщенным раствором бикарбоната натрия (30 мл), сушат над безводным сульфатом магния, органику концентрируют при пониженном давлении, и сырой продукт очищают с помощью хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 10:90 EtOAc/CH2Cl2, до получения 54 мг указанного в заглавии соединения. ТСХ: Rf = 0,46 3:97 метанол/CH2Cl2. ВЭЖХ: Rт = 16,48 мин; /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 160
А. Соединение XXII /A = трет.-бутоксикарбонил, D' =циклогексилметил, E= 4-ацетамидофенил/. К раствору соединения, полученного в примере 158A (400 мг, 1,06 ммоля), в CH2Cl2 (10 мл) добавляют 4-ацетамидобензолсульфохлорид (0,75 г, 3,2 ммоля), затем добавляют насыщенный раствор бикарбоната натрия (3 мл) и 0,18 г твердого бикарбоната натрия. Полученную смесь перемешивают при комнатной температуре в течение ночи. Этот раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, организму концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя CH2Cl2, затем 1:99 метанол/ CH2Cl2 в качестве системы растворителей до получения 290 мг указанного в заглавии соединения в виде белого твердого вещества. TCX:Rf = 0,14, 3:97 метанол /CH2Cl2, /1H/-ЯМР /CDCl3/ соответствует структуре.
В. Соединение XXII /A=H,D'=циклогексиметил, E=4-ацетамидофенил, гидрохлорид/. К соединению, полученному в примере 160А (0,29 г, 0,51 ммоля) добавляют 30% вес/вес HCl в EtOAc (10 мл). Полученную смесь перемешивают в течение 3 часов при комнатной температуре. Этот раствор концентрируют при пониженном давлении до получения 0,28 г белого твердого вещества, которое используют непосредственно в последующей реакции. TCX:Rf = 0,10, 3:97 метанол/CH2Cl2.
С. Соединение 160. К раствору соединения, полученного в примере 160B (100 мг, 0,20 ммоля), в CH2Cl2 (8 мл) добавляют триэтиламин (2 мл, 1,44 ммоля) затем соединение, полученное в примере 48A (67 мг, 0,30 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 6 часов. Этот раствор разбавляют CH2Cl2 (200 мл), промывают насыщенным раствором бикарбоната натрия (30 мл), сушат над безводным сульфатом магния, органику концентрируют при пониженном давлении и сырой продукт очищают на хроматографической колонке среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 10: 90 EtOAc/CH2Cl2, затем 20:80 EtOАc/CH2Cl2, до получения 56,8 мг белого твердого вещества. TCX:Rf = 0,17, 3:97 метанол/ CH2Cl2, ВЭЖХ: Rт = 14,65 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 161
А. 4-морфолинсульфонилхлорида. Раствор 4,6 г сульфурилхлорида в ацетонитриле обрабатывают, прикапывая 996 мг морфолина при комнатной температуре в атмосфере азота. После завершения добавления полученную смесь кипятят с обратным холодильником в течение 16 часов, охлаждают до комнатной температуры, концентрируют в вакууме до получения указанного в заглавии продукта в виде масла красного цвета. TCX:Rf = 0,65 CH2Cl2.
/1H/-ЯМР /CDCl3/ соответствует структуре.
В. Соединение XXII /A = трет.-бутоксикарбонил, D'=изобутил, E=морфолинил/. Раствор 98 мг соединения, полученного в примере 39A, в смеси 4:1 CH2Cl2 насыщенный водный NaHCO3 обрабатывают последовательно при комнатной температуре в атмосфере азота 270 мг соединения, полученного в примере 161A, 122 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента CH2Cl2, а затем с помощью препаративной ВЭЖХ до получения 22 мг указанного в заглавии продукта в виде маслянистого твердого вещества. TCX: Rf = 0,46, 20% диэтиловый эфир /CH2Cl2. ВЭЖХ:Rт = 15,50 мин, /1H/-ЯМР /CDCl3/ соответствует структуре.
C. Соединение XXII /A=H, D'=изобутил, E=морфолинил, гидрохлорид/. Раствор 22 мг соединения, полученного в примере 161B, в этилацетате обрабатывают газообразным HCl при -20oC. Затем через смесь барботируют азот в течение 15 минут, и растворитель удаляют в вакууме до получения маслянистой полутвердой массы, которую используют непосредственно в последующей реакции.
D. Соединение 161. Раствор соединения, полученного в примере 161C, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота к раствору 16 мг соединения, полученного в примере 48A, в 18 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 21 мг указанного в заглавии продукта в виде маслянистого твердого вещества. TCX: Rf = 0,22 20% диэтиловый эфир /CH2Cl2, ВЭЖХ:Rт = 13,01 мин.
1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 162
Соединение 162. Раствор 30 мг соединения, полученного в примере 166A, обрабатывают газообразным хлористым водородом для удаления защиты, и полученное соединение подвергают взаимодействию с соединением, полученным в примере 155A по способу примера 155B. После концентрирования смеси в вакууме и соответствующей обработки, остаток очищают с помощью хроматографии в толстом слое силикагеля, используя в качестве элюента 5% MeOH/ /CH2Cl2, затем с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1 TFA в качестве элюента до получения 6,2 мг указанного в заглавии соединения. TCX:Rf = 0,65, 5% MeOH/CH2Cl2. ВЭЖХ:Rт = 15,93 мин. /1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 163
Соединение 163. Порцию 120,3 мг соединения, полученного в примере 153B, подвергают взаимодействию с соединением, полученным в примере 82A, по способу примера 82B. После обработки и концентрирования в вакууме остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя в качестве элюента 50% EtOAc в CH2Cl2, затем с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 40% до 100% ацетонитрил /вода для элюирования, до получения 44,3 мг указанного в заглавии соединения. TCX:Rf = 0,18, 50% EtOAc/CH2Cl2/ ВЭЖХ:Rт = 13,13 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 164.
A. N-гидросисукцинимидил-/2-фенил/этилкарбонат. Раствор 306 мг фенэтилового спирта и 535 мг N,N-дисукцинимидилкарбоната в ацетонитриле обрабатывают при комнатной температуре в атмосфере азота, 810 мг N,N-диизопропилэтиламина. Полученную смесь перемешивают в течение 60 часов и концентрируют в вакууме. Остаток помещают в этилацетат и промывают насыщенным NaHCO3, насыщенным NaCl, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения указанного в заглавии продукта в виде масла желтого цвета. TCX: Rf = 0,40 /5% метанол в CH2Cl2/. /1H/-ЯМР /CDCl3/ соответствует структуре.
B. Соединение 164. Раствор 81 мг соединения, полученного в примере 164A, в CH2Cl2, добавляют при комнатной температуре в атмосфере азота к раствору 41 мг соединения, полученного в примере 40a, 45 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 4 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния и концентрируют в вакууме. Остаток обрабатывают с помощью препаративной ВЭЖХ до получения 18 мг указанного в заглавии продукта. TCX:Rf=0,83 /5: 10:85 NH4OH/CH3OH/CH2Cl2/ ВЭЖХ:Rт = 15,78 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 165
Соединение 165. Раствор 36 мг соединения, полученного в примере 51D, в смеси 4:1 CH2Cl2/ насыщенный водный NaHCO3 обрабатывают последовательно при комнатной температуре в атмосфере азота, 20 мг р-толуолсульфонилхлорида и 18 мг бикарбаната натрия. Полученную смесь перемешивают в течение 3 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 5% диэтиловый эфир CH2Cl2 до получения 38 мг указанного в заглавии продукта в виде белого твердого вещества. TCX:Rf = 0,15, 5% диэтиловый эфир/CH2Cl2. ВЭЖХ:Rт = 15,27 мин.
/1H/-ЯМР /CDCl3/ соответствует структуре.
Пример 166
А. Соединение XXII/A = трет.-бутоксикарбонил, D'=циклопентилметил, E= 4-метоксифенил/. К раствору соединения, полученного в примере 114B (1,8 г, 4,96 ммоля) в 10 мл CH2Cl2 добавляют 4-метоксибензолсульфонилхлорид/ 2,10 г, 9,93 ммоля/, затем добавляют насыщенный раствор бикарбоната натрия (3 мл) и 0,83 г твердого бикарбоната натрия. Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над сульфатом магния, концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя CH2Cl2, затем 1:99 метанол /CH2Cl2; затем 2:98 метанол/ /H2Cl2 в качестве системы растворителей, до получения 1,49 г указанного в заглавии соединения в виде твердого вещества белого цвета.
TCX:Rf = 0,37, 3:97 метанол/СH2Cl2;
/1H/-ЯМР /CDCl3/ соответствует структуре.
B. Соединение XXII /A=H,D'=циклопентилметил, E=4-гидроксифенил/. Раствор соединения, полученного в примере 166A /1,11 г, 2,08 ммоля/ в 20 мл СH2Cl2 добавляют к раствору трехбромистого бора в СH2Cl2/ 1,0М, 10,4 мл/. Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор выливают в 40 мл насыщенного раствора бикарбоната натрия. Водный слой экстрагируют 250 мл СH2Cl2, затем экстрагируют 250 мл EtOAc. Объединенную органику сушат над сульфатом магния, концентрируют при пониженном давлении, и сырой продукт очищают с помощью хроматографии среднего давления на колонке, используя градиентную систему растворителей:СH2Cl2, затем 1:99 метанол/СH2Cl2, затем 9:98 метанол/СH2Cl2, затем 1:5:95 конц, NH4OH/метанол/СH2Cl2, до получения 0,38 г указанного в заглавии соединения.
TCX:Rf - 0,18, 3:97 метанол/СH2Cl2.
/1H/-ЯМР /CDCl3/ соответствует структуре.
C. Соединение 166. К соединению, полученному в примере 166B (300 мг, 0,69 ммоля), в СH2Cl2 (5 мл) добавляют триэтиламин (0,12 мл, 8,6 ммоля), затем медленно добавляют за 3 часа соединение, полученное в примере 82A (0,21 г, 0,77 ммоля) в виде раствора в СH2Cl2 (5 мл). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор разбавляют 250 мл CH2Cl2, промывают водой, сушат над безводным сульфатом магния, и органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью хроматографии среднего давления на колонке, используя градиентную систему растворителей: CH2Cl2, затем 1:99 метанол/CH2Cl2, затем 2:98 метанол /CH2Cl2 в качестве элюента до получения 110 мг твердого вещества белого цвета. TCX: Rf = 0,14 /3: 97 метанол/CH2Cl2/, ВЭЖХ: Rт = 12,69 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 167
Соединение 167. Раствор 102 мг соединения, полученного в примере 51D, в смеси 4: 1 CH2Cl2 /насыщенный водный NaHCO3, обрабатывают последовательно, при комнатной температуре в атмосфере азота 65 мг p-нитробензолсульфонилхлорида и 51 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 20% диэтиловый эфир/ CH2Cl2, до получения 124 мг указанного в заглавии соединения в виде твердого вещества белого цвета. TCX: Rf = 0,36, 20% диэтиловый эфир /CH2Cl2. ВЭЖХ: Rт = 15,15 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 168
Соединение 168. Раствор 124 мг соединения, полученного в примере 167, в этилацетате обрабатывают при комнатной температуре 13 мг 10% палладия на угле. Полученную смесь перемешивают в течение 14 часов в атмосфере азота, фильтруют через слой Целита, концентрируют в вакууме. Остаток обрабатывают с помощью препаративной ВЭЖХ до получения 82 мг указанного в заглавии продукта в виде белого твердого вещества. TCX: Rf = 0,10, 20% эфир /CH2Cl2/. ВЭЖХ: Rт = 13,16 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 169
Соединение 169. К раствору соединения, полученного в примере 166B /80 мг, 0,18 ммоля/, в CH2Cl2 (15 мл) добавляют насыщенный раствор бикарбоната натрия (5 мл), затем соединение, полученное в примере 48A (55 мг, 0,24 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 5 часов. Раствор разбавляют 300 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя CH2Cl2, затем 1:99 метанол/ CH2Cl2 в качестве системы растворителей, до получения 56 мг указанного в заглавии соединения в виде твердого вещества белого цвета. TCX: Rf = 0,24, 3:97 метанол /CH2Cl2. ВЭЖХ: Rт = 14,29 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 170
A. Соединение XXII /A = трет.-бутоксикарбонил, D' = циклопентилметил, E = 4-нитрофенил/. К раствору соединения, полученного в примере 114B (250 мг, 0,69 ммоля), в CH2Cl2 (15 мл) добавляют насыщенный раствор бикарбоната натрия (5 мл), затем твердый бикарбонат натрия (0,12 г, 1,37 ммоля) и 4-нитробензолсульфонил хлорид (200 мг, 0,9 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 1: 99 метанол /CH2Cl2 до получения 360 мг указанного в заглавии соединения оранжевого цвета. TCX: Rf = 0,45, 3 : 97 метанол /CH2Cl2. /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = циклопентилметил, E = 4 - нитрофенил, гидрохлорид/. К соединению, полученному в примере 170A (360 мг, 0,66 ммоля), добавляют 10% вес/вес HCl в EtOAc (15 мл). Полученную смесь перемешивают в течение 3 часов при комнатной температуре. Этот раствор концентрируют при пониженном давлении до получения 310 мг указанного в заглавии соединения в виде твердого вещества оранжевого цвета, которое используют непосредственно в последующей реакции. TCX: Rf = 0,70, 1:10:90: NН4OH/метанол/CH2Cl2.
C. Соединение 170. К раствору соединения, полученного в примере 170B (310 мг, 0,64 ммоля), в CH2Cl2 (15 мл) добавляют насыщенный раствор бикарбоната натрия (5 мл), затем добавляют твердый бикарбонат натрия (0,11 г, 1,3 ммоля) и соединение, полученное в примере 48A (0,18 г, 0,77 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор разбавляют 150 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния и концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя в качестве системы растворителей: CH2Cl2, затем 1:99 метанол/CH2Cl2, до получения 0,32 г указанного в заглавии соединения в виде твердого вещества белого цвета. TCX: Rf = 0,28, 3: 97 метанол/CH2Cl2, ВЭЖХ: Rт = 16,06 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 171
Соединение 171. Раствор соединения, полученного в примере 170C (0,19 г, 0,34 ммоля), в EtOAc (10 мл) обрабатывают при комнатной температуре 50 мг 10% палладия на угле и гидрируют в течение 72 часов при несколько избыточном давлении водорода. Полученную смесь фильтруют и концентрируют в вакууме, а неочищенный продукт очищают с помощью жидкостной хроматографии среднего давления, CH2Cl2, затем 1:99 метанол /CH2Cl2, затем 3:97 метанол /CH2Cl2, затем 10: 90 метанол /CH2Cl2 в качестве системы растворителей, до получения 97 мг указанного в заглавии соединения в виде твердого вещества белого цвета. TCX: Rf = 0,25 3:97 метанол /CH2Cl2, ВЭЖХ: Rт = 14,28 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 172
A. Соединение XXII /A = трет.-бутоксикарбонил, D' = циклопентилметил, E = 2,4-динитрофенил/. К раствору соединения, полученного в примере 114B (500 мг, 1,38 ммоля), в CH2Cl2 добавляют насыщенный раствор бикарбоната натрия (5 мл), затем твердый бикарбонат натрия (0,23 г, 2,76 ммоля) и 2,4-динитробензолсульфонилхлорид (440 мг, 1,65 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 2 часов. Этот раствор разбавляют 200 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентную систему растворителей: CH2Cl2, затем 1: 99 метанол /CH2Cl2 до получения 700 мг указанного в заглавии соединения в виде твердого вещества коричневого цвета. TCX: Rf = 0,48, 3:97 метанол /CH2Cl2. /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение XXII /A = H, D' = циклопентилметил, E = 2,4 - динитрофенил, гидрохлорид/. К соединению, полученному в примере 172A (700 мг, 1,18 ммоля) добавляют 10% вес/вес HCl в EtOAc (20 мл). Полученную смесь перемешивают в течение 3 часов при комнатной температуре. Этот раствор концентрируют при пониженном давлении до получения 590 мг указанного в заглавии соединения в виде твердого вещества коричневого цвета, которое используют без дополнительно очистки, TCX: Rf = 0,55, 1:10:90: NH4OH/метанол/CH2Cl2.
C. Соединение 172. К раствору соединения, полученного в примере 172B (590 мг, 1,11 ммоля) в CH2Cl2 (15 мл) добавляют насыщенный раствор бикарбоната натрия (5 мл), затем твердый бикарбонат натрия (0,19, 2,2 ммоля), и соединение, полученное в примере 48A (0,31 г, 1,3 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 24 часов. Этот раствор разбавляют 150 мл CH2Cl2, органику выделяют, сушат над безводным сульфатом магния, и органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя градиентное элюирование CH3OH/CH2Cl2, до получения продукта в виде белого твердого вещества (0,59 г). ВЭЖХ: Rт = 16,36 мин, /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 173
Соединение 173. Раствор соединения, полученного в примере 172C (0,20 г, 0,33 ммоля) в 10 мл EtOAc обрабатывают при комнатной температуре 50 мг 10% палладия на угле и гидрируют в течение 72 часов при несколько избыточном давлении водорода. Полученную смесь фильтруют и концентрируют в вакууме, а сырой продукт очищают с помощью жидкостной хроматографии среднего давления, используя в качестве системы растворителей: CH2Cl2, затем 1:99 метанол /CH2Cl2, затем 3: 97 метанол /CH2Cl2 и 10:90 метанол /CH2Cl2 до получения 120,2 мг указанного в заглавии соединения в виде твердого вещества светло-коричневого цвета. TCX: Rf = 0,17, 3:97 метанол /CH2Cl2, ВЭЖХ: Rт = 13,47 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 174
A. 4-бензилоксибензолсульфонилхлорид. К 0,87 г диметилформамида при 0oC в атмосфере азота добавляют 1,61 г сульфорилхлорида. Полученную смесь перемешивают в течение 15 минут и обрабатывают 2,0 г бензилфенилового эфира. Полученную смесь нагревают при 100oC в течение 1,5 часа, охлаждают до около 40oC, выливают на лед, экстрагируют CH2Cl2, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 10% этилацетат в гексане в качестве элюента до получения 0,78 г указанного в заглавии соединения в виде белого твердого вещества. TCX: Rf = 0,46, 10% этилацетат в гексане. /1H/-ЯМР/CDCl3/ соответствует структуре.
B. Соединение 174. Раствор 30 мг соединения, полученного в примере 51D, в смеси 4:1 CH2Cl2/ насыщенный водный NaHCO3, обрабатывают последовательно при комнатной температуре в атмосфере азота, 24 мг соединения, полученного в примере 174A, и 18 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 20% диэтиловый эфир /CH2Cl2 в качестве элюента до получения 14 мг указанного в заглавии продукта в виде твердого белого цвета. TCX: Rf = 0,43, 20% диэтиловый эфир /CH2Cl2. ВЭЖХ: Rт = 17,01 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 175
Соединение 175. Раствор 11 мг соединения, полученного в примере 174B, в этилацетате обрабатывают при комнатной температуре 2 мг 10% палладия на угле. Полученную смесь перемешивают в течение 14 часов в атмосфере водорода, фильтруют через слой Целита, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 10% метанол в CH2Cl2 в качестве элюента до получения 9 мг указанного в заглавии продукта в виде твердого продукта белого цвета. ТСХ: Rf = 0,38, 10% метанол в CH2Cl2. ВЭЖХ: Rт = 13,37 мин. /1H-ЯМР /CDCl3 соответствует структуре.
Пример 176
A. 1,2-бензодиоксол-5-сульфонилхлорид. К 3,50г диметилформамида при 0oC в атмосфере азота добавляют 6,47 г сульфурилхлорида. Полученную смесь перемешивают в течение 15 минут и обрабатывают 5,32 г 1,3-бензодиоксола. Полученную смесь нагревают при 120oC в течение 45 минут, охлаждают до примерно 40oC, выливают на лед, экстрагируют CH2Cl2, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 40% CH2Cl2 в гексане в качестве элюента, до получения 2,7 г указанного в заглавии соединения в виде твердого вещества желтого цвета. ТСХ: Rf = 0,37, 40% CH2Cl2 в гексане.
/1H-ЯМР /CDCl3/ соответствует структуре.
B. Соединение XXII /A = трет.-бутокси, D' = изобутил, E = 3,4-бензодиоксол/. Раствор 49 мг соединения, полученного в примере 39A, в смеси 4:1 CH2Cl2 /насыщенный водный NaHCO3 обрабатывают последовательно при комнатной температуре в атмосфере азота 45 мг соединения, полученного в примере 176A, и 28 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя 20% диэтиловый эфир/CH2Cl2 в качестве элюента до получения 71 мг указанного в заглавии продукта в виде воскообразного твердого продукта. ТСХ: Rf = 0,65, 20% диэтиловый эфир/CH2Cl2. /1H-ЯМР /CDCl3/ соответствует структуре.
C. Соединение XXII /A = H, D' = изобутил, E = 3,4-бензодиоксол, гидрохлорид/. Раствор 71 мг соединения, полученного в примере 176B, в этилацетате обрабатывают -20oC газообразным HCl. HCl барботируют через смесь в течение 20 минут, причем за это время температура повышается до 20oC. Затем через смесь барботируют азот в течение 15 минут, растворитель удаляют в вакууме до получения 66 мг указанного в заглавии продукта в виде твердого вещества белого цвета, которое используют непосредственно в последующей реакции.
D. Соединение 176. Раствор 18 мг соединения, полученного в примере 176C, в CH2Cl2 добавляют при комнатной температуре в атмосфере азота к раствору 13 мг соединения, полученного в примере 48A, и 14 мг N,N-диизопропилэтиламина в CH2Cl2. Полученную смесь перемешивают в течение 16 часов, разбавляют CH2Cl2, промывают насыщенным NaHCO3 и насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученный остаток очищают с помощью хроматографии низкого давления на силикагеле, используя в качестве элюента 5% диэтиловый эфир/ CH2Cl2, до получения 9 мг указанного в заглавии продукта в виде твердого вещества белого цвета. ТСХ: Rf = 0,14, 5% диэтиловый эфир/CH2Cl2. ВЭЖХ: Rт = 15,52 мин.
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 177
A. /4-метоксифенил/-метил-4-нитрофенилкарбонат. К раствору 1,50 г р-нитрофенилхлорформата в 30 мл CH2Cl2 при 0oC добавляют последовательно 0,77 мл 4-метоксибензилового спирта и 0,82 мл 4-метилморфолина. После перемешивания в течение получаса при комнатной температуре полученную смесь разбавляют CH2Cl2, промывают водой, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, до получения бледно-желтого твердого вещества, которое тщательно растирают со смесью CH2Cl2/гексан и фильтруют до получения 1,51 г указанного в заглавии соединения. ТСХ: Rf = 0,40, 20% EtOAc/гексан.
B. Соединение 177. К раствору 96,7 мг соединения, полученного в примере 141A, в 2 мл CH2Cl2 последовательно добавляют 90 мкл диизопропилэтиламина и 81,3 мг соединения, полученного в примере 178A. После перемешивания в течение 24 часов полученную смесь разбавляют CH2Cl2, промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя в качестве элюента 5% метанол в CH2Cl2, до получения 104,8 мг указанного в заглавии соединения. ТСХ: Rf = 0,4, 20% EtOAc/гексан. ВЭЖХ: Rт = 17,66 мин.
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 178
A. /3-метоксифенил/-метил-4-нитрофенилкарбонат. Получают по способу примера 177A, за исключением того, что в реакции с р-нитрофенилхлорформатом используют 3-метоксибензиловый спирт, до получения указанного в заглавии соединения в виде бледно-желтого твердого вещества. ТСХ: Rf = 0,40, 20% EtOAc/гексан.
B. Соединение 178. К раствору 97,8 мг соединения, полученного в примере 141A, в 2 мл CH2Cl2 добавляют последовательно 91 мкл диизопропилэтиламина и 82,2 мг соединения, полученного в примере 179A. После перемешивания в течение 24 часов полученную смесь разбавляют CH2Cl2, промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 5% метанол в CH2Cl2 в качестве элюента, до получения 25,7 мг указанного в заглавии соединения. ТСХ: Rf = 0,4, 20% EtOAc/гексан, ВЭЖХ: Rт = 17,75 мин.
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 179
A. /2-метоксифенил/-метил-4-нитрофенилкарбонат. Получают тем же способом, что и в примере 177A, за исключением того, что в реакции с р-нитрофенилхлорформатом используют 2-метоксибензиловый спирт, до получения указанного в заглавии соединения в виде бледно-желтого твердого продукта. ТСХ: Rf = 0,40 20% EtOAc/гексан.
B. Соединение 179. К раствору 97,8 мг соединения, полученного в примере 141A, в 2 мл CH2Cl2 добавляют последовательно 99 мкл диизопропилэтиламина и 89,2 мг соединения, полученного в примере 179A. После перемешивания в течение 24 часов полученную смесь разбавляют CH2Cl2, промывают водой и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной тонкослойной хроматографии, используя 5% метанол в CH2Cl2 в качестве элюента, до получения 107,0 мг указанного в заглавии соединения. ТСХ: Rf = 0,4, 20% EtOAc/гексан, ВЭЖХ: Rт = 17,58 мин.
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 180
A. 2,3-дигидробензофуран-5-сульфонилхлорид. К 3,35 г диметилформамида при 0oC в атмосфере азота добавляют 6,18 г сульфурилхлорида. Полученную смесь перемешивают в течение 15 минут и обрабатывают 4,69 г 2,3-дигидробензофурана. Полученную смесь нагревают при 100oC в течение 1,5 часа, охлаждают до около 40oC, выливают на лед, экстрагируют CH2Cl2, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток помещают в этилацетат, охлаждают до 5oC в течение 16 часов, и полученные кристаллы розового цвета собирают вакуумным фильтрованием до получения 6,12 г указанного в заглавии продукта. ТСХ: Rf = 0,41, 10% этилацетат в гексане.
/1H-ЯМР /CDCl3/ соответствует структуре.
B. Соединение 180. Раствор 32 мг соединения, полученного в примере 140D, в смеси 4: 1 CH2Cl2 /насыщенный водный NaHCO3 обрабатывают последовательно при комнатной температуре в атмосфере азота, 22 мг соединения, полученного в примере 180A, и 18 мг бикарбоната натрия. Полученную смесь перемешивают в течение 14 часов, разбавляют CH2Cl2, промывают насыщенным NaCl, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на силикагеле, используя диэтиловый эфир/CH2Cl2 в качестве элюента до получения 20 мг указанного в заглавии продукта, в виде белого твердого вещества. ТСХ: Rf = 0,52, 20% диэтиловый эфир/CH2Cl2. ВЭЖХ: Rт = 15,49 мин.
/1H-ЯМР /CDCl3/ соответствует структуре.
Пример 181
Соединение 181. К раствору соединения, полученного в примере 140D (150 мг, 0,4 ммоля), в CH2Cl2 (10 мл) добавляют насыщенный раствор бикарбоната натрия (5 мл), затем твердый бикарбонат натрия (0,1 г, 1,2 ммоля) и 4-цианобензолсульфонилхлорид (0,1 г, 0,48 ммоля). Полученную смесь перемешивают при комнатной температуре в течение 4 часов. Этот раствор разбавляют 200 мл CH2Cl2 органику выделяют, сушат над безводным сульфатом магния, и органику концентрируют при пониженном давлении. Сырой продукт очищают с помощью жидкостной хроматографии среднего давленя, используя CH2Cl2, затем 1:99 метанол/CH2Cl2 в качестве системы растворителей, до получения 0,19 г (выход 86%) указанного в заглавии соединения в виде твердого вещества белого цвета. ТСХ: Rf = 0,40, 3:97 метанол/CH2Cl2, ВЭЖХ: Rт = 15,02 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 182
Соединение 182. Это соединение получают из соединения, полученного в примере 114D, и соединения, полученного в примере 48A, по способу примера 88. После обработки и очистки с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент элюента от 35% до 100% CH3CN/H2O с 0,1% TFA, получают указанное в заглавии соединение, ТСХ: Rf = 0.25, 4% МeOH/CH2Cl2. ВЭЖХ: Rт = 16,6 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 183
Соединение 183. Это соединение получают из соединения, полученного в примере 84, за счет обработки газообразным HCl и последующим взаимодействием с соединением, полученным в примере 48A, по способу примера 132. После обработки и очистки с помощью кристаллизации из EtOAc получают 33,0 мг указанного в заглавии в виде белого твердого вещества. ТСХ: Rf = 0,25, 4% MeOH/CH2Cl2. ВЭЖХ: Rт = 17.71 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 184
A. /N-трет. бутоксикарбонил-/R/-3-пирролидинил-N-гидроксисукцинимидилкарбонат. К раствору 1,0 г /R/-3-гидроксипирролидина в 50 мл тетрагидрофурана добавляют последовательно при комнатной температуре 3,75 г ди-трет-бутилдикарбоната и 1 мл 2н гидроксида натрия. Полученную смесь перемешивают в течение 1 часа, фильтруют и концентрируют в вакууме. Полученное соединение подвергают взаимодействию с N,N-дисукцинимидилкарбонатом по способу примера 155A. После обработки и очистки с помощью хроматографии в толстом слое силикагеле, используя в качестве элюента EtOAc, получают указанное в заглавии соединение в виде твердого вещества белого цвета. /1H/-ЯМР (CDCl3) соответствует структуре.
B. Соединение 184. Раствор 350 мг соединения, полученного в примере 166A, обрабатывают газообразным HCl для удаления защиты, и полученное соединение подвергают взаимодействию с соединением, полученным в примере 184A, по способу примера 88. После концентрирования полученной смеси в вакууме и последующей обработки остаток очищают с помощью хроматографии в толстом слое силикагеля, используя 7% MeOH/CH2Cl2 в качестве элюента, до получения 120 мг указанного в заглавии соединения, ТСХ: Rf = 0,45, 5% MeOH/CH2Cl2. ВЭЖХ: Rт = 16,97 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 185
Соединение 185. Раствор 120 мг соединения, полученного в примере 184B, в EtOAc (25 мл) при 0oC обрабатывают безводным газообразным HCl в течение 10 минут, оставляют выстаиваться в течение 12 часов, причем температура при этом повышается до комнатной. После концентрирования в вакууме получают 110 мг указанного в заглавии соединения. ТСХ: Rf = 0,35, 10% MeOH/89% - CH2Cl2/ 1% NH4OH. ВЭЖХ: Rт = 13,72 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 186
A. Соединение XXX //син,анти//OH, A карбобензилокси, R3=/S/-втор.-бутил, R3'= H, D'=бензил, A'=трет.бутоксикарбонил/. Раствор 1,37 г соединения, полученного в примере 1B, в 150 мл метиленхлорида обрабатывают 1,03 г Cbz-Ile, 523 мг HOBT•H2O и 742 мг ЕДС. Полученную смесь перемешивают в течение 18 часов, разбавляют 3 объемами диэтилового эфира и промывают последовательно водой, насыщенным NaHCO3, 10% раствором KOH и рассолом. После сушки над сульфатом магния и концентрирования в вакууме остаток очищают с помощью хроматографии на колонке с силикагелем, используя градиент от 1% до 1,5% MeOH в метилхлориде в качестве элюента, до получения 2,10 г указанного в заглавии соединения в виде белой пены. ТСХ: Rf = 0,51, 5% метанол/CH2Cl2.
B. Соединение XXX//син,анти/-OH, A=карбобензилокси, R3=/S/-втор.бутил, R3'= H, A'=H/, гидрохлорид. Раствор 650 мг соединения, полученного в примере 186A, в 12 мл этилацетата охлаждают в бане лед/вода и обрабатывают медленным потоком газообразного HCl в течение приблизительно 6 мин при интенсивном перемешивании. Полученную смесь закрывают и перемешивают еще 10 минут, затем продувают потоком азота в течение 15 минут и концентрируют в вакууме до получения белого твердого вещества, которое используют без дополнительной очистки. ТСХ: Rf = 0,18, 95:5:0,5 CH2Cl2/метанол/ концентрированный NH4OH.
C. Соединение 186. Раствор 20 мг соединения, полученного в примере 186B, в 0,8 мл метиленхлорида охлаждают в смеси лед/метанол /пример 15oC/, затем обрабатывают 13,8 мкл DJEA, затем 7,6 мг альфа-толуолсульфонилхлорида. Полученную смесь перемешивают в течение 15 часов, медленно нагревая до комнатной температуры. Полученную смесь концентрируют в вакууме до небольшого объема, помещают на препаративную пластину толщиной 0,5 мм и элюируют 3,5% MeOH/CH2Cl2. Полосу, содержащую целевой диастереоизомер, выделяют и элюируют 8% MeOH/CH2Cl2, до получения 4,8 мг указанного в заглавии соединения. ТСХ: Rf = 0,42, 15% диэтиловый эфир/CH2Cl2. ВЭЖХ: Rт = 17,81. ЯМР/CDCl3/: 0,78 (ДД. 6H), 0.84 (м, 1H), 1,07 (м, 1H), 1,76 - 1,86 (м, 2H), 2,72 (м, 2H), 3,14 (с, 2H), 3,49 (дд, 1H), 3,87 (дд, 1H), 3,58 (м, 1H), 4,01 (д, 1H), 4,14 (д, 1H), 4,26 (д, 1H), 4,35 (д, 1H), 4,90 (м, 1H), 5,08 (с, 2H), 5,97 (д, 1H), 7,08 (д, 2H), 7,17 (т. 1H), 7,20 - 7,40 (м, 17H).
Пример 187
Соединение 187. 100 мг соединения, полученного в примере 54A, обрабатывают 1 мл 90% водной TFA и оставляют выстаиваться в течение 12 часов. Полученную смесь концентрируют в вакууме, а остаток помещают в 10 мл сухого CH2Cl2, обрабатывают 65 мг N-Cbr-L-изолейцина (0,235 ммоля), 50 мкл DJEA (0,27 ммоля), 30 мг HOBt (0,2 ммоля) и 42 мг 1-/3-диметиламинопропил/3-этилкарбодиимидгидрохлорида (0,22 ммоля). Полученную смесь перемешивают в течение 3 часов, разбавляют CH2Cl2, промывают последовательно водой, насыщенным NaHCO3 раствором и рассолом. После сушки над сульфатом магния и концентрирования в вакууме полученную смесь очищают хроматографически на колонке с силикагелем, используя 5% CH3OH в CH2Cl2 в качестве элюента, до получения указанного в заглавии соединения, часть которого очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента до получения 36,0 мг соединения, со степенью чистоты 99%. ТСХ: Rf = 0,25, 5% CH3OH в CH2Cl2, ВЭЖХ: Rт = 16,45 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 188
Соединение 188. Раствор 51 мг соединения, полученного в примере 187A, в 15 мл метанола, гидирируют при слегка позитивном давлении водорода в присутствии 10 мг 10% палладия/OH/2 в течение 14 часов. После фильтрования и концентрирования в вакууме неочищенную смесь помещают в 10 мг CH2Cl2 и обрабатывают 0,203 мл DJEA, и 19,0 мг 2-хиноксалоилхлорида. Полученную смесь перемешивают в течение 6 часов, затем разбавляют CH2Cl2 и промывают водой. После сушки над сульфатом магния и концентрирования в вакууме порцию этой смеси очищают с помощью препаративной ВЭЖХ с обращенной фазой C18, используя линейный градиент от 35% до 100% CH3CN/H2O с 0,1% TFA в качестве элюента до получения 2,1 мг указанного в заглавии соединения. ТСХ: Rf = 0,25, 6% CH3OH в CH2Cl2. ВЭЖХ: Rт = 16,21 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 189
A. Соединение XXII /D'=изобутил, A=Н, E=4-ацетамидофенил, соль трифторуксусной кислоты/. К раствору 89,3 мг (0,167 ммоля) соединения, полученного в примере 39B, в CH2Cl2 (1 мл) при 0-5oC добавляют трифторметансульфоновую кислоту (1 мл). После перемешивания в течение 0,5 часа полученную смесь концентрируют в вакууме, и полученную смолу желтого цвета используют без последующей обработки.
B. Соединение 189. Раствор соединения, полученного в примере 189A (0,167 ммоля), в CH2Cl2 обрабатывают последовательно при комнатной температуре в атмосфере азота 44,2 (0,217 ммоля) N-Boc- α- -аминоизомасляной кислоты, 0,044 мл (0,251 ммоля) диизопропиламина, 27,1 мг (0,201 ммоля) 1-гидроксибензотриазолгидрата, 38,5 мг (0,201 ммоля) 1-/3-диметиламинопропил/-3-этил-карбодиимидгидрохлорида. Полученную смесь перемешивают в течение 16 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают водой, 0,5н соляной кислотой, промывают бикарбонатом натрия, насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя от 10% до 35% градиентное элюирование этилацетат/CH2Cl2, до получения 69,3 мг указанного в заглавии продукта в виде белого твердого продукта. ТСХ: Rf = 0,46, 60% этилацетат/CH2Cl2/. ВЭЖХ: Rт = 15,0 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 190
A. Соединение XXXI /A=H, R3=метил, R3'=метил, D'=изобутил, E=4-амидофенил, гидрохлорид/. К раствору 60,1 мг соединения, полученного в примере 189B, в CH2Cl2 (1 мл) при 0-5oC добавляют 1 мл трифторметансульфоновой кислоты. После перемешивания в течение 0,75 часа полученную смесь концентрируют в вакууме, и полученное белое вещество используют без дополнительной очистки в последующей реакции.
B. Соединение 190. К раствору 37 мг (0,059 ммоля) соединения, полученного в примере 190A, в CH2Cl2 (1 мл) добавляют последовательно при комнатной температуре в атмосфере азота, 15,4 мг (0,089 ммоля) 1-гидроксибензотриазолгидрата, и 17,8 мг (0,089 ммоля) ЕДС. Полученную смесь перемешивают в течение 16 часов, затем концентрируют в вакууме. Остаток помещают в EtOAc и промывают насыщенным рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии на колонке с силикагелем, используя 50% EtOAc в CH2Cl2 в качестве элюента до получения 32,5 мг указанного в заглавии продукта. ТСХ: Rf = 0,35, 50% EtOAc/CH2Cl2, ВЭЖХ: Rт = 15,65 мин /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 191
A. /2S, 3R/-S-амино-1-хлор-2-гидрокси-4-фенилбутан/. Раствор 2,24 г (6,71 ммоля) /1S, 2RS/-N-1/-бензил-3-хлор-2-гидроксипропил/-бензилоксикарбониламина в 5 мл метанола добавляют при комнатной температуре в атмосфере азота к суспензии 0,22 г (10 вес.%) 10% палладия на угле в 60 мл метанола, и гидрируют в течение 24 часов при несколько избыточном давлении водорода. Полученную смесь фильтруют и концентрируют в вакууме до получения 1,34 г смешанных диастереоизомерных продуктов. ТСХ: Rf = 0,33, 10% CH3OH/CH2Cl2.
B. /2S/-2-бензилоксикарбониламино-N1-//1S, 2RS/-1-бензил-3-хлор-2-гидроксипропил/-N4-тритилсукцинамид.
Раствор 1.34 г (6,71 ммоля) соединения, полученного в примере 191A, в 60 мл дихлорметана обрабатывают последовательно при комнатной температуре в атмосфере азота, 3,58 г (7,05 ммоля) Cbz- Nδ тритил-аспарагина. 0,95 г (7,05 ммоля) 1-гидроксибензотриазолгидарата. 1,35 г (7,05 ммоля) ЕДС. Полученную смесь перемешивают в течение 24 часов, а затем концентрируют в вакууме. Остаток помещают в этилацетат и промывают водой, насыщенным NaHCO3, насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 10% смесь этилацетат/дихлорметан в качестве элюента до получения 3,08 г смешанных диастереоизомерных продуктов. ТСХ: Rf = 0,75, 0,83, 40% EtOAc/CH2Cl2, /1H/-ЯМР (CDCl3) соответствует структуре.
C. /2S/-2-амино N1-//1S, 2RS/-1-бензил-3-хлор-2-гидроксипропил/-N4-тритилсукцинамид. Раствор 2.80 г (4,06 ммоля) соединений, полученных в примере 191B, в 5 мл метанола, добавляют при комнатной температуре в атмосфере азота к суспензии 0,28 г (10 вес.%) 10% палладия на угле в 100 мл метанола и гидрируют в течение 24 часов при слегка избыточном давлении водорода. Полученную смесь фильтруют и концентрируют в вакууме до получения 2,26 г смешанных диастереоизомерных продуктов. ТСХ: Rf = 0,42, 10% CH3OH/CH2Cl.
D. /2S/-2-//1S, 2RS/-1-бензил-3-хлор-2-гидроксипропил/-N1-//хинолин-2-карбонил/- амино-N4-тритилсукцинамид. Раствор 2,26 г (4,06 ммоля) соединений, полученных в примере 191C, в 60 мл дихлорметана обрабатывают последовательно при комнатной температуре в атмосфере азота 0,74 г (4,27 ммоля) хинальдиновой кислоты, 0,58 г (4,27 ммоля) 1-гидроксибензотриазолгидрата, и 0,82 г (4,27 ммоля) ЕДС. Спустя 24 часа добавляют 30 мл дихлорметана. Полученную смесь промывают водой, 5% NaHCO3 раствором, насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток растворяют в 50% смеси этилацетат/гексан, и фильтруют через слой силикагеля. После удаления растворителей, получают 2,30 г смешанных диастереоизомерных продуктов. ТСХ: Rf = 0,53, 0,58, 40% EtOAc/CH2Cl2; /1H/-ЯМР (CDCl3) соответствует структуре.
E. /2S/-2-//1S, 2RS/-1-бензил-2-гидрокси-3-иодопропил/-N1-//-хинолин-2- карбонил/-амино-N4-тритилсукцинамид.
Раствор 1,05 г (1,48 ммоля) соединений, полученных в примере 191D, и 0,36 г (2.37 ммоля) иодида натрия в 15 мл метилэтилкетона нагревают при кипячении с обратным холодильником в течение 24 часов. Полученную смесь охлаждают до комнатной температуры, а затем концентрируют в вакууме. Остаток помещают в дихлорметан, промывают водой, насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 1,3 г смешанных диастереоизомерных продуктов. ТСХ: Rf = 0,58, 0.65, 40% EtOAc/CH2Cl2; /1H/-ЯМР (CDCl3) соответствует структуре.
F. /2S/-2-//1S, 2 син, анти/-3-/2-метилпропил/амино-1-бензил-2- гидроксипропил-/N1-//хинолин-2-карбонил/-амино-N4- тритилсукцинамид.
Раствор 207,6 мг (0,26 ммоля) соединений, полученных в примере 191E, и 0,5 мл (5,17 ммоля) изобутиламина в 9 мл ацетонитрила в запаяной ампуле при кипячении с обратным холодильником в течение 24 часов. После охлаждения до комнатной температуры полученную смесь концентрируют в вакууме. Остаток помещают в дихлорметан и промывают водой, насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 209,2 мг смеси диастереоизомерных продуктов. ТСХ: Rf = 0,11 10% CH3OH/CH2Cl2; /1H/-ЯМР (CDCl3) соответствует структуре.
C. Соединение XIV //син.анти/-OH, A=хинолин-2-карбонил, D'=изобутил/. Раствор 192,9 мг (0,26 ммоля) соединений, полученных в примере 191F, 0,07 мл (0,388 ммоля) диизопропилэтиламина в 5 мл дихлорметана обрабатывают 112,9 мг (0,517 ммоля) ди-трет. -бутилдикарбоната. Спустя 24 часа полученную смесь разбавляют дихлорметаном, промывают водой, 5% NaHCO3, 0,5н HCl, насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя 40% этилацетат/дихлорметан в качестве элюента до получения 147,3 мг смешанных диастереоизомерных продуктов. ТСХ: Rf = 0,60, 0,67, 40% EtOAc/CH2Cl2; /1H/-ЯМР (CDCl3) соответствует структуре.
H. Соединение 191. Раствор 147,3 мг (0,174 ммоля) соединений, полученных в примере 191G, в 2 мл дихлорметана обрабатывают 2 мл трифторуксусной кислоты. Спустя 4 часа полученную смесь концентрируют в вакууме. ТСХ: Rf = 0,11, 10% CH3OH/CH2Cl2. К раствору полученного соединения в 2 мл дихлорметана последовательно добавляют 0,5 мл насыщенного NaHCO3, небольшое количество твердого NaHCO3 и 67 мг (0,226 ммоля) смеси 4-ацетамидо-3-фторбензолсульфонилхлорида и 3-ацетамидо-4-фторбензолсульфонилхлорида. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют один раз дихлорметаном. Объединенные органические слои промывают насыщенным NaCl, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя смесь 2% метанол/дихлорметан в качестве элюента до получения 64 мг диастереоизомеров и региоизомеров, которые далее очищают с помощью препаративной ВЭЖХ до получения 18,9 мг смешанных региоизомеров, содержащих соединения 191 в виде белого твердого продукта. ТСХ: Rf = 0,14, 5% CH3OH/CH2Cl2; ВЭЖХ, Rт = 13,36 мин. /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 193
Соединение 193. Раствор 81,2 мг (0,096 ммоля) полученного в примере 9/192A диастереоизомера с низким Rf, в 3 мл дихлорметана обрабатывают 3 мл трифторуксусной кислоты. Спустя 4 часа полученную смесь концентрируют в вакууме. ТСХ: Rf = 0,11, 10% CH3OH/CH2Cl2. К раствору 20,6 мг (0,0431 ммоля) полученного остатка в 1 мл дихлорметана последовательно добавляют 0,3 мл насыщенного NaHCO3, небольшое количество твердого NaHCO3 и 12,4 мг (0,053 ммоля) 4-ацетамидобензолсульфонилхлорида. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют один раз дихлорметаном. Объединенный органический слой промывают рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ до получения 8.3 мг указанного в заглавии соединения в виде белого твердого продукта: ТСХ: Rf = 0,10, 5% CH3OH/CH2Cl2, ВЭЖХ, Rт = 12,7 мин; /1H/-ЯМР (CDCl3) соответствует структуре.
Пример 194
Соединение 194. К раствору 13,0 мг (0,026 ммоля) продукта, полученного в примере 193 после удаления защиты трифторуксусной кислотой, в 1 мл дихлорметана последовательно добавляют 0,3 мл насыщенного NaHCO3, небольшое количество твердого NaHCO3 и 8,4 мг (0,033 ммоля) 5-/изоксазол/3-ил/тиофен-2-сульфонилхлорида. Спустя 3 часа полученную смесь разбавляют дихлорметаном. Два слоя разделяют, и водный слой экстрагируют дихлорметаном. Объединенный органический слой промывают рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью препаративной ВЭЖХ, до получения 5,1 мг указанного в заглавии продукта в виде твердого вещества белого цвета; ТСХ: Rf = 0,27, 5% CH3OH/CH2Cl2; ВЭЖХ, Rт = 14,4 мин; /1H/-ЯМР (CDCl2) соответствует структуре.
Пример 195
A. Соединение XXII /A/-S/-3-тетрагидрофурил, D'=циклопентилметил, A'-трет. -бутоксикарбонил/. К раствору 264 мг соединения, полученного в примере 140D, в 10 мл CH2Cl2, добавляют 0,14 мл диизопропилэтиламина и 175 мг ди-трет-бутилпропилэтиламина и 175 мг ди-трет. -бутилпропилкарбоната. После перемешивания в течение 4 часов полученную смесь разбавляют 50 мл CH2Cl2, промывают 0,5 н HCl и рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме до получения 364 мг указанного в заглавии соединения в виде белого твердого продукта, который используют без последующей очистки. ТСХ: Rf= 0,58, 40% EtOAc/CH2Cl2/
B. Раствор 334 мг соединения, полученного в примере 195A, в 5 мл этанола, гидрируют при давлении 2,109 кг/см2 в присутствии 80 мг оксида платины /IV/ в течение 24 часов. Полученную смесь фильтруют и концентрируют. Остаток очищают с помощью хроматографии низкого давления на хроматографической колонке, используя в качестве элюента 20% EtOAc в CH2Cl2 до получения 268 мг указанного в заглавии соединения. ТСХ: Rf = 0,55, 40 EtOAc/CH2Cl2.
/1H/-ЯМР/CDCl3/ соответствует структуре.
C. Раствор 268 мг соединения, полученного в примере 195 B, в 10 мл EtOAc обрабатывают безводным газообразным HCl в течение 5 минут. Реакционную смесь продувают азотом, затем концентрируют в вакууме и полученное твердое вещество белого цвета используют без дополнительной очистки в последующих реакциях.
D. Соединение 195. К раствору 233 мг неочищенного соединения, полученного в примере 195C, в 10 мл CH2Cl2 добавляют 2 мл водного бикарбоната натрия и 149 мг 4-метилоксибензолсульфонилхлорида. Спустя 3 часа полученную смесь разбавляют CH2Cl2, промывают бикарбонатом натрия, рассолом, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии низкого давления на колонке с силикагелем, используя в качестве элюента 0% - 20% EtOAc/CH2Cl2, до получения 225 мг указанного в заглавии соединения в виде твердого вещества белого цвета. ТСХ: Rf = 0,40. 20% EtOAc/CH2Cl2; ВЭЖХ: Rт = 15,65 мин. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 196
A. /1S,2S/-N-/1-изобутил-3-хлор-2-гидроксипропил/бензилоксикарбониламин. К раствору N-Cbz-лейцинхлорметилкетона (2,0 г) в 20 мл метанола добавляют при 0oC 1,0 г боргидрида натрия, и полученную смесь перемешивают при комнатной температуре в течение 24 часов. Раствор концентрируют при пониженном давлении и остаток разделяют между 20 мл насыщенного водного NH4Cl и 500 мл диэтилового эфира. Органическую фракцию выделяют, сушат над сульфатом магния и концентрируют в вакууме, а остаток очищают с помощью хроматографии на силикагеле, до получения 1,8 г белого твердого вещества.
B. /1S/-1-1/S//карбобензилокси/амино/-2-изобутилоксиран. К раствору соединения, полученного в примере 196 /300 мг/ в абсолютированном этаноле добавляют 67 мг порошка KOH. Полученную смесь перемешивают в течение 3 часов при комнатной температуре, фильтруют через диатомовую землю и концентрируют в вакууме. Остаток растворения в диэтиловом эфире, сушат над сульфатом магния, и концентрируют до получения 230 мг бесцветного масла, которое используют непосредственно в последующей реакции.
C. /2R, 3S/-N3-карбобензилокси-N1-изобутил-1,3- диамино-2-гидрокси-b-метилгексан. Порцию в 230 мг соединения, полученного в примере 196B, суспендируют в 5 мл изобутиламина, и полученную смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь концентрируют в вакууме до получения указанного в заглавии продукта в виде 179 мг белого твердого вещества, которое используют непосредственно в последующей реакции.
Соединение 1 /A = трет.-бутоксикарбонил, x = O, D = изобутил, E = 4-метоксифенил, /S/-гидрокси/. По способу примера 81, раствор соединения, полученного в примере 196C /170 мг/, в CH2Cl2 подвергают взаимодействию с 4-метоксибензол хлоридом (150 мг) в присутствии водного NaHCO3. После обработки и хроматографической очистки на силикагеле получают 90 мг продукта в виде белого твердого вещества.
E. Соединение I /A = H, x = O, D = изобутил, E = 4-метоксифенил, /S/-гидрокси/. Раствор соединения, полученного в примере 196 D (90 мг), в этаноле обрабатывают 50 мг 10% палладия на угле, и полученную смесь перемешивают в атмосфере водорода. После завершения реакции полученную смесь фильтруют и концентрируют в вакууме до получения 60 мг указанного в заглавии соединения, которое используют непосредственно в последующей реакции.
F. Соединение 196. Продукт реакции соединения примера 196E (60 мг) в CH2Cl2 подвергают взаимодействию с продуктом примера 48A (150 мг), получая после водной обработки, сушки над сульфатом магния, фильтрования и концентрирования в вакууме, остаток, который очищают с помощью хроматографии на силикагеле, используя смесь метанол/CH2Cl2 в качестве элюента, указанное в заглавии соединение в виде 40 мг белого твердого продукта. /1H/-ЯМР/CDCl3/ соответствует структуре.
Пример 197
Были измерены константы ингибирования соединений, перечисленных в таблице VI против HIV-1 протеазы по способу Pennington et al.
Были измерены также антивирусные эффективности соединений в CCRM-CEM клетках по способу Meek et.al. В таблице далее Ki и ИК90 выражены в нМ.
В таблице VIII использована следующая классификация:
A: ингибирует HIV репликацию в концентрации 100 нМ или менее.
B: ингибирует HIV репликацию в концентрации от 101 до 1000 нМ.
C: ингибирует HIV репликацию в концентрации от 1001 до 10000 нМ.
D: ингибирует репликацию HIV в концентрации от 10000 до 40000 нМ.
ND: тест не проводился
Как показано в таблицах VII и VIII все тестированные соединения демонстрируют ингибирующую и антивирусную активности. Более того, некоторые из этих соединений демонстрируют уровни активности, значительно превосходящие ровни активности для известных ингибиторов HIV протеазы.
Хотя был описан ряд вариантов изобретения, очевидно, что основные конструкции могут быть изменены, с тем чтобы обеспечить другие варианты, в которых использованы продукты и процессы настоящего изобретения. Поэтому следует учитывать, что объем изобретения определяется прилагаемой формулой изобретения, а не конкретными его вариантами, которые были представлены в примерах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОДАВЛЕНИЯ МНОЖЕСТВЕННОЙ ЛЕКАРСТВЕННОЙ УСТОЙЧИВОСТИ | 1995 |
|
RU2165410C2 |
| ПРОИЗВОДНЫЕ 1-(2-ОКСОАЦЕТИЛ)ПИПЕРИДИН- ИЛИ ПИРРОЛИДИН-2-КАРБОНОВЫХ КИСЛОТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ | 1993 |
|
RU2158258C2 |
| ИНГИБИТОРЫ КАСПАЗ | 1999 |
|
RU2274642C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
| ДИГИДРОДИАЗЕПИНЫ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2007 |
|
RU2475488C2 |
| ИНГИБИТОРЫ СЕТР | 2006 |
|
RU2513107C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОТИЕНИЛПИПЕРАЗИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2157371C2 |
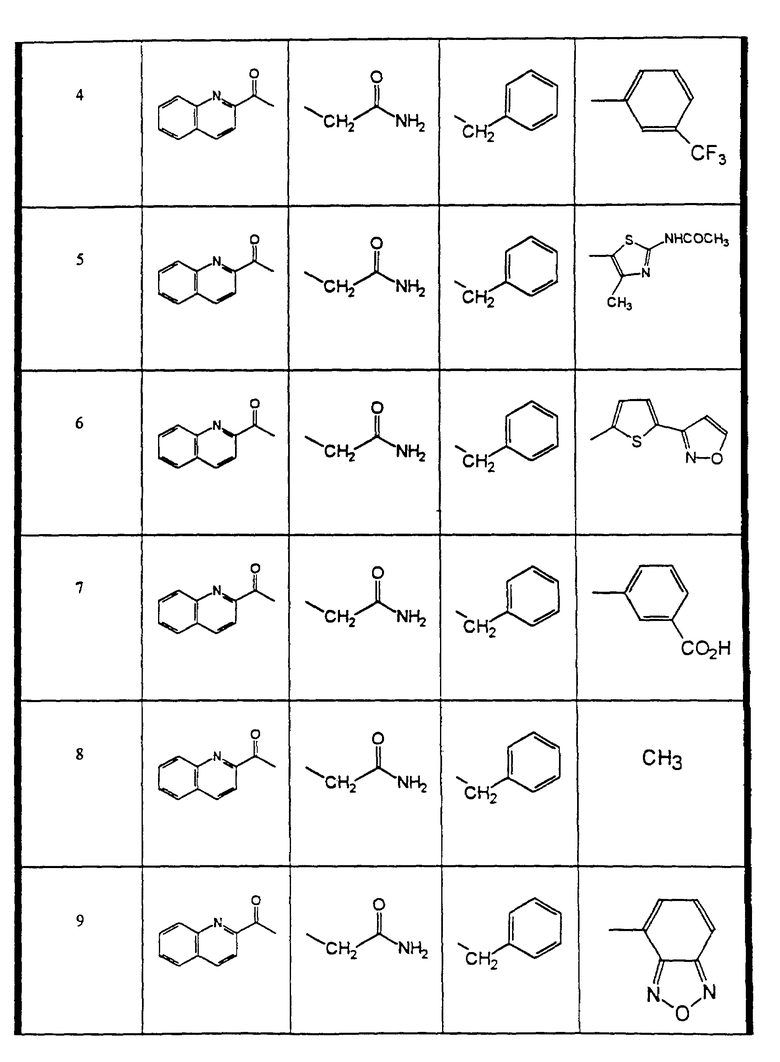
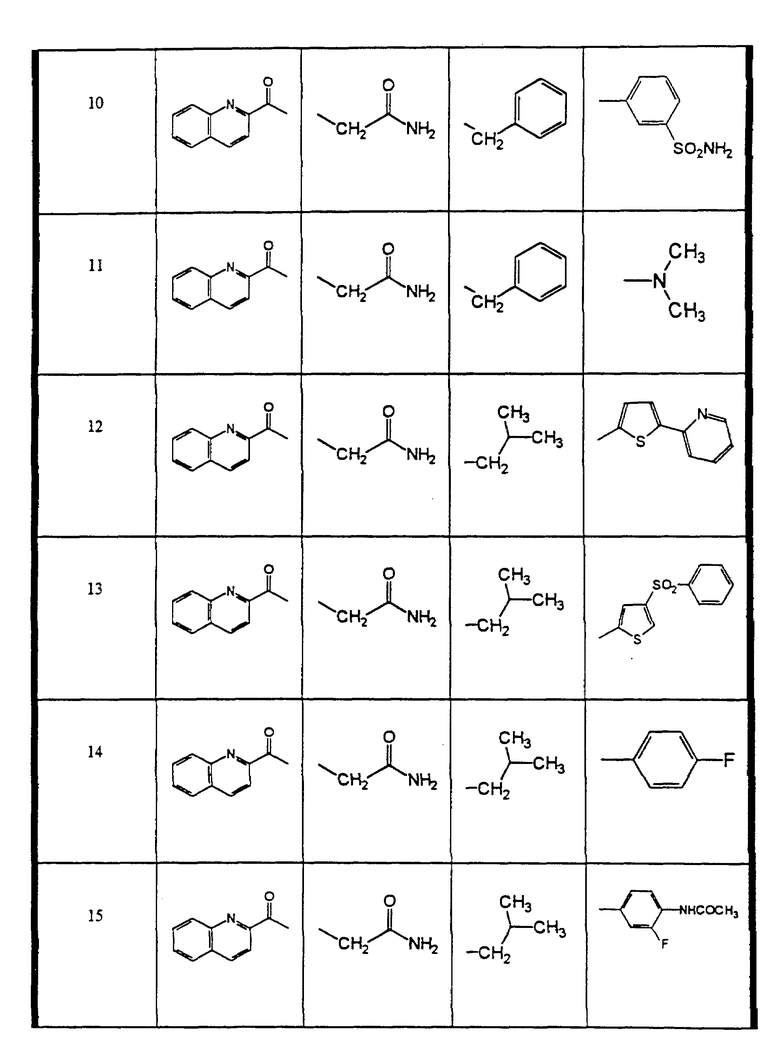
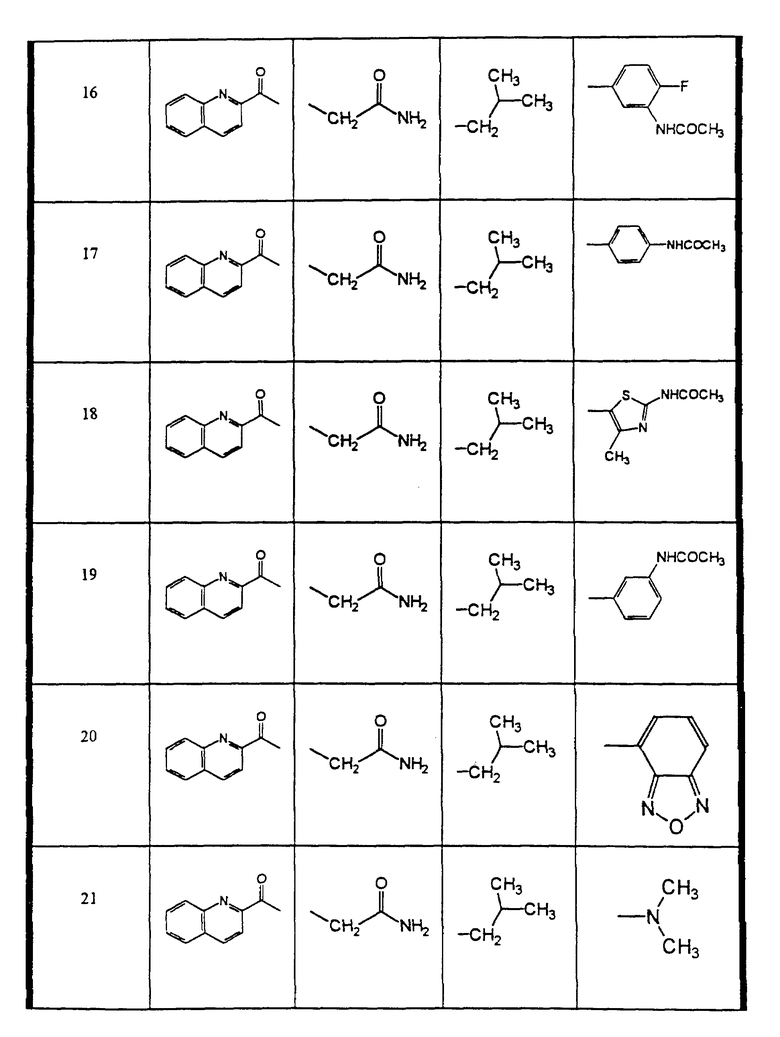
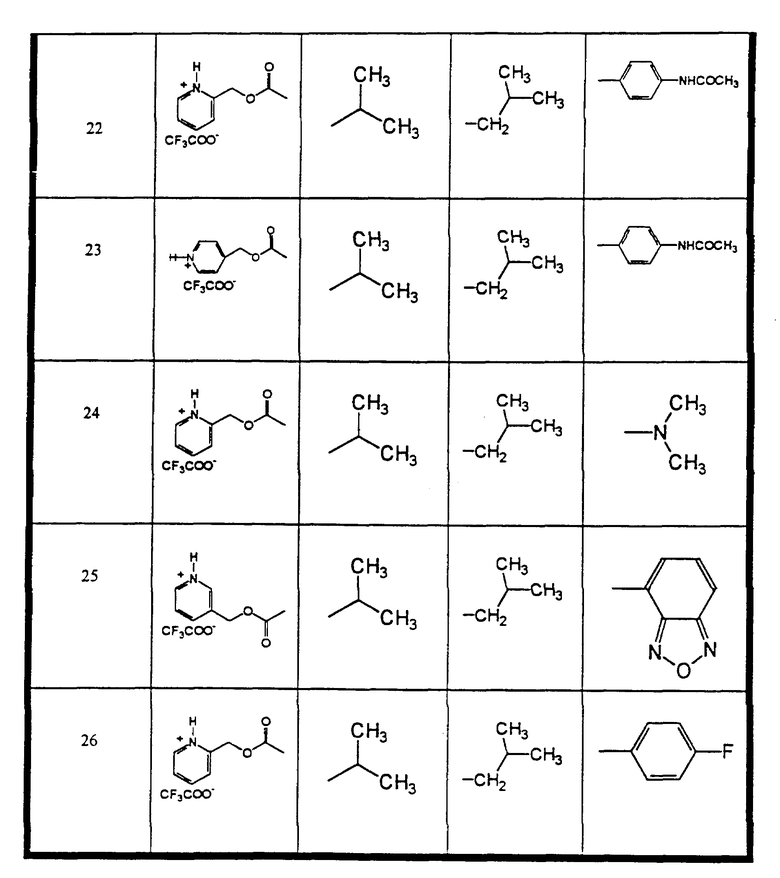
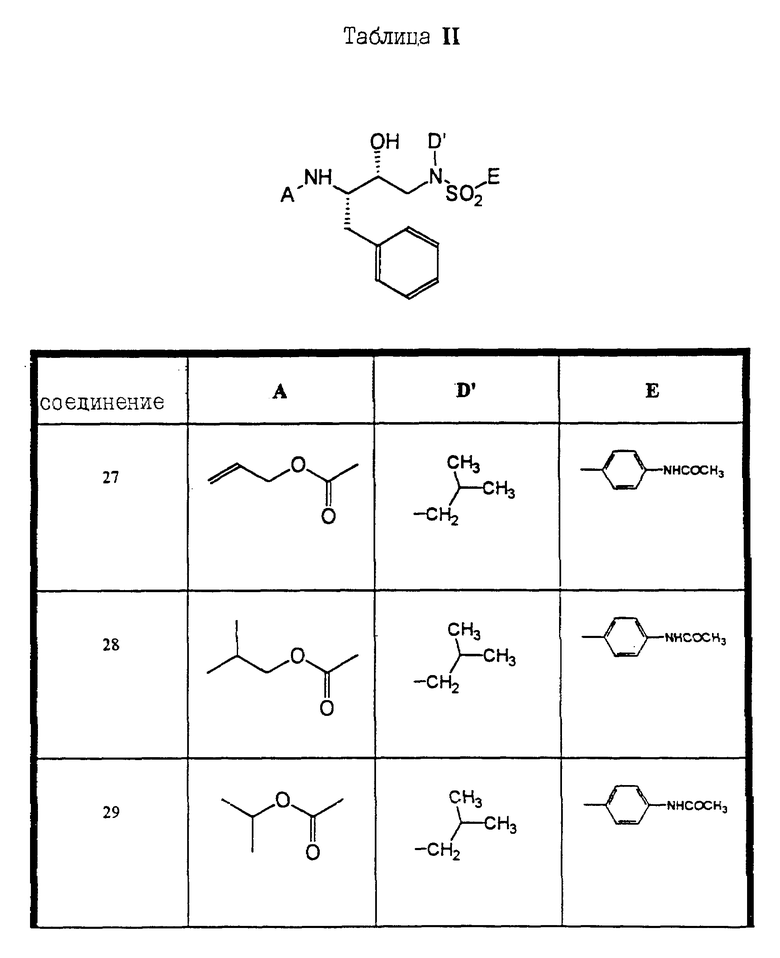
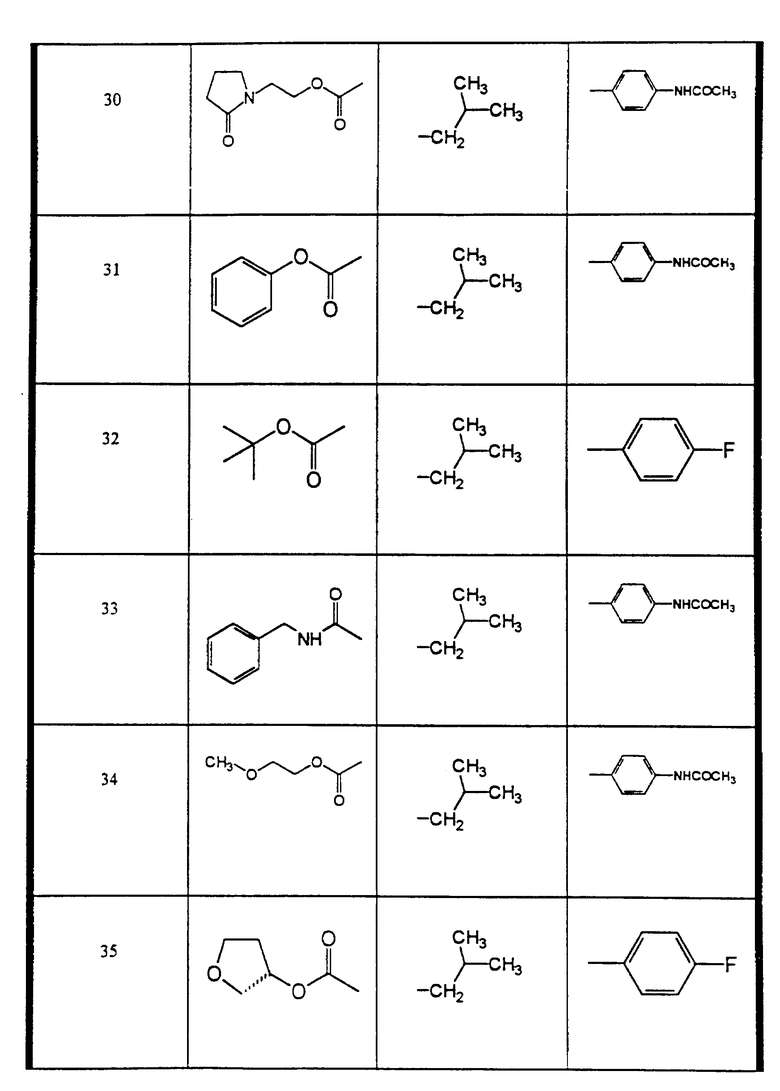
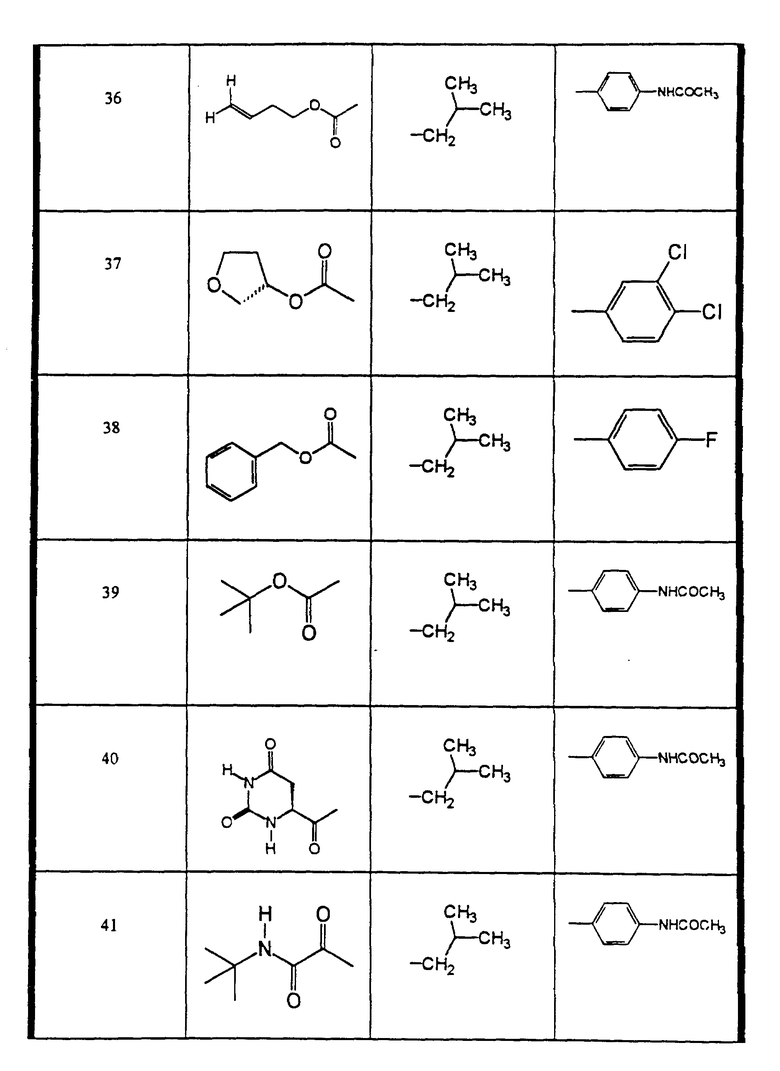
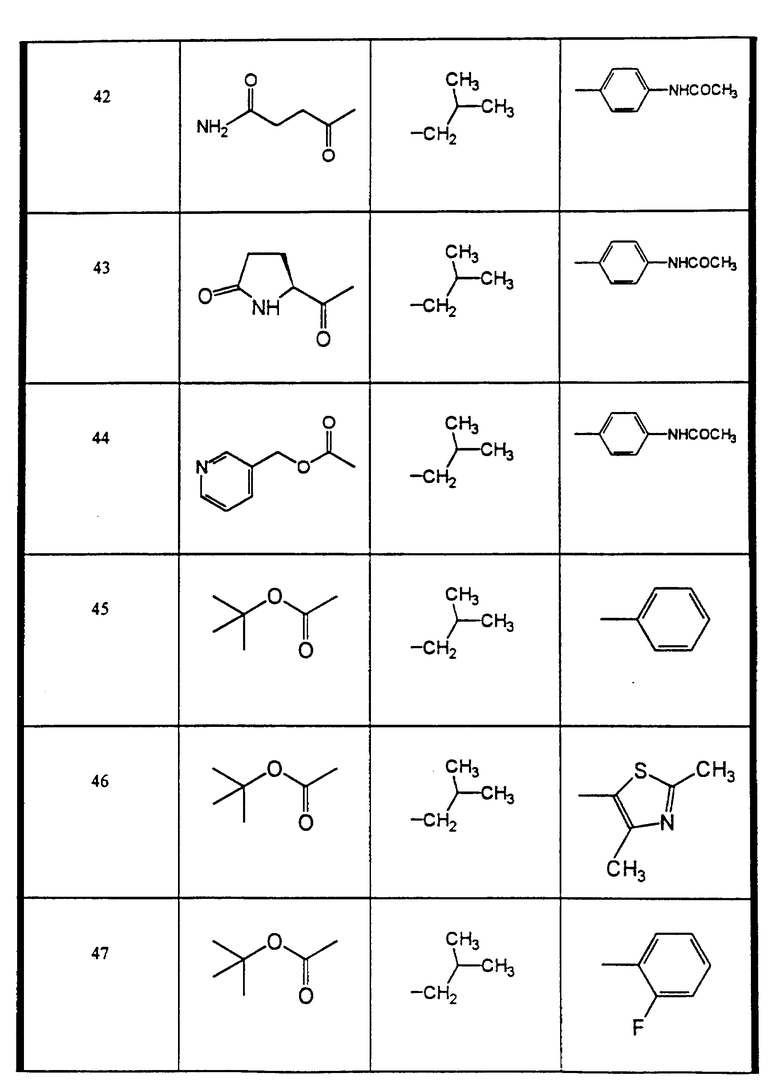
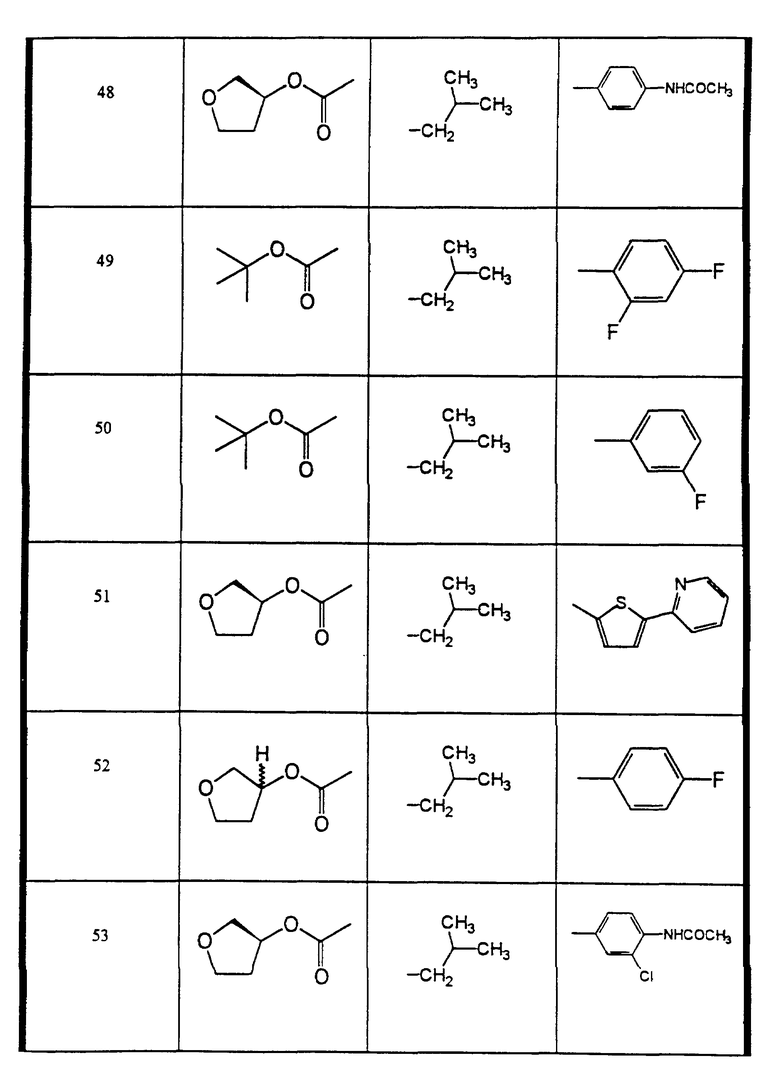
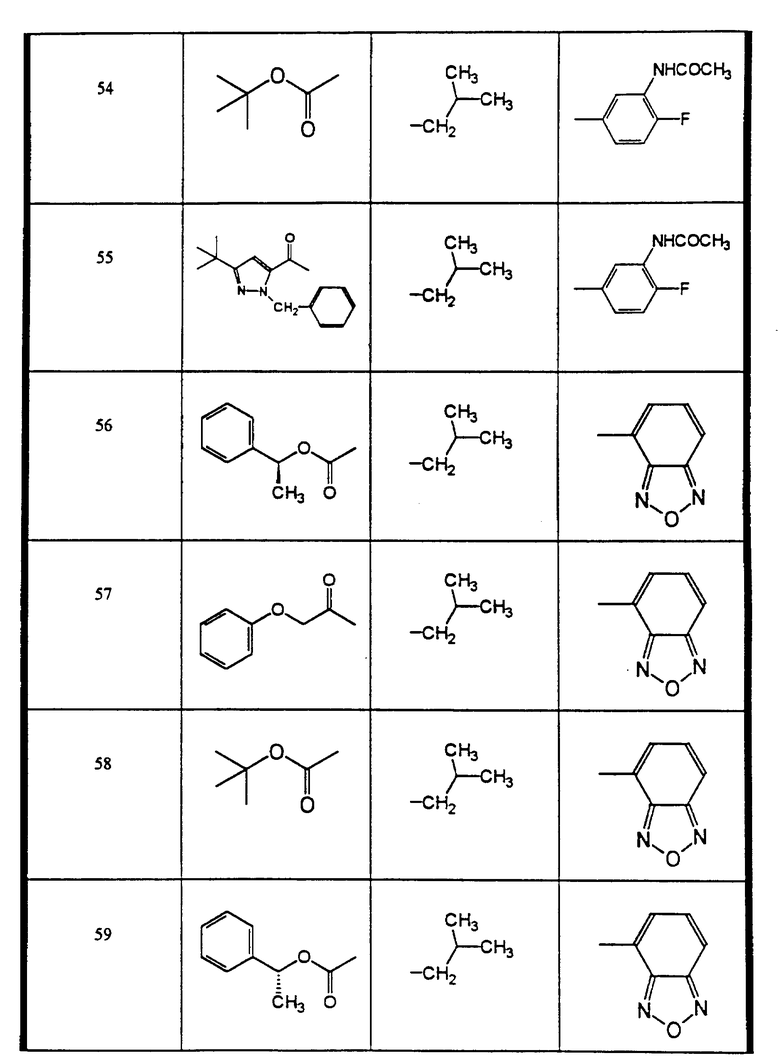
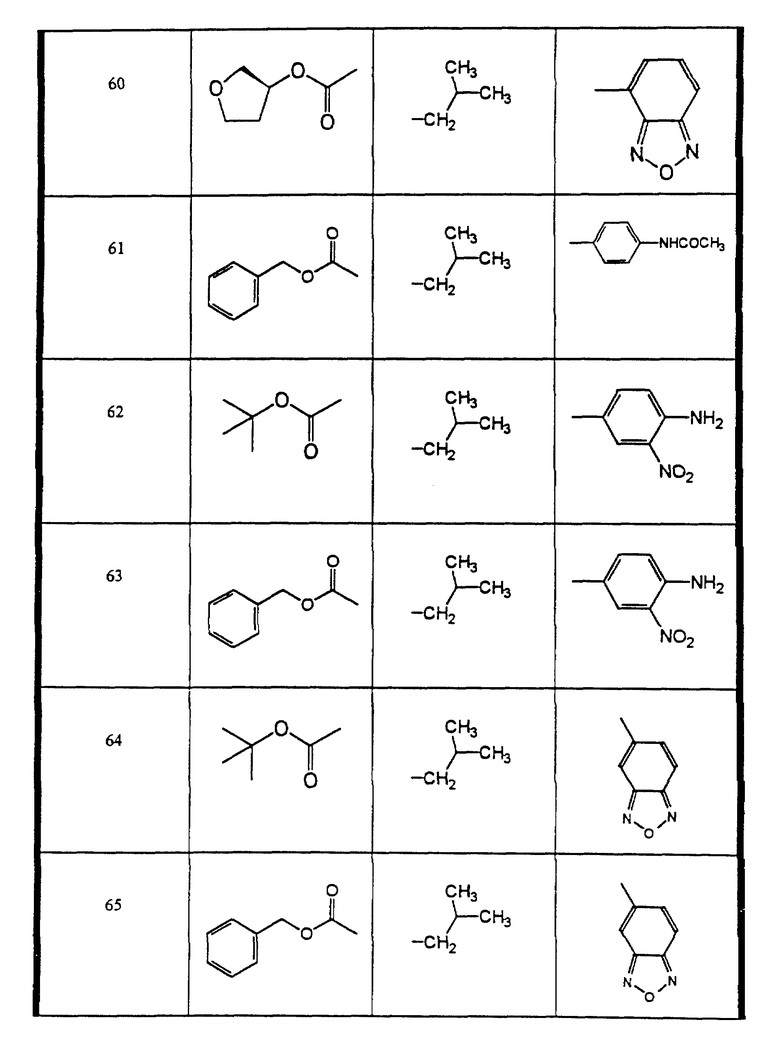
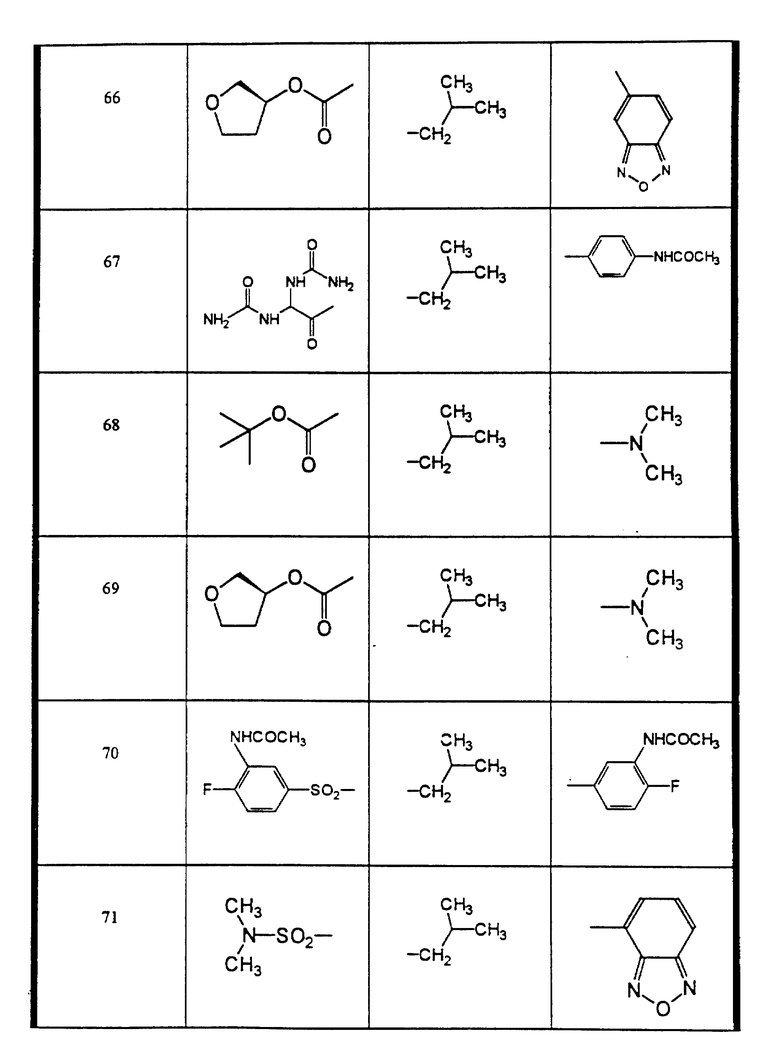
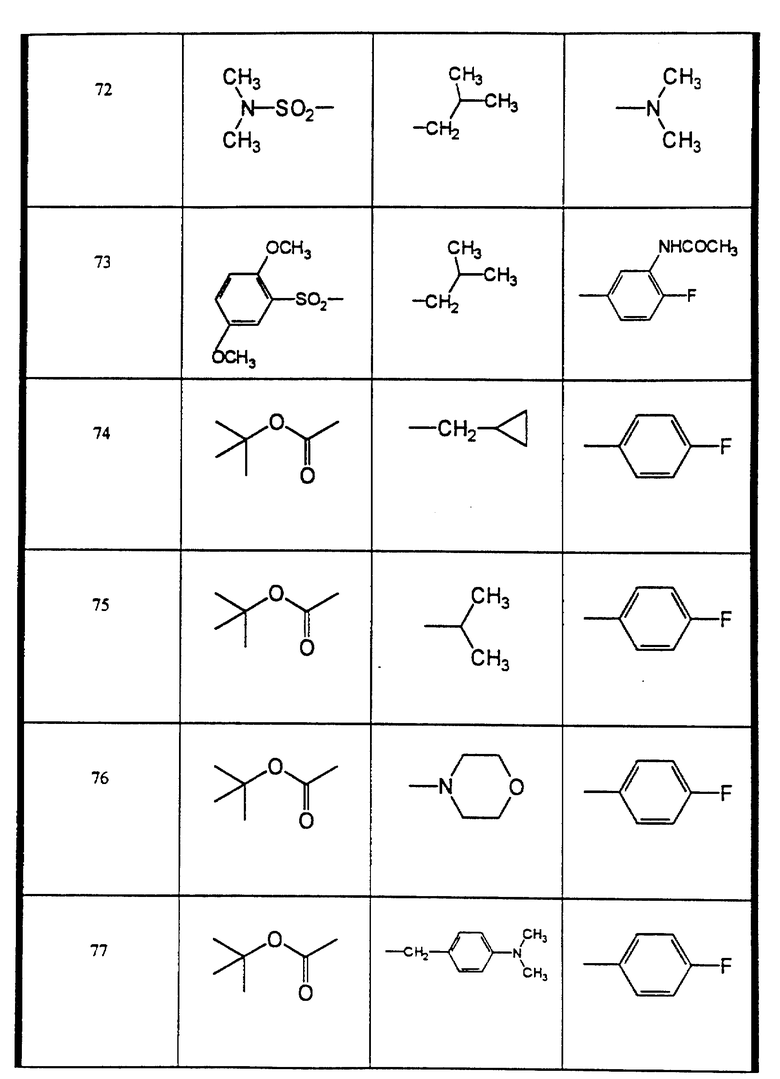
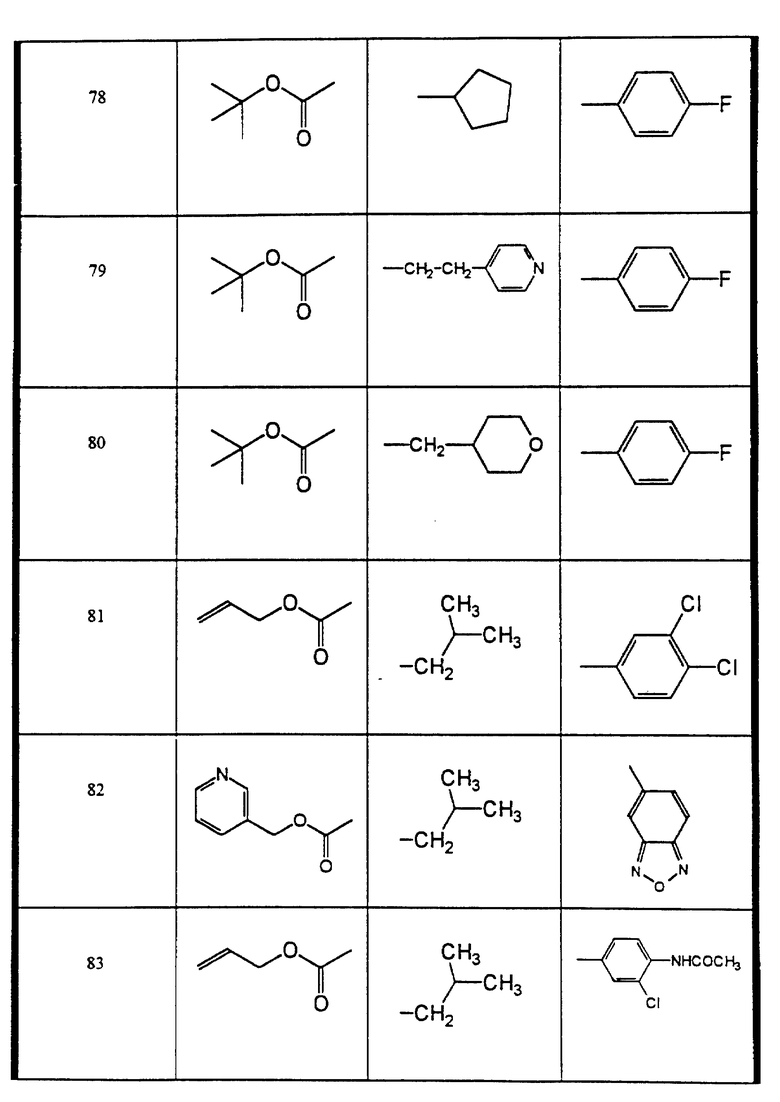
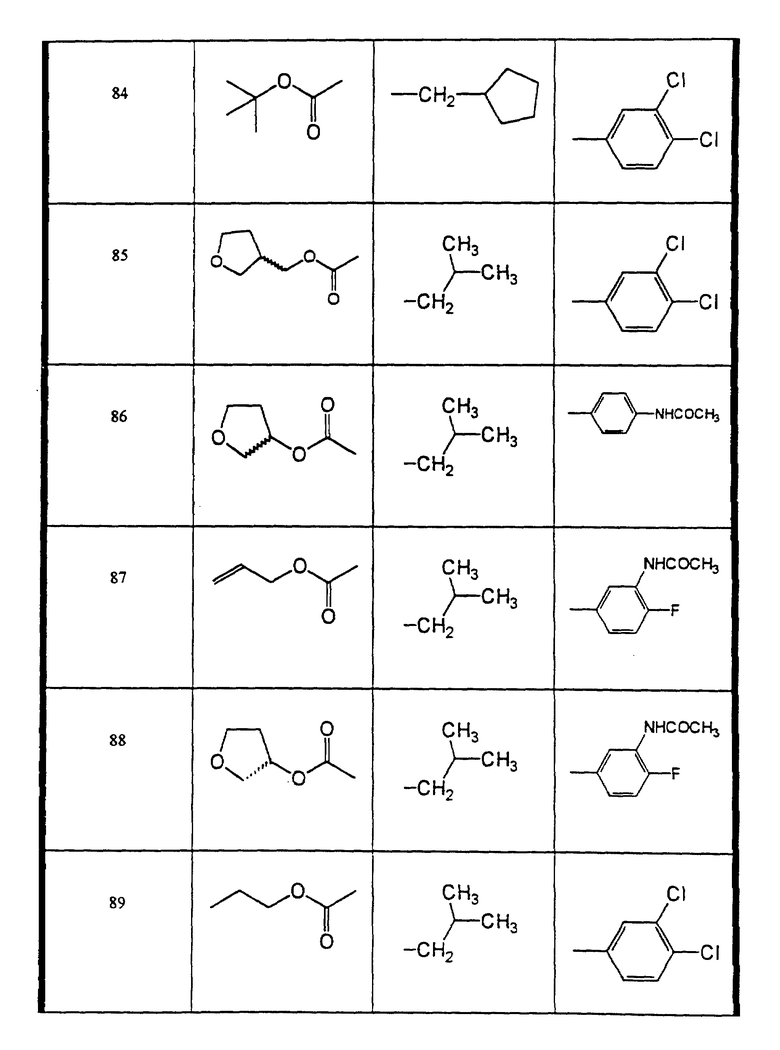
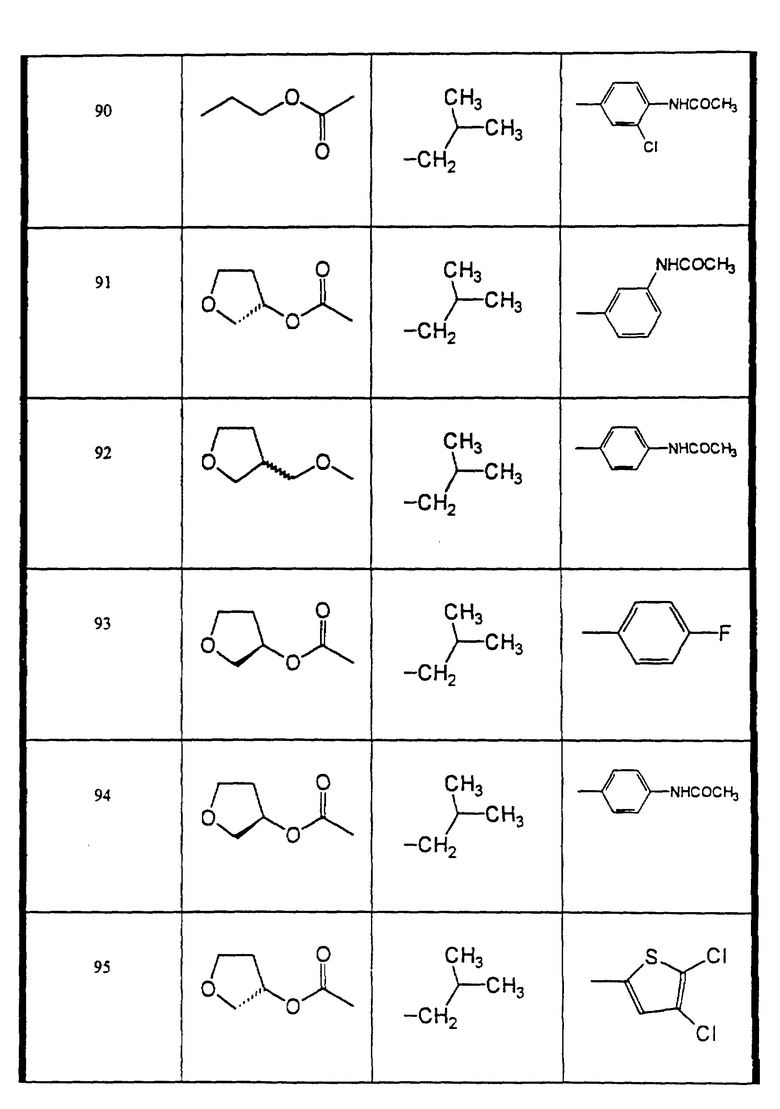
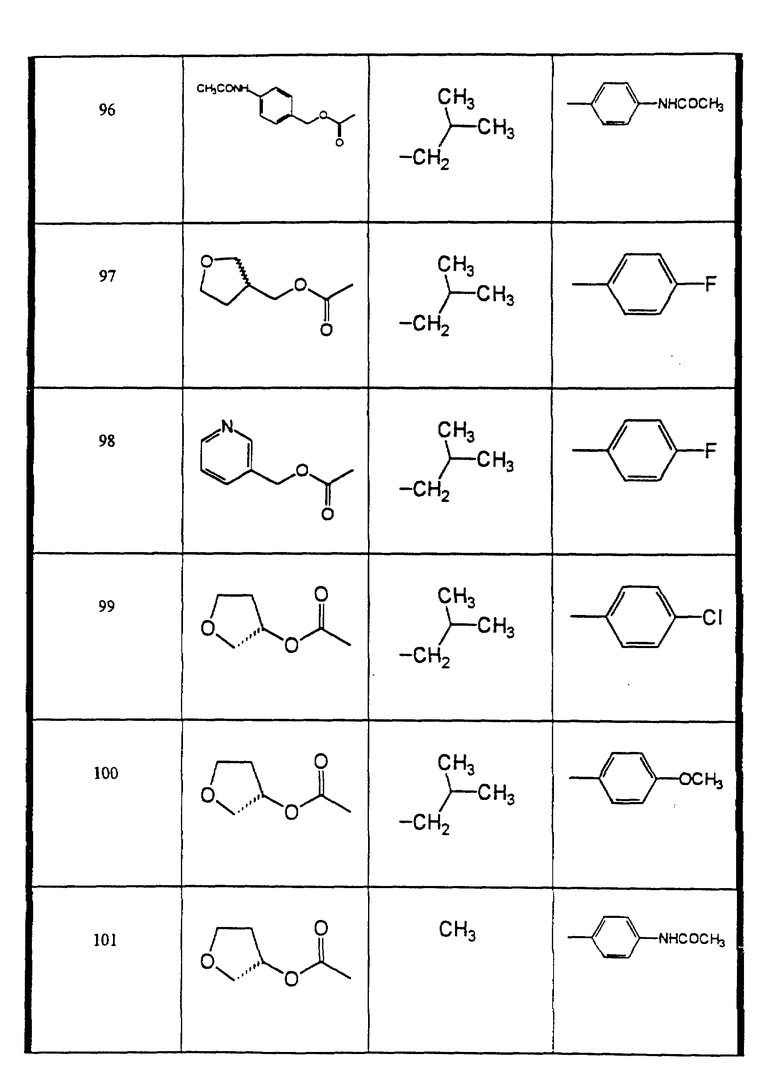
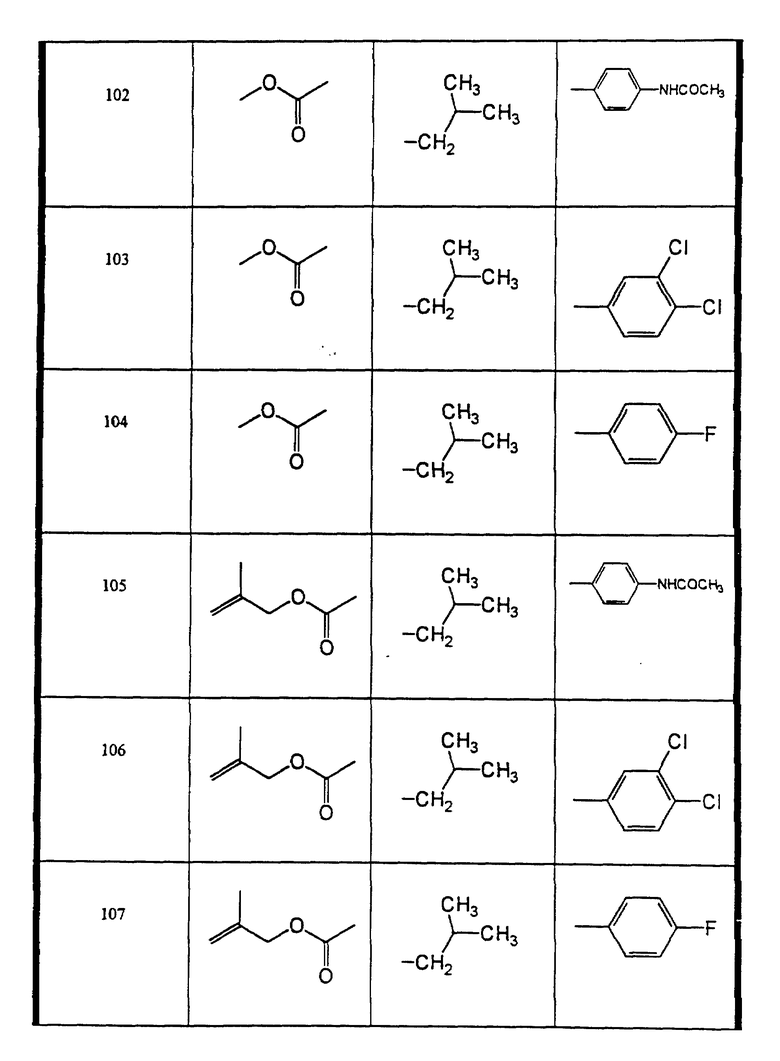
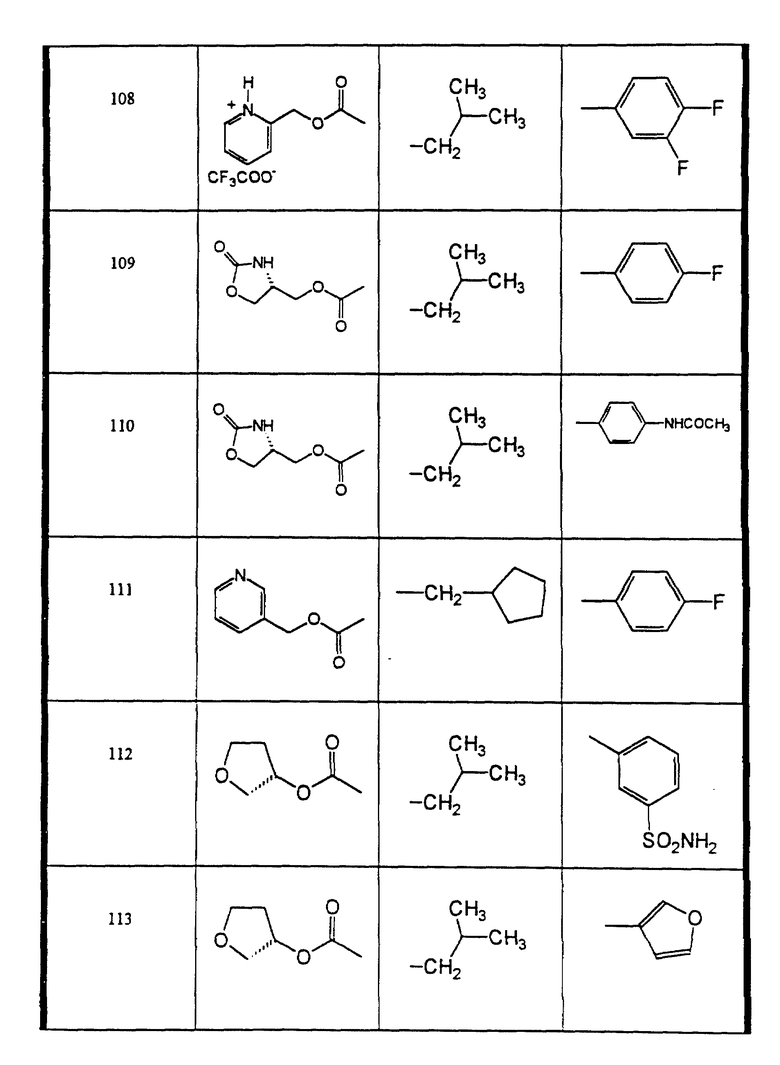
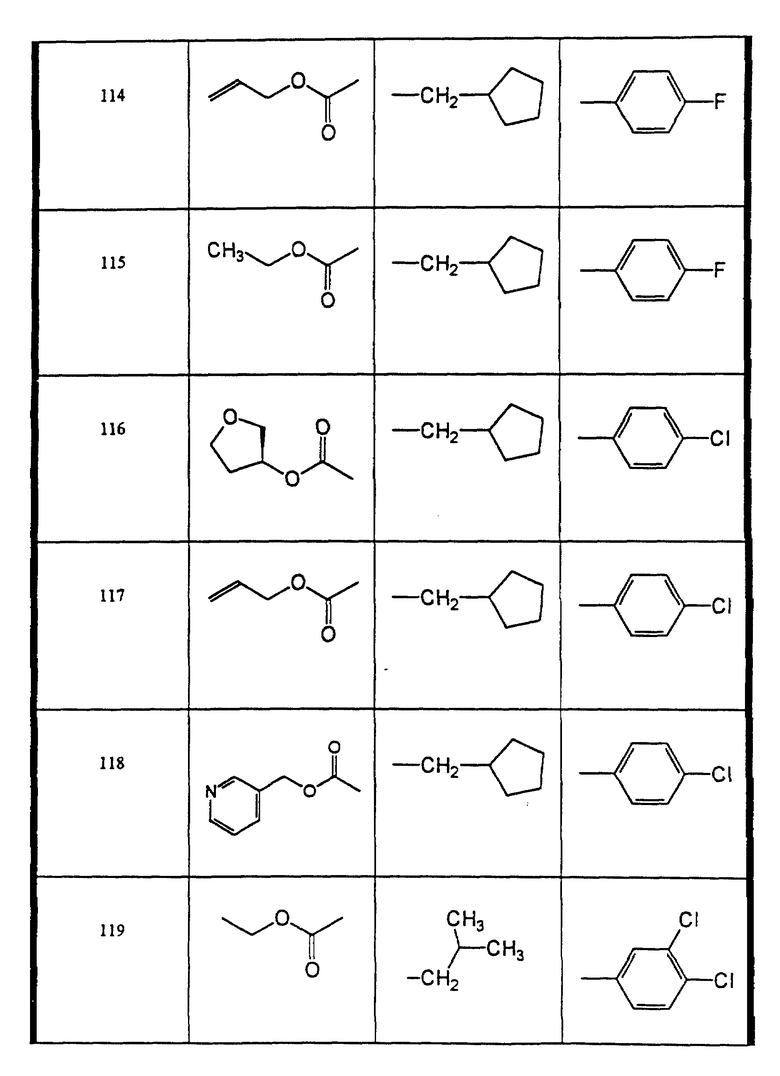
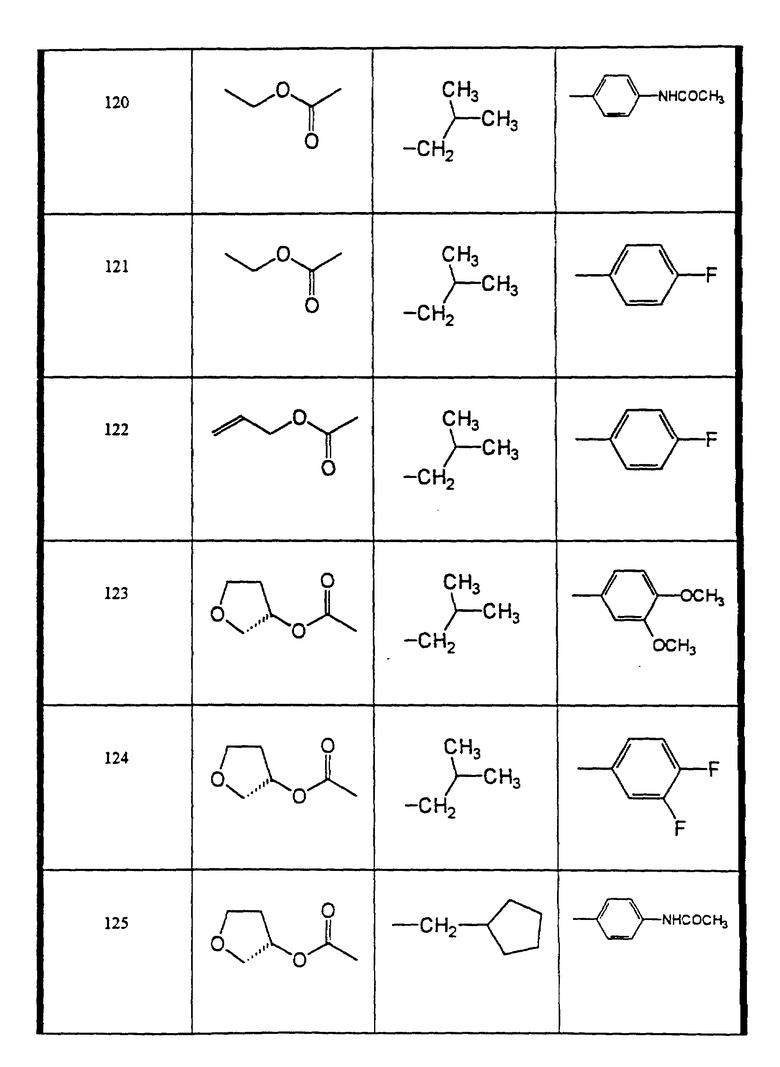
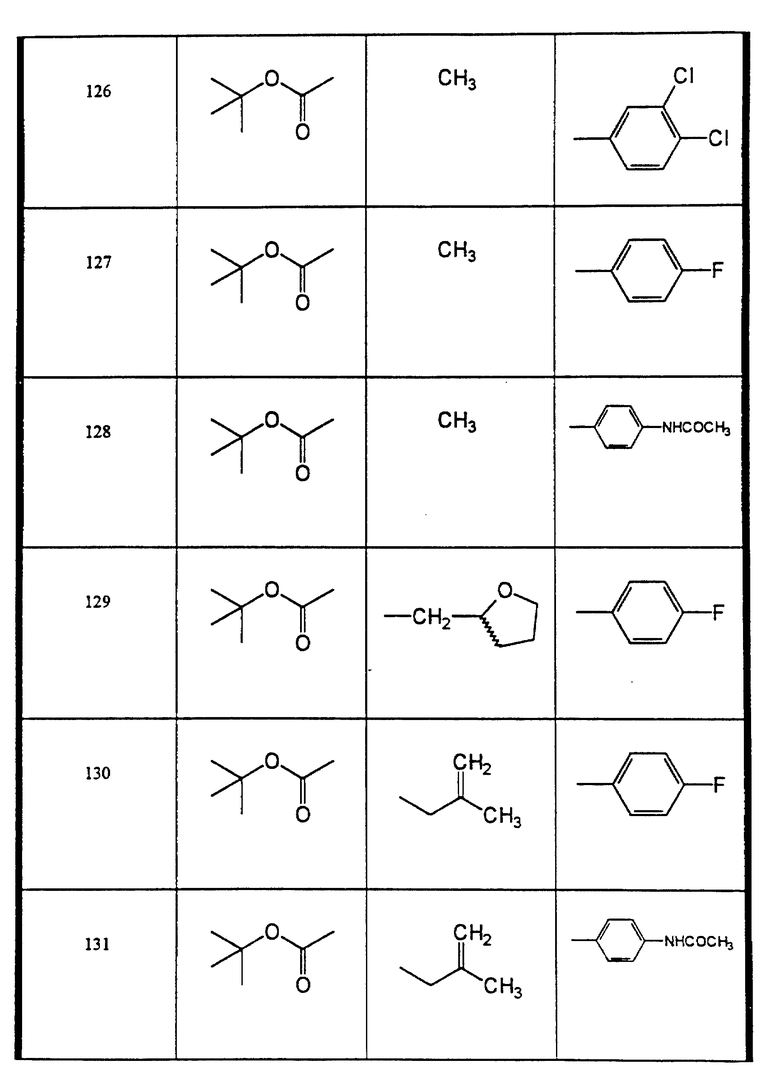
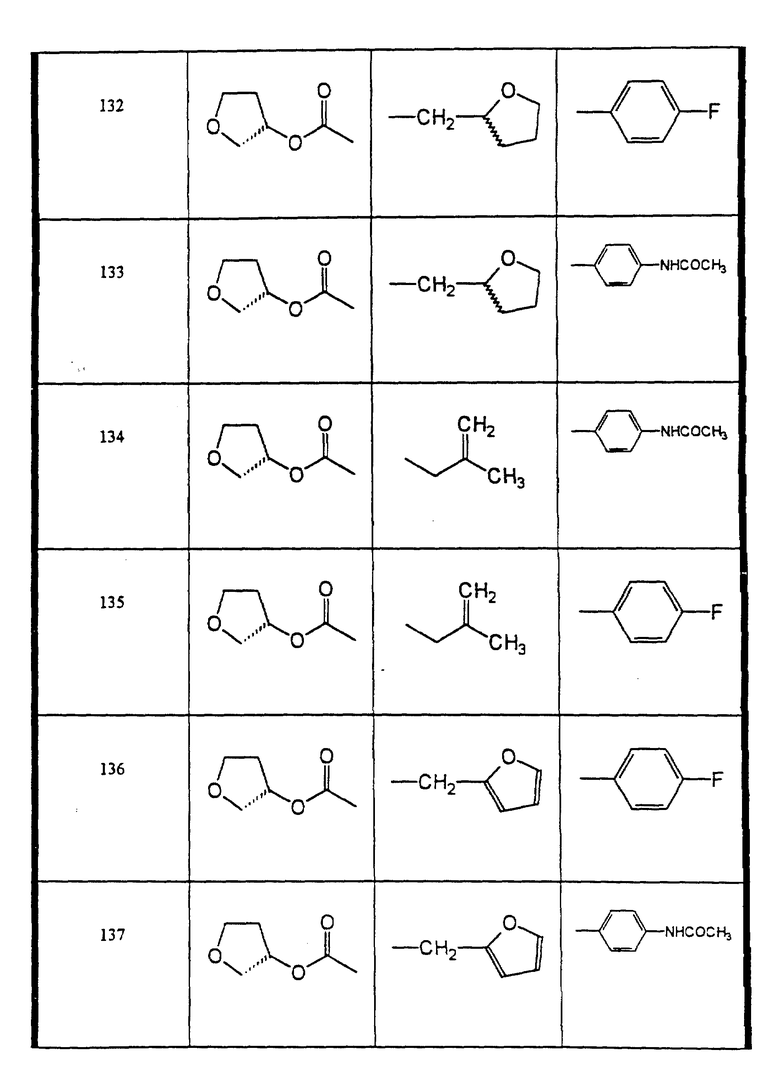
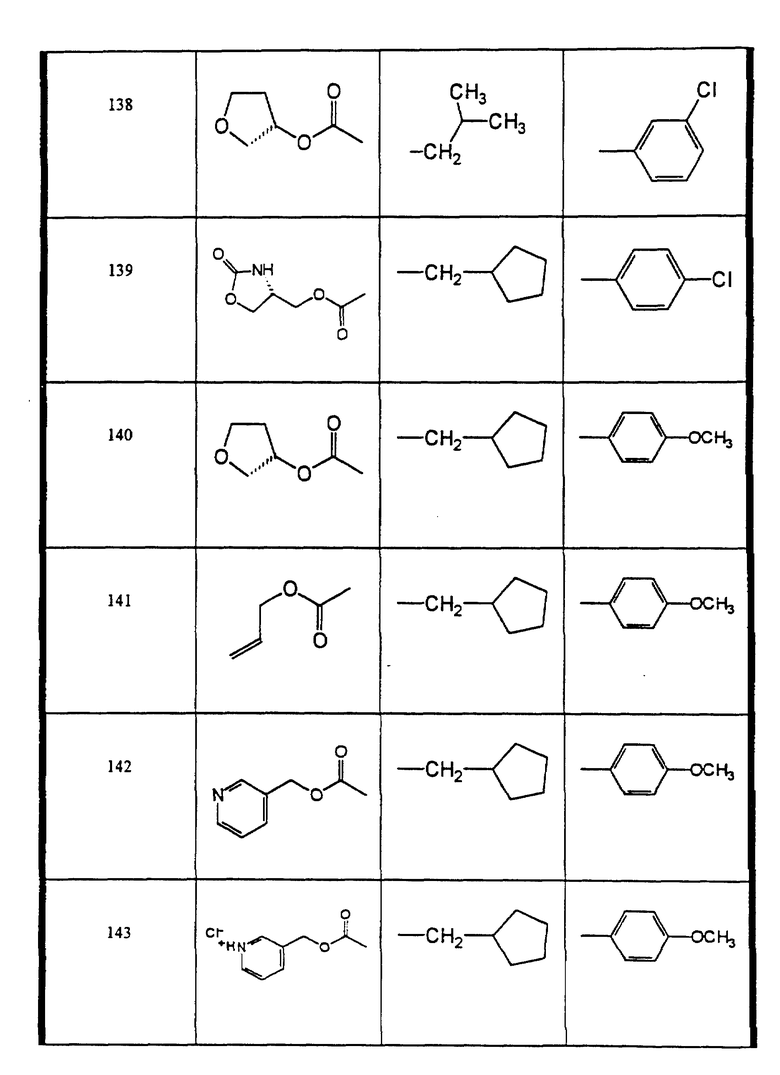
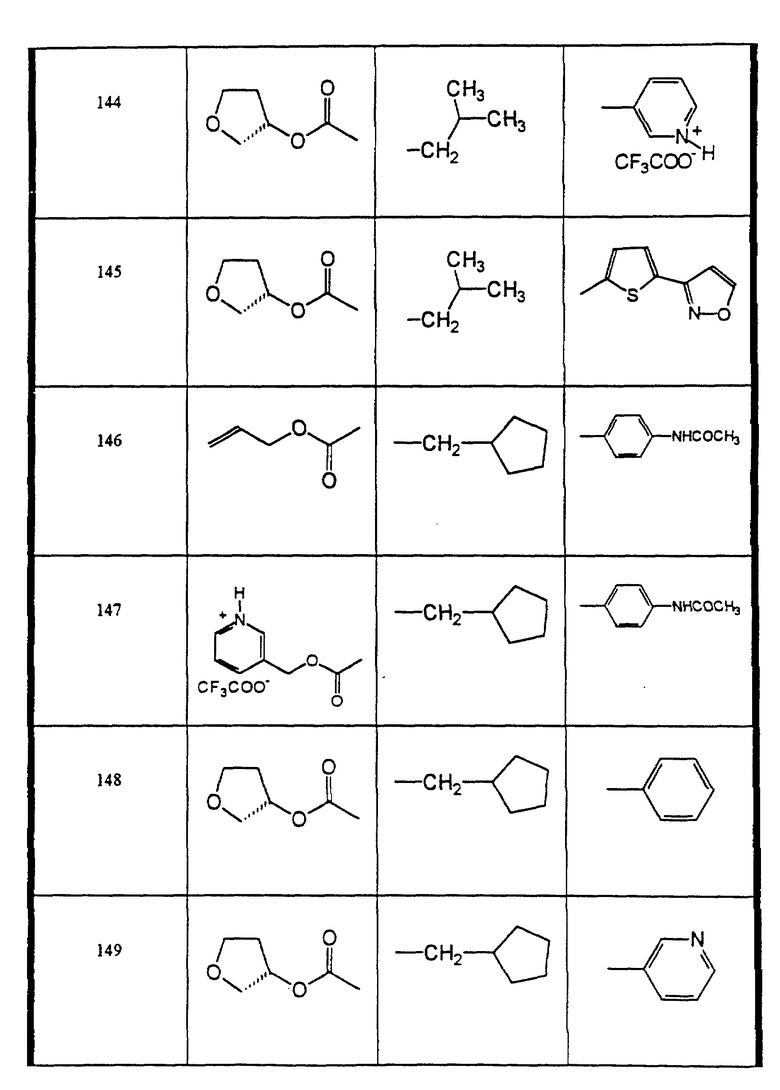
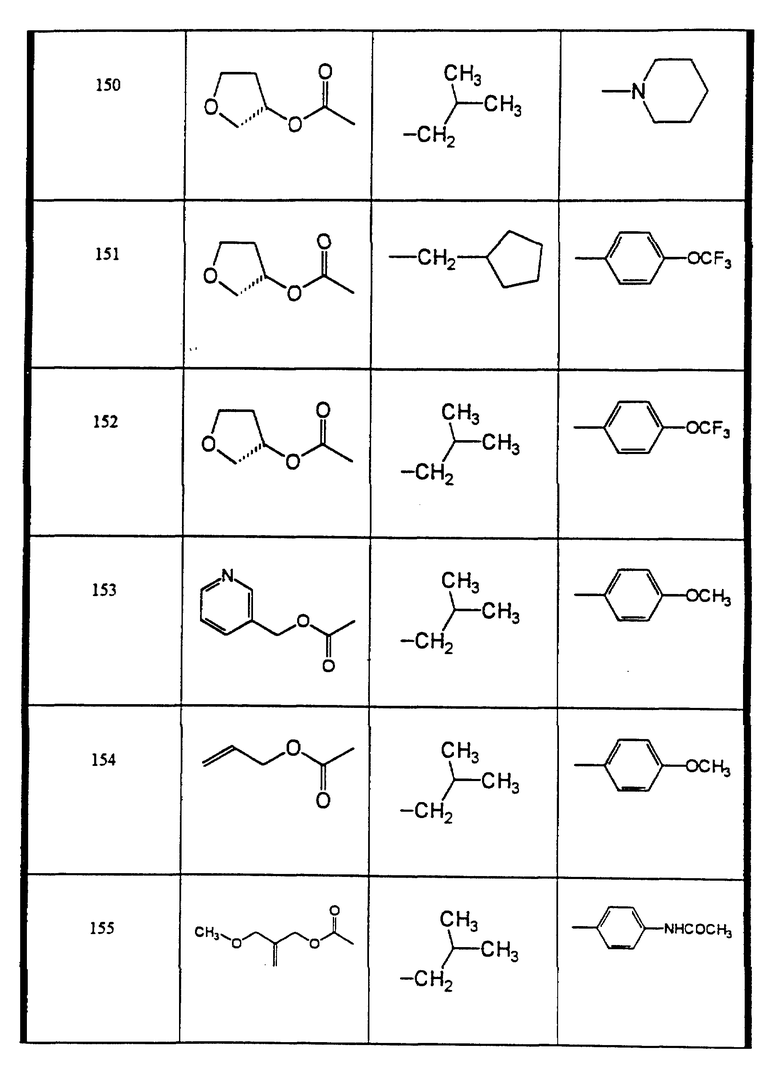
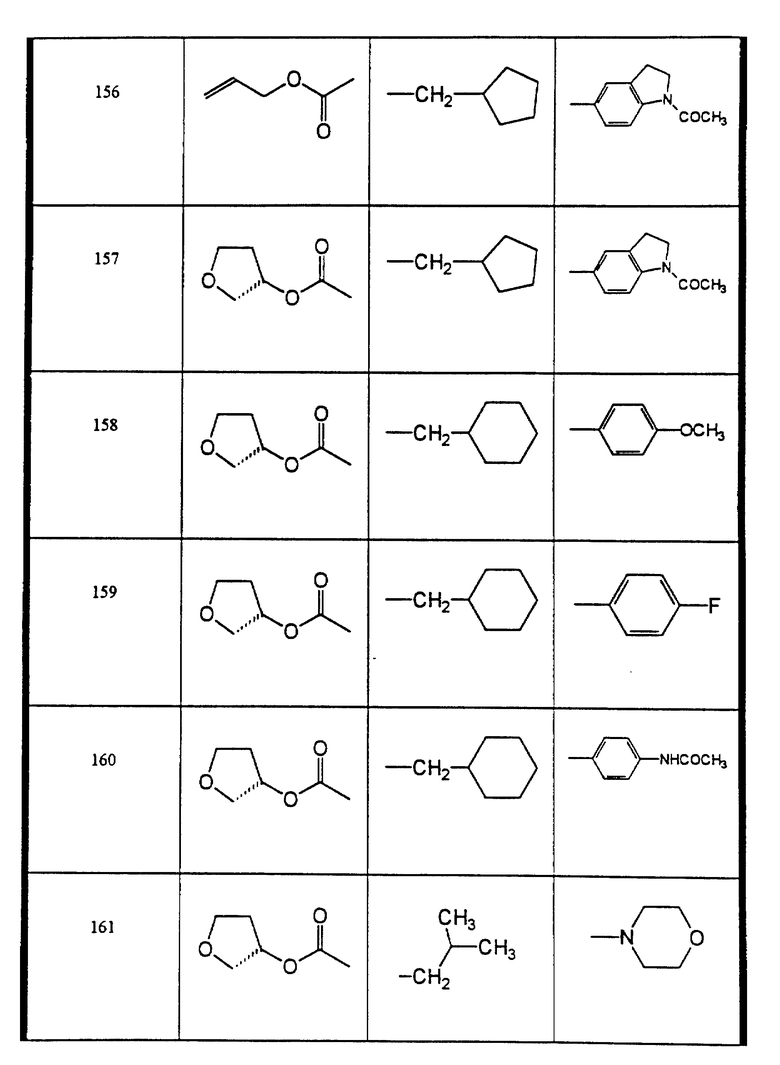
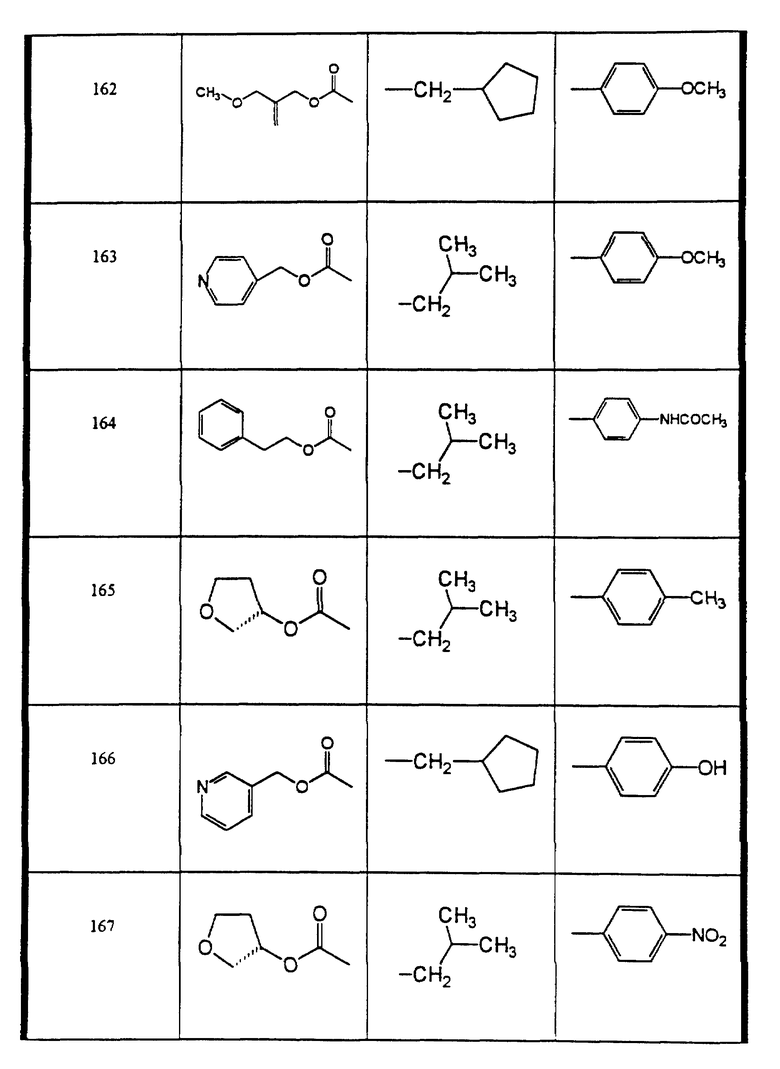
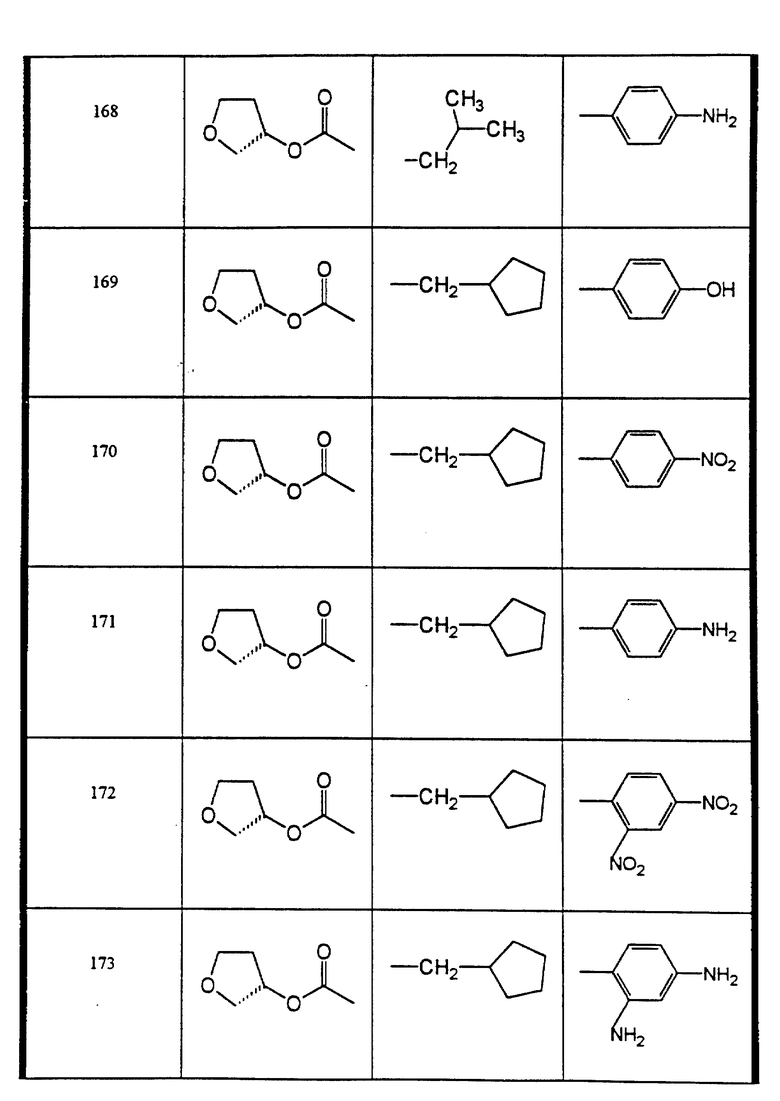
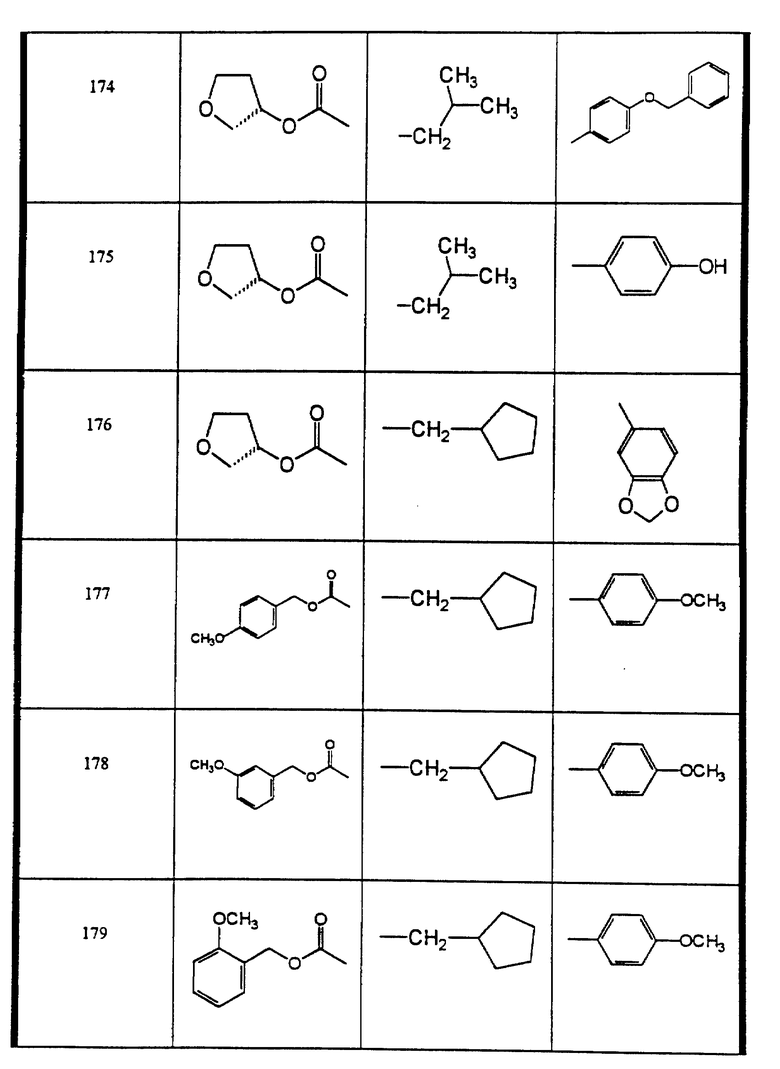
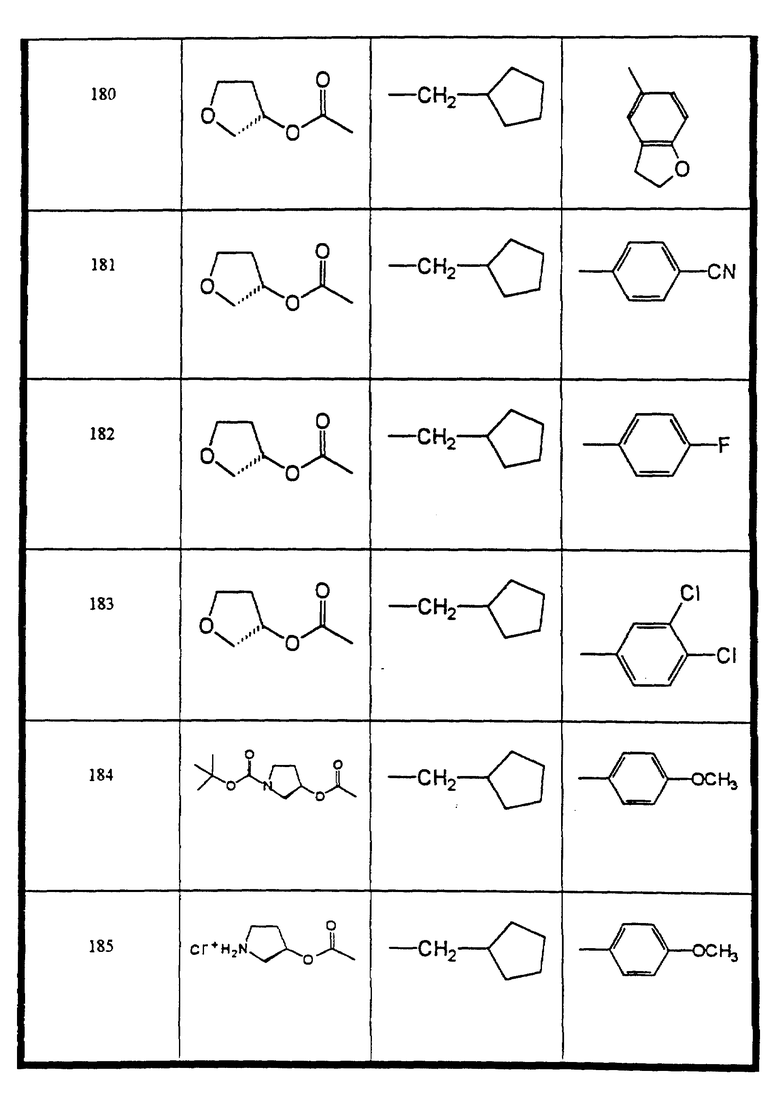
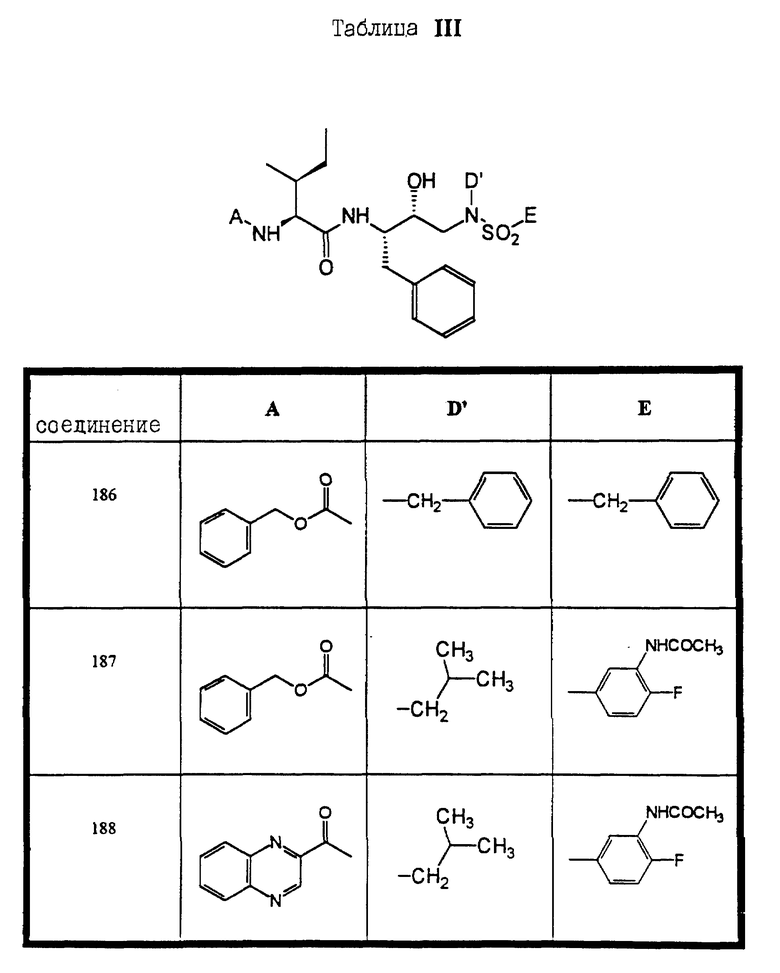
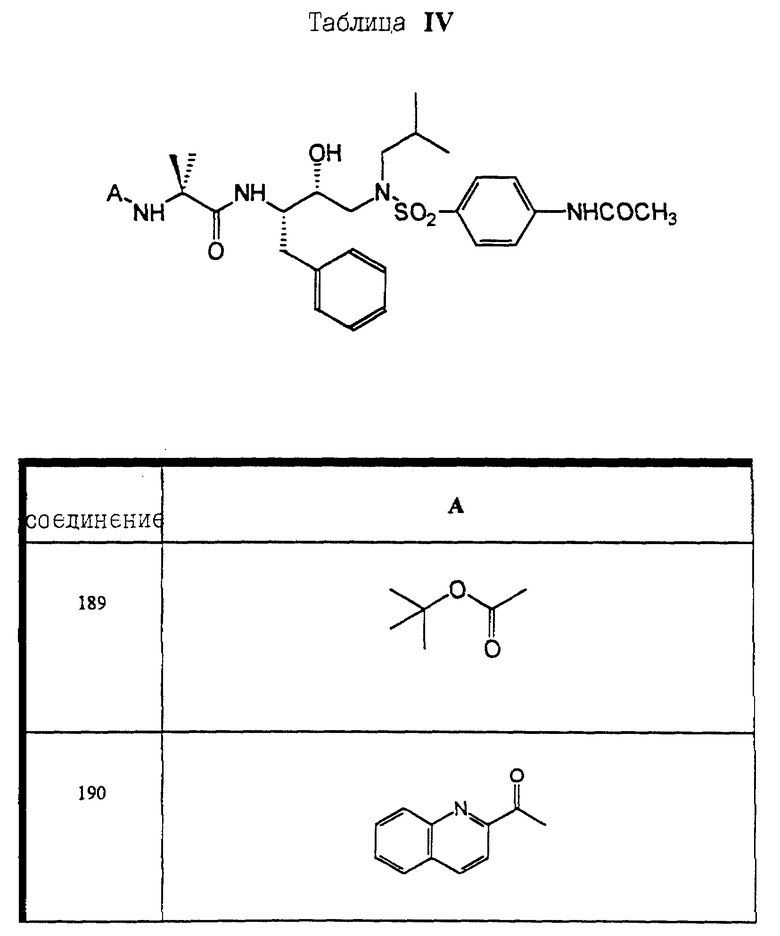
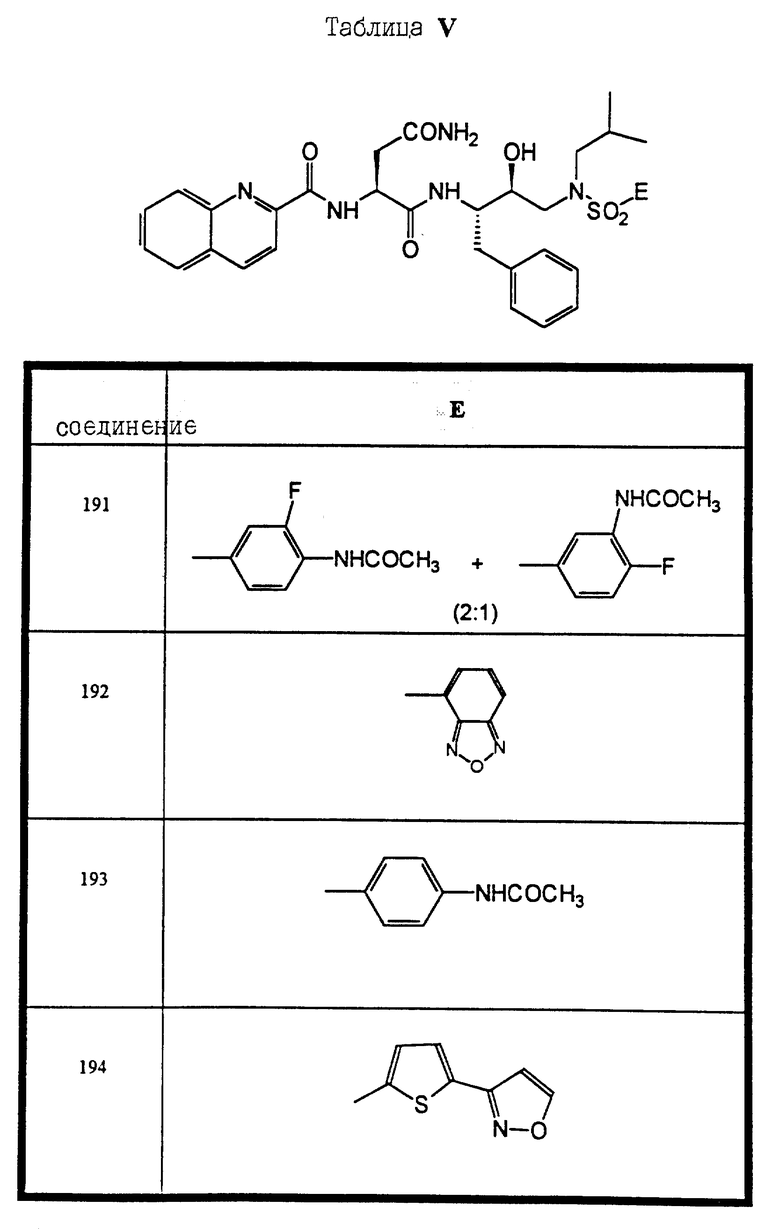
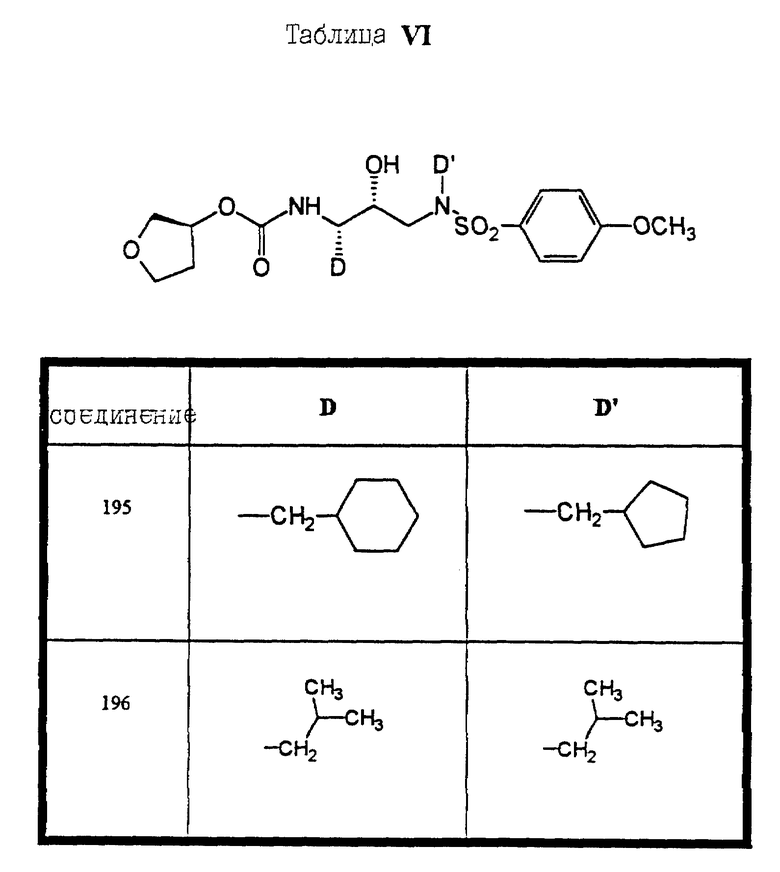




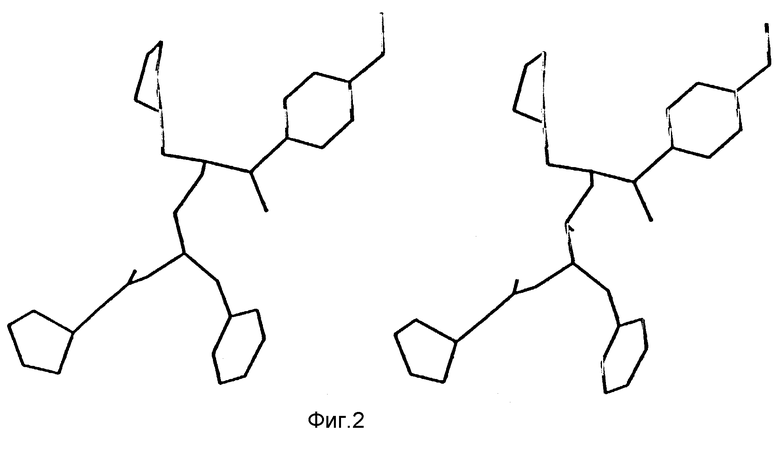
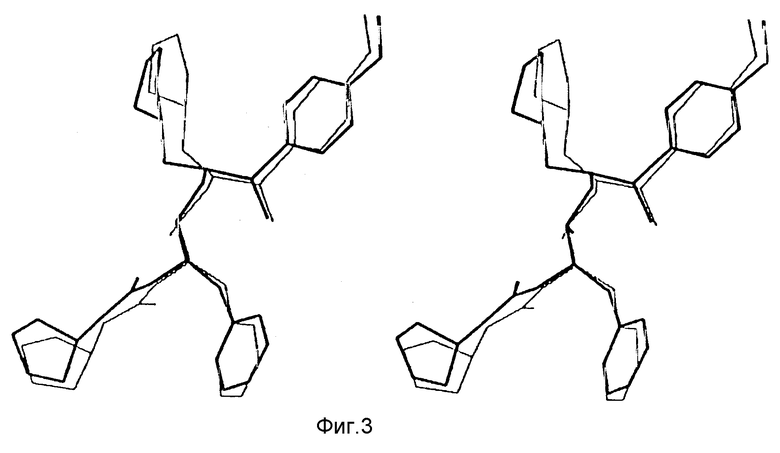
Изобретение относится к новому классу сульфонамидов, которые являются ингибиторами аспартил-протеазы формулы I. Изобретение относится к новому классу H1У аспартил-протеазных ингибиторов, отличающемуся специфическими структурными и физико-химическими характеристиками. Изобретение относится также к фармацевтическим композициям, содержащим эти соединения. Соединения и фармацевтические композиции изобретения особенно подходят для ингибирования Н1У-1 и Н1У-2 протеазной активности, и, соответственно, их можно с выгодой использовать в качестве вирусных агентов против Н1У-1 и Н1У-2 вирусов. Изобретение относится также к способам ингибирования активности Н1У аспартил-протеазы, за счет использования соединений настоящего изобретения, и способам скринирования соединений по их анти-HIУ активности. 5 с. и 21 з.п. ф-лы, 3 ил., 8 табл.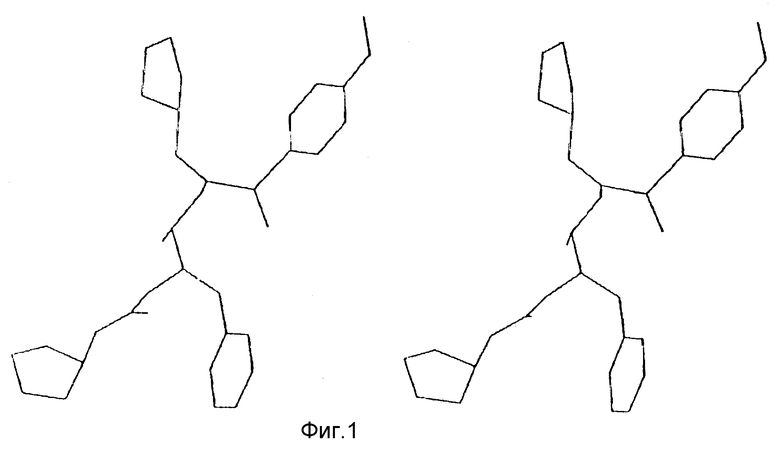
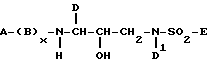
где A выбран из группы, включающей H; Het; -R1-Het; -R1 - C1 - C6-алкил, который может быть необязательно замещен одной или несколькими группами, выбранными из гидрокси, C1 - C4-алкокси, Het, -O-Het, -NR2-CO-N(R2) (R2) и -CO- N(R2) (R2); и -R1-C2 - C6-алкенил, который может быть необязательно замещен одной или несколькими группами, выбранными из гидрокси и C1 - C4 алкокси;
каждый R1, независимо, выбран из группы, состоящей из -C(O)-, -S(O)2-, -O-C(O)-, -NR2-S(O)2-, -NR2-C(O)- и -NR2 - C(O)-C(O)-;
каждый Het независимо, выбран из группы, включающей C6 - C10-арил; 5 - 7 членный насыщенный или ненасыщенный гетероцикл, содержащий один или несколько гетероатомов, выбранных из N, N(R2), O и S, где указанный гетероцикл может быть необязательно конденсирован с бензольным кольцом и где любой член из указанных Het может быть необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей оксо, -OR2, -R2, -N(R2) (R2), -CN, -CO2R2, -S(O)2-N(R2) (R2), -N(R2)-C(O)-R2, -C(O)-R2, -OCF3, -S(O)nAr,
метилендиокси, галоген, -CF3 и -NO2,
каждый R2, независимо, выбран из группы, состоящей из H и C1 - C3 алкила, необязательно замещенного Ar;
B, если присутствует, представляет -N(R2)-C(R3)(R3)-, C(O)-; x = 0 или 1;
каждый R3, независимо, выбран из группы, включающей H, Het, C1 - C6 алкил, C2 - C6-алкенил и C3 - C6-циклоалкил, где любой член из указанных R3, за исключением H, может быть, необязательно, замещен одним или несколькими -C(O)-NH-R2;
D и D', независимо, выбраны из группы, включающей Ar; C1 - C4 алкил, который может быть необязательно замещен одной или несколькими группами, выбранными из C3 - C6-циклоалкила, -OR2 и Ar; C2 - C4-алкенил и C3 - C6-циклоалкил;
каждый Ar, независимо, выбран из группы, включающей фенил, 3 - 6 членное карбоциклическое кольцо и 5 - 6 членное гетероциклическое кольцо, содержащие один или несколько гетероатомов, выбранных из O, N, S и N(R2), где указанное карбоциклическое или гетероциклическое кольцо может быть насыщенным или ненасыщенным и необязательно замещено одной или несколькими группами, выбранными из группы, содержащей -OR2, -R2; и -N(R2) (R2);
E выбран из группы, включающей Het; Het - Het; -NR2R3; C1 - C6 алкил, который может быть, необязательно, замещен одной или несколькими группами, выбранными из R4 и Het;
и каждый R4, независимо, выбран из группы, включающей -OR2, -C(O)-NHR2, -S(O)2-NHR2, галоген, -NR2-C(O)-R2 и -CN.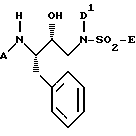
где A, D' и E имеют указанные в п.1 значения.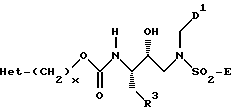
где x, Het, R3, D' и E имеют указанные в п.1 значения.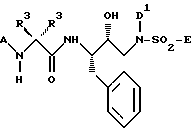
где A, R3, D' и E имеют указанные в п.1 значения.
каждый R1, независимо, выбран из группы, включающей -C(O)-, -S-(O)2-, -O-C(O)- и -NR2-S(O)2-;
каждый Het, независимо, выбран из группы, включающей C6 - C10 арил и 5 - 7 членный насыщенный или ненасыщенный гетероцикл, содержащий один или несколько гетероатомов, выбранных из N, O и S, который может быть необязательно конденсирован с бензольным кольцом, где любой член из указанных Het может быть необязательно замещен одним или несколькими заместителями, выбранными из группы, включающей оксо, -OR2, -R2, -N(R2)2, -CN, -CO2R2 и -S(O)2-N(R2)2; каждый R2, независимо, выбран из группы, состоящей из H и C1 - C3 алкила;
B, если присутствует, представляет -NH-CH(R3)-C(O)-;
x = 0 или 1;
R3 выбран из группы, включающей Het, C1 - C6 алкил, C2 - C6 алкенил, C3 - C6 циклоалкил, где любой член указанного R3 может быть необязательно замещен одним или несколькими -C(O)-NH-R2;
D и D', независимо, выбраны из группы, включающей Ar; C1 - C4 алкил, который может быть необязательно замещен C3 - C6 циклоалкилом или Ar; C2 - C4 алкенил и C3 - C6 циклоалкил;
Ar выбран из группы, включающей фенил; 3 - 6 членное карбоциклическое кольцо и 5 - 6 членное гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из O, N и S, где указанное карбоциклическое или гетероциклическое кольцо может быть насыщенным или ненасыщенным и, необязательно, замещено одной или несколькими группами, выбранными из OR2, -R2 и -N(R2)2;
E выбран из группы, включающей Het, -NR2R5, C1 - C6 алкил, который может быть необязательно замещен одним или несколькими R4 или Het;
каждый R4, независимо, выбран из группы, включающей -OR2, -C(O)-NHR2, -S(O)2-NHR2, галоген и CN;
каждый R5, независимо, выбран из группы, состоящей из H и R3.
D' выбран из группы, включающей C1 - C3 алкил, который может быть, необязательно, замещен одной или несколькими группами, выбранными из C3 - C6 циклоалкила, -OR2 и Ar; и C3 алкенил.
(S)-N-1-(3-((3-ацетиламино-4-фторбензолсульфонил)-бензиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил-2-((хинолин-2-карбонил)-амино) сукцинамид и (S)-N-1-(3-((4-ацетиламино-3-фторбензолсульфонил)-бензиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)-амино)-сукцинамид (соединение 2);
(S)-N-1-(3-((5-ацетиламино-3-метилтиазол-2-сульфонил)бензиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)-амино)сукцинамид (соединение 5);
(S)-N-1-(1-бензил-3-(бензил-(5-изоксазол-3-илтиофен-2-сульфонил)-амино)-(1S, 2 син)-2-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 6);
(S)-N-1-(3-((бензо(1,2,5)оксадиазол-4-сульфонил)бензиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 9);
N-1-(1-(S)-бензил-3-(бензил-(3-сульфамоилбензолсульфонил)-амино-2-(син)-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 10);
(S)-N-1-(1-(S)-бензил-2-(син-гидроксил-3-(изобутил-(5-пиридин-2-илтиофен-2-сульфонил)амино)пропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 12);
(S)-N-1-(3-((4-бензосульфонилтиофен-2-сульфонил)изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 13);
(S)-N-1-(1-(S)-бензил-3-((4-фторбензолсульфонил)-изобутиламино)-2-(син)-гидроксипропил)-2-((хинолин-2-карбонил)амино)-сукцинамид (соединение 14);
(S)-N-1-(3-((4-ацетиламино-3-фторбензолсульфонил)-изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)-амино)сукцинамид (соединение 15);
(S)-N-1-(3-((3-ацетиламино-4-фторбензолсульфонил)-изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил-2-((хинолин-2-карбонил)-амино)сукцинамид (соединение 16);
(S)-N-1-(1-(S)-бензил-3-((4-ацетиламинобензолсульфонил)изобутиламино)-2-(син)-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 17);
(S)-N-1-(3-((5-ацетиламино-3-метилтиазол-2-сульфонил)изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил-2-((хинолин-2-карбонил)-амино)сукцинамид (соединение 18;
(S)-N-1-(3-((3-ацетиламинобензолсульфонил)изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 19);
(S)-N-1-(3-((бензо(1,2,5)оксадиазол-4-сульфонил)-изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 20);
N-1-((1S, 2 син)-1-бензил-2-гидрокси-3-(1-изобутил-3,3-диметилсульфамид)пропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 21);
N-1-(3-((4-ацетиламинобензолсульфонил)изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-(пиридин-2-илметоксикарбониламино)-3-S-метилбутирамид (соединение 22);
N-1-(3-((4-ацетиламинобензолсульфонил)изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-(пиридин-4-илметоксикарбониламино)-3-S-метилбутирамид (соединение 23);
N-1-(3-((4-фторбензолсульфонил)изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-(пиридин-2-илметоксикарбониламино)-3-S-метилбутирамид (соединение 26);
4-фтор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 35);
3,4-дихло-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 37);
N-(4-(((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-илметоксикарбониламино)бутил)изобутилсульфамоил)фенил)-ацетамид (соединение 44);
(1,1-диметилэтоксикарбониламино)-(2 син, 3S)-2-гидрокси-4-фенилбутил)изобутиламид 2,4-диметилтиазол-5-сульфокислоты (соединение 46);
N-(4-(((2 син, 3S)-2-гидрокси-4-фенил-3-(S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутилсульфамоил)фенил)ацетамид (соединение 48);
4-фтор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((R)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-N-изобутилбензолсульфонамид (соединение 52);
((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-илметоксикарбониламино)бутил)изобутиламид бензо(1,2,5)оксадиазол-5-сульфоновой кислоты (соединение 82);
N-(4-(((2 син, 3S)-2-гидрокси-4-фенил-3-((R)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутилсульфамоилфенил)ацетамид и N-(4-(((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутилсульфамоил)фенил)ацетамид (соединение 86);
N-(2-фтор-5-(((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутилсульфамоил)фенил)ацетамид (соединение 88);
N-(3-(((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутилсульфамоил)-фенилацетамид (соединение 91);
4-фтор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((R)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединение 93);
N-(4-(((син)-2-гидрокси-(S)-4-фенил-3-((тетрагидрофуран)-(R)-3-ил)оксикарбониламино)бутил)изобутилсульфамоил)фенил)ацетамид (соединение 94);
4-фтор-N-(2 син, 3S)-2-гидрокси-4-фенил-3-((тетрагидрофуран-(R)-3-илметоксикарбониламино)бутил)-N-изобутилбензолсульфонамид и 4-фтор-N-(2 син, 3S)-2-гидрокси-4-фенил-3-((тетрагидрофуран)-(S)-3-илметоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединения 97);
4-фтор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-ил-метоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединение 98);
4-хлор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутилбензолсульфонамид (соединение 99);
N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутил-4-метоксибензолсульфонамид (соединение 100);
4-фтор-N-(2-(син)-гидрокси-3-((2-оксазолидон-(S)-4-ил)метоксикарбониламино)-4-(S)-фенилбутил)-N-изобутилбензолсульфонамид (соединение 109);
1-амид-3-((2 син, 3S)-2-гидрокси-4-фенил-3-(3-(S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутиламид бензол-1,3-дисульфоновой кислоты (соединение 112);
(2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-изобутиламид фуран-3-сульфоновой кислоты (соединение 113);
N-((3-аллилоксикарбониламино)-(2 син, 3S)-2-гидрокси-4-фенилбутил)-N-циклопентилметил-4-фторбензолсульфонамид (соединение 114);
N-циклопентилметил-N-((3-этоксикарбониламино)-(2 син, 3S)-2-гидрокси-4-фенилбутил)-4-фторбензолсульфонамид (соединение 115);
4-хлор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)бензолсульфонамид (соединение 116);
4-хлор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-илметоксикарбонил)бутил)бензолсульфонамид (соединение 118);
N-(4-(циклопентилметил)-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-сульфамоил)-фенил)ацетамид (соединение 125);
3-хлор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединение 138);
4-хлор-N-циклопентилметил-N-(2-(син)-гидрокси-3-((2-оксазолидон-4-(S)-илметил)оксикарбониламино)-4-фенилбутил)-бензолсульфонамид (соединение 139);
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)тетрагидрофуран-3-илоксикарбониламино)бутил)-4-метоксибензолсульфонамид (соединение 140);
N-((3-аллилоксикарбониламино)-(2 син, 3S)-2-гидрокси-4-фенилбутил)-N-циклопентилметил-4-метоксибензолсульфонамид (соединение 141);
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-(3-пиридин-3-илметоксикарбониламино)бутил)-4-метоксибензолсульфонамид (соединение 142);
трифторуксусная соль ((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутиламида пиридин-3-сульфоновой кислоты (соединение 144);
((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутиламид 5-изоксазол-3-илтиофен-2-сульфоновой кислоты (соединение 145);
N-(4-((3-(аллилоксикарбониламино)-(2 син, 3S)-2-гидрокси-4-фенилбутил)-циклопентилметилсульфамоил)фенил)ацетамид (соединение 146);
N-(4-(циклопентилметил)-((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-илметоксикарбониламино)бутил)сульфамоил)-фенил)ацетамид (соединение 147);
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)тетрагидрофуран-3-илоксикарбониламино)бутил)бензолсульфонамид (соединение 148);
циклопентилметил-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)тетрагидрофуран-3-илоксикарбониламино)бутил)амид пиридин-3-сульфоновой кислоты (соединение 149);
((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутиламид пиперидин-1-сульфоновой кислоты (соединение 150);
N-4-((2-(син)-гидрокси-3-((2-метоксиметилаллилоксикарбониламино)-4-(S)-фенилбутил)изобутилсульфамоил)фенил)-ацетамид (соединение 155);
((аллилоксикарбониламино)-(2 син, 3S)-2-гидрокси-4-фенил-бутил)-циклопентилметиламид 1-ацетил-2,3-дигидро-1H-индол-6-сульфоновой кислоты (соединение 156);
циклопентилметил-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)амид 1-ацетил-2,3-дигидро-1H-индол-6-сульфоновой кислоты (соединение 157);
N-циклогексилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)тетрагидрофуран-3-илоксикарбониламино)бутил)-4-метоксибензолсульфонамид (соединение 158);
N-циклогексилметил-4-фтор-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)бензолсульфонамид (соединение 159);
N-4-(Циклогексилметил)-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)сульфамоилфенил)ацетамид (соединение 160);
N-(2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-4-илметилоксикарбониламино)бутил)-N-изобутил-4-метоксибензолсульфонамид (соединение 163);
N-((2 син, 3S)-2-гидрокси-4-фенил-3-((син)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутил-4-метилбензолсульфонамид (соединение 165);
N-циклопентилметил-4-гидрокси-N-((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-илметилоксикарбониламино)бутил)-бензолсульфонамид (соединение 166);
N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутил-4-нитробензолсульфонамид (соединение 167);
4-амино-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединение 168);
N-циклопентилметил-4-гидрокси-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-бензолсульфонамид (соединение 169);
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-4-нитробензолсульфонамид (соединение 170);
4-амино-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)-4-бензолсульфонамид (соединение 171);
2,4-диамино-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-бензолсульфонамид (соединение 173);
4-гидрокси-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединение 175);
4-фтор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)бензолсульфонамид (соединение 182);
3,4-дихлор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-бензолсульфонамид (соединение 183);
бензилоксикарбонил-(L)-изолейцин-N-(5-((3-амино)-(2 син, 3S)-2-гидрокси-4-фенилбутил)изобутилсульфамоил)-2-фторфенил)-ацетамид (соединение 187) и
N-((2 син, 3S)-4-циклогексил-2-гидрокси-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-циклопентилметил-4-метоксибензолсульфонамид (соединение 195).
(S)-N-1-(1-(S)-бензил-2-(син)-гидроксил-3-(изобутил-(5-пиридин-2-илитиофен-2-сульфонил)амино)пропил)-2-((хинолин-2-карбонид)амино)сукцинамид (соединение 12);
(S)-N-1-(1-(S)-бензил-3-((4-фторбензолсульфонил)-изобутиламино)-2-(син)-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 14);
(S)-N-1-(3-((4-ацетиламино-3-фторбензолсульфонил)-изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 15);
(S)-N-1-(3-((бензо(1,2,5)оксадиазол-4-сульфонил)-изобутиламино)-(1S, 2 син)-1-бензил-2-гидроксипропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 20);
N-1-((1S, 2 син)-1-бензил-2-гидрокси-3-(1-изобутил-3,3-диметилсульфамид)пропил)-2-((хинолин-2-карбонил)амино)сукцинамид (соединение 21);
N-(4-(((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)изобутилсульфамоил)фенил)ацетамид (соединение 48);
N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутил-4-метоксибензолсульфонамид (соединение 100);
4-хлор-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)бензолсульфонамид (соединение 116);
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-4-метоксибензолсульфонамид (соединение 140);
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-(3-пиридин-3-илметоксикарбониламино)бутил-4-метокси-бензолсульфонамид (соединение 142);
N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)бензолсульфонамид (соединение 148);
N-циклогексилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-4-метоксибензолсульфонамид (соединение 158);
N-(4-(циклогексилметил)-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-сульфамоилфенил)ацетамид (соединение 160);
N-циклопентилметил-4-гидрокси-N-((2 син, 3S)-2-гидрокси-4-фенил-3-(пиридин-3-илметоксикарбониламино)-бутил)бензолсульфонамид (соединение 166);
4-амино-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединение 169);
4-амино-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)-бутил)бензолсульфонамид (соединение 171);
2,4-диамино-N-циклопентилметил-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-бензолсульфонамид (соединение 173);
4-гидрокси-N-((2 син, 3S)-2-гидрокси-4-фенил-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-изобутилбензолсульфонамид (соединение 175) и
N-((2 син, 3S)-4-циклогексил-2-гидрокси-3-((S)-тетрагидрофуран-3-илоксикарбониламино)бутил)-N-циклопентилметил-4-метоксибензолсульфонамид (соединение 195).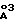 , если соединение связано с активным сайтом указанной ВИЧ аспартил-протеазы, причем указанное пространство перекрывается с объемом пространства, который должен быть заполнен нативным субстратом указанной ВИЧ аспартил-протеазы или ее негидролизуемым изостером; 5) энергия деформации связывания соединения с указанной ВИЧ аспартил-протеазой не более, чем 10 ккал/мол и 6) нейтральный или благоприятный вклад энтальпии от суммы всех электростатических взаимодействий соединения и протеазы, если соединение связано с указанной ВИЧ аспартил-протеазой.
, если соединение связано с активным сайтом указанной ВИЧ аспартил-протеазы, причем указанное пространство перекрывается с объемом пространства, который должен быть заполнен нативным субстратом указанной ВИЧ аспартил-протеазы или ее негидролизуемым изостером; 5) энергия деформации связывания соединения с указанной ВИЧ аспартил-протеазой не более, чем 10 ккал/мол и 6) нейтральный или благоприятный вклад энтальпии от суммы всех электростатических взаимодействий соединения и протеазы, если соединение связано с указанной ВИЧ аспартил-протеазой.
Z1 - Q1 - L1 - M - L2 - Q2 - Z2,
где Q1 и Q2, независимо, представляют акцепторные фрагменты водородной связи, способные связываться с атомами водорода "щитковой" молекулы воды ВИЧ аспартил-протеазы, при условии, что по крайней мере один из Q1 или Q2 поляризуется сильнее, чем поляризуется карбонил;
M представляет связывающий водород фрагмент, который может быть либо донором, либо акцептором водородной связи, способный одновременно образовывать водородные связи с Asp 25 и Asp 25' указанной ВИЧ аспартил-протеазы;
L1 и L2, независимо, представляют ациклические или циклические линкерные фрагменты и
каждый из Z1 и Z2 может, необязательно, присутствовать, и, если присутствует, он независимо выбран из группы, которая занимает объем пространства, перекрывающийся с объемом пространства, который должен быть заполнен нативным субстратом указанной ВИЧ аспартил-протеазы.
Z1 - Q1 - L1 - M - L2 - Q2 - Z2,
где Q1 и Q2 представляют независимо акцепторный фрагменты, образующие водородные связи с атомами водорода "щитковых" молекул воды ВИЧ аспартил-протеазы, при условии, что по крайней мере один из Q1 или Q2 поляризуется более сильно, нежели карбонил;
M представляет фрагмент, образующий водородную связь, который может быть либо донором, либо акцептором водородной связи, способным к одновременному образованию водородных связей с Asp 25 и Asp 25' указанной ВИЧ аспартил-протеазы;
L1 и L2, независимо, представляют ациклические или циклические линкерные фрагменты и
каждый из Z1 и Z2 может необязательно присутствовать, и если присутствует, то его независимо выбирают из группы, которая занимает объем пространства, перекрывающийся с объемом пространства, который должен быть заполнен нативным субстратом указанной ВИЧ аспартил-протеазы.
| WO 9208701, 29.05.92 | |||
| Power et al., "Nucleotide Sequence of SRV-1, a Type D Simian Acquired Immune Deficiency Syndrome Retrovizus", Science, 231, p | |||
| Устройство для ориентированного радиоприема | 1924 |
|
SU1567A1 |
| Балаян М.С | |||
| Приобретенный иммунодефицитный синдром | |||
| - Клиническая медицина, т | |||
| Способ крашения тканей | 1922 |
|
SU62A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
