Предметом данного изобретения являются новые производные сордарина, обладающие антигрибной активностью, способы их получения, содержащие их фармацевтические составы и их применение в медицине, в частности для профилактики или лечения заболеваний животных, включая людей, вызываемых грибной инфекцией.
Известно [1] получение антибиотика SL2266 путем культивирования штамма NRRL 3196 гриба вида Sordaria araneosa. И известно, что SL2266, названный позднее сордарином, обладает фунгистатической активностью. Эта же исследовательская группа описала [2] деградацию сордарина до сордарицина. Известно также [3] получение антибиотика зофимарина, обладающего антигрибной активностью.
Сордарин, сордарицин и зофимарин могут быть представлены приводимой ниже формулой (A)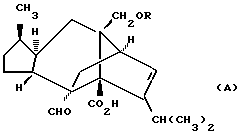
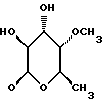
где OR как
описывает сордарин;
OR как OH описывает сордарицин; и
OR как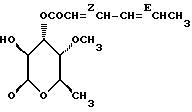
описывает зофимарин.
Несмотря на то, что сордарин и зофимарин проявляют антигрибную активность, оба эти соединения являются лишь умеренно активными и имеют ограниченный спектр действия при тестировании против набора грибных организмов. Мы предлагаем новую группу фунгицидных производных сордарина, которые обладают превосходной антигрибной активностью и широким спектром действия. Так, например, согласно первому аспекту настоящего изобретения мы предлагаем соединения формулы (I)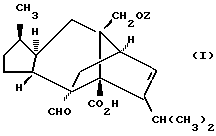
где Z является тетрагидропираногруппой, выбираемой из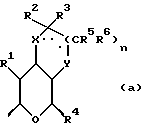
и их фармацевтически приемлемые соли и сольваты (например, гидраты) или их метаболически лабильные производные,
где R1 представляет собой водород, галоген, гидроксил, C1-4алкоксил или ацилоксил;
R2 и R3 могут независимо представлять собой водород, C1-6алкил или C1-4алкоксиC1-4алкил, либо R2 и R3 могут вместе с угле родным атомом, к которому они присоединены, представлять C=O, C=S или C3-8циклоалкил;
R4 представляет собой водород или CH2R7 (где R7 является водородом, гидроксилом, C1-4алкоксилом или группой OCOR, в которой R8 является C1-4алкилом или арилом);
R5 и R6 каждый может независимо представлять собой водород, C1-6алкил или C1-4алкоксиC1-4алкил либо R5 и R6 могут вместе с углеродным атомом, к которому они присоединены, представлять C=O, C=S или C3-8циклоалкил;
n = 0 или 1;
X и Y каждый независимо может представлять собой кислород, серу или CR9R10 (где R9 и R10 каждый может независимо представлять собой водород, C1-6алкил; C1-4алкоксил или C1-4алкоксиC1-4алкил либо R9 и R10 могут вместе с атомом углерода, к которому они присоединены, представлять собой C=O, C=S, C3-8циклоалкил или C= CHR11, где R11 является водородом или C1-4алкилом); либо, если X и Y являются кислородом, а n = 0, то -Y-CR2R3 или -X-CR2R3 соответственно могут также представлять собой -N=CR3- или -NR12-CR2R3- (где CR2 и R3 являются C=O, a R12 является C1-4алкилацильной группой COR13, где R13 является C1-6алкилом) либо если Y является кислородом, а n = 0, то X может быть представлен группой CR11 (где R11 имеет определенные выше значения), которая присоединена к пирановому кольцу посредством двойной связи;
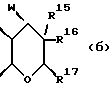
R15 представляет собой водород, галоген, азид, C1-6алкил, гидроксил, C1-6алкоксил (возможно замещенный 1 или 2 гидроксилами либо их кеталем или 1 или 2 C1-3алкоксильными группами), арилC1-4алкоксил, C3-6алкенилоксил, группу OCOR18 (где R18 является арил C1-4алкоксилом или C1-10алкильной группой, возможно содержащей одну или две двойных связи) либо C1-6алкоксикарбонил C1-4алкоксил, a R16 представляет собой водород, либо R15 и R16 могут вместе с атомом углерода, к которому они присоединены, представлять C=O или C=CH2;
R17 представляет собой CH2R19, где R19 является водородом, гидроксилом, C1-14алкоксилом или группой OCOR20, в которой R20 является C1-4алкилом); и
W представляет собой атом кислорода или серы либо CH2 группу;
а пунктирная линия в группе (a) показывает возможное присутствие дополнительной связи.
Пригодные фармацевтически приемлемые соли соединений формулы (I) включают соли неорганических оснований, такие как соли щелочных металлов (например, соли натрия и калия) и соли аммония и соли органических оснований. Пригодные соли органических оснований включают аминные соли, такие как соли триалкиламина (например, триэтиламин), диалкиламина (например, дициклогексиламин), возможно замещенного бензиламина (например, фенилбензиламин или п-бромбензиламин), прокаина, этаноламина, диэтаноламина, N-метилглюкозамина и три(гидроксиметил)метиламина и соли аминокислот (например, соли лизина и аргинина).
Ссылки далее на соединение формулы (I) включают это соединение и его фармацевтические приемлемые соли.
Другие соли, не являющиеся фармацевтически приемлемыми, могут быть использованы для получения соединений формулы (I) и они формируют следующий аспект изобретения.
Метаболически лабильные производные соединений формулы (I) являются соединениями, которые в организме превращаются в соединения формулы (I). Примеры таких производных включают обычные метаболически лабильные сложные эфиры, образованные из свободной карбоновой кислоты в молекуле.
Следует понимать, что настоящее изобретение заключает в себе любые отдельные изомеры, включая оптические изомеры соединений, описываемых формулой (I), также как и их смеси, включая их полностью или частично рацемические смеси.
Используемый здесь термин "алкил" как группа или часть C1-4алкоксильной группы может представлять собой прямую или разветвленную цепь. Приемлемые примеры включают метил, этил, n-пропил, i-пропил, n-бутил, s-бутил и t-бутил, n-гексил и n-октил.
Используемый здесь термин "арил" как группа или часть группы обозначает фенил или гетероарил, замещенный возможно одним или более (например, 1, 2 или 3) атомами или группами, выбираемыми из галогена, гидроксила, C1-6алкила, C1-6алкоксила или C1-4алкоксикарбонила. Гетероарильная группа может быть 5- или 6-членным гетероароматическим кольцом, содержащим один или более гетероатомов, выбираемых из азота, кислорода и серы. Приемлемые примеры гетероарильных групп включают пиридил, фурил, тиенил и пирролил.
Термин "галоген" обозначает здесь фтор, хлор, бром или иод.
Если R1 является ацилоксильной группой, то она может представлять собой, например, группу OCOR13, где R13 определен выше.
Примеры C3-8циклоалкильных групп включают циклопептильные и циклогексильные группы.
Примеры X групп включают кислород, CR9R10, где R9 и R10 каждый являются водородом, C1-4алкоксилом или C1-4алкилом, либо CR9R10 представляет собой группу C=O или C=CHR11, например C=CH2, либо X представляет собой CR11.
Примеры приемлемых Y групп включают кислород или CR9R10, где R9 является водородом, C1-4алкоксилом или C1-4алкилом, а R10 является водородом или C1-4алкилом.
Если R18 является ненасыщенной C1-10алкильной группой, она может, в частности, представлять собой C5-8алкильную группу с прямой или разветвленной цепью, содержащую две двойных связи, например -CH=ZCH-CH=ECHCH3.
Если R15 является C1-6алкоксильной группой, замещенной гидроксилом или алкоксилом, то она может быть, например, 2,3-дигидроксипропоксилом и полученным из него ацетонкеталем или 2,3-диметокси-пропоксильной группой.
R16 является предпочтительно атомом водорода с R15, расположенным в α- конфигурации.
R1 может представлять собой, например, атом водорода или гидроксильную группу.
R2 может представлять собой, например, водород или C1-4алкил (например, метил), а R3 может представлять собой, например, водород, C1-4алкил (например, метил, этил или n-пропил) или C1-4алкоксиC1-4алкил (например, метоксиэтил), либо CR2R3 может представлять собой C=O, C=S или C3-8циклоалкил (например, циклопентил).
R4 может представлять собой, например, метил или C1-4алкоксиметил (например, метоксиметил).
R5 и R6 каждый могут независимо представлять собой, например, водород или C1-4алкил (например, метил).
Примеры кольцевых систем, описываемых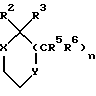
включают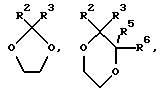
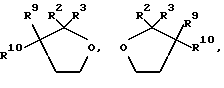
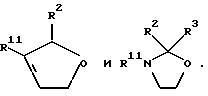
Примеры отдельных R15 групп включают галоген (например, фтор), азид, гидроксил, C1-4алкоксил, бензилоксил, бензилокси- карбонилоксил, C1-4алкоксикарбонилоксил и C1-4алкоксиC1-4алкоксил (например, метоксиэтоксил), C1-4алкоксикарбонил - C1-4алкоксил, C1-4алкилкарбонилоксил, 2,3-дигидроксипропоксил и их ацетонкеталь, 2,3-диметоксипропоксил, C3-6алкеноксил, например аллилоксил или 3-метилаллилоксил, либо R15 и R16 и атом углерода, к которому они присоединены, представляют собой C=O или C=CH2.
R17 может представлять собой, например, метил или C1-4алкоксиметил (в частности, метоксиметил) либо гидроксиметил.
R15 предпочтительно представляет собой C1-4алкоксил, C3-4алкенилоксил, бензилоксил или OCOR4 (где R4 является C1-4алкильной группой), особенно расположенные в α- конфигурации.
R17 предпочтительно представляет собой метил.
W предпочтительно представляет собой кислород.
Особой группой соединений по изобретению являются соединения формулы (I), у которых Z является группой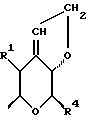
или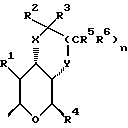
где один из X и Y является кислородом, а другой является кислородом или группой CR9R10. В пределах этой группы более конкретно R1 является гидроксилом или водородом; X является кислородом, либо CR9R10, где R9 является водородом, C1-3алкоксилом, C1-4алкилом, a R10 является водородом, или CR9R10 представляет собой группу C=O или C=CHR11, Y является кислородом или CHR9, где R9 является H или C1-4алкилом, R2 и R3 каждый независимо является водородом, C1-4алкилом, например метилом, пропилом или C1-4алкоксиалкилом, в частности метоксиметилом, либо R2 и R3 вместе с атомом углерода, к которому они присоединены, представляют собой циклопентильную группу или группу C=O либо C=S; R5 и R6 каждый предпочтительно являются водородом.
Если R1 представляет собой гидроксил или C1-4алкоксил, то R1 часть предпочтительно расположена в аксиальной конфигурации. Однако R1 предпочтительно представляет собой атом водорода.
R2 и R3 могут каждый в отдельности представлять собой водород или C1-4алкил.
R4 предпочтительно представляет собой метил.
R5 и R6 каждый предпочтительно является водородом.
n предпочтительно равно нулю.
Конкретные кольцевые системы, описываемые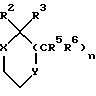
включают,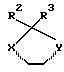
где R2 и R3 описаны ранее, а X и Y каждый независимо являются кислородом или CR9R10 (где R9 и R10 определены ранее), при условии, что по меньшей мере либо X либо Y являются кислородом или кольцом.
Среди кольцевых систем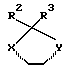
предпочтительными кольцами являются те, у которых один из X или Y представляет собой кислород, а другой представляет собой CR9R10 (где R9 и R10 каждый независимо представляет собой водород или C1-4алкил (например, метил) или CR9R10 представляют собой группу C=O либо C=CH2), либо X и Y оба представляют собой кислород; R2 и R3 представляют собой водород, C1-4алкил или группу CO.
Следует понимать, что настоящее изобретение охватывает все комбинации конкретных и предпочтительных вышеописанных групп.
Следующей особой группой соединений по изобретению являются соединения формулы (I), где Z является группой (a), и где R1 является водородом, a R4 является метилом;
n = 0, a R2 и R3 являются водородом или C1-4алкилом, X и Y являются кислородом, либо Y является кислородом, а X является группой CHR9, где R9 является водородом или C1-4алкилом, C=O, C=CH2, либо X является CH, Y является кислородом, n равно нулю, а R2 и R3 являются водородом.
Следующей особой группой соединений по изобретению являются соединения формулы (I), где Z является группой (б)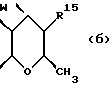
и их фармацевтически приемлемые соли и сольваты (например, гидраты), где W является кислородом или серой, а R15 - как определено выше. Более конкретно, W является кислородом, а R15 является группой, выбираемой из C1-4алкоксила, бензилоксила или OCOR4 (где R4 является C1-4алкильной группой, например изопропилом или t-бутилом), либо C3-4алкенилоксилом, C1-4алкоксикарбонилалкоксилом.
Конкретные соединения согласно настоящему изобретению включают:
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,7R,9R]-2,8-Диокса-9-метил-4-метилен- цис-бицикло[3.4.0] -нон-7-ил-окси-метил] -4-формил-4,4a,5,6,7, 7a, 8,3a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновую кислоту.
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-дидезокси-3,4-O-изопропилиден- β- D-аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновую кислоту;
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4R,7R,9R]-2,8-диокса-4,9-диметил-цис-бицикло [3.4.0] -нон-7-ил-окси-метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновую кислоту;
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-[[1S, 4S,6R,8R]-2,7-диокса-4,6-диметил-цис-бицикло [3.4.0] -нон-8-ил-окси-метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновую кислоту;
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-ангидро-6-деокси-4-O-пропил- β- D- маннопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-5-инда-цен-3a(1H)-карбоновую кислоту;
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-ангидро-6-деокси-4-O-метил-β--D- маннопиранозилоксиметил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновую кислоту;
и их фармацевтически приемлемые соли и сольваты (например, гидраты) или их метаболически лабильные производные.
Соединения формулы (I) являются очень активными фунгицидами, пригодными для борьбы с грибными инфекциями у животных, включая людей. Например, они могут применяться для лечения грибных инфекций, вызываемых такими организмами, как виды Candida (например, Candida albicans, Candida glabrata, (Torulopsis glabrata), Candida tropicalis, Candida parapsilosis и Candida pseudotropicalis), Cryptococcus neoformans, Pneumocystis carinii, Aspergillus sp. (например, Aspergillus flavus и Aspergillus fumigatus), Coccidioides (например, Coccidioides immitis), Paracoccidioides (например, Paracoccidioides brasiliensis), Histoplasma (например, Histoplasma capsulatum) или Blastomyces (например, Blastomyces dermatitidis). Они также могут быть применены для лечения других грибных инфекций, вызываемых видами Candida, Trichophyton, Microsporum или Epidermophyton (например, Trichophyton mentographytes, Trichophyton rubrum, Microsporum canis или Epidermophyton floccosum), или при инфекциях слизистых оболочек, вызываемых Candida albicans.
Соединения формулы (I) могут также быть применены для лечения других инфекций, вызываемых видами нитевидных грибов, таких как Geotrichum (например, Geotrichum clavatum), Trichosporon (например, Trichosporon beigelii), Blastoschizomyces (например, Blastoschizomyces capitatus), Sporothrix (например, Sporothrix schenckii), Scedosporium (например, Scedosporium apiosperum), Cladosporium (например, Cladosporium carrionii) и Pityrosporum ovale.
Соединения формулы (I) могут также быть применены для лечения инфекций, вызываемых такими простейшими, как Toxoplasma, Cryptosporidium, Leishmania, Tripanosoma, Giardia и Trichomonas.
In vitro определение антигрибной активности соединений по изобретению проводили в жидкой или на твердой среде, используя способ антигрибного двукратного серийного разведения для определения минимальной ингибирующей концентрации (МИК) антигрибного агента, которая ингибировала развитие роста через 24-48 часов инкубирования при 37oC. На практике серии агаровых чашек или кювет для микроразведений питательной среды, содержавшие двукратные разведения тестируемого антигрибного агента, инокулировали стандартной культурой клинически целесообразного патогена, например Candida albicans. Чашки с агаром или кюветы для микроразведений питательной среды затем анализировали на наличие или отсутствие роста гриба и фиксировали соответствующие значения МИК.
МИК значения (определяемые как самая низкая антигрибная концентрация, которая убивает по меньшей мере 99,9% исходного инокулюма в жидкой среде) могут также быть определены путем субкультивирования 0,01 и 0,1 мкл питательной среды из свободной от препарата контрольной лунки, первой лунки, в которой отмечали рост, и каждой прозрачной лунки на агаровых чашках.
In vivo определение соединений формулы (I) может быть осуществлено на сериях дозовых уровней путем их введения (например, подкожно, перорально, внутрибрюшинно или внутривенно) мышам или крысам, инокулированным штаммом Candida albicans. Необработанные животные погибают в интервале от 3 до 9 суток, при этом регистрируют дозовый уровень, при котором тестируемое соединение обеспечивает 50%-ную защиту против летального действия инфекции.
С точки зрения их антигрибной активности, соединения формулы (I) рекомендуют для лечения различных грибных инфекций у людей и животных. Такие инфекции включают поверхностные, кожные, подкожные и системные микотические инфекции, такие как инфекции дыхательных путей, желудочно-кишечного тракта, сердечно-сосудистые инфекции, инфекции мочевых путей, инфекции ЦНС, кандидоз и хронический кандидоз слизистых оболочек (в частности, молочницу и вагинальный кандидоз) и кожные инфекции, вызываемые грибами, кожный и кожно- слизистый кандидоз, дерматомикозы, включая дерматофитии, эпидермофитию стопы, паронихию, отрубе-видный лишай, эритразму, интертригинозный дерматит, грибные высыпания, кандидозный вульвит, кандидозный баланит и внешний отит. Они также могут применяться в качестве профилактических средств для предотвращения системных и локальных грибных инфекций. Применение в качестве профилактических средств может быть целесообразным, например, как часть схемы избирательного обеззараживания кишечника для предотвращения инфекции у пациентов с нарушениями иммунитета (в частности, у пациентов, больных СПИДом, проходящих курс противораковой терапии или пациентов, перенесших операции по пересадке органов и тканей). Предотвращение чрезмерного развития грибов в ходе лечения антибиотиками также может быть желательным при некоторых болезненных синдромах или ятрогенных состояниях.
Несмотря на то, что допустимо, чтобы для применения в терапии соединения по изобретению могли быть введены в виде неочищенного соединения, предпочтительно все же представлять активный ингредиент в виде фармацевтического препарата. Изобретение таким образом предлагает далее фармацевтический препарат, содержащий соединения формулы (I) и их фармацевтически приемлемые соли совместно с одним или более фармацевтически приемлемых носителей и, возможно, другими терапевтическими и/или профилактическими ингредиентами. Носитель (и) должен быть "приемлемым" в том смысле, что он должен быть совместимым с другими ингредиентами препарата и не являться вредным для его реципиента.
Составы по изобретению включают составы в форме, специально предназначенной для перорального, трансбуккального, парентерального введения, введения в виде имплантата, для ректального, местного, офтальмического или генито-уринарного введения, либо в форме, пригодной для введения посредством ингаляций или вдувания.
Таблетки или капсулы для перорального введения могут содержать обычные эксципиенты, такие как связывающие агенты, например сироп, гуммиарабик, желатин, сорбит, трагакант, крахмальный клей или поливинилпирролидон; наполнители, например лактозу, сахар, микрокристаллическую целлюлозу, кукурузный крахмал, фосфат кальция или сорбит; лубриканты, например стеарат магния, стеариновую кислоту, тальк, полиэтиленгликоль или кремний; дезинтегранты, например картофельный крахмал или крахмальный гликолят натрия или кросскармеллозу натрия; или смачивающие агенты, такие как лаурилсульфат натрия. Таблетки, к числу которых относятся жевательные, рассасывающиеся или шипучие таблетки, могут быть покрыты оболочкой согласно способам, известным из уровня техники. Жидкие препараты для перорального введения могут иметь форму, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров, либо могут иметь форму сухого продукта, соединяемого с водой или другим пригодным носителем перед использованием. Такие жидкие препаративные формы могут содержать обычные добавки, такие как суспендирующие агенты, например сироп сорбита, метилцеллюлозу, сироп глюкоза/сахар, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрогенизированные пищевые жиры; эмульгирующие агенты, например лецитин, моноолеат сорбитана или гуммиарабик; неводные носители (которые могут включать пищевые масла), например миндальное масло, фракционированное кокосовое масло, масляные сложные эфиры, пропиленгликоль или этиловый спирт; и консерванты, например метил- или пропил-p-гидроксибензоаты или сорбиновую кислоту.
Состав для трансбуккального введения может иметь форму таблеток или пастилок, приготовленных обычным способом.
Состав согласно изобретению может быть изготовлен в форме для парентерального введения путем инъекции или продолжительного вдувания. Препараты для инъецирования могут находиться в виде разовых доз в ампулах, или в многодозовых контейнерах с добавлением консервантов. Составы могут иметь такие формы, как суспензии, растворы либо эмульсии в масляных или водных носителях, и могут содержать формирующие агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. С другой стороны, активный ингредиент может находиться в порошкообразной форме, соединяемой перед применением с подходящим носителем, например стерильной непирогенной водой.
Составы по изобретению для введения посредством ингаляции обычно имеют форму аэрозольного распылителя из упаковок с повышенным давлением с применением соответствующего пропелланта, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, углекислого газа или другого пригодного газа, или из распылителя. В случае аэрозоля под давлением дозировочная единица может быть определена посредством установки клапана для введения измеренного количества.
С другой стороны, составы согласно изобретению для введения посредством ингаляции могут иметь форму сухих порошковых составов, например порошковой смеси соединения и подходящей порошкообразной основы, такой как лактоза или крахмал, либо в виде модифицированной физической формы данного лекарственного вещества без добавок. Порошковый состав может находиться в виде единичной дозировочной формы, например в капсулах или упаковках, в частности, из желатина, или в виде пакетов, из которых порошок может быть введен с помощью ингалятора или вдувателя.
Составы могут иметь форму суппозитория, например содержащего обычную суппозиторную основу или вагинального суппозитория, например содержащего обычную основу для вагинальных суппозиториев.
Составы могут также изготовляться для местного применения в форме мазей, кремов, гелей, лосьонов, шампуней, порошков (включая распыляемые порошки), вагинальных суппозиториев, тампонов, распылителей, жидкостей для смачивания, аэрозолей, капель (например, глазных, ушных или носовых капель) или примочек. Мази и кремы могут, например, быть изготовлены из водной или масляной основы с добавлением подходящих уплотняющих и/или желирующих агентов. Мази для введения в глаз могут быть изготовлены стерильным образом с применением стерилизованных компонентов. Примочки могут, например, быть изготовлены для применения в ветеринарии в маслосодержащих органические растворителях, возможно с формирующими агентами, например стабилизирующими и солюбилизирующими агентами. Вагинальные суппозитории и тампоны для вагинального введения могут быть изготовлены с применением обычных способов и, если это целесообразно, могут содержать пенообразующий носитель. Такие составы также могут содержать другие активные ингредиенты, такие как кортикостероиды, антибиотики или антипаразитарные средства, как целесообразно.
Жидкие препараты для внутриносового введения могут иметь форму растворов или суспензий и могут содержать обычные эксципиенты, такие как регулирующие тонус агенты, например хлорид натрия, декстрозу или маннит; консерванты, например бензальконийхлорид, тиомерсаль, фенилэтиловый спирт; и другие составляющие агенты, такие как суспендирующие, забуферивающие, стабилизирующие, диспергирующие и/или корригирующие агенты.
Чрескожное введение может быть осуществлено посредством применения подходящей системы, которая стимулирует абсорбцию активного соединения через кожу и которая как правило состоит из препарата-основы, заключенного в клейкий пластырь, содержащий оболочки, мембраны и выделяющие слои. Такие системы могут содержать усилители абсорбции, такие как спирты, или действовать путем усиления ионотофореза.
Состав согласно изобретению может быть изготовлен в виде препарата пролонгированного действия. Такие препараты длительного действия могут вводиться посредством имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Так, например, соединение по изобретению может быть скомпановано с подходящими полимерными или гидрофобными материалами (например, эмульсия в приемлемом масле) или ионообменными смолами, либо в виде слаборастворимых производных, например в виде слаборастворимой соли.
Когда составы содержат дозировочные единицы, каждая единица предпочтительно должна содержать от 0,001 мг до 1000 мг, преимущественно от 0,01 мг до 400 мг, активного ингредиента, если соединение по изобретению должно вводиться перорально. Суточная доза, применяемая для лечения взрослого человека, предпочтительно должна варьировать от 0,001 мг до 5000 мг активного ингредиента, наиболее предпочтительно - от 0,01 мг до 2000 мг, которые могут быть введены в виде 1-4 разовых доз, например в зависимости от пути введения и от состояния пациента и заболевания, подлежащего лечению.
Соединение может быть введено путем внутривенного вливания, используя, например, до 50 мг/кг/сутки активного ингредиента. Продолжительность лечения должна определяться степенью ответа, а не просто количеством суток.
Соединения по изобретению могут применяться также в сочетании с другими терапевтическими средствами, и изобретение поэтому в следующем аспекте предлагает сочетание, включающее соединение по изобретению вместе с другим терапевтически активным агентом.
Так, например, соединения по изобретению могут применяться в сочетании с одним или более других антигрибных агентов, таких как полиеновое производное (Амфотерицин Б, Нистатин, липидный препарат Амфотерицина Б), производное азола (Флуконазол, Интраконазол, Кетоконазол, Миконазол, Клотримазол, ZD-08070, UK-109496), 5-Фторцитозин, производное Пневмокандина или Эхинокандина, так же как Цилофунгин, LY-303366, L-733560, и/или один или более иммуномодулирующих агентов, таких как интерферон (ИФН -γ ), интерлейкин (ИЛ-1, ИЛ-2, ИЛ-3 и ИЛ-8) и факторы, стимулирующие колонии [(G)-КСФ, (M)-КСФ и (GM)-КСФ], и дефенсины.
Наиболее приемлемые для применения с соединениями по изобретению соединения включают Интраконазол, Флуцитозин, Флуконазол или Амфотерицин Б.
Если соединения по изобретению вводят в сочетании с другим антигрибным агентом, то соединения по изобретению и другой грибной агент могут быть введены в рекомендованной максимальной клинической дозе или в меньших дозах.
Упомянутые выше сочетания могут быть предложены для применения в форме фармацевтического препарата и такие фармацевтические препараты, содержащие описанное выше сочетание вместе с их фармацевтически приемлемым носителем, составляют следующий аспект изобретения. Отдельные компоненты таких сочетаний могут быть введены либо последовательно, либо одновременно в виде отдельных или объединенных фармацевтических препаратов.
Если соединение по изобретению применяют в сочетании со вторым терапевтическим агентом против того же самого состояния, то доза каждого соединения может отличаться от дозы, в которой используют каждое соединение по-отдельности. Целесообразные дозы должны определяться специалистом.
Согласно следующему аспекту настоящего изобретения, мы предлагаем соединение формулы (I) или его физиологически приемлемую соль, либо фармацевтический состав, содержащий соединение формулы (I) или его физиологически приемлемую соль, как определено выше, для применения в терапии, в частности для лечения грибных инфекций у животных (особенно людей).
Согласно следующему аспекту настоящего изобретения мы предлагаем применение соединения формулы (I) или его физиологически приемлемой соли для изготовления лекарственного препарата для лечения грибных инфекций у людей и животных.
Согласно следующему аспекту настоящего изобретения, мы предлагаем способ лечения организма человека или животного с целью борьбы с грибными заболеваниями, этот способ включает введение указанному организму эффективного количества соединения формулы (I) или его физиологически приемлемой соли.
Специалистам следует иметь в виду, что ссылки на лечение относятся как к профилактике, так и к лечению установленных состояний или инфекций.
Соединения по изобретению могут быть получены описанным ниже способом.
Так, общий способ (A) для получения соединения формулы (I), где Z является группой (a), включает взаимодействие соединения формулы (II)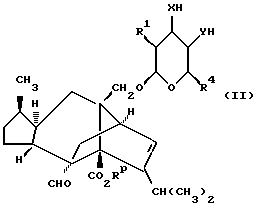
в которой R1 и R4 определены в формуле (I) ранее, Rp является карбоксилзащитной , группой, а X и Y определены выше в формуле (I) за тем исключением, что X и/или Y не могут являться CR9R10,
для образования требуемой циклической системы с последующим удалением карбоксилзащитной группы.
Согласно первой реализации способа (A), соединение формулы (Ia), в которой R1-R4 определены выше в формуле (I), n = 0 а X и Y являются оба кислородом, может быть получено путем обработки диола формулы (III)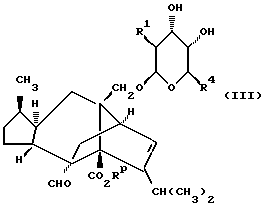
в которой R1 и R4 определены выше в формуле (I), a Rp является карбоксилзащитной группой,
соединением (L)2CR2R3 (в котором L является приемлемой уходящей группой), с последующим удалением карбоксилзащитной группы. Если как R2 так и R3 оба являются водородом, то реакцию образования кольца удобно осуществлять путем обработки соединения формулы (III) дигалометаном (в частности, дибромметаном) в присутствии сильного основания, такого как гидроксид щелочного металла (например, гидроксид натрия), предпочтительно в условиях фазового перехода, применяя, например, соль тетраалкиламмония (в частности, тетрабутиламмонийбромид), при температуре, близкой к температуре окружающей среды. Если по меньшей мере либо R2, либо R3 является C1-6алкильной группой или C1-4алкоксиC1-4алкильной группой, либо CR2R3 является C3-8циклоалкильной группой, то реакцию образования кольца удобно осуществлять посредством обработки соединения формулы (III) кеталем (RO)2CR2R3 (где R является C1-6алкильной группой, например метилом), предпочтительно в присутствии приемлемой кислоты, такой как п-толуолсульфоновая кислота или пиридиний-п-толуолсульфонат, и в приемлемом растворителе, таком как кетон (например, ацетон), нитрил (например, ацетонитрил), или галогенизированный углеводород (например, дихлорметан) примерно при комнатной температуре. Соединения формулы (Ia), в которой X и/или Y представляют собой серу, аналогично могут быть получены из промежуточных соединений, соответствующих соединениям формулы (III), в которой одна или обе диольных гидроксильных группы заменены тиолом. Если CR2R3 представляет собой C=O или C=S, то реакцию образования кольца удобно осуществлять посредством взаимодействия соединения формулы (III) с карбонилдиимидазолом или тиокарбонилдиимидазолом в соответствующем растворителе, таком как углеводород (например, толуол) или эфир (например, тетрагидрофуран), с дефлегмацией. Если же CR2R3 представляет собой C=S, то реакцию образования кольца удобно осуществлять посредством обработки соединения формулы (III) оксидом олова (например, оксидом дибутилолова) в углеводородном растворителе (например, толуоле в состоянии дефлегмации), с последующим добавлением галотионформиата (например, фенилхлортионоформиата) в углеводородном растворителе (например, толуоле) при температуре, близкой к комнатной.
Согласно следующей реализации способа (A), соединение формулы (I), в которой R1 и R4 те же, как определено в приведенной выше формуле (I), а либо R2, либо R3 представляют собой C1-6алкил, а оставшийся радикал представляет собой водород, C1-6алкил или C1-4алкоксиC1-4алкил, (CR5R6)n представляет собой CR5R6, где R5 и R6 те же, как определено в формуле (I), приведенной выше, а X и Y оба представляют собой кислород, может быть получено посредством обработки диола формулы (III) оксидом олова (например, оксидом дибутилолова) в углеводородном растворителе (например, толуоле в состоянии дефлегмации), с последующим добавлением аллилгалида ГалCR5R6CR2=CHR14 (где R14 является водородом или C1-6алкилом, а Гал является галогеном, например бромом) и соли фтора (например, тетрабутиламмонийфторида) в соответствующем растворителе, таком как эфир (например, тетрагидрофуран), и прогревания смеси при температуре в интервале приблизительно от 40oC до 80oC с образованием соединения формулы (IV)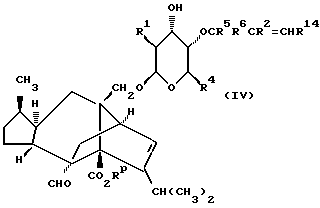
которое может быть превращено в желаемое соединение формулы (Ia) посредством циклизации, включающей внутримолекулярное электрофильное присоединение, индуцируемое ртутной солью (например, трифторацетатом ртути) с последующим гидридным восстановлением, например с помощью гидрида триалкилолова (например, гидрида трибутилолова), а затем удалением карбоксилзащитной группы. Реакцию удобно проводить при температуре, близкой к комнатной, в присутствии подходящего растворителя, такого как эфир (например, тетрагидрофуран).
В другой реализации способа (A) соединение формулы (Ia), в которой R1-R4 те же, как определено выше в формуле (I), n = 0, а либо X, либо Y представляют собой кислород, а оставшийся радикал является CR9R10 (где один из R9 или R10 представляет собой C1-6алкил, а другой представляет собой водород, C1-6алкил или C1-4алкоксиC1-4алкил, либо CR9R10 представляет собой C=CHR11), может быть получено посредством обработки соединения формулы (III) оксидом олова (например, оксидом дибутилолова) в углеводородном растворителе (например, толуоле в состоянии дефлегмации), и возможно в присутствии соли фтора (например, тетрабутиламмонийфторида), а затем либо добавлением к нему аллилгалида ГалCR2R3CR9=CHR14 или акинилгалида ГалCR2R3C≡CR11 (где R11, R14 и Гал те же, как определено выше), либо добавлением аллилгалида или алкинилгалида после защиты одной из диольных гидроксильных групп с образованием соединения формулы (V)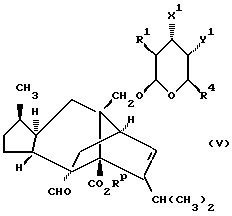
где X1 представляет собой OH, a Y1 представляет собой OCR2R3CR9=CHR14 или OCR2R3C≡CR11 , либо X представляет собой OCR2R3CR9=CHR14 или OCR2R3C≡CR11 , a Y1 представляет собой защищенную гидроксильную группу.
Циклизация может быть завершена посредством первоначального удаления гидроксилзащитной группы, если она имеется, а затем активацией свободной гидроксильной группы, например путем образования S-алкилдитиокарбоната (в частности, S-метилдитиокарбоната), затем замыкания кольца при радикальных условиях, например путем обработки раствора активированного промежуточного соединения в углеводородном растворителе (например, толуоле) при дефлегмации донором водорода, например гидридным восстанавливающим агентом (в частности, гидридом триакилолова, таким как гидрид трибутилолова), в присутствии активирующего агента [например азобис(изобутиронитрила)], а затем удалением карбоксилзащитной группы. Образование S-алкилдитиокарбоната можно легко осуществить посредством обработки соединения формулы (V), в которой один из радикалов X1 и Y1 является OH, а другой является OCR2R3CR9=CHR14 или OCR2R3C≡CR11 , сильным основанием щелочного металла (например, гидридом натрия), в присутствии имидазола и в соответствующем растворителе, таком как эфир (например, тетрагидрофуран) при пониженной температуре (например, около 0oC), а затем добавления дисульфида углерода и алкилгалида (например, метилиодида), при температуре, близкой к комнатной.
В следующей реализации способа A соединение формулы (I), в которой Y является кислородом, n = 0, a R2 и R3 являются водородом, X является группой CR9R10, может быть получено посредством взаимодействия соединения формулы (V), в которой X1 является гидроксилом, с трифенилфосфином, иодом и имидазолом в соответствующем растворителе, таком как тетрагидрофуран, с получением в результате желаемого 4-иодопроизводного (Va). Затем может быть получена желаемая кольцевая система в радикальных условиях, таких как взаимодействие с гидридом триалкилолова в углеводородном растворителе при нагревании.
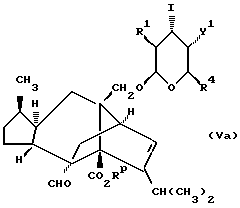
В следующей реализации способа A соединения формулы (I), в которой Y является кислородом, X является NR12, а группа CR2R3 является C=O, могут быть получены в результате реакции соединения иода формулы (Va) с гидридом натрия и изоцианатом R12NCO в апротонном растворителе, таком как тетрагидрофуран, с последующим удалением карбоксилзащитной группы.
В следующей реализации способа (A) соединение формулы (I), в которой R1 и R4 те же, как определено в приведенной выше формуле (I), R2 и R3 каждый независимо представляют собой водород, C1-6алкил или C1-4алкоксиC1-4алкил, либо CR2R3 представляет собой C3-8циклоалкил, n = 0, а один из радикалов X или Y представляет собой -NR12- (где R12 является таким же, как определено в приведенной выше формуле (I)), а другой представляет собой кислород, может быть получено путем N-ациллирования соединения формулы (VI)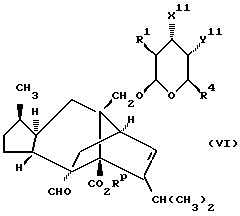
в которой один из радикалов X11 и Y11 является OH, а другой является NH2,
или его защищенного производного, например путем применения такого ацилгалида, как ацилхлорид, в соответствующих условиях с последующей обработкой амида альдегидом или кетоном R2R3C=O либо диалкилацеталем R2R3C(OАлкил)2 в присутствии соответствующей кислоты, такой как п-толуолсульфоновая кислота или п-толуолсульфонат пиридиния, и в соответствующем растворителе, таком как галогенизированный углеводород (например, дихлорметан), при температуре, близкой к комнатной, а после этого удаления любых присутствующих защитных групп.
В другой реализации способа (A) соединение формулы (Ia), в которой R1 и R4 тоже, как определено в приведенной выше формуле (I), n = 0, -X-CR2R3 или -Y-CR2R3 представляют собой -N= CR3- (где R3 является C1-6алкилом или C1-4алкоксиC1-4алкилом), а оставшийся X или Y является кислородом, может быть получено путем обработки соединения формулы (VI) иминоэфиром R3C(=NH)OR (где R является C1-6алкильной группой, например метилом).
Согласно общему способу Б соединения формулы (I), в которой Z является группой (a), a Y является кислородом, n = 0, R2 и R3 являются водородом, X является CH, могут быть получены в результате реакции соединения формулы (VII)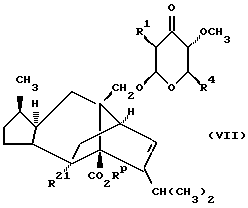
в которой R21 является группой CHO или ее защищенным производным, Rp является защищенной карбоксильной группой, с диалкилом диазометилфосфонатом в присутствии основания, такого как трет-бутоксид калия, в апротонном растворителе, таком как эфир (например, тетрагидрофуран), с последующим удалением карбоксил- защитной группы Rp и, если это необходимо, альдегидзащитной группы, 4-кетотетрагидропирановое производное (VII) может быть получено путем окисления соответствующего 4-гидроксильного производного обычным способом, например посредством Swern окисления.
Общий способ (B) получения соединения формулы (I), в которой Z является группой (б), a W является кислородом, включает циклизацию соединения формулы (VIII)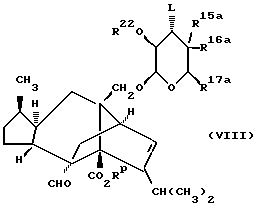
в которой R15a, R16a и R17a имеют те же значения, которые определены для R15, R16 и R17 в приведенной ранее формуле (I), либо являются их защищенными производными, Rp является водородом или карбоксилзащитной группой, L является соответствующей уходящей группой, такой как алкильная или арилсульфонилоксильная группа, а R22 является водородом, либо OR22 является такой же группой, которая определена для L,
с последующим, если это необходимо, удалением любых присутствующих защитных групп.
Реакцию циклизации удобно осуществлять посредством обработки соединения формулы (VIII) сильным основанием, таким как гидрид щелочного металла (например, гидридом натрия), в соответствующем растворителе, таком как диметилформамид или эфир (например, тетрагидрофуран), обычно при комнатной температуре. Кроме того, можно применять натрий в спиртовом растворителе (например, метаноле), особенно если OR22 является такой же группой, которая определена для L. В этом случае основную систему добавляют к раствору соединения формулы (VIII) в соответствующем растворителе, таком как галогенизированный углеводород (например, дихлорметан), реакцию проводят обычно при температуре от комнатной до температуры дефлегмации.
Другой общий способ (Г) получения соединения формулы (I), в которой W является серой, включает обработку соединения формулы (IX)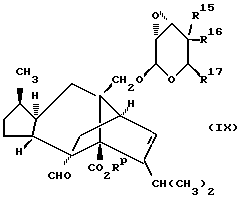
в которой R15, R16 и R17 имеют те же значения, которые определены для них в приведенной выше формуле (I),
донором серы. Так например реакцию удобно осуществлять путем обработки соединения формулы (IX) 5,5-диметил-2-тиоло-2-тиоксо-1,2,3- диоксафосфоринаном в таком растворителе, как диметилформамид, предпочтительно в присутствии соответствующего основания, такого как триалкиламин (например, триэтиламин), при повышенной температуре (например, от 80 до 120oC).
Соединение формулы (IX) может быть легко получено из соединения формулы (VIII), в которой L является гидроксильной группой, a OR22 представляет собой приемлемую уходящую группу, такую как алкильная или арилсульфонилоксильная группа, применяя условия, описанные в способе (В), с последующим, если это необходимо, удалением любых присутствующих защитных групп.
Другой общий способ (Д) включает реакцию взаимного превращения изомеров, когда соединения формулы (I) получают из другого соединения формулы (I) или его защищенного производного.
Согласно первой реализации способа (Д) карбоксилзащищенное производное соединения формулы (I), в котором Z является группой (a) и в которой R1 является гидроксилом, может быть превращено в соответствующее соединение формулы (I), в котором R1 является водородом, посредством процесса, включающего (i) образование S-алкилдитиокарбоната согласно способу, описанному ранее, и (ii) удаление этой группы путем обработки раствора промежуточного соединения в углеводородном растворителе (например, толуоле) при повышенной температуре (например, от 80 до 120oC) гидридным восстанавливающим агентом (например, гидридом триакилолова, таким как гидрид трибутилолова), а затем удаление карбоксилзащитной группы.
В следующей реализации способа (Д) соединение формулы (I), в которой Z является группой (a), в которой R1 является C1-4алкилоксиметилом и/или R4 является C1-4алкоксиметилом, может быть получено путем алкилирования защищенного производного соединения формулы (I), в которой R1 является гидроксилом и/или R4 является гидроксиметилом, а все лабильные группы (например, карбоксильные и гидроксильные группы) являются защищенными, с последующим удалением присутствующих защитных групп. Алкилирование удобно проводить посредством исходной реакции с основанием сильного щелочного металла (например, гидрида натрия), а затем с алкилгалидом (например, метилиодидом). Реакция может быть осуществлена в соответствующем растворителе, таком как эфир (например, тетрагидрофуран), при температуре в пределах интервала приблизительно от 0 до 30oC.
В другой реализации способа (Д) соединение формулы (I), в которой R1 является ацилоксилом и/или R4 является CH2OCOR8, может быть получено путем ациллирования защищенного производного соединения формулы (I), в которой R1 является гидроксилом и/или R4 является CH2OH и все лабильные группы (например, карбоксильные и гидроксильные группы) являются защищенными, с последующим удалением любых присутствующих защитных групп. Реакцию ациллирования можно проводить традиционными способами, например путем обработки карбоновой кислотой в присутствии активирующего агента, такого как дициклогексилкарбодиимид, и соответствующего основания, такого как диметиламинопиридин, либо применяя кислый галид (например, кислый хлорид), возможно в присутствии соответствующего основания, такого как пиридин или 4-диметиламинопиридин.
Согласно следующей реализации способа (Д), соединение формулы (I), в которой R1 является гидроксильной группой в экваториальной конфигурации, может быть получено из защищенного производного соединения формулы (I), в которой R1 является гидроксильной группой в аксиальной конфигурации и все лабильные группы (например, карбоксильные, гидроксильные и CHO группы) являются защищенными, с последующим удалением любых присутствующих защитных групп. Реакцию изомеризации можно легко осуществить посредством двуступенчатого способа, включающего (i) окисление 2'-аксиальной OH в оксогруппу путем обработки подходящей окисляющей системой [например, оксидом хрома в присутствии пиридина в таком растворителе как галогенизированный углеводород (в частности, дихлорметан), содержащем уксусный ангидрид] и (ii) восстановление оксогруппы до экваториальной ОН группы с помощью подходящего восстанавливающего агента, такого как бороводород (например, бороводород натрия). Восстановление может быть легко осуществлено в подходящем растворителе, таком как спирт (например, водный метанол), при температуре в интервале приблизительно от 0 до 10oC.
В другой реализации способа (Д) соединение формулы (I), в которой R1 является атомом галогена, может быть получено из защищенного производного соединения формулы (I), в которой R1 является гидроксильной группой, с последующим удалением любых присутствующих защитных групп. При инверсии конфигурации имеет место реакция замещения. Так, например, аксиальная OH группа
легко может быть превращена в экваториальный атом иода посредством добавления иода к раствору исходного материала в соответствующем растворителе, таком как углеводород (например, толуол), в присутствии трифенилфосфина и иода, а затем прогревания смеси (например, с дефлегмацией).
Аналогично может быть введен атом фтора путем обработки соответствующим фторирующим агентом, таким как трифторид диэтиламиносеры (ДАСТ), в подходящем растворителе, таком как галогенизированный углеводород (например, дихлорметан) или ароматический углеводород (например, толуол), при температуре, близкой к комнатной.
В следующей реализации способа Д соединение формулы (I), где Y является кислородом, n = 0, R1 и R2 являются водородом, а X является CH, или его защищенное производное может быть превращено в соответствующее соединение, у которого X становится CH2, в результате восстановления водородом в присутствии соответствующего катализатора, например палладия на угле, в подходящем растворителе с последующим удалением карбоксилзащитной группы.
В другой реализации способа Д соединение формулы (I), где Y является кислородом, n = 0, R2 и R3 являются водородом, а X является группой C=CH2, может быть превращено в соответствующее соединение, у которого X становится группой C= O в результате окисления например с помощью четырехокиси осмия и периодата натрия.
Аналогичным образом соединения формулы (I), где Y является кислородом, n = 0, R2 и R3 являются водородом, а X является группой C=CH2, могут быть превращены в соответствующее соединение формулы (I), в которой R2 и R3 и атом углерода, к которому они присоединены, превращаются в группу C=O в результате окисления например с помощью трехокиси хрома в пиридине.
Согласно следующей реализации способа (Д), карбоксилзащищенное производное соединения формулы (I), в которой R15 является гидроксилом, может быть превращено в соответствующее соединение формулы (I), в которой R15 является галогеном, посредством стандартной реакции замещения с последующим удалением карбоксилзащитной группы. Так например, замещение гидроксила атомом фтора легко может быть осуществлено с инверсией конфигурации путем добавления трифторида диэтиламиносеры (ДАСТ) к раствору исходного материала в таком растворителе, как галогенизированный углеводород (например, дихлорметан). Реакция легко проходит при комнатной температуре.
В следующей реализации способа (Д) соединение формулы (I), в которой R15 является C1-6алкоксильной или возможно замещенной алкоксильной группой и/или R17 является C1-4алкоксиметилом, может быть получено путем алкилирования защищенного производного соединения формулы (I), в которой R15 и/или R17 содержит свободную гидроксильную группу и любые лабильные группы (например, карбоксильные и гидроксильные группы) являются защищенными, если это целесообразно, с последующим удалением присутствующих защитных групп. Алкилирование легко может быть осуществлено в результате исходной реакции с основанием сильного щелочного металла (например, гидридом натрия), а затем с алкилгалидом (например, метилибромидом). Реакция может быть проведена в соответствующем растворителе, таком как эфир (например, тетрагидрофуран), при температуре в пределах интервала приблизительно от 0oC до 50oC. Если это целесообразно, может присутствовать соль аммония, такая как тетраалкиламмонийгалид (например, тетрабутиламмонийиодид).
С другой стороны, прямая или разветвленная алкильная группа легко может быть введена в две стадии, первая стадия включает алкенилирование с помощью подходящего алкенилгалида в присутствии такого основания, как карбонат (например, карбонат цезия), в таком растворителе, как диметилформамид, при температуре, близкой к комнатной, а вторая стадия включает процесс галогенизации в присутствии палладиевого катализатора (например, 10%-ный палладий на угле) при температуре, близкой к комнатной.
В другой реализации способа (Д) соединения формулы (I), в которой R15 является CH2OCOR20 или R1 является OCOR18, может быть получено путем ацилирования защищенного производного соединения формулы (I), в которой R17 является CH2OH или R15 является гидроксилом, а любые лабильные группы (например, карбоксильные или гидроксильные группы) являются защищенными, с последующим удалением любых присутствующих защитных групп. Реакция ацилирования может быть проведена обычным способом, например посредством обработки карбоновой кислотой в присутствии активирующего агента, такого как дициклогексилкарбодиимид, и соответствующего основания, такого как диметиламинопиридин, либо путем использования галогенангидрида (например, хлорангидрида кислоты), возможно в присутствии подходящего основания, такого как пиридин или диметиламинопиридин.
Согласно следующей реализации способа (Д), соединение формулы (I), в которой CR15R16 представляет собой C=O, может быть получено посредством окисления защищенного производного соединения формулы (I), в которой R15 является гидроксильной группой, а все лабильные группы (например, карбоксильные и гидроксильные группы) являются защищенными, с последующим удалением любых присутствующих защитных групп. Реакция окисления легко может быть осуществлена путем добавления соответствующего окисляющего агента, такого как диметилсульфоксид, в присутствии трифторуксусного ангидрида. Окисление легко проходит в присутствии подходящего растворителя, такого как галогенизированный углеводород (например, дихлорметан), при повышенной температуре (например, приблизительно от 40 до 80oC).
Соединения формулы (I), где CR15R16 представляет собой группу CH=CH2, могут быть получены в результате реакции соответствующего соединения формулы (I) или его защищенного производного, где CR15R16 является группой C=O, с алкилтрифенил-фосфонийгалидом и алкиллитием в апротонном растворителе, таком как тетрагидрофуран.
Соединения формулы (I), где R15 является азидной группой, могут быть получены из соответствующего соединения формулы (I) или его защищенного производного, где R15 является гидроксильной группой, в результате реакции с толуолсульфонилгалидом с последующей обработкой образовавшегося толуолсульфонатного производного азидом щелочного металла, например азидом лития, в апртонном растворителе, например дихлорметане.
Многие из вышеупомянутых способов для получения желаемого соединения формулы (I) требуют удаления одной или более защитных групп в качестве заключительной стадии. Так, следующий общий способ (Е) включает снятие защиты с защищенного производного соединения формулы (I). Подходящие для применения в нем карбоксилзащитные группы и гидроксилзащитные группы включают любые обычные защитные группы [4,5]. Примеры приемлемых карбоксилзащитных групп включают арилалкилные группы, такие как дифенилметильные, п-метоксибензильные и силильные группы (например, триметилсилилэтил или t-бутилдиметилсилил). Примеры приемлемых гидроксилзащитных групп включают арилалкильные группы, такие как п-метоксибензильные и эфирные группы, такие как бензилоксикарбонил. Альдегидные группы обычно могут быть защищены в форме циклических кеталей.
Защитные группы могут быть удалены стандартными способами. Так, дифенилметильная группа может быть легко удалена с помощью трифторуксусной кислоты или путем гидрогенолиза в присутствии палладиевого катализатора (например, 10%-ный палладий на угле). Бензилоксикарбонильная группа легко может быть удалена посредством гидрогенолиза в присутствии палладиевого катализатора (например, 10%-ный палладий на угле). П-метоксибензильная группа может быть легко удалена с помощью 2,3-дихлор-5,6-дициа-но-1,4-бензохинона. Силильные группы, такие как триметилсилилэтил или t-бутилдиметилсилил могут быть легко удалены с помощью ионов фтора. Группы циклических кеталей могут быть превращены в альдегидную группу посредством добавления соответствующей кислоты, такой как соляная кислота.
Соединения формулы (II) могут быть легко получены посредством реакции соединения формулы (X)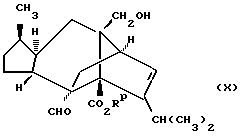
где Rp является карбоксилзащитной группой,
с соединением формулы (XI)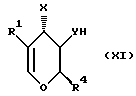
где R1, R4, X и Y такие же, как определено в приведенной выше формуле (II) или его защищенным производным с последующим удалением, если это необходимо, любых присутствующих защитных групп. Эта реакция может быть легко осуществлена путем нагревания (X) и (XI) при температуре в интервале приблизительно от 40oC до 80oC в соответствующем растворителе, таком как углеводород (например, толуол), в присутствии кислоты, такой как бромоводородная кислота-трифенилфосфин.
Если XH и YH в формуле (XI) представляют собой гидроксильные группы, то они могут быть легко защищены как ацилоксильные (например, ацетоксильные) группы, с удалением защитных групп после реакции с соединением формулы (X). Удаление ацетоксилзащитных групп легко может быть осуществлено путем добавления подходящего основания, такого как алкококсид (например, метоксид натрия), в соответствующем растворителе, таком как спирт (например, метанол) при температуре, близкой к комнатной.
Вышеупомянутая реакция наиболее пригодна для получения соединений формулы (II), в которой R4 не является метильной группой. Если R4 является метильной группой, то может быть более целесообразным получать соединения формулы (II) из 4'- диметил-сордарина, соединения формулы (XII)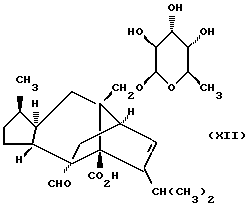
Так, например, соединения формулы (III), в которой R4 представляет собой метил, могут быть получены путем защиты карбоксильной группы в (XII) соответствующими способами, с последующим, если это необходимо, превращением 2'-аксиальной гидроксильной группы в другую R1 группу посредством способов взаимного превращения, описанных выше. Может оказаться необходимым защитить 3' и 4' гидроксильные группы при манипуляциях с 2-аксиальной гидроксильной группой. Защиту легко можно осуществить путем образования изопропилиденовой группы посредством процесса, описанного в первой реализации способа (A). Последующее удаление этой группы для обеспечения диольной функции может быть достигнуто путем обработки такой кислотой, как неорганическая кислота (например, соляная кислота).
Если карбоксильная группа в соединении формулы (XII) защищена дифенилметильной группой, то реакция защиты легко может быть осуществлена путем обработки раствора соединения формулы (XII) в спиртовом растворителе (например, метаноле) и/или в галогенизированном углеводороде (например, дихлорметане) дифенилдиазометаном, который удобно добавлять в виде раствора в галогенизированном углеводородном растворителе (например, дихлорметане).
Соединения формулы (II), где X и Y представляют собой кислород или серу, также могут быть получены путем декарбоксилирования соответствующего защищенного производного соединения формулы (I), в которой CR2R3 представляет собой C= O, а n равно нулю. Декарбоксилирование может быть легко осуществлено посредством добавления алкоксильного соединения (например, CH3ONa) в соответствующем растворителе, таком как спирт (например, метанол).
Соединения формулы (X) могут быть легко получены из сордарицина, применяя обычный способ защиты карбоксильной группы. Так, например, если Rp в соединении формулы (X) представляет собой триметилсилилэтил, то эта группа может быть введена посредством обработки сордарицина O-[2-(триметилсилил)этил] -N,N'- диизопропилизомочевиной, обычно в соответствующем растворителе, таком как эфир (например, тетрагидрофуран) при повышенной температуре (например, с дефлегмацией). Если Rp представляет дифенилметил, то эта группа может быть введена способом, описанным выше.
Если гидроксильная группа в карбоксилзащищенном производном сордарина или соединении формулы (XII) защищена п-метоксибензильной группой, то эта группа может быть введена посредством реакции с оксидом олова (например, оксидом дибутилолова) в углеводородном растворителе (например, толуоле в состоянии дефлегмации) с последующим добавлением п-метоксибензилгалида в присутствии соли фтора (например, тетрабутиламмонийфторида). Если эта группа защищена бензилоксикарбонильной группой, то она может быть введена посредством реакции с бензилгалоформиатом в присутствии соответствующего аминного основания, такого как 4-диметиламинопиридин, в таком растворителе, как галогенизированный углеводород (например, дихлорметан) или ацетонитрил.
Образование группы L в формуле (VIII), где L является алкил- или арилсульфонилоксильной группой и/или группой R22O, где R22O является алкил- или арилсульфонилоксильной группой, может быть осуществлено в результате реакции соответствующего защищенного производного сордарина или соединения формулы (XII) с алкил- или арилсульфонилгалидом в присутствии соответствующего растворителя, такого как пиридин, и возможно также содержащего аминное основание (например, 4-диметиламинопиридин), или в галогенизированном углеводородном растворителе (например, дихлорметане) в присутствии соответствующего аминного основания (например, 4-диметиламинопиридине). Реакция может быть легко осуществлена при комнатной температуре. Другие группы могут быть введены аналогичным образом посредством обычных способов.
Следует отметить, что может оказаться целесообразным превращение гидроксильной группы в желаемую R15 группу перед циклизацией согласно способу (В). Так, соответствующим образом защищенное производное 4'-деметилсордарина или соединение формулы (II), в которой R15a является гидроксилом, посредством обычных способов могут быть подвергнуты обработке, в результате которой 4'-гидроксильная группа превращается в желаемую группу R1. Так, например, превращение в C1-6алкоксильную, C1-4алкоксиC1-4алкоксильную или арилC1-4алкоксильную группу может быть осуществлено посредством обычного алкилирования, например согласно различным методам, описанным здесь выше. Удаление гидроксильной группы для получения соединения, в котором R15 является водородом, легко может быть осуществлено в две стадии, включающие (i) образование S-алкилдитиокарбоната путем обработки основанием сильного щелочного металла (например, гидридом натрия), в присутствии имидазола, и в соответствующем растворителе, таком как эфир (например, тетрагидрофуран), при пониженной температуре (например, около 0oC), а затем добавления дисульфида углерода и алкилгалида (например, метилиодида) при температуре, близкой к комнатной, и (ii) удаления этой группы посредством обработки раствора промежуточного соединения в углеводородном растворителе (например, толуоле) при повышенной температуре (например, приблизительно от 80 до 120oC) с гидридным восстанавливающим агентом (например, гидридом триалкилолова, таким как гидрид три-бутилолова), возможно в присутствии активирующего агента [например азобис(изобутиронитрила)] . Превращение в C2-6алкильную группу может легко быть осуществлено посредством (i) образования S-алкилдитиокарбоната, как описано ранее, (ii) замещения этой группы алкенильной группой в результате реакции с соединением триалкилалкенилолова при условиях, описанных выше для удаления s-алкилдитиокарбонатной группы и (iii) восстановления алкенильной группы в алкильную группу посредством, например, гидрогенизации в присутствии подходящего палладиевого катализатора (например, 10%-ного палладия на угле).
Основные соли соединений формулы (I) могут быть легко образованы посредством обработки соединения формулы (I) соответствующей солью или основанием. Так например, соли могут быть легко получены путем обработки соединения формулы (I) солью или основанием, выбранным из гидроксида, гидрокарбоната, карбоната или ацетата калия или натрия (например, гидроксидом калия, гидрокарбонатом калия, гидрокарбонатом натрия или ацетатом калия), ацетата аммония, ацетата кальция и L-лизина, если это целесообразно. Соль может, например, быть получена путем добавления соответствующей соли или основания (если необходимо в виде водного раствора) к раствору или суспензии соединения формулы (1) в подходящем растворителе, таком как спирт (например, метанол) или диоксане при температуре, например, от 0oC до 80oC, а обычно при комнатной температуре.
Фармацевтически приемлемые соли могут также быть получены стандартными методами из других солей, включая другие фармацевтически приемлемые соли соединений формулы (I).
Метаболически лабильные сложные эфиры соединений формулы (I) могут быть образованы с присутствующей в этих соединениях карбоксильной группой, и они могут быть получены стандартными способами. Аналогично метаболически лабильные сложные эфиры могут также быть образованы с любой свободной гидроксильной группой, присутствующей в молекуле.
Новое соединение формулы (XII) может быть легко получено в результате описанного ниже ферментационного процесса либо посредством деметилирования сордарина в результате процесса биотрансформации.
Ферментационный процесс включает культивирование микроорганизма, способного продуцировать соединение формулы (XII) с последующим выделением соединения формулы (XII) из культуры.
Микроорганизмы, способные продуцировать соединение формулы (XII) обычно являются мутантными штаммами Sordaria araneosa, которые могут быть с легкостью идентифицированы посредством скриннинга выживших в результате мутагенеза организмов путем анализирования опытного образца, полученного после ферментации микроорганизма, применяя стандартную методологию. В частности, микрорганизмом, пригодным для использования, является мутантный штамм Sordaria araneosa, депонированный в коллекции типовых культур CAB International Mycological Institute, Genetic Resource Reference Collection, Bakeham Lane, Egham, Surrey TW20 9TY, England. Этот штамм был получен институтом 10 июня 1994 года и ему впоследствии был присвоен каталожный номер IMI 362184 и даты приема и подтверждения жизнеспособности - 13 июня и 21 июня 1994 года, соответственно. Данный институт является Международным Органом по Депонированию согласно Будапештскому Договору. Характеристики, уже идентифицированные к настоящему времени для IMI 362184, приведены в Примере 74.
В следующем своем аспекте настоящее изобретение предлагает микрорганизм IMI 362184 как таковой и его мутанты.
Мутанты IMI 362184 могут возникать спонтанно или могут продуцироваться различными известными способами [6] . Такие способы включают ионизирующее облучение, химические методы, например обработку N-метил-N'-нитро-N-нитрозогуанидином (НТГ), нагревание, генетические методы, такие как рекомбинация и трансформация, и селективные методы для спонтанных мутантов.
Получение соединения формулы (XII) посредством ферментации может быть осуществлено обычными способами, например путем культивирования соответствующего организма в присутствии ассимилируемых источников углерода, азота и минеральных солей с последующим выделением искомого продукта.
Ассимилируемые источники углерода, азота и минералов могут вводиться в виде либо простых, либо комплексных источников питания. Источники углерода обычно включают глюкозу, мальтозу, крахмал, глицерин, мелассу, декстрин, лактозу, сахарозу, фруктозу, галактозу, мезо-инозит, D-маннит, соевое масло, карбоновые кислоты, аминокислоты, глицериды, спирты, алканы и растительные масла. Источники углерода обычно составляют от 0,5 до 10% от веса ферментационной среды. Фруктоза, глюкоза и сахароза представляют собой предпочтительные источники углерода.
Источники азота обычно включают отходы (жмых) бобов, кукурузный экстракт, барду, дрожжевые экстракты, отходы семян хлопчатника, пептоны, жмых молотого арахиса, солодовый экстракт, мелассы, казеин, смеси аминокислот, аммоний (газ или раствор), аммонийные соли или нитраты. Также могут быть использованы мочевина и другие амиды. Источники азота обычно составляют от 0,1 до 10% от веса ферментационной среды.
Питательные минеральные соли, которые могут быть введены в культуральную среду, включают используемые обычно соли, способные образовывать ионы натрия, калия, аммония, железа, магния, цинка, никеля, кобальта, марганца, ванадия, хрома, кальция, меди, молибдена, бора, фосфатные, сульфатные, хлоридные и карбонатные ионы.
Культивирование микроорганизма обычно осуществляют при температуре от 20 до 40oC, предпочтительно от 20 до 35oC, особенно при 25oC, желательно с аэрацией и перемешиванием, например встряхиванием и перемешиванием с помощью мешалки. Среда может изначально быть инокулирована небольшим количеством мицелия и/или спор. Полученный вегетативный инокулюм может быть перенесен в ферментационную среду либо в одну или более посевных установок, если будет иметь место дальнейший рост перед переносом в основную ферментационную среду. Ферментацию обычно проводят в интервале pH от 3,5 до 9,5, предпочтительно от 4,5 до 7,5. Может оказаться необходимым добавлять в ферментационную среду основание или кислоту для поддержания pH в рамках требуемого интервала. Приемлемые основания, которые могут быть добавлены, включают гидроксиды щелочных металлов, такие как водный гидроксид натрия или водный гидроксид калия. Приемлемые кислоты включают минеральные кислоты, такие как соляная, серная или фосфорная кислота.
Ферментация может продолжаться в течение 4-30 суток, предпочтительно около 5-15 суток. Для контроля избыточного пенообразования может присутствовать пеногаситель, добавляемый по мере необходимости. Источники углерода и/или азота также могут добавляться в ферментационную среду по мере необходимости.
Соединение формулы (XII) ассоциировано главным образом с клетками и может быть переведено в раствор либо посредством добавления кислоты и смешивающегося с водой органического растворителя, или, что более предпочтительно, путем добавления основания (например, гидроксида натрия). Клетки могут быть отделены от этих растворов либо посредством центрифугирования с последующей фильтрацией, либо посредством мембранной фильтрации. Жидкость может быть затем подвергнута обработке кислотой, такой как серная кислота, до тех пор, пока pH не станет ниже 6 (например, около 4,5).
Соединение формулы (XII) может быть выделено и очищено с помощью различных способов фракционирования, например абсорбции-элюции, осаждения, кристаллизации, экстракции растворителем и фракционирования жидкость-жидкость, которые могут быть скомбинированы различным образом.
Было установлено, что абсорбция на твердой подложке с последующей элюцией является наиболее приемлемой для выделения и очистки соединения формулы (IX).
Приемлемые твердые подложки включают кремний; нефункциональные макросетчатые адсорбционные смолы, например такие перекрестносшитые стирол-дивинил-бензольные полимерные смолы, как CG161 и Amberlite XAD-2, XAD-4, XAD-16 или XAD-1180 смолы (Rohm & Haas Limited) или Kastell S112 (Montedison); замещенный стирол-дивинил-бензольный полимер, такой как Diaion SP207 (Mitsubishi); анионообменник [например IRA-958 или MacroPrep High Q (BioRad)] , совместимый с органическим растворителем перекрестносшитый декстран, такой как Sephadex LH20 (Pharmacia UK Limited), или подложки с обращенной фазой, такие как сшитый с углеводородом кремний, например C18-сшитый кремний.
Соединение формулы (XII) может также быть выделено и очищено посредством применения такого анионообменника, как LA 2.
Выбор приемлемых растворителей для элюции соединения формулы (XII) будет конечно зависеть от природы адсорбента. При использовании полимерной смолы, такой как XAD-16, наиболее приемлемыми могут быть смешиваемые с водой растворители, такие как метанол, ацетон, изпропанол или ацетонитрил в различных пропорциях в воде.
Наличие соединения формулы (XII) в ходе процессов экстракции/выделения можно контролировать традиционными способами, такими как жидкостная хроматография высокого разрешения (ЖХВР) или УФ спектроскопия, либо посредством использования оптического вращения или другого свойства соединения.
Если соединение формулы (XII) получают в форме раствора в органическом растворителе, например после очистки посредством абсорбции/элюции, растворитель может быть удален традиционными способами, например выпариванием, получая в результате искомое соединение. Если требуется, соединение может быть очищено далее хроматографическими методами, такими как противоточная хроматография с применением экстрактора в виде змеевика, такого как многослойный змеевиковый экстрактор, либо жидкостная хроматография высокого разрешения или надкритическая жидкостная хроматография на таких адсорбентах, как углерод, алюминий, ванадий, полимерные смолы или кремний, со связанными фазами или без них. Выбор приемлемых растворителей/элюентов для хроматографической очистки/разделения соединения формулы (XII) будет конечно зависеть от природы адсорбента. При использовании C8 связанного кремния наиболее приемлемы смеси ацетонитрила и воды. С другой стороны, соединение может быть очищено далее посредством экстракции растворителем, например используя соответствующий органический растворитель, такой как кетон (в частности, ацетон или метилэтилкетон), галогенизированный углеводород, спирт (например, метанол), диол (например, пропан-1,2-диол или бутан-1,3-диол), либо сложный эфир (например, метилацетат или этилацетат). Кроме того, растворы соединения (XII) могут быть очищены далее посредством обработки адсорбентами, которые селективно удаляют примеси при добавлении в соответствующих количествах (например, ДЕАЕ-целлюлоза), или посредством кристаллизации (например, из смеси ацетонитрила и воды), либо путем использования сочетания указанных процессов.
Биотрансформация сордарина в 4'-деметилсордарин, соединение формулы (XII), может быть осуществлена путем инкубации сордарина в культуре, содержащей соответствующий организм и источники углерода и азота, включая источники, описанные ранее, с последующим выделением соединения формулы (XII) из культуры.
Микроорганизмы, способные деметилировать сордарин по 4'-положению, могут быть с легкостью идентифицированы путем применения мелкомасштабного теста и анализа полученного опытного образца, применяя стандартную методологию, например, применяя ЖХВР. Примеры микроорганизмов, которые были идентифицированы как деметиляторы сордарина, включают Streptomyces capreolus ATCC 31963, Streptomyces avermitilis ATCC 31272, Streptomyces armentosus NRRL.3176, Streptomyces antibioticus ATCC 31771,
Streptomyces rimosus ATCC 23955, Streptomyces platensis ATCC 29778, Streptomyces mashuensis ATCC 23934, Streptomyces eurythermus ATCC 14975, Nocardia orientalis ATCC 43491 и Cunninghamella echinulata var elegans 36112.
Культивирование организма обычно проводят при температуре от 20 до 40oC, предпочтительно от 20 до 35oC, особенно около 28oC, и обычно желательно при аэрации и перемешивании, например путем встряхивания или перемешивания с помощью мешалки. Среда может быть изначально инокулирована небольшим количеством мицелия и/или спор. Полученный вегетативный инокулюм может быть перенесен в ферментационную среду, либо в одну или более посевных установок, где происходит дальнейший рост (например, в течение 1-3 дней) перед переносом в основную ферментативную среду. Основная ферментативная среда также будет содержать сордарин, а ферментацию обычно проводят при pH в интервале от 3,5 до 9,5, предпочтительно от 4,5 до 7,5. Может оказаться необходимым добавлять в ферментационную среду основание или кислоту для поддержания pH в рамках требуемого интервала. Приемлемые основания, которые могут быть добавлены, включают гидроксиды щелочных металлов, такие как водный гидроксид натрия или гидроксид калия. Пригодные кислоты включают минеральные кислоты, такие как соляная, серная или фосфорная. Ферментация может продолжаться в течение периода от 2 до 5 суток, предпочтительно около 3 суток. Для контроля избыточного пенообразования может присутствовать пеногаситель, который добавляют по мере необходимости. Источники углерода и азота также могут добавляться в ферментационную среду по мере необходимости.
Разделение и выделение соединения формулы (XII) из ферментационной питательной среды может быть осуществлено традиционными методами, описанными ранее. Если необходимо понизить pH жидкости ниже pH 6 (например, до примерно 2,5), то это может быть легко достигнуто путем добавления такой кислоты, как ортофосфорная кислота.
Следует отметить, что биотрансформация может быть осуществлена несколькими различными способами. Например, клетки могут быть выращены и собраны перед добавлением раствора сордарина, например, в буфере, истощенной ферментационной среде или воде. Вероятно также, что могут быть выделены и использованы соответствующие ферменты (с соответствующими коферментами) либо эти ферменты могут быть клонированы и переэкспрессированы.
Как уже отмечалось ранее, сордарин и сордарицин являются известными соединениями, которые могут быть получены способами, описанными в литературе. Так, например, описано [1] получение сордарицина путем культивирования Sordaria araneosa NRRL 3196 (депонированной также в ATCC как ATCC 36386). Конкретные примеры получения сордарина с применением сходных методов описаны ниже.
Сордарицин может быть легко получен в ферментационных условиях, сходных с условиями, описанными для получения сордарина с помощью Sordaria araneosa NRRL 3196 или ее подходящего мутанта, с выделением искомого соединения соответствующими хроматографическими средствами. Один такой мутант был депонирован в коллекции типовых культур CAB International Mycological Institute, Genetic Resource Reference Collection, Bakehem Lane, Egham, Surrey TW20 9TY, England. Этот штамм был получен и принят институтом 11 августа 1994 года, затем ему был присвоен каталожный номер IMI 362947 с датой подтверждения жизнеспособности 19 августа 1994 года. Данный институт является Международным Органом по Депонированию согласно Будапештскому Договору. Характеристики, идентифицированные для IMI 362947, приведены в Примере 75.
Настоящее изобретение предусматривает в своем следующем аспекте микроорганизм IMI 362947 как таковой и его мутанты.
Способы получения мутантов IMI 362947 и их генетического материала обычно схожи со способами, описанными выше для обработки IMI 362184.
Сордарицин может также быть получен из сордарина посредством биотрансформации. Биотрансформация может быть легко осуществлена путем инкубации сордарина в культуре, содержащей подходящий организм и источники углерода и азота, включая упомянутые выше источники, с последующим выделением сордарицина из культуры.
Микроорганизмы, способные превращать сордарин в сордарицин, могут быть с легкостью идентифицированы с помощью мелкомасштабного теста и анализирования опытного образца, полученного стандартным способом, например с использованием ЖХВР. Мы идентифицировали один такой микроорганизм и депонировали его в National Collections of Industrial and Marine Bacterial Limited (NCIMB), 23 St. Machar Drive, Aberdeen AB2 IRY, Scotland. Штамм был получен NCIMB 4 августа 1994 года и в тот же день был занесен в депозит для целей патентной процедуры, и была подтверждена жизнеспособность микроорганизма. Микроорганизму, который является видом Coryneform, обладающим характеристиками, приведенными в Примере 76, был присвоен каталожный номер NCIMB 40675. NCIMB является Международным Органом по Депонированию, согласно Будапештскому Договору.
Изобретение таким образом предусматривает в еще одном своем аспекте микроорганизм NCIMB 40675 как таковой и его мутанты.
Согласно следующему аспекту настоящего изобретения, мы предусматриваем генетический материал NCIMB 40675 и его мутанты, которые принимают участие в биоконверсии сордарина в сордарицин.
Способы получения мутантов NCIMB 40675 и их генетического материала схожи со способами, описанными выше для работы с IMI 362184.
Культивирование NCIMB 40675 обычно проводят при температуре от 20 до 40oC, предпочтительно от 20 до 35oC, особенно около 28oC, и желательно при аэрации и перемешивании, например, посредством встряхивания или перемешивания с помощью мешалки. Среда изначально может быть инокулирована небольшим количеством мицелия и/или спор. Полученный вегетативный мицелий может быть перенесен в ферментационную среду или в одну или более посевных установок, если имеет место дальнейший рост (например, около 1-3 дней) перед переносом в основную ферментационную среду. Основная ферментационная среда содержит также сордарин, а ферментацию проводят обычно в интервале pH от 3,5 до 9,5, предпочтительно около 7,5. Может оказаться необходимым добавлять в ферментационную среду основание или кислоту для поддержания pH в пределах заданного интервала. Приемлемые основания, которые могут быть добавлены, включают гидроксиды щелочных металлов, такие как водный гидроксид натрия или гидроксид калия. Приемлемые кислоты включают минеральные кислоты, такие как соляная, серная или фосфорная. Ферментацию можно проводить в течение периода от 4 до 8 суток, предпочтительно около 6 суток. Для контроля избыточного пенообразования может присутствовать пеногаситель, добавляемый по мере необходимости. Источники углерода и азота также могут добавляться в ферментационную среду по мере необходимости.
Следует отметить, что биотрансформацию можно осуществлять несколькими различными способами. Например, клетки могут выращиваться и собираться перед добавлением раствора сордарина, например в буфере, истощенной ферментационной среде или воде. Вероятно также, что могут быть выделены и использованы соответствующие ферменты, либо эти ферменты могут быть клонированы и переэкспрессированы.
Разделение и выделение сордарицина из ферментационной среды может быть осуществлено описанными ранее традиционными способами. Если требуется понизить pH жидкости примерно до pH 6, то этого легко можно достигнуть путем добавления такой кислоты как ортофосфорная.
Следует понимать, что описанные выше для получения сордарицина процессы ферментации и биоконверсии составляют следующие аспекты настоящего изобретения.
Приводимые ниже примеры иллюстрируют аспекты настоящего изобретения, но никоим образом не ограничивают его.
ПРЕПАРАТ 1
Получение сордарина
Культуру Sordaria araneosa NRRL 3196 (ATCC36386) выращивали на агаризованной среде до тех пор, пока не будет наблюдаться зрелый рост. Агаровые блоки диаметром 6 мм, содержащие культуру, переносили в стерильную воду или среду на основе сердечно-мозговой вытяжки (Oxoid) + 10%-ный глицерин и хранили при температуре окружающей среды или при -140oC, соответственно. Суспензию, содержавшую 2 таких агаровых блока, применяли для инокуляции колбы Erlenmyer емкостью 250 мл, содержавшей 50 мл среды FS.
Среда FS - г/л
Пептон (Oxoid L34) - 10
Солодовый экстракт (Oxoid L39) - 21
Глицерин (Glycerine CP) - 40
Junlon 110 (Honeywell & Stein) - 1
Дистиллированная вода
Культуру инкубировали при 25oC в течение 5 дней на роторной качалке, работавшей со скоростью 250 об/мин и диаметром орбитального движения 50 мм. Аликвоты (2 мл) развившегося инокулюма применяли для инокуляции колб Erienmyer емкостью 250 мл, содержавших среду FS (50 мл), которые инкубировали, как описано выше. 80 мл развитого инокулюма, выросшего в колбах при перемешивании, применяли для инокуляции двух 7-литровых ферментаторов, содержавших 5 л среды FS. Ферментацию проводили при температуре 25oC. Культуру перемешивали со скоростью 400 об/мин и аэрировали при расходе 2 л/мин. После 3 суток ферментации, 10 л культуры применяли для инокуляции 780- литрового ферментатора, содержавшего 500 л среды SM55VAR.
SM55VAR - г/л
Glucidex 32D (Roqette Frere) - 74
Пептон (Oxoid L37) - 10
Proflo (Traders & Protein) - 30
Свекольная меласса - 15
MgSO4•7H2O (BDH) - 5
CaCO3 (BDH) - 5
FeSO4•7H2O (Sigma) - 2
ZnSO4•7H2O (BDH) - 0,04
L-триптофан (Sigma) - 2
PPG2000 (K & K Greef) - 0,5
Силикон 1520 (Dow Corning) - 0,04
Дистиллированная вода
Ферментацию проводили при температуре 25oC. Питательную среду перемешивали при 300-350 об/мин и аэрировали при 200 л/мин. Для поддержания положительной остаточной концентрации глюкозы к культуре добавляли 70%-ный (в/об) Meritose (Tunnel Refineries) раствор. Дистиллированную воду добавляли для поддержания культурального объема на уровне 500 л. Для контроля пенообразования добавляли пеногаситель PPG2000. Все экстракты питательной среды (в водном ацетонитриле + 1%-ная трифторуксусная кислота) анализировали на присутствие сордарина с помощью ЖХВР с обращенной фазой. Культуру собирали через 11 суток, когда экстракт образца питательной среда показывал титр сордарина 0,6 г/л. Ферментационную среду делали 0.1М относительно гидроксида натрия и после инкубации в течение ночи при температуре окружающей среды фильтровали через Dicallite на роторном вакуумном фильтре (1%-ный Dicallite добавляли в среду для облегчения фильтрации). Фильтрат доводили до pH 6-7 концентрированной серной кислотой и раствор наносили на XAD16 смолу (10 объемов фильтрата/объем смолы). Адсорбент промывали водой и смесью ацетон: вода (1:3), получая в обоих случаях прозрачный элюент перед элюцией сордарина смесью ацетон: вода (3:1; собирали 2 объема колонки). Скорость протока в ходе процесса составляла в среднем 1-2 объема колонки/час. Элюат концентрировали до небольшого объема (8,5 л). Концентрат доводили до pH 3 фосфорной кислотой и оставляли на ночь при температуре окружающей среды для выпадения сордарина в осадок. Супернатант декантировали, затем центрифугировали, и супернатант выбрасывали. Осадки после центрифугирования и отстаивания переносили в 75%-ный водный ацетонитрил, получая в результате 3,9 л темно-коричневого раствора. К нему при перемешивании добавляли 1,0 л 0,2М NH4H2PO4, раствор доводили до pH 4,0 фосфорной кислотой, получая конечный объем 5,0 л с примерным составом 60% ацетонитрила - 0,1М NH4H2PO4. Полученный неочищенный раствор сордарина (5,0 л) подвергали препаративной ЖХВР за 10 инъекций (каждая 450-550 мл) на колонке (15x25 см) из 7-миллиметрового Kroasil C8 в подвижной фазе состава: 50% ацетонитрила - 0,1M NH4H2PO4, pH 4 (50 л ацетонитрила доводили до 100 л водой, содержавшей 575 г NH4H2PO4 и 40 мл H3PO4), скорость протока 600 мл/мин, контроль по УФ-поглощению ( λ 210 мм).
Собирали фракцию, элюированную между 15,4 и 19,2 мин. Объединенные фракции от 10 инъекций (23 л) разбавляли водой до 50 л, и этот раствор снова наносили на Kromasil колонку со скоростью 28 л/час. Колонку промывали водой (25 л), а затем элюировали 90%-ным ацетонитрилом (10 л). Элюат упаривали до остатка объемом 1300 мл, который сушили замораживанием, получая в результате соединение, указанное в заголовке, в виде светло-желтого порошка (105,6 г). МС и ЯМР анализы продукта показали, его эквивалентность с аутентичным образцом сордарина.
ПРЕПАРАТ 2
Получение калиевой соли сордарина
Sordaria araneosa NRRL3196 (ANCC36386) выращивали в среде на основе сердечно-мозговой вытяжки (Oxoid) + 10%-ный глицерол при -140oC, как описано в Препарате 1. Суспензию, содержавшую 2 агаровых высечки, использовали для инокуляции колбы Erlenmyer емкостью 250 мл, содержавшей 50 мл среды FS. Культуру инкубировали при 25oC в течение 5 суток на роторной качалке, работавшей со скоростью 250 об/мин и диаметре орбитального движения 50 мм. Аликвоты (2 мл) развитого инокулюма использовали для инокуляции колб Erlenmyer емкостью 250 мл, содержавших среду FS (50 мл), которые инкубировали, как описано выше. 80 мл развитого инокулюма, выросшего в колбах при перемешивании, использовали для инокуляции каждого из двух ферментаторов емкостью 7 л, содержавших 5 л среды FS. Ферментацию проводили при температуре 25oC. Культуру перемешивали при 400 об/мин и аэрировали при 2 л/мин. После 3 суток ферментации 10 л культуры применяли для инокуляции ферментатора емкостью 780 л, содержавшего 500 л среды SD1.
SDI - г/л
Meritose (Tunnel refineries) - 22
Лактоза - 20
Glucidex 32D (Roquette Frere) - 30
Arkasoy (The British Arkady Co) - 20
CSL - 20
FeCl3•6H2O - 0,05
NH4H2PO4 - 5
ZnSO4•7H2O (BDH) - 0,1
PPG2000 - 0,5
Дистиллированная вода
Ферментацию проводили при температуре 25oC. Культуральную среду перемешивали со скоростью 350 об/мин и аэрировали при 200 л/мин. Для поддержания объема культуры 500 мл добавляли воду. Экстракты питательной среды тестировали на наличие сордарина с помощью ЖХВР с обращенной фазой. Культуру собирали через 6 суток, когда титр сордарина в экстракте образца среды составлял 1,3 г/л. Образец собранной среды объемом 50 л делали 0,1М относительно гидроксида натрия и хранили при 4oC в течение ночи. Клетки удаляли посредством вакуумной фильтрации через слой Dicalite, добавляя дополнительно к среде с целью фильтрации 2%-ный Dicallite. Фильтрат доводили до pH 6 концентрированной серной кислотой. Экстракт питательной среды прокачивали через слой Amberchrom CG161 смолы со скоростью 320 мл/мин (0,64 объемов слоя в минуту). Эффлюент анализировали с помощью ЖХВР. Спустя 45 минут (эквивалентно прокачанному объему 14,4 л) сордарин начинал проходить и прокачивание прекращали. Адсорбент промывали водой (2 л), затем 25%-ным об/об ацетоном в воде (2 л). Сордарин элюировали 75%-ным об/об ацетоном в воде (1,5 л). 75%-ный об/об ацетоновый элюат упаривали приблизительно до 200 мл с помощью роторного испарителя при 40oC. Добавляли 200 мл бутан-1-ол и упаривание продолжали до получения вязкого масла (содержавшего некоторое количество бутан-1-ола). Масляный остаток экстрагировали горячим метанолом (2x500 мл). Экстракты объединяли, фильтровали (бумага Whatman номер 1), а затем выпаривали при 40-45oC, получая в результате вязкое масло. Его экстрагировали горячим ацетоном (2x500 мл). Ацетоновые экстракты объединяли, фильтровали и упаривали до вязкого масла. Добавляли пропан-2-ол (350 мл) и масло растворяли при 45oC, получая прозрачный коричневый раствор. В коническую колбу емкостью 500 мл вливали раствор калий-2-этиленгексаноата (39%-ный в/в раствор в пропан-2-оле, 54 г). Сордарин-содержащий раствор в пропан-2-оле вливали в эту коническую колбу, компоненты перемешивали и оставляли на 4 часа при комнатной температуре. В раствор вносили затравку калиевой соли сордарина (около 5 мг) и заткнутую колбу инкубировали в течение 3 суток при 4oC. Образовавшееся беловатое вещество отфильтровывали под вакуумом (шлаковая воронка N 4) и полученный слежавшийся осадок промывали пропан-2-олом (около 20-30 мл). Вещество переносили в кристаллизационную чашку и сушили под вакуумом над P2O5 в течение 16 часов, получая в результате соединение, указанное в заголовке (10,5 г).
ПРЕПАРАТ 3
Получение калиевой соли сордарина
500 литров культуральной среды получали, как в Препарате 1. Питательную среду делали 0,1М относительно гидроксида натрия и спустя четверо суток при 0oC фильтровали через слой Dicalite на роторном вакуумном фильтре. К питательной среде добавляли 1%-ный Dicalite для облегчения фильтрации. pH фильтрата доводили от pH 9,6 до pH 7,5 концентрированной серной кислотой. Фильтрат (10 л) доводили до pH 6 с помощью H3PO4 и наносили на слой (200 мл) XAD-16 смолы, находящейся в воде при скорости протока 1-1,5 объемов слоя/ч. Слой смолы промывали водой (1 объем слоя), а затем 25%-ным водным изопропанолом (2,5 объема слоя) перед элюцией чистым изопропанолом. После протока 50 мл изопропанола элюат собирали в виде фракций объемом 100 мл. Содержащие сордарин фракции 2-6 включительно объединяли и упаривали до половины исходного объема. Добавляли изопропанол, доводя объем снова до 500 мл, затем упаривание до половины исходного объема повторяли для удаления остаточной воды посредством азеотропной дистилляции. После третьего упаривания до половины объема остаток фильтровали, а фильтр промывали изопропанолом. К объединенному фильтрату и промывным водам (400 мл) добавляли раствор 2-этиленгексаноата (8 г) в изопропаноле (100 мл). В смесь вносили затравку калиевой соли сордарина и оставляли на несколько суток при 4oC до тех пор, пока не начиналась медленная кристаллизация. Кристаллы отфильтровывали на шлаковом фильтре N 3, промывали небольшим количеством охлажденного на льду изопропанола и сушили под вакуумом, получая в результате соединение, указанное в заголовке, в виде бледно-коричневого порошка (4,85 г).
ПРЕПАРАТ 4
Получение 4'-деметилсордарина
(i) IMI 362184 поддерживали в питательной среде на основе сердечно-мозговой вытяжки (Oxoid) + 10%-ный глицерин при -140oC, как описано в Препарате 1. Суспензию, содержавшую 2 агаровых высечки, применяли для инокуляции колбы Erlenmyer объемом 250 мл, содеравшую 50 мл среды FS. Культуру инкубировали при 25oC в течение 5 суток на роторной качалке, работавшей со скоростью 250 об/мин при диаметре орбитального вращения 50 мм. Аликвоты (2 мл) развитого инокулюма использовали для инокуляции следующих колб Erlenmyer емкостью 250 мл, содеравших среду FS (50 мл) и инкубировали их, как описано выше. 80 мл развитого инокулюма, выросшего в колбах при перемешивании, применяли для инокуляции каждого из двух ферментаторов емкостью 7 л, содеравших 5 л среды FS. Ферментацию проводили при температуре 25oC. Культуру перемешивали со скоростью 400 об/мин и аэрировали со скоростью протока 2 л/мин. После 3 суток ферментации 10 л культуры использовали для инокуляции ферментатора емкостью 780 л, содержавшего 500 л среды SM55VAR (как описано в Препарате 1). Ферментацию проводили при температуре 25oC. Питательную среду перемешивали со скоростью 350 об/мин и аэрировали со скоростью протока 500 л/мин. Для поддержания положительной остаточной концентрации глюкозы к культуре добавляли 70%-ный в/об раствор Meritose (Tunnel Refineries). Для поддержания объема культуры на уровне 500 л добавляли дистиллированную воду. Экстракты питательной среды тестировали на наличие 4'-деметилсордарина с помощью ЖХВР с обращенной фазой. Культуру собирали через 10 суток, когда экстракт образца питательной среды показывал титр 4'-деметилсордарина, равный 0,8 г/л. Ферментационную среду доводили до 0,1М относительно гидроксида натрия и после инкубации в течение 1 часа при температуре окружающей среды фильтровали через Dicalite на роторном вакуумном фильтре (1%-ный Dicalite добавляли к питательной среде для облегчения фильтрации). Фильтрат доводили до pH 4,5 концентрированной серной кислотой и наносили раствор на XAD16 смолу (20 г продукта/л смолы). Адсорбент промывали 0,1%-ной фосфорной кислотой (10 объемов колонки) и смесью ацетонитрил:вода 1:4 (6 объемов колонки) перед элюцией продукта смесью ацетонитрил:вода (1:1; 2 объема колонки). Скорость протока в ходе процесса составляла 1-2 объемов колонки/час. Элюат концентрировали до сухого состояния с добавлением бутан-1-ола, твердую фазу экстрагировали при 60oC метанолом (12 л), затем ацетоном (10 л). Нерастворимый материал отделяли на каждой стадии посредством фильтрации через стеклянный шлак N 3, а экстракты концентрировали до сухого состояния, как и ранее. Вещество кристаллизовали из смеси ацетонитрил:вода (3:7) перед перекристаллизацией из того же самого растворителя и сушили до постоянного веса над P2O5, получая в результате соединение, указанное в заголовке (244,0 г), которое при протонном ЯМР анализе показало эквивалентность с аутентичным образцом 4'-деметилсордарина.
(ii) Повторяли процесс ферментаци, описанный в (i), и после того, как питательную среду доводили до 0,1М относительно гидроксида натрия, ее подвергали ультрафильтрации через ETNA 10A мембрану (10 кДальтон размер пор). После диафильтрации с водой отфильтрованная жидкость представляла собой прозрачный раствор. После доведения до pH 5,2 с помощью концентрированной серной кислоты фильтрат наносили на колонку со смолой XAD16 со скоростью протока 2 колонки в час, получая в результате конечную нагрузку 32 г 4'-деметилсордарина на литр смолы. Колонку промывали со скоростью 2 объема колонки в час, сначала 0,1%-ной об/об фосфорной кислотой, а затем 20%-ной об/об смесью ацетонитрил/вода (10 объемов колонки каждого компонента). Колонку элюировали 65%-ной смесью ацетонитрил:вода со скоростью протока 2 объема колонки в час. Головной погон величиной 0,75 объемов колонки отбрасывали. 85% нанесенного 4'-деметилсордарина извлекали следующей порцией элюата объемом 1,6 объемов колонки. Обогащенный элюат обрабатывали при перемешивании в течение 5 минут 2%-ной в/об DE52 целлюлозой, которую удаляли посредством фильтрации. Аликвоту DE52 обработанного элюата концентрировали до 62% исходного объема (43%-ный ацетонитрил) с помощью роторного испарителя. Концентрированный элюат выдерживали в течение 15 часов при 4oC для кристаллизации, а образовавшуюся твердую фазу собирали посредством фильтрации через стеклянный шлак. Кристаллы промывали объемом 30%-ной об/об смеси ацетонитрил:вода, равным четырем объемам слоя, и сушили при 30oC под вакуумом. Соединение, указанное в заголовке (1,94 г), извлекали из сконцентрированного элюата в виде бледно-серого вещества.
ПРЕПАРАТ 5
Скриннинг микроорганизмов, способных деметилировать сордарин по 4' положению
Микроорганизмы, способные деметилировать сордарин по 4' положению, могут быть идентифицированы посредством культивирования их при 28oC (бактерии) или 25oC (грибы) в 10 мл объемах SB1 (бактерии) или FB1 (грибы) в конических колбах объемом 50 мл, перемешивавшихся со скоростью 250 об/мин. Через 2-ое суток добавляли сордарин до конечной концентрации 0,5 мг/мл (из 200 мг/мл сток-раствора в 80%-ном этаноле), а колбы затем инкубировали в течение еще 3 суток. Образец культуры объемом 500 мкл смешивали в пробирке Эппендорф с 500 мкл смеси 80%-ный ацетонитрил/2%-ная трифторуксусная кислота и оставляли для экстракции при комнатной температуре на 30 минут. Экстрактивный супернатант, полученный после центрифугирования образцов в микроцентрифуге, анализировали посредством изократической ЖХВР на присутствие 4'-деметилсордарина. 4'-деметилсордарин элюировали за 3,35 мин, используя 35%-ный ацетонитрил в воде в качестве подвижной фазы, скорость протока 2 мл/мин, применяя колонку Spherisorb C6 (5 мкм, 15 см x 4,6 мм). С помощью этого метода были идентифицированы в качестве деметиляторов сордарина следующие микроорганизмы:
Streptomyces capreolus - ATCC 31963;
Streptomyces avermitilis - ATCC 31272;
Streptomyces armentosus - NRRL 3176;
Streptomyces antibioticus - ATCC 31771;
Streptomyces rimosus - ATCC 23955;
Streptomyces platensis - ATCC 29778;
Streptomyces mashuensis - ATCC 23934;
Streptomyces eurythermus - ATCC 14975;
Nocardia orientalis - ATCC 43491;
Cunninghamella echinulata var elegans - ATCC 36112.
Среда SB1 - г/л
Arkasoy - 25
Дрожжевой экстракт - 5
KH2PO4 - 5
Глюкоза - 20
Дистиллированная вода
pH 7
Среда FB1 - г/л
Соевое масло - 30
Arkasoy - 10
Дрожжевой экстракт - 5
K2HPO4 - 5
Глюкоза - 20
Дистиллированная вода
pH 5,5
ПРЕПАРАТ 6
Получение 4'-деметилсордарина посредством биотрансформации сордарина
0,3 мл споровой суспензии Strepcomyces capreolus ATCC 31963 (в 15%-ном об/об глицерине, хранимой при -20oC) инокулировали в 30 мл SB1 среды в Erlenmyer колбе емкостью 250 мл для получения посевной культуры, которую инкубировали при 28oC и 250 об/мин на роторной качалке. Спустя 4 суток 0,5 мл культуры использовали для инокуляции 35 мл SB1 в колбе емкостью 250 мл, которую выращивали в течение 48 часов при 28oC, 250 об/мин. На этой стадии культуру в виде аликвот объемом 10 мл переносили в Erlenmyer колбы емкостью 50 мл, в которые добавляли 5 мг сордарина (из 200 мг/мл сток-раствора в этаноле). Инкубацию продолжали еще в течение 3 суток. Ко всему объему среды добавляли 80%-ный об/об ацетонитрил в воде (14 мл) и выдерживали смесь при комнатной температуре и периодическом перемешивании. Через 30 минут клетки удаляли центрифугированием. Ацетонитрил удаляли посредством упаривания, а pH водного раствора доводили до 2,5 с помощью ортофосфорной кислоты. Раствор пропускали через колонку, содержавшую Amberlite XAD-16 смолу (объем слоя 5 мл). Адсорбент промывали последовательно водой (10 мл), 10%-ным об/об ацетонитрилом в воде (20 мл), 30%-ным об/об ацетонитрилом в воде (10 мл), 50%-ным об/об ацетонитрилом в воде (20 мл) и 90%-ным об/об ацетонитрилом в воде (10 мл). Фракции анализировали посредством ЖХВР; 4'-деметилсордарин локализовался в элюате 50%-ного об/об ацетонитрила в воде. Фракцию, содержавшую 4'-деметилсордарин, упаривали до сухого состояния под вакуумом при комнатной температуре, а остаток перерастворяли в 35%-ном ацетонитриле в воде (15 мл), 4'-деметилсордарин очищали посредством ЖХВР:
Колонка - Spherisorb 5 микрон C6 25 см x 2,5 см;
Скорость протока - 25 мл/мин;
Обнаружение - УФ при 210 нм;
Подвижная фаза - 350 мл ацетонитрила доводили до 1000 мл 0,05 М дигидрофосфатом аммония в воде, pH доводили до 2,5 ортофосфорной кислотой;
Вводимый объем - 4,5 мл.
4'-деметилсордарин элюировали через 10 минут при этих условиях. Объединенные фракции от четырех циклов ЖХВР разбавляли 1:1 водой, а затем снова наносили на силикагель (после промывки его ацетонитрилом и повторного уравновешивания водой). Адсорбент промывали водой (200 мл), а адсорбированный продукт элюировали 95%-ным об/об ацетонитрилом в воде (200 мл). Ацетонитрил/водный элюат упаривали для удаления органического растворителя, а водный раствор сушили замораживанием, получая в результате 4'-деметилсордарин (1,5 мг), в виде белого порошка.
δ (1H, CDCl3); 9,74 (s,1H); 6,08 (шир, 3,1H); 4,70 (d, 1,5, 1H); 4,16 (d, 9,5, 1H); 4,08 (dd, 4,5, 3,5, 1H); 3,88 (dd, 4,5, 1,5, 1H), 3,75 (dq, 8,5, 6,1, 1H); 3,62 (d, 9,5, 1H); 3,68 (dd, 8,5, 3,5, 1H); 2,65 (m, 1H); 2,34 (m,1H); 1,32 (d, 6, 3H); 1,30 (d, 12,5, 1H); 1,23 (m, 1H); 1,04 (d, 7, 3H); 0,99 (d, 7, 3H); 0,81 (d, 7, 3H)
ПРЕПАРАТ 7
Получение сордарицина
Проводили процесс согласно описанию к Препарату 1 включительно до стадии препаративной ЖХВР. Собирали фракцию, элюированную между 21,4 и 25 мин. Объединенные фракции от 10 введений (22 л) разбавляли равным объемом воды и этот раствор вновь прокачивали через Kromasil колонку при скорости протока 28 л/ч. Колонку промывали водой (20 л), а затем элюировали 90%-ным ацетонитрилом (4,5 л). Этот элюат объединяли с соответствующей фракцией элюата (4,5 л) из аналогичной ферментации, которую осуществляли тем же способом. Объединенные элюаты в 90%-ном ацетонитриле концентрировали с помощью роторного испарителя до тех пор, пока не начиналась кристаллизация продукта (объем около 1,6 л). Остаток нагревали на водяной бане с температурой 60oC, добавляя минимальный объем ацетонитрила для получения прозрачного раствора. Раствор охлаждали и хранили при 4oC. Кристаллы отфильтровывали на шлаковом фильтре N 3 и сушили под вакуумом, получая в результате коричневое вещество. Его подвергали повторной кристаллизации из смеси ацетонитрил:вода (40:60), а продукт фильтровали, промывали 25%-ным ацетонитрилом и сушили под вакуумом, получая в результате соединение, указанное в заголовке (10,7 г). МС и ЯМР анализы продукта показали его эквивалентность аутентичному образцу сордарицина.
ПРЕПАРАТ 8
Биотрансформация сордарина в сордарицин
0,2 мл суспензии NCIMB 40675 использовали для инокуляции колбы Erlenmyer емкостью 250 мл, содержавшей 50 мл питательной среды (Oxoid), обогащенной 0,2%-ным дрожжевым экстрактом. Культуру инкубировали при 28oC в течение 29 часов на роторной качалке, работавшей со скоростью 250 об/мин при орбитальном движении 50 мм. Аликвоты (1 мл) развитого инокулюма использовали для инокуляции следующих Erlenmyer колб объемом 250 мл, содержавших 50 мл питательной среды в удвоенной концентрации (Oxoid), обогащенной 0,1%-ным дрожжевым экстрактом. Их инкубировали в течение приблизительно 24 часов. 62 таких колбы объединяли, получая 2,65 литра развитой культуры, которую добавляли к 30 литрам ферментационного фильтрата, содержавшего сордарин, pH доводили до 7,5, как описано в описании к Препарату 1. Реакцию проводили в ферментаторе емкостью 70 литров при 30oC, при перемешивании со скоростью 200 об/мин и аэрации 0,5 об/мин. pH поддерживали на уровне 7,5 посредством добавления 1N соляной кислоты. Спустя 6 суток приблизительно 16 г сордарина превращалось в сордарицин. Среду после биотрансформации фильтровали через Dicalite на вакуумном фильтре, фильтрационный слой промывали водой, получая 31,5 л фильтрата, pH 8,2. Его доводили до pH 6,0 с помощью H3PO4 и прокачивали через слой (25 см х 25 см) Amberchrom CG161 смолы при скорости протока 290 мл/мин. Слой Amberchrom промывали 0,1%-ной H3PO4 (2 л) и 25%-ным ацетонитрилом (4 л), а затем элюировали 60%-ным CH3CN (2 л). Элюат концентрировали на роторном испарителе до момента начала кристаллизации, затем остаток нагревали на водяной бане при 60oC, добавляя минимальное количество ацетонитрила и получая в результате прозрачный раствор. Его охлаждали и выдерживали при 4oC в течение ночи. Кристаллы отфильтровывали, промывали 25%-ным ацетонитрилом и сушили под вакуумом, получая коричневый порошок. Неочищенный продукт подвергали перекристаллизации из 1 л смеси ацетонитрил:вода (40:60), фильтровали, промывали и сушили, как описано ранее, получая в результате сордарицин в виде бледно-коричневых игольчатых кристаллов (6,15 г).
ПРЕПАРАТ 9
Биотрансформация сордарина в сордарицин
С поверхности агара петлей снимали выросшую культуру NCIMB 40675, и инокулировали ею Erlenmyer колбы объемом 250 мл, содержащие 50 мл питательной среды (Oxoid), обогащенной 0,2%-ным дрожжевым экстрактом. Культуру инкубировали в течение 26 часов при 25oC на роторной качалке, работавшей со скоростью 250 об/мин и диаметром движения 50 мм. Аликвоты развитой культуры объемом 1 мл использовали для инокуляции Erlenmyer колб емкостью 250 мл, как описано выше. В колбы добавляли чистый сордарин в 80%-ном этаноле до конечной концентрации 1,25 мг/мл. Колбы инкубировали в течение 8 суток, затем их содержимое объединяли, получая 365 мл культуральной среды, содержавшей сордарицин. Смесь после биоконверсии центрифугировали для удаления NCIMB 40675 клеток, а супернатант (350 мл) декантировали. К супернатанту добавляли Whatman Partisil P40 DS-3 (5 мл, предварительно увлажненного ацетонитрилом), pH смеси доводили до 4,0 с помощью H3PO4. Partisil отфильтровывали на воронке Buchner и элюировали сначала ацетонитрилом (100 мл), а затем 75%-ным ацетонитрилом (100 мл). Объединенные элюаты упаривали до водного остатка объемом 10-15 мл. Его нагревали на водяной бане при температуре 60oC и добавляли ацетонитрил до получения прозрачного раствора, затем нагревание продолжали и добавляли воду до тех пор, пока раствор не начинал мутнеть. Смесь охлаждали и хранили при 4oC в течение нескольких суток. Кристаллы отфильтровывали на шлаковом фильтре N 3, промывали водой и сушили под вакуумом над P2O5, получая в результате сордарицин в виде белых игольчатых кристаллов (92 мг).
ПРЕПАРАТ 10
Получение сордарицина
Замороженную ампулу IMI 362947 использовали для инокуляции 50 мл посевной среды FS в Erlenmyer колбе емкостью 250 мл. Эту колбу инкубировали при 25oC в течение 6 суток на роторной качалке, работавшей со скоростью 250 об/мин и диаметром орбитального движения 50 мм. Аликвоты (1 мл) развитого инокулюма использовали для инокуляции Erlenmyer колб емкостью 250 мл, содержавших 50 мл среды SM55/A для культивирования в качалочных колбах.
SM55/A - г/л
Мальтодекстрин MD30E (Roquette Frere) - 120
Свекольная меласса - 15
Пептон (Oxoid L37) - 10
Proflo (Traders Protein) - 30
L-Триптофан (Sigma) - 2
ZnSO4•7H2O (BDH) - 0,04
FeSO4•7H2O (Sigma) - 2
CaCO3 (BDH) - 5
MgSO4•7H2O (BDH) - 5
Готовят на дистиллированной воде и автоклавируют при 121oC в течение 120 минут.
Эти культуры инкубировали, как описано выше, в течение 7 суток. Содержимое колб объединяли и порцию объемом 40 мл обрабатывали 1N гидроксидом натрия (4 мл) при периодическом перемешивании при комнатной температуре в течение 45 минут. Затем клетки удаляли посредством центрифугирования при 3000 об/мин в течение 20 минут, отбирали супернатант и доводили его pH до 3 с помощью 1N соляной кислотой. Раствор пропускали через Bond Elut колонку (размер 1 г; C18), адсорбент промывали водой (20 мл), затем элюировали 90%-ным об/об ацетонитрилом в воде (15 мл). Элюат упаривали до сухого состояния при комнатной температуре, а остаток переносили в 50%-ный об/об ацетонитрил в воде (9 мл). Сордарицин очищали посредством препаративной ЖХВР:
Колонка - Spherisorb 5 микрон ODS-2 (25 см x 2,1 см);
Скорость протока - 25 мл/мин;
Подвижная фаза - 550 мл ацетонитрила, доведенного до 1000 мл водой, с 1 мл трифторуксусной кислоты на литр подвижной фазы;
Обнаружение - 210 нм;
Вводимый объем - 4,5 мл.
Сордарицин элюировали в этих условиях через 6 мин. Фракции, содержавшие сордарицин, объединяли, ацетонитрил удаляли с помощью роторного испарителя при комнатной температуре, а водный раствор сушили замораживанием, получая в результате соединение, указанное в заголовке (8,9 мг), в виде белого порошка.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси- β- альтропиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору 4'-деметилсордарина (10 г) в метаноле (200 мл) при комнатной температуре по каплям добавляли 0,35М раствор дифенилдиазометана (90 мл) в метиленхлориде, смесь перемешивали в течение 6 часов. Растворитель выпаривали до сухого состояния, а остаток хроматографировали на колонке быстрого разделения с силикагелем, применяя в качестве элюента смесь гексан:этилацетат (3: 1), получая в результате соединение, указанное в заголовке (12,6 г), в виде бледно-желтой пены.,
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,98 (s, 1H, 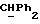 ), 6,05 (dd, 1H, H-2, J= 1,5 и 3,6 Гц), 4,65 (d, 1H, H-1, J=1,5 Гц), 4,09, 3,76 (2d, 2H, 8a-
), 6,05 (dd, 1H, H-2, J= 1,5 и 3,6 Гц), 4,65 (d, 1H, H-1, J=1,5 Гц), 4,09, 3,76 (2d, 2H, 8a- 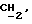 J= 9 Гц), 4,01 (m, 1H, H-2'), 3,84 (m, 1H, H-3'), 3,75 (m, 1H, H-5'), 3,69 (m, 1H, H-4'), 2,73 (t, 1H, H-1, J=4,2 Гц).
J= 9 Гц), 4,01 (m, 1H, H-2'), 3,84 (m, 1H, H-3'), 3,75 (m, 1H, H-5'), 3,69 (m, 1H, H-4'), 2,73 (t, 1H, H-1, J=4,2 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 2
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(6-дезокси-3,4-O-изопропилиден- β- D-альтропиранозилокси) метил] -4-формил-4,4a, 5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)- (1H)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 1 (650 мг) в 15 мл смеси диметоксипропан: ацетон (1:2) добавляли п-толуолсульфоновую кислоту (10 мг). Раствор перемешивали при комнатной температуре в течение 1,5 часов, затем добавляли карбонат калия (1,0 г), перемешивание продолжали еще в течение 30 минут и выпаривали растворитель до сухого состояния. Сырую смесь фракционировали между этилацетатом (50 мл) и водой (25 мл), водную фазу экстрагировали этилацетатом (2x50 мл), органическую фазу промывали рассолом, сушили над сульфатом магния и упаривали до сухого состояния. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат:гексан (1:3), получая в результате соединение, указанное в заголовке (600 мг), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,24 (m, 10H, 2Ph), 6,98 (s, 1H, CHPh2), 6,06 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,57 (d, 1H, H-1', J=3,0 Гц), 4,30 (dd, 1H, H-3', J=3,6 и 5,7 Гц), 4,07 (d, 1H, 8aCH2, J=9,0 Гц), 3,95-3,93 (m, 1H, H-2'), 3,85 (dd, 1H, H-4', J=5,7 и 9,3 Гц), 3,75 (d, 1H, 8aCH2, J=9,0 Гц), 3,44 (dq, 1H, H-5', Jd=6,3 Гц), 2,73 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 3
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(6-дезокси-3,4-O-(2-пентилиден)- β- D- альтропиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a, -октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К суспензии Промежуточного соединения 1 (500 мг) и п-толуолсульфоновой кислоты (5 мг) в дихлорметане (5 мл) добавляли 2,2-диметоксипентан (2 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли карбонат калия (150 мг) и продолжали перемешивание в течение 30 минут. Растворитель выпаривали до сухого состояния, а остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат:гексан (15: 85) и (30:70), получая в результате соединение, указанное в заголовке (474 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,24 (m, 1H, 2Ph), 6,98 (s,1H, 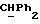 ), 6,06 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,57 (d, 1H, H-1', J=2,1 Гц), 4,30 (dd, 1H, H-3', J=3,9 и 6,0 Гц), 4,04 (d,1H, 8aCH2, J=9,0 Гц), 3,94-3,91 (m, 1H, H-2'), 3,86 (dd, 1H, H-4', J=6,3 и 9,6 Гц), 3,73 (d, 1H, 8aCH2, J= 9,0 Гц), 3,45 (dq, 1H, H-5, Jd=9,0 Гц, Jd=6,0 Гц), 2,72 (t, 1H, H-1, J=3,6 Гц).
), 6,06 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,57 (d, 1H, H-1', J=2,1 Гц), 4,30 (dd, 1H, H-3', J=3,9 и 6,0 Гц), 4,04 (d,1H, 8aCH2, J=9,0 Гц), 3,94-3,91 (m, 1H, H-2'), 3,86 (dd, 1H, H-4', J=6,3 и 9,6 Гц), 3,73 (d, 1H, 8aCH2, J= 9,0 Гц), 3,45 (dq, 1H, H-5, Jd=9,0 Гц, Jd=6,0 Гц), 2,72 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 4
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(6-дезокси-3,4-O-(4-метокси-2-бутилиден) - β- D-альтропиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
К суспензии Промежуточного соединения 1 (350 мг) и п-толуолсульфоновой кислоты (5 мг) в дихлорметане (4 мл) добавляли 2,2,4-триметоксибутан (1 мл). Смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли карбонат калия (150 мг) и продолжали перемешивание в течение 30 минут. Растворитель выпаривали до сухого состояния, а остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат:гексан (3: 7) и (4:6), получая в результате соединение, указанное в заголовке (290 мг) в эпимерном соотношении 3:1.
δ (1H, CDCl3) сигналы для основного компонента: 9,73 (s, 1H, CHO), 7,45-7,20 (m, 10H, 2Ph), 6,98 (s, 1H,  ), 6,06 (dd, 1H, H-2, J=1,5H 3,6 Гц), 4,57 (d, 1H, H-1', J=2,1 Гц), 4,31 (dd, 1H, H-3', J=3,9 и 6,0 Гц), 4,05 (d, 1H, 8aCH2, J=9,0 Гц), 3,95-3,90 (m, 1H, H-2'), 3,88 (dd, 1H, H-4', J=6,0 и 9,3 Гц), 3,73 (d, 1H, 8aCH2, J=9,0 Гц), 3,51 (t, 2H, CH2, J=7,2 Гц), 3,50-3,45 (m, 1H, H-5'), 3,33 (s, 3H, CH3O), 2,72 (t, 1H, H-1, J=3,6 Гц).
), 6,06 (dd, 1H, H-2, J=1,5H 3,6 Гц), 4,57 (d, 1H, H-1', J=2,1 Гц), 4,31 (dd, 1H, H-3', J=3,9 и 6,0 Гц), 4,05 (d, 1H, 8aCH2, J=9,0 Гц), 3,95-3,90 (m, 1H, H-2'), 3,88 (dd, 1H, H-4', J=6,0 и 9,3 Гц), 3,73 (d, 1H, 8aCH2, J=9,0 Гц), 3,51 (t, 2H, CH2, J=7,2 Гц), 3,50-3,45 (m, 1H, H-5'), 3,33 (s, 3H, CH3O), 2,72 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 5
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(6-дезокси-3,4-O-циклопентилиден-β- D- альтропиранозилокси)]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К суспензии Промежуточного соединения 1 (160 мг) в смеси дихлорметан: 1,1-диметоксициклопентан (3: 1, 4 мл) добавляли п-толуолсульфоновую кислоту (5 мг) и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли карбонат калия и продолжали перемешивание в течение 30 минут. Растворитель выпаривали до сухого состояния, а остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат:гексан (1:4), получая в результате соединение, указанное в заголовке (142 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,25 (m, 10H, 2Ph), 6,98 (s, 1H, CHPh2), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,55 (d, 1H, H-1', J=1,8 Гц), 4,17 (dd, 1H, H-3', J=3,0 и 5,1 Гц), 4,08 (d, 1H, 8aCH2, J=9,0 Гц), 3,95 (dt, 1H, H-2', Jd=1,8 Гц, Jt=3,0 Гц), 3,81 (dd, 1H, H-4', J= 5,1 и 9,3 Гц), 3,76 (d, 1H, 8aCH2, J=9,0 Гц), 3,42 (dq, 1H, H-5', Jd= 9,3 Гц, Jq=6,3 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 6
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a[(6-дезокси-3,4-O-изопропилиден-2-O-(метилтио) тиокарбонил- β- D-альтропиранозилокси)метил] -4-формил-4, 4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано- s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Промежуточное соединение 2 (100 мг) и имидазол (1 мг) растворяли в сухом тетрагидрофуране (4 мл) в атмосфере азота. Добавляли гидрид натрия (5 мг) и полученную суспензию перемешивали при комнатной температуре в течение 0,5 часов. Добавляли дисульфид углерода (27 мкл), продолжали перемешивание в течение еще 20 минут и добавляли метилиодид (18 мкл). Спустя 2 часа реакцию останавливали посредством добавления 1N хлорида аммония (20 мкл). Смесь экстрагировали этилацетатом (3x25 мл), органическую фазу промывали рассолом, сушили над сульфатом магния и выпаривали растворитель до сухого состояния. Остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат: гексан (1:9), получая в результате соединение, указанное в заголовке (110 мг).
δ (1H, CDCl3: 9,71 (s, 1H, CHO), 7,44-7,25 (m, 10H, 2Ph), 6,96 (s, 1H, CHPh2), 6,01 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 6,90 (dd, 1H, H-2', J=2,4 и 5,4 Гц), 4,85 (d, 1H, H-1', J=2,4 Гц), 4,53 (dd, 1H, H-3', J=5,4 и 6,3 Гц), 4,00 (dd, 1H, H-4', J=6,3 и 8,7 Гц), 3,93 (d, 1H, 8aCH2, J=9,3 Гц), 3,65 (dq, 1H, H-5', Jd=8,7, Jq=6,3 Гц), 2,68( t, 1H, H-1, J=3,9 Гц), 2,59 (s, 3H, CH3S).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 7
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(2,6-Дидезокси-3,4-O-изопропилиден- β- D- аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
(i) Промежуточное соединение 6 (95 мг) растворяли в сухом толуоле (5 мл) в атмосфере азота и прогревали при 110oC. Спустя 1,5 часа при перемешивании по каплям добавляли раствор гидрида трибутилолова (64 мкл) в сухом толуоле (5 мл). Прогревание продолжали еще в течение 1,5 часов, за тем добавляли метанол (2 мл) и растворитель выпаривали до сухого состояния. После хроматографии быстрого разделения на силикагеле остатка с элюцией смесью этилацетат:гексан (1:9) получали соединение, указанное в заголовке (42 мг).
δ (1H, CDCl3: 9,73 (s, 1H, CHO), 7,44- 7,25 (m, 10H, 2Ph), 6,98 (1H, s, CHPh2), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,54 (dd, 1H, H-1', J=2,7 и 9,3 Гц), 4,39 (dt, 1H, H-3', Jd=2,7 Гц, Jt=3,6 Гц), 4,04 (d, 1H, 8aCH2, J=9,0 Гц), 3,67 (d, 1H, 8aCH2, J=9,0 Гц), 3,65 (dd, 1H, H-4', J=3,6 и 8,7 Гц), 3,44 (dq, 1H, H-5', Jd= 6,3 Гц, Jq=8,7 Гц), 2,75 (t, 1H, J=3,9 Гц).
(ii) Раствор гидрида трибутилолова (5,5 мл) в сухом толуоле (150 мл) дегазировали током аргона в течение 1 часа и нагревали с дефлегмацией. Затем спустя 2 часа добавляли раствор промежуточного соединения 6 (6 г) в сухом толуоле (50 мл). Прогревание продолжали до завершения реакции (1,5 часа). В результате удаления растворителя при пониженном давлении получали сырой продукт, который подвергали хроматографии быстрого разделения на силикагеле, элюируя гексаном и смесью гексан:этилацетат (20:1 до 15:1), получая в результате соединение, указанное в заголовке (4 г).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 8
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a[(2,6-Дидезокси-3,4-O-(4-метокси-2-бутилиден)- β- -D-аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору промежуточного соединения 4 (600 мг) в сухом тетрагидрофуране (10 мл) при 0oC в атмосфере азота добавляли гидрид натрия (30 мг) и имидазол (5 мг). Раствор перемешивали в течение 10 минут и добавляли сероуглерод (147 мкл). Через 20 минут добавляли метилиодид (127 мкл) продолжали перемешивание еще в течение 30 минут. Добавляли 1N хлорид аммония (20 мл) и экстрагировали смесь этилацетатом (3x25 мл), органическую фазу промывали рассолом, сушили над сульфатом магния и выпаривали растворитель до сухого состояния. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат: гексан (15:85), получая в результате белую пену. Эту пену растворяли в сухом толуоле (10 мл) в атмосфере азота и спустя 2 часа при 115oC по каплям добавляли раствор гидрида трибутилолова (0,46 мл) в толуоле (10 мл). Прогревание продолжали в течение 2 часов. Растворитель выпаривали до сухого состояния, а остаток очищали посредством хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат:гексан (15:85), получая в результате соединение, указанное в заголовке (260 мг) в эпимерном соотношении 4:1.
δ (1H, CDCl3): 9,63 (s, 1H, CHO), 7,44-7,20 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,53 (dd, 1H, H-1', J=2,7 и 8,7 Гц), 4,42 (dt, 1H, H-3', Jd=3,0 Гц, Jf=4,2 Гц), 4,03 (d, 1H, 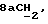 , J=9,0 Гц), 3,68 (dd, 1H, H-4', J=5,1 и 9,3 Гц), 3,66 (d, 1H, J= 9,0 Гц), 3,52 (t, 2H, CH2O, J=7,2 Гц), 3,40 (dq, 1H, H-5', Jd=9,3 Гц, Jq=6,3 Гц), 3,34 (s, 3H, CH3O), 2,74 (t, 1H, H-1, J=4,2 Гц).
, J=9,0 Гц), 3,68 (dd, 1H, H-4', J=5,1 и 9,3 Гц), 3,66 (d, 1H, J= 9,0 Гц), 3,52 (t, 2H, CH2O, J=7,2 Гц), 3,40 (dq, 1H, H-5', Jd=9,3 Гц, Jq=6,3 Гц), 3,34 (s, 3H, CH3O), 2,74 (t, 1H, H-1, J=4,2 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 9
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a[(6-дезокси-3,4-O-тиокарбонил- β- D- альтропиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Смесь промежуточного соединения 1 (0,500 г) и тиокарбонилдиимидазола (0,270 г) в тетрагидрофуране (10 мл) прогревали с дефлегмацией в течение 6 часов. Растворитель выпаривали под вакуумом, получая в результате желтый остаток, который хроматографировали на колонке для быстрого разделения с силикагелем, элюируя смесью гексан:этилацетат (3:1), получая в результате соединение, указанное в заголовке (0,263 г).
δ (1H, CDCl3): 9,71 (s, 1H, CHO), 7,44-7,26 (m, 10H, 2XPh), 6,97 (s, 1H, 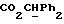 ), 6,06 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,93 (dd, 1H, H-3', J=3,9 и 7,2 Гц), 4,63 (d, 1H, H-1', J=2,1 Гц), 4,61 (dd, 1H, H-4', J=7,2 и 9,3 Гц), 4,12 (m, 1H, H-2'), 4,08 (d, 1H, 8aCH2, JAB=9,3 Гц), 3,74 (d, 1H, 8aCH2, JAB=9,3 Гц), 3,64 (m, 1H, H-5'), 2,69 (m, 1H, H-1).
), 6,06 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,93 (dd, 1H, H-3', J=3,9 и 7,2 Гц), 4,63 (d, 1H, H-1', J=2,1 Гц), 4,61 (dd, 1H, H-4', J=7,2 и 9,3 Гц), 4,12 (m, 1H, H-2'), 4,08 (d, 1H, 8aCH2, JAB=9,3 Гц), 3,74 (d, 1H, 8aCH2, JAB=9,3 Гц), 3,64 (m, 1H, H-5'), 2,69 (m, 1H, H-1).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 10
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a[(2,6-Дидезокси- β- D-аллопиранозилокси)метил] -4- формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору промежуточного соединения 7 (1,5 г) в смеси тетрагидрофурана (30 мл) и метанола (15 мл) при комнатной температуре и при интенсивном перемешивании по каплям добавляли IN раствор соляной кислоты (15 мл). Сразу по окончании реакции (ТСХ контроль) добавляли насыщенный бикарбонат натрия (50 мл) и этилацетат (200 мл) и смесь разделяли. Органический слой промывали водой (2x100 мл) и сушили над сульфатом магния. В результате удаления растворителя получали остаток, который подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью гексан: этилацетат (5:1) и (2:1), получая в результате соединение, указанное в заголовке (1,1 г), в виде белой пены.
δ (1H, CDCl3: 9,73 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,64 (dd, 1H, H-1', J=2,1 и 9,6 Гц), 4,11 (m, 1H, H-3'), 4,06 (d, 1H, 8aCH2, JAB=9,3 Гц), 3,70 (m, 2H, H-5' и 8aCH2), 3,34 (m, 1H, H-4'), 2,75 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 11
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a[(2,6-Дидезокси-3,4-O-тиокарбонил- β- D- аллопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
Суспензию Промежуточного соединения 10 (160 мг) и оксида дибутилолова (124,5 мг) в сухом толуоле (15 мл) прогревали с дефлегмацией в течение 2 часов в колбе, снабженной конденсатором Dean-Stark, а затем оставляли при комнатной температуре в атмосфере азота. К полученному раствору добавляли фенил-хлортионформиат (69 мкл) в сухом толуоле (5 мл). ТСХ контроль показал, что реакция завершалась через 6 часов. Затем растворитель удаляли при пониженном давлении, а сырую смесь подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью гексан:этилацетат (20:1) и (15:1), получая в результате соединение, указанное в заголовке (150 мг), в виде бесцветного масла.
δ (1H, CDCl3): 9,72 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,97 (s,1H, CHPh2), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,10 (m, 1H, H-3'), 4,61 (dd, 1H, H-1', J=2,4 и 8,4 Гц), 4,44 (dd, 1H, H-4', J=6,6 и 9,0 Гц), 4,04 и 3,67 (2d, 2H, 8aCH2, JAB=9 Гц), 3,62 (m, 1H, H-5'), 2,70 (t, 1H, H-1, J=2,0 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 12
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Аллил-2,6-дидезокси- β- D- аллопиранозилокси)метил] -4-формил-4-4a,5,6,7,7a,8,8a-октагидpo-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Суспензию Промежуточного соединения 10 (400 мг) и оксида трибутилолова (240 мг) в сухом толуоле (50 мл) прогревали с дефлегмацией в колбе, снабженной конденсатором Dean-Stark, а затем оставляли при комнатной температуре в атмосфере азота (приблизительно 10 мл азеотропной смеси удаляли). Последовательно добавляли аллилбромид (71 мкл) и тетрабутиламмонийфторид (1М раствор в тетрагидрофуране, 0,95 мл) и прогревали смесь при 50oC в течение 24 часов. В результате удаления растворителя получали остаток, который подвергали хроматографи быстрого разделения на силикагеле с элюцией смесью ацетон:гексан (1:20) и (1:15), получая в результате соединение, указанное в заголовке (300 мг), в виде бесцветного масла.
δ (1H, CDCl3): 9,72 (s, 1H, CHO), 7,52-7,2 (m, 1H, 2Ph), 6,98 (s, H, CHPh2), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,89 (m, 1H,  ), 5,30-5,20 (m, 2H,
), 5,30-5,20 (m, 2H,  ), 4,64 (dd, 1H, H-1', J=2,1 и 9,6 Гц), 4,19 (m, 1H, H-3'), 4,15-3,95 (m, 3H,
), 4,64 (dd, 1H, H-1', J=2,1 и 9,6 Гц), 4,19 (m, 1H, H-3'), 4,15-3,95 (m, 3H, 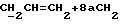 ), 3,80-3,60 (m, 2H, H5'+8aCH2), 3,06 (dd, 1H, H-4', J=3,3 и 9,3 Гц), 2,75 (t, 1H, H-1, J=3,9 Гц), 2,38 (bd, 1H, OH).
), 3,80-3,60 (m, 2H, H5'+8aCH2), 3,06 (dd, 1H, H-4', J=3,3 и 9,3 Гц), 2,75 (t, 1H, H-1, J=3,9 Гц), 2,38 (bd, 1H, OH).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 13
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2,6-Дидезокси-4-O-(2-метил-2-пропенил)- β- D-аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
Суспензию Промежуточного соединения 10 (500 мг) и оксида трибутилолова (300 мг) в сухом толуоле (50 мл) прогревали с дефлегмацией в колбе, снабженной конденсатором Dean-Stafk, а затем оставляли при комнатной температуре в атмосфере азота (приблизительно 10 мл азеотропной смеси удаляли). Последовательно добавляли 3-Бром-2-метил-пропен (202 мкл) и тетрабутиламмонийфторид (1М раствор в тетрагидрофуране, 0,90 мл), смесь прогревали при 50oC в течение 24 часов. После удаления растворителя получали остаток, который подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью ацетон: гексан (1:20) и (1:15), получая в результате соединение, указанное в заголовке (340 мг), в виде бесцветного масла.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,50-7,20 (m, 1H, 2Ph), 6,98 (s, 1H, CHPh2), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,96 и 4,91 (2m, 2H, =CH2), 4,65 (dd, 1H, H-1', J= 1,8 и 9,6 Гц), 4,20 (m, 1H, H-3'), 4,05 и 3,68 (2d, 2H, 8aCH2, JAB=9,3 Гц), 4,00 и 3,87 (2d, 2H, OCH2, JAB=12 Гц), 3,75 (m, 1H, H-5'), 3,06 (dd, 1H, H-4', J=3 и 9,6 Гц), 2,75 (t, 1H, H-1, J=3,6 Гц), 2,37 (bd, 1H, OH), 1,76 (bs, 3H, 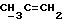 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 14
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,7R,9R]-2,5,8-Триокса-3,7-диметил-цис- бицикло[4,4,0] -дек-9-ил-окси-метил] -4-фор-мил-4,4a, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил 1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору промежуточного соединения 12 (200 мг) в без водном тетрагидрофуране (10 мл) при комнатной температуре в атмосфере азота небольшими порциями добавляли твердый трифторацетат ртути (213 мг). Реакционную смесь оставляли на 30 минут при комнатной температуре, а затем при интенсивном перемешивании добавляли гидрид трибутилолова (135 мкл). Спустя 30 минут добавляли этилацетат (50 мл) и фильтровали суспензию через броунмиллерит, получая в результате бесцветный раствор, который промывали водой (3x100 мл) и сушили над сульфатом магния. В результате удаления растворителя получали масло, которое подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью ацетон:гексан (1:15), в результате чего получали соединение, указанное в заголовке (120 мг), в виде 1:1 смеси эпимеров при C-3'.
δ (1H, CDCl3) включает: 4,73 (dd, 1H, H-9', J=3,0 и 6,3 Гц), 4,62 (dd, 1H, Н-9', J=2,1 и 9,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 15
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,7R,9S]-2,5,8-Триокса- 3,3,7-триметил-цис-бицикло[4,4,0] -дек-9-ил-окси-ме-тил] -4- формил-4,4a,5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3а(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 13 (325 мг) в безводном тетрагидрофуране (10 мл) при комнатной температуре в атмосфере азота добавляли небольшими порциями твердый трифторацетат ртути (303 мг). Реакционную смесь оставляли при комнатной температуре в течение 30 минут, а затем при интенсивном перемешивании добавляли гидрид трибутилолова (192 мкл). Спустя 30 минут добавляли этилацетат (50 мл) и фильтровали полученную суспензию через броунмиллерит, получая при этом бесцветный раствор, который промывали водой (3x100 мл) и сушили над сульфатом магния. После удаления растворителя получали масло, которое подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью ацетон: гексан (1:20) и (1:15), получая в результате соединение, указанное в заголовке (210 мг), в виде бесцветного масла.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,50-7,20 (m, 1H, 1Ph), 6,98 (s, 1H, CHPh2), 6,03 (dd, 1H, H-2, J=1,2 Гц, J=3,3 Гц), 4,54 (dd, 1H, H-9', J= 2,1 и 9,6 Гц), 4,25 (m, 2H, H-7' и Н-1'), 4,05 и 3,66 (2d, 2H, 8aCH2, JAB=9 Гц), 3,41 и 3,18 (2d, 2H, OCH2, JAB=11,7 Гц), 3,25 (dd, 1H, H6', J=3,3 и 10,2 Гц), 2,75 (t, 1H, H-1, J=3,3 Гц), 1,25 и 1,16 (2s, 2CH3).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 16
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2,6-Дидезокси-4-O-p-метоксибензил- β- D-аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7а,8,8а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
Суспензию Промежуточного соединения 10 (500 мг) и оксида трибутилолова (374 мг) в сухом толуоле (50 мл) прогревали с дефлегмацией в колбе, снабженной конденсатором Dean Stark, а затем оставляли при комнатной температуре в атмосфере азота (приблизительно 10 мл азеотропной смеси удаляли). Последовательно добавляли п-Метоксибензилхлорид (135 мкл) и тетрабутиламмонийфторид (1М раствор в тетрагидрофуране, 1,5 мл) и прогревали полученную смесь при 60oC в течение 24 часов. После удаления растворителя получали остаток, который подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью ацетон:гексан (1:20) и (1:15), получая в результате соединение, указанное в заголовке (300 мг), в виде бесцветного масла.
δ (1H, CDCl3): 9,72 (s, 1H, CHO), 7,5-7,2 (m, 12H, 2Ph и 2H орто), 6,97 (s, H, CHPh2), 6,88 (m, 2H, 2H мета), 6,04 (dd, 1H, H-2, J=1,5 Гц, J=3,6 Гц), 4,64 (dd, 1H, H-1', J=2,4 и 9,9 Гц), 4,55 и 4,50 (2d, 2H, CH2Ph, JAB= 11,2 Гц), 4,18 (m, 1H, Н-3'), 4,04 и 3,66 (2d, 2H, 8aCH2, JAB=9 Гц), 3,80 (s, 3H, CH3O), 3,74 (m, 1H, H-5'), 3,12 (dd, 1H, H-4', J=3,0 и 9,3 Гц), 2,75 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 17
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(3-O-Аллил-2,6-дидезокси-4-O-p- метоксибензил- β- D-аллопиранозилокси)метил]-4-формил-4,4а,5, 6,7,7а,8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3а(1H)-карбоновой кислоты дифенилметиловый эфир
К интенсивно перемешивавшемуся раствору Промежуточного соединения 16 (400 мг) в безводном тетрагидрофуране (10 мл) в атмосфере азота при 0oC добавляли маленькими порциями гидрид натрия (48 мг). После завершения добавления смесь оставляли при комнатной температуре в течение 1 часа, а затем добавляли аллилбромид (191 мкл). Спустя 24 часа реакцию гасили добавлением хлорида аммония (1N раствор в воде, 100 мл), а смесь экстрагировали этилацетатом (200 мл). Органический слой промывали водой (1x100 мл), сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате масло, которое подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью этилацетат:гексан (1:20) и (1:15), получая в результате соединение, указанное в заголовке (400 мг), в виде бесцветного масла.
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 7,5-7,2 (m, 12H, 2Ph и 2H орто), 6,98 (s, H, CHPh2), 6,86 (m, 1H, 2H мета), 6,04 (dd, 1H, H2, J=1,2 и 3,3 Гц), 5,95 (m, 1H, 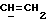 ), 5,28 и 5,15 (2m, 2H,
), 5,28 и 5,15 (2m, 2H, 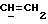 ), 4,60 (m, 2H, Н-1' и CH2Ph), 4,42 (d, 1H, CH2Ph, JAB=11,4 Гц), 4,13 (m, 2H,
), 4,60 (m, 2H, Н-1' и CH2Ph), 4,42 (d, 1H, CH2Ph, JAB=11,4 Гц), 4,13 (m, 2H, 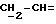 ), 4,02 и 3,68 (2d, 2H, 8aCH2, JAB=9,6 Гц), 3,89 (m, 2H, Н-5' и Н-3'), 3,80 (s, 3H, CH3O), 3,09 (dd, 1H, H-4', J=3,0 и 9,3 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц).
), 4,02 и 3,68 (2d, 2H, 8aCH2, JAB=9,6 Гц), 3,89 (m, 2H, Н-5' и Н-3'), 3,80 (s, 3H, CH3O), 3,09 (dd, 1H, H-4', J=3,0 и 9,3 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 18
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(3-O-Аллил-2,6-дидезокси- β- D- аллопиранозилокси)метил-4-формил-4,4a, 5,6,7,7а, 8,8а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 17 (2,3 г) в дихлорметане (200 мл) перемешивали с водой (20 мл) и добавляли 2,3-дихлор-5,6- дицианохинон (0,79 г). После перемешивания в течение ночи при комнатной температуре смесь фильтровали через слой броунмиллерита с помощью дополнительного количества дихлорметана. Фазу дихлорметана промывали водным бикарбонатом натрия, а затем раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали до сухого состояния. Остаток хроматографировали на силикагеле, используя смесь дихлорметан: метанол (49:1) в качестве элюента, получая в результате соединение, указанное в заголовке (1,95 г), в виде белой пены.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,45-7,24 (m, 1H, Ph2), 6,99 (s, 1H, 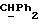 ), 6,05 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 5,95 (m, 1H,
), 6,05 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 5,95 (m, 1H, 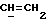 ), 5,26 (m, 2H,
), 5,26 (m, 2H, 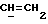 ), 4,53 (dd, 1H, H-1', J=1,8 и 9,6 Гц), 4,19 и 3,96 (2m, 2H,
), 4,53 (dd, 1H, H-1', J=1,8 и 9,6 Гц), 4,19 и 3,96 (2m, 2H, 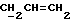 ), 4,03 и 3,69 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,80 (q, 1H, Н-3', J= 3,3 Гц), 3,60 (dq, 1H, H-5', J=9,6 и 6,3 Гц), 3,23 (ddd, 1H, Н-4', J=10,8, 9,6 и 3,3 Гц), 2,74 (t, 1H, H-1, J=3,6 Гц), 2,25 (d,1H, OH, J=10,8 Гц).
), 4,03 и 3,69 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,80 (q, 1H, Н-3', J= 3,3 Гц), 3,60 (dq, 1H, H-5', J=9,6 и 6,3 Гц), 3,23 (ddd, 1H, Н-4', J=10,8, 9,6 и 3,3 Гц), 2,74 (t, 1H, H-1, J=3,6 Гц), 2,25 (d,1H, OH, J=10,8 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 19
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(3-O-Аллил-2,6-дидезокси-4-O- (метилтио)тиокарбонил- β- D-аллопиранозилокси)метил] -4-формил- 4,4a, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3а(1H)-карбоновой кислоты дифенилметиловый эфир
К охлажденному раствору Промежуточного соединения 18 (1,9 г) в безводном тетрагидрофуране (50 мл) в атмосфере азота добавляли малыми порциями (240 мг) гидрид натрия и каталитическое количество имидазола. Затем удаляли ледяную баню и оставляли реакционную смесь при комнатной температуре с перемешиванием в течение 1 часа. Затем последовательно добавляли сероуглерод (1,5 мл) и метилиодид (1,25 мл). После завершения реакции (ТСХ контроль) сырую смесь вливали в этилацетат (500 мл) и гасили 0,1 N водным хлоридом аммония (250 мл). Органический слой затем промывали рассолом (1x500 мл) и водой (1x500 мл), сушили над безводным сульфатом натрия и концентрировали до сухого состояния. Масляный остаток подвергали хроматографии быстрого разделения на силикагеле, используя в качестве элюента смесь гексан:дихлорметан (3:5) и получая в результате соединение, указанное в заголовке (1,85 г), в виде белой пены.
δ (1H, CDCl3):
9,74 (s, 1H, CHO), 7,45-7,24 (m, 1H, Ph2), 6,99 (s,1H,  ), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,88 (m, 1H,
), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,88 (m, 1H, 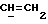 ), 5,41 (dd, 1H, H-4', J= 2,7 и 9,6 Гц), 5,22 (m, 2H,
), 5,41 (dd, 1H, H-4', J= 2,7 и 9,6 Гц), 5,22 (m, 2H, 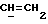 ), 4,70 (dd, 1H, H1', J=2,1 и 9,3 Гц), 4,20-4,02 (m, 5H, H-5',
), 4,70 (dd, 1H, H1', J=2,1 и 9,3 Гц), 4,20-4,02 (m, 5H, H-5', 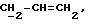 H-3' и 8aCH2(1H)), 3,71 (d, 1H, 8a-CH2, J=9,3 Гц), 2,75 (t, 1H, H-1, J=3,9 Гц), 2,58 (s, 3H, 3CH3).
H-3' и 8aCH2(1H)), 3,71 (d, 1H, 8a-CH2, J=9,3 Гц), 2,75 (t, 1H, H-1, J=3,9 Гц), 2,58 (s, 3H, 3CH3).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 20
(a) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4S,6R,8R]-2,7-Диокса-4,6-диметил- цис-бицикло[3,4,0] -нон-8-ил-окси-метил] -4-формил-4,4a, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир и
(б) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S, 4R,6R,8R]-2,7-Диокса- 4,6-диметил-цис-бицикло[3.4.0]-нон-8-ил-окси-метил]-4-формил-4, 4a,5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано- s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 19 (1,8 г) в сухом толуоле (250 мл) дегазировали током аргона в течение 1 часа, а затем нагревали с дефлегмацией. В течение приблизительно 2 часов, пока продолжалось нагревание и продувание аргоном, с помощью шприца добавляли каталитическое количество азобис(изобутиронитрил)а и гидрида трибутилолова (0,95 мл), растворенных в сухом толуоле (50 мл). После того, как добавление было завершено, реакционную смесь прогревали с дефлегмацией в течение 1 часа и затем охлаждали, концентрировали до сухого состояния и подвергали хроматографии быстрого разделения на силикагеле, используя в качестве элюентов гексан, а затем смесь гексан: этилацетат (9: 1). Промежуточное соединение 20 (а) (680 мг; Rf=0,31 в смеси гексан:этилацетат 4:1) получали в виде белой пены, а Промежуточное соединение 20(б) (180 мг; Rf=0,35 в смеси гексан:этилацетат 4:1) выделяли в виде вещества белого цвета.
(а) δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,28 (m, 1H, Ph2), 6,99 (s, 1H, CHPh2), 6,05 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,46 (dd, 1H, H-8', J= 2,4 и 9,6 Гц), 4,14-4,05 (m, 3H, 8a-CH2(1H), H-1' и CH2-3'(1H), 3,67 (d, 1H, 8a-CH2, J= 9,3 Гц), 3,36-3,25 (m, 2H, Н-6' и CH2-3'(1H)), 2,77 (t, 1H, H-1, J= 3,9 Гц), 1,05 (d, 3H, 4'-CH3, J=6,9 Гц).
(б) δ (1H, CDCl3): 9,72 (s, 1H, CHO), 7,45-7,24 (m, 1H, Ph2), 6,99 (s, 1H, 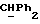 ), 6,05 (dd, 1H, H-2, J=1,50 и 3,6 Гц), 4,42 (dd, 1H, H-8', J= 2,1 и 9,6 Гц), 4,20 (m, 1H, Н-1'), 4,07 и 3,67 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,97 (t, 1H, CH2-3', J=8,4 Гц), 3,54 (m, 1H, Н-6'), 3,46 (dd, 1H, CH2-3', J=10,5 Гц), 2,77 (t, 1H, H-1, J=3,9 Гц), 2,53 (m, 1H, Н-4'), 1,02 (d, 3H, 6-CH3).
), 6,05 (dd, 1H, H-2, J=1,50 и 3,6 Гц), 4,42 (dd, 1H, H-8', J= 2,1 и 9,6 Гц), 4,20 (m, 1H, Н-1'), 4,07 и 3,67 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,97 (t, 1H, CH2-3', J=8,4 Гц), 3,54 (m, 1H, Н-6'), 3,46 (dd, 1H, CH2-3', J=10,5 Гц), 2,77 (t, 1H, H-1, J=3,9 Гц), 2,53 (m, 1H, Н-4'), 1,02 (d, 3H, 6-CH3).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 21
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Аллил-2,6-дидезокси-3-O-(метилтио) тиокарбонил- β- D-аллопиранозилокси)метил]-4-формил-4,4a,5,6,7, 7a,8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 12 (380 мг) в безводном тетрагидрофуране (10 мл) в атмосфере азота при 0oC добавляли малыми порциями твердый гидрид натрия (48 мг) и каталитическое количество имидазола. После завершения добавления охлаждающую баню удаляли и реакция продолжалась в течение 1 часа при комнатной температуре. Последовательно добавляли дисульфид углерода (300 мкл) и метилиодид (250 мкл). Спустя 4 часа смесь вливали в этилацетат (100 мл) и гасили водным 1N хлоридом аммония (50 мл). Органический слой промывали рассолом (1x100 мл) и водой (1x100 мл), сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате желтое масло, которое затем подвергали хроматографии быстрого разделения на силикагеле, используя в качестве элюента смесь этилацетат:гексан (1:25) и (1:20) и получая в результате соединение, указанное в заголовке (350 мг), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,50- 7,20 (m, 1H, 2Ph), 6,98 (s, H, CHPh2), 6,24 (m, 1H, H-3'), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,85 (m, 1H, 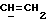 ), 5,30- 5,20 (m, 2H,
), 5,30- 5,20 (m, 2H,  ), 4,56 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,15- 3,75 (m, 4H,
), 4,56 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,15- 3,75 (m, 4H,  =CH2+8aCH2+H-5'), 3,69 (d, 1H, 8aCH2, J=9,3 Гц), 3,21 (dd, 1H, H-4', J=3,3 и 9,3 Гц), 2,72 (t, 1H, H-1, J= 3,89 Гц), 2,59 (s, 3H, SCH3).
=CH2+8aCH2+H-5'), 3,69 (d, 1H, 8aCH2, J=9,3 Гц), 3,21 (dd, 1H, H-4', J=3,3 и 9,3 Гц), 2,72 (t, 1H, H-1, J= 3,89 Гц), 2,59 (s, 3H, SCH3).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 22
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси-3-O- (метилтио)тиокарбонил-4-O-(2-метил-2-пропенил)- β- D- аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7а,8,8а- октагидро-7-метил-3-(1-метил этил)-1,4-мeтaнo-s-индaцeн-3a(1H)- кapбoнoвoй кислоты дифенилметиловый эфир
К охлажденному раствору Промежуточного соединения 13 (700 мг) в безводном тетрагидрофуране (30 мл) в атмосфере азота добавляли малыми порциями гидрид натрия (85 мг) и каталитическое количество имидазола. Затем ледяную баню удаляли, а реакционную смесь оставляли при комнатной температуре при перемешивании с помощью магнитной мешалки в течение 1 часа. Последовательно добавляли сероуглерод (0,54 мл) и метилиодид (0,45 мл). После окончания реакции (ТСХ контроль) сырую смесь вливали в этилацетат (200 мл) и гасили 0,1N водным хлоридом аммония (120 мл). Органический слой затем промывали рассолом (1x200 мл) и водой (1x200 мл), сушили над безводным сульфатом натрия и концентрировали до сухого состояния. Полученный таким образом масляный остаток хроматографировали на силикагеле, применяя в качестве элюента смесь гексан: этилацетат (19: 1) и получая в результате соединение, указанное в заголовке (600 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,26 (m, 1H, Ph2), 6,98 (s, 1H, 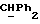 ), 6,24 (m, 1H, H-3'), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,92 и 4,88 (2m, 2H,
), 6,24 (m, 1H, H-3'), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,92 и 4,88 (2m, 2H, 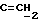 ), 4,55 (dd, 1H, H-1', J=2,1 и 9,6 Гц), 4,03 и 3,69 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,95 и 3,82 (2d, 2H,
), 4,55 (dd, 1H, H-1', J=2,1 и 9,6 Гц), 4,03 и 3,69 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,95 и 3,82 (2d, 2H, 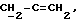 J=12,3 Гц), 3,86 (m, 1H, Н-5'), 3,19 (dd, 1H, H-4', J=3 и 9,3 Гц), 2,72 (t, 1H, H-1, J=3,9 Гц), 2,58 (s,3H, 3CH3), 1,70 (s, 3H,
J=12,3 Гц), 3,86 (m, 1H, Н-5'), 3,19 (dd, 1H, H-4', J=3 и 9,3 Гц), 2,72 (t, 1H, H-1, J=3,9 Гц), 2,58 (s,3H, 3CH3), 1,70 (s, 3H, 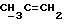 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 23
(а) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4R,7R,9R]-2,8-Диокса-4,9- димeтил-циc-бициклo[3.4.0] -нон-7-ил-окси-метил] -4-формил-4, 4a,5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир и
(б) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4S,7R,9R]-2,8-Диокса-4,9-димeтил- циc-бициклo[3.4.0] -нон-7-ил-окси-метил] -4-формил-4,4a, 5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
МЕТОД А
Раствор Промежуточного соединения 21 (1,5 г) в сухом толуоле (150 мл) дегазировали аргоном в течение 90 минут, а затем нагревали с дефлегмацией. К этому кипящему раствору добавляли гидрид трибутилолова (847 мкл) и азобис(изобутиронитрил) (33 мг). Нагревание продолжали еще в течение 30 минут. После удаления растворителя получали сырую смесь, которую дважды подвергали хроматографии быстрого разделения на силикагеле, применяя в качестве элюента смесь гексан:этилацетат (20:1) и получая в результате соединение, указанное в заголовке (а) (15,0 мг; Rf=0,6 в смеси гексан:этилацетат 4;1) и соединение, указанное в заголовке (б) (260 мг; Rf=0,4 в смеси гексан:этилацетат 4: 1) в виде бесцветных масел.
(а) δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,46 (dd, 1H, H-7', J=3 и 7,5 Гц), 4,06-3,95 (m, 2H, H-3' и 8aCH2), 3,78-3,60 (m, 2H, H-1' и 8aCH2), 3,38-3,26 (m, 2H, H-9' и Н-3'), 2,75 (t, 1H, H-1, J=3,6 Гц), 0,98 (d, 3H, 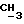 CH, J=6,9 Гц).
CH, J=6,9 Гц).
(б) δ (1H, CDCl3): 9,74 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,04 (dd, 1H, H-1, J=1,5 и 3,6 Гц), 4,74 (t, 1H, H-7', J=3 Гц), 3,94 и 3,61 (2d, 2H, 8aCH2, JAB=9,3 Гц), 3,88 и 3,76 (2d, 2H, H-3'), 3,66 (m, 1H, H-9'), 3,43 (t, 1H, H-1', J=7,8 Гц), 2,72 (t, 1H, H-1, J=3,6 Гц), 2,46 (2m, 2H, 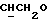 и H-5'), 0,97 (d, 3H,
и H-5'), 0,97 (d, 3H, 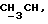 J=6,9 Гц).
J=6,9 Гц).
МЕТОД Б
Раствор Промежуточного соединения 98 (685 мг) в о-ксилене (15 мл) дегазировали аргоном в течение 60 минут, а затем нагревали с дефлегмацией. Добавляли гидрид трибутилолова (379 мкл) и продолжали нагревание еще в течение 15 минут. После охлаждения смесь распределяли между диэтиловым эфиром (150 мл) и водой (100 мл). Органический слой промывали насыщенным водным раствором фторида калия до тех пор, пока не прекращалось выпадение осадка фторида трибутилолова, фильтровали и выпаривали до сухого состояния, получая в результате остаток, который дважды подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (20:1) и получая в результате соединение, указанное в заголовке (а) (103 мг) и соединение, указанное в заголовке (б) (235 мг).
МЕТОД В
Раствор гидрида трибутилолова (684 мкл) в сухом толуоле (25 мл) дегазировали током аргона в течение 1 часа с дефлегмацией, а затем спустя 2 часа с помощью шприца добавляли раствор промежуточного соединения 98 (610 мг) в сухом толуоле (30 мл). Нагревание продолжали в течение 3,5 часов. В результате удаления растворителя при пониженном давлении получали сырой продукт, который подвергали хроматографии быстрого разделения на силикагеле, элюируя гексаном и смесью гексан:этилацетат от (10:1) до (8:1), получая в результате соединение, указанное в заголовке (а) (357 мг).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 24
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,7R,9R]-2,8-Диокса-4,4,8- триметил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил] -4-формил-4, 4a, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 22 (580 мг) в сухом толуоле (150 мл) дегазировали током аргона в течение 1 часа, а затем нагревали с дефлегмацией. В течение 2 часов, пока продолжалось нагревание и продувание аргоном, с помощью шприца добавляли каталитическое количество азобис(изобутиронитрил)а и гидрид трибутилолова (0,27 мл), растворенных в сухом толуоле (20 мл). После завершения добавления реакционную смесь нагревали еще в течение 3 часов, а затем последовательно охлаждали, концентрировали и хроматографировали на силикагеле, применяя в качестве элюентов сначала гексан, а затем смесь гексан: этилацетат (4:1) и получая в результате соединение, указанное в заголовке (100 мг), в виде белой пены.
δ (1H, CDCl3): 9,74 9,74 (s, 1H, 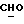 ), 7,45-7,26 (m, 1H, Ph2), 6,99 (s, 1H,
), 7,45-7,26 (m, 1H, Ph2), 6,99 (s, 1H, 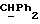 ), 6,04 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,71 (t, 1H, H-7', J=3,3 Гц), 3,93 и 3,61 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,85 (t, 1H, H-1', J=8,7 Гц), 3,56 (m, 1H, H-9'), 3,55 и 3,43 (2d, 2H, CH2-3', J=8,4 Гц), 2,73 (m, 1H, H-1), 1,08 и 0,99 (2s, 6H, 4'-CH3).
), 6,04 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,71 (t, 1H, H-7', J=3,3 Гц), 3,93 и 3,61 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,85 (t, 1H, H-1', J=8,7 Гц), 3,56 (m, 1H, H-9'), 3,55 и 3,43 (2d, 2H, CH2-3', J=8,4 Гц), 2,73 (m, 1H, H-1), 1,08 и 0,99 (2s, 6H, 4'-CH3).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 25
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[(6-дезокси- β- D-альтропиранозилокси) метил] -4-(1,3-диоксолан-2-ил)-4,4а,5,6,7,7а,8,8а-октагидро-7-метил- 3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К перемешиваемому раствору Промежуточного соединения 1 (2,5 г) в сухом ацетонитриле (100 мл) при комнатной температуре добавляли этиленгликоль (30 мл), триметилортоформиат (2 мл) и каталитическое количество п-толуолсульфоновой кислоты. Реакционную смесь перемешивали в течение 3 часов, затем разбавляли этилацетатом (300 мл) и тщательно промывали 5%-ным водным бикарбонатом натрия и рассолом. Органический слой сушили над безводным сульфатом натрия, концентрировали и очищали посредством хроматографии на силикагеле, применяя в качестве элюента смесь гексан:этилацетат (3:1) и получая в результате соединение, указанное в заголовке (2,6 г).
δ (1H, CDCl3): 7,46-7,24 (m, 1H, Ph2), 6,94 (s, 1H, 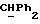 ), 5,83 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,07 (s, 1H,
), 5,83 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,07 (s, 1H, 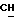 бета), 4,65 (d,1 H, H-1', J= 1,8 Гц), 4,07-4,04 (m, 2H, 8a-CH2(1H) и Н-3'), 3,88- 3,69 (m, 8h,
бета), 4,65 (d,1 H, H-1', J= 1,8 Гц), 4,07-4,04 (m, 2H, 8a-CH2(1H) и Н-3'), 3,88- 3,69 (m, 8h, 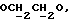 8a-CH2(1H), Н-2', Н-4' и Н-5'), 2,51 (t, 1H, H-1, J=4,5 Гц), 2,37 (d, 1H, OH, J=2,7 Гц), 2,28 (d, 1H, OH, J=3,6 Гц), 2,04 (d, 1H, OH, J= 5,7 Гц).
8a-CH2(1H), Н-2', Н-4' и Н-5'), 2,51 (t, 1H, H-1, J=4,5 Гц), 2,37 (d, 1H, OH, J=2,7 Гц), 2,28 (d, 1H, OH, J=3,6 Гц), 2,04 (d, 1H, OH, J= 5,7 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 26
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-изопропилиден- β- D- альтропиранозилокси)метил]-4-(1,3-диоксолан-2-ил)-4,4a, 5,6,7,7a,8,8а-октагидро-7-метил-3- (1-метилэтил) -1,4-метано-s- индацен-3а(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 25 (200 мг) в сухом ацетонитриле (6 мл) в атмосфере азота обрабатывали 2,2-диметоксипропаном (0,6 мл) и каталитическим количеством п-толуолсульфоната пиридина. После 4 часов при комнатной температуре смесь обрабатывали насыщенным водным раствором однозамещенного карбоната натрия (20 мл) и дважды экстрагировали этилацетатом (20 мл). Объединенные органические фазы промывали водой и рассолом, сушили над сульфатом магния, фильтровали и упаривали. Остаток очищали посредством хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (4:1), соответствующие фракции объединяли и упаривали, получая в результате соединение, указанное в заголовке (205 мг), в виде белой пены.
δ (1H, CDCl3: 7,47-7,2 (m, 1H, 2Ph), 6,93 (s, 1H, 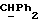 ), 5,83 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 1,3-диоксолан), 4,56 (d, 1H ,H-1', J=2,1 Гц), 4,3 (dd, 1H, H-3', J=3,6 и 5,7 Гц), 4,04 (d, 1H, A часть от 8aCH2, JAB= 9 Гц), 3,95-3,75 (m, 7H, H- 2', H-4',
), 5,83 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 1,3-диоксолан), 4,56 (d, 1H ,H-1', J=2,1 Гц), 4,3 (dd, 1H, H-3', J=3,6 и 5,7 Гц), 4,04 (d, 1H, A часть от 8aCH2, JAB= 9 Гц), 3,95-3,75 (m, 7H, H- 2', H-4', 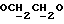 и B часть от 8aCH2), 3,44 (dq, 1H, H-5', J=6 и 9 Гц), 2,65 (m, 1H,
и B часть от 8aCH2), 3,44 (dq, 1H, H-5', J=6 и 9 Гц), 2,65 (m, 1H, 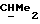 ), 2,51 (bt, 1H, H-1, J=3,6 Гц), 2,35 (d, 1H, OH2'), 1,47, 1,36 (2s, 6H, 2CH3 кеталь изопропилидена).
), 2,51 (bt, 1H, H-1, J=3,6 Гц), 2,35 (d, 1H, OH2'), 1,47, 1,36 (2s, 6H, 2CH3 кеталь изопропилидена).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 27
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-изопропилиден- β- D- аллопиранозилокси)метил]-4-(1,3-диоксолан-2-ил)-4,4а,5,6,7,7a,8,3а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
Пиридин (0,67 мл) добавляли к смеси оксида хрома (CrO3) (412 мг) в дихлорметане (25 мл) при 0oC. Спустя 15 минут добавляли уксусный ангидрид (0,40 мл), а еще через 10 минут при той же самой температуре добавляли раствор Промежуточного соединения 26 (750 мг) в дихлорметане (25 мл). Смесь перемешивали в течение 2 часов, фильтровали через колонку с силикагелем с верхним слоем из сульфата магния, фильтрат выпаривали до сухого состояния при пониженном давлении, в результате чего получали сироп. Его растворяли в смеси метанол: вода (10:1, 11 мл), а затем при 0oC добавляли боргидрид натрия (40 мг). Смесь выдерживали при 0oC в течение 2 часов, подкисляли до pH 6-7 раствором 1N соляной кислоты, а затем выпаривали до сухого состояния при пониженном давлении, получая в результате белое вещество, которое дважды очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя последовательно смесью гексан:этилацетат (6:1), а затем смесью (2: 1). Фракции, содержавшие искомый продукт, объединяли и выпаривали под вакуумом, получая в результате соединение, указанное в заголовке (585 мг).
δ 1H, CDCl3): 7,24-7,46 (m, 1H, 2Ph), 6,92 (s, 1H, 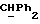 ), 5,87 (dd, 1H, H-2, J=3,9 и 1,5 Гц), 5,09 (s, 1H,
), 5,87 (dd, 1H, H-2, J=3,9 и 1,5 Гц), 5,09 (s, 1H, 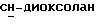 ), 4,52 (t, 1H, H3', J= 4,5 Гц), 4,45 (d, 1H, H-1', J=7,8 Гц), 4,07 (d, 1H, 8aCH2, J=9 Гц), 3,74-3,86 (m, 6H, 2CH2-диоксолан, H-4', 8aCH2), 3,67 (m, 1H, H-2'), 2,65 (m, 1H, Н-14), 2,58 (t, 1H, H-11).
), 4,52 (t, 1H, H3', J= 4,5 Гц), 4,45 (d, 1H, H-1', J=7,8 Гц), 4,07 (d, 1H, 8aCH2, J=9 Гц), 3,74-3,86 (m, 6H, 2CH2-диоксолан, H-4', 8aCH2), 3,67 (m, 1H, H-2'), 2,65 (m, 1H, Н-14), 2,58 (t, 1H, H-11).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 28
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 4-Формил-4,4a,5,6,7,7a,8,8a-октагидро-8a- гидроксиметил-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты 2-(триметилсилил) этиловый эфир
Раствор сордарицина (6,6 г) и O-[2-(триметилсилил)этил]- N,N'-диизопропилизомочевины (9,3 г) в безводном тетрагидрофуране (150 мл) нагревали с дефлегмацией в течение 3 часов. Затем смесь охлаждали до комнатной температуры, фильтровали и концентрировали до сухого состояния. Остаток растворяли в этилацетате (750 мл) и интенсивно промывали 1N соляной кислотой, 5%-ным водным бикарбонатом и рассолом. Органический слой сушили над безводным сульфатом натрия, концентрировали под вакуумом и очищали посредством колоночной хроматографии быстрого разделения на силикагеле, применяя в качестве элюента раствор (10%-ный) этилацетата в гексане и получая в результате соединение, указанное в заголовке (6,8 г) в виде белой пены.
δ (1H, CDCl3): 9,67 (s, 1H, CHO), 6,07 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,27 (m, 2H, 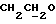 ), 3,93 и 3,49 (2m, 2H, 8a-CH2), 2,75 (bs, 1H, OH), 2,54 (t, 1H, H-1), 1,05 (m, 2H,
), 3,93 и 3,49 (2m, 2H, 8a-CH2), 2,75 (bs, 1H, OH), 2,54 (t, 1H, H-1), 1,05 (m, 2H, 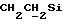 ), 0,07 (s, 9H, (CH3)3Si);
), 0,07 (s, 9H, (CH3)3Si);
δ (13C, CDCl3): 204,9 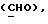 173,6
173,6 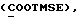 148,2
148,2 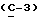 , 130,6
, 130,6 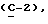 66,9 (8a-CH2O), 64,0
66,9 (8a-CH2O), 64,0 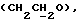 177,7
177,7 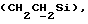 1,6 (CH3)3Si). 1[7].
1,6 (CH3)3Si). 1[7].
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 29
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(3,4,6-Три-O-ацетил-2-дезокси- β- D- аллопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7а, 8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты триметилсилилэтиловый эфир
К раствору Промежуточного соединения 28 (63 мг) и 3,4,6-три- O-ацетил D-аллала1 (20 мг) в сухом толуоле (1 мл) добавляли бромистоводородный трифенилфосфин (9 мг). Реакционную смесь прогревали при 60oC в течение 2 часов и вливали в 1М раствор бикарбоната натрия (100 мл). Добавляли этилацетат (100 мл), органический слой промывали водой, сушили над безводным сульфатом магния и фильтровали. Растворитель упаривали до сухого состояния, а остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (10:1), получая в результате соединение, указанное в заголовке (16 мг), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,03 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,48 (m, 1H, H-3'), 4,89 (dd, 1H, H-4', J=3,3 и 9,6 Гц), 4,65 (dd, 1H, H-1', J=2,4 и 8,7 Гц), 4,23 (m, 4H, H-6', 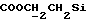 ), 4,02 (ddd, 1H, H-5', J=3, 5,4 и 9,6 Гц), 3,92 и 3,69 (2d, 2H, 8aCH2, J=9,6 Гц), 2,75 (t, 1H, H-1), 2,28 (m, 1H,
), 4,02 (ddd, 1H, H-5', J=3, 5,4 и 9,6 Гц), 3,92 и 3,69 (2d, 2H, 8aCH2, J=9,6 Гц), 2,75 (t, 1H, H-1), 2,28 (m, 1H, 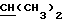 ). 1[8].
). 1[8].
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 30
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2-дезокси-3,4-O-изопропилиден-6- O-метил- β- D-аллопиранозилокси)метил] -4-формил-4,4a,5,6, 7,7а,8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано- s-индацен-3a(H)-карбоновой кислоты 2-(триметилсилил)этиловый эфир
К перемешивавшемуся раствору Промежуточного соединения 29 (275 мг) в абсолютном метаноле (10 мл) добавляли 2 или 3 капли 1М раствору метоксида натрия в метаноле. После перемешивания в течение 1,5 часов реакционную смесь концентрировали, разводили толуолом (10 мл) и концентрировали до получения желтого сиропа. Его растворяли в ацетоне (5 мл) и 2,2-диметоксипропане (60 мкл) и добавляли моногидрат п-толуолсульфоновой кислоты (75 мг). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, нейтрализовали карбонатом натрия, фильтровали и концентрировали фильтрат до сухого состояния. Масляный остаток растворяли в этилацетате (20 мл), промывали водой и рассолом, сушили и концентрировали, получая в результате масло. Раствор этого масла в сухом тетрагидрофуране (5 мл) при 0oC обрабатывали гидридом натрия (10 мг) и перемешивали в течение 30 минут перед добавлением метилиодида (100 мкл). Реакционную смесь в течение 3 часов постепенно нагревали до комнатной температуры. Реакцию гасили метанолом (1 мл), смесь затем концентрировали, остаток трижды очищали посредством колоночной хроматографии быстрого разделения, применяя последовательно смеси гексан:этилацетат (10: 1), (6: 1) и (4:1), соответствующие фракции объединяли и растворители выпаривали, в результате чего получали соединение, указанное в заголовке (50 мг), в виде бесцветного масла.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,04 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,61 (dd, 1H, H-1', J=2,4 и 8,4 Гц), 4,43 (m, 1H, H-3'), 4,34-4,10 (m, 2H, CH2-SEM защитная группа), 3,66 и 3,89 (2d, 2H, 8aCH2, J=9,3 Гц), 3,87 (m, 1H, H-4'), 3,48-3,64 (m, 3H, 2H-6' и Н-5'), 3,24 (s, 3H, 6'OMe), 2,73 (t, 1H, Н-1, J=3,6 Гц), 1,34 и 1,35 (2s, 6H, метильные группы изопропилидена).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 31
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-((E)-2-Бутенил)-2,6-дидезокси- β- D-аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8а-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Суспензию промежуточного соединения 10 (1,5 г) и оксида дибутилолва (0,9 г) в сухом толуоле (150 мл) нагревали с дефлегмацией в течение 2 часов в колбе, снабженной Dean-Stark конденсатором, а затем оставляли при комнатной температуре в атмосфере азота (приблизительно 20 мл азеотропной смеси удаляли). Последовательно добавляли кротилбромид (преимущественно транс, 0,37 мл), тетрабутиламмонийфторид (3,6 мл 1М раствора в тетрагидрофуране)
и 4 A молекулярные сита (активированный порошок), смесь прогревали при 60oC в течение 24 часов. Молекулярные сита отфильтровывали. После удаления растворителя получали остаток, который подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью гексан: ацетон (95:5), получая в результате соединение, указанное в заголовке (1 г), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,44-7,23 (m, 10H, 2Ph), 6,98 (s, 1H, 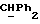 ), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,75-5,52 (m,2H,
), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,75-5,52 (m,2H, 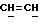 ), 4,64 (dd, 1H, H-1', J= 2,1 и 9,6 Гц), 4,16 (m, 1H, H-3'), 4,98 (m, 3H, 8a-CH2, (1H) и
), 4,64 (dd, 1H, H-1', J= 2,1 и 9,6 Гц), 4,16 (m, 1H, H-3'), 4,98 (m, 3H, 8a-CH2, (1H) и 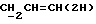 ), 3,74 (m, 1H, H-5'), 3,67 (d, 1H, 8a-CH2(1H), J=9,3 Гц), 3,05 (dd, 1H, H-4', J=3,0 и 9,6 Гц), 2,75 (t, 1H, H-1, J=3,6 Гц), 2,40 (d, 1H, OH, J=2,1 Гц), 1,72 (m, 3H,
), 3,74 (m, 1H, H-5'), 3,67 (d, 1H, 8a-CH2(1H), J=9,3 Гц), 3,05 (dd, 1H, H-4', J=3,0 и 9,6 Гц), 2,75 (t, 1H, H-1, J=3,6 Гц), 2,40 (d, 1H, OH, J=2,1 Гц), 1,72 (m, 3H, 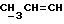 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 32
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси-4-O-пропаргил- β- D- аллопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7а, 8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
Суспензию промежуточного соединения 10 (2 г) и оксида дибутилолова (1,2 г) в сухом толуоле (150 мл) нагревали с дефлегмацией в колбе, снабженной Dean-Stark конденсатором, а затем оставляли при комнатной температуре в атмосфере азота (приблизительно 20 мл азеотропной смеси удаляли). Последовательно добавляли пропаргилбромид (0,55 мл 80%-ного (вес.) раствора в толуоле), тетрабутиламмонийфторид (4,8 мл 1М раствора в тетрагидрофуране) и 4A молекулярные сита (активированный порошок), полученную смесь прогревали при 60oC в течение 24 часов. Молекулярные сита отфильтровывали. После удаления растворителя получали остаток, который дважды подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью гексан: ацетон (96:4), получая в результате соединение, указанное в заголовке (1,25 г), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,44-7,26 (m, 10H, 2Ph), 6,98 (s, 1H, 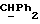 ), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,65 (dd, 1H, H-1', J=2,1 и 9,6 Гц), 4,29 (10, 1H, H-3'), 4,24 и 4,22 (2d, 2H,
), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,65 (dd, 1H, H-1', J=2,1 и 9,6 Гц), 4,29 (10, 1H, H-3'), 4,24 и 4,22 (2d, 2H, 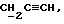 J=2,4 Гц), 4,05 и 3,68 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,75 (m, 1H, H-5'), 3,25 (dd, 1H, H-4', J= 3,0 и 9,3 Гц), 2,75 (t, 1H, H-1, J=3,9 Гц), 2,46 (t,1H,
J=2,4 Гц), 4,05 и 3,68 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,75 (m, 1H, H-5'), 3,25 (dd, 1H, H-4', J= 3,0 и 9,3 Гц), 2,75 (t, 1H, H-1, J=3,9 Гц), 2,46 (t,1H, 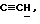 J=2,4 Гц).
J=2,4 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 33
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-(3-метил-2-бутенил)-2,6-дидезокси- β- D-аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8а-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновой кислоты дифенилметиловый эфир
Суспензию промежуточного соединения 10 (1 г) и трибутилиноксида (600 мг) в 150 мл сухого толуола нагревали с дефлегмацией в течение 2 часов в колбе, снабженной Dean-Stark конденсатором (25 мл азеотропной смеси удаляли), а затем охлаждали до комнатной температуры. Затем добавляли 2-Метил-4-бром-2-бутен (276 мкл) и тетрабутиламмонийфторид (1М раствор в тетрагидрофуране, 2,4 мл) и нагревали смесь до 50oC в течение 24 часов. После удаления растворителя получали сырой продукт, который подвергали хроматографии быстрого разделения (этилацетат:гексан, 1:20, 1:15 и 1:10), получая в результате 770 мг соединения, указанного в заголовке, в виде пены (выход 69%).
δ 1H, CDCl3): 9,73 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,98 (s, 1H, CHPh2), 6,05 (dd, 1H, H2, J=1,5 и 3,3 Гц), 5,33 (m, 1H,  ), 4,64 (dd, 1H, H1', J=2,1 и 9,6 Гц), 4,20 (m, 1H, H3'), 4,25-3,95 (m, 3H,
), 4,64 (dd, 1H, H1', J=2,1 и 9,6 Гц), 4,20 (m, 1H, H3'), 4,25-3,95 (m, 3H,  ), 3,75-3,65 (m, 2H, H5'+8aCH2), 3,03 (dd, 1H, H4', J=3,3 и 9,6 Гц), 2,75 ( t, 1H, H1, J=3,9 Гц), 2,44 (d, 1H, OH, J=2,1 Гц).
), 3,75-3,65 (m, 2H, H5'+8aCH2), 3,03 (dd, 1H, H4', J=3,3 и 9,6 Гц), 2,75 ( t, 1H, H1, J=3,9 Гц), 2,44 (d, 1H, OH, J=2,1 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 34
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(4-O-((E)-2-Бутенил)-2,6-дидезокси-3-O- метилтио)тиокарбонил- β- D-аллопиранозилокси)метил]-4-формил-4,4a, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метил-этил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К охлажденному раствору промежуточного соединения 31 (1 г) в безводном тетрагидрофуране (20 мл) в атмосфере азота добавляли малыми порциями гидрид натрия (0,156 г) и каталитическое количество имидазола. Затем ледяную баню удаляли и реакционную смесь оставляли при комнатной температуре при перемешивании в течение 1 часа. Последовательно добавляли сероуглерод (0,58 мл) и метилиодид (0,8 мл). После завершения реакции (ТСХ контроль) сырую смесь вливали в этилацетат (400 мл) и гасили ее 1N водным хлоридом аммония (500 мл). Органический слой затем тщательно промывали 1N соляной кислотой (200 мл), 5%-ным водным бикарбонатом натрия (200 мл) и рассолом (200 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Полученный таким образом остаток подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью дихлорметан: гексан (6: 4), получая в результате соединение, указанное в заголовке (1,04 г), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45- 7,26 (m, 10H, 2Ph), 6,98 (s, 1H, 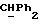 ), 6,23 (dd, 1H, H-3', J=3,0 и 6,3 Гц), 6,05 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 5,75- 5,40 (m, 2H,
), 6,23 (dd, 1H, H-3', J=3,0 и 6,3 Гц), 6,05 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 5,75- 5,40 (m, 2H, 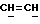 ), 4,55 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,05- 3,80 (m, 4H, 8a-CH2(1H),
), 4,55 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,05- 3,80 (m, 4H, 8a-CH2(1H), 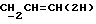 и H-5'), 3,75 (d, 1H, 8a- CH2(1H), J= 9,3 Гц), 3,20 (dd, 1H, H-4',J=3,0 и 9,3 Гц), 2,72 (t, 1H, H-1, J=3,9 Гц), 2,58 (s, 3H, CH3S), 1,68 (m, 3H,
и H-5'), 3,75 (d, 1H, 8a- CH2(1H), J= 9,3 Гц), 3,20 (dd, 1H, H-4',J=3,0 и 9,3 Гц), 2,72 (t, 1H, H-1, J=3,9 Гц), 2,58 (s, 3H, CH3S), 1,68 (m, 3H, 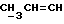 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 35
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси-3-O-(метилтио) тиокарбонил-4-O-пропагил- β- D-аллопиранозилокси)метил] -4- формил-4,4а, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3а(1H)-карбоновой кислоты дифенилметиловый эфир
К охлаждавшемуся раствору промежуточного соединения 32 (1,25 г) в безводном тетрагидрофуране (25 мл) в атмосфере азота добавляли малыми порциями гидрид натрия (0,2 г) и каталитическое количество имидазола. Затем ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Последовательно добавляли дисульфид углерода (0,6 мл) и метилиодид (0,75 мл). После завершения реакции (ТСХ контроль) сырую смесь вливали в этилацетат (400 мл) и гасили 1N водным хлоридом аммония (500 мл). Органический слой затем интенсивно промывали 1N соляной кислотой (200 мл), 5%-ным водным бикарбонатом натрия (200 мл) и рассолом (200 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью гексан:этилацетат 97:3 и 95:5, получая в результате соединение, указанное в заголовке (1,2 г), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,26 (m, 10H, 2Ph), 6,99 (s, 1H, 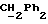 ), 6,28 (q, 1H, H-3', J=3,3 Гц), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,57 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,19 и 4,17 (2d, 2H,
), 6,28 (q, 1H, H-3', J=3,3 Гц), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,57 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,19 и 4,17 (2d, 2H, 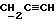 , J=2,4 Гц), 4,03 и 3,70 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,86 (dq, 1H, H-5', J=6,3 и 9,3 Гц), 3,50 (dd, 1H, H-4', J=3,0 и 9,3 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц), 2,58 (s, 3H, CH3S), 2,42 (t, 1H,
, J=2,4 Гц), 4,03 и 3,70 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,86 (dq, 1H, H-5', J=6,3 и 9,3 Гц), 3,50 (dd, 1H, H-4', J=3,0 и 9,3 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц), 2,58 (s, 3H, CH3S), 2,42 (t, 1H, 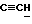 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 36
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-(3-метил-2- бутенил)-2,6-дидезокси-3-O-(метилтио)тиокарбонил- β- D- аллопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилиетиловый эфир
К раствору промежуточного соединения 33 (770 мг) в сухом тетрагидрофуране (15 мл) добавляли при 0oC в атмосфере азота гидрид натрия (105 мг) и имидазол (30 мг). Смесь перемешивали в течение 1 часа при комнатной температуре, затем последовательно добавляли сероуглерод (300 мкл) и метилиодид (311 мкл). После перемешивания в течение 1 часа реакцию гасили добавлением 0,1N хлорида аммония (50 мл) и дважды проводили экстракцию этилацетатом (2x100 мл), сушили над сульфатом магния и концентрировали, получая в результате масло, которое подвергали хроматографии быстрого разделения (силикагель, этилацетат: гексан 1: 25 и 1:20), получая в результате 780 мг соединения, указанного в за головке (90%), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,5- 7,2 (m, 10H, 2Ph), 6,98 (s, 1H, CHPh2), 6,26 (m, 1H, H-3'), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,26 (m, 1H, 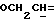 ), 4,54 (dd, 1H, H1', J=1,8 и 9,6 Гц), 4,10-3,75 (m, 4H,
), 4,54 (dd, 1H, H1', J=1,8 и 9,6 Гц), 4,10-3,75 (m, 4H, 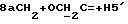 ), 3,69 (d, 1H, 8aCH2, J=9,6 Гц), 3,18 (dd, 1H, H4', J=3 и 9,3 Гц), 2,72 (t, 1H, H1, J=3,9 Гц), 2,58 (s, 3H, SCH3).
), 3,69 (d, 1H, 8aCH2, J=9,6 Гц), 3,18 (dd, 1H, H4', J=3 и 9,3 Гц), 2,72 (t, 1H, H1, J=3,9 Гц), 2,58 (s, 3H, SCH3).
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ 37 и 38
а) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[1S,4R,7R,9R]-2,8-Диокса- 4-этил-9-метил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил]]-4- формил-4,4a,5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
б) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[1S,4S,7R,9R]-2,8- Диокса-4-этил-9-метил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил] ] -4- формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор промежуточного соединения 34 (1,04 г) в сухом толуоле (50 мл) дегазировали током аргона в течение 1 часа при дефлегмации. Добавляли гидрид трибутилолова (0,55 мл) и каталитическое количество азобис(изобутиронитрила). Реакционную смесь далее подвергали дефлегмации в течение 20 минут и последовательно охлаждали, концентрировали, а затем дважды подвергали хроматографии быстрого разделения на силикагеле с элюцией гексаном и смесью гексан:этилацетат (94:6). В результате получали Промежуточное соединение 37 (220 мг, Rf=0,32 в смеси гексан:этилацетат 4:1) в виде белой пены и промежуточное соединение 38 (444 мг, Rf=0,27 в смеси гексан:этилацетат 4:1) в виде белого вещества.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 37
δ (1H, CDCl3): 9,75 (s, 1H, CHO, 7,45-7,26 (m, 10H, 2Ph), 6,99 (s, 1H, 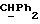 ), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,50 (dd, 1H, H-7, J=2,7 и 6,9 Гц), 4,06 и 3,38 (2dd, 2H, CH2-3', J=8,4 и 7,5 Гц), 4,01 и 3,65 (2d, 2H, 8a-CH2, J=9,0 Гц), 3,71 (dd, 1H, H-1', J=7,5 и 9,0 Гц), 3,34 (m, 1H, H-9'), 2,75 (t, 1H, H-1), 0,93 (t, 3H,
), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,50 (dd, 1H, H-7, J=2,7 и 6,9 Гц), 4,06 и 3,38 (2dd, 2H, CH2-3', J=8,4 и 7,5 Гц), 4,01 и 3,65 (2d, 2H, 8a-CH2, J=9,0 Гц), 3,71 (dd, 1H, H-1', J=7,5 и 9,0 Гц), 3,34 (m, 1H, H-9'), 2,75 (t, 1H, H-1), 0,93 (t, 3H, 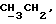 J=7,5 Гц).
J=7,5 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 38
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 7,45-7,26 (m, 10H, 2Ph), 6,99 (s, 1H, 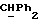 ), 6,04 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,73 (t, 1H, H-7', J=3,0 Гц), 3,92 (m, 2H, 8a-CH3(1H) и CH3-3'(1H)), 3,75 (m, 1H, H-9' и H-1'), 3,62 (d, 1H, 8a-CH2, J= 9,3 Гц), 3,48 (m, 1H, CH2-3'(1H)), 2,71 (t, 1H, H-1, J=3,6 Гц), 2,55 и 2,23 (2m, 3H, H-4', H-5' и 3-CH), 0,96 (m, 3H,
), 6,04 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,73 (t, 1H, H-7', J=3,0 Гц), 3,92 (m, 2H, 8a-CH3(1H) и CH3-3'(1H)), 3,75 (m, 1H, H-9' и H-1'), 3,62 (d, 1H, 8a-CH2, J= 9,3 Гц), 3,48 (m, 1H, CH2-3'(1H)), 2,71 (t, 1H, H-1, J=3,6 Гц), 2,55 и 2,23 (2m, 3H, H-4', H-5' и 3-CH), 0,96 (m, 3H, 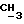 CH2).
CH2).
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ 39 и 40
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4R,7R,9R)-2,8-Диокса-4-(1-метилэтил)- 9-метил-цис-бицикло[3.4.0]-нон-7-ил-оксиметил)]-4-формил-4, 4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир и [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4S,7R,9R)-2,8-Диокса-4-(1-метилэтил)-9-метил- цис-бицикло[3.4.0] -нон-7-ил-оксиметил)]-4-формил-4,4a,5,6,7,7a,8, 8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
Раствор промежуточного соединения 36 (780 мг) в сухом толуоле (50 мл) дегазировали аргоном в течение 1 часа, а затем нагревали с дефлегмацией. Добавляли гидрид трибутилолова (367 мкл) и каталитическое количество a,a'-азоизобутиронитрила (20 мг) и смесь подвергали дефлегмации в течение 2 часов, затем охлаждали до комнатной температуры. После удаления растворителя получали остаток, который подвергали хроматографии быстрого разделения (силикагель, гексан:этилацетат 25:1, 20:1, 18:1 и 15:1), получая в результате 280 мг промежуточного соединения 39 (выход 41%) и 300 мг промежуточного соединения 40 (выход 44%) в виде масел (Rf 0,5 и 0,3 в смеси гексан:этилацетат в/в 3:1, соответственно).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 39
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,5-7,2 (m, 10H, 1Ph), 6,99 (s,1H, CHPh2), 6,05 (dd, 1H, H2, J=1,2 и 3,6 Гц), 4,57 (dd, 1H, H7', J=2,7 и 5,7 Гц), 4,05-3,90 (m, 2H, H3'+8aCH2), 3,73 (dd, 1H, H1', J=8,1 и 9,3 Гц), 3,62 (d, 1H, 8aCH2, J=9 Гц), 3,50-3,45 (m, 2H, H3'+H9'), 2,74 (t, 1H, H1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 40
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,03 (dd, 1H, H2, J=1,5 и 3,3 Гц), 4,74 (t, 1H, H7', J=3 Гц), 4,0-3,8 (m, 8aCH2+H9'+H3'), 3,73 (dd, 1H, H1', J=3,3H 4,5 Гц), 3,63 (d, 1H, 8aCH2, J= 9,6 Гц), 3,52 (dd, 1H, H3', J=8,1 и 10,5 Гц), 2,70 (t, 1H, H1, J= 3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 41
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[1S,7R,9R]-2,8-Диокса-4-метилен,9- метил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил] -4-формил-4,4a, 5,6,7,7 a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано- s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор промежуточного соединения 35 (1,02 г) в сухом толуоле (50 мл) дегазировали током аргона в течение 1 часа при дефлегмации. Добавляли гидрид трибутилолова (0,55 мл) и каталитическое количество азобис(изобутиронирил)а. Затем реакционную смесь подвергали дефлегмации в течение 1 часа, после чего охлаждали, концентрировали, а затем дважды подвергали хроматографии быстрого разделения на силикагеле с элюцией гексаном и смесью гексан:этилацетат (94: 6). Таким образом получали чистое соединение, указанное в заголовке (380 мг), и выделяли его в виде белой пены.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,44-7,28 (m, 10H, 2Ph), 6,99 (s, 1H, 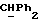 ), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,07 и 5,02 (2dd, 2H,
), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,07 и 5,02 (2dd, 2H,  J= 2,4 и 4,8 Гц), 4,49-4,31 (m, 3H, H-7' и CH2-3'), 3,95 и 3,66 (2d, 2H, 8a-CH2, J= 9,3 Гц), 3,80 (dd, 1H, H-1', J=7,5 и 9,3 Гц), 3,26 (m, 1H, H-9'), 3,02 (m, 1H, H-5'), 2,76 (t, 1H, H-1, J=3,9 Гц);
J= 2,4 и 4,8 Гц), 4,49-4,31 (m, 3H, H-7' и CH2-3'), 3,95 и 3,66 (2d, 2H, 8a-CH2, J= 9,3 Гц), 3,80 (dd, 1H, H-1', J=7,5 и 9,3 Гц), 3,26 (m, 1H, H-9'), 3,02 (m, 1H, H-5'), 2,76 (t, 1H, H-1, J=3,9 Гц);
δ (13C, CDCl3): 204,4  171,0
171,0 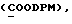 148,8 и 148,3 (C-3 и C-4'), 139,5 и 139,4 (2 CIV Ph), 130,7 (C-2), 128,6, 128,5, 128,2, 128,0, 127,7 и 127,0
148,8 и 148,3 (C-3 и C-4'), 139,5 и 139,4 (2 CIV Ph), 130,7 (C-2), 128,6, 128,5, 128,2, 128,0, 127,7 и 127,0 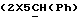 103,9
103,9 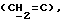 98,3 (C-7'), 80,4 и 69,5 (C-1' и C-9'), 78,5 (CHPh2), 73,9 и 70,6
98,3 (C-7'), 80,4 и 69,5 (C-1' и C-9'), 78,5 (CHPh2), 73,9 и 70,6 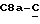 и C-3').
и C-3').
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 42
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,7R,9R]-2,8-Диокса-9-метил-4-метилен-3- оксо-цис-бицикло[3.4.0]-нон-7-ил-окси-метил]-4-формил-4,4a,5,6,7, 7а, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К перемешивавшемуся раствору оксида хрома (VI) (0,23 г) и сухого пиридина (0,37 мл) в сухом дихлорметане (20 мл) при 0oC добавляли промежуточное соединение 41 (0,15 г) в сухом дихлорметане (3 мл). Перемешивание при комнатной температуре продолжали в течение 6 часов, затем реакционную смесь прямо подвергали хроматографии быстрого разделения на силикагеле, используя дихлорметан в качестве элюента и получая в результате соединение, указанное в заголовке (65 мг), в виде белой пены.
δ (1H, CDCl3): 9,74 (s, 1H, 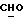 ), 7,44-7,25 (m, 10H, 2Ph), 6,98 (s,1H,
), 7,44-7,25 (m, 10H, 2Ph), 6,98 (s,1H, 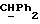 ), 6,37 и 5,64 (2d, 2H,
), 6,37 и 5,64 (2d, 2H, 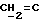 ), 6,04 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,47 (dd, 1H, H-7', J=3,6 и 6,0 Гц), 4,29 (m, 1H, H-1'), 4,00 и 3,64 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,40 (m, 1H, H-5'), 3,30 (m, 1H, H-9'), 2,73 (t, 1H, H-1, J=3,9 Гц).
), 6,04 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,47 (dd, 1H, H-7', J=3,6 и 6,0 Гц), 4,29 (m, 1H, H-1'), 4,00 и 3,64 (2d, 2H, 8a-CH2, J=9,3 Гц), 3,40 (m, 1H, H-5'), 3,30 (m, 1H, H-9'), 2,73 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 43
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,7R,9R]-2,8-диокса-9-метил-4-оксо-цис- бицикло[3.4.0] -нон-7-ил-окси-метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метил-этил)-1,4-метано- s-индацен-3а(1H)-карбоновой кислоты дифенилметиловый эфир
МЕТОД А
К охлаждавшейся на льду смеси Промежуточного соединения 41 (105 мг) и N-метилморфолин N-оксида (43 мг) в системе ацетон: вода (8:1,9 мл) добавляли четырехокись осмия (2,5%-ный вес. раствор в 2-метил-2-пропаноле, 0,075 мл). Затем ледяную баню удаляли, а смесь перемешивали при комнатной температуре в течение 24 часов. Реакцию гасили бисульфитом натрия, перемешивали в течение 1 часа и выпаривали растворитель. Сырой остаток разбавляли этилацетатом (100 мл) и последовательно промывали водой (30 мл), 1N соляной кислотой (30 мл), нас. водн. бикарбонатом натрия (30 мл) и водой (30 мл), затем сушили и упаривали. Сырой продукт растворяли в смеси диоксан:вода (2:1, 6 мл) и охлаждали до 0oC. Небольшими порциями добавляли периодат натрия (0,13 г) и перемешивали смесь в течение 6 часов при комнатной температуре, затем концентрировали и фракционировали сырой продукт между водой (60 мл) и этилацетатом (60 мл). Органическую фазу сушили и концентрировали, а полученный таким образом остаток очищали посредством хроматографии быстрого разделения на силикагеле (гексан:этилацетат 85:15), получая в результате соединение, указанное в заголовке (0,045 г).
МЕТОД Б
К раствору трифторуксусного ангидрида (0,43 мл) в сухом дихлорметане (2,5 мл) добавляли диметилсульфоксид (0,22 мл) при -60oC в атмосфере азота при интенсивном перемешивании. Спустя 10 минут добавляли раствор Промежуточного соединения 79 (470 мг) в сухом дихлорметане (2,5 мл) и перемешивали смесь при -60oC в течение 2 часов. В течение 10 минут по каплям добавляли триэтиламин (1,4 мл) и доводили температуру до -20oC. Затем добавляли воду (20 мл) и перемешивали смесь при комнатной температуре в течение 1 часа. Смесь разделяли и экстрагировали водный слой дихлорметаном (100 мл). Объединенные органические слои промывали 1N соляной кислотой (100 мл), насыщенным бикарбонатом натрия (100 мл) и рассолом (100 мл), сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате масло, которое растворяли в дихлорметане и обрабатывали триэтиламином (1 мл) в течение ночи. Растворитель удаляли, а полученное масло подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан 1:25, 1:20 и 1:15), получая в результате 400 мг соединения, указанного в заголовке (выход 86%).
δ (1H, CDCl3): 9,71 (s, 1H, 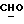 ), 7,44-7,26 (m, 10H, 2Ph), 6,97 (s, 1H,
), 7,44-7,26 (m, 10H, 2Ph), 6,97 (s, 1H, 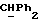 ), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,18-4,11 (m, 3H, H-7', H-1' и H-3' (1H), 4,00 и 3,62 (2d, 1H, 8a-CH2, J=9,3 Гц), 3,91 (d, 1H, H-3', J=18 Гц), 3,30 (m, 1H, H-9'), 2,85 (bt, 1H, H-5', J=7,8 Гц), 2,75 (t, 1H, H-1, J= 3,9 Гц).
), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,18-4,11 (m, 3H, H-7', H-1' и H-3' (1H), 4,00 и 3,62 (2d, 1H, 8a-CH2, J=9,3 Гц), 3,91 (d, 1H, H-3', J=18 Гц), 3,30 (m, 1H, H-9'), 2,85 (bt, 1H, H-5', J=7,8 Гц), 2,75 (t, 1H, H-1, J= 3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 44
[R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-{[4-O- (Транс-2- бутенил)-6-дезокси- β- D-альтропиранозилокси] метил}-4-формил- 4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Суспензию промежуточного соединения 1 (5 г) и оксида дибутилолова (2,3 г) в сухом толуоле (300 мл) подвергали дефлегмации в течение 3 часов в колбе, снабженной Dean-Stark конденсатором, в атмосфере азота, а затем охлаждали до 60oC. Последовательно добавляли молекулярные сита (4А, порошок), кротилбромид (2,4 мл) и 1М раствор в тетрагидрофуране тетрабутиламмонийфторида (23 мл), смесь прогревали в течение 1 часа при 60oC и выдерживали при комнатной температуре в течение 12 часов. Растворитель упаривали до сухого состояния, а остаток подвергали хроматографии на колонке быстрого разделения с силикагелем с элюцией гексаном и смесью гексан: ацетон (9:1), получая в результате соединение, указанное в заголовке (3,2 г), в виде белой пены.
δ (1H, CDCl3); 9,73 (s, 1H, CHO), 7,45- 7,2 (m, 10H, 2Ph), 6,98 (s, 1H, 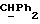 ), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,8-5,65 (m, 1H,
), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,8-5,65 (m, 1H, 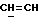 ), 5,65-5,5 (m, 1H,
), 5,65-5,5 (m, 1H, 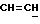 ), 4,63 (d, 1H, H-1', J=0,9 Гц), 4,2-3,8 (m, 5H, H-2', H-3', 8aCHa и
), 4,63 (d, 1H, H-1', J=0,9 Гц), 4,2-3,8 (m, 5H, H-2', H-3', 8aCHa и 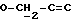 ), 3,8-3,65 (m, 2H, H-5', 8aCHb), 3,6 (dd, 1H, H-4', J=3 и 9,3 Гц), 2,74 (t, 1H, H-1, J=3,9 Гц), 1,71 (dd, 3H, CH3-C=C, J= 1,2 и 6,3 Гц).
), 3,8-3,65 (m, 2H, H-5', 8aCHb), 3,6 (dd, 1H, H-4', J=3 и 9,3 Гц), 2,74 (t, 1H, H-1, J=3,9 Гц), 1,71 (dd, 3H, CH3-C=C, J= 1,2 и 6,3 Гц).
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ 45, 46 и 47
а) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-{[2,3-Ангидро-4-O-(транс- 2-бутенил)-6-дезокси- β- D-маннопиранозилокси] метил} -4-формил- 4,4a,5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
б) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4S,6S,7R,9R)-2,8- Диокса-4-этил-6-гидрокси-9-метил-цис-бицикло[3.4.0] -нон-7-ил- оксиметил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
в) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4R,6S,7R,9R)-2,8- Диокса-4-этил-6-гидрокси-9-метил-цис-бицикло[3.4.0] -нон-7- ил-оксиметил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Промежуточное соединение 44 (1,225 г), трифенилфосфин (1,38 г) и имидазол (0,36 г) подвергали дефлегмации в толуоле (35 ил) при перемешивании, а затем по каплям в течение 2 часов
добавляли раствор иода (0,89 г) в толуоле (15 мл). Добавляли тетрабутиламмонийиодид (0,2 г) и продолжали дефлегмацию в течение 1 часа. Реакционную смесь охлаждали и фракционировали между этилацетатом (100 мл) и 1N водной соляной кислотой (50 мл). Органический слой последовательно промывали 1N водной соляной кислотой, водой, водным раствором метабисульфита натрия, водой и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали под вакуумом. Остаток подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью гексан:этидацетат (7:1), получая в результате 2:1 смесь двух соединений с очень близкими значениями Rf в системе гексан:этилацетат (1,08 г) (Rf= 0,4 в системе гексан:этилацетат 4:1). Смесь растворяли в сухом толуоле (25 мл), раствор дегазировали током аргона в течение 1 часа, а затем нагревали с дефлегмацией. Добавляли гидрид трибутилолова (0,36 мл) и продолжали дефлегмацию в течение 20 минут. После охлаждения добавляли четыреххлористый углерод (2 мл) и перемешивали раствор при комнатной температуре в течение 1 часа. Затем добавляли разбавленный раствор иода в эфире до тех пор, пока сохранялось слабое окрашивание. Затем растворитель удаляли под вакуумом, а остаток переносили в диэтиловый эфир и промывали несколько раз насыщенным водным раствором фторида натрия до тех пор, пока не прекращалось образование осадка фторида трибутилолова. Органический слой сушили и упаривали, получая в результате остаток, который дважды подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан:этилацетат (95: 5) и получая в результате соединение, указанное в заголовке (а) Промежуточное соединение 47 (230 мг, Rf=0,8 в системе дихлорметан: этилацетат 9:1), указанное в заголовке (б) промежуточное соединение 45 (275 мг, Rf= 0,5 в системе дихлорметан: этилацетат 9:1) и указанное в заголовке (в) промежуточное соединение 46 (200 мг, Rf=0,4 в системе дихлорметан: этилацетат
9:1).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 45
δ (1H, CDCl3): 9,72 (s, 1H, CHO), 7,46-7,23 (m, 10H, 2Ph), 6,98 (s, 1H, 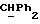 ), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,61 (d, 1H, H-7', J=3,3 Гц), 4,03 (d, 1H, 8a-CHa, J= 9,3 Гц), 3,90 (dd, 1H, Ha-3', J=6,9 и 8,7 Гц), 3,82-3,69 (m, 3H, H-6', H-1' и H-9'), 3,66 (d, 1H, 8a-CHb, J=9,6 Гц), 3,61 (dd, 1H, Hb-3', J=6,6 и 8,4 Гц), 2,69 (t, 1H, H-1, J=3,9 Гц).
), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,61 (d, 1H, H-7', J=3,3 Гц), 4,03 (d, 1H, 8a-CHa, J= 9,3 Гц), 3,90 (dd, 1H, Ha-3', J=6,9 и 8,7 Гц), 3,82-3,69 (m, 3H, H-6', H-1' и H-9'), 3,66 (d, 1H, 8a-CHb, J=9,6 Гц), 3,61 (dd, 1H, Hb-3', J=6,6 и 8,4 Гц), 2,69 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 46
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 7,45-7,22 (m, 10H, 2Ph), 6,99 (s, 1H, 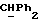 ), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,50 (d, 1H, H-7', J=2,1 Гц), 4,1-4,02 (m, 2H, 8aCHa и Ha-3'), 3,84 (dd, 1H, H-1', J=7,5 и 8,7 Гц), 3,76-3,66 (m, 2H, 8aCHb и H-6'), 3,5-3,38 (m, 2H, H-9' и Hb-3'), 2,72 (t, 1H, H-1, J=3,9 Гц), 0,94 (t, 3H,
), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,50 (d, 1H, H-7', J=2,1 Гц), 4,1-4,02 (m, 2H, 8aCHa и Ha-3'), 3,84 (dd, 1H, H-1', J=7,5 и 8,7 Гц), 3,76-3,66 (m, 2H, 8aCHb и H-6'), 3,5-3,38 (m, 2H, H-9' и Hb-3'), 2,72 (t, 1H, H-1, J=3,9 Гц), 0,94 (t, 3H, 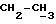 J=7,5 Гц).
J=7,5 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 47.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,45-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, 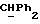 ), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,85-5,7 (m, 1H,
), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,85-5,7 (m, 1H, 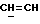 ), 5,65-5,5 (m, 1H,
), 5,65-5,5 (m, 1H, 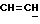 ), 4,67 (s, 1H, H-1'), 4,2-4,05 (m, 2H, OCHa-C=C и 8aCHa), 4,05-3,9 (m, 1H, OCHb-C=C), 3,78 (d, 1H, 8aCHb, J=9 Гц), 3,3-3,15 (m, 3H, H-3', H-4' и H5'), 3,12 (d, 1H, H-2', J=3,9 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц).
), 4,67 (s, 1H, H-1'), 4,2-4,05 (m, 2H, OCHa-C=C и 8aCHa), 4,05-3,9 (m, 1H, OCHb-C=C), 3,78 (d, 1H, 8aCHb, J=9 Гц), 3,3-3,15 (m, 3H, H-3', H-4' и H5'), 3,12 (d, 1H, H-2', J=3,9 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 48
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6- дезокси-4-O-метил- β- D-альтропиранозилокси) метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
В раствор сордарина (10,0 г) в дихлорметане (150 ил) по каплям добавляли раствор дифенилдиазометана в дихлорнетане (0,35 М, 85 мл). Полученный раствор перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли при пониженном давлении, а остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (4: 1) и (2:1). Фракции объединяли и упаривали, получая в результате соединение, указанное в заголовке (11,8 г), в виде белой пены.
δ (1H, CDCl3): 10,00 (s, 1H, CHO), 7,63 (m, 10H, 2Ph), 7,26 (s, 1H, 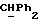 ), 6,30 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,92 (d, 1H, H-1', J=0,9 Гц), 4,84 (t, 1H, H-3', J=3,3 Гц), 4,03 и 4,35 (2d, 2H, 8aCH2, J=9 Гц), 3,88 (m, 1H, H-2'), 3,70 (dq, 1H, H-5', J=6,3 и 9,3 Гц), 3,41 (s, 3H, 4'-OMe), 3,20 (dd, 1H, H-4', J=3,3 и 9 Гц), 2,75 (t, 1H, H-1).
), 6,30 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,92 (d, 1H, H-1', J=0,9 Гц), 4,84 (t, 1H, H-3', J=3,3 Гц), 4,03 и 4,35 (2d, 2H, 8aCH2, J=9 Гц), 3,88 (m, 1H, H-2'), 3,70 (dq, 1H, H-5', J=6,3 и 9,3 Гц), 3,41 (s, 3H, 4'-OMe), 3,20 (dd, 1H, H-4', J=3,3 и 9 Гц), 2,75 (t, 1H, H-1).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 49
а) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-деэокси-4-O-метил-2,3- ди-О-тозил- β- D-альтропиранозилокси)метил] -4-формил-4,4a,5, 6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано- s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир и
б) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-4-O-метил-2-O-тозил- β- D- альтропиранрзилокси)метил]4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Промежуточное соединение 48 (2 г) и 4-диметиламинопиридин (500 мг) растворяли в сухом пиридине (30 мл). По каплям добавляли раствор тозилхлорида (960 мг) в сухом пиридине (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 суток. Растворитель выпаривали под вакуумом, получая в результате остаток желтого цвета, который хроматографировали на колонке с силикагелем, элюируя смесью гексан:этилацетат (4:1) и (1:1), получая в результате соединение, указанное в заголовке (а), с большим значением Rf в виде белой пены (1,65 г) и соединение, указанное в заголовке (б), с меньшим значением Rf и также в виде белой пены (1,17 г).
(а) δ (1H, CDCl3): 9,71 (s, 1H, CHO), 7,85-7,91 (m, 4H, орто-Ts), 7,26-7,41 (m, 14H, мета-Ts и 2Ph), 6,95 (s, 1H, 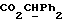 ), 5,87 (dd, 1H, H-2, J= 1,2 и 3,3 Гц), 4,92 (dd, 1H, H-3', J=3,3 и 4,2 Гц), 4,65 (dd, 1H, H-2', J= 1,2 и 4,5 Гц), 4,61 (d, 1H, H-1', J=1,2 Гц), 3,50 и 3,94 (2d, 2H, 8aCH2, J=9 Гц), 3,68 (dq, 1H, H-5', J=6,6 и 9 Гц), 3,16 (dd, 1H, H-4', J=3 и 9 Гц), 2,78 (s, 3H, 4'-OMe), 2,45 и 2,47 (2s, 6H, 2Ts).
), 5,87 (dd, 1H, H-2, J= 1,2 и 3,3 Гц), 4,92 (dd, 1H, H-3', J=3,3 и 4,2 Гц), 4,65 (dd, 1H, H-2', J= 1,2 и 4,5 Гц), 4,61 (d, 1H, H-1', J=1,2 Гц), 3,50 и 3,94 (2d, 2H, 8aCH2, J=9 Гц), 3,68 (dq, 1H, H-5', J=6,6 и 9 Гц), 3,16 (dd, 1H, H-4', J=3 и 9 Гц), 2,78 (s, 3H, 4'-OMe), 2,45 и 2,47 (2s, 6H, 2Ts).
(б) δ (1H, CDCl3): 9,71 (s, 1H, CHO), 7,85 (m, 2H, орто-Ts), 7,26-7,41 (m, 12H, мета-Ts и 2Ph), 6,95 (s, 1H, 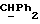 ), 5,87 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,61 (d, 1H, H-1', J=1,5 Гц), 4,55 (dd, 1H, H-2', J=1,2 и 4,2 Гц), 4,37 (m, 1H, H-3'), 3,50 и 3,95 (2d, 2H, 8aCH2, J=9 Гц), 3,70 (dq, 1H, H-5', J=8,4 и 6,3 Гц), 3,23 (dd, 1H, H-4', J=3,3 и 8,7 Гц), 2,42 (s, 3H, Ts).
), 5,87 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,61 (d, 1H, H-1', J=1,5 Гц), 4,55 (dd, 1H, H-2', J=1,2 и 4,2 Гц), 4,37 (m, 1H, H-3'), 3,50 и 3,95 (2d, 2H, 8aCH2, J=9 Гц), 3,70 (dq, 1H, H-5', J=8,4 и 6,3 Гц), 3,23 (dd, 1H, H-4', J=3,3 и 8,7 Гц), 2,42 (s, 3H, Ts).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 50
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-O-метил- β- D- маннопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a- -октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
При охлаждении до 0oC в безводный метанол добавляли натрий (0,5 г), затем Промежуточное соединение 49 а (670 мг) в сухом дихлорметане (20 мл). Смесь перемешивали в течение 5 суток при комнатной температуре, и 4 суток с дефлегмацией. Смесь фильтровали и выпаривали растворитель. Остаток очищали посредством хроматографии быстрого разделения с элюцией смесью гексан:этилацетат (3: 1), получая в результате соединение, указанное в заголовке (330 мг).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,26-7,44 (m, 10H, 2Ph), 6,99 (s, 1H, 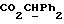 ), 6,08 (dd, 1H, H-2, J=1,5 и 3 Гц), 4,68 (s, 1H, H-1'), 3,78 и 4,11 (2d, 2H, 8aCH2, J=9 Гц), 3,49 (s, 3H, 4'-OMe), 3,26 (d, 1H, H-2', J=3,9 Гц), 3,18 (m, 1H, H-5'), 3,12 (d, 1H, H-3', J=3,6 Гц), 3,08 (m, 1H, H-4'), 2,86 (m, 1H, H-1').
), 6,08 (dd, 1H, H-2, J=1,5 и 3 Гц), 4,68 (s, 1H, H-1'), 3,78 и 4,11 (2d, 2H, 8aCH2, J=9 Гц), 3,49 (s, 3H, 4'-OMe), 3,26 (d, 1H, H-2', J=3,9 Гц), 3,18 (m, 1H, H-5'), 3,12 (d, 1H, H-3', J=3,6 Гц), 3,08 (m, 1H, H-4'), 2,86 (m, 1H, H-1').
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 51
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-O-метил- β- D- аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
Раствор гидрида натрия (25,9 мг) суспендировали в сухом диметилформамиде (5 мл). По каплям добавляли Промежуточное соединение 49 (б) (470 мг) в сухом диметилформамиде и перемешивали смесь при комнатной температуре в течение 40 минут. Реакционную смесь при перемешивании вливали в охлаждавшуюся льдом воду (20 мл), а полученный раствор обрабатывали этилацетатом (3x20 мл). Органические слои объединяли и сушили над безводным сульфатом магния. Смолистый остаток, полученный после выпаривания растворителей, очищали посредством колоночной хроматографии быстрого разделения с элюцией смесью гексан:этилацетат (5: 1), получая в результате соединение, указанное в заголовке, в виде белой пены (332 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,26-7,44 (m, 10H, 2Ph), 6,98 (s, 1H, CO2CHPh2), 6,08 (dd, 1H, H-2, J=1,5 и 3 Гц), 4,60 (s, 1H, H-1'), 3,76 и 4,03 (2d, 2H, 8aCH2, J=9,3 Гц), 3,51 (m, 4H, H-4' и 4'-ОМе), 3,33 (m, 3H, H-2', H-3' и H-5'), 2,80 (m, 1H, H-1).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 52
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-O-метил- β- D- альтропиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновая кислота
К раствору Промежуточного соединения 51 (100 мг) в этилацетате (15 мл) добавляли в атмосфере азота 10%-ный палладий на угле (100 мг). Смесь встряхивали в аппарате Parr при 15 пси водорода в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали посредством хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (3:1) и дихлорметан: метанол (15: 1). Соответствующие фракции объединяли, растворитель удаляли и получали в результате соединение, указанное в заголовке (70 мг) в виде пены.
δ (1H, CDCl3): 9,77 (s, 1H, CHO), 6,09 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,67 (d, 1H, H-1', J=0,9 Гц), 3,58 и 4,17 (2d, 2H, 8aCH2, J=9,6 Гц), 3,51 (m, 4H, H-3' и 4'-ОМе), 3,38 (m, 1H, H-5'), 3,34 (d, 1H, H-2', J= 4,2 Гц), 3,30 (dd, 1H, H-4',J=9 и 1,5 Гц), 2,65 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 53
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-О-Аллил-6-дезокси- β- D-альтропиранозилокси) метил] -4-формил-4,4a, 5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Суспензию Промежуточного соединения 1 (2 г) и оксида дибутилолова (870 мг) в сухом толуоле (25 мл) подвергали в атмосфере азота дефлегмации в течение 2 часов в колбе, снабженной Dean-Stark конденсатором, а затем оставляли при комнатной температуре. Последовательно добавляли аллилбромид (0,3 мл) и 1М раствор тетрабутиламмонийфторида (3,5 мл), смесь прогревали в атмосфере азота при 40oC в течение 24 часов. Растворитель упаривали до сухого состояния, а остаток хроматографировали на колонке быстрого разделения с силикагелем, элюируя смесью гексан: ацетон (10:1) и (9:1), получая в результате соединение, указанное в заголовке (1,7 г), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,98 (s, 1H, 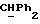 ), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,89 (m, 1H,
), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,89 (m, 1H, 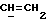 ), 5,25 (m, 2H,
), 5,25 (m, 2H, 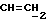 ), 4,64 (d, 1H, 1H, H-1', J= 1,5 Гц), 4,15 (t, 1H, H-3', J=3,3 Гц), 4,09 (m, 3H,
), 4,64 (d, 1H, 1H, H-1', J= 1,5 Гц), 4,15 (t, 1H, H-3', J=3,3 Гц), 4,09 (m, 3H, 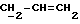 ) и
) и 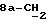 ), 3,87 (m , 1H, H-2'), 3,74 (m, 2H, H-5' и
), 3,87 (m , 1H, H-2'), 3,74 (m, 2H, H-5' и 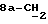 ), 3,38 (dd, 1H, H-4', J=3,3 и 9 Гц), 2,74 (t, 1H, H-1, J=3,9 Гц).
), 3,38 (dd, 1H, H-4', J=3,3 и 9 Гц), 2,74 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 54
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(4-O-Бензил-6-дезокси- β- D-альтропиранозилокси) метил] -4-формил-4,4a, 5,6,7,7a,8,8a-октагидpo-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Интенсивно перемешивавшуюся смесь Промежуточного соединения 1 (6,0 г) и оксида дибутилолова (3,55 г) в сухом толуоле (80 мл) подвергали в атмосфере азота дефлегмации в течение 3 часов в колбе, снабженной Dean-Stark конденсатором. Реакционную смесь охлаждали до 0oC, а затем обрабатывали бензилбромидом (1,2 мл) и тетрабутиламмонийфторидом (1М в тетрагидрофуране, 10 мл). После прогревания при 40oC в течение 18 часов в атмосфере азота реакционную смесь концентрировали при пониженном давлении, а остаток очищали посредством хроматографии быстрого разделения, элюируя смесью гексан:этилацетат (6:1) и (4: 1). Соответствующие фракции объединяли, а растворители выпаривали, получая в результате соединение, указанное в заголовке (3,73 г), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,24- 7,44 (m, 15H, 3Ph), 6,98 (s, 1H, CO2CHPh2), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,65 (d, 1H, H-1', J=1,2 Гц), 4,57 (AB система, 4'OCH2Ph, J=11,1 Гц), 4,21 (m, 1H, H-3'), 3,76 и 4,07 (2d, 2H, 8aCH2, J= 9 Гц), 3,87 (m, 1H, H-2'), 3,76 (m, 1H, H-5'), 3,49 (dd, 1H, H-4', J=3,3 и 9 Гц), 2,74 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 55
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2-О-Бензилоксикарбонил-6- дезoкcи-4-O-метил- β- D-aльтpoпиpaнoзилoкcи)мeтил] -4- фopиил-4,4a,5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1Н)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 48 (3 ммоль) в сухом дихлорметане (15 мл) при 0oC в атмосфере азота добавляли 4-диметиламинопиридин (6,3 ммоль). После перемешивания в течение 15 минут смесь охлаждали до -20oC и по каплям добавляли раствор бензилхлорформиата (3,6 ммоль) в сухом дихлорметане (15 мл). Спустя 2 часа растворитель выпаривали, а остаток очищали посредством хроматографии быстрого разделения, используя в качестве элюента смесь гексан:этилацетат (3:1) и получая в результате соединение, указанное в заголовке (1,2 г).
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 7,35 (m, 15H, 3xPh), 6,96 (s, 1H, 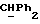 ), 5,99 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,18 (AB система, 2H,
), 5,99 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,18 (AB система, 2H, 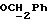 J= 12 Гц), 5,00 (dd, 1H, H-2', J=1,5 и 4,2 Гц), 4,70 (d, 1H, H-1', J=1,8 Гц), 4,13 (m, 1H, H-3'), 4,01 и 3,66 (dd, 1H, 8a-CH2, J=9 Гц), 3,75 (m, 1H, H-5'), 3,40 (s, 3H, OMe), 3,17 (dd, 1H, H-4', J=3,3 и 8,4 Гц), 2,62 (t, 1H, H-1, J=3,6 Гц), 2,43 (d, 1H, OH, J=2,4 Гц), 2,21 (m, 1H,
J= 12 Гц), 5,00 (dd, 1H, H-2', J=1,5 и 4,2 Гц), 4,70 (d, 1H, H-1', J=1,8 Гц), 4,13 (m, 1H, H-3'), 4,01 и 3,66 (dd, 1H, 8a-CH2, J=9 Гц), 3,75 (m, 1H, H-5'), 3,40 (s, 3H, OMe), 3,17 (dd, 1H, H-4', J=3,3 и 8,4 Гц), 2,62 (t, 1H, H-1, J=3,6 Гц), 2,43 (d, 1H, OH, J=2,4 Гц), 2,21 (m, 1H, 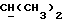 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 56
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Аллил-2-O-бензилокси-карбонил-6-дезокси- β- D-альтропиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 53 (1,4 г) в сухой метиленхлориде (30 мл) при 0oC добавляли диметиламинопиридин (513 мг) и перемешивали смесь в течение 15 минут. Затем в течение 15 минут по каплям добавляли бензилхлорформиат (0,35 мл) в сухом метиленхлориде (15 мл) и перемешивали смесь в течение 4 часов. Затем смесь разбавляли метанолом (1 мл) и метиленхлоридом (20 мл) и промывали водой, 1N соляной кислотой и рассолом. Органический слой сушили над безводным сульфатом магния, фильтровали и упаривали растворитель до сухого состояния. Остаток очищали посредством колоночной хроматографии быстрого разделения, элюируя смесью гексан:этилацетат (7:1) и (5:1) и получая в результате соединение, указанное в заголовке (1,1 г).
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 6,96 (s, 1H, 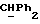 ), 5,99 (dd, 1H, H-2, J= 1,5 и 3,6 Гц), 5,88 (m, 1H,
), 5,99 (dd, 1H, H-2, J= 1,5 и 3,6 Гц), 5,88 (m, 1H, 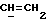 ), 5,20 (m, 4H,
), 5,20 (m, 4H, 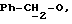
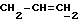 ), 4,99 (dd, 1H, H-2', J=1,5 и 4,2 Гц), 4,70 (d, 1H, H-1', J=1,5 Гц), 4,12 (m, 1H, H-3'), 4,03 (m, 3H,
), 4,99 (dd, 1H, H-2', J=1,5 и 4,2 Гц), 4,70 (d, 1H, H-1', J=1,5 Гц), 4,12 (m, 1H, H-3'), 4,03 (m, 3H, 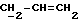 и
и 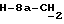 ), 3,76 (m, 1H, H-3'), 3,64 (d, 1H,
), 3,76 (m, 1H, H-3'), 3,64 (d, 1H, 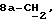 8,7 Гц), 3,34 (dd, 1H, H-4', J=3,3 и 8,7 Гц), 2,61 (t, 1H, H-1, J=3,9 Гц).
8,7 Гц), 3,34 (dd, 1H, H-4', J=3,3 и 8,7 Гц), 2,61 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 57
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Бензил-2-O-бензилоксикарбонил-6-дезокси- β- D-альтропиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 54 (3,7 г) в сухом дихлорметане (25 мл) при 0oC в атмосфере азота добавляли 4-диметиламинопиридин (1,85 г). После перемешивания в течение 15 минут смесь охлаждали до -20oC и по каплям добавляли раствор бензилхлорформиата (0,68 мл) в сухом дихлорметане (10 мл) до тех пор, пока продукт не обнаруживали посредством ТСХ (гексан:этилацетат 4: 1). Растворитель затем упаривали до сухого состояния, а остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат (6:1) и получая в результате соединение, указанное в заголовке (3,6 г), в виде белой пены.
δ (1H, CDCl3); 9,70 (s, 1H, CHO), 7,26- 7,42 (m, 20H, 4Ph), 6,96 (s, 1H, 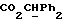 ), 5,99 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,18 (AB система, 2′-OCO2CH2Ph, J=12,3 Гц), 5,00 (dd, 1H, H-2', J=1,5 и 4,2 Гц), 4,71 (d, 1H, H-1', J=1,5 Гц), 4,56 (AB система, 4-
), 5,99 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,18 (AB система, 2′-OCO2CH2Ph, J=12,3 Гц), 5,00 (dd, 1H, H-2', J=1,5 и 4,2 Гц), 4,71 (d, 1H, H-1', J=1,5 Гц), 4,56 (AB система, 4- 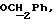 J=11, 1 Гц), 4,15 (m, 1H, H-3'), 3,66 и 4,00 (2d, 2H, 8aCH2, J=9 Гц), 3,81 (dq, 1H, H-5', J= 6,6 и 9 Гц), 3,43 (dd, 1H, H-4', J=3,3 и 9 Гц), 2,60 (t, 1H, H-1, J=3,6 Гц).
J=11, 1 Гц), 4,15 (m, 1H, H-3'), 3,66 и 4,00 (2d, 2H, 8aCH2, J=9 Гц), 3,81 (dq, 1H, H-5', J= 6,6 и 9 Гц), 3,43 (dd, 1H, H-4', J=3,3 и 9 Гц), 2,60 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 58
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2-О-Бензилоксикарбонил-6- дезокси-4-О-метил-3-О-тозил- β- D-альтропиранозилокси)метил]-4- формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 55 (3 ммоль) и 4-диметиламинопиридина (5,1 ммоль) в сухом дихлорметане (60 мл) по каплям обрабатывали раствором тозилхлорида (4,5 ммоль) в сухом дихлорметане (20 мл). Спустя 24 часа растворитель выпаривали, а остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат (5:1) и получая
в результате соединение, указанное в заголовке (1,8 г).
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 7,84 и 7,32 (d, m, 2H, 17H, Ar), 6,96 (s, 1H, 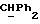 ), 5,94 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,17 (AB система, 2H,
), 5,94 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,17 (AB система, 2H,  J= 12 Гц), 5,10 (m, 1H, H-3'), 4,88 (dd, 1H, H-2', J=3 и 5,4 Гц), 4,68 (d, 1H, H-1', J=2,1 Гц), 3,97 и 3,63 (2d, 2H, 8a-CH2, J=9 Гц), 3,76 (dd, 1H, H-5', J=6,3 и 8,1 Гц), 3,18 (dd, 1H, H-4', J=3 и 8,1 Гц), 3,05 (s, 3H, OMe), 2,51 (t, 1H, H-1, J=3,6 Гц), 2,43 (s, 3H,
J= 12 Гц), 5,10 (m, 1H, H-3'), 4,88 (dd, 1H, H-2', J=3 и 5,4 Гц), 4,68 (d, 1H, H-1', J=2,1 Гц), 3,97 и 3,63 (2d, 2H, 8a-CH2, J=9 Гц), 3,76 (dd, 1H, H-5', J=6,3 и 8,1 Гц), 3,18 (dd, 1H, H-4', J=3 и 8,1 Гц), 3,05 (s, 3H, OMe), 2,51 (t, 1H, H-1, J=3,6 Гц), 2,43 (s, 3H,  ), 2,01 (m, 1H,
), 2,01 (m, 1H, 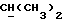 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 59
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Аллил-2-O-бензилокси-карбонил-6-дезокси- 3-O-тозил- β- D-альтропиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновой кислоты дифенилметиловый
К раствору Промежуточного соединения 56 (1,1 г) и диметиламинопиридина (300 мг) в сухом метиленхлориде (40 мл) по каплям добавляли раствор тозилхлорида (400 мг) в сухом метиленхлориде (20 мл) и перемешивали реакционную смесь в течение 3 суток при комнатной температуре. Растворитель упаривали до сухого состояния, а остаток Хроматографировали на колонке быстрого разделения с силикагелем, элюируя смесью гексан:этилацетат (9:1) и (6:1) и получая в результате соединение, указанное в заголовке (820 мг).
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 6,95 (s, 1H, 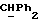 ), 5,94 (dd, 1H, H-2, J= 1,5 и 3,9 Гц), 5,61 (m, 1H,
), 5,94 (dd, 1H, H-2, J= 1,5 и 3,9 Гц), 5,61 (m, 1H, 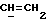 ), 5,12 (m, 4H,
), 5,12 (m, 4H, 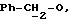
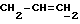 ), 4,84 (dd, 1H, H-2', J=3 и 5,1 Гц), 4,69 (d, 1H, H-1, J=1,8 Гц), 3,96 и 3,63 (2d, 2H,
), 4,84 (dd, 1H, H-2', J=3 и 5,1 Гц), 4,69 (d, 1H, H-1, J=1,8 Гц), 3,96 и 3,63 (2d, 2H, 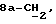 J= 9,0 Гц), 3,76 (m, 4H, H-3', H5',
J= 9,0 Гц), 3,76 (m, 4H, H-3', H5', 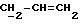 ), 3,38 (dd, 1H, H-4', J=3 и 8,1 Гц), 2,50 (t, 1H, H-1, J=3,9 Гц), 2,42 (s, 3H,
), 3,38 (dd, 1H, H-4', J=3 и 8,1 Гц), 2,50 (t, 1H, H-1, J=3,9 Гц), 2,42 (s, 3H, 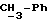 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 60
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Бензил-2-O-бензилоксикарбонил- 6-дезокси-3-O-тозил- β- D-альтропиранозилокси)метил]-4-формил- 4,4a, 5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый
К раствору Промежуточного соединения 57 (3,5 г) и диметиланинопиридина (1,3 г) в сухом метиленхлориде (80 мл) по каплям добавляли раствор тозилхлорида (2 г) в сухом метиленхлориде (50 мл) и перемешивали смесь в течение 3 суток при комнатной температуре. Растворитель выпаривали, а остаток хроматографитровали на колонке быстрого разделения с силикагелем, применяя в качестве элюента смесь гексан:этилацетат (9:1) и получая в результате соединение, указанное в заголовке (3,14 г).
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 6,95 (s, 1H, 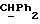 ), 5,95 (dd, 1H, H-2, J-1,2 и 3,3 Гц), 5,16 (m, 3H,
), 5,95 (dd, 1H, H-2, J-1,2 и 3,3 Гц), 5,16 (m, 3H, 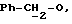 H-2'), 4,93 (dd, 1H, H-3', J-3 и 4,8 Гц), 4,71 (d, 1H, H-1', J=1,8 Гц), 4,25 (AB система, 2H, O-CH2Ph, J= 11,5 Гц), 3,96, 3,63 (2d, 2H,
H-2'), 4,93 (dd, 1H, H-3', J-3 и 4,8 Гц), 4,71 (d, 1H, H-1', J=1,8 Гц), 4,25 (AB система, 2H, O-CH2Ph, J= 11,5 Гц), 3,96, 3,63 (2d, 2H, 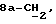 J-9,0 Гц), 3,83 (m, 1H, H-5'), 3,45 (dd, 1H, H-4', J= 3 и 8,4 Гц), 2,51 (t, 1H, H-1, J-3,9 Гц), 2,35 (s, 3H, CH3-Ph).
J-9,0 Гц), 3,83 (m, 1H, H-5'), 3,45 (dd, 1H, H-4', J= 3 и 8,4 Гц), 2,51 (t, 1H, H-1, J-3,9 Гц), 2,35 (s, 3H, CH3-Ph).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 61
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-деэокси-4-О-метил-3-О-тозил- β- D- альтропиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 58 (1,9 ммоль) в этилацетате (60 мл) в атмосфере азота добавляли 10%-ный палладий на угле (100 мг). Смесь встряхивали в Parr аппарате при 15 пси водорода в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а раствор обрабатывали 0,35М раствором дифенилдиазометана (20 мл). Спустя 2 часа растворитель выпаривали, а остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат (3:1) и получая в результате соединение, указанное в заголовке. (1,3 г).
δ (1H, CDCl3): 9,72 (s, 1H, CHO), 7,85 и 7,32 (d,m, 2H, 12H, Ar), 6,98 (s, 1H, 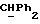 ), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,84 (dd, 1H, H-3', J=3 и 4,2 Гц), 4,64 (d, 1H, H-1', J-1,5 Гц), 4,06 и 3,75 (2d, 2H, 8a-CH2, J=9,3 Гц), 4,02 (m, 1H, H-2'), 3,70 (m, 1H, H-5'), 3,16 (dd, 1H, H-4', J=3 и 9 Гц), 2,93 (s, 3H, OMe), 2,69 (t, 1H, H-1, J=3,9 Гц), 2,45 (s, 3H,
), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,84 (dd, 1H, H-3', J=3 и 4,2 Гц), 4,64 (d, 1H, H-1', J-1,5 Гц), 4,06 и 3,75 (2d, 2H, 8a-CH2, J=9,3 Гц), 4,02 (m, 1H, H-2'), 3,70 (m, 1H, H-5'), 3,16 (dd, 1H, H-4', J=3 и 9 Гц), 2,93 (s, 3H, OMe), 2,69 (t, 1H, H-1, J=3,9 Гц), 2,45 (s, 3H, 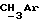 ), 2,30 (d, 1H, OH, J=3 Гц), 2,23 (m, 1H,
), 2,30 (d, 1H, OH, J=3 Гц), 2,23 (m, 1H, 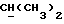 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 62
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-4-O-пропил-3-O-тозил- β- D-альтропиранозилокси)метил] -4-формил-4,4a,5,6,7,7а,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 59 (800 мг) в этилацетате (200 мл) в атмосфере азота добавляли 10%-ный палладий на угле (300 мг). Смесь встряхивали в Parr аппарате при 20 пси водорода в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток растворяли в метиленхлориде (50 мл) и по каплям добавляли 0,35М раствор дифенилдиазометана (6 мл). Смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали, а остаток хроматографировали на колонке быстрого разделения с силикагелем, элюируя смесью гексан: этилацетат (8:1) и (3:1) и получая в результате соединение, указанное в заголовке (630 мг).
δ (1H, CDCl3): 9,72 (s, 1H, CHO), 6,98 (s, 1H, 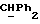 ), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,84 (dd, 1H, H-2', J=3 и 4,2 Гц), 4,64 (d, 1H, H-1', J= 1,5 Гц), 4,05 (m, 2H, H-3' и
), 6,06 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,84 (dd, 1H, H-2', J=3 и 4,2 Гц), 4,64 (d, 1H, H-1', J= 1,5 Гц), 4,05 (m, 2H, H-3' и 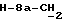 ), 3,73 (m, 2H, H-5' и
), 3,73 (m, 2H, H-5' и 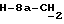 ), 3,29 (dd, 1H, H-4', J=2,7 и 9 Гц), 3,01 (t, 2H,
), 3,29 (dd, 1H, H-4', J=2,7 и 9 Гц), 3,01 (t, 2H, 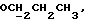 J=7 Гц), 2,68 (t, 1H, H-1, J=3,9 Гц), 2,44 (s, 3H, p-Ts).
J=7 Гц), 2,68 (t, 1H, H-1, J=3,9 Гц), 2,44 (s, 3H, p-Ts).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 63
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-О-Бензил-6-дезокси-3-О-тозил- β- D- альтропиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 60 (1,0 г) в этилацетате (70 мл) в атмосфере азота добавляли 10%-ный палладий на угле. Смесь встряхивали в Parr аппарате при 15 пси водорода в течение 4 часов при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали посредством Хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (3:1) и (1:1). Соответствующие фракции объединяли, а растворитель выпаривали, получая в результате соединение, указанное в заголовке (530 мг).
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 7,81 и 7,10-7,30 (2m, 9H, Ts и Ph), 6,08 (dd, 1H, H-2, J-1,5 и 3,3 Гц), 4,91 (dd, 1H, H-3', J=3,3 и 4,5 Гц), 4,68 (d, 1H, H-1', J-1,5 Гц), 4,12 (m, 3H, 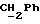 и H-2'), 4,06 и 3,64 (2d, 2H,
и H-2'), 4,06 и 3,64 (2d, 2H, 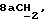 J= 9,3 Гц), 3,46 (m, 1H, H-4'), 2,65 (m, 1H, H-1), 2,36 (s, 3H, Ts).
J= 9,3 Гц), 3,46 (m, 1H, H-4'), 2,65 (m, 1H, H-1), 2,36 (s, 3H, Ts).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 64
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2.3-Ангидро-6-дезокси-4-O- метил- β- D-маннопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a,8.8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
Суспензию гидрида натрия (3,2 ммоль) в сухом диметилформамиде (6 мл) в атмосфере азота обрабатывали раствором Промежуточного соединения 61 (1,6 ммоль) в сухом диметилформамиде (6 мл). Спустя 40 минут добавляли смесь вода: этилацетат (III, 60 мл). Органическую фазу выпаривали, а остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат (4:1) и получая в результате соединение, указанное в заголовке (960 мг), имевшее протонный ЯМР спектр, идентичный спектру Промежуточного соединения 50.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 65,
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-O-пропил- β- D-маннопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7а,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К суспензии гидрида натрия (35 мг) в сухом диметилфорамиде (10 мл) по каплям добавляли раствор Промежуточного соединения 63 (600 мг) в сухом диметилформамиде (15 мл). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 1 часа, а затем обрабатывали 1N хлоридом аммония (50 мл) и этилацетатом (70 мл). Органический слой промывали водой и рассолом, сушили над безводным сульфатом магния и упаривали до сухого состояния. Остаток хроматографировали на колонке быстрого разделения с силикагелем, элюируя смесью гексан: этилацетат (8:1) и (6:1) и получая в результате соединение, указанное в заголовке (450 мг), в виде белой пены.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,99 (s, 1H, 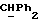 ), 6,08 (dd, 1H, H-2, J=1,5 и 4,8 Гц), 4,68 (s, 1H, H-1'), 4,11 и 3,79 (2d, 2H, 8a-CH2, J=9,0 Гц), 3,68 (m, 1H, H-5'), 3,45 (m, 1H, H-4'), 3,24 и 3,12 (2d, 2H, H-2', H-3', J= 3,9 Гц), 3,19 (t, 2H,
), 6,08 (dd, 1H, H-2, J=1,5 и 4,8 Гц), 4,68 (s, 1H, H-1'), 4,11 и 3,79 (2d, 2H, 8a-CH2, J=9,0 Гц), 3,68 (m, 1H, H-5'), 3,45 (m, 1H, H-4'), 3,24 и 3,12 (2d, 2H, H-2', H-3', J= 3,9 Гц), 3,19 (t, 2H, 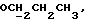 J=7 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц).
J=7 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 66
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3-O-тозил- β- D- альтропиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновая кислота
К суспензии 10%-ного палладия на угле (800 мг) в этилацетате (60 мл) в колбе для гидрогенизации в атмосфере азота добавляли раствор Промежуточного соединения 60 (1,0 г) в этилацетате (30 мл) и смесь метанол; 1N соляная кислота (3:1; 4 мл). Колбу встряхивали в Parr аппарате для гидрогенизации при давлении водорода 30 пси в течение 18 часов. Реакционную смесь фильтровали, а фильтрат упаривали до сухого состояния. Остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан: метанол (20:1). Соответствующие фракции объединяли, а растворитель выпаривали, получая в результате соединение, указанное в заголовке (380 мг).
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 7,84 и 7,38 (2d, 4H, Ts), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,75 (dd, 1H, H-3', J=3,3 и 4,5 Гц), 4,61 (d, 1H, H-1', J=1,2 Гц), 4,05 (d, 1H, H-8aCH2a, J=9,3 Гц), 3,92 (dd, 1H, H-2', J=1,2 и 4,5 Гц), 3,65 (m, 3H, H-5', H-4' и H-8aCH2b), 2,63 (m, 1H, H-1), 2,46 (s, 3H, Ts).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 67
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси- β D- маннопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8, 8а-октагидpo-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновой кислоты дифенилметиловый эфир
Раствор из Примера 55 (585 мг) в сухом дихлорметане (20 мл) обрабатывали по каплям фиолетовым раствором дифенилдиазометана в дихлорметане (0,35 М, 8 мл). Полученный раствор перемешивали при комнатной температуре в течение 14 часов. Растворитель удаляли при пониженном давлении, а остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью гексан: этилацетат (6: 1) и (4:1). Соответствующие фракции объединяли и упаривали, получая в результате соединение, указанное в заголовке (764 мг), в виде белой пены.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,26-7,44 (m, 10H, 2Ph), 7,00 (s, 1H, 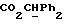 ), 6,09 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,71 (s, 1H, H-1'), 3,79 и 4,12 (2d, 2H, 8aCH2, J=9,3 Гц), 3,63 (dd, 1H, H-4', J=5,7 и 8,7 Гц), 3,21 (m, 1H, H-5'), 3,15 и 3,24 (2d, 2H, H-2' и H-3', J=3,6 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц).
), 6,09 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,71 (s, 1H, H-1'), 3,79 и 4,12 (2d, 2H, 8aCH2, J=9,3 Гц), 3,63 (dd, 1H, H-4', J=5,7 и 8,7 Гц), 3,21 (m, 1H, H-5'), 3,15 и 3,24 (2d, 2H, H-2' и H-3', J=3,6 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 68
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-4,6-дидезокси-4-фторо- β- D- талопиранозилокси)метил]-4-формил-4,4a,5,6,7,7а,8,8a-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 67 (300 мг) в дихлорметане (25 мл) обрабатывали трифторидом диэтиламиносеры (0,16 мл), полученную смесь перемешивали в течение ночи при комнатной температуре. Смесь охлаждали до 0oC, гасили путем добавления метанола (15 мл), а затем концентрировали при пониженном давлении. После хроматографии быстрого разделения на силикагеле остатка с элюцией смесью гексан:этилацетат (6:1) и (4:1) получали соединение, указанное в заголовке (160 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,26 (m, 10H, 2Ph), 6,98 (s, 1H, 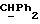 ), 6,09 (dd, 1H, H-2, J=1,5 и 3,9 Гц), 5,09 (d, 1H, H-1', J=1,5 Гц), 4,75 и 4,58 (2dq, 1H, H-5', J5'F=48,6 Гц; J=6,6 Гц, J=3,9 Гц), 4,18 и 4,11 (2d, 1H, H-4', J4'F=23,1 Гц, J=3,9 Гц), 3,97 и 3,81 (2d, 2H, 8aCH2, J=9,3 Гц), 3,75 и 3,70 (2m, 2H, H-2' и H-3'), 2,84 (t, 1H, H-1).
), 6,09 (dd, 1H, H-2, J=1,5 и 3,9 Гц), 5,09 (d, 1H, H-1', J=1,5 Гц), 4,75 и 4,58 (2dq, 1H, H-5', J5'F=48,6 Гц; J=6,6 Гц, J=3,9 Гц), 4,18 и 4,11 (2d, 1H, H-4', J4'F=23,1 Гц, J=3,9 Гц), 3,97 и 3,81 (2d, 2H, 8aCH2, J=9,3 Гц), 3,75 и 3,70 (2m, 2H, H-2' и H-3'), 2,84 (t, 1H, H-1).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 69
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-O-(2-метоксиэтил)- β- D-маннопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К суспензии гидрида натрия (24 мг) в сухом тетрагидрофуране (20 мл) при 0oC в атмосфере азота добавляли Промежуточное соединение 67 (300 мг) и полученную смесь перемешивали в течение 30 минут. Добавляли 2-Бромэтилметиловый эфир (300 мг) и 1N тетрабутиламмонийиодид (2 мл) и прогревали смесь в течение 2 суток при 40oC. Реакцию гасили 1N хлоридом аммония (10 мл) и разбавляли смесь этилацетатом (30 мл). Органический слой промывали водой и рассолом, сушили над безводным сульфатом магния, а растворитель упаривали до сухого состояния. Остаток хроматографировали на колонке быстрого разделения с силикагелем, элюируя смесью гексан:этилацетат (5:1) и получая в результате соединение, указанное в заголовке, в виде бесцветного масла.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,98 (s, 1H, 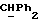 ), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,67 (s, 1H, H-1'), 4,10 и 3,78 (2d, 2H, 8a-CH2, J=9,0 Гц), 4,09-4,00 (m, 2H, H-4', H-5'), 3,90-3,53 (m, 4H,
), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,67 (s, 1H, H-1'), 4,10 и 3,78 (2d, 2H, 8a-CH2, J=9,0 Гц), 4,09-4,00 (m, 2H, H-4', H-5'), 3,90-3,53 (m, 4H, 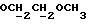 ), 3,40 (s, 3H,
), 3,40 (s, 3H, 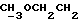 ), 3,29 и 3,11 (2d, 2H, H-3' и H-2', J=3,9 и 3,9 Гц), 2,86 (t, 1H, H-1, J=3,7 Гц).
), 3,29 и 3,11 (2d, 2H, H-3' и H-2', J=3,9 и 3,9 Гц), 2,86 (t, 1H, H-1, J=3,7 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 70
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2. 3-Ангидро-6-дезокси-4-O-(2-метилпроп-2-енил)- β- D-маннопиранозилокси)метил] -4-формил-4,4a,5,6,7,7а, 8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Смесь промежуточного соединения 67 (0,3 ммоль), карбоната цезия (0,3 ммоль) и 3-бром-2-метил-пропена (0,5 ммоль) в сухом диметилформамиде (1,5 мл) перемешивали в течение 3 суток при комнатной температуре. После разбавления диэтиловым эфиром (30 мл) смесь промывали водой. Растворитель выпаривали, а остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат (4:1) и получая в результате соединение, указанное в заголовке (62 мг).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,35 (m, 10H, 2xPh), 6,99 (s, 1H, 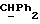 ), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,00 и 4,93 (m,m, 1H, 1H, CH2= C), 4,70 (s, 1H, H-1'), 4,12 и 3,79 (2d, 2H, 8a-CH2, J=9,3 Гц), 4,12 и 3,96 (d, d, 1H, 1H, CH2O, J=12 Гц), 3,26 (m, 3H, H-3', H-4' и H-5'), 3,13 (d, 1H, H-2', J=3,9 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц), 2,23 (m, 1H,
), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,00 и 4,93 (m,m, 1H, 1H, CH2= C), 4,70 (s, 1H, H-1'), 4,12 и 3,79 (2d, 2H, 8a-CH2, J=9,3 Гц), 4,12 и 3,96 (d, d, 1H, 1H, CH2O, J=12 Гц), 3,26 (m, 3H, H-3', H-4' и H-5'), 3,13 (d, 1H, H-2', J=3,9 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц), 2,23 (m, 1H, 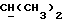 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 71
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-O-(1-метилэтил) карбонил- β- D-маннопиранозилокси)метил]-4-формил-4,4a,5, 6,7,7а, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 67 (160 мг) в сухом
дихлорметане (5 мл) добавляли 4-диметиламинопиридин (70 мг) и изобутирилхлорид (60 мкл). Смесь перемешивали в течение ночи при комнатной температуре, а затем концентрировали с получением масла желтого цвета, которое хроматографировали на колонке быстрого разделения с силикагелем, применяя в качестве растворителя смесь гексан:этилацетат (5:1) и получая в результате соединение, указанное в заголовке (162 мг), в виде белой пены.
δ (1H, CDCl3): 9,75 (5, 1H, CHO), 7,26-7,44 (m, 10H, 2Ph), 6,99 (s, 1H, 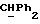 ), 6,08 (dd, 1H, H-2), 5,75 (s, 1H, H-1'), 4,68 (d, 1H, H-4', J=9 Гц), 4,12 и 3,80 (2d, 2H,
), 6,08 (dd, 1H, H-2), 5,75 (s, 1H, H-1'), 4,68 (d, 1H, H-4', J=9 Гц), 4,12 и 3,80 (2d, 2H, 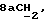 J=9 Гц), 3,43 (dq, 1H, H-5', J=6,6 и 9 Гц), 3,15 (s, 2H, H-2' и H-3'), 2,86 (m, 1H, H-11), 2,59 (m, 1H, 4'-
J=9 Гц), 3,43 (dq, 1H, H-5', J=6,6 и 9 Гц), 3,15 (s, 2H, H-2' и H-3'), 2,86 (m, 1H, H-11), 2,59 (m, 1H, 4'- 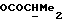 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 72
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6- дезокси-4-O-(2,2-диметилпропионил)- β- D-маннопиранозилокси) метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Смесь Промежуточного соединения 67 (0,3 ммоль) и 4-диметиламинопиридина (0,6 ммоль) в сухом дихлорметане (15 мл) обрабатывали хлорангидридом триметилуксусной кислоты (0,45 ммоль). Спустя 2,5 часа смесь промывали водой. Органическую фазу выпаривали, а остаток очищали посредством колоночной хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат (5: 1) и получая в результате соединение, указанное в заголовке (190 мг).
δ (1H, CDCl3): 9,76 (s, 1H, CHO), 7,83 (m, 10H, 2xPh), 7,00 (s, 1H, 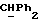 ), 6,08 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,77 (s, 1H, H-1'), 4,67 (d, 1H, H4', J= 9 Гц), 4,13 и 3,81 (d,d, 1H, 1H, 8a-CH2, J=9 Гц), 3,43 (m, 1H, H5'), 3,13 (m, 2H, H-2' и H-3'), 2,86 (t, 1H, H-1, J=3,9 Гц), 2,24 (m, 1H,
), 6,08 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,77 (s, 1H, H-1'), 4,67 (d, 1H, H4', J= 9 Гц), 4,13 и 3,81 (d,d, 1H, 1H, 8a-CH2, J=9 Гц), 3,43 (m, 1H, H5'), 3,13 (m, 2H, H-2' и H-3'), 2,86 (t, 1H, H-1, J=3,9 Гц), 2,24 (m, 1H, 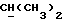 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 73
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-4-O-бензилокси-карбонил-6- дезокси- β- D-маннопираноэилокси)метил]-4-форнил-4,4a,5,6,7,7a,8, 8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 67 (0,3 ммоль) и 4-диэтиламинопиридина (0,8 ммоль) в сухом дихлорметане (15 мл) обрабатывали по каплям бензилоксикарбонилхлоридом (0,7 ммоль) и перемешивали в течение 3 часов. После промывания водой и рассолом растворитель выпаривали, а остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан: этилацетат (5:1) и получая в результате соединение, указанное в заголовке (185 мг).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 7,32 (m, 15H, 3xPh), 6,99 (s, 1H, 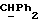 ), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,20 (m, 2H,
), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,20 (m, 2H, 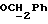 ), 4,72 (s, 1H, H-1'), 4,57 (d, 1H, H-4', J=8,4 Гц), 4,11 и 3,79 (2d, 2H, 8a-CH2, J=9 Гц), 3,43 (m, 1H, H-5'), 3,27 и 3,15 (2d, 2H, H-2' и H-3', J=3,6 Гц), 2,85 (t, 1H, H-1, J=3,9 Гц), 2,24 (m, 1H,
), 4,72 (s, 1H, H-1'), 4,57 (d, 1H, H-4', J=8,4 Гц), 4,11 и 3,79 (2d, 2H, 8a-CH2, J=9 Гц), 3,43 (m, 1H, H-5'), 3,27 и 3,15 (2d, 2H, H-2' и H-3', J=3,6 Гц), 2,85 (t, 1H, H-1, J=3,9 Гц), 2,24 (m, 1H, 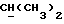 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 74
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-оксо- β- D- маннопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7а,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор трифторуксусного ангидрида (0,1 мл) в сухом дихлорметане (5 мл) при -60oC обрабатывали по каплям диметилсульфоксидом (0,06 мл) и раствором Промежуточного соединения 67 (0,39 ммоль) в сухом дихлорметане (5 мл). Спустя 60 минут добавляли триэтиламин (0,24 мл) и перемешивали смесь при -20oC в течение 2 часов. Смесь разбавляли дихлорметаном (20 мл) и промывали водой. После удаления растворителя остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат (4:1) и получая в результате соединение, указанное в заголовке (171 мг).
δ (1H,CDCl3): 9,76 (s, 1H, CHO), 7,36 (m, 10H, 2xPh), 6,59 (s, 1H, 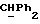 ), 6,09 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,84 (s, 1H, H-1'), 4,11 и 3,84 (2d, 2H, 8a-CH2, J=9 Гц), 3,92 (q, 1H, H-5', J=6,9 Гц), 3,59 и 3,37 (2d, H-2' и H-3', J=4,2 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц), 2,26 (m, 1H,
), 6,09 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,84 (s, 1H, H-1'), 4,11 и 3,84 (2d, 2H, 8a-CH2, J=9 Гц), 3,92 (q, 1H, H-5', J=6,9 Гц), 3,59 и 3,37 (2d, H-2' и H-3', J=4,2 Гц), 2,86 (t, 1H, H-1, J=3,9 Гц), 2,26 (m, 1H, 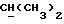 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 75
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2-O-Бензоил-6-дезокси-4-O-метил- β- D- альтропиранозилокси)метил]-4-формил-4,4a,5,6,7,7а,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-мeтaнo-s-индaцен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 48 (15,88 г) и диметиламинопиридина (8,83 г) в сухом дихлорметане (200 мл) при - 20oC в атмосфере азота по каплям добавляли бензоилхлорид (2 мл) в сухом дихлорметане (10 мл). После перемешивания в течение 45 минут охлаждающую баню убирали и осторожно добавляли 1N соляную кислоту (10 мл). Две фазы разделяли, а органический слой разбавляли дихлорметаном (600 мл) и промывали 1N соляной кислотой (500 мл), 10%-ным бикарбонатом натрия (500 мл) и рассолом (500 мл). Растворитель удаляли до сухого состояния и получали в результате масло, которое подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан в/в 1:20 и 1: 7), получая в результате 8,75 г соединения, указанного в заголовке выход 48%), в виде пены.
δ 1H, CDCl3): 9,66 (s, 1H, CHO), 8,15-8,05, 7,60- 7,2 (dd, 15H, 3Ph), 6,95 (s, 1H, CHPh2), 5,93 (bd, 1H, H2, J=2,1 Гц), 5,35 (dd, 1H, H2', J=1,5 и 4,8 Гц), 4,82 (d, 1H, H1', J=1,8 Гц), 4,25 (m, 1H, H3'), 3,99 (d, 1H, 8aCH2, J= 9 Гц), 3,86 (dq, 1H, H5', J=6,3 и 8,1 Гц), 3,68 (d, 1H, 8aCH2, J=9 Гц), 3,43 (s, 3H, OCH3), 3,22 (dd, 1H, H4', J=3 и 8,1 Гц), 2,62 (t, 1H, H1, J=3,9 Гц), 2,45 (d, 1H, OH, J=2,4 Гц), 2,19 (m, 1H, CH(CH3)2).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 76
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2-О-Бензоил-3,6-дидезокси-4-O-метил-3-оксо- β- D-аллопиранозилокси)метил] -4-фориил-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты дифенилметиловый эфир
К раствору трифторуксусного ангидрида (1,29 мл) в сухом дихлорметане (15 мл) в течение 10 минут в атмосфере азота при -60oC добавляли ДМСО (0,66 мл). К образовавшейся в результате суспензии добавляли раствор Промежуточного соединения 75 (4,45 г) в сухом дихлорметане (30 мл) и перемешивали смесь при -60oC в течение 1 часа. Добавляли по каплям триэтиламин (5,76 мл), доводили температуру до -20oC, а затем к образовавшемуся раствору желтого цвета добавляли воду (6 мл) и перемешивали смесь при комнатной температуре в течение 1 часа. Добавляли дихлорметан (200 мл) и воду (200 мл) и разделяли фазы. Органический слой сушили над сульфатом магния и концентрировали до сухого состояния, получая сырой продукт, который растворяли в Дихлорметане (10 мл) и обрабатывали триэтиламином (2 мл) в течение ночи. После удаления растворителя получали масло, которое подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан в/в 1:15 и 1:10), получая в результате 2,6 г соединения, указанного в заголовке (выход 76%) в виде пены.
δ (1H, CDCl3): 9,68 (s, 1H, CHO), 8,2-8,0, 7,6- 7,2 (2m, 15H, 3Ph), 6,96 (s, 1H, CHPh2), 5,76 (dd, 1H, H2, J=1,2 и 3,3 Гц), 5,77 (dd, 1H, H2, J=1,2 и 3,3 Гц), 5,39 (dd, 1H, H2', J=1,2 и 8,1 Гц), 4,53 (d, 1H, H1', J= 8,1 Гц), 4,09 (d, 1H, 8aCH2, J=9 Гц), 3,75 (d, 1H, 8aCH2, J=9 Гц), 3,7-3,4 (m, 5H, H4'+OCH3+H5'), 2,63 (t, 1H, H1, J=4,2 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 77
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3,6-Тридезокси-4-О-метил-3-оксо- β- D-аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
К хорошо дегазированному раствору (Ar, 15 минут) гидрида трибутилолова (5,96 мл) в сухом толуоле (120 мл) посредством шприца в течение 1 часа при 90oC добавляли раствор Промежуточного соединения 76 (5,7 г) и a,a'-азоизобутиронитрила (420 мг) в сухом толуоле. Нагревание продолжали до завершения реакции, а затем смесь охлаждали до комнатной температуры. Добавляли воду (40 мл) и фторид калия (1,5 г) и перемешивали смесь в течение ночи. Сырой продукт, полученный после удаления растворителя, смешивали с диэтиловым эфиром в течение 1 часа, твердую фазу удаляли посредством фильтрации. Эфирный раствор концентрировали до сухого состояния, а полученное масло подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан 1:15 и 1;10), получая в результате 2,22 г соединения, указанного в заголовке (выход 61%), в виде пены после вакуумной сушки.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,98 (s, 1H, CHPh2), 6,04 (dd, 1H, H2, J=1,5 и 3,6 Гц), 4,45 (dd, 1H, H1', J=2,7 и 8,7 Гц), 4,06 (d, 1H, 8aCH2, J=9 Гц), 3,70 (d, 1H, 8aCH2, J=9 Гц), 3,52 (s, 3H, OCH3), 3,5-3,3 (m, 2H, H4'+H5'), 2,7-2,5 (m, 3H, H1+H2'), 2,24 (m, 1H, CH(CH3)2),
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 78
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S, 7R,9R)-2,8-Диокса-9-метил- бицикло[3.4.0] -нон-4-ен-7-ил-оксиметил] -4-формил-4,4a,5,6,7, 7a,8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-мeтaнo-s- индaцeн-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К интенсивно перемешивавшемуся раствору Промежуточного соединения 77 (260 мг) и диэтилдиазометилфосфата (110 мг) в сухом тетрагидрофуране при 0oC в атмосфере азота медленно добавляли суспензию трет-бутоксида калия (112 мг) в сухом тетрагидрофуране (1 мл) (при этом наблюдали немедленное выделение газа). Спустя 10 минут смесь разбавляли в дихлорметане (100 мл) и промывали 1N соляной кислотой (2х100 мл), насыщенным бикарбонатом натрия (2x100 мл), рассолом (2x100 мл) и сушили над сульфатом магния. После удаления растворителя получали масло, которое очищали посредством хроматографии быстрого разделения (силикагель, этилацетат:гексан в/в 1:15 и 1:12), получая в результате 200 мг соединения, указанного в заголовке (выход 77%), в виде пены.
δ (1H, CDCl3); 9,75 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,05 (dd, 1H, H2, J=1,2 и 3,3 Гц), 5,45 (m, 1H, H4'), 4,65 (m, 2H, H3'), 4,25-4,15 (m, 2H, H7'+H1'), 4,07 (d, 1H, 8aCH2, J=9 Гц), 3,71 (d, 1H, 8aCH2, J= 9 Гц), 3,19 (dq, 1H, H9', J=6 и 8,4 Гц), 2,77 (t, 1H, H1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 79
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4R,7R,9R)-2,8-Диокса-4-гидрокси- 9-метил-цис-бицикло[3.4.0]-нон-7-ил-оксиметил]-4-гидроксиметил- 4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 78 (620 мг) в безводном тетрагидрофуране (5 мл) в атмосфере азота добавляли 9-борабицикло [3,3,1]нонан (0,5М раствор в тетрагидрофуране, 6 мл). Смесь прогревали при 50oC в течение 1 часа. Затем при 50oC в течение 3 часов добавляли дополнительный объем раствора 9-борабицикло[3, 3,1]нонана для завершения реакции и оставляли при перемешивании в течение 1 часа при комнатной температуре. Осторожно добавляли этиловый спирт (2 мл) (выделение газа) и перемешивали раствор в течение 1 часа. Затем последовательно при 0oC добавляли 3N гидроксид натрия (5 мл) и перекись водорода (35% в/в, 5 мл) и перемешивали смесь при комнатной температуре, при 0oC и при 70oC в течение ночи. Полученный раствор охлаждали до комнатной температуры и концентрировали до половины исходного объема, затем его разбавляли 1N соляной кислотой (100 мл) и дважды экстрагировали этилацетатом (2x200 мл). Органический слой промывали 1N соляной кислотой (100 мл), бикарбонатом натрия (2x100 мл) и рассолом (100 мл), сушили над сульфатом натрия и концентрировали до сухого состояния, получая в результате сироп, который подвергали хроматографии быстрого разделения (ацетон:гексан 1: 10 и 1:5), получая в результате 475 мг соединения, указанного в заголовке (выход 74%), в виде пены.
δ (1H, CDCl3): 7,5-7,2 (m, 10H, 2Ph), 7,04 (s, 1H, CHPh2), 5,90 (dd, 1H, H2, J=1,5 и 3,6 Гц), 4,75 (d,1H, 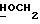 J=10,5 Гц), 4,62 (dd, 1H, H7', J=2,7 и 5,1 Гц), 4,27 (m, 1H, H4'), 4,16 (d, 1H, 8aCH2, J=9 Гц), 4,09 (dd, 1H, H3', J=6 и 9,6 Гц), 3,91 (t, 1H, H3', J=6,9 Гц), 3,7-3,5 (m, 3H, 8aCH2+H1'+H9'), 3,35 (d, 1H, HOCH2, J=11,5 Гц), 3,22 (dd, 1H, HOCH2, J=11,1 и 12 Гц), 2,57 (t, 1H, H1, J=4,5 Гц), 2,48 (m, 1H, H5'), 2,30 (m, 1H, CH(CH3)2).
J=10,5 Гц), 4,62 (dd, 1H, H7', J=2,7 и 5,1 Гц), 4,27 (m, 1H, H4'), 4,16 (d, 1H, 8aCH2, J=9 Гц), 4,09 (dd, 1H, H3', J=6 и 9,6 Гц), 3,91 (t, 1H, H3', J=6,9 Гц), 3,7-3,5 (m, 3H, 8aCH2+H1'+H9'), 3,35 (d, 1H, HOCH2, J=11,5 Гц), 3,22 (dd, 1H, HOCH2, J=11,1 и 12 Гц), 2,57 (t, 1H, H1, J=4,5 Гц), 2,48 (m, 1H, H5'), 2,30 (m, 1H, CH(CH3)2).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 80
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S, 7R,9R)-2,8-Диокса-9-бицикло[3.4.0] -нон-4-ен-7-ил-оксиметил] -4-[трет-бутил-диметилсилилоксиметил] - 4,4a, 5,6,7,7a,8,8а-октагидро-7-метил-3-(1-метил-этил)-1,4-метано- s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 78 (200 мг) в тетрагидрофуране (5 мл) добавляли твердый бороводородистый натрий (18,9 мг) и воду (1 мл), смесь перемешивали при 0oC в течение 30 минут, а затем 1 час при комнатной температуре. Смесь осторожно гасили добавлением воды (2 мл) при 0oC и фракционировали между этилацетатом (100 мл) и водой (100 мл). Органический слой сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате сироп, который растворяли в сухом диметилформамиде (5 мл) и обрабатывали в течение ночи при комнатной температуре в атмосфере азота имидазолом (204 мг) и трет-бутил- диметил-силилхлоридом (316 мг). Раствор вливали в 100 мл смеси 1N хлорида аммония и этилацетата (в/в 1:1) и перемешивали в течение 1 часа. Органический слой промывали 1N хлоридом аммония (100 мл) и рассолом (100 мл), сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате пену, которую подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан в/в 1:20), получая в результате 215 мг соединения, указанного в заголовке (общий выход 92%).
δ (1H, CDCl3): 7,5-7,2 (m, 10H, 2Ph), 6,94 (s, 1H, CHPh2), 5,96 (dd, 1H, H2, J= 1,2 и 3,6 Гц), 5,54 (m, 1H, H4'), 4,65 (m, 2H, H3'), 4,25- 4,15 (m, 3H, SiOCH2+H7'+H1'), 4,1 (d, 1H, 8aCH2, J=9,3 Гц), 3,69 (d, 1H, SiOCH2, J=9 Гц), 3,30 (d, 1H, 8aCH2, J=9,3 Гц), 3,19 (dq, 1H, H9', J=6 и 9 Гц), 2,68 (dd, 1H, H6', J=2 и 13,2 Гц), 2,57 (t, 1H, H1, J=4,2), 2,3 (m, 2H, CH(CH3)2+H6'), -0,017 и -0,03]. (2s, 6H, (CH3)2Si).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 81
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S, 4R, 7R, 9R)-2,8-Диокса-4гидрокси- 9-метил-цис-бицикло[3.4.0] -нон-7-ил-оксиметил] -4-[трет-бутил- диметилсилил-оксиметил] -4,4a, 5,6,7,7a,8,8а-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 80 (700 мг) в сухом тетрагидрофуране (5 мл) в атмосфере азота добавляли 9-борабицикло [3,3,1] нонан (0,5М раствор в тетрагидрофуране, 6 мл). Смесь нагревали до 50oC в течение 1 часа. В течение 3 часов для завершения реакции при 50oC добавляли дополнительный объем 9- борабицикло[3,3,1] нонана, затем смесь охлаждали до комнатной температуры и осторожно добавляли этиловый спирт (2 мл). Спустя 1 час при 0oC последовательно добавляли 3N гидроксид натрия (5 мл) и перекись водорода (35% в/в, 5 мл), смесь перемешивали при комнатной температуре в течение 1 часа и в течение ночи при 70oC. Раствор охлаждали до комнатной температуры и концентрировали до половины исходного объема, затем разбавляли ее 1N соляной кислотой (100 мл) и дважды экстрагировали этилацетатом (2x2100 мл). Органический слой промывали 1N соляной кислотой (100 мл), насыщенным бикарбонатом натрия (100 мл) и рассолом (100 мл), сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате сироп, который подвергали хроматографии быстрого разделения (ацетон:гексан 1:20 и 1:15), получая в результате 550 мг (выход 77%) соединения, указанного в заголовке, в виде белой пены.
δ (1H, CDCl3): 7,5-7,2 (m, 10H, 2Ph), 6,94 (s, 1H, CHPh2), 5,95 (m, 1H, H2), 4,57 (dd, 1H, H7', J= 3 и 5,1 Гц), 4,30-4,15 (m, 2H, H4'+CH2OSi), 4,10-3,95 (m, 2H, H3'+8aCH2), 3,87 (t, 1H, H3', J=6,9 Гц), 3,7- 3,5 (m, 3H, H1+8aCH2+H9'), 3,30 (d, 1H, CH2OSi, J=9,3 Гц), 2,53 (t, 1H, H1, J= 3,9 Гц), 2,46 (m, 1H, H5'), 2,32 (m, 1H, CH(CH3)2), -0,016 и -0,029 (2s, 6H, (CH3)2Si).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 82
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S, 4R,7R,9R)-2,8-Диокса-4-метокси-9-метил- цис-бицикло[3.4.0] -нон-7-ил-оксиметил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 81 (537 мг) в безводном тетрагидрофуране (5 мл) малыми порциями в атмосфере азота при 0oC добавляли гидрид натрия (75 мг). После перемешивания в течение 10 минут добавляли метилоидид (374 мкл) и оставляли смесь при комнатной температуре до завершения реакции. Смесь гасили добавлением воды (100 мл), а затем экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом (100 мл) и концентрировали до сухого состояния, получая в результате пену, которую растворяли в безводном ТГФ (5 мл) и при 4 0oC в течение 24 часов обрабатывали тетрабутиламмонийфторидом (1,1М раствор в тетрагидрофуране, 2 мл). Полученный раствор концентрировали до сухого состояния, а остаток растворяли в АцОЭт и промывали водой (2x100 мл), сушили над сульфатом магния и концентрировали, получая в результате сироп, который растворяли в сухом дихлорметане 85 мл). Добавляли хлорхроматпиридиний (215 мг) и перемешивали смесь при комнатной температуре до завершения реакции. Суспензию вливали в хорошо перемешивавшуюся смесь дихлорметана и метабисульфита натрия (10%-ный водный раствор). Органический слой промывали 1N соляной кислотой (200 мл) и 1N гидроксидом натрия (200 мл), сушили над сульфатом магния и кон центрировали до сухого состояния, получая в результате масло, которое подвергали хроматографии быстрого разделения на силикагеле (этилацетат:гексан 1:15, 1:10 и 1:8), получая в результате 255 мг соединения, указанного в заголовке, в виде масла (общий выход 57%).
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,04 (dd, 1H, H2, J=1,2 и 3,6 Гц), 4,60 (m, 1H, H7'), 4,10-3,95 (m, 2H, H4'+8aCH2), 3,90-3,65 (m, 3H, H3'+H1'), 3,65-3,55 (m, 2H, 8aCH2+H9'), 3,35 (5, 3H,OCH3), 2,73 (t, 1H, H1, J=3,6 Гц), 2,54 (m, 1H, H5'), 22,4 (m, 1H, CH(CH3)2).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 83
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-4-O-метил- β- D-альтропиранозилокси) метил] -4-(1,3-диоксолан-2-ил)-4,4a, 5,6,7,7a, 8, 8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 48 (9,7 ммоль) в сухом метаноле (40 мл) обрабатывали этиленгликолем (110 мл), триметилортоформиатом (3,25 мл) и каталитическим количеством п-толуолсульфоновой кислоты. Смесь прогревали при 40oC в течение 3 часов. После охлаждения смесь вливали в смесь этилацетат: водный гидрокарбонат натрия (1:1; 500 мл), водный слой тщательно экстрагировали этилацетатом. Объединенные органические слои промывали водой и рассолом и сушили. После удаления растворителя остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан: этилацетат (6: 1) и получая в результате соединение, указанное в заголовке (6,1 г).
δ (1H, CDCl3): 7,43 и 7,30 (m,m, 4H, 6H, 2xPh), 6,94 (s, 1H, 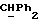 ), 5,83 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,07 (s, 1H, 4-CH), 4,63 (d, 1H, H1', J= 1,5 Гц), 4,20 (t, 1H, H-3, J=3,3 Гц), 4,07 (d, 1H, 8a-CHa, J=9,3 Гц), 3,84 (m, 6H, H-2', 8a-CHb и OCh2CH2O), 3,71 (m, 1H, H-5'), 3,42 (s, 3H, OCH3), 3,21 (dd, 1H, H-4', J=3 и 9 Гц), 2,63 (m, 1H,
), 5,83 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,07 (s, 1H, 4-CH), 4,63 (d, 1H, H1', J= 1,5 Гц), 4,20 (t, 1H, H-3, J=3,3 Гц), 4,07 (d, 1H, 8a-CHa, J=9,3 Гц), 3,84 (m, 6H, H-2', 8a-CHb и OCh2CH2O), 3,71 (m, 1H, H-5'), 3,42 (s, 3H, OCH3), 3,21 (dd, 1H, H-4', J=3 и 9 Гц), 2,63 (m, 1H, 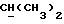 ), 2,54 (t, 1H, H-1, J=3,9 Гц).
), 2,54 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 84
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2-O-Бензоил-6-дезокси-4-O-метил- β- D-альтропиранозилокси)метил] -4-[1,3-диоксолан-2-ил)-4, 4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1, 4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 83 (12,12 г) и диметиламинопиридина (6,3 г) в сухом дихлорметане (155 мл) по каплям при -20oC в атмосфере азота добавляли бензоилхлорид (1,40 мл) в 50 мл сухого дихлорметана. После перемешивания в течение 2 часов смесь оставляли нагреваться до комнатной температуры, раствор промывали 0,1N соляной кислотой (500 мл), насыщенным бикарбонатом натрия (500 мл) и рассолом (500 мл). После удаления растворителя получали остаток, который подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан в/в 1:4), получая в результате 5,89 г соединения, указанного в за головке (42%), в виде пены.
δ (1H, CDCl3): 8,14-8,05, 7,6-7,2 (2m, 15H, 3Ph), 6,91 (s, 1H, CHPh2), 5,70 (bd, 1H, H2, J=2,1 Гц), 5,34 (dd, 1H, H2', J=2,1 и 5,1 Гц), 5,01 (s, 1H, OCHO), 4,82 (d, 1H, H1', J=1,8 Гц), 4,26 (m, 1H, H3'), 3,98 (d, 1H, 8aCH2, J=9,3 Гц), 3,92-3,74 (m, 5H, H5'+OCH2CH2O), 3,72 (d, 1H, 8aCH2, J=9,3 Гц), 3,42 (s, 3H, OCH3), 3,23 (dd, 1H, H4', J=3,3 и 8,1 Гц), 2,57 (m, 1H, CH(CH3)2), 2,44-2,38 (m, 2H, OH+H1).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 85
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2-О-Бензоил-3,6-дидезокси- 4-O-метил-3-оксо- β- D-аллопиранозилокси)метил] -4-[1,3- диоксолан-2-ил)-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенигметиловый
К раствору трифторуксусного ангидрида (0,45 мл) в сухом дихлорметане (5 мл) в течение 10 минут при -60oC в атмосфере азота добавляли ДМСО (0,23 мл). К полученной таким образом суспензии добавляли раствор Промежуточного соединения 84 (1,2 г) в сухом дихлорметане (5 мл), смесь перемешивали при -60oC в течение 1 часа. Добавляли по каплям триэтиламин (1,24 мл), доводили температуру до -20oC, затем к полученному раствору желтого цвета добавляли воду (2 мл) и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли дихлорметан (200 мл) и воду (250 мл) и разделяли фазы. Органический слой сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате сырой продукт, который растворяли в дихлорметане (10 мл) и обрабатывали триэтиламином (2 мл) в течение ночи. После удаления растворителя получали масло, которое подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан в/в 1:15 и 1:10), получая в результате 1,1 г соединения, указанного в заголовке (выход 92%), виде пены, которую затем сушили под вакуумом.
δ 1H, CDCl3): 8,2-8,0, 7,6-7,2 (2m, 15H, 3Ph), 6,90 (s, 1H, CHPh2), 5,57 (dd, 1H, H2, J=1,2 и 3,6 Гц), 5,37 (dd, 1H, H2', J=1,5 и 8,1 Гц), 5,05 (s, 1H, OCHO), 4,52 (d, 1H, H1', J=8,1 Гц), 4,08 (d, 1H, 8aCH2, J= 8,7 Гц), 3,9-3,7 (m, 5H, OCH2CH2O+8aCh2), 3,6-3,4 (m, 5H, H4'+OCH3+H5'), 2,62 (m, 1H, CH(CH3)2), 2,43 (t, 1H, H1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 86
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3,6-Тридезокси-4-О-метил- 3-оксо- β- D-аллопиранозилокси)метил]-4-[1,3-диоксолан-2- ил)-4,4a,5,6,7,7a, 8,8а-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К хорошо дегазированному раствору (Ar, 15 минут) гидрида трибутилолова (263 мкл) в сухом толуоле (10 мл) при 90oC в течение 1 часа посредством шприца добавляли раствор Промежуточного соединения 85 (250 мг) и a,a'-азоизобутиронитрила (16 мг) в сухом толуоле (5 мл). Нагревание продолжали до завершения реакции, затем смесь охлаждали до комнатной температуры. Добавляли воду (5 мл) и фторид калия (100 мг) и перемешивали смесь в течение ночи. Сырой продукт, полученный после удаления растворителя, смешивали с диэтиловым эфиром в течение 1 часа, твердую фазу удаляли посредством фильтрации. Эфирный раствор концентрировали до сухого состояния, а полученное масло подвергали хроматографии быстрого разделения (силикагель, этилацетат: гексан 1: 15 и 1:10), получая в результате 168 мг соединения, указанного в заголовке (выход 80%), в виде пены при вакуумной сушке.
δ (1H, CDCl3): 7,6-7,1 (m, 10H, 2Ph), 6,92 (s, 1H, CHPh2), 5,80 (dd, 1H, H2, J=0,9 и 3,6 Гц), 5,08 (s, 1H, OCHO), 4,44 (dd, 1H, H1', J=2,7 и 8,7 Гц), 4,05 (d, 1H, 8aCH2, J=9,3 Гц), 3,9-3,7 (m, 5H, OCH2CH2O+8aCH2), 3,50-3,30 (m, 2H, H5'+H4'), 2,5-2,1 (m, 3H, H2'+CH(CH3)2), 2,50 (t, 1H, H1, J=4,5 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 87
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S, 7R,9R)-2,8-Диокса-9-метил- бицикло[3.4.0] -нон-4-ен-7-ил-оксиметил] -4-[1,3-диоксолан-2- ил] -4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метaнo-s-индaцeн-3a(1H)-кapбoнoвoй кислоты дифенилметиловый эфир
К интенсивно перемешивавшемуся раствору Промежуточного соединения 86 (160 мг) и диэтил-диазометил-фосфоната (63 мг) в сухом тетрагидрофуране медленно добавляли при 0oC в атмосфере азота суспензию трет-бутоксида калия (100 мг) в сухом тетрагидрофуране (2,3 мл) (наблюдали немедленное выделение газа). Спустя 10 минут смесь разбавляли дихлорметаном (100 мл) и промывали 1N соляной кислотой (2x100 мл), насыщенным бикарбонатом натрия (2x100 мл), рассолом (2x100 мл) и немедленно сушили над сульфатом магния. После удаления растворителя получали масло, которое подвергали хроматографии быстрого разделения (силикагель, этилацетат:гексан в/в 1:15 и 1:12), получая в результате 110 мг соединения, указанного в заголовке (выход 70%), в виде пены.
δ 1H, CDCl3): 7,5-7,2 (m, 10H, 2Ph), 6,94 (s, 1H, CHPh2), 5,83 (dd, 1H, H2, J= 1,2 и 3,3 Гц), 5,53 (m, 1H, H4'), 5,06 (s, 1H, OCHO), 4,65 (m, 2H, H3'), 4,25-4,10 (m, 2H, H7'+H1'), 4,05 (d, 1H, 8aCH2, J=9,3 Гц), 3,90- 3,70 (m, 5H, OCH2CH2O+8aCH2), 3,18 (dq, 1H, H9', J=6 и 8,7 Гц), 2,75- 2,50 (m, 3H, H6'+CH(CH3)2+H1)
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 88
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(4-О-Аллил-3,6-дидезокси-3-иодо- β- D-маннопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7а, 8, 8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 53 (GM 2008) (103 мг), трифенилфосфина (197 мг) и имидазола (51 мг) в сухом тетрагидрофуране (5 мл) обрабатывали порциями иода (95 мг). После перемешивания в течение 1 часа при комнатной температуре раствор подвергали дефлегмации до тех пор, пока не был потреблен весь исходный материал (ТСХ анализ гексан:этилацетат 4:1). Реакционную смесь охлаждали и фракционировали между этилацетатом (50 мл) и 1N водной соляной кислотой (30 мл). Органический слой промывали последовательно 1N водной соляной кислотой, водой, водный раствором метабисульфита натрия, водой и рассолом, затем сушили (Na2SO4), фильтровали и концентрировали. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью гексан: этилацетат (9:1) и получая в результате соединение, указанное в заголовке (70 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,46-7,22 (m, 10H, 2Ph), 6,98 (s, 1H, 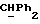 ), 6,03 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 6,02-5,9 (m, 1H,
), 6,03 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 6,02-5,9 (m, 1H, 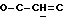 ), 5,35-5,15 (m, 2H,
), 5,35-5,15 (m, 2H, 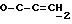 ), 4,4-4,32 (m, 1H, O-CHa-C=C), 4,35 (d, 1H, H-1', J= 0,6 Гц), 4,18-3,96 (m, 4H, O-CHb-C=C, H-2', H-3', 8aCHa), 3,75 (d, 1H, 8a-CHb, J=9 Гц), 3,45 (dd, 1H, H-4', J=9 и 10,2 Гц), 3,34 (dq, 1H, H-5', J-5,7 и 8,7 Гц), 2,72 (t, 1H, H-1, J=3,9 Гц).
), 4,4-4,32 (m, 1H, O-CHa-C=C), 4,35 (d, 1H, H-1', J= 0,6 Гц), 4,18-3,96 (m, 4H, O-CHb-C=C, H-2', H-3', 8aCHa), 3,75 (d, 1H, 8a-CHb, J=9 Гц), 3,45 (dd, 1H, H-4', J=9 и 10,2 Гц), 3,34 (dq, 1H, H-5', J-5,7 и 8,7 Гц), 2,72 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ 89а и 89б
a) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4S,6S,7R,9R)-2,8-Диокса-4,9-диметил-6- гидрокси-цис-бицикло[3.4.0] -нон-7-ил-окси-метил]-4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
б) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4R,6S,7R,9R)-2,8-Диокса-4,9-диметил- 6-гидрокси-цис-бицикло[3.4.0] -нон-7-ил-окси-метил]-4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор промежуточного соединения 88 (80 мг) в сухом толуоле (2 мл) дегазировали током аргона в течение 60 минут, а затем нагревали с дефлегмацией. Добавляли гидрид трибутилолова (41 мкл) и продолжали дефлегмацию еще в течение 15 минут. Растворитель удаляли под вакуумом, а остаток переносили в диэтиловый эфир (50 мл) и промывали насыщенным водным раствором фторида калия до тех пор, пока не прекращалось выпадение осадка фторида трибутилолова. Органический слой сушили и упаривали, получая остаток, который очищали посредством препаративной ТСХ (силикагель, дихлорметан:этилацетат 95:5), получая в результате соединение, указанное в заголовке (а) Промежуточное соединение 89а (35 мг, Rf=0,4 дихлорметан:этилацетат 9:1) и соединение, указанное в заголовке (б) Промежуточное соединение 89б (24 мг, Rf=0,3 дихлорметан:этилацетат 9:1).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 89а
δ 1H, CDCl3): 9,73 (s, 1H, CHO), 7,46-7,22 (m, 10H, 2Ph), 6,99 (s, 1H, 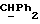 ), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,63 (d, 1H, H-7', J=3,3 Гц), 4,02 (d, 1H, 8aCHa, J=9 Гц), 3,86 (dd,1 H, Ha-3', J=6,6 и 8,7 Гц), 3,83-3,6 (m, 4H, H-6', H-1', H-9' и 8a-CHb), 3,52 (dd, 1H, Hb-3', J=5,7 и 8,7 Гц), 2,69 (t, 1H, H-1, J=3,9 Гц).
), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,63 (d, 1H, H-7', J=3,3 Гц), 4,02 (d, 1H, 8aCHa, J=9 Гц), 3,86 (dd,1 H, Ha-3', J=6,6 и 8,7 Гц), 3,83-3,6 (m, 4H, H-6', H-1', H-9' и 8a-CHb), 3,52 (dd, 1H, Hb-3', J=5,7 и 8,7 Гц), 2,69 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 89б
δ 1H, CDCl3): 9,74 (s, 1H, CHO), 7,45-7,22 (m, 10H, 2Ph), 6,99 (s, 1H, 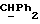 ), 6,06 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,48 (d, 1H, H-7', J-2,1 Гц), 4,1-4,0 (m, 2H, 8a-CHa и Ha-3'), 3,85 (dd, 1H, H-1', J=7,8 и 9 Гц), 3,76-3,68 (m, 1H, 8a-CHb и H-6'), 3,46-3,3 (m, 2H, H-9' и Hb-3'), 2,73 (t, 1H, H-1, J=3,9 Гц).
), 6,06 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,48 (d, 1H, H-7', J-2,1 Гц), 4,1-4,0 (m, 2H, 8a-CHa и Ha-3'), 3,85 (dd, 1H, H-1', J=7,8 и 9 Гц), 3,76-3,68 (m, 1H, 8a-CHb и H-6'), 3,46-3,3 (m, 2H, H-9' и Hb-3'), 2,73 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 90
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси- β- D-альтропиранозилокси)метил] - 4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору 4'-деметилсордарина (10 г) в метаноле (20 мл) добавляли по каплям при комнатной температуре раствор дифенилдиазометана (90 мл) в метиленхлориде и перемешивали смесь в течение 6 часов. Растворитель упаривали до сухого состояния, а остаток хроматографировали на колонке для быстрого разделения на силикагеле, элюируя смесью н-гексан:этилацетат (3:1) и получая в результате соединение, указанное в заголовке (12,6 г), в виде пены бледно-желтого цвета.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,98 (s, 1H, 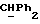 ), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,65 (d, 1H, H-1', J=1,5 Гц), 4,09, 3,76 (2d, 2H, 8a-
), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,65 (d, 1H, H-1', J=1,5 Гц), 4,09, 3,76 (2d, 2H, 8a- 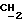 J= 9 Гц), 4,01 (m, 1H, H-2'), 3,84 (m, 1H, H-3'), 3,75 (m, 1H, H-5'), 3,69 (m, 1H, H-4'), 2,73 (t, 1H, H-1, J=4,2 Гц).
J= 9 Гц), 4,01 (m, 1H, H-2'), 3,84 (m, 1H, H-3'), 3,75 (m, 1H, H-5'), 3,69 (m, 1H, H-4'), 2,73 (t, 1H, H-1, J=4,2 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 91
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-изопропилиден- β- D- альтропиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a-октагидро- 7-метил-3-(1-метилэтил)-(1H)-1,4-метано-s-индацен-3a (1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 90 (650 мг) в 15 мл смеси 2,2-диметоксипропан: ацетон (1:2) добавляли п-толуолсульфоновую кислоту (10 мг). Раствор перемешивали при комнатной температуре в течение 1,5 часов, затем добавляли карбонат калия (1,0 г), продолжали перемешивание еще в течение 30 минут и упаривали растворитель до сухого состояния. Сырую смесь фракционировали между этилацетатом (50 мл) и водой (25 мл), водную фазу экстрагировали этилацетатом (2x50 мл), органическую фазу промывали рассолом, сушили над сульфатом магния и упаривали до сухого состояния, остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат: гексан (1: 3) и получая в результате соединение, указанное в заголовке (600 мг), в виде пены белого цвета.
δ 1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,24 (m, 10H, 2Ph), 6,98 (s, 1H,
CHPh2), 6,06 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,57 (d, 1H, H-1', J=3,0 Гц), 4,30 (dd, 1H, H-3', J=3,6 и 5,7 Гц), 4,07 (d,1H, 8aCH2, J=9,0 Гц), 3,95-3,93 (m, 1H, H-2'), 3,85 (dd, 1H, H-4', J=5,7 и 9,3 Гц), 3,75 (d, 1H, 8aCH2, J= 9,0 Гц), 3,44 (dq, 1H, H-5', Jd=9,3 Гц, Jq=6,3 Гц), 2,73 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 92
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-изопропилирен-2-O-(метилтио) тиокарбонил- β- D-альтропиранозилокси)метил)-4-формил-4,4a,5, 6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3a(1H)-карбоновой кислоты дифенилметиловый эфир
Промежуточное соединение 91 (100 мг) и имидазол (1 мг) растворяли в сухом тетрагидрофуране (4 мл) в атмосфере азота. Добавляли гидрид натрия (5 мг) и перемешивали суспензию при комнатной температуре в течение 0,5 часов. Добавляли сероуглерод (2,7 мл), перемешивание продолжали еще в течение 20 минут и добавляли метилиодид (18 мл). Спустя 2 часа реакцию останавливали добавлением 1N хлорида аммония (20 мл). Смесь экстрагировали этилацетатом (3x25 мл), органическую фазу промывали рассолом, сушили над сульфатом магния и упаривали растворитель до сухого состояния. Остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью этилацетат: гексан (1:9) и получая в результате соединение, указанное в заголовке (110 мг).
δ 1H, CDCl3): 9,71 (s, 1H, CHO), 7,44-7,25 (m, 10H, 2Ph), 6,96 (s, 1H, CHPh2), 6,01 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 6,90 (dd, 1H, H-2', J=2,4 и 5,4 Гц), 4,85 (d, 1H, H-1', J=2,4 Гц), 4,53 (dd, 1H, H-3', J=5,4 и 6,3 Гц), 4,00 (dd, 1H, H-4', J=6,3 и 8,7 Гц), 3,93 (d, 1H, 8aCH2, J=9,3 Гц), 3,65 (dq, 1H, H-5', Jd=8,7, Jq=6,3 Гц), 2,68 (t, 1H, H-1, J=3,9 Гц), 2,59 (8, 3H, CH3S).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 93
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси-3,4-O-изопропилиден- β- D- аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Промежуточное соединение 92 (95 мг) растворяли в сухом толуоле (5 мл) в атмосфере азота и прогревали при 110oC. Через 1,5 часа при перемешивании по каплям добавляли раствор гидрида трибутилолова (64 мл) в сухом толуоле (5 мл). Нагревание продолжали еще в течение 1,5 часов, добавляли метанол (2 мл) и упаривали растворитель до сухого состояния. После хроматографии быстрого разделения остатка на силикагеле с элюцией смесью этилацетат:гексан (1:9) получали соединение, указанное в заголовке (42 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,44-7,25 (m, 10H, 2Ph), 6,98 (1H, s, CHPh2), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,54 (dd, 1H, H-1', J=2,7 и 9,3 Гц), 4,39 (dt, 1H, H-3', Jd=2,7 Гц, Jt=3,6 Гц), 4,04 (d, 1H, 8aCH2, J=9,0 Гц), 3,67 (d, 1H, 8aCH2, J=9,0 Гц), 3,65 (dd, 1H, H-4', J=3,6 и 8,7 Гц), 3,44 (dq, 1H, H-5', Jd=6,3 Гц, Jq=8,7 Гц), 2,75 (t, 1H, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 94
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси- β- D-аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 93 (1,5 г) в смеси тетрагидрофурана (30 мл) и метанола (15 мл) при комнатной температуре с интенсивным перемешиванием по каплям добавляли 1N раствор соляной кислоты (15 мл). Сразу после окончания реакции (ТСХ контроль) добавляли насыщенный бикарбонат натрия (50 мл) и этилацетат (200 мл) и фракционировали смесь. Органический слой промывали водой (2x100 мл) и сушили над сульфатом магния. После удаления растворителя получали остаток, который подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (5:1) и (2:1) и получая в результате соединение, указанное в заголовке (1,1 г), в виде белой пены.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,5-7,2 (m, 10H, 2Ph), 6,99 (s, 1H, CHPh2), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,64 (dd, 1H, H-1', J-2,1 и, 9,6 Гц), 4,11 (m, 1H, H-3'), 4,06 (d, 1H, 8aCH2, JAB=9,3 Гц), 3,70 (m, 2H, H-5' и 8aCH2), 3,34 (m, 1H, H-4'), 2,75 (t, 1H, H-1, J=3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 95
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3,6-Тридезокси-3-иод- β- D-глюкопиранозилокси) метил]-4-формил-4,4a,5,6,7,7a,8,8а-октагидро-7-метил-3-(1- метилэтил)-1,4-мeтaнo-s-индaцeн-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Промежуточное соединение 94 (2,0 г), трифенилфосфин (3,4 г) и имидазол (186 мг) подвергали дефлегмации в толуоле (50 мл) при перемешивании, а затем обрабатывали иодом, добавляя его небольшими порциями. Дефлегмацию продолжали в течение 4 часов. Реакционную смесь охлаждали и через отдельную воронку декантировали в избыток водного гидрокарбоната натрия и тиосульфата натрия. Смесь встряхивали до полного потребления иода. Толуольную фазу промывали водой, сушили над сульфатом магния и концентрировали. Остаток очищали посредством колоночной хроматографии быстрого разделения, элюируя смесью гексан: этилацетат (15: 1) и (5:1) и получая в результате соединение, указанное в заголовке (1,65 г).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 7,26-7,44 (m, 10H, 2Ph), 6,98 (s, 1H, 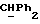 ), 6,04 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,25 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,08 (m, 1H, H-4') , 3,67 и 4,01 (2d, 2H, 8aCH2, J=9 Гц), 3,40 (m, 1H, H-3'), 3,32 (m, 1H, H-5'), 2,74 (t, 1H, H-1, J=4,2 Гц), 2,32 и 2,53 (2m, 2H, 2H-2').
), 6,04 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,25 (dd, 1H, H-1', J=1,8 и 9,3 Гц), 4,08 (m, 1H, H-4') , 3,67 и 4,01 (2d, 2H, 8aCH2, J=9 Гц), 3,40 (m, 1H, H-3'), 3,32 (m, 1H, H-5'), 2,74 (t, 1H, H-1, J=4,2 Гц), 2,32 и 2,53 (2m, 2H, 2H-2').
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 96
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3,6-Тридезокси-3-этиламино-3-N, 4- O-карбонил- β- D-аллопираноэил)окси)метил] -4-формил- 4,4a, 5,6,7,7а, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор промежуточного соединения 95 (200 мг) в сухом тетрагидрофуране (15 мл) обрабатывали гидридом натрия (24 мг, 1,0 моль). Через 15 минут добавляли этилизоцианат (0,04 мл) и реакционную смесь перемешивали с дефлегмацией в течение 6 часов. После погашения водой (3 мл) смесь экстрагировали этилацетатом (3x5 мл), объединенные органические растворы обрабатывали рассолом (1x5 мл) и сушили над сульфатом магния, фильтровали и концентрировали под вакуум до получения сиропа. Его очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат 3: 1, получая в результате чистое соединение, указанное в заголовке (75 мг).
δ (1H, CDCl3): 9,74 (s, 1H, 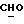 ), 7,44-7,26 (m, 10H, 2Ph), 6,98 (s, 1H, -CHPh2), 6,03 (dd, 1H, H-2, J=3,3 и 1,2 Гц), 4,60 (dd, 1H, H-1', J=5,1 и 3,9 Гц), 4,12 (m, 2H, H-3', H-4'), 4,01 (d, 1H, H-8a, J=9,3 Гц), 3,64 (d, 1H, H-8a, J= 9,3 Гц), 3,61 (dq, 1H, H-5', J=8,7 и 6,3 Гц), 3,49 (m, 1H,
), 7,44-7,26 (m, 10H, 2Ph), 6,98 (s, 1H, -CHPh2), 6,03 (dd, 1H, H-2, J=3,3 и 1,2 Гц), 4,60 (dd, 1H, H-1', J=5,1 и 3,9 Гц), 4,12 (m, 2H, H-3', H-4'), 4,01 (d, 1H, H-8a, J=9,3 Гц), 3,64 (d, 1H, H-8a, J= 9,3 Гц), 3,61 (dq, 1H, H-5', J=8,7 и 6,3 Гц), 3,49 (m, 1H, 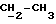 ), 3,13 (m, 1H,
), 3,13 (m, 1H, 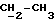 ), 2,70 (t, 1H, H-1, J=3,9 Гц), 2,24 (m, 1H
), 2,70 (t, 1H, H-1, J=3,9 Гц), 2,24 (m, 1H 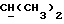 ), 1,14 (t, 3H
), 1,14 (t, 3H 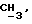 J=7,2 Гц).
J=7,2 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 97
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4R,6S,7R,9R)-2,8-Диокса-4,9-диметил-6- (метилтио)-тиокарбонилокси-цис-бицикло[3.4.0] -нон-7-ил-окси- метил] -4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифеиилметиловый эфир
Промежуточное соединение 89б (250 мг) и имидазол (каталитическое количество) в атмосфере азота растворяли в сухом тетрагидрофуране (10 мл). Добавляли гидрид натрия (45 мг) и перемешивали суспензию при комнатной температуре в течение 20 минут. Добавляли сероуглерод (140 мкл), перемешивание продолжали в течение 20 минут и добавляли метилиодид (150 мкл). Спустя 2 часа реакцию гасили добавлением по каплям этилацетата (10 мл), а затем воды (5 мл). Смесь фракционировали между этилацетатом (50 мл) и водой (50 мл), органический слой промывали водой и рассолом, а затем сушили и выпаривали. Остаток очищали посредством хроматографии быстрого разделения на силикагеле, элюируя смесью гексан: этилацетат от (10:1) до (8:1), получая в результате соединение, указанное в заголовке (211 мг).
δ (1H, CDCl3): 9,72 (s, 1H, CHO), 7,46-7,22 (m, 10H, 2Ph), 6,97 (s, 1H, 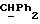 ), 6,02 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,80 (dd, 1H, H-6', J=2,1 и J= 6,3 Гц), 4,72 (d, 1H, H-7', J=2,1 Гц), 4,05 (dd, 1H, Ha-3', J=7,2 и 8,4 Гц), 3,98-3,86 (m, 2H, 8aCHa и H1'), 3,66-3,55 (m, 2H, 8aCHb и H-9'), 3,34 (t, 1H, Hb-3', J=8,4 Гц), 2,73 (t, 1H, H1, J-3,9 Гц), 2,59 (s, 3H, SCH3).
), 6,02 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,80 (dd, 1H, H-6', J=2,1 и J= 6,3 Гц), 4,72 (d, 1H, H-7', J=2,1 Гц), 4,05 (dd, 1H, Ha-3', J=7,2 и 8,4 Гц), 3,98-3,86 (m, 2H, 8aCHa и H1'), 3,66-3,55 (m, 2H, 8aCHb и H-9'), 3,34 (t, 1H, Hb-3', J=8,4 Гц), 2,73 (t, 1H, H1, J-3,9 Гц), 2,59 (s, 3H, SCH3).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 98
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Аллил-3-иодо-2,3,6-тридезокси- β- D- глюкопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a, 8,8a-oктaгидpo-7-мeтил-3-(1-мeтилэтил)-1,4-мeтaнo-s-индaцeн-3a (1H)-карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 12 (67 мг), трифенилфосфина (105 мг) и имидазола (28 мг) в сухом тетрагидрофуране (5 мл) обрабатывали небольшими порциями иода (51 мг). Смесь перемешивали при комнатной температуре в течение 1,5 часов и фракционировали между этилацетатом (30 мл) и 1N водной соляной кислотой (30 мл). Органический слой промывали последовательно 1N водной соляной кислотой, водой, водным раствором метабисульфита натрия, водой и рассолом, затем сушили, фильтровали и концентрировали. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью гексан:этилацетат (9: 1) и получая в результате соединение, указанное в заголовке (68 мг).
δ 1H, CDCl3): 9,73 (s, 1H, CHO), 7,45-7,22 (m, 10H, 2Ph), 6,97 (s, 1H, 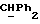 ), 6,07-5,9 (m, 2H, H-2 и
), 6,07-5,9 (m, 2H, H-2 и 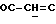 ), 5,37-5,14 (m, 2H,
), 5,37-5,14 (m, 2H, 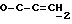 ), 4,45-4,35 (m, 1H,
), 4,45-4,35 (m, 1H, 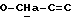 ), 4,25-4,1 (m, 2H, H-1' и
), 4,25-4,1 (m, 2H, H-1' и 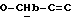 ), 4,1-3,95 (m, 2H, H-3' и 8a-CHa), 3,66 (d, 1H, 8a-CHb, J=9 Гц), 3,29 (dq, 1H, H-5', J= 6 и 8,7 Гц), 3,15 (dd, 1H, H-4', J=8,7 и 9,9 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц).
), 4,1-3,95 (m, 2H, H-3' и 8a-CHa), 3,66 (d, 1H, 8a-CHb, J=9 Гц), 3,29 (dq, 1H, H-5', J= 6 и 8,7 Гц), 3,15 (dd, 1H, H-4', J=8,7 и 9,9 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 99
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(2,3-Ангидро-6-дезокси- β- D-маннопиранозилокси)метил-4-(1,3-диоксолан-2-ил)-4,4a, 5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор промежуточного соединения 67 (3,2 ммоль) в сухом ацетонитриле (40 мл) обрабатывали этиленгликолем (39 мл), триметилортоформиатом (1,1 мл) и п-толуолсульфоновой кислотой (30 мг). Спустя 5 часов смесь тщательно экстрагировали этилацетатом. После удаления растворителя остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан: этилацетат 3:1 и получая в результате соединение, указанное в заголовке (2,08 г).
δ (1H, CDCl3): 7,43 и 7,29 (m,m, 4H, 6H, 2xPh), 6,95 (s, 1H, 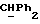 ), 5,86 (d, 1H, H-2, J=3,3 Гц), 5,08 (s, 1H, 4-CH), 4,69 (s, 1H, H-1'), 4,11 (d, 1H, 8a-CHa, J=9,3 Гц), 3,81 (m, 5H, 8a-CHb и
), 5,86 (d, 1H, H-2, J=3,3 Гц), 5,08 (s, 1H, 4-CH), 4,69 (s, 1H, H-1'), 4,11 (d, 1H, 8a-CHa, J=9,3 Гц), 3,81 (m, 5H, 8a-CHb и 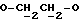 ), 3,63 (m, 1H, H-5'), 3,23 и 3,15 (d,d, 1H, 1H, H-2' и H-3', J=3,6 Гц), 3,20 (m, 1H, H-4'), 2,63 (m, 2H, H-1 и
), 3,63 (m, 1H, H-5'), 3,23 и 3,15 (d,d, 1H, 1H, H-2' и H-3', J=3,6 Гц), 3,20 (m, 1H, H-4'), 2,63 (m, 2H, H-1 и 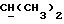 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 100
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(2,3-Ангидро-4-оксо-6-дезокси- β- D-маннопиранозилокси)метил-4-(1,3-диоксолан-2-ил)- 4,4a,5,6,7,7a,8,8а-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор трифторуксусного ангидрида (230 мкл) в сухом дихлорметане (10 мл) при -60oC обрабатывали по каплям диметилсульфоксидом (138 мкл) и раствором Промежуточного соединения 99 (0,9 ммоль) в сухом дихлорметане (10 мл). Спустя 60 минут добавляли триэтиламин (554 мкл) и смесь перемешивали при -20oC в течение 2 часов. Затем ее разбавляли дихлорметаном и промывали водой. После удаления растворителя остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат 4:1 и получая в результате соединение, указанное в заголовке (214 мг).
δ (1H, CDCl3): 7,44 и 7,30 (m,m, 4H, 6H, 2xPh), 6,94 (s, 1H, 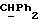 ), 5,87 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,10 (s, 1H, 4-CH), 4,81 (s, 1H, H-1'), 4,09 (d, 1H, 8a-CHa, J=8,7 Гц), 3,88 (m, 5H, 8a-CHb и
), 5,87 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,10 (s, 1H, 4-CH), 4,81 (s, 1H, H-1'), 4,09 (d, 1H, 8a-CHa, J=8,7 Гц), 3,88 (m, 5H, 8a-CHb и 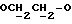 ), 3,58 и 3,36 (d,d, 1H, 1H, H-2' и H-3', J=3,9 Гц), 2,65 (m, 2H, H-1 и
), 3,58 и 3,36 (d,d, 1H, 1H, H-2' и H-3', J=3,9 Гц), 2,65 (m, 2H, H-1 и 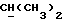 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 101
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(2,3-Ангидро-4,6-дидезокси-4- метилен- β- D-маннопиранозилокси)метил-4-(1,3-диоксолан-2- ил)-4,4a,5,6,7,7a, 8,8a-oктaгидpo-7-метил-3-(1-мeтилэтил)-1, 4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Раствор метилтрифенилфосфонийбромида (0,3 мноль) в сухом тетрагидрофуране (10 мл) в атмосфере азота при -78oC обрабатывали н-Бутиллитием 2,44М (0,15 мл). Через 15 минут медленно добавляли раствор Промежуточного соединения 100 (0,21 ммоль) в сухом тетрагидрофуране (5 мл), затем смесь медленно нагревали до комнатной температуры. Смесь разбавляли этилацетатом и промывали хлоридом аммония (IN) и рассолом. После удаления растворителя остаток очищали Посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат 4:1 и получая в результате соединение, указанное в заголовке (91 мг).
δ (1H, CDCl3): 7,45 и 7,29 (m,m,4 H,6 H, 2xPh), 6,95 (s, 1H, 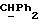 ), 5,87 (m, 1H, H-2), 5,35 и 5,22 (d,d, 1H, 1H, CH2=C, J=2,4 Гц), 5,08 (s, 1H, 4-CH), 4,68 (s, 1H, H-1'), 4,10 (m, 2H, 8a-CHa и H-5'), 3,82 (m, 5H, 8a-CHb и
), 5,87 (m, 1H, H-2), 5,35 и 5,22 (d,d, 1H, 1H, CH2=C, J=2,4 Гц), 5,08 (s, 1H, 4-CH), 4,68 (s, 1H, H-1'), 4,10 (m, 2H, 8a-CHa и H-5'), 3,82 (m, 5H, 8a-CHb и 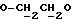 ), 3,57 и 3,35 (d,d, 1H, 1H, H-2' и H-3', J=3,9 Гц), 2,63 (m, 2H, H-1 и
), 3,57 и 3,35 (d,d, 1H, 1H, H-2' и H-3', J=3,9 Гц), 2,63 (m, 2H, H-1 и 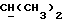 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 102
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(2,3-Ангидро-6-дезокси- β- D-талопиранозилокси) метил-4-(1,3-диоксолан-2-ил)-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 100 (1,13 ммоль) в сухом тетрагидрофуране (45 мл) в атмосфере азота при -78oC медленно добавляли L-селектрид 1N (2 мл). Спустя 1 час смесь разбавляли этилацетатом (100 мл), нагревали до комнатной температуры и промывали хлоридом аммония (1N), водой и рассолом. После удаления растворителя остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан:этилацетат 3:1 и получая в результате соединение, указанное в заголовке (431 мг).
δ (1H, CDCl3): 7,44 и 7,31 (m,m, 4H, 6H, 2xPh), 6,95 (s, 1H, 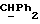 ), 5,85 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 4-CH), 4,92 (s, 1H, H-1'), 4,11 (d, 1H, 8a-CHa, J=9,3 Гц), 3,83 (m, 5H, 8a-CHb и
), 5,85 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 4-CH), 4,92 (s, 1H, H-1'), 4,11 (d, 1H, 8a-CHa, J=9,3 Гц), 3,83 (m, 5H, 8a-CHb и 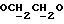 ), 3,62 (m, 1H, H-4'), 3,54 (dd, 1H, H-3', J=3,6 и 5,1 Гц), 3,40 (m, 1H, H-5'), 3,24 (d, 1H, H-2, J=3,9 Гц).
), 3,62 (m, 1H, H-4'), 3,54 (dd, 1H, H-3', J=3,6 и 5,1 Гц), 3,40 (m, 1H, H-5'), 3,24 (d, 1H, H-2, J=3,9 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 103
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(2,3-Ангидро-4-О-третбутил-карбонил-6- дезокси- β- D-талопиранозилокси)метил-4-(1,3-диоксолан- 2-ил)-4,4a, 5,6,4,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3a(1H)-карбоновой кислоты дифенилметиловый эфир
Смесь Промежуточного соединения 102 (0,2 ммоль) и 4-диметиламинопиридина (0,4 ммоль) в сухом дихлорметане (15 мл) обрабатывали хлорангидридом триметилуксусной кислоты (0,3 ммоль). Спустя 30 минут промывали водой и очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан: этилацетат 5:1 и получая в результате соединение, указанное в заголовке (95 мг).
δ (1H, CDCl3): 7,44 и 7,30 (m,m, 4H, 6H, 2xPh), 6,95 (s, 1H, 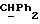 ), 5,85 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 4-CH), 4,82 (t, 1H, H4', J= 3,9 Гц), 4,60 (s, 1H, H-1'), 4,12 (d, 1H, 8a-CHa, J=9,6 Гц), 3,83 (m, 5H, 8a-CHb и
), 5,85 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 4-CH), 4,82 (t, 1H, H4', J= 3,9 Гц), 4,60 (s, 1H, H-1'), 4,12 (d, 1H, 8a-CHa, J=9,6 Гц), 3,83 (m, 5H, 8a-CHb и 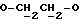 ), 3,65 (m, 1H, H-5'), 3,53 (t, 1H, H-3', J = 4,2 Гц), 3,17 (d, 1H, H-2", J=4,2 Гц), 2,63 (m, 2H, H-1 и
), 3,65 (m, 1H, H-5'), 3,53 (t, 1H, H-3', J = 4,2 Гц), 3,17 (d, 1H, H-2", J=4,2 Гц), 2,63 (m, 2H, H-1 и  ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 104
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-(4-О-Аллил-2,3-ангидро-6-дезокси- β- D-талопиранозилокси)метил-4-(1,3-диоксолан-2-ил)-4,4a, 5,6,7,7a, 8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 102 (0,3 ммоль) в сухом тетрагидрофуране (10 мл) в атмосфере азота добавляли гидрид натрия (0,6 ммоль) и каталитическое количество тетрабутиламмонийиодида. Спустя 30 минут добавляли аллилбромид (0,9 ммоль) и перемешивали смесь в течение ночи при комнатной температуре. Затем ее разбавляли этилацетатом (10 мл) и промывали хлоридом аммония (1N) и рассолом. После удаления растворителя остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь гексан: этилацетат 4:1 и получая в результате соединение, указанное в заголовке (163 мг).
δ (1H, CDCl3): 7,44 и 7,29 (m, m, 4H, 6H, 2x Ph), 6,95 (s, 1H,  ), 5,96 (m, 1H, H-2''), 5,85 (dd, 1H, H-2, J = 1, 2 и 3,6 Гц), 5,30 и 5,12 (m, m, 1H, 1H, CH2-3''), 5,06 (s, 1H, 4-CH), 4,58 (s, 1H, H-1'), 4,31 (m, 1H, H-4'), 4,10 (m, 2H, H-5' и 8a-CHa), 3,83 (m, 5H, 8a-CHb и
), 5,96 (m, 1H, H-2''), 5,85 (dd, 1H, H-2, J = 1, 2 и 3,6 Гц), 5,30 и 5,12 (m, m, 1H, 1H, CH2-3''), 5,06 (s, 1H, 4-CH), 4,58 (s, 1H, H-1'), 4,31 (m, 1H, H-4'), 4,10 (m, 2H, H-5' и 8a-CHa), 3,83 (m, 5H, 8a-CHb и 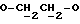 ), 3,53 (m, 2H, CH2O), 4,23 (t, 1H, H-3', J=4,2 Гц), 3,19 (d, 1H, H-2', J = 4,2 Гц), 2,65 (t, 1H, H-1, J = 4,2 Гц), 2,61 (n, 1H,
), 3,53 (m, 2H, CH2O), 4,23 (t, 1H, H-3', J=4,2 Гц), 3,19 (d, 1H, H-2', J = 4,2 Гц), 2,65 (t, 1H, H-1, J = 4,2 Гц), 2,61 (n, 1H, 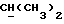 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 105
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-(2,3-Ангидро-6-дезокси-4-O-тозил β- D-талопиранозилокси)метил-4-(1,3-диоксолан-2-ил)-4,4a,5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)
-карбоновой кислоты дифенилметиловый эфир
Раствор полученного соединения 102 (0,45 моль) в сухом дихлорметане (7 мл) обрабатывали 4-диметиламинопиридином (1,32 ммоль) и тозилхлоридом (0,9 ммоль) и перемешивали в течение ночи при комнатной температуре. Затем ее промывали водой и рассолом. После удаления растворителя остаток очищали посредством хроматографии, применяя в качестве элюента смесь гексан:этилацетат 3: 1. Соответствующие фракции выпаривали, получая в результате соединение, указанное в заголовке (277 мг).
δ (1H CDCl3) : 7,86, 7,42 и 7,29 (m, m, 10H, Ar), 6,93 (s, 1H,  ), 5,82 (m, 1H, H-2), 5,06 (s, 1H, 4-CH), 4,68 (t, 1H, H-3', J = 4,2 Гц), 4,55 (s, 1H, H-1'), 4,07 (d, 1H, 8a-CH2, J = 8,7 Гц), 3,81 (m, 5H, 8a-CHb и
), 5,82 (m, 1H, H-2), 5,06 (s, 1H, 4-CH), 4,68 (t, 1H, H-3', J = 4,2 Гц), 4,55 (s, 1H, H-1'), 4,07 (d, 1H, 8a-CH2, J = 8,7 Гц), 3,81 (m, 5H, 8a-CHb и 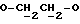 ), 3,62 (m, 1H, H-5'), 3,30 (t, 1H, H-2', J = 4,2 Гц), 3,15 (d, 1H, H-4', J = 3,9 Гц), 2,61 (m, 2H, H-1 и
), 3,62 (m, 1H, H-5'), 3,30 (t, 1H, H-2', J = 4,2 Гц), 3,15 (d, 1H, H-4', J = 3,9 Гц), 2,61 (m, 2H, H-1 и 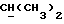 ).
).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 106
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-(2,3-Ангидро-6-дезокси-4-азидо-4,6-дидезокси- β- D-маннопиранозилокси)метил-4-(1,3-диоксолан-2-ил)-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
Раствор Промежуточного соединения 105 (0,15 ммоль) и азида лития (0,45 ммоль) в сухом диметилформамиде (3 мл) прогревали в течение ночи при 100oC. реакционную смесь разбавляли этилацетатом (20 мл) и промывали водой и рассолом. После удаления растворителя остаток очищали посредством хроматографии, применяя в качестве элюента смесь гексан:этилацетат. Соответствующие фракции упаривали, получая в результате соединение, указанное в заголовке (53 мг).
δ (1H CDCl3): 7,46 - 7,23 (m, 10H, Ph2), 6,94 (s, 1H, 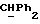 ), 5,86 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 4-CH), 4,67 (s, 1H, H-1'), 4,10 (d, 1H, 8a-CHa, J = 8,7 Гц), 3,83 (m, 4H, 8a-CHb и
), 5,86 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,08 (s, 1H, 4-CH), 4,67 (s, 1H, H-1'), 4,10 (d, 1H, 8a-CHa, J = 8,7 Гц), 3,83 (m, 4H, 8a-CHb и 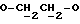 ), 3,40 (d, 1H, H-4', J = 9,3 Гц), 3,31 и 3,15 (d,d,H-2' и h-3', J = 3,6 Гц), 3,23 (m, 1H, H-5'), 2,65 (m, 2H, H и
), 3,40 (d, 1H, H-4', J = 9,3 Гц), 3,31 и 3,15 (d,d,H-2' и h-3', J = 3,6 Гц), 3,23 (m, 1H, H-5'), 2,65 (m, 2H, H и 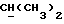 ).
).
ПРОМЕЖУТОСНОЕ СОЕДИНЕНИЕ 107
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-[(4-O-Аллил,2,3-ангидро-6-дезокси- β- D-маннопиранозил)оксиметил] -4-формил-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
СПОСОБ А
К раствору Промежуточного соединения 21 (650 мг) в 50 мл сухого ТГФ при 0oC добавляли 90 мл гидрида натрия. Смесь перемешивали в атмосфере при 0oC в течение 30 минут. Затем добавляли 875 мг аллилбромида и каталитическое количество тетрабутиламмонийиодида (50 мг). Реакция заканчивалась после 5 часов перемешивания при комнатной температуре. Сырой продукт обрабатывали хлоридом аммония 1N и этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным MgSO4 и упаривали до сухого состояния. Остаток хроматографировали на колонке с силикагелем, применяя в качестве элюента смесь н-гексан:этилацетат 7:1 и получая в результате 590 мг соединения, указанного в заголовке, в виде белой пены.
СПОСОБ Б
К раствору Промежуточного соединения 53 (3 г) в сухом метиленхлориде (200 мл) при комнатной температуре добавляли 2,35 г диметиламинопиридина. Затем при перемешивании медленно добавляли раствор 3,66 г тозилхлорида в 100 мл метиленхлорида. Смесь перемешивали в течение 3 суток при комнатной температуре до окончания реакции. Сырой продукт промывали 1N соляной кислотой, насыщенным раствором бикарбоната натрия и рассолом. Органический слой сушили над безводным сульфатом магния, фильтровали и обрабатывали 20 мл раствора метолксила в метаноле (25 вес.%). Эту смесь перемешивали при комнатной температуре в течение 1 суток. Сырой продукт обрабатывали 1N соляной кислотой и промывали водой и рассолом. Органический слой сушили над безводным сульфатом магния и упаривали. Остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь н-гексан:этилацетат 8:1 и получая в результате соединение, указанное в заголовке, в виде белой пены (общий выход 45%).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,99 (s, 1H, 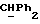 ), 6,08 (dd, 1H, H-12), J = 1,2 и 3,3 Гц), 5,90 (m, 1H,
), 6,08 (dd, 1H, H-12), J = 1,2 и 3,3 Гц), 5,90 (m, 1H, 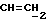 ), 5,28 (m, 2H,
), 5,28 (m, 2H, 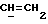 ), 4,69 (d, 1H, H-1, J = 1,2 Гц), 4,23 (m, 1H, H-5'), 4,11 (d, 1H, H-19a, J = 9 Гц), 4,06 (m, 1H, H-4'), 3,79 (d, 1H, H-19b, J = 9 Гц), 3,26 (d, 1H, H-2', J = 3,9 Гц), 3,12 (m, 2H,
), 4,69 (d, 1H, H-1, J = 1,2 Гц), 4,23 (m, 1H, H-5'), 4,11 (d, 1H, H-19a, J = 9 Гц), 4,06 (m, 1H, H-4'), 3,79 (d, 1H, H-19b, J = 9 Гц), 3,26 (d, 1H, H-2', J = 3,9 Гц), 3,12 (m, 2H, 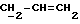 ), 3,13 (d, 1H, H-3', J = 3,9 Гц), 2,86 (t, 1H, H-11, J = 3,6 Гц).
), 3,13 (d, 1H, H-3', J = 3,9 Гц), 2,86 (t, 1H, H-11, J = 3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 108
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-[2,3-ангидро-6-дезокси-4-O-(2,3-дигидроксипропил) β- D-маннопиранозил)оксиметил] -4-формил-4,4a, 5,6,7,7a, 8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
К раствору Промежуточного соединения 107 (2 г) в 50 мл ацетона и 5 мл при 0oC добавляли N-оксид-N-метилморфолина. К этой смеси медленно добавляли 0,2 мл раствора четырехоксида осмия (2,5 вес.% в изобутаноле). Гидроксилирование GM 218045 завершалось после перемешивания в течение 3 суток при комнатной температуре. Сырой продукт обрабатывали гидросульфидом натрия, фильтровали через броунмиллерит и упаривали до сухого состояния. Остаток растворяли в этилацетате и промывали водой и рассолом. Органический слой сушили над безводным сульфатом магния, упаривали до сухого состояния и хроматографировали на колонке с силикагелем в системе н-гексан:этилацетат, 1: 1 и метиленхлорид:метанол, 50:1. Соединение, указанное в заголовке (смесь энантиомера 50:50) получали в виде белой пены (1,75 г).
δ 1H CDCl3): 9,75 (s, 1H, CHO), 6,99 (s, 1H, 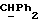 ), 6,08 (dd, 1H, H-12, J = 1,5 и 3,6 Гц), 4,69 (s, 1H, H-1'), 4,10 (d, 1H, H-19a, J = 9,3 Гц), 3,90 - 3,58 (m, 5H,
), 6,08 (dd, 1H, H-12, J = 1,5 и 3,6 Гц), 4,69 (s, 1H, H-1'), 4,10 (d, 1H, H-19a, J = 9,3 Гц), 3,90 - 3,58 (m, 5H, 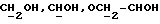 в 4'), 3,79 (d, 1H, H-19d, J = 9,3 Гц), 3,27, 3,13 (d,d 1H, 1H, H-2' и H-3', J = 3,9 Гц), 3,28 - 3,22 (m, 2H, H-4' и H-5'), 2,85 (t, 1H, H-11, J = 3,6 Гц).
в 4'), 3,79 (d, 1H, H-19d, J = 9,3 Гц), 3,27, 3,13 (d,d 1H, 1H, H-2' и H-3', J = 3,9 Гц), 3,28 - 3,22 (m, 2H, H-4' и H-5'), 2,85 (t, 1H, H-11, J = 3,6 Гц).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 109
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-[(2,3-ангидро-6-дезокси-4-O-[(2,3-O-изопропилиден)-2,3-дигидроксипропил] - β- D-маннопиранозил)оксиметил]-4-формил-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновой кислоты дифенилметиловый эфир
К раствору промежуточного соединения 108 (350 мг) в сухом ацетоне (20 мл) добавляли 250 мкл 2,2-диметоксипропана и каталитическое количество п-толуолсульфоновой кислоты (30 мг). Смесь перемешивали при комнатной температуре в течение 2 часов в атмосфере азота. Сырой продукт упаривали до сухого состояния, а остаток снова растворяли в этилацетате (70 мл) и промывали водой и рассолом. Органический слой концентрировали и очищали на колонке с силикагелем в системе н-гексан:этилацетат 6:1, получая в результате соединение, указанное в заголовке (смесь изомеров 50:50), в виде белой пены (290 мг, выход 80%).
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 6,99 (s, 1H, 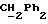 ), 6,08 (dd, 1H, H-12, J = 0,9 и 3,3 Гц), 4,67 (d, 1H, H-1', J = 2,1 Гц), 4,32 - 4,22 (m, 1H,
), 6,08 (dd, 1H, H-12, J = 0,9 и 3,3 Гц), 4,67 (d, 1H, H-1', J = 2,1 Гц), 4,32 - 4,22 (m, 1H, 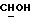 в 4'), 4,12 - 4,03 (m, 2H, H-19a и
в 4'), 4,12 - 4,03 (m, 2H, H-19a и 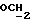 в 4'), 3,82 - 3,51 (m, 4H, H-19b,
в 4'), 3,82 - 3,51 (m, 4H, H-19b, 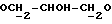 ), 3,29 и 3,12 (d,d, 1H, 1H, H-2' и H-3', J = 3,9 Гц), 3,26 - 3,20 (m, 2H, H-4' и H-5'), 2,85 (t, 1H, H-11, J = 3,9 Гц), 1,42 и 4,36 (d,d, 3H, 3H
), 3,29 и 3,12 (d,d, 1H, 1H, H-2' и H-3', J = 3,9 Гц), 3,26 - 3,20 (m, 2H, H-4' и H-5'), 2,85 (t, 1H, H-11, J = 3,9 Гц), 1,42 и 4,36 (d,d, 3H, 3H 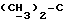 изопропилиден).
изопропилиден).
ПРИМЕР 1
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-[(6-дезокси-3,4-O-изопропилиден)- β- D-маннопиранозил)метил] -4-формил-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H) -карбоновая кислота
К суспензии 10%-ного палладия на угле (80 мг) в этилацетата (10 мл) в атмосфере азота добавляли раствор Промежуточного соединения 2 (180 мг) в этилацетате (10 мл) и смесь гидрогенизировали при комнатной температуре при давлении водорода 30 пси в течение 1 часа. Катализатор отфильтровывали, растворитель упаривали до сухого состояния. После хроматографии быстрого разделения на силикагеле остатка с элюцией смесью дихлорметан:метанол (20:1) получали соединение, указанное в заголовке (128 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J = 1,5 и 3,3 Гц), 4,61 (d, 1H, H-1', J = 2,1 Гц), 4,30 (dd, 1H, H-3', J = 3,6 и 5,4 Гц), 4,04 (d, 1H, 8aCH2, J = 9,3 Гц), 3,95 (dd, 1H, H-2', J = 2,1 и 3,6 Гц), 3,85 (dd, 1H, H-4', J = 5,4 и 9,0 Гц), 3,66 (d, 1H, J = 9,3 Гц), 3,49 (dg, 1H, H-5', Jd = 9,0 Гц, Jq = 6,3 Гц), 2,69 (t, 1H, H-1, J=3,6 Гц); δ (13C, CDCl3): 204,8(CHO), 175,4 (CO2H), 148,3 (C-3), 130,7 (C-2), 109,3 (Cq.изоп), (98,9(C-1'), 76,9(C-4'), 75,8(C-2'), 74,2 (8aCH2), 71,7(C-3'), 68,6(C-5'), 65,6(C-8a), 58,9(C-4).
ПРИМЕР 2
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-изопропилиден- β- D-альтропиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a,-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты натриевая соль
Соединение из примера 1 (246 мг) растворяли в диоксане (2 мл) и добавляли 0,5 М бикарбоната натрия (0,99 мл). Раствор перемешивали в течение 15 минут и сушили замораживанием, получая в результате соединение, указанное в заголовке (256 мг).
δ (1H, CDCl3): 9,88 (s, 1H, CHO), 5,95 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,51 (d, 1H, H-1', J=1,8 Гц), 4,26 (dd, 1H, H-3', J=4,8 и 6,0 Гц), 4,01 (d, 1H, 8aCH2, J=9,6 Гц), 3,87-3,80 (m, 2H, H-2' и H-4'), 3,74 (d, 1H, 8aCH2, J = 9,6 Гц), 3,50-3,42 (m, 1H, H-5'), 2,66 (t, 1H, H-1, J=3,9 Гц).
ПРИМЕР 3
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-(2-пентилиден)- β- D-альтропиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a-(1H)-карбоновая кислота
К суспензии 10%-ного палладия на угле (30 мг) в этилацетате (10 мл) в атмосфере азота добавляли раствор Промежуточного соединения 3 (140 мг) в этилацетате и гидрогенизировали полученную смесь при давлении водорода 30 пси в течение 45 минут. Катализатор отфильтровывали, а раствор упаривали до сухого состояния. После хроматографии быстрого разделения на силикагеле с применением в качестве элюента смеси дихлорметан:метанол (30:1) получали соединение, указанное в заголовке (75 мг), в эпимерном соотношении 26:74.
δ (1H, CDCl3), 9,82 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J-1,5 и 3,3 Гц), 4,60 (d, 1H, H-1', J=1,8 Гц), 4,30 (dd, 1H, H-3', J = 4,2 и 6,0 Гц), 3,98 (d, 1H, 8aCH2, J= 9,3 Гц), 3,94 (dd, 1H, H-2', J=1,8 и 4,2 Гц), 3,87 (dd, 1H, H-4', J=6,0 и 9,3 Гц), 3,65 (d, 1H, 8aCH2, J=9,3 Гц), 3,47 (dq, 1H, H-5', Jd= 9,3 Гц, Jq= 6,3 Гц), 2,70 (t, 1H, H-1, J=3,9 Гц); δ (13C, CDCl3); 204,5 (CHO), 176,5 (CO2H), 148,2 (C-3), 130,6 (C-2), 110,8 (Cq ацетал), 98,8 (C-1'), 76,6 (C-4'), 75,6 (C-2'), 73,9 (8aCH2), 71,3 (C-3'), 68,9 (C-5'), 65,5 (C8a), 58,8 (C-4).
ПРИМЕР 4
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-(4-метокси-2-бутилиден)- β- -D-альтропиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К суспензии 10%-ного палладия на угле (100 мг) в этилацетате (15 мл) добавляли раствор Промежуточного соединения 4 (260 мг) в этилацатате (10 мл) и гидрогенизировали полученную смесь при давлении азота 30 пси в течение 45 минут при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан:метанол (30:1) и получая в результате соединение, указанное в заголовке (172 мг).
δ (1H CDCl3): 9,71 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,60 (d, 1H', J = 2,4 Гц), 4,31 (dd, 1H, H-3', J = 4,2 и 6,0 Гц), 3,98 (d, 1H, 8aCH2, J= 9.3 Гц), 3,94 (dd, 1H, H-2', J=2,4 и 4,2 Гц), 3,88 (dd, 1H, H-4', J=6,0 и 9,3 Гц), 3,65 (d, 1H, 8aCH2, J=9,3 Гц), 3,50 (t, 1H, CH2O, J= 7,2 Гц), 3,50-3,40 (m, 1H, H-5'), 2,69 (t, 1H, H-1, J=3,9 Гц); δ (13C, CDCl3): 204,5 (CHO), 176,2 (CO2H), 148,2 (C-3), 130,6 (C-2), 109,8 (C-2''), 98,8 (C-1'), 76,7 (C-4'), 75,7 (C-2'), 73,8 (8aCH2), 71,2 (C-2'), 68,8 (C-5'), 68,4 (CH2O), 65,6 (C8a), 58,9 (C-4), 58,5 (CH3O).
ПРИМЕР 5
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-Дезокси-3,4-O-циклопентилиден- β- D-альтропиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метил-3-(1-метилэтил)-1,4-метано-s-метано-s-индацен-3a-(1H)- карбоновая кислота
К суспензии 10%-ного палладия на угле (50 мг) в этилацетате (10 мл) добавляли раствор Промежуточного соединения 5 (232 мг) в этилацетате и полученную смесь гидрогенизировали при комнатной температуре при давлении водорода 35 пси в течение 1,5 часов. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан:метанол (30:1) и получая в результате соединение, указанное в заголовке (67 мг).
δ (1H, CDCl3): 9,71 (s, 1H, CHO), 6,07 (dd, 1H, H-2, J=1,5 и 3,3 Гц), 4,59 (d, 1H, H-1', J=2,1 Гц), 4,17 (dd, 1H, H-3', J=3,6 и 5,4 Гц), 4,54 (d, 1H, 8aCH2, J= 9,3 Гц), 3,97 (dd, 1H, H-2', J=2,1 и 3,6 Гц), 3,81 (dd, 1H, H-4', J=5,4 и 9,0 Гц), 3,70 (d, 1H, 8aCH2, J=9,3 Гц), 3,44 (dq, 1H, H-5', Jd = 9,0 Гц, Jq = 6,3 Гц), 2,69 (t, 1H, H-1, J=3,3 Гц).
ПРИМЕР 6
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1,6-Дидезокси-3,4-O-изопропилиден- β- D-аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a-(1H)-карбоновая кислота
К раствору Промежуточного соединения 7 (1,2 г) в этилацетате (60 мл) в атмосфере азота порциями добавляли 10%-ный палладий на угле (600 мг). Смесь встряхивали в Parr аппарате при давлении водорода 40 пси при комнатной температуре. Катализатор отфильтровывали и промывали дополнительным количеством этилацетата. Растворитель упаривали до сухого состояния, а остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан: метанол (25:1) и получая в результате соединение, указанное в заголовке (0,79 г).
δ (1H, CDCl3): 9,81 (s, 1H, CHO), 6,05 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,63 (dd, 1H, H-1', J=2,4 и 8,4 Гц), 4,38 (dt, 1H, H-3', Jd=3,0 Гц, Jt= 5,1 Гц), 4,29 (d, 1H, 8aCH2, J=9,3 Гц), 3,64 (dd, 1H, H-4', J=5,1 и 9,3 Гц), 4,43 (dq, 1H, H-5', Jd=9,3 Гц), Jq=6,9 Гц), 3,40 (d, 1H, 8aCH2, J= 9.3 Гц), 2,53 (t, 1H, H-1, J= 3,0 Гц); δ (13C, CDCl3): 204,7 (CHO), 174,6 (CO2H), 148,2 (C-3), 130,6 (C-2), 108,9 (C(Ch2)2), 98,3 (C-1'), 76,6 (C-4'), 73,7 (8aCH2), 72,5 (C-3'), 71,7 (C-5'), 67,0 (C8a), 65,2, 59,0 (C-4).
ПРИМЕР 7
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси-3,4-O-(4-метокси-2-бутилиден)- β- D-аллопиранозилокси)метил] -4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К суспензии 10%-ного палладия на угле (300 мг) в этилацетате (10 мл) добавляли раствор Промежуточного соединения 8 (246 мг) в этилацетате (10 мл) и гидрогенизировали полученную смесь при комнатной температуре при давлении водорода 45 пси в течение 1 часа. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали посредством хроматографии быстрого разделения на силикагеле, элюируя дихлорметаном и смесью дихлорметан: метанол (30:1) и получая в результате соединение, указанное в заголовке (182 мг), в виде пены, в эпимерном соотношении 4:1.
δ (1H, CDCl3) сигналы основного компонента: 9,81 (s, 1H, CHO), 6,06 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,62 (dd, 1H, H-1', J=2,7 и 8,1 Гц), 4,41 (dt, 1H, H-3', Jd=3,3 Гц, Jt = 2,5 Гц), 4,24 (d, 1H, 8aCH2, J=9,0 Гц), 3,68 (dd, 1H, H-4', J=5,7 и 9,0 Гц), 3,50 (t, 2H, CH2, J=7,5 Гц), 3.46 - 3,38 (m, 1H, H-5'), 3,33 (s, 3H, CH2O), 2,54 (t, 1H, H-1, J= 4,2 Гц); δ (13C, CDCl3): 204,5 (CHO), 175,1 (CO2H), 148,2 (C-3), 130,7 (C-2), 109,4 (C-2''), 98,3 (C-1'), 76,4 (C-4'), 73,6 (8aCH2), 71,9 (C-3'), 71,4 (C-5'), 68,6 (CH2O), 65,2 (C8a), 58,9 (C-4), 58,6 (CH3O).
ПРИМЕР 8
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси-3,4-O-изопропилиден- β- D-аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты натриевая соль
Соединение из Примера 6 (376 мг) растворяли в диоксане (3 мл) и добавляли 0,5N натрий бикарбонат (1,50 мл). Полученный раствор перемешивали в течение 30 минут при комнатной температуре, а затем сушили замораживанием, получая в результате соединение, указанное в заголовке (392 мг).
δ (1H, CDCl3): 9,84 (s, 1H, CHO), 5,94 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,52 (dd, 1H, H-1', J=2,4 и 9,0 Гц), 4,41 - 4,37 (m, 1H, H-3'), 4,02 (d, 1H, 8aCH2CH, J=9,6 Гц), 3,74 (d, 1H, 8aCH2, J=9,6 Гц), 3,57 (dd, 1H, H-4', J=5,4 и 10,0 Гц), 3,42 (dq, 1H, H-5', Jd=6,3 Гц, Jq=6,3 Гц), 2,56 (t, 1H, H-1, J= 3,6 Гц).
ПРИМЕР 9
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-O-тионокарбонил- β- D-альтропиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-актагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a (1H)-карбоновая кислота
2%-ный раствор трифторуксусной кислоты в дихлорметане (20 мл) добавляли к раствору Промежуточного соединения 9 (0,220 г) в дихлорметане (5 мл) при 0oC. Реакционную смесь перемешивали при 0oC в течение 1 часа, а затем промывали водой (2х20 мл) и сушили (сульфат магния). Растворитель выпаривали, получая в результате остаток, который подвергали хроматографии на колонке с силикагелем, элюируя смесью дихлорметан:метанол (от 40:1 до 20:1) и получая соединение, указанное в заголовке (0,1 г).
δ (1H, CDCl3): 9,68 (s, 1H, CHO), 6,09 (dd, 1H, H-2, J = 1,5 и 3,6 Гц), 4,94 (dd, 1H, H-3', J=3,9 и 7,2 Гц), 4,62 (m, 2H, H-1'), 4,14 (dd, 1H, H-2', J= 2,1 и 3,9 Гц), 4,00 (d, 1H, 8aCH2, JAB=9.3 Гц), 3,72 (d, 1H, 8aCH2, JAB= 9.3 Гц), 3,68 (m, 1H, H-5'), 2,71 (m, 1H, H-1); δ (13C, CDCl3): 204,4 (CHO), 190,2 (CS), 176,4 (CO2H), 148,4 (C-3), 130,6 (C-2), 98,2 (C-1'), 82,3 и 80,1 (C-3' и C-4'), 74,5 (8aCH2), 71,7 (C-3a), 69,2 и 66,4 (C-2' и C-5'), 65,5 (C-8a), 58,8 (C-4), 46,0 (C-1)
ПРИМЕР 10
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,6-Дидезокси-3,4-O-карбонил- β- -D-аллопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 10 (125 мг) в дефлегмирующем сухом толуоле (15 мл) добавляли в атмосфере азота небольшими порциями избыток карбонилдиимидазола. Сразу после завершения реакции (ТСХ контроль) растворитель удаляли при пониженном давлении, а сырую смесь растворяли в дихлорметане (200 мл), промывали 1N соляной кислотой (3х100 мл), рассолом (1x100
мл) и водой (1х100 мл), затем сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате масло, которое применяли без дальнейшей очистки. Масло растворяли в этилацетате (15 мл) и встряхивали (15 мл) в и встряхивали в Parr аппарате при давлении азота 20 пси в присутствии 10%-ного палладия на активированном угле (50 мг) в течение 1 часа при комнатной температуре. После отфильтровывания катализатора и удаления растворителя получали сироп, который очищали посредством препаративной ТСХ (силикагель, метанол: дихлорметан 1: 15), получая в результате соединение, указанное в заголовке (50 мг), выделенное из фракции с Rf 0,5.
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 6,07 (ss, 1H, H-2, J = 1,2 и 3,3 Гц), 4,93 (m, 1H, H-3'), 4,63 (dd, 1H, H-1', J = 2,4 и 7,8 Гц), 4,24 (dd, 1H, H4', J = 6,6 и 9,3 Гц), 4,05 и 3,57 (2d, 2H, 8aCH2, JAB = 9 Гц), 3,63 (m, 1H, H-5'), 2,66 (t, 1H, H-1, J = 3,9 Гц); δ (13C,CDCl3): 204,6 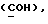 17,6
17,6 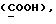 154,0
154,0 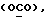 148,2
148,2  130,7
130,7  97,5
97,5 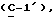 76,6
76,6 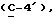 74,7
74,7 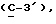 69,9
69,9 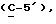 31,7
31,7 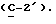
ПРИМЕР 11
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2,6-Дидезокси-3,4-O-тиокарбонил- β- D-аллопираносилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8a-актагидро-7-метил- 3(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 11 (130 мг) в сухом дихлорметане (5 мл) при 0oC добавляли трифторуксусную кислоту (250 мкл). Через 2 часа раствор разбавляли дихлорметаном (50 мл), промывали водой (2х100 мл), сушили над сульфатом магния и концентрировали до сухого состояния, получая в результате масло, которое очищали посредством препаративной ТСХ (силикагель, метанол: дихлорметан 1:15), получая в результате соединение, указанное в заголовке (68 мг), выделенное из фракции с Rf 0,6.
δ (1H, CDCl3): 9,70 (s, 1H, CHO), 6,07 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,08 (m, 1H, H-3'), 4,63 (dd, 1H, H-1', J = 2,7 и 8,4 Гц), 4,45 (dd, 1H, H-4', J = 6,6 и 9,0 Гц), 4,03 и 3,59 (2d, 2H, 8aCH2, J AB = 9,3 Гц), 3,64 (m, 1H, H-5'), 2,67 (t, H-1, J=3,0 Гц); δ (13, CDCl3): 204,5 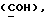 190,9
190,9 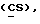 176,3
176,3 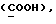 148,2
148,2 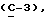 130,7
130,7 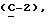 97,5
97,5 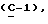 80,3
80,3 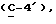 79,6 (C-3'), 69,5(C-5'), 31,3(C-2').
79,6 (C-3'), 69,5(C-5'), 31,3(C-2').
ПРИМЕР 12
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2,6-Дидезокси-3,4-O-метилен- β- D-аллопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил- 3(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
Раствору Промежуточного соединения 10 (450 мг) в дибромметане (10 мл) при интенсивном перемешивании добавляли тетрабутиламмонийбромид (40 мг) и 50%-ный водный гидроксид натрия (100 мл). Спустя 12 часов смесь гасили добавлением 1N соляной кислоты (100 мл) и экстрагировали дихлорметаном (100 мл). Органический слой промывали 1N соляной кислотой (1х100 мл), сушили на сульфатом магния и концентрировали до сухого состояния, получая в результате белое вещество, которое применяли без дальнейшей очистки. Это вещество повторяли в этилацетате (15 мл) и встряхивали в Parr аппарате при давлении водорода 20 пси в присутствии 10%-ного палладия на активированном угле (50 мг) в течение 1 часа при комнатной температуре. После отфильтровывания катализатора и удаления растворителя масляный остаток подвергали хроматографии быстрого разделения на силикагеле с элюцией смесью ацетон:гексан (1:10) и (1: 5), получая в результате соединение, указанное в заголовке (130 мг), в виде масла).
δ (1H, CDCl3): 9,80 (s, 1H, CHO), 6,05 (dd, 1H, H-2, J = 1,2 и 3,3 Гц), 5,12 и 4,86 (2m, 2H, OCH2O), 4,60 (dd, 1H, H-1', J = 2,4 и 8,7 Гц), 4,20 и 3,44 (2d, 2H, 8aCH2, JAB = 9,3 Гц), 4,13 (m, 1H,H-3'), 3,67 (dd, 1H, H-4'), J = 5,1 и 8,7 Гц), 3,38 (m, 1H, H-5'), 2,58 (t, 1H, H-1, J = 3,9 Гц); δ (13C, CDCl3): 204,6 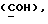 174,8
174,8 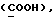 148,3
148,3 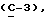 130,6
130,6 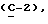 98,1
98,1 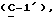 94,9
94,9 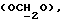 75,4
75,4 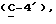 73,9
73,9 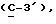 70,3
70,3 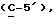 32,3
32,3 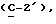
ПРИМЕР 13
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2,6-Дидезокси-3,4-O-метилен- β- D-аллопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил- 3(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 12 (200 мг) в метаноле (5 мл) по каплям добавляли 1,106N раствор гидроксида натрия в воде (3,98 мл). Спустя 30 минут растворитель удаляли при пониженном давлении, а остаток растворяли в диоксане (5 мл) и лиофилизировали, получая соединение, указанное в заголовке (209 мг).
δ (1H, CDCl3): 9,87 (s, 1H, CHO), 6,54 (dd, 1H, H-2, J = 1,5 и 3,6 Гц), 5,10 и 4,82 (2m, 2H, OCH2O), 4,51 (dd, 1H, H-1', J = 2,4 и 8,7 Гц), 4,12 (m, 1H, H-3'), 4,03 и 3,73 (2d, 2H, 8aCH2, JAB = 9,9 Гц), 3,61 (m, 1H, H-4'), 3,35 (m, 1H, H-5'), 2,56 (t, 1H, H-1); δ (13C, CD3OD): 200,2  169,0
169,0  142,9
142,9 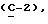 120,6
120,6 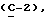 90,3
90,3  86,4
86,4  67,5
67,5 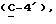 66,2
66,2 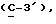 61,8
61,8 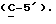
ПРИМЕР 14
(а) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,3R,7R, 9R]-2,5,8-Триокса-3,7-диметил-цис-бицикло[4.4.0] -дек-9-ил-окси-метил] - 4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил- 3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
(б) [1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S, 3R, 7R, 9R]-2,5,8-Триокса-3,7-диметил-цис-бицикло[4.4.0] -дек-9-ил-окси-метил] - 4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил- 3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
Промежуточное соединение 14 (120 мг) в этилацетате (15 мл) встряхивали в Parr аппарате при давлении водорода 20 пси в присутствии 10%-ного палладия на угле (50 мг) в течение 1 часа при комнатной температуре. После отфильтровывания катализатора и выпаривания растворителя при пониженном давлении получали сироп, который подвергали хроматографии быстрого разделения на силикагеле, применяя в качестве элюента смесь ацетон:гексан (1:10), (1:7) и (1:5), получая в результате соединение, указанное в заголовке (а) (35 мг; Rf = 0,5 в системе гексан:ацетон 4:1) и соединение, указанное, в заголовке (б) (40 мг; Rf = 0,4 в системе гексан:ацетон 4:1), в виде бесцветных масел.
(а) δ (1H, CDCl3): 9,80 (s, 1H, CHO), 6,04 (dd, 1H, H-2, J = 1,2 и 3,3 Гц), 4,62 (dd, 1H, H-9', J = 2,1 и 9,9 Гц), 4,20 (m, 2H, 8aCH2 + H-7'), 3,97 (m, 1H, H-1'), 3,73 (m, 1H, 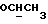 ), 3,40 (m, 2H, 8aCH2 + OCH2), 3,27 (d, 1H, OCH2, JAB = 10,8 Гц), 3,18 (dd, 1H, H-6', J = 3,3 и 10,2 Гц); δ (13C, CDCl3): 204,6
), 3,40 (m, 2H, 8aCH2 + OCH2), 3,27 (d, 1H, OCH2, JAB = 10,8 Гц), 3,18 (dd, 1H, H-6', J = 3,3 и 10,2 Гц); δ (13C, CDCl3): 204,6 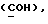 , 174,6
, 174,6 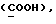 148,3
148,3 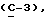 130,6
130,6 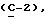 97,9
97,9 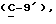 , 73,5
, 73,5 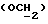 73,2
73,2 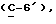 72,1
72,1 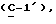 71,9
71,9  65,4
65,4 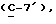 31,9
31,9 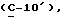 23,7
23,7 
(б) δ (1H, CDCl3): 9,79 (s, 1H, CHO), 6,05 (dd, 1H, H-2, J = 1,5 и 3,6 Гц), 4,73 (dd, 1H, H-9', J = 3,0 и 6,3 Гц), 4,26 (m, 1H, H-1'(1), 4,15 (m, 2H, 8aCH2 + H-7'), 3,98 (m, 1H,  ), 3,79 (dd, 1H, OCH2, J = 3,1 и 11,4 Гц), 3,41 (d, 1H, 8aCH2, J = 9,6 Гц), 3,35 (m, 1H, OCH2, J = 3,1 и 6,3 Гц), 3,27 (dd, 1H, H-6', J = 5,7 и 11,7 Гц), 2,55 (t, 1H, H-1); δ (13C, CDCl3): 204,6
), 3,79 (dd, 1H, OCH2, J = 3,1 и 11,4 Гц), 3,41 (d, 1H, 8aCH2, J = 9,6 Гц), 3,35 (m, 1H, OCH2, J = 3,1 и 6,3 Гц), 3,27 (dd, 1H, H-6', J = 5,7 и 11,7 Гц), 2,55 (t, 1H, H-1); δ (13C, CDCl3): 204,6 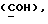 174,6
174,6 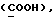 148,3
148,3 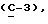 130,6
130,6 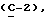 99,2
99,2 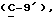 74,7
74,7  73,6
73,6 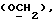 69,3
69,3 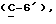 66,3
66,3 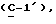 65,4
65,4 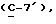 31,9
31,9 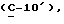 22,6
22,6 
ПРИМЕР 15
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S, 3R,7R, 9R]-2,5,8-Триокса-3,3,7-триметил-цис-бицикло[4.4.0] -дек-9-ил-окси-метил] - 4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил- 3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 15 (200 мг) в этилацетате (15 мл) в атмосфере азота добавляли 10%-ный палладий на угле (50 мг). Смесь встряхивали в Parr аппарате при давлении водорода 20 пси в течение 1 часа при комнатной температуре. После отфильтровывания катализатора и выпаривания растворителя получали остаток, который подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью ацетон: гексан (1:10) и (1:15) и получая в результате соединение, указанное в заголовке (140 мг), в виде белой пены.
δ (1H, CDCl3): 9,81 (s, 1H, CHO), 6,03 (dd, 1H, H-2, J = 1,5 Гц), 4,62 (dd, 1H, H-1', J = 2,1 Гц, J = 9,6 Гц), 4,35 - 4,15 (m, 3H, 8aCH2, H-7' и H-1'), 3,40 (m, 2H, 8aCH2 и OCH2), 3,25 - 3,15 (m, 2H, H-6' и OCH2), 2,52 (t, 1H, H-1), 1,33 и 1,13 (2s, 2CH3); δ (13C, CDCl3): 204,7  174,1
174,1 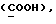 148,3
148,3 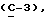 130,6
130,6 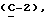 98,0
98,0 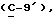 73,4
73,4 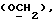 73,3
73,3 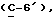 65,1
65,1 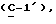 64,7
64,7 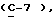 31,9
31,9 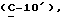 21,1
21,1 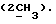
ПРИМЕР 16
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4S,6R, 8R]-2,7-Диокса-4,6-диметил-цис-бицикло[3.4.0] -нон-9-ил-окси-метил] - 4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил- 3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 20(а) (680 мг) в этилацетате (100 мл) в атмосфере азота добавляли 10%-ный палладий на угле (200 мг). Смесь встряхивали в Parr аппарате при давлении водорода 25 пси в течение 2 часов при комнатной температуре. Остаток очищали посредством хроматографии быстрого разделения, элюируя смесью дихлорметан: метанол (49:1) и получая в результате соединение, указанное в заголовке (450 мг), в виде белой пены.
δ (1H, CDCl3): 9,86 (s, 1H, CHO), 6,04 (dd, 1H, H-2, J = 1,5 и 3,6 Гц), 4,57 (dd, 1H, H-8', J = 2,4 и 9,3 Гц), 4,04 (d, 1H, 8a-H2, J = 9,6 Гц), 4,08 (m, 2H, H-1' и CH2-3'(1H)), 3,27 (m, 3H, H-6', CH2-3' (1H), 8a-CH2 (1H)), 2,47 (t, 1H, H-1, J= 3,9 Гц), 1,02 (d, 3H, 4'-CH3, J = 6,6 Гц); δ (13C, CDCl): 204,7 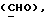 173,8 (COOH), 148,4(C-3), 130,5(C-2), 98,5 (C-8'), 75,2(C-1'), 74,3
173,8 (COOH), 148,4(C-3), 130,5(C-2), 98,5 (C-8'), 75,2(C-1'), 74,3 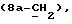 73,3(C-3'), 71,9 (C-6'), 51,2 (C-5'), 36,0 (C-4'), 32,9(C-9'), 19,4 (4'-CH3).
73,3(C-3'), 71,9 (C-6'), 51,2 (C-5'), 36,0 (C-4'), 32,9(C-9'), 19,4 (4'-CH3).
ПРИМЕР 17
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4S,6R, 8R]-2,7-Диокса-4,6-диметил-цис-бицикло[3.4.0] -нон-8-ил-окси-метил] - 4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил- 3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 16 (450 мг) в метаноле (100 мл) по каплям добавляли водный гидроксид натрия (0,099 N, 9,5 мл). Растворитель удаляли при пониженном давлении, а остаток растворяли в воде (80 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (470 мг).
δ (1H, CD3OD): 9,86 (s, 1H, CHO), 5,94 (dd, 1H, H-2, J = 1,5 и 3,6 Гц), 4,43 (dd, 1H, H-8', J = 2,1 и 9,3 Гц), 4,13 - 4,02 (m, 3H, 8a-CH2(1H), H-1', CH2-3'(1H), 3,74 (d, 1H, 8a-CH2, J = 9,9 Гц), 3,34 - 3,23 (m, 2H, CH2-3'(1H), H-6'), 2,56 (t, 1H, H-1, J = 3,9 Гц), 2,34 (m, 1H, H-4'), 0,99 (d, 3H, 4'-CH3, J = 6,9 Гц); δ (13C, CD3OD): 209,7 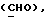 178,4 (COO-Na+), 152,4 (C-3), 130,1 (C-2), 100,1 (C-8'), 77,2 (C-1'), 77,0 (C-3'), 75,3 (8a-CH2), 73,0 (C-6'), 52,8 (C-5'), 37,2 (C-4'), 34,5 (C-9'), 19,6 (4'-CH3).
178,4 (COO-Na+), 152,4 (C-3), 130,1 (C-2), 100,1 (C-8'), 77,2 (C-1'), 77,0 (C-3'), 75,3 (8a-CH2), 73,0 (C-6'), 52,8 (C-5'), 37,2 (C-4'), 34,5 (C-9'), 19,6 (4'-CH3).
ПРИМЕР 18
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4R,6R,8R]-2,7-Диокса-4,6-диметил-цис-бицикло[3.4.0] -нон-8-ил- окси-метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 23(a) (150 мг) в этилацетате (15 мл) в атмосфере азота добавляли 10%-ный палладий на угле (50 мг). Суспензию встряхивали в Parr аппарате при давлении водорода 25 пси в течение 2 часов при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток дважды подвергали хроматографии быстрого разделения на силикагеле, применяя в качестве элюентов последовательно смеси дихлорметан : метанол (49 : 1) и гексан : ацетон (4 : 1) и получая в результате соединение, указанное в заголовке (90 мг), в виде белой пены.
δ (1H CDCl3) : 11,5 (bs, 1H, 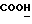 ), 9,87 (s, 1H,
), 9,87 (s, 1H, 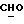 ), 6,04 (dd, 1H, H-2, J = 1,2 и 3,3 Гц), 4,53 (dd, 1H, H-8', J = 2,4 и 9,9 Гц), 4,45 и 3,26 (2d, 2H, 8a-CH2, J = 9,3 Гц), 4,19 (q, 1H, H-1', J = 3,3 Гц), 3,96 (t, 1H, CH2-3, J = 8,4 Гц), 3,58 (m, 1H, H-6'), 3,43 (dd, 1H, CH2-3', J = 10,2 Гц), 2,55 (m, 1H, H04'); δ (13C, CDCl3) : 204,6 (CHO), 174,4 (COOH), 148,3 (C-3), 130,6 (C-2), 97,9 (C-8'), 78,7 (C-1'), 73,3 (C-3'), 72,9
), 6,04 (dd, 1H, H-2, J = 1,2 и 3,3 Гц), 4,53 (dd, 1H, H-8', J = 2,4 и 9,9 Гц), 4,45 и 3,26 (2d, 2H, 8a-CH2, J = 9,3 Гц), 4,19 (q, 1H, H-1', J = 3,3 Гц), 3,96 (t, 1H, CH2-3, J = 8,4 Гц), 3,58 (m, 1H, H-6'), 3,43 (dd, 1H, CH2-3', J = 10,2 Гц), 2,55 (m, 1H, H04'); δ (13C, CDCl3) : 204,6 (CHO), 174,4 (COOH), 148,3 (C-3), 130,6 (C-2), 97,9 (C-8'), 78,7 (C-1'), 73,3 (C-3'), 72,9 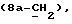 68,7 (C-6'), 46,1 (C-5'), 36,6 (C-4'), 34,02 (C-9'), 21,8 (6'-CH3), 13,6 (4'-CH3).
68,7 (C-6'), 46,1 (C-5'), 36,6 (C-4'), 34,02 (C-9'), 21,8 (6'-CH3), 13,6 (4'-CH3).
ПРИМЕР 19
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4R,7R,9R]-2,8-Диокса- 4,9-диметил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил] -4-фтормил-4, 4a5,6,7,7a, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано- s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 23(a) (150 мг) в этилацетате (15 мл) в атмосфере азота добавляли 10%-ный палладий на угле (50 мг). Смесь встряхивали в Parr аппарате при давлении водорода 20 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Полученный таким образом остаток очищали посредством хроматографии быстрого разделения, элюируя смесью ацетон : гексан (1 : 3), получая в результате соединение, указанное в заголовке (100 мг), в виде кристаллического вещества.
δ (1H, CDCl3) : 9,86 (s, 1H, CHO), 6,05 (dd, 1H, H-2, J = 1,2 и 3,3 Гц), 4,63 (dd 1H, H-7', J = 3,3 и 6,6 Гц), 4,37 (d, 1H, 8aCH2, JAB = 9,6 Гц), 3,98 и 3,67 (2dd, 2H, H-3', J = 6,6 и 8,1 Гц), 3,40 - 3,20 (m, 3H, 8aCH2, H-9' и H-1'), 2,46 (t, 1H, H-1, J = 3,9 Гц), 1,04 (d, 3H, 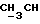 ).
).
ПРИМЕР 20
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4R,7R,9R]-2,8-Диокса-4,9-диметил-цис- бицикло[3.4.0] -7-ил-окси-метил] -4-формил-4,4a, 5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3a(1H)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 19 (100 мг) в метаноле (5 мл) по каплям добавляли гидрокси натрия (0,16 N раствор в воде, 1,88 мл). Спустя 30 минут растворитель удаляли при пониженном давлении, а остаток растворяли в воде (3 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (104 мг).
δ (1H, CD3OD) : 9,88 (s, 1H, CHO), 5,93 (dd, 1H, H-2, J = 1,2 и 3,3 Гц),
4,49 (m, 1H, H-7'), 4,02 (m, 2H, H-3' и 8aCH2), 3,72 (d, 1H, 8aCH2, JAB = 9,3 Гц), 2,56 (t, 1H, H-1, J = 3,6 Гц); δ (13C, CD3OD) : 209,7 (CHO), 178,4 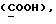 152,4 (C-3), 130,1 (C-2), 99,9 (C-7'), 82,1 (C-1'), 76,5
152,4 (C-3), 130,1 (C-2), 99,9 (C-7'), 82,1 (C-1'), 76,5 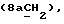 72,2 (C-9'), 37,5 (C-5'), 15,2
72,2 (C-9'), 37,5 (C-5'), 15,2 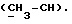
ПРИМЕР 21
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4S,7R,9R]-2,8-Диокса-4,9-диметил-цис-бицикло[3.4.0] -нон-7- ил-окси-метил] -4-формил-4, 4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного сечения 23(б) (260 мг) в этилацетате добавляли 10%-ный палладий на угле (50 мг). Смесь встряхивали в Parr аппарате при давлении водорода 20 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Затем остаток очищали посредством препаративной TCX (силикагель, метанол : дихлорметан 1 : 20), получая в результате соединение, указанное в заголовке (185 мг), в виде масла с Ff = 0,6.
δ (1H, CDCl3) : 9,85 (s, 1H, CHO), 6,04 (dd, 1H, H-2, J = 1,5 т 3,6 Гц), 4,88 (t, 1H, H-7', 2,7 Гц), 4,27 и 3,28 (2d, 2H, 8aCH2, JAB = 9,9 Гц), 3,85 и 3,73 (2m, 2H, OCH2), 3,60 (m, 1H, H-9'), 3,37 (dd, 1H, H-1', J = 7,2 и 8,4 Гц), 2,47 (m, 3H, 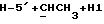 ); δ(13C,CDCl3): 204,4
); δ(13C,CDCl3): 204,4 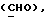 173,7
173,7 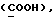 148,2 (C-3), 130,5 (C-2), 98,7 (C-7'), 81,2 (C-1'), 72,8
148,2 (C-3), 130,5 (C-2), 98,7 (C-7'), 81,2 (C-1'), 72,8 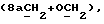 69,9 (C-9'), 25,4 (C-6'), 12,4
69,9 (C-9'), 25,4 (C-6'), 12,4 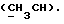
ПРИМЕР 22
[1R-(1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[[1S, 7R,9R]-2,8-Диокса-4,4, 9-триметил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил] -4-формил-4, 4a,5,6,7,7a8,8a-октагидро-7-метил-3-(1-метилэтил)-(1,4-метано-s- индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 24 (100 мг) в этилацетате (40 мл) в атмосфере азота добавляли 10%-ный палладий на угле (50 мг). Смесь встряхивали в Parr встряхивали в Parr аппарате при давлении водорода 25 пси в течение 2 часов при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток дважды подвергали хроматографии быстрого разделения, применяя в качестве элюентов последовательно 1,5%-ный метанол в дихлорметане и 15%-ный ацетон в гексане и получая в результате соединение, указанное в заголовке (40 мг), в виде белой пены.
δ (1H, CDCl3) : 9,85 (s, 1H, CHO), 6,05 (dd, 1H, H-2, J = 1,2 и 3,6 Гц), 4,84 (t, 1H, H-7', J = 3 Гц), 4,23 и 3,29 (2d, 2H, 8a-CH2, J = 9,6 Гц), 3,81 (t, 1H, H-1', J = 8,7 Гц), 3,52 (dq, 1H, H-9', J = 9,3 и 6,3 Гц), 3,50 и 3,42 (2d, 2H, CH2-3', J = 8,4 Гц), 2,48 (t, 1H, H-1, J = 4,2 Гц), 1,09 и 0,98 (2s, 6H, 4'-CH3); δ (13C, CDCl3): 204,5 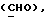 174,0
174,0 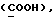 148,2
148,2 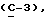 130,5 (C-2), 98,7 (C-7'), 81,01 (C-1'), 78,8 (C-3'), 72,7
130,5 (C-2), 98,7 (C-7'), 81,01 (C-1'), 78,8 (C-3'), 72,7 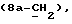 70,2 (C-9'), 41,3 (C-4'), 40,4 (C-5'), 27,3 (4'-CH3), 26,5 (C-6'), 22,8 (4'-CH3).
70,2 (C-9'), 41,3 (C-4'), 40,4 (C-5'), 27,3 (4'-CH3), 26,5 (C-6'), 22,8 (4'-CH3).
ПРИМЕР 23
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(6-дезокси-3,4-о-изопропилиден-β-D-аллопиранозилокси)-метил] -4-формил-4,4a, 5,6,7,7a,8,8a- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3a(1H)- карбоновая кислота
К раствору Промежуточного соединения 27 (125 мг) в метаноле (25 мл) в атмосфере азота добавляли 10%-ный палладий на угле (100 мг). Смесь встряхивали в течение 1,5 часов при комнатной температуре. Катализатор отфильтровывали, к фильтрату при 0oC добавляли 1N соляную кислоту (0,5 мл). После перемешивания в течение 1 часа смесь нефтрализовали 10%-ным гидроксикарбонатом натрия. Растворитель упаривали до сухого состояния, а масляный остаток очищали посредством колоночной хроматографии быстрого разделения, элюируя смесью гексан : этилацетат (4 : 1) и дихлорметан : метанол (20 : 1) и получая в результате соединение, указанное в заголовке (62 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,10 (dd, 1H, H-2, J = 1,2 и 3,3 Гц), 4,51 (t, 1H, H-2', J = 4,5 Гц), 4,48 (d, 1H, H-1', J = 7,5 Гц), 3,65 и 4,07 (2d, 2H, 8aCH2, J = 9,3 Гц), 3,78 (dd, 1H, H-4', J = 9 и 5,1 Гц), 3,69 (dd, 1H, H-3', J = 7,5 и 5,1 Гц), 3,50 (m, 1H, H-5', J = 6 и 9 Гц), 2,59 (t, 1H, H-1, J = 3,9 Гц), 2,32 (m, 1H, CHMe2); δ 13C, CDCl3): 204,6 (CHO), 176,1 (CO2H), 148,2 (C-3), 130,8 (C-2), 110,4 (четвертичный C Изопропилиден), 100,9 (C-1'), 78,34 (C-4'), 75,1 (C-2'), 74,7 (8аCH2), 72,5 (C-4a), 71,7 (C-3'), 69,3 (C-5'), 65,6 (C-8a), 58,9 (C-4), 46,3 (C-1).
ПРИМЕР 24
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a-[(2-дезокси-3,4-о-изопропилиден-6-O-метил-β-D-аллопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a-(1H)-карбоновая кислота
Раствор Промежуточного соединения 30 (50 мг) в тетрагидрофуране (5 мл) при комнатной температуре обрабатывали тетрабутиламмонийфторидом (1,0M раствор в тетрагидрофуране, 200 мл). Спустя 24 часа реакционную смесь концентрировали до сухого состояния, а полученное масло желтого цвета очищали посредством колоночной хроматографии быстрого разделения, элюируя смесью дихлорметан : метанол (15 : 1) и получая в результате соединение, указанное в заголовке (18 мг).
δ (1H, CDCl3): 9,80 (s. 1H, CHO), 5,98 (dd, 1H, H-2, J = 1,2 и 3,3 Гц), 4,64 (dd, 1H, H-1', J = 2,7 и 8,1 Гц), 4,42 (m, 1H, H-3'), 3,68 и 4,01 (2d, 2H, 8aCH2, J = 9 Гц), 3,90 (m, 1H, H-4'), 3,45 - 3,65 (m, 3H, H-5' и 2H-6'), 3,22 (s, 3H, 6'-OCH3), 2,60 (m, 1H, H-1'), 1,34 и 1,35 (2s, 6H, метильные группы изопропилидена); δ (13C, CDCl3): 206,3 (CHO), 175,7 (CO2H), 149,7 (C-3), 129,7 (C-2), 108,9 (C-четвертичный изопропилиден), 98,2 (C-1'), 74,8 (C-3a), 74,8 (C-4), 74,4 (8aCH2), 72,6 (C-3'), 71,3 (C-5'), 65,1 (C-8a), 61,1 (C-6'), 58,4 (C-4), 48,4 (6'-OMe), 46,1 (C-1), 32,8 (C-2').
ПРИМЕР 25
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4R,7R,9R]-2,8-Диокса-4-этил-9-метил-цис-бицикло[3.4.0] -нон-7-ил- окси-метил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору промежуточного соединения 37 (200 мг) в этилацетатен (40 мл) в атмосфере азота добавляли 10%-ный палладий на угле (50 мг). Смесь встряхивали в Parr аппарате при давлении водорода 30 пси при комнатной температуре в течение 2 часов. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Полученный таким образом остаток подвергали хроматографии быстрого разделения на силикагеле, применяя в качестве элюента смесь дихлорметан : метанол (98 : 2) и получая в результате соединение, указанное в скобках (125 мг).
δ (1H, CDCl3): 9,86 (s, 1H, CHO), 6,05 (dd, 1H, H-2, J = 1,5 и 3,3 Гц), 4,66 (dd, 1H, H-7', J = 3,0 и 6,0 Гц), 4,32 и 3,29 (2d, 2H, 8a-CH2, J = 9,6 Гц), 4,02 (dd, 1H, CH2-3', (1H), J = 7,2 и 8,4 Гц), 3,67 (dd, 1H, H-1', J = 7,8 и 9,3 Гц), 3,41 - 3,22 (m, 2H, H-9' и CH2-3' (1H)), 2,48 (t, 1H, H-1, J = 3,9 Гц), 0,92 (t, 3H, 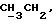 J = 7,5 Гц); δ (13C, CDCl3): 204,5 (CHO), 174,2 (COOH), 148,3 (C-3), 130,5 (C-2), 98,5 (C-7'), 80,3 (C-1'), 73,0 и 73,0 (
J = 7,5 Гц); δ (13C, CDCl3): 204,5 (CHO), 174,2 (COOH), 148,3 (C-3), 130,5 (C-2), 98,5 (C-7'), 80,3 (C-1'), 73,0 и 73,0 ( 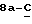 и С-3'), 70,9 (C-9').
и С-3'), 70,9 (C-9').
ПРИМЕР 26
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4R,7R,9R]-2,8- Диокса- 4-этил-9-метил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил] -4-формил -4,4a,5,6,7,7а, 8, 8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1Н)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 25 (88 мг) в метаноле (10 мл) по каплям добавляли 0,099N водный гидроксид натрия (1,8 мл). Растворитель удаляли при пониженном давлении, а остаток растворяли в воде (50 мл) и лиофилизировали, получая соединение, указанное в заголовке (92 мг).
δ (1H DMSO-d6): 9,77 (s, 1H, CHO), 5,78 (m, 1H, H-2), 4,40 (brdd, 1H, H-7'), 3,92 (m, 1H, H-3'), 3,87 (d, 1H, 8a-CH2(1H), J=9,6 Гц), 3,50 (m, 2H, 8a-CH2(1H) и H-1'), 3,38-3,21 (m, 2H, H-3' и Н-9').
ПРИМЕР 27
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S,4S,7R,9R]-2,8-Диокса-4-этил-9-метил-цис-бицикло [3.4.0] -нон-7-ил-окси-метил] -4-формил -4,4a,5,6,7,7a, 8,8а-октагидро-7-метил-3-(1- метилэтил)-1,4-метано-s-индацен-3а(1Н)-карбоновая кислота
К раствору промежуточного соединения 38 (420 мг) в этилацетате (80 мл) в атмосфере азота добавляли 10%-ный палладий на угле (100 мг). Смесь встряхивали в Parr аппарате при давлении водорода 30 пси при комнатной температуре в течение 1,5 часов. Катализатор отфильтровывали, растворитель выпаривали до сухого состояния, полученный таким образом остаток очищали посредством хроматографии быстрого разделения на силикагеле, применяя в качестве элюента смесь дихлорметан: метанол (98:2) и получая в результате соединение, указанное в заголовке (280 мг).
δ (1H, CDCl3): 9,86 (s, 1H, CHO), 6,05 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,89 (m, 1H, H-7'), 4,29 и 3,29 (2d, 2H, 8a-CH2, J=9,9 Гц), 3,90 (dd, 1H, H-3', J=7,5 и 8,7 Гц), 3,75 (m, 1H, H-1'), 3,64 (m, 1H, H-9'), 3,43 (t, 1H, H-3', J= 8,7 Гц), 2,46 (t, 1H, H-1, J=3,6 Гц), 0,93 (t, 3H, 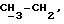 J=7,2 Гц); δ (13C, CDCl3): 204,4 (CHO), 173,7 (COOH), 148,2 (C-3), 130,5 (С-2), 98,8 (C-7'), 81,3 (C-1'), 72,8 и 70,9 ((8а-C) и C-3'), 69,8 (C-9').
J=7,2 Гц); δ (13C, CDCl3): 204,4 (CHO), 173,7 (COOH), 148,2 (C-3), 130,5 (С-2), 98,8 (C-7'), 81,3 (C-1'), 72,8 и 70,9 ((8а-C) и C-3'), 69,8 (C-9').
ПРИМЕР 28
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8а- [[1S, 4S,7R, 9R]-2,8-Диокса-4-этил-9-метил-цис-бицикло[3.4.0]-нон-7-ил-окси-метил [4-формил -4,4а,5,6,7,7a, 8,8а-октагидро-7 -метил- 3- (1-метилэтил) -1,4-метано-s-индацен-3а(1Н)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 27 (262 мг) в метаноле (20 мл) по каплям добавляли 0,099N водный гидроксид натрия (5,3 мл). Растворитель удаляли при пониженном давлении, а остаток растворяли в воде (100 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (272 мг).
δ (1H, DMSO-d6: 9,77 (s, 1H, 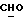 ), 5,79 (dd, 1H, H-2 J=1,2 и 3,6 Гц), 4,57 (m, 1H, H-7'), 3,83 и 3,45 (2d, 2H, 8a-CH2, J=9,6 Гц), 3,76 и 3,33 (2m, 2H, CH2-3'), 3,52 (m, 2Н, Н-1' и Н-9').
), 5,79 (dd, 1H, H-2 J=1,2 и 3,6 Гц), 4,57 (m, 1H, H-7'), 3,83 и 3,45 (2d, 2H, 8a-CH2, J=9,6 Гц), 3,76 и 3,33 (2m, 2H, CH2-3'), 3,52 (m, 2Н, Н-1' и Н-9').
ПРИМЕР 29
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[[1S,4R,7R,9R]-2,8-Диокса- 4-(1-метилэтил)-9-метил-цис-бицикло [3.4.0] -нон-7-ил- оксиметил) 1-4-формил-4,4а, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3a(1H)карбоновая кислота
К раствору промежуточного соединения 39 (280 мг) в этилацетате (15 мл) в атмосфере азота добавляли палладий (10%) на угле (50 мг). Смесь встряхивали в Parr аппарате (PH2= 20 пси) в течение 1 часа при комнатной температуре, после фильтрации катализатора и выпаривания растворителя получали остаток, который очищали посредством хроматографии быстрого разделения (силикагель, гексан: ацетон в/в 10: 1 и 5:1), получая в результате 180 мг соединения, указанного в заголовке, в виде масла (выход 84%).
δ (1H, CDCl3): 12-11 (b, 1H, COOH), 9,88 (s, 1H, CHO), 6,053 (dd, 1H, H2, J=1,5 и 3,6 Гц), 4,77 (m, 1H, H7'), 4,37 (d, 1H, 8aCH2, J=9,3 Гц), 3,95 (dd, 1H, H3', J=7,2 и 8,7 Гц), 3,67 (dd, 1H, H1', J=8,7 и 9 Гц), 3,5-3,36 (m, 2H, H3'+H9'), 3,23 (d, 1H, 8aCH2 J=9,3 Гц), 2,43 (t, 1H, H1, J=3,9 Гц).
ПРИМЕР 30
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[[1S,4R,7R,9R]-2,8-Диокса-4-(1-метилэтил)-9-метил-цис-бицикло [3.4.0] -нон-7-ил-оксиметил)]-4-формил-4,4a, 5,6,7,7а, 8, 8а-октагидро-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3a(1Н)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 29 (170 мг) в метаноле (15 мл) добавляли 0,0956N раствор гидроксида натрия (3,45 мл). После перемешивания в течение 1 часа растворитель удаляли до сухого состояния, получая вещество, которое растворяли в 3 мл воды и сушили замораживанием.
δ (1H, CDCl6): 9,78 (s, 1H, CHO), 5,80 (dd, 1H, H2, J=1,2 и 3,6 Гц), 4,46 (dd, 1H, H7', J=2,7 и 5,1 Гц), 3,9-3,8 (m, 2H, H3'+8aCH2), 3,54 (dd, 1Н, H1', J= 8,1 и 9 Гц), 3,47 (d, 1H,8aCH2, J=9,3 Гц); δ (13C, DMSO-d6): 206,4 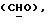 174,3 (COO-), 151,3
174,3 (COO-), 151,3 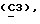 127,6
127,6 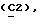 97,1 (C7'), 80,7 (C1').
97,1 (C7'), 80,7 (C1').
ПРИМЕР 31
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4S,7R,9R)-2,8-Диокса-4-(1-метилэтил) -9-метил-цис-бицикло [3.4.0]-нон-7-ил-оксиметил)]4-формил-4, 4a, 5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4 -метано-s-индацен-3а(1Н)-карбоновая кислота
К раствору соединения из Примера 40 (280 мг) в этилацетате (15 мл) в атмосфере азота добавляли палладий (10%) на угле (50 мг). Смесь встряхивали в Parr аппарате (PH2= 20 пси) в течение 1 часа при комнатной температуре. После фильтрации катализатора и выпаривания растворителя получали остаток, который подвергали хроматографии быстрого разделения (силикагель, гексан: ацетон в/в 10:1 и 5:1), получая в результате 190 мг соединения, указанного в заголовке, в виде кристаллического вещества (выход 89%).
δ (1H, CDCl3): 9,88 (s, 1H, CHO), 6,05 (dd, 1H, H2, J=1,5 и 3,3 Гц), 4,92 (1,1H, H7', J=2,7 Гц), 4,33 (d, 1H,8aCH2, J=10,2 Гц), 3,94 (t, 1H, H3', J= 8,1 Гц), 3,8-3,7 (m, 2H, H1'+H9'), 3,47 (dd, 1H, H3', J=8,4 и 10,8 Гц), 3,29 (d, 1H, 8aCH2, J=10,5 Гц).
ПРИМЕР 32
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[1S,4S,7R,9R)-2,8-Диокса-4-(1-метилэтил) -9-метил-цис-бицикло [3.4.0]-нон-7-ил-оксиметил)]-4-формил-4, 4a, 5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен- 3а(1Н)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 31 (165 мг) в метаноле (15 мл) добавляли 0,0956N раствор гидроксида натрия (3,35 мл). После перемешивания в течение 1 часа растворитель удаляли до сухого состояния, получая в результате вещество, которое растворяли в 3 мл воды и сушили замораживанием.
δ (1H, CDCl3): 9,77 (s, 1H, CHO), 5,79 (dd, 1H, H2, J=0,9 и 3,3 Гц), 4,58 (bs, 1H, H7'), 3,85-3,72 (m, 2Н, H3'+8aCH2), 3,71- 3,56 (m, 2H, H9'+ H1'), 3,45 (d, 1H, 8aCH2, J=9,9 Гц); δ (13C, CDCl3): 206,3 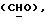 174,3(COO-), 151,4
174,3(COO-), 151,4 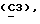 127,5
127,5 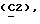 96,7 (C7'), 80,8 (C1').
96,7 (C7'), 80,8 (C1').
ПРИМЕР 33
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[[1S,7R,9R]-2,8-Диокса-9-метил-4-метилен-цис-бицикло [3.4.0] - нон-7-ил-окси-метил) 1-4-формил-4,4а,5,6,7,7а, 8,8а-октагидро- 7-метил-3-(1-метилэтил)-1.4-метано-5-индацен-3а(1Н)-карбоновая кислота
К охлаждаемому льдом раствору промежуточного соединения 41 (330 мг) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (0,2 мл). Смесь перемешивали в течение 20 минут при 0oC и затем разбавляли дихлорметаном (150 мл), промывали три раза водой (100 мл). Органический слой сушили над безводным сульфатом натрия и концентрировали до сухого состояния, получая в результате масло, которое подвергали хроматографии быстрого разделения на силикагеле, применяя в качестве элюента смесь дихлорметан: метанол (98:2). Соответствующие фракции затем собирали и подвергали RP-18 под давлением среды (около 10 бар), применяя линейный градиент ацетонитрила (от 60 до 75%) в воде и получая в результате соединение, указанное в заголовке.
δ (1H, CDCl3): 9,87 (s, 1Н, 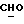 ), 6,04 (d, 1H, H-2, J=3,3 Гц), 5,08 и 5,02 (2m, 2H,
), 6,04 (d, 1H, H-2, J=3,3 Гц), 5,08 и 5,02 (2m, 2H, 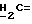 ), 4,50 (dd, 1H, H-7', J=2,7 и 8,4 Гц), 4,48- 4,29 (m, 3H,
), 4,50 (dd, 1H, H-7', J=2,7 и 8,4 Гц), 4,48- 4,29 (m, 3H, 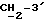 и 8а-CH2(1Н)), 3,76 (dd, 1H, H-1', J-7,2 и 9,3 Гц), 3,27 (m, 1H, H-9'), 3,02 (m, 1H, H-5'), 2,45 (m, 1Н, Н-1); δ (13C, CDCl3): 205,1
и 8а-CH2(1Н)), 3,76 (dd, 1H, H-1', J-7,2 и 9,3 Гц), 3,27 (m, 1H, H-9'), 3,02 (m, 1H, H-5'), 2,45 (m, 1Н, Н-1); δ (13C, CDCl3): 205,1 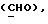 176,0
176,0 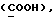 148,6 (C-3), 130,4 (C-2), 104,2
148,6 (C-3), 130,4 (C-2), 104,2 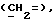 98,2 (C-7'), 80,1 (C-1'), 73,4 и 70,6 (
98,2 (C-7'), 80,1 (C-1'), 73,4 и 70,6 ( 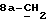 и C-3'), 69,8 (C-9').
и C-3'), 69,8 (C-9').
ПРИМЕР 34
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a- [[1S,7R,9R]-2,8-Диокса-9- метил-4-метилен-дис-бицикло [3.4.0] -нон-7-ил-окси-метил) 1- 4-формил-4,4а, 5, 6,7,7a, 8,8а-октагидро-7-метил-3- (1- метилэтил) -1,4-метано-s-индацен-3а(1Н)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 33 (140 мг) в метаноле (15 ил) по каплям добавляли 0,945N водный гидроксид натрия (3,06 мл). Растворитель удаляли при пониженном давлении, а остаток растворяли в воде (60 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (145 мг).
δ (1H, DMSO-d6): 9,76 (5,1H, 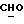 ), 5,76 (d, 1H, H-2, J=3,3 Гц), 5,04 (m, 2Н
), 5,76 (d, 1H, H-2, J=3,3 Гц), 5,04 (m, 2Н 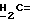 ), 4,34-4,20 (m, 3H, H-7' и CH2-3'), 3,84 и 3,50 (2d, 2H, 8a-CH2, J= 9,9 Гц), 3,62 (dd, 1H, H-1', J=7,5 и 9,3 Гц), 3,15 (m, 1H, H-9'), 2,89 (m, 1H, H-5').
), 4,34-4,20 (m, 3H, H-7' и CH2-3'), 3,84 и 3,50 (2d, 2H, 8a-CH2, J= 9,9 Гц), 3,62 (dd, 1H, H-1', J=7,5 и 9,3 Гц), 3,15 (m, 1H, H-9'), 2,89 (m, 1H, H-5').
ПРИМЕР 35
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[[1S,7R,9R]-2,8-Диокса- 9-метил-4-метилен-3-оксо-цис-бицикло [3.4.0] -нон-7-ил- окси-метил)] 4-формил-4,4а, 5,6,7,7а, 8,8а-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3а(1Н)-карбоновая кислота
К раствору промежуточного соединения 42 (62 Мг) в сухом дихлорметане (1,1 мл) добавляли трифторуксусную кислоту (0,05 мл). Смесь перемешивали при 0oC в течение 20 минут, затем разбавляли дихлорметаном (50 мл) и промывали водой (3х25 мл). Органический слой сушили над безводным сульфатом натрия и концентрировали, получая в результате масло, которое дважды подвергали хроматографии быстрого разделения на силикагеле, применяя в качестве элюента смесь дихлорметан:метанол (98:2). Таким образом получали соединение, указанное в заголовке, в виде пены бледно- желтого цвета (25 мг).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,35 и 5,63 (2d, 2H, H2C=C, J=3,3 Гц),
6,04 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,52 (t, 1H, H-7', J=4,8 Гц), 4,24 (t, 1H, H-4', J=8,7 Гц), 4,09 и 3,46 (2d, 2H, 8a CH2, J=9,3 Гц), 3,38 (m, 1H, H-5'), 3,30 (m, 1H, H-9'), 2,59 (t, 1H, H-1, J=3,9 Гц); δ (13C, CDCl3): 204,5 (CHO), 175,3 (COOH), 169,7(C-3'), 148,2 (C-3), 136,8 (C-4'), 130,6 (C-2), 122,4 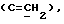 97,7(C-7'), 77,8(C-1') и 71, 2 (C-9').
97,7(C-7'), 77,8(C-1') и 71, 2 (C-9').
ПРИМЕР 36
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[[1S, 7R,9R]-2,8-Диокса-9-метил-4-метилен-3-оксо- цис-бицикло[3.4.0] -нон-7-ил-окси-метил)] -4-формил-4,4a, 5,6,7,7a, 8,8a- октагидро-7-метил-3-(1-метил-этил)-1,4-метано-s-индацен-3a(1H)- карбоновой кислоты натриевая соль
К раствору соединения из примера 35 (25 мг) в метаноле (10 мл) по каплям добавляли 0,0945N водный гидроксид натрия (0,53 мл). Растворитель удаляли при пониженном давлении, а остаток растворяли в воде (25 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (26 мг).
δ (1H, DMSO-d6): 9,75 (s, 1H, CHO), 6,15 и 5,82 (2d, 2H, 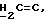 J=3.3 Гц), 5,76 (brd, 1H, H-2), 4,31 (dd, 1H, H-7', J=2,4 и 7,8 Гц), 4,24 (t, 1H, H-1', J=8,7 Гц), 3,84 и 3,52 (2d, 2H, 8a-CH2, J=9,9 Гц), 3,38 (m, 1H, H-5'), 3,21 (m, 1H, H-9').
J=3.3 Гц), 5,76 (brd, 1H, H-2), 4,31 (dd, 1H, H-7', J=2,4 и 7,8 Гц), 4,24 (t, 1H, H-1', J=8,7 Гц), 3,84 и 3,52 (2d, 2H, 8a-CH2, J=9,9 Гц), 3,38 (m, 1H, H-5'), 3,21 (m, 1H, H-9').
ПРИМЕР 37
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ]8a-[(1S,7R, 9R]-2,8-Диокса-9-метил-4-оксо-цис-бицикло[3.4.0] -нон-7-ил-окси-метил]- 4-формил-4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метил-этил)-1,4- метано-s-индацен-3a(1H)-карбоновая кислота
К раствору промежуточного соединения 43 (45 мг) в этилацетате (15 мл) в атмосфере азота добавляли 10%-ный палладий на угле (25 мг). Смесь встряхивали в Parr аппарате при давлении водорода 25 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Полученный таким образом остаток очищали посредством хроматографии быстрого разделения на силикагеле, применяя в качестве элюента смесь гексан:этилацетат (65:35) и получал в результате соединение, указанное в заголовке (25 мг).
δ (1H, CDCl3): 9,77 (s, 1H, CHO), 6,05(dd, 1H, H-2, J=2, J=1,5 и 3,6 Гц), 4,17 (m, 4H, H-3'(1H), 8a-CH2(1H), H-7' и H-1'), 3,90 (d, 1H, H-3'(1H), J= 17,7 Гц), 3,42 (d, 1H, 8a-CH2(1H), J=9 Гц), 3,31 (m, 1H, H-9'), 2,85 (m, 1H, H-5'), 2,58 (t, 1H,H-3, J=3,9 Гц); δ (13C, CDCl3): 214,4(C-4), 204,8 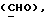 148,3 (C-3), 130,6(C-2), 98,9(C-7'), 78,6 и 70,3 (C-1' и C-9'), 73,6 и 69,1
148,3 (C-3), 130,6(C-2), 98,9(C-7'), 78,6 и 70,3 (C-1' и C-9'), 73,6 и 69,1 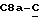 и С-3').
и С-3').
ПРИМЕР 38
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ]-8a-[[1S,4S,6S,7R, 9R]-2,8-Диокса-4-этил-6-гидрокси-9-метил-цис-бицикло[3.4.0] -нон-7-ил-окси-метил] -4-формил-4,4a, 5,6,7a, 8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору Промежуточного соединения 45 (123 мг) в этилацетате (50 мл) в атмосфере азота добавляли 10%-ный палладий на угле (65 мг) и гидрогенизировали полученную смесь при давлении водорода 15 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан: метанол от (19:1) до (18:2), получая в результате соединение, указанное в заголовке (90 мг).
δ (1H, CDCl3): 9,67(s, 1H, CHO), 6,07 (d,1H,H-2, J=3,3 Гц), 4,64 (d, 1H, h-7', J=3,6 Гц), 4,02 (d,1H, 8aCHa, J=9,9 Гц), 3,89 (dd, 1H, Ha-3' J=6,9 и 9 Гц), 3,85-3,7 (m, 3H, H-6', H-1' и H-9'), 3,68-3,5 (m, 2H, Hb-3' и 8a-CHb), 2,65 (t, 1H, h-1, J= 3,6 Гц); δ (13C, CDCl3): 204,9 (CHO), 176,3 (CO2H), 148,9(C-3), 130,1 (C-2), 99,2 (C-7'), 80,5, 70,1 и 65,5 (C-6', C-9'), 73,7 и 71,0 ( 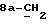 и C-3'), 13,4
и C-3'), 13,4 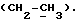
ПРИМЕР 39
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a[(1S, 4R, 6S, 7R, 9R]-2,8-Диокса-4-этил-6-гидрокси-9-метил-цис-бицикло [3.4.0] -нон-7-ил-оксиметил] -4-формил-4,4a, 5,6,7,7a, 8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3a(1H)-карбоновая кислота
К раствору промежуточного соединения 46 (102 мг) в этилацетате (25 мл) в атмосфере азота добавляли 10%-ный палладий на угле (60 мг), полученную смесь гидрогенизировали при давлении водорода 15 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан:метанол (25:1), получая в результате соединение, указанное в заголовке (75 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,2, J=1,2 и 3,6 Гц), 4,53 (d,1H, H-7', J=1,8 Гц), 4,05 (dd, 1H, Ha-3',J=7,2 и 8,4 Гц), 4,02 (d, 1H, 8a-CHa, J=9,3 Гц), 3,83 (dd, 1H,H-1', J=7,2 и 8,7 Гц), 3,72 (dd, 1H, H-6', J= 2,1 и 4,8 Гц), 3,64 (d, 1H, 8a-CHb, J=9,3 Гц), 3,54-3,36 (m, 2H, H-9' и Hb-3'), 2,68 (t, 1H, H-1, J=3,9 Гц), 0,96 (t, 3H,  J=7,5); δ 13C, CDCl3): 204,7 (CHO), 176,1 (CO2H), 148,5(C-3), 130,5 (C-2), 98,8(C-7'), 79,2, 70,7 и 69,1(C-6', C-1' и C-9'), 73,7 и 72,7 (
J=7,5); δ 13C, CDCl3): 204,7 (CHO), 176,1 (CO2H), 148,5(C-3), 130,5 (C-2), 98,8(C-7'), 79,2, 70,7 и 69,1(C-6', C-1' и C-9'), 73,7 и 72,7 ( 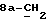 и C-3'), 12,7
и C-3'), 12,7 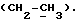
ПРИМЕР 40
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S, 4R, 6S, 7R, 9R]-2,8-Диокса- -4-этил-6-гидрокси-9-метил-цис-бицикло [3.4. О] 1-нон- 7-ил-окси-метил]-4-формил-4,4а, 5,6,7,7а,8,8а-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1Н)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 39 (490 мг) в метаноле (50 мл) по каплям добавляли 0,095N водный раствор гидроксида натрия (10 мл). Спустя 30 минут растворитель удаляли при пониженном давлении, а остаток растворяли в воде (10 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (475 мг).
δ (1H, (CD3)2)SO): 9,77 (s, 1H, CHO), 5,8 (d, 1H, H-2, J=2,4 Гц), 4,41 (bs, 1H, -ОН), 4,31 (d,1 H, H-7', J=2,1 Гц), 3,94-3,82 (m, 2H, 8aCHa и Ha-3'), 3,66 (t, 1H, H-1', J=8,1 Гц), 3,54-3,34 (m, 3H, 8aCHb, H-6' и Н-9'), 3,24 (t, 1H, Hb-3, J=8,1 Гц), 2,55 (t, 1H, H-1, J=3,6 Гц).
ПРИМЕР 40
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[1S,4R,6S,7R,9R]-2,8- Диокса-4-этил-6-гидрокси-9-метил-дис-бицикло [3.4.О]-нон-7- ил-окси-метил]-4-формил-4,4а, 5,6,7,7а, 8,8а-октагидро-7- метил-3-(1-метилэтил) -1,4-метано-s-индацен-3а (1Н)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 39 (490 мг) в метаноле (50 мл) по каплям добавляли 0,095N водный раствор гидроксида натрия (10 мл). Спустя 30 минут растворитель удаляли при пониженном давлении, а остаток растворяли в воде (10 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (475 мг).
δ (1H, (CD3)2SO): 9,77 (s, 1H, CHO), 5,8 (d, 1H, H-2, J=2,4 Гц), 4,41 (bs, 1H, -ОН), 4,31 (d, 1H, H-7', J=2,1 Гц), 3,94-3,82 (m, 2Н, 8aCHa и Ha-3'), 3,66 (t, 1H, H-1', J-8,1 Гц), 3,54-3,34 (m, 3H, 8aCHb, H- 6' и Н-9'), 3,24 (t, 1H, Hb-3',J=8,1 Гц), 2,55 (t, 1H, H-1, J=3,6 Гц).
ПРИМЕР 41
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[[1S,7R,9R]2,8-Диокса-9-метил-4-оксо-цис-бицикло [3.4.0] -нон-7-ил-оксиметил] 1-4-формил-4, 4а, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3а(1Н)-карбоновой кислоты натриевая соль
К раствору промежуточного соединения 43 (350 мг) в этилацетате (15 мл) в атмосфере азота добавляли палладий (10%-ный) на угле (50 мг). Смесь встряхивали в Parr аппарате (PH2=25 пси) в течение 1 часа при комнатной температуре. После фильтрации катализатора и выпаривания растворителя получали остаток, который подвергали хроматографии быстрого разделения (силикагель, дихлорметан: метанол в/в 100:1, 70:1 и 50:1), получая в результате 200 мг пены, которую растворяли в метаноле (10 мл) и обрабатывали 0,0947М гидроксидом натрия (4,54 мл). Растворитель удаляли под вакуумом, а полученное вещество растворяли в воде (15 мл) и сушили замораживанием.
δ (1H, DMSO-d6): 9,76 (s, 1H, CHO), 5,80 (dd, 1H, H2, J=0,9 и 3,3 Гц), 4,20-3, 95(m, 4H, H7'+H1+H3'), 3,89 (d, 1H, 8aCH2, J=9,6 Гц), 3,47(d, 1H, 8aCH2=9,6 Гц), 2,92(m, 1H, H5').
ПРИМЕР 42
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8a-[(1S,4R,7R,9R)-2,8-Диокса-4-метокси-9-метил-дис-бицикло [3.4.0]-нон-7-ил-оксиметил 1-4-формил-4,4а,5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3а(1Н)-карбоновой кислоты натриевая соль
К раствору промежуточного соединения 82 (250 мг) в этилацетата (15 мл) в атмосфере азота добавляли палладий (10%-ный) на угле (50 мг). Смесь встряхивали в Parr аппарате (PH2=25 пси) в течение 1 часа при комнатной температуре. После фильтрации катализатора и выпаривания растворителя получали остаток, который подвергали хроматографии быстрого разделения (силикагель, дихлорметан: метанол 100: 1, 70:1, 50:1 и 40:1), получая в результате 120 мг сиропа, который растворяли в метаноле (15 мл) и обрабатывали 0,0947 М гидроксидом натрия (2,51 мл). После удаления растворителя получали полутвердое вещество, которое растворяли в воде (15 мл) и сушили охлаждением.
δ (1H, DMSO-d6): 9,77 (s, 1H, CHO), 5,80 (dd, 1H, H2,J=1,2 и 3,3 Гц), 4,47 (t, 1H ,H7', J=3,9 Гц), 3,96-3,84 (m, 2Н,H3'+8аСН2), 3,80 (m, 1H, Н4'), 3,65-3,40 (m, 4H, H9'+8aCH2+H3'+H1'), 3,23 (s, 3H, OCH3), 2,46-2,2 (m,4Н, Н1+Н6'+Н5'+CH(CH3)2; δ(13C,DMSO-d6): 206,4 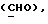 174,4
174,4 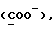 151,3
151,3 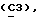 127,6
127,6 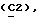 96,9
96,9 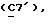 84,1 (C4'), 56,6
84,1 (C4'), 56,6 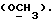
ПРИМЕР 43
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ)] 8а-[[1S,7R,9R)-2,8-Диокса-9-метил-бицикло [3.4.0] -нон-4-ен-7-ил-оксиметил]-4-формил-4,4а,5,6,7,7a, 8, 8а-октагидро-7-метил-3- (1-метилэтил) -1,4-метано-s-индацен-3а(1Н)-карбоновая кислота
К раствору промежуточного соединения 87 (120 мг) в сухом дихлорметане (20 мл) при 0oC в атмосфере азота добавляли трифторуксусную кислоту (100 мкл). Спустя 5 минут смесь разбавляли дихлорметаном (100 мл) и промывали водой (100 мл) и рассолом (100 мл), затем сушили над сульфатом магния и концентрировали, получая сырой продукт, который растворяли в смеси тетрагидрофуран: метанол (в/в 2:1, 15 мл) и обрабатывали 1N соляной кислотой (5 мл). После завершения реакции ее разбавляли водой (100 мл) и дважды экстрагировали дихлорметаном. Органический слой сушили над сульфатом магния, получая масло, которое очищали посредством препаративной ТСХ (силикагель; ацетон: гексан 1: 3), получая в результате 62 мг соединения; указанного в заголовке (выход 73%), в виде пены белого цвета.
δ (1H, CDCl3): 9,79 (s, 1H, CHO), 6,06 (dd, 1H, H2, J=1,2 и 3,3 Гц), 5,56 (m, 1H, H4'), 4,65-4,65 (m, 2H, H3'), 4,3-4,0 (m, 3H, H7+8aCH2+ H1'), 3,50 (d, 1H, 8aCH2, J=9 Гц), 3,18 (dq, 1H, H9', J=6,3 и 9 Гц), 2,72 (m, 1H, H6'), 2,62 (t, 1H, H1, J=3 Гц); δ (13C, CDCl3): 204,6 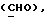 175,2
175,2 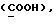 148,3
148,3 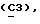 136,3 (C5'), 130,6 (C2), 119,15 (C4'), 101,5 (C7'), 86,5 (C1').
136,3 (C5'), 130,6 (C2), 119,15 (C4'), 101,5 (C7'), 86,5 (C1').
ПРИМЕР 44
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(1S,7R,9R)-2,8-Диокса-9-метил-бицикло[3.4.0] -нон- 4-ен-7ил-оксиметил-4-(формил-4,4a, 5,6,7,7a,8,8а-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 43 (50 мг в метаноле (5 мл) добавляли 0,0966N гидроксид натрия (1,09 мл). Смесь перемешивали в течение 15 минут и удаляли растворитель до сухого состояния. Остаток растворяли в воде (1 мл) и сушили замораживанием.
δ (1-H, DMSO-d6): 9,78 (s, 1H, CHO), 5,80 (bd, 1H, H2, J=3,6 Гц), 5,64 (bs, 1H, H4'), 4,6-4,4 (m, 2H, H3'), 4,1 (dd, 1H, H7', J=2,7 и 9,3 Гц), 4,05-3,90 (m, 2H, H1'+8aCH2), 3,56 (d, 1H, 8aCH2, J=9,6 Гц), 3,06( dq, 1H, H9', J= 6,3 и 8,7 Гц); δ (13C, DMSO-d6): 215,9 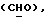 183,9
183,9 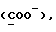 160,9
160,9 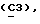 146,1(C5', 137,2(C2), 128,3(C4'), 110,9 (C7'), 95,7(C1').
146,1(C5', 137,2(C2), 128,3(C4'), 110,9 (C7'), 95,7(C1').
ПРИМЕР 45
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(1S,7R,9R)-Диокса-9-метил-цис-бицикло[3.4.0] - нон-7-ил-оксиметил]-4-формил-4,4a,5,6,7,7a,8,8а-октагидро-7-метил- 3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К раствору промежуточного соединения 87 (100 мг в этилацетате (15 мл) в атмосфере азота добавляли палладий (10%-ный) на угле (50 мг). Смесь встряхивали в Parr аппарате (PH2=20 пси) в течение 1 часа при комнатной температуре. После фильтрации катализатора и выпаривания растворителя получали остаток, который растворяли в смеси тетрагидрофуран: метанол (в/в 2:1, 15 мл) и обрабатывали 1N соляной кислотой (5 мл). После окончания реакции добавляли дихлорметан (100 мл) и воду (100 мл) и разделяли два слоя. Органический слой сушили над сульфатом магния и концентрировали, получая масло, которое очищали посредством препаративной ТСХ (гексан: ацетон в/в 3:1), получая в результате 20 мг соединения, указанного в заголовке, в виде прозрачного масла (общий выход 30%).
δ (1H, CDCl3): 9,86 (s, 1H, CHO), 6,05 (dd, 1H, H2, J=1,5 и 3,6 Гц), 4,07 (dd, 1H, H7', J=3 и 5,4 Гц), 4,33 (d, 1H, 8aCH2, J=9,6 Гц), 3,90 (m, 1H, H3'), 3,69 (m, 1H, H3'), 3,61 (dd, 1H, H1', J=8,1 и 9 Гц), 3,39 (dq, 1H, H9', J= 6 и 9 Гц), 3,28 (d, 1H, 8aCH2, J=9,6 Гц), 2,6-2,4 (m, 2H, H5'+H1), 2,32 (m, 1H, CH(CH3)2).
ПРИМЕР 46
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(1S,7R,9R)-2,8-Диокca-4,9-диметил-6-гидрокси-цис- бицикло[3.4.0] -нон-4-ен-7-ил-оксиметил]-4-формил-4,4a, 5,6,7,7a, 8,8а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен3а(1H)-карбоновой кислоты натриевая соль
К раствору Промежуточного соединения 89б (90 мг) в этилацетате (25 мл) в атмосфере азота добавляли 10%-ный палладий на угле (50 мг), а полученную смесь гидрогенизировали при давлении водорода 40 пси в течение 45 минут при комнатной температуре. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Остаток подвергали хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан: этилацетат (10:1), получая в результате соединение, указанное в заголовке (60 мг).
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,53 (d, 1H, H-7', J=2,1 Гц), 4,09 (d, 1H, 8a-CHa, J=9,6 Гц), 4,02 (dd, 1H, Ha-3', J= 7,5 и 8,4 Гц), 3,84 (dd, 1H, H-1', J=7,8 и 9 Гц), 3,74 (dd, 1H, H-6', J= 2,1 и 5,1 Гц), 3,60 (d, 1H, 8a-CHb, J=9,3 Гц), 3,42 (dq, 1H, H-9', J=6 и 9 Гц), 3,34 (t, 1H, Hb-3', J=8,7 Гц), 2,66 (t, 1H, J=3,9 Гц).
ПРИМЕР 47
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(1S,4R,6S,7R,9R)-2,8-Диокса-4,9-димeтил-6-гидpoкcи-цис-бициклo [3.4.0] -нон-7-ил-окси-метил]-4-формил-4,4а, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а (1H)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 46 (258 мг) в метаноле (20 мл) по каплям добавляли 0,103N водный раствор гидроксида натрия (4,98 мл). Спустя 15 минут растворитель удаляли при пониженном давлении, а остаток растворяли в воде (10 мл) и лиофилизировали, получая в результате соединение, указанное в заголовке (250 мг).
δ (1H, (CD3)2SO): 9,77 (5, 1H, CHO), 5,79 (d, 1H, H-2, J=3,6 Гц), 4,29 (d, 1H, H-7', J=1,8 Гц), 3,94-3,8 (m, 2H, 8a-CHa и Ha-3'), 3,67 (t, 1H, H-1', J=8,4 Гц), 3, 55-3, 25(m, 3H, 8a-CHb, H-6',H-9'), 3,14 (t, 1H, Hb-3', J= 8,4 Гц), 2,54 (t, 1H, H-1, J=3,6 Гц).
ПРИМЕР 48
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(((2,3,6-Тридезокси-3- этиламино-3-N, 4-O-кapбoнил- β- D-aллoпиpaнoзил)oкcи)метил) - 4-формил-4,4а,5,6,7,7a, 8,8а-октагидро-7-метил-3-(1-метилэтил)- 1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К раствору промежуточного соединения 96 (180 мг) в этилацетате (15 мл) в атмосфере азота добавляли палладий (10%-ный) на угле (30 мг). Смесь встряхивали в Parr аппарате (PH2=20 пси) в течение 2 часов при комнатной температуре. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесью гексан: этилацетат 1:1, получая в результате чистый GM 233039Х (70 мг).
δ (1H, CDCl3): 9,76 (s, 1H, 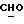 ), 6,05 (dd, 1H, H-2, J=3,0 и 1,2 Гц), 4,64 (dd, 1H, H-1', J=5,7 и 3,3 Гц), 4,11 (m, 2H, H-3',H-4'), 4,04 (d, 1H, H-8a, J=9,3 Гц), 3,62 (dq, 1H, H-5', J=8,4 и 6,0 Гц), 3,51 (d, 1H, H-8a, J= 9,3 Гц), 3,49 (m, 1H,
), 6,05 (dd, 1H, H-2, J=3,0 и 1,2 Гц), 4,64 (dd, 1H, H-1', J=5,7 и 3,3 Гц), 4,11 (m, 2H, H-3',H-4'), 4,04 (d, 1H, H-8a, J=9,3 Гц), 3,62 (dq, 1H, H-5', J=8,4 и 6,0 Гц), 3,51 (d, 1H, H-8a, J= 9,3 Гц), 3,49 (m, 1H,  ), 3,13 (m, 1H,
), 3,13 (m, 1H,  ), 2,62 (t, 1H, H-1, J= 3,6 Гц), 2,32 (m, 1H,
), 2,62 (t, 1H, H-1, J= 3,6 Гц), 2,32 (m, 1H, 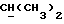 ), 1,15 (t, 3H,
), 1,15 (t, 3H, 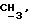 J=7,2 Гц); δ (13С, CDCl3): 204,4
J=7,2 Гц); δ (13С, CDCl3): 204,4 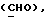 175,7
175,7 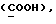 157,6
157,6 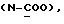 148,3 (C-З), 130,5 (C-2), 97,2 (C-1'), 74,2 (C-4'), 73,4
148,3 (C-З), 130,5 (C-2), 97,2 (C-1'), 74,2 (C-4'), 73,4 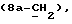 69,4 (C-5'), 51,2
69,4 (C-5'), 51,2 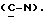
ПРИМЕР 49
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[((2,3-Ангидро-6-дезокси-4-O-метил- β- D-маннопиранозилокси)метил]-4-формил-4,4а,5,6,7,7а,8,8a-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 50 (140 мг) в этилацетате (20 мл) в атмосфере азота добавляли 10%-ный палладий на угле (100 мг). Смесь встряхивали в Parr аппарате при давлении водорода 15 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Остаток очищали посредством хроматографии быстрого разделения на силикагеле, элюируя смесью гексан: этилацетат (5:1) и дихлорметан: метанол (20:1). Соответствующие фракции объединяли, а растворитель удаляли, получая в результате соединение, указанное в заголовке (85 мг), в виде пены.
δ (1H, CDCl3): 9,76 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,74 (s, 1H, H-1'), 3,63 и 4,13 (2d, 2H, 8aCH2, J=9,3 Гц), 3,48 (m, 4H, H-4' и 4'-OMe), 2,26 (d, 1H, H-2', J=3,9 Гц), 3,19 (m, 1H, H-5'), 3,13 (d, 1H, H-3', J=3,9 Гц), 3,06 (d, 1H, H-4', J=8, 7 Гц), 2,74 (t, 1H, H-1, J=3,6 Гц); δ (13C, CDCl3): 204,8 (CHO), 175,6 (CO2H), 148,2 (C-3), 130,8 (C-2), 98,0 (C-1'), 76,5 (C-4'), 74,3 (8aCH2), 74,4 (C-3a), 72,5 (C-5'), 65,6 (C-8a), 59,0 (C-4), 58,2 (4'-OMe), 53,6 (C-2'), 50,3 (C-3'), 46,4 (C-1).
ПРИМЕР 50
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8а-[(2,3-Ангидро-6-дезокси-4-O-метил- β- D-маннопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7a, 8,8a -октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а (1H)-карбоновой кислоты натриевая соль
Соединение из Примера 49 (310 мг) растворяли в метаноле (10 мл) и добавляли водный раствор гидроксида натрия (0,0976N, 6,69 мл). Смесь перемешивали в течение 2 часов при комнатной температуре. Растворитель удаляли, а остаток растворяли в воде (2 мл) и сушили замораживанием, получая в результате соединение, указанное в заголовке (324 мг), в виде пены белого цвета.
δ (1H, CDCl3): 9,87 (s, 1H, CHO), 5,98 (dd, 1H-2, J=1,5 и 3,6 Гц), 4,69 (s, 1H, H-1'), 3,85 и 4,07 (2d, 2H, 8aCH2, J=9,6 Гц), 3,47 (s, 3H, 4'-ОМе), 3,26 (d, 1H, H-2', J=3,9 Гц), 3,19 (dq, 1H, H-5', J=5,7 и 9 Гц), 3,12 (d, 1H, H-3', J=3,9 Гц), 2,97 (dd, 1H, H-4', J=0,6 и 8,7 Гц), 2,66 (t, 1H, H-1, J= 3,6 Гц); δ (13C, CDCl3): 209,6 (CHO), 178,6 (CO2H, 152,4 (C-3), 130,2 (C-2), 100,1 (C-1'), 78,2 (C-4'), 77,9 (8aCH2), 73,8 (C-3a), 73,9 (C-5'), 65,6 (C-8а), 59,2 (C-4), 58,2 (4'-OMe), 54,6 (C-2'), 51,9 (C-3'), 46,4 (C-1).
ПРИМЕР 51
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Эпитио-4-O-метил-2,3,6-тридезокси- β- D-маннопиранозилокси)метил]-4-формил-4,4а,5,6,7,7a,8,8а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)- карбоновая кислота
Смесь Промежуточного соединения 52 (0,67 ммоль), 5,5-диметил- 2-тиоло-2-тиоксо-1,3,2-диоксафосфоринана1 (3,37 ммоль) и триэтиламина (4,02 ммоль) в сухом диметилформамиде (5 мл) прогревали в течение 48 часов при 100oC. После охлаждения смесь вливали в водный эфир. Органическую фазу выпаривали, а остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь дихлорметан: метанол (30:1), получая в результате соединение, указанное в заголовке (180 мг).
δ (1H, CDCl3): 9,77 (5, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,95 (d, 1H, H-1', J=1,8 Гц), 4,15 и 3,65 (d,d, 1H, 1H, 8a-CH2), J=9,3 Гц), 3,33 (d, 1H, H-4', J=8,7 Гц), 3,18 (m, 1H, H-5'), 3,14 (m, 2H, H-2' и H-3'), 2,70 (m, 1H, H-1), 2,30 (m, 1H, 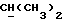 ); δ (13C, CDCl3); 204,7 (CHO), 175,6 (CO2H), 148,3 (C-3), 130,7 (C-2), 98,5 (C-1'), 79,1 (C-4'), 74,1 (8а-CH2), 73,5 (C-5'), 46,6 (C-1), 35,6 (C-2'), 35,2 (C-3'), 27,5
); δ (13C, CDCl3); 204,7 (CHO), 175,6 (CO2H), 148,3 (C-3), 130,7 (C-2), 98,5 (C-1'), 79,1 (C-4'), 74,1 (8а-CH2), 73,5 (C-5'), 46,6 (C-1), 35,6 (C-2'), 35,2 (C-3'), 27,5 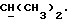 1[9]
1[9]
ПРИМЕР 52
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a- [(2,3-Ангидро-6-дезокси-4-O- пропил- β- -D-маннопиранозилокси)метил]-4-формил-4,4а, 5,6,7,7а,8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 65 (400 мг) в этилацетате (100 мл) в атмосфере азота добавляли 10%-ный палладий на угле. Смесь встряхивали в Parr аппарате при давлении водорода 20 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель выпаривали до сухого состояния. Остаток очищали на колонке с силикагелем, элюируя метиленхлоридом и смесью метиленхлорид: метанол (25:1), получая в результате соединение, указанное в заголовке (300 мг), в виде пены белого цвета.
δ (1H, CDCl3): 9,76 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,72 (s, 1H, H-1'), 4,13 и 3,65 (2d, 2H, 8a-CH2, J=9 Гц), 3,67 (m, 1H, H-5'), 3,44 (m, 1H, H-4'), 3,25 и 3,12 (2d, 2H, H-2', H-3', J=4,2 Гц), 3,20 (m, 2H, 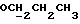 ), 2,74 (t, 1H, H-1, J=3,9 Гц); δ (13C, CDCl3): 204,8 (CHO), 175,8 (CO2H), 130,8 (C-2), 148,2 (C-3), 98,0 (C-1'), 74,9 (C-4'), 74,3
), 2,74 (t, 1H, H-1, J=3,9 Гц); δ (13C, CDCl3): 204,8 (CHO), 175,8 (CO2H), 130,8 (C-2), 148,2 (C-3), 98,0 (C-1'), 74,9 (C-4'), 74,3 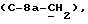 , 72,6 (C-5'), 72,4
, 72,6 (C-5'), 72,4 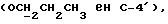 65,6 (C-8а), 59,0 (C-3), 54,2 (C-2'), 50,3 (C-3'), 18,6 (C-6').
65,6 (C-8а), 59,0 (C-3), 54,2 (C-2'), 50,3 (C-3'), 18,6 (C-6').
ПРИМЕР 53
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-6-дезокси-4-O- пропил- β- -D-маннопиранозилокси)метил] 4-формил-4,4а,5,6,7,7а,8, 8а-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а (1H)-карбоновой кислоты натриевая соль
К раствору соединения из Примера 52 (400 мг) в сухом метаноле (30 мл) при комнатной температуре по каплям добавляли 0,0976N раствор гидроксида натрия (8,15 ил), смесь перемешивали в течение 1 часа. Растворитель упаривали до сухого состояния, а твердый остаток растворяли в минимальном объеме воды (10-15 мл). Этот раствор затем лиофилизировали, получая в результате соединение, указанное в заголовке (417 мг), в виде вещества белого цвета.
δ (1H, CDCl3): 9,89 (s, 1H, CHO), 5,97 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,70 (s, 1H, H-1'), 4,09 и 3,86 (2d, 2H, 8a-CH2, J=9,7 Гц), 3,70 (m, 1H, H-5'), 3,44 (m, 1H, H-4'), 3,24 и 3,12 (2d, 2H, H-2' и H-3', J=4 Гц), 3,20 (m, 2H, 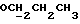 ), 2,65 (t, 1H, H-1, J=3,9 Гц); δ (13C, CDCl3): 209,6 (CHO), 178,38 (CO2Na), 152,4 (C-3), 130,1 (C-2), 100,1 (C-1'), 77,9
), 2,65 (t, 1H, H-1, J=3,9 Гц); δ (13C, CDCl3): 209,6 (CHO), 178,38 (CO2Na), 152,4 (C-3), 130,1 (C-2), 100,1 (C-1'), 77,9 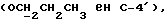 76,6 (C-4'), 73,5 (C-5'), 73,1
76,6 (C-4'), 73,5 (C-5'), 73,1  66,6 (C-3'), 18,9 (C-6').
66,6 (C-3'), 18,9 (C-6').
ПРИМЕР 54
[1R (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-4-О-бензил-6-дезокси- β- D-маннопиранозилокси)метил]-4-формил-4,4а,5,6,7,7a,8,8а-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К суспензии гидрида натрия (22 мг) в сухом диметилформамиде (5 мл) по каплям добавляли раствор Промежуточного соединения 63 (200 мг) в сухом диметилформамиде (7 мл) и перемешивали полученную смесь при комнатной температуре в течение 1 часа. Реакционную смесь нейтрализовали 1N соляной кислотой и добавляли этилацетат (30 мл). Органический слой промывали водой и рассолом, сушили над безводным сульфатом магния, фильтровали и упаривали до сухого состояния. Остаток подвергали хроматографии на колонке быстрого разделения с силикагелем, применяя в качестве элюентов смеси метиленхлорид: метанол (50: 1) и (35:1) и получая в результате соединение, указанное в заголовке, в виде пены белого цвета (150 мг).
δ (1H, CDCl3): 9,86 (s, 1H, CHO), 7,31 (m, 5H,  ), 6,08 (dd, 1H, H-2, J= 1,2 и 3,3 Гц), 4,75 (d, 1H,
), 6,08 (dd, 1H, H-2, J= 1,2 и 3,3 Гц), 4,75 (d, 1H, 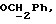 J=11,4 Гц), 4,73 (s, 1H, H-1'), 4,56 (d, 1H,
J=11,4 Гц), 4,73 (s, 1H, H-1'), 4,56 (d, 1H, 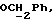 J=11,4 Гц), 4,17 и 3,62 (2d, 2H,
J=11,4 Гц), 4,17 и 3,62 (2d, 2H,  J=9,0 Гц), 3,32 и 3,14 (2d, 2H, H-3' и H-2', J=3,7 Гц), 3,30 (m, 2H, H-4', H-5), 2,71 (t, 1H, H-1); δ (13C, CDCl3): 204,9 (CHO), 175,8 (COOH), 137,2, 128,14, 128,11 (6C-Ph), 148,3 (C-3), 130,8 (C-2), 98,0 (C-1'), 74,4
J=9,0 Гц), 3,32 и 3,14 (2d, 2H, H-3' и H-2', J=3,7 Гц), 3,30 (m, 2H, H-4', H-5), 2,71 (t, 1H, H-1); δ (13C, CDCl3): 204,9 (CHO), 175,8 (COOH), 137,2, 128,14, 128,11 (6C-Ph), 148,3 (C-3), 130,8 (C-2), 98,0 (C-1'), 74,4  74,3 (C-4'), 72,6
74,3 (C-4'), 72,6 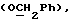 72,5 (C-5'), 65,5 (C-8a), 54,0 (C-2'), 50,3 (C-3'), 18,6 (C-6').
72,5 (C-5'), 65,5 (C-8a), 54,0 (C-2'), 50,3 (C-3'), 18,6 (C-6').
ПРИМЕР 55
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-6-дезокси- β- -D-маннопиранозилокси) метил] -4-формил-4,4а,5,6,7,7а,8,8а-октагидpo-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К суспензии сухого гидрида натрия (75 мг) в сухом диметилформамиде (5 мл) в атмосфере азота добавляли раствор Промежуточного соединения 66 (500 мг) в сухом диметилформамиде (5 мл). Реакционную смесь перемешивали в атмосфере азота в течение 3 часов, а затем гасили реакцию добавлением воды и нейтрализовали до pH 6,5-7 с помощью 0,5H водной соляной кислоты. Смесь концентрировали, а остаток фракционировали в системе вода: этилацетат (1:1; 100 мл). Водную фазу экстрагировали этилацетатом (2x20 мл), органические фазы объединяли и обрабатывали рассолом, затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме до сиропа. Его очищали посредством хроматографии быстрого разделения на силикагеле, элюируя смесью дихлорметан: метанол (40: 1), получая в результате соединение, указанное в заголовке (172 мг), в виде пены белого цвета.
δ (1H, CDCl3): 9,76 (s, 1H, CHO), 6,09 (dd, 1H, H-12, J=1,5 и 3,6 Гц), 4,77 (s, 1H, H-1'), 3,61 и 4,21 (2d, 2H, 8aCH2, J=9 Гц), 3,60 (d, 1H, H-4' J=8,7 Гц), 3,21 (dq, 1H, H-5', J=9 и 6 Гц), 3,16 и 3,27 (2d, 2H, H-2'и H-3', J= 3,6 Гц), 2,70 (t, 1H, H-1), δ (13C, CDCl3); 205,5 (CHO), 174,6 (CO2H), 148,4 (C-3), 130,8 (C-2), 97,8 (C-1'), 74,1 (8aCH2), 73,9 (C-4'), 73,7 (C-3a), 67,2 (C-Ba), 65,5 (C-8a), 59,0 (C-4), 56,5 (C-2'), 50,4 (C-3'), 46,5 (C-1).
ПРИМЕР 56
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[2,3-Ангидро-4,6-дидезокси-4-фтop- β- D-тaлoпиpaнoзилoкcи)метил] -4-формил-4,4a,5,6,7,7a,8,8a -октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а (1H)-карбоновая кислота
К раствору Промежуточного соединения 68 (130 мг) в этилацетате (30 ил) в атмосфере азота добавляли 10%-ный палладий на угле (100 мг). Смесь встряхивали в Parr аппарате при давлении водорода 15 пси в течение 1 часа при комнатной температуре. Катализатор отфильровывали, а растворитель упаривали до сухого состояния. Остаток хроматографировали на колонке для быстрого разделения с силикагелем, применяя в качестве элюентов смеси гексан: этилацетат (4: 1) и дихлорметан: метанол (20:1) и получая в результате соединение, указанное в заголовке, в виде пены белого цвета (82 мг).
δ (1H, CDCl3): 9,79 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 5,13 (s, 1H, H-1'), 4,75 и 4,58 (2dq, 1H, H-5', J5'F=48,3 Гц, J=6,6 Гц, J= 3,9 Гц), 4,18 и 4,11 (2d, 1H, H-4', J=3,9 Гц и J4'F=23,1 Гц), 4,07 и 3,61 (2d, 2H, 8aCH2, J=9,3 Гц), 3,77 (m, 2H, H-2' и H-3'), 2,67 (t, 1H, H-1); δ (13C, CDCl3): 204,6 (CHO), 175,3 (CO2H), 148,2 (C-3), 130,7 (C-2), 101,7 (C-1'), 90,2 (d, C-4', JC4'-F=157 Гц), 80,3 (d, C-5', JC5'-F=21,4 Гц), 74,3 (C-8aCH2), 73,4 (C-3a), 65,4 (C-8a), 58,9 (C-4), 55,9 (C-2'), 55,1 (d, C-3', JC3'-F=6,3 Гц), 16,4 (d, C-6', JC6'-F=21 Гц).
ПРИМЕР 57
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[2,3-Ангидро-6-дезокси-4-O-(2-метоксиэтил)- β- D- маннопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8а-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 69 (140 мл) в этилацетате (50 мл) в атмосфере азота добавляли 10%-ный палладий на угле (70 мг). Смесь встряхивали Parr аппарате при давлении водорода 20 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали на колонке для быстрого разделения с силикагелем, элюируя дихлорметаном и смесью дихлорметан: метанол (40:1), получая в результате соединение, указанное в заголовке (74 мг), в виде пены белого цвета.
δ (1H, CDCl3): 9,78 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,74 (s, 1H, H-1'), 4,23 и 3,60 (2d, 2H, 8a-CH2, J=9 Гц), 3,86- 3,50 (m, 4H, 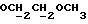 , 3,38 (s, 3H,
, 3,38 (s, 3H,  3,30 и 3,13 (2d, 2H, H-2' и H-3', J= 3,9 Гц), 3,26-3,22 (m, 2H, H-4' и H-5'), 2,68 (t, 1H, H-1, J=3,7 Гц); δ (13C, CDCl3): 204,8 (CHO), 175,2 (COOH), 148,3 (C-3), 130,8 (C-2), 97,9 (C-1'), 74,2
3,30 и 3,13 (2d, 2H, H-2' и H-3', J= 3,9 Гц), 3,26-3,22 (m, 2H, H-4' и H-5'), 2,68 (t, 1H, H-1, J=3,7 Гц); δ (13C, CDCl3): 204,8 (CHO), 175,2 (COOH), 148,3 (C-3), 130,8 (C-2), 97,9 (C-1'), 74,2  72,4 (C-5'), 71,7 и 69,9
72,4 (C-5'), 71,7 и 69,9 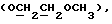 54,1 (C-2'), 50,3 (C-3'), 46,5
54,1 (C-2'), 50,3 (C-3'), 46,5 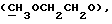 18,5 (C-6').
18,5 (C-6').
ПРИМЕР 58
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-6-дезокси-4-O-(2-метилпропил)- β- -D-маннопиранозилокси)метил]-4-формил-4,4a,5,6,7,7а,8,8а-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 70 (0,14 нмоль) в этилацетате (30 мл) в атмосфере азота добавляли 10%-ный палладий на угле *50 мг). Смесь встряхивали в Parr аппарате при давлении водорода 25 пси в течение 30 минут при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь дихлорметан: метанол и получая в результате соединение, указанное в заголовке (49 мг).
δ (1H, CDCl3): 9,76 (s, 1H, CHO), 6,09 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,73 (s, 1H, H-1'), 4,12 и 3,65 (2d, 2H, 8а-CH2 J=9,3 Гц), 3,49 (m, 1H, H-5'), 3,22 (m, 3H, H-2' и CH2-О), 3,13 (m, 2H, H-3' и H- 4'), 2,75 (t, 1H, H-1, J=3,9 Гц), 2,33 (m, 1H, 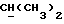 ); δ (13C, CDCl3): 204,8 (CHO), 176,0 (CO2H), 148,2 (C-3), 130,8 (C-2), 98,0 (C-1'), 77,6 (C-1''), 75,2 (C-4'), 74,3 (8a-CH2), 72,9 (C-3a), 72,6 (C-5'), 65,6 (C-8a), 59,0 (C-4), 54,1 (C-2'), 50,4 (C-3'), 46,4 (C-1), 41,7 (C-4a), 41,3 (C-7a).
); δ (13C, CDCl3): 204,8 (CHO), 176,0 (CO2H), 148,2 (C-3), 130,8 (C-2), 98,0 (C-1'), 77,6 (C-1''), 75,2 (C-4'), 74,3 (8a-CH2), 72,9 (C-3a), 72,6 (C-5'), 65,6 (C-8a), 59,0 (C-4), 54,1 (C-2'), 50,4 (C-3'), 46,4 (C-1), 41,7 (C-4a), 41,3 (C-7a).
ПРИМЕР 59
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-6-дезокси-4-O-(1-метилэтил) карбонил- β- D-маннопиранозилокси)метил]-4-формил-4,4a, 5,6,7,7a, 8,8а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 71 (140 мг) в этилацетате (20 мл) в атмосфере азота добавляли 10%-ный палладий на угле (120 мг). Смесь встряхивали в Parr аппарате при давлении водорода 20 пси в течение 1 часа при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали посредством колоночной хроматографии быстрого разделения на силикагеле, элюируя смесями гексан:этилацетат (4:1) и дихлорметан: метанол (20: 1) и получая в результате соединение, указанное в заголовке (103 мг), в виде пены белого цвета.
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 6,09 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,78 (s, 1H, H-1'), 4,66 (d, 1H, H-4', J=9 Гц), 4,10 и 3,60 (2d, 2H, 8aCH2, J= 9 Гц), 3,42 (dq, 1H, H-5', J=9 и 3 Гц), 3,15 (s, 2H, H-2' и H-3'), 2,78 (m, 1H, H-1), 2,58 (m, 1H, 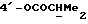 ); δ (13C, CDCl3): 204,9 (CHO), 176,1, 175,9 (CO2 и 4'OCOR), 148,2 (C-3), 130,9 (C-2), 97,9 (C-1'), 74,5 (C-8aCH2), 72,6 (C-3a),71,4 (C-4'), 67,8 (C- 5'), 66,0 (C-8а), 59,0 (C-4), 18,5 и 18,8
); δ (13C, CDCl3): 204,9 (CHO), 176,1, 175,9 (CO2 и 4'OCOR), 148,2 (C-3), 130,9 (C-2), 97,9 (C-1'), 74,5 (C-8aCH2), 72,6 (C-3a),71,4 (C-4'), 67,8 (C- 5'), 66,0 (C-8а), 59,0 (C-4), 18,5 и 18,8 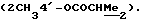
ПРИМЕР 60
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-6-дезокси-4-O-(2,2-диметилпропионил)- β- D- маннопиразанозилокси)метил]-4-формил-4,4а,5,6,7,7а, 8,8а- октагидро-7-метил-3-(1-метилэтил) -1,4-метано-s-индацен-3а(1H) - карбоновая кислота
К раствору Промежуточного соединения 72 (0,21 ммоль) в этилацетате (60 мл) в атмосфере азота добавляли 10%-ный палладий на угле. Смесь встряхивали в Parr аппарате при давлении водорода 20 пси в течение 30 минут при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь дихлорметан: метанол (40:1) и получая в результате соединение, указанное в заголовке (101 мг).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,10 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 4,80 (s, 1H, H-1'), 4,64 (d, 1H, H-4', J=9 Гц), 4,11 и 3,70 (2d, 2H, 8a-CH2, J= 9,3 Гц), 3,43 (m, 1H, H-5'), 3,13 (m, 2H, H-2' и H-3'), 2,78 (t, 1H, H-1, J= 3,9 Гц), 2,34 (m, 1H, 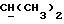 ); δ (13C,CDCl3): 204,9 (CHO), 177,3 и 176,5 (2•CO2), 148,2 (C-3), 130,9 (C-2), 98,0 (C-1'), 74,5 (8а-CH2), 72,6 (C-2), 71,3 (C-5'), 67,9 (C-4'), 65,7 (C-8a), 59,0 (C-4), 54,3 (С-2'), 50,1 (C-3'), 46,3 (C-1), 41,8 (C-4a), 41,3 (C-8), 38,8 (C-tBu), 32,0 (C-5), 30,9 (C-7), 18,5 (tBu).
); δ (13C,CDCl3): 204,9 (CHO), 177,3 и 176,5 (2•CO2), 148,2 (C-3), 130,9 (C-2), 98,0 (C-1'), 74,5 (8а-CH2), 72,6 (C-2), 71,3 (C-5'), 67,9 (C-4'), 65,7 (C-8a), 59,0 (C-4), 54,3 (С-2'), 50,1 (C-3'), 46,3 (C-1), 41,8 (C-4a), 41,3 (C-8), 38,8 (C-tBu), 32,0 (C-5), 30,9 (C-7), 18,5 (tBu).
ПРИМЕР 61
[R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) 8a[(2,3-Ангидро-4-О-бензилоксикарбонил-6-дезокси- β- D -маннопиранозилокси)метил]-4-формил-4,4a,5,6,7,7a,8,8а-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
Раствор Промежуточного соединения 73 (0,22 ммоль) в дихлорметане (10 мл) при 0oC обрабатывали трифторуксусной кислотой (0,1 мл). Спустя два часа смесь промывали водой, а органическую фазу выпаривали. Остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь дихлорметан: метанол (40:1) и получая в результате соединение, указанное в заголовке (60 мг).
δ ( 1H,CDCl3): 9,74 (s,1H,CHO), 7,37 (m,5H,Ph), 6,09 (dd,1H,H-2,J=1,5 и 3,6 Гц), 5,20 (m,2H,  ), 4,75 (s,1H,H-1'), 4,55 (d,1H,H-4', J=9 Гц), 4,08 и 3,70 (2d,2H,8а-CH2, J=9 Гц), 3,47 (m,1H,H-5'), 3,28 и 3,16 (d,d,1H, 1H, H-2' и H-3' Гц), 2,79 (t,1H,H-1, J=3,9 Гц), 2,33 (m,1H,
), 4,75 (s,1H,H-1'), 4,55 (d,1H,H-4', J=9 Гц), 4,08 и 3,70 (2d,2H,8а-CH2, J=9 Гц), 3,47 (m,1H,H-5'), 3,28 и 3,16 (d,d,1H, 1H, H-2' и H-3' Гц), 2,79 (t,1H,H-1, J=3,9 Гц), 2,33 (m,1H,  ); δ (13C, CDCl3): 204,8 (CHO), 176,4 (CO2H), 154,1 (CO3), 148,1 (C-3), 134,6 (Cipso), 130,9 (C-2), 128,8, 128,7, 128,5, 128,4 (Ph), 97,8 (C-1'), 74,5 (8а-CH2), 72,5 (C-3a), 71,6 (C-5'), 71,1 (8C-4'), 70,3
); δ (13C, CDCl3): 204,8 (CHO), 176,4 (CO2H), 154,1 (CO3), 148,1 (C-3), 134,6 (Cipso), 130,9 (C-2), 128,8, 128,7, 128,5, 128,4 (Ph), 97,8 (C-1'), 74,5 (8а-CH2), 72,5 (C-3a), 71,6 (C-5'), 71,1 (8C-4'), 70,3 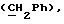 65,7 (C-8a), 58,9 (C-4), 53,9 (C-2'), 50,0 (C-3'), 46,2 (C-1), 41,7 (C-4a), 41,3 (C-7a).
65,7 (C-8a), 58,9 (C-4), 53,9 (C-2'), 50,0 (C-3'), 46,2 (C-1), 41,7 (C-4a), 41,3 (C-7a).
ПРИМЕР 62
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-6-дезокси- 4-оксо- β- D-маннопиранозилокси)метил] -4-формил-4,4a, 5,6,7,7а, 8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s- индацен-3а(1H)-
карбоновая кислота
К раствору Промежуточного соединения 74 (0,26 ммоль) в этилацетате (30 мл) в атмосфере азота добавляли 10%-ный палладий на угле (50 мг). Смесь встряхивали в Parr аппарате при давлении водорода 25 пси в течение 1,5 часов при комнатной температуре. Катализатор отфильтровывали, а растворитель упаривали до сухого состояния. Остаток очищали посредством хроматографии быстрого разделения, применяя в качестве элюента смесь дихлорметан: метанол (40: 1), соответствующие фракции собирали и выпаривали, получая в результате соединение, указанное в заголовке (38 мг).
δ (1H, CDCl3), 9,72 (s, 1H, CHO), 6,12 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,87 (s, 1H, H-1'), 4,10 и 3,76 (2d, 2H, 8aCH2, J=9 Гц), 3,83 (q, 1H, H-5', J=6,6 Гц), 3,60 и 3,38 (d,d, 1H, 1H,3' и 2', J=3,9 Гц), 2,82 (t, 1H, H-1, J= 3,6 Гц), 2,36 (m, 1H, 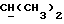 ); δ (13C, CDCl3): 204,9 (CHO), 202,9 (CO-4'), 176,2 (CO2H), 148,2 (C-3), 130,9 (C-2), 96,1 (С-1'), 76,6 (С-5'), 74,7 (8aCH2), 72,1 (C-3a), 65,7 (C-8a), 58,9 (C-4), 54,2 и 53,5 (C-2' и C-3'), 46,1 (C-1), 41,8 (C-4a), 41,3 (C-7a).
); δ (13C, CDCl3): 204,9 (CHO), 202,9 (CO-4'), 176,2 (CO2H), 148,2 (C-3), 130,9 (C-2), 96,1 (С-1'), 76,6 (С-5'), 74,7 (8aCH2), 72,1 (C-3a), 65,7 (C-8a), 58,9 (C-4), 54,2 и 53,5 (C-2' и C-3'), 46,1 (C-1), 41,8 (C-4a), 41,3 (C-7a).
ПРИМЕР 63
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро-4,6-дидезокси- -4-метилен- β- D-маннопиранозилокси)метил] -4-формил-4,4а,5,6,7,7а,8,8а-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
Раствор промежуточного соединения 101 (0,17 ммоль) в дихлорметане (15 мл) при 0oC обрабатывали трифторуксусной кислотой (0,15 мл). Спустя 2 часа его обрабатывали водой (10 мл). Органическую фазу выпаривали, а остаток растворяли в этаноле (15 мл) и 1N соляной кислоте (1,5 мл) и перемешивали при 0oC в течение еще 2 часов. Растворитель удаляли, а остаток очищали, применяя в качестве элюента смесь дихлорметан: метанол 20:1. Соответствующие фракции упаривали, получая в результате соединение, указанное в заголовке (24 мг).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,2 и 3,3 Гц), 5,20 и 5,10 (s,s, 1H, 1H, CH2=C), 4,82 (d, 1H, H-1, J=2,1 Гц), 4,42 (m, 2H, H-5' и H-3'), 4,19 и 3,56 (d,d, 1H,1 H, 8a-CH2, J=9,6 Гц), 3,65 (m,1H,H-2'), 2,63 (t, 1H, H-1, J= 3,9 Гц), 2,36 (m,1H, 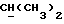 ); δ (13C, CDCl3): 204,7 (CHO), 175,0 (CО2H), 148,5 (C-3), 145,6 (C-4'), 130,5 (C-2), 112,1
); δ (13C, CDCl3): 204,7 (CHO), 175,0 (CО2H), 148,5 (C-3), 145,6 (C-4'), 130,5 (C-2), 112,1 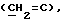 99,5 (C-1'), 74,0 (8a-CH2), 73,0 (C-5'), 72,8 (C-3a), 72,2 и 71,6 (C-2' и C-3').
99,5 (C-1'), 74,0 (8a-CH2), 73,0 (C-5'), 72,8 (C-3a), 72,2 и 71,6 (C-2' и C-3').
ПРИМЕР 64
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-4-O-третбутилкарбонил-6-дезокси- β- -D-талопиранозилокси)метил]-4-формил-4,4а,5,6,7,7a,8,8а-октагидро- 7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-За(1H)-карбоновая кислота
Смесь промежуточного соединения 103 (0,1 ммоль) и воды (2 мл) при 0oC обрабатывали трифторуксусной кислотой (3 мл). Через 90 минут смесь вливали в смесь этиловый эфир: вода 1:1 (30 мл). Органическую фазу упаривали, а остаток очищали хроматографически, применяя в качестве элюента смесь дихлорметан: метанол 40: 1, получая в результате соединение, указанное в заголовке (32 мг).
δ (1H, CDCl3): 9,77 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 4,75 (dd, 1H, H-4', J=3,6 и 4,8 Гц), 4,68 (3, 1H, H-1'), 4,17 (d, 1H, 8a-CHa, J= 9,3 Гц), 3,66 (m, 2H, 8a-CHb и H-5'), 3,57 (t, 1H, H-3', J=4,2 Гц), 3,17 (d, 1H, H-2', J= 3,6 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц), 2,33 (m, 1H, 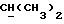 ); δ (13C, CDCl3): 204,8 (CHO), 178,5 и 175,7 (2•CO2), 148,3 (C-3), 130,8 (C-2), 97,9 (C-1'), 74,1 (8a-CH2), 73,0 (C-3а), 71,5 (C-5'), 66,0 (C-4'), 65,6 (C-8a), 59,0 (C-4), 51,3 (C-2'), 50,7 (C-3').
); δ (13C, CDCl3): 204,8 (CHO), 178,5 и 175,7 (2•CO2), 148,3 (C-3), 130,8 (C-2), 97,9 (C-1'), 74,1 (8a-CH2), 73,0 (C-3а), 71,5 (C-5'), 66,0 (C-4'), 65,6 (C-8a), 59,0 (C-4), 51,3 (C-2'), 50,7 (C-3').
ПРИМЕР 65
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[2,3-Ангидро-4-O-(транс-2-бутенил) -6-дезокси β- -D-маннопиранозилокси]метил]-4-формил-4,4а,5,6,7,7а,8,8а-октагидро- 7-метил] -3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
Раствор промежуточного соединения 47 (103 мг) в сухой дихлорметане (5 мл) при 0oC обрабатывали трифторуксусной кислотой (100 мкл). Спустя 1 час смесь фракционировали между этилацетатом (50 мл) и насыщенным водным раствором сульфата натрия (50 мл). Органический слой промывали насыщенным водным раствором сульфата натрия, водой и рассолом, затем сушили и выпаривали. Остаток дважды хроматографировали на силикагеле, элюируя смесью гексан: этилацетат от (2:1) до (1:1), получая в результате соединение, указанное в заголовке (50 мг).
δ (1H, CDCl3): 9,76 (s, 1H, CHO), 6,08 (dd, 1H, H-2, J=1,5 и 3,6 Гц), 5,82-5,68 (m, 1H, 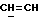 ), 5,62- 5,48 (m, 1H,
), 5,62- 5,48 (m, 1H,  ), 4,72 (s, 1H, H-1'), 4,2-4,1 (m, 2H, O-CHа-C=C и 8а-CHa), 4,02-3,92 (m, 1H, OCHb-C=C), 3,63 (d, 1H, 8a-CHb, J= 9,3 Гц), 3,3-3,15 (m, 3H, H-3', H-4' и H-5'), 3,12 (d, 1H, H-2', J= 3,9 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц), 1,72 (dd, 3H, H3C=C, J=1,2 и 6,6 Гц); δ (13C, CDCl3): 204,8 (CHO), 175,6 (CO2H), 148,2 (C-3), 130,8 (C-2), 131,1 и 126,6
), 4,72 (s, 1H, H-1'), 4,2-4,1 (m, 2H, O-CHа-C=C и 8а-CHa), 4,02-3,92 (m, 1H, OCHb-C=C), 3,63 (d, 1H, 8a-CHb, J= 9,3 Гц), 3,3-3,15 (m, 3H, H-3', H-4' и H-5'), 3,12 (d, 1H, H-2', J= 3,9 Гц), 2,73 (t, 1H, H-1, J=3,9 Гц), 1,72 (dd, 3H, H3C=C, J=1,2 и 6,6 Гц); δ (13C, CDCl3): 204,8 (CHO), 175,6 (CO2H), 148,2 (C-3), 130,8 (C-2), 131,1 и 126,6 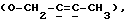 , 97,9 (C-1'), 74,2
, 97,9 (C-1'), 74,2 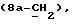 71,2
71,2 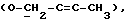 73,5 и 72,5 (C-4' и C-5')
73,5 и 72,5 (C-4' и C-5')
ПРИМЕР 66
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-(2,3-Ангидро-6-дезокси-4-O-пропил- β- D-талопиранозилокси)метил] -4-формил-4,4а,5,6,7,7a,8,8a -октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а (1H)карбоновая кислота
К раствору промежуточного соединения 104 (150 мг, 0,21 ммоль) в этаноле (50 мл), этилацетате (10 мл) и 1N соляной кислоте (1 мл) в атмосфере азота добавляли палладий (10%-ный) на угле (30 мг). Смесь встряхивали в Parr аппарате (PH2=35 пси) в течение 3,5 часов при комнатной температуре. Катализатор фильтровали, а растворитель выпаривали до сухого состояния. Остаток очищали посредством хроматографии, применяя в качестве элюента смесь дихлорметан: метанол 30:1 и получая соединение, указанное в заголовке (60 мг).
δ (12H, CDCl3): 9,83 (s, 1H, CHO), 6,06 (dd, 1H, H-2, J=1,2 и 3,6 Гц), 4,75 (s, 1H, H-1'), 4,44 и 3,42 (dd, 2H, 8a-CH2, J=9,3 Гц), 4,37 (t, 1H, H-3', J=2,7 Гц), 4,10 (m, 1H, H-4'), 3,74- 3,65 (m, 2H, CH2O), 3,91 (m, 1H, H-5'), 3,19 (m, 1H, H-2'), 2,51 (t, 1H, H-1, J=3,6 Гц); δ (13C, CDCl3): 204,7 (CHO), 174,9 (CO2H), 148,6 (C-3), 130,4 (C-2), 97,9 (C-1'), 78,7 (C-4'), 73,9 (8a-CH2), 73,3 (CH2O), 69,8 (C-5'), 69,3 (C-3'), 65,3 (C-8а), 59,0 (C-4), 54,6 (C-2'), 47,0 (C-1).
ПРИМЕР 67
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а (2,3-Ангидро-4-азидо-4,6-дидезокси- β- D-маннопиранозилокси)метил-4-формил-4,4а, 5,6,7,7a, 8,8a-октагидро-7-метил-3- (1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
Раствор промежуточного соединения 106 (0,06 ммоль) в сухом дихлорметане (5 мл) при 0oC обрабатывали трифторуксусной кислотой (70 мкл). Спустя 2 часа его промывали водой и рассолом. Растворитель выпаривали, а остаток очищали хроматографически, применяя в качестве элюента смесь дихлорметан:метанол 30: 1 и получая в результате соединение, указанное в заголовке, (12 мг).
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,09 (d, 1H, H-2, J=3,6 Гц), 4,73 (s, 1H, H-1'), 4,12 и 3,67 (d,d, 1H, 1H, 8a-CH2, J=9 Гц), 3,39 (d, 1H, H-4', J=9 Гц), 3,33 и 3,17 (d,d, 1H, 1H, H-2' и H-3', J=3,6 Гц), 3,22 (m, 1H, H-5'), 2,76 (t, 1H, H-1, J=3,6 Гц), 2,34 (m, 1H, 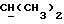 ); δ (13C, CDCl3): 205,1 (CHO), 175,3 (CO2H), 148,4 (C-3), 130,8 (C-2), 97,8 (C-1'), 74,6 (8a-CH2), 72,7 (C-5'), 68,0 (C-3а), 65,7 (C-8а), 59,0 (C-4), 58,7 (C-4), 54,4 (C-2'), 50,1 (C-3'), 46,3 (C-1).
); δ (13C, CDCl3): 205,1 (CHO), 175,3 (CO2H), 148,4 (C-3), 130,8 (C-2), 97,8 (C-1'), 74,6 (8a-CH2), 72,7 (C-5'), 68,0 (C-3а), 65,7 (C-8а), 59,0 (C-4), 58,7 (C-4), 54,4 (C-2'), 50,1 (C-3'), 46,3 (C-1).
ПРИМЕР 68
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(4-O-Аллил, 2,3-ангидро, 6-дезокси- β--D-маннопиранозил)оксиметил] -4-формил-4,4a,5,6,7,7a,8,8а-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
Раствор промежуточного соединения 107 (250 мг) в смеси 10 мл метиленхлорида и 0,2 мл трифторуксусной кислоты перемешивали при 0oC в течение 4 часов. Сырой продукт выпаривали до сухого состояния, а остаток очищали посредством хроматографии быстрого разделения в системе метиленхлорид: метанол 50:1, получая в результате 140 мг соединения, указанного в заголовке, в виде пены белого цвета.
δ (1H, CDCl3): 9,77 (s, 1H, CHO), 6,08 (dd, 1H, H-12, J=1,2 и 3,3 Гц), 5,90 (m, 1H, 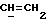 ), 5,28 (m, 2H,
), 5,28 (m, 2H, 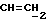 ), 4,75 (s, 1H, H-1'), 4,28 (m, 1H, H-5'), 4,21 (d, 1H, H-19a, J=9,6 Гц), 4,05 (m, 1H, H-4'), 4,05 (m, 1H, H-4'), 3,60 (d, 1H, H-19b, J=9,6 Гц), 3,27, 3,14 (d,d, 1H, 1H, H-2', H-3', J= 3,9 Гц), 3,25 (m, 2H,
), 4,75 (s, 1H, H-1'), 4,28 (m, 1H, H-5'), 4,21 (d, 1H, H-19a, J=9,6 Гц), 4,05 (m, 1H, H-4'), 4,05 (m, 1H, H-4'), 3,60 (d, 1H, H-19b, J=9,6 Гц), 3,27, 3,14 (d,d, 1H, 1H, H-2', H-3', J= 3,9 Гц), 3,25 (m, 2H, 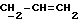 ), 2,68 (t, 1H, H-11, J=4,2 Гц); δ (13C, CDCl3): 205,07 (CHO), 174,6 (CO2H), 148,4 (C-13), 133,7
), 2,68 (t, 1H, H-11, J=4,2 Гц); δ (13C, CDCl3): 205,07 (CHO), 174,6 (CO2H), 148,4 (C-13), 133,7 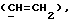 129,7 (C-12), 118,0
129,7 (C-12), 118,0 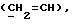 , 97,8 (C-1'), 74,17 (C-19), 74,04 (C-4'), 72,5 (C-5'), 71,0
, 97,8 (C-1'), 74,17 (C-19), 74,04 (C-4'), 72,5 (C-5'), 71,0 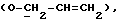 54,1 (C-2'), 50,2 (C-3'), 27,5 (C-14), 22,7 (0-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
54,1 (C-2'), 50,2 (C-3'), 27,5 (C-14), 22,7 (0-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
ПРИМЕР 69
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро,6-дезокси, 4-O-третбутоксикарбонилметил- β- D-маннопиранозил)оксиметил] -4-формил-4,4а, 5,6,7,7а, 8,8а-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 21 (200 мг) в 15 мл сухого ТГФ при 0oC добавляли 30 мг гидрида натрия. Смесь перемешивали при 0oC в атмосфере азота в течение 30 минут. Затем добавляли 260 мкл трет-бутил-бромацетата и перемешивали смесь при комнатной температуре в течение 3 суток. Сырой продукт обрабатывали хлоридом аммония 1N и этилацетатом. Органический слой промывали водой и рассолом и упаривали до сухого состояния. Остаток гидрогенизировали стандартным способом, а сырой продукт очищали посредством хроматографии быстрого разделения в системе метиленхлорид:метанол 50:1, получая в результате 140 мг соединения, указанного в заголовке, в виде пены белого цвета.
δ (1H, CDCl3): 9,74 (s, 1H, CHO), 6,08 (dd, 1H, H-12, J-1,5 и 3,9 Гц), 4,74 (s, 1H, H-1'), 4,21 (dd, 1H, H-4', J=3,6 и 6,0 Гц), 4,16 (d, 1H, H-19a, J= 9 Гц), 4,12 (s, 2H, 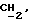 -CO2C(CH3)3), 3,63 (d, 1H, H-19b, J=9 Гц), 3,36, 3,14 (d, d, 1H, 1H, H-2', H-3', J=3,9 Гц), 3,30 (m, 1H, H-5'), 2,71 (t, 1H, H-11, J= 3,9 Гц); δ (13C, CDCl3): 205,7 (CHO), 174,9 (CO2C(CH3)3), 168,8
-CO2C(CH3)3), 3,63 (d, 1H, H-19b, J=9 Гц), 3,36, 3,14 (d, d, 1H, 1H, H-2', H-3', J=3,9 Гц), 3,30 (m, 1H, H-5'), 2,71 (t, 1H, H-11, J= 3,9 Гц); δ (13C, CDCl3): 205,7 (CHO), 174,9 (CO2C(CH3)3), 168,8  148,4 (C-13), 130,8 (C-12), 97,8 (C-1'), 82,2
148,4 (C-13), 130,8 (C-12), 97,8 (C-1'), 82,2  75,6 (C-4'), 72,4 (C-5'), 68,2
75,6 (C-4'), 72,4 (C-5'), 68,2 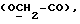 59,0 (C-3), 53,9 (C-2'), 50,3 (C-3'), 27,5 (C-14), 22,7 (C-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
59,0 (C-3), 53,9 (C-2'), 50,3 (C-3'), 27,5 (C-14), 22,7 (C-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
ПРИМЕР 70
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8а-[(2,3-Ангидро-6-дезокси-4-O-этил- β-D-маннопиранозил)оксиметил] -4-формил- 4,4a,5,6,7,7a,8,8a-октагидро-7-метил-3-(1-метилэтил)-1,4- метано-s-индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 21 (290 мг) в 5 мл сухого диметилформамида при 0oC в атмосфере азота добавляли 25 мг гидрида натрия. Смесь перемешивали в течение 30 минут при 0oC, а затем добавляли 1 ммоль этилиодида. Реакция завершалась после 16-часового перемешивания при комнатной температуре. Сырой продукт обрабатывали хлоридом аммония 1N и этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным сульфатом магния и упаривали до сухого состояния. Остаток гидрогенизировали стандартным способом, а сырой продукт очищали посредством хроматографии быстрого разделения в системе метиленхлорид: метанол, 50:1, получая в результате 180 мг соединения, указанного в заголовке, в виде пены белого цвета.
δ (1H, CDCl3): 9,73 (s, 1H, CHO), 6,08 (d, 1H, H-12, J=3,3 Гц), 4,73 (s, 1H, H-1'), 4,16 (d, 1H, H-19a, J=9,3 Гц), 3,78, 3,54 (m,m, 1H, 1H, 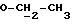 ), 3,63 (s, 1H, H-19b, J=9,3 Гц), 3,25, 3,13 (d,d, 1H, 1H, H-2', H-3', J=3,9 Гц), 3,19 (m, 2H, H-4', H-5'), 2,71 (t, 1H, H-11, J=3,3 Гц); δ (13C,CDCl3): 201,8 (CHO), 175,0 (CO2H), 148,2 (C-13), 130,7 (C-12), 97,9 (C-1'), 74,6 (C-4'), 74,3 (С-19), 72,5 (C-5'), 54,3 (C-2'), 50,3 (C-3'), 27,5 (C-14), 22,7 (C-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
), 3,63 (s, 1H, H-19b, J=9,3 Гц), 3,25, 3,13 (d,d, 1H, 1H, H-2', H-3', J=3,9 Гц), 3,19 (m, 2H, H-4', H-5'), 2,71 (t, 1H, H-11, J=3,3 Гц); δ (13C,CDCl3): 201,8 (CHO), 175,0 (CO2H), 148,2 (C-13), 130,7 (C-12), 97,9 (C-1'), 74,6 (C-4'), 74,3 (С-19), 72,5 (C-5'), 54,3 (C-2'), 50,3 (C-3'), 27,5 (C-14), 22,7 (C-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
ПРИМЕР 71
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро, 6-дезокси-4-O- (2,3-дигидроксипропил) - β-D-маннопиранозил)оксиметил] ] -4-формил-4,4a, 5,6,7,7a, 8,8а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано -s-индацен-3а(1H) -карбоновая кислота
К раствору Промежуточного соединения 108 (700 мг) в этаноле (100 мл) в атмосфере азота добавляли палладий (10%-ный) на угле (350 мг). Смесь встряхивали в Parr аппарате (PH2=20 пси) в течение 1 часа при комнатной температуре. Катализатор фильтровали а растворитель выпаривали до сухого состояния. Остаток очищали на колонке с силикагелем, элюируя метиленхлоридом и смесью метиленхлорид: метанол 25:1, получая в результате 425 мг соединения, указанного в заголовке (смесь изомеров 50:50), в виде пены белого цвета.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,08 (dd, 1H, H-12, J=1,2 и 3,6 Гц), 4,73 (s, 1H, H-1'), 3,98 (d, 1H, H-19a, J=9,6 Гц), 3,81 (d, 1H, H-19b, J= 9,6 Гц), 3,81-3,68 (m, 2H, 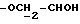 в C-4'), 3,55-3,31 (m, 3H,
в C-4'), 3,55-3,31 (m, 3H, 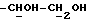 в C-4'), 3,30-3,11 (m, 4H, H-2', H-3', H-4', H-5'), 2,76 (t, 1H, H-11, J=3,6 Гц); δ (13C, CDCl3): 207,70 (CHO), 178,2 (CO2H), 150,49 (C-13), 131,44 (C-12), 99,81 (C-1'), 77,07 и 76,96 (
в C-4'), 3,30-3,11 (m, 4H, H-2', H-3', H-4', H-5'), 2,76 (t, 1H, H-11, J=3,6 Гц); δ (13C, CDCl3): 207,70 (CHO), 178,2 (CO2H), 150,49 (C-13), 131,44 (C-12), 99,81 (C-1'), 77,07 и 76,96 ( 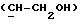 в C-4'), 76,57 (C-19), 73,55 (C-4'), 72,86 и 72,69 (
в C-4'), 76,57 (C-19), 73,55 (C-4'), 72,86 и 72,69 ( 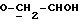 в C-4'), 72,24 (C-5'), 55,01 (C-3'), 51,89 (C-2'), 23,2 (C-20), 21,6 (C-15), 19,4 (C-6'), 17,9 (C-17).
в C-4'), 72,24 (C-5'), 55,01 (C-3'), 51,89 (C-2'), 23,2 (C-20), 21,6 (C-15), 19,4 (C-6'), 17,9 (C-17).
ПРИМЕР 72
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a-[(2,3-Ангидро, 6-дезокси-4-O-((2,3-диметоксипропил)- β-D- маннопиранозил)оксиметил] -4-Формил-4,4а, 5,6,7,7а,8,8а- октагидро-7-метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H) - карбоновая кислота
К раствору Промежуточного соединения 108 (350 мг) в сухом ТГФ (20 мл) в атмосфере азота добавляли гидрид натрия (40 мг), смесь перемешивали в течение 30 минут, затем добавляли метилиодид (0,5 мл) и реакция проходила при перемешивании при комнатной температуре в течение двух суток в атмосфере азота. Сырой продукт обрабатывали 1N хлоридом аммония и этилацетатом. Органический слой промывали водой и рассолом и выпаривали до сухого состояния. Остаток гидрогенизировали стандартным способом без предварительной очистки, получая в результате соединение, указанное в заголовке (смесь изомеров 50: 50), в виде белой пены (225 мг).
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,08 (d, 1H, H-12, J=3,3 Гц), 4,73 (d, 1H, H-1', J= 1,5 Гц), 4,18 (d, 1H, H-19b, J=9 Гц), 3,62 (d, 1H, H-19b, J=9 Гц), 3,48 и 3,47 (s,s, 3H, 3H,  , 3,36-3,20 (m, 4H, H-2', H-3', H-4', H-5'), 2,70 (t, 1H, H-11, J=3,6 Гц); δ (13C,CDCl3): 205,2 (CHO), 174,7 (CO2H), 148,4 (C-11), 130,7 (C-12), 97,9 (C-1'), 79,0
, 3,36-3,20 (m, 4H, H-2', H-3', H-4', H-5'), 2,70 (t, 1H, H-11, J=3,6 Гц); δ (13C,CDCl3): 205,2 (CHO), 174,7 (CO2H), 148,4 (C-11), 130,7 (C-12), 97,9 (C-1'), 79,0  в C-4'), 75,6 и 75,7 (C-4'), 72,4 (C-5'), 71,6 и 71,5
в C-4'), 75,6 и 75,7 (C-4'), 72,4 (C-5'), 71,6 и 71,5  в C-4'), 70,1 и 69,8 (
в C-4'), 70,1 и 69,8 ( 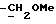 в C-4'), 59,2, 57,9 и 57,8 (
в C-4'), 59,2, 57,9 и 57,8 ( 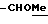 и
и 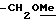 в C-4'), 53,9 и 50,2 (C-2' и C-3'), 22,7 (C-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
в C-4'), 53,9 и 50,2 (C-2' и C-3'), 22,7 (C-20), 21,2 (C-15), 18,6 (C-6'), 17,4 (C-17).
ПРИМЕР 73
[1R- (1α,3aβ,4β,4aβ,7β,7aα,8aβ) ] 8a- [(2,3-Ангидро, 6-дезокси-4-O-[(2,3-O-изопропилиден) -2,3-дигидроксипропил]- β-D-маннопиранозил)оксиметил] -4-формил-4,4а, 5,6,7,7а, 8,8а-октагидро-7- метил-3-(1-метилэтил)-1,4-метано-s-индацен-3а(1H)-карбоновая кислота
К раствору Промежуточного соединения 109 (250 мг) в этилацетате (50 мл) в атмосфере азота добавляли палладий (10%-ный) на угле (100 мг). Смесь встряхивали в аппарате (PH2=20 пси) в течение 1 часа при комнатной температуре. Катализатор фильтровали, а растворитель выпаривали до сухого состояния. Остаток очищали посредством хроматографии быстрого разделения в системах н-гексан: этилацетат 1:1 и метиленхлорид: метанол 50:1, получая в результате соединение, указанное в заголовке (смесь
изомеров 50:50), в виде пены белого цвета.
δ (1H, CDCl3): 9,75 (s, 1H, CHO), 6,08 (dd, 1H, H-12, J=1,5 и 3,6 Гц), 4,71 (d, 1H, H-1', J=0,9 Гц), 4,29-4,24 (m, 1H, 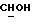 в 4'), 4,15-4,01 (m, 3H, H-19a и
в 4'), 4,15-4,01 (m, 3H, H-19a и 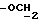 , 3,82-3,49 (m, 4H, H-19b,
, 3,82-3,49 (m, 4H, H-19b, 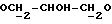 3,29 и 3,13 (d, d, 1H, 1H, H-2' и H-3', J=3,9 Гц), 3,27- 3,18 (m, 2H, H-4'и H-5'), 2,74 (t, 1H, H-11, J= 3,6 Гц), 1,41 и 1,35 (d,d, 3H, 3H,
3,29 и 3,13 (d, d, 1H, 1H, H-2' и H-3', J=3,9 Гц), 3,27- 3,18 (m, 2H, H-4'и H-5'), 2,74 (t, 1H, H-11, J= 3,6 Гц), 1,41 и 1,35 (d,d, 3H, 3H, 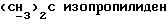 ; δ (13C, CDCl3): 204,8 (CHO), 165,7 (CO2H, 148,2 (C-11), 130,8 (C-12), 97,9 (C-1'), 75,7 и 75,6 (
; δ (13C, CDCl3): 204,8 (CHO), 165,7 (CO2H, 148,2 (C-11), 130,8 (C-12), 97,9 (C-1'), 75,7 и 75,6 ( 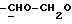 в C-4'), 74,4 (C-4'), 72,8 и 72,4 (C-5'), 72,0 и 71,6
в C-4'), 74,4 (C-4'), 72,8 и 72,4 (C-5'), 72,0 и 71,6 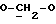 в C-4'), 54,0 и 53,8 (C-3'), 50,3 (C-2'), 22,6 (C-20), 21,1 (C-15), 18,6 (C-6'), 17,4 (C-17).
в C-4'), 54,0 и 53,8 (C-3'), 50,3 (C-2'), 22,6 (C-20), 21,1 (C-15), 18,6 (C-6'), 17,4 (C-17).
ПРИМЕР 74
Характеристика IMI 362184
IMI 362184 является мутантом Sordaria araneosa (ATCC 36386, NRRL 3196), выделенным после обработки мутагеном N-метил-N'- нитро-N-нитрозогуанидином аскоспор этого штамма. Характеристики IMI 362184 в основном сходны с характеристиками, описанными [1] для NRRL 3196, с тем исключением, что IMI 362184 продуцирует 4' -деметилсордарин в качестве основного продукта в тех же условиях, которые применяют для продуцирования сордарина штаммом NRRL 3196.
ПРИМЕР 75
Характеристика IMI 362947
IMI 362947 является мутантом Sordaria araneosa (ATCC 36386, NRRL 3196), выделенным после обработки мутагеном N-метил-N'-нитро- N-нитрозогуанидином аскоспор этого штамма. Характеристики IMI362947 в основном сходны с характеристиками, описанными [1] для NRRL 3196, с тем исключением, что IMI 362947 не способен быстро продуцировать аскоспоры при культивировании на arape. Этот штамп также отличается от NRRL 3196 тем, что он продуцирует сордарицин в качестве основного продукта в тех же условиях, которые применяют для продуцирования сордарина штаммом NRRL 3196.
ПРИМЕР 76
Характеристика NCIMB 40675
NCIMB 40675 является аэробной. Грамм-положительной неподвижной несимметричной палочкой, которая продуцирует лимонно-желтые, полупрозрачные, круглые, чистые, выпуклые колонии с диаметром между 0,5-1 мм при росте в течение 48 часов при 28oC на триптозно-соевом агаре с добавлением 2% (в/об) дрожжевого экстракта. Организм хорошо растет при температурах до 37oC, но не при 45oC. Метахроматических гранул не наблюдали, штамм является каталаза-положительным, оксидаза-отрицательным и не утилизирует ферментативно глюкозу. Данный штамм может утилизировать следующие источники углерода: α- D-глюкозу, D-фруктозу, п-гидроксифенил-уксусную кислоту, D-маннит, метилпируват, лактамид, D-трегалозу и сахарозу. Указанный организм может только слабо утилизировать D-глюконовую кислоту, пировиноградную кислоту и салицин в качестве единственных источников углерода. Морфология колоний и микроскопическая морфология соответствует морфологии булавовидных бактерий. Род Corynebacteriym был исключен на том основании, что пептидоглюкан из NCIMB 40675 содержит орнитин вместо мезо-изомера 2,6-диаминопимелиновой кислоты или диамино-масляной кислоты. Также этот организм содержит комплексную смесь жирных кислот с разветвленными цепями, нетипичную для видов Corynebacterium, а именно 12-метилтетрадекановую, 14-метилгексадеконовую и 14-метилпентадекановую кислоты. Присутствие α- разветвленных- β- гидроксилированных жирных кислот не было определено. На основании этих результатов NCIMB 40675 наиболее близко сходен с одним из следующих родов актинобактерий: Aureo-bacterium, Curtobacterium или Cellulomonas.
Для уточнения таксонометрического положения NCIMB 40675 было проведено сравнение неполной последовательности из 1100 пар оснований гена 16S рРНК с 24 другими видами, представляющими набор актинобактерий и родственных родов. Результаты этих анализов показывают, что имеется близкое родство между NCIMB 40675 и родом Aureobacterium и Curtobacterium, но не Cellulomonas или Corynebacterium. Более точная идентификация штамма может быть проведена путем выполнения филогенетического анализа сравнения различных областей гена 16S рРНК для различных видов Aureobacterium и Curtobacterium в дополнение к последующим хемотаксономическим и физиологическим тестам.
Фармацевтические Примеры
1. Стандартная пероральная таблетка
Лекарственное вещество - 100 мг
Микрокристаллическая целлюлоза - 160 мг
Натрий-кросскармелоза - 20 мг
Стеарат магния - 5 мг
Лекарственное вещество смешивают с микрокристаллической целлюлозой, натрий-кросскармелозой и стеаратом магния, а затем прессуют в таблетки.
Жевательная пероральная таблетка
Лекарственное вещество - 100 мг
Ксилит - 865 мг
Экстракт мяты перечной (пепперминт) - 5 мг
Аспартам - 10 мг
Поливинилпирролидон - 15 мг
Стеарат магния - 5 мг
Лекарственное вещество, ксилит, аспартам и поливинилпирролидон смешивают вместе и гранулируют с водой, а затем сушат. Эту гранулу смешивают с экстрактом мяты перечной и стеаратом магния, а затем формуют в таблетки.
Водный раствор для перорального введения
Лекарственное вещество - 100 мг
Гидроксипропилметилцеллюлоза - 150 мг
Пропилгидроксибензоат натрия - 1 мг
Метилгидроксибензоат натрия - 2 мг
Апельсиновый ароматизатор - 10 мг
Сахарин натрия - 5 мг
Сахароза - 800 мг
Соответствующие буферы - Сколько требуется
Очищенная вода до - 5 мл
Растворяют лекарственное вещество и все эксципиенты в большей части очищенной воды и перемешивают. Доводят до объема и перемешивают. Для поддержания pH в области максимальной стабильности могут быть добавлены соответствующие буферы.
Неводная суспензия для перорального введения
Лекарственное вещество - 100 мг
Аспартам - 50 мг
Грейпфрутовый ароматизатор - 25 мг
Маннит - 800 мг
Коллоидный кремний - 10 мг
Фракционированное кокосовое масло - 5 мл
Диспергируют лекарственное вещество и маннит в объеме фракционированного кокосового масла посредством высокоскоростного перемешивания. Добавляют оставшийся ингредиенты и перешивают. Доводят объем фракционированным кокосовым маслом и перемешивают.
5. Мазь
Лекарственное вещество - 200 мг
Мягкий белый парафин - 9800 мг
Плавят мягкий белый парафин, добавляют лекарство и перемешивают. Продолжают перемешивание до тех пор, пока мазь не начнет застывать.
6. Инъекция
Лекарственное вещество - 40 мг
Соответствующие буферы - сколько требуется
Соответствующие антиоксиданты - сколько требуется
Соответствующие хелатирующие агенты - сколько требуется
Вода для инъекций до - 2 мл
Растворяют лекарственное вещество в большей части воды для инъекций. Для поддержания pH в области оптимальной стабильности могут быть добавлены соответствующие забуферивающие агенты. Для повышения стабильности инъекции могут быть добавлены соответствующие антиоксиданты и хелатирующие агенты. Доводят до отметки водой для инъекций. Заполняют ампулы или флаконы, а затем стерилизуют путем автоклавирования. Можно стерилизовать фильтрацией и заполнять емкости асептически.
Антигрибная активность
Соединения формулы (I) тестировали на антигрибную активность посредством стандартного скриннинга in vitro и определяли для каждого соединения минимальную ингибирующую концентрацию (МИК; мкг/мл) против набора клинически значимых патогенов. Ниже приведены результаты, полученные для некоторых соединений по изобретению.
Соединения по изобретению в целом нетоксичны в терапевтически применяемых дозировках. Например, соединение из примера 8 при введении перорально в дозе 50 мг/кг обеспечивало защиту самцов мышей, инфицированных C. albicans 4711E. Значение ЛД50 для соединения из примера 8 на самцах мышей составляло свыше 1000 мг/кг перорально.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ ВИТАМИНА D, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2173682C2 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ АМИНОДИГИДРОТИАЗИНА | 2009 |
|
RU2476431C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СИГМА РЕЦЕПТОРА | 2007 |
|
RU2451015C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУКЦИНИМИДНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2298554C2 |
| 5-ГЕТЕРОЦИКЛО-1,5-БЕНЗОДИАЗЕПИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1995 |
|
RU2152939C1 |
| 3-АЗАБИЦИКЛО[3.1.0]ГЕКСИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2510396C2 |
| (6S,9AS)-N-БЕНЗИЛ-6-[(4-ГИДРОКСИФЕНИЛ)МЕТИЛ]-4,7-ДИОКСО-8-({ 6-[3-(ПИПЕРАЗИН-1-ИЛ)АЗЕТИДИН-1-ИЛ]ПИРИДИН-2-ИЛ} МЕТИЛ)-2-(ПРОП-2-ЕН-1-ИЛ)-ОКТАГИДРО-1H-ПИРАЗИНО[2,1-C][1,2,4]ТРИАЗИН-1-КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2669805C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СИГМА РЕЦЕПТОРОВ | 2011 |
|
RU2582338C2 |
| СПОСОБ ПОЛУЧЕНИЯ 17-(3-ГИДРОКСИПРОПИЛ)-17-ГИДРОКСИСТЕРОИДОВ | 2008 |
|
RU2466137C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И БЕНЗОМОРФОЛИНА В КАЧЕСТВЕ МОДУЛЯТОРА МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2009 |
|
RU2517181C2 |
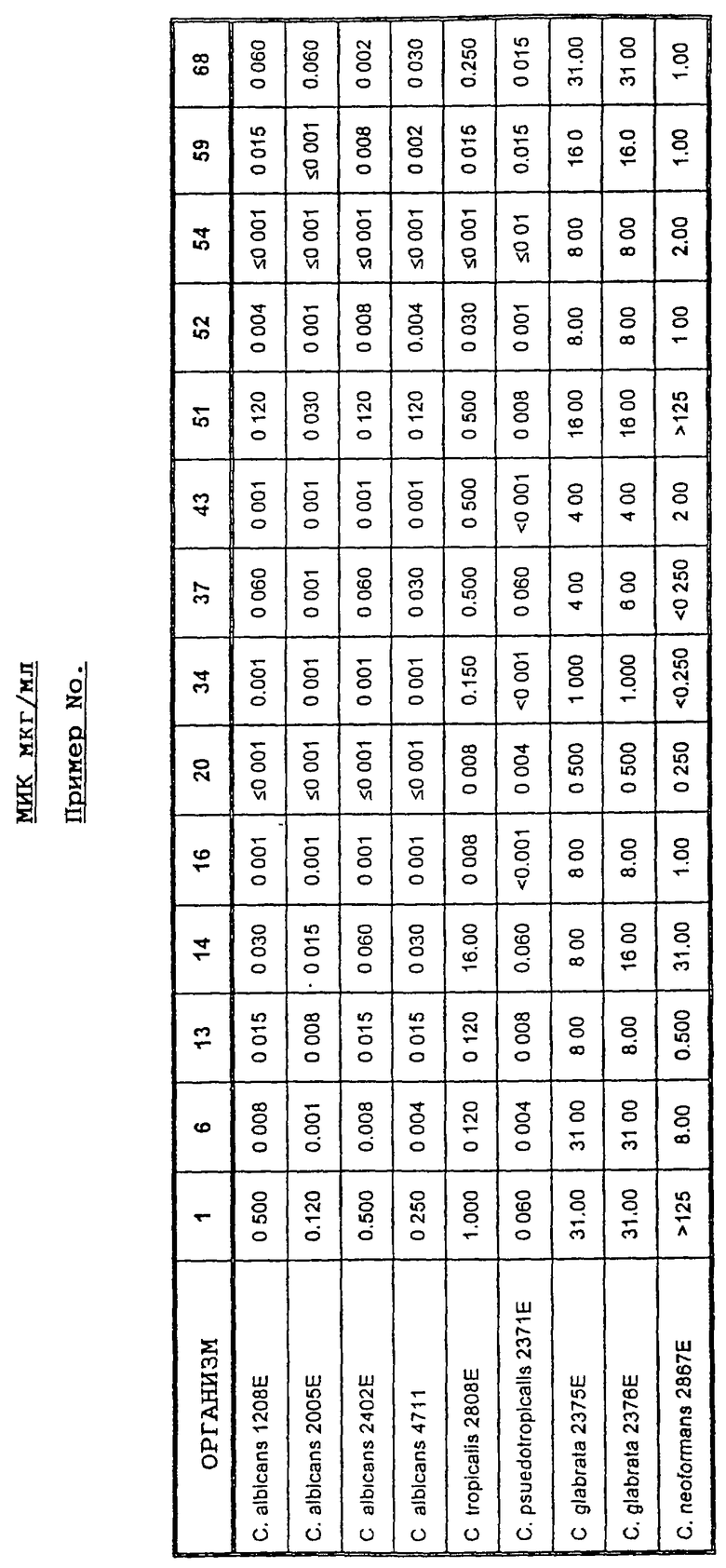
Описываются новые производные сордарина общей формулы I, где Z является тетрагидропираногруппой, выбранной из формул (а) и (б). Значения радикалов X, Y, W, R1-R17 указаны в формуле изобретения. Они обладают противогрибковой активностью. Описывается фармацевтический состав на их основе. 3 с. и 10 з. п. ф-лы, 1 табл.
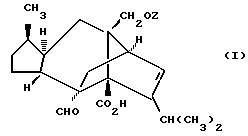
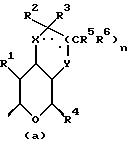
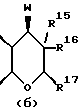
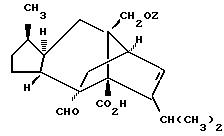
где Z является тетрагидро-пираногруппой, выбранной из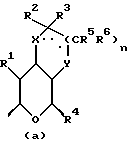
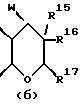
и его фармацевтически приемлемые соли и сольваты (например гидраты) или метаболически лабильные производные,
где R1 представляет собой водород, гидроксил;
R2 и R3 каждый может независимо представлять собой водород С1-6 алкил или С1-4 алкокси С1-4 алкил, или R2 и R3 могут вместе с атомом углерода, к которому они присоединены, представлять собой С=О, С=S или С3-8 циклоалкил;
R4 представляет собой водород или CH2R7 (где R7 является водородом, С1-4 алкокси);
R5 и R6 каждый может независимо представлять собой водород;
n = 0 или 1;
X и Y каждый может независимо представлять собой кислород или CR9R10, (где R9 и R10 каждый может независимо представлять собой водород, С1-6 алкил, С1-4 алкокси или R9 и R10 могут вместе с атомом углерода, к которому они присоединены, представлять собой С=О, С=СНR11, где R11 представляет собой водород; либо если X или Y является кислородом, а n = 0, то -Y-CR2R3 или -X-CR2R3-, соответственно, могут также представлять собой -NR12, где R12 является С1-4 алкилом; либо, если Y является кислородом, а n = 0, то Х может представлять собой группу СR11, которая присоединена к пирановому кольцу двойной связью;
R15 представляет собой водород, галоген, азидогруппу, С1-6 алкокси (возможно замещенный одним или двумя гидрокси или его кеталем или одной или двумя С1-3 алкоксильными группами), арил С1-4 алкокси, С3-6 алкенилокси, группу ОСОR18, где R18 является арил С1-4 алкоксильной или C1-10 алкильной группой; либо С1-6 алкоксикарбонил С1-4 алкокси;
R16 представляет собой водород, или
R15 и R16 могут вместе с атомом углерода, к которому они присоединены, представлять собой С=О или C=СH2;
R17 представляет собой CH2R19, где R19 является водородом;
W представляет собой атом кислорода или серы;
пунктирная линия в группе (а) показывает возможное присутствие дополнительной связи.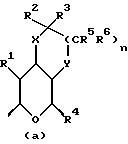
или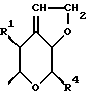
где один из X или Y является кислородом, а другой является кислородом или группой СR5R10.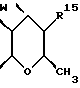
где W является кислородом.
| Способ получения защищенной 4,6-0-алкилиден- - -глюкопиранозы | 1969 |
|
SU447886A1 |
| JP 06157582 A, 03.06.94 | |||
| JP 62040292 A, 21.02.92. | |||
