Изобретение относится к O6-замещенным производным гуанина, способу их получения и к их применению для лечения опухолевых клеток. Изобретение, в частности, относится к производным гуанина, имеющим гетарильные или нафтилалкильные заместители в O6-положении, при этом указанные соединения обладают способностью снижать активность O6-алкилгуанин-ДНК алкилтрансферазы (АТазы) в опухолевых клетках.
Предпосылки изобретения
Было предложено использовать О6-алкилпроизводные гуанина, обладающие способностью снижать активность О6-алкилгуанин-ДНК алкилтрансферазы, для усиления эффективности химико-терапевтических алкилирующих агентов, которые используют для уничтожения опухолевых клеток. Появляется все больше доказательств того, что токсическое и мутагенное воздействие алкилирующих агентов на клетки млекопитающих в значительной степени является следствием алкилирования О6-положения гуанина в ДНК. Восстановлению О6-алкилгуанина способствует АТаза - белок-восстановитель, который воздействует на О6-алкилированные остатки гуанина, осуществляя стехиометрический перенос алкильной группы в остатки цистеина, расположенные в активном сайте белка-восстановителя, в автоинактивационном процессе. Важная роль АТазы при защите клеток от биологического воздействия алкилирующих агентов наиболее наглядно была продемонстрирована путем переноса и экспрессии генов АТазы или комплементарных ДНК в клетки с дефицитом АТазы: тем самым повышается устойчивость к действию различных агентов, в основном тех, которые метилируют или хлорэтилируют ДНК. Хотя механизм уничтожения клеток под действием О6-метилгуанина в клетках с дефицитом АТазы до сих пор не прояснен, уничтожение клеток под действием О6-хлорэтилгуанина происходит за счет образования межцепочечных сшивок к остатку цитозина противоположной цепочки посредством циклического этангуанинового промежуточного соединения, и именно этот процесс подавляется за счет стимулируемого АТазой удаления хлорэтильной группы или образования комплекса.
Использование О6-метилгуанина и О6-бутилгуанина для ослабления активности АТазы описано (Dolan et al., Cancer Res., 1989, 46, p. 4500; Dolan et al., Cancer Chemother. Pharmacol, 1989, 25, p. 103). Предложено использовать производные О6-бензилгуанина для ослабления активности АТазы, с целью сделать клетки, экспрессирующие АТазу, более восприимчивыми к цитотоксическому воздействию хлорэтилирущих агентов (Maschel et al., J. Med. Chem., 1992, 35, p. 4486). В Патенте США 5091430 и международной патентной заявке WO 91/13898 (Moschem et al.) приводится способ уменьшения содержания О6-алкилгуанин-ДНК трансферазы в опухолевых клетках реципиента, который заключается в назначении реципиенту эффективного количества композиции, содержащей О6-бензилированные производные гуанина следующей формулы: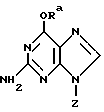
где Z обозначает атом водорода, или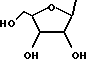
или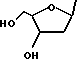
Ra обозначает бензильную группу или замещенную бензильную группу.
Бензильная группа может содержать в орто-, мета- или пара-положении такие заместители, как атом галогена, нитро, арил, такой как фенил или замещенный фенил, алкил, содержащий 1-4 атома углерода, алкокси, содержащий 1-4 атома углерода, алкенил, содержащий вплоть до 4 атомов углерода, алкинил, содержащий вплоть до 4 атомов углерода, амино, моноалкиламино, диалкиламино, трифторметил, гидрокси, гидроксиметил и SOnRb, где n равно 0, 1, 2 или 3, а Rb обозначает атом водорода, алкил, содержащий 1-4 атома углерода, или арил. В статье Mi-Joung Chae et al., J. Med. Chem., 1994, 37, pp. 342-347 - опубликована после даты приоритета настоящей заявки на изобретение - описываются тестовые испытания аналогов О6-бензилгуанина, содержащих в положении 9 бензильного кольца все более возрастающие по размеру заместители. Соединением N 6 там является O6-(2-пиридилметил)гуанин, который в указанной заявке обозначают как О6-(2-пиколил)гуанин. Однако в разделе Результаты и Обсуждение результатов на страницах 342-343 статьи Chae et al. не подчеркивается, что Соединение 6 может представлять интерес, а включено в группу "остальные соединения", которые "обладают промежуточной активностью" (страница 343, строки 12-15 по тексту статьи). Авторы подтвердили ранее сделанные ими наблюдения (J. Med. Chem., 1992, 35, р. 4466), что лишь аллильные или бензильные заместители в O6-положении гуанина эффективно дезактивируют АТазу (страница 343, строки 21-23 по тексту статьи).
О6-Бензилгуанин имеет ограниченное использование в качестве дозактиватора АТазы. Он более устойчив, чем это необходимо, что приводит к тому, что он долгое время сохраняется в животном, которому он введен. Это соединение потенциально токсично как само по себе, так и в сочетании с хлорэтилирующими агентами, что также нежелательно и может быть связано с временем выживания.
Соединения по настоящему изобретению обладают отличными от О6-бензилгуанина инактивирующими свойствами по отношению к АТазе, а в ряде случае их активность до 8 раз выше, чем активность О6-бензилгуанина. Исследованы также различные полупериоды существования и токсические свойства. Таким образом, объектом настоящего изобретения являются новые соединения, полезные для ослабления активности АТазы, с целью усиления воздействия химико-терапевтических средств, таких как хлорэтилирующие или метилирующие антиопухолевые агенты.
Другой целью настоящего изобретения являются фармацевтические композиции, содержащие соединения, которые полезны для ослабления активности АТазы. Еще одной целью настоящего изобретения является способ ослабления активности АТазы в опухолевых клетках. Наконец, еще одной целью настоящего изобретения является способ лечения опухолевых клеток реципиента.
Описание изобретения
Таким образом, настоящее изобретение относится к производным О6-алкилгуанидина формулы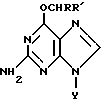
где Y представляет водород, рибозил или дезоксирибозил, которые могут быть замещены гидрокси или C1-C4алкоксигруппами, R' представляет водород, C1-C20алкил или гидроксиC1-C20алкил;
R обозначает
(i) циклическую группу, имеющую пятичленное гетероциклическое кольцо, содержащее по меньшей мере один гетероатом, выбранный из О, N или S, необязательно конденсированное с кольцом бензола, пиридина или нафталина, или шестичленное гетероциклическое кольцо, содержащее по меньшей мере один атом N, необязательно с атомом S, необязательно конденсированное с одним или двумя бензольными кольцами, где указанная циклическая группа может быть необязательно замещена в гетероциклическом(их) кольце(ах) и/или карбоциклическом(их) кольце(ах) группами, выбранными из C1-C5алкила, галогена, циано, нитро, азидо, C1-C5алкокси, арила, SOnR"", где R"" представляет C1-C5алкил и n равно 0, 1, 2, COOR5, где R5 представляет H или C1-C5алкил или N-оксиды,
(ii) нафтил, необязательно замещенный галогеном или C1-C5алкокси, или их фармацевтически приемлемые соли.
В частности, изобретение относится к соединению формулы I, где R содержит гетероциклическое кольцо, конденсированное с бензольным кольцом, и где О6-алкилгуаниновая группа присоединена к R либо в гетероциклическом, либо в бензольном кольце.
Предпочтительным является соединение формулы I, где R обозначает пятичленное гетероциклическое кольцо, содержащее один атом серы.
Например, R может быть выбран из тиофенового кольца, фуранового кольца или их замещенных производных.
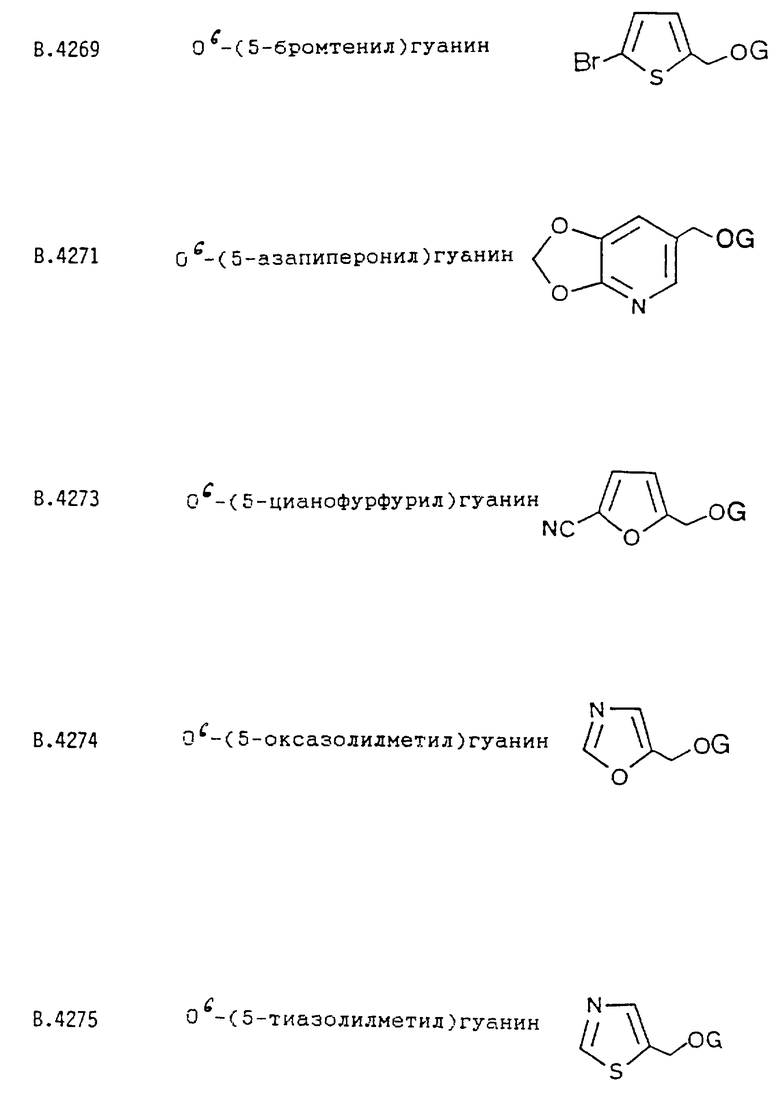
Предпочтительным также является соединение формулы I, где R является циклической группой, замещенной галогеном, циано, SOnR"", где R"" представляет C1-C5алкил и n = 0, 1 или 2, или -COOR5, где R5 представляет C1-C5алкил. Например, R может быть выбран из тиофенового кольца, фуранового кольца и их замещенных производных, которые выбраны из их бром- и цианопроизводных. Из них предпочтительным является соединение формулы I, где -CHRR' является О6-тенилом или его бром-замещенным производным.
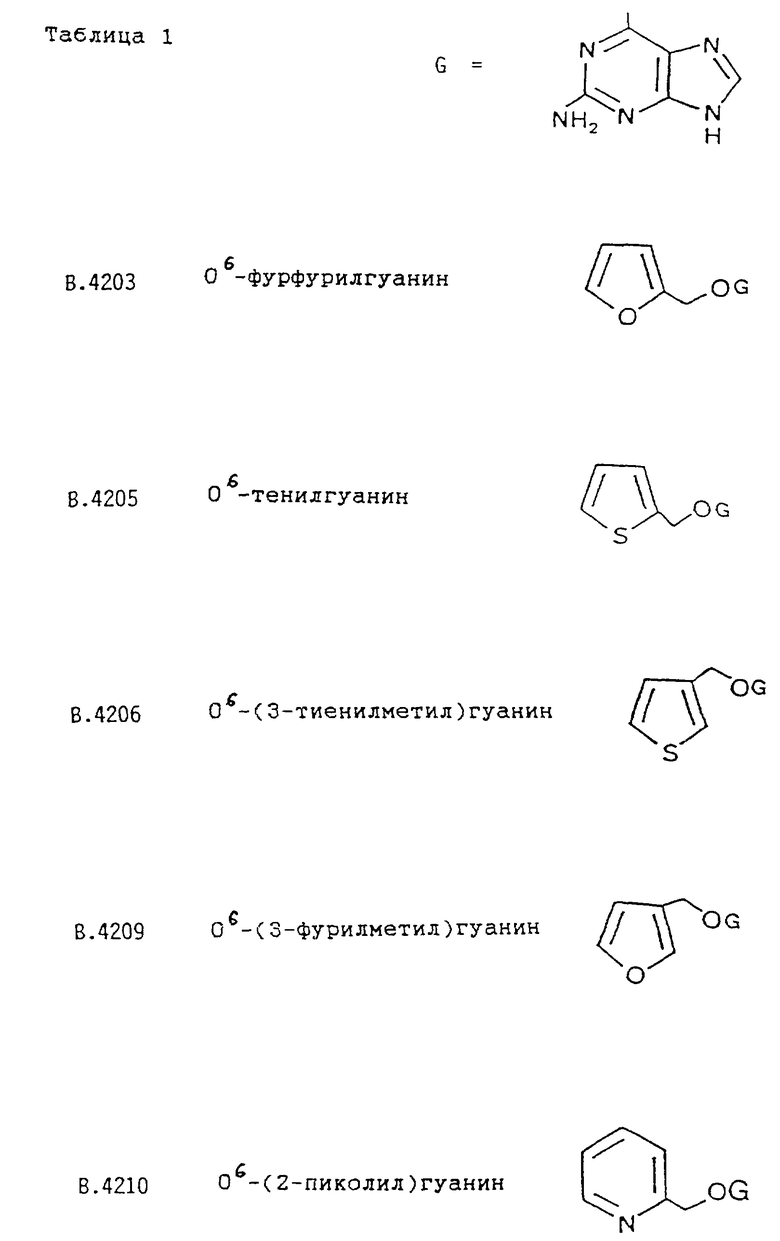
Предпочтительным также является соединение формулы I, выбранное из
О6-тенилгуанина;
О6-(3-тенилметил)гуанина;
О6-пиперонилгуанина;
О6-фурфурилгуанина;
О6-(3-фурилметил)гуанина;
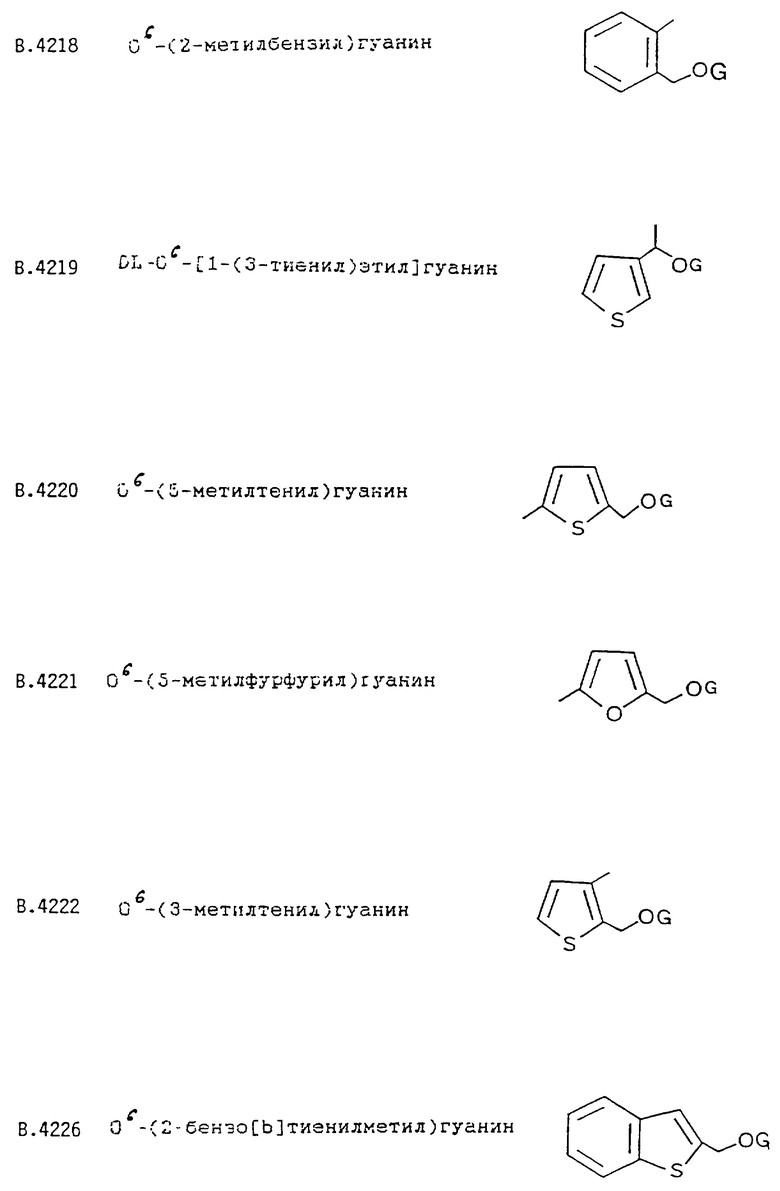
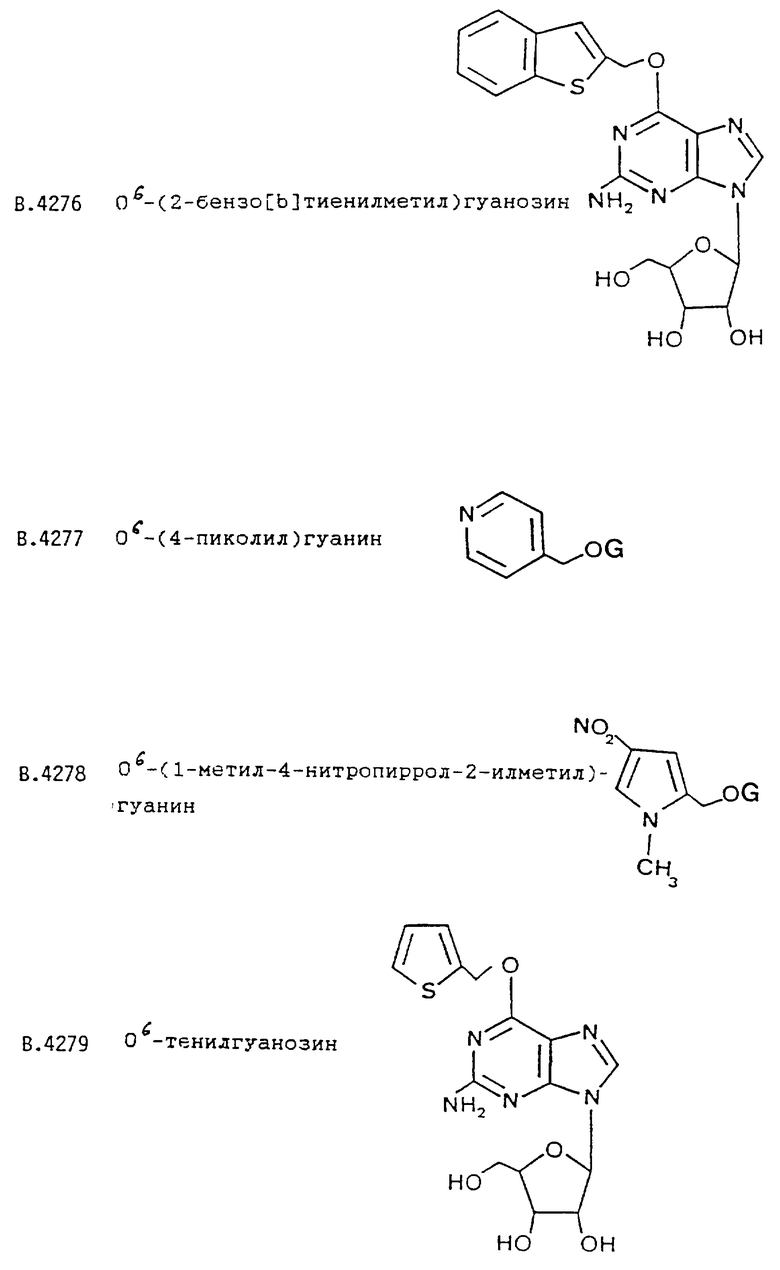
О6-(2-бензо[b]тиенилметил)гуанина;
O6-(2-бензофуранилметил)гуанина;
О6-(5-тиазолилметил)гуанина;
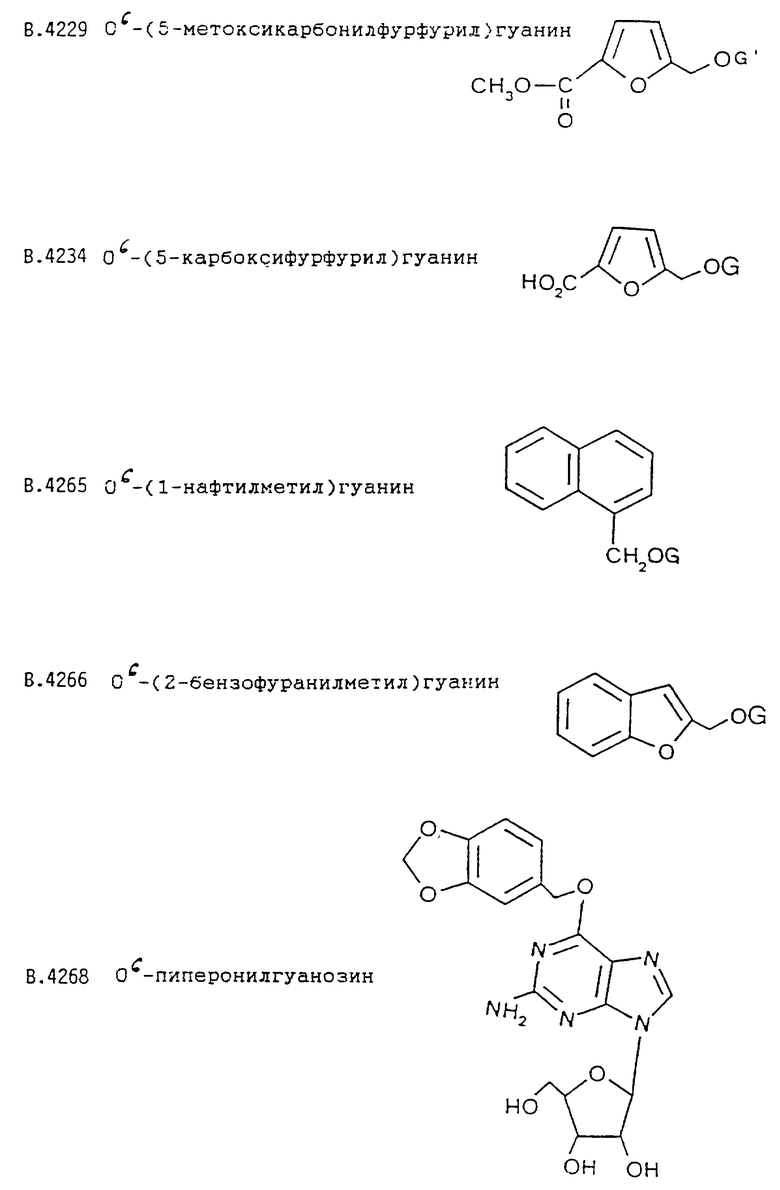
О6-(5-метоксикарбонилфурфурил)гуанина;
О6-(5-бромтенил)гуанина;
О6-(5-цианофурфурил)гуанина;
О6-(2-бензо[b]тиенилметил)гуанозина;
О6-(4-пиколил)гуанина;
О6-(2-нафтилметил)гуанина.
Предложенные соединения формулы I могут использоваться для получения фармацевтической композиции, обладающей способностью ослаблять активность О6-алкилгуанин-ДНК алкилтрансферазы в опухолевых клетках. Предпочтительным является О6-тенилгуанин или его замещенное производное.
Предложена также фармацевтическая композиция, обладающая способностью ослаблять активность О6-алкилгуанин-ДНК алкилтрансферазы у хозяина, содержащая производное гуанина и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве производного гуанина она содержит эффективное количество соединения формулы I или его фармацевтически приемлемую соль по любому из предшествующих пунктов.
Фармацевтическая композиция может дополнительно содержать алкилирующий агент, который может быть, например, выбран из 1,3-бис(2-хлорэтил)-1-нитрозомочевины (BCNU) и темозоломида.
Кроме того, предложен способ получения производных О6-алкилгуанина формулы I, описанных выше, заключающийся в том, что осуществляют следующие стадии: взаимодействие гидрата натрия с раствором RR'OH, где R и R' указаны выше, в органическом растворителе, добавление хлорида 2-амино-N,N,N-триметил-1H-пурин-6-аминия или 2-амино-6-хлорпуринбензола, обработку слабой кислотой и эфиром и экстракцию целевого продукта.
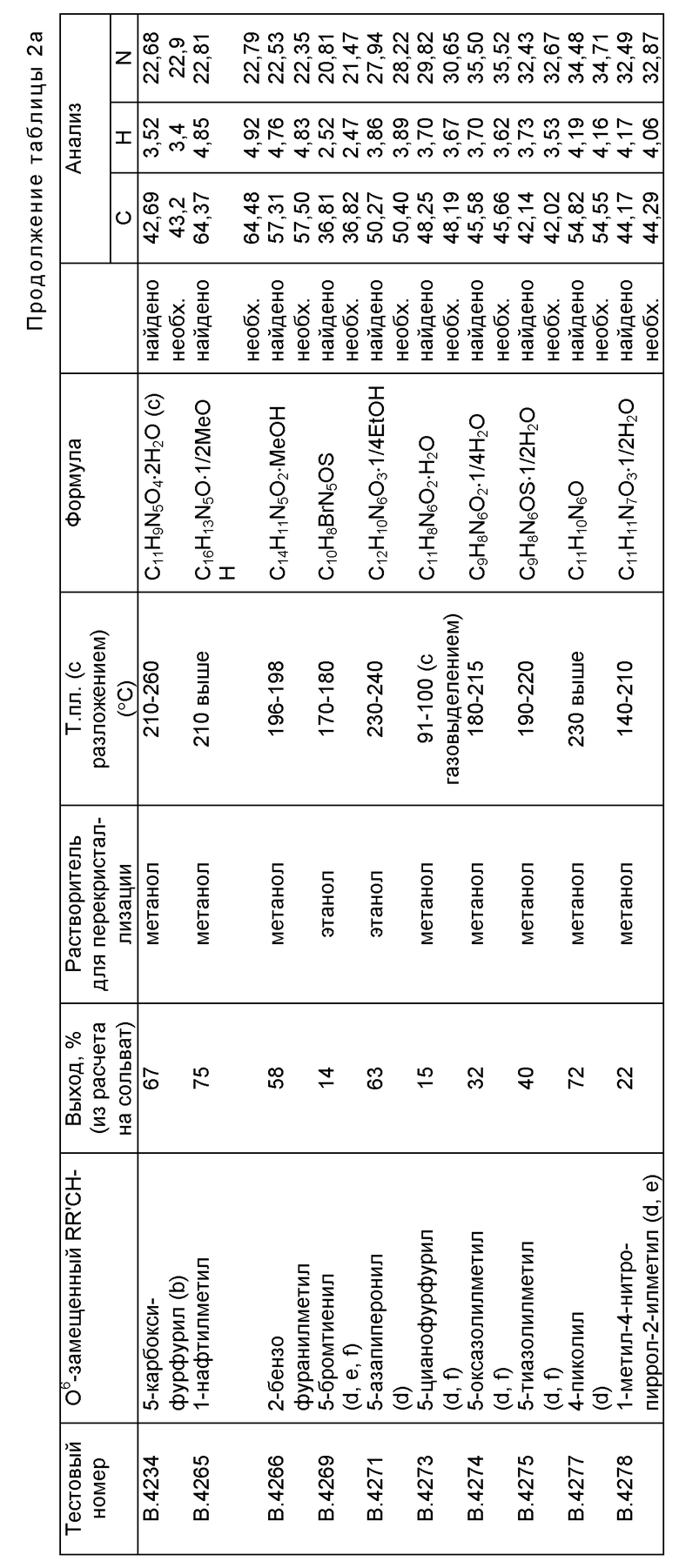
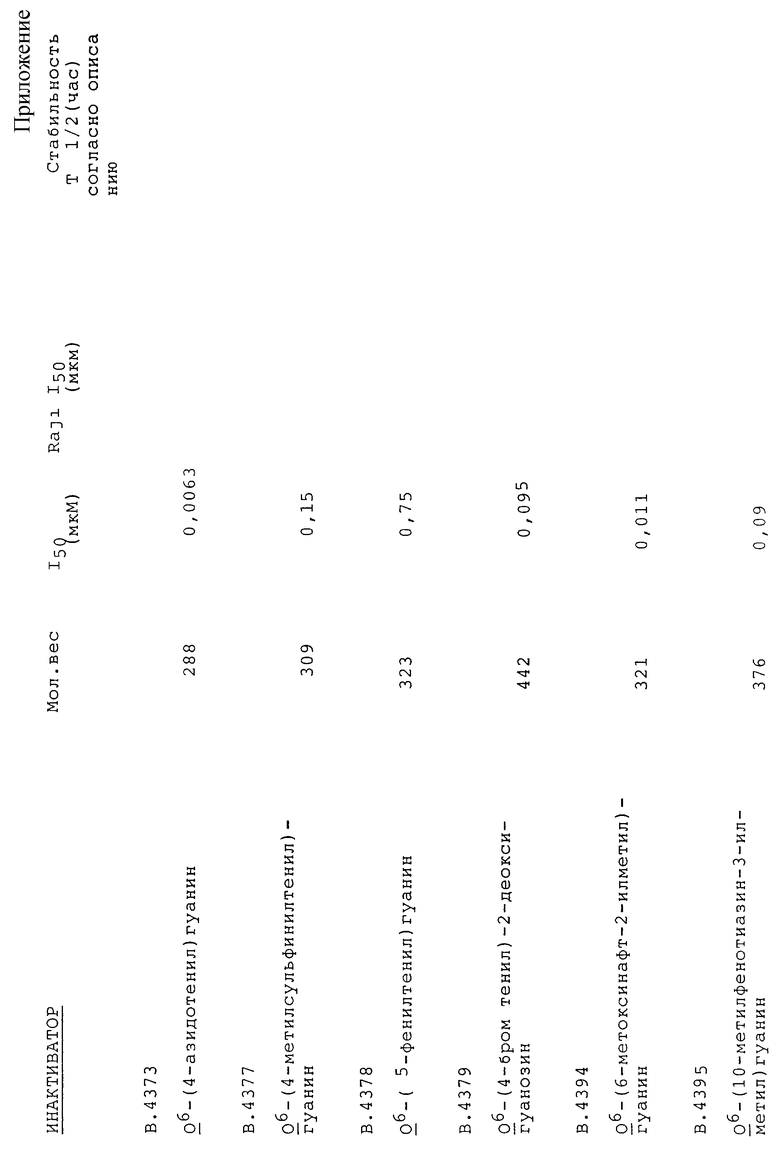
Примеры соединений по настоящему изобретению (вместе с соединениями В. 4214 и В.4218, которые не входят в объем изобретения) представлены в таблице 1.
Среди соединений, приведенных в таблице 1, соединения B.4214 и В.4218 не являются соединениями по настоящему изобретению. Соединение В.4210 (т.е. соединение формулы I, в котором R обозначает 2-пиридил, R' обозначает H, а Y обозначает H) не является предпочтительным соединением по настоящему изобретению.
Наиболее предпочтительные соединения по настоящему изобретению включают:
В.4205 О6тенилгуанин
В.4206 О6-(3-тиенилметил)гуанин
В.4212 О6-пиперонилгуанин
В.4226 О6-(2-бензо[b]тиенилметил)гуанин
B.4266 О6-(2-бензофуранилметил)гуанин
В.4275 О6-(5-тиазолилметил)гуанин
и соединения, замещенные в гетероцикле группы R атомами галогена, цианогруппой или сложноэфирной группой, в том числе
В.4229 О6-(5-метоксикарбонилфурфурил)гуанин
В.4269 О6-(5-бpoмтенил)гуaнин
В.4273 О6-(5-цианофурфурил)гуанин.
Другие предпочтительные соединения включают
В.4209 О6-(3-фурилметил)гуанин
B.4276 О6-(2-бензо[b]тиенилметил)гуанозин
B.4277 О6-(4-пиколил)гуанин.
Наиболее предпочтительными соединениями по изобретению являются те соединении, которые инактивируют АТазу в условиях in vitro и/или в клетках млекопитающих и/или опухолевых ксенотрансплантатах более эффективно, чем О6-бензилгуанин (BeG), и которые стимулируют клетки млекопитающих и/или опухолевые ксенотрансплантаты на уничтожение клеток или стимулируют ингибирующее воздействие нитрозомочевины и/или метилирующих агентов более эффективно, чем BeG. Предпочтительные соединения должны обладать по сравнению с BeG меньшей токсичностью по отношению к нормальным тканям и/или к организму в целом при совместном использовании с указанными агентами. Предпочтительные соединения сами по себе не должны быть токсичными или проявлять токсичность большую, чем минимальная токсичность в дозах, необходимых для инактивации АТазы; кроме того, не должны быть токсичными любые продукты гидролиза предпочтительного соединения, которое не является химически стойким. Хотя изобретение не ограничивается какими-либо теоретическими выводами, может оказаться, что предпочтительные соединения должны быть менее стойкими, чем BeG, так чтобы они могли претерпевать спонтанное разрушение вскоре после того, как достигнута максимальная степень инактивации АТазы: в этом случае любое воздействие процессов обмена на агент, приводящее к образованию токсических производных, могло бы быть сведено к минимуму. Предпочтительные соединения должны в меньшей степени повышать чувствительность костного мозга человека или других типов нормальных клеток к токсическому воздействию алкилирующих агентов, с тем чтобы они не усиливали бы известное токсическое воздействие или не приводили бы к появлению новой токсичности указанных агентов в нормальных тканях человека.
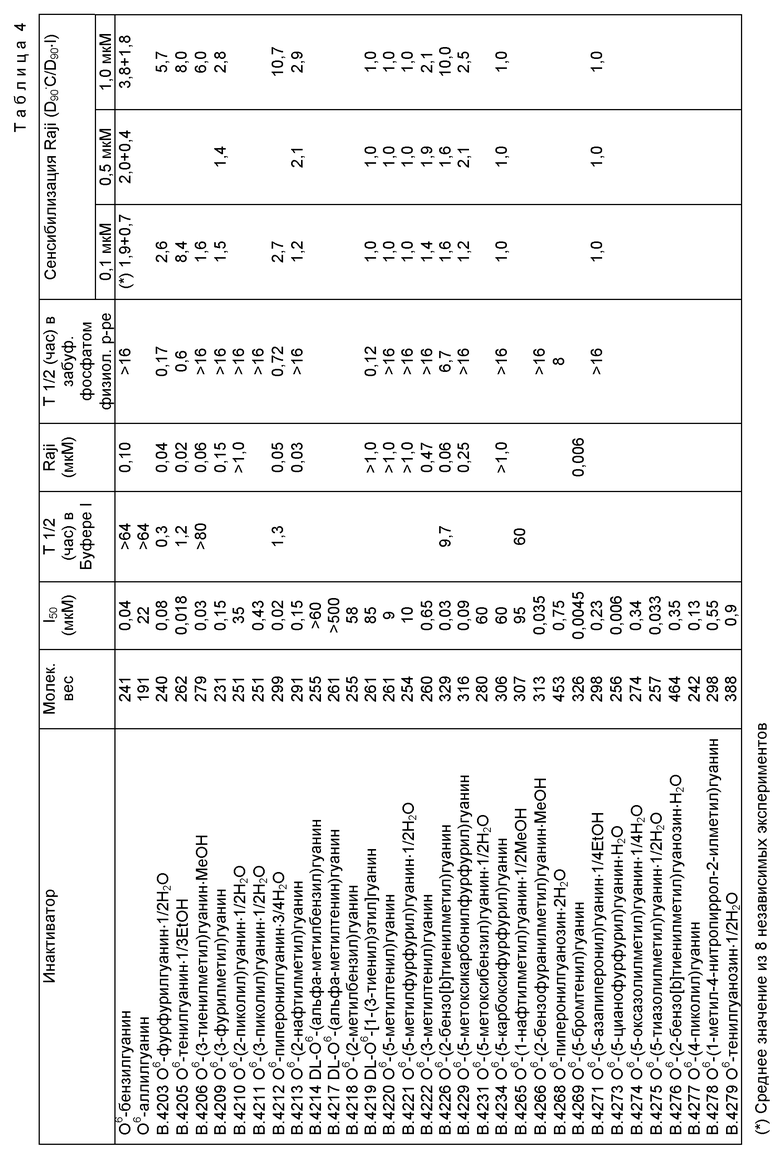
Предпочтительные соединения по настоящему изобретению включают соединения, имеющие относительно низкие значения I50, которые приведены ниже в таблице 4 (в частности, ниже 1,0 мкМ, более предпочтительно менее 0,04 мкмМ), и/или соединения, имеющие относительно короткий по лупериод существования в Буфере 1 (пример проведения анализа в условиях in vitro) и/или забуференном фосфатом солевом физиологическом растворе (пример условий физиологической среды), которые приведены ниже в таблице 4 (в частности, менее 20 час в Буфере I или менее 16 час в забуференном фосфатом физиологическом растворе).
Относительно короткий полупериод существования может служить свидетельством того, что соединение по изобретению менее устойчиво, чем О6-бензилгуанин вследствие реакционноспособности группы RR'CH- и имеет тенденцию к распаду при гидролизе в физиологической среде.
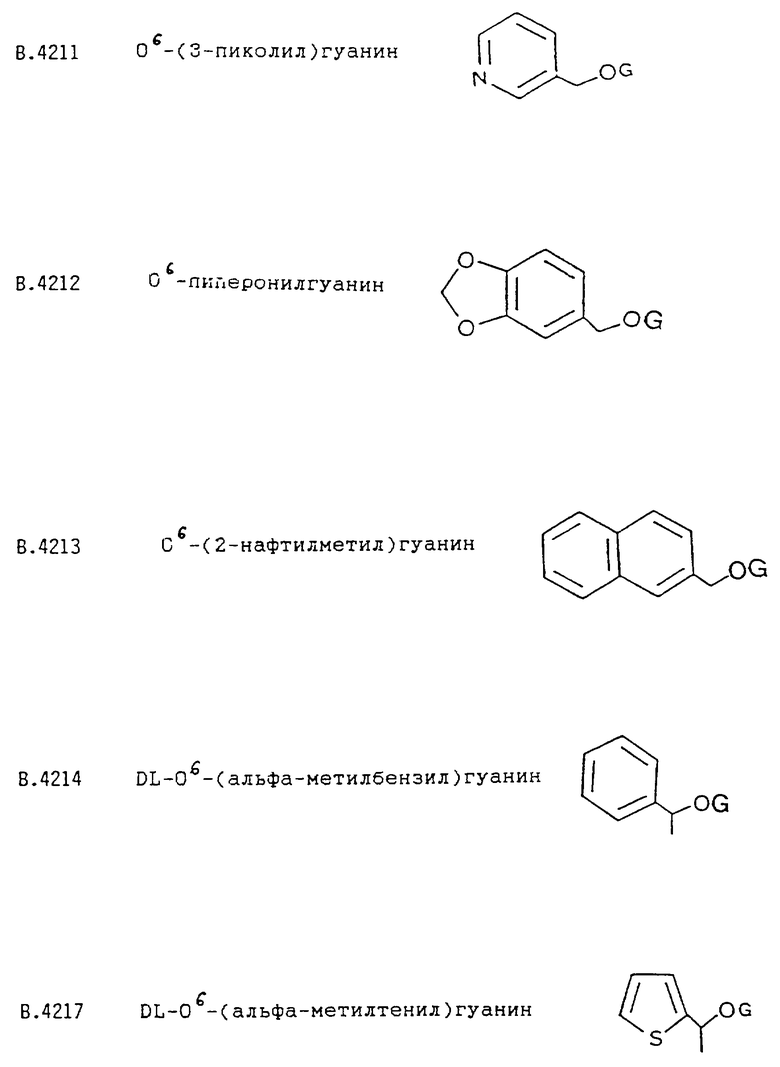
Влияние группы RR'CH - в соединениях формулы I, позволяющее им действовать как ингибиторы АТазы, определяется электронными, стерическими и физико-химическими факторами. Стерические факторы могут быть связаны со свойствами ближайшего окружения сайта цистеинового рецептора АТазы. Преимущественно, R' обозначает атом водорода. Было показано, что второй атом углерода, присоединенный к О6 (как в DL производных B.4214 или B.4217), значительно ослабляет инактивирующую способность, вероятность, вследствие большого размера заместителя.
Циклическая группа R не содержит метильную группу в вицинальном положении (как в В.4222), хотя влияние вицинального заместителя, очевидно, значительно меньше в том случае, когда R представляет собой гетероцикл, чем в случае нафтильных изомеров В.4213 и В.4265.
Физико-химические факторы, такие как стабильность, растворимость и способность к распределению в системе вода - липид важны при выборе соединений для использования в условиях in vivo и влияют на состав, поглощение и перенос соединений. На выбор соединений может оказать влияние их различная способность к распределению в различных тканях.
В одном из вариантов осуществления настоящего изобретения заявляется фармацевтическая композиция, содержащая соединение формулы I, где значения Y, R и R' указаны ранее, или его фармацевтически приемлемую соль и фармацевтически приемлемый наполнитель. Композиция необязательно может также содержать алкилирующий агент, такой как хлорэтилирующий или метилирующий агент.
В другом варианте осуществления настоящего изобретения заявляется способ ослабления активности АТазы у реципиента, заключающийся в назначении реципиенту эффективного количества композиции, содержащей соединение формулы I, где значения Y, R и R' указаны ранее, или его фармацевтически приемлемую соль, в частности, указанной выше композиции. Указанный способ иначе можно определить как способ ослабления стимулируемой АТазой активности по восстановлению ДНК у реципиента.
В изобретении далее заявляется способ лечения опухолевых клеток у реципиента, заключающийся в назначении реципиенту эффективного количества композиции, содержащей соединение формулы I, где значения Y, R и R' указаны ранее, или его фармацевтически приемлемую соль, в частности, указанной выше композиции, и назначении реципиенту эффективного количества композиции, содержащей алкилирующий агент. Способ может применяться для лечения опухолей, которые чувствительны к действию алкилирующих агентов, в частности меланобластомы и глиомы и других, устойчивость которых к действию одних лишь алкилирующих агентов может быть преодолена путем использования активатора по настоящему изобретению.
В изобретении заявляется также способ получения соединений формулы I, включающий следующие стадии: взаимодействие гидрида натрия с раствором RR'CHOH (где значения R и R' указаны ранее) в органическом растворителе, преимущественно, при комнатной температуре или температуре ниже комнатной, добавление хлорида 2-амино-N,N,N-триметил-1H-пурин-6-аминия или 2-амино-6-хлорпуринрибозида; обработку реакционной смеси слабой кислотой в эфире; и экстракцию целевого продукта.
Краткое описание чертежей
Изобретение более подробно поясняется со ссылкой на прилагаемые чертежи, из которых:
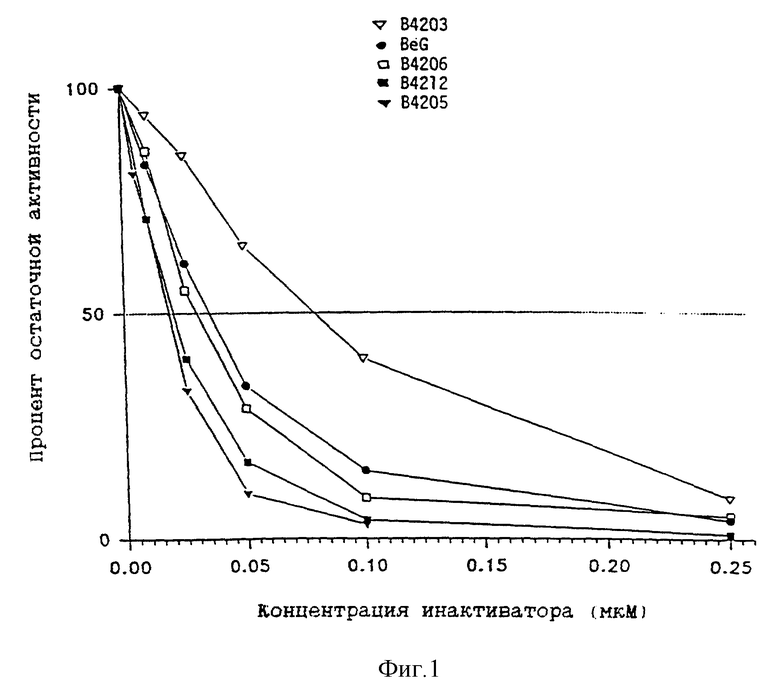
На фиг. 1 графически представлена величина остаточной активности очищенной рекомбинантной АТазы человека с последующим выдерживанием в инкубаторе с различными концентрациями разнообразных дезактиваторов. Каждая точка показывает среднее из трех значений. Линия, соответствующая 50%-ной активности, используется для вычисления величины I50, т.е. концентрации дезактиватора, и необходимой для снижения на 50% активности АТазы.
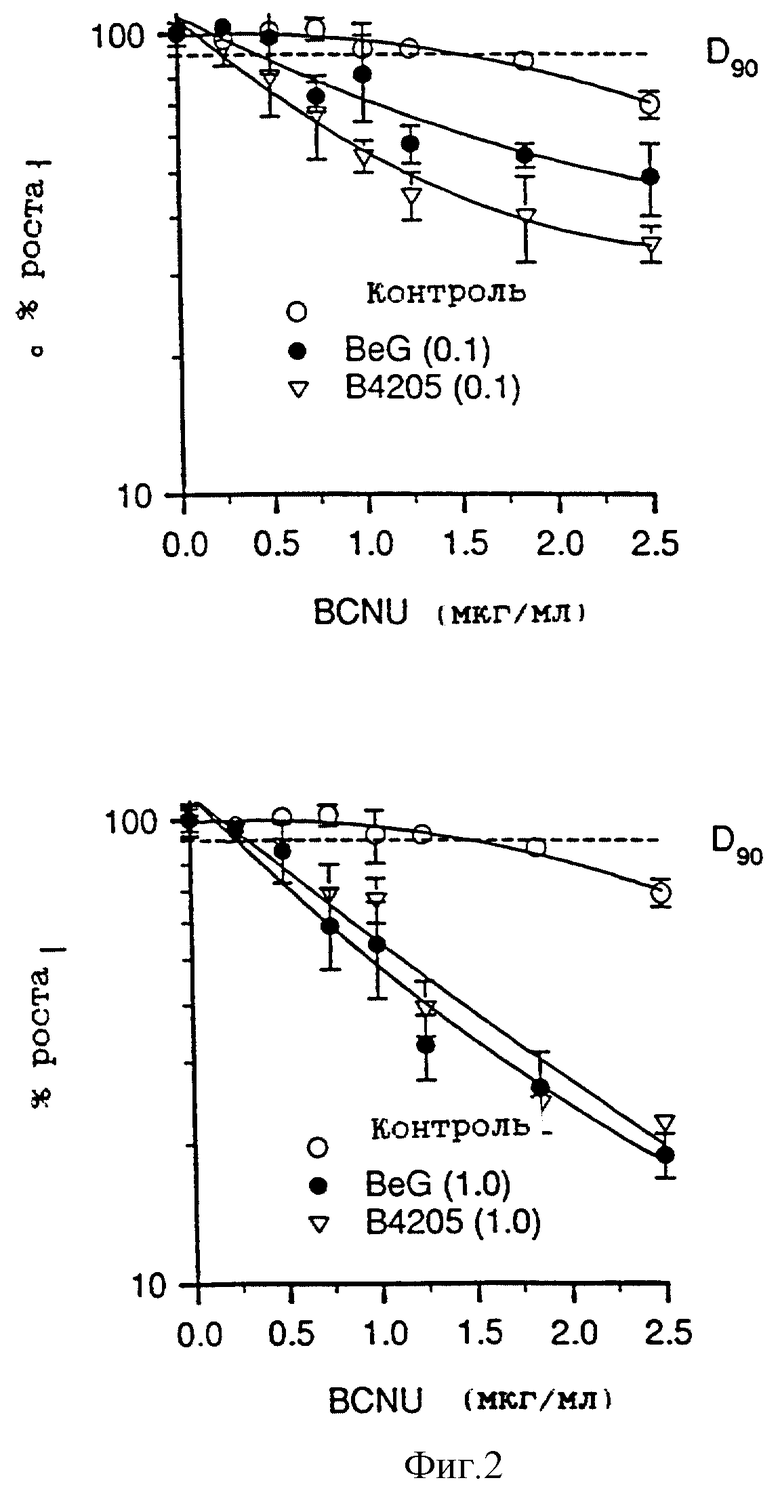
На фиг. 2 приведены два графика зависимости роста клеток от концентрации алкилирующего агента (мкг/мл), которые показывают сенсибилизирующее воздействие О6-бензилгуанина (BeG) и О6-тенилгуанина (В.4205) при двух различных концентрациях (0,1 и 1,0 мкМ) на BCNU при сенсибилизации клеток Raji. Линия, соответствующая 90% роста, используется для вычисления величин D90, т. е. дозы BCNU, при которой наблюдается 90% роста по сравнению с не подвергавшимися воздействию контрольными образцами, т.е. соответствует росту ингибирования на 10%.
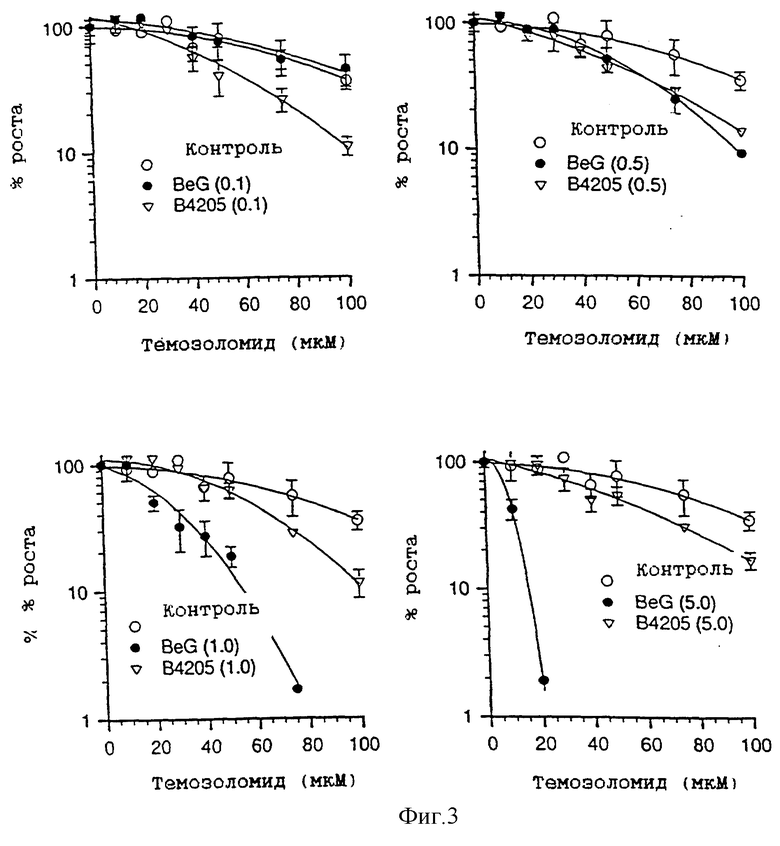
На фиг. 3 приведены четыре графика зависимости роста клеток, выраженного в процентах, от концентрации алкилирующего агента (мкМ), которые показывают влияние четырех различных концентраций (0,1, 0,5, 1,0 и 5,0 мкМ) BeG и В. 4205 на темозоломид при сенсибилизации клеток Raji.
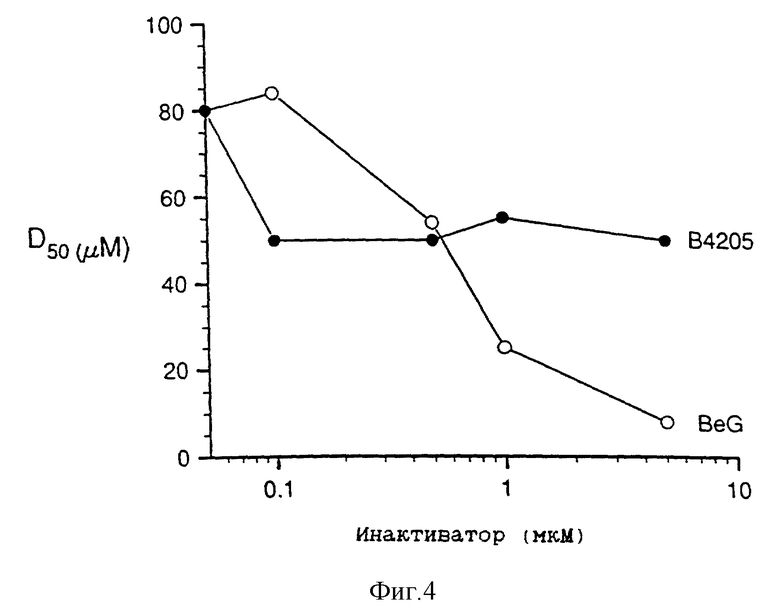
Фиг. 4 представляет собой график, полученный экстраполяцией значений, приведенных на фиг. 3, для величин D50 (мкМ) (т.е. дозы темозоломида, при которой наблюдается 50%-ный рост, по сравнению с не подвергавшимися воздействию контрольными образцами) в зависимости от концентрации инактиватора (мкМ).
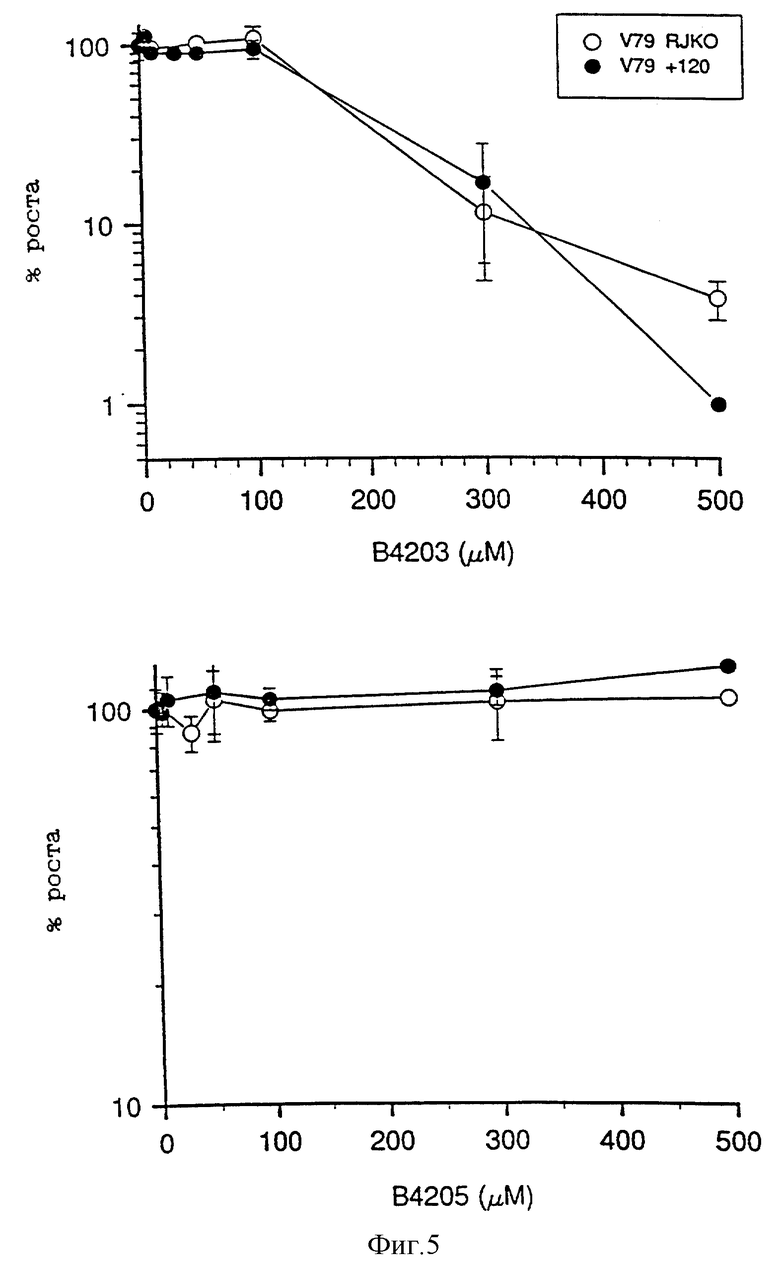
На фиг. 5 приведены два графика зависимости роста клеток, выраженного в процентах, от концентрации инактиватора, которые показывают увеличение ингибирующего воздействия О6-фурфурилгуанина (В.4203) и В.4205 на клетки V79 китайского хомячка (RKJO) и их сублинии (+120).
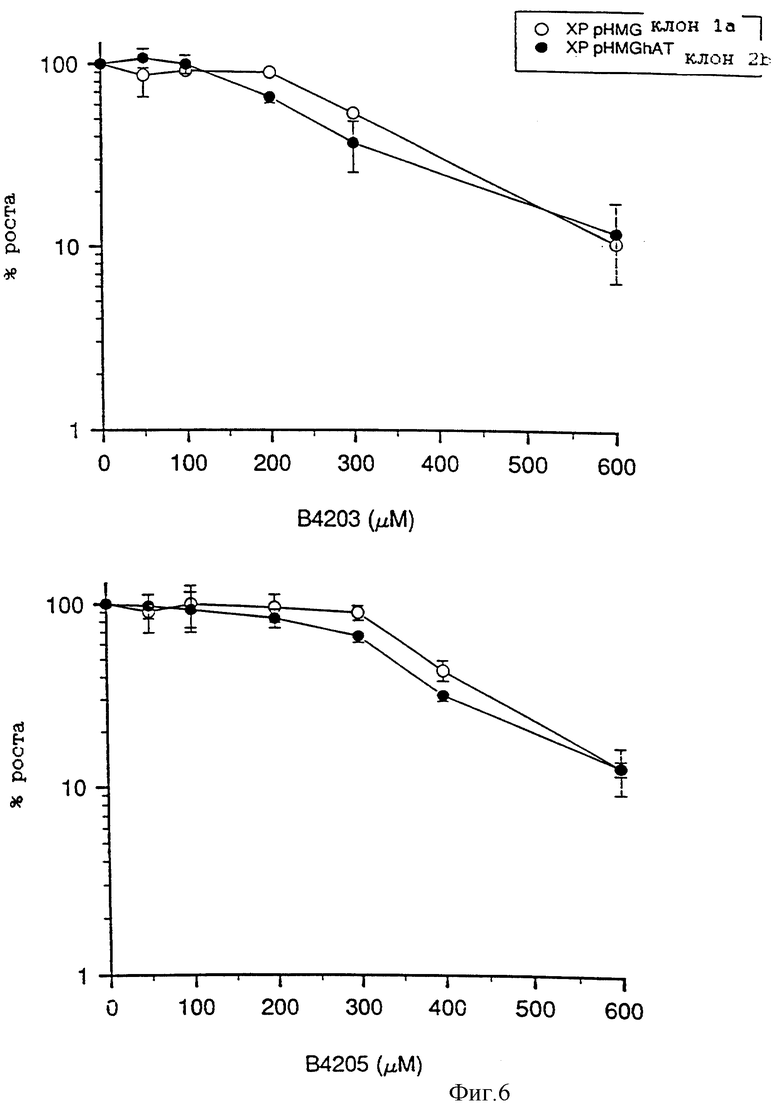
На фиг. 6 приведены два графика зависимости роста клеток, выраженного в процентах, от концентрации инактиватора, которые показывают увеличение ингибирующего воздействия В.4203 и В.4205 на субклоны Xeroderma pigmentosum.
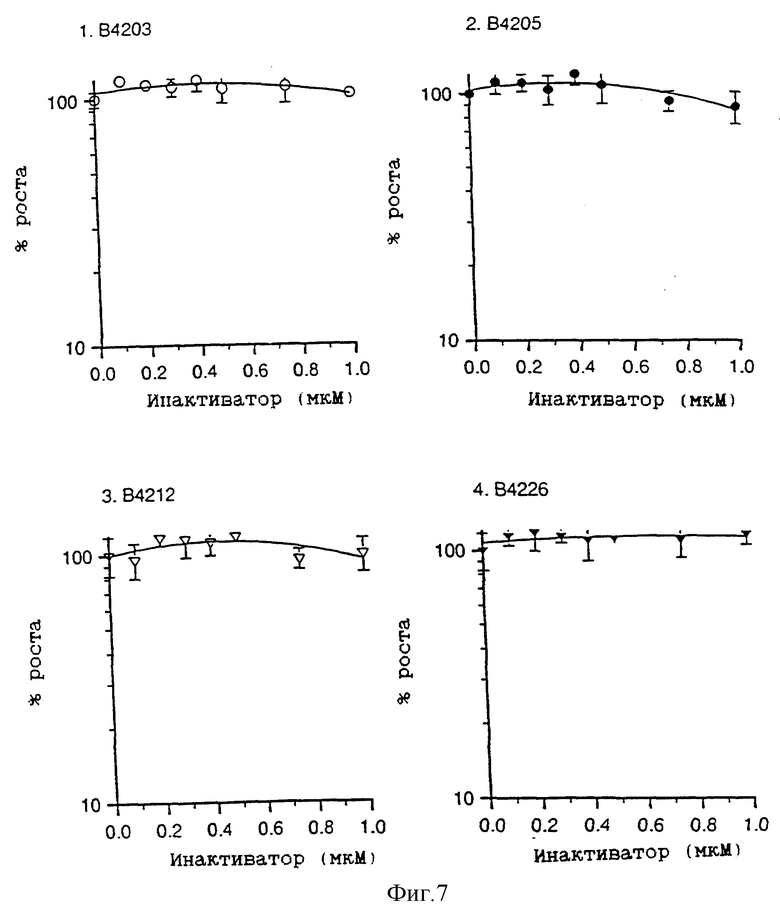
На фиг. 7 приведены четыре графика зависимости роста клеток, выраженного в процентах, от концентрации инактиватора (мкМ), которые показывают воздействие продуктов распада инактиваторов В.4203, В.4205 и В.4212 (О6-пиперонилгуанин) и В. 4226 (О6-[2-бензо(b)тиенилметил]гуанин) на рост клеток Raji.
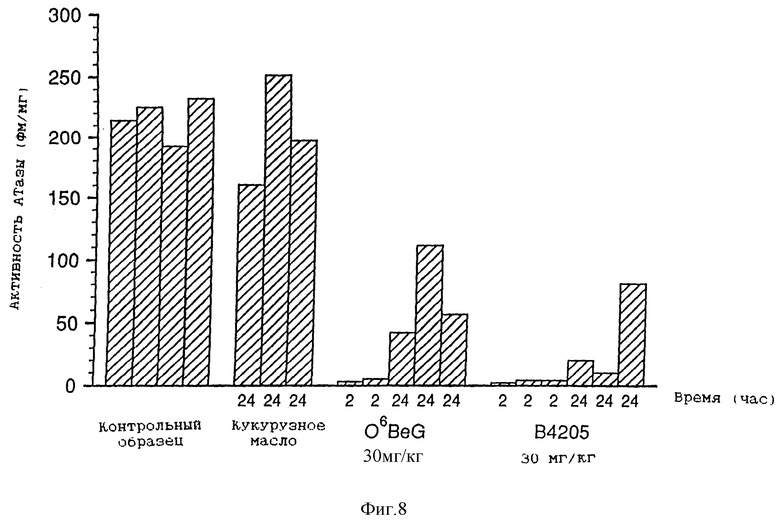
Фиг. 8 представляет собой диаграмму зависимости активности АТазы (фм/мг) от времени (в часах), которая показывает ослабление активности АТазы в ксенотрансплантатах А375 у голых мышей для не подвергавшихся воздействию контрольных образцов, обработанных кукурузным маслом, и экстрактов, обработанных BeG (30 мг/кг) и В.4205 (30 мг/кг).
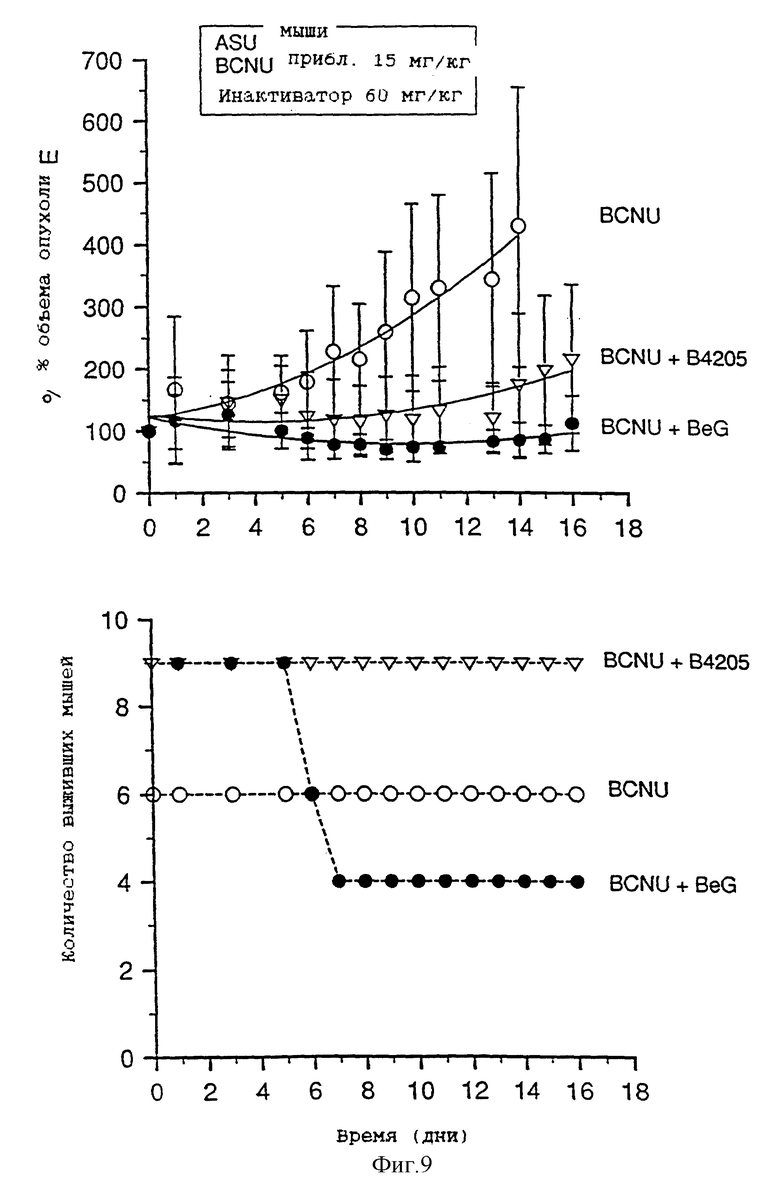
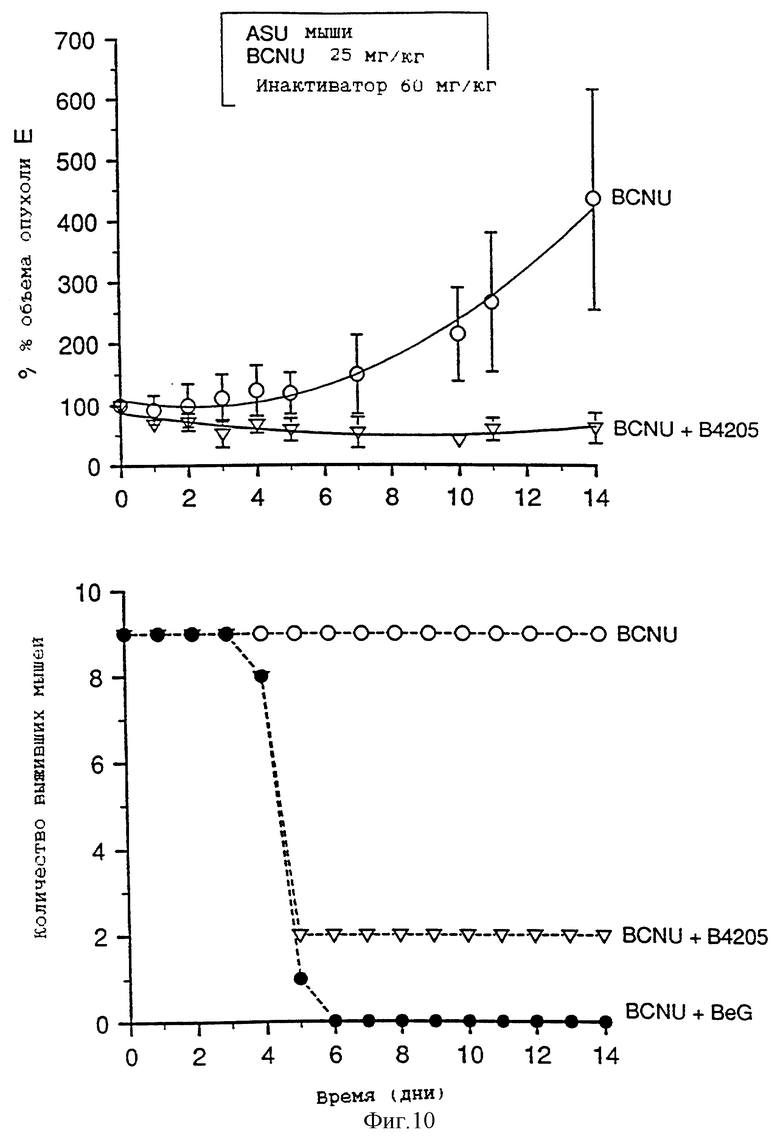
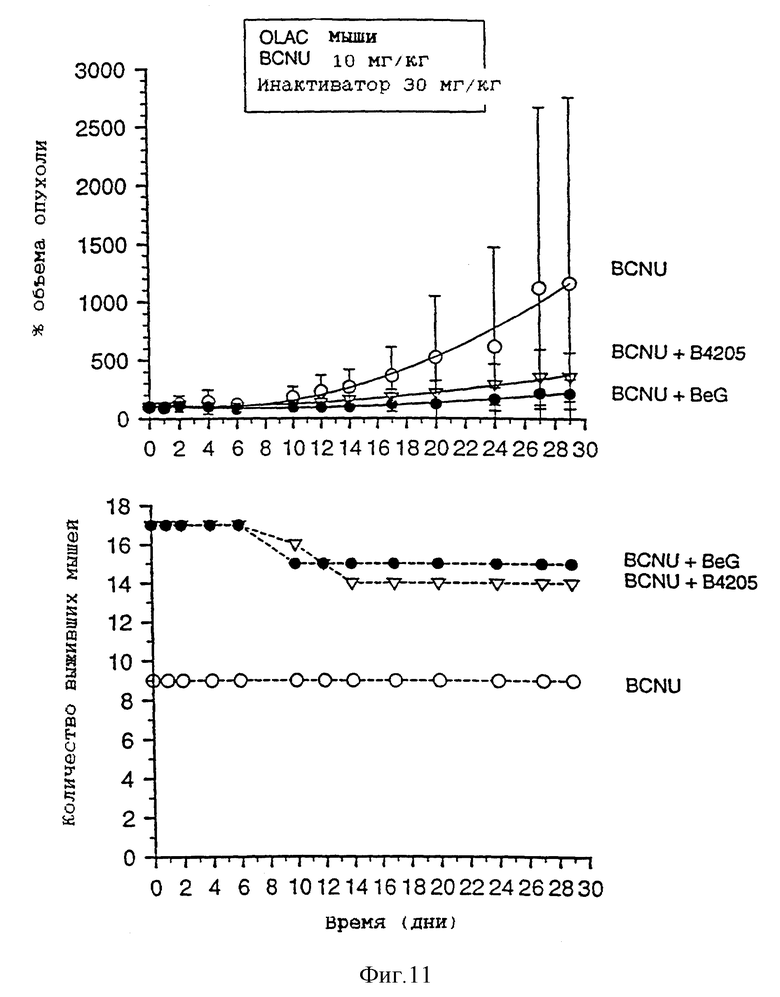
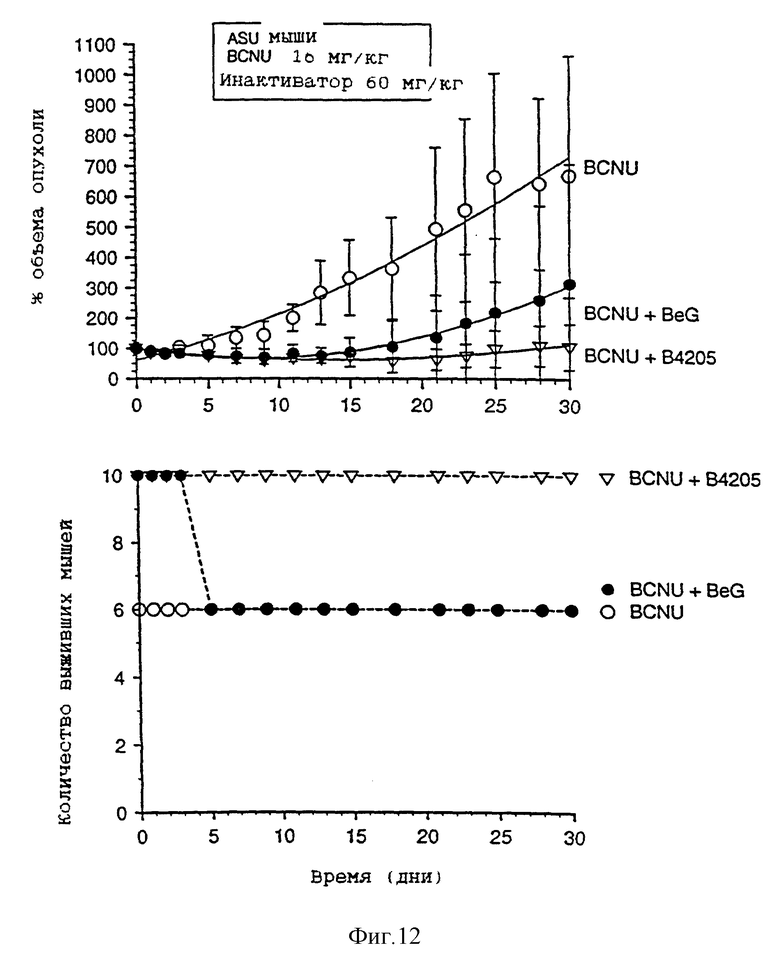
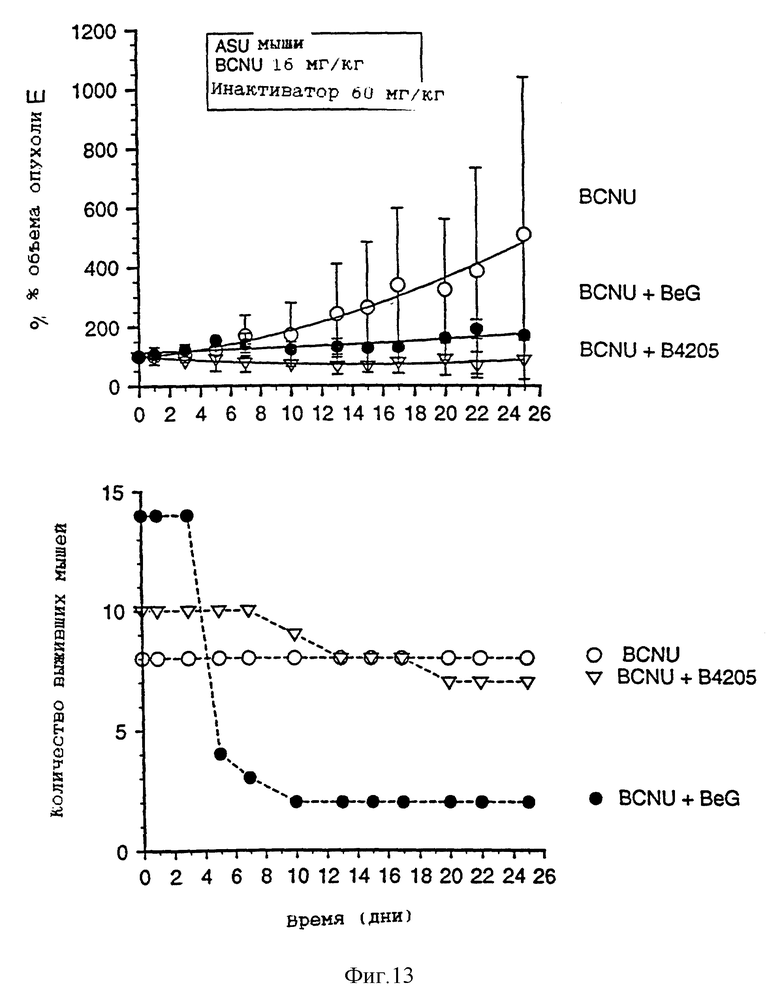
На фиг. 9-13 приведены графики результатов исследования ксенотрансплантатов. На каждом фигуре верхний график показывает зависимость объема опухоли, выраженного в процентах, от времени (в днях) для ксенотрансплантатов А375 у голых мышей, подвергавшихся воздействию одного BCNU, BeG в сочетании с BCNU и В.4205 в сочетании с BCBU. Нижний график на каждой фигуре показывает количество выживших мышей в каждой подвергшейся воздействию группе в зависимости от времени (в днях) после обработок, приведенных на верхнем графике.
Чертежи имеют следующие отличия (табл. 2)
На фиг. 14 приведены три графика зависимости процента выживания от концентрации темозоломида (мкМ), которые показывают выживание клеток костного мозга с последующей обработкой инактиватором (10 мкМ) или ДМСО (контрольные образцы) в сочетании с возрастающими дозами темозоломида.
Способы осуществления изобретения
Производные О6-гетарилалкилгуанина, обладающие инактивирующими способностями по отношению к АТазе, могут быть получены путем адаптации стандартных методик, которые представлены ниже.
Хлорид 3-амино-N,N,N-триметил-1H-пурин-6-аминия получают в соответствии с методикой приведенной в статье Kiburis et al., J.Chem. Soc. (С), 1971, 3942. Подробные условия проведения реакции этой четвертичной соли с бензилоксидом натрия (с образованием О6-бензилгуанина), не указанные в статье Mc Coss, Chen and Folman, Fetrahedron Zett., 1985, 26, 1815, приведены в Европейской патентной заявке 184473 (Mc Coss, Folman, Waqner and Hannah), однако они не приемлемы для относительно чувствительных аналогов, поэтому заявителями была разработана приведенная ниже стандартная методика.
Стандартная методика получения О6-гетарилалкилгуанинов (Формула I, Y = Н)
Гидрид натрия (60% в масле; 0,8 г, 20 ммол) добавляют к раствору RR'CHOH (56 ммоль, прибл. 5 мл) в ДМСО (5 мл) и смесь перемешивают при комнатной температуре в течение 1 час. Для твердых спиртов или спиртов с большим молекулярным весом вместо 5 мл можно использовать до 10 мл ДМСО. Добавляют хлорид 2-амино-N, N, N-триметил-1H-пурин-6-аммония (2,29 г, 10 ммоль) и перемешивание продолжают еще в течение 1 час. Изменение в УФ-поглощении заканчивается (λmax 312 ---> 284 нм) и практически прозрачный раствор обрабатывают уксусной кислотой (1,7 мл). После охлаждения и разбавления эфиром (300 мл) смесь оставляют стоять (2 час) и твердое вещество (А) отфильтровывают. Растирают с водой и получают целевое соединение. Вторую фракцию можно получить после упаривания фильтрата эфир-ДМСО и растирания остатка последовательно с эфиром и водой. Иначе продукт можно экстрагировать из твердого вещества (А) теплым ацетонитрилом. Перекристаллизованные соединения дают одно пятно по данным тонкослойной хроматографии (ТСХ) (бензол:метанол, 4:1) и подтверждаются элементным анализом и данными ЯМР-спектроскопии. Часто они содержат в своем составе кристаллизационный растворитель. Температуры плавления и аналитические данные представлены в Таблице 2, данные УФ и 1H ЯМР-спектроскопии приведены в таблице 3. Спектры 1H ЯМР получены на спектрометрах Bruker WP80 и MSL 300.
Эту стандартную методику (вариации указаны в виде символов в таблице 2) используют для получения соединений, приведенных в таблицах 2а и 3а. В соединениях В. 4217 и В.4219 R' обозначает метил; в остальных соединения R' обозначает H. О6-Бензилгуанин и Соединения В.4214, В.4218 и В.4231, приведенные ниже в таблице 4, также получены по этой стандартной методике для проведения сравнительных испытаний.
Вариации, указанные символами в таблице 2, означают следующее:
a) Для получения этого соединения используют 5 ммоль гидрида натрия на 1 ммоль четвертичной соли. Если используют стандартное количество (2 ммоль), то при разработке реакционной смеси может быть регенерировано 40% четвертичной соли.
b) Это соединение получают гидролизом метилового эфира В.4229 (145 мг, 0,5 ммоль) в 2-метоксиэтаноле (2,5 мл) и воде (2,5 мл) обработкой 2М раствором едкого натра (2,5 мл) в течение 4 час при комнатной температуре. Нейтрализуют уксусной кислотой (0,32 мл, 5,5 ммоль), осторожно упаривают, растирают с водой (3 мл) и отфильтровывают, получая твердое вещество, из которого после экстракции метанолом получают кислоту В.4234.
с) Необходимые цифры основаны на моногидрате смеси 4 частей натриевой соли кислоты и 3 частей кислоты, что требует для Na 4,3%. Найдено: Na 4,44%.
d) Вместо стандартного количества 5,6 ммоль используют 3 ммоль спирта RCH2OH на 1 моль четвертичной соли.
е) Для спиртов, которые слишком чувствительны к действию гидрида натрия в ДМСО при комнатной температуре, DCH2ONa получают в ДМФА (2,5 мл при минус 10oC; 3 ммоль RCH2OH; 2 ммоль гидрида натрия). Через 15-20 минут добавляют 1 ммоль четвертичной соли и перемешивание продолжают в течение 2-3 час при комнатной температуре.
f) Продукты экстрагируют ацетонитрилом.
Получение рибозидов (Формула I, Y = рибозил)
Раствор алкоксида, полученного по приведенной выше стандартной методике из гидрида натрия (60% в масле; 120 мг, 3 ммоль) и RR'CHOH (4,6 ммоль) в сухом ДМСО (2 мл), в течение 1 час обрабатывают 2-амино-6-хлорпуринрибозидом (302 мг, 1 ммоль) и перемешивают в течение 5 минут при комнатной температуре, а затем 15 минут при температуре 60 - 65oC. О завершении реакции свидетельствует изменение УФ-спектра (λmax 311 ---> 284 нм). Охлаждение и тщательное растирание с эфиром (100 + 15 мл) с последующей фильтрацией позволяет получить твердое вещество, которое обрабатывают водой (10 мл). Доводят pH с 11 до 8, недолго пропуская углекислый газ. Неорганические вещества отделяют фильтрованием и высушенный остаток экстрагируют метанолом при комнатной температуре (четырьмя порциями по 10 мл, содержащими каплю пиридина). После испарения практически всего количества метанола и добавления небольшого количества эфира получают продукт, чистый по данным ТСХ (бензол:метанол, 4:1). Его перекристаллизовывают из метанола, содержащего следовые количества пиридина, растворяя и упаривая при температуре ниже 40oC, иногда в конце добавляют немного эфира.
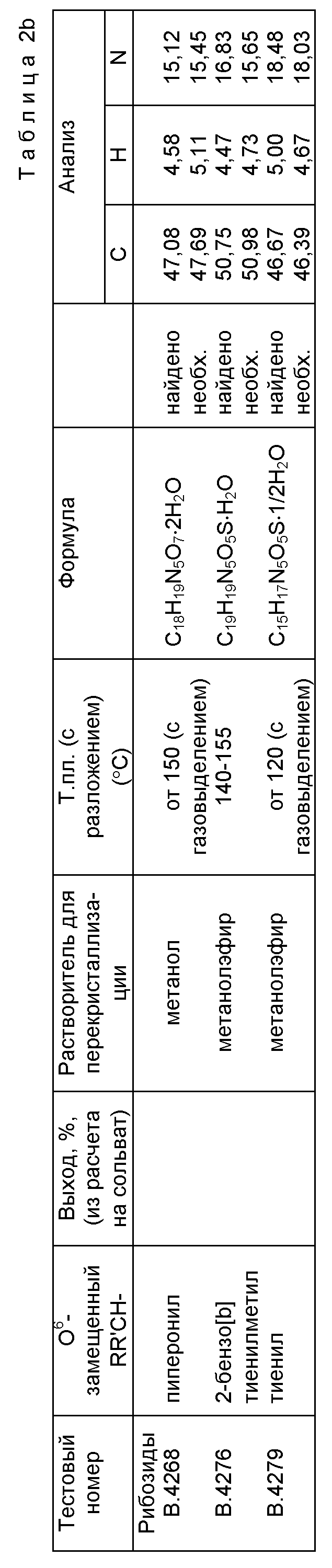
Указанную методику используют для получения соединений, представленных в таблицах 2b и 3b. Следовые количества примесей трудно удалить, однако данные ЯМР-спектроскопии показывают, что чистота нуклеозидов составляет приблизительно 90%. Выходы составляют порядка 30-40%.
Исходные спирты RR'CHOH обычно получают восстановлением соответствующих альдегидов, большинство которых производится промышленно с помощью боргидрида натрия. Другой подход используют для получения предшественников сложного эфира В.4229 и нитрила В. 4273. Сахарозу превращают [W.N. Haworth and W.G.M. Jones, J. Chem. Soc., 1944, 667] через промежуточно образующийся 5-хлорметилфурфураль в 5-гидроксиметилфурфураль. Окислением [Т.Reichstein, Helv. Chim.Acta, 9, 1926, 1066] этого альдегида получают карбоновую кислоту, которую этерифицируют по методу Bocchi et al. [V. Bocchi, G. Casnati, A. Dossena and R. Marchelli, Synthesis. 1979, 961] , получая метиловый эфир 5-(гидроксиметил) фурановой кислоты [Р.A. Finan, J. Chem. Soc., 1963, 3917; J. A. Moore and J.E. Kelly, J. Polym. Sci., Polym. Chem. Ed., 22, 1984, 863], необходимый для синтеза В. 4229. Оксим [J. Kiermayer, Chemiker-Zeitunq, 19, 1895, 1004] альдегида дегидратируют по методу Carotti et al. [A. Carotti, F. Campagna and R. Ballini, Synthesis. 1979, 56]; сырой продукт перед проведением экстракции дихлорметаном предварительно обрабатывают метанольным раствором концентрированного водного раствора аммиака. После перегонки получают 5-цианофурфуриловый спирт [A.J. Floyd, R.G. Kinsman, Y. Roshan-Ali and D.W. Brown, Tetrahedron. 39, 1983, 3881], необходимый для синтеза В.4273.
При синтезе В.4266 по реакции Вильсмейера [V.T. Suu, N.P. Buu-Hoi and N. D. Xuong, Bull. Soc. Ghim. France, 1962, 1875] из бензофурана получают 2-альдегид, который восстанавливают в требуемый спирт [Т. Reichstein and L. Reichstein Helv. Chim. Acta. 13, 1930, 1275], а при синтезе В.4226 вначале получают литиевое производное, которое обрабатывают диметилформамидом и получают 2-альдегид, а из него - спирт [Н. Takeshita, H. Mametsuka and H. Motomura. J.Heterocycl.Chim., 23, 1986, 1211].
При синтезе В.4274 диметиловый эфир винной кислоты окисляют [F.J. Wolf and J. Weijlard, Orq. Synth. Coll. Vol. 4, 1963, 124] в метиловый эфир глиоксиловой кислоты, который взаимодействует [А.М. van Leusen, B.E. Hoogenboom and H. Siderius, Tetrahedron Lett., 1972, 2369] с тозилметилизоцианидом с образованием метилового эфира оксазол-5-карбоновой кислоты [A.Maguestiau, R. Flammang and F.B. Abdelouahab, Heterocycles, 29, 1989, 103]. Его восстанавливают алюмогидридом лития по методу Fallab [S. Fallab, Helv. Chim. Acta, 35, 1952, 215] и получают спирт [A.S. Kende, K. Kawamura and R.J. DeVita, J. Amer. Chem. Soc.. 112, 1990, 4070].
При синтезе В.4275 из броммалонового альдегида [S. Trofimenko, J. Orq. Chem. , 28, 1963, 3243] и тиомочевины получают 2-аминотиазол-5-карбоксальдегид [I. Sawhney and J.R.H. Wilson (Shell Internationale), EP. 395, 174, 1989 (Chem. Abs.. 114, 143410s)]. Его дезаминируют амилнитритом [I.Sawhney and J. R. H. Wilson (Shell Internationale), EP. 395, 174, 1989 (Chem.Abs., 114, 143410s)] с последующим восстановлением боргидридом натрия и получают 5-гидроксиметилтиазол [S. Fallab, Helv. Chim. Acta. 35, 1952, 215].
При синтезе В.4271 5-азапиперониловый спирт (т. пл. 82-84oC) найдено: C 54,60; H 4,58; N 9,09; вычислено для C7H7NO3: C 54,90; H 4,61; N 9,15%) получают из соответствующего альдегида [F. Dallacker, P. Fechter and V. Mues, Z. Naturforsch., 34b, 1979, 1729].
При синтезе В.4278 1-метилпиррол-2-карбоксальдегид нитруют [Р. Fournari, Bull. Soc. Chim. France, 1963, 488] и восстанавливают до спирта [L. Grehn, Chem.Scr., 16, 1980, 72] с помощью боргидрида натрия.
В качестве конкретного примера приводится методика получения О6-тенилгуанина (В.4205):
Получение О6-тенилгуанина
Раствор тенилового спирта (3,18 мл, 33,6 ммоль) в ДМСО (3 мл) обрабатывают гидридом натрия (60% в масле; 0,48 г, 12 ммоль), проводя вначале осторожное перемешивание. Через 1 час добавляют хлорид 2-амино-N,N,N-триметил-1Н-пурин-6-аминия (1,37 г, 6 ммоль) и перемешивание продолжают еще в течение 1 час. Добавляют уксусную кислоту (1,0 мл), недолго охлаждают и разбавляют смесь эфиром (180 мл). Через 1-2 час выделяют твердое вещество (2,09 г). После упаривания эфира и отгонки ДМСО и избытка тенилового спирта (т.пл. 48-57oC/0,2 мм рт. ст.) получают остаток, из которого растиранием с эфиром получают вторую порцию твердого вещества (0,36 г). Полученные порции твердого продукта объединяют, растирают с водой (6 мл) и получают продукт (1,335 г, 90%), который показывает одно большое пятно по данным ТСХ (бензол:метанол, 4: 1) и лишь следовые количества примеси. После растворения в горячем этаноле (30 мл), осветления путем фильтрации через целит и упаривания (до 10 мл) с помощью ротационного испарителя получают B.4205 (1,125 г, 71%, содержит 1/3 EtOH на моль в виде сольвата).
Соединения формулы I, в которых Y обозначает R"XCHR'" (секо-нуклеозиды), могут быть получены аналогично реакции O6-бензилгуанина со сложными эфирами альфа-хлоркарбоновых кислот (MacCoss et al., Fetrahedron Lett.; Европейская патентная заявка 184473) или алкилбромидами (в частности, Kjellberg, Liljenberg and Johansson, Fetrahedron Lett., 1986, 27, 877; Moschel, McDougall, Dolan, Stine and Pegg, J. Med. Chem., 1992, 35, 4486).
Типичные компоненты "сахара", соответствующие R"XCHR'", которые приводят к секо-нуклеозидам, получают по методикам, описанным, в частности, в статье McCormick and Mcblhinney, J. Chem Soc., Perkin Frans, 1, 1983, 93; Zucey, McCormick and Mcblhinney, J. Chem. Soc., Perkin Frans. 1, 1990, 795.
Соединения формулы I, в которых Y обозначает рибозил или дезоксирибозил (нуклеозиды), могут быть получены по методикам, аналогичным синтезу рибозида и 2-дезоксирибозида O6-бензилгуанина (Moschel et al., 1992; Gao, Fathi, Gaffney et al., J. Org. Chem., 1992, 57, 6954; Moschel, Hudgins and Dipple, J. Am. Chem. Soc., 1981, 103, 5489 (см. выше получение рибозидов).
Промышленное использование
Количество применяемого соединения по настоящему изобретению варьируют в зависимости от эффективного количества, требуемого для лечения опухолевых клеток. Подходящей дозой является доза, приводящая к созданию в опухолевой клетке, лечение которой приводят, такой концентрации соединения по настоящему изобретению, которая приведет перед проведением терапии с использованием алкилирующего агента к ослаблению активности АТазы, в частности приблизительно 1 - 2000 мг/кг, предпочтительно 1 - 800 мг/кг, наиболее предпочтительно 1 - 120 мг/кг.
Фармацевтическая композиция по настоящему изобретению может быть приготовлена в обычных формах вместе с обычными наполнителями, как указано в Международной заявке WO 91/13898, описание которой приводится здесь для справок. Композиция может содержать инактиватор по настоящему изобретению вместе с алкилирующим агентом; или же композиция может состоять из двух частей, одна из которых содержит инактиватор, а другая содержит алкилирующий агент. Способ назначения реципиенту соединения по настоящему изобретению также может быть обычным, как, например, описано в заявке WO 91/13898. При назначении пациенту инактиватора по настоящему изобретению фармацевтическая композиция может содержать соответствующий инактиватор в подходящем носителе, таком как 40%-ный полиэтиленгликоль 400 в физиологическом солевом растворе, или в физиологическом солевом растворе или в 3%-ном этаноле (в физиологическом растворе) - для внутривенных инъекций или в виде порошка в форме соответствующих капсул - для орального назначения.
Алкилирующие агенты можно назначать в соответствии с известными методиками и в виде обычных дозировочных форм, как описано, например, в заявке WO 91/13898, или предпочтительно в виде единичной дозы сразу же или в течение 24 час, однако, преимущественно, в течение приблизительно 2 час после назначения инактивирующего АТазу агента в дозах, меньших, чем дозы, которые применяют при обычных режимах лечения. Уменьшение дозы может оказаться необходимым, поскольку, как ожидается, инактиваторы обычно увеличивают токсичность алкилирующих агентов. Примерами хлорэтилирующих агентов являются 1,3-бис(2-хлорэтил) - 1-нитрозомочевина (BCNU), 1-(2-хлорэтил)-3-циклогексил-1- нитрозомочевина (CCNU), фотемустин, митозоломид и кломезон и агенты, приведенные в публикациях McCormick, McElhinney, McMury and Maxwell, J.Chem. Soc. , Perkin Fraus, 1, 1991, 877; Billy, Double, McCormick, McElhinney, Radacic, Pratesi and Dumont, Anti-Cancer Dnig Des., 1993, 8, 115. Примеры метилирующих агентов включают темозоломид (Патент Великобритании 2104522) и декарбазин, прокарбазин и стрептозоцин.
Методы
Клонирование комплементарной ДНК и сверхэкспрессия АТазы человека уже описаны ранее [Fan, C.-Y., Potter, P.M. Rafferty, J.A. Watson, A.J., Cawkwell, L., Searle, P.F., O'Connor, P.J. and Margison, G.P. (1991) Nucleic Acids Res. 18, 5723-5727] . Очистку рекомбинантных белков проводят либо аффинной хроматографией на колонке ДНК-целлюлоза, как описано Wilkinson et al. [Wilkinson, M. C. , Potter P.M., Cawkwell, L., Georgiadis, P., Patel, D., Swann, P. F. and Margison, G.P. (1989) Nucleic Acids Res. 17, 8475-8484.] [Wilkinson, M. C., Cooper, D.P., Southan, C., Potter, P.M. & Margison, G.P. (1990) Nucleic Acids Res., 18, 13-16], или ион-обменной хроматографией на DEAE-целлюлозе. В последнем случае белок АТазу перед тем как разместить в колонке DEAE-целлюлоза частично очищают осаждением сульфатом аммония (30-60%) и диализом по отношению к раствору, содержащему 10 мM Fris - HCl с pH 7,5, 1 мМ дитиотреитола, 2 мМ ЭДТК, 10% глицерина. Затем АТазу элюируют в градиенте 0-0,1 М NaCl. Очищенная АТаза человека сохраняет свою активность, если хранить ее в концентрированном виде при температуре минус 20oC в буфере 1 [50 мМ Fris - HCl (pH 8,3)/3 мМ дитиотреитол/1 мМ ЭДТК] и может быть разморожена и вновь заморожена несколько раз без существенной потери своей активности.
Выдерживание в инкубаторе с инактиватором и анализ с АТазой
Испытуемые соединения растворяют в ДМСО с конечной концентрацией 10 мМ и разбавляют непосредственно перед использованием в Буфере 1, содержащем 1 мг/мл бычьего сывороточного альбумина (IBSA). Рекомбинантную АТазу разбавляют в IBSA и оттитровывают, чтобы реакция проводилась в условиях, лимитируемых АТазой, а не субстратом. При проведении каждого анализа фиксированные количества АТазы (60-75 фмоль) выдерживают в инкубаторе с различными количествами O6-бензилгуанина или испытуемым соединением с общим объемом 200 мкл IBSA, содержащего 10 мкг ДНК вилочковой железы теленка, в течение 1 час при температуре 37oC. Добавляют [3H] -метилированную ДНК в качестве субстрата (100 мкл содержит 6,7 мкг ДНК и 100 фмоль O6-метилгуанина) выдерживают в инкубаторе 1 час при температуре 37oC, пока реакция не завершится. После кислотного гидролиза ДНК, как описано ранее [Morten, J.E.N. & Margison, G.P. (1988) Carcinogenesis 9, 45-49], [3H]-метилированный белок выделяют и определяют его количество методом сцинтилляции в жидкости. Обычно проводят по два анализа образцов, а эксперименты повторяют несколько раз. I50 является концентрацией инактиватора, требуемой для снижения активности АТазы на 50%.
Клеточная культура и получение экстрактов
Клетки млекопитающих выращивают в стандартных условиях. Например, клетки Raji (линия лимфобластоидных клеток человека из лимфомы Беркитта) выращивают в суспензии культуры в среде RPMI, в которую добавлено 10% сыворотки лошади. Клетки А375М являются клетками меланомы человека, от которых получают вышеописанные ксенотрансплантаты и затем вводят их подкожно голым мышам; клетки WiDr представляют собой клеточную линию раковых клеток толстой кишки человека; клетки Hamster +120 представляют собой отобранную с помощью митозоломида сублинию клеточной линии V 79 фибропласта легких китайского хомячка, которую обозначают RJ KO; клетки Yoshita представляют собой клеточную линию карпиносаркомы крыс, a Yrbus представляют ее сублинию, устойчивую к действию бусульфана; клетки XP представляют собой трансформированную под действием SV 40 клеточную линию фибропластов, полученную из кожи пациента, больного Xeroderma pigmentosum; клетки pHMGhAT2b являются клоном указанных клеток, которые заражают вектором экспрессии клеток млекопитающих, содержащим комплементарную ДНК АТазы человека, а клетки pHMG1a представляют собой клон, который заражают лишь вектором экспрессии (т.е. не содержит ДНК АТазы человека). Таблетки клеток вновь суспендируют в холодном (4oC) Буфере 1, содержащем 2 мкг/мл лейпептина, подвергают воздействию ультразвука в течение 10 секунд с интервалами 12 микросекунд между импульсами. После охлаждения льдом клетки подвергают воздействию ультразвука еще в течение 10 секунд с импульсами 18 микросекунд. Сразу же после обработки ультразвуком добавляют 0,01 объемов раствора 6,7 мг/мл фенилметансульфонилфторида в этаноле и обработанную ультразвуком смесь центрифугируют с ускорением 15000g в течение 10 минут при температуре 4oC, чтобы отделить клеточные остатки в виде таблетки. Жидкость над осадком сохраняют для определения активности АТазы (см. далее).
Устойчивость инактиваторов при 37oC
Инактиваторы (10 мМ в ДМСО) разбавляют до концентрации 0,1 мМ в предварительно нагретом девазированном Буфере 1 (1 мМ ЭДТК, 50 мМ Fris, pH 8,3) или забуференном фосфатом солевом растворе (pH 7-7,2). Забуференный фосфатом солевой физиологический раствор представляет собой раствор, содержащий 0,8% хлорида натрия, 0,02% хлорида калия, 0,15% дигидрофосфата натрия, 0,02% дигидрофосфата калия с pH 7,2. Образцы немедленно переносят в спектрофотометр CARY 13 (блок кювет находится при температуре 37oC) и сканируют на подходящей длине волны (в соответствии со спектральными свойствами соединения) в течение до 80 час с интервалами 3-10 минут. Результаты выражают в виде изменения процента поглощения в зависимости от времени, а значения T1/2 (полупериод существования) получают из них экстраполяцией.
Ингибирование активности АТазы в клетках млекопитающих
Клетки разбавляют до концентрации 106/мл в среде для выращивания культуры, содержащей либо соответствующие концентрации инактиватора, либо эквивалентный объем носителя (ДМСО). После выдерживания в инкубаторе в течение 2 час при температуре 37oC клетки отбирают центрифугированием, дважды промывают забуференным фосфатом солевым раствором и полученную таблетку из клеток (1-2 х 107 на таблетку) хранят при температуре минус 20oC.
Активность АТазы определяют, как указано ранее, в двух сериях клеточных экстрактов, подвергнутых воздействию ультразвука, и выражают в виде процента остаточной активности по отношению к активности, имеющейся в необработанных контрольных образцах (например, 350-450 фм/мг в клетках Raji). Значения I50 (т. е. концентрацию инактиватора, требуемую для снижения активности АТазы на 50%) получают из них экстраполяцией.
Сенсибилизация клеток Raji и A375M к действию BCNU и темозоломида
Чувствительность клеток Raji к цитотоксическому воздействию BCNU и темозоломида после предварительной обработки в течение 2 час инактиватором изучают с помощью анализа ХТТ [Scudiero, D.A., Shoemaker R.J., Paull R.D., Monks A. , Tierney S. , Nofziger Т.Н., Currens M.J., Seniff D, & Boyd M.R. (1988), Cancer Research 48, 4827-4833]. Если коротко, то клетки помещают в количестве 1000 на ячейку на планшеты с 96 ячейками и перед добавлением среды, содержащей либо инактиватор в соответствующей концентрации, либо эквивалентный объем носителя, выдерживают в инкубаторе в течение 30 минут при температуре 37oC. Через 2 час в инкубаторе при 37oC добавляют среду, содержащую либо возрастающие дозы BCNU, темозоломида или эквивалентное количество носителя, и дают клеткам расти в течение 6 дней. По окончании указанного срока добавляют раствор ХТТ и выдерживают клетки в инкубаторе еще в течение 4 час при температуре 37oC. Полученный в качестве продукта реакции формазан красно-оранжевого цвета количественно определяют, измеряя поглощение на длине волны 450 нм с помощью аппарата для прочтения микротитрапионных планшетов.
Чувствительность клеток A375M к цитотоксическому воздействию BCNU изучают с помощью анализа МТТ [Carmichael J., DeGraff W.C., Gazdar A.F., Minna J. D. , Mitchell J.B. (1987). Evaluation of a tetrazolium based semiautomated colorimetric assay : assessment of chemosensitivity testing. Cancer Res. 47: 936-942] , который имеет следующие отличия от вышеописанного анализа ХТТ. Клеткам A375M (1000 на ячейку) дают расти в течение 24 час, затем обрабатывают инактиватором, а еще через 2 час - с помощью BCNU. Через 6 дней добавляют раствор МТТ (4 мг/мл) и выдерживают клетки в инкубаторе в течение 3 час при температуре 37oC. Среду удаляют и полученные розовые кристаллы формазана растворяют в ДМСО (120 мкл) и жизнеспособные клетки определяют, изменяя поглощение на длине волны 450 нм с помощью аппарата для прочтения микротитрационных планшетов.
Из полученных данных вычисляют процент роста клеток по отношению в контрольным ячейкам для интервала доз BCNU или темозоломида как в присутствии, так и в отсутствии инактиватора. Чувствительность клеток Raji к BCNU (D90. C/D90.I) определяют как частное от деления величины D90 (т.е. дозы, при которой наблюдается 90% роста по отношению к не подвергавшимся воздействию контрольным образцам, или ингибирование роста на 10%), вычисленной из результатов по использованию одного BCNU (D90. C), и величины D90 для BCNU плюс инактиватор (D90. I ). Значение, равное единице (I), указывает, таким образом, на отсутствие сенсибилизации под влиянием инактиватора. Чувствительность клеток Raji к темозоломиду и чувствительность клеток A375M к BSNU определяют из соответствующих значений D50 (т.е. дозы, при которой наблюдается 50%-ное ингибирование роста).
Чувствительность клеток млекопитающих к действию инактиваторов или продуктов их гидролиза
Для исследования влияния самих инактиваторов на клетки Xeroderma pigmentosum и клетки V79 китайского хомячка на них в течение 2 час при температуре 37oC воздействуют возрастающими концентрациями (вплоть до 600 мкМ) выбранного инактиватора. В некоторых случаях инактиваторы предварительно подвергают гидролизу в течение 20 час при температуре 37oC, а затем добавляют к клеткам Raji, чтобы изучить возможную степень ингибирования роста клеток продуктами распада. Через 6 дней степень роста клеток определяют, как это описано ранее.
Сенсибилизация клеток костного мозга к действию темозоломида (анализ GM-CFC)
Для анализа гранулоцит/макрофаг колониеобразующих клеток (анализа GM-CFC) получают первичные образцы костного мозга человека от пациентов, которым проводят кардиоторакальные операции. После удаления из образцов эритроцитов клетки помещают в количестве 1-2 х 105/мл в среду Искова 300mOsM, содержащую 10 мкМ инактиватора или эквивалентный объем ДМСО, 20% сыворотки плода коровы, 10% кондиционной среды 5637 как источника факторов роста и 0,3% очищенного агара, в чашки Петри, содержащие соответствующие дозы темозоломида, и выдерживают в инкубаторе при температуре 37oC в атмосфере, содержащей 5% диоксида углерода и 95% воздуха. Через 9 дней определяют колонии, включающие более 50 клеток. Колонии являются клетками-предшественниками гранулоцит/макрофаговой линии дифференцировки (huGM-CFC). Выживаемость определяют в виде процента от количества колоний при нулевой концентрации темозоломида.
Изучение ксенотрансплантатов
Животные
Мужские особи крыс BALB-C с врожденным отсутствием вилочковой железы (nu/nu) с весом 25-35 г получают из выращиваемой в условиях помещения колонии из Патерсоновского института по исследованию рака (Christie Hospital NHS Frust, Wilmslow Road, Manchester M20 9BX, England) (мыши ASU). Животных до проведения эксперимента помещают в стерильных условиях. Для одного из экспериментов мышей получают от Harlan-Olac, Harlan UK Ltd, Shawis Farm, Blackthorn, Bicester, Oxon, OX6 OPT. England (мыши OLAC). Вес этих мышей составляет 17-23 г; впоследствии оказалось, что эти мыши более чувствительны к токсическим эффектам при комбинированной (инактиватор и BCNU) обработке. По этой причине используют меньшие дозы.
Клетки
Клетки A375M (меланомы человека) выращивают в модифицированной Иглом среде Дульбекко, содержащей 10% сыворотки плода коровы. Клетки получает для in vivo инокуляции, выдерживая в инкубаторе с 0,01% трипсина.
Опухоли
Клетки (106) в 100 мкл забуференного фосфатом физиологического солевого раствора вводят подкожно в правый бок nu/nu мышам в возрасте 8-10 недель. Дают этим клеткам в течение 3-4 недель развиться в опухоль для пересадки экспериментальным животным. Блоки из опухолей с размером 2 мм х 2 мм х 2 мм имплантируют подкожно в правый бок. Указанные животные через 7-10 дней готовы для проведения экспериментов с инактиватором.
Лечение опухоли
Мышам nu/nu внутрибрюшинно вводят либо 30 или 60 мг/кг O6--бензилгуанина или B.4205 и соответствующее количество носителя для контроля за 60-90 минут до введения 10-25 мг/кг BCNU (внутрибрюшинно). O6-Бензилгуанин и B.4205 готовят в виде раствора с концентрацией 5 мг/кг в кукурузном масле, а BCNU - в забуференном фосфатом солевом растворе + 3% этанола (2 мг/мл).
Измерения опухолей
Измерения ксеноимплантатов проводят через каждые 1-2 дня работниками с помощью цифрового циркуля. Объем опухоли вычисляют по формуле (высота х ширина х длина) π /6. Экспериментальных животных также взвешивает каждые 1-2 дня. Измерения продолжают до тех пор, пока опухоли у контрольных животных достигают максимального допустимого объема (т.е. 1 см х 1 см х 1 см).
Результаты
В таблицах 4, 5 и 6 представлены физические, биохимические свойства и полученные в условиях in vivo данные для каждого инактиватора. Приведенные тестовые испытания поясняются в разделе Методы.
На фиг. 1 приведены результаты анализа по инактивации АТазы в условиях in vitro с использованием четырех соединений. Соединение B.4206 несколько более эффективно, чем BeG, однако B.4212 и B.4205 в условиях проведения анализа показывают значительно более лучшие результаты. B.4203 не столь аффективно как BeG.
На фиг. 2 показано, что концентрация 0,1 мкМ инактиватора B.4205 более эффективна, чем BeG при сенсибилизации клеток Raji к возрастанию ингибирующего действия BCNU; при концентрации 1,0 мкМ B.4205 и BeG одинаково эффективны.
Фиг. 3 показывает, что при концентрации инактиватора, равной 0,1 мкМ, B. 4205 более эффективно, чем BeG, сенсибилизирует клетки Raji к возрастающему ингибирующему воздействию темозоломида, однако с увеличением дозы инактиватора сенсибилизация с помощью BeG становится более эффективной, а сенсибилизация с помощью B. 4205 остается на прежнем уровне. Отсутствие подобной ответной реакции при действии B.4205 и явная зависимая от дозы BeG ответная реакция хорошо видны из фиг. 4.
Фиг. 5 показывает, что несмотря на некоторый рост ингибирования клеток V79 под действием B.4203 при дозах более 100 мкМ (т.е. по крайней мере в 100 раз больших, чем доза I50 для этого соединения), таких эффектов для B.4205 не наблюдается даже при максимальных исследованных концентрациях.
Фиг. 6 показывает, что клетки XP чувствительны к росту ингибирующего действия B. 4203, однако эти клетки более чувствительны, чем клетки V79, к действию B.4205. Однако дозы, требуемые для повышения ингибирования, по крайней мере в 100 раз превышают дозы, необходимые для инактивации АТазы. Можно сделать вывод, что потенциальное ингибирующее действие указанных инактиваторов не будет вносить ощутимый вклад в сенсибилизацию клеток к возрастающему ингибирующему воздействию алкилирующих агентов.
На фиг. 7 показано, что заметный рост ингибирующего воздействия не наблюдается в клетках Raji, когда указанным соединениям позволяют пройти гидролиз (см. Методы), прежде чем их добавляют к клеткам без дальнейшего добавления алкилирующих агентов. Можно поэтому ожидать, что в указанных экспериментальных условиях продукты разложения не будут вносить вклад в увеличение ингибирования, которое наблюдается при использовании этих средств в сочетании с алкилирующими агентами.
Фиг. 8-13 более подробно показывают результаты изучения ксенотрансплантатов. Ослабление и восстановление активности АТазы вслед за действием O6-бензилгуанина или B.4205 измеряют в экстрактах ксенотрансплантатов опухоли A375 (фиг. 8), полученных из тканей животных, умерщвленных через 2 или 24 часа после назначения инактиватора. Фиг. 9-13 указывают на сенсибилизацию ксенотрансплантатов A375 в голых мышах как к O6-бензилгуанином, так и B.4205 в сочетании с BCNU, как это описано в разделе Методы. На каждом рисунке верхний график показывает увеличение объема опухоли, выраженное в процентах, в процессе проведения эксперимента. Нижний график показывает количество животных в каждой испытуемой группе, которые выживают после воздействия. Графики показывают, что B.4205 сравним с O6-бензилгуанином (BeG) в уменьшении объема опухоли к является значительно менее токсичным в сочетании с BCNU, чем BeG в сочетании с BCNU. У пациентов-людей кожную злокачественную меланому лечат с помощью BCNU, в частности в США (C.M. Balch, A. Houghto & L. Peters (1989) "Cutancous Melanoma", in: Cancer: Principles and Practise of Oncology, V.T. De Vita, S. Helman & S.A. Rosenberg (eds.). pp. 1499-1542, Lipicott: Philadelphia), так что ксенотрансплантаты меланомы человека, выращенные в голых мышах, являются животной модельной системой, которая является клинически высоко надежной.
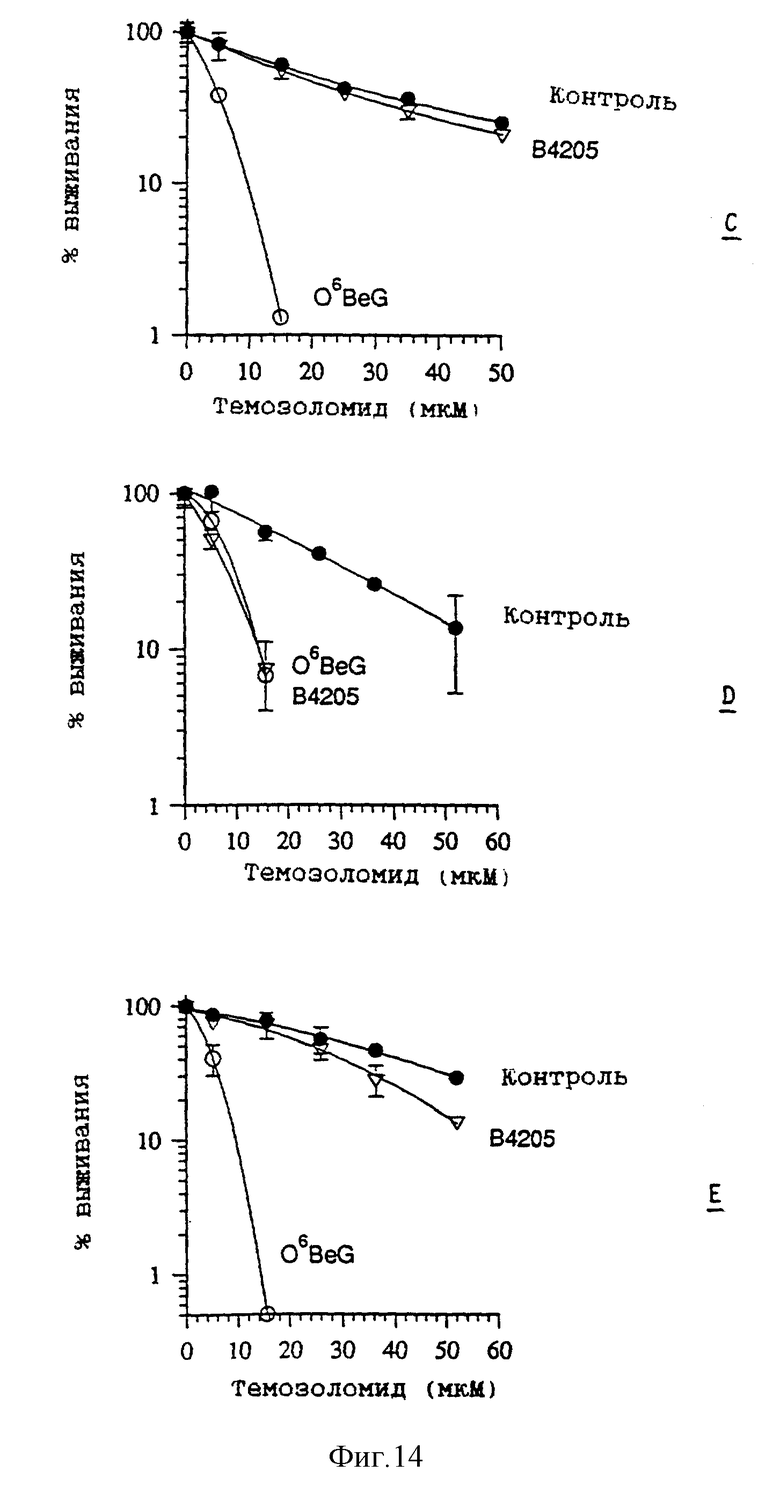
Фиг. 14 показывает результаты испытаний по сенсибилизации клеток костного мозга человека к действию темозоломида, как описано выше в разделе Методы. Клетки костного мозга получают от трех пациентов, обозначенных как C, D и E соответственно. Кривые выживания показывают отношение к цитотоксическому воздействию комбинированного воздействия инактиватор/темозоломид. Желательно, чтобы этот эффект снижался. Результаты для пациентов C и E показывают, что B. 4205 оказывает меньшее сенсибилизирующее воздействие, чем O6-бензилгуанин (O6BeG); для пациентов D отличий не наблюдается, однако этот результат можно рассматривать как допустимое отклонение при проведении научных экспериментов с человеческим материалом.
В таблице 7 показано токсическое действие самого инактиватора на клетки костного мозга, полученные от пяти пациентов A-E, в том числе тех же пациентов C-E, данные для которых приведены на фиг. 14. Результаты для B.4205 сравнимы с результатами для O6-бензилгуанина. Отметим, что клетки пациента B почти вдвое более чувствительны к действию как BeG, так и B.4205, по сравнению с другими образцами.
Краткие итоги
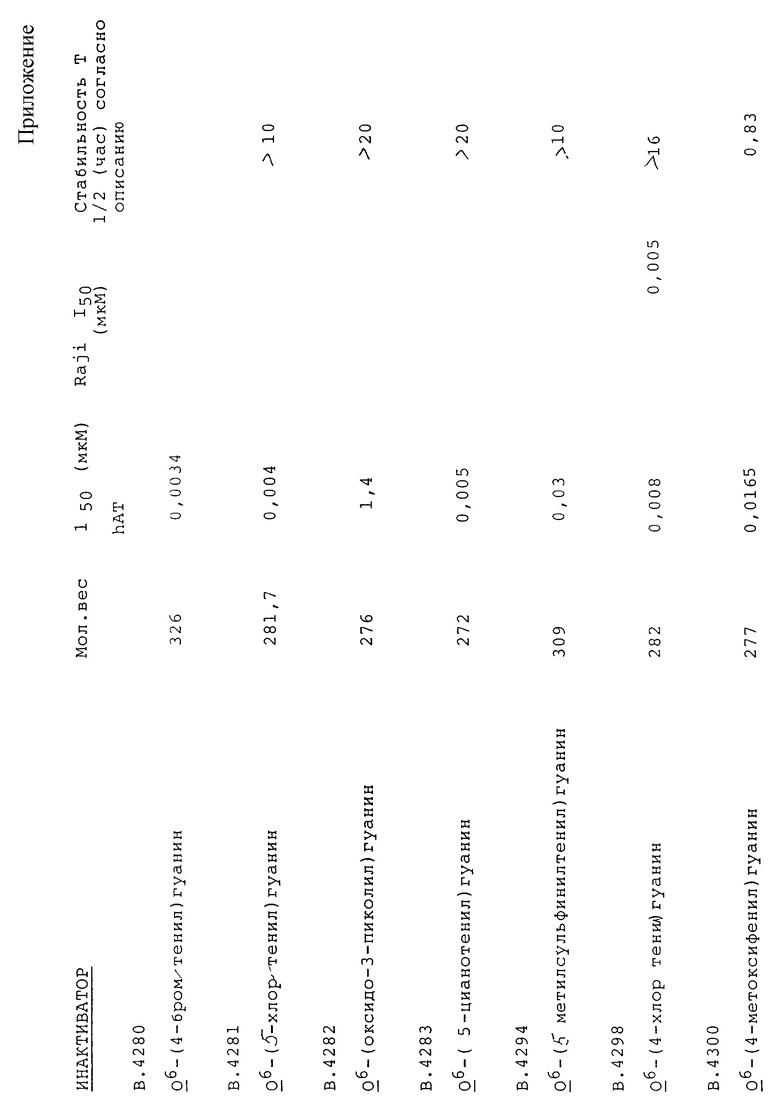
1) Все соединения испытывают на их способность инактивировать рекомбинантную АТазу человека в стандартных условиях при проведении in vitro анализов. В указанных условиях два из соединений (B.4214 и B.4217) не инактивируют АТазу вплоть до наиболее высоких используемых концентраций, однако это соединения, в которых R' обозначает метил. Остальные соединения инактивируют АТазу, при этом значения I50 составляют от 0,0045 до 95 мкмоль. Восемь соединений (B. 4205, B. 4206, B.4212, B.4226, B.4266, B.4269, B.4273, B.4275) имеют значения I50 меньшие, чем для BeG (таблица 4).
2) Инактиваторы претерпевают гидролиз в водных растворах с различными скоростями (полупериоды существования от 0,17 до > 80 час), однако это не связано с их эффективностью как инактиваторов АТазы (таблица 4).
3) Соединения, которые эффективны при инактивации АТазы в условиях in vitro (I50 < 1,0 мкМ), инактивируют также АТазу в клетках человека, при этом величины I50 в общем случае лишь незначительно выше (в среднем в 1,5 раза), чем значения, найденные при использовании рекомбинантного белка в in vitro анализе (таблица 4).
4) B.4205 с равной эффективностью инактивирует АТазу в клетках человека, крысы и китайского хомячка (значения I50 составляют 0,02-0,03). Для B.4203 указанный интервал составляет 0,04-0,12. В клеточных линиях использование B. 4205 и B.4203 почти в семь раз эффективнее, чем BeG (таблица 5).
5) B. 4203 и B.4205 токсичны по отношению к изученным клеткам XP, а B. 4203 токсичен по отношению к клеткам V79, однако при концентрациях, которые по крайней мере в 100 раз превосходят концентрации, при которых наблюдается сенсибилизация к действию BCNU (фиг. 5 и 6).
6) Соединения, эффективные при инактивации АТазы в условиях in vitro (I50 < 1,0 мкМ) и в клетках Raji (I50 < 1,0 мкМ), сенсибилизируют клетки Raji и A375M к возрастающему ингибирующему воздействию BCNU в процессе проведения соответствующих тестовых испытаний (таблица 4).
7) При концентрациях инактиватора, равных 0,1 мкМ, B.4205 более эффективен, чем BeG , при сенсибилизации клеток Raji по отношению к возрастающему ингибирующему воздействию темозоломида (фиг. 13).
8) Анализ в условиях in vitro может использоваться для предсказания, какие из соединений наиболее вероятно могут оказаться эффективными для сенсибилизации клеток млекопитающих к возрастающему ингибирующему воздействию BCNU и аналогичных цитотоксических агентов.
9) B. 4205 аналогичен BeG или несколько более эффективен, чем BeG, при сенсибилизации ксенотрансплантатов меланомы человека, выращенных в голых мышах, к возрастающим ингибирующим воздействиям BCNU (фиг. 9-13).
10) B.4205 столь же эффективен, как и BeG, при инактивации АТазы в ксенотрансплантатах меланомы человека, выращенных в голых мышах (фиг. 8).
11) Анализ в условиях in vitro и/или анализ на ослабление АТазы в ксенотрансплантатах может использоваться для предсказания того, какие соединения с наибольшей вероятностью будут эффективными сенсибилизаторами ксенотрансплантатов меланомы к возрастающему воздействию BCNU и аналогичных цитотоксических агентов.
12) В отличие от BCNU, который вызывает смертность до 70% подвергнутых воздействию животных, B.4205 оказывает очень слабый эффект на чувствительность голых мышей, имеющих ксенотрансплантаты меланомы человека, к острым токсическим эффектам BCNU в условиях проведения испытаний (фиг. 9-13).
13) BeG сенсибилизирует GM-CFC в трех образцах костного мозга человека, подвергнутых испытанию на токсическое воздействие темозоломида, однако в двух из этих образцов сенсибилизация под действием B.4205 оказывается слабой или вообще не наблюдается. Указанный анализ может поэтому использоваться для предсказания возможных миелосупрессивных эффектов инактиваторов АТазы при использовании в клинических условиях в сочетании с BCNU и родственными соединениями (фиг. 14).








Описываются новые производные О6-гуанидина общей формулы I, где Y представляет водород, рибозил или дезоксирибозил, которые могут быть замещены гидрокси или С1-C4алкоксигруппами; R1 представляет водород, С1-20алкил или гидрокси С1-20алкил; R обозначает (i) циклическую группу, имеющую пятичленное гетероциклическое кольцо, содержащее по меньшей мере один гетероатом, выбранный из О, N или S, необязательно конденсированное с кольцом бензола, пиридина или нефталина, или шестичленное гетероциклическое кольцо, содержащее по меньшей мере один атом N, необязательно с атомом S, необязательно конденсированное с одним или двумя бензольными кольцами, где указанная циклическая группа может быть необязательно замещена в гетероциклическом(их) кольце(ах) и/или карбоциклическом(их) кольце(ах) группами, выбранными из С1-С5алкила, галогена, циано, нитро, азидо, С1-С5алкокси, арила, SOnR"", где R"" представляет С1-С5алкил и n равно 0, 1, 2, COOR5, где R5 представляет Н или С1-С5алкил или N-оксиды. Указанные соединения проявляют способность снижать активность О6-алкилгуанин-ДНК алкилтрансферазы в опухолевых клетках. Их активность до 8 раз выше, чем известных аналогов. Описывается также композиция на основе соединений формулы I. 3 с. и 11 з.п. ф-лы, 14 ил., 7 табл.
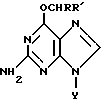
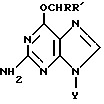
где Y представляет водород, рибозил или дезоксирибозил, которые могут быть замещены гидрокси или С1-С4алкоксигруппами;
R' представляет водород, С1-20алкил или гидроксиС1-20алкил;
R обозначает (i) циклическую группу, имеющую пятичленное гетероциклическое кольцо, содержащее по меньшей мере один гетероатом, выбранный из О, N или S, необязательно конденсированное с кольцом бензола, пиридина или нафталина, или шестичленное гетероциклическое кольцо, содержащее по меньшей мере один атом N, необязательно с атомом S, необязательно конденсированное с одним или двумя бензольными кольцами, где указанная циклическая группа может быть необязательно замещена в гетероциклическом(их) кольце(ах) и/или карбоциклическом(их) кольце(ах) группами, выбранными из С1-С5алкила, галогена, циано, нитро, азидо, С1-С5алкокси, арила, SOnR'''', где R'''' представляет С1-С5алкил и n равно 0, 1, 2, СООR5, где R5 представляет Н или С1-С5алкил или N-оксиды, (ii) нафтил, необязательно замещенный галогеном или С1-С5алкокси,
или их фармацевтически приемлемые соли.
Приоритет по пунктам:
08.06.1993 - по пп. 1 - 4, 12, 14 и 8, 9, 11, 10 по соединениям, зависящим от пп. 1 - 4;
23.05.1994 - по п.13 для BCNU;
08.06.1994 - по пп. 5 - 7 и пп. 8 - 11 по соединениям, зависящим от пп. 5 - 7, и п.13 для темозоломида.
| Огнетушитель | 0 |
|
SU91A1 |
| СПОСОБ ПРЕОБРАЗОВАНИЯ ГЕОПАТОГЕННЫХ ЗОН В БЛАГОПРИЯТНЫЕ НА ОГРОМНЫХ ТЕРРИТОРИЯХ | 1998 |
|
RU2139107C1 |
| A.P.MARTINEZ et al | |||
| ''Potential Antimor agents''- J.Med | |||
| Chem., 1977, v.20, N 3, p.341 - 344 | |||
| B.PAUL et al | |||
| ''Ingibitor of nucleozide transport''- J.Med | |||
| Chem., 1975, v.18, p.968 - 973 | |||
| Способ получения производных гуанина или их кислотно-аддитивных фармацевтически приемлемых солей | 1988 |
|
SU1634138A3 |

