Изобретение относится к усовершенствованному способу получения макролидного полусинтетического антибиотика, производного эритромицина A, а именно к способу получения N-метил-11-аза-10-дезоксо-10-дигидроэритромицина-азитромицина (I), который обладает широким спектром антимикробной активности.
Известны способы получения азитромицина N-метилированием 11-аза-10-дезокси-10-дигидроэритромицина (далее азаэритромицин). Описано два способа N-метилирования азаэритромицина (V, схема 1). Первый, путем предварительного окисления азаэритромицина перекисью водорода с образованием N-оксида, с последующим алкилированием иодистым метилом в присутствии акцептора иодистого водорода и каталитическим восстановлением полученного алкильного производного над Pd/C или над Ni-Ренея (патент США N 4474468, кл. C 07 H 17/08, 1984 [1] . И второй, действием на азаэритромицин формальдегида в присутствии муравьиной кислоты в среде хлорированного углеводорода, такого как хлороформ или четыреххлористый углерод (патент США N 4517359, кл. C 07 H 17/08, 1981 [2] ). Для выделения целевого продукта проводят реэкстракцию реакционной массы водой при pH 5,0, затем продукт вновь экстрагируют 4-х кратным объемом хлороформа при pH 7,5, экстракт высушивают и выпаривают досуха. Выход азитромицина, выделенного таким образом, составляет 83,0% ([2], пример 1).
Второй способ наиболее близок настоящему изобретению и является его прототипом.
Исходный азаэритромицин (V) согласно всем известным способам, получают из эритромицина (II) по следующей схеме (схема 1):
- оксимилирование эритромицина (II) с получением оксима эритромицина (III);
- превращение оксима III в иминоэфир эритромицина (IV) путем перегруппировки Бекмана;
- восстановление иминоэфира IV до азаэритромицина (V).
Оксимилирование эритромицина, согласно всем известным способам, проводят в среде безводного метанола действием гидроксиламина, который используют либо в виде свободного основания, частично нейтрализованного кислотой (Европейский патент N 0342990, кл. C 07 H 17/086, 1989 [3]), либо в виде гидрохлорида в присутствии акцептора хлористого водорода; эритромицин реагирует с гидроксиламином солянокислым в присутствии карбоната бария в абсолютном метаноле при температуре кипения (патент Англии N 1100504, кл. C 07 H 21/00, 1968, [4]); исходное соотношение эритромицин/метанол составляет 1:5, время кипячения реакционной массы - 22 ч, для выделения продукта реакционную массу фильтруют, концентрируют в вакууме до 1/3 объема, при охлаждении концентрата кристаллизуется оксим эритромицина, выход оксима III составляет 48,3% ([4], пример 1) - прототип по п. 2 формулы изобретения.
Перегруппировку Бекмана оксима эритромицина (III) по описанным способам осуществляют действием хлорангидридов арилсульфоновых кислот в смеси ацетон-вода (J. Chem. Soc. Perkin. Traus, 1986, c. 1881-90 [6], патент США N 4328334, кл. C 07 H 17/08, 1982 [7]. Согласно патенту СССР N 1447288, кл. C 07 H 17/08, 1983 [5] оксим III подвергают перегруппировке Бекмана действием 2-4 молярного избытка п-толуолсульфохлорида, п-иодбензолсульфохлорида или п-ацетиламинобензолсульфохлорида в присутствии 2-8 молярного избытка бикарбоната натрия или триэтиламина при температуре 0-5oC в смеси ацетон-вода. По примеру 2 [5] для выделения продукта реакции IV ацетон упаривают в вакууме, водный остаток pH-градиентно экстрагируют хлороформом, экстракт, полученный при pH 8,0, высушивают и выпаривают досуха. Выход полученного таким образом иминоэфира IV составляет 74,3%.
Восстановление иминоэфира IV по известным способам осуществляют каталитическим гидрированием при давлении водорода 65-70 ати в ледяной уксусной кислоте, как растворителе, используя в качестве катализатора благородные металлы или их окислы, такие как Pd/C или PtO2, выход азаэритромицина (V) при этом составляет 80,0% (патент США N 4328334, кл. C 07 H 17/08, 1982 [7]), электрохимическим способом в электролизере с синтетической диафрагмой на ртутном катоде со свинцовым анодом в присутствии иодистого тетрабутиламмония с использованием в качестве католита уксусной кислоты, а в качестве анолита водного раствора ацетата натрия, выход азаэритромицина по этому способу составляет 81,5% (патент СССР N 1530096, кл. C 07 H 17/08, 1990 [8]); химическим восстановлением комплексными гидридами металлов, такими как натрия борогидрид в абсолютном метаноле при температуре около 4oC [7]. Согласно примеру 4 [7] для восстановления иминоэфира IV используют 20-ти кратный молярный избыток натрия борогидрида, для выделения продукта восстановления - азаэритромицина (V), реакционную массу после барботажа CO2 фильтруют, фильтрат упаривают досуха, остаток растворяют в хлороформе, добавляют воду, проводят pH-градиентную экстракцию, экстракт высушивают, упаривают досуха, сухой остаток экстрагируют эфиром, экстракт упаривают досуха. Выход полученного таким образом азаэритромицина составляет 60,5%.
Целью изобретения является упрощение процесса (упрощение технологии, снижение энергоемкости, улучшение показателей пожаро- и экобезопасности) и повышение выхода целевого продукта.
Поставленная цель достигается осуществлением следующего способа.
Эритромицин оксимилируют гидроксиламином солянокислым в присутствии карбоната натрия в среде этанол-вода (20-40% воды), при исходном соотношении эритромицин/растворитель 1:(1,4-1,7) при температуре 50-80oC, для выделения продукта оксимилирования используют экстракционный метод, для чего реакционную массу экстрагируют хлорированным углеводородом при pH 9-11. Выход оксима эритромицина составляет 75-80%.
Описанное выше осуществление данной стадии позволяет значительно повысить концентрацию реагирующих веществ, что является существенным, т.к. реакция оксимилирования эритромицина всегда сопровождается процессом дегидратации эритромицина с образованием ангидроэритромицина; эта побочная реакция является мономолекулярной и, следовательно, скорость ее не зависит от концентрации, тогда как скорость целевой реакции увеличивается пропорционально концентрации реагирующих веществ. Таким образом увеличение концентрации реагирующих веществ приводит к уменьшению содержания примеси ангидроэритромицина в реакционной массе и повышению выхода оксима эритромицина. Кроме того, исключается использование метанола-токсичного и пожароопасного растворителя и необходимость упаривания реакционной массы в конце реакции, что позволяет существенно снизить энергоемкость процесса. Использование экстракционного метода выделения оксима III позволяет легко и полностью отделить (а при необходимости регенерировать) избыток гидроксиламина, тогда как по известному способу [4] избыток гидроксиламина в значительной степени разлагается при длительном кипячении реакционной массы, а частично сокристаллизуется с оксимом III после концентрирования реакционной массы и охлаждения концентрата, что загрязняет продукт реакции.
Для проведения перегруппировки Бекмана под действием хлорангидрида арилсульфоновой кислоты используют двухфазную систему растворителей: хлорированный углеводород - вода. Это позволяет по окончании реакции легко отделить примеси и осадить иминоэфир эритромицина из водного раствора в кристаллическом виде. По окончании реакции при отделении водной фазы отделяются все неорганические примеси, при последующей реэкстракции иминоэфира в воду при pH 4,5-5,5 в хлороформе остаются все примеси органического характера. Из водного реэкстракта после установления pH 9-11 при температуре (60±5)oC кристаллизуется чистый иминоэфир IV. Использование двухфазной системы растворителей позволяет исключить необходимость упаривания ацетона из реакционной массы по окончании реакции, а также ступенчатую экстракцию примесей. Продукт выделяют кристаллизацией, вместо нетехнологичной упарки "досуха". Выход иминоэфира по данному способу составляет 82,0%.
При восстановлении иминоэфира IV комплексными борогидридами металлов в качестве растворителя используется вода, реакцию проводят при температуре окружающей среды (16-20)oC, восстановитель применяют в количестве 1,0-1,2 моль на моль иминоэфира. Нами было обнаружено, что при действии борогидридов металлов или при предварительной обработке иминоэфира IV борной кислотой образуются водорастворимые боратные комплексы иминоэфира, благодаря чему становится возможным проведение реакции восстановления в водном растворе при pH 9-11, что является оптимальной областью при работе с комплексными борогидридами металлов. Поэтому для полноты восстановления иминоэфира IV в этих условиях достаточно эквимолярного количества восстановителя. При этом продуктом реакции восстановления является боратный комплекс азаэритромицина, который используют для получения азитромицина путем метилирования и последующего гидролиза боратного комплекса, или боратный комплекс азаэритромицина гидролизуют перед метилированием. И в том и в другом случае боратный комплекс гидролизуют обработкой разбавленной минеральной или органической кислотой при pH 2,5, обычно, в течение 5-15 мин при температуре 18-20oC. Предпочтительно использовать для гидролиза оксикислоты, т.к. их присутствие препятствует протеканию обратной реакции комплексообразования. Азаэритромицин (V) выделяют после гидролиза комплекса экстракцией хлороформом. Выход азаэритромицина составляет 85-88%, считая на иминоэфир IV.
Азитромицин (I) получают из азаэритромицина (V) или его боратного комплекса путем метилирования в хлороформе действием формальдегида и муравьиной кислоты, продукт метилирования реэкстрагируют водой при pH 4,5-5,5, при необходимости проводят гидролиз боратного комплекса азитромицина минеральной или органической кислотой при pH 2,5, целевой продукт осаждают из водного раствора при pH 9-11 и температуре 50-80oC (выход азитромицина составляет 76-78%, считая на иминоэфир эритромицина) и очищают перекристаллизацией из смеси ацетон-вода или этанол-вода в соотношениях от (2:1) до (1:2). Использование для очистки азитромицина смеси ацетон-вода или этанол-вода в широком диапазоне соотношений позволяет регулировать качество получаемого продукта: при использовании более водных смесей повышается выход целевого продукта, но частично сокристаллизуются примеси, тогда как уменьшение количества воды в кристаллизационной смеси снижает выход целевого продукта, но обеспечивает получение препарата высокой степени чистоты. Выход на стадии перекристаллизации составляет 70-94%.
Таким образом предлагаемый способ получения азитромицина заключается в следующем:
Эритромицин (II) оксимилируют действием гидроксиламина солянокислого в присутствии карбоната натрия в среде этанол-вода (20-40% воды) при исходном соотношении эритромицин/растворитель 1:(1,4-1,7) (масса/объем) и температуре (50-80%)oC, продукт реакции - оксим эритромицина (III) выделяют из реакционной массы экстракцией хлорированным углеводородом при pH 9-11. Оксим III подвергают перегруппировке Бекмана действием хлорангидрида арилсульфоновой кислоты в двухфазной смеси растворителей: вода-хлорированный углеводород, полученный иминоэфир эритромицина (IV) выделяют из водного раствора кристаллизацией при pH 9-11 и температуре (60±5)oC. Иминоэфир IV восстанавливают действием комплексного борогидрида металла, взятого в 0-0,2 молярном избытке, в водной среде, полученный боратный комплекс азаэритромицина выделяют экстракцией хлороформом и, в виде боратного комплекса или после гидролиза комплекса действием минеральной или органической кислоты, предпочтительно оксикислоты, при pH 2,5, метилируют в хлороформе формальдегидом в присутствии муравьиной кислоты, продукт метилирования реэкстрагируют водой при pH 3,5-5,5, при необходимости проводят гидролиз боратного комплекса, и азитромицин выделяют осаждением из водного раствора при pH 9-11 и температуре (50-30)oC. Азитромицин очищают перекристаллизацией из смеси ацетон-вода или этанол-вода в соотношении от (2:1) до (1:2).
Данный способ позволяет получить азитромицин с показателями качества, удовлетворяющими требованиями Фармакопеи США и ВФС РФ, и выходом 35-40%, считая на эритромицин.
Другой целью изобретения является повышение выхода и упрощение процесса получения оксима эритромицина (III).
Ближайшим аналогом-прототипом предлагаемого способа является способ получения оксима эритромицина по патенту [4].
Поставленная цель достигается тем, что при оксимилировании эритромицина (II) гидроксиламином солянокислым в присутствии карбоната натрия в качестве растворителя используется смесь этанол-вода (20-40% воды), что позволяет создать высокие концентрации реагирующих веществ (исходное соотношение эритромицин/растворитель составляет 1:1,4 - 1:1,7 масса/объем), реакцию проводят при температуре (50-80)oC, при этом процесс оксимилирования заканчивается за 2-0,3 ч, оксим эритромицина (III) выделяют экстракцией хлорированным углеводородом, таким как хлористый метилен или хлороформ. Выход оксима III составляет 75-80%.
Третья цель изобретения состоит в повышении выхода азаэритромицина (V), получаемого восстановлением иминоэфира эритромицина (IV) комплексным борогидридом металла.
Ближайшим аналогом-прототипом - предлагаемого способа является способ получения азаэритромцина [7], пример 4.
Поставленная цель достигается тем, что иминоэфир эритромицина (IV) восстанавливают в водном растворе действием комплексного борогидрида металла, взятого в 0-0,2 молярном избытке при температуре окружающей среды, продукт восстановления - боратный комплекс азаэритромицина подвергают гидролизу действием минеральной или органической кислоты, предпочтительно оксикислоты, при pH 2,5, азаэритромицин (V) выделяют экстракцией хлороформом при pH 9-11. Выход азаэритромицина составляет 85-88%.
Способ может быть проиллюстрирован следующими примерами.
Пример 1. Получение оксима эритромицина (III).
Эритромицин (200 г) растворяют в 210 мл этанола при нагревании до 60oC. К полученному раствору эритромицина добавляют раствор, содержащий 36,0 г углекислого натрия и 90,0 г гидроксиламина солянокислого в 90 мл воды, с температурой 60oC. Получают водно-спиртовой раствор (30% воды) с исходным соотношением эритромицин/растворитель 1: 1,5 г/мл. Реакционную массу перемешивают при температуре 60oC в течение 1 ч, затем переносят в экстрактор, содержащий 1,5 л воды и 1,5 хлороформа. При перемешивании добавляют 20% раствор едкого натра до установления значения pH водной фазы в интервале 9-11. Слои разделяют, хлороформенный экстракт промывают 300 мл воды. Получают 2,15 л хлороформенного раствора, содержащего 66,0 мг/мл оксима эритромицина (поляриметрически). Выход 77,3%.
Кристаллический оксим эритромицина выделяют после упаривания хлороформенного раствора до объема 400 мл и охлаждения концентрата до (8-10)oC в течение 16 ч. Получают 134,0 г бесцветных кристаллов с т.пл. 157-159oC, что соответствует данным литературы (Tetrahedren Letters, 1970, c. 157-160 [9]). Выход 72,8%.
Пример 2. Получение иминоэфира эритромицина (IV).
В стеклянный реактор загружают хлороформенный раствор оксима эритромицина (пример 1), добавляют раствор 54,0 г двууглекислого натрия в 2 л воды. Смесь охлаждают до (5±2)oC и при перемешивании добавляют из капельной воронки раствор п-толуолсульфохлорида в хлороформе (64,0 г п-ТСХ в 250 мл хлороформа). Добавление реагента продолжается в течение 1 ч. Реакционную массу выдерживают при перемешивании и температуре (7-10)oC еще 2 ч, затем слои разделяют. К хлороформенному слою добавляют 1,5 л воды и разбавленную соляную кислоту до установления значения pH водной фазы в пределах 4,5-5,5. Массу перемешивают 30 мин, контролируя значение pH водной фазы, которое должно оставаться в указанных пределах. Затем слои разделяют. К водному реэкстаркту при перемешивании приливают 120 мл конц. аммиака, при этом pH устанавливается в пределах 9-11. Кристаллизационную массу нагревают при перемешивании до (60±5)oC и выдерживают при этой температуре 15 мин, продолжая перемешивание. Осадок иминоэфира IV отфильтровывают и промывают на фильтре 400 мл воды с температурой ~60oC. Продукт высушивают до постоянного веса. Получают 115,0 г мелкокристаллического белого порошка, т.пл. 129-130oC, [α]
Пример 3. Получение азаэритромицина (V).
В стеклянный реактор помещают 100,0 г иминоэфира эритромицина (пример 2) и 1,2 л воды. К суспензии при перемешивании добавляют из капельной воронки 20% серную кислоту до полного растворения иминоэфира (pH 5,6-6,4). К раствору, продолжая перемешивание, приливают раствор 13,8 г калия борогидрида в 100 мл воды. Реакционную массу оставляют при комнатной температуре на 16-18 ч для завершения реакции восстановления. Реакционная масса представляет собой бесцветный раствор с pH 9,0-11,0. По окончании реакции к раствору добавляют 1,3 л хлороформа, перемешивают в течение 30 мин, слои разделяют. К хлороформенному слою (Раствор A), содержащему боратный комплекс азаэритромицина, добавляют 1,0 л воды и 20% раствор серной кислоты до установления значения pH водной фазы (2,5±0,1). Реакционную массу выдерживают при этом значении pH в течение 15 мин при температуре (18-20)oC, затем добавляют 130 мл 20% раствора едкого натра, смесь перемешивают 30 мин, после чего слои разделяют. При этом pH водной фазы находится в интервале 9-11. Хлороформный экстракт азаэритромицина сушат сульфатом натрия. Получают 1,22 л хлороформенного раствора азаэритромицина, с содержанием основного вещества 65 мг/мл (поляриметрически) - Раствор Б. Экстракт упаривают досуха, получают 87,3 г аморфного порошка. Содержание азаэритромицина 90,0% [α]
Пример 4. Получение азитромицина.
А) Раствор A (пример 3), содержащий боратный комплекс азаэритромицина, помещают в колбу, снабженную обратным холодильником, добавляют 18,0 мл формальдегида (40%) и 9,7 мл муравьиной кислоты (99%). Реакционную массу нагревают до кипения и кипятят в течение 6 ч. Затем массу охлаждают, при необходимости добавляют 20% раствор серной кислоты до установления значения pH водной фазы в интервале 4,5-5,5. Слои разделяют, к водному слою добавляют лимонную кислоту (19,5 г) до pH (2,5±0,1), раствор перемешивают 15 мин, затем добавляют 1,4 л хлороформа и 20% раствор едкого натра до установления значения pH водной фазы в интервале 9-11. Слои разделяют. К хлороформенному слою приливают 1,4 л воды и 20% раствор серной кислоты до pH 4,5-5,5. Смесь перемешивают 30 мин, контролируя pH водной фазы. Слои разделяют, водный слой помещают в кристаллизатор, добавляют 1,5 л воды и 82,0 мл конц. аммиака. При этом pH кристаллизационной массы устанавливается в интервале 9-11. Кристаллизационную массу нагревают до температуры около 70oC, перемешивают при этой температуре 15 мин и фильтруют. Осадок промывают на фильтре 300 мл воды с температурой около 70oC и высушивают до постоянного веса. Получают 75,5 г белого мелкокристаллического порошка, массовая доля азитромицина 86,0% (микробиологический метод). Выход 74,8%, считая на иминоэфир эритромицина (IV).
Б. Раствор Б (пример 3), содержащий азаэритромицин, переносят в колбу, снабженную обратным холодильником, добавляют 9,7 мл муравьиной кислоты и 18,0 мл формалина (40%). Реакционную массу нагревают до кипения и кипятят в течение 6 ч. Затем массу охлаждают, добавляют 1,4 л воды и 20% раствор серной кислоты до установления значения pH водной фазы 4,5-5,5. Смесь перемешивают 30 мин, контролируя pH водной фазы, которое должно оставаться в пределах 4,5-5,5. Слои разделяют. Водный слой помещают в кристаллизатор и добавляют 1,5 л воды и 82 мл конц. аммиака. При этом pH кристаллизационной массы устанавливается в пределах 9-11. Кристаллизацию азитромицина проводят, как описано в п. А. Получают 76,2 г. Выход 75,3%, считая на иминоэфир эритромицина.
Пример 5. Перекристаллизация азитромицина.
К 75,5 г азитромицина, полученного по примеру 4, добавляют 225 мл ацетона и суспензию перемешивают до полного растворения азитромицина. Полученный раствор фильтруют через мембрану "Владипор" МФЦ N 2, мембрану промывают 75,0 мл ацетона. Объединенный фильтрат переносят в кристаллизатор и при перемешивании добавляют воду до начала кристаллизации. Кристаллизационную массу перемешивают в течение 1 ч, затем из капельной воронки добавляют воду, так чтобы общее количество добавленной воды составило 150 мл, а соотношение ацетон-вода в кристаллизационной массе 2:1. Массу перемешивают еще два часа при комнатной температуре для завершения кристаллизации, затем фильтруют. Осадок промывают на фильтре смесью ацетон-вода (2:1). Продукт высушивают до постоянного веса, получают 66,8 г белого негигроскопичного кристаллического вещества, с массовой долей основного вещества 94,0% и с содержанием воды 4,8%. Выход азитромицина составляет 37,5%, считая на эритромицин.
Удельное вращение: минус 41o
Специфические примеси: менее 0,5%
Сульфатная зола: следовые количества
pH водной суспензии: 8,6
Содержание ацетона: 0,10%
Дифрактограмма продукта соответствует стандартному образцу азитромицина.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХЛОРИДА ОКСИМА ЭРИТРОМИЦИНА А | 2001 |
|
RU2199546C2 |
| 9А-АЗАЛИДНЫЕ ФРАГМЕНТЫ МАКРОЛИДНЫХ АНТИБИОТИКОВ КЛАССА АЗАЛИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2130936C1 |
| НОВЫЕ КЕТОАЗОЛИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2211222C2 |
| 8-ФТОРАНТРАЦИКЛИНГЛИКОЗИДЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2095365C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИМА ЭРИТРОМИЦИНА | 2004 |
|
RU2256665C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ МАКРОЛИДОВ И КЕТОЛИДОВ, И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2608390C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1,1-ДИОКСИДА ПЕНИЦИЛЛАНОВОЙ КИСЛОТЫ И ЕЕ СОЛИ | 1993 |
|
RU2064931C1 |
| ПРОСТОЙ СПОСОБ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2711358C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ ГИДРОКСИЛАММОНИЯ | 1996 |
|
RU2159211C2 |
| СПОСОБ СИНТЕЗА АРОМАТИЧЕСКИХ ОКСИМОВ | 2016 |
|
RU2720240C2 |
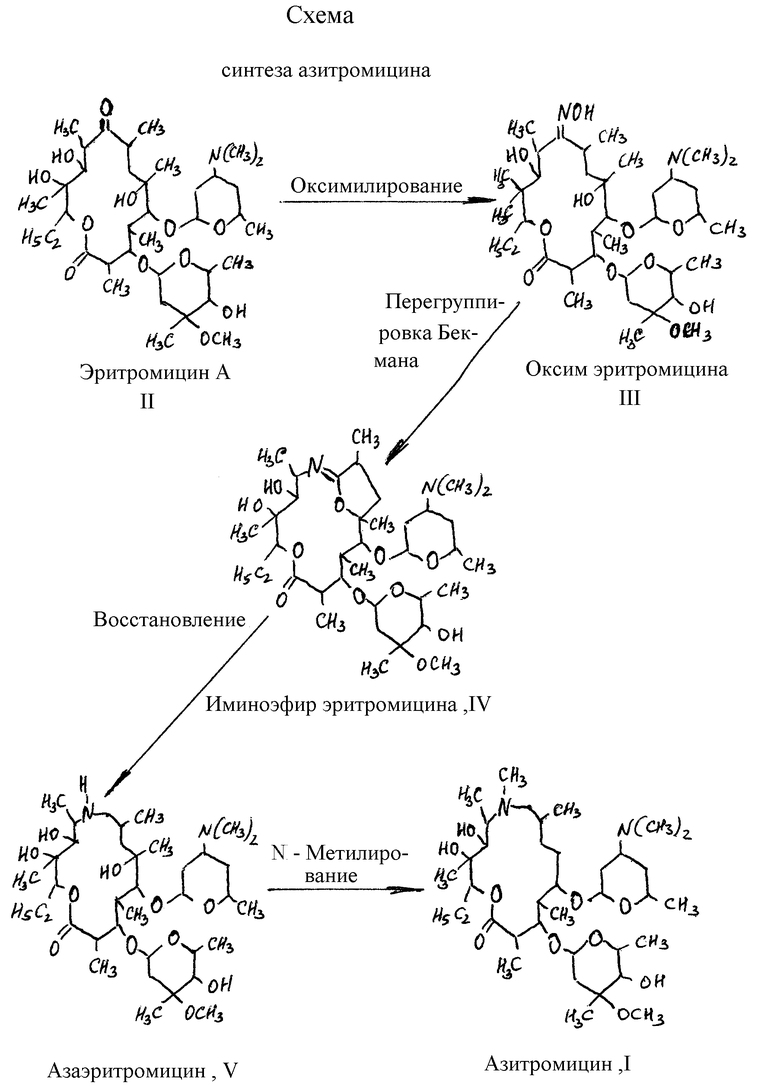
Описывается способ получения азитромицина (I). Эритримицин (II) оксимилируют действием гидроксиламина солянокислого в присутствии карбоната натрия в среде этанол - вода (20-40% воды) при исходном соотношении эритромицин/растворитель, г/мл, 1:1,4 - 1:1,7 и температуре 50-80oС, продукт оксимилирования - оксим эритромицина (III) выделяют из реакционной массы экстракцией хлорированным углеводородом при рН 9-11 и подвергают перегруппировке Бекмана действием хлорангидрида арилсульфоновой кислоты в двухфазной системе растворителем - вода/хлорированный углеводород, образовавшийся иминоэфир эритромицина (IV) выделяют из водного раствора кристаллизацией при рН 9-11 и температуре 60±5oC, с последующим восстановлением иминоэфира IV в водной среде при комнатной температуре действием комплексного борогидрида металла, взятого в 0-0,2 молярном избытке, продукт восстановления - азаэритромицин (V) в виде боратного комплекса минеральной или органической кислотой, предпочтительно оксикислотой, при рН 2,5, метилируют в хлороформе формальдегидом в присутствии муравьиной кислоты, продукт метилирования выделяют реэкстракцией водой при рН 4,5-5,5, при необходимости проводят гидролиз боратного комплекса, целевой продукт осаждают из водного раствора при рН 9-11 и температуре 50-80oС, с последующей перекристаллизацией из смеси ацетон - вода или этанол - вода, взятых в соотношении от 2:1 до 1:2. Описываются также способы получения промежуточных соединений. Технический результат - упрощение процесса и повышение выхода целевого продукта. 3 с.п. ф-лы.
| US 4474468, 1984 | |||
| US 4517359, 1981 | |||
| 0 |
|
SU342990A1 | |
| US 4328334, 1982 | |||
| Способ получения 11-аза-10-деоксо-10-дигидроэритромицина А | 1986 |
|
SU1530096A3 |
| Способ получения 7,16-диокса-2-аза-10-О-кладинозил-12-О-десозаминил-4,5-дигидрокси-6-этил-3,5,9,11,13,15-гексаметилбицикло @ 11,2,1 @ гексадека -1(2)-ен- 8-она | 1984 |
|
SU1447288A3 |
| Устройство для измерения частоты первой гармоники квазипериодических сигналов | 1982 |
|
SU1100504A1 |
