Настоящее изобретение относится к новым производным ацилированных аминоалкилимидазолов и триазолов, способу их получения и фармацевтической композиции на их основе.
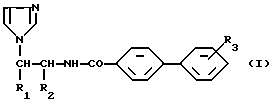
Более конкретно настоящее изобретение касается алкилированных аминоалкилимидазолов общей формулы (I)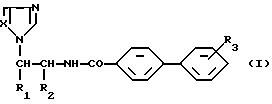
где:
либо
R1 представляет собой фенильную группу, возможно монозамещенную атомом галогена, или 1-нафтильную группу, а
R2 обозначает атом водорода;
либо
R1 обозначает атом водорода, а
R2 представляет собой пиридильную или 2-(5-хлор)пиридильную группу;
R3 обозначает атом галогена или алкоксигруппу, содержащую от 1 до 4 атомов углерода, находящиеся в свободном виде или в виде фармацевтически приемлемой соли.
Предпочтительным является соединение формулы I, в котором либо R1 представляет собой фенильную группу, возможно монозамещенную атомом хлора в положении 4, a R2 обозначает атом водорода, либо R1 обозначает атом водорода, a R2 представляет собой 2-(5-хлор)пиридильную группу, либо R1 обозначает 1-нафтильную группу, a R2 представляет собой атом водорода, находящиеся в свободном виде или в виде фармацевтически приемлемой соли.
Особенно предпочтительно соединение формулы I, представляющее собой N-[4-(4-хлорфенил)бензоил] -2-(1H-имидазол-1-ил)-2- фенил-1-аминоэтан в виде рацемата или 2(S)- или 2(R)-энантиомерных форм, находящиеся в свободном виде или в виде соли.
Из указанной группы алкилированных аминоалкилимидазолов формулы I следует выделить соединение, где R1 представляет собой атом водорода, а радикалы R2 и R3 обозначают соответственно:
- 2-(5-хлор)пиридил и 4-хлор, в рацемической или (+)- или (-)-энантиомерной форме, или
-2-пиридил и 4-хлор, или
-3-пиридил и 4-хлор, или
-2-(5-хлор)пиридил и 4-этокси или
-2-(5-хлор)пиридил и 4-н-бутокси;
или
R2 представляет водород, R3 представляет хлор и R1 представляет 4-хлорфенил в рацемической или (+)S-энантиомерной форме, или представляет 1-нафтил, в свободном виде или в виде соли.
Предлагаемые согласно изобретению алкилированные аминоалкилимидазолы общей формулы I обладают способностью избирательно ингибировать гидроксилазы 25-гидроксивитамина D3.
Известные близкие по структуре имидазолы или триазолы, описанные в DE-A-408127, проявляют антимикотическую активность и могут быть использованы в качестве фунгицидов. Действие известных соединений никак не связано с избирательным ингибированием гидроксилаз 25-гидроксивитамина D3, атакующих боковую цепь кальцитриола и, соответственно, в отличие от соединений настоящего изобретения не катаболизируют проявляющие гормональную активность метаболиты витамина D3 (например, кальцитриол).
Соединения, описываемые в приведенной ссылке, содержат незамещенную метиленовую группу, связанную с имидазольным или триазольным фрагментом, с последующим фенилзамещенным метиленовым звеном, в то время как соединения согласно настоящему изобретению либо содержат арил- или гетероарилзамещенную метиленовую группу, связанную с имидазольным или триазольным фрагментом, с последовательно присоединенным заместителем - незамещенной метиленовой группой, или содержат незамещенную метиленовую группу, присоединенную к имидазольному фрагменту, с последовательно присоединенным заместителем - гетероарилзамещенным метиленом.
В случае, если фенильная группа замещена, то она предпочтительно замещена в положении 4. Если замещена пиридильная группа, то она предпочтительно замещена в положении 5.
Понятие "атом галогена" соответствует атому фтора, хлора, брома или иода, предпочтительно - хлора. Алкоксирадикалы в предпочтительном случае независимо друг от друга имеют от 1 до 4 атомов углерода, в частности, - 1 или 2, в особенности - 1.
Понятие "пиридильная группа" предпочтительно соответствует 2- или 4-пиридильной группе. Понятие "нафтильная группа" предпочтительно обозначает 1-нафтильную группу.
Согласно настоящему изобретению предлагается также способ получения алкилированных аминоалкилимидазолов общей формулы I, заключающийся в том, что соединение формулы II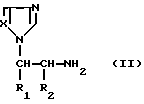
где заместители R1 и R2 имеют значения, указанные выше, подвергают ацилированию для введения соответствующей бифенил-4-карбонильной группы с последующим выделением целевого продукта в свободном виде или в виде соли (способ a).
Однако предлагаемые согласно настоящему изобретению соединения можно получить и другим способом. В частности соединения, отвечающие формуле: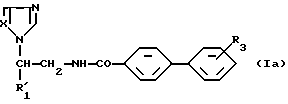
в которой R1' соответствует радикалу R1, но не является атомом водорода, а X означает группу CH или N, могут быть получены взаимодействием соединения, отвечающего формуле
или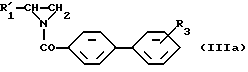
с соответствующими соединениями, описываемыми формулой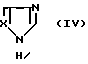
и выделяют образующиеся при этом соединения, отвечающие формуле (I), в свободном виде или в виде соли.
Предусмотренный настоящим изобретением способ можно осуществлять традиционным образом.
Осуществляя вариант способа a), можно, например, приводить соединение, отвечающее формуле (II), во взаимодействие с приемлемым ацилгалогенидом в растворителе, включая органическое или неорганическое основание, которое в то же время может служить связывающим кислоту агентом, таким как пиридин, возможно с добавлением ускорителя ацилирования, такого как 4-диметиламино-пиридин. Указанную реакцию также можно осуществлять, приводя соединение, описываемое формулой (II), во взаимодействие с приемлемым активированным эфиром или с активированным производным ацила, например, смешанного ангидрида или имидазола, получаемого из N,N-карбонилдиимидазола.
Указанную реакцию можно осуществлять, например, с использованием 2-пиридилтиолового эфира, такого как 2-пиридилтиоловый эфир 4'-хлорбифенил-4-карбоновой кислоты, в инертном растворителе, таком как амид динизшего алкила карбоновой кислоты, например диметилформамиде, предпочтительно при комнатной температуре.
Осуществить вариант способа б) можно, смешивая соединение, описываемое формулой (III) или (IlIa), с соединением, отвечающим формуле (IV), и нагревая полученную смесь до повышенной температуры, например до 120oC.
Указанные исходные вещества либо известны, либо могут быть получены в соответствии с известными способами, либо по аналогии с известными способами, либо по аналогии со способами, приведенными в настоящем описании, в том числе в примерах.
Соединения, отвечающие формуле (II), можно получить, например, в соответствии со следующей схемой реакций: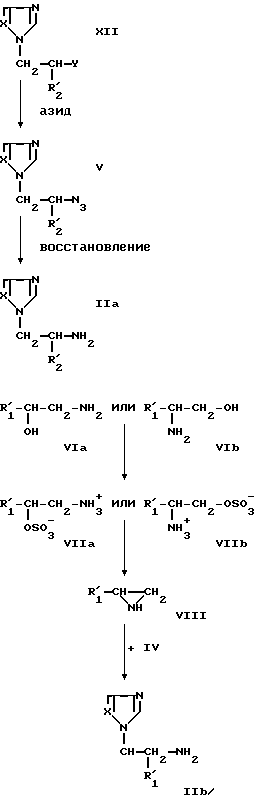
В представленной выше схеме радикал R2' соответствует радикалу R2, однако не может быть атомом водорода; Y представляет собой атом галогена, предпочтительно хлора, а остальные заместители таковы, как описано выше.
Исходные вещества, отвечающие формуле (III), могут быть получены, например, в соответствии со следующей схемой реакций: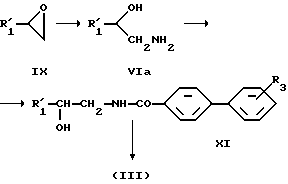
Исходные вещества, описываемые формулой (IlIa), могут быть получены, например, путем ацилирования соответствующего соединения, отвечающего формуле (VIII).
Соединения, описываемые формулой (I), являются хиральными по атому углерода, несущему группу R1 или R2, отличную от атома водорода, в результате чего они могут существовать в виде рацематов, чистых энантиомеров [(R) и (S)] или их смесей. Настоящее изобретение охватывает все стереоизомеры, а также рацемические смеси. Указанные изомеры могут быть выделены или разделены с использованием традиционных способов, например, с помощью хроматографии.
Получение чистых энантиомеров соединений, соответствующих формуле (I), предпочтительно осуществляют с использованием чистых энантиомеров в качестве исходных веществ в вариантах способа а) и б). Раскрытие кольца чистых энантиомеров азиридиновых соединений, отвечающих формуле (VIII) или (IlIa), при реакции с соединением, описываемым формулой (IV), происходит с превращением их конфигурации с получением соединения, отвечающего формуле (IIб) или (Iа) с противоположной конфигурацией. Кроме того, превращение указанной конфигурации происходит также при реакции, ведущей к получению соединений, описываемых формулой (III), из соединений, отвечающих формуле (XI), а также на последующей стадии, ведущей к получению соединений, соответствующих формуле (Iа). На всех остальных стадиях, соответствующих вышеописанным реакциям, указанная конфигурация остается неизмененной. Таким образом, использование чистых энантиомеров исходных веществ приводит к образованию конечных продуктов, характеризующихся противоположной конфигурацией, в том случае, если на одной из используемых стадий реакций происходит превращение конфигурации на соответствующем хиральном центре, но приводит к образованию конечных продуктов, с неизмененной конфигурацией, если не используют хотя бы одну или обе стадии, на которых происходит превращение конфигурации на соответствующем хиральном центре.
Отдельные стадии указанных реакций можно осуществлять традиционным образом.
Предусмотренное настоящим изобретением соединение может находиться в свободном виде или в виде соли, такой как соль, образующаяся при добавлении кислоты. Предусмотренное настоящим изобретением соединение, находящееся в свободном виде, можно обычным известным способом перевести в соль, такую как соль, образующуюся в результате добавления кислоты, например соответствующий гидрохлорид или нитрат; или наоборот.
Изобретение также относится к фармацевтической композиции, обладающей способностью избирательно ингибировать гидроксилазы 25-гидроксивитамина D3, содержащей соединение формулы I по п.1 в свободном виде или в виде его фармацевтически приемлемой соли и, по меньшей мере, один фармацевтически приемлемый носитель или разбавитель.
В настоящем описании используют следующие сокращения:
25(OH)D3: 25-гидроксивитамин D3
1,25(OH)2D3: 1,25-дигидроксивитамин D3 (кальцитриол)
24,25(OH)2D3: 24,25-дигидроксивитамин D3
1,24,25(OH)3D3: 1,24,25-тригидроксивитамин D3
1,23,25(OH)3D3: 1,23,25-тригидроксивитамин D3
ДМФ диметилформамид
ДМСО диметилсульфоксид
HPLC высокоразрешающая жидкостная хроматография
ТГФ тетрагидрофуран
т.к. температура кипения
p/мин разложение в мин
т.пл. температура плавления
ЭФР эпидермальный фактор роста
ФСТ фетальная сыворотка теленка
ССРХ сбалансированный солевой раствор Хэнка
СРК среда для роста кератиноцитов (Clonetics, San Diego, CA, USA)
Все температуры приведены в градусах Цельсия. Следующие ниже примеры иллюстрируют настоящее изобретение, но не имеют лимитирующего характера.
Пример 1:
(1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)-N-[4-(4-хлорфенил)бензоил]-1- аминоэтан
(вариант способа а))
К раствору 0,44 г 1-амино-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1- ил)этана в 1 мл диметилформамида добавляют раствор 0,645 г 2-пиридилтиолового эфира 4'-хлорбифенил-4-карбоновой кислоты в 2 мл сухого диметилформамида. Полученную реакционную смесь перемешивают при комнатной температуре в атмосфере аргона в течение 18 ч. Указанную реакционную смесь выливают в холодный насыщенный солевой раствор и экстрагируют этилацетатом. Собранные органические фазы промывают солевым раствором, высушивают над Na2SO4, фильтруют и выпаривают растворитель (= способ получения A). Обрабатывая сырой продукт диэтиловым эфиром, получают указанное в заголовке соединение в виде бесцветных кристаллов; т.пл. 143-146oC.
Используя оптически активные энантиомеры в качестве исходного вещества и осуществляя обработку по аналогии с вышеописанной, получают соответствующие оптически активные энантиомеры указанного в заголовке соединения:
(+)-энантиомер: [α]
(-)-энантиомер: [α]
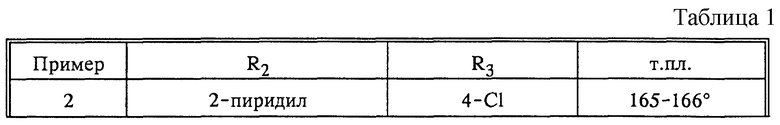
По аналогии с примером 1, в соответствии с вариантом способа а) получают следующие соединения, отвечающие формуле (I; X=CH, R1=H) в виде рацемата (табл.1).
Пример 7:
N-[4-(4-хлорфенил)бензоил]-2-(1H-имидазол-1-ил)- 2(S)-фенил-1-аминоэтан
(вариант способа а))
Раствор 0,96 г неочищенного (S)-2-фенил-2-(1H-имидазол-1-ил)-1- аминоэтана в 5 мл N,N-диметилформамида при перемешивании приводят во взаимодействие с 1 г 2-пиридилтиоловым эфиром 4'-хлорбифенил-4- карбоновой кислоты. Через 6 часов останавливают реакцию, выливая указанную смесь в холодную воду (0o, 50 мл). Образовавшийся осадок экстрагируют этилацетатом, полученную органическую фазу промывают солевым раствором, высушивают над сульфатом магния и выпаривают. Полученный кристаллический остаток растирают в порошок с этанолом, фильтруют с отсасыванием и промывают холодным этанолом. Высушивают образующиеся кристаллы с получением чистого указанного в заголовке соединения; т.пл. 176-177o; [α]
(R)-энантиомер указанного в заголовке соединения получают аналогичным образом, используя в качестве исходного вещества (R)-2-фенил-2-(1H-имидазол-1-ил)-1-аминоэтан; т.пл. 176-179o; [α]
Пример 8:
N-[4-(4-хлорфенил)бензоил]-2-(1H-имидазол-1-ил)-2(S)-фенил-1-аминоэтан
(вариант способа б))
0,398 г N-(4'-хлорбифенил-4-карбонил)-2(R)-фенилазиридина смешивают с 0,15 г имидазола, после чего нагревают при температуре 110o в течение 16 ч. После охлаждения указанной смеси образовавшийся твердый осадок растворяют в 1,5 мл ДМФ при нагревании. Полученный раствор выливают в ледяную воду. Выпавшее в осадок твердое вещество собирают отсасыванием, промывают водой и высушивают. Полученное вещество очищают с помощью хроматографии на силикагеле (элюирование смесью толуола и этанола в соотношении 8:1). Получают указанное в заголовке соединение в виде белых кристаллов; т.пл. 176-179o; [α]
1H-ЯМР (DMCO-d6) δ: 8,82 (t, J=5,3 Гц, 1H), 7,87-7,84 (m, 3H), 7,78-7,74 (m, 4H), 7,55 (d, J=5,5 Гц, 2H), 7,41-7,30 (m, 6H), 6,92 (s, 1H), 5,70 (dd, J=6,0, 9,1 Гц, 1H), 4,16-3,94 (m, 2H).
Пример 9:
N-[4-(4-хлорфенил)бензоил]-2-(1H-имидазол-1-ил)-2(R)-(фенил-1- аминоэтан
(вариант способа б))
380 мг 2-(4'-хлорбифенил-4-ил)-5(S)-фенил-4,5-дигидрооксазола и 775 мг имидазола выдерживают при температуре 120o в течение 23 ч. Добавляют этилацетат и воду, полученную органическую фазу трижды промывают водой, высушивают над сульфатом магния и концентрируют в вакууме. После добавления толуола получают указанное в заголовке соединение в виде бесцветных кристаллов; т. пл. 176-179o (перекристаллизация из толуола); [α]
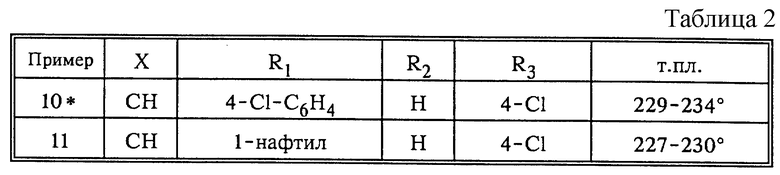
По аналогии с примером 8, используя вариант способа б), получают рацематы следующих соединений, соответствующих формуле (I) ( табл. 2), где
* 2(S)-энантиомер соединения из примера 10 получают из соответствующего 4,5-дигидрооксазола по аналогии с описанием, приведенным в примере 9; т.пл. 197-204o; [α]
Пример 12:
N-[4-(4-хлорфенил)бензоил]-2-(1H-имидазол-1-ил)-2-фенил-1-аминоэтан
(вариант способа а))
0,94 г 2-фенил-2-(1H-имидазол-1-ил)-1-аминоэтана ацилируют 0,17 г 2-пиридилтиоловым эфиром 4'-хлорбифенил-4-карбоновой кислоты по аналогии с примером 1. С помощью хроматографии на силикагеле (элюирование смесью дихлорметана, метанола, водного раствора NH3 и гептана в соотношении 7:1:0,1:5) получают указанное в заголовке соединение в виде бесцветных кристаллов; т. пл. 209-212o.
* Указанные исходные вещества могут быть получены следующим образом:
А. 1-Амино-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан
а. 2- Ацетил-5-хлорпиридин
К суспензии 56 г 2-бром-5-хлорпиридина в 560 мл сухого диэтилового эфира при температуре -78o и в атмосфере аргона добавляют раствор 179 мл н-бутиллития (15%-ный раствор в гексане) таким образом, чтобы температура смеси не превышала -72o. Сразу после этого добавляют раствор 25,7 мл N,N-диметилацетамида в сухом ТГФ. Полученную смесь перемешивают в течение 1 часа при той же температуре, после чего разлагают, осторожно добавляя 100 мл 3н соляной кислоты, а затем - 100 мл воды при сильном охлаждении. Осуществляя вариант способа а) (см. пример 1) и проводя очистку с помощью хроматографии на силикагеле (элюирование смесью толуола и этилацетата в соотношении 20:1), получают указанное соединение в виде белых игл; т.пл. 58-65o.
б. 2-Бромацетил-5-хлорпиридин гидробромид
К раствору 10 г 2-ацетил-5-хлорпиридина в 150 мл 48%-ной водной бромоводородной кислоты при перемешивании по каплям при температуре 80o добавляют 4 мл брома, растворенного в 40 мл бромоводородной кислоты. Полученную смесь перемешивают в течение еще 3 часов при температуре 80o. Затем указанный раствор выпаривают в вакууме. Образовавшийся остаток растирают с ацетоном, фильтруют с отсасыванием, промывают ацетоном и высушивают в вакууме с получением желтых кристаллов указанного в заголовке соединения, которое используют на следующем этапе без дополнительной очистки.
в. 2-[2'-(1H-имидазол-1-ил)ацетил]-5-хлорпиридин
15 г гидробромида 2-бромацетил-5-хлорпиридина растворяют в 50 мл сухого дихлорметана, добавляют 9,7 г имидазола и перемешивают полученную смесь в течение 24 часов при комнатной температуре. Выпаривают растворитель в вакууме, после чего образовавшийся остаток подвергают хроматографии на силикагеле (элюирование смесью дихлорметана, метанола и гептана в соотношении 7: 1:4) с получением указанного в заголовке соединения в виде твердого вещества кремового цвета; т.пл. 122-129o.
г. 1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан-1-ол
К раствору 7,8 г 2-[2'-(1H-имидазол-1-ил)ацетил]-5-хлорпиридина в 40 мл метанола при температуре 0o по частям добавляют 2,0 г боргидрида натрия. Полученную смесь перемешивают в течение 1 часа при температуре 0o. Для завершения реакции указанную смесь разлагают, осторожно добавляя 1н соляную кислоту при температуре 0o, а затем - 1н гидроксид натрия до pH 9. Растворители выпаривают в вакууме. Полученный остаток перемешивают с водой и дихлорметаном, водную фазу дважды экстрагируют дихлорметаном, а объединенные органические экстракты промывают солевым раствором, высушивают и выпаривают. Получают указанное в заголовке соединение в виде твердого вещества кремового цвета; т. пл. > 210o (с разложением). Возможна очистка с помощью хроматографии на силикагеле.
д. 1-Хлор-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан
1 г 1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан-1-ола растворяют в 2 мл толуола, добавляют 6 мл тионилхлорида и перемешивают полученную реакционную смесь в течение 30 минут при комнатной температуре. Отгоняют растворитель и избыток тионилхлорида, растворяют образовавшийся остаток в 2н NaOH, экстрагируют этилацетатом, высушивают над Na2SO4 и выпаривают с получением указанного в заголовке соединения в виде бесцветного масла.
е. 1-Азидо-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан
0,92 г 1-хлор-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этана в атмосфере аргона добавляют к раствору 0,37 г азида лития в 2 мл сухого диметилформамида и перемешивают полученную реакционную смесь при температуре 60o в течение 18 ч. Осуществляя вариант способа а) (см. пример 1), получают указанное в заголовке вещество в виде желтоватого масла.
ж. 1-Амино-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан
Раствор 0,83 г 1-азидо-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил) этана в 4 мл сухого пиридина перемешивают в атмосфере H2S в течение 3 ч. Выпаривают реакционную массу, полученный остаток вновь растворяют в 5 мл разбавленной уксусной кислоты (уксусная кислота и вода в соотношении 1:4) и экстрагируют этилацетатом. Выпаривают объединенные органические фазы с получением указанного в заголовке продукта в виде желтоватого масла.
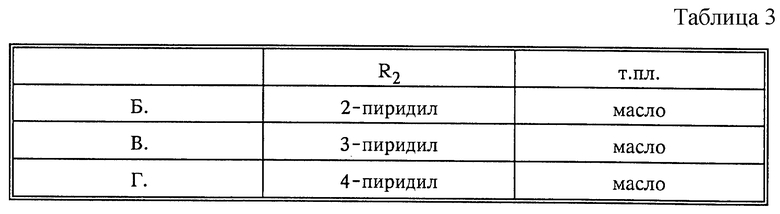
По аналогии с описанием, приведенным выше в пункте A, получают следующие исходные вещества формулы (II) в виде вязких желтоватых масел, причем указанные масла используют без дополнительной очистки (X=CH, R1=H) (табл. 3).
Д. S-(5)-2-фенил-2-(1H-имидазол-1-ил)-1-аминометан
а. Кислый сульфат (R)-2-амино-2-фенил-этанол-O-сульфата
Раствор 10,65 г (R)-(-)-2-амино-2-фенилэтанола в 30 мл воды нейтрализуют 48%-ной серной кислотой до изменения окраски метилового красного (4,2 мл), после чего добавляют равный объем серной кислоты. Затем с помощью ротационного испарителя при температуре бани сначала при 70-90o, а к концу 130o, а также при давлении 0,1 торр удаляют воду до постоянного веса. Образовавшейся смеси позволяют постепенно затвердеть, после чего ее измельчают в ступке. Полученное вещество можно использовать на следующем этапе без дополнительной очистки.
б. (R)-(-)-2-фенилазиридин
16,2 г тонко измельченного кислого сульфата (R)-2-амино-2- фенил-этанол-O-сульфата при температуре 0o по частям добавляют к перемешиваемому раствору 2н гидроксида натрия. Полученную реакционную смесь выдерживают при температуре 100o в течение 3 ч. Для осуществления реакции указанную смесь охлаждают до комнатной температуры и 4 раза экстрагируют эфиром. Полученную органическую фазу промывают солевым раствором, высушивают над сульфатом магния и выпаривают. Осуществляя при пониженном давлении перегонку образовавшегося остатка, получают указанное в заголовке соединение в виде бесцветного масла; т.к. 46o (0,1 торр); [α]
в. (S)-2-фенил-2-(1H-имидазол-1-ил)-1-аминоэтан
1 г (R)-(-)-2-фенилазиридина и 1,14 г имидазола выдерживают при температуре 120o в течение 24 часов. После охлаждения полученную реакционную смесь можно использовать для ацилирования без дополнительной очистки. Пригодный для анализа образец можно получить с помощью хроматографии на силикагеле;
1H-ЯМР (CDCl3) δ: 7,66 (s, 1H), 7,41-7,31 (m, 3H), 7,24-7,18 (m, 2H), 7,11 (t, J=1 Гц, 1H), 7,03 (t, J=1 Гц, 1H), 5,17 (m, 1H), 3,43 (m, 2H), 2,60 (s, b; 2H).
E. N-(4'-хлорбифенил-4-карбонил)-2(R)-фенилазиридин
К раствору 0,232 г 4'-хлорбифенил-4-карбоновой кислоты в 3 мл сухого ДМФ при перемешивании добавляют 0,18 г N,N'-карбонилдиимидазола при комнатной температуре в атмосфере аргона. Через 1 час по каплям добавляют раствор 0,12 г фенилазиридина в 0,5 мл сухого ДМФ. Полученную реакционную смесь перемешивают в течение 21 часа, после чего осуществляют реакцию в соответствии с примером 1. Получают указанное в заголовке соединение в виде светло-желтого масла, которое используют на следующем этапе без дополнительной очистки.
Ж. 2-(4'-Хлорбифенил-4-ил)-5(S)-фенил-4,5-дигидрооксазол
а. 2-Амино-1(R)-фенилэтанол
Раствор 15,8 г R(-)-фенилоксирана в 150 мл этанола охлаждают до температуры -60o и добавляют 75 мл аммиака. Полученную реакционную смесь перемешивают в течение 70 часов при комнатной температуре в закрытом стальном реакторе. Указанные растворители выпаривают и добавляют 300 мл воды и 60 мл толуола. Отделяя водную фазу и удаляя растворитель в вакууме, получают 14,14 г бесцветного твердого вещества, содержащего 86% 2-амино-1(R)-фенилэтанола, 9% 2-амино-2-фенилэтанола и 5% 2-(2-гидрокси-2-фенилэтиламино)-1- фенилэтанола (судя по результатам 1H-ЯМР). Указанную смесь используют на следующем этапе без дополнительной очистки. С помощью хроматографии на силикагеле (элюирование смесью дихлорметана, метанола и 28%-ного водного раствора гидроксида аммония в соотношении 100:10:1) получают пригодный для анализа образец; т.пл. 64,6 (с возгонкой); [α]
1H-ЯМР (DMCO-d6) δ: 7,37-7,22 (m, 5H), 4,44 (dd, J=4,4, 7,7 Гц, 1H), 2,70/2,57 (ABX, J=11,3, 4,4, 7,7 Гц, 2H), 2,60 (bs; 3H).
б. 4'-Хлорбифенил-4-карбонил-[2(R)-гидрокси-2-фенилэтил]амид
Раствор 1,2 г 4'-хлорбифенил-4-карбоновой кислоты и 715 мкл триэтиламина в 20 мл сухого ДМФ охлаждают до температуры -20o и обрабатывают 0,710 мл изобутилхлороформата. Полученную реакционную среду перемешивают в течение еще 20 минут, после чего добавляют раствор 955 мг неочищенного (74%-ного) 2-амино-1(R)- фенилэтанола в 5 мл ДМФ.
Указанной смеси позволяют нагреться до комнатной температуры и перемешивают в течение 16 ч. Полученную суспензию выливают на 100 мл ледяной воды, в агломератную воронку собирают образующийся осадок, после чего трижды промывают его водой и один раз - этанолом. Высушивая до постоянного веса, получают указанное в заголовке соединение в виде светло-желтых кристаллов;
т.пл. 229,3o; [α]
1H-ЯМР (DMCO-d6) δ: 8,62 (t, J=5,6 Гц, 1H), 7,95/7,78/7,55 (3d, J=8,5 Гц, 8H), 7,41-7,22 (m, 5H), 5,55 (d, J=4,4 Гц, 1H), 4,81 (ddd, J=4,4, 4,7, 8,1 Гц, 1H), 3,56-3,46/3,40-3,29 (2m, 2H).
в. 2-(4'-Хлорбифенил-4-ил)-5(S)-фенил-4,5-дигидрооксазол
Раствор 1,24 4'-хлорбифенил-4-карбонил-[2(R)-гидрокси-2-фенилэтил] амида в 20 мл пиридина охлаждают на ледяной бане и обрабатывают раствором 921 мг ангидрида метансульфоновой кислоты в дихлорметане. Полученную реакционную смесь перемешивают в течение еще 16 часов при температуре 5o, добавляют этилацетат и насыщенный водный раствор кислого карбоната натрия, отделяют органическую фазу, высушивают и концентрируют в вакууме. Используя вакуумную флеш-хроматографию на силикагеле (элюирование смесью толуола и этилацетата в соотношении 3: 1) получают указанное в заголовке соединение в виде светло-желтых кристаллов; т.пл. 121,5o (перекристаллизация из этанола);
[α]
1H-ЯМР (CDCl3) δ: 8,09/7,63/7,43 (4d, J=8,5 Гц, 8H), 7,44-7,36 (m, 5H), 5,68 (dd, J=7,9, 10,1 Гц, 1H), 4,51/4,02 (ABX, J=14,9, 10,1, 7,9 Гц, 2H).
З. (+) и (-)-1-амино-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан
а. 1-[(1S)-камфаноил]амино-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан
К раствору 0,38 г амино-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил) этана (рацемат, см. выше пункт A) в 4 мл сухого дихлорметана добавляют 0,26 г (0,36 мл) триэтиламина. Полученную смесь перемешивают и охлаждают до температуры -79o. Затем добавляют 0,44 г хлорида (1S)-(-)- камфановой кислоты таким образом, чтобы температура реакционной смеси не превышала -74o. Продолжают перемешивание еще в течение 90 минут без охлаждения. Для осуществления реакции указанную смесь выливают на ледяную воду, трижды экстрагируют дихлорметаном и промывают солевым раствором. После высушивания полученной смеси над сульфатом натрия выпаривают растворитель. Получают смесь двух диастереоизомерных амидов, которые разделяют с помощью хроматографии на силикагеле (элюирование смесью дихлорметана, метанола и гептана в соотношении 10: 1: 5). Получают два указанных в заголовке соединения в виде белых кристаллов.
- диастереоизомер B: т.пл. 195-199o
- диастереоизомер A: т.пл. 151-160o.
б. (+) и (-)-1-амино-1-(5-хлор-2-пиридил)-2-(1H-имидазол-1-ил)этан
0,16 г диастереоизомера B камфаноила и 0,8 мл 35%-ной перхлорной кислоты выдерживают в стеклянном автоклаве в течение 12 ч при температуре 120o. После разведения с помощью 2 мл воды указанную смесь экстрагируют этилацетатом (который в дальнейшем отбрасывают). Полученную водную фазу подщелачивают с помощью 2 н. гидроксида натрия и трижды экстрагируют этилацетатом. Органическую фазу высушивают над сульфатом магния и выпаривают до сухого состояния. Осуществляя хроматографию на силикагеле (элюирование смесью дихлорметана, метанола и гептана в соотношении 10:1:5 - 7:1:4), получают (+)-энантиомер указанного в заголовке соединения (изомер B) в виде светло-желтого масла:
1H-ЯМР (CDCl3) δ: 8,57 (d, J=2,5 Гц, 1H), 7,61 (dd, J=2,5, J=8,3 Гц, 1H), 7,38 (s, 1H), 7,13 (d, J2=8,3 Гц, 1H), 7,02 (s, 1H), 6,83 (s, 1H), 4,32-4,14 (m, 3H), 1,73 (s,b, 2H).
По аналогии с приведенным выше описанием, гидролизом диастереоизомера A камфаноила получают (-)-энантиомер указанного в заголовке соединения (изомер A).
И. 2-Фенил-2-(1H-имидазол-1-ил)-1-аминоэтан
Указанное соединение получают из 2-фенилазиридина по аналогии с описанием, приведенным в пункте Д)в). Осуществляя хроматографию на силикагеле (элюирование смесью дихлорметана, метанола и водного раствора NH3 в соотношении 10: 1: 0,1), получают указанное в заголовке соединение в виде бесцветного вязкого масла.
Соединения, соответствующие формуле (I) и свободном виде или в виде фармакологически приемлемой соли, в дальнейшем кратко называются "активными ингредиентами", проявляют фармакологическую активность, и могут быть использованы в качестве фармацевтических средств. Указанные соединения проявляют сильное избирательное действие ингибитора гидроксилаз 25-гидроксивитамина D3, атакующих боковую C20-27 цепь кальцитриола, в частности, 25(OH)D3-24-гидроксилазы, и поэтому катаболизируют проявляющие гормональную активность метаболиты витамина D3 (например, кальцитриол), оказывая при этом гораздо более слабое влияние непосредственно на синтез кальцитриола, осуществляемый с участием 25(OH)D3-1-гидроксилазы.
Избирательное подавление можно продемонстрировать, например, с помощью следующих тестов.
Активность 1-гидроксилазы.
Указанную активность можно непосредственно определять в конфлюэнтных культурах кератиноцитов человека, в то время как для определения активности гидроксилазы, атакующей боковую C20-27 цепь, необходимо проводить 16-часовую предобработку указанных конфлюэнтных культур с помощью 1,25(OH)2D3 (10 нМ), что способствует усилению изучаемых активностей (оба метода описаны в Bikie D.D. и др. J.Clin Invest, 78, 557 (1986) [1]).
Токсичность предлагаемых согласно изобретению соединений была изучена на соединении примера 7.
R-соединение из примера 7 при его оральном и дермальном введении крысам в течение 2 и 4 недель не дало никаких патологических изменений в плазме и моче, связанных с уровнем кальция или эндокринологическим статусом. Не проявлялись никакие ненормальные морфологические изменения, в частности, ни в отношении пролиферации кератиноцитов, ни в дифференциации кожных покровов.
Культуры кератиноцитов:
Нормальные кератиноциты человека выделяют из свежей кожи взрослого человека, полученной при удалении молочной железы и сразу же используемой в стерильных условиях. Выделение и культивирование при отсутствии сыворотки, а также без питающей подложки проводят в соответствии с модифицированными протоколами, использованными Bikie D. D. и др. Biochemistry, 25 (1986), 1545-1548 [2] . Указанные условия выбирают для того, чтобы предотвратить неконтролируемые эффекты метаболитов витамина D, возможно присутствующих в сыворотке и/или питающей подложке. Отделив эпидермис от дермы со стерильной дерматомой, полученный эпидермис инкубируют в 0,25%-ном растворе трипсина в течение 45 минут при температуре 37o. После этого отбирают полученные клетки и помещают их в 50 мл среды ССРХ, содержащей 10%-ную ФСТ для того, чтобы остановить дальнейшее расщепление трипсином, а также центрифугируют при 2000 об/мин в течение 2 минут. Осадок, состоящий из полученных таким образом клеток, суспендируют в СРК, определенной среде, не содержащей сыворотку, характеризующейся низкой (0,06 мМ) концентрацией кальция, а также включающей в себя 0,1 нг/мл ЭФР, 5 мкг/мл инсулина, 0,5 мкг/мл гидрокортизона, экстракт гипофиза быка и антибиотики (гентамицин, амфотерицин) с получением первичной культуры. После культивирования в инкубаторе в течение 24 ч при температуре 37o в условиях повышенной концентрации углекислого газа (95% O2, 5% CO2) удаляют неприкрепившиеся клетки, промывают используемую в работе флягу и добавляют в нее свежую СРК. В дальнейшем указанную культуральную среду меняют через день и пассируют клетки до достижения ими 80-90%-ного слоя (обычно через 6-10 дней после засева). При пересеве удаляют старую СРК, а прикрепленные кератиноциты подвергают короткой (5 мин) обработке 0,125%-ным трипсином, после чего помещают в ССРХ + ФСБ, центрифугируют и, наконец, ресуспендируют в СРК таким образом, что из 1 чашки первичной культуры получают 3 чашки клеток первого пассажа. Для того, чтобы увеличить конечное количество клеток, в дальнейшем кератиноциты культивируют в соответствии с приведенным выше описанием и обычно используют их на стадии второго пассажа.
а. Избирательные ингибиторы гидроксилаз витамина D3:
Конфлюэнтные культуры кератиноцитов человека, поддерживаемые в СРК при низкой (0,06 мМ) концентрации кальция, проявляют высокую способность продуцировать кальцитриол с помощью 1-гидроксилазы, при этом, однако, почти полностью отсутствуют активности, характерные для последующего метаболизма по пути окисления боковой цепи. Таким образом, подавление 1-гидроксилазы в сплошных культурах определяют непосредственно, а подавление последующего метаболизма после усиления активности соответствующих гидроксилаз, например, 24-гидроксилазы, определяют, проводя ночную (17 ч) предобработку 10 нМ кальцитриола, как описано Bikie в [1].
Активности 1-гидроксилазы и ферментов, обслуживающих конкретные этапы последующего метаболизма (например, образования 1,24,25(OH)3D3 + 1,24oксо, 25(OH)2D3) и все еще сохраняющих сходные с кальцитриолом активности, образование метаболитов 3-эпикальцитриола, полярных метаболитов, сохраняющихся в водной фазе и свидетельствующих о расщеплении боковой цепи, а также их подавление исследуемыми соединениями определяют с помощью окисления 3H-25(OH)D3 (Amersham; специфическая активность приблизительно равна 0,61 мКи/нМ) следующим образом.
Конфлюэнтные кератиноциты человека в 1 мл СРК, а также в 6-луночных планшетах инкубируют в виде двух реплик при температуре 37o с 3H-25(OH)D3 в течение 1 часа (в случае 1-гидроксилазы), 4 часа (в случае последующего метаболизма), а также в течение промежутков времени в пределах от 1 до 24 часов без или с использованием прединкубации с 10 нМ кальцитриола, а также в отсутствии или в присутствии исследуемых ингибиторных соединений, добавляемых в концентрации в пределах от 0 до 10 мкМ.
После этого указанную реакцию останавливают, добавляя 1 мл метанола на лунку, отбирают клетки, переносят в тестерную пробирку вместе с супернатантом и дважды промывают (1 мл метанола и 0,8 мл воды). Неизмененный 3H-25(OH)D3, а также большую часть образующихся продуктов полностью экстрагируют из объединенных растворов и клеточного осадка с помощью последующей экстракции с использованием 2,1 мл и 1 мл CHCl3 при комнатной температуре. Определяют активность 3H в фазе CHCI3 и воде, а также суммарную активность 3H. Инкубирование в течение более длительных промежутков времени (> 4 часов) приводит к резкому увеличению активности 3H в водной фазе за счет восокополярных метаболитов, а также к существенному снижению активности 3H в летучих продуктах, что почти полностью определяется расщеплением по боковой цепи, при котором удаляется 3H-метка в C26/27.
Затем выпаривают объединенные CHCl3 экстракты в атмосфере аргона при температуре 35o, растворяют образовавшийся остаток в 0,4 мл этанола, а аликвоту полученного раствора подвергают анализу с помощью HPLC на колонке ZorbaxSil (Dupont; 4,6 x 250 мм) с использованием нелинейного градиента смеси гексана и 2-пропанола в соотношении от 97:3 до 85:15 при малой скорости, равной 2 мл/мин, и общем времени пробега, равном 70 мин. Субстрат и отдельные метаболиты привязывают к полученным пикам с помощью сравнения их с немеченными стандартами, используемыми в сопряженной хроматографии. Выявляют степень образования различимых продуктов в присутствии определенного ингибитора, используемого в концентрациях в пределах от 0 до 10 мкМ, и с помощью графика Диксона (1/пробег, отнесенный к концентрации ингибитора) рассчитывают силу подавления (IC50). Избирательность можно оценить, сравнивая величины IC50, полученные для конкретного ингибитора определенных последующих процессов и 1-гидроксилазы.
б. Включение 3H-тимидина в кератиноциты человека - подавляющие пролиферацию эффекты метаболитов витамина D в присутствии ингибиторов их катаболизма.
Включение 3H-тимидина измеряют в 96-луночных планшетах следующим образом: кератиноциты, находящиеся в 200 мл СРК с низкой концентрацией 104 кальция, засевают с исходной плотностью 10 клеток на лунку (второй пассаж) и выдерживают в инкубаторе с повышенным содержанием углекислого газа (95% O2, 5% CO2) в течение 24 часов при температуре 37o. Затем добавляют исследуемые соединения, растворенные в 1 мкл этанола в концентрации в пределах от 0 до 10 мкМ, в присутствии и в отсутствии 25(OH)D3 или 1 α 25(OH)2D3 (каждый в концентрациях в пределах от 0 до 7 нМ), по 3 повторности на каждую концентрацию. После каждой тройки лунок помещают бланки, содержащие только указанный растворитель или метаболит витамина D50. Добавляют 5-10 бланков на чашку в отсутствии растворителя. Еще через 24 часа добавляют 50 мкл 3H-тимидина (1 мкКи) в СРК, продолжают инкубацию в течение еще 17 часов и, наконец, останавливают ее, собирая и лизируя клетки.
Сбор осуществляют на Filtermate 196 - Harvester (Packard-Canberra). На первом этапе полученный супернатант наносят на 96-луночный чашечный фильтр и трижды промывают водой. Количественный анализ указанных чашечных фильтров, осуществляемый в соответствии с приведенным ниже описанием, четко показывает, что не происходит какой-либо потери клеток, включающих активность 3H. Затем слипшиеся клетки, находящиеся в указанных инкубированных чашках, освобождают с помощью обработки 100 мкл 0,125%-ного трипсина в ФСБ при температуре 37o в течение 5 минут, собирают их на новый чашечный фильтр и трижды промывают водой. Добавляют 50 мкл сцинцилляционной смеси (MicroScint O, Packard) и подсчитывают активность 3H с помощью Microplate Scincilation Counter (Topcount, Packard Canberra).
Данные представлены в виде средних значений ± SEM (средняя квадратичная ошибка) (n= 3). Значения IC50, полученные при подавлении пролиферации метаболитами витамина D в присутствии различных концентраций ингибиторов метаболизма по боковой цепи и наоборот, определяют, строя график зависимости величины, обратной темпу пролиферации, от одной из концентраций ингибитора, в то время как концентрации остальных ингибиторов остаются неизменными (способ Диксона). Вне зависимости от механизма подавления значения lC50 могут быть сняты с указанного графика в точках его пересечения с осью X.
Предусмотренные настоящим изобретением соединения проявляют в вышеуказанных тестах а) и б) ингибиторную активность при использовании в концентрации от приблизительно 0,01 мкМ до приблизительно 10 мкМ.
Итак, предусмотренные настоящим изобретением соединения рекомендуются для использования в качестве избирательных ингибиторов гидроксилаз 25-гидроксивитамина D3, атакующих боковую C20-27 цепь витамина D3, для лечения заболеваний, связанных с пролиферацией или дифференцировкой в тканях, зависимых от витамина D, в частности, для лечения состояний, при которых необходимо избирательно подавить катаболизм гормона кальцитриола, не оказывая влияния на его синтез из его предшественника 25(OH)D3, в результате чего повышают длительно сохраняющиеся уровни эндогенного гормона и тем самым достигают благоприятных эффектов в плане пролиферации и дифференцировки, иммунной функции и кальциевого гомеостаза (например, в случае кожи), сверхпролиферирующих и воспалительных заболеваний (таких как псориаз и экземические заболевания), дегенеративных изменений соединительной ткани (как в случае патологии, так и в результате нормального старения), предотвращения выпадения волос и восстановления роста волос, а также, рассматривая эффекты, оказываемые не только на кожу, - в плане подавления опухолей, повышения устойчивости к аллотрансплантатам, при ревматоидных артритах и восстановлении костей.
В случае перечисленных показаний необходимая доза должна безусловно зависеть, например, от используемого активного ингредиента, предусмотренного настоящим изобретением, от хозяина, от способа применения и желаемой цели.
Тем не менее, на животных для достижения удовлетворительных результатов рекомендуется использовать суточную дозу в пределах от приблизительно 1 мг/кг до приблизительно 20 мг/кг веса тела животного. В случае более крупных млекопитающих, например, человека, рекомендуемая суточная доза находится в пределах от приблизительно 70 мг до приблизительно 1400 мг предусмотренного настоящим изобретением активного ингредиента, используемого традиционным образом, например, при дробном введении доз вплоть до 2 или 4 раз в день при внутреннем или системном воздействии, а также в пределах от менее, чем 0,5%, например, от 0,1% до приблизительно 2% при использовании в качестве крема или раствора для местного применения.
Предусмотренные настоящим изобретением соединения можно смешивать с традиционными фармацевтически приемлемыми разбавителями и носителями, а также, возможно, с дополнительными эксципиентами.
Указанные соединения можно применять традиционным способом, в частности, либо энтерально, например, перорально, в том числе в форме таблеток или капсул, либо локально, например, в форме лосьонов, гелей, кремов, жидкостей для пульверизации, а также растворов, таких как растворы для глаз и носа или аэрозоли, предназначенные для местного лечения кожи и слизистых мембран, таких как глаз, дыхательный тракт, влагалище, ротовая и носовая полости.
Указанные агенты можно применять непосредственно или в комбинации с низкими дозами кальцитриола, либо со стандартными средствами, такими как циклоспорин A (SandimmunR) для усиления подавления иммунного ответа и воспалительных эффектов; с гораздо более низкими дозами отдельных лекарств, что должно обычно ослаблять нежелательные побочные эффекты.
Предусмотренные настоящим изобретением агенты могут служить для терапии вместе с глюкокортикоидами (например, дексаметазоном, бетаметазоном, преднизолоном) и другими агентами, подавляющими иммунитет, а также могут служить дополнением при лечении кальцитонином и паратироидным гормоном.
В случае обоих назначений предпочтительными агентами являются энантиомерные соединения N-[4-(4-хлорфенил)бензоил] -2-(1H-имидазол-1-ил)-2(S)-фенил-1-аминометан (пример 8), а также его (R)-энантиомер (пример 9). Указанные соединения являются избирательными ингибиторами гидролаз 25-гидроксивитамина D3, атакующих боковую С20-27 цепь витамина D3, не оказывая при этом влияния на его синтез из его предшественника 25(OH)D3. Например, показано, что в вышеупомянутых тестах а) и б) в случае подавления метаболизма по боковой цепи они характеризуются величиной IC50 в пределах от 10 нМ до 50 нМ, в частности, соответственно для (S)- и (R)- энантиомера:
в тесте а): 40 нМ и 12 нМ, соответственно, судя по стабилизации уровней 3-эпи 1,25(OH)2D3, а также 45 нМ и 19 нМ, судя по стабилизации суммарных уровней 1,25(OH)2D3, 1,24,25(OH)3D3 и 1,24оксо,25(OH)2D3, однако величина IC50 равна соответственно 530 нМ и 620 нМ в случае подавления 1-гидроксилазы;
в тесте б): величина IC50 равна соответственно 30 нМ и 35 нМ, исходя из концентрации, необходимой для увеличения вдвое снижающего пролиферацию эффекта, производимого 1 α, 25(OH)2D3, а также 11 нМ и 12 нМ в случае 25(OH)D3.
Таким образом, указанные агенты могут быть рекомендованы для лечения псориаза при их использовании в концентрации от приблизительно 0,1% до приблизительно 0,5% в случае местного применения по отношению к крупным млекопитающим, например людям, с использованием приемов, сходных с применяемыми традиционно.
Настоящее изобретение предусматривает также фармацевтические композиции, предназначенные для, например, местного применения и содержащие предусмотренный настоящим изобретением агент вместе с по меньшей мере одним фармацевтически приемлемым носителем или разбавителем. Указанные составы могут быть произведены традиционным способом, посредством смешивания предусмотренного настоящим изобретением активного ингредиента с по меньшей мере одним фармацевтически приемлемыми носителями или разбавителями или дополнительными эксципиентами. Единичная дозировочная форма содержит, например, от приблизительно 20 мг до приблизительно 700 мг активного вещества и зависит от показателей веса тела.
Примером таких фармацевтических композиций с использованием активного ингредиента согласно изобретению могут быть мази.
Эта мазь может быть получена путем смешения обычным способом R-изомера из примера 6 (содержание его составляет 0,6 вес.%) со следующими добавками:
петролатум в качестве мазевой основы,
цетиловый спирт в качестве загустителя,
стеарат глицерина в качестве эмульгатора,
гексиленгликоль в качестве растворителя и олеиловый спирт в качестве растворителя.
Количество активного ингредиента обычно берется таковым, чтобы соответствовать его эффективно действующему значению.
Предпочтительным является использование указанных составов для лечения псориаза.
Предусмотренные настоящим изобретением соединения проявляют более выраженную и избирательную активность, подавляющую гидроксилазу 25 (OH)D3, нежели можно предположить для близких структурных аналогов.

Описываются ацилированные аминоалкилимидазолы общей формулы I, в которой либо R1 представляет собой фенильную, возможно монозамещенную атомом галогена или 1-нафтильную группу, а R2 обозначает атом водорода; либо R1 соответствует атому водорода, a R2 представляет собой пиридальную или 2-(5-хлор)пиридильную группу, R3 -атом галогена или С1-4-алкоксигруппу, находящиеся в свободной форме или в форме соли. Соединения формулы I, обладающие способностью избирательно ингибировать гидроксилазы 25-гидроксивитамина D3. Описывается также способ их получения и фармацевтическая композиция на основе соединения формулы I. 3 c. и 3 з.п.ф-лы, 3 табл.
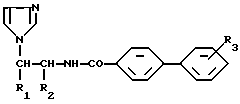
где либо R1 представляет собой фенильную группу, возможно монозамещенную атомом галогена, или 1-нафтильную группу;
R2 обозначает атом водорода;
либо R1 обозначает атом водорода;
R2 представляет собой пиридильную или 2-(5-хлор) пиридильную группу;
R3 обозначает атом галогена или алкоксигруппу, содержащую 1 - 4 атомов углерода,
находящиеся в свободном виде или в виде фармацевтически приемлемой соли.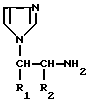
где заместители R1 и R2 имеют значения, указанные в п.1,
подвергают ацилированию для введения соответствующей бифенил-4-карбонильной группы с последующим выделением целевого продукта в свободном виде или в виде соли.
| Способ получения производных имидазола или их солей | 1979 |
|
SU865125A3 |
| Способ получения производных имидазола или их фармацевтически приемлемых солей присоединения кислот | 1987 |
|
SU1572415A3 |
| Моющее средство для мытья посуды | 1972 |
|
SU436851A1 |

