Изобретение относится к комбинациям противовирусных агентов. Более конкретно настоящее изобретение относится к комбинациям аналогов 1,3-оксатиоланнуклеозида с другими противовирусными агентами, например агентами, обладающими активностью против ВИЧ.
Вирус иммунодефицита человека (ВИЧ) вызывает ряд патологических состояний, включая синдром приобретенного иммунодефицита (СПИД) и хронические неврологические расстройства. Нуклеозиды, такие, как AZT, ddC, и ddI, обладают способностью к in vitro - ингибированию репликации ВИЧ и, очевидно, оказывают свое противовирусное действие на вирус-кодированную обратную транскриптазу после их клеточного метаболизма в 5-трифосфат-производные.
AZT позволяет снизить заболеваемость и смертность среди пациентов со СПИДом. Однако ВИЧ-инфекция клеток приводит к интеграции клеточного генома в хромосому хозяина, что обуславливает необходимость применения AZT в течение длительного периода времени. Такая продолжительность AZT-терапия обычно оказывает токсическое действие на костный мозг, а также приводит к появлению AZT-резистентных штаммов ВИЧ-1. Аналогично у некоторых пациентов, больных СПИДом и проходивших ddC-терапию, обнаруживалось развитие периферической невропатии, а у пациентов, лечившихся с применением ddI, обнаруживалось развитие панкреатита и периферической невропатии.
Использование комбинаций противовирусных соединений может способствовать получению эквивалентного противовирусного эффекта с более низкой токсичностью, либо получению более высокой эффективности лекарственного средства, в случае, если эти соединения обладают синергическим действием. Использование в целом более низких доз может также способствовать снижению частоты появления резистентных к лекарственному средству штаммов ВИЧ. Для оценки действия комбинаций различных соединений в различных аналитических системах было использовано множество методов. Все эти методы имеют определенные ограничения, а некоторые методы, например, были применены совсем не к тем системам, для которых они были предназначены. Эти методы показали, что AZT обладает синергической противовирусной активностью in vitro в сочетании с агентами, которые действуют на стадии репликации ВИЧ-1, а не на стадии обратной транскрипции, например, такими, как рекомбинантный растворимый кастаноспермин СД4 и рекомбинантный интерферон-альфа. Однако в этой связи следует отметить, что комбинации соединений могут способствовать увеличению цитотоксичности. Например, AZT и рекомбинантный интерферон-альфа оказывают повышенное цитотоксическое действие на нормальные клетки-предшественники костного мозга человека.
Были исследованы также комбинации AZT с другими нуклеозидами. Например, ddC нейтрализует цитотоксичное действие высоких доз AZT на костный мозг, не влияя при этом на его противовирусную активность. ddI и AZT обнаруживают несколько повышенную избирательность к комбинации, где синергическое противовирусное действие превалирует по отношению к суммарному токсическому действию на нормальные клетки-предшественники костного мозга.
Соединение формулы (I)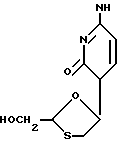
которое также известно как BCH-180 или N ЕРВ-21, было описано в литературе как противовирусное средство, в частности, как средство против вируса иммунодефицита человека (ВИЧ), являющегося возбудителем СПИДа (5-я Конференция Анти-СПИД, Монреаль Канада, 5-9 июня 1989 г., Abstracts T.C.O.I и M. C. P. 63; публ. европейской патентной заявки N 0382562). Соединение формулы (I) представляет собой рацемическую смесь двух энантиомеров формул (I-1) и (I-2)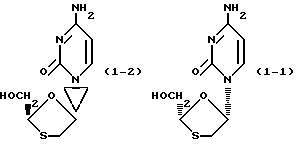
Хотя энантиомеры соединения формулы (I) обладают идентичной эффективностью против ВИЧ, однако, один из этих энантиомеров [(-)-энантиомер] имеет значительно более низкую цитотоксичность, чем другой энантиомер (+)-энантиомер.
(-)-энантиомер имеет химическое название (-)цис-4-амино-1-(2-гидросиметил-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2-он. Стереохимически он абсолютно соответствует соединению формулы (I-1), которое имеет следующее название: (2R, цис)-4-амино-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2-он. Это соединение известно как 3ТС.
Авторами настоящей заявки было установлено, что соединение формулы (I), а особенно его (-)-энантиомер, неожиданно обнаруживает преимущественные свойства в сочетании с известными ингибиторами ВИЧ-репликации. В частности, соединение формулы (I) обнаруживается синергическое противовирусное действие и/или пониженное цитотоксическое действие при его использовании в комбинации с известными ингибиторами ВИЧ-репликации.
Поэтому в одном из своих вариантов настоящее изобретение относится к комбинации, содержащей соединение формулы (I) или его фармацевтически приемлемое производное и ингибитор репликации ВИЧ.
Указанным ингибитором может быть любой ингибитор ВИЧ-репликации независимо от механизма его ингибирования ВИЧ-репликации. Такими ингибиторами являются, например, соединения, ингибирующие обратную транскриптазу ВИЧ, протеазу ВИЧ, ТАТ и т.п.
Примерами таких ингибиторов могут служить 3'-азидо-3'-дезокситимидин (AZT, зидовудин); 2',3'-дидезоксицитидин (ddC); 2',3'-дидезоксиинозин (ddI); N'[I(S)-бензил-3-[4a(S), 8a(S)-3(S)-(трет-бутилкарбамоил) декагидроизохинолин-2-ил-2(R)-гидроксипропил] N''-(хинолин-2-илкарбонил)-L-аспарагинамид (Ro 31-8949); и (+)-S-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)-имидазо(4,51-j к) (1,4)-бензодиазепин-2-(1H)тион(R-82150; TIBO) или их фармацевтически приемлемое производное.
Предпочтительно, если соединение формулы (I) присутствует в форме его (-)-энантиомера (ЗТС).
Предпочтительно также, если ингибитор ВИЧ-репликации выбирают из AZT, ddI, Ro 31-8959 или R-82150(TIBO).
Особенно предпочтительным ингибитором ВИЧ-репликации является ddI, а более предпочтительным AZT.
Если соединение формулы (I) присутствует в форме (-)-энантиомера, то это означает, что оно в основном не содержит соответствующего (+)-энантиомера, то есть оно содержит не более чем около 5% мас. (+)-энантиомера, предпочтительно не более чем около 2%, а более предпочтительно менее чем около 1% мас. (+)-энантиомера.
Термин "фармацевтически приемлемое производное" означает любые фармацевтически приемлемые соли, сложные эфиры, или соли таких сложных эфиров исходного соединения или любого другого соединения, которое после его введения реципиенту преобразуется в исходное соединение (непосредственно или опосредованно) или в его метаболит или остаток, обладающий противовирусной активностью.
Каждому специалисту ясно, что соединение формулы (I) может быть модифицировано в отношении функциональных групп как в основной части, так и в гидроксиметильный группе оксатиаланового кольца в целях получения его фармацевтически приемлемых производных. Модификация во всех указанных функциональных группах также входит в объем настоящего изобретения. Однако особый интерес представляют фармацевтически приемлемые производные, полученные путем модификации 2-гидроксиметильной группы оксатиоланового кольца.
Предпочтительными сложными эфирами соединения формулы (I) являются соединения, в которых водород 2-гидроксиметильной группы является замещенным ацильной функциональной группой 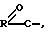 где некарбонильную часть R сложного эфира выбирают из водорода, прямого или разветвленного алкила (например, метила, этила, н-пропила, т-бутила, н-бутила), алкоксиалкила (например, метоксиметила), аралкила (например, бензила), арилоксиалкила (например, феноксиметила), арила (например, фенила, необязательно замещенного галогеном, C1-4-алкилом или C1-4-алкокси); сульфонатные сложные эфиры, такие как алкил- или аралкилсульфонил (например, метансульфонил); сложные эфиры аминокислот (например, L-валил или L-изолейцил); и сложные эфиры моно-, ди- или трифосфорной кислоты.
где некарбонильную часть R сложного эфира выбирают из водорода, прямого или разветвленного алкила (например, метила, этила, н-пропила, т-бутила, н-бутила), алкоксиалкила (например, метоксиметила), аралкила (например, бензила), арилоксиалкила (например, феноксиметила), арила (например, фенила, необязательно замещенного галогеном, C1-4-алкилом или C1-4-алкокси); сульфонатные сложные эфиры, такие как алкил- или аралкилсульфонил (например, метансульфонил); сложные эфиры аминокислот (например, L-валил или L-изолейцил); и сложные эфиры моно-, ди- или трифосфорной кислоты.
В описанных выше сложных эфирах, если это не оговорено особо, все алкильные части содержат в основном 1-16 атомов углерода, а в частности 1-4 атомов углерода. А все арильные части, присутствующие в указанных сложных эфирах, в основном представляют собой фенильную группу.
В частности, сложными эфирами могут быть C1-16-алкиловый сложный эфир, незамещенный бензиловый сложный эфир, или бензиловый сложный эфир, замещенный, по крайней мере, одним атомом галогена (брома, хлора, фтора или иода) C1-6-алкилом, C1-6-алкокси, нитро- или трифторометильной группами.
Фармацевтически приемлемые соли соединения формулы (I) могут быть получены из фармацевтически приемлемых неорганических или органических кислот и оснований. Примерами подходящих кислот являются соляная, бромистоводородная, серная, азотная, перхлорная, фумаровая, малеиновая, фосфорная, гликолевая, молочная, салициловая, янтарная, толуол-п-сульфоновая, винная, уксусная, лимонная, метасульфоновая, муравьиная, бензойная, малоновая, нафталин-2-сульфоновая, и бензолсульфоновая кислоты. Другие кислоты, такие как щавелевая кислота, которые сами по себе не являются фармацевтически приемлемыми, могут быть использованы в качестве промежуточных соединений при получении соединения настоящего изобретения и их фармацевтически приемлемых кислых аддитивных солей.
Солями, полученными из соответствующих оснований, являются соли щелочных металлов (например, натрия), щелочно-земельных металлов (например, магния), аммония и NP4+ (где R является C1-4-алкилом).
В сочетании со вторым компонентом, соединение формулы (I) либо усиливает действие второго компонента (синергический эффект), либо снижает цитотоксическое действие второго компонента, либо обладает и тем и другим из указанных свойств.
Преимущественное действие соединений формулы (I) и вторых противовирусных агентов может быть реализовано в широких пределах соотношений, например, 1: 250 - 250:1, предпочтительно 1:50 - 50:1, а более предпочтительно около 1: 10-10:1. Обычно каждое соединение может быть использовано в данной комбинации в количестве, при котором оно обладает противовирусной активностью, будучи использованным отдельно.
Ожидается, что комбинации настоящего изобретения могут быть использованы против вирусных инфекций или вирус-ассоциированных опухолей у человека, а поэтому способ использования указанных комбинаций для ингибирования инфекционности вируса или опухолевого роста in vitro или in vito также входит в объем настоящего изобретения.
В соответствии с этим, в другом своем варианте настоящее изобретение относится к способу лечения вирусных инфекций у млекопитающих, включая человека, заключающемуся в совместном введении противовирусного соединения формулы (I) и нигибитора ВИЧ-репликации. Способы терапии, предусматривающие введение комбинации соединения формулы (I) и более, чем одно из других противовирусных агентов, либо вместе, либо во множестве парных комбинаций также входят в объем настоящего изобретения.
При этом следует отметить, что соединение формулы (I) и второй противовирусный агент могут быть введены либо одновременно, либо последовательно, либо в комбинации. При последовательном введении промежуток времени между введением соединения формулы (I) и вторым активным ингредиентом не должен быть слишком большим, так как при этом может быть утерян синергический эффект комбинации. Предпочтительно, если введение осуществляют одновременно.
Следует также отметить, что используемое в настоящем описании понятие "лечение" относится как к профилактике заболеваний, так к лечению уже установленных инфекций или симптомов.
Кроме того, следует отметить, что количество комбинаций настоящего изобретения, необходимое для лечения, может варьироваться не только в зависимости от конкретно выбранного соединения, но также и в зависимости от способа введения, заболевания, возраста и состояния пациента, и может быть определено в каждом конкретном случае лечащим врачом или ветеринаром. Однако в основном подходящие дозы составляют в пределах от около 1 до около 750 мг/кг, например, от около 10 до около 75 мг на кг веса тела в день, а в частности, от 3 до около 120 мг на кг веса тела реципиента в день, предпочтительно в пределах 6-90 мг/кг/день, а наиболее предпочтительно 15-60 мг/кг/день каждого из активных ингредиентов комбинации.
Нужные дозы могут быть введены в виде разовых доз или в виде дробных доз, вводимых через определенные интервалы времени, например, два, три, четыре или более раз в день.
Комбинация, вводимая в унифицированной стандартной лекарственной форме, содержит, например, 10-1500 мг, обычно 20-1000 мг, а предпочтительно 50-700 мг каждого активного ингредиента на данную лекарственную форму.
В идеальном случае указанные комбинации должны быть введены в таком количестве, чтобы пиковые концентрации каждого из активных соединений в плазме достигали от около 1 до около 75 мМ, предпочтительно около 2-50 мМ, а наиболее предпочтительно от около 3 до около 30 мМ. Такие концентрации могут быть достигнуты, например, путем внутривенной инъекции 0,1-5% раствора активных ингредиентов, например (но необязательно) солевого раствора, либо путем перорального введения в виде болюса, содержащего от около 1 до 100 мг каждого активного ингредиента. Желательные уровни активных соединений в крови могут поддерживаться посредством непрерывного влияния, содержащего от около 0,01 до 5,0 мг/кг/час, либо периодического вливания, содержащего от около 0,4 до около 15 мг/кг каждого активного ингредиента.
Хотя при использовании в терапевтических целях активные ингредиенты комбинации настоящего изобретения могут быть введены в виде неочищенных химических соединений, однако, предпочтительно, если эти комбинации вводятся в виде фармацевтических препаратов.
Поэтому настоящее изобретение также относится к фермацевтическому препарату, содержащему соединение формулы (I) или его фармацевтически приемлемое производное и ингибитор ВИЧ-репликации в сочетании с одним или несколькими фармацевтически приемлемыми носителями и необязательно с другими терапевтическими и/или профилактическими ингредиентами. Термин "приемлемый", используемый в отношении носителя, означает, что данный носитель является совместимым с другими ингредиентами препарата и не оказывает неблагоприятного воздействия на реципиента.
Фармацевтические препараты могут быть изготовлены в формах, предназначенных для перорального, рентального, назального, местного (трансбуккального и подъязычного), вагинального, или парентерального (включая, внутримышечное, подкожное, и внутривенное) введения, или в формах, предназначенных для введения путем ингаляции или инсуффляции. Эти фармацевтические препараты могут быть изготовлены в виде дискретных унифицированных лекарственных форм любым из известных способов, обычно применяемых в фармацевтической практике. Все эти способы включают в себя стадию смешивания активного соединения с жидкими носителями или тонко измельченными твердыми носителями или теми и другими, а затем, если это необходимо, стадию формирования продукта в нужную лекарственную композицию.
Фармацевтические композиции, предназначенные для перорального введения, могут быть изготовлены в виде дискретных стандартных лекарственных форм, таких, как капсулы, облатки или таблетки, каждая из которых содержит заранее определенное количество активного ингредиента, а также в виде порошков, гранул, растворов, суспензий или эмульсий. Активный ингредиент может быть также введен в виде болюса, электуария или пасты. Таблетки и капсулы для перорального введения могут содержать стандартные наполнители, такие, как связывающие агенты, замасливатели, дизентеграторы или смачивающие агенты. Таблетки могут быть покрыты в соответствии со стандартной техникой. Пероральные жидкие препараты могут быть изготовлены в виде, например, водных или масляных суспензий, растворов, эмульсий, сиропов или элексиров, либо они могут быть изготовлены в виде сухих продуктов, которые могут быть затем перед непосредственным использованием разведены водой или другим подходящим разбавителем. Указанные жидкие препараты могут содержать стандартные добавки, такие, как суспендирующие агенты, эмульгирующие агенты, безводные наполнители (например, пригодные в пищу масла) или консерванты.
Соединения настоящего изобретения могут быть также введены парентерально (например, путем инъекции, в частности, инъекции ударной дозы, или путем непрерывной инфузии), и в этих целях они могут быть изготовлены в виде стандартных ампул, готовых заполненных шприцев, флаконов с небольшими или многократными дозами для вливания; причем указанные препараты могут содержать добавки в виде консервантов. Композиции настоящего изобретения могут быть изготовлены в виде суспензий, растворов или эмульсий в масляных или водных наполнителях, и могут, кроме того, содержать такие добавки, как суспендирующие стабилизирующие и/или диспергирующие агенты. Альтернативно активный ингредиент может быть получен в виде порошка путем асептического выделения стерильного твердого материала или путем лиофилизации из раствора; причем этот порошок непосредственно перед его использованием может быть смешан с соответствующим носителем, таким, как стерильная апирогенная вода.
Соединения настоящего изобретения, предназначенные для наружного применения путем нанесения на эпидермис, могут быть изготовлены в виде мазей, кремов или лосьонов, либо в виде трансдермальных пластырей. Например, мази и кремы могут быть изготовлены на водной или масляной основе с добавлением подходящего загущающего и/или гелеобразующего агентов. Лосьоны могут получены на водной или масляной основе, и кроме того, могут также содержать один или несколько эмульгирующих агентов, стабилизирующих агентов, диспергирующих агентов, суспендирующих агентов, загущающих агентов или окрашивающих агентов.
Препараты, предназначенные для местного применения в области рта, такие, как таблетки, обычно содержат активный ингредиенты на ароматизирующей основе, например, в сахарозе, аравийской или трагакантовой камеди; препараты, такие как пастилки, обычно содержат активный ингредиент в инертном наполнителе, таком, как желатини глицерин, или сахароза и аравийская камедь, а препараты, такие, как полоскания для рта, обычно содержат активный ингредиент в подходящем жидком носителе.
Фармацевтические препараты, предназначенные для ректального введения, где носитель является твердым веществом, предпочтительно изготавливать в виде суппозиториев в унифицированных формах. Эти суппозитории могут быть изготовлены с использованием подходящего носителя, например, масло-какао и других материалов, которые обычно используются в этих целях, путем смешивания активного ингредиента с отвержденным или расплавленным носителем (или носителями) с последующим охлаждением и формованием из расплава.
Препараты, предназначенные для вагинального введения, могут быть изготовлены в виде пессариев, тампонов, гелей, паст, пенистых препаратов или препаратов для распыления, содержащих помимо активного ингредиента подходящие стандартные носители.
Лекарственные препараты для интраназального введения соединений настоящего изобретения могут быть изготовлены в виде жидких растворов для распыления, диспергируемых порошков или в виде капель.
Капли могут быть получены на водной или безводной основе и помимо активного ингредиента содержать один или несколько диспергирующих агентов, солюбилизирующих агентов или суспендирующих агентов. Жидкие растворы для распыления могут изготовлены в виде аэрозольных упаковок.
Для введения соединения настоящего изобретения путем ингаляции могут быть использованы инсуффляторы, ингаляторы или аэрозольные распылители, либо другие обычно используемые в фармацевтической практике средства. Аэрозольные препараты могут включать в себя соответствующие распыляющие агенты, такие, как дихлородифтороментан, трихлорофторометан, дихлоротетрафтороэтан, двуокись углерода или другой подходящий газ. В случае использования аэрозольных упаковок стандартная доза может быть определена путем измерения количества лекарственного средства, проходящего через клапан упаковки.
Альтернативно соединения настоящего изобретения, предназначенные для введения путем ингаляции или инсуффляции, могут быть изготовлены в виде сухих порошковых композиций, например, порошкообразных смесей активного соединения и соответствующего порошкового наполнителя, такого, как лактоза или крахмал. Порошковые композиции могут быть получены в виде унифицированных стандартных форм, таких, как капсулы или ампулы, либо, например, желатиновые упаковки или водные составы, из которых порошок может быть введен в организм с помощью ингалятора или инсуффлятора.
Если необходимо, вышеуказанные композиции могут быть получены в виде препаратов с пролонгированным высвобождением активного ингредиентов.
Фармацевтические композиции настоящего изобретения могут также содержать другие активные ингредиенты, такие, как противомикробные агенты или консерванты.
Соединение формулы (I) может быть получено в соответствии с описанием, приведенным в публикации заявки на Европатент N 0382526.
Отдельные энантиомеры этого соединения могут быть получены из их рацемических смесей путем разделения стандартными способами, обычно используемыми для разделения рацематов. В частности, они могут быть получены из известного рацемата путем хиральной ВЭЖХ, путем ферменто-опосредованного энантиоселективного катаболизма с использованием соответствующего фермента, такого, как цитидиндеаминаза, либо путем селективного ферментного расщепления соответствующего производного с использованием 5-нуклеотида. Способы получения ЭТС описаны в публикации заявки на Европатент N W 091/17159.
Представленные ниже примеры иллюстрируют настоящее изобретение, но не ограничивает его объема.
Промежуточное соединение 1
5-метокси-1,3-оксатиолан-2-метанол, бензоат
Раствор хлорида цинка (1,6 г) в горячем метаноле (15 мл) добавляли к размешанному раствору меркаптоацетальдегида, диметилацеталя (34,2 г), и бензилоксиацетальдегида (48,3 г) в толуоле (1300 мл), и полученный раствор нагревали с обратным холодильником в атмосфере азота в течение 50 минут. Охлажденную смесь концентрировали, разбавляли некоторым количеством толуола и фильтровали через Кизельгур (Kieselguhr). Объединенные фильтраты и толуол промывали водным насыщенным раствором бикарбоната натрия (х2) и солевым раствором, затем осушали сульфатом магния и выпаривали до получения маслообразного продукта, который подвергали колоночной хроматографии на силикагеле (2 кг, Мееск 9385), жлюируя хлороформом, в результате чего получали целевой продукт в виде маслообразной смеси (45,1 г) аномеров около 1:1; 1H-ЯМР (ДМСО-d6) 3,1-3,3 (4H); 3,42 (6H); 4,4-4,6 (4H); 5,41 (1H); 5,46 (1H); 5,54(1Н), 5,63 (1H); 7,46 (4H); 7,58 (2H); 8,07 (4H); λмакс CHB3 1717,6 см-1.
Промежуточное соединение 2
(±)-цис-1-(2-бензоилоксиметил-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2,4-дион.
Смесь тонкоизмельченного урацила (9,62 г), гексаметилдизилазана (50 мл) и сульфата аммония (30 мг) нагревали с обратным холодильником до тех пор, пока не был получен прозрачный раствор. Этот раствор охлаждали, выпаривали и получали бесцветное маслообразное вещество, которое растворяли в ацетонитриле (100 мл) в атмосфере азота. Полученный раствор добавляли к размешанному и охлажденному льдом раствору 5-метокси-1,3-оксатиалан-2-метанола, бензоата (промежуточного соединения 1) (19,43 г) в ацетонитриле (600 мл), а затем добавляли триметилсилилтрифторометансульфонат (14,7 мл). После этого ледяную баню удаляли, и раствор нагревали с обратным холодильником в атмосфере азота в течение 45 минут. После охлаждения и выпаривания остаток очищали с помощью колоночной хроматографии на силикагеле (1 кг, MeZck 9385), элюируя хлороформом/эталоном, 9:1. Нужные фракции охлаждали и выпаривали с получением неочищенного остатка. Этот остаток подвергали фракционированной кристаллизации из минимального количества горячего метанола (ок. 1200 мл), в результате чего получали целевое соединение (6,32 г) в виде белых кристаллов. 1H-ЯМР (ДМСО-d6) δ 11,36 (1H, шир.с); 7,50-8,0 (6H, м); 6,20 (1H, т); 5,46 (2H, м); 4,62 (2H, м); 3,48 (1H, м); 3,25 (1H, м).
Промежуточное соединение 3
(±)-(цис)-4-амино-1-(2-бензоилоксиметил-1,3-оксатиолан-5-ил)-(1H)- пиримидин-2-он.
Метод (а)
Суспензию цетозина (20,705 г) и сульфата аммония несколько граммов в гексаметилдизилазане (110 мл) размешивали и с обратным холодильником в течение 2,5 час в атмосфере азота. Растворитель удаляли путем выпаривания, а твердый остаток растворяли в сухом ацетонитриле (350 мл). Этот раствор переносили (используя гибкую иглу) в размешанный охлажденный льдом раствор 5-метокси-1,3-оксатиолан-2-метанола, бензоата (промежуточное соединение 1) (43,57 г) в ацетонитриле (650 мл) в атмосфере азота. Затем добавляли триметилсилилтрифторометансульфонат (33 мл), и полученный раствор оставляли нагреваться до комнатной температуры (1,5 часа), после чего раствор нагревали с обратным холодильником в течение ночи. Оставшуюся смесь концентрировали, разводили насыщенным водным раствором бикарбоната натрия (50,0 мл), и экстрагировали этилацетатом (3х500 мл). Объединенные экстракты промывали водой (2х250) мл и солевым раствором (250 мл), затем осушали сульфатом магния и выпаривали до получения пенистого продукта, который подвергали колоночной хроматографии на силикагеле (600 г, Me ск 7734), элюируя смесями этилацетата и метанола, в результате чего получали смесь аномеров (ок. 1:1, 31,59 г). Полученную смесь кристаллизовали из воды (45 мл) и этанола (9,0 мл) и получали твердое вещество (10,23 г), которое перекристаллизовывали из этанола (120 мл) и воды (30 мл), в результате чего получали целевой продукт в виде белого твердого вещества (9,26 г); λ макс. (MeOH) 229,4 мм (E1% 610); 272,4 мм (E1%...? [очевидно, в тексте оригинала пропуск - (прим. пер.)] 293; 1H-ЯМР (ДМСО-d6) δ 3,14 (1H); 3,50 (1H); 4,07 (2H); 5,52 (1H); 5,66 (1H); 6,28 (1H); 7,22 (2H); 7,56 (2H); 7,72 (2H); 8,10 (2H).
Метод (b)
Оксихлорид фосфора (7,0 мл) по капле добавляли к размешанной охлажденной льдом суспензии 1,2,4-триазола (11,65 г) в ацетонитриле 120 мл), а затем, поддерживая внутреннюю температуру ниже 15oC, по капле добавляли триэтиламин (22.7 мл). Через 10 минут медленно добавляли раствор (±)-(цис)-1-(2-бензоилоксиметил-1,3-оксатиолан-1-ил)-(1H)-пиримидин-2,4- диона (промежуточное соединение 2) (6,27 г) в ацетонитриле (330 мл). Затем продолжали размешивать в течение ночи при комнатной температуре. Смесь охлаждали в ледяной бане, после чего медленно добавляли триэтиламин (30 мл), а затем воду (21 мл). Полученный раствор выпаривали, а остаток распределяли между насыщенным раствором бикарбоната натрия (400 мл и хлороформом (3х200 мл). Объединенные хлороформные экстракты осушали сульфатом магния, фильтровали и выпаривали, в результате чего получали неочищенный остаток (9,6 г). Этот остаток растворяли в 1,4-диоксане (240 мл), и добавляли концентрированный водный раствор аммиака (примерно 0,880 г, 50 мл). Через полтора часа раствор выпаривали, а остаток растворяли в метаноле. В результате этого образовывался твердый осадок, который отфильтровывали. Маточные растворы очищали с помощью колоночной хроматографии на силикагеле (MeZск 9386, 600 г). Соответствующие фракции объединяли и выпаривали, в результате чего получали целевое соединение в виде желтовато-коричневого твердого вещества (2,18 г), идентичного веществу, полученному методом (а).
Промежуточное соединение 4
(±)-(цис)-4-амино-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-(1H)- пиримидин-2-он.
Суспензию (цис)-4-амино-1-(2-бензоилоксиметил-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2-она (промежуточное соединение 3) (8,19 г) и амберлитовой смолы (Amberlitee IRA-400(OH) (8,24 г) в метаноле (250 мл) размешивали и нагревали с обратным холодильником в течение 1,25 часа. Твердое вещество удаляли путем фильтрации, а затем промывали метанолом. Объединенные фильтраты выпаривали. Остаток растирали с этилацетатом (80 мл). Полученное белое твердое вещество собирали путем фильтрации и получали целевой продукт (5,09 г). 1H-ЯМР (ДМСО-d6) 3,04 (1H); 3,40 (1H); 3,73 (2H); 5,18 (1H); 5,29 (1H); 5,73 (1H); 6,21 (1H); 7,19 (2H); 7,81 (1H).
Пример 1
(-)-цис-4-амино-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-(1H)-пиримидин-2-он
(1) Три 50 - миллилитровые колбы с питательным бульоном (Oxoid Ltd) инокулировали (каждую) E. coli (АТСС 23848), которые с помощью специальной петли соскребали с чашки с питательным агаром. Эти колбы инкубировали в течение ночи при 37oC в шейкере при 250 об/мин, после чего каждую колбу использовали для инокуляции 41 СДД-среды (глутаминовая кислота, 3 г/л, MgSO4, 0,2 г/л; K2SO4, 2,5 г/л; NaCl, 2,3 г/л; Na2HPO42H2O, 1,1 г/л; NaH2PO42H2O, 0,6 г/л, цитидин, 1,2 г/л) в 7-литровом ферментере. Культуры подвергали ферментации при 750 об/мин, 37oC, с аэрацией при 41 мин. После культивирования в течение 24 часов клетки собирали путем центрифугирования (5000 г, 30 мин) и получали 72 г мокрого веса. Клеточный осадок ресуспендировали в 300 мл 20 мМ Трис-HCl-буфера (pH 7,5), и клетки лизировали путем обработки ультразвуком (4х45 сек). Клеточный дебрис удаляли путем центрифугирования (30000 г, 30 мин), а белок в супернатанте осаждали путем добавления сульфата аммония до насыщения 75%. Осадок собирали путем центрифугирования (30000 г, 30 мин), а остаток ресуспендировали в 25 мл HEPES-буфера (100 мМ, pH 7,0), содержащего сульфат аммония (насыщение 75%). Супернатант отбрасывали, осадок растворяли в Трис-HCl-буфере (pH 7,0 100 мМ) до исходного объема.
(II) Промежуточное соединение 4 (115 мг) растворяли в воде (100 мл) и размешивали. Затем добавляли ферментный раствор (0,5 мл) и полученную смесь поддерживали при постоянном pH путем постоянного добавления HCl (25 мМ). Конверсию контролировали с помощью хиральной ВЭЖХ, которая показала, что (+) - энантиомер субстрата был преимущественно деаминирован. Через 22 часа (+)-энантиомер субстрата (комн. тем-ра, 12,5 мин.) был полностью удален, а pH раствора доводили до 10,5 путем добавления конц. гидроксида натрия.
Полученный раствор элюировали на колонке с сефадексом ОАЕ (А25, Pharmaiia; 30х1,6 см), предварительно уравновешенной до pH 11. Колонку промывали водой 200 мл, а затем HCl (0,1 М). Нужные фракции собирали (40 мл) и анализировали с помощью образенно-фазвой ВЭЖХ. Фракции 5-13, содержащие непрореагировавший (-)-энантиомер субстрата, объединяли и доводили до pH 7.5 с помощью HCl. Фракцию 47, содержащую деаминированный продукт, доводили до pH 7,5 с помощью разб. NaOH. Анализ с помощью хиральной ВЭЖХ показал, что этот продукт является смесью, содержащей один энантиомер (комн. тем-ра 10,2 мин) в качестве главного компонента, и другой энантиомер (комн. тем-ра, 8,5 мин) в качестве второстепенного компонента.
(III) Повторяли процедуру стадии (II), но в более крупном масштабе. Соединение примера 1 (363 мг) в 250 мл воды инкубировали с ферментным раствором (0.5 мл), полученным как описано в стадии I. Затем через 18 и 47 часов добавляли аликвоты (0,5 мл) фермента. Реакционную смесь размешивали в течение 70 часов, а затем оставляли еще на 64 часов для отстаивания. Анализ с помощью хиральной ВЭЖХ показал, что (+)-энантиомер субстрата был полностью деаминирован, и полученный раствор доводили до pH 10,5 с помощью NaOH.
Полученный раствор загружали в ту же самую колонку OAE и элюировали, как описано в стадии (I). Фракции 2-6, содержащие смесь остаточного субстрата и деминированный продукт, объединяли. Фракции 7-13, содержащие остаточный субстрат (2-)-энантиомер субстрата, объединяли и доводили до pH 7,5. Фракции 25-26, содержащие деаминированный продукт, объединяли и нейтрализовали.
Полученные фракции 2-6 снова элюировали на то же самой колонке OAE. Фракции 3-11 из этой второй колонки содержали непрореагировавший (-)-энантиомер субстрата. Фракции 70 содержала деаминированный продукт.
(IV) Выделенные субстратные фракции, полученные в стадиях (II) и (III), объединяли и доводили до pH 7,5. Этот раствор элюировали через колонку ХАД-16 (40х2,4 см), помещенную в воду. Эту колонку промывали водой, а затем элюировали смесью ацетона и воды (1:4 по объему соответственно). Фракции, содержащие нужный (-)-энантиомер, объединяли и осушили вымораживанием, в результате чего получали белый порошок (190 мл).
При этом использовали следующие методы ВЭЖХ:
1. Обращенно-фазовая аналитическая ВЭЖХ
Колонка: главный патрон
Сферисорб ODS-2 (5 мкМ)
150х4,6 мм
Элюент: первичный кислый фосфат аммония (50 мМ) +5% MeCN
Поток: 1,5 мл/мин
Детекция: УФ, 270 нм
Время удерживания: BCH-189 5,5 минут
деаминированный BCH-189 8,1 минут
2. Хиральная аналитическая ВЭЖХ
Колонка: Cyclobond Acetye, 250х4,6 мм
Элюент: 0,2% ацетат триэтиламония (pH 7,2)
Поток: 1,0 мл/мин.
Детекция: УФ, 270 нм
Время удерживания: BCH-189 11,0 и 12,5 минут
Деаминированный BCH-189 8,5 и 10,2 минут
(биоконверсию контролировали путем слежения за потерей пика при 12,5 мин, и за аккумулированием продукта при 10,2 мин).
Пример 2
3.1. Противовирусная активность соединений, взятых отдельно или в сочетании
Соединения серийно разводили с 2-кратным уменьшением в 96-луночных планшетах для микротитрования. Титрование методом "шахматной доски" проводили путем смешивания 25 мл аликвот от каждого разведения соединения, взятого отдельно или в комбинации (до конечного объема 50 мл в новых 90-луночных планшетах для микротитрования). Аликвоты клеток МТ-4 (106 клеток/мл) в ростовой среде RPMI 1640 инфицировали ВИЧ-1 (штаммом PI) при множественности заражения 2•10-3 инфекц. доза/клетку. Вирус адсорбировали при комнатной температуре в течение 90 минут, после чего клетки промывали в ростовой среде RPMI 1640 для удаления адсорбированного вируса, и ресуспендировали при 106 кл. /мл в ростовой среде RPMI 1640. 50 мл инфицированной клеточной суспензии инкубировали в лунках, содержащих соединение или только ростовую среду, 50 мл ложноинфицированной клеточной суспензии инокулировали в лунки, не содержащие соединения. После этого планшеты инкубировали при 37oC в течение 7 дней в смеси 5% CO2/воздуха.
После инкубирования, во все лунки добавляли 10 мл 3-[4,5-диметилтиазол-2-ил] -2,5-дифенилтетразолийбромида (МТТ) при 7.5 мг/мл, и планшеты инкубировали при 37oC еще 90 минут. Затем добавляли 150 мл 10% (об/об) тритона Х-100 в изопропаноле, и клетки ресуспендировали. После выдерживания 15 минут при комнатной температуре планшеты анализировали с помощью планшетридера (Multiskan MC, F. low Laboratories, irvine, ИК) при 405 нм. Конверсия желтого МТТ в его формазановое производное была максимальной в неинфицированных необработанных клетках, и отсутствовала в необработанных инфицированных клетках.
Для каждого соединения, взятого отдельно, строили кривые "доза-ответ" (IC50-величины), и такие кривые строили для реципрокного титрования каждого соединения при фиксированной концентрации второго соединения. Затем строили изоболограммы всех комбинаций соединений, дающих IC50-величины.
На рис. 1-5 представлены изоболограммы для ЭТС в комбинации с AZT, ddC, ddI, Ro 31-8959 и R-82150 (TIBO), соответственно. Если величины IC 50% комбинации соединений лежат на линии, объединяющей величины IC 50% каждого соединения, взятого независимо, то эти два соединения действуют аддитивно. Если комбинация IC 50% лежит слева от этой линии, то соединения действуют синергически.
Кривые "доза-ответ" для ЭТС в комбинации с AZT, ddC, ddI, Ro 31-8959 и R-82150 (TIBO) представлены на рис. 1-5 соответственно.
При определении противовирусных активностей комбинаций соединений, токсического действия этих комбинаций не наблюдалось.
Пример 3
Цитотоксичность соединений, взятых отдельно и в комбинации
В этих экспериментах цитотоксичности ЭТС, AZT и ddC, взятых отдельно и в комбинации (при мг/мл-отношениях 1:1, 1:5 и 5:1) сравнивали для неинфицированных лимфоцитов периферической крови и клеточной линии покоящихся Т-лимфоцитов.
Цитотоксичность измеряли с помощью анализа с использованием [3H]-тимидина. Типичные кривые "доза-ответ", полученные для каждого соединения или их комбинации (1:1) для лимфоцитов периферической крови представлены на рис. 6 и 7.
Пример 4
Испытания in vitro соединений, активных против ВИЧ и, в частности, комбинации (2R, цис)-4-амино-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-1H-пиримидин- 2-она, известного как ЗТС, с ингибиторами репликации ВИЧ, включая 3'-азидо-3'-деокситимидин (AZT).
Соединения первоначально серийно разбавлялись в 96-луночных титрационных планшетах. Титрование в двух направлениях осуществляли путем смешения аликвот каждого соединения по отдельности или в комбинации. Аликвоты клеток МТ-4 в ростстимулирующей среде RPM1 в течение 90 минут при комнатной температуре инфицировали ВИЧ-1 штаммом RF при множественности инфицирования 2•103 инфекционных доз на клетку. Аликвоты промытых инфицированных клеток добавляли в лунки используемого планшета для испытания. Планшеты инкубировали в течение 7 дней при 37oC в 5% CO2/воздух атмосфере.
После инкубации в каждую лунку добавляли МТТ (бромид 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия) в количестве 7,5 мг/мл и инкубировали дополнительно 90 минут при 37oC. Кристаллы формазана растворяли с использованием Triton-X100 в изопропаноле и измеряли оптическую плотность при 540 нм.
Кривые доза-ответ строили для каждого компонента по отдельности (значения IC50) и для взаимных титрований каждого соединения при фиксированной концентрации второго соединения. Определяли комбинационные индексы.
Значения < 1 указывают на синергизм.
Результаты трех экспериментов представлены в Таблице 1. Результаты показывают, что все испытанные комбинации, которые находятся для ЗТС:AZT в диапазоне от 1,8:1 до 167:1, обладали синергетическим эффектом.
Пример 5
Проведение клинических испытаний соединений, активных против ВИЧ, в Соединенных Штатах. Исследование, связанное с воздействием комбинации (2R, цис)-4-амино-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-1H-пиримидин-2-она (ламивудин) и 3'-азидо-3'-деокситимидина (зидовудин) по сравнению с зидовудиновой монотерапией и ламивудиновой монотерапией.
Исследование было двойным слепым, рандомизированным (случайная выборка), многоцентровым, плацебо-контролируемым испытанием, продолжавшимся 24 недели, со слепой фазой продолжения в течение дополнительных 28 недель, осуществлявшееся в 26' местах в Северной Америке. Пациентам по случайной выборке было предназначено получать один из четырех курсов лечения: 200 мг зидовудина каждые 8 часов плюс плацебо, походящее на ламивудин (только зидовудиновая группа); 300 мг ламивудина каждые 12 часов плюс плацебо, походящее на зидовудин (только ламивудиновая группа); 150 мг ламивудина каждые 12 часов плюс 200 мг зидовудина каждые 8 часов (группа низкодозовой комбинационной терапии); или 300 мг ламивудина каждые 12 часов плюс 200 мг эидовудина каждые 8 часов (группа высокодозовой комбинированной терапии). Всего 366 пациентов случайным образом были назначены в группы этих четырех курсов лечения.
Первичными оцененными результативными критериями в группах лечения были отклонения от основной линии в уровнях клеток ВИЧ-1 РНК и CD4+. Представлены доказательства, что комбинации числа CD4 и ВИЧ-1 РНК представляет собой показатель прогрессирования к СПИДу и смерти и что вызванное лечением повышение числа CD4 и снижение ВИЧ-1 РНК коррелирует с уменьшением риска прогрессирования болезни ВИЧ.
Результаты CD-4 и ВИЧ-1 РНК после 24 недель и 52 недель показаны в Таблице 2. Эти результаты ясно демонстрируют, что воздействие испытанной комбинации больше, чем сумма воздействия ламивудиновой и зидовудиновой монотерапий; другими словами, комбинация является синергетической. Особенно эффектно, что средние отклонения числа CD4 для пациентов, получавших комбинационную терапию и монотерапию, расходятся (смотри фиг. 1). Иными словами, числа CD4 для пациентов в группах комбинированной терапии продолжали улучшаться, тогда как числа CD4 у пациентов, которые получали только ламивудиновую монотерапию или зидовудиновуюю монотерапию, снижались до базовой линии или ниже ее. Более того, уровни ВИЧ-1 РНК оставались подавленными в значительно большей степени у тех пациентов, которые получали комбинационную терапию, тогда как в двух группах с монотерапией они возвращались к уровню первоначальной базовой линии. Ясно, что такое не могло бы произойти, если бы эффект комбинации был просто аддитивным.
Подпись к рисунку:
Ось Y: Среднее отклонение в числе CD4 (клеток/мм3)
Ось X: Недели
Пациенты, для которых могла быть проведена оценка
(Прим. переводчика: числа расположены и соответствуют числу недель на графике)
Только зидовудин (- -) 53 82 82 80 73 71 68 59 63 60 60 57 56 46
Только ламивудин (- -) 71 74 75 68 68 67 65 62 57 56 62 51 48
Низкодозовая
комбинационная
терапия (- -) 92 84 75 77 71 68 71 68 60 58 58 53 46
Высокодозовая
комбинационная
терапия (- -) 94 87 78 77 76 66 61 60 58 58 49 47 42
Фигура 1: Среднее (± среднеквадратичная ошибка) отклонение от основной линии в абсолютном числе CD4+, соответствующее неделям проведения исследования.
Число пациентов, показанное для каждой недели для каждой из четырех групп лечения, является числом, которое могло быть оценено на этот момент. Через 24 недели число пациентов указывает числа, доступные для исследования для каждой точки анализа; числа не указывают случаев отмены исследования. Некоторые пациенты еще не завершили продленную фазу исследования ко времени проведения данного анализа.

Изобретение предназначено для лечения вирусных инфекций. Предложенная комбинация, обладающая противовирусным действием, содержит (2R,цис)-4-амино-1-/2-гидроксиметил-1,3-оксатиолан--5-ил)-1Н-пиримидин-2-он(ЗТС) или его фармацевтически приемлемое производное и 3'-азидо-3'-дезокситимидин (АZT) или его фармацевтически приемлемое производное. Эта комбинация проявляет синергизм в отношении противовирусного действия. Фармацевтическая композиция включает указанную комбинацию и фармацевтически приемлемый носитель. Способ лечения млекопитающего, включая человека, инфицированного или восприимчивого к ВИЧ-инфекции, осуществляют путем одновременного введения указанной комбинации из двух соединений - ЗТС к АZТ или их производных или последовательного введения этих соединений. Использование изобретения позволит снизить токсичность лекарства и повысить эффективность его действия. 3 с. и 7 з.п. ф-лы, 1 ил., 2 табл.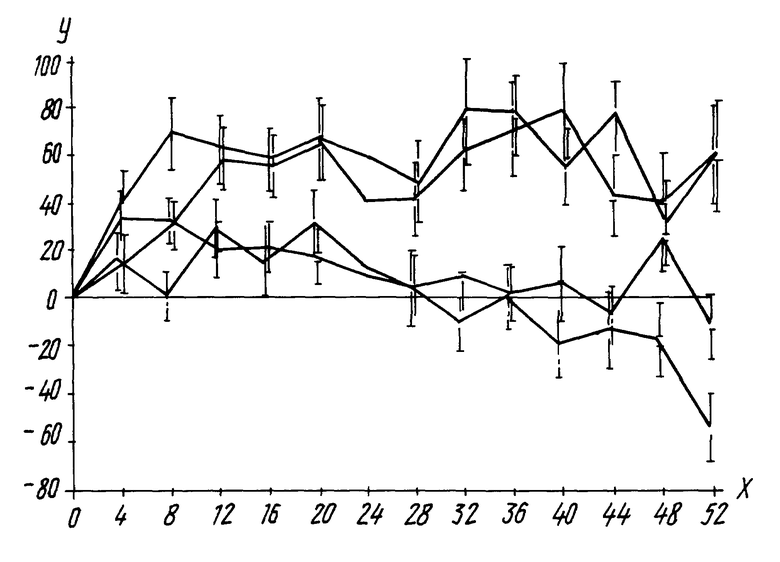
| КЛАПАН ЦЕНТРАЛИЗОВАННОЙ СИСТЕМЫ ПОДКАЧКИ ШИН ТРАНСПОРТНОГО СРЕДСТВА | 0 |
|
SU382526A1 |
| SU 5900949 A, 1979. | |||
