Описание
Настоящее изобретение относится к новым соединениям, которые поддерживают, повышают или восстанавливают чувствительность клеток к терапевтическим или профилактическим средствам. Изобретение относится также к фармацевтическим композициям, содержащим эти соединения. Эти соединения и фармацевтические композиции настоящего изобретения, в частности, хорошо пригодны для лечения клеток, устойчивых к действию многих лекарственных средств, для предотвращения развития устойчивости ко многим лекарственным средствам и для терапии ракового заболевания, устойчивого ко многим лекарственным средствам.
Предпосылки к созданию изобретения
Основной проблемой в эффективности химиотерапии является развитие клеток, которые под действием химиотерапевтического лекарственного средства становятся устойчивыми ко многим структурно несвязанным лекарственным средствам и терапевтическим агентам. Появление такой устойчивости ко многим лекарственным средствам часто имеет место в присутствии избыточной экспрессии мембранного P-гликопротеина 170 KDа (др-170). Белок др-170 кроме раковых клеточных линий присутствует в клеточных мембранах некоторых здоровых тканей. Он гомологичен бактериальным транспортным белкам (Hait et al., Cancer Communications, Vol. 1(1), 35(1989), West, TIBS, Vol. 15, 42(1990). Белок действует в качестве транспортного насоса, придающего устойчивость к лекарственным средствам путем активного вытеснения токсичных химикатов. Хотя механизм действия этого насоса неизвестен, предполагают, что белок др-170 действует путем выталкивания веществ, которые обладают определенными общими химическими или физическими характеристиками, например, обладают гидрофобностью, содержат карбонильные группы или конъюгат глутатиона (смотри West).
Для подавления устойчивости ко многим лекарственным средствам и восстановления восприимчивости к лекарственным средствам вводили различные химические средства. Хотя некоторые лекарственные средства улучшают чувствительность MDR-клеток к химиотерапевтическим средствам, однако, лечение ими часто сопровождается нежелательными клиническими побочными действиями (смотри Hait et al.). Например, хотя циклоспорин A (CsA), широко признанный иммунодепрессант, может сенсибилизировать клетки некоторых карцином к химиотерапевтическим средствам (Slater et al., Br.J.Cancer, Vol. 4, 235 (1986)), концентрации его, необходимые для достижения этого действия, вызывают значительную иммунодепрессию у пациента, имеющего иммунную систему, уже ослабленную химиотерапией (смотри Hait et al.). Аналогично блокаторы транспорта кальция и ингибиторы калмодулина сенсибилизируют устойчивые ко многим лекарственным средствам (MRD) клетки, но как те, так и другие, вызывают нежелательные физиологические эффекты (смотри Hait et al., Br.J.Cancer, Vol. 56, 55 (1987)).
Недавние достижения привели к появлению средств, которые, как сообщается, имеют потенциально более высокую клиническую эффективность в сенсибилизации MDR-клеток. Эти средства включают аналоги CsA, которые не оказывают иммунодепрессивное действие, например, II-метиллейцинциклоспорин (II-met-leu CsA) (смотри Hait et al., Twentymann et al.), или средства, которые могут быть эффективными при низких дозах, например, иммунодепрессант FK-506 (Epand and Epand, Anti-Cancer Drug Deseng 6, 189 (1991)). Несмотря на эти достижения, остается необходимость в эффективных средствах, которые можно применять для ресенсибилизации MDR- клеток к терапевтическим или профилактическим средствам или для предотвращения развития устойчивости ко многим лекарственным средствам.
Краткое изложение существа изобретения
Настоящее изобретение предлагает новые соединения, которые полезны для поддержания, повышения или восстановления восприимчивости к лекарственному средству устойчивых к действию многих лекарственных средств (MDR) клеток, композиции, содержащие эти соединения, и способы применения их. Соединения настоящего изобретения можно применять отдельно или в комбинации с другими терапевтическими или профилактическими средствами для поддержания, повышения или восстановления терапевтического или профилактического действия лекарственных средств в клетках, особенно MDR-клетках, или для предупреждения развития MDR-клеток. В соответствии с одним примером осуществления настоящего изобретения эти новые соединения, композиции и способы преимущественно применяют для того, чтобы содействовать или усилить режим химиотерапии для лечения или профилактики рака и других заболеваний.
Настоящее изобретения предлагает также способы получения соединений этого изобретения и промежуточные продукты, применяемые в этих способах.
Подробное описание изобретения
Настоящее изобретение относится к новому классу соединений, характеризующихся способностью предотвращать устойчивость ко многим лекарственным средствам или поддерживать, повышать или восстанавливать чувствительность к лекарственному средству в клетках, устойчивых ко многим лекарственным средствам ("MDR"). Более конкретно, эти соединения представлены формулой (I):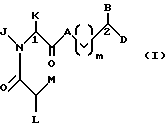
в которой A представляет собой CH2, кислород, NH или N-(C1-C4-алкил);
в которой B и D независимо представляют собой:
(i) водород, Ar, (C1-C10)-алкил нормального или разветвленного строения, (C2-C10)-алкенил или алкинил нормального или разветвленного строения, (C5-C7)-циклоалкилзамещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)-алкенил или алкинил нормального или разветвленного строения, (C5-C7)-циклоалкенилзамещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)-алкенил или алкинил нормального или разветвленного строения или Ar-замещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)--алкенил или алкинил нормального или разветвленного строения, причем в каждом случае любая одна из CH2-групп цепей этих алкилов, алкенилов или алкинилов возможно может быть замещена на гетероатом, выбранный из группы, состоящей из O, S, SO, SO2, N и NR, где R выбран из группы, состоящей из водорода, (C1-C4)-алкила нормального или разветвленного строения, (C2-C4)-алкенила или алкинила нормального или разветвленного строения и мостикового (C1-C4)-алкила, причем мостик образован между атомом азота и атомом углерода этой содержащей гетероатом цепи и сам образует ядро, которое возможно конденсировано с группой Ar; или
(ii) группу формулы: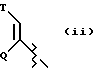
в которой Q представляет собой водород, (C1-C6)-алкил нормального или разветвленного строения или (C2-C6)-алкенил или алкинил нормального или разветвленного строения; в которой T представляет собой Ar или замещенный 5-7-членный циклоалкил с заместителями в положениях 3 и 4, независимо выбранными из группы, состоящей из оксогруппы, водорода, гидроксигруппы, группы O-(C1-C4)-алкил и O-(C2-C4)-алкенил; в которой Ar представляет собой карбоциклическую ароматическую группу, выбранную из группы, состоящей из фенила, 1-нафтила, 2-нафтила, инденила, азуленила, флуоренила, антраценила, или моно- и бициклические гетероциклические системы, состоящие из 5- или 6-членных ядер, которые могут содержать в одном или обоих ядрах всего 1-4 гетероатома, независимо выбранных из атомов кислорода, азота и серы, причем такие циклические системы включают гетероциклические ароматические группы, выбранные из группы, состоящей из 2-фурила, 3-фурила, 2-тиенила, 3-тиенила, 2-пиридила, 3-пиридила, 4-пиридила, пирролила, оксазолила, тиазолила, имидазолила, пирзолила, 2-пиразолинила, пиразолидинила, изоксазолила, изотиазолила, 1,2,3-оксадиазолила, 1,2,3-триазолила, 1,3,4-тиадиазолила, пиридазинила, пиримидинила, пиразинила, 1,3,5-триазинила, 1,3,5-тритианила, индолизинила, индолила, изоиндолила, 3H-индолила, индолинила, бензо[b]фуранила, бензо[b] тиофенила, 1H-индазолила, бензимидазолила, бензтиазолила, пуринила, 4H-хинолизинила, хинолинила, изохинолинила, циннолинила, фталазинила, хиназолинила, хиноксалинила, 1,8-нафтиридинила, птеридинила, карбазолила, акридинила, феназинила, фенотиазинила и феноксазинила;
в которой Ar может содержать 1-3 заместителя, которые независимо выбраны из группы, состоящей из водорода, галогена, гидроксигруппы, нитрогруппы, трифторметила, трифторметоксигруппы, (C1-C6)-алкила нормального или разветвленного строения, (C2-C6)-алкенила нормального или разветвленного строения, группы O-(C1-C4)-алкил нормального или разветвленного строения, O-(C2-C4)-алкенил нормального или разветвленного строения, O-бензил, O-фенил, 1,2-метилендиоксигруппы, аминогруппы, карбоксигруппы, N-(C1-C5)-алкил или алкенил нормального или разветвленного строения) карбоксамидогрупп, N,N-ди-(C1-C5-алкил нормального или разветвленного строения или C2-C5-алкенил нормального или разветвленного строения)карбоксамидогрупп, N-морфолинокарбоксамидогруппы, N-бензилкарбоксамидогруппы, N-тиоморфолинокарбоксамидогруппы, N-пиколиноилкарбоксамидогруппы, группы O-X, CH2-(CH2)q-X, O-(CH2)q-X, (CH2)q-O-X и CH= CH-X, где X представляет собой 4-метоксифенил, 2-пиридил, 3-пиридил, 4-пиридил, пиразил, хинолил, 3,5-диметилизоксазоил, изоксазоил, 2-метилтиазоил, тиазоил, 2-тиенил, 3-тиенил или пиримидил и q является числом 0-2;
в которой L представляет собой водород или U; M представляет собой кислород или CH-U, причем, если L является водородом, то M представляет собой CH-U, если M является кислородом, то L представляет собой U;
в которой U представляет собой водород, O-(C1-C4)-алкил нормального или разветвленного строения или O-(C2-C4)-алкенил нормального или разветвленного строения, (C1-C6)-алкил нормального или разветвленного строения или (C2-C6)-алкенил нормального или разветвленного строения, (C5-C7)-циклоалкил или (C5-C7)-циклоалкенил, замещенный (C1-C4)-алкилом нормального или разветвленного строения или (C2-C4)-алкенилом нормального или разветвленного строения, [(C1-C4)-алкил или (C2-C4)-алкенил]-Y или Y;
в которой Y выбран из группы, состоящей из фенила, 1-нафтила, 2-нафтила, инденила, азуленила, флуоренила, антраценила, 2-пирролинила, 3-пирролинила, пирролидинила, 1,3-диоксолила, 2-имидазолинила, имидазолидинила, 2H-пиранила, 4-пиранила, пиперидила, 1,4-диоксанила, морфолинила, 1,4- дитианила, тиоморфолинила, пиперазинила, хинуклидинила и указанных выше гетероциклических ароматических групп;
где Y может содержать 1-3 заместителя, которые независимо выбраны из группы, состоящей из водорода, галогена, гидроксигруппы, нитрогруппы, трифторметила, трифторметоксигруппы, (C1-C6)-алкила нормального или разветвленного строения, (C2-C6)-алкенила нормального или разветвленного строения, O-(C1-C4)-алкил нормального или разветвленного строения, O-(C2-C4)-алкенил нормального или разветвленного строения, O-бензил, O-фенил, 1,2-метилендиоксигруппы, аминогруппы и карбоксигруппы;
в которой J представляет собой водород, (C1-C2)-алкил или бензил; K представляет собой (C1-C4)-алкил нормального или разветвленного строения, бензил или циклогексилметил или J и K вместе образуют 5-7-членное гетероциклическое ядро, которое может содержать гетероатом, выбранный из O, S, SO и SO2; и
в которой m является числом 0-3.
Стереохимия в положениях 1 и 2 (формула I) может быть независимо R или S.
Предпочтительно, по меньшей мере, один из B и D независимо является нормальной цепью, имеющей на конце арил, т.е. группой, представленной формулой -(CH2)r-(X)-(CH2)s-Ar, в которой
r является числом 0-4;
s является числом 0-1;
Ar имеет указанные выше значения и
каждый X независимо выбран из группы, состоящей из CH2, O, S, SO, SO2, N и NR, где R выбран из группы, состоящей из водорода, (C1-C4)-алкила нормального или разветвленного строения, (C2-C4)-алкенила и алкинила нормального или разветвленного строения и мостикового алкила, причем мостик образован между атомом азота и группой Ar.
В соответствии с одним примером осуществления настоящего изобретения гетероциклические ароматические группы выбраны из группы, состоящей из фурана, тиофена, пиррола, пиридина, индолизина, индола, изоиндола, бензо[b] фурана, бензо[b] тиофена, 4H-хинолизина, хинолина, изохинолина, 1,2,3,4-тетрагидрохинолина, изоксазола и 1,2,3,4-тетрагидроизохинолина.
В соответствии с другим примером осуществления настоящего изобретения, по меньшей мере, один из B и D выбран из группы, состоящей из (C2-C10)-алкинила нормального или разветвленного строения, (C5-C7)-циклоалкилзамещенного (C2-C6)-алкинила нормального или разветвленного строения, (C5-C7)-циклоалкенилзамещенного (C2-C6)-алкинила нормального или разветвленного строения и Ar-замещенного (C2-C6)-алкинила нормального или разветвленного строения.
В объеме настоящего изобретения находятся также соединения формулы (I), у которых, по меньшей мере, один из B и D выбран из группы, состоящей из Ar', Ar'-замещенного (C1-C6)-алкила нормального или разветвленного строения и Ar'-замещенного (C2-C6)-алкенила или алкинила нормального или разветвленного строения, причем Ar' является группой Ar, имеющей 1-3 заместителя, которые независимо выбраны из группы, состоящей из N-(C1-C5-алкил или C2-C5-алкенил нормального или разветвленного строения) карбоксидов, N,N-ди-(C1-C5-алкил или C2-C5-алкенил нормального или разветвленного строения) карбоксамидов, N-морфолинокарбоксамида, N-бензилкарбоксамида, N-тиоморфолинокарбоксамида, N-пиколиноилкарбоксида, группы O-X, CH2-(CH2)q-X, O-(CH2)q-X, (CH2)q-O-X и CH=CH-X, в которой X представляет собой 4-метоксифенил, 2-пиридил, 3-пиридил, 4-пиридил, пиразил, хинолил, 3,5-диметилизоксазоил, изоксазоил, 2-метилтиазоил, тиазоил, 2-тиенил, 3-тиенил или пиримидил и q является числом 0-2.
Примеры некоторых предпочтительных соединений формулы (I), у которых J и K, соединяясь, образуют 5-7-членное гетероциклическое ядро, приведены в таблице 1 и далее иллюстрируются в примерах*.
* Должно быть принято, что касается особенности настоящего изобретения по применению описанных в нем соединений в композициях или способах лечения или предотвращения устойчивости ко многим лекарственным средствам, то эти соединения представлены формулой (I), указанной выше. Что касается особенности настоящего изобретения по новым соединениям, описанным в нем, то эти соединения представлены формулой (I), приведенной выше, за исключением того, что B и D не могут быть водородом.
Наиболее предпочтительными соединениями настоящего изобретения являются 4-пиридин-3-ил-1-(3-пиридин-3-ил)пропил)-бутиловый эфир (S)-1-(2-оксо-2-(3,4,5-триметоксифенил)ацетил)пиперидин-2-карбоновой кислоты и 4-пиридин-3-ил-1-(3-(пиридин-3-ил)пропил)бутиловый эфир (R)-1-(2-оксо-2-(3,4,5-триметоксифенил)ацетил)пиперидин-2-карбоновой кислоты, их фармацевтически пригодные производные и их смеси.
В применяемом определении соединения настоящего изобретения, включающие соединения формулы (I), включают их фармацевтически пригодные производные. "Фармацевтически пригодное производное" обозначает любую фармацевтически пригодную соль, эфир или соль такого эфира соединения изобретения или любое другое соединение, которое при введении пациенту способно образовать (непосредственно или не непосредственно) соединение изобретения, или его метаболит или остаток, характеризующий способность поддерживать, повышать или восстанавливать чувствительность MDR-клеток к действию терапевтических или профилактических средств или предотвращать развитие устойчивости ко многим лекарственным средствам.
Соединения настоящего изобретения, представленные формулой (I), можно получить любым обычным способом. Предпочтительно эти соединения химически синтезируют из легко доступных исходных соединений, например, альфа-аминокислот. Модулярные и конвергентные способы также предпочтительны для синтеза этих соединений. По способу конвергенции, например, большие части конечного продукта соединяют вместе в последних стадиях синтеза, что лучше, чем получать продукт постепенным присоединением небольших частей к растущей молекулярной цепи.
Схема 1 иллюстрирует характерный пример конвергентного способа синтеза соединений формулы (I'), являющихся предпочтительной подгруппой соединений формулы (I), у которых A представляет собой кислород. Этот способ предусматривает этерификацию защищенной альфа-аминокислоты формулы (X), в которой P является защитной группой, спиртом формулы (XI). Защищенные альфа-аминокислоты являются хорошо известными соединениями, многие из них коммерчески доступны. Например, обычные защитные группы и обычные способы защиты аминокислот описаны в T.W.Greene, P.G.M.Wuts, Protective Groups in Organic Chemistry, 2nd, Ed., John Wiley and Sons, New York (1991). Для защиты атома азота в соединениях формулы (X) предпочтительны алкоксикарбонильные группы, наиболее предпочтительны трет-бутоксикарбонил (Boc), бензилоксикарбонил (Cbz), аллилоксикарбонил (Alloc) и триметилсилилэтоксикарбонил (Teoc).
После этерификации соединения формулы (XII) освобождают от защитной группы в подходящих условиях удаления защитных групп (смотри Greent, supra) и свободную аминогруппу соединения формулы (XIII) затем ацилируют соединением формулы (XI) или активированным производным его для получения соединения формулы (I'). Способы активирования карбоксигрупп в карбоновых кислотах, например, соединениях формулы (XIV), хорошо известны и многие активирующие средства коммерчески доступны.
Спирты формулы (XI), у которых m является O (XI'), можно также получить стандартным способом, например, как показано на схемах 2 и 3. Реакцией металлорганического реагента формулы (XV) и альдегида формулы (XVI) получают спирты формулы (XI') (схема 2).
Схема 1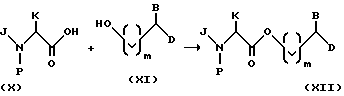
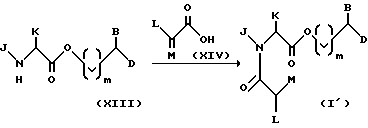
Схема 2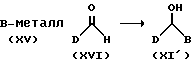
Схема 3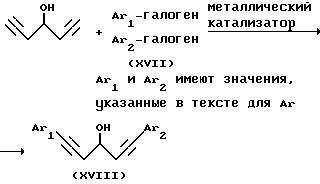
Альтернативно (схема 3) 1,6-гептадиин-4-ол можно конденсировать путем катализируемой металлом реакции с ароматическими галогенидами формулы (XVII) для получения спирта формулы (XVII). Последующим гидрированием этого спирта можно получить спирт формулы (XI') из предпочтительного подкласса спиртов формулы (XI).
Таким образом, настоящее изобретение предлагает способ получения соединений формулы (I'), включающий стадии:
(a) этерификации защищенной аминокислоты формулы (X) спиртом формулы (XI) для образования промежуточного продукта формулы (XII);
(b) удаления аминозащитной группы у промежуточного продукта формулы (XII) для получения аминоэфира формулы (XIII); и
(c) ацилирования свободной аминогруппы соединения формулы (XIII) соединением формулы (XIV) или его активированным производным.
Тем, кто работает в данной области, должно быть понятно, что способами, иллюстрированными синтетическими схемами 1, 2 и 3, можно легко получить большой ряд соединений формулы (I). Те же способы можно применять для синтеза многих других конечных продуктов путем изменения изменяемых групп в исходных соединениях.
Например, соединения формулы (I'') (не приведены), у которых A представляет собой NH или N-(C1-C4-алкил), можно синтезировать реакцией пептидного взаимодействия между карбоновой кислотой формулы (X) и амином формулы (X''') (не приведена) для образования амида формулы (XII'). Эта стадия аналогична первой реакции этерификации схемы I. Стадии превращения (XII') в (I'') также аналогичны стадиям превращения (XII) в (I') схемы I.
Оптически активные соединения формулы (I) можно также получить с применением оптически активных исходных соединений, благодаря чему отпадает необходимость в расщеплении энантиомеров и разделении диастереомеров на последней стадии синтеза.
Работающим в данной области должно быть понятно также, что указанные выше синтетические схемы не предусматривают предоставление исчерпывающего перечня всех средств, при помощи которых можно синтезировать соединения или промежуточные продукты настоящего изобретения. Другие способы или модификации указанных выше общих схем должны быть ясны для специалистов данной области.
Соединения настоящего изобретения можно модифицировать путем присоединения соответствующих функциональных групп с целью усиления селективных биологических свойств. Такие модификации известны в данной области и включают те модификации, которые повышают биологическое проникновение в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), повышают пероральную доступность, повышают растворимость, чтобы соединение можно было вводить инъекцией, изменяют метаболизм и изменяют скорость экскреции соединения.
Соединения настоящего изобретения характеризуются способностью повышать, восстанавливать или поддерживать восприимчивость MDR-клеток к действию цитотоксичных соединений, таких например, которые обычно применяют в химиотерапии. Благодаря этой способности соединения изобретения успешно применяют в качестве химиосенсибилизирующих средств, которые повышают эффективность химиотерапии у пациентов, которые поражены устойчивыми к лекарственным средствам раковыми заболеваниями, опухолями, метастазами или другими заболеваниями. Кроме того, соединения изобретения способны поддерживать восприимчивость к терапевтическим или профилактическим средствам в невосприимчивых клетках. Следовательно, соединения изобретения пригодны для лечения или профилактики устойчивости пациента ко многим лекарственным средствам. Применяемый в описании термин "пациент" относится к животным, включая человека. Термин "клетка" относится к клеткам млекопитающих, включая клетки человека.
Применяемые в описании термины "сенсибилизирующее средство", "сенсибилизатор", "химиосенсибилизирующее средство", "химиосенсибилизатор" и "MDR-модификатор" обозначают соединение, обладающее способностью повышать или восстанавливать восприимчивость MDR-клеток или поддерживать восприимчивость невосприимчивых клеток к одному или нескольким терапевтическим или профилактическим средствам. Термины "MDR-сенсибилизация" и "сенсибилизация" и "ресенсибилизация" относятся к действию такого соединения в поддержании, повышении или восстановлении восприимчивости к лекарственным средствам.
В соответствии с одним примером осуществления настоящего изобретения соединения настоящего изобретения полезны для повышения, восстановления или поддержания восприимчивости к лекарственным средствам и способны также связываться с белком FKBP-12 или другими, родственными FK-506 связывающими белками, например FKBP-13, FKBP-26 и FKBP-52. В проводимых in vitro испытаниях (данные не приведены) эти соединения демонстрируют способность связываться с FKBP-12. Таким образом, это изобретение включает также класс химиосенсибилизирующих средств, не являющихся FK-506 и характеризующихся способностью связываться со связывающим белком FK-12 или родственными связывающими белками FK, фармацевтические композиции, включающие такие средства и физиологически пригодный вспомогательный компонент, носитель или наполнитель, и способы применения этих композиций для лечения или предупреждения устойчивости пациента ко многим лекарственным средствам.
Предпочтительными соединениями, пригодными для применения при предупреждении или восстановлении устойчивости ко многим лекарственным средствам, являются те соединения, которые не проявляют заметной иммунодепрессивности при клинически пригодных или профилактически или терапевтически активных дозах, т. е. эффект, если он вообще имеется, иммунодепрессии такого соединения не превосходит величину активности его по сенсибилизации пациента. Такую иммунодепрессивную способность можно установить испытаниями in vitro, предложенными в заявках на патент США N 07/547 814 (теперь патент США 5192773), 07/704734, 07/697785 и 07/881152, которые включены сюда в качестве ссылок.
Соединения настоящего изобретения можно применять в форме фармацевтически пригодных солей, полученных из неорганических или органических кислот и оснований. Такие соли с кислотами включают: ацетат, адипат, альгинат аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорасульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глицерофосфат, полусульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат и удеканоат. Соли с основаниями включают соли аммония, соли щелочных металлов, например, соли натрия и калия, соли щелочноземельных металлов, например, соли кальция и магния, соли с органическими основаниями, например, соли дициклогексиламина, N-метил-D-глюкамина, и соли с аминокислотами, например, аргинином, лизином и т.д. Азотсодержащие основные группы можно также кватернизовать такими агентами, как низшие алкилгалогениды, например, метил-, этил-, пропил- и бутилхлоридами, -бромидами и иодидами; диалкилсульфаты, например, диметил-, диэтил, дибутил- и диамилсульфатами, высшие галогениды, например децил-, лаурил, миристил- и стеарилхлоридами, -бромидами и -иодидами; аралкилгалогениды, например, бензил- и фенетилбромидами и т.д. При этом получают растворимые или диспергируемые в воде или масле продукты.
Соединения настоящего изобретения можно вводить перорально, парентерально, ингаляцией аэрозоля, местно, ректально, через нос, трансбуккально, вагинально или через имплантированный резервуар в дозированных формах, содержащих обычные нетоксичные фармацевтически пригодные носители, вспомогательные компоненты и наполнители. Применяемый термин "парентеральный" включает способы чрезкожной, внутривенной, внутримышечной, интраартикулярной, внутрисуставной, интрагрудинной, подоболочечной, внутрипочечной, внутрираневой и внутричерепной инъекции или инфузии.
Фармацевтические композиции настоящего изобретения содержат любое из соединений изобретения или их фармацевтически пригодных солей с любым фармацевтически пригодным носителем, вспомогательным компонентом или наполнителем. Фармацевтически пригодные носители, вспомогательные компоненты и наполнители, которые можно применять в фармацевтических препаратах настоящего изобретения, включают, но не ограничиваются ими, ионообменные смолы, оксид алюминия, стеарат алюминия, лецитин, белки сыворотки крови, например, альбумин сыворотки крови человека, буферные вещества, например, фосфаты, глицин, сорбиновую кислоту, сорбат калия, частично глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, например, сульфат протамина, вторичный кислый фосфат натрия, первичный кислый фосфат калия, хлористый натрий, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натриевую соль карбоксиметилцеллюлозы, полиакрилаты, воски, блок-полимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и ланолин.
В соответствии с настоящим изобретением фармацевтические композиции могут быть в форме стерильного инъецируемого препарата, например, стерильной инъецируемой водной или маслянистой суспензии. Эту суспензию можно приготовлять по известным методикам с применением подходящих диспергирующих или смачивающих средств и суспендирующих средств. Стерильный инъецируемый препарат может быть также стерильным инъецируемым раствором или суспензией в нетоксичном парентерально пригодном разбавителе или носителе, например, раствором в 1,3-бутандиоле. Среди пригодных наполнителей и растворителей, которые можно применять, указываются вода, раствор Рингера и изотонический раствор хлористого натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно применяют стерильные, нелетучие масла. Для этой цели можно применять любое мягкое нелетучее масло, включая синтетические моно- и диглицериды. Жирные кислоты, например, олеиновая кислота и ее глицеридные производные, пригодны для получения инъецируемых препаратов, для этого пригодны также природные фармацевтически пригодные масла, например, оливковое масло или касторовое масло, особенно их полиоксиэтилированные аналоги. Эти масляные растворы или суспензии могут содержать также в качестве разбавителя или диспергатора высший спирт, например Ph. Helv. или аналогичный спирт.
Фармацевтические препараты настоящего изобретения можно перорально вводить в любой перорально пригодной дозированной форме, включая, но не ограничиваясь ими, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального введения применяемые обычно носители включают лактозу и кукурузный крахмал. Обычно добавляют также смызывающие вещества, например, стеарат магния. Для получения перорального препарата в капсулированной форме пригодные разбавители включают лактозу и высушенный кукурузный крахмал. Когда для перорального применения нужны водные суспензии, активный компонент комбинируют с эмульгирующими и суспендирующими агентами. При желании, можно также добавить обычные подслащивающие вещества, корригенты или окрашивающие средства.
Альтернативно фармацевтические препараты настоящего изобретения можно применять в форме суппозиториев для ректального введения. Их можно получить смешиванием активного компонента с подходящим, нераздражающим наполнителем, который является твердым при комнатной температуре, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке, высвобождая лекарственное средство. Такие материалы включают какао-масло, пчелиный воск и полиэтиленгликоли.
Фармацевтические препараты настоящего изобретения можно также вводить местно, особенно когда объект лечения включает участки или органы, легко доступны для местного применения, включая заболевания глаз, кожи или низшей части кишечника. Подходящие препараты для местного введения легко получают для каждого из этих участков или органов.
Местное применение препарата для нижней части кишечника можно осуществить с использованием ректальных суппозиторий (смотри выше) или пригодной клизмы. Можно также применять местно-чрескожные пластыри.
Для местного применения фармацевтические композиции можно приготовить в виде подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или нескольких носителях. Носители для местного введения соединений настоящего изобретения включают, но не ограничиваются ими, минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгируемый воск и воду. Альтернативно фармацевтические композиции можно приготовлять в форме пригодного лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически пригодных носителях. Пригодные носители включают, но не ограничиваются ими, минеральное масло, моностеарат сорбитана, полисорбат 60, цетиловые эфиры воска, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
Для офтальмологического применения фармацевтические композиции можно приготовлять в форме микронных суспензий в изотоническом стерильном солевом растворе с регулированным pH или, что предпочтительно, в форме растворов в изотоническом стерильном солевом растворе с установленным pH, возможно, с добавлением консерванта, например, хлористого бензилалкония. Альтернативно для офтальмологического применения фармацевтические композиции можно приготовить в виде мази, например, в вазелине.
Фармацевтические композиции настоящего изобретения можно также вводить через нос распылением аэрозоля или ингаляцией. Такие композиции получают способами, хорошо известными для получения фармацевтических композиций, их можно получить в виде растворов в солевом растворе, применяя бензиловый спирт или другие пригодные консерванты, промоторы абсорбции для повышения биопригодности, фторкарбоны и/или другие обычные растворяющие или диспергирующие средства.
Количество активного компонента, который можно комбинировать с носителями для получения композиции в виде разовых доз, будет изменяться в зависимости от пациента, которому вводят препарат, и способа введения препарата. Должно быть понятно, однако, что определенная доза и режим лечения для конкретного пациента будут зависеть от различных факторов, включая активность применяемого специфического соединения, возраста, массы тела, общего здоровья, пола, диеты, времени введения, скорости экскреции, комбинации лекарственных средств, решения лечащего врача и остроты конкретного заболевания, которое лечат этим препаратом. Количество активного компонента может также зависеть от терапевтического или профилактического средства, если оно присутствует, с которым активный компонент вводят совместно. Применяемый термин "фармацевтически эффективное количество" относится к количеству, эффективному для предотвращения появления устойчивости ко многим лекарственным средствам или поддержания, повышения или восстановления восприимчивости MDR-клеток к лекарственному средству.
Пригодны пределы доз активного компонента между около 0,01 и около 100 мг/кг тела в день, предпочтительно между около 0,5 и около 50 мг/кг тела в день. Типичная композиция будет содержать между около 5% и около 95% активного соединения (масса/масса). Предпочтительно такие композиции содержат между около 20% и около активного соединения.
Когда соединения этого изобретения применяют в комбинационной терапии с другими средствами, их можно вводить пациенту последовательно или одновременно. Альтернативно фармацевтические или профилактические композиции в соответствии с настоящим изобретением могут содержать комбинацию соединения настоящего изобретения и другого терапевтического или профилактического средства.
Например, соединения можно вводить отдельно или в комбинации с одним или несколькими терапевтическими средствами, такими как химиотерапевтические средства (например, актиномицином D, доксорубицином, винкристином, винбластином, этопозидом, амзакрином, митоксантроном, тениразидом, таксолом и колхицином) и/или химиосенсибилизурующие средства (например, циклоспорином A и его аналогами, фенотиазинами и тиоксантерами) для повышения восприимчивости MDR- клеток пациента к этому средству или средствам.
Для того чтобы настоящее изобретение можно было понять более полно, предложены следующие примеры. Эти примеры приведены только для целей иллюстрации и не должны рассматриваться никоим образом как ограничение объема изобретения.
Примеры
Общие способы
Спектры протонного ядерного магнитного резонанса (1H ЯМР) регистрировали на спектрометре Bruker AMX 500 при частоте 500 MHz. Химические сдвиги представляли в частях на миллион (δ) относительно Me4Si (δ 0.0). Аналитическую жидкостную хроматографию высокого разрешения проводили на жидкостном хроматографе Waters 600E или Hewlett Packard 1050.
Пример 1
Синтез (S)-1,7-дифенил-4-гептанил-N-(3,4,5-триметоксифенилглиоксил)пипеколата (3)
4-фенил-1-масляный альдегид (119). В раствор 3,2 мл (20,8 ммоля) 4-фенил-1-бутанола (Aldrich Chemical Co) в 20 мл CH2Cl2 при 0oC добавляли 3,2 г порошкообразных молекулярных сит 3  и затем 5,37 г (24,9 ммоля) хлорхромата пиридиния (PCC). Полученную суспензию перемешивали при 0oC в течение 1 часа, в это время добавляли дополнительно 2,16 г (10,0 ммоля) PCC и реакционную смесь нагревали до комнатной температуры. После перемешивания при комнатной температуре 0,5 ч реакционную смесь разбавляли простым эфиром и фильтровали через целит, получая 2,5 г неочищенного продукта. Флаш-хроматографией (элюирование 5%-ным этилацетатом в гексане) выделили 700 мг альдегида 119. 1H ЯМР-спектр его согласуется со структурой.
и затем 5,37 г (24,9 ммоля) хлорхромата пиридиния (PCC). Полученную суспензию перемешивали при 0oC в течение 1 часа, в это время добавляли дополнительно 2,16 г (10,0 ммоля) PCC и реакционную смесь нагревали до комнатной температуры. После перемешивания при комнатной температуре 0,5 ч реакционную смесь разбавляли простым эфиром и фильтровали через целит, получая 2,5 г неочищенного продукта. Флаш-хроматографией (элюирование 5%-ным этилацетатом в гексане) выделили 700 мг альдегида 119. 1H ЯМР-спектр его согласуется со структурой.
Бромид 3-фенил-1-прропилмагния (120). В суспензию 736 мг (30,3 ммоля) магниевых стружек в 50 мл ТГФ при комнатной температуре добавляли 50 мкл 1,2-дибромэтана и затем по каплям 5,5 г (25,1 ммоля) 1-бром-3-фенилпропана (Aldrich Chemical Со.). После перемешивания при комнатной температуре 0,5 ч супернатант переносили при помощи трубки в сосуд для хранения объемом 100 мл и затем применяли в виде 0,5 М раствора реактива Гриньяра 120 в ТГФ.
1,7-Дифенил-4-гептанол (121). В раствор 700 мг (4,7 ммоля) 4-фенил-1-бутаналя (119) в 5,0 мл ТГФ при 0oC добавляли 10,0 мл (5,0 ммолей) бромистого 3-фенил-1-пропилмагния (120) и полученную смесь перемешивали при 0oC 0,5 ч. В смесь затем добавляли по каплям насыщенный раствор NH4Cl и разбавляли простым эфиром. Фазы разделяли и органический слой промывали водой и соляным раствором и затем сушили над MgSO4. После концентрирования получали 1,12 г спирта 121 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(S)-Boc-1-Пипеколил-1,7-дифенил-4-гептаниловый эфир (122). В раствор 164 мг (0,72 ммоля) Boc-L-пипеколиновой кислоты в 5,0 мл CH2Cl2 при комнатной температуре добавляли 174 мг (0,65 ммоля) спирта 121, 140 мг (0,72 ммоля) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЕДС) и каталитическое количество N, N-диметиламинопиридина (DMAP). Реакционную смесь перемешивали при комнатной температуре 0,5 ч и затем переносили непосредственно в колонку с силикагелем. Элюированием 10%-ным этилацетатом в гексане получили 76,2 мг эфира 122 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(S)-1,7-Дифенил-4-гептанилпипеколат (123). В раствор 47 мг (0,10 ммоля) эфира 122 в 1,0 мл CH2Cl2 при комнатной температуре добавляли 1,0 мл трифторуксусной кислоты. После перемешивания при комнатной температуре 0,5 ч полученный раствор нейтрализовали добавлением по каплям насыщенного раствора K2CO3. Слои разделяли и органическую фазу промывали водой, сушили над MgSO4 и концентрировали, получая 23 мг амина 123 в виде масла. 1H-ЯМР-спектр согласуется со структурой.
3,4,5-Триметоксибензоилмуравьиная кислота (124). В раствор 9,2 г (43,4 ммоля) 3,4,5-триметоксиацетофенона (Aldrich Chemical Co) в 35 мл пиридина добавляли 6,3 г ( 56,7 ммоля) диоксида селена и полученный раствор нагревали с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит и концентрировали, получая темно-коричневое масло, которое растворяли в этилацетате и промывали 1,0 N HCl и затем насыщенным раствором NaHCO3. Щелочной водный слой разбавляли эфиром и подкисляли концентрированной HCl. Слои разделяли и органическую фазу промывали соляным раствором и затем сушили над Na2SO4, получая 8,4 г кислоты 124 в виде бледно-желтого твердого вещества. 1H-ЯМР-спектр согласуется со структурой.
(S)-1,7-Дифенил-4-гептанил-N-(3,4,5-триметоксифенилглиоксил)пипеколат (3). В раствор 23 мг (0,06 ммоля) амина 123 в 1,0 мл CH2Cl2 при комнатной температуре добавляли 21,8 мг (0,09 ммоля) кислоты 124 и затем 17,9 мг (0,09 ммоля) EDC и полученный раствор перемешивали при комнатной температуре 0,5 ч и затем непосредственно перенесли в колонку с силикагелем. Элюированием 15%-ным этилацетатом в гексане выделили 8,4 мг амина 3 в виде смеси ротамеров (поворотных изомеров). 1H ЯМР-спектр (500 МГц, CDCl3) δ 7,35-7,06 (м), 5,32 (ш. с), 5,00 (ш.с), 4,88 (ш.с), 4,58 (д), 4,31 (ш.с), 3,95 (с), 3,89 (с), 3,44 (д), 3,21 (т), 3,04 (т), 2,54 (ш.с), 2,51 (ш.с), 2,42 (ш.с), 2,30 (д), 2,15 (д), 1,83-1,21 (м).
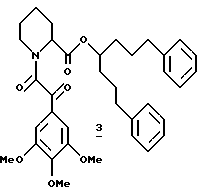
Пример 2
Синтез (R и S)-1-(3-феноксифенил)-4-фенил-1-бутил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколата (4)
3-феноксибензальдегид (125). В раствор 1,8 мл (10,3 ммоля) 3-феноксибензилового спирта (Aldrich Chemical Co) в 20 мл CH2Cl2 при комнатной температуре добавляли 1,5 г порошкообразных молекулярных сит 4  и 2,5 г активированного MnO2. Полученную суспензию перемешивали при комнатной температуре 0,5 ч, в это время добавляли дополнительно 2,5 г MnO2. После перемешивания 0,5 ч при комнатной температуре реакционную смесь фильтровали через целит, получая 1,84 г альдегида 125 в виде масла. 1H-ЯМР-спектр согласуется со структурой.
и 2,5 г активированного MnO2. Полученную суспензию перемешивали при комнатной температуре 0,5 ч, в это время добавляли дополнительно 2,5 г MnO2. После перемешивания 0,5 ч при комнатной температуре реакционную смесь фильтровали через целит, получая 1,84 г альдегида 125 в виде масла. 1H-ЯМР-спектр согласуется со структурой.
(R и S)-1-(3-Феноксифенил)-4-фенил-1-бутанол (126).
Спирт 126 получали из 190 мг (0,96 ммоля) альдегида 125 и 2,0 мл (1,0 ммоля) реактива Гриньяра 120 в 2,0 мл ТГФ, как описано выше для синтеза спирта 121 в примере 1. Флаш-хроматографией (элюирование 10%-ным этилацетатом в гексане) выделили 108 мг рацемического спирта 126. 1H ЯМР-спектр согласуется со структурой.
(S)-N-(3,4,5-Триметоксифенил)глиоксилпипеколиновая кислота (127). Во взвесь 953,3 мг (3,4 ммоля) тартратной соли (S)-пипеколиновой кислоты (Egbertson, M. and Danishefsky, S.J. Org Cnem, 1989, 54, 11) в 7,0 мл CH2Cl2 добавляли 3,9 мл (22,39 ммоля) диизопропилэтиламина и 2,4 мл (18, ммоля) хлортриметилсилана и полученный раствор перемешивали 0,5 ч при 0oC. В другой реакционной колбе в раствор 820 мг (3,4 ммоля) кислоты 124 в 7,0 мл добавляли 450 мкл (5,2 ммоля) хлористого оксалила и три капли ДМФ. После прекращения выделения газа все содержимое второй колбы добавляли в первую реакционную колбу и образованную смесь перемешивали 1 ч при комнатной температуре. Реакционную смесь концентрировали, растворяли в эфире и промывали 0,5 N HCl и затем насыщенным раствором NaHCO3. Щелочную водную фазу подкисляли концентрированной HCl и экстрагировали эфиром. Эфирные экстракты промывали водой, соляным раствором, сушили над MgSO4 и концентрировали, получая 490 мг кислоты 127. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-(3-Феноксифенил)-4-фенил-1-бутил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколат (4). В раствор 29,4 мг (0,08 ммоля) кислоты 127 в 2,0 мл CH2Cl2 при комнатной температуре добавляли 11 мкл (0,13 ммоля) хлористого оксалила и три капли ДМФ и реакционную смесь перемешивали 0,5 ч при комнатной температуре и затем концентрировали и суспензировали в 1,0 мл бензола. К этой суспензии добавляли 32,0 мг (0,1 ммоля) спирта 126 и 13,4 мг (0,1 ммоля) цианида серебра. Полученную смесь нагревали с обратным холодильником в течение ночи, охлаждали до комнатной температуры и концентрировали. Флаш-хроматографией (элюирование 10%-ным этилацетатом в гексане) выделили 8,8 мг эфира 4 в виде смеси диастереомеров. 1H ЯМР-спектр (500 МГц, CDCl3) δ 7,34 - 7,19 (м), 7,18 - 7,03 (м), 7,02 - 6,84 (м), 6,83 - 6,72 (м), 5,73 (к), 5,69 - 5,55 (м), 5,38 (т), 4,55 (ш.д.), 4,35 (дд), 3,94 (с), 3,92 (с), 3,89 (с), 3,83 (с), 3,73 (с), 3,63 (с), 3,84 - 3,35 (м), 3,20 (т), 3,10 (т), 2,60 (к), 2,40 (дд), 1,95 - 1,91 (м), 1,90 - 1,45 (м).
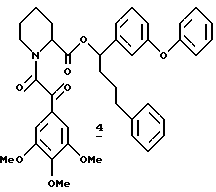
Пример 3
Синтез (R и S)-6-фенил-1-(3-пиридил)-3-гексил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколата (7)
3-(3-Пиридил)-1-пропилальдегид (128). В раствор 2,3 г (5,46 ммоля) периодината Десс-Мартина (Dess, D. B., Martin, J.C.J. Org. Chem. 1983, 48, 4155) в 10 мл CH2Cl2 при 0oC добавляли 470 мкл (3,65 ммоля) 3-(3-пиридил)-1-пропанола и полученную смесь оставляли для нагревания от 0oC до комнатной температуры в течение периода 1,5 ч. В этот раствор добавляли 6,0 г (38,22 ммоля) Na2S2O3 в насыщенном растворе NaHCO3 и реакционную смесь перемешивали 15 мин при комнатной температуре. Продукт экстрагировали CH2Cl2, сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование смесью гексана и ацетона 3: 1) выделили альдегид 128 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-6-Фенил-1-(3-пиридил)-3-гексанол (129). Спирт 129 получали из 125 мг (0,92 ммоля) альдегида 128 и 2,0 мл (1,0 ммоля) соединения 120 в 2,0 мл ТГФ, как описано выше для синтеза спирта 121 в примере 1. Получили 221 мг неочищенного спирта 129. 1H ЯМР-спектр согласуется со структурой.
(S)-Вос-Пипеколил-(R и S)-6-фенил-1-(3-пиридил)-3-гексиловый эфир (130). Эфир 130 получали из 125 мг (0,49 ммоля) спирта 129, 93 мг (0,41 ммоля) Вос-пипеколиновой кислоты, 94 мг (0,49 ммоля) EDC и каталитического количества DMAP в 1,0 мл CH2Cl2 и 1,0 мл ДМФ, как описано выше для синтеза 122 в примере 1. Флаш-хроматографией (элюирование смесью гексана и этилацетата 2: 1) выделили 105 мг диастереомерного эфира 130 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-6-фенил-1-(3-пиридил)-3-гексил-(S)пипеколат (131). Амин 131 синтезировали обработкой 95 мг (0,20 ммоля) эфира 130 1,0 мл трифторуксусной кислоты в 3,0 мл CH2Cl2, как описано выше для получения амина 123 в примере 1. Получали 58 мг диастереомерного амина 131 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-6-Фенил-1-(3-пиридил)-3-гексил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколат (7). Эфир 7 получали из 54 мг (0,15 ммоля) аминов 131, 50 мг (0,22 ммоля) кислоты 124 и 42 мг (0,22 ммоля) EDC в 3,0 мл CH2Cl2, как описано выше в синтезе эфира 3 в примере 1. Флаш-хроматографией (элюирование смесью этилацетата и гексана 1:1) выделили 73 мг диастереомерногго эфира 7 в виде смеси ротамеров. 1H ЯМР-спектр (500 МГц, CDCl3) δ 8,48 - 8,42 (м), 7,50 - 7,41 (м), 7,32 (д), 7,27 - 7,03 (м), 5,38 (д), 5,06 - 5,01 (м), 4,97 - 4,93 (м), 4,60 (ш.д), 3,92 (с), 3,88 (с), 3,86 (с), 3,84 (с), 3,82 (с), 3,79 (с), 3,46 (ш.д), 3,27 (ш.т), 2,73 - 2,68 (м), 2,38 - 2,29 (м), 1,98 - 1,76 (м), 1,75 - 1,60 (м), 1,56 - 1,51 (м), 1,38 - 1,20 (м).
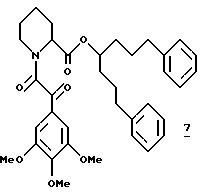
Пример 4
Синтез (R и S)-(E)-1-[транс-(4-гидроксициклогексил)]-2-метил-6- фенил-3-гекс-1-енил-(S)-N-(3,4,5-триметоксифенилглиоксил)пипеколата (8)
цис- и транс-4-(трет-Бутилдиметилсилилокси)циклогексан-1-ол (132) и (133). В раствор 3,43 г (21,7 ммоля) цис- и транс-метил-4-гидроксициклогексанкарбоксилата (Noyce, D.S., Denney, D.B.J. Am. Chem. Soc. Vol, 74, 5912 (1952)) в 45 мл хлористого метилена при 0oC добавляли 3,0 мл (26,0 ммоля) 2,6-лутидина и затем 5,5 мл (23,0 ммоля) трет-бутилдиметилсилилтрифторметансульфоната. Ледяную баню убирали и реакционную смесь перемешивали 2 ч при 25oC, после чего раствор выливали в насыщенный раствор бикарбоната натрия. Слои разделяли и органический слой промывали насыщенным раствором сульфата меди и водой и затем сушили над MgSO4, получая 5,9 г неочищенных метиловых эфиров. Раствор 5,72 г (21,0 ммоля) этой смеси в 45 мл безводного ТГФ обрабатывали 400 мг (10,5 ммоля) алюмогидрида лития. Реакционную смесь перемешивали 0,5 ч при 25oC, в нее затем медленно добавляли насыщенный раствор соли Rochelle's. Смесь разбавляли эфиром, слои разделяли и водный слой промывали 2 раза этилацетатом. Объединенный органический экстракт сушили над MgSO4 и концентрировали, получая 4,9 г диастереомерных спиртов. Флаш-хроматографией (элюирование смесью этилацетата и гексана 1:5) выделили 650 мг изомера 132, 1,10 г изомера 133 и 2,40 г смеси обоих. Данные для 132: 1H ЯМР-спектр (300 МГц, CDCl3) δ 3,99 - 3,92 (м), 3,46 (д), 1,72 - 1,58 (м), 1,57 - 1,36 (м), 0,86 (с), 0,08 (с). Данные для 133: 1H ЯМР-спектр (300 МГц, CDCl3) δ 3,47 (дддд), 3,38 (д), 1,86 - 1,67 (м), 1,47 - 1,16 (м), 1,05 - 0,77 (м), 0,72 (с), 0,02 (с).
(E)-Этил-3-[транс-(4-трет-бутилдиметилсилилоксициклогексил)] -2- метилпроп-2-еноат (134). В раствор хлористого оксалила (785 мкл, 9,0 ммоля) в 10 мл хлористого метилена при -78oC добавляли диметилсульфоксид (1,3 мл, 18,0 ммоля). Полученный раствор перемешивали 5 мин и затем добавляли 1,1 г (4,5 ммоля) спирта 133 в 10 мл хлористого метилена. Реакционную смесь перемешивали при -78oC 45 мин и в это время добавляли 3,8 мл (27,0 ммоля) триэтиламина. Раствор оставляли для нагревания до комнатной температуры. Реакцию прекращали добавлением 1,0 N HCl и водный слой экстрагировали тремя порциями хлористого метилена. Объединенный органический экстракт сушили над MgSO4 и выпаривали досуха, получая 1,0 г промежуточного альдегида. Раствор этого альдегида обрабатывали непосредственно 710 мг (1,95 ммоля) (карбэтоксиэтилиден)трифенилфосфорана в 5,0 мл хлористого метилена. Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выливали в воду. Слои разделяли и водный слой экстрагировали два раза хлористым метиленом. Объединенный органический слой сушили над MgSO4 и концентрировали, получая енолят 134, содержащий небольшое количество Z-изомера. 1H ЯМР-спектр согласуется со структурой.
(E)-3-[транс-(4-трет-Бутилдиметилсилилоксициклогексил)] -2- метилпроп-2-ен-1-ол (135). В раствор 860 мг (2,6 ммоля) енолята 134 в 5,0 мл безводного тетрагидрофурана при 25oC добавляли 50 мг (1,3 ммоля) алюмогидрида лития и полученную смесь перемешивали 30 мин. Реакцию прерывали медленным добавлением насыщенного раствора соли Rochelle's и разбавляли этилацетатом. Слои разделяли и водный слой экстрагировали двумя порциями этилацетата. Объединенный органический экстракт промывали водой и соляным раствором и сушили над MgSO4. Выпариванием и флаш-хроматографией (элюирование 15%-ным этилацетатом в гексане) выделили 370 мг аллилового спирта 135. 1H ЯМР-спектр согласуется со структурой.
(E)-3-[транс-(4-трет-Бутилдиметилсилилоксициклогексил)] -2- метилпроп-2-ен-1-аль (136). В раствор хлористого оксалила (105 мкл, 1,2 ммоля) в 1,0 мл хлористого метилена при -78oC добавляли диметилсульфоксид (170 мкл, 2,4 ммоля). Полученный раствор перемешивали 5 мин и затем добавляли 170 мг (0,6 ммоля) спирта 135 в 1,0 мл хлористого метилена. Реакционную смесь перемешивали 45 мин при -78oC, в это время добавляли 500 мкл (3,6 ммоля) триэтиламина и раствор оставляли для нагревания до комнатной температуры. Реакцию прекращали 1,0 N HCl и водный слой экстрагировали тремя порциями хлористого метилена. Объединенный органический экстракт сушили над MgSO4 и выпаривали досуха, получая неочищенный альдегид 136, который применяли непосредственно в следующей реакции. 1H ЯМР-спектр согласуется со структурой.
(R и S)-(E)-1-[транс-(4-трет-Бутилдиметилсилилоксициклогексил)] -2-метил-6-фенилгекс-1-ен-3-ол (137). Спирт 137 получали из неочищенного альдегида 136 и 1,5 мл (0,75 ммоля) соединения 120 в 2,0 мл ТГФ, как описано выше для синтеза спирта 121 в примере 1. Получили 220 мг неочищенного диастереомерного спирта 137. Флаш-хроматографией (элюирование 20%-ным этилацетатом в гексане) выделили 146 мг спирта 137 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-(E)-1-[транс-(4-трет-Бутилдиметилсилилоксициклогексил)]-2-метил-6- фенил-3-гекс-1-енил-(S)-N-(3,4,6-триметоксифенилглиоксил) пипеколат (138). В раствор 75,7 мг (0,22 ммоля) кислоты 127 в 2,5 мл хлористого метилена при комнатной температуре добавляли 30 мкл (0,34 ммоля) хлористого оксалила и три капли ДМФ и реакционную смесь перемешивали 0,5 ч при комнатной температуре и затем концентрировали и суспензировали в 1,0 мл бензола. В эту суспензию добавляли 43,4 мг (0,11 ммоля) спирта 137 и 28,8 мг (0,22 ммоля) цианида серебра. Полученную смесь нагревали с обратным холодильником в течение ночи, охлаждали до комнатной температуры и концентрировали. Флаш-хроматографией (элюирование 4% ацетона в гексане) выделили 17,5 мг эфира 138 в виде смеси диастереомеров. 1H ЯМР-спектр согласуется со структурой.
(R и S)-(E)-1-[транс-(4-Гидроксициклогексил)]-2-метил-6-фенил- 3-гекс-1-енил-(S)-N-(3,4,6-триметоксифенилглиоксил)пипеколат (8). В раствор 17,5 мг (0,02 ммоля) эфира 138 в 1,0 мл CH3CN при комнатной температуре добавляли 10 капель раствора (95: 5) CH3CN и 5% HF и полученную смесь перемешивали при комнатной температуре 0,5 ч. Реакционную смесь нейтрализовали насыщенным раствором K2CO3 и экстрагировали эфиром. Эфирные слои промывали водой, сушили над MgSO4 и концентрировали, получая 7,2 мг неочищенного продукта. Флаш-хроматографией (элюировали 15% ацетона в гексане) выделили 4,9 мг диастереомерного спирта 8 в виде смеси ротамеров. 1H ЯМР-спектр (500 МГц, CDCl3) 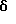 7,38 - 7,02 (м), 5,35 - 5,01 (м), 4,62 - 4,53 (м), 4,28 (т), 3,95 (с), 3,89 (с), 3,87 (с), 3,86 (с), 3,85 (с), 3,81 (с), 3,55 (м), 3,45 (м), 3,20 (м), 3,10 - 2,90 (м), 2,60 - 2,45 (м), 2,32 (т), 2,10 (т), 1,95 (д), 1,85 - 1,40 (м), 1,39 - 1,02 (м).
7,38 - 7,02 (м), 5,35 - 5,01 (м), 4,62 - 4,53 (м), 4,28 (т), 3,95 (с), 3,89 (с), 3,87 (с), 3,86 (с), 3,85 (с), 3,81 (с), 3,55 (м), 3,45 (м), 3,20 (м), 3,10 - 2,90 (м), 2,60 - 2,45 (м), 2,32 (т), 2,10 (т), 1,95 (д), 1,85 - 1,40 (м), 1,39 - 1,02 (м).
Пример 5
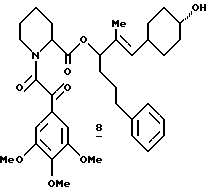
Синтез (R и S)-5-(3-индолил)-1-фенил-2-пентил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколат (II)
N-Метил-N-метокси-4-(3-индолил)бутирамид (139). Во взвесь 1,75 г (8,61 ммоля) 3-индолмасляной кислоты (Aldrich Chemical Co.) в ацетонитриле при комнатной температуре добавляли 7,0 мл (40,2 ммоля) N,N-диизопропилэтиламина, 3,8 г (21,5 ммоля) гидрохлорида N,N-диметилгидроксиламина и 4,19 г (9,5 ммоля) гексафторфосфата бензотриазол-1-илокситри(диметиламино) фосфония (реагент ВОР) и полученную смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали досуха. Остаток растворяли в этилацетате и промывали водой, 0,5 N HCl, насыщенным раствором NaHCO3 и соляным раствором и сушили над MgSO4 и концентрировали. Флаш-хроматографией (градиентное элюирование 2-10% эфира в хлористом метилене) выделили 2,0 г амида 139. 1H ЯМР-спектр согласуется со структурой.
Бензил-3-(3-индолил)пропилкетон (140). В раствор 147 мг (0,60 ммоля) амида 139 в 4,0 мл ТГФ при -78oC добавляли 1,31 мл (1,31 ммоля) хлористого бензилмагния (1,0 M в диэтиловом эфире) и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали 3 ч. Реакцию прерывали добавлением 5% KHSO4 и экстрагировали эфиром. Объединенный эфирный слой промывали соляным раствором и сушили над MgSO4. Флаш-хроматографией (элюирование 25% эфира в гексане) выделили 108 мг кетона 140. 1H ЯМР-спектр согласуется со структурой.
(R и S)-5-(3-индолил)-1-фенил-2-пентанол (141). Во взвесь 105 мг (0,38 ммоля) кетона 140 в 3,0 мл MeOH при 0oC добавляли 30 мг (0,79 ммоля) твердого NaBH4 и образованную суспензию перемешивали 3 ч. В реакционную смесь добавляли 5% KHSO4 и экстрагировали этилацетатом. Объединенный органический экстракт промывали соляным раствором и сушили над MgSO4. Флаш-хроматографией (элюирование 4% эфира в хлористом метилене) выделили 81 мг спирта 141 в виде белого твердого вещества. 1H ЯМР-спектр согласуется со структурой.
(S)-Вос-Пипеколил-(R и S)-5-(3-индолил)-1-фенил-2-пентиловый эфир (142). Эфир 142 получали из 80 мг (0,29 ммоля) спирта 141, 82 мг (0,36 ммоля) (S)-Вос-пипеколиновой кислоты, 66 мг (0,34 ммоля) EDC и каталитического количества 4-пирролидинопиридина в 2,0 мл хлористого метилена (смесь перемешивали в течение ночи при комнатной температуре), как описано выше для синтеза эфира 122 в примере 1. Флаш-хроматографией (элюирование смесью эфира, хлористого метилена, гексана, 4:10:26) выделили 108 мг диастереомерного эфира 142 в виде белой пены. 1H ЯМР-спектр согласуется со структурой.
Гидрохлорид (R и S)-5-(3-индолил)-1-фенил-2-пентил-(S) -пипеколата (143). Безводный HCl барботировали в раствор 103 мг (0,21 ммоля) эфира 142 в 10 мл этилацетата при -20oC в течение 10 мин и затем реакционную смесь продували N2. Концентрированием выделяли 108 мг неочищенного амина 143 в виде гидрохлорида. 1H ЯМР-спектр согласуется со структурой.
(R и S)-5-(3-Индолил)-1-фенил-2-пентил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколат (II). Во взвесь 108 мг неочищенного гидрохлорида амина 143 в CH3CN при комнатной температуре добавляли 91 мкл (0,52 ммоля) N,N-диизопропилэтиламина, 76 мг (0,31 ммоля) кислоты 124 и 111 мг (0,25 ммоля) реагента ВОР и полученную смесь перемешивали два дня при комнатной температуре и затем концентрировали досуха. Остаток растворяли в 75 мл этилацетата и затем последовательно промывали водой, 5% KHSO4, насыщенным раствором NaHCO3 и соляным раствором и затем сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование 4% эфира в хлористом метилене) выделили 56,7 мг диастереомерного амида II в виде ротамерной смеси. 1H ЯМР-спектр (500 МГц, CDCl3) δ 7,98 (д), 7,56 (т), 7,38 - 6,73 (м), 5,38 - 5,14 (м), 3,90 (м), 3,38 (ш. т), 3,10 (ш. т.) 2,97 - 2,60 (м), 2,31 (д), 2,10 (д), 1,98 - 1,17 (м), 0,8 (м), Rf 0,51 (10% эфира в хлористом метилене).
Пример 6
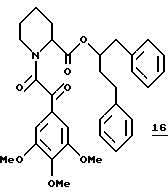
Синтез (R и S)-2-бензил-4-фенил-1-бутил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколата (16)
(R и S)-2-Бензил-4-фенил-1-масляная кислота (144).
В раствор 1,06 г (6,43 ммоля) 4-фенилмасляной кислоты в 20 мл ТГФ при 0oC добавляли 193 мг (6,43 ммоля) твердого NaH (80% в минеральном масле). После перемешивания при 0oC в течение 0,5 ч добавляли 3,2 мл (6,473 ммоля) комплекса диизопропиламид лития-ТГФ (2,0 M) и полученный красный раствор перемешивали при 0oC 45 мин. В эту смесь добавляли 765 мкл (6,43 ммоля) бромистого бензила и раствор перемешивали в течение ночи при комнатной температуре. В реакционную смесь медленно добавляли насыщенный раствор NaHCO3 и затем промывали эфиром. Щелочные экстракты подкисляли твердым KHSO4 и экстрагировали этилацетатом. Объединенный органический экстракт промывали соляным раствором, сушили над MgSO4 и концентрировали, получая 484 мг кислоты 144. 1H ЯМР-спектр согласуется со структурой.
(R и S)-2-Бензил-4-фенил-1-бутанол (145). В раствор 469 мг (1,84 ммоля) кислоты 144 в 3,0 мл ТГФ при -78oC добавляли 2,03 мл (2,03 ммоля) алюмогидрида лития (1,0 M в ТГФ) и полученный раствор оставляли для нагревания до комнатной температуры и перемешивали в течение ночи. В реакционную смесь медленно добавляли соль Rochelle's и экстрагировали эфиром. Объединенный эфирный экстракт промывали водой и соляным раствором и сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование 2% эфира в хлористом метилене) выделили 264 мг спирта 145. 1H ЯМР-спектр согласуется со структурой.
(S)-Вос-Пипеколил-(R и S)-2-бензил-4-фенил-1-бутиловый эфир (146). Эфир 146 получали из 264 мг (1,10 ммоля) спирта 145, 302 мг (1,32 ммоля) (S)-Вос-L-пипеколиновой кислоты, 235 мг (1,32 ммоля) EDC и каталитического количества 4-пирролидинопиридина в 2,0 мл хлористого метилена (смесь перемешивали 3 дня при комнатной температуре), как описано выше для синтеза эфира 122 в примере 1. Флаш-хроматографией (элюирование смесью эфира, хлористого метилена и гексана, 1:5:14) выделили 375 мл диастереомерного эфира 146. 1H ЯМР-спектр согласуется со структурой.
Гидрохлорид (R и S)-2-бензил-4-фенил-1-бутил-(S)-пипеколата (147). Безводный HCl барботировали в раствор 375 мг (0,83 ммоля) эфира 146 в 10 мл этилацетата при -20oC в течение 10 мин и затем реакционную смесь продували N2. Концентрированием продукта получили неочищенный амин 147 в виде гидрохлорида. 1H ЯМР-спектр согласуется со структурой.
(R и S)-2-Бензил-4-фенил-1-бутил-(S)-N-(3,4,5- триметоксифенилглиоксил)пипеколат (16). Во взвесь 54 мг (0,14 ммоля) неочищенного гидрохлорида амина 147 в 2,0 мл CH3CN при комнатной температуре добавляли 60 мкл (0,35 ммоля) N, N-диизопропилэтиламина, 50 мг (0,21 ммоля) кислоты 124 и 73 мг (0,16 ммоля) реагента ВОР и полученную смесь перемешивали 3 дня при комнатной температуре и затем концентрировали досуха. Остаток растворили в 75 мл этилацетата и затем промыли последовательно водой, 5% раствором KHSO4, насыщенным раствором NaHCO3 и соляным раствором и затем сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование 2% эфира в хлористом метилене) выделили 52,7 мг диастереомерного амида 16 в виде ротамерной смеси. 1H ЯМР-спектр (500 МГц, CDCl3) δ 7,21 - 7,01 (м), 5,41 (ш.с), 4,21 (дд), 4,08 (дд), 4,12 (д), 3,88 (д), 3,95 (с), 3,91 (с), 3,49 (д), 3,39 (дт), 2,80 - 2,62 (м), 2,38 (ш.т), 2,09 (ш.с), 1,87 - 1,20 (м). Rf 0,9 (метанол, эфир, хлористый метилен, 1:3:26).
Пример 7
Синтез (R и S)-1-фенил-7-(2-пиридил)-4-гептил-(S)-N-(третбутилглиоксил) пипеколат (21)
(E и Z)-3-(1,3-Диоксан-2-ил)-1-(2-пиридил)-1-пропен (148 и 149). В суспензию 4,6 г (10,2 ммоля) бромистого [2-(1,3-диоксан-2-ил)этил]трифенилфосфония (Aldrich Chemical Co. ) в 50 мл ТГФ при 0oC добавляли 6,4 мл (10,2 ммоля) н-бутиллития (1,6 M в гексанах) и полученный красный раствор перемешивали при 0oC 0,5 ч. В этот раствор добавляли 880 мкл (9,3 ммоля) 2-пиридинкарбоксальдегида (Aldrich Chemical Co.). Полученную смесь перемешивали 1 ч при комнатной температуре и затем выливали в воду и экстрагировали эфиром. Объединенный эфирный экстракт сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование смесью гексана и этилацетата, 3:1) выделили 0,43 г E-3-(1,3-диоксан-2-ил)-1-(2-пиридил)-1-пропена (148) и 1,12 г Z-3-(1,3-диоксан-3-ил)-1-(2-пиридил)-1-пропена (149). 1H ЯМР-спектры согласуются со структурами.
1-(1,3-Диоксан-2-ил)-3-(2-пиридил)пропан (150). Через суспензию 800 мг (4,2 ммоля) олефина 149 и 100 мг 10% палладия на угле барботировали равномерный поток водорода в течение периода 10 мин. Реакционную смесь затем фильтровали через целит и концентрировали, получая 805 мг ацеталя 150 в виде бесцветного масла. 1H ЯМР-спектр согласуется со структурой.
4-(2-Пиридил)-1-масляный альдегид (151). Раствор 420 мг (2,2 ммоля) ацеталя 150 в 4,0 мл ТГФ и 3,0 мл 4 N HCl перемешивали при комнатной температуре 1,5 ч и затем нейтрализовали медленным добавлением твердого NaHCO3. Реакционную смесь экстрагировали этилацетатом, сушили над MgSO4 и концентрировали, получая 288 мг альдегида 151. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-Фенил-7-(2-пиридил)-4-гептанол (152). Спирт 152 получали из 288 г (1,93 ммоля) альдегида 151 и 2,3 мл (2,3 ммоля) 120 в 3,0 мл ТГФ, как описано выше для синтеза спирта 121 в примере 1. Получили 520 мг неочищенного спирта 152. 1H ЯМР-спектр согласуется со структурой.
(S)-Вос-Пипеколил-(R и S)-1-фенил-7-(2-пиридил)-4-гептиловый эфир (153). Эфир 153 получали из 520 мг (1,93 ммоля) спирта 152, 442 мг (1,93 ммоля) (S)-Вос-L-пипеколиновой кислоты, 370 мг (1,93 ммоля) EDC и каталитического количества DMAP в 4,0 мл хлористого метилена и 4,0 мл ДМФ, как описано выше для синтеза 122 в примере 1. Флаш-хроматографией (элюирование смесью гексана и этилацетата, 3:1) выделили 740 мг диастереомерного эфира 153 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-Фенил-7-(2-пиридил)-4-гептил-(S)-пипеколат (154). Амин 154 синтезировали обработкой 740 мг (1,54 ммоля) эфира 153 2,0 мл трифторуксусной кислоты в 5,0 мл хлористого метилена, как описано выше для получения 123 в примере 1. Получили 580 мг диастереомерного амина 154 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-Фенил-7-(2-пиридил)-4-гептанол-(S)-N- метилоксалилпипеколат (155). В раствор 48 мг (0,13 ммоля) амина 154 в 1,0 мл хлористого метилена при 0oC добавляли 33 мкл (0,19 ммоля) N,N-диизопропилэтиламина и 14 мкл (0,15 ммоля) хлористого метилоксалила и полученный раствор нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором NH4Cl и соляным раствором, сушили над MgSO4 и затем концентрировали. Флаш-хроматографией (элюирование 25-30% этилацетата в гексане) выделили 49 мг диастереомерного амида 155 в виде смеси ротамеров. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-Фенил-7-(2-пиридил)-4-гептанол-(S)-N-(трет-бутилглиоксил)пицеколат (21). В раствор амида 155 в 1,2 мл ТГФ при -78oC добавляли по каплям трет-бутиллитий до полного расхода исходного соединения (определяли ТСХ). В реакционную смесь добавляли насыщенный раствор NH4Cl и экстрагировали этилацетатом. Объединенный органический экстракт промывали соляным раствором, сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование 30% этилацетата в гексане) выделили диастереомерный амид 21 в виде смеси ротамеров. 1H- ЯМР-спектр (500 МГц, CDCl3) δ 8,50 (5), 7,57 (5), 7,20-7,05 (м), 5,23 (д), 5,18 (д), 4,56 (д), 4,44 (ш.д), 4,13 (д), 3,69 (ш. д), 3,37-3,28 (м), 3,13-3,00 (м), 2,85-2,70 (м), 2,65-2,54 (м), 2,38-2,15 (м), 1,82-1,65 (м), 1,56-1,44 (м), 1,55-1,30 (м), 1,27 (с), 1,21 (с).
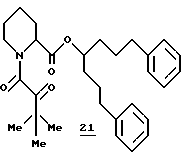
Пример 8
Синтез (R и S)-1-(3-фенилпропил)-4-пиридин-3-ил-бутилового эфира (S)-1-[2-Оксо-2-(3,4,5-триметоксифенил)ацетил]пиперидин-2-карбоновой кислоты (9)
(E и Z)-3-(1,3-Диоксан-2-ил)-1-(3-пиридил)-1-пропен (156). В суспензию 9,9 г (22,4 ммоля) бромистого [2-(1,3-диоксан-2-ил)этил]трифенилфосфония (Aldrich Chemical Co. ) в 50 мл ТГФ при 0oC добавляли 14,0 мл (22,4 ммоля) бутиллития (1,6 М в гексане) и полученный красный раствор перемешивали 0,5 ч при 0oC. В этот раствор добавляли 1,8 мл (18,7 ммоля) 3-пиридинкарбоксальдегида (Aldrich Chemical Co.) и реакционную смесь перемешивали при комнатной температуре 1,5 ч и затем выливали в воду и экстрагировали эфиром. Объединенный эфирный экстракт сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование смесью гексана и этилацетата, 2:1) выделили 3,3 г алкена 156 в виде смеси олефиновых изомеров. 1H-ЯМР-спектр согласуется со структурой.
1-(1,3-Диоксан-2-ил)-3-(3-пиридил)пропан (157). Через раствор 3,2 г (16,7 ммоля) олефина 156 и 300 мг 10% палладия на угле барботировали равномерный поток водорода в течение 10 мин. Реакционную смесь затем фильтровали через целит и концентрировали, получая 2,8 г ацеталя 157 в виде бесцветного масла. 1H ЯМР-спектр согласуется со структурой.
4-(3-Пиридил)-1-масляный альдегид (158). Раствор 1,5 г (7,8 ммоля) ацеталя 157 в 10,0 мл ТГФ и 10,0 мл 4N HCl перемешивали в течение ночи при комнатной температуре и затем нейтрализовали медленным добавлением твердого NaHCO3. Реакционную смесь экстрагировали этилацетатом, сушили над MgSO4 и концентрировали, получая 1,1 г альдегида 158. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-Фенил-7-(3-пиридил)-4-гептанол (159). Спирт 159 получали из 1,1 г (7,4 ммоля) альдегида 158 и 8,1 мл (8,1 ммоля) соединения 120 в 30,0 мл ТГФ, как описано выше дл синтеза 121 в примере 1. Получили 1,9 г неочищенного спирта 159. 1H ЯМР-спектр согласуется со структурой.
(S)-Boc-Пипеколил-(R и S)-1-фенил-7-(3-пиридил)-4-гептиловый эфир (160). Эфир 160 получали из 1,65 г (6,12 ммоля) спирта 159, 1,54 г (6,73 моля) (S)-Boc-пипеколиновой кислоты, 1,29 г (6,73 ммоля) EDC и каталитического количества DMAP в 8,0 мл хлористого метилена и 8,0 мл ДМФ, как описано выше для синтеза 122 в примере 1. Флаш-хроматографией (элюирование смесью гексана и этилацетата, 2:1) выделили 1,42 г диастереомерного эфира 160 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-Фенил-7-(3-пиридил)-4-гептил-(S)-пипеколат (161). Амин 161 синтезировали обработкой 1,42 г (2,95 ммоля) эфира 160 2,0 мл трифторуксусной кислоты в 8,0 мл хлористого метилена, как описано выше для синтеза 123 в примере 1. Получили 1,02 г диастереомерного амина 161 в виде масла. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-фенил-7-(3-пиридил)-4-гептил-(S)-N-(3,4,5-триметоксифенилглиоксил)пипеколат (9). Эфир 9 получали из 995 мг (2,61 ммоля) амина 161, 645 мг (2,87 ммоля) кислоты 124 и 551 мг (2,87 ммоля) EDC в 6,0 мл хлористого метилена, как описано выше для синтеза эфира 3 в примере 1. Флаш-хроматографией (элюирование смесью ацетона и гексана, 3:1) выделили 976 мг диастереомерного амида 9 в виде смеси ротамеров. 1H ЯМР-спектр согласуется со структурой.
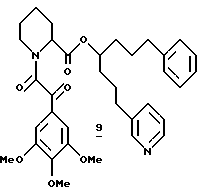
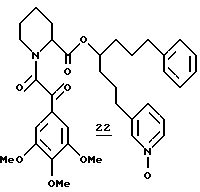
Пример 9
Синтез N-оксида (R и S)-1-фенил-7-(3-пиридил)-4-гептил-(S)-N- (3,4,5-триметоксифенилглиоксил)-пипеколата (22)
N-Оксид (R и S)-1-фенил-7-(3-пиридил)-4-гептил-(S)-N- (3,4,5-триметоксифенилглиоксил)пипеколата (22). В раствор 15 мг (0,02 ммоля) амида 9 в 2,0 мл хлористого метилена при комнатной температуре добавляли 9,3 мкл (0,03 ммоля) 55% 3-хлорпероксибензойной кислоты и полученный раствор перемешивали в течение ночи при комнатной температуре. Флаш-хроматографией (элюирование 100%-ным ацетоном) выделили 12,6 мг N-оксида 22 в виде смеси ротамеров. 1H ЯМР-спектр (500 МГц, CDCl3) δ 8,10 (м), 7,46-7,02 (м), 5,88 (д), 5,80 (д), 5,06-5,00 (м), 4,95-4,89 (м), 4,61 (м), 4,31 (дд), 3,87 (с), 3,84 (с), 3,83 (с), 3,81 (с), 3,78 (с), 3,50 (ш.д), 3,27 (ддд), 3,12 (ддд), 3,00 (ддд), 2,67-2,49 (м), 2,32 (ш.д), 1,86-1,78 (м), 1,55-1,50 (м), 1,39-1,22 (м).
Пример 10
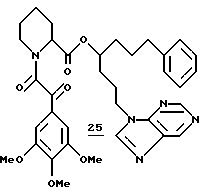
Синтез (R и S)-1-фенил-7-пуринил-4-гептил-(S)-N-(3,4,5-триметоксифенилглиоксил)пипеколата (25)
4-Хлорфенилмасляный альдегид (162). В раствор 19,1 г (0,16 моля) 4-хлор-1-бутанола (Aldrich Chemical Co.) в 50 мл хлористого метилена при 0oC добавляли 1,0 г порошкообразных молекулярных сит 4  и 38,7 г (0,18 моля) бихромата пиридиния и полученную суспензию перемешивали при 0oC 45 мин. Реакционную смесь разбавляли эфиром, фильтровали через целит и концентрировали. Остаток перегоняли в вакууме (т.кип. 45-55oC), получая 5,0 г альдегида 162 в виде масла. 1H-ЯМР-спектр согласуется со структурой.
и 38,7 г (0,18 моля) бихромата пиридиния и полученную суспензию перемешивали при 0oC 45 мин. Реакционную смесь разбавляли эфиром, фильтровали через целит и концентрировали. Остаток перегоняли в вакууме (т.кип. 45-55oC), получая 5,0 г альдегида 162 в виде масла. 1H-ЯМР-спектр согласуется со структурой.
(R и S)-1-Хлор-7-фенил-4-гептанол (163). Спирт 163 получали из 182 мг (1,7 ммоля) альдегида 162 и 1,9 мл (1,9 ммоля) соединения 120 в 20,0 мл ТГФ, как описано выше для синтеза 121 в примере 1. Получили 128 мг спирта 163 (флаш-хроматографией в 100%-ном хлористом метилене). 1H ЯМР-спектр согласуется со структурой.
(S)-Boc-Пипеколил-(R и S)-1-хлор-7-фенил-4-гептиловый эфир (164). Эфир 164 получали из 128 мг (0,56 ммоля) спирта 163, 156 мг (0,68 ммоля) (S)-Boc-пипеколиновой кислоты, 1380 мг (0,68 ммоля) EDC и каталитического количества 4-пирролидинопиридина в 2,0 мл хлористого метилена, как описано выше для синтеза 122 в примере 1. Флаш- хроматографией (элюирование смесью эфира, хлористого метилена и гексана, 1:5:14) выделили 159 мг диастереомерного эфира 164. 1H ЯМР-спектр согласуется со структурой.
(S)-Boc-Пипеколил-(R и S)-1-фенил-7-пуринил-4-гептиловый эфир (165). В раствор 34 мг (0,28 ммоля) пурина в 3,0 мл ДМФ при комнатной температуре добавляли 8,4 мг (0,28 ммоля) твердого NaH (80% в минеральном масле) и полученный раствор перемешивали при комнатной температуре 10 мин. В эту реакционную смесь добавляли 62 мг (0,14 ммоля) эфира 164 и 10 мг NaCl и перемешивали при комнатной температуре в течение ночи и затем концентрировали досуха. Остаток растворяли в этилацетате, промывали последовательно водой, насыщенным раствором NaHCO3 и соляным раствором и затем сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование 15%-ным раствором смеси NH4OH, MeOH и CH2Cl2 в соотношении 5:10:85 в CH2Cl2) выделили 56 мг замещенного пурина 165 в виде масла. 1H ЯМР-спектр согласуется со структурой.
Гидрохлорид (R и S)-1-фенил-7-пуринил-4-гептил-(S)-пипеколата (166). Безводный HCl барботировали в раствор 53,7 мг (0,10 ммоля) эфира 165 в 10 мл этилацетата при -20oC в течение 10 мин и затем реакционную смесь продували N2. Концентрированием получили неочищенный амин 166 в виде гидрохлорида. 1H ЯМР-спектр согласуется со структурой.
(R и S)-1-фенил-7-пуринил-4-гептил-(S)-N-(3,4,5-триметоксифенилглиоксил)пипеколат (25). Во взвесь неочищенного гидрохлорида амина 166 в CH3CN при комнатной температуре добавляли 45 мкл (0,26 ммоля) N,N-диизопропилэтиламина, 37 мг (0,15 ммоля) кислоты 124 и 54 мг (0,12 ммоля) реагента ВОР и полученную смесь перемешивали при комнатной температуре в течение двух дней и затем концентрировали досуха. Остаток растворяли в 75 мл этилацетата и затем промывали последовательно водой, 5%-ным KHSO4, насыщенным NaHCO3 и соляным раствором и затем сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование смесью метанола, диэтилового эфира и хлористого метилена, 1: 4:36) выделили 26,5 мг диастереомерного амида 25 в виде ротамерной смеси. 1H ЯМР-спектр (500 МГц. CDCl3) δ 9,11 (с), 8,95 (м), 8,09 (м), 7,36-7,05 (м), 5,31 (м), 4,28 (м), 3,90 (м), 3,46 (ш.т), 3,20 (м), 2,58 (м), 2,28 (ш, д), 2,17-1,18 (м), Rf 0,1 (30% эфира в хлористом метилене).
Пример 11
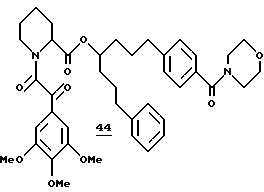
Синтез (R и S)-4-[4-(морфолин-4-карбонил)фенил] - 1-(3-фенилпропил)бутилового эфира (S)-1-[2-оксо-2- (3,4,5-триметоксифенил)ацетил]пиперидин-2-карбоновой кислоты (44)
Метиловый эфир 4-формилбензойной кислоты (167). В суспензию 9,6 г (63,6 ммоля) 4-карбоксибензальдегида (Aldrich Chemical Co.) в 100 мл хлористого метилена при 0oC добавляли избыток триметилсилилдиазометана и полученную смесь перемешивали при 0oC 1 час. Смесь выливали в насыщенный водный раствор NaHCO3 и экстрагировали три раза этилацетатом. Объединенный органический экстракт сушили над MgSO4, фильтровали и концентрировали, получая 4,3 г эфира 176 в виде масла. 1H ЯМР-спектр согласуется со структурой.
Метиловый эфир (E и Z)-4-(3-(1,3-диоксолан-2-ил)пропенил)бензойной кислоты (168). Олефин получали из 4,3 г (26,2) ммоля) альдегида 167, 13,94 г бромистого [1-(1,3-диоксан-2-ил)этил]трифенилфосфония и 12,6 мл (32,0 ммоля) н-бутиллития в 75 мл ТГФ, как описано выше для синтеза 156 в примере 8. Флаш-хроматографией (элюирование 10% этилацетата в гексане) выделили 3,27 г олефина 168. 1H ЯМР-спектр согласуется со структурой.
Метиловый эфир 4-[3-(1,3-диоксан-2-ил)пропил] бензойной кислоты (169). Олефин 169 (3,21 г, 12,9 ммоля) гидрировали над 328 мг 10% Pd/C в 50 мл этилового спирта, как описано для получения соединения 157 в примере 8. После фильтрования и выпаривания получили 2,85 г соединения 169 в виде масла. 1H ЯМР-спектр согласуется со структурой.
[4-(3-(1,3-диоксанил-2)пропилфенил]метанол (170). В раствор 2,85 г (11,4 ммоля) эфира 169 в 25 мл ТГФ при 0oC добавляли 4,4 мл (24,7 ммоля) гидрида диизобутилалюминия и полученную смесь перемешивали при 0oC 15 мин. Реакцию прекращали насыщенным раствором смешанного тартрата калия и натрия и экстрагировали три раза этилацетатом. Объединенный органический слой сушили над MgSO4, фильтровали и концентрировали, получая 2,58 г неочищенного спирта 170 в виде масла. 1H ЯМР-спектр согласуется со структурой.
2-[3-(4-трет-Бутилдифенилсилилоксиметилфенил)пропил] -1,3-диоксолан (171). В раствор 2,58 г (11,6 ммоля) спирта 170 и 1,19 г (17,5 ммоля) имидазола в 50 мл хлористого метилена добавляли 3,4 мл (13,1 ммоля) трет-бутилхлордифенилсилана и полученную смесь перемешивали 1 час при комнатной температуре. Смесь затем разбавляли этилацетатом и промывали 0,5 N HCl. Органический слой сушили над MgSO4, фильтровали и концентрировали. Флаш-хроматографией (элюирование 5% этилацетата в гексане) выделили 5,5 г соединения 171. 1H ЯМР-спектр согласуется со структурой.
4-(4-трет-Бутилдифенилсилилоксиметилфенил)масляный альдегид (172). В раствор 5,5 г (11,9 ммоля) диоксолана 171 в 40 мл ТГФ при комнатной температуре добавляли 40 мол 4,0 N HCl и полученный раствор перемешивали 1 час. Смесь затем нейтрализовали твердым K2CO3, экстрагировали этилацетатом и концентрировали. Неочищенную смесь растворяли в 25 мл хлористого метилена, в который добавляли 600 мг (8,8 ммоля) имидазола и 1,9 мл (7,3 ммоля) трет-бутилхлордифенилсилана. Полученную смесь перемешивали при комнатной температуре в течение ночи и затем выливали в 0,5 N HCl и экстрагировали этилацетатом. Экстракт сушили над MgSO4, фильтровали и концентрировали. Флаш-хроматографией (элюирование 8% этилацетата в гексане) выделили 2,12 г альдегида 172 в виде масла. 1H ЯМР-спектр согласуется со структурой.
1-(4-трет-Бутилдифенилсилилоксиметилфенил)-7-фенилгептан-4-ол (173). Спирт 173 получали из 2,12 г (5,0 ммоля) соединения 172 и 9,0 мл (9 ммоля) соединения 120 в 50 мл ТГФ, как описано выше для синтеза 121 в примере 1. Флаш-хроматографией (элюирование 10% этилацетата в гексане) выделили 3,3 г спирта 173. 1H ЯМР-спектр согласуется со структурой.
(R и S)-2-[4-(4-трет-Бутилдифенилсилилоксиметилфенил)-1-(3-фенилпропил) бутиловый] -1-трет-бутиловый эфир (S)-пиперидин-1,2-дикарбоновой кислоты (174). Эфир 174 получали из 3,3 г (6,15 ммоля) спирта 173, 1,7 г (7,4 ммоля) (S)-Вос-пипеколиновой кислоты, 1,4 г (7,3 ммоля) EDC и каталитического количества DMAP в 35 мл хлористого метилена, как описано выше для синтеза 122 в примере 1. Флаш-хроматографией (элюирование 5% этилацетата в гексане) выделили 2,4 г эфира 174. 1H ЯМР-спектр согласуется со структурой.
1-трет-Бутиловый-(R и S)-2-[4-(4-гидроксиметилфенил)-1-(3-фенилпропил)бутиловый] эфир (S)-пиперидин-1,2-дикарбоновой кислоты (175). В 750 г (1,0 ммоля) эфира 174 в 10 мл ТГФ добавляли 1,1 мл (1,1 ммоля) раствора фтористого тетрабутиламмония (1,0 M в ТГФ) и полученную смесь перемешивали при комнатной температуре 15 мин. Смесь разбавляли этилацетатом, промывали 5% KHSO4, сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюирование 20% этилацетата в гексане) выделили 308 мг спирта 175. 1H ЯМР-спектр согласуется со структурой.
1-трет-Бутиловый (R и S)-2-[4-(4-карбоксифенил)-1-(3-фенилпропил)бутиловый] эфир (S)-пиперидин-1,2-дикарбоновой кислоты (176). В раствор 326 мг (0,64 ммоля) спирта 175 в 3,0 мл ацетона добавляли 0,5 мл (1,27 ммоля) реагента Джонса и полученную смесь перемешивали при комнатной температуре 1 час и затем фильтровали через целит и концентрировали. Флаш-хроматографией (элюирование 2% MeOH в хлористом метилене) выделили 155 мг кислоты 176. 1H ЯМР-спектр согласуется со структурой.
Трифторацетатная соль (R и S)-4-(4-карбоксифенил)-1-(3-фенилпропил)бутилового эфира (S)-пиперидин-2-карбоновой кислоты (177). В раствор 155 мг (0,3 ммоля) кислоты 176 в 3,0 мл хлористого метилена добавляли 500 мкл трифторуксусной кислоты и полученный раствор перемешивали при комнатной температуре 3 часа, в это время летучую часть удаляли в вакууме. Неочищенный остаток суспензировали в 5,0 мл сухого бензола и летучую часть удаляли, получая безводную пробу кислоты 177.
(R и S)-4-(4-карбоксифенил)-1-(3-фенилпропил)бутиловый эфир (S)-1-[2-оксо-2-(3,4,5-триметоксифенил)ацетил] пиперидин-2-карбоновой кислоты (178). В суспензию 159 мг (0,3 ммоля) соли 177 в 2,5 мл хлористого метилена при 0oC добавляли 110 мкл (0,63 ммоля) N,N-диизопропилэтиламина и затем 40 мкл (0,31 ммоля) хлортриметилсилана и полученную смесь перемешивали при 0oC в течение 30 мин. В этот раствор добавляли 85 мг (0,44 ммоля) EDC и 106 мг (0,44 ммоля) кислоты 124 и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли этилацетатом и промывали 0,5 N HCl, водой, соляным раствором, сушили над MgSO4 и концентрировали. Флаш-хроматографией (элюировали 30% MeOH в хлористом метилене) получили 97 мг продукта 178 в виде смеси ротамеров. 1H ЯМР-спектр согласуется со структурой.
(R и S)-4-[4-(морфолин-4-карбонил)фенил] -1-(3-фенилпропил) бутиловый эфир (S)-1-[2-оксо-2-(3,4,5-триметоксифенил)ацетил] пиперидин-2-карбоновой кислоты (44). В раствор 11,2 мг (17 мкмоля) кислоты 178 в 1,0 мл хлористого метилена добавляли 4,1 мг (21,4 мкмоля) EDC и 1,8 мг (20,7 мкмоля) морфолина и полученный раствор перемешивали при комнатной температуре в течение ночи. Флаш-хроматографией (элюирование 20% MeOH в хлористом метилене) выделили 7,6 мг амида 44 в виде смеси ротамеров. 1H ЯМР-спектр (500 МГц, CDCl3) δ 7,32 (д), 7,30 (д), 7,26 (с), 7,21 - 7,08 (м), 5,33 (м), 5,01 (м), 4,92 (м), 3,92 (с), 3,89 (с), 3,88 (с), 3,87 (с), 3,86 (с), 3,85 (с), 3,81-3,53 (м), 3,42 (ш. д. ), 3,29-3,21 (м), 3,05 (м), 2,61 (с), 2,42 (дд), 2,31 (д), 2,12 (м), 1,83 (м), 1,73 -1,42 (м), 2,42-1,20 (м).
Далее см. пример 12: Данные ЯМР-спектров (прилагаются в конце описания).
Пример 13
Испытания на сенсибилизацию MDR-клеток
Для испытания способности соединений в соответствии с настоящим изобретением к повышению антипролиферативной активности лекарственного средства можно применять клеточные линии, которые известны как устойчивые к конкретному лекарственному средству. Эти клеточные линии включают, но не ограничиваются ими, клеточные линии L1210, P388D, CHO и MCF7. Альтернативно устойчивые клеточные линии можно развивать. Клеточные линии подвергают действию лекарственного средства, к которому они устойчивы, или испытываемого соединения. Затем измеряют жизнеспособность клеток и сравнивают с жизнеспособностью клеток, которые подвергали действию лекарственного средства в присутствии испытываемого соединения.
Мы проводили испытания с применением клеток лейкемии мышей L1210, трансформированных ретровирусами pHa MDRI/A, имеющими кДНК MDRI, как описано Pastan et al., Proc. Natl. Acad. Sci., Vol. 85, 4486-4490 (1988). Устойчивую линию, меченую L1210VMDRC. 06, получили от M.M.Gottesman of the National Cancer Institute. Эти устойчивые к лекарственным средствам трансфектанты отбирали культивированием клеток в 0,06 мг/мл колхицина.
Испытания на устойчивость ко многим лекарственным средствам проводили путем посева клеток (2•103, 1•104 или 5•104 клеток/лунку) в 96 лунках микротитрационного планшета и подвергания их действию доксорубицина с пределом концентрации 50 нм - 10 мкМ в присутствии или отсутствие соединений-модификаторов устойчивости ко многим лекарственным средствам (MDR-ингибиторов) настоящего изобретения (1, 2,5 или 10 мкМ), как описано в Ford et al., Canser Res., Vol. 50, 1748-1756, (1990). После культивирования в течение 3 дней жизнеспособность клеток количественно определяли с применением красителей MTT (Mossman) или XTT для установления митохондриальной функции. Все определения проводили в 4-8 повторениях. Смотри также Mossman T., J. Immunol. Methods, Vol. 65, 55-63 (1983).
Результаты получали сравнением IC50 только для доксорубицина с IC50 для доксорубицина +MDR-ингибитора. Подсчитывали MRD-отношение (IC50Dox/IC50Dox+ингибитор) и целые числа применяли для сравнения эффективности соединений.
Во всех анализах соединения настоящего изобретения испытывали на присущую антипролиферативную или цитотоксичную активность. Результаты суммированы в приведенной ниже таблице 2. Как указано в таблице 2, соединения обычно вызывали менее 10% цитотоксичности при концентрациях 10 мкМ или выше.
Соединения формулы (I) испытывали также на MDR-сенсибилизирующую активность с применением других MDR-клеточных линий, включая несколько клеточных линий человека, например, клетки миеломы (8226/DOX6, 8226/DOX40, MDR10V, MR20), клетки меланомы (VCR4.5, VBL3.0, COL-1), клетки GM369T, клетки карциномы молочной железы MCF-7, бронхогенной адренокарциномы A549, меланомы LOX, P388/ADR и P388VMDRC.04) и различных химиотерапевтических лекарственных средств (например, доксорубицина, винкристина, винбластина, таксола, колхицина и этопозида). Результаты, аналогичные тем, которые приведены в таблице 2, были получены в этих испытаниях (данные не приведены). Они дополнительно показывают эффективность соединений настоящего изобретения в сенсибилизации клеток при устойчивости их ко многим лекарственным средствам.
Примеры фармацевтических композиций, дополнительно содержащих хемосенсибилизатор.
Пример 14
Соединение настоящего изобретения, такое, как 4-пиридин-3-ил-1-(3-пиридин-3-ил)пропил)бутиловый эфир (S)-1-(2-оксо-2-(3,4,5-триметоксифенил)ацетил)-2-карбоновой кислоты (0,5 г), циклоспорин A (25 мг) и карбонат кальция (4,5 г) измельчают и смешивают вместе с получением гомогенного порошка. Полученный порошок затем помещают в желатиновые капсулы для перорального введения, содержащие 25 мг эфира каждая.
Пример 15
Соединение настоящего изобретения, такое, как 4-пиридин-3-ил-1-(3-пиридин-3-ил)пропил)бутиловый эфир (R)-1-(2-оксо-2-(3,4,5-триметоксифенил)ацетил)-2-карбоновой кислоты (5,0 г), циклоспорин A (25 мг) и сухой кукурузный крахмал (5,0 г) и стеарат магния (3,0 г) измельчают и смешивают вместе с получением гомогенного порошка. Полученный порошок затем помещают в желатиновые капсулы для перорального введения, содержащие 200 мг эфира каждая.
Пример 16
Соединение настоящего изобретения (250 мг) и циклоспорин A (25 мг) смешивают с лактозой (200 мг) и натрийкрахмалгликолятом (25 мг). Смесь затем гранулируют в воде и сушат. Последующее добавление стеарата магния (5 мг) и прессование полученной смеси дают таблетки для перорального введения.
Хотя мы описали ряд примеров осуществления настоящего изобретения, очевидно, что наши основные построения можно изменить для того, чтобы предложить другие примеры осуществления изобретения, которые применяют продукты, способы их получения и способы применения настоящего изобретения. Следовательно, следует признать, что объем этого изобретения нужно определять предлагаемой формулой изобретения, а не конкретными примерами осуществления изобретения, которые представлены путем приведения примеров.




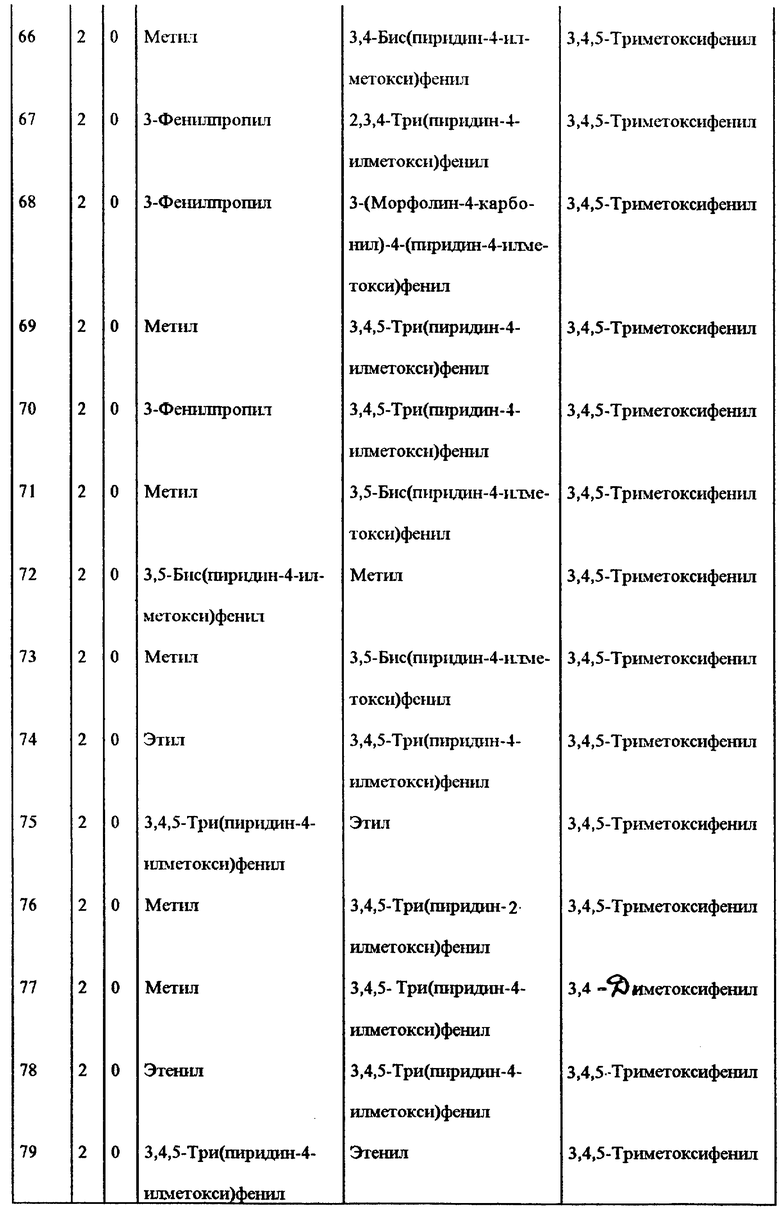
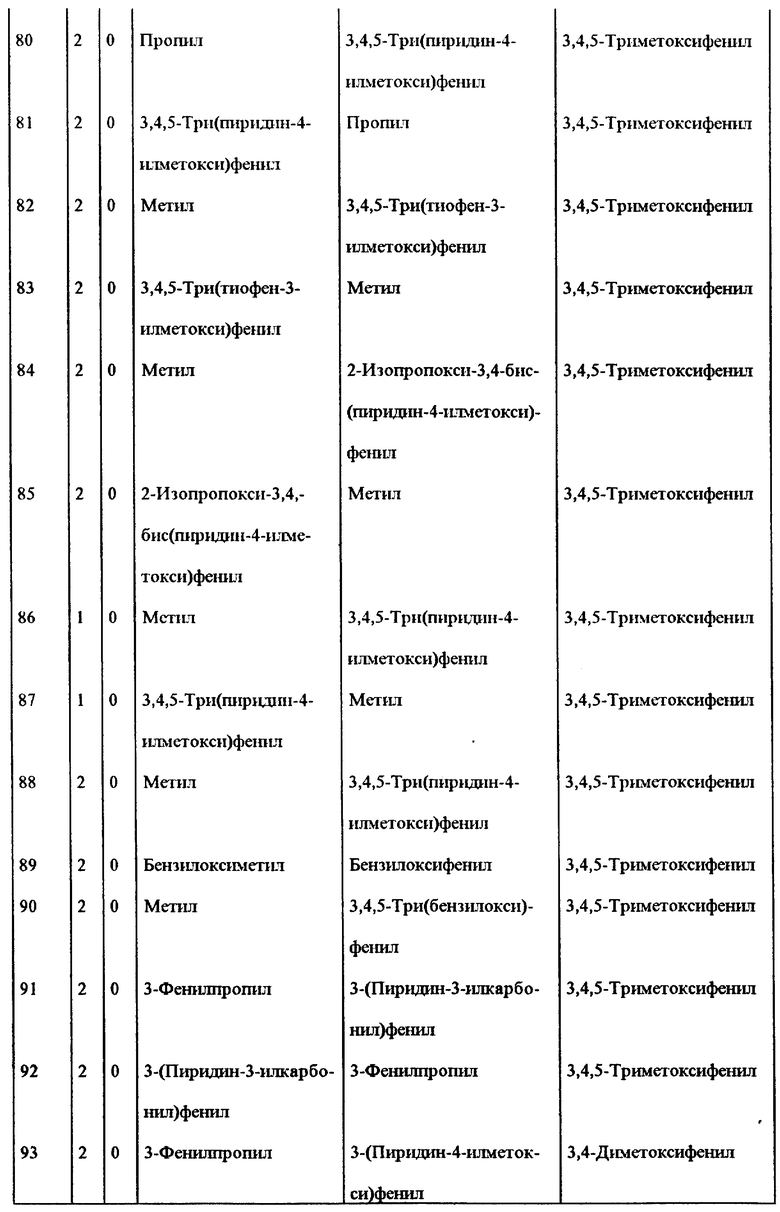
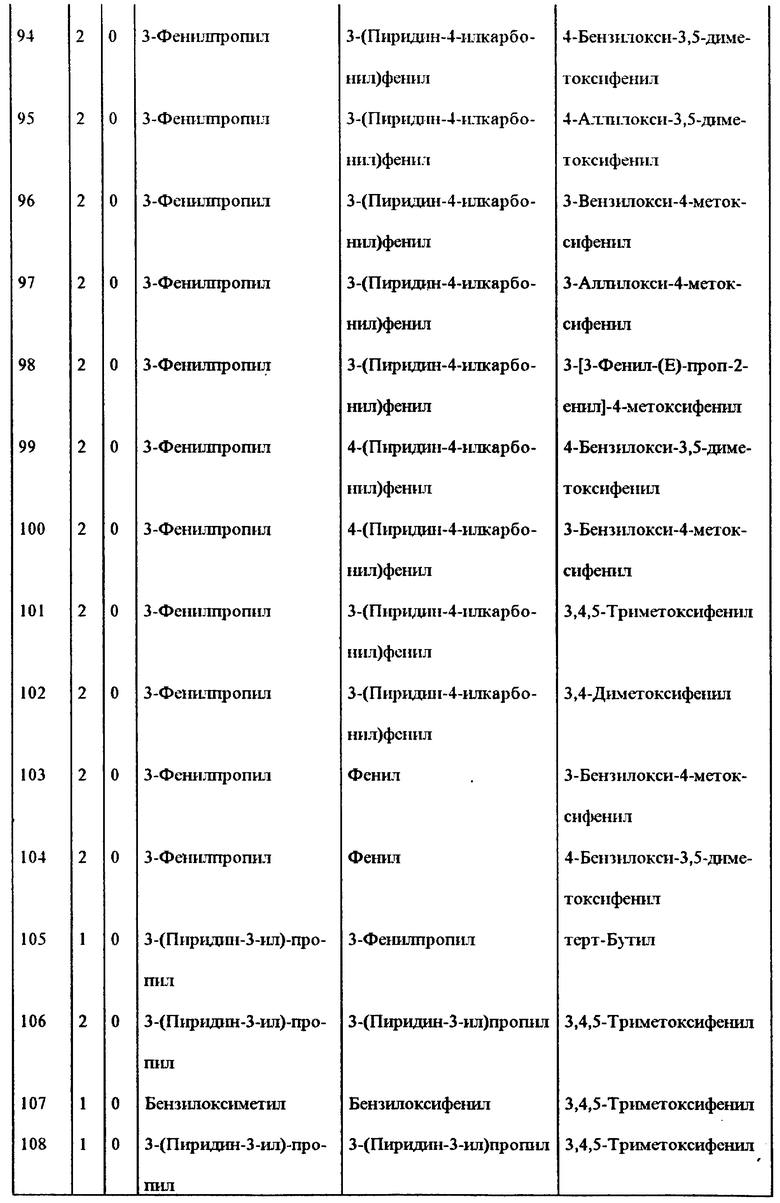
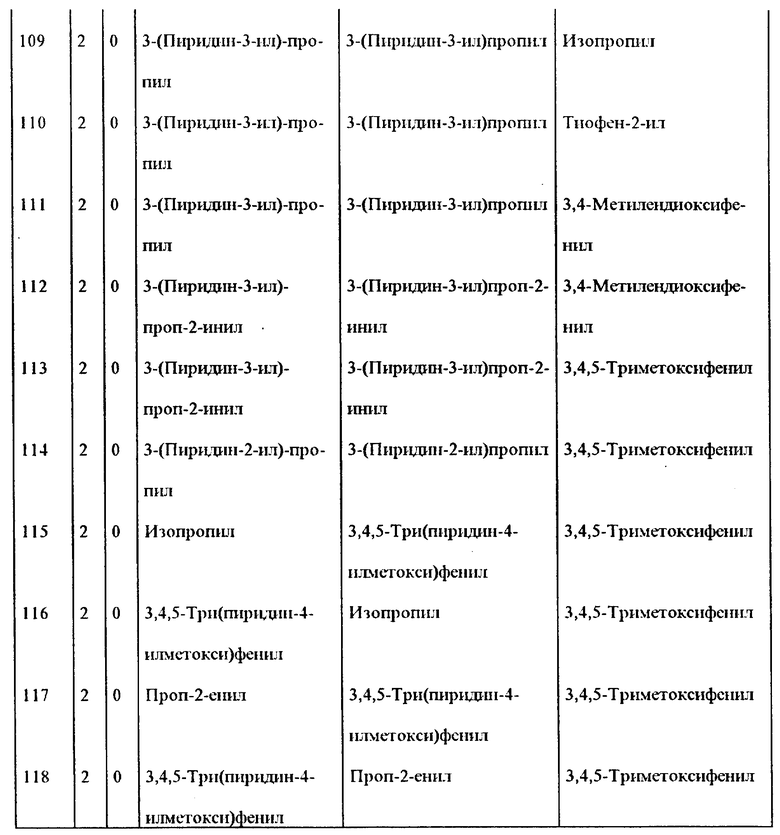
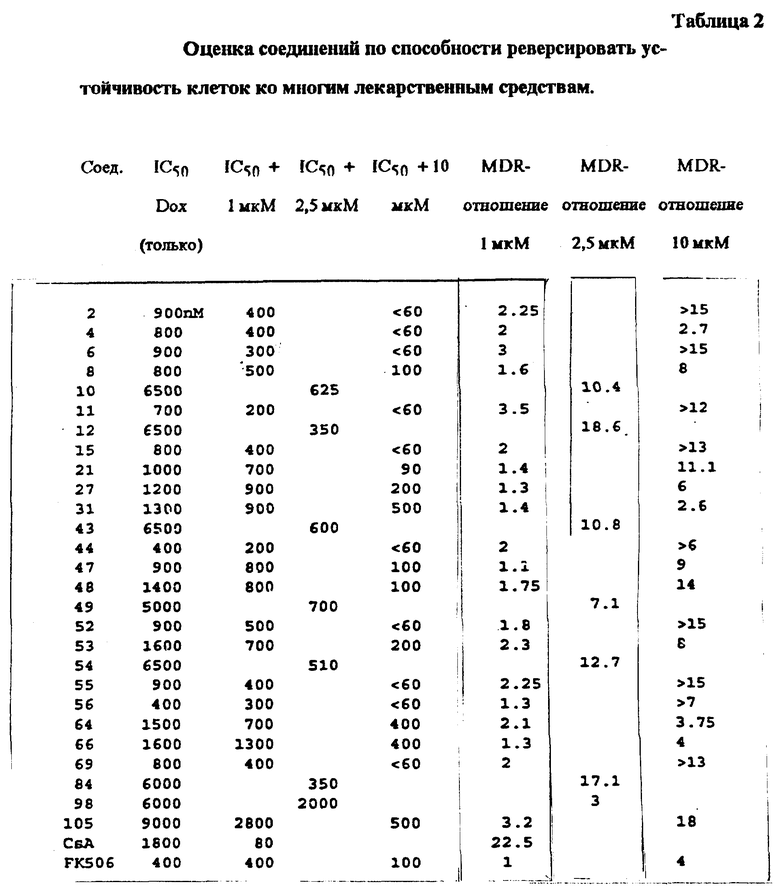






















Описываются новые производные 1-(2-оксоацетил)пиперидин- или пирролидин-2-карбоновых кислот общей формулы (I), где А представляет кислород, В и D независимо представляют собой Ar, (C1-C10)-алкил нормального или разветвленного строения, (C2-C10)-алкенил или (C2-C10)-алкинил нормального или разветвленного строения, Ar - замещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)-алкенил или (C2-C6)-алкинил нормального или разветвленного строения, причем в каждом случае любая одна из CH2-групп указанных алкилов, алкенилов или алкинилов необязательно может быть замещена на атом кислорода или группу формулы (ii), в которой Q представляет собой водород или (C1-C6)-алкил нормального или разветвленного строения, Т представляет Ar или 5-7- членный циклоалкил, замещенный в 4 положении гидроксигруппой, при условии, что по меньшей мере один из B и D независимо выбран из группы, включающей (C2-C10)-алкинил нормального или разветвленного строения, Ar - замещенный (C2-C6)-алкинил нормального или разветвленного строения, где Ar означает фенил, 2-тиенил, 2-, 3- или 4-пиридил, бензимидазолил, 3H-индолил, пуринил, 7-азаиндолил, Ar может необязательно содержать 1-3 заместителей, независимо выбранных из группы, включающей гидроксигруппу, гидроксиметильную группу, O-(C1-C4)-алкенил нормального или разветвленного строения, O-фенил, O-бензил, карбоксигруппу, N,N-ди((C1-C5)-алкил нормального или разветвленного строения) карбоксамид, N-бензилкарбоксамид, морфолинкарбонил, тиоморфолинкарбонил, N-метил или N-бензилпиперазинокарбонил, или группу формулы -O-(CH2)q-X, где X представляет собой 2, 3- или 4-пиридил, пиразинил или 2- или 3-тиенил, q равно целому числу 0 - 2, где L представляет собой U, M представляет кислород, U представляет собой (C1-C6)-алкил нормального или разветвленного строения или Y, где Y может необязательно содержать 1-3 заместителей, которые независимо выбраны из группы, состоящей из O-(C1-C4)-алкила нормального или разветвленного строения. O-бензила, O-фенила, O-(C2-C4)-алкенила нормального или разветвленного строения, 1,2-метилендиоксигруппы или 3-фенил(E)проп.-2-енила, n = 1 или 2, m представляет целое число 0 - 3. Новые соединения формулы I поддерживают, повышают или восстанавливают чувствительность клеток к терапевтическим или профилактическим средствам. Изобретение относится также к фармацевтическим композициям, содержащим эти соединения. Эти соединения и фармацевтические композиции хорошо пригодны для лечения клеток, устойчивых к действию многих лекарственных средств, для предотвращения развития устойчивости ко многим лекарственным средствам и для терапии ракового заболевания, устойчивого ко многим лекарственным средствам. Описывается также способ лечения и способ получения соединений формулы I. 5 с. и 8 з.п.ф-лы, 2 табл.
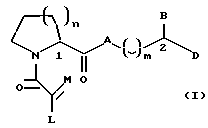
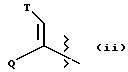
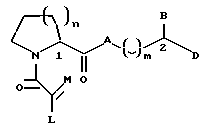
где A представляет кислород;
B и D независимо представляют собой Ar, (C1-C10)-алкил нормального или разветвленного строения, (C2-C10)-алкенил или (C2-C10)-алкинил нормального или разветвленного строения, Ar-замещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)-алкенил или (C2-C6)-алкинил нормального или разветвленного строения, причем в каждом случае любая одна из CH2-групп указанных алкилов, алкенилов или алкинилов необязательно может быть замещена на атом кислорода; или группу формулы ii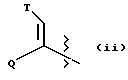
в которой Q представляет собой водород или (C1-C6)-алкил нормального или разветвленного строения, T представляет Ar или 5 - 7-членный циклоалкил, замещенный в 4 положении гидроксигруппой;
при условии, что по меньшей мере один из B и D независимо выбран из группы, включающей (C2-C10)-алкинил нормального или разветвленного строения, Ar-замещенный (C2-C6)-алкинил нормального или разветвленного строения, где Ar означает фенил, 2-тиенил, 2-, 3- или 4-пиридил, бензимидазолил, 3H-индолил, пуринил, 7-азаиндолил и Ar может необязательно содержать 1 - 3 заместителей, независимо выбранных из группы, включающий гидроксигруппу, гидроксиметильную группу, О-(C1-C4)-алкил нормального или разветвленного строения, О-(C2-C4)-алкенил нормального или разветвленного строения, О-фенил, О-бензил, карбоксигруппу, N,N-ди((C1-C5)-алкил нормального или разветвленного строения) карбоксамид, N-бензилкарбоксамид, морфолинкарбонил, тиоморфолинкарбонил, N-метил или N-бензилпиперазинокарбонил, или группу формулы -O-(CH2)q-X, где X представляет собой 2, 3- или 4-пиридил, пиразинил или 2- или 3-тиенил и q равно целому числу 0 - 2;
где L представляет собой U;
M представляет кислород;
U представляет собой (C1-C6)-алкил нормального или разветвленного строения, или Y, где Y означает фенил, 2-тиенил или 3-тиенил и Y может необязательно содержать 1 - 3 заместителей, которые независимо выбраны из группы, состоящей из О-(C1-C4)-алкила нормального или разветвленного строения, О-бензила, О-фенила, О-(C2-C4)-алкенила нормального или разветвленного строения, 1,2-метилендиоксигруппы или 3-фенил[E]проп-2-енила;
n равно 1 или 2;
m представляет целое число 0 - 3.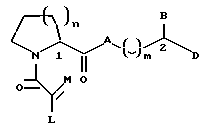
в которой A представляет кислород;
B и D независимо представляют собой Ar, (C1-C10)-алкил нормального или разветвленного строения, (C2-C10)-алкенил или алкинил нормального или разветвленного строения, или Ar-замещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)-алкенил или алкинил нормального или разветвленного строения, причем в каждом случае любая одна из CH2-групп указанных алкилов, алкенилов или алкинилов необязательно может быть замещена на атом кислорода; или группу формулы ii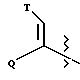
в которой Q представляет собой водород, (C1-C6)-алкил нормального или разветвленного строения;
T представляет собой Ar или замещенный 5 - 7-членный циклоалкил, замещенный в положении 4 гидроксигруппой;
при условии, что по меньшей мере один из B и D независимо выбран из группы, состоящей из Ar', Ar'-замещенного (C1-C6)-алкила нормального или разветвленного строения и Ar'-замещенного (C2-C6)-алкенила нормального или разветвленного строения; где Ar' представляет собой фенил, 2-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, бензилимидазол, 3H-индолил, пуринил и 7-азаиндолил и Ar' имеет от 1 до 3 заместителей, из которых первый заместитель выбран из группы, включающей N, N-ди-(C1-C5)-алкил нормального или разветвленного строения) карбоксамид, морфолинкарбонил, N-бензилкарбоксамид, тиоморфолинкарбонил, N-метил или N-бензилпиперазинкарбонил, или группы -O-(CH2)q-X, где X представляет собой 2-пиридил, 3-пиридил, 4-пиридил, пиразил, 2-тиенил или 3-тиенил;
q равно целому числу 0 - 2;
второй и третий заместители, если присутствуют, выбраны из группы, включающей гидроксил, гидроксиметил, О-(C1-C4)-алкил нормального или разветвленного строения, О-(C2-C4)-алкенил нормального или разветвленного строения, О-бензил, О-фенил, карбоксил, N,N-ди-(C1-C5-алкил нормального или разветвленного строения)карбоксамид, морфолинкарбонил, N-бензилкарбоксамид, тиоморфолинкарбонил, N-метил- или N-бензилпиперазинкарбонил или группы формулы O-(CH2)q-X, где X представляет собой 2-пиридил, 3-пиридил, 4-пиридил, пиразил, 2-тиенил или 3-тиенил, q равно целому числу 0 - 2;
L представляет собой U;
M представляет кислород;
U представляет собой (C1-C6)-алкил нормального или разветвленного строения, или Y;
Y выбран из группы, состоящей из фенила, 2-тиенила или 3-тиенила; Y может содержать 1 - 3 заместителей, которые независимо выбраны из группы, включающей О-(C1-C4)-алкил нормального или разветвленного строения, О-(C2-C4)-алкенил нормального или разветвленного строения, О-бензил, О-фенил, 1,2-метилендиокси или 3-фенил[E]-проп-2-енил;
n = 1 или 2;
m равно целому числу 0 - 3.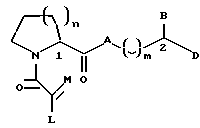
в которой A представляет собой кислород;
B и D независимо представляют собой; (i) Ar, (C1-C10)-алкил нормального или разветвленного строения, (C2-C10)-алкенил или алкинил нормального или разветвленного строения, (C5-C7)-циалоалкил-замещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)-алкенил или алкинил нормального или разветвленного строения или Ar-замещенный (C1-C6)-алкил нормального или разветвленного строения, (C2-C6)-алкенил или алкинил нормального или разветвленного строения, причем в каждом случае любая одна из CH-групп этих алкилов, алкенилов или алкинилов необязательно может быть замещена атомом кислорода; или группу формулы (ii)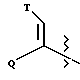
в которой Q представляет собой водород, (C1-C6)-алкил нормального или разветвленного строения;
T представляет собой Ar или замещенный 5 - 7-членный циклоалкил, замещенный в положении 4 гидроксигруппой, где Ar представляет собой фенил, 2-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 3H-индолил, бензимидазолил, пуринил, 7-азаиндолил, Ar может содержать 1 - 3 заместителей, которые независимо выбраны из группы, включающей гидрокси, гидроксиметил; О-(C1-C4)-алкил нормального или разветвленного строения, О-(C2-C4)-алкенил нормального или разветвленного строения, О-бензил, О-фенил, карбоксил, N,N-ди-((C1-C5)-алкил нормального или разветвленного строения)карбоксамид, морфолинкарбонил, N-бензилкарбоксамид, тиоморфолинкарбонил, N-метил или N-бензилпиперазинкарбонил или группу O-(CH2)q-X, где X представляет собой 2-пиридил, 3-пиридил, 4-пиридил, пиразил, 2-тиенил или 3-тиенил; q равно целому числу 0 - 2;
L представляет собой U;
M представляет собой кислород;
U представляет собой (C1-C6)-алкил нормального или разветвленного строения или Y;
Y выбран из группы, включающей фенил, 2-тиенил или 3-тиенил; Y может содержать 1 - 3 заместителей, которые независимо выбраны из группы, включающей О-(C1-C4)-алкил нормального или разветвленного строения, О-(C2-C4)-алкенил нормального или разветвленного строения, О-бензил, О-фенил, 1,2-метилендиокси или 3-фенил[E]-проп-2-енил;
n = 1 или 2;
m равно целому числу 0 - 3.
-(CH2)r-(X)-(CH2)s-Ar,
в которой r равно целому числу 0 - 4;
s равно целому числу 0 - 1;
Ar имеет значения, указанные в п.1, каждый из X представляет кислород.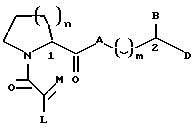
в которой A представляет O и B, D, L, m и n имеют значения, определенные в п.1, отличающийся тем, что включает
(а) этерификацию защищенной аминокислоты общей формулы X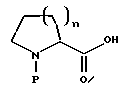
где P означает защитную группу,
спиртом формулы XI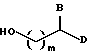
в которой B, D и m имеют значения, определенные в п.1,
с образованием промежуточного соединения общей формулы XII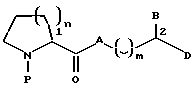
в которой B, D, P, m и n имеют вышеуказанные значения;
A представляет O;
(б) удаление Y соединений общей формулы XII аминозащитной группы с получением аминоэфира общей формулы XIII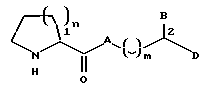
в которой B, D, m и n имеют вышеуказанные значения;
A представляет O;
(в) ацилирование свободной аминогруппы соединения общей формулы XIII соединением общей формулы XIV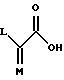
в которой L и M имеет вышеуказанные значения,
или его активированным производным.
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| СПОСОБ РЕЛЕЙНОЙ ЗАЩИТЫ ЭНЕРГООБЪЕКТА | 2002 |
|
RU2247456C2 |
| ХОЛОДОВ Л.Е., Яковлев В.П | |||
| Клиническая фармакинетика | |||
| - М.: Медицина, 1985, с.394 - 398. | |||

