ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/346664, поданной 20 мая 2010 года, содержание которой включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Изобретение, описанное в настоящей заявке, относится к способам получения макролидных антибактериальных агентов. В частности, изобретение относится к способам получения макролидов и кетолидов из эритромицина А.
УРОВЕНЬ ТЕХНИКИ И КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Макролидные антибиотики, характеризующиеся большим лактонным кольцом, к которому присоединены один или более дезоксисахаров, как правило, кладиноз и дезозамин, представляют собой противомикробные лекарственные средства, которые обладают активностью в отношении аэробных и анаэробных грамположительных кокков и предназначены для лечения инфекций дыхательных путей и мягких тканей. Макролиды, которые относятся к поликетидному классу природных продуктов, действуют путем обратимого связывания с субъединицей 50S бактериальных рибосом, блокируя синтез белков и предотвращая рост и размножение бактерий. Несмотря на то, что это действие является, главным образом, бактериостатическим, в более высоких концентрациях макролиды могут обладать бактерицидным действием. Эритромицин и его полусинтетические производные (азитромицин и кларитромицин) входят в число представленных на рынке макролидных антибиотиков.
Кетолиды, которые представляют собой полусинтетические производные 14-членного макролида эритромицина А, относятся к классу лекарственных средств, применяемых для лечения инфекций дыхательных путей. Эти лекарственные средства являются эффективными в отношении резистентных к макролидам бактерий благодаря своей способности связываться с двумя участками бактериальных рибосом. К этой группе антибиотиков принадлежат телитромицин и цетромицин.
Приобретенная резистентность бактерий к макролидам возникает, главным образом, за счет посттранскрипционного метилирования бактериальной рибосомы 23S, что приводит к перекрестной резистентности к макролидам, линкозамидам и стрептограминам. Хоть и редко, но приобретенная резистентность также может возникать в результате продуцирования деактивирующих лекарственные средства ферментов, таких как эстеразы или киназы, а также в результате продуцирования активных АТФ-зависимых эффлюксных белков, которые транспортируют макролиды из клетки. Значительная часть пневмококков резистентна к доступным в настоящее время антибиотикам. Соответственно, существует потребность в новых макролидных и кетолидных антибиотиках, а также в способах их получения.
В частности, в публикации международной заявки на патент WO 2004/080391 и публикации эквивалентной заявки на патент США №2006/0100164, содержание которых включено в настоящую заявку посредством ссылки, описано семейство макролидных и кетолидных антибиотиков, включая фторкетолидные антибиотики, формулы (I)
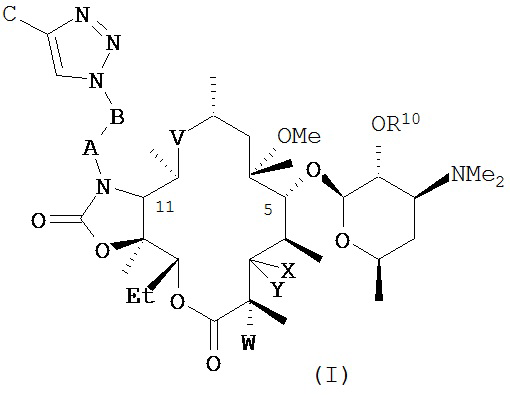
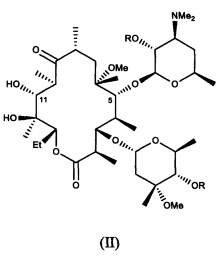
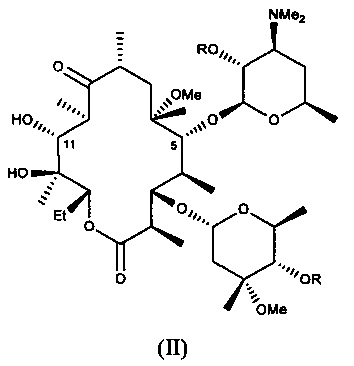
и фармацевтически приемлемые соли указанных соединений, где R10, X, Y, V, W, А, B и C описаны в заявке, Me обозначает метил, a Et обозначает этил. Известным, но не ограничивающим, примером соединения формулы (I) является солитромицин, также обозначаемый как ОР-1068 и/или СЕМ-101. Получение СЕМ-101 и родственных соединений описано в WO 2009/055557, содержание которой включено в настоящую заявку посредством ссылки. Исходным веществом, применяемым согласно WO 2009/055557 A1 для получения макролидных антибактериальных агентов, является кларитромицин. В способах, описанных в указанной заявке, кларитромицин превращают в производное кларитромицина, в котором гидроксильные группы фрагментов сахаров защищены ацильными группами, например, в кларитромицина дибензоат, также известный как 2',4''-ди-O-бензоил-6-O-метилэритромицин А, с получением соединений формулы (II).
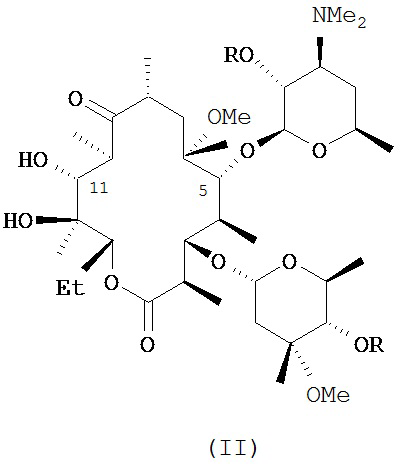
и их фармацевтически приемлемых солей, где R описан в заявке.
Кларитромицин представляет собой полусинтетический антибактериальный агент, в котором 6-гидроксигруппа эритромицина А превращена в 6-метоксигруппу для исключения нежелательного взаимодействия с карбонильной группой в положении 9 макролидного кольца с обеспечением тем самым стабилизации антибиотика. Кларитромицин получают при помощи различных способов. В наиболее распространенных способах получения в качестве исходного вещества применяют эритромицин A, который превращают в оксим, а затем в защищенное 9-оксимное производное эритромицина А, используемое в качестве промежуточного соединения, при этом данные способы также включают разнообразные стадии введения и снятия защиты для гидроксильных и диметильных групп присоединенных фрагментов сахаров до и после метилирования 6-гидроксигруппы макролидного кольца (см., например, патент США 6515116 для обзора указанных способов; альтернативный подход, включающий защиту дезозаминильной аминогруппы в виде N-оксида, описан в патенте США 6809188). Для эффективного получения производного кларитромицина, в котором гидроксильные группы фрагментов сахаров защищены ацильными группами, а следовательно, и гидроксильные группы фрагментов сахаров конечного макролидного антибактериального агента защищены ацильными группами, необходимо получение дизащищенного производного эритромицина А, не требующее проведения стадий введения и удаления защитных групп, применяемых согласно известным способам получения кларитромицина. В настоящей заявке описаны способы прямого получения производных кларитромицина формулы (II) из эритромицина А, в которых гидроксильные группы фрагментов сахаров защищены ацильными группами, при этом предложенные способы включают меньшее число стадий. Также в настоящей заявке описаны способы получения соединений формулы (I) из соединений формулы (II).
ПОДРОБНОЕ ОПИСАНИЕ
В одном из иллюстративных вариантов реализации изобретения описаны способы получения соединений формулы (I)
и их фармацевтически приемлемых солей, где:
R10 представляет собой водород, ацил или фрагмент пролекарства;
X представляет собой H; a Y представляет собой OR7, где R7 представляет собой моносахарид, дисахарид, алкил, арилалкил или гетероарилалкил, каждый из которых возможно замещен, или ацил или C(O)NR8R9; где каждый R8 и R9 независимо выбран из группы, состоящей из водорода, гидрокси, алкила, гетероалкила, алкокси, арила, арилалкила, гетероарила и гетероарилалкила, каждый из которых возможно замещен, и диметиламиноалкила, ацила, сульфонила, уреидо и карбамоила; или R8 и R9 совместно с присоединенным к ним атомом азота образуют возможно замещенный гетероцикл; или X и Y совместно с присоединенным к ним атомом углерода образуют карбонил;
V представляет собой C(O), C(=NR11), CH(NR12, R13) или N(R14)CH2; где N(R14) присоединен к С-10 атому углерода; где R11 представляет собой гидрокси или алкокси; каждый R12 и R13 независимо выбран из группы, состоящей из водорода, гидрокси, алкила, алкокси, гетероалкила, арила, арилалкила, гетероарила и гетероарилалкила, каждый из которых возможно замещен, и диметиламиноалкила, ацила, сульфонила, уреидо и карбамоила; R14 представляет собой водород, гидрокси, алкил, алкокси, гетероалкил, арил, арилалкил, гетероарил или гетероарилалкил, каждый из которых возможно замещен, или диметиламиноалкил, ацил, сульфонил, уреидо или карбамоил;
W представляет собой Н, F, Cl, Br, I или OH;
A представляет собой CH2, C(O), C(O)O, C(O)NH, S(O)2, S(O)2NH или C(O)NHS(O)2;
B представляет собой (CH2)n, где n представляет собой целое число от 0 до 10; или ненасыщенную углеродную цепь, содержащую от 2 до 10 атомов углерода; и
C представляет собой водород, гидрокси, алкил, алкокси, гетероалкил, арил, арилалкил, гетероарил или гетероарилалкил, каждый из которых возможно замещен, или ацил, ацилокси, сульфонил, уреидо или карбамоил.
В другом иллюстративном варианте реализации описаны способы получения соединений формулы (II)
и их фармацевтически приемлемых солей, где R представляет собой ацильную группу. В другом варианте реализации R представляет собой пространственно затрудненную ацильную группу, такую как бензоил.
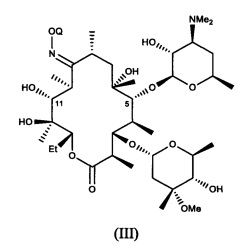
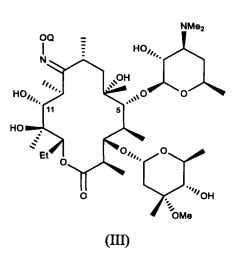
В другом варианте реализации описаны способы, включающие стадию (а) приведения соединения формулы (III)
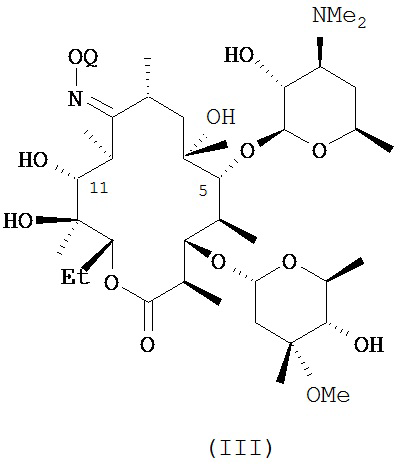
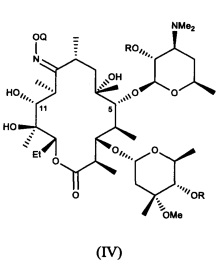
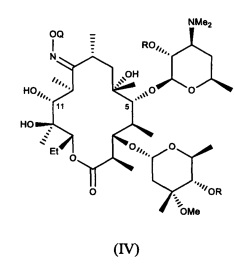
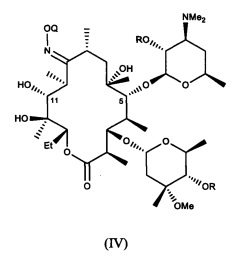
или его соли присоединения кислоты, в контакт с ацилирующим реагентом с получением соединения формулы (IV)
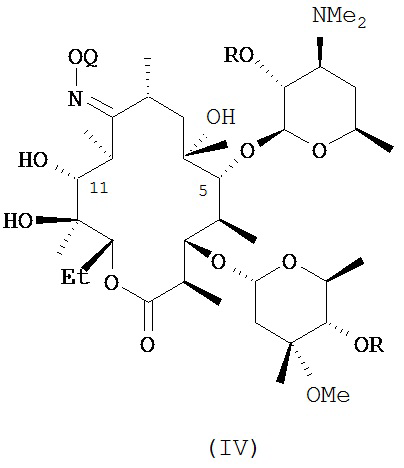
или его соли присоединения кислоты; где в каждом случае Q в комбинации с атомом кислорода оксима образует ацеталь или кеталь, или Q представляет собой тропил, a R представляет собой ацильную группу. В другом варианте реализации стадия (а) способа включает применение основания.
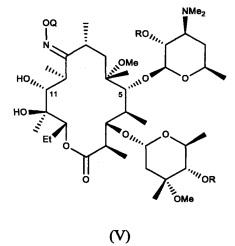
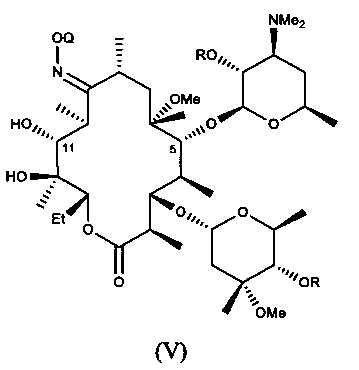
В другом иллюстративном варианте реализации описаны способы, включающие стадию (b) приведения соединения формулы (IV), описанного в настоящей заявке, или его соли присоединения кислоты в контакт с метилирующим агентом с получением соединения формулы (V)
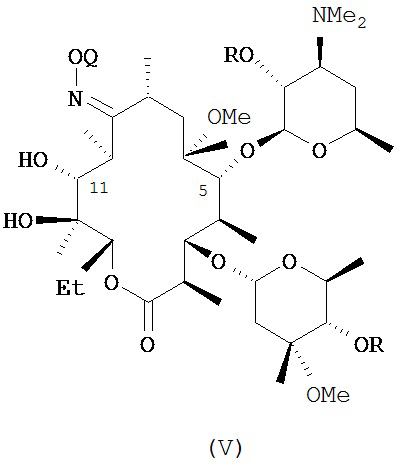
или его соли присоединения кислоты, где Q и R описаны для альтернативных вариантов реализации в настоящей заявке. В другом варианте реализации стадия (b) способа включает применение основания. В другом варианте реализации стадия (b) способа включает применение апротонного полярного растворителя.
В другом иллюстративном варианте реализации описаны способы, включающие стадию (с) приведения соединения формулы (V), описанного в настоящей заявке, или его соли присоединения кислоты в контакт с деоксимирующими агентом с получением соединения формулы (II)
или его соли присоединения кислоты, где R описан для альтернативных вариантов реализации в настоящей заявке.
Следует понимать, что любые стадии (а), (b) и (с) можно объединять в дополнительные варианты реализации. Также следует понимать, что модификации любых стадий (a), (b) и (с), описанных в настоящей заявке, можно объединять без ограничений в дополнительные варианты реализации. Например, другой иллюстративный способ включает стадию ацилирования (а), дополнительно включает стадию метилирования (b) и дополнительно включает стадию деоксимирования (с). Другой иллюстративный способ включает стадию метилирования (b) и дополнительно включает стадию деоксимирования (с). Другой иллюстративный способ включает стадию ацилирования (а), дополнительно включает стадию метилирования (b), дополнительно включает стадию деоксимирования (с) и дополнительно включает стадии, описанные в WO 2009/055557, для превращения соединений формулы (V) в соединения формулы (I).
В другом варианте реализации описаны способы получения соединений формулы (IV), например, соединений формулы (IV), где R представляет собой бензоил, или солей присоединения кислоты, где предложенные способы включают стадию приведения соединения формулы (III), описанного в настоящей заявке, или его соли присоединения кислоты в контакт с ацилирующим агентом, например с бензоилангидридом, также называемым ангидридом бензойной кислоты, для получения соединения формулы (IV) или его соли присоединения кислоты. В одном из вариантов данную стадию осуществляют в присутствии основания.
В другом варианте реализации описаны способы получения соединений формулы (V) или солей присоединения кислоты, где способы включают стадию приведения соединения формулы (IV), описанного в настоящей заявке, или его соли присоединения кислоты, в контакт с метилирующим агентом с получением 6-О-метилсодержащего соединения формулы (V), описанного в настоящей заявке, или его соли присоединения кислоты. В одном из вариантов данную стадию осуществляют в присутствии основания. В другом варианте данную стадию осуществляют в апротонном полярном растворителе. В другом варианте данную стадию осуществляют в присутствии основания и в полярном апротонном растворителе.
В другом варианте реализации описаны способы получения соединений формулы (II), включая соединения формулы (II), где R представляет собой бензоил, или солей присоединения кислоты, где способы включают стадию приведения соединения формулы (V), описанного в настоящей заявке, или его соли присоединения кислоты, в контакт с деоксимирующим агентом, с получением соединения формулы (II) или его соли присоединения кислоты.
В другом иллюстративном варианте реализации любого из предложенных выше способов Q представляет собой О-защитную группу. В одном из вариантов Q в комбинации с атомом кислорода оксима образует ацеталь или кеталь, или Q представляет собой тропил. В другом иллюстративном варианте реализации R представляет собой ацильную группу. В другом иллюстративном варианте реализации Q представляет собой О-защитную группу. В одном из вариантов Q в комбинации с атомом кислорода оксима образует ацеталь или кеталь, или Q представляет собой тропил, a R представляет собой ацильную группу.
В другом иллюстративном варианте реализации любого из предложенных выше способов Q представляет собой C(RA)(RC)(ORB), где
RA представляет собой группу формулы CH2RD, где RD представляет собой водород, (1-3С)алкил или (1-6С)алкокси;
RB представляет собой (1-6С)алкил, (5-7С)циклоалкил, фенил или арилалкил; и
RC представляет собой водород, (1-4С)алкил, фенил или арилалкил;
или в качестве альтернативы RB и RD совместно образуют этилен, пропилен или триметиленовую группу; или
RB и RD совместно образуют (3-5С)алкандиильную группу, которая может быть дополнительно замещена одним - тремя (1-3С)алкильными заместителями; или
RB и RC совместно образуют (3-4С)алкандиильную группу.
В другом варианте реализации любого из предложенных выше способов Q представляет собой 2-метокси-2-пропил, 1-метоксициклогексил или 1-изопропоксициклогексил. В другом варианте реализации любого из предложенных выше способов Q представляет собой 2-метокси-2-пропил.
Соединения формулы (III), описанные в настоящей заявке, можно получать путем приведения 9-оксима эритромицина А в контакт с соответствующим соединением формулы RE-C(RA)(RC)(ORB), в котором RE представляет собой (1-6С)алкокси, или в котором RA и RE совместно образуют группу формулы CHRD, присоединенную при помощи двойной связи. Данную стадию можно осуществлять в присутствии кислотного катализатора, например в присутствии гидрохлорида пиридина. В другом варианте данную стадию осуществляют с применением 2-метоксипропена с получением соединения формулы (III), в котором Q представляет собой 2-метокси-2-пропил. В другом варианте стадию осуществляют в дихлорметане при температуре, равной примерно от 0°C до комнатной температуры, в присутствии гидрохлорида пиридина с применением избытка 2-метоксипропена. В другом варианте Q представляет собой тропил, и соединения формулы (III) можно получать путем реакции 9-оксима эритромицина А с тетрафторборатом тропилия в апротонном полярном растворителе.
В другом варианте реализации любого из описанных в настоящей заявке способов R представляет собой пространственно затрудненную ацильную группу, например, бензоильную группу. В другом варианте реализации любого из описанных в настоящей заявке способов R не является ацетилом. Не желая быть связанными теорией, полагают, что применение пространственно затрудненной группы R может приводить к усовершенствованию способа и/или к улучшению чистоты выделяемого продукта. Было обнаружено, что пространственно незатрудненные ацильные группы, например ацетильные группы, в положении С-5 сахарида могут мигрировать в другие положения макролида, например, из 2'-гидроксигруппы дезозаминового фрагмента к аминогруппе боковой цепи. Применение пространственно затрудненных групп R снижает и/или исключает указанную миграцию, что приводит к усовершенствованию способа и/или к улучшению чистоты выделяемых продуктов.
В другом варианте реализации любого из описанных в настоящей заявке способов R представляет собой бензоил.
В другом варианте реализации любого из описанных в настоящей заявке способов стадию (а) осуществляют с применением ацилирующего агента, который представляет собой ангидрид, галогенангидрид или активированный сложный эфир соответствующей ацильной группы R. В другом варианте реализации любого из описанных в настоящей заявке способов ацилирующий агент представляет собой ангидрид, содержащий ацильную группу R. В другом варианте реализации любого из описанных в настоящей заявке способов применяют примерно от 2 до примерно 6 эквивалентов ацилирующего агента на один эквивалент соединения формулы (III). В другом варианте реализации любого из описанных в настоящей заявке способов на стадии (а) применяют основание, например третичный амин. В другом варианте реализации любого из описанных в настоящей заявке способов основание представляет собой триэтиламин, диизопропилэтиламин или 4- метилморфолин или их комбинацию. В другом варианте реализации любого из описанных в настоящей заявке способов применяют примерно от 1 до 4 эквивалентов основания на один эквивалент соединения формулы (III). В другом варианте реализации любого из описанных в настоящей заявке способов ацилирование проводят в присутствии примерно от 0,5 до примерно 2,5 эквивалентов катализатора ацилирования на один эквивалент соединения формулы (III). В другом варианте реализации любого из описанных в настоящей заявке способов катализатор ацилирования представляет собой 4-диметиламинопиридин.
В другом варианте реализации любого из описанных в настоящей заявке способов метилирующий агент представляет собой метилбромид, метилйодид, диметилсульфат, метил п-толуолсульфонат или метил-метансульфонат. В другом варианте реализации метилирующий агент представляет собой метилйодид. В другом варианте реализации способа, описанного в настоящей заявке, в комбинации с метилирующим агентом применяют основание, такое как гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия или т-бутоксид калия или их смеси. В другом варианте реализации основание, которое применяют в комбинации с метилирующим агентом, представляет собой гидроксид калия. В другом варианте реализации стадию метилирования осуществляют в апротонном полярном растворителе, таком как диметилсульфоксид, диметилформамид, 1-метил-2-пирролидон, их смесь, или смесь любого из указанных растворителей с одним или более растворителями, выбранными из тетрагидрофурана, 2-метилтетрагидрофурана, 1,2-диметоксиэтана, ацетонитрила и этилацетата. В другом варианте реализации любого из описанных в настоящей заявке способов стадию метилирования осуществляют при температуре, составляющей примернот от -15°C до примерно 60°C. В другом варианте реализации способов, описанных в настоящей заявке, касающемся метилирования соединения формулы (IV), стадию метилирования осуществляют при температуре от примерно 0°C до примерно 30°C.
Неожиданно обнаружено, что стадия метилирования соединений формулы (IV), где R представляет собой бензоил, проходит без расщепления или по существу без расщепления бензоатной сложноэфирной группы, входящей в состав соединений формулы (IV).
Для иллюстрации, удаление группы Q, например, путем удаления О-защитной группы, и/или удаление оксимной группы при С-9 с образованием кетона, например, путем деоксимирования, можно проводить при помощи любых традиционных способов и/или реагентов. Иллюстративные способы деоксимирования включают, но не ограничиваются ими, гидролитические, окислительные и восстановительные условия. В одном из вариантов реализации деоксимирующий агент содержит восстановитель. Иллюстративные варианты реализации деоксимирующих агентов включают, но не ограничиваются ими, неорганические соединения оксидов серы, такие как гидросульфит натрия, пиросульфат натрия, тиосульфат натрия, сульфит натрия, гидросульфит натрия, метабисульфит натрия, бисульфит натрия, дитионат натрия, гидросульфит калия, тиосульфат калия и метабисульфит калия и их смеси. В другом варианте реализации любого из описанных в настоящей заявке способов деоксимирующий агент представляет собой метабисульфит натрия или бисульфит натрия или их комбинацию. Следует понимать, что удаление О-защитной группы можно проводить перед деоксимированием; или удаление О-защитной группы и деоксимирование можно проводить в одну («в одном реакторе») стадию путем обработки, например последовательной, параллельной, одновременной или синхронной обработки, кислотой, такой как муравьиная кислота, и деоксимирующим агентом.
В другом варианте реализации любого из описанных в настоящей заявке способов стадию превращения С-9 оксима в карбонил осуществляют путем приведения в контакт соединения формулы (V), где деоксимирующий агент содержит муравьиную кислоту и метабисульфит натрия, в водном растворе спирта при температуре в диапазоне от температуры окружающей среды примерно до температуры кипения растворителя.
Неожиданно обнаружено, что удаление О-защитной группы и удаление оксима в соединении формулы (V), в котором R представляет собой бензоил, можно проводить без расщепления или по существу без расщепления бензоатной сложноэфирной группы, входящей в состав соединений формулы (V).
Следует понимать, что различные подвиды, частицы и соединения, описанные в настоящей заявке, можно получать при помощи различных вариантов реализаций способов, описанных в настоящей заявке. Например, в другом варианте реализации любого из способов, предложенных в настоящем описании, V представляет собой C(O); и/или
R7 представляет собой аминосахар или галогенсахар; или
R7 представляет собой 4-нитрофенилацетил или 2-пиридилацетил; или
X и Y совместно с присоединенным к ним атомом углерода образуют карбонил; и/или
A представляет собой CH2; и/или
B представляет собой алкенилен; и/или
B представляет собой (CH2)n, где n составляет от 2 до 6, от 2 до 5 или от 2 до 4, или от 2 до 3, или 3; и/или
C представляет собой аминофенил; или
C представляет собой 3-аминофенил; и/или
W представляет собой фтор; или
W представляет собой водород; и/или
R10 представляет собой водород или ацил; или
R10 представляет собой водород; или
R10 представляет собой бензоил.
В другом варианте реализации любого из описанных в настоящей заявке способов соединение формулы (I) представляет собой СЕМ-101 или его фармацевтически приемлемую соль, сольват или гидрат. Соединение СЕМ-101 имеет регистрационный номер Chemical Abstracts 760981-83-7, структура соединения представлена ниже:
Используемый в настоящем описании индивидуально или в комбинации термин «алкил» относится к возможно замещенному линейному, возможно замещенному разветвленному или возможно замещенному циклическому алкильному радикалу, содержащему от 1 примерно до 30 атомов углерода, более предпочтительно от 1 до 12 атомов углерода. Примеры алкильных радикалов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, трет-амил, пентил, гексил, гептил, октил и т.д. «Низший алкил» представляет собой алкил с короткой цепью, например алкил, содержащий от 1 примерно до 6 атомов углерода.
Термин «алкокси», используемый индивидуально или в комбинации, относится к радикалу простого алкильного эфира, алкил-О, где алкил определен выше. Примеры алкокси радикалов включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси и т.д.
Термин «алкенил», используемый индивидуально или в комбинации, относится к возможно замещенному линейному, возможно замещенному разветвленному или возможно замещенному циклическому алкенильному углеводородному радикалу, содержащему одну или более двойных связей углерод-углерод и от 2 до примерно 30 атомов углерода, более предпочтительно от 2 до примерно 18 атомов углерода. Примеры алкенильных радикалов включают этенил, пропенил, бутенил, 1,4-бутадиенил и т.д. Термин также может включать циклические алкенильные структуры. «Низший алкенил» относится к алкенилу, содержащему от 2 до примерно 6 атомов углерода.
Термин «ацилокси» относится к сложноэфирной группе OC(O)-R, где R представляет собой Н, алкил, алкенил, алкинил, арил или арилалкил, где алкил, алкенил, алкинил и арилалкильные группы могут быть замещенными.
Термин «ацил» включает алкил, арил, гетероарил, арилалкил или гетероарилалкильные заместители, присоединенные к соединению через карбонильную функциональную группу (например, CO-алкил, CO-арил, CO-арилалкил или CO-гетероарилалкил и т.д.).
Термин «гетероалкил», в целом, относится к цепи атомов, которая содержит атомы углерода и по меньшей мере один гетероатом. Иллюстративные гетероатомы включают азот, кислород и серу.
Используемый в настоящем описании термин «арил» включает моноциклические и полициклические ароматические карбоциклические и ароматические гетероциклические группы, каждая из которых может быть замещенной. Используемый в настоящем описании термин «гетероарил» включает ароматические гетероциклические группы, каждая из которых может быть замещенной. Иллюстративные карбоциклические ароматические группы, описанные в настоящей заявке, включают, но не ограничиваются ими, фенил, нафтил и т.д. Иллюстративные гетероциклические ароматические группы включают, но не ограничиваются ими, пиридинил, пиримидинил, пиразинил, триазинил, тетразинил, хинолинил, хиназолинил, хиноксалинил, тиенил, пиразолил, имидазолил, оксазолил, тиазолил, изоксазолил, изотиазолил, оксадиазолил, тиадиазолил, триазолил, бензимидазолил, бензоксазолил, бензтиазолил, бензизоксазолил, бензизотиазолил и т.д.
Термин «арилалкил» относится к алкильной группе, замещенной одной или более незамещенными или замещенными моноциклическими или полициклическими арильными группами. Иллюстративные арилалкильные группы включают бензил, дифенилметил, тритил, 2-фенилэтил, 1-фенилзтил, 2-пиридилметил, 4,4'-диметокситритил и т.д.
Термин «алкиларил» относится к арильной группе, замещенной алкильной группой.
Термин «сульфонил» относится к SO2-R, где R представляет собой H, алкил или арил.
Термин «сахарид» включает моносахариды, дисахариды и полисахариды, каждый из которых может быть замещенным. Термин также включает сахара и дезоксисахара, возможно замещенные амином, амидом, уреилом, галогеном, нитрилом или азидными группами. Иллюстративные примеры включают глюкозамин, N-ацетилглюкозамин, дезозамин, форозамин, сиаловую кислоту и т.д.
Термин «активированный сложный эфир» включает производные карбоновых кислот, в которых атом водорода гидроксигруппы замещен на остаток, что приводит к получению хорошей уходящей группы, включая 4-нитрофениловый эфир и активированный сложный эфир или ангидрид, полученный из реагента для реакции сочетания.
В другом варианте реализации описаны соединения формулы (IV)
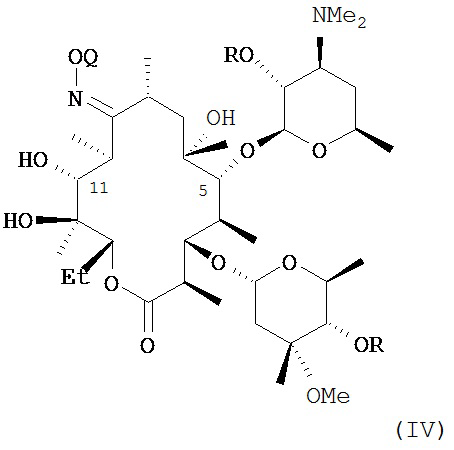
и их соли присоединения кислоты; где Q и R описаны в различных вариантах реализации в настоящей заявке.
В другом варианте реализации описаны соединения формулы (V)
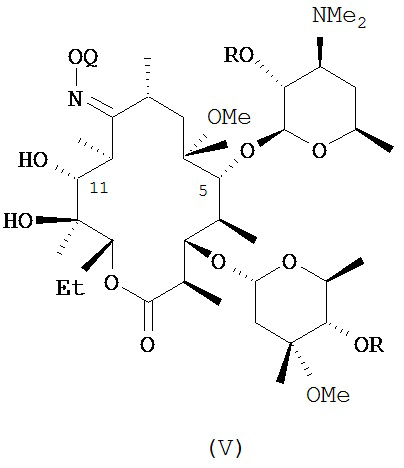
и их соли присоединения кислоты; где Q и R описаны в различных вариантах реализации в настоящей заявке.
Очевидно, что так как каждое соединение формул (I), (II), (III), (IV) и (V) содержит диметиламиногруппу во фрагменте дезозамина, то соединения могут образовывать соли присоединения кислоты. Соответственно, следует понимать, что любая соль присоединения кислоты соединения формул (I), (II), (III), (IV) и (V), подходящая для применения для фармацевтического производства или для получения свободного основания, которое подходит для фармацевтического производства, описана в настоящей заявке и включена в объем изобретения, описанного в настоящей заявке.
Для каждого из выше- и нижепредложенных вариантов реализации следует понимать, что формулы включают и представляют не только все фармацевтически приемлемые соли соединений, но также включают любые и все гидраты и/или сольваты соединения представленной формулы. Очевидно, что некоторые функциональные группы, такие как гидрокси, амин и т.д., образуют комплексы и/или координированные соединения с водой и/или различными растворителями, в различных физических формах соединений. Соответственно, следует понимать, что предложенные выше формулы включают и представляют различные гидраты и/или сольваты. Для каждого из выше- и нижепредложенных вариантов реализации также следует понимать, что формулы включают и представляют каждый возможный изомер, например стереоизомер и геометрический изомер по отдельности или в виде любых возможных смесей. Для каждого из выше- и нижепредложенных вариантов реализации также следует понимать, что формулы включают и представляют любые и все кристаллические формы, частично кристаллические формы и некристаллические формы и/или аморфные формы соединений. Например, иллюстративная морфология кристаллов описана в находящейся в настоящее время на рассмотрении международной заявке PCT/US2011/029424, содержание которой включено в настоящую заявку по всей полноте посредством ссылки.
ПРИМЕРЫ
В предложенных ниже примерах дополнительно проиллюстрированы конкретные варианты реализации изобретения; тем не менее предложенные ниже иллюстративные примеры не следует рассматривать как ограничивающие изобретение. Сокращения, используемые в примерах, включают: ДХМ - дихлорметан; DMAP - 4-диметиламинопиридин; ДМСО - диметилсульфоксид; ЭА - этилацетат; 1Н ЯМР - спектроскопия протонного ядерного магнитного резонанса; MeOH - метанол; MM - молекулярная масса; КТ - комнатная температура (температура окружающей среды); ТГФ - тетрагидрофуран; ТСХ - тонкослойная хроматография.
ПРИМЕР. Синтез 9-оксима эритромицина А (1)
Смесь эритромицина A (15 г, 20,4 ммоль), NH2OH⋅HCl (7,3 г, 105 ммоль) и триэтиламина (7 г, 69 ммоль) в MeOH (23 мл) кипятили с обратным холодильником в течение ночи. В результате реакции образовывалось белое твердое вещество. Реакционную смесь концентрировали до небольшого объема. В полученный остаток добавляли разбавленный водный раствор NH4OH при 0°C до достижения значения pH смеси примерно 10-11. Из смеси дополнительно осаждалось белое твердое вещество. Смесь фильтровали, собранное твердое вещество промывали водой и сушили в вакууме с получением 14,2 г соединения 1 в виде белого гранулированного твердого вещества с выходом 93%. Анализ ТСХ (ДХМ:MeOH:NH4OH=90:10:1) полученного соединения 1 показывал наличие небольшого количества дополнительного соединения (нижнее пятно), соответствующего Z-изомеру. Массовый анализ полученного соединения 1 показывал пик с ММ=749, соответствующий титульному соединению. Данные 1Н ЯМР анализ продукта соответствовали титульному соединению, а также показывали получение смеси соединения (1) и соли HCl. Продукт применяли без очистки.
ПРИМЕР. Крупномасштабное получение соединения (1)
Эритромицин (250 г, 0,34 моль) и гидрохлорид гидроксиламина (80,3 г, 1,15 моль) в метаноле (325 мл) кипятили с обратным холодильником в присутствии триэтиламина (45 г, 0,44 моль). Прохождение реакции отслеживали путем ТСХ с применением смеси толуол/триэтиламин (8:2) в качестве элюента. После завершения (примерно через 24 часа) реакционную массу постепенно охлаждали и перемешивали при 0-5°C в течение 1 часа, фильтровали и промывали холодным метанолом (100 мл). Влажное твердое вещество (265 г) суспендировали в изопропиловом спирте (350 мл) и нагревали до 50-55°C, затем добавляли водный аммиак (650 мл) в течение 2 часов. Раствор перемешивали в течение 1 часа при 50-55°C и постепенно охлаждали до 10-15°C и выдерживали в течение 2 часов. Твердое вещество отфильтровывали и промывали водой, сушили при 80-85°C в течение 12 часов для выделения 186 г. Получение примерно 3% соответствующего изомерного Z-оксима наблюдали при помощи ВЭЖХ. Получение повторяли с применением соответствующих масштабируемых количеств других реагентов.
ПРИМЕР. Синтез соединения формулы (III), Q=2-метокси-2-пропил (9)
В раствор (1) (3 г, 4 ммоль) в безводном дихлорметане (ДХМ, 21 мл) добавляли 2-метоксипропен (1,5 г, 20,8 ммоль), затем гидрохлорид пиридина (0,72 г, 6,2 ммоль) при 0°C. После завершения добавления реакционную смесь перемешивали при КТ в течение 30 минут. Конверсию отслеживали путем анализа ТСХ реакционной смеси (ДХМ:MeOH:NH4OH=90:10:1). Если конверсия была неполной, то смесь охлаждали до 0°C и добавляли еще 0,5 г 2-метоксипропена (6,9 ммоль). Смесь дополнительно перемешивали при 0°C в течение 0,5 часа. Если конверсия была неполной, то добавляли еще 0,5 г 2-метоксипропена (6,9 ммоль), а затем 0,1 г гидрохлорида пиридина (0,86 ммоль) в реакционную смесь при 0°C. Реакционную смесь дополнительно перемешивали при 0°C в течение 15 минут. После достижения полной конверсии реакционную смесь разбавляли насыщенным водным раствором NaHCO3. Слой в ДХМ отделяли, а водный слой экстрагировали в ДХМ. Объединенные слой в ДХМ промывали солевым раствором, сушили над MgSO4, концентрировали насухо с получением 3,3 г неочищенного продукта в виде белой пены с количественным выходом. Массовый анализ продукта давал значение ММ=821, соответствующее титульному соединению, а также показывал очень небольшой пик с молекулярной массой 861. Данные спектра 1Н ЯМР продукта соответствовали титульному соединению и небольшим количествам 2-метоксипропан-2-ола и пиридина. Продукт применяли без дополнительной очистки.
ПРИМЕР. Синтез соединения формулы (IV), Q=2-метокси-2-пропил, R=бензоил (10)
В раствор (9) (4,1 г, 5 ммоль) в этилацетате (65 мл) добавляли ангидрид бензойной кислоты (4,5 г, 20 ммоль), затем триэтиламин (1,26 г, 12,5 ммоль) и DMAP (0,9 г, 7,4 ммоль) при КТ. Полученную смесь перемешивали при КТ в течение 36 часов. Реакционную смесь разбавляли насыщенным водным раствором NaHCO3. Слой в ЭА отделяли, а водный слой экстрагировали в ЭА. Объединенные слои в ЭА промывали солевым раствором, сушили над MgSO4, фильтровали для удаления осушителя и концентрировали насухо. Полученный остаток подвергали очистке путем колоночной хроматографии на силикагеле (ДХМ:MeOH:NH4OH=97:3:0,3) с получением 4,2 г 10 с выходом 80% в виде белого твердого вещества. Массовый анализ очищенного продукта давал значение ММ=1029, соответствующее титульному соединению. Данные 1Н ЯМР соответствовали указанному в заголовке соединению.
ПРИМЕР. Крупномасштабное получение соединения (9)
Оксим эритромицина (1) (200 г, 0,26 моль) растворяли в ДХМ (1,4 л) и объем уменьшали до 1 л путем перегонки при атмосферном давлении. После охлаждения реакционной массы до 0-5°C добавляли 2-метоксипропен (80 г, 1,1 моль) и гидробромид пиридина (50 г, 0,31 моль) и перемешивали в течение 3 часов при 20-25°C. Массовый анализ подтверждал наличие (9). Без выделения полученного продукта добавляли ангидрид бензойной кислоты (211 г, 0,93 моль), триэтиламин (54 г, 0,53 моль), DMAP (48,8 г, 0,40 моль) и реакцию проводили в течение 24 часов при 30°C. Прохождение реакции отслеживали путем ТСХ и анализировали путем масс-спектрометрии. После завершения добавляли насыщенным бикарбонат натрия (1 л) и перемешивали в течение 15 минут и оставляли отстаиваться. Слои разделяли, органический слой концентрировали. Выделяли 190 г вещества с чистотой 48-51%. Получение проводили с применением соответствующих масштабируемых количеств других реагентов.
Неочищенные продукты, полученные в последовательных партиях, объединяли (450 г) и растворяли в ЭА (4,5 л) с получением прозрачного раствора, который промывали насыщенным водным бикарбонатом натрия (2,2 л), водой (2,2 л) и солевым раствором (2,2 л) и концентрировали. Выделенный продукт перекристаллизовывали из смеси IPE/н-гексан с получением 360 г (84%).
ПРИМЕР. Синтез соединения формулы (V), Q=2-метокси-2-пропил, R=бензоил (11)
Раствор (10) (3,8 г, 3,7 ммоль) в безводном ТГФ (15 мл) и безводном ДМСО (15 мл) охлаждали до 0°C. Добавляли порошковый KOH (0,46 г, 8,2 ммоль), затем метилйодид (1,06 г, 7,5 ммоль) при 0°C. Полученную реакционную смесь перемешивали при 0°C в течение 5 минут, затем смесь превращалась в густую пасту, и перемешивание останавливали. Смесь нагревали до КТ в течение 5 минут, смесь сохраняла вид густой пасты, разбавляли 15 мл ТГФ и 15 мл ДМСО с получением свободно текучей суспензии. Смесь дополнительно перемешивали при КТ в течение 0,5 часов, разбавляли насыщенным водным раствором NaHCO3 и экстрагировали в этилацетате. Экстракт в этилацетате промывали солевым раствором, сушили над MgSO4 и концентрировали насухо. Выделенный остаток очищали путем колоночной хроматографии на силикагеле (ДХМ:MeOH:NH4OH=97:3:0,3) с получением 2,83 г (11) в виде белого твердого вещества с выходом 73%. Массовый анализ давал значение ММ=1043, соответствующее титульному соединению, а также небольшой пик с ММ=1057. Данные 1Н ЯМР соответствовали указанному в заголовке соединению.
ПРИМЕР. Крупномасштабное получение соединения (11)
Бензоилсодержащий оксим (10) (100 г, 0,09 моль) растворяли в толуоле (1,8 л) и раствор перегоняли в вакууме для удаления толуола (300 мл), охлаждали до 15°C и разбавляли ДМСО (1,5 л). После охлаждения до 5°C добавляли метилйодид (20,5 г, 0,14 моль), затем KOH (10,8 г, 0,19 моль) и реакцию проводили в течение 3 часов. Реакцию останавливали путем добавления 40% диметиламина (22 г), температуру реакционной массы повышали до КТ и смесь разбавляли водой (500 мл) при перемешивании. Слои разделяли и водный слой экстрагировали в толуоле (500 мл). Объединенные органические слой промывали водой (2 л), а органический слой концентрировали путем перегонки в вакууме. Выделенный продукт перемешивали в IPE (500 мл) в течение 5 часов и фильтровали с получением 82 г указанного в заголовке соединения, которое применяли без дополнительной очистки. Получение проводили с применением соответствующих масштабируемых количеств других реагентов.
ПРИМЕР. Синтез кларитромицина дибензоата, формула (II), R=бензоил
В раствор соединения (11) (800 мг, 0,78 ммоль) в этаноле (8 мл) и воде (8 мл) добавляли метабисульфит натрия (740 мг, 3,89 ммоль) при КТ. pH полученной смеси доводили до 2-3 путем добавления муравьиной кислоты (1,5 мл). Смесь нагревали при 60°C в течение 1 часа. Конверсию отслеживали путем масс-спектрометрии. Если реакцию считали незавершенной, или если анализ показывал наличие большого количества промежуточного оксима со снятой защитой (ММ=971), то добавляли еще 2 г метабисульфита натрия (10,5 ммоль). Смесь дополнительно перемешивали при 60°C в течение 7 часов, затем охлаждали до КТ. В результате прохождения реакции образовывался белый осадок. Реакционную смесь нейтрализовали разбавленным водным раствором NaHCO3 до pH 8-9, полученную смесь фильтровали. Выделенное белое твердое вещество сушили в вакууме с получением 760 мг кларитромицина дибензоата. Неочищенный продукт объединяли с веществами, полученными в других примерах (примерно 200 мг) и очищали путем колоночной хроматографии на силикагеле с получением 730 мг кларитромицина дибензоата с выходом 79%. Массовый анализ давал значение ММ=956, соответствующее указанному в заголовке соединению, и небольшой пик ММ=970, который соотносили с сопутствующими примесями в соединении (11). Данные 1Н ЯМР соответствовали титульному соединению.
ПРИМЕР. Крупномасштабное получение кларитромицина дибензоата
Метилированный оксим (11) (80 г, 0,07 моль) растворяли в абсолютном спирте (400 мл). Добавляли воду (400 мл), затем бисульфит натрия (72 г, 0,69 моль) и муравьиную кислоту (21 г). Реакционную массу кипятили с обратным холодильником в течение 6 часов, охлаждали до КТ и разбавляли водой (400 мл). Реакционную массу охлаждали до 10-15°C и медленно добавляли 25% NaOH (160 мл). Смесь перемешивали в течение 2 часов и фильтровали. Выделенное твердое вещество промывали водой (500 мл) и растворяли в этилацетате (400 мл). Органический слой промывали водой (400 мл), затем солевым раствором (400 мл), а затем концентрировали. Выделенное вещество кристаллизовали из этилацетата (1,7 Т) с получением 40,8 г (95% чистота). В качестве альтернативы выделенное вещество кристаллизовали из IPA/IPE с получением вещества с 89-90% чистотой. Получение проводили с применением соответствующих масштабируемых количеств других реагентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С (HCV) | 2006 |
|
RU2405783C2 |
| ПАРЕНТЕРАЛЬНЫЕ СОСТАВЫ ДЛЯ ВВЕДЕНИЯ МАКРОЛИДНЫХ АНТИБИОТИКОВ | 2013 |
|
RU2658050C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОКИСЛОТНЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2643146C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ РАСЩЕПЛЯЕМЫЕ ФЕРМЕНТАМИ ОПИОИДНЫЕ ПРОЛЕКАРСТВА С МОДИФИЦИРОВАННЫМ КЕТОНОМ И ИХ ДОПОЛНИТЕЛЬНЫЕ ИНГИБИТОРЫ | 2010 |
|
RU2600736C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛИРОВАННЫХ ЦИКЛОПЕНТИЛПИРИМИДИНОВЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2643811C2 |
| НЕКОТОРЫЕ ХИМИЧЕСКИЕ СТРУКТУРЫ, КОМПОЗИЦИИ И СПОСОБЫ | 2009 |
|
RU2513636C2 |
| ИНГИБИТОРЫ ПРОТЕИНФОСФАТАЗЫ-1 И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2527561C2 |
| ПЕПТИДНЫЕ АНТИБИОТИКИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2428429C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2456296C2 |
| СПОСОБ СИНТЕЗА ГИДРАЗИНА, КОТОРЫЙ МОЖНО ПРИМЕНЯТЬ ПРИ ЛЕЧЕНИИ ВИРУСА ПАПИЛЛОМЫ | 2013 |
|
RU2649006C2 |
Изобретение, описанное в настоящей заявке, относится к способам получения макролидных антибактериальных агентов. В частности, изобретение относится к способам получения макролидов и кетолидов из эритромицина А. Способ получения соединения формулы (I) или его фармацевтически приемлемой соли, в которой R10 представляет собой водород или ацил; X представляет собой Н; a Y представляет собой OR7; где R7 представляет собой кладинозу, алкил или арилалкил, или ацил или C(O)NR8R9, где каждый R8 и R9 независимо выбран из группы, состоящей из водорода, гидрокси, алкила, алкокси, арила, арилалкила, диметиламиноалкила, ацила, сульфонила, уреидо и карбамоила; или R8 и R9 совместно с присоединенным к ним атомом азота образуют гетероцикл; или X и Y совместно с присоединенным к ним атомом углерода образуют карбонил; V представляет собой С(О), C(=NR11), CH(NR12, R13) или N(R14)CH2; где N(R14) присоединен к С-10 атому углерода; где R11 представляет собой гидрокси или алкокси; каждый R12 и R13 независимо выбран из группы, состоящей из водорода, гидрокси, алкила, алкокси, арила, арилалкила, диметиламиноалкила, ацила, сульфонила, уреидо и карбамоила; R14 представляет собой водород, гидрокси, алкил, алкокси, арил, арилалкил, диметиламиноалкил, ацил, сульфонил, уреидо или карбамоил; W представляет собой Н, F, Cl, Br, I или ОН; А представляет собой СН2, С(О), С(O)O, C(O)NH, S(O)2, S(O)2NH или C(O)NHS(O)2; В представляет собой (СН2)n, где n представляет собой целое число от 0 до 10; или ненасыщенную углеродную цепь, содержащую от 2 до 10 атомов углерода; и С представляет собой водород, гидрокси, алкил, алкокси, арил, арилалкил, ацил, ацилокси, сульфонил, уреидо или карбамоил; где алкил независимо в каждом случае представляет собой C1-С30 алкил; арил в каждом случае независимо выбран из группы, состоящей из фенила, необязательно замещенного аминогруппой, нафтила, пиридинила, пиримидинила, пиразинила, триазинила, тетразинила, хинолинила, хиназолинила, хиноксалинила, тиенила, пиразолила, имидазолила, оксазолила, тиазолила, изоксазолила, изотиазолила, оксадиазолила, тиадиазолила, триазолила, бензимидазолила, бензоксазолила, бензтиазолила, бензизоксазолила и бензизотиазолила; ацил в каждом случае независимо выбран из группы, состоящей из С(O)алкила и С(O)арила, С(O)арилалкила; включает (b) приведение соединения формулы (IV) или его соли присоединения кислоты, где R представляет собой пространственно затрудненную ацильную группу; a Q представляет собой 2-метокси-2-пропил, 1-метоксициклогексил или тропил; в контакт с метилирующим агентом с получением соединения формулы (V) или его соли присоединения кислоты; и (с) приведение соединения формулы (V) или его соли присоединения кислоты в контакт с деоксимирующим агентом с получением соединения формулы (II) или его соли присоединения кислоты; и превращение соединения формулы (II) или его соли присоединения кислоты в соединение формулы (I) или его фармацевтически приемлемую соль. 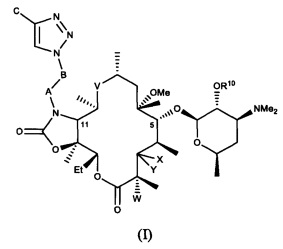
4 н. и 28 з.п. ф-лы, 9 пр.
1. Способ получения соединения формулы (I)
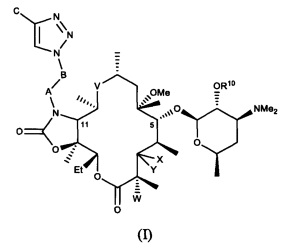
или его фармацевтически приемлемой соли, где:
R10 представляет собой водород или ацил;
X представляет собой Н; a Y представляет собой OR7; где R7 представляет собой кладинозу, алкил или арилалкил, или ацил или C(O)NR8R9, где каждый R8 и R9 независимо выбран из группы, состоящей из водорода, гидрокси, алкила, алкокси, арила, арилалкила, диметиламиноалкила, ацила, сульфонила, уреидо и карбамоила; или R8 и R9 совместно с присоединенным к ним атомом азота образуют гетероцикл; или X и Y совместно с присоединенным к ним атомом углерода образуют карбонил;
V представляет собой С(О), C(=NR11), CH(NR12, R13) или N(R14)CH2; где N(R14) присоединен к С-10 атому углерода; где R11 представляет собой гидрокси или алкокси; каждый R12 и R13 независимо выбран из группы, состоящей из водорода, гидрокси, алкила, алкокси, арила, арилалкила, диметиламиноалкила, ацила, сульфонила, уреидо и карбамоила; R14 представляет собой водород, гидрокси, алкил, алкокси, арил, арилалкил, диметиламиноалкил, ацил, сульфонил, уреидо или карбамоил;
W представляет собой Н, F, Cl, Br, I или ОН;
А представляет собой СН2, С(О), С(O)O, C(O)NH, S(O)2, S(O)2NH или C(O)NHS(O)2;
В представляет собой (СН2)n, где n представляет собой целое число от 0 до 10; или ненасыщенную углеродную цепь, содержащую от 2 до 10 атомов углерода; и С представляет собой водород, гидрокси, алкил, алкокси, арил, арилалкил, ацил, ацилокси, сульфонил, уреидо или карбамоил;
где алкил независимо в каждом случае представляет собой C1-С30 алкил;
арил в каждом случае независимо выбран из группы, состоящей из фенила, необязательно замещенного аминогруппой, нафтила, пиридинила, пиримидинила, пиразинила, триазинила, тетразинила, хинолинила, хиназолинила, хиноксалинила, тиенила, пиразолила, имидазолила, оксазолила, тиазолила, изоксазолила, изотиазолила, оксадиазолила, тиадиазолила, триазолила, бензимидазолила, бензоксазолила, бензтиазолила, бензизоксазолила и бензизотиазолила;
ацил в каждом случае независимо выбран из группы, состоящей из С(O)алкила и С(O)арила, С(O)арилалкила;
способ, включающий:
(b) приведение соединения формулы (IV)
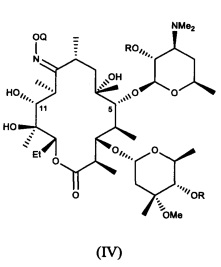
или его соли присоединения кислоты, где R представляет собой пространственно затрудненную ацильную группу; a Q представляет собой 2-метокси-2-пропил, 1-метоксициклогексил или тропил;
в контакт с метилирующим агентом с получением соединения формулы (V)
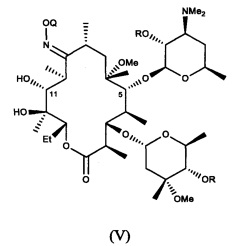
или его соли присоединения кислоты; и
(с) приведение соединения формулы (V) или его соли присоединения кислоты в контакт с деоксимирующим агентом с получением соединения формулы (II)
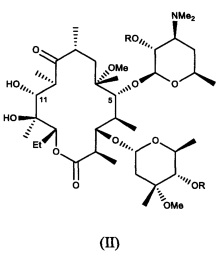
или его соли присоединения кислоты; и
превращение соединения формулы (II) или его соли присоединения кислоты в соединение формулы (I) или его фармацевтически приемлемую соль.
2. Способ по п. 1, дополнительно включающий (а) приведение соединения формулы (III)
или его соли присоединения кислоты в контакт с ацилирующим агентом с получением соединения формулы (IV) или его соли присоединения кислоты.
3. Способ по п. 1 или 2, отличающийся тем, что V представляет собой С(О).
4. Способ по п. 1 или 2, отличающийся тем, что W представляет собой фтор.
5. Способ по п. 1 или 2, отличающийся тем, что W представляет собой водород.
6. Способ по п. 1 или 2, отличающийся тем, что X и Y совместно с присоединенным к ним атомом углерода образуют карбонил.
7. Способ по п. 1 или 2, отличающийся тем, что А представляет собой СН2.
8. Способ по п. 1 или 2, отличающийся тем, что В представляет собой алкенилен.
9. Способ по п. 1 или 2, отличающийся тем, что В представляет собой (СН2)n, где n представляет собой целое число от 2 до 4.
10. Способ по п. 1 или 2, отличающийся тем, что С представляет собой 3-аминофенил.
11. Способ по п. 1 или 2, отличающийся тем, что R10 представляет собой бензоил.
12. Способ по п. 1 или 2, отличающийся тем, что R10 представляет собой водород.
13. Способ по п. 1 или 2, отличающийся тем, что соединение формулы (I) представляет собой соединение формулы:
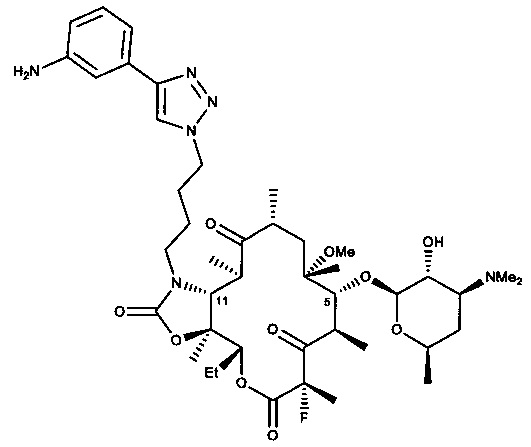
или его фармацевтически приемлемую соль.
14. Способ получения соединения формулы (II)
или его соли присоединения кислоты, где:
R представляет собой пространственно затрудненную ацильную группу;
способ, включающий:
(b) приведение соединения формулы (IV)
или его соли присоединения кислоты, где Q представляет собой 2-метокси-2-пропил, 1-метоксициклогексил или тропил, в контакт с метилирующим агентом с получением соединения формулы (V)
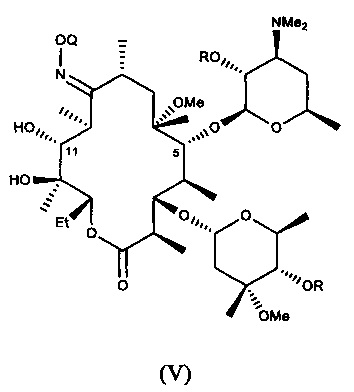
или его соли присоединения кислоты; и
(с) приведение соединения формулы (V) или его соли присоединения кислоты в контакт с деоксимирующим агентом с получением соединения формулы (II).
15. Способ по п. 14, дополнительно включающий (а) приведение соединения формулы (III)
или его соли присоединения кислоты в контакт с ацилирующим агентом с получением соединения формулы (IV) или его соли присоединения кислоты.
16. Способ по пп. 1, 2, 14 или 15, отличающийся тем, что Q представляет собой 2-метокси-2-пропил.
17. Способ по пп. 1, 2, 14 или 15, отличающийся тем, что R представляет собой бензоил.
18. Способ по п. 2 или 15, отличающийся тем, что (а) проводят в присутствии основания.
19. Способ по п. 18, отличающийся тем, что основание, применяемое в (а), представляет собой третичный амин.
20. Способ по п. 2 или 15, отличающийся тем, что (а) проводят в присутствии 4-диметиламинопиридина.
21. Способ по п. 2 или 15, отличающийся тем, что ацилирующий агент представляет собой ангидрид.
22. Способ по пп. 1, 2, 14 или 15, отличающийся тем, что (b) проводят в присутствии основания.
23. Способ по п. 22, отличающийся тем, что основание, применяемое в (b), представляет собой гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия или трет-бутоксид калия, или их смеси.
24. Способ по пп. 1, 2, 14 или 15, отличающийся тем, что (b) проводят в апротонном полярном растворителе.
25. Способ по п. 24, отличающийся тем, что апротонный полярный растворитель, применяемый на стадии (b), представляет собой диметилсульфоксид, диметилформамид, 1-метил-2-пирролидон, их смеси, возможно дополнительно в виде смеси с одним или более из тетрагидрофурана, 2-метилтетрагидрофурана, 1,2-диметоксиэтана, ацетонитрила или этилацетата.
26. Способ по пп. 1, 2, 14 или 15, отличающийся тем, что метилирующий агент, применяемый на стадии (b), представляет собой метилбромид, метилйодид, диметилсульфат, метил-п-толуолсульфонат или метил-метансульфонат.
27. Способ по пп. 1, 2, 14 или 15, отличающийся тем, что деоксимирующий агент, применяемый на стадии (с), содержит восстановитель.
28. Способ по пп. 1, 2, 14 или 15, отличающийся тем, что деоксимирующий агент, применяемый на стадии (с), содержит муравьиную кислоту и метабисульфит натрия.
29. Соединение формулы (IV)
или его соль присоединения кислоты, где Q представляет собой 2-метокси-2-пропил, 1-метоксициклогексил или тропил, a R представляет собой пространственно затрудненную ацильную группу.
30. Соединение формулы (V)
или его соль присоединения кислоты, где Q представляет собой 2-метокси-2-пропил, 1-метоксициклогексил или тропил, a R представляет собой пространственно затрудненную ацильную группу.
31. Соединение по п. 29 или 30, отличающееся тем, что Q представляет собой 2-метокси-2-пропил.
32. Соединение по п. 29 или 30, отличающееся тем, что R представляет собой бензоил.
| Цилиндрическая зубчатая передача | 1983 |
|
SU1167375A1 |
| WO 2006050941 A1, 18.05.2006 | |||
| WO 2006050942 A1, 18.05.2006 | |||
| Z.J | |||
| ZHU ET AL | |||
| Structure-activity relationship of macrolides against Mycobacterium tuberculosis, Tuberculosis, 2008, vol.88, p.S49-S63 | |||
| WO 2009055557 A1, 30.04.2009 | |||
| US 4990602 A, 05.02.1991 | |||
| СПОСОБ ПОЛУЧЕНИЯ КЛАРИТРОМИЦИНА В ВИДЕ КРИСТАЛЛОВ ФОРМЫ II | 2001 |
|
RU2230748C2 |

