Изобретение относится к области медицины и может быть использовано в контрольно-аналитических лабораториях для стандартизации и контроля качества лекарственных средств.
В настоящее время особо остро стоит проблема обеспечения населения эффективными высококачественными лекарственными средствами. Одним из важных факторов, способствующих улучшению качества лекарственных средств, является систематическое повышение эффективности методов их контроля и стандартизации.
Спектрофотометрия в видимой и УФ-области спектра относится к числу методов, получивших наибольшее распространение в анализе лекарственных средств. Объектами настоящего исследования являются перспективные лекарственные средства, применяемые для лечения сосудистых поражений головного мозга - производные пурина N-гликозидной структуры - аденозин, фосфаден и рибоксин.
Известен способ спектрофотометрического определения аденозина и фосфадена, заключающийся в приготовлении водных растворов испытуемых веществ с последующим их фотометрированием при длинах волн 259,5 и 257 нм соответственно и расчетом результатов с использованием значений молярного и удельного показателей поглощения для аденозина и фосфадена соответственно /ТУ 64-01-12-10-89" Аденозин 1,5-водный для аденозинтрифосфорной кислоты", ФС 42-1960-83 "Фосфаден"/.
Наиболее близким и принятым нами за прототип является способ определения рибоксина путем приготовления водных растворов испытуемого вещества и стандартного образца рибоксина соответственно с последующим их спектрофотометрированием при длине волны 249 нм и расчетом результатов по стандартному образцу рибоксина /ФС 42-2069-83 "Рибоксин"/.
Рекомендованный нормативно-технической документацией спектрофотометрический метод количественного определения фосфадена и аденозина недостаточно воспроизводим на разных приборах, так как для расчета содержания фосфадена используется удельный, а аденозина - молярный показатели поглощения. Это является причиной погрешностей их спектрофотометрического определения, достигающих ± 6,1%.
Для устранения погрешности градуировки и повышения точности спектрофотометрического анализа используют стандартные образцы. Однако выпуск стандартных образцов аденозина, фосфадена, а также и рибоксина является дорогостоящим. Поэтому способ определения с использованием таких стандартных образцов будет мало доступным для многих лабораторий.
Техническим результатом предлагаемого способа является повышение воспроизводимости результатов определения и уменьшение погрешности анализа.
Технический результат достигается путем приготовления раствора определяемого вещества и стандартного образца свойств с последующим их спектрофотометрированием и расчетом результатов.
Новым в достижении технического результата является то, что в качестве растворителя для приготовления испытуемых растворов используют 0,1 М раствор соляной кислоты.
Новым является и то, что в качестве стандартного образца используют дихромат калия, вводя в формулу расчета результатов коэффициент пересчета.
Изучаемые вещества изменяют спектр поглощения в зависимости от pH среды. Исходя из значений констант ионизации и свойств аденозина, фосфадена и рибоксина, авторы доказали, что оптимальным растворителем для их спектрофотометрического определения является 0,1 М раствор соляной кислоты. Оптимальный растворитель обеспечивает стабилизацию испытуемых растворов, что повышает воспроизводимость результатов определения и уменьшает погрешности анализа.
В данном растворителе спектры поглощения аденозина и фосфадена характеризуются максимумом поглощения при 258 нм, рибоксина - при 249 нм.
Исходя из установленной авторами зависимости, согласно которой в качестве стандартных образцов свойств могут применяться вещества, для которых интервал между аналитической длиной волны и максимумом поглощения этого стандартного образца не превышает половины полуширины его полосы поглощения, в качестве стандартного образца в предлагаемом способе используют дихромат калия, так как оптимальные области поглощения, в которых его можно использовать в качестве стандартного образца свойств 249-262 и 340-359 нм. Дихромат калия выпускается серийно промышленностью как химически чистое вещество, имеется ГОСТ, регламентирующий его качество, раствор дихромата калия устойчив в оптимальных растворителях длительное время. Использование дихромата калия в предлагаемом способе приводит к уменьшению погрешности анализа.
Сопоставительный анализ показывает, что заявляемое техническое решение отличается тем, что в качестве растворителя для приготовления испытуемых растворов используют 0,1 М раствор соляной кислоты, а в качестве стандартного образца - дихромат калия, вводя в формулу расчета результатов коэффициент пересчета, что соответствует критерию изобретения "новизна".
Новая совокупность признаков обеспечивает повышение воспроизводимости результата определения, уменьшение погрешности анализа, а также позволяет снизить стоимость анализа, что соответствует критерию "промышленная применимость".
При анализе известных решений было выявлено, что в них отсутствуют сведения о влиянии отличительных признаков на достижение поставленного технического результата, следовательно, изобретение соответствует критерию "изобретательский уровень".
Способ осуществляют следующим образом.
Готовят раствор стандартного образца дихромата калия для анализа аденозина, фосфадена, рибоксина. Для этого точную массу дихромата калия /0,1500 г/ помещают в мерную колбу вместимостью 100 мл, растворяют в 0,1 М растворе соляной кислоты и доводят раствор до метки этим же растворителем, перемешивают. 1 мл приготовленного раствора дихромата калия помещают в мерную колбу вместимостью 50 мл и доводят 0,1 М раствором соляной кислоты до метки, перемешивают. Затем проводят количественное определение аденозина, фосфадена и рибоксина в субстанции. Для этого точную массу препарата /0,0500 г/ помещают в мерную колбу вместимостью 100 мл, растворяют в горячей воде, доводят этой водой до метки и перемешивают. Раствор охлаждают. 1 мл приготовленного раствора переносят в мерную колбу вместимостью 50 мл и доводят 0,1 М раствором соляной кислоты до метки, перемешивают. Измеряют оптическую плотность испытуемого раствора при длине волны 258 нм /для аденозина и фосфадена/ и при 249 нм /для рибоксина/ относительно 0,1 М раствора соляной кислоты на спектрофотометре в кюветах с длиной рабочего слоя 10 мм. Параллельно измеряют оптическую плотность раствора стандартного образца дихромата калия при длине волны 258 нм /при анализе аденозина и фосфадена/ и при длине волны 249 нм /при анализе рибоксина/ относительно 0,1 М раствора соляной кислоты на спектрофотометре в кювете с длиной рабочего слоя 10 мм.
Расчет результатов количественного определения проводят по формуле:
где Дх и Дст - оптические плотности определяемого вещества и стандартного образца соответственно;
aх и aст - точные навески определяемого вещества и стандартного образца соответственно;
100 - коэффициент для пересчета в проценты;
Kпер. - коэффициент пересчета, который рассчитывают следующим образом: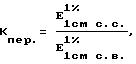
где E
E
Kпер. для аденозина - 0,264;
Kпер. для фосфадена - 0,344;
Kпер. для рибоксина - 0,360.
Содержание аденозина должно быть не менее 98,0%, фосфадена в пересчете на сухое вещество должно быть не менее 98,0% и не более 102,0%, рибоксина в пересчете на сухое вещество должно быть не менее 96,0% и не более 102,0%.
Предлагаемый способ поясняется следующими примерами. Готовят растворы испытуемого образца и стандартного вещества выше описанным способом. Измеряют на спектрофотометре оптические плотности приготовленных растворов. Далее ведут расчет результатов по формуле, используя коэффициент пересчета.
При определении фосфадена получили следующие результаты: Дx = 0,426, Дст. = 0,443, ax = 0,05225, aст. = 0,14980, влажность 4,79%, X = 99,6%. При п = 6,  S = 0,50,
S = 0,50,  εα= 0,47 /P = 95%/, A% = 0,47.
εα= 0,47 /P = 95%/, A% = 0,47.
При определении аденозина получили следующие результаты: Дx = 0,620, Дст. = 0,453, ax = 0,05626, aст. = 0,15595, X = 100,2%. При п = 6,  S = 0,56,
S = 0,56, 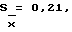 εα= 0,50 /P = 95%/, A% = 0,50.
εα= 0,50 /P = 95%/, A% = 0,50.
При определении рибоксина получили следующие результаты: Дx = 0,396, Дст. = 0,432, aх = 0,0500, aст. = 0,1505, X = 100,0%. При п = 6,  S = 0,37,
S = 0,37, 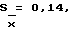 εα= 0,34 /P = 95%/, A% = 0,34.
εα= 0,34 /P = 95%/, A% = 0,34.
Данные примеры подтверждают, что содержание аденозина, фосфадена и рибоксина соответствует требованиям нормативного документа.
Предлагаемый способ с использованием стандартного образца дихромата калия оказался оптимальным и для количественного определения фосфадена и рибоксина в таблетках и растворе для инъекций, а также позволил с достаточной точностью установить однородность дозирования таблеток фосфадена и провести контроль теста "растворения" таблеток фосфадена и рибоксина.
Методики количественного определения фосфадена и рибоксина в лекарственных формах отличаются от методики количественного определения фосфадена и рибоксина в субстанции только приготовлением испытуемого раствора.
Для количественного определения фосфадена в таблетках по 0,05 г и рибоксина в таблетках по 0,2 г, покрытых оболочкой, берут точную массу порошка /0,2500 г/ растертых таблеток фосфадена или одну таблетку рибоксина, растертую в порошок, и количественно переносят с помощью горячей воды в мерные колбы вместимостью 100 мл и 200 мл соответственно, взбалтывают в течение 15 минут, доводят объем раствора этой же водой до метки, перемешивают, охлаждают и фильтруют через бумажный фильтр, отбрасывая первые 15 - 20 мл фильтрата. 1 мл полученного фильтрата переносят в мерные колбы вместимостью 50 мл и 100 мл соответственно и доводят объем раствора 0,1 М раствором соляной кислоты до метки, перемешивают.
Содержание фосфадена в таблетках по 0,05 г должно быть 0,045 - 0,055 г, рибоксина в таблетках по 0,2 г должно быть 0,19 - 0,21 г.
Количественное определение рибоксина и фосфадена в таблетках поясняется следующими примерами.
При анализе таблеток рибоксина по 0,2 г получены результаты: Дx = 0,452, Дст. = 0,456, aст. = 0,0515, X = 0,2042.
При анализе таблеток фосфадена по 0,05 г получены результаты: Дx = 0,419, Дст. = 0,24, ax = 0,24835, aст. = 0,1042, Pср. = 0,2083, X = 0,0525.
Данные примеры подтверждают, что содержание рибоксина и фосфадена в таблетках соответствует требованиям нормативного документа.
Для количественного определения рибоксина в 2% растворе для инъекций и фосфадена в 2% растворе для инъекций, объем лекарственной формы 5 мл и 1 мл соответственно помещают в мерную колбу вместимостью 100 мл и доводят объем раствора 0,1 М раствором соляной кислоты до метки, перемешивают.
Содержание рибоксина и фосфадена в 1 мл препарата в граммах /X/ должно быть 0,019 - 0,021.
При анализе рибоксина в 2% растворе для инъекций получены результаты: Дx = 0,407, Дст. = 0,429, aст. = 0,1500, X = 0,0205.
При анализе фосфадена в 2% растворе для инъекций получены результаты: Дx = 0,431, Дст. = 0,414, aст. = 0,14415, X = 0,0206.
Для определения однородности дозирования таблеток фосфадена по 0,05 г одну таблетку помещают в мерную колбу вместимостью 100 мл и растворяют в горячей воде, доводят той же водой до метки, перемешивают, охлаждают и фильтруют. 1 мл фильтрата помещают в мерную колбу вместимостью 50 мл и доводят объем раствора 0,1 М раствором соляной кислоты до метки.
Допустимые нормы отклонения от номинала не более ± 15% для десяти проанализированных таблеток.
При анализе десяти таблеток фосфадена по 0,05 г получили максимальное отклонение от номинального содержания +4,2 и -5,2%.
Для контроля теста растворения таблетки рибоксина и фосфадена за основу брали унифицированную методику /ГФ XI изд., С. 159-160/. В качестве среды растворения использовали 0,1 М раствор соляной кислоты, время растворения для таблеток рибоксина установили 45 минут, для таблеток фосфадена - 25 минут.
При анализе таблеток рибоксина по 0,2 г и таблеток фосфадена по 0,05 г, 5 мл фильтрата помещают в мерную колбу вместимостью 100 мл и 25 мл соответственно, доводят объем раствора 0,1 М раствором соляной кислоты до метки и перемешивают.
Согласно ГФ XI изд. С. 159-160, в среду растворения должно перейти не менее 75% действующего вещества от содержания в лекарственной форме.
При анализе таблеток рибоксина по 0,2 г высвобождение действующего вещества составило 82,5; 92,7; 91,6; 91,3; 92,5% для пяти таблеток соответственно.
При анализе таблеток фосфадена по 0,05 г высвобождение действующего вещества составило 92,3; 89,4; 94,7; 95,6; 96,8% для пяти таблеток соответственно.
Таким образом, предлагаемый способ определения лекарственных средств производных пурина N-гликозидной структуры с использованием стандартного образца свойств позволяет повысить воспроизводимость результатов определения, уменьшить погрешность анализа и снизить его стоимость.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ КИСЛОТЫ НИКОТИНОВОЙ И КСАНТИНОЛА НИКОТИНАТА | 2000 |
|
RU2193191C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ АНТИБИОТИКОВ ГРУППЫ ХЛОРАМФЕНИКОЛА С ИСПОЛЬЗОВАНИЕМ СТАНДАРТНОГО ОБРАЗЦА СВОЙСТВ | 2000 |
|
RU2187802C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ДРОТАВЕРИНА | 2012 |
|
RU2514002C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЦИННАРИЗИНА | 2009 |
|
RU2424515C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДИКЛОФЕНАКА НАТРИЯ | 2006 |
|
RU2333488C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ НАТРИЯ ПАРА-АМИНОСАЛИЦИЛАТА | 2008 |
|
RU2399049C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПИРАЗИНАМИДА | 2008 |
|
RU2394226C2 |
| Способ количественного определения ацикловира | 2017 |
|
RU2655775C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ИБУПРОФЕНА | 2006 |
|
RU2333490C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПИРАЗИДОЛА | 2006 |
|
RU2334983C2 |
Изобретение относится к медицине и может быть использовано в контрольно-аналитических лабораториях для стандартизации и контроля качества лекарственных средств. В качестве растворителя для приготовления испытуемых растворов используют 0,1 М раствор соляной кислоты, а в качестве стандартного образца - раствор дихромата калия для аденозина, фосфадена и рибоксина. Затем растворы определяемого вещества N-гликозидной структуры и стандартного образца свойств спектрофотометрируют при длине волны 258 нм для аденозина и фосфадена и при 249 нм для рибоксина. В формулу расчета результатов количественного определения вводят коэффициент пересчета. Способ обеспечивает повышение воспроизводимости результатов определения и уменьшение погрешности анализа.
Способ определения лекарственных средств производных пурина N-гликозидной структуры с использованием стандартного образца свойств путем приготовления растворов определяемого вещества и стандартного образца свойств с последующим их спектрофотометрированием и расчетом результатов, отличающийся тем, что в качестве растворителя для приготовления испытуемых растворов используют 0,1 М раствор соляной кислоты, а в качестве стандартного образца - дихромат калия, вводя в формулу расчета результатов коэффициент пересчета.
| Способ определения содержания воды в образцах гемоглобина | 1986 |
|
SU1515108A1 |
| УСТРОЙСТВО ДЛЯ ОБЛУЧЕНИЯ БИОЛОГИЧЕСКОЙ ЖИДКОСТИ | 1991 |
|
RU2008041C1 |

