В центральной нервной системе (ЦНС) млекопитающих передача нервных импульсов контролируется взаимодействием между нейротрансмиттером, который выделяется передающим нейроном, и поверхностным рецептором принимающего нейрона, что вызывает возбуждение этого принимающего нейрона.
L -глутамат, который является наиболее распространенным нейротрансмиттером в ЦНС, служит промежуточным звеном на основном пути передачи возбуждения у млекопитающих и носит название возбудительной аминокислоты (ВАК). Рецепторы, которые реагируют на глутамат, называют рецепторами возбудительных аминокислот (ВАКрецепторами) см. работы Watkins and Evans, Ann. Rev. Pharmacol. Toxicol., 21, 165(1981); Monaghan, Bridges and Cotmon в Ann. Rev. Pharmacd. Toxicol. , 29, 365 (1989); Watkins, Krogspaard-Larsen and Honore, Trans. Pharm. Sci.,11, 25 (1990). Возбудительные аминокислоты имеют огромное физиологическое значение, играя роль в самых разнообразных физиологических процессах. В частности, в долговременном сохранении способностей к обучению и запоминанию, в развитии синаптической гибкости, двигательном контроле, дыхании, сердечно- сосудистой регуляции, эмоциональных состояниях и сенсорном восприятии.
Излишнее или неуместное стимулирование рецепторов возбудительных аминокислот приводит к повреждению или утрате нервной клетки по механизму, известному как эксцитотоксичность. Полагают, что такой процесс является промежуточным звеном перерождения нейронов в самых различных условиях. Медицинское следствие такого нейронного перерождения обуславливает важность терапевтической цели, каковой является ослабление этих процессов нейрологического перерождения.
Рецепторы возбудительных аминокислот подразделяют на два основных типа. Рецепторы, которые непосредственно связаны с открыванием катионных каналов клеточных мембран нейронов, носят название "ионотропных". Рецепторы этого типа подразделяют по меньшей мере на три подтипа, которые определяются деполяризационными действиями селективных агонистов N-метил-D-аспартата (NMDA), альфа-амино-3-окси-, 5- метилизоксазол-4-пропионовой кислоты (АМПК) и каиновой кислоты (КК). Рецептором второго основного типа является G-белок или вторичный мессенджерсвязанный "метаботропный" рецептор возбудительной аминокислоты. Этот второй тип связан с многочисленными вторыми мессенджерными системами, что приводит к улучшенному фосфоинозитидному гидролизу, активации фосфолипазы D, увеличению или уменьшению ЦАМФ-образования и изменяет функционирование ионных каналов [см. работу Schoepp and Conn в Trends in Pharmacol. Sci. , 14, 13 (1993)] . По-видимому, рецепторы обоих типов не только служат промежуточным звеном нормальной синаптической передачи возбуждения вдоль пути возбуждения, но также принимают участие в модификации синаптических связей в процессе развития и всей жизни [см. работы Schoepp, Backaert and Sladecrek, Trends in Pharmacol. Sci., 11,508 (1990); McDonald and Johnson, Brain Research Reviews 15,41 (1990)].
Метаботропные глутаматные рецепторы представляют собой исключительно гетерогенное семейство глутаматных рецепторов, которые связаны с многочисленными вторично-мессенджерными путями. Обычно роль этих рецепторов заключается в модуляции пресинаптического выделения глутамата и в постсинаптической чувствительности нейронной клетки к глутаматному возбуждению. Метаботропные глутаматные рецепторы (мГлуР) фармакологически подразделяют на два подтипа. Рецепторы одной группы положительно связаны с фосфолипазой С, которая вызывает гидролиз клеточных фосфоинозитидов (Р1). Рецепторы этой первой группы называют P1-связанными метаботропными глутаматными рецепторами. Рецепторы второй группы отрицательно связаны с аденилциклазой, которая предотвращает инициированное форсколином аккумулирование циклического аденозинмонфосфата (сАМР) [см. работу Schoepp and Conn Trends in Phamacol. Sci., 14, 13 (1993)] . Рецепторы, входящие в эту вторую группу, называют САМР-связанными метаботропными глутаматными рецепторами. Агонисты САМР-связанных метаботропных глутаматных рецепторов должны быть полезными при лечении острых и хронических неврологических заболеваний и психиатрических болезней.
Недавно были открыты соединения, которые воздействуют на метаботропные глутаматные рецепторы, но никак не влияют на ионотропные глутаматные рецепторы. (1S, 3R)-1- аминоциклопентан-1,3-дикарбоновая кислота (1S, 3Р-АЦПД) является агонистом P1-связанных и САМР-связанных метаботропных глутаматных рецепторов [см. работы Schoepp, Johnson, True, and Monn в Eur. J.Pharmacol., 207, 351 (1991); Schoepp, Johnson and Monn. J.Neurochem 58, 1184 (1992)]. Недавно в качестве селективного агониста САМР-связанного метаботропного глутаматного рецептора был описан (2S, 3S, 4S-2-(карбоксициклопропил)-глицин (L-КЦГ-1), однако в повышенных концентрациях это соединение проявляет активность при P1-связанных метаботропных рецепторах [см.работы NaRagawa и др. в Eur. J.Pharmacol., 184, 205 (1990); Hayashi, et al. в Br.J.Pharmacol., 107, 539 (1992); Schoepp et al. в J.Neurochem., 63, стр. 769-772 (1994)].
В соответствии с настоящим изобретением предлагаются соединения, которые селективно воздействуют на отрицательно-связанные сАМР-связанные метаботропные глутаматные рецепторы. Более конкретно настоящее изобретение относится к соединениям формулы I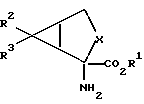
где X обозначает группу (CH2)n; R2 обозначает группу СО2R4, а R3 обозначает водородный атом или же R2 обозначает водородный атом, а R3 обозначает группу CO2R4; каждым из R1и R4 независимо от другого обозначен водородный атом, C1 - C10 алкил. C1-C10, алкенил, арил или арилалкил; а n равно 1; или к их фармацевтически приемлемым солям.
В соответствии с настоящим изобретением предлагаются также фармацевтические композиции, включающие в себя соединение формулы 1 в сочетании с одним или несколькими фармацевтическими приемлемыми носителями, разбавителями или наполнителями.
Соединения, предлагаемые в соответствии с данным изобретением, могут быть получены в методе воздействия на сАМР-связанные метаботропные глутаматные рецепторы, а также в методах лечения неврологических расстройств или психиатрических заболеваний, которые связаны с возбудительными амикислотными рецепторами, согласно которым предусмотрено введение в организм соединения формулы 1. Примеры неврологических расстройств, которые лечат соединением формулы 1, включают в себя церебральные нарушения, являющиеся следствием хирургической операции с искусственным кровообращением и трансплантации, церебральную ишемию (например, в результате приступа и остановки сердца); травму спинного мозга; травму головы; болезнь Альгеймера; хорею Хантингтона; боковой амиотрофический склероз; спровоцированную СПИДом деменцию; спазмы мышц; мигрени; недержание мочи; судороги; перинатальную кислородную недостаточность; гипоглуикемическое нейронное расстройство; толерантность к лекарственному средству, отмену и прекращение приема лекарств, например опиатов, бензодиазепинов, никотина, кокаина или этанола, прекращение курения; повреждение глаза и ретинопатию; расстройства сознания; болезнь неясного происхождения и болезнь Паркинсона, вызванную лекарствами; рвоту; отек головного мозга; хронические боли; расстройства сна; синдром Туретта; расстройства, проявляющиеся в недостатке внимания, и запоздалую дискенезию. Примеры психиатрических расстройств, которые лечат соединением формулы 1, включают шизофрению, страх и связанные с ним заболевания (например, приступы панического состояния и заболевания, связанные со стрессом), депрессию, биполярные расстройства, психоз и навязчивые компульсивные расстройства.
В соответствии с настоящим изобретением предлагаются также соединения, которые могут быть использованы для синтеза соединений формулы 1. Более конкретно, настоящее изобретение относится к соединениям формулы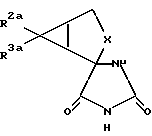
где X обозначает группу (CH2)n; n обозначает 1; R2a обозначает группу СО2R4a, а R3a обозначает водородный атом, или R2a обозначает водородный атом, а R3a обозначает группу CO2R4a; а R4a обозначает водородный атом или карбоксиблокирующую группу; и их солям.
В соответствии с настоящим изобретением предлагается также способ получения соединения формулы 1 или его фармацевтически приемлемой соли, который включает:
1) гидролиз соединения формулы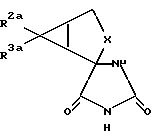
где X обозначает группу (CH2)n; n обозначает 1; R2a обозначает группу CO2R4a, а R3a обозначает водородный атом, или R2a - обозначает водородный атом, а R3a - обозначает группу CO2R4a; а R4a обозначает водородный атом или карбоксиблокирующую группу;
2) реакция соединения формулы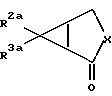
где значения символов X, R2a и R3a определены выше, с цианидом щелочного металла и аммониевой солью и гидролиз полученного промежуточного продукта как указано выше (1) или
3) гидролиз соединения формулы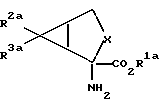
где R1a обозначает карбоксизащитную группу, а значения символов X, R2a и R3a определены выше, и
4) необязательное удаление карбоксизащитной группу и
5) необязательная этерификация одной или обеих карбоксильных групп и
6) необязательное разделение диастереомеров и/или выделение энантиомеров и
7) необязательное получение фармацевтически приемлемой соли соединения формулы 1.
Термин "C1-C10алкил" служит для обозначения прямой, разветвленной или циклической цепи, содержащей от одного до десяти углеродных атомов. Типичные прямые или разветвленные С1-С10 алкильные группы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор.бутил, трет. бутил, н-пентил, изопентил, неопентил, н-гексил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,2-диметилбутил, 2,3-диметилбутил, 3,3-диметилбутил, гептил, н-октил, 2,2-диметилгексил, 2,5- диметилгексил, 2-метилгептил, 4-метилгептил, 2,2,4-триметилпентил, 2,3,4-триметилпентил, нонил, 3,5,5-триметилгексил, децил, 3,7-диметилоктил и тому подобное. Значения термина "C1-C10алкил" включают в себя также значения терминов "C1-C6алкил" и "C1-C4алкил". К типичным циклическим алкильным группам относятся циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и тому подобное. Класс типичных C1-C6-алкильных групп включает в себя метил, этил, н-пропил. изопропил, н-бутил, изобутил, втор.бутил, трет. бутил, н-пентил, изопентил, неопентил и н-гексил.
Термин "C1-C10алкенил" служит для обозначения прямых или разветвленных ненасыщенных алкильных цепей, содержащих от двух до десяти углеродных атомов и имеющих от одной до нескольких улерод-углеродных двойных связей, в частности диенов и триенов. Этот класс включает в себя также как E7, так и Z-изомеры. К типичным радикалам этого класса относятся винил, аллил, алленил, 1-бутенил, 2-бутенил, 2-метил-1-пропенил, 3-бутенил., 2-метил-2-пропенил. бутадиенил, 1-пентенил, 2-пентенил, 2-метил-2-бутенил., 4-пентенил, 3-метил-2-бутенил. 3-метил-1,2- бутадиенил, 3-гексенил, 2-гексенил, 4-метил-3-пентенил, 4- гексенил, 5-гексенил, 3-метил-1-пентен-3-ил, 4-метил-3З-пентенил, 6-метил-5-гептен-2-ил, 7-октенил. 1-октен/3-ил, 3-ноненил. 2,4- диметил-2,6-гептан-диенил. 3,7-диметил-6-октенил, 5-деценил, 9- деценил, 2,6-диметил-7-октенил и тому подобное. Значения термина "C2-C10алкенил" охватывают также значения термина "C2-C6алкенил".
Термин "стереоизомерное соединение" использован для обозначения оптического изомера соединения формулы 1. К типичным стереоизомерным соединениям относятся 1S, 2S, 5R, 6S-, изомер, 1R, 2R, 5S, 6R- -изомер, 1S, 2R, 5R, -6R-изомер, 1R, 2S, 5S - 6R-изомер, 1S, 2S, 5R, 6R-изомер. 1R, 2R, 5S, 6S-изомер, 1S, 2R, 5R-6R-изомер и 1R, 2S, 5S, 6S-изомер.
Термин "диастереомерное соединение" служит для обозначения смеси двух стереоизомеров (non - superimposable) соединения формулы 1. Класс типичных диастереомерных соединений включает в себя 1SR, 2SR, 5SR, 6SR -смесь, 1SR, 2RS, 5RS, 6SR - смесь, 1SR, 2SR, 5RS, 6RS-смесь и 1SR, 2RS 5RS, 6RS-смесь. Предпочтительным диастереомерным соединением является 1SR, 2SR-5RS, 6SR-смесь. Предпочтительным этантиомером служит 1S, 2S-5R, 6S-изомер.
Термин "карбоксизащитная группа" использован здесь для обозначения одного из сложноэфирных производных карбоксильной группы, которую обычно применяют для блокирования или защиты карбоксильной группы, когда реакции проводят с участием других функциональных групп. Защита карбоксильных групп в общем описана в работах McOmie, Protecting Groups in Organic Chemistery, Plenum Press, NY, 1973; и Greene and Wuts, Protecting Groups in Organic Synthesis издание 2-е, Tohn Wiley and Sons, NY, 1991. К примерам таких карбоксизащитных групп относятся метил, этил, метоксиметил, метилтиометил. трифенилметил, бензил. 4-нитробензил, 4-метоксибензил, 3,4- диметоксибензил. 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, бензгидрил, трет. бутил, трет.-амил, тритил, триметилсилил, трет. бутил-диметилсилил, аллил, 1-(триметилсилилметил)-проп-1-ен-3-ил и тому подобное. Особенно предпочтительными карбоксизащитными группами являются C1-C6алкильные группы, в частности метил и этил. Термин "защищенный карбоксил" относится к карбоксильной группе, у которой имеется карбоксизащитная группа.
Термин "азотзащищающая группа" использован здесь для обозначения заместителей аминогрупп, которые обычно применяют для блокирования или защиты функциональной аминогруппы, когда реакцию проводят с участием других функциональных групп. Защита аминогрупп в общем описана в работах McOmie, Protecting Groups in Organic Chemistry, Plenum Press, NY, 1973; и Greene and Wuts, Protecting Groups in Organic Synthesis издание 2-е, John Willey and Sons, NY, 1991. К примерам азоблокирующих групп относятся бензил, трет. бутил, аллил, трифенилметил, трет.-бутилдиметилсилил, трифенилсилил, формил, тритил, фталимидогруппа, трихлорацетил. хлорацетил, фталоил, 2-нитрофеноксиацетил, бензилоксикарбонил. метоксикарбонил, 2-метилбензилоксикарбонил, трет.-бутоксикарбонил, аллилоксикарбонил, 2,2,2-трихлорэтоксикарбонил и тому подобное. Термин "защищенный амин" служит для обозначения первичного или вторичного амина, содержащего азотблокирующую группу.
Термин "C1-C4алкоксигруппа служит для обозначения таких групп, как метокси-, этокси-, н-пропокси-, изопропокси-н-бутокси-, втор-бутокси- и трет. бутоксигруппы. Термин "атом галогена" служит для обозначения атома фтора, хлора, брома или иода.
Термин "алкоксикарбонил" служит для обозначения карбоксильной группы, включающей в себя C1-C6алкильную группу, связанную через кислородный атом с карбонильным углеродным атомом. К типичным представителям групп этого типа относятся метоксикарбонил, этоксикарбонил, н-пропоксикарбонил, н-бутоксикарбонил. трет.-бутоксикарбонил и тому подобное.
Предпочтительной алкоксикарбонильной группой является метоксикарбонил.
Используемый здесь термин "замещенный фенил" служит для обозначения фенильной группы, замещенной одним или двумя заместителями, выбираемыми из группы, которая включает атомы галогена, окси-, циано-, нитрогруппы, C1-C6алкил, C1-C4алкоксигруппы, алкоксикарбонил, защищенный карбоксил, карбоксиметил, оксиметил. амин, защищенный амин, аминометил и трифторметил. К примерам замещенной фенильной группы относятся 4- хлорфенил, 2,6-дихлорфенил, 2,5-дихлорфенил, 3,4-дихлорфенил, 3-хлорфенил, 3-бромфенил, 4-бромфенил, 3, 4-дибромфенил, 3-хлор-4-фторфенил, 2-фторфенил, 4-оксифенил, 3-оксифенил, 2,4-диоксифенил, 3-нитрофенил, 4-нитрофенил, 4-цианофенил, 4-метилфенил, 4- этилфенил, 4-этоксифенил, 4-карбоксифенил, 4-(оксиметил)-фенил, 4-аминофенил, 4-пропил-фенил, 4-бутилфенил, 4-трет.бутилфенил, 3-фтор-2-метилфенил, 2,3-дифторфенил, 2,6-дифторфенил, 2,6-диметилфенил, 2-фтор-5-метилфенил, 2,4,6-трифторфенил, 2-трифторметилфенил, 2-хлор-5-трифторметилфенил, 2,4-бис-(трифторметил)-фенил, 3,5-бис(трифторметил)-фенил, 2-метоксифенил, 3-метоксифенил, 3,5-диметоксифенил, 4-окси-3-метилфенил, 3,5-диметил-4-оксифенил, 4-окси-3-(оксиметил)-фенил, 2-амино-5-метилфенил, 4-амино-3-трифторметилфенил, 3-амино-4- оксифенил, 2-метил-4-нитрофенил, 4-метокси-2-нитрофенил, 2,4- динитрофенил, 3-циано-4-нитрофенил и тому подобные.
Термин "арил" служит для обозначения таких групп, как фенил, замещенный фенил и нафтил. Термин "арилалкил" обозначает C1-C4 алкильную группу, у которой имеется одна или несколько арильных групп. Типичными примерами групп этого последнего типа является бензил, 2-нитробензил, 4-нитробензил, 1-фенилэтил, 2-фенилэтил, 3-фенилпропил. 4-фенилбутил, 2- метил-2-фенилпропил, (4-хлорфенил)-метил, (2,6-дихлорфенил) - метил, бис-(2,6-дихлорфенил)-метил, (4-оксифенил)-метил, (2, 4- динитрофенил)-метил, трифенилметил, (4-метоксифенил)- дифенилметил, бис-(4-метоксифенил)-метил, альфа-нафтилдифенилметил, бис-(2-нитрофенил)-метил и тому подобное.
Термин "влияющий" относится к соединению формулы 1, которое действует как агонист при рецепторе возбудительной аминокислоты. Термин "рецептор возбудительной аминокислоты" относится к метаботропному глутаматному рецептору, то есть рецептору, который связан с клеточными эффекторами через СТР-связывающие белки. Термин "САМР"- связанный метаботропный глутаматный рецептор" относится к метаботропному рецептору, который связан с ингибированием активности аденилатциклазы.
Термин "неврологическое расстройство" относится как к острым, так и хроническим "нейродегенеративным состояниям", включая сюда церебральные нарушения, являющиеся следствием хирургической операции с искусственным кровообращением и трансплантации, церебральную ишемию (например, приступ в результате остановки сердца), травму спинного мозга, травму головы, болезнь Альгеймера, хорею Хантингтона, боковой амиотрофический склероз, спровоцированную СПИДом деменцию, перинатальную кислородную недостаточность, гипогликемическое нейронное расстройство, повреждение глаза и ретинопатию, расстройства сознания, болезнь неясного происхождения и болезнь Паркинсона, вызванную лекарствами. Этот термин также охватывает другие неврологические состояния, которые вызваны глутаматной дисфункцией, включая сюда спазмы мышц, мигрени, недержание мочи, лекарственную толерантность, абстиненцию и прекращение приема лекарств, например опиатов, бензодиазепинов, никотина, кокаина или этанола, прекращение курения, рвоту, отек головного мозга, хронические боли, расстройства сна, судороги, синдром Туретта, расстройства, проявляющиеся в недостатке внимания и запоздалую дискинезию.
Термин "психиатрические расстройства" охватывает как острые, так и хронические психиатрические состояния, включая сюда шизофрению, страх и связанные с ним заболевания (например, приступы панического состояния и связанные со стрессом сердечно-сосудистые заболевания), депрессию, биполярные расстройства, психоз и навязчивые компульсивные расстройства.
Рамками настоящего изобретения охватываются фармацевтически приемлемые соли соединений формулы 1. Эти соли могут существовать в сочетании с кислотной или основной частью молекулы и могут существовать в форме кислотно-аддитивных солей, солей присоединения первичного, вторичного, третичного или четвертичного аммониевого основания, солей щелочных металлов или щелочноземельных металлов. Обычно кислотно-аддитивные соли получают реакцией кислоты с соединением формулы 1. Соли щелочных металлов и соли щелочно-земельных металлов обычно получают реакцией соединения в форме соли гидроокиси желаемого металла и соединения формулы 1, у которого R1 и/или R4 обозначает водородный атом.
К кислотам, которые обычно применяют для получения таких солей, относятся минеральные кислоты, в частности соляная, бромистоводородная, иодистоводородная, серная и фосфорная кислоты, а также такие органические кислоты, как паратолуолсульфо, метансульфокислоты, щавелевая, пара-бромфенил-сульфокислота, угольная, янтарная, лимонная, бензойная и уксусная кислоты и родственные им минеральные и органические кислоты. Таким образом, класс этих фармацевтически приемлемых солей включает в себя сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, аммоний, кислые фосфаты, вторичные кислые фосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капраты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацаты, фумараты, гиппураты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты. динитробензоаты, оксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, альфа-оксибутираты, гликоляты, малеаты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1- сульфонаты, нафталин-2-сульфонаты, манделаты, соли магния, тетраметиламмония, калия, триметиламмония, натрия, метиламмония, кальция и тому подобные соли.
Соединения формулы 1, предлагаемые по настоящему изобретению, обладают четырьмя аcимметричными углеродными атомами. Асимметричными центрами служат замещенный углеродный атом, несущий амино- и карбоксильные группы, углеродный атом, по месту которого присоединены группы R2 и R3, и два углеродных атома по месту соединения сконденсированных колец. Асимметричные углеродные атомы находятся соответственно в положениях 2, 6, 1 и 5. Таким образом, соединения настоящего изобретения могут существовать как практически чистые оптические изомеры, смесь двух энантиомеров (включая сюда рацемические модификации) и смесь двух диастереомеров. Когда R2 обозначает группу СО2R4, а каждый из R', R3 и R4 обозначает водородный атом, биологически активный и наиболее предпочтительный стереоизомер, как это определяют испытаниями с рецепторным связыванием, характеризуется положительным оптическим вращением (AD). X-лучевой однокристаллический анализ позволил установить структуру этого наиболее предпочтительного энантиомера, что дало возможность выявить относительную стереохимическую конфигурацию, приведенную ниже: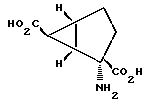
Абсолютную стереохимическую конфигурацию этого наиболее предпочтительного энантиомера определяют как 1S, 2S, 5R, 6S. Таким образом, рамками настоящего изобретения охватываются стереоизомерные соединения формулы 1, характеризующиеся этой самой предпочтительной стереохимической конфигурацией, смеси энантиомеров, содержащие компоненты с такой предпочтительной стереохимической конфигурацией (включая сюда рацематы), и смеси диастереомеров, содержащие компоненты с такой предпочтительной стереохимической конфигурацией.
Хотя, как полагают, все соединения формулы 1 по настоящему изобретению селективно воздействуют на отрицательно соединенные САМР-связанные метаботропные глутаматные рецепторы, некоторые соединения изобретения для такого применения предпочтительнее. В предпочтительном варианте R2 обозначает группу CO2R4, R3 обозначает водородный атом, а каждым из R1 и R4 независимо от другого обозначен водородный атом, С1-С6-алкил, арил или арилалкил. Типичные соединения этой предпочтительной группы соединений формулы 1 охватывают 2-аминодицикло[3.1.0] гексан-2,6-дикарбонову кислоту, диметил-2-аминодицикло[3.1.0] гексан-2,6-дикарбоксилат, диэтил-2- аминодицикло[3.1.0] гексан-2,6-дикарбоксилат, дибутил-2- аминодицикло [3.1.0]гексан-2,6-дикарбоксилат, дигексил-2- аминодицикло[3.1.0] гексан-2,6-дикарбоксилат, дифенил-2- аминодицикло [3.1.0] гексан-2,6-дикарбоксилат и дибензил- -2-аминодицикло[3.1.0] -гексан-2,6-дикарбоксилат. Некоторые соединения настоящего изобретения более предпочтительны для использования при воздействии на САМР-связанные метаботропные глутаматные рецепторы. В более предпочтительном варианте каждым из R1 и R4 независимо от другого обозначен водородный атом, C1-C4алкил, арил или арилалкил. К типичным соединениям этой более предпочтительной группы соединений относятся 2-аминодицикло[3.1.0] -гексан-2,6-дикарбоновая кислота, диметил-2-аминодицикло[3.1.0] гексан-2,6-дикарбоксилат, диэтил-2-аминодицикло[3.1.0] гексан-2,6-дикарбоксилат, дибутил- 2-аминодицикло [3.1.0]-гeкcaн-2,6-дикapбoкcилaт, дифенил-2-аминодицикло [3.1.0] гексан-2,6-дикарбоксилат и дибензил-2- аминодицикло [3.1.0]гексан-2,6-дикарбоксилат.
Некоторые соединения являются наиболее предпочтительными для использования при воздействии на САМР-связанные метаботропные глутаматные рецепторы. В наиболее предпочтительном варианте каждый из R1 и R4 независимо от другого обозначает водородный атом или C1-С4алкил. К типичным соединениям этой группы наиболее предпочтительных соединений относятся 2-аминодицикло[3.1.0] -гексан-2,6-дикарбоновая кислота, диметил-2-аминодицикло [3.1.0] гексан-2,6-дикарбоксилат, диэтил- 2-аминодицикло [3.1.0.]-гексан-2,6-дикарбоксилат, дибутил-2- аминодицикло[3.1.0]гексан-2, 6-дикарбоксилат и дипропил-2-аминодицикло[3.1.0]гексан-2,6-дикарбоксилат.
Наиболее предпочтительными соединениями формулы (1) являются (+) - 2-аминодицикло [3.1.0]-гексан-2,6-дикарбоновая кислота, ее С1-С4алкиловые, аралкиловые и ариловые сложные эфиры и их фармацевтически приемлемые соли.
Хотя предполагают, что все соединения формулы X по настоящему изобретению могут быть использованы для синтеза соединений формулы 1, некоторые из таких соединений предпочтительны. В предпочтительном варианте R2a обозначает группу CO2R4a, а R3a обозначает водородный атом. В более предпочтительном варианта R4a обозначает водородный атом или С1-С6алкильную группу, например этильную группу.
Соединения формулы 1 по настоящему изобретению обычно синтезируют циклопропанированием 2-циклоалкен-1-она, то есть соединения формулы II, у которого значения символа X определены выше для соединений формулы 1. Соединения формулы 1, у которых R3 обозначает водородный атом, а R2 обозначает группу СО2R4, получают так, как это показано на схеме 1.
Схема I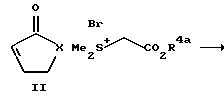
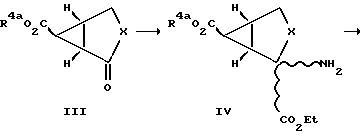
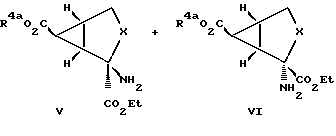
Обычно проводят реакцию соединения формулы II с метилдиметилсульфонийбромидом с защищенной карбоксильной группой с получением дициклического соединения формулы III, у которых R4a обозначает карбоксилзащитную группу. Реакцией Штреккера или Бухерера-Бергса проводят превращение этого соединения в аминокислоту с последующими гидролизом и этерификацией с получением соединений формулы IV в виде смеси изомеров. Эту изомерную смесь разделяют с получением соединений формулы V и формулы VI. Эти соединения далее гидролизуют с получением соединений формулы I, у которых R2 обозначает группу CO2R4, а R1 и R4 обозначают водородные атомы.
Более конкретно проводят реакцию 2-циклоалкен-1-она с (защищенный карбоксил) метилдиметилсульфонийбромидом с получением бициклического промежуточного продукта формулы III. Это циклопропанидирование удобно проводить в органическом растворителе в присутствии аминового основания. Класс приемлемых для такой реакции растворителей включает в себя ацетонитрил, дихлорметан, бензол, толуол и ксилол, предпочтительнее ацетонитрил или дихлорметан. К аминовым основаниям, приемлемым для использования в ходе проведения этой реакции, относятся ненуклеофильные основания, в частности такие, как 1,8-диазадицикло [5.4.0]ундец-7-ен, пиридин и коллидин. Предпочтительным аминовым основанием для использования в ходе проведения такой реакции является 1,8- диазадицикло [5.4.0]ундeц-7-eн. В предпочтительном варианте проводят реакцию карбоэтоксиметилдиметилсульфонийбромида с аминовым основанием с получением in situ этил-(диметилсульфуранилиден) -ацетата. Образовавшуюся смесь обрабатывают 2-цик- лоалкен-1-оном. К примерам 2-циклоалкен-1-онов относятся 2- циклопентен-1-он, 2-циклогексен-1-он, 2-циклогептен-1-он и 2- циклооктен-1-он. Такую реакцию обычно проводят при температуре приблизительно от 25 до 50oC, предпочтительнее при температуре примерно от 25 до 30oC. Обычно такую реакцию завершают в течение приблизительно от 18 ч до трех дней.
Бициклический промежуточный продукт III превращают в бициклическую аминокислоту реакцией Штреккера или Бухерера-Бергса с последующим гидролизом промежуточных продуктов Краух и Кунц, Organic Name Reactions, 76 (1964), см. содержащиеся в работе ссылки. В предпочтительном варианте проводят реакцию бициклического кетона III с водным раствором цианида калия или цианида натрия и карбонатом аммония с получением гидантоиновых промежуточных продуктов. Такую реакцию обычно проводят в спиртовом растворителе, в частности в таком, как этанол или метанол. Реакцию обычно ведут при температуре приблизительно от 25oC до температуры кипения растворителя с обратным холодильником, предпочтительнее примерно при 50oC. По истечении примерно 18-часового периода эту реакцию обычно завершают. Изомерные гидантоины можно выделять и очищать согласно изложенному ниже. В предпочтительном варианте смесь изомерных гидантоинов гидролизуют с использованием гидроокиси натрия и в дальнейшем подвергают сложной этерификации без выделения или очистки до соединения формулы V или формулы VI. Этот гидролиз как правило проводят при температуре кипения растворителя с обратным холодильником в течение приблизительно от 15 до 20 ч.
Продукты гидролиза, смесь изомерных соединений формулы I, у которых R1 обозначает водородный атом, перед разделением диастереомеров и энантиомеров предпочтительнее этерифицировать. Когда в процессе гидролиза карбоксизащитную группу удаляют, в дальнейшем получают эфир двухосновной кислоты. Раствор карбоновой кислоты или дикарбоновой кислоты в спирте, например в метаноле, этаноле, изопропаноле или н-бутаноле, обрабатывают хлористым тионилом и кипятят с обратным холодильником. Как правило перед добавлением хлористого тионила раствор продукта гидролиза охлаждают приблизительно до 0oC. Этерификацию обычно завершают по истечении примерно 48 ч.
Диастереомерные продукты, соединения формулы V и формулы VI, разделяют с применением стандартных методик. К предпочтительным методам разделения относятся кристаллизация и/или хроматография. Соединение формул V и VI можно селективно кристаллизовать получением кислотно-аддитивных солей, в частности оксалата. Эту соль получают обработкой этилацетатного раствора, содержащего смесь соединений формул V и VI, щавелевой кислотой и этанолом. Для преимущественной кристаллизации одного из диастереомеров можно добавлять дополнительное количество этанола. Осуществление такой процедуры позволяет получить кристаллический материал, обогащенный одним из изомеров, и фильтрат (маточный раствор), обогащенный другим изомером. Такие соединения можно подвергать дальнейшей очистке с использованием хроматографии, в частности хроматографии на силикагеле.
При необходимости соединения формул V и VI гидролизуют и карбоксилзащитную группу удаляют, получая соединения формулы I, у которых R1 и R4 обозначают водородные атомы. Эти соединения, как правило, гидролизуют обработкой раствора соединения формулы V или формулы VI в органическом растворителе, в частности в тетрагидрофуране, водным основанием, в частности гидроокисью натрия. Такой гидролиз как правило проводят при комнатной температуре, для его завершения требуется приблизительно 18 ч. Карбоксилзащитные группы удаляют в соответствии со стандартными методами синтеза (см. MсOmie и Greene and Wuts).
Энантиомеры каждой пары диастереомеров промежуточных соединений V и VI разделяют с применением стандартных методик разделения [см.Jacques, Collet. and Wilen, Enantiomers, Racemates, and Resolutions. (1981)]. По предпочтительному методу разделения энантиомеров предусмотрено получение диастереомерных солей между рацемическими модификациями и оптически активными (хиральными) разделительными агентами (Jaques, Collet and Wilen, глава 5). Соединения формул V и VI, у которых R4a обозначает карбоксилзащитную группу, можно разделять с использованием кислотных хиральных разделительных агентов. Примеры подходящих кислотных хиральных разделительных агентов включают в себя (+)-камфорную кислоту, (+)- и (-)-дибензоилвинную кислоту, диацетонкетоглюконовую кислоту, лазалоцидную, (+) и (-) -миндальную кислоту, (+)- и (-)-яблочную кислоту, (+) - и (-)-хинную кислоту, (+)- и (-)-винную кислоту, (+)-ди-п-толуол- D-винную кислоту и (-)-ди-п-толуол-L-винную кислоту. Предпочтительными кислотными разделительными агентами для разделения соединений формул V и VI, у которых R4a обозначает карбоксилзащитную группу, являются (+)-ди-п-толуоил-D-винную кислоту и (-)-ди-п-толуоил-L-винную кислоту. Соединения формул V и VI, у которых R4a обозначает водородный атом, можно разделять с использованием основных хиральных разделительных агентов. Примером основного хирального разделительного агента служит (8)-1-фенил- этиламин.
По другому варианту бициклическое соединение формулы III превращают в смесь диастереомерных гидантоинов так, как это показано на схеме II.
Схема II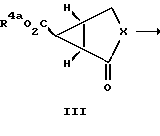
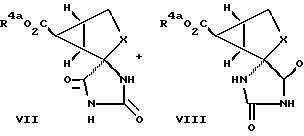
Проводят реакцию бициклического промежуточного продукта III, полученного, как описано с раствором цианистого калия или цианистого натрия и карбонатом аммония, получая диастереомерные гидантоиновые промежуточные соединения формулы VII и формулы VIII. Эту реакцию как правило ведут в смеси воды с таким спиртом, как метанол или этанол. Реакцию ведут при температуре приблизительно от 55 до 60oC и ее обычно завершают по истечении примерно от 18 ч до 4 дней. Диастереомерные продукты разделяют с применением стандартных методик, в частности, кристаллизацией и/или хроматографией. В предпочтительном варианте соединения формул VII и VIII разделяют кристаллизацией.
Соединения формул VII и VIII, у которых R4a обозначает водородный атом, можно разделять с использованием основного хирального разделительного агента. Примером основного хирального разделительного агента служит (R)-1-фенилэтиламин.
Гидантоиновое промежуточное соединение формул VII или VIII превращают в соединение формулы I, у которого R1 и R4 обозначают водородные атомы, гидролизом. Гидантоиновую группу и сложноэфирную группу гидролизуют с использованием такого водного основания, как гидроокись натрия, или водной кислоты, в частности соляной кислоты. Этот гидролиз, как правило, проводят при температуре приблизительно от 100 до 150oC. Полученное соединение формулы I очищают с использованием ионообменной хроматографии.
Соединения формулы I, у которых R2 обозначает водородный атом, a R3 обозначает группу CO2R4a, получают так, как показано на схеме реакции III.
Схема III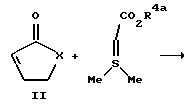
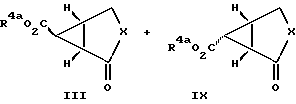
Проводят реакцию 2-циклоалкен-1-она и диметилсульфуранилиденацетата с защищенным карбоксилом с получением изомерных бициклических промежуточных продуктов III и 2Y. Это циклопропанирование проводят в органическом растворителе при температуре приблизительно от 45 до 85oC. Класс подходящих растворителей охватывает бензол, толуол, ксилол и ацетонитрил. В предпочтительном варианте реакцию проводят в бензоле при 50oC. Диастереомерные продукты разделяют с использованием хроматографии на силикагеле. Конверсию соединения формулы IX в соединения формулы I проводят с использованием методик, которые описаны выше для конверсии соединений формулы III.
Соединения формулы III, у которых X обозначает метилен, можно получить так, как это представлено на схеме реакций IV.
Схема IV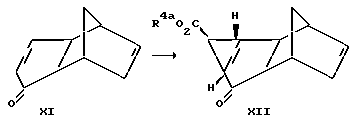
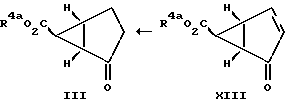
Проводят реакцию соединения формулы XI с (защищенный карбоксил)метилдиметилсульфоний бромидом с получением соединения формулы XII, у которого R4a обозначает карбоксилзащитную группу. Эту реакцию обычно проводят в соответствии с методом, аналогичным тому, который описан выше для циклопропанировании соединения формулы II. Далее, полученное соединение формулы XII превращают в соединение формулы XIII выдержкой при повышенной температуре, например при температуре в интервале от 160 до 500oC, предпочтительнее от 180 до 300oC. В результате такой выдержки из соединения формулы XII высвобождается циклопентадиен. Эту стадию удобно осуществлять в атмосфере инертного газа, в частности азота, и в среде инертного органического растворителя, например дихлорбензола. Далее превращают полученное соединение формулы XIII в соединение формулы III восстановлением, например гидрогенизацией в присутствии палладия на угле. Такое восстановление удобно проводить при температуре в интервале от 0 до 50oC. Класс приемлемых для такого восстановления растворителей включает спирты, в частности этанол, сложные эфиры, в частности этилацетат, ароматические углеводороды, в частности толуол, и такие амиды, как диметилформамид.
Совершенно очевидно, что использование оптически активного соединения формулы XI в качестве исходного материала позволяет получить оптические активное соединение формулы III.
Соединения формулы XIII являются, как полагают, новыми; они являются дополнительным аспектом изобретения.
Соединения формулы XI (включая их оптически активные формы) могут быть получены в соответствии с методом, описанным в работах Klunder et al., Tetrahedron Lett.,1986, 27, 2543 и Takano et al., Synlett 1991, 636.
Соединения формулы I, у которых R1 и R4 обозначают C1-C10, C2-C10 алкенил, арил или арилалкил, получают из соответствующих соединений, у которых R1 и R4 обозначают водородные атомы. Эти соединения обычно получают с применением стандартных методик синтеза. В типичном примере проводят реакцию соединений формулы I, у которых R1 и R4 обозначают водородные атомы, с С1-С10 алкиловым, С2-С10, алкениловым, ариловым или арилалкиловым спиртом в присутствии кислоты с получением соответствующего сложного эфира. Эту реакцию, как правило, проводят с избытком спирта в присутствии каталитического количества концентрированной серной кислоты.
Соединения формулы I, у которых значения R1 и R4 неодинаковы, могут быть получены из двухосновной кислоты, у которой R1 и R4 водородные атомы, с использованием стандартной методики органического синтеза. Так, например, с этой целью можно прибегнуть к химическим приемам, которые были разработаны для селективной функционализации карбоксильных групп глутаминовой и аспарагиновой кислот. По другому варианту, путем выбора карбоксилзащитной группы для соединения формулы X, которая стойка в условиях проведения гидролиза для гидантоиновой группы, можно селективно манипулировать с карбоксильными группами.
Соединения формулы I по настоящему изобретению представляют собой агонисты некоторых метаботропных рецепторов возбудительных аминокислот. Конкретно, соединения формулы I являются агонистами отрицательно соединенных САМР-связанных метаботропных глутаматных рецепторов. Таким образом, другой аспект настоящего изобретения относится к способу воздействия на рецептор возбудительной аминокислоты млекопитающих, который состоит во введении в организм млекопитающего, для которого требуется модулированная нейротрансмиссия возбудительной аминокислоты, фармацевтически эффективного количества соединения формулы I. Термин "фармацевтически эффективное количество" используют для обозначения такого количества соединения по изобретению, которое способно воздействовать на рецепторы возбудительных аминокислот. Благодаря такому воздействию соединение по изобретению проявляет действие агониста. Когда соединение по изобретению действует как агонист, взаимодействие этого соединения с EaA-рецептором напоминает реакцию взаимодействия такого рецептора с его естественным лигандом (то есть с L-глутаматом).
Конкретная доза соединения, вводимого в организм, в каждом случае определяется конкретными обстоятельствами, включая конкретное вводимое в организм соединение, способ введения в организм, состояние пациента, когда применяют такую обработку, и тому подобными соображениями. Такие соединения можно вводить самыми различными способами, включая сюда пероральный, ректальный через кожу, подкожно, внутривенно, внутримышечно или интраназальный путь. Кроме того, такое соединение можно вводить в организм непрерывным вливанием. Обычная ежедневная доза активнодействующего соединения настоящего изобретения представляет собой приблизительно от 0,001 до 100 мг/кг. Предпочтительные ежедневные дозы составляют приблизительно от 0,05 до 50 мг/кг, предпочтительнее примерно от 0,1 до 20 мг/кг.
Было установлено, что излишнее или неуместное стимулирование трасмиссии возбудительной аминокислоты приводит к воздействию на различные физиологические функции. Полагают, что соединения формулы 1 по настоящему изобретению обладают способностью лечить у млекопитающих различные неврологические расстройства, связанные с таким состоянием, включая сюда острые неврологические расстройства, в частности такие, как церебральные нарушения, являющиеся следствием хирургической операции с искусственным кровообращением и трансплантации, церебральная ишемия (например, в результате приступа и остановки сердца), травма спинного мозга, травма головы, перинатальная кислородная недостаточность и гипогликемическое нейронное расстройство. Соединения формулы 1, как полагают, обладает способностью излечивать разнообразные хронические неврологические расстройства, в частности такие, как болезнь Альгеймера, хорея Хантингтона, боковой амиотрофический склероз, спровоцированная СПИДом деменция, повреждение глаза и ретинопатия, расстройства сознания, болезнь неясного происхождения и болезнь Паркинсона, вызванную лекарствами.
Соединения формулы 1 по настоящему изобретению обладают также, как полагают, способностью излечивать у млекопитающих самые разнообразные другие неврологические расстройства, которые связаны с глутаматной дисфункцией, включая сюда спазмы мыши; судороги; мигрени; недержание мочи, психоз; лекарственную толерантность, отмену и прекращение приема лекарства (например, опиатов, бензодиазепинов, никотина, кокаина или этанола); прекращение курения; страх и связанные с ним заболевания (например, приступы панического состояния и заболевания, связанные со стрессом); рвоту; отек головного мозга; хронические боли; расстройства сна; синдром Туретта; расстройства, проявляющиеся в недостатке внимания, и запоздалую дискинезию.
Соединения настоящего изобретения являются агонистами сАМР-связанных метаботропных глутаматных рецепторов. Эти соединения отрицательно соединяются через рецептор с аденилциклазой, ингибирующей образование циклического аденозинмонофосфата. Соединения формулы I по настоящему изобретению обладают, следовательно, способностью лечить самые разнообные психиатрические заболевания, в частности такие, как шизофрения, страх и связанные с ним заболевания (например, приступы панического состояния и заболевания, связанные со стрессом), депрессия, биполярные расстройства, психоз и навязчивые компульсивные расстройства.
Для демонстрации способности соединений формулы I влиять на рецепторы возбудительных аминокислот были проведены эксперименты. Сродство таких соединений к метаботропным глутаматным рецепторам было продемонстрировано селективным замещением чувствительного к (1S, 3R)-1 аминоциклопентан-1,3-дикарбоновой кислоты [3H] глутамата, связывающегося с мембранами клеток головного мозга крысы. Связывание [3H] глутамата ([3H]глу) осуществляли с помощью необработанных мембран переднего мозга крысы согласно описанному Schoepp and True [см. работы Schoepp and True, Neurosciense Lett, 145, 100-104 (1992); Wright, MeDonald, and Shoepp, J.Neurochem., 63, 938-945 (1994). Концентрация соединения формулы I, которая ингибирует 50%-ное связывание (ИК50) или процентное замещение [3H] глу при концентрации соединения формулы I 10 мкм или 100 мкМ, представлена в табл. 1.
Все соединения. 3, 6, 7 и 8 представляют собой дикарбоновые кислоты. В общем было установлено, что в ходе проведения испытания на связывание рецептора сложноэфирные производные (те соединения формулы 1, у которых одним или обоими символами R1 и R4 обозначены не водородные атомы), оказались неактивными. Однако полагают, что такие соединения in vivo способны гидролизоваться до соответствующей кислоты и, следовательно, могут выполнять функции пролекарств. Необходимо иметь в виду, что в соответствии с настоящим изобретением предлагаются активнодействующие дикарбоновые кислоты, а также все пролекарственные формы, которые способны in vivo образовывать активнодействующую кислоту.
Соединения формулы 1 эффективны при воздействии на САМР-связанные метаботропные глутаматные рецепторы. Типичные соединения испытали на их способность ослаблять стимулируемое форсколином образование cAMP в гиппокампе крысы и коре головного мозга крысы с использованием методик, которые описаны Schoepp and Johnson L [см. Schoepp and Johonson Neurochem Int., 22, 277-283 (1993)]. Результаты таких экспериментов сведены в табл. II
Способность соединений формулы 1 лечить страх или связанные с ним расстройства может быть продемонстрирована с использованием потенцированного страхом вздрагивания в сверхкаскадных лабиринтных моделей страха, описанных соответственно в работах Davis Psychopharmacology 62: 1; 1979 и Lister Psychopharmacol., 92:180-185; 1987.
В модели потенцированного страхом вздрагивания на животных воздействуют таким нейтральным раздражителем, как свет (условный раздражитель), совместно с таким раздражителем, вызывающим отрицательную реакцию, как шок (безусловный раздражитель). После выработки условного рефлекса, когда на животных воздействуют громким звуковым раздражителем, в том случае, если раздражителю, вызывающему испуг, предшествует свет, проявляется усиленный рефлекс вздрагивания.
Сверхкаскадная лабиринтная модель основана на естественных отрицательных эмоциях, которые у грызунов вызывают высота и открытые пространства.
Диазепам и буспиронгидрохлорид, которые клинические зарекомендовали себя как транквилизаторы, эффективны для ослабления страха (усиленного рефлекса вздрагивания), связанного с воздействием светом на потенцированную страхом модель вздрагивания, и ослабления боязни открытых пространств в сверхкаскадной лабиринтной модели.
Из питомника, Harlan Sprague-Dawley Кумбрерленд, штат Индиана, США, получали самцов крыс Long Evans (весом по 180-400 г) и самцов швейцарских мышей NIH (весом по 18-35 г) и животных акклиматизировали в течение по меньшей мере 3 дней до начала испытаний. Животных содержали при 23±2oC и относительной влажности от 30 до 70%. Их кормили апробированным на "Purina" кормом для грызунов и им давали воду неограниченно. Дневной режим состоял из 12 ч светового периода и 12 ч темноты, причем темнота наступала примерно в 18 ч 00 мин.
Перед применением испытываемые соединения растворяли в носителе из очищенной воды и нейтрализовали раствор 5н. гидроокисью натрия до величины pH 7-8. Диазепам (фирмы "Сигма кемикал компани", Сент-Луис, штат Миссури) суспендировали в очищенной воде с помощью добавляемого по каплям продукта Tween 80. Животным контрольной группы давали соответствующий носитель.
Потенцированное страхом вздрагивание. Для выработки условного рефлекса, а также для получения и фиксирования рефлексов вздрагивания пользовались камерами SL-LAB (фирмы "Сан Диего инструментс", Сан-Диего, штат Калифорния). Для достижения потенцированных рефлексов вздрагивания использовали методику выработки условного рефлекса по методу Павлова. На первые 2 дня крыс помещали в темные старт-камеры, в которых были установлены шоковые сетки. По истечении 5-минутного периода акклиматизации на каждое животное воздействовали 1-миллиамперным электрошоком (500 мкс), которому предшествовало 5-секундное воздействие светом (15 Вт), остававшемуся включенным на время электрошока. В течение каждой процедуры выработки условного рефлекса 10-кратно воздействовали светом и электрошоком, животным через желудочный зонд вводили раствор испытываемого соединения в воде и продолжали процедуры испытаний на вздрагивание. С целью свести к минимуму влияние начальной быстрой фазы привыкания к раздражителю в начале процедуры осуществляли блок из 10 последовательных воздействий звуковым раздражителем, вызывающим испуг (110 дБ, без сочетания со светом). За этим следовали 20 альтернативных опытов с использованием только звука или звука, которому предшествовал свет. Исключая начальный блок опытов, амплитуды рефлексов вздрагивания для опыта каждого типа (только один звук против света + звук) на каждом животном для всей процедуры испытания усредняли. Представленные данные отражают разницу между результатами, полученными с использованием только одного звука и света + звук. Все параметры сведены в табл. III.
Автоматизированный сверхкаскадный лабиринт. Конструкция сверхкаскадного лабиринта была основана на конструкции, которая была внедрена для мышей Листером (1987). Весь лабиринт был изготовлен из прозрачного плексигласа. Этот лабиринт состоял из двух открытых участков (30 х 5 х 0,25 см) и двух закрытых участков (30 х 5 х 15 см). Для придания структурности пол в каждом лабиринте был рифленым. Участки проходили от центральной платформы и поворачивали относительно любого другого под углом 90 градусов. Лабиринт был приподнят на высоту 45 см над полом и освещался красным светом. Вдоль каждого из участков лабиринта были смонтированы индивидуальные инфракрасные фотоэлементы с целью контролировать деятельность на закрытом, открытом участках или тыкание носом. Мышей индивидуально помещали на центральную платформу лабиринта, фиксировали результаты подсчетов числа закрытых участков, открытых участков и тыканий носом (тыкание головой только в открытый участок из закрытого участка лабиринта) и использовали данные как меру входов на участки и времени, потраченном на различных секциях лабиринта в течение пятиминутного периода испытаний.
Пероральное введение в организм соединения 6 приводило к существенному усилению деятельности на открытых участках в дозах 1, 3 и 10 мг/кг. Число тыканий носом значительно увеличивалось при дозе 3 мг/кг. При любой дозе соединения 6 подсчитанные параметры деятельности на закрытых участках заметных изменений не претерпевали.
Способность соединений формулы 1 защищать теплокровных животных от эффектов синдрома отмены или прекращения принятия лекарств может быть продемонстрирована с использованием звуковой модели испуга. В этой модели животным дают лекарство (никотин или диазепам), а затем давать лекарство прекращают. Эта отмена лекарства вызывает усиление реакции вздрагивания на звуковые раздражители. Затем в организм животных вводят испытываемые соединения с целью определить, способны ли они ослаблять повышенную реакцию вздрагивания.
Крыс Long Evans (200-400 г, Harlan Spraque Dawley, Колубус, штат Индиана) содержали индивидуально в контролируемой окружающей среде с 12-часовым циклом свет-темнота и свободным доступом к корму (Purina Rodent Chow) в воде. Животных анестерировали изофлураном и подкожно им имплантировали осмотические насосы Олзета (фирмы "Олзет корпорейшн").
При введении в организм испытываемое соединение растворяли в носителе, которым служила очищенная вода, и нейтрализовывали раствор 5н. гидроокисью натрия до величины pH 7-8. Диазепам (фирмы "Сигма кемикал компани", Сент-Луис, штат Миссури) суспендировали в носителе, который состоял из 40% полиэтиленгликоля 300, 10% этанола, 2% бензилового спирта, 1% продукта 80 и 47% очищенной воды. Никотин (фирмы "Ресерч байокемикалс инк.", Нейтик, штат Массачусетс) растворяли в солевом растворе. Контрольным животным давали соответствующий носитель.
Отмена никотина. Насосы заполняли с таким расчетом, чтобы они выделяли никотин (6 мг/кг/день подкожно), диазепам (10 мг/кг/день подкожно), испытываемое соединение (0, 1, 3, 10 мг/кг подкожно) или носитель. Спустя 12 дней после подкожной имплантации насосов крыс анестезировали изофлураном и насосы удаляли. Во время отмены лекарства (после удаления насоса) с использованием старт-камер фирмы "Сан-Диего инструментс" (Сан-Диего, штат Калифорния) регистрировали звуковую реакцию вздрагивания индивидуальных животных (пиковая амплитуда, Vмакс). Методики запугивания животных заключались в 5-минутном периоде адаптации при уровне звукового фона 70±2 дБ, непосредственно за которым следовало 25-кратное воздействие звукового раздражителя (уровень звука - 120±2 дБ, продолжительность импульса - 50 мкс) с 8-секундными интервалами между импульсами. Далее в отношении каждой процедуры пиковые амплитуды вздрагивания для всех 25 импульсов раздражителя усредняли и все данные приводили как значения для всей методики. Звуковые реакции вздрагивания оценивали ежедневно спустя 1, 2, 3, 4 или 5 дней после отмены лекарства. Базовую линию реакции вздрагивания оценивали перед удалением насоса на 12-й день.
По сравнению с животными контрольной группы, которым давали воду, звуковая реакция вздрагивания заметно возрастала в течение первых трех дней после прекращения хронического употребления животными никотина. Животные, которым дозу никотина заменяли внутрибрюшинным введением в организм доз 0,03 мг/кг или больше, не проявляли той же самой усиленной реакции вздрагивания, что и у животных, которым никакой замены никотина не производили. Предварительная обработка соединением 6 также обеспечивала блокаду зависимой от дозирования реакции вздрагивания, вызываемой отменой. По сравнению с животными контрольной группы, которым давали никотин (ED50 = 0,7 мг/кг внутрибрюшинно) существенное ослабление усиленного испуга бывало очевидным при пероральном введении дозы соединения 6, равной 3 мг/кг.
Отмена диазепама. В течение первых четырех дней после прекращения постоянного введения в организм диазепама в сравнении с животными контрольной группы, которым давали только носитель, реакция вздрагивания крыс на звуковой раздражитель заметно возрастала. Замещение внутрибрюшинного дозирования диазепама при 3 и 10 мг/кг не блокировало усиленной реакции вздрагивания и в некоторых случаях еще больше повышала способность реагировать, указывая на толерантность. Замещение у крыс, которым внутрибрюшинно вводили по 30 мг/кг диазепама, ежедневно за 60 мин до оценки реакции вздрагивания приводило к проявлению повышенной способности реагировать после прекращения введения в организм диазепама в период с 1-го по 4-й день в сравнении с животными контрольной группы, которым давали диазепам. Предварительная обработка соединением 6 блокировала ожидаемое усиление реакции вздрагивания, которое следовало после прекращения введения в организм диазепама. Дозы 0,1 и 0,3 мг/кг перорально вводимого соединения 6 значительно ослабляли усиленное вздрагивание в сравнении с реакцией, что отмечали у животных контрольной группы (ED50 = 0,1 мг/кг перорально).
Перед введением в организм на основе соединений настоящего изобретения предпочтительнее готовить композиции. Таким образом, еще одним аспектом настоящего изобретения является фармацевтическая композиция, которая включает в себя соединение формулы 1 в сочетании с одним или несколькими фармацевтически приемлемыми носителями, разбавителями или наполнителями. Предлагаемые фармацевтические композиции готовят по известным методикам с использованием хорошо известных и легко доступных компонентов. В процессе приготовления композиций настоящего изобретения активнодействующий компонент обычно смешивают с носителем или разбавляют носителем, или же вводят в носитель, причем препарат может быть приготовлен в форме капсулы, саше, находиться в бумажном или выполненном из другого материала контейнере. В том случае, когда носитель выполняет роль разбавителя, он может представлять собой твердый, полутвердый или жидкий материал, который действует как носитель, лекарственная среда или среда для активнодействующего компонента. Такие композиции могут быть приготовлены в форме таблеток, пилюль, порошков, лепешек, саше, крахмальных капсул, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозольных препаратов, мазей, содержащих, например, до 10 вес.% активнодействующего вещества, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций, пластырей, подкожных имплантатов и порошков в стерильной упаковке.
Некоторые примеры подходящих носителей, наполнителей и разбавителей включают в себя лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, камедь, гуммиарабик, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, водный сироп, метилцеллюлозу, метил- и пропилоксибензоаты, тальк, стеарат магния, стеариновую кислоту и минеральное масло. Такие композиции могут дополнительно содержать смазывающие агенты, смачивающие агенты (поверхностно-активные вещества), эмульгаторы и суспендирующие агенты, консерванты, подслащивающие вещества, вкусовые и ароматизирующие добавки. Композиции по изобретению могут быть приготовлены с таким расчетом, чтобы обеспечивать быстрое, постоянное или замедленное выделение активнодействующего вещества после введения в организм пациента с применением методик, которые известны в технике.
Такие композиции предпочтительнее готовить в одноразовой дозированной форме, причем каждая доза включает в себя приблизительно от 1 до 500 мг, предпочтительнее примерно от 5 до 200 мг, активнодействующего компонента. Термин "одноразовая дозированная форма" использован для ссылки на физически дискретную единицу, приемлемую для использования в качестве одноразовых доз для людей и других млекопитающих, причем каждая такая единица содержит предварительно определенное количество активнодействующего вещества, рассчитанное на достижение желаемого терапевтического эффекта, в сочетании с приемлемым фармацевтическим носителем, разбавителем или наполнителем. Нижеприведенные примеры композиций приведены только с иллюстративными целями, и ни в коем случае не предназначены для ограничения объема изобретения (см. состав композиций 1-8 в конце текста после таблиц).
Соединения настоящего изобретения и способы их синтеза дополнительно проиллюстрированы с помощью нижеприведенных примеров. Эти примеры не следует считать ограничивающими объем настоящего изобретения. Все эксперименты проводили под избыточным давлением сухого аэота или аргона. Во всех случаях, за исключением специально оговоренных, все растворители и реактивы были в готовом виде и их применяли без дополнительной обработки. Сухой тетрагидрофуран (ТГФ) перед использованием обрабатывали перегонкой с натрием или натрийбензофенонкетилом. Спектр протонного ядерного магнитного резонанса (1H-ЯМР) получали с помощью спектрометра GE QE-300 при частоте 300.15 МГц, спектрометра Брукера АМ-500 при частоте 500 МГц или спектрометра Брукера АС-200Р при частоте 200 МГц. Масс-спектроскопию с бомбардировкой свободными атомами (МСБСА) проводили с помощью прибора VG-ZAB-2SE. Масс-спектроскопию (МСФ) области десорбции осуществляли с применением прибора VG 70SE или Varian MAT 731. Оптическое вращение измеряли с помощью поляриметра Перкина-Элмера 241. Хроматографическое разделение в приборе Waters Prep 500 LC обычно проводили с использованием линейного градиента растворителей, которые указаны в тексте. Момент завершения реакции обычно определяли по данным тонкослойной хроматографии (ТСХ). Тонкослойную хроматографию проводили с использованием пластин Kieselgel 60 F254 фирмы "Э.Мерк" толщиной 0,25 мм и с остальными размерами 5 см х 10 см. Пятна определяли с использованием УФ- и химического обнаружения (пластины, вымоченные в растворе церий (IV) аммониймолибдата 75 мг молибдата аммония и 4 г сульфата четырехвалентного церия в 500 мл 10%-ной водной серной кислоты и затем нагретые на горячей плите).
Флэш-хроматографию проводили как описано в Still et al., J.Org.Chem., 43, 2923. Элементный анализ на углерод, водород и азот проводили в элементном анализаторе 440 фирмы "Контрол экуипмент корпорейшн" или осуществляли в аналитическом центре Universidad Complutense (Facultad de Farmacia, Мадрид, Испания). Точки плавления определяли в открытых стеклянных капиллярах на приборе для определения температуры плавления с горячей воздушной баней Gallencamp или на приборе для определения температуры плавления Buchi и не корректировали. Числа в скобках, приведенные после названия соединения, служат ссылкой на порядковый номер соединения.
Пример получения 1. - Карбоэтоксиметилдиметилсульфонийбромид
Раствор 265 г этилбромацетата и 114 г диметилсульфида в 500 мл ацетона перемешивали при комнатной температуре. По истечении трех дней фильтрованием реакционной смеси выделяли соединение, указанное в заголовке примера. Температура плавления: 88-90oC.
Пример 1. (1SR, 5RS, 6SR)-этил-2-oкcoдициклo[3.1.0] гексан-6-карбоксилат
Суспензию 45,5 г карбоэтоксиметилдиметилсульфонийбромида в 350 мл толуола обрабатывали 30,2 г 1,8-диазадицикло [5.-4.0]ундец-7-ена. Образовавшуюся смесь перемешивали при комнатной температуре. По истечении одного часа реакционную смесь обрабатывали 19,57 г 2-циклопентен-1-она. По истечении дополнительных 18 ч реакционную смесь добавляли в 1н. раствор хлористого натрия в соляной кислоте. Полученную смесь экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью зтилацетат с гексанами с линейным градиентом от 10 до 50% этилацетата, в результате чего получали 22.81 г соединения, указанного в заголовке примере. Температура плавления: 36-38oC.
Масс-спектр: m/z = 168 (М+).
Данные элементного анализа для C9H12O3:
Вычислено: С - 64,27, H - 7,19
Найдено: С - 64,54, H - 7,11
Пример 2. (1SR, 2RS, 5RS, 6SR)-диэтил-2-аминодицикло [3.1.0]гексан-2,6-дикарбоксилат (1) и (1SR, 2SR, 5RS, 6SR)-диэтил-2-aминoдициклo[3.1.0] гексан-2,6-дикарбоксилат (2)
Раствор 22,81 г соединения, полученного по примеру 1, в 200 мл этанола обрабатывали водным раствором 9,71 г цианистого калия и 21,2 г карбоната аммония в 200 мл воды. Образовавшуюся смесь нагревали до температуры приблизительно 50oC. По истечении примерно 18 ч реакционной смеси давали остыть до комнатной температуры и ее обрабатывали 16,2 г гидроокиси натрия. Полученную смесь выдерживали при температуре кипения с обратным холодильником. По истечении приблизительно 18 ч реакционной смеси давали остыть до комнатной температуры, а затем ее охлаждали до 0oC. Добавлением концентрированной соляной кислоты величину pH холодной смеси доводили до 1. Эту смесь концентрировали досуха в вакууме. Остаток растворяли в этаноле, охлаждали до 0oC и обрабатывали 80,6 г хлористого тионила. Образовавшуюся смесь выдерживали при температуре кипения с обратным холодильником. По истечении приблизительно 48 ч реакционную смесь концентрировали в вакууме досуха. Остаток обрабатывали 1н. раствором, гидроокиси натрия и полученную смесь экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты сушили над карбонатом калия, фильтровали и концентрировали в вакууме, получая 24,6 г смеси соединений, указанных в заголовке примера.
Пример 3. - (1SR, 2SR, 5RS, 6SR)-диэтил-2-аминодицикло [3.1.0]гексан-2,6-дикарбонат (2)
Раствор 20,71 г соединений, полученных согласно вышеизложенному в примере 2, в 200 мл этилацетата обрабатывали раствором 15,46 г щавелевой кислоты в 50 мл этанола. Образовавшуюся смесь перемешивали при комнатной температуре. По истечении одного часа реакционную смесь обрабатывали дополнительными 50 мл этанола. По истечении 18 ч смесь фильтровали и фильтрат выпаривали в вакууме досуха. Остаток обрабатывали 1н. раствором гидроокиси натрия и полученную смесь экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты промывали солевым раствором, сушили над карбонатом калия, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью хлористого метилена с 5%-ной гидроокисью аммония и метанолом (97: 3), получая 15,41 г соединения, указанного в заголовке примера. Масс-спектр: m/z = 242 (М+Н).
Данные элементного анализа для C12H19NO4:
Вычислено: С - 59,74, H - 7,94, N - 5,81
Найдено: С - 59,78, H- 8,13 N - 5,77
Пример 4. - (1SR, 2SR, 5RS, 6SR)-2-аминодицикло [3.1.0]гексан-2,6-дикарбоновая кислота (3)
Раствор 3,1 г соединения, полученного по вышеизложенному в примере 3, в 25 мл 2н. раствора гидроокиси натрия и 25 мл тетрагидрофурана перемешивали при комнатной температуре. По истечении приблизительно 18 ч в вакууме удаляли тетрагидрофуран и величину pH образовавшегося раствора доводили до 9. Указанное в заголовке соединение очищали с помощью ионообменной хроматографии (BiO-Rad AGl-X8), элюируя 50%-ным водным раствором уксусной кислоты, с получением 2,12 г продукта. Температура плавления: > 250oC (с разложением). Масс-спектр: m/z = 186 (М+Н).
Данные элементного анализа для C8H11NO4:
Вычислено: С - 51,89, H - 5,99, N - 7,56
Найдено: С - 51,74, H - 6,15, N - 7,45
Пример 5. - Гидрохлоридная соль (1SR, 2SR, 5RS, 6SR)-диэтил-2-aминoдициклo[3.1.0]гексан-2,6-дикapбoкcилaтa (2).
Раствор 2,41 г соединения, полученного в примере 3, в 75 мл диэтилового эфира перемешивали при комнатной температуре с одновременным пропусканием газообразной соляной кислоты над поверхностью раствора до тех пор, пока не прекращалось дальнейшее образование соли. После дополнительных пяти минут соль удаляли фильтрованием, промывали холодным диэтиловым эфиром и сушили в вакууме при 60oC в течение приблизительно 18 ч, получая 2,75 г соединения, указанного в заголовке примера. Температура плавления: 189-191oC.
Масс-спектр: m/z = 242 (М+Н).
Данные элементного анализа для C12H20ClNO4:
Вычислено; С - 51,89, H - 7,26, N - 5,04
Найдено: С - 52,03, H - 7,48, N - 5,06
Пример 6. -(-)-Диэтил-2-аминодицикло[3.1.0]гексан-2,6-дикарбоксилат (4)
Раствор 6,56 г рацемической смеси соединений, полученных в примере 3, в 100 мл этилацетата обрабатывали раствором 12,0 г (+)-ди-П- толуоил-D-винной кислоты в 100 мл этилацетата. После выдержки в спокойном состоянии в течение ночи при комнатной температуре кристаллический твердый продукт удаляли фильтрованием и сушили, получая 14,7 г вещества. Охлаждением фильтрата до 0oC получали дополнительное количество кристаллического твердого продукта. Объединенные кристаллические твердые продукты растворяли в горячем этилацетате, содержавшем 2-пропанол в количестве, достаточном для полного растворения. После охлаждения до 0oC кристаллический твердый материал выделяли фильтрованием, получив 2,3 г твердого продукта, энантиомерный избыток которого составлял ≥ 95%. Разделением соли между водным раствором бикарбоната натрия и этилацетатом продукт получали в форме свободного основания. Органическую фазу отделяли, сушили над карбонатом калия, фильтровали и концентрировали в вакууме, получая 0,77 г соединения, указанного в заголовке примера.
Масс-спектр: m/z = 242 (М+Н).
Оптическое вращение: αD= -5,15° (с = 1, EtOH).
Данные элементного анализа для C12H19NO4:
Вычислено: С - 59,74, H - 7,94, N - 5,81
Найдено: С - 59,68, H - 8,13, N - 5,58
Пример 7. - (+)-Диэтил-2-аминодицикло[3.1.0.]гексан-2,6-дикарбоксилат (5)
Маточные растворы из эксперимента примера 6 объединяли и концентрировали в вакууме. Превращение кислотно-аддитивной соли в свободное основание осуществляли разделением между водным раствором бикарбоната натрия и этилацетатом. Органическую фазу отделяли, сушили над карбонатом калия и концентрировали в вакууме с получением 3,7 г маслоподобного продукта. Этот маслоподобный продукт обрабатывали 7,14 г (-)-П-толуоил-L-винной кислоты в 100 мл этилацетата. После выдержки в спокойном состоянии при комнатной температуре в течение ночи кристаллы собирали фильтрованием и сушили. Кристаллические твердые частицы растворяли в горячем этилацетате, содержавшем 2-пропанол в количестве, достаточном для полного растворения. После охлаждения до 0oC кристаллы выделяли фильтрованием с получением 2,25 г соединения, указанного в заголовке примера, энантиомерный избыток которого составлял ≥ 95%. Соединение, указанное в заголовке примера, в форме свободного основания получали практически идентично изложенному выше в количестве 0,74 г.
Масс-спектр; m/z = 242 (М+Н).
Оптическое вращение: αD= -7,22° (с = 1, EtOH)
Данные элементного анализа для С12H19ClNO4:
Вычислено: С - 59,74, H - 7,94, N - 5,81
Найдено: С - 59,81, H - 7,88, N - 5,76.
Пример 8. -(+)-2-аминодицикло[3.1.0]гексан- 2,6-дикарбоновая кислота (6)
Раствор 0,69 г соединения, полученного в примере 6, в 10 мл тетрагидрофурана обрабатывали 10 мл 1н. раствора гидроокиси натрия и образовавшуюся смесь интенсивно перемешивали при комнатной температуре. По истечении нескольких дней соединение, указанное в заголовке примера, выделяли анионообменной хроматографией (BiO-Rad AGl-X8), элюируя 50%-ным водным раствором уксусной кислоты, с получением 0,53 г продукта, указанного в заголовке примера.
Масс-спектр: m/z = 186 (М+Н).
Оптическое вращение: αD= 21,32° (с = 1, 1н. соляная кислота).
Данные элементного анализа для C8H11NO4•1,25H2O:
Вычислено; С - 46,26, H - 6,55, N - 6,74
Найдено: С - 46,68, H - 6,47, N - 6,49
Пример 9. -(-)-2-aминoдициклo[3.1.0]гексан-2,6-дикapбоновая кислота (7)
Соединение, указанное в заголовке примера, получали в основном идентично вышеизложенному в примере 8 из соединения, получение которого описано в примере 7 (0,59 г). По истечении нескольких дней соединение, указанное в заголовке примера, выделяли анионообменной хроматографией (BiO-Rad AGl-X8), элюируя 50%-ным водным раствором уксусной кислоты, с получением 0,45 г продукта, указанного в заголовке примера.
Масс-спектр; m/z=186 (М+Н).
Оптическое вращение αD= -22,72° (с = 1, 1н.соляная кислота).
Данные элементного анализа для C8H11NO4•H2O:
Вычислено: С - 47,29, H - 6,45, N - 6,89
Найдено: С - 47,50, H - 6,62, N - 6,31
Пример 10. - (1SR, 2SR, 5RS, 6RS)-2-аминодицикло-[3.1.0]- гексан-2,6-дикарбоновая кислота (8)
Соединение, указанное в заголовке примера, получили из (1SR, 2SR, 5RS, 6RS)-диэтил-2-аминодицикло[3.1.0] гексан-2,6- дикарбоксилата в основном согласно вышеизложенному в примерах 3 и 4.
Пример 11. - Гидрохлорид (+)-диэтил-2-аминодицикло[3.-1.0]гексан-2,6-дикарбоксилата
Поток безводного газообразного хлористого водорода при 0oC пропускали над поверхностью раствора соединения примера 6 в 75 мл безводного диэтилового эфира до тех пор, пока не прекратилось выпадение белого осадка. Образовавшуюся суспензию перемешивали при комнатной температуре в течение 2 ч. Далее реакционную смесь разбавляли 100 мл диэтилового эфира и в вакууме отфильтровывали твердый материал. Этот твердый материал промывали 250 мл диэтилового эфира и при температуре 70oC сушили в вакууме в течение 4 ч, получая 2,32 г (8,4 ммол., 77%) соединения, указанного в заголовке примера. Температура плавления = 138-140oC.
Масс-спектр: = 242 М+1.
Данные элементного анализа для C12H20ClNO4:
Вычислено: С - 51,89, H - 7,26, N - 5,04
Найдено: С - 51,61, H - 7,32, N - 4,99
[α] = +35,52o (с = 0,09, вода).
Пример 12. - Этиловый эфир (+)-[1R-(1a, 1aα,1bβ, 2b, 5a, -5aβ,6aα)- 1, 1a, 1b, 2,5,5a,6,6a - октагидро-6-оксо-2,5-метанциклопром[a]инден-1-карбоновой кислоты
Суспензию 8,46 г (36,9 ммол.) карбоэтоксиметилдиметилсульфонийбромида в 27 мл ацетонитрила при комнатной температуре в токе азота обрабатывали 5,52 мл (36,9 ммол. ) 1,8-диазадицикло[5.4.0]ундец-7-ена. После перемешивания в течение 1 ч образовавшуюся желтую смесь обрабатывали 3,60 г (24,6 ммол.) (3aR) -3aα.4,7,aα-тетрагидро-4α,7α- метан-1Н-инден-1-она в твердом состоянии и отдельными порциями в течение 3 мин. Коричневую реакционную смесь перемешивали при комнатной температуре в течение 15 ч. Реакцию гасили добавлением 13 мл 5%-ной соляной кислоты, смесь разбавляли 50 мл солевого раствора и промывали 3 порциями по 50 мл метилтерт.бутилового эфира. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением 5,98 г коричневого маслоподобного продукта. В результате хроматографии (100 г силикагеля для флэш-хроматографии смесь гексанов с этилацетатом в соотношении вначале 8:1, а затем 2:1 сырого маслообразного продукта получали 5,0 г (88%-ный выход) соединения, указанного в заголовке примера в форме бесцветного маслоподобного продукта, которое, как определяли жидкостной хроматографией высокого давления, представляло собой единственный диастереомер. [α]
Данные элементного анализа для C14H16O3:
Вычислено: С - 72,39, H - 6,94
Найдено: С - 72,53, H - 7,08
Пример 13. Этиловый эфир (+)-[1(R), 5(S), 6(R)]-дицикло [3.1.0]гекс-3-ен-2-он-6-карбоновой кислоты
Раствор 4,89 г (21,1 ммол.) продукта примера 12 в 14 мл сухого диметилсульфоксида выдерживали при температуре кипения с одновременными перемешиванием и продувкой азотом (барботер под поверхностью) с целью отгонки высвобождавшегося циклопентадиена в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли 100 мл метил-трет.бутилового эфира и один раз промывали 50 мл воды. Водный слой один раз промывали 25 мл метил-трет. бутилового эфира и объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением светло-желтого твердого материала. Этот сырой циклопентенон кристаллизовали из смеси гексанов с метилтрет. бутиловым эфиром, получая 1,91 г (55%-ный выход) соединения, указанного в заголовке примера. Температура плавления: 96-98oC; [α]
Данные элементного анализа для C9H10,O3;
Вычислено: С - 65,05, H - 6,07
Найдено; С - 64,78, H - 6,24
Пример 14. - Этиловый эфир (-)-[1(R), 5(S), 6(R)] - дицикло[3.1.0]гексан-2-он-6-карбоновой кислоты
Раствор 1,73 г (10,4 ммол.) продукта примера 13 в 35 мл 95%-ного этанола в токе азота обрабатывали 87 мг (5 вес.%) 10%-ного палладия на угле. Колбу продували водородом и перемешивание продолжали в атмосфере водорода (баллонное давление) в течение 5 ч, причем за этот промежуток времени добавляли дополнительно 35 мг (2 вес.%) 10%-ного палладия на угле. Смесь перемешивали в водородной атмосфере в течение еще 50 мин. Колбу продували азотом и катализатор удаляли фильтрованием через целит, промывая этилацетатом. Фильтрат и промывные жидкости концентрировали в вакууме, получая 1,75 г желтого твердого материала. Этот сырой твердый материал кристаллизовали из смеси гексанов с метил-трет. бутиловым эфиром, получая 1,38 г (79%-ный выход) соединения, указанного в заголовке примера. Температура плавления: 63-65oC; [α]
Данные элементного анализа для C9H12O3:
Вычислено: С - 64,27, H - 7,19
Найдено: С - 64,10, H - 7,31
Пример 15. - Этиловый эфир (+)-[1(R), 2(P), 5(S), 6(R), 5'(R)]-2-спиро-5'-гидантоиндицикло[3.1.0]гексан-6-карбоновой кислоты
Смесь 1,20 г (7,13 ммол.) продукта примера 14 с 511 мг (7,85 ммол.) цианистого калия и 1,37 г (7,13 ммол.) карбоната аммония в 7,1 мл 95%-ного этанола и 2,9 мл воды перемешивали при 36oC в течение 10 ч и при комнатной температуре 13 ч. Мутную желтую реакционную смесь охлаждали до 0oC и разбавляли 7,8 мл холодной воды. После перемешивания в течение 1,5 ч белый твердый продукт собирали и дважды промывали по 5 мл холодной воды. Твердый материал сушили в вакууме, получая 1,17 г (69%-ный выход) соединения, указанного в заголовке примера, в виде единственного диастереомера, как это определяли высокоэффективным жидкостным хроматографическим анализом. Температура плавления: 247-249oC.
[α]
Данные элементного анализа для C11H14N2O4:
Вычислено: С - 55,46, H - 5,92, N- 11,76
Найдено: С - 55,76, H - 5,95, N - 11,84
Пример 16. -(-)-[1(R), 2(R), 5(S), 6(R)]-2-аминодицикло[3.1.0]гексан-2,6-дикарбоновая кислота
Раствор 976 мг (4,10 ммол.) продукта примера 15 в 8,2 мл 3н раствора гидроокиси натрия выдерживали при температуре кипения с обратным холодильником при перемешивании в течение 24 ч. После охлаждения до комнатной температуры реакционную смесь переносили непосредственно в ионообменную колонну (заполненную 50 г ацетатной смолы BiO-Rad AG1-X8, подготовленной промывкой 50 мл 1н. раствора гидроокиси натрия, а затем 50 мл воды, за которой после введения реакционной смеси следовали 50 мл 1н. раствора гидроокиси натрия), элюируя смесью воды с уксусной кислотой в соотношении 1:1, отбирая в результате 50-миллилитровые фракции. Фракции 2 и 3, которые содержали продукт, объединяли и концентрировали в вакууме, получая 770 мг белого твердого материала. Этот твердый материал суспендировали в 4 мл воды и фильтровали, один раз промывая 4 мл воды. Твердый продукт сушили в вакууме при 40oC, получая в виде белого порошка 634 мг (76%-ный выход) соединения, указанного в заголовке примера. ИК (бромид калия): 3235 (br, S), 2971 (m), 2016 (br, W), 1694 (m), 1613 (S), 1509 (m), 1237 (m) см-1; 1H-ЯМР-спектр-(трифторуксусная кислота -d, δ): 2,76-2,74 (m, 1H), 2,65-2,52 (m, 3H), 2,38-2,31 (m, 2H), 1,96-1,88 (m, 1H) 13C-ЯМР-спектр (трифторуксусная кислота-d, δ): 179,43, 175,63, 69,53, 34,92, 31,75, 31,66, 27,63, 23,04. Образец для анализа получали кристаллизацией из воды. Температура плавления: 277-280oC (с разложением); [α]
Пример 17. 2-оксодицикло [3.1.0]гексан-6-карбоновая кислота
Смесь 60 г этил-2-оксодицикло[3.1.0]гексан-6- карбоксилата с 300 мл 1н. раствора гидроокиси натрия перемешивали при 25-30oC. По истечении 2,5 ч добавляли концентрированную соляную кислоту для доведения величины pH до 0,8-1,2. Образовавшийся раствор экстрагировали этилацетатом. Экстракты сушили над сульфатом магния, фильтровали и концентрировали с получением 49,1 г (98%-ный выход) сырого материала. В результате перекристаллизации из 100 мл этилацетата получали соединение, указанное в заголовке примера, с температурой плавления 123,5- 128oC.
Масс-спектр: m/z = 140 (М+).
Данные элементного анализа для C7H8O3:
Вычислено: С - 60,00, H - 5,75
Найдено: С - 60,14, H - 5,79
Пример 18. - Соль 2-оксодицикло [3.1.0]гексан-6-карбоновой кислоты и (S)-1-фенилэтиламина
Раствор 14 г соединения, полученного в примере 17, в 140 мл 25%-ного раствора этанола в этилацетате объединяли с 1 экв. (S)-1-фенилэтиламина. После перемешивания в течение ночи выпавшую в осадок соль выделяли фильтрованием и сушили с получением 11,87 г (45,4%) целевой соли. Превращение соли, частично растворенной 2-оксодицикло[3.1.0] гексан-6-карбоновой кислоты по методу примера 17 и анализ показали, что энантиомерный избыток для такой соли составлял 68%. Этот энантиомерный избыток определяли превращением в метиловый эфир диазометаном с последующей хиральной жидкостной хроматографией высокого давления на колонке Chiralpak AS при 40oC, элюируя смесью 10% изопропанола с 90% гексана со скоростью 1 мл/мин и обнаружении при длине волны 210 нм.
Пример 19. -(+)-2-оксодицикло[3.1.0]гексан-6-карбоновая кислота
Смесь 1,31 г продукта примера 18 и 10 мл 1н. соляной кислоты перемешивали в течение 5 мин и экстрагировали этилацетатом. Экстракты сушили над сульфатом натрия, фильтровали и концентрировали с получением 0,61 г соединения, указанного в заголовке примера, с температурой плавления 110-115oC. Согласно определению с помощью хиральной жидкостной хроматографии высокого давления (по методу примера 18 энантиомерный избыток для продукта составлял 68%).
Масс-спектр: m/z = 141 (М+Н).
Оптическое вращение: αD= 49,85o
Пример 20. -(-)-2-спиро-5'-гидантоиндицикло[3.1.0] гексан-6-карбоновая кислота
Раствор 1 экв. соединения, полученного по вышеизложенному в примере 19 (68%-ный энантиомерный избыток), 1,25 экв. цианистого калия и 2,5 экв. карбоната аммония совмещали между собой и перемешивали при 25oC в смеси этанола с водой в течение 40 ч. Смесь подкисляли 6н. соляной кислотой, концентрировали, разбавляли водой и фильтровали, получая смесь диастереомеров в соотношении 90: 10, температура плавления которой составляла 286-290oC, с достижением 79%-ного выхода. Эту диастереомерную смесь перекристаллизовывали из смеси изопропанола с водой, получая с 48%-ным выходом соединение, указанное в заголовке примера, с достижением 100%-ной диастереомерной и 100%-ной энантиомерной чистоты, энантиомерное соотношение определяли хиральной жидкостной хроматографией высокого давления на колонке Chiracel OD-H размерами 4,6 х 150 мм, злюируя смесью 15% изопропанола с 85% гексана со скоростью потока 1 мл/мин при 40oC и с обнаружением при длине волны 220 нм, диастереомерное соотношение определяли жидкостной хроматографией высокого давления на колонке Zorbax SB-Phenyl при 40oC, элюируя смесью буфера с ацетонитрилом в соотношении 90:10, со скоростью потока при элюировании 2 мл/мин и с обнаружением при длине волны 220 нм (буфером служил 0,1М раствор моногидрата вторичного кислого фосфата натрия, величину pH которого добавлением фосфорной кислоты доводили до 2.1).
Масс-спектр: m/z = 211 (М+H).
Оптическое вращение: αD= -25,98
Данные элементного анализа для C9H10N2O4:
Вычислено: С - 51,43, H - 4,79, N - 13,33
Найдено: С - 51,38, H - 4,80, N - 13,26.
Пример 21. - Этил-2-спиро-5'-гидантоиндицикло/3.1.0/-гексан-6-карбоксилат
Смесь 5,05 г этил-2-оксодицикло/3.1.0/гексан-6-карбоксилата с 2,15 г цианистого калия, 5,77 г карбоната аммония, 30 мл этанола 2B-3 и 12 мл воды перемешивали при 35oC до завершения реакции, что определяли жидкостной хроматографией высокого давления. По истечении 15 ч реакционную смесь охлаждали до 0oC и в смесь добавляли 33 мл воды. По истечении 2 ч выдержки при 0oC осадок выделяли фильтрованием и сушили, получая 5,23 г (73%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 217-220oC.
Масс-спектр: m/z = 238,1 (М+).
Данные элементного анализа для C11H14N2O4:
Вычислено: С - 55,46, H - 5,92, N - 11,76
Найдено: С - 55,74, H - 5,88, N - 11,50
Пример 22. 2-спиро-5'-гидантоиндицикло[3.1.0]гексан-6-карбоновая кислота
Смесь 16,32 г продукта примера 21 и 137 мл 2н. раствора гидроокиси натрия перемешивали при 25oC. По истечении 1 ч для доведения величины pH до 1,0 добавляли концентрированной соляной кислоты. Образующийся осадок выделяли фильтрованием и сушили, получая 13,70 г (95%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 277-279oC.
Масс-спектр: m/z = 210,1 (М+).
Данные элементного анализа для C9H10N2O4:
Вычислено: С - 51,43, H - 4,79. N - 13,33
Найдено: С - 51,70, H - 4,93, N - 13,43
Пример 23. (S)-1-фенилэтиламиновая соль 2-спиро-5'-гидантоиндицикло [3.1.0]гексан-6-карбоновой кислоты
Смесь 1,05 г продукта примера 22 с 16,6 мл раствора ацетона в воде в соотношении 1,6: 1 перемешивали при 25oC, одновременно добавляя 1,53 г R-(+)-1-фенилэтиламина. Эту смесь перемешивали в течение 2 ч при комнатной температуре. Кристаллы отфильтровывали, промывали ацетоном и сушили, получая 0,74 г (45%-ный выход) соединения, указанного в заголовке примера, при температуре 205-212oC.
Оптическое вращение: αD= -31,88 (с = 1, метанол).
Пример 24. -(-)-2-спиро-5'-гидантоиндицикло[3.1.0]гексан-6- карбоновая кислота
Смесь 0,74 г продукта примера 23 с 10 мл воды перемешивали при 25oC, одновременно доводя величину pH с 6,81 до 1,0 добавлением 1н. соляной кислоты. Реакционную смесь перемешивали в течение 1 ч и продукт собирали фильтрованием и сушили, получая 0,35 г (75%-ный выход) соединения, указанного в заголовке примера, с температурой плавления 310oC (с разложением).
Масс-спектр: m/z = 210,1 (М+).
Оптическое вращение: αD= -24,22 (с=1, метанол).
Данные элементного анализа для C9H10N2O4:
Вычислено: С - 51,43, H - 4,80, N- 13,33
Найдено: С - 51,67, H - 4,87, N - 13,61
Пример 25. -(+)-2-аминодицикло[3.1.0]гексан-2,6-карбоновая кислота
Раствор 184 г (-)-2-спиро-5'-гидантоиндицикло[3.1.0]- гексан-6-карбоновой кислоты и 1750 мл 3н. раствора гидроокиси натрия выдерживали при температуре кипения с обратным холодильником до завершения реакции, о чем судили по данным жидкостной хроматографии высокого давления. По истечении 28 ч раствор охлаждали до комнатной температуры и фильтровали через стекловолокно, удаляя следовые количества нерастворимого материала. Добавлением концентрированной соляной кислоты величину pH раствора доводили до 3,0. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и два часа при 0oC. Выпадавший в осадок продукт собирали фильтрованием, промывали 170 мл холодной воды и сушили, получая 152,3 г (86%-ный выход) соединения, указанного в заголовке примера.
Масс-спектр: m/z = 186,1 (М+1).
Оптическое вращение: αD= = -23,18o (с = 1, 1н. соляная кислота).








Описываются новые соединения формулы I, где X обозначает (CH2)n; R2 обозначает CO2R4; R3 обозначает водород или R2 обозначает водород и R3 обозначает CO2R4; R1 и R4 каждый независимо обозначает водород, C1 - C10-алкил, C2 - C10-алкенил, арил или арилалкил; n равно 1, или их фармацевтически приемлемые соли. Вышеуказанные соединения воздействуют на некоторые рецепторы возбудительных аминокислот и могут быть использованы для лечения неврологических расстройств и психиатрических расстройств. Кроме того, описывается фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы I и промежуточные соединения, используемые для получения соединения (I). 3 с. и 8 з.п. ф-лы, 3 табл.
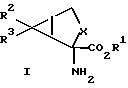
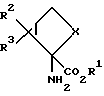
где Х обозначает (СН2)n;
R2 обозначает CO2R4 и R3 обозначает водород, или R2 обозначает водород и R3 обозначает CO2R4;
R1 и R4 каждый независимо обозначают водород, С1-С10-алкил, С2-С10-алкенил, арил или арилалкил;
n равно 1,
или фармацевтически приемлемые соли.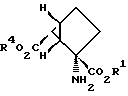
3. Соединение по п.1 или 2, отличающееся тем, что R1 и R4 каждый независимо обозначают водород, С1-С4-алкил, арил или арилалкил, или его фармацевтически приемлемая соль.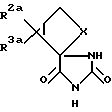
где Х обозначает (CH2)n;
n равно 1;
R2а обозначает СО2R4а и R3а обозначает водород, или R2а обозначает водород и R3a обозначает СО2R4а;
R4а обозначает карбоксизащитную группу.
| EP, 0500443, A1, 26.08.1992 | |||
| SU, 398539, A, 27.09.1973 | |||
| EP, 0376072, A3, 04.07.1990. |

