Изобретение относится к способу получения N-(2- нитроксиэтил)-никотинамида формулы I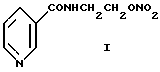
известного также под названием никорандил. N-(2- нитроксиэтил)никотинамид является принципиально новым высокоэффективным антиангинальным лекарственным средством класса органических нитратов. Его различные лекарственные формы находят все более широкое применение в мировой медицинской практике [Am. J. Cardiology, 1989, 63, N 21, 1-85 (15 публикаций по материалам симпозиума "Новое в лечении стенокардии и сердечной недостаточности", Сан-Франциско, 17.10.1987); Машковский М.Д. Лекарственные средства. Тринадцатое издание, новое. Харьков, Олма пресс, т. I, 1998, стр. 396, 429]. N-(2-нитроксиэтил)никотинамид в России не производится.
В основе известных способов получения N- (2-нитроксиэтил)-никотинамида лежат две различные химические реакции. Так, в японском патенте [яп. пат. N 58-17463, заявл. 2.04.1976, опубл. 7.04.1983] и последующих патентах, полученных патентообладателем [яп. пат. N 58-17463, заявл. 2.04.1976, опубл. 7.04.1983] в ФРГ [пат. ФРГ N 2714713 от 2.04.1978, опубл. 20.10.1977], Бельгии [пат. Бельгии N 853144 от 1.04.1977, опубл. 1.08.1977], США [пат. США N 4200640 от 7.09.1978, опубл. 23.04.1980] и СССР [пат. СССР N 999965 от 1.04.1977, опубл. 23.02.1983], описан способ его получения по реакции производных никотиновой кислоты (хлорангидрид, ангидрид, сложный эфир и др.) с 2-нитроксиэтиламином (NH2CH2CH2ONO2). 2-Нитроксиэтиламин вводится в реакцию в виде азотнокислой соли, которая представляет собой достаточно мощное взрывчатое вещество с детонационными характеристиками и чувствительностью к удару, превосходящими тротил [Jahresber. chem. -techn. Reichsanstalt. , 1933-1935, 244].
Это делает взрывоопасным производство N-(2-нитроксиэтил)никотинамида по такому способу.
Другие способы получения N-(2-нитроксиэтил)никотинамида [яп. пат. N 58-17463, заявл. 2.04.1976, опубл. 7.04.1983, пат. Бельгии N 853144 от 1.04.1977, опубл. 1.08.1977, пат. СССР N 999965 от 1.04.1977, опубл. 23.02.1983, яп. заявка N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990, пат. СССР N 942593 от 28.07.1978, опубл. 10.07.1982, пат. Польши N 162496 от 31.01.1990, опубл. 31.12.1993] основаны на взаимодействии N-(2-гидроксиэтил)-никотинамида формулы II (или его гидрохлорида, гидронитрата)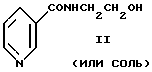
с концентрированной дымящей азотной кислотой с последующим выделением целевого продукта формулы I при подщелачивании безводным карбонатом натрия (или калия) реакционной массы, предварительно разбавленной водой.
При концентрации исходной азотной кислоты ниже 90% выход целевого продукта заметно снижается: при 85%-ной концентрации он ниже 30%, а при 70%-ной концентрации выделить целевой продукт не удается [яп. заявка, N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990].
В качестве наиболее близкого по технической сущности аналога - прототипа нами выбран польский патент [пат. Польши N 162496 от 31.01.1990, опубл. 31.12.1993] , в котором, по сравнению с другими аналогами [яп. пат. N 58-17463, заявл. 2.04.1976, опубл. 7.04.1983, пат. Бельгии N 853144 от 1.04.1977, опубл. 1.08.1977, пат. СССР N 999965 от 1.04.1977, опубл. 23.02.1983, яп. заявка N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990, пат. СССР N 942593 от 28.07.1978, опубл. 10.07.1982], изменена стадия очистки целевого продукта. Сущность способа получения N-(2-нитроксиэтил)-никотинамида состоит в прибавлении 1 вес. ч. N-(2-гидроксиэтил)-никотинамида формулы II к 3 вес. ч. дымящей азотной кислоты при температуре от -10 до - 5oC, дополнительном перемешивании реакционной массы при температуре 0-5oC в течение 1 ч, выливании реакционной массы в смесь воды со льдом, подщелачивании полученного раствора кристаллическим карбонатом натрия (или калия) и отфильтровывании продукта-сырца. Очистка сырца от примесей проводилась отмыванием холодной водой с последующей сушкой при температуре не выше 30oC. Полученный при такой очистке продукт имеет т. пл. 92oC и, по мнению авторов [пат. Польши N 162496 от 31.01.1990, опубл. 31.12.1993], не нуждается в дополнительной очистке. В японской заявке [яп. заявка N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990], где при очистке использовались органические растворители, продукт имел т. пл. 92,5-93,5oC.
Поскольку способность концентрированной азотной кислоты образовывать с органическими горючими взрывчатые растворы широко известна, нами по методике [Eremenko L. T. , Nesterenko D.A. Proc. 20th Intern. Pyrotechnics Seminar, July, 25-29, 1994, p.p. 265-277] была проверена детонационная способность гомогенных реакционных масс, полученных при смешении 1 вес. ч. N-(2- гидроксиэтил)никотинамида с 3 вес. ч. азотной кислоты двух концентраций: 94 и 100%. Перед испытанием на детонационную способность нитромассы перемешивали 30 мин при температуре около 0oC. Обе массы оказались детонационноспособными. Система на основе 94%-ной азотной кислоты имеет относительный детонационный импульс на уровне тротила, система на основе 100%-ной азотной кислоты - на 6% выше. Таким образом, процесс нитрования N-(2- гидроксиэтил)никотинамида концентрированной азотной кислотой [яп. пат. N 58-17463, заявл. 2.04.1976, опубл. 7.04.1983, пат. Бельгии N 853144 от 1.04.1977, опубл. 1.08.1977, пат. СССР N 999965 от 1.04.1977, опубл. 23.02.1983, яп. заявка N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990, пат. СССР N 942593 от 28.07.1978, опубл. 10.07.1982, пат. Польши N 162496 от 31.01.1990, опубл. 31.12.1993] также является взрывоопасным. Естественным следствием высокой химической активности концентрированной азотной кислоты является необходимость проведения процесса нитрования при низких температурах (до -10oC). Кроме того, проведение процессов нейтрализации избытка азотной кислоты и последующего перевода соли целевого продукта в его основную форму с использованием карбонатов натрия (или калия) связано с выделением углекислого газа, вспениванием массы и увеличением объема используемой аппаратуры. Указанные недостатки усугубляются использованием больших избытков концентрированной азотной кислоты: мольное отношение азотной кислоты к субстрату лежит в пределах 8-14 [яп. пат. N 58-17463, заявл. 2.04.1976, опубл. 7.04.1983, пат. Бельгии N 853144 от 1.04.1977, опубл. 1.08.1977, пат. СССР N 999965 от 1.04.1977, опубл. 23.02.1983, яп. заявка, N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990, пат. СССР N 942593 от 28.07.1978, опубл. 10.07.1982, пат. Польши N 162496 от 31.01.1990, опубл. 31.12.1993].
Целью изобретения является разработка взрывобезопасного, более дешевого и технологичного способа получения N-(2-нитроксиэтил)-никотинамида (I) путем нитрования N-(2-гидроксиэтил)никотинамида (II).
Поставленная цель достигается тем, что N-(2-гидроксиэтил)никотинамид вводят во взаимодействие со смесью азотной и серной кислот определенного состава, обеспечивающего образование недетонационноспособной реакционной массы, при температуре 0-25oC, перемешивают при этой температуре 0,5-1 ч, выливают в ледяную воду, нейтрализуют водным раствором аммиака. Образовавшийся осадок отфильтровывают, отмывают от примесей холодной водой, сушат в вакууме и получают N-(2- нитроксиэтил)-никотинамид с выходом 82-88% и т. пл. 91-92oC. При необходимости N-(2-нитроксиэтил)никотинамид перекристаллизовывают из дистиллированной воды. После сушки получают продукт с т. пл. 92,5-93,5oC. Состав смесей азотной и серной кислот, который обеспечивает недетонационноспособность реакционной массы в условиях испытаний [Eremenko L. T., Nesterenko D.A. Proc. 20th Intern. Pyrotechnics Seminar, July, 25-29, 1994, p. p. 265-277] и вместе с тем позволяет получать качественный целевой продукт с высоким выходом, может варьироваться в определенных пределах. При этом содержание азотной кислоты в системе азотная кислота-серная кислота-вода не должно превышать 45 вес. %, а количество воды должно быть достаточным для получения гомогенной и недетонационноспособной реакционной массы. Использование таких смесей позволяет существенно снизить расход концентрированной дымящей азотной кислоты; они могут быть приготовлены, например, на основе значительно более дешевой 65-70%-ной азотной кислоты. Кроме того, использование серно-азотных кислотных смесей позволяет снизить мольное отношение азотной кислоты к субстрату до 3-4 и проводить нитрование при более высоких температурах, от 0oC до комнатной, в течение 0,5-1 ч.
Кроме того, поставленная цель достигается заменой на стадии нейтрализации карбонатов натрия (или калия) водным раствором аммиака. Это позволяет избежать газовыделения и получать более растворимые в воде соли аммония, вследствие чего уменьшается объем используемой аппаратуры и в условиях высаливания обеспечивается достаточная полнота выделения целевого продукта.
Весьма существенным новым признаком изобретения является упрощение процесса очистки N-(2- нитроксиэтил)никотинамида путем замены используемых пожароопасных и токсичных органических растворителей [яп. пат. N 58-17463, заявл. 2.04.1976, опубл. 7.04.1983, пат. ФРГ N 2714713 от 2.04.1978, опубл. 20.10.1977, пат. Бельгии N 853144 от 1.04.1977, опубл. 1.08.1977, пат. США N 4200640 от 7.09.1978, опубл. 23.04.1980, пат. СССР N 999965 от 1.04.1977, опубл. 23.02.1983, Jahresber. chem.-techn. Reichsanstalt., 1933-1935, 244, яп. заявка N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990, пат. СССР N 942593 от 28.07.1978, опубл. 10.07.1982] на воду. Это позволяет получать N-(2-нитpoкcиэтил)-никoтинaмид с т. пл. 92,5-93,5oC.
Изобретение характеризуется нижеприведенными примерами, которые лишь иллюстрируют некоторые варианты его применения, но не ограничивают его возможностей. В частности, нитрующие смеси любого заданного состава, содержащие азотную кислоту, серную кислоту и воду, могут быть приготовлены самыми различными способами с учетом экономической и технологической целесообразности.
Пример 1. К 3,48 вес. ч. нитрующей смеси, содержащей 33,5% азотной кислоты, 52,9% серной кислоты и 13,6% воды, при перемешивании и температуре 8-12oC постепенно прибавляют 1 вес. ч. N-(2-гидроксиэтил)- никотинамида, перемешивают еще 30 мин при этой температуре. Полученный раствор выливают в смесь воды со льдом и нейтрализуют водным раствором аммиака до слабощелочной среды. Выпавшие кристаллы отфильтровывают и промывают холодной водой. После высушивания при комнатной температуре получают 1,05 вес. ч. N-(2- нитроксиэтил)никотинамида (выход 83%) с т. пл. 91-92oC. После перекристаллизации из воды и высушивания в вакууме т. пл. 92,5-93,5oC.
Пример 2. В условиях примера 1 к 4,31 вес. ч. смеси, содержащей 34,5% азотной кислоты, 54,5% серной кислоты и 11% воды, при температуре 15-18oC прибавляют 1 вес. ч. N-(2- гидроксиэтил)никотинамида, перемешивают еще 1 ч при этой температуре и выделяют 1,07 вес. ч. N-(2-нитроксиэтил)никотинамида (выход 84%), т. пл. 91-92oC. После очистки в условиях примера 1 т. пл. 92,5-93,5oC.
Пример 3. В условиях примера 2 реакционную массу получают при температуре 20-25oC, перемешивают еще 30 мин при этой температуре и выделяют 1,06 вес. ч. N-(2-гидроксиэтил)никотинамида (выход 83%), т. пл. 91-92oC. После очистки в условиях примера 1 т. пл. 92,5-93,5oC.
Пример 4. В условиях примера 1 к 4,83 вес. ч. смеси, содержащей 24,2% азотной кислоты, 65,5% серной кислоты и 10,4% воды, прибавляют 1 вес. ч. N-(2-гидроксиэтил)никотинамида и выделяют 1,12 вес. ч. N-(2-нитроксиэтил)-никотинамида (выход 88%), т. пл. 91-92oC. После очистки в условиях примера 1 т. пл. 92,5-93,5oC.
Пример 5. В условиях примера 1 к 5,02 вес. ч смеси, содержащей 23,1% азотной кислоты, 62,9% серной кислоты и 14% воды, прибавляют 1 вес. ч N-(2-гидроксиэтил)никотинамида и выделяют 1,05 вес. ч. N-(2-нитроксиэтил)никотинамида (выход 83%), т. пл. 91-92oC. После очистки в условиях примера 1 т. пл. 92,5-93,5oC.
Пример 6. В условиях примера 1 к 4,46 вес. ч. смеси, содержащей 44,4% азотной кислоты, 42,2% серной кислоты и 13,4% воды, при температуре 0-5oC прибавляют 1 вес. ч. N-(2- гидроксиэтил)никотинамида, перемешивают еще 1 ч при этой температуре и выделяют 1,05 вес. ч. N-(2-нитроксиэтил)никотинамида (выход 84%), т. пл. 91-92oC. После очистки в условиях примера 1 т. пл. 92,5-93,5oC.
Таким образом, использование предлагаемого технического решения позволяет достичь поставленной цели и получать 2-нитроксиэтилникотинамид формулы I взрыво-и пожаробезопасным, более технологичным и дешевым способом. Так, при прочих равных условиях, N-(2-нитроксиэтил)никотинамид, получаемый по предлагаемому способу, только по стоимости исходного химического сырья в 1,5 раза дешевле, чем при получении по способу-прототипу [пат. Польши N 162496 от 31.01.1990, опубл. 31.12.1993] и существенно дешевле по сравнению с другими аналогами [пат. Бельгии N 853144 от 1.04.1977, опубл. 1.08.1977, пат. СССР N 999965 от 1.04.1977, опубл. 23.11.1983, яп. заявка N 2-207072 (A) от 6.02.1989, опубл. 16.08.1990, пат. СССР N 942593 от 28.07.1978, опубл. 10.07.1982] . При приготовлении смесей азотная кислота-серная кислота-вода используется азотная кислота различной концентрации (в том числе и относительно дешевая азотная кислота концентрации 65-70%), причем общий расход азотной кислоты значительно сокращается. Такие смеси более стабильны и в целом более дешевые, чем дымящая азотная кислота. Кроме того, любой из выбранных составов может быть приготовлен различными способами с учетом экономических и технических аспектов. Использование аммиака на стадии нейтрализации исключает нежелательные процессы газообразования и вспенивания и позволяет получать аммиачные соли, утилизация которых создает принципиальные возможности для дальнейшего удешевления целевого продукта и организации всего процесса его получения в целом без образования вредных для окружающей среды отходов производства. И, наконец, предложен простой, пожаробезопасный и более дешевый способ очистки целевого продукта путем перекристаллизации из воды.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-(2-НИТРОКСИЭТИЛ)НИКОТИНАМИДА ИЗ ЭТИЛНИКОТИНАТА | 2005 |
|
RU2341517C2 |
| АМИДОАЛКАНОЛНИТРАТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2147301C1 |
| НИТРАТЫ СПИРТОВ, СОДЕРЖАЩИЕ АМИДНУЮ, ДИНИТРОМЕТИЛЕНОВУЮ И НИТРОАМИННУЮ ГРУППЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, 3-(АЦИЛАМИНОДИНИТРОАЛКИЛ)-ТЕТРАГИДРО-1,3-ОКСАЗОЛЫ И - ОКСАЗИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2146243C1 |
| НИТРОКСИСУКЦИНАТ 2,4,6-ТРИМЕТИЛ-3-ОКСИПИРИДИНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2003 |
|
RU2250210C1 |
| НИТРАТЫ N-АЛКАНОЛСУКЦИНАМИДОВ ИЛИ ИМИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, N-АЛКАНОЛСУКЦИНАМИДЫ ИЛИ ИМИДЫ | 1996 |
|
RU2102380C1 |
| ПРОИЗВОДНЫЕ ТЕТРАХЛОРИДА ПЛАТИНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2245328C1 |
| ПРОИЗВОДНЫЕ ЯНТАРНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ СИНТЕЗА И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 1997 |
|
RU2125040C1 |
| 2,4,6-ТРИАЗИДОТОЛУОЛ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2430080C2 |
| БИТУМНО-РЕЗИНОВАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1998 |
|
RU2164927C2 |
| НИТРОКСИСУКЦИНАТ 2-ЭТИЛ-6-МЕТИЛ-3-ОКСИПИРИДИНА (ВАРИАНТЫ ИСПОЛЬЗОВАНИЯ) И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2008 |
|
RU2394815C2 |
Описывается способ получения N-(2-нитроксиэтил)никотинамида нитрованием N-(2-гидроксиэтил)никотинамида. Нитрование проводят нитрующей смесью, содержащей азотную и серную кислоты и воду. Технический результат - разработка взрывобезопасного, более дешевого и технологичного способа. 7 з.п. ф-лы.
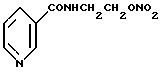
нитрованием N-(2-гидроксиэтил)никотинамида формулы II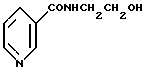
отличающийся тем, что соединение формулы II подвергают взаимодействию с нитрующей смесью, содержащей азотную и серную кислоты и воду.
| 0 |
|
SU162496A1 | |
| Способ получения азотнокислого эфира N-/2-оксиэтил/ никотинамида или его солей | 1978 |
|
SU942593A3 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| НИТРАТЫ N-АЛКАНОЛСУКЦИНАМИДОВ ИЛИ ИМИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, N-АЛКАНОЛСУКЦИНАМИДЫ ИЛИ ИМИДЫ | 1996 |
|
RU2102380C1 |

