ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому производному фталазинонкетона формулы (I), к способам его получения, к фармацевтической композиции, содержащей это производное, и к его применению в качестве терапевтического агента, в частности, в качестве ингибитора поли(АДФ-рибоза)полимеразы (PARP).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Химиотерапия и радиотерапия являются двумя общепринятыми способами лечения рака. Оба способа лечения могут индуцировать однонитевой и/или двунитевой разрыв ДНК, что приводит к цитотоксичности, следовательно, целевые раковые клетки будут погибать вследствие хромосомного повреждения. Важным результатом в ответ на сигнал повреждения ДНК является активация сигнала в регуляторном сайте, цель которого состоит в том, чтобы защитить клетки от митоза в случае повреждения ДНК, предотвращая посредством этого повреждение клетки. В большинстве случаев опухолевые клетки проявляют дефекты сигнала регуляции в клеточном цикле и обладают высокой скоростью пролиферации. Поэтому можно предсказать, что опухолевые клетки обладают специфичными механизмами репарации ДНК, которые могут реагировать быстро, чтобы репарировать хромосомное повреждение, релевантное для регуляции пролиферации, посредством этого спасая клетки от цитотоксических эффектов некоторых способов лечения и поддерживая их жизнь.
В клиническом применении эффективная концентрация химиотерапевтического лекарственного средства или терапевтическая интенсивность излучения может бороться с этим механизмом репарации ДНК, чтобы обеспечить эффект уничтожения в отношении целевых опухолевых клеток. Однако в опухолевых клетках может развиваться толерантность к лечению за счет усиления их механизмов репарации повреждений ДНК, и они выживают после летального повреждения ДНК. В целях преодоления этой толерантности обычно необходимо увеличить дозировку терапевтического лекарственного средства или интенсивность излучения. Данный подход приведет к побочным эффектам в отношении нормальной ткани вблизи повреждений, и, следовательно, сделает курс лечения осложненным тяжелыми побочными реакциями, в результате, повышая риск лечения. В то же время, постоянно возрастающая толерантность снизит терапевтический эффект, поэтому можно сделать вывод, что цитотоксичность агентов, повреждающих ДНК, можно улучшить на пути специфичности к опухолевым клеткам посредством контроля механизма репарации, стимулируемого сигналом повреждения ДНК.
PARP (поли(АДФ-рибоза)полимеразы), характеризующиеся активностью поли-АДФ-рибозилирования, состоят из надсемейства 18 ядерных ферментов и цитоплазматических ферментов. Такой эффект поли-АДФ-рибозилирования может регулировать активность белка-мишени и взаимодействие между белками, а также регулировать многие другие фундаментальные биологические процессы, включая репарацию ДНК и клеточную гибель. Кроме того, геномная стабильность также связана с поли-АДФ-рибозилированием (см. D'Amours et al. Biochem. J, 1999, 342, 249).
Активность PARP-1 составляет примерно 80% суммарной клеточной активности PARP. PARP-1 вместе с PARP-2, который является наиболее сходным с PARP-1, являются членами семейства PAPR, обладающими способностью к репарации повреждения ДНК. Как сенсорный и сигнальный белок повреждения ДНК, PARP-1 может быстро обнаруживать сайты повреждения ДНК и непосредственно связываться с ними, а затем индуцировать агрегацию различных белков, необходимых для репарации ДНК, обеспечивая посредством этого репарацию повреждения ДНК. Когда в клетках отсутствует PARP-1, PARP-2 может осуществлять репарацию повреждения ДНК вместо PARP-1.
Исследования показали, что по сравнению с нормальными клетками экспрессия белков семейства PARP в солидных опухолях в целом усилена. Кроме того, показано, что опухоли (такие как рак молочной железы и рак яичника), в которых отсутствует ген, связанный с репарацией ДНК (такой как BRCA-1 или BRCA-2), крайне чувствительны к ингибиторам PARP-1. Это позволяет предположить потенциальное применение ингибиторов PARP в качестве единственного агента, который может быть назван трижды негативным раком молочной железы (см. Plummer, E.R. Curr. Opin. Pharmacol. 2006, 6, 364; Ratnam, et al; Clin. Cancer Res. 2007, 13, 1383). В то же время, поскольку механизм репарации повреждения ДНК является основным механизмом ответа опухолевых клеток на толерантность, производимую лечением химиотерапевтическими лекарственными средствами и ионизирующим излечением, PARP-1 считают эффективной мишенью для освоения новых способов терапии рака.
Ингибиторы PARP прежде разработаны и сконструированы с использованием никотинамида NAD+, который можно использовать в качестве каталитического субстрата PARP, в качестве матрицы для разработки их аналогов. Как конкурентные ингибиторы NAD+, эти ингибиторы конкурируют с NAD+ за каталитические сайты PARP, посредством этого предотвращая синтез цепи поли(АДФ-рибоза). PARP без модификации поли(АДФ-рибозилирование) не может диссоциировать из сайтов повреждения ДНК, что приведет другие белки, вовлеченные в репарацию, в сайт повреждения, предотвращая посредством этого осуществление процесса репарации. Следовательно, под действием цитотоксических лекарственных средств или излучения ингибитор PARP, в конечном счете, уничтожит опухолевые клетки с повреждением ДНК.
Кроме того, NAD+, который потребляется в качестве каталитического субстрата PARP, является существенным фактором в процессе синтеза АТФ клеток. При высоком уровне активности PARP внутриклеточные уровни NAD+ значительно снизятся, воздействуя посредством этого на внутриклеточный уровень АТФ. Вследствие недостатка внутриклеточного содержания АТФ клетки не могут достигать программируемого АТФ-зависимого процесса клеточной гибели и могут только переходить к некрозу, специфичному механизму апоптоза. В процессе некроза будет высвобождаться множество воспалительных цитокинов, производя посредством этого токсические эффекты в отношении других органов и тканей (Horvath ЕМ et al. Drug News Perspect, 2007, 20, 171-181). Следовательно, ингибиторы PARP можно также применять для лечения ряда заболеваний, обусловленных этим механизмом, включая нейродегенеративные заболевания (такие как болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона), диабет, сопутствующие заболевания при ишемии или процессе ишемии-реперфузии, таком как инфаркт миокарда и острая почечная недостаточность, заболевания системы кровообращения, такие как септический шок, и воспалительные заболевания, такие как хронический ревматизм, и т.д. (см. Tentori L, et al. Pharmacol. Res., 2002, 45, 73-85; Horvath ЕМ et al. Drug News Perspect., 2007, 20, 171; Faro R, et al. Ann. Thorac. Surg., 2002, 73, 575.; Kumaran D, et al. Brain Res., 2008, 192, 178).
В настоящее время существует серия заявок на патенты в отношении фталазинонкетонового ингибитора PARP, включающая WO 2002036576, WO 2004080976 и WO2006021801.
Хотя раскрыта серия ингибиторов PARP для лечения опухолей, все еще существует потребность в разработке новых соединений с лучшими результатами эффективности и фармакокинетики. После продолжительных исследований авторы настоящего изобретения разработали серию соединений формулы (I) и обнаружили, что соединение, имеющее такую структуру, проявляет отличный эффект и функцию.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение было направлено на разработку производных фталазинонкетона формулы (I) и их таутомеров, энантиомеров, диастереомеров, рацематов и фармацевтически приемлемых солей, а также их метаболитов, метаболических предшественников или пролекарств:
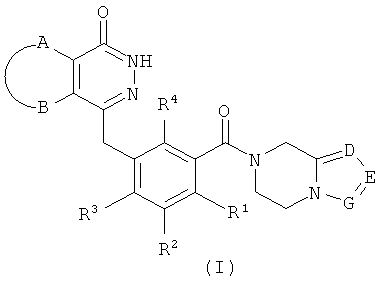
где:
А и В вместе с присоединенными атомами углерода образуют циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, гетероциклил, арил или гетероарил каждый независимо и необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, алкоксила, циклоалкила, гетероциклила, арила, гетероарила, -C(O)OR5, -OC(O)R5, -O(СН2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7;
R1, R2, R3 или R4 каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, циано и алкоксила, где алкил или алкоксил каждый независимо и необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидроксила, алкила и алкоксила;
D, Е или G каждый независимо выбран из группы, состоящей из азота и C(R8);
R5 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил или гетероарил каждый независимо и необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, алкоксила, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
R6 или R7 каждый независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где алкил, циклоалкил, гетероциклил, арил или гетероарил каждый независимо и необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, алкоксила, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
либо R6 и R7 вместе с присоединенным атомом N образуют гетероциклил, где гетероциклил содержит один или более чем один гетероатом N, О или S(O)m, и гетероциклил необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, алкоксила, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и алкоксикарбонила;
R8 выбран из группы, состоящей из водорода, алкила, галогена, гидроксила, циано, алкоксила, циклоалкила, гетероциклила, арила, гетероарила, бензила, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -(CH2)nNR6R7, -C(O)R5, -NHC(O)R5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7, где алкил, алкоксил, циклоалкил, гетероциклил, арил, гетероарил или бензил каждый независимо и необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из алкила, галогена, гидроксила, алкоксила, циклоалкила, гетероциклила, арила, гетероарила, оксо, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -C(O)NR6R7;
m выбрано из группы, состоящей из 0, 1 и 2; и
n выбрано из группы, состоящей из 0, 1 и 2.
Предпочтительной формой осуществления является соединение формулы (I) или его фармацевтически приемлемая соль, где А и В вместе с присоединенными атомами углерода образуют арил, где предпочтительно арил представляет собой фенил.
Предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль, где R1 представляет собой атом водорода.
Предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль, где R1 представляет собой атом галогена, предпочтительно атом фтора.
Предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль, где R1 представляет собой атом галогена, предпочтительно атом фтора.
Предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль, где R1, R2, R3 или R4 каждый независимо выбран из водорода.
Предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль, где R8 выбран из группы, состоящей из водорода, алкила, галогена, циано, -C(O)OR5, -(CH2)nNR6R7 и -C(O)NR6R7, где алкил необязательно замещен одним или более чем одним атомом галогена.
Предпочтительно соединение формулы (I) или его фармацевтически приемлемая соль, где R8 представляет собой трифторметил.
Соединение формулы (I) может содержать асимметрические атомы углерода, следовательно, оно может существовать в форме оптически чистого диастереомера, диастереомерной смеси, диастереомерного рацемата, смеси диастереомерного рацемата или в виде мезосоединения. Настоящее изобретение включает все эти формы. Диастереомерную смесь, диастереомерный рацемат или смесь диастереомерного рацемата можно выделить общепринятыми способами, такими как колоночная хроматография, тонкослойная хроматография и высокоэффективная жидкостная хроматография.
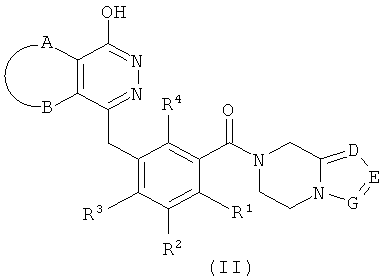
Эквивалент может быть понят обычным специалистом в данной области техники таким образом, что соединение формулы (I) может также иметь таутомеры. Таутомерные формы соединения (I) включают, но не ограничены, структуру представленную приведенной ниже формулой (II):
Соединения по изобретению включают, но не ограничены ими, приведенные ниже:
или их фармацевтически приемлемые соли.
Данное изобретение также относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, включающему стадии:
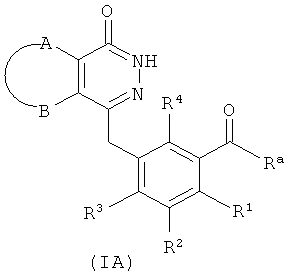
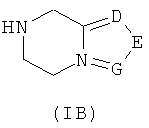
необязательного гидролиза соединения формулы (IA) до карбоновой кислоты, взаимодействия карбоновой кислоты с соединением формулы (IB) или его солью в присутствии конденсирующего агента, такого как бензотриазол-N,N,N',N'-тетраметилмочевины гексафторфосфат, в щелочных условиях с получением соединения формулы (I);
где:
R3 выбран из группы, состоящей из гидроксила, галогена и алкоксила;
А, В, D, Е, G и R1-R4 являются такими, как указано для формулы (I). В другом аспекте настоящее изобретение относится к применению соединений формулы (I) или их фармацевтически приемлемых солей для получения ингибиторов PARP.
В другом аспекте настоящее изобретение относится к способу ингибирования PARP, включающему введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединений формулы (I) или их фармацевтически приемлемых солей.
В другом аспекте настоящее изобретение относится к применению соединений формулы (I) или их фармацевтически приемлемых солей для получения адъюванта при лечении рака или лекарственного средства, вызывающего чувствительность опухолевых клеток к ионизирующему излучению или химиотерапии.
В другом аспекте настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям для применения в качестве адъюванта при лечении рака или лекарственного средства, вызывающего чувствительность опухолевых клеток к ионизирующему излучению или химиотерапии.
В другом аспекте настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям для применения в качестве ингибиторов PARP.
В другом аспекте настоящее изобретение относится к применению соединений формулы (I) или их фармацевтически приемлемых солей для получения лекарственного средства для лечения рака, где рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака поджелудочной железы, рака простаты, рака печени и рака толстой кишки, где лекарственное средство дополнительно вводят совместно с терапевтически эффективным количеством лекарственного средства, выбранного из группы, состоящей из темозоломида, адриамицина, таксола, цисплатина, карбоплатина, дакарбазина, топотекана, иринотекана, гемцитабина и бевацизумаба.
В другом аспекте настоящее изобретение относится к способам лечения рака, включающим введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединений формулы (I) или их фармацевтически приемлемых солей, где рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака поджелудочной железы, рака простаты, рака печени и рака толстой кишки, где лекарственное средство дополнительно вводят совместно с терапевтически эффективным количеством лекарственного средства, выбранного из группы, состоящей из темозоломида, адриамицина, таксола, цисплатина, карбоплатина, дакарбазина, топотекана, иринотекана, гемцитабина и бевацизумаба.
В другом аспекте настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям для применения в качестве лекарственного средства для лечения рака, где рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака поджелудочной железы, рака простаты, рака печени и рака толстой кишки, где лекарственное средство дополнительно вводят совместно с терапевтически эффективным количеством лекарственного средства, выбранного из группы, состоящей из темозоломида, адриамицина, таксола, цисплатина, карбоплатина, дакарбазина, топотекана, иринотекана, гемцитабина и бевацизумаба.
Кроме того, настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединений формулы (I) или их фармацевтически приемлемых солей по настоящему изобретению и фармацевтически приемлемый носитель или наполнитель. Настоящее изобретение также относится к данной фармацевтической композиции для применения в качестве ингибитора PARP, либо в качестве адъюванта при лечении рака или лекарственного средства, вызывающего чувствительность опухолевых клеток к ионизирующему излучению или химиотерапии, либо в качестве лекарственного средства для лечения рака. Настоящее изобретение также относится к применению данной фармацевтической композиции для получения ингибитора PARP. Настоящее изобретение также относится к применению данной фармацевтической композиции для получения адъюванта при лечении рака или лекарственного средства, вызывающего чувствительность опухолевых клеток к ионизирующему излучению или химиотерапии. Настоящее изобретение также относится к применению данной фармацевтической композиции для получения лекарственного средства для лечения рака, где рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака поджелудочной железы, рака простаты, рака печени и рака толстой кишки, где лекарственное средство дополнительно вводят совместно с терапевтически эффективным количеством лекарственного средства, выбранного из группы, состоящей из темозоломида, адриамицина, таксола, цисплатина, карбоплатина, дакарбазина, топотекана, иринотекана, гемцитабина и бевацизумаба.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
"Алкил" относится к насыщенной алифатической углеводородной группе, включающей С1-С20 прямоцепочечные и разветвленные группы. Предпочтительно алкильная группа представляет собой алкил, имеющий от 1 до 12 атомов углерода. Репрезентативные примеры включают, но не ограничены ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втop-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их изомеры разветвленной цепи. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода. Репрезентативные примеры включают, но не ограничены ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.д. Алкильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя могут быть замещены в любой доступной точке присоединения, предпочтительно группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, оксо, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -С(O)NR6R7.
"Циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе и имеет от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода. Репрезентативные примеры моноциклического циклоалкила включают, но не ограничены ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.д. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо и кольцо с внутренним мостиком.
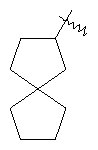
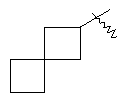
"Спиро-циклоалкил" относится к 5-20-членной полициклической группе с кольцами, соединенными через один общий атом углерода (называемый спиро-атомом), где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы. Предпочтительно спиро-циклоалкил является 6-14-членным, более предпочтительно является 7-10-членным. В соответствии с числом общих спиро-атомов спиро-циклоалкил делят на моно-спироциклическое кольцо, ди-спироциклическое кольцо или поли-спироциклическое кольцо, предпочтительно он относится к моно-спироциклическому кольцу или ди-спироциклическому кольцу. Более предпочтительно спиро-циклоалкил представляет собой 4-членное/4-членное, 4-членное/5-членное, 4-членное/6-членное, 5-членное/5-членное, or 5-членное/6-членное моноциклическое спиро-кольцо. Репрезентативные примеры спиро-циклоалкила включают, но не ограничены ими, приведенные ниже группы:
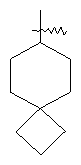
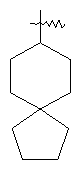
 и
и 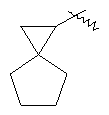 .
.
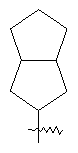
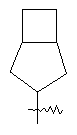
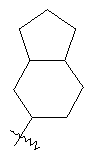
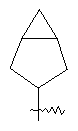
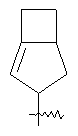
"Конденсированный циклоалкил" относится к 5-20-членной полициклической углеводородной группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы. Предпочтительно конденсированная циклоалкильная группа является 6-14-членной, более предпочтительно 7-10-членной. В соответствии с числом колец-членов конденсированный циклоалкил делят на конденсированный циклоалкил с бициклическим кольцом, трициклическим кольцом, тетрациклическим кольцом или пол и циклическим кольцом, предпочтительно он относится к конденсированному циклоалкилу с бициклическим кольцом или трициклическим кольцом. Более предпочтительно конденсированный циклоалкил представляет собой конденсированный циклоалкил с 5-членным/6-членным или 5-членным/6-членным бициклическим кольцом. Репрезентативные примеры конденсированного циклоалкила включают, но не ограничены ими, приведенные ниже группы:
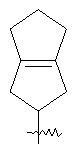
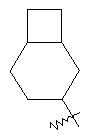 и
и 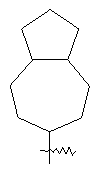 .
.
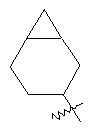
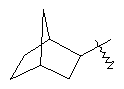
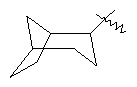
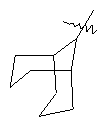
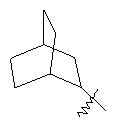
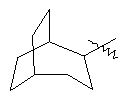
"Циклоалкил с внутренним мостиком" относится к 5-20-членной полициклической углеводородной группе, где каждые два кольца в системе имеют два общих разъединенных атома углерода. Эти кольца могут иметь одну или более чем одну двойную связь, но не имеют полностью конъюгированной пи-электронной системы. Предпочтительно циклоалкил с внутренним мостиком является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов циклоалкил с внутренним мостиком делят на бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо с внутренним мостиком, предпочтительно он относится к бициклическому, трициклическому или тетрациклическому циклоалкилу с внутренним мостиком, более предпочтительно относится к бициклическому или трициклическому циклоалкилу с внутренним мостиком. Репрезентативные примеры циклоалкила с внутренним мостиком включают, но не ограничены ими, приведенные ниже группы:
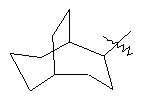
 и
и 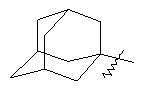 .
.
Циклоалкил может быть конденсирован с кольцом арила, гетероарила или гетероциклического алкила, где кольцом, соединенным с исходной структурой, является циклоалкил. Репрезентативные примеры циклоалкила с внутренним мостиком включают, но не ограничены ими, инданилуксусную кислоту, тетрагидронафталин, бензоциклогептил и т.д. Циклоалкил может быть необязательно замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляют собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, оксо, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -OC(O)NR6R7 и -C(O)NR6R7.
"Алкенил" относится к алкилу, определенному, как описано выше, который имеет по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную двойную связь, например, винил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенил и т.д. Алкенильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляют собой одну или более чем одну группу независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -С(O)NR6R7.
"Алкинил" относится к алкилу, определенному, как описано выше, который имеет по меньшей мере два атома углерода и по меньшей мере одну углерод-углеродную тройную связь, например, этинил, 1-пропинил, 2-пропинил, 1-, 2- или 3-бутинил и т.д. Алкинильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляют собой одну или более чем одну группу независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -C(O)NR6R7.
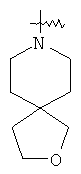
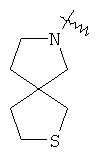
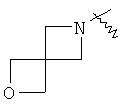
"Гетероциклил" относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более чем один гетероатом, выбранный из группы, состоящей из N, О или S(O)m (где m равно 0, 1 или 2), в качестве кольцевых атомов, но исключая -O-O-, -O-S- или -S-S-в кольце, где остальными кольцевыми атомами являются атомы С. Предпочтительно гетероциклил является 3-12-членным, имеющим от 1 до 4 гетероатомов, более предпочтительно 3-10-членным. Репрезентативные примеры моноциклического гетероциклила включают, но не ограничены ими, пирролидил, пиперидил, пиперазинил, морфолинил, сульфоморфолинил, гомопиперазинил и т.д. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо и кольцо с внутренним мостиком. "Спиро-гетероциклил" относится к 5-20-членному полициклическому гетероциклилу с кольцами, соединенными через один общий атом углерода (называемый спиро-атомом), где кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О или S(O)p (где p равно 0, 1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы. Предпочтительно спиро-гетероциклил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом общих спиро-атомов спиро-гетероциклил делят на моно-спиро-гетероциклил, ди-спиро-гетероциклил или поли-спиро-гетероциклил, предпочтительно он относится к моно-спиро-гетероциклилу и ди-спиро-гетероциклилу. Более предпочтительно спиро-гетероциклил представляет собой 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 6-членный/5-членный или 5-членный/6-членный моно-спиро-гетероциклил. Репрезентативные примеры спиро-гетероциклила включают, но не ограничены ими, приведенные ниже группы:
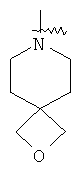
 и
и 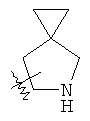 .
.
"Конденсированный гетероциклил" относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и где кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О или S(O)p (где p равно 0, 1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С. Предпочтительно конденсированный гетероциклический алкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов конденсированный гетероциклил делят на конденсированный гетероциклил с бициклическим кольцом, трициклическим кольцом, тетрациклическим кольцом или полициклическим кольцом, предпочтительно он относится к конденсированному гетероциклилу с бициклическим кольцом или трициклическим кольцом. Более предпочтительно конденсированный гетероциклил представляет собой конденсированный гетероциклил с 5-членным/5-членным или 5-членным/6-членным бициклическим кольцом. Репрезентативные примеры конденсированного гетероциклила включают, но не ограничены ими, приведенные ниже группы:
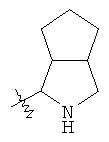
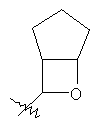
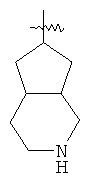
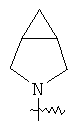
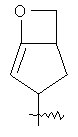
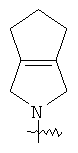
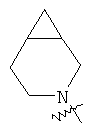
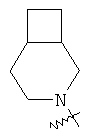
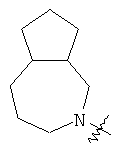
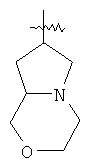
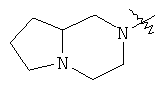 и
и 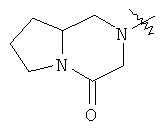 .
.
"Гетероциклил с внутренним мостиком" относится к 5-14-членной полициклической гетероциклической алкильной группе, где каждые два кольца в системе имеют два общих разъединенных атома углерода, кольца могут иметь одну или более чем одну двойную связь, но не имеют полностью конъюгированной пи-электронной системы, и кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О и S(O)m (где m равно 0, 1 или 2), в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С. Предпочтительно гетероциклил с внутренним мостиком является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов гетероциклил с внутренним мостиком делят на гетероциклил с внутренним мостиком с бициклическим кольцом, трициклическим кольцом, тетрациклическим кольцом или полициклическим кольцом, предпочтительно он относится к гетероциклилу с внутренним мостиком с бициклическим кольцом, трициклическим кольцом или тетрациклическим кольцом, более предпочтительно к гетероциклилу с внутренним мостиком с бициклическим кольцом или трициклическим кольцом. Репрезентативные примеры гетероциклила с внутренним мостиком включают, но не ограничены ими, приведенные ниже группы:
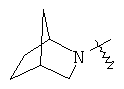
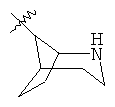
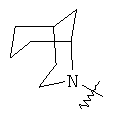
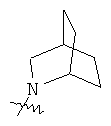
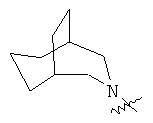
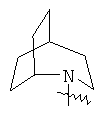
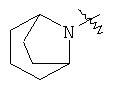 и
и  .
.
Кольцо гетероциклила может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцом, соединенным с исходной структурой, является гетероциклил. Репрезентативные примеры включают, но не ограничены ими, приведенные ниже группы:
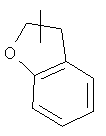
 и
и  , и т.д.
, и т.д.
Гетероциклил может быть необязательно замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляют собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, оксо, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -C(O)NR6R7.
"Арил" относится к 6-14-членной полностью углеродной моноциклической кольцевой или к полициклической конденсированной кольцевой ("конденсированная" кольцевая система означает, что каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом в системе) группе и имеет полностью конъюгированную пи-электронную систему. Предпочтительно арил является 6-10-членным, таким как фенил и нафтил. Арил может быть конденсирован с кольцом гетероарила, гетероциклила или циклоалкила, где кольцом, соединенным с исходной структурой, является арил. Репрезентативные примеры арила включают, но не ограничены ими, приведенные ниже группы:
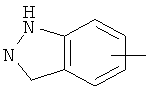
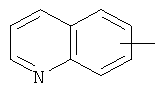
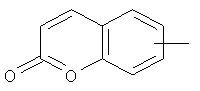 и
и 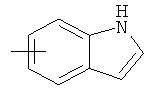 .
.
Арильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляют собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -C(O)NR6R7.
"Гетероарил" относится к гетероарильной системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N, в качестве кольцевых атомов и имеющей от 5 до 14 кольцевых атомов. Предпочтительно гетероарил является 5-10-членным. Более предпочтительно гетероарил является 5- или 6-членным. Примеры гетероарильных групп включают фурил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразолил и тому подобное. Гетероарил может быть конденсирован с кольцом арила, гетероциклила или циклоалкила, где кольцом, соединенным с исходной структурой, является гетероарил. Репрезентативные примеры гетероарила включают, но не ограничены ими, приведенные ниже группы:
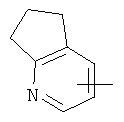
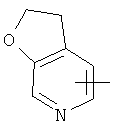
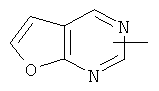
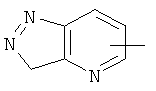
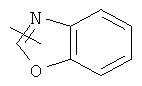
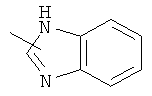
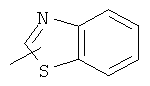
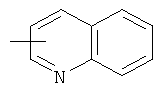 и
и  .
.
Гетероарильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляют собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -C(O)NR6R7.
"Алкоксил" относится как к группе -О-(алкил), так и к группе -O-(незамещенный циклоалкил), где алкил определен выше. Репрезентативные примеры включают, но не ограничены ими, метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и тому подобное. Алкоксил может быть необязательно замещенным или незамещенным. Когда он замещен, заместитель предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, -C(O)OR5, -OC(O)R5, -O(CH2)nC(O)OR5, -C(O)R5, -NHC(O)R5, -NR6R7, -ОС(O)NR6R7 и -C(O)NR6R7.
"Гидрокси" относится к группе -ОН.
"Атом галогена" относится к атому фтора, хлора, брома или йода.
"Амино" относится к группе -NH2.
"Циано" относится к группе -CN.
"Нитро" относится к группе -NO2.
"Бензил" относится к группе -СН2-(фенил).
"Оксо" относится к группе =O.
"Карбоксил" относится к группе -С(O)ОН.
"Алкоксикарбонил" относится к группе -С(O)O(алкил) или (циклоалкил), где алкил и циклоалкил определены выше.
"Необязательный" или "необязательно" означает, что описанное впоследствии событие или обстоятельство может произойти, но необязательно происходит, и что описание включает случаи, где событие или обстоятельство может произойти или не произойти. Например, "гетероциклическая группа, необязательно замещенная алкилом" означает, что алкильная группа может присутствовать, но необязательно присутствует, и что описание включает случай гетероциклической группы, замещенной алкилом, и гетероциклической группы, не замещенной алкилом.
"Замещенный" относится к одному или более чем одному атому водорода в группе, предпочтительно к атомам водорода в количестве вплоть до 5, более предпочтительно от 1 до 3, независимо замещенным соответствующим числом заместителей. Разумеется, заместители существуют в их любом возможном химическом положении. Специалист в данной области техники способен определить, возможно или невозможно замещение, не вкладывая избыточных усилий, с помощью эксперимента или теории. Например, комбинация аминогруппы или гидроксильной группы, имеющей свободный атом водорода, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые), может быть нестабильной.
"Фармацевтическая композиция" относится к смеси одного или более чем одного из соединений, описанных в настоящем изобретении, или их физиологически/фармацевтически приемлемых солей или пролекарств и других химических компонентов, таких как физиологически/фармацевтически приемлемые носители и наполнители. Целью фармацевтической композиции является облегчение введения соединения в организм, которое является благоприятным для всасывания активного ингредиента и, следовательно, проявления биологической активности.
m, n и R5-R7 являются такими, как определено в соединениях формулы (I).
СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Чтобы выполнить цель изобретения, в настоящем изобретении применено приведенное ниже техническое решение:
Способ получения соединений формулы (I) по изобретению или их фармацевтически приемлемых солей, включающий стадии:
необязательного гидролиза соединения формулы (IA) до карбоновой кислоты, затем взаимодействия карбоновой кислоты с соединением формулы (IB) или его солью в присутствии конденсирующего реагента, такого как бензотриазол-N,N,N',N'-тетраметилмочевины гексафторфосфат, в щелочных условиях с получением соединения формулы (I);
где:
Ra выбран из гидроксила, галогена или алкоксила;
А, В, D, Е, G и R1-R4 определены, как в формуле (I).
Вышеописанную реакцию конденсации проводят между кислотным соединением и аминным соединением в присутствии конденсирующего агента в основных условиях, где конденсирующий агент выбран из группы, состоящей из N,N'-дициклогексилкарбодиимида, N,N'-диизопропилкарбодиимида и O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (TBTU), предпочтительно O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторбората (TBTU); щелочные условия обеспечивают органическим или неорганическим основанием, где органическое основание выбрано из группы, состоящей из диизопропилэтиламина, пиридина, триэтиламина, гексагидропиридина, N-метилпиперазина, 4-диметиламинопиридина и т.д., предпочтительно диизопропилэтиламина; где используемый растворитель выбран из группы, состоящей из толуола, бензола, дихлорметана, тетрагидрофура на, хлороформа, N,N-диметилформамида или смеси вышеописанных растворителей, предпочтительно N,N-диметилформамида; температуру реакции контролируют от -80°С до 100°С, предпочтительно от 0°С до 60°С; время реакции обычно контролируют от 1 минуты до 72 часов, предпочтительно от 15 минут до 24 часов.
ПРЕДПОЧТИТЕЛЬНЫЕ ФОРМЫ ОСУЩЕСТВЛЕНИЯ
Приведенные ниже примеры служат для иллюстрации изобретения, но эти примеры не следует рассматривать как ограничивающие объем изобретения.
Примеры
Структуру соединения идентифицировали с помощью ЯМР (ядерного магнитного резонанса) и/или МС (масс-спектрометрии). Химические сдвиги ЯМР (δ) приведены в 10-6 (млн-1). ЯМР определяли с помощью прибора Bruker AVANCE-400. Растворители представляли собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта.
МС определяли с помощью масс-спектрометра FINNIGAN LCQAd (ИЭР, ионизация электрораспылением) (изготовитель: Thermo, тип: Finnigan LCQ advantage MAX).
ВЭЖХ (высокоэффективную жидкостную хроматографию) определяли на спектрометре жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire C18 150×4,6 мм) и на спектрометре жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150×4,6 мм).
IC50 определяли с помощью NovoStar ELIASA (BMG Co., German);
В качестве тонкослойного силикагеля использовали пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин, используемых в ТСХ, составлял от 0,15 мм до 0,2 мм, а размер пластин, используемых для очистки продукта тонкослойной хроматографией, составлял от 0,4 мм до 0,5 мм.
При колоночной хроматографии в качестве носителя, как правило, использовали силикагель Yantai Huanghai с размером ячеек от 200 до 300 меш.
Известный исходный материал по изобретению может быть получен общепринятым способом синтеза в данной области техники или приобретен у фирмы ABCR GmbH & Со. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc или Dari Chemical Company и т.д.
Если в примерах не указано иное, нижеописанные реакционные смеси помещали в атмосферу аргона или в атмосферу азота.
Термин "атмосфера аргона" или "атмосфера азота" относится к тому, что реакционная колба оборудована баллоном, содержащим 1 л аргона или азота.
При реакциях гидрогенизации реакционную систему обычно помещали в вакуум и заполняли водородом, и вышеуказанную операцию повторяли три раза.
Микроволновые реакции проводили с помощью микроволнового реактора СЕМ Discover-S 908860.
Если в примерах не указано иное, раствор, используемый в нижеописанных реакциях, относится к водному раствору.
Если в примерах не указано иное, температура, используемая в нижеописанных реакциях, составляет комнатную температуру.
Комнатная температура была наиболее точной температурой реакции, и составляла от 20°С до 30°С.
За ходом реакций следили с помощью тонкослойной хроматографии (ТСХ), хроматографическая система растворителей включала: А: систему дихлорметана и метанола, В: систему н-гексана и этилацетата, С: систему петролейного эфира и этилацетата, D: ацетон. Соотношение объемов растворителей регулировали в соответствии с полярностью соединений.
Система элюирования очистки соединений колоночной хроматографией итонкослойной хроматографией включала: А: систему дихлорметана и метанола, В: систему н-гексана и этилацетата, где соотношение объемов растворителей регулировали в соответствии с полярностью соединений, и иногда также добавляли небольшое количество щелочного реагента, такого как триэтиламин, или кислотного реагента, такого как уксусная кислота.
Пример 1
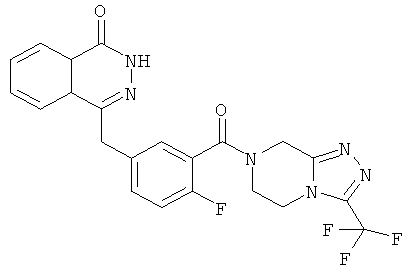
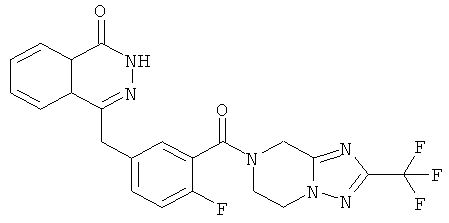
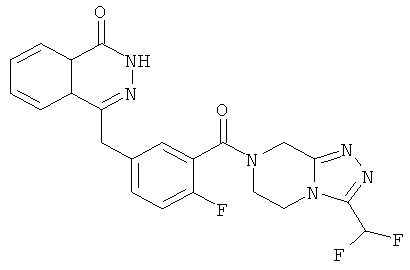
4-[[4-Фтор-3-[3-(трифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
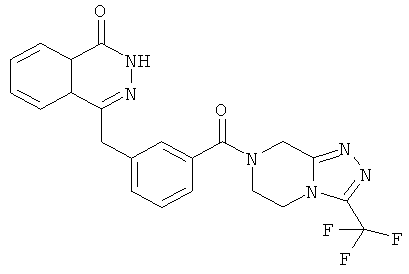
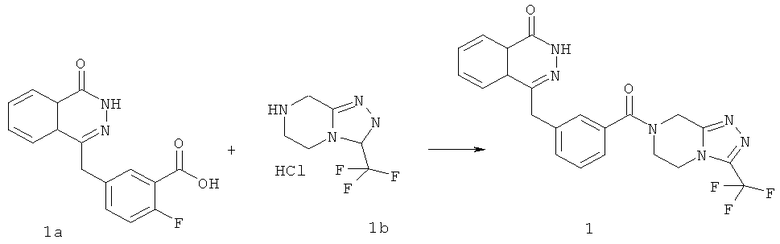
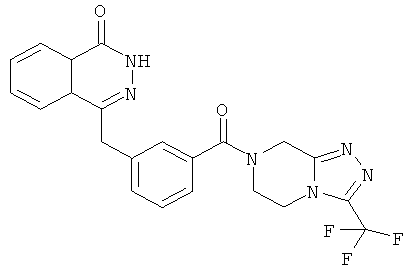
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (150 мг, 0,50 ммоль, полученную известным способом, раскрытым "заявкой на патент WO 2004080976") растворяли в 2 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (284 мг, 0,75 ммоль), 3-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразина гидрохлорида 1b (138 мг, 0,60 ммоль, полученного известным способом, раскрытым "заявкой на патент WO 2004080958") и N,N-диизопропилэтиламина (0,2 мл, 1 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[4-фтор-3-[3-(трифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 1 (25 мг, выход 10,6%) в виде белого твердого вещества.
MC m/z (ИЭР):473,2 [М+1]
1Н ЯМР (400 МГц, CDCl3):δ 10.04 (br. s, 1Н), 8.48 (d, 1H), 7.80 (m, 3H), 7.55 (m, 1H), 7.40 (m, 1H), 7.15 (m, 1H), 4.29 (s, 2H), 4.23 (m, 2H), 3.74 (m, 2H), 3.20 (m, 2H)
Пример 2
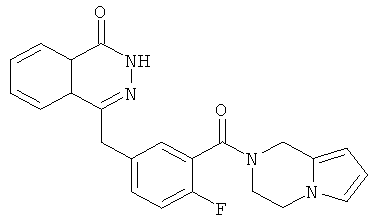
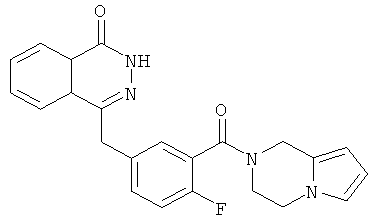
4-[[3-(3,4-Дигидро-1Н-пирроло[1,2-а]пиразин-2-карбонил)-4-фтор-фенил]метил]-2Н-фталазин-1-он
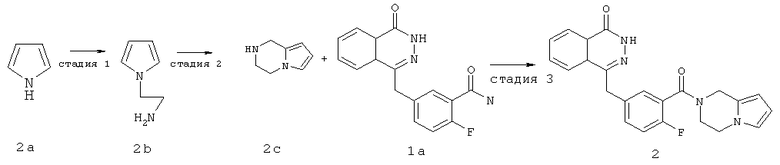
Стадия 1
2-пиррол-1-ил-этанамин
Пиррол 2а (12 г, 17,90 ммоль) растворяли в 150 мл ацетонитрила с последующим добавлением 2-хлорэтиламина гидрохлорида (24,60 г, 21,20 ммоль), гидроксида натрия (0,50 г, 4 ммоль) и тетрабутиламмония гидросульфата (2,40 г, 7 ммоль). После перемешивания в течение 4 часов в условиях кипячения с обратным холодильником реакционную смесь нагревали до 50°С и подвергали взаимодействию в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении с получением 2-пиррол-1-ил-этанамина 2b (8 г, выход 41,0%) в виде светло-желтого масла.
Стадия 2
1,2,3,4-тетрагидропирроло[1,2-а]пиразин
2-пиррол-1-ил-этанамин 2b (2 г, 18 ммоль) растворяли в 40 мл этанола с последующим добавлением раствора формальдегида (40%, 1,5 мл, 18 ммоль) и медленным добавлением по каплям 1 мл трифторуксусной кислоты. Реакционную смесь нагревали до 50°С в течение 15 минут, затем охлаждали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, добавляли к ней 50 мл этилацетата, промывали насыщенным раствором бикарбоната натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 1,2,3,4-тетрагидропирроло[1,2-а]пиразина 2c (1,60 г, выход 72,7%) в виде светло-желтого масла.
Стадия 3
4-[[3-(3,4-дигидро-1Н-пирроло[1,2-а]пиразин-2-карбонил)-4-фтор-фенил]метил]-2Н-фталазин-1-он
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1a (300 мг, 1 ммоль) растворяли в 3 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (568 мг, 1,50 ммоль), 1,2,3,4-тетрагидропирроло[1,2-а]пиразина 2c (210 мг, 1,50 ммоль) и N,N-диизопропилэтиламина (350 мкл, 2 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[3-(3,4-дигидро-1Н-пирроло[1,2-а]пиразин-2-карбонил)-4-фтор-фенил]метил]-2H-фталазин-1-она 2 (15 мг, выход 3,7%) в виде белого твердого вещества.
МС m/z (ИЭР):403,1 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.19 (br. s, 1H), 8.51 (d, 1H), 7.82 (m, 3Н), 7.41 (m, 2Н), 7.13 (m, 1H), 6.65 (m, 1H), 6.24 (m, 1H), 5.81 (m, 1H), 4.97 (s, 1H), 4.59 (s, 1H), 4.33 (s, 2Н), 4.13 (m, 1Н),4.00(m, 1H), 3.71 (m, 1Н), 2.85(m, 1H)
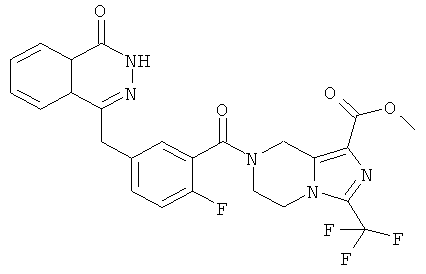
Пример 3
Метил-7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксилат
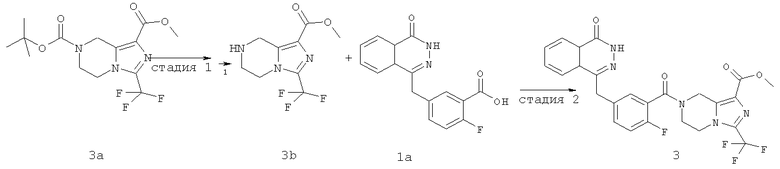
Стадия 1
Метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат
O7-трет-бутил-O1-метил-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1,7-дикарбоксилат 3а (600 мг, 1,72 ммоль, полученный известным способом, раскрытым "заявкой на патент WO 2009082881") растворяли в 20 мл раствора хлорида водорода в 1,4-диоксане (2 М). После перемешивания в течение 5 часов реакционную смесь концентрировали при пониженном давлении и добавляли к ней 50 мл дихлорметана. К реакционной смеси добавляли по каплям насыщенный раствор бикарбоната натрия до pH 8. Органическую фазу отделяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 3b (430 мг) в виде белого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 2
Метил-7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксилат
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1a (300 мг, 1 ммоль) растворяли в 2 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (568 мг, 1,50 ммоль), сырого метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 3b (300 мг, 1,50 ммоль) и N,N-диизопропилэтиламина (0,4 мл, 2 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением метил-7-[2-фтор-5-[(4-оксо-ЗН-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксилата 3 (120 мг, выход 23,0%) в виде светло-желтого твердого вещества.
МС m/z(ИЭР):530,1 [М+1]
1Н ЯМР (400 МГц, CDCl3):δ 10.48 (br. s, 1Н), 8.52 (d, 1H), 7.87 (m, 3Н), 7.43 (m, 2Н), 7.30 (m, 1Н), 5.02 (m, 2Н), 4.34 (s, 2h), 4.17 (m, 2Н), 3.99 (m, 2Н), 3.00 (s, 3Н)
Пример 4
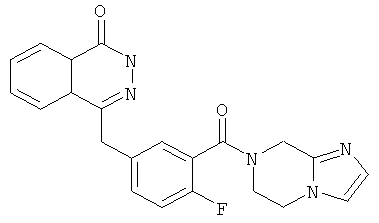
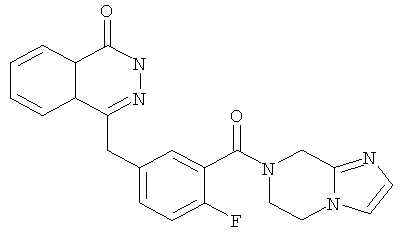
4-[[3-(6,8-дигидро-5Н-имидазо[1,2-а]пиразин-7-карбонил)-4-фтор-фенил]метил]-2Н-фталазин-1-он
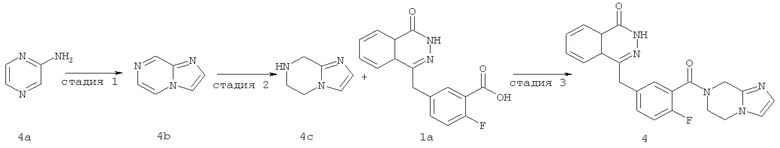
Стадия 1
имидазо[1,2-а]пиразин
(5 г, 52 ммоль) растворяли в 40% растворе 2-хлорацетальдегида (15 мл, 78 ммоль) с последующим добавлением бикарбоната натрия (6,60 г, 78 ммоль). После перемешивания в течение 48 часов при 100°С реакционную смесь охлаждали до комнатной температуры, добавляли к ней 100 мл насыщенного раствора карбоната калия и экстрагировали дихлорметаном (100 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением имидазо[1,2-а]пиразина 4b (3 г, выход 50,0%) в виде коричневого твердого вещества.
MC m/z (ИЭР):120,1 [М+1]
Стадия 2
5,6,7,8-тетрагидроимидазо[1,2-а]пиразин
Имидазо[1,2-а]пиразин 4b (500 мг, 4,20 ммоль) растворяли в 5 мл 2-метоксиэтанола с последующим добавлением диоксида платины (100 мг, 0,36 ммоль), и реактор продували водородом три раза. После перемешивания в течение 12 часов реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением 5,6,7,8-тетрагидроимидазо[1,2-а]пиразина 4с (200 мг, выход 38,7%) в виде желтого масла.
МС m/z (ИЭР):124,1 [М+1]
Стадия 3
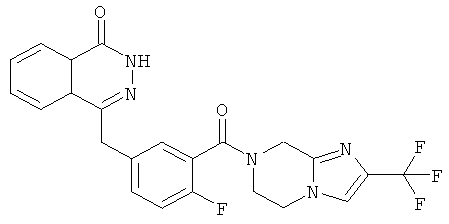
4-[[3-(6,8-дигидро-5H-имидазо[1,2-а]пиразин-7-карбонил)-4-фтор-фенил]метил]-2H-фталазин-1-он
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (323 мг, 1,08 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (614 мг, 1,63 ммоль), 5,6,7,8-тетрагидроимидазо[1,2-а]пиразина 4с (200 мг, 1,63 ммоль) и N,N-диизопропилэтиламина (0,4 мл, 2,16 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[3-(6,8-дигидро-5H-имидазо[1,2-а]пиразин-7-карбонил)-4-фтор-фенил]метил]-2H-фталазин-1-она 4 (10 мг, выход 2,3%) в виде белого твердого вещества.
МС m/z (ИЭР):404.1 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.07 (br. s, 1H), 8.53 (d, 1H), 7.96 (m, 1Н), 7.83 (m, 3Н), 7.51 (m, 1H), 7.30 (m, 2Н), 6.01 (t, 1H), 4.73 (d, 2H), 4.35 (s, 2H), 1.60 (m, 2Н), 1.34 (m, 2Н)
Пример 5
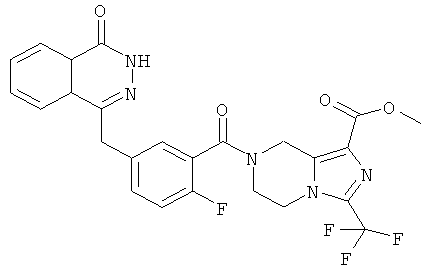
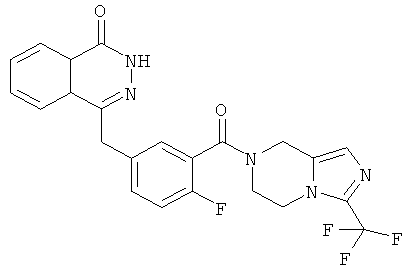
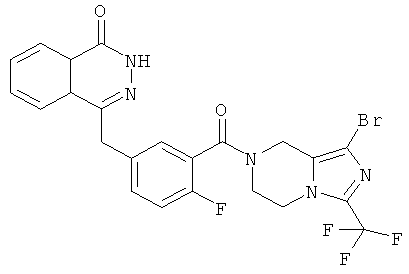
4-[[4-фтор-3-[3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2Н-фталазин-1-он
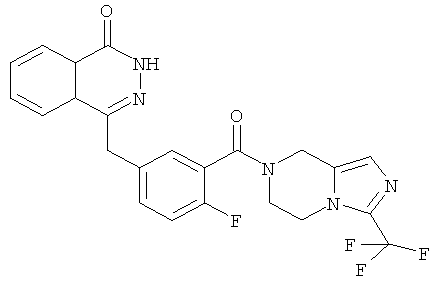
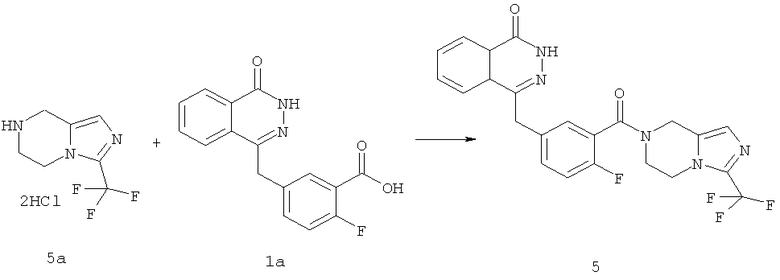
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (500 мг, 1,68 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (955 мг, 2,52 ммоль), 3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразина гидрохлорида 5а (457 мг, 2 ммоль, полученного известным способом, раскрытым "заявкой на патент WO 2009082881"), и N,N-диизопропилэтиламина (0,6 мл, 3,36 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[4-фтор-3-[3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 5 (400 мг, выход 50,5%) в виде белого твердого вещества.
МС m/z (ИЭР):472,1 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.81 (br. s, 1H), 8.49 (m, 1H), 7.79 (m, 3H), 7.42 (m, 2H), 7.08 (m, 1H),5.00(m, 1H),4.64(m, 1H), 4.32 (m, 2H), 4.16 (m, 3H), 3.75 (m, 1H),3.49(s, 1H)
Пример 6
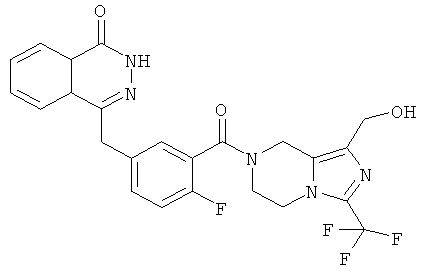
4-[[4-фтор-3-[1-(гидроксиметил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
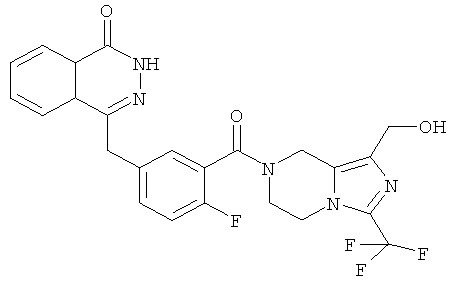

Стадия 1
[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанол
Метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 3b (315 мг, 1,26 ммоль) растворяли в 10 мл этанола с последующим добавлением боргидрида натрия (240 мг, 6,33 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли по каплям 2 М соляную кислоту до тех пор, пока в реакционной смеси больше не образовывался газ. Реакционную смесь концентрировали при пониженном давлении с получением сырого [3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанола 6a (230 мг) в виде белого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 2
4-[[4-фтор-3-[1-(гидроксиметил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (372 мг, 1,25 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением N-гидроксибензотриазола (85 мг, 0,63 ммоль), [3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанола 6а (277 мг, 1,25 ммоль), 1-этил-(3-диметил-аминопропил)карбодиимида гидрохлорида (359 мг, 1,88 ммоль) и триэтиламина (0,3 мл, 2,5 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением
4-[[4-фтор-3-[1-(гидроксиметил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 6 (400 мг, выход 64,0%) в виде белого твердого вещества.
МС m/z(ИЭР):502,2 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.81 (br. s, 1H), 8.47 (s, 1H), 7.83-7.75 (m, 3Н), 7.42-7.36 (m, 2Н), 7.14-7.12 (m, 1H), 5.31 (s, 1H), 5.04 (s, 1H), 4.69 (d, 1H), 4.50 (s, 1H), 4.32-4.25 (m, 4Н), 4.16-4.10 (m, 1H), 2.05 (s, 1H)
Пример 7
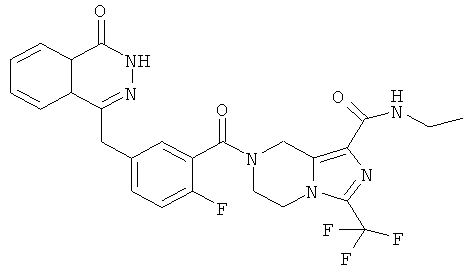
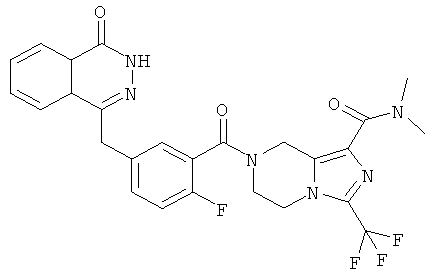
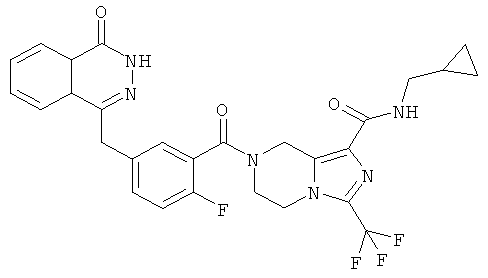
N-этил-7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамид
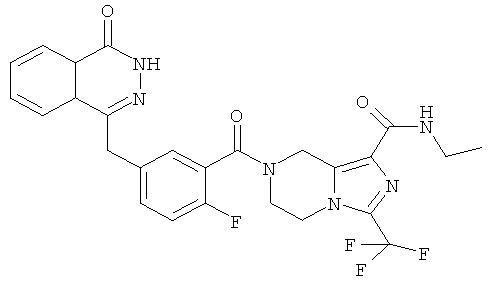
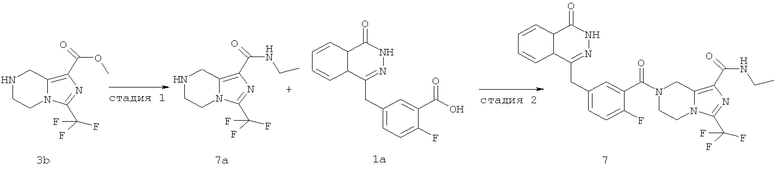
Стадия 1
N-этил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамид
Метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 3b (1 г, 4 ммоль) растворяли в 40 мл раствора этиламина (60%). После перемешивания при 50°С в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением сырого N-этил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 7а (1,15 г) в виде белого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):263,1 [М+1]
Стадия 2
N-этил-7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксамид
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1а (250 мг, 0,84 ммоль) растворяли в 20 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (480 мг, 1,26 ммоль), сырого N-этил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 7а (242 мг, 0,92 ммоль) и N,N-диизопропилэтиламина (0,3 мл, 1,68 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли 50 мл H2O и экстрагировали дихлорметаном (50 мл×3). Органическую фазу объединяли, концентрировали при пониженном давлении, добавляли к ней 100 мл этилацетата, последовательно промывали насыщенным раствором бикарбоната натрия (40 мл), насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением N-этил-7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксамида 7 (200 мг, выход 43,9%) в виде белого твердого вещества.
МС m/z (ИЭР):543,2 [М+1]
1Н ЯМР (400 МГц, CDCl3):δ 11.38 (br. s, 1 H), 8.47 (m, 1 Н), 7.84 (m, 3Н), 7.37 (m, 2Н), 7.19 (m, 1Н), 5.10 (s, 2Н), 4.30 (s, 2Н), 4.29 (m, 4Н), 3.47 (m, 2Н), 1.27 (m, 3Н)
Пример 8
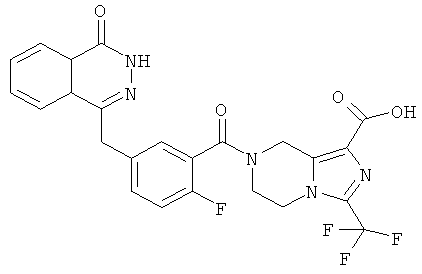
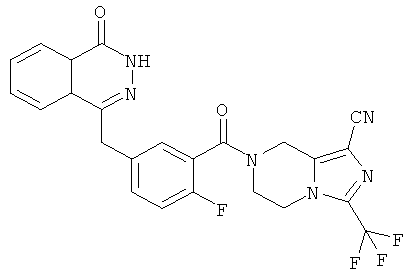
7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоновая кислота
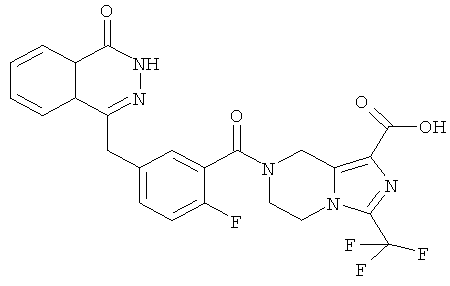
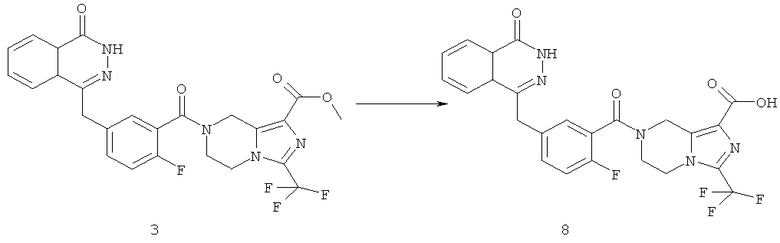
Метил-7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксилат 3 (30 мг, 0,057 ммоль) растворяли в 1,5 мл смешанного растворителя из тетрагидрофурана, метанола и воды (об./об./об.=1:1:1) с последующим добавлением гидроксида натрия (10 мг, 0,25 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли по каплям концентрированную соляную кислоту до pH 2. Реакционную смесь экстрагировали дихлорметаном (15 мл×2). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоновой кислоты 8 (10 мг, выход 34,4%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):516.5[М+1]
1Н ЯМР (400 МГц, CD3OD):δ 8.36 (d, 1Н), 7.93 (d, 1Н), 7.83 (m, 2Н), 7.60 (d, 1H), 7.29 (m, 1Н), 6.97 (t, 1Н), 4.32 (s, 2Н), 3.41 (m, 6Н)
Пример 9
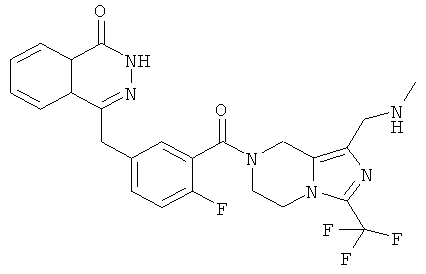
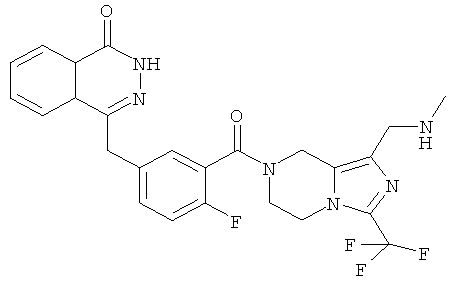
4-[[4-фтор-3-[1-(метиламинометил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2Н-фталазин-1-он
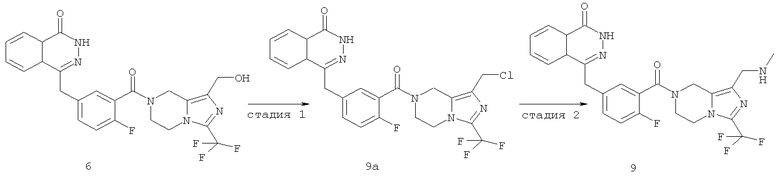
Стадия 1 4-[[3-[1-(хлорметил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2H-фталазин-1-он
4-[[4-Фтор-3-[1-(гидроксиметил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2Н-фталазин-1-он 6 (200 мг, 0,40 ммоль) растворяли в 5 мл тионилхлорида. Реакционную смесь нагревали до образования флегмы в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении, добавляли к ней 10 мл Н2О, экстрагировали дихлорметаном (10 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 4-[[3-[1-(хлорметил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2Н-фталазин-1-она 9а (200 мг, выход 96,6%) в виде желтого твердого вещества.
МС m/z (ИЭР):520,1 [М+1]
Стадия 2
4-[[4-фтор-3-[1-(метиламинометил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
4-[[3-[1-(Хлорметил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2H-фталазин-1-он 9а (372 мг, 1,25 ммоль) растворяли в 5 мл ацетонитрила с последующим добавлением 0,6 мл 2 М раствора метиламина в тетрагидрофуране и карбоната калия (159 мг, 1,15 ммоль). Реакционную смесь нагревали до образования флегмы в течение 6 часов. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении и очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[4-фтор-3-[1-(метиламинометил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2Н-фталазин-1-она 9 (20 мг, выход 10,1%) в виде желтого твердого вещества.
МС m/z (ИЭР):515,2 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 11.87 (br. s, 1Н), 8.35-8.42 (m, 1Н), 7.72-7.81 (m, 3Н), 7.35-7.43 (m, 1Н), 6.96-7.06 (m, 1Н), 5.01-5.02 (m, 1Н), 3.99-4.28 (m, 6Н), 3.71-3.72 (m, 1Н), 3.47 (s, 1 H), 2.74 (d, 3H), 2.03-2.05 (m, 1 H)
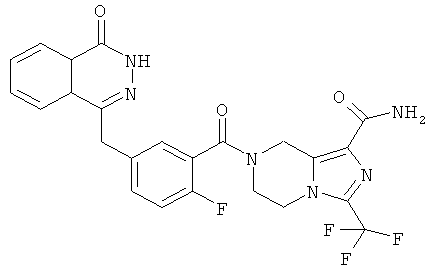
Пример 10
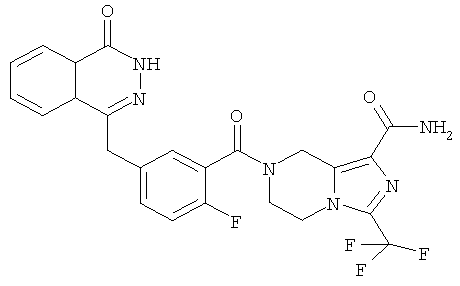
7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамид
Стадия 1
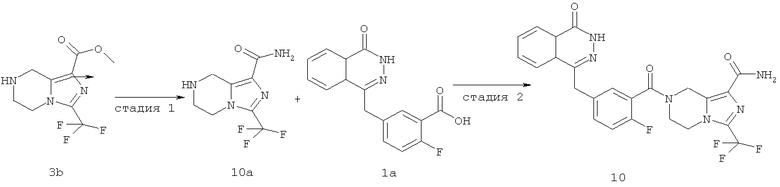
3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамид
Метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 3b (250 мг, 1 ммоль) и 10 мл гидроксида аммония добавляли в 20 мл герметичную пробирку. Реакционную смесь нагревали до 100°С и подвергали взаимодействию в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении с получением сырого 3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 10а (240 мг) в виде белого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):235,1 [М+1]
Стадия 2
7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамид
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (150 мг, 0,50 ммоль) растворяли в 10 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (285 мг, 0,75 ммоль), сырого 3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 10а (130 мг, 0,55 ммоль) и N,N-диизопропилэтиламина (0,2 мл, 1 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли 50 мл Н2О, экстрагировали дихлорметаном (60 мл×3). Органическую фазу объединяли, концентрировали при пониженном давлении, добавляли к ней 100 мл этилацетата, последовательно промывали Н2О (40 мл) и насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксамида 10 (50 мг, выход 20,0%) в виде белого твердого вещества.
МС m/z (ИЭР):515,1 [М+1]
1H ЯМР(400 МГц, CDCl3):δ 8.49 (m, 1Н), 7.85 (m, 3Н), 7.33 (m, 2Н), 7.15 (m, 1Н), 5.07 (s, 2Н), 4.30 (s, 2Н), 4.23 (m, 4Н)
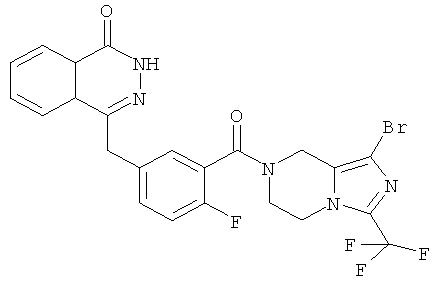
Пример 11
4-[[3-[1-бром-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2H-фталазин-1-он
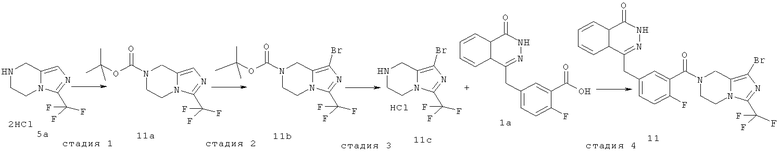
трет-Бутил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат
3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин гидрохлорид 5а (2,20 г, 8,30 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением триэтиламина (4,6 мл, 33,20 ммоль) и ди-трет-бутилдикарбоната (2,70 г, 12,50 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли 50 мл H2O, экстрагировали дихлорметаном (50 мл×3). Органическую фазу объединяли, последовательно промывали насыщенным раствором хлорида аммония (40 мл) и насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением трет-бутил-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбоксилата 11а (2,20 г, выход 91,7%) в виде светло-коричневого твердого вещества.
МС m/z (ИЭР):292,1 [М+1]
Стадия 2
трет-Бутил-1-бром-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат
трет-Бутил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат 11а (370 мг, 1,27 ммоль) растворяли в 30 мл тетрагидрофурана с последующим добавлением N-бромсукцинимида (453 мг, 2,54 ммоль) при -78°С. После перемешивания в течение 1 часа реакционную смесь нагревали до комнатной температуры и подвергали взаимодействию в течение 12 часов. К реакционной смеси добавляли 50 мл H2O, экстрагировали этилацетатом (60 мл×3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого трет-бутил-1-бром-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилата 11b (510 мг) в виде светло-желтого масла. Продукт использовали непосредственно в следующей реакции без очистки.
MC m/z (ИЭР):372,0 [М+1]
Стадия 3
1-бром-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразина гидрохлорид
Сырой трет-бутил-1-бром-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат 11b (470 мг, 1,27 ммоль) растворяли в 50 мл 2 М раствора хлорида водорода в 1,4-диоксане. После перемешивания в течение 4 часов реакционную смесь концентрировали при пониженном давлении с получением 1-бром-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразина гидрохлорида 11с (220 мг, выход 56,5%) в виде светло-желтого масла.
Стадия 4
4-[[3-[1-бром-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2H-фталазин-1-он
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1а (210 мг, 0,70 ммоль) растворяли в 30 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (360 мг, 0,95 ммоль), 1-бром-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразина гидрохлорида 11с (214 мг, 0,70 ммоль) и N,N-диизопропилэтиламина (0,4 мл, 2,10 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли 50 мл Н2О, экстрагировали дихлорметаном (80 мл×3). Органическую фазу объединяли, концентрировали при пониженном давлении, добавляли к ней 100 мл этилацетата, последовательно промывали насыщенным раствором карбоната натрия (40 мл), Н2О (40 мл) и насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[3-[1-бром-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2H-фталазин-1-она 11 (185 мг, выход 48,0%) в виде белого твердого вещества.
МС m/z (ИЭР):552,0 [М+1]
1H ЯМР(400 МГц, CDCl3):δ 8.48 (m, 1H), 7.73 (m, 3H), 7.31 (m, 2Н), 7.11 (m, 1Н), 4.89 (s, 2Н), 4.49 (s, 2Н), 4.48 (m, 4Н)
Пример 12
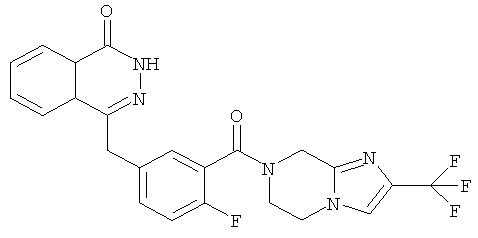
4-[[4-фтор-3-[2-(трифторметил)-6,8-дигидро-5H-имидазо[1,2-а]пиразин-7-карбонил]фенил] метил]-2H-фталазин-1-он
Стадия 1
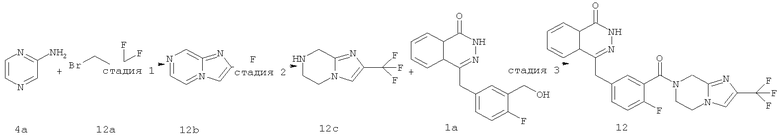
2-(трифторметил)имидазо[1,2-а]пиразин
Пиразин-2-амин 4а (5,25 г, 55,20 ммоль) растворяли в 120 мл этанола с последующим добавлением 3-бром-1,1,1-трифтор-пропан-2-она 12a (5,7 мл, 55,20 ммоль). Реакционную смесь нагревали до образования флегмы в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении, добавляли к ней 100 мл этилацетата и 100 мл насыщенного раствора бикарбоната натрия и отделяли. Водную фазу экстрагировали этилацетатом (50 мл×3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования В с получением 2-(трифторметил)имидазо[1,2-а]пиразина 12b (2,40 г, выход 22,8%) в виде желтого твердого вещества.
МС m/z (ИЭР):188,0 [М+1]
Стадия 2
2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин
2-(Трифторметил)имидазо[1,2-а]пиразин 12b (2,40 г, 12,55 ммоль) растворяли в 100 мл метанола с последующим добавлением Pd-C (10%, 480 мг), и реактор продували водородом три раза. После перемешивания в течение 12 часов реакционную смесь фильтровали, и фильтрационный кек промывали метанолои. Фильтрат концентрировали при пониженном давлении с получением 2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина 12с (2,30 г, выход 95,8%) в виде желтого масла.
Стадия 3
4-[[4-фтор-3-[2-(трифторметил)-6,8-дигидро-5H-имидазо[1,2-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (500 мг, 1,68 ммоль) растворяли в 10 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (830 мг, 2,52 ммоль), 2-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,2-а]пиразина 12с (384 мг, 2 ммоль) и N,N-диизопропилэтиламина (1 мл, 5 ммоль). После перемешивания в течение 12 часов полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением 4-[[4-фтор-3-[2-(трифторметил)-6,8-дигидро-5H-имидазо[1,2-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 12 (200 мг, выход 25,0%) в виде белого твердого вещества.
МС m/z (ИЭР):472,1 [М+1]
1Н ЯМР (400 МГц, CDCl3):δ 10.29 (br. s, 1Н), 8.47 (m, 1Н), 7.80 (m, 3Н), 7.37 (m, 2Н), 7.25 (m, 1Н), 6.50 (m, 1Н), 4.67 (s, 2Н), 4.28 (m, 2Н), 4.14 (m, 2Н), 3.73 (m, 2Н)
Пример 13
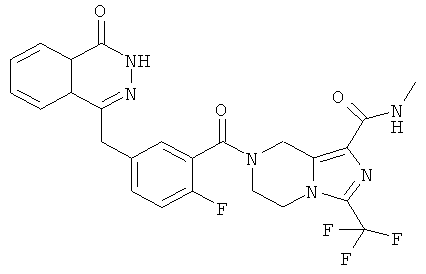
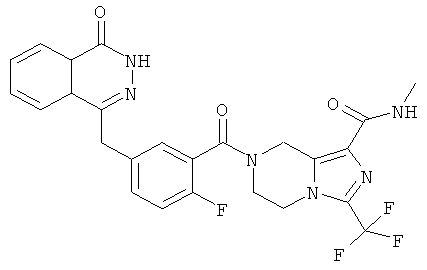
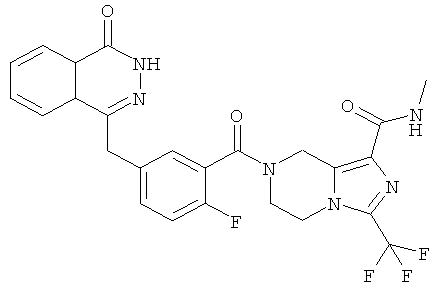
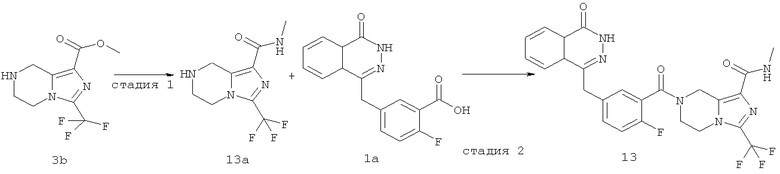
7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-N-метил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамид
Стадия 1
N-метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-a]пиразин-1-карбоксамид
Метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 3b (500 мг, 2 ммоль), растворенный в 8 мл раствора метиламина (от 20% до 30%), добавляли в 20 мл герметичную пробирку. После перемешивания при 60°С в течение 6 часов реакционную смесь концентрировали при пониженном давлении с получением сырого
N-метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 13а (498 мг) в виде белого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):249,1 [М+1]
Стадия 2
7-[2-сртор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-N-метил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамид
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (598 мг, 2 ммоль) растворяли в 10 мл N,N-диметилформамида с последующим добавлением 1-гидроксибензотриазола (135 мг, 1 ммоль), сырого N-метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 13а (498 мг, 2 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорида (573 мг, 3 ммоль) и N,N-диизопропилэтиламина (774 мг, 6 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, добавляли к ней 30 мл H2O, экстрагировали этилацетатом (50 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-N-метил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамида 13 (650 мг, выход 61,0%) в виде белого твердого вещества.
МС m/z (ИЭР):529,1 [М+1]
1H ЯМР (400 МГц, CD3OD):δ 8.36-8.34 (t, 1H), 7.96-7.94 (d, 1H), 7.86-7.81 (m, 2Н), 7.50-7.45 (m, 2Н), 7.22-7.15 (dd, 1H), 5.23 (s, 1H), 4.95 (s, 1H), 4.39 (d, 2Н), 4.32 (d, 1H), 4.21 (s, 1H), 4.14 (s, 1H), 3.76 (s, 1H), 2.85 (d, 3H)
Пример 14
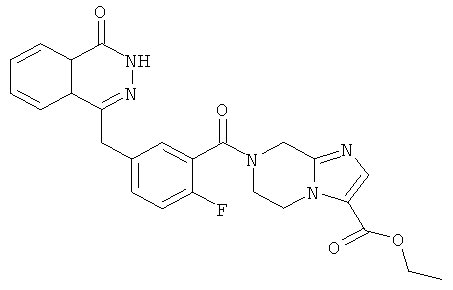
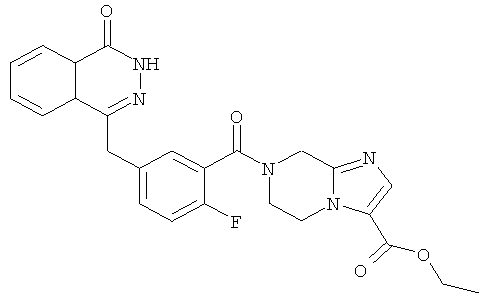
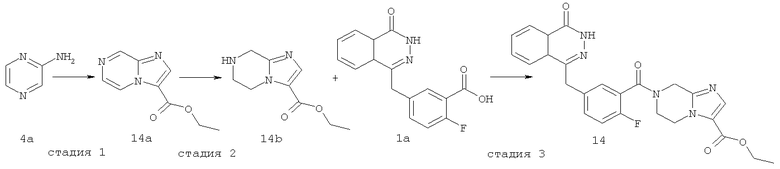
Этил-7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-6,8-дигидро-5H-имидазо[1,2-а]пиразин-3-карбоксилат
Стадия 1
Этил-имидазо[1,2-а]пиразин-3-карбоксилат
Пиразин-2-амин 4а (1 г, 10 ммоль) растворяли в 50 мл этиленгликоля диметилового эфира с последующим добавлением 50 мл метанола и 3-бром-2-оксо-пропионата (2,30 г, 12 ммоль). После перемешивания в течение 4 часов при комнатной температуре реакционную смесь охлаждали до 0°С и перемешивали в течение 30 минут до осаждения твердого вещества. Реакционную смесь фильтровали, и фильтрационный кек промывали эфиром (10 мл×3). Твердое вещество растворяли в 50 мл безводного этанола, и раствор кипятили с обратным холодильником в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении, добавляли к ней 100 мл дихлорметана, последовательно промывали насыщенным раствором карбоната натрия (40 мл) и насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением этил-имидазо[1,2-а]пиразин-3-карбоксилата 14а (0,55 г, выход 28,9%) в виде коричневого твердого вещества.
МС m/z (ИЭР):192,1 [М+1]
Стадия 2
Этил-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-3-карбоксилат
Этил-имидазо[1,2-а]пиразин-3-карбоксилат 14а (550 мг, 2,76 ммоль) растворяли в 30 мл метанола с последующим добавлением Pd-C (10%, 100 мг), и реактор продували водородом три раза. После перемешивания в течение 3 часов реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением этил-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-3-карбоксилата 14b (480 мг, выход 87,6%) в виде желтого масла.
МС m/z (ИЭР):196,1 [М+1]
Стадия 3
Этил-7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-6,8-дигидро-5H-имидазо[1,2-а]пиразин-3-карбоксилат
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1а (300 мг, 1 ммоль) растворяли в 20 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (570 мг, 1,50 ммоль), этил-5,6,7,8-тетрагидроимидазо[1,2-а]пиразин-3-карбоксилата 14b (200 мг, 1 ммоль) и N,N-диизопропилэтиламина (0,3 мл, 2 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли 50 мл Н2О, экстрагировали дихлорметаном (80 мл×3). Органическую фазу объединяли, концентрировали при пониженном давлении, добавляли к ней 100 мл этилацетата, последовательно промывали насыщенным раствором карбоната натрия (40 мл), H2O (40 мл), насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением этил-7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-6,8-дигидро-5H-имидазо[1,2-а]пиразин-3-карбоксилата 14 (280 мг, выход 58,6%) в виде белого твердого вещества.
MC m/z(ИЭР):476,1 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.53 (br. s, 1Н), 8.46 (m, 1H), 7.76 (m, 3H), 7.59 (s, 1H), 7.36 (m, 2H), 7.08 (m, 1H), 4.69 (s, 2H), 4.37 (m, 2H), 4.31 (s, 2H), 4.27 (m, 4H), 1.26 (t, 3H)
Пример 15
4-[[3-[3-(трифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
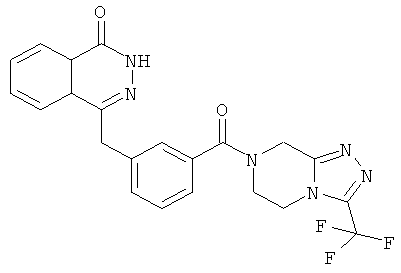
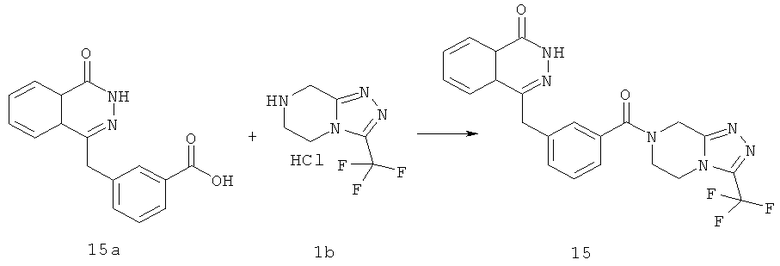
3-[(4-Оксо-3H-фталазин-1-ил)метил]бензойную кислоту 15а (300 мг, 1,07 ммоль, полученную известным способом, раскрытым "заявкой на патент WO 2004080976") растворяли в 10 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (730 мг, 1,93 ммоль), 3-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а] пиразина гидрохлорида 1b (269 мг, 1,40 ммоль) и N,N-диизопропилэтиламина (0,9 мл, 5,30 ммоль). После перемешивания в течение 12 часов к реакционной смеси добавляли 15 мл Н2О, экстрагировали этилацетатом (20 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[3-[3-(трифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 15 (100 мг, выход 20,6%) в виде белого твердого вещества.
МС m/z (ИЭР):455,1 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.30 (br. s, 1Н), 8.49 (d, 1H), 8.02 (m, 1Н), 7.78 (m, 3Н), 7.43 (m, 3Н), 5.31 (s, 2Н), 4.35 (s, 2Н), 4.21 (m, 2Н), 4.12 (m, 2Н)
Пример 16
4-[[3-(6,8-дигидро-5H-[1,2,4]триазоло[1,5-а]пиразин-7-карбонил)-4-фтор-фенил]метил]-2H-фталазин-1-он
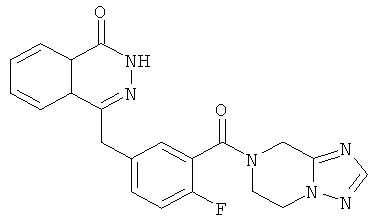
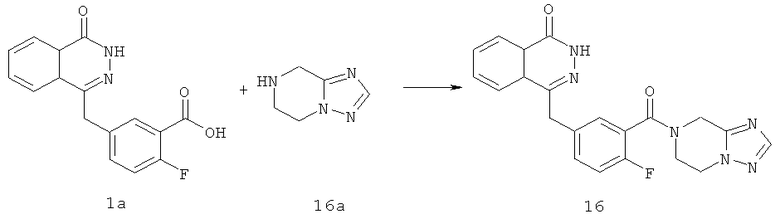
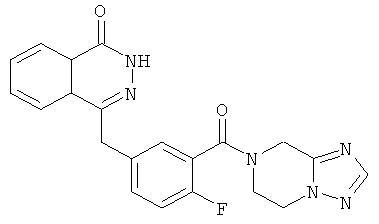
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (360 мг, 1,20 ммоль) растворяли в 10 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (600 мг, 1,80 ммоль), 5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-а]пиразина 16а (150 мг, 1,20 ммоль, полученного известным способом, раскрытым "заявкой на патент WO2009090055") и N,N-диизопропилэтиламина (0,4 мл, 2,40 ммоль). После перемешивания в течение 20 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[3-(6,8-дигидро-5H-[1,2,4]триазоло[1,5-а]пиразин-7-карбонил)-4-фтор-фенил]метил]-2H- фталазин-1-она 16 (100 мг, выход 21,0%) в виде желтого твердого вещества.
МС m/z (ИЭР):405,1 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.47 (br. s, 1Н), 8.51-8.49 (m, 1Н), 7.99-1.77 (m, 4Н), 7.42-7.30 (m, 2Н), 7.30-7.12 (m, 1Н), 4.76 (m, 2Н), 4.37-4.28 (m, 4Н), 3.77-3.73 (m, 2Н)
Пример 17
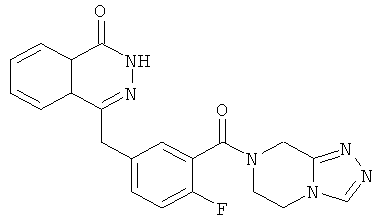
4-[[3-(6,8-дигидро-5Н-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил)-4-фтор-фенил]метил]-2H-фталазин-1-он
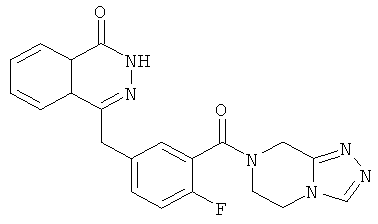
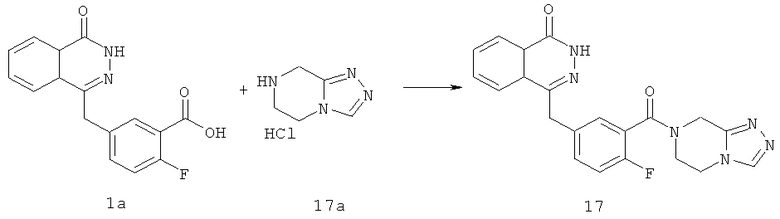
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1a (170 мг, 0,57 ммоль) растворяли в 10 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (323 мг, 0,85 ммоль), 5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразина гидрохлорида 17а (100 мг, 0,63 ммоль, полученного известным способом "Journal of Medicinal Chemistry, 2005, 48 (1), 141-151") и N,N-диизопропилэтиламина (302 мг, 1,70 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[3-(6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил)-4-фтор-фенил]метил]-2H-фталазин-1-она 17 (50 мг, выход 21,7%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):405,1 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 10.87 (br. s, 1Н), 8.46-8.45 (m, 1H), 8.18 (s, 1H), 7.80-7.76 (m, 3Н), 7.40-7.38 (m, 2Н), 7.12-7.07 (m, 1Н), 4.79 (m, 2Н), 4.31-4.20 (m, 4Н), 3.75-3.62 (m, 2Н)
Пример 18
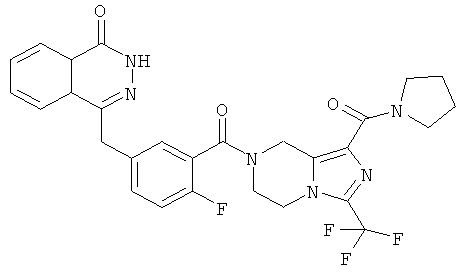
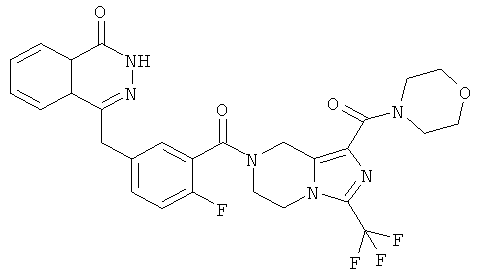
4-[[4-фтор-3-[1-(пирролидин-1-карбонил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2Н-фталазин-1-он
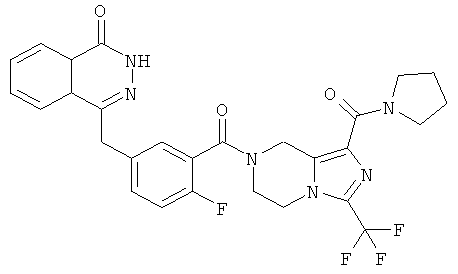
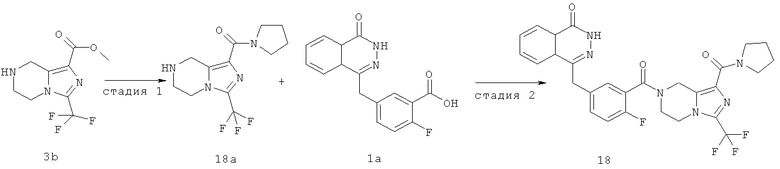
Стадия 1
пирролидин-1-ил-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанон
Пирролидин (560 мг, 8 ммоль), метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 3b (400 мг, 1,60 ммоль) и 0,4 мл H2O смешивали в герметичной пробирке. После перемешивания при 50°С в течение 4 часов реакционную смесь концентрировали при пониженном давлении с получением сырого пирролидин-1-ил-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанона 18а (460 мг) в виде светло-желтого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):289,1 [М+1]
Стадия 2
4-[[4-фтор-3-[1-(пирролидин-1-карбонил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (417 мг, 1,40 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (1 г, 2,80 ммоль), сырого пирролидин-1-ил-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанона 18а (400 мг, 1,40 ммоль) и N,N-диизопропилэтиламина (0,7 мл, 4,20 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, добавляли к ней 20 мл H2O, экстрагировали этилацетатом (10 мл×3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением
4-[[4-фтор-3-[1-(пирролидин-1-карбонил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 18 (150 мг, выход 18,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):569,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6):δ 12.57 (br. s, 1H), 8.26 (d, 1H), 7.83-7.93 (m, 3H), 7.46-7.50 (m, 2H), 7.26-7.31 (m, 1 H), 5.07 (s, 1 H), 4.84 (s, 1 H), 4.27-4.34 (m, 2H), 4.26-4.27 (m, 1H), 4.07-4.17 (m, 2H), 3.89-3.92 (m, 2H), 3.66-3.68 (m, 1H), 3.48-3.49 (m, 1H), 3.36-3.38 (m, 1H), 1.76-1.91 (m, 4H)
Пример 19
4-[[4-фтор-3-[2-(трифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
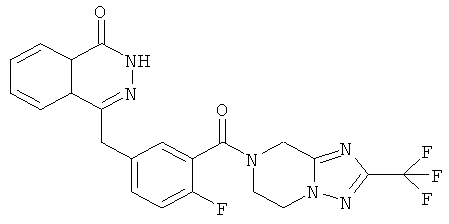
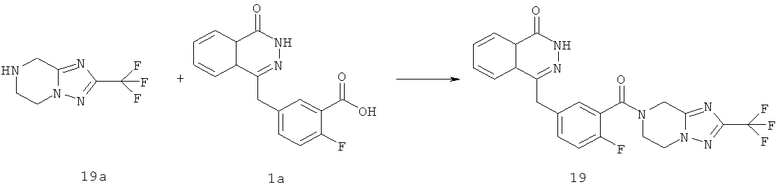
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1а (780 мг, 2,65 ммоль) растворяли в 15 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (1,80 г, 4,77 ммоль), 2-(трифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[1,5-а]пиразина 19а (560 мг, 2,92 ммоль, полученного известным способом, раскрытым "заявкой на патент WO2009025784") и N,N-диизопропилэтиламина (1,4 мл, 7,95 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, добавляли к ней 30 мл H2O, экстрагировали этилацетатом (30 мл×3). Органическую фазу объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[4-фтор-3-[2-(трифторметил)-6,8-дигидро-5Н-[1,2,4]триазоло[1,5-а]пиразин-7-карбонил]фенил]метил]-2Н-фталазин-1-она 19 (205 мг, выход 16,4%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):473,1 [М+1]
1Н ЯМР (400 МГц, CDCl3):δ 10.67 (br. s, 1H), 8.48 (s, 1H), 7.77 (m, 3H), 7.42 (m, 2H), 7.11 (t, 1H), 5.10 (s, 1H), 4.75 (s, 1H),4.39 (s, 2H), 4.32 (d, 3H), 3.88 (s, 1H)
Пример 20
4-[[4-фтор-3-[1-(морфолин-4-карбонил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
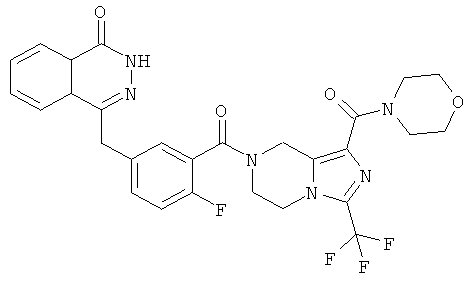
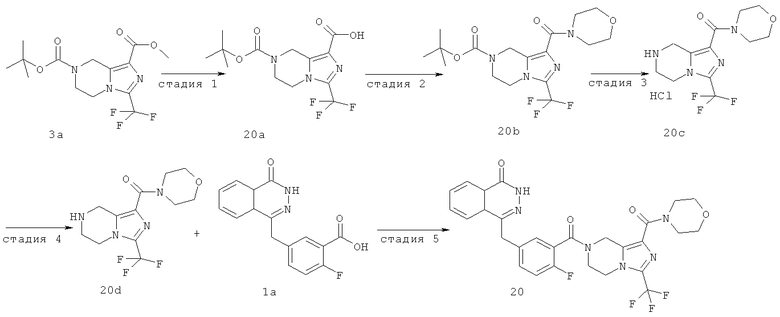
Стадия 1
7-трет-Бутоксикарбонил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоновая кислота
O7-трет-бутил-O1-метил-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1,7-дикарбоксилат 3а (4,10 г, 12 ммоль) растворяли в смешанном растворителе из 15 мл тетрагидрофурана и метанола (об/об=2:1) с последующим добавлением 20 мл 2 М раствора гидроксида натрия. После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, к ней добавляли по каплям 1 М соляную кислоту до pH реакционной смеси от 5 до 7. Реакционную смесь фильтровали, и фильтрационный кек высушивали в вакууме с получением 7-трет-бутоксикарбонил-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоновой кислоты 20а (2 г, выход 50,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):334,1 [М+1]
Стадия 2
трет-Бутил-1-(морфолин-4-карбонил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбоксилат
7-трет-Бутоксикарбонил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоновую кислоту 20а (330 мг, 1 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (756 мг, 2 ммоль), морфолина (174 мг, 2 ммоль) и N,N-диизопропилэтиламина (0,5 мл, 3 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, добавляли к ней 20 мл насыщенного раствора хлорида аммония, экстрагировали дихлорметаном (20 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением трет-бутил-1-(морфолин-4-карбонил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбоксилата 20b (400 мг, выход 100,0%) в виде желтого твердого вещества.
МС m/z (ИЭР):405,1 [М-1]
Стадия 3
морфолино-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанона гидрохлорид
трет-Бутил-1-(морфолин-4-карбонил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат 20b (470 мг, 1,27 ммоль) растворяли в 20 мл 2 М раствора хлорида водорода в 1,4-диоксане. После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением сырого морфолино-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанона гидрохлорида 20с (300 мг) в виде светло-желтого масла. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 4
морфолино-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанон
Сырой морфолино-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил] метанона гидрохлорид 20с (330 мг, 1 ммоль) растворяли в 10 мл этилацетата с последующим добавлением карбоната калия (10 г, 72 ммоль). После перемешивания в течение 4 часов реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением сырого морфолино-[3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-ил]метанона 20d (300 мг) в виде светло-желтого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 5
4-[[4-фтор-3-[1-(морфолин-4-карбонил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1а (390 мг, 1,30 ммоль) растворяли в 10 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (983 мг, 2,60 ммоль), сырого морфолино-[3-(трифторметил)-5,6,7,8- тетрагидроимидазо[1,5-а]пиразин-1-ил]метанона 20d (400 мг, 1,30 ммоль) и N,N-диизопропилэтиламина (0,7 мл, 3,90 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[4-фтор-3-[1-(морфолин-4-карбонил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 20 (150 мг, выход 20,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):585,2 [М+1]
1H ЯМР (400 МГц, ДМСО-d6):δ 12.58 (br. s, 1Н), 8.27 (d, 1H), 7.83-7.98 (m, 3Н), 7.48-7.50 (m, 2Н), 7.27-7.32 (m, 1H), 5.07 (s, 1H), 4.82 (s, 1H), 4.27-4.35 (m, 2Н), 4.26-4.27 (m, 1H), 4.07-4.12 (m, 3Н), 3.59-3.66 (m, 6Н), 3.17-3.18 (m, 2Н)
Пример 21
N-метил-7-[3-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамид
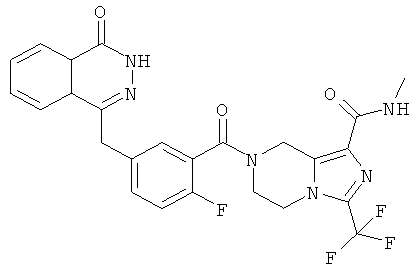
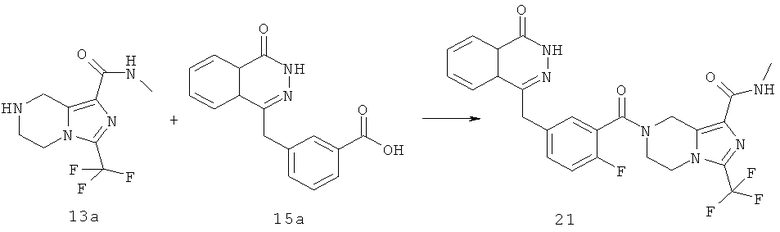
3-[(4-Оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 15а (186 мг, 0,67 ммоль) растворяли в 20 мл N,N-диметилформамида с последующим добавлением 1-гидроксибензотриазола (98 мг, 0,73 ммоль), сырого N-метил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 13а (150 мг, 0,61 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорида (173 мг, 0,91 ммоль) и триэтиламина (253 мкл, 1,82 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, добавляли к ней 50 мл H2O и экстрагировали этилацетатом (50 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением N-метил-7-[3-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамида 21 (280 мг, выход 90,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):511,2 [М+1]
1H ЯМР (400 МГц, CDCl3):δ 11.80 (br. s, 1Н), 8.49 (d, 1H), 7.89 (m, 2Н), 7.79 (t, 1H), 7.52 (m, 2Н), 7.43 (m, 2Н), 5.26 (s, 2Н), 4.35 (s, 2Н), 4.22 (m, 4Н), 3.01 (m, 3Н)
Пример 22
7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-N,N-диметил-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксамид
Стадия 1
трет-бутил-1-(диметилкарбамоил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат
7-Трет-бутоксикарбонил-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоновую кислоту 20а (330 мг, 1 ммоль) растворяли в 5 мл of N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (756 мг, 2 ммоль), диметиламина гидрохлорида (156 мг, 2 ммоль) и N,N-диизопропилэтиламина (387 мг, 3 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, добавляли к ней 50 мл этилацетата и последовательно промывали насыщенным раствором хлорида аммония (30 мл), насыщенным раствором хлорида натрия (20 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого трет-бутил-1-(диметилкарбамоил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбоксилата 22а (362 мг) в виде светло-желтого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):363,1 [М+1]
Стадия 2
N,N-диметил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида гидрохлорид
Сырой трет-бутил-1-(диметилкарбамоил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат 22а (362 мг, 1 ммоль) растворяли в 3 мл 2 М раствора хлорида водорода в 1,4-диоксане. После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением сырого N,N-диметил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида гидрохлорида 22b (262 мг) в виде светло-желтого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 3
N,N-диметил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамид
N,N-диметил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида гидрохлорид 22b (234 мг, 0,80 ммоль) растворяли в 10 мл этилацетата с последующим добавлением карбоната калия (10 г, 72 ммоль). После перемешивания в течение 4 часов реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением сырого N,N-диметил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 22с (200 мг) в виде светло-желтого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 4
7-[2-фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензоил]-N,N-диметил-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксамид
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (300 мг, 1 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (756 мг, 2 ммоль), N,N-диметил-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 22с (200 мг, 0,80 ммоль) и N,N-диизопропилэтиламина (0,5 мл, 3 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-N,N-диметил-3- (трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамида 22 (45 мг, выход 11,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):543,1 [М+1]
1H ЯМР (400 МГц, ДМСО-d6):δ 12.58 (br. s, 1H), 8.27 (d, 1H), 7.83-7.96 (m, 3Н), 7.49-7.51 (m, 2Н), 7.27-7.31 (m, 1H), 4.80 (s, 1H), 4.35 (s, 2H), 4.26-4.27 (m, 1H), 4.05-4.07 (m, 1H), 3.66-3.67 (m, 1H), 3.30-3.39 (m, 6Н), 2.88-2.97 (m, 2H)
Пример 23
4-[[3-[3-(дифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2H-фталазин-1-он
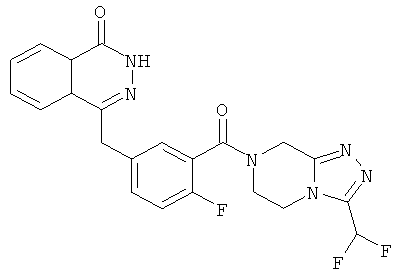
Стадия 1
2,2-дифтор-N'-пиразин-2-ил-ацетогидразидадифторацетат
Пиразин-2-илгидразин 23а (1 г, 9 ммоль) добавляли в бутылку в форме баклажана (25 мл) с последующим добавлением по каплям дифторуксусного ангидрида (4 г, 22,98 ммоль) при 0°С. После перемешивания при комнатной температуре в течение 3 часов реакционную смесь концентрировали при пониженном давлении с получением сырого 2,2-дифтор-N'-пиразин-2-ил-ацетогидразида дифторацетата 23b (2 г) в виде коричневого масла. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 2
3-(дифторметил)-[1,2,4]триазоло[4,3-а]пиразин
2,2-Дифтор-N'-пиразин-2-ил-ацетогидразида дифторацетат 23b (2 г, 0,01 моль) растворяли в 10 мл полифосфорной кислоты. После перемешивания при 140°С в течение 7 часов реакционную смесь охлаждали до 50°С и перемешивали в еще в течение 12 часов. Реакционную смесь наливали в горячем виде в 50 мл ледяной воды, добавляли по каплям 30% водный аммиак до pH реакционной смеси от 7 до 8 и экстрагировали этилацетатом (30 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток растворяли в 30 мл этилацетата и добавляли активированный углерод. После перемешивания в течение 30 минут смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением 3-(дифторметил)-[1,2,4]триазоло[4,3-а]пиразина 23с (460 мг, выход 30%) в виде желтого твердого вещества.
МС m/z (ИЭР):171 [М+1]
Стадия 3
3-(дифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразин
3-(Дифторметил)-[1,2,4]триазоло[4,3-а]пиразин 23с (460 мг, 2,70 ммоль) растворяли в 10 мл метанола с последующим добавлением Pd-C (10%, 46 мг), и реактор продували водородом три раза. После перемешивания в течение 3 часов реакционную смесь фильтровали, и фильтрационный кек промывали метанолом (10 мл). Фильтрат концентрировали при пониженном давлении с получением сырого 3-(дифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразина 23d (400 мг) в виде светло-желтого масла. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):175,0 [М+1]
Стадия 4
4-[[3-[3-(дифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2Н-фталазин-1-он
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (685 мг, 2,30 ммоль) растворяли в 10 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (1,10 г, 3,45 ммоль), сырого 3-(дифторметил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразина 23d (400 мг, 2,30 ммоль) и N,N-диизопропилэтиламина (1,2 мл, 6,90 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[3-[3-(дифторметил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]-4-фтор-фенил]метил]-2H-фталазин-1-она 23 (200 мг, выход 20,0%) в виде белого твердого вещества.
МС m/z (ИЭР):454,6 [М+1]
Пример 24
N-(циклопропилметил)-7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоксамид
Стадия 1
трет-Бутил-1-(циклопропилметилкарбамоил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбоксилат
7-трет-бутоксикарбонил-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбоновую кислоту 20а (330 мг, 1 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (756 мг, 2 ммоль), циклопропилметиламина (142 мг, 2 ммоль) и N,N-диизопропилэтиламина (0,5 мл, 3 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении и добавляли к ней 50 мл этилацетата и последовательно промывали насыщенным раствором хлорида аммония (15 мл×3) и насыщенным раствором хлорида натрия (10 мл). Органическую фазу собирали, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого трет-бутил-1-(циклопропилметилкарбамоил)-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-7-карбоксилата 24а (300 мг) в виде коричнево-красного масла. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):389,1 [М+1]
Стадия 2
N-(циклопропилметил)-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида гидрохлорид
Сырой трет-бутил-1-(циклопропилметилкарбамоил)-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-7-карбоксилат 24а (300 мг, 0,77 ммоль) растворяли в 20 мл 2 М раствора хлорида водорода в 1,4-диоксане. После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении с получением сырого N-(циклопропилметил)-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида гидрохлорида 24b (250 мг) в виде светло-желтого масла. Продукт использовали непосредственно в следующей реакции без очистки.
МС m/z (ИЭР):289,1 [М+1]
Стадия 3
N-(циклопропилметил)-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамид
N-(циклопропилметил)-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида гидрохлорид 24b (250 мг, 0,77 ммоль) растворяли в 10 мл дихлорметана с последующим добавлением карбоната калия (320 мг, 2,30 ммоль). После перемешивания в течение 4 часов реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением сырого N-(циклопропилметил)-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 24с (250 мг) в виде желтого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 4
N-(циклопропилметил)-7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксамид
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (300 мг, 1 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (756 мг, 2 ммоль), сырого N-(циклопропилметил)-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 24с (250 мг, 0,87 ммоль) и N,N-диизопропилэтиламина (0,5 мл, 3 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением
N-(циклопропилметил)-7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбоксамида 24 (150 мг, выход 30,0%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР):569,2 [М+1]
Пример 25
7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5Н-имидазо[1,5-а]пиразин-1-карбонитрил
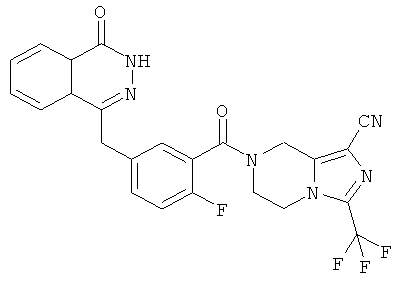
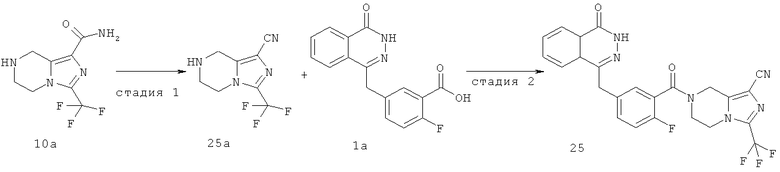
Стадия 1
3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбонитрил
3-(Трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамид 10а (100 мг, 0,43 ммоль) растворяли в 5 мл оксихлорида фосфора. Реакционную смесь нагревали до образования флегмы в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении, добавляли к ней 10 мл насыщенного раствора карбоната натрия и экстрагировали этилацетатом (25 мл×3). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого 3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбонитрила 25а (100 мг) в виде коричневого твердого вещества. Продукт использовали непосредственно в следующей реакции без очистки.
MC m/z (ИЭР):217,0 [M+1]
Стадия 2
7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбонитрил
2-Фтор-5-[(4-оксо-3Н-фталазин-1-ил)метил]бензойную кислоту 1а (210 мг, 0,70 ммоль) растворяли в 5 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (350 мг, 0,92 ммоль), сырого 3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбонитрила 25а (100 мг, 0,46 ммоль) и N,N-диизопропилэтиламина (250 мкл, 1,18 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 7-[2-фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензоил]-3-(трифторметил)-6,8-дигидро-5H-имидазо[1,5-а]пиразин-1-карбонитрила 25 (50 мг, выход 21,9%) в виде белого твердого вещества.
МС m/z (ИЭР):496,6 [М+1]
Пример 26
4-[[4-фтор-3-[3-(2,2,2-трифторэтил)-6,8-дигидро-5Н-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
Стадия 1
3-(2,2,2-трифторэтил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразин
3-(2,2,2-трифторэтил)-[1,2,4]триазоло[4,3-а]пиразин 26а (464 мг, 2,29 ммоль, полученный известным способом "Journal of Medicinal Chemistry, 2005, 48 (1), 141-151") растворяли в 20 мл метанола с последующим добавлением Pd-C (10%, 200 мг), и реактор продували водородом три раза. После перемешивания в течение 3 часов реакционную смесь фильтровали, и фильтрат концентрировали при пониженном давлении с получением сырого
3-(2,2,2-трифторэтил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразина 26b (480 мг) в виде бесцветного масла. Продукт использовали непосредственно в следующей реакции без очистки.
Стадия 2
4-[[4-фтор-3-[3-(2,2,2-трифторэтил)-6,8-дигидро-5Н-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-он
2-Фтор-5-[(4-оксо-3H-фталазин-1-ил)метил]бензойную кислоту 1а (801 мг, 2,69 ммоль) растворяли в 25 мл N,N-диметилформамида с последующим добавлением O-(1-бензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата (1,27 г, 3,36 ммоль), сырого 3-(2,2,2-трифторэтил)-5,6,7,8-тетрагидро-[1,2,4]триазоло[4,3-а]пиразина 26b (460 мг, 2,24 ммоль) и N,N-диизопропилэтиламина (0,8 мл, 4,48 ммоль). После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении, добавляли к ней 30 мл H2O и экстрагировали этилацетатом (30 мл×3). Органическую фазу объединяли, концентрировали при пониженном давлении, добавляли к ней 30 мл этилацетата, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюирования А с получением 4-[[4-фтор-3-[3-(2,2,2-трифторэтил)-6,8-дигидро-5H-[1,2,4]триазоло[4,3-а]пиразин-7-карбонил]фенил]метил]-2H-фталазин-1-она 26 (240 мг, выход 22,1%) в виде белого твердого вещества.
МС m/z (ИЭР):486,6 [М+1]
ТЕСТОВЫЕ ПРИМЕРЫ
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
Пример 1 Анализ на определение ферментативной активности PAPR
Приведенный ниже скрининговый анализ in vitro используют для определения активности соединений по настоящему изобретению в отношении ингибирования ферментативной активности PAPR.
Нижеописанный анализ предназначен для определения активности соединений по настоящему изобретению в отношении ингибирования ферментативной активности PAPR путем использования набора для анализа ингибирования гомологичной поли(аденозиндифосфат-рибоза)полимеразы Trevigen HT F (TREVIGEN HT F homogeneous PARP Inhibition Assay Kit, No. 4690-096-K). Анализы основаны на количестве NAD+, необходимом для потребления в процессе репарации ДНК, который также используется в другой реакции для катализа субстрата без флуоресцентной активности до молекул с высокой флуоресцентной активностью. Таким образом, уровень NAD+ в реакционной системе можно исследовать путем измерения степени усиления флуоресцентного сигнала, а затем вычислять степень ингибирования тестируемого соединения в отношении ферментативной активности PARP.
Инструкции к набору для анализа ингибирования гомологичной поли(аденозиндифосфат-рибоза)полимеразы Trevigen HT F можно использовать в качестве ссылки на подробную процедуру анализов, а также приготовления реагентов, таких как реакционная смесь (reaction mix), циклическая реакционная смесь (cycling mix), буферный раствор (buffer) и тому подобное.
Методы анализа кратко изложены ниже: Тестируемые соединения растворяли в диметилсульфоксиде, а затем разводили 1× буфером до концентрации, желаемой в эксперименте. 25 мкл 200 нМ раствора NAD+ добавляли в 96-луночный круглодонный планшет с последующим добавлением 1 мкл раствора тестируемых соединений, и ставили контроль для дублируемых лунок. Затем в каждую лунку добавляли 25 мкл реакционной смеси, содержащей ДНК, фермент PARP и реакционный буфер. После инкубации в течение 30 минут при комнатной температуре в каждую лунку добавляли 50 мкл циклической реакционной смеси и инкубировали в темноте при комнатной температуре в течение периода от 15 до 40 минут. Затем в каждую лунку добавляли 50 мкл останавливающего раствора, и значения флуоресценции каждой лунки считывали на приборе ELIASA (Ex (возбуждение) 544 нм, Em (испускание) 590 нм). Коэффициент ингибирования тестируемого соединения в отношении ферментативной активности PARP можно вычислить с помощью уравнения стандартной кривой NAD+.
Значения IC50 соединений можно вычислить с помощью коэффициента ингибирования при различных концентрациях.
Вывод: Предпочтительные соединения по настоящему изобретению обладают значительной активностью ингибирования в отношении пролиферационного ингибирования киназы PARP-1.
Пример 2 Анализ ингибирования клеточной пролиферации
Нижеописанный анализ предназначен для определения активности соединений по настоящему изобретению при ингибировании пролиферации трижды негативного фенотипа линии клеток рака молочной железы MDA-MB-436 in vitro.
Нижеописанный клеточный анализ in vitro предназначен для определения активности тестируемых соединений при ингибировании пролиферации трижды негативного фенотипа линии клеток рака молочной железы. Активность ингибирования представлена значением IC50.
Методы анализа кратко изложены ниже: Клетки MDA-MB-436 высевали в 96-луночный культуральный планшет при соответствующей концентрации клеток (например, 3000 клеток/мл среды) путем использования среды DMEM (модифицированной Дульбекко среды Игла) F12 с 10% ФСТ (фетальной сыворотки теленка) (и то, и другое приобретено у фирмы Gibco) в качестве полной среды. В условиях 37°С и 5% диоксида углерода клетки культивировали в инкубаторе с постоянной температурой и выращивали в течение ночи. Тестируемые соединения растворяли в диметилсульфоксиде, а затем разводили культуральной средой без ФСТ до концентрации, желаемой в анализах. После прикрепления клеток к стенкам культуральную среду заменяли свежей культуральной средой, в которой содержались тестируемые соединения при серийных концентрациях (обычно от 7 до 9 концентраций). Затем планшеты с клетками культивировали непрерывно в течение 72 часов в условиях 37°С и 5% диоксида углерода. Спустя 72 часа активность тестируемых соединений при ингибировании клеточной пролиферации определяли путем использования метода набора ССК8 (Cell Counting kit-8, № СК04, приобретенного у фирмы Dojindo).
Значения IC50 тестируемых соединений вычисляли по данным коэффициентов ингибирования тестируемых соединений при различных концентрациях.
Вывод: Предпочтительные соединения по настоящему изобретению обладают значительной ингибиторной активностью при ингибировании пролиферации клеток MDA-MB-436.
ФАРМАКОКИНЕТИЧЕСКИЙ АНАЛИЗ
Тестовый Пример 1: Фармакокинетический анализ соединений Примера 7, Примера 13 и Примера 19 по изобретению.
1. Реферат
Соединения Примера 7, Примера 13 и Примера 19 вводили внутрижелудочно или путем внутривенной инъекции крысам для определения концентрации лекарственного средства в плазме с различные моменты времени методом ЖХ/МС/МС (жидкостной хроматографии с масс-спектрометрией/масс-спектрометрии), используя крыс линии SD в качестве подопытных животных. У крыс оценивали и исследовали фармакокинетическое поведение соединений по настоящему изобретению.
2. Протокол
2.1 Образцы
Соединения Примера 7, Примера 13 и Примера 19
2.2 Подопытные животные
24 здоровых взрослых крысы линии SD, самцов и самок поровну, приобретали у фирмы SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., CO, №сертификата: SCXK (Shanghai) 2003-0002.
2.3 Приготовление тестируемых соединений
Группа внутрижелудочного введения: точное количество тестируемых соединений взвешивали и растворяли в 0,5 мл ДМСО, разводили физиологическим раствором до 10 мл с получением 1,5 мг/мл.
Группа введения путем внутривенной инъекции: точное количество тестируемых соединений взвешивали и добавляли в 0,5% раствор КМЦ-Na (натриевой соли карбоксиметилцеллюлозы) с получением суспензии 1,5 мг/мл.
2.4 Введение
После ночного голодания 24 здоровым взрослым крысам линии SD, самцам и самкам поровну, вводили соединения внутрижелудочно в дозе 15,0 мг/кг и в объеме введения 10 мл/кг.
2.5 Сбор образцов
Группа внутрижелудочного введения: образцы крови (0,2 мл) брали из глазничного синуса перед введением и через 0,25 часа, 0,5 часа, 1,0 час, 1,5 часа, 2,0 часа, 3,0 часа, 4,0 часа, 6,0 часов, 7,0 часов, 9,0 часов, 12,0 часов и 24,0 часа после введения, хранили в гепаринизированных пробирках и центрифугировали в течение 20 минут при 3500 об/мин для отделения плазмы. Образцы плазмы хранили при -20°С. Крыс кормили через 2 часа после введения.
Группа введения путем внутривенной инъекции: образцы крови (0,2 мл) брали из глазничного синуса перед введением и через 2 минуты, 15 минут, 0,5 часа, 1,0 час, 2,0 часа, 3,0 часа, 4,0 часа, 6,0 часов, 8,0 часов, 12,0 часов и 24,0 часа после введения, хранили в гепаринизированных пробирках и центрифугировали в течение 20 минут при 3500 об/мин для отделения плазмы. Образцы плазмы хранили при -20°С.
3. Ход работы
К 20 мкл чистой плазмы крыс, взятой в различные моменты времени после введения, добавляли 50 мкл раствора внутреннего стандарта и 140 мкл метанола и смешивали в течение 3 минут с помощью вортекса. Смесь центрифугировали в течение 10 минут при 13500 об/мин. 20 мкл супернатанта анализировали с помощью ЖХ-МС/МС. Основные фармакокинетические параметры вычисляли с помощью программного обеспечения DAS 2.0.
4. Результаты фармакокинетических параметров Фармакокинетические параметры соединений по настоящему изобретению представлены ниже:
Вывод: Соединения примеров по настоящему изобретению обладают лучшими фармакокинетическими данными и значительно улучшенными фармакокинетическими свойствами.
АНАЛИЗ ПРОТИВООПУХОЛЕВОГО ЭФФЕКТА
Тестовый Пример 2: этот анализ предназначен для определения противоопухолевого эффекта соединений по настоящему изобретению у мышей.
1. Цель
Терапевтический эффект соединений по настоящему изобретению, вводимых в комбинации с темозоломидом (TMZ), в отношении перевитых опухолей карциномы толстой кишки человека SW620 или клеток рака молочной железы человека МХ-1 у бестимусных мышей оценивали путем использования бестимусных мышей BALB/cA в качестве подопытных животных.
2. Тестируемое лекарственное средство
Соединения Примера 1 и Примера 19
3. Подопытные животные
Бестимусных мышей BALB/cA, SPF (без специфичных патогенов), 16-20 г, самок, приобретали у фирмы SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., CO. №сертификата: SCXK (Shanghai) 2008-0016.
4. Экспериментальные методы
4.1 Бестимусных мышей адаптировали к окружающей среде лаборатории в течение трех суток.
4.2 В правый бок бестимусных мышей подкожно инокулировали клетки карциномы толстой кишки человека SW620. После того, как опухоли вырастали до 339±132 мм3, мышей случайным образом делили на группы (d0).
Бестимусных мышей подкожно инокулировали клетками рака молочной железы человека МХ-1. После того, как опухоли вырастали до 100-200 мм3, мышей случайным образом делили на группы (d0).
4.3 Дозировка и режимы дозирования представлены в приведенной ниже таблице.
Объем опухолей и массу мышей измеряли и записывали от 2 до 3 раз в неделю. Объем опухоли (V) вычисляли по приведенному ниже уравнению:
V=1/2×a×b2
где: a, b представляют собой длину и ширину соответственно.
Противоопухолевый коэффициент (%)=(С-Т)/С(%)
где: Т, С представляют собой объем опухоли подопытной группы (тестируемые соединения) и группы чистого контроля в конце эксперимента, соответственно.
5. Дозировка, режимы дозирования и результаты
Вывод: диапазон данных противоопухолевого коэффициента (%) показан, как описано ниже: "+": 50%-60%; "++": 60%-80%; "+++": 80%-100%. Тестируемые соединения по настоящему изобретению, вводимые в комбинации с темозоломидом (TMZ), обладали значительными противоопухолевыми коэффициентами в отношении клеток карциномы толстой кишки SW620 и клеток карциномы молочной железы МХ-1, которые все были выше 60%.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРАЗИНА ИЛИ ИМИДАЗОДИАЗЕПИНА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ2 | 2008 |
|
RU2540074C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| СОЛИ ПРОИЗВОДНЫХ ТЕТЕРАГИДРО-ИМИДАЗО[1,5-A]ПИРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2010 |
|
RU2523543C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ПИРРОЛОПИРАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2673064C2 |
| БИЦИКЛИЧЕСКИЕ 6-АЛКИЛИДЕНПЕНЕМЫ В КАЧЕСТВЕ ИНГИБИТОРОВ β-ЛАКТАМАЗ | 2003 |
|
RU2339640C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| АНТАГОНИСТЫ ЕР4 | 2016 |
|
RU2761341C2 |
Изобретение относится к производному фталазинонкетона, представленному формулой (I), в которой радикалы и символы имеют значения, приведенные в формуле изобретения, способу его получения, фармацевтической композиции, содержащей это производное, его применению в качестве ингибитора поли(АДФ-рибоза)полимеразы (PARP). 6 н. и 7 з.п. ф-лы, 26 пр.
1. Соединения формулы (I) или их фармацевтически приемлемые соли:
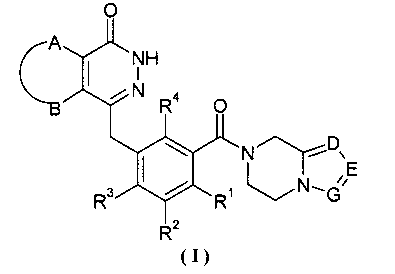
где:
A и B вместе с присоединенными атомами углерода образуют фенил;
R1 представляет собой водород или галоген;
R2, R3 и R4 являются водородом;
D, E или G каждый независимо выбран из группы, состоящей из азота и C(R8);
R5 выбран из группы, состоящей из водорода и C1-6алкила;
R6 или R7 каждый независимо выбран из группы, состоящей из водорода и C1-6алкила, где C1-6алкил необязательно замещен одним или более чем одним C3-10циклоалкилом;
либо R6 и R7 вместе с присоединенным атомом N образуют гетероциклил, выбранный из пирролидинила и морфолинила;
R8 выбран из группы, состоящей из водорода, C1-6алкила, галогена, циано, C3-10циклоалкила, -C(O)OR5, -(CH2)nNR6R7, -NR6R7 и -C(O)NR6R7, где C1-6алкил необязательно замещен одной или более чем одной группой, выбранной из группы, состоящей из галогена, гидроксила и C3-10циклоалкила;
n выбрано из группы, состоящей из 0, 1 и 2.
2. Соединения формулы (I) или их фармацевтически приемлемые соли по п. 1, где R1 представляет собой водород.
3. Соединения формулы (I) или их фармацевтически приемлемые соли по п. 1, где R1 представляет собой галоген.
4. Соединения формулы (I) или их фармацевтически приемлемые соли по п. 1, где R1 представляет собой фтор.
5. Соединения формулы (I) или их фармацевтически приемлемые соли по п. 1, где R8 выбран из группы, состоящей из водорода, C1-6алкила, галогена, циано, -C(O)OR5, -(CH2)nNR6R7 и -C(O)NR6R7, где C1-6алкил необязательно замещен одним или более чем одним атомом галогена.
6. Соединения или их фармацевтически приемлемые соли по п. 5, где R8 представляет собой трифторметил.
7. Соединения формулы (I) или их фармацевтически приемлемые соли по любому из пп. 1-6, где соединения выбраны из группы, состоящей из:
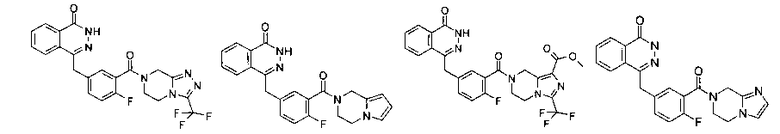
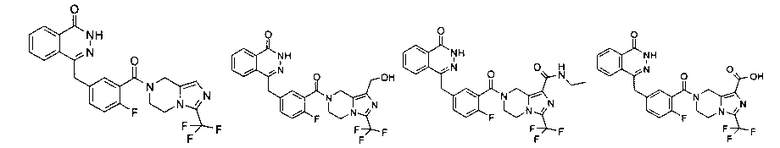
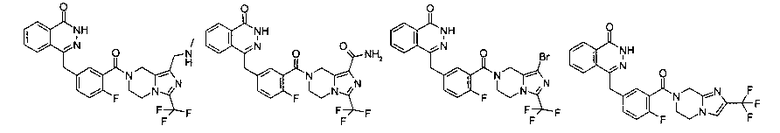
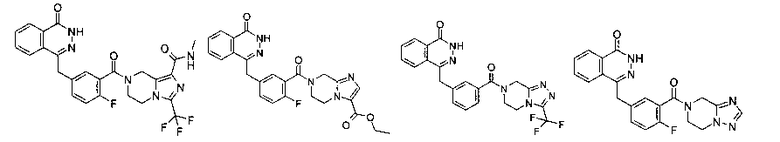
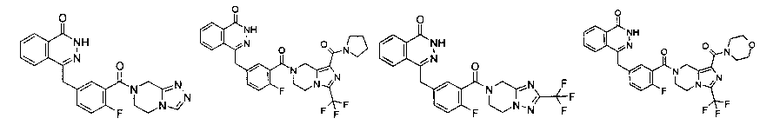
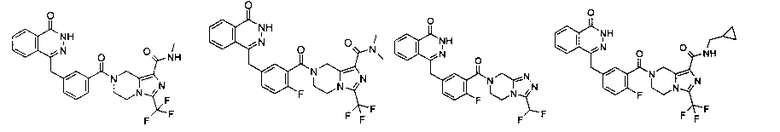
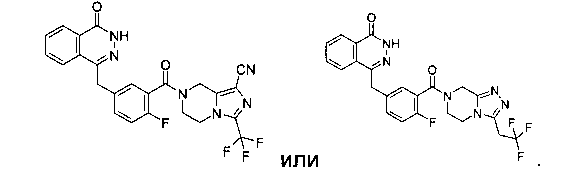
8. Способ получения соединений формулы (I) или их фармацевтически приемлемых солей по п. 1, включающий стадии:
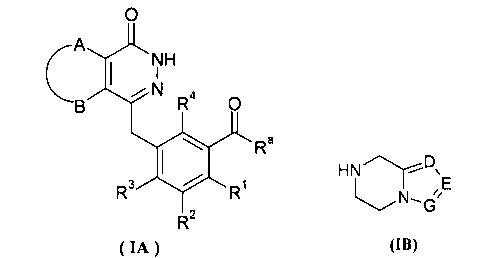
необязательного гидролиза соединения формулы (IA) до карбоновой кислоты, взаимодействия карбоновой кислоты с соединением формулы (IB) или его солью с получением соединения формулы (I);
где:
Ra выбран из группы, состоящей из галогена, гидроксила и C1-6алкоксила;
A, B, D, E, G и R1-R4 такие, как указано в п. 1.
9. Фармацевтическая композиция, обладающая ингибирующим действием в отношении поли(АДФ-рибоза)полимеразы (PARP), содержащая терапевтически эффективное количество соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп. 1-7 и фармацевтически приемлемый носитель или наполнитель.
10. Применение соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп. 1-7 или фармацевтической композиции по п. 9 для получения ингибитора PARR.
11. Применение соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп. 1-7 или фармацевтической композиции по п. 9 для получения адъюванта при лечении рака или лекарственного средства, вызывающего чувствительность опухолевых клеток к ионизирующему излучению или химиотерапии.
12. Применение соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп. 1-7 или применение фармацевтической композиции по п. 9 для получения лекарственного средства для лечения рака, где рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака поджелудочной железы, рака простаты, рака прямой кишки, рака печени и рака толстой кишки.
13. Применение по любому из пп. 11 и 12, где лекарственное средство дополнительно вводят совместно с терапевтически эффективным количеством темозоломида.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Приспособление для удаления золы из зольных камер гидравлическим путем | 1927 |
|
SU9469A1 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИРИДАЗИНА И ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2003 |
|
RU2292337C2 |
| ЕА 200971100 А1, 30.01.2010. | |||

