Изобретение относится к области обработки раковых клеток человека с использованием производного тетразинона, в частности к фармацевтической композиции с антиопухолевой активностью и способу ее получения.
Известно применение темозоломида, представляющего собой 8-карбамоил-3-метилимидазо[5,1-d] -1,2,3,5-тетразин-4-(3H)-он, в качестве антиопухолевого активного вещества (см. Newlands и др., J. Cancer, 65, стр. 287, 1992 г.).
Задача изобретения заключается в повышении антиопухолевой активности темозоломида.
Указанная задача решается предлагаемой фармацевтической композицией с антиопухолевой активностью, содержащей темозоломид и дополнительно ингибитор O6-алкилгуанин-ДНК-алкилтрансферазы в отношении от 1 : 1 до 1 : 20000, а также, по меньшей мере, один фармацевтически приемлемый носитель.
Предлагаемую композицию получают путем смешивания темозоломида и указанного ингибитора в указанном соотношении по меньшей мере с одним фармацевтически приемлемым носителем. Данный способ является дополнительным объектом изобретения.
Согласно изобретению токсичность темозоломида можно значительно повысить путем его использования в сочетании с усилителем, представляющим собой ингибитор O6-алкилгуанин-ДНК-алкилтрансферазы (далее: АТаза). В частности, путем использования ингибиторов АТазы, например, O6-бензилгуанина (далее: БГ), можно повысить токсичность темозоломида, например, в клеточной линии MAWI, до 300-кратной величины, если его используют в нижеуказанных дозах согласно настоящему изобретению, что позволяет использовать темозоломид для хемотерапии видов рака в человеке, которые до сих пор не были чувствительными в отношении такой терапии или лишь в незначительной степени. В ряде опухолевых клеточных линий человека токсичность темозоломида после воздействия в течение одного часа взаимосвязана с их содержанием АТазы. Таким образом, метилирование темозоломидом положения O6 гуанина в ДНК приводит к цитотоксичному повреждению. Чувствительность раковых клеток человека к темозоломиду можно определять путем исследования их относительного производства АТазы. При этом клетка, производящая большое количество АТазы, является менее чувствительной к темозоломиду, чем клетка, производящая минимальное количество АТазы.
Путем предварительной обработки клеток одной дозой БГ можно достичь небольшого повышения (менее чем в 4 раза) токсичности темозоломида. В опытах на колониях выявилось, что человеческие фибробласты, трансфецированные кДНК АТазы, после предварительной обработки с использованием БГ являются более устойчивыми к воздействию темозоломида, чем контрольные трансфецированные фибробласты, несмотря на удаление АТазы. Невероятно, что причина этого заключается в разном транспорте темозоломида. Данное явление может быть признаком ресинтеза АТазы phAT-фибробластами для снижения действия предварительной обработки ингибитором.
Однако в клеточной линии MAWI неожиданно наблюдалась значительная потенциация токсичности темозоломида (примерно до 300-кратной величины) путем обработки с использованием БГ в течение пяти дней. Подобная степень повышения наблюдалась в клетках MCF-7, которые также содержат большое количество АТазы, но лишь небольшое действие показали клетки U373, содержащие небольшое количество АТазы. На основе исследований клеточных линий MAWI и MCF-7 можно полагать, что при длительном наличии ингибитора АТазы в растущей степени повреждается ДНК.
Дачу ингибитора АТазы предпочтительно повторяют в течение нескольких или многих дней и предпочтительно ее осуществляют перед дачей темозоломида. Дозы можно дать в течение 1, 2, 3, 4 или 5 дней, причем предпочтительно терапию осуществляют в течение 4 или 5 дней.
Кроме того, предпочтительно темозоломид дают пациенту повторно в течение нескольких дней, и перед дачей каждой дозы темозоломида дают ингибирующее АТазу количество ингибитора АТазы, чем достигается выраженное повышение токсичности темозоломида в отношении раковых клеток человека, например, примерно в 300 раз в случае клеточной линии MAWI.
Согласно дальнейшему предпочтительному варианту изобретения ингибитор АТазы дают в количестве, достаточном для сенсибилизации опухоли в живом организме, не вызывая не желаемой сенсибилизации здоровой ткани, когда ингибитор АТазы используют вместе с темозоломидом.
Количество ингибитора АТазы, даваемое в рамках настоящего изобретения, колеблется в зависимости от эффективного количества, требуемого для обработки опухолевых клеток. Пригодной дозой является доза, приводящая к концентрации ингибитора АТазы в обрабатываемых опухолевых клетках, вызывающей исчезновение активности АТазы, при этом доза может составлять, например, примерно 1 - 2000 мг на кг веса тела, предпочтительно примерно 10 - 800 мг на кг веса тела, если ингибитор дают перед хемотерапией.
В качестве опухолей, для обработки которых темозоломид особенно пригоден, можно назвать карциномы, меланомы, саркомы, лимфомы и лейкозы, в частности, астроцитомы, глиомы, злокачественные меланомы, фунгоидную гранулему, саркому Юингс, хронический лимфоцитный лейкоз, а также опухоли в легких и в груди. Особенно сильное повышение активности темозоломида путем использования ингибитора АТазы наблюдают в опухолевых клетках в груди, в клетках астроцитом, и в опухолях в толстой и прямой кишках.
Обычно доза темозоломида составляет 0,1 - 200, предпочтительно 1-20 мг на кг веса тела в день, или, в отношении поверхности тела, примерно 40 - 400, предпочтительно примерно 150 - 300 мг на м2 в день.
Степень потенциации ингибитором АТазы зависит от количества АТазы, обычно имеющегося в раковых клетках конкретного типа. Активность темозоломида в отношении раковых клеток, содержащих большее количество АТазы, потенцируется сильнее путем предварительной дачи ингибитора АТазы, чем в отношении клеток, содержащих меньшее количество АТазы.
В рамках настоящего изобретения пригодны ингибиторы АТазы, которые обладают такой активностью, например, О6 -алкилгуанины как, например, О6-метилгуанин, алкенилгуанины как, например, О6-аллилгуанин, О6-арилгуанины как, например, О6-бензилгуанин, и О6-бензилированные гуанин, гуанозин и 2'-деоксигуанозин. Для использования в рамках настоящего изобретения, в частности, пригоден О6-бензилгуанин.
В конкретном случае доза ингибитора АТазы зависит от количества АТазы, обычно имеющегося в подлежащих обработке раковых клетках, возраста и состояния пациента и используемого ингибитора АТазы.
Темозоломид особенно предпочтительно дают в виде повторных доз в последовательных днях, причем эффекта сильной потенциации согласно изобретению достигают при предпочтительном режиме, согласно которому ингибирующую АТазу дозу ингибитора АТазы дают перед дачей каждой дозы темозоломидаи или одновременно с ней. Предпочтительно в качестве ингибитора АТазы используют О6-бензилгуанин, который дают вместе с темозоломидом, причем последний дают в количестве 150 - 300 мг/м2 в день в четырех или пяти дозах в течение четырех или пяти дней (общая доза - 750-1500 мг/м). Предпочтительно каждую дозу ингибитора АТазы дают 2 - 8 часов перед дачей очередной дозы темозоломида, чем достигают наиболее сильной потенциации токсичности темозоломида, что приводит к наиболее эффективной обработке опухоли пациента. Особенно предпочтительно такую обработку повторяют по истечении примерно четырех недель.
Альтернативный режим дачи темозоломида вместе с О6-бензилгуанином представляет собой непрерывный режим, причем оба активных вещества дают ежедневно в течение четырех или больше дней. Такую комбинированную терапию можно продлить непрерывно по мере необходимости, до уменьшения опухоли.
Кроме того, путем исследований производства АТазы в конкретной раковой клетке можно определять потенциацию токсичности темозоломида в данной клетке. Согласно предпочтительному варианту изобретения содержание АТазы в определенной клеточной линии определяют, например, путем метода, описанного Lee и др. (Cancer Res. , 51, стр. 619, 1991 г.), и исследуют возможную чувствительность к темозоломиду. На основе такого исследования затем можно найти пригодное соотношение темозоломида и ингибитора АТазы, которое дают в рамках соответствующего режима.
Предлагаемая фармацевтическая композиция может иметься в виде обычного жидкого или твердого препарата, который содержит по меньшей мере один инертный фармацевтически приемлемый носитель.
В качестве твердых препаратов можно назвать, например, порошки, таблетки, поддающиеся диспергированию грануляты, капсулы, в том числе крахмальные капсулы, и суппозитории. Порошки и таблетки могут содержать примерно 5-70% активного вещества. Пригодные твердые носители известны. В качестве таких носителей можно назвать, например, карбонат и стеарат магния, тальк, сахар и лактозу. Таблетки, порошки, крахмальные и прочие капсулы могут содержать по одной дозе активного вещества, и они пригодны для оральной дачи.
Для получения суппозиториев сперва расплавляют воск с низкой точкой плавления, например, смесь глицеридов жирных кислот или какаовое масло, а затем в нем гомогенно диспергируют активное вещество путем размешивания. Расплавленную гомогенную смесь разливают в формы обычного размера, дают охлаждаться, причем препарат застывает.
В качестве жидких препаратов можно назвать, например, растворы, суспензии и эмульсии как, например, растворы в воде или смеси воды и пропиленгликоля, пригодные для парентеральной инъекции.
Также пригодны твердые препараты, предназначенные для перевода в жидкий препарат непосредственно перед дачей, в частности, оральной или парентеральной дачей. Твердые препараты можно переводить в растворы, суспензии или эмульсии.
Предлагаемые соединения можно также дать трансдермально, причем в качестве препаратов, пригодных для трансдермальной дачи, можно назвать кремы, лосьоны, аэрозоли и эмульсии, которые также можно использовать в известных стандартных трансдермальных пластырях или трансдермальных пластырях продленного действия.
Предпочтительно фармацевтическая композиция имеется в виде дозировочных единиц. Каждая дозировочная единица включает отдельные дозы препарата, содержащие пригодное количество активного вещества, например, эффективное количество, обеспечивающее достижение желаемого эффекта.
Количество активного вещества в отдельной дозе может составлять примерно 0,1 - 1000 мг, предпочтительно примерно 1 - 500 мг в зависимости от конкретного случая применения.
Конкретную применяемую дозу можно варьировать в зависимости от пациента и серьезности его состояния. Специалист в состоянии определять пригодную дозу в конкретном случае. При желании общую суточную дозу можно дать в виде нескольких отдельных доз.
Темозоломид можно дать путем известных методов, описанных, например, Wassermann и др. (Cancer, 36, стр. 1258-1268, 1975 г.). В случаях, где подходит оральная дача, предпочитают оральную дачу в количестве 40 - 400 мг/м2 в сутки, в частности 150 - 300 мг/м2, в 1 - 5 дозах, в частности, 4 - 5 дозах, предпочтительно в течение 4 - 5 последовательных дней. Для непрерывной терапии пригодна внутривенная дача 25 - 250 мг/м2 в сутки. Оральная дача пригодна в рамках схемы повторной дачи.
Как уже указывалось, ингибитор АТазы можно дать или отдельно перед темозоломидом, или же одновременно с ним. В тех случаях, где желательна одновременная дача, ингибитор АТазы и темозоломид могут иметься в объединенном виде для облегчения дачи. При этом пригодны вышеприведенные препараты, причем предпочитают препараты, предназначенные для оральной или внутривенной дачи.
Темозоломид и ингибитор АТазы можно включать в один набор, в котором темозоломид и ингибитор АТазы имеются в виде отдельных препаратов для дачи определенным путем, при этом приложены указания по применению содержащихся в наборе препаратов. Например, набор с препаратами, предназначенными для оральной дачи, содержит пригодный для оральной дачи препарат темозоломида, и, отдельно, пригодный для оральной дачи препарат ингибитора АТазы.
Врач будет в состоянии варьировать размер дозы в рамках хемотерапии на конкретном пациенте.
В нижеследующих примерах приведены несколько из возможных препаратов предлагаемой фармацевтической композиции, которые можно дать или отдельно, или одновременно.
Пример 1. Предназначенный для оральной дачи препарат, мг на капсулу:
Темозоломид - 100
Лактоза - 213
Микрокристаллическая целлюлоза - 30
Лаурилсульфат натрия - 20
Кукурузный крахмал - 25
Стеарат магния 2
Размешивают темозоломид, лактозу, микрокристаллическую целлюлозу, лаурилсульфат натрия и кукурузный крахмал, полученную смесь пропускают через сито N 80, добавляют стеарат магния, повторно размешивают и подают в составные желатиновые капсулы пригодного размера.
Пример 2. Предназначенный для оральной дачи препарат, мг на капсулу:
О6-бензилгуанин - 100
Лактоза - 213
Микрокристаллическая целлюлоза - 30
Лаурилсульфат натрия - 20
Кукурузный крахмал - 25
Стеарат магния - 2
Размешивают О6-бензилгуанин, лактозу, микрокристаллическую целлюлозу, лауриловый сульфат натрия и кукурузный крахмал, полученную смесь пропускают через сито N 80, добавляют стеарат магния, повторно размешивают и подают в составные желатиновые капсулы пригодного размера.
Пример 3. Предназначенный для оральной дачи препарат, мг на капсулу:
Темозоломид - 100
О6-бензилгуанин - 100
Лактоза - 213
Микрокристаллическая целлюлоза - 30
Лаурилсульфат натрия - 20
Кукурузный крахмал - 25
Стеарат магния - 2
Размешивают темозоломид, О6-бензилгуанин, лактозу, микрокристаллическую целлюлозу, лаурилсульфат натрия и кукурузный крахмал, полученную смесь пропускают через сито N 80, добавляют стеарат магния, повторно размешивают и подают в составные желатиновые капсулы пригодного размера.
Пример 4. Предназначенный для внутривенной дачи препарат, мг на мл:
Темозоломид - 100
Бисульфит натрия - 3,2
Динатриевая соль этилендиаминотетрауксусной кислоты - 0,1
Вода для инъекции - До 1 мл
Пример 5. Предназначенный для внутривенной дачи препарат, мг на мл:
Темозоломид - 100
О6-бензилгуанин - 100
Бисульфит натрия - 3,2
Динатриевая соль этилендиаминотетрауксусной кислоты - 0,1
Вода для инъекции - До 1 мл
Положительный эффект иллюстрируется нижеследующим примером.
Пример 6.
Материал и метод
Используют культурную среду, продаваемую фирмой ICN Biochemicals Ltd. (г. High Wycombe, Объединенное Королевство), и фетальную телячью сыворотку фирмы Gibco Ltd. (г. Paisley, Объединенное Королевство). О6-бензилгуанин (БГ) поставил др. R.C. Moschel (NCI- Frederick Cancer Research & Development Center, г. Frederick, Мэрилэнд, США). Темозоломид производили May and Baker Ltd. (г. Dagenham, Объединенное Королевство), и его хранили в качестве раствора в диметилсульфоксиде при температуре -70oC. Все остальные химические вещества произведены фирмой Sigma Chemical Co. Ltd. (г. Poole, Объединенное Королевство).
Исследования по цитотоксичности
Клеточные линии выращивали обычным методом в качестве монослоев в буферной среде ДМЕМ, содержащей 10% фетальной телячьи сыворотки, 25 ммоль HEPES, глутамин и пенициллин/стрептомицин. Цитотоксичность исследовали в свободной от HEPES среде в атмосфере 5% двуокиси углерода. В 96 плиток, снабженных углублениями, подали по 750 - 1000 клеток на углубление, инкубировали в течение ночи, в течение двух часов обрабатывали с 33 мкмоль БГ или без него. Затем в течение часа в той же среде добавили темозоломид, причем конечная концентрация диметилсульфоксида не превышала 1%. Клетки выращивали в течение 7 дальнейших дней в свежей среде, после чего содержание протеина определяли с помощью анализа согласно Skehan и др. (J. Natl. Cancer Inst., 82, стр. 1107, 1990 г.). Исследования роста выявили, что во время опыта клетки находились в фазе логарифмического роста. В рамках схемы повторной дачи темозоломида клетки обрабатывали последовательно в течение 24 часов в день, каждый день используя свежую среду. Опыты осуществляли по меньшей мере по два раза.
В минимальной поддерживающей среде выращивали трансфецированные кДНК АТазы клетки человека соответственно контрольные клетки XP (клетки xeroderma pigmentosa) [см. Fan и др., Nucleic Acid Res., 18, стр. 5723, 1990 г.], и в углубления плиток подали по 1000 клеток. Инкубировали в течение трех часов, затем добавляли темозоломид, свежерастворенный в минимальной поддерживающей среде, и плитки инкубировали в течение пяти дней. Клетки, которые переживали, исследовали путем опыта по Morten и др. (Carcinogenesis, 13, стр. 483, 1992 г.). В опытах с использованием БГ трижды подали по 300 клеток в углубления плиток и оставили связываться в течение пяти часов. Затем три часа перед обработкой темозоломидом, свежеразбавленным в минимальной поддерживающей среде, содержащей 10 мкмоль БТ, добавили БГ (10 мкмоль в минимальной поддерживающей среде). По истечении семи дней колонии анализировали известными приемами и подвергали считыванию.
Опыт с использованием О6-алкилгуанин-ДНК-трансферазы
Данный опыт осуществляли согласно методу, описанному Lee и др. (Cancer Res. , 51, стр. 619, 1991 г.). При этом разные количества клеточных экстрактов при температуре 37oC в течение 2 часов инкубировали вместе с ДНК, содержащей О6-метилгуанин, маркированный [3H] в метильной группе, в 300 мкл буфера 1, содержащего 1 мг на мл бычьего сывороточного альбумина. После инкубации последовательно быстро добавляли бычий сывороточный альбумин (100 мкл раствора 10 мг на мл буфера 1) и надхлорную кислоту (100 мкл 4 м раствора). Добавляют еще 2 мл 1 М надхлорной кислоты и полученную смесь нагревают при температуре 75oC в течение 40 минут для превращения ДНК в растворимый в кислоте материал. Протеин, содержащий метилированную АТазу, затем собирают путем центрифугирования и промывают 4 мл 1 М надхлорной кислоты, после чего повторно суспендируют в 300 мкл 0,01 м гидроокиси натрия, растворяют в 3 мл водной сцинтиляционной среды (марки Ecosint A фирмы National Diagnostics) и подвергают считыванию. Содержание протеина в клетках определяли с помощью набора BioRad с использованием в качестве стандарта бычьего сывороточного альбумина. Активность АТазы выражали в фмоль переведенного в протеин метила на мг общего содержания протеина в экстракте.
Поглощение клетками маркированного [14C] темозоломида
8-карбамоил-3-[14C] метилимидазо[5,1-d] -1,2,3,5-тетразин-4-(3H)-он (специфическая активность 26,3 мкюри/ммоль) было поставлено др. John Slack (Aston Molecules Ltd., г. Birmingham, Объединенное Королевство). Клеточные суспензии, содержащие клетки в концентрации 5 х 106 на мл, уравновешивали при температуре 4oC и обрабатывали с использованием 200 мкмоль маркированного лекарства. 106 клеток пипеткой подали в пробирки Эппендорфа и центрифугировали вместе с 250 мкл смеси силиконовых масел в соотношении 4 : 1 (торговый продукт "Three-in-One" фирмы Dow Corning). Водный слой аэрировали и масляный слой осторожно промывали с использованием 300 мкл соляного раствора. После центрифугирования аэрировали оба слоя, содержащий клетки остаток растворяли в пригодном солюбилизаторе, например торговом продукте Protosol, представляющем собой гидроокись четвертичного аммония в толуоле, и подавали в сцинтилляционные пробирки, содержащие Optiphase, представляющий собой 95-99%-ный диизопропилнафталин.
Результаты опытов по определению цитотоксичности
Результаты приведены ниже в табл. 1.
Клетки в культурной среде ткани перед обработкой отдельной дозой темозоломида подвергали или не подвергали воздействию О6-бензилгуанина (БГ).
1. КТ50[-БГ]/КТ50[+БГ]
2. Результаты, полученные путем анализа согласно Wassermann и др. (Int. J. Radiat. Oncol. Biol. Phys., 15, стр., 699, 1988 г.).
Данные табл. 1 графически показаны на фиг. 1, на которой опухолевые клеточные линии человека приведены по порядку повышения уровня АТазы: ZR-75-1, U87MG, U373, LS174T, LOVO, MCF-7 и MAWI. Эти данные показывают взаимосвязь между чувствительностью (определяемой через концентрацию, приводящую к 50%-ному росту, или KT50) опухолевых клеточных линий к темозоломиду (r = коэффициент взаимосвязи = 0,87) и их содержанием АТазы.
Клеточные линии, предварительно обработанные нетоксичной дозой БГ, показали чувствительность относительно темозоломида, составляющую 3,5-кратное чувствительности необработанных клеточных линий.
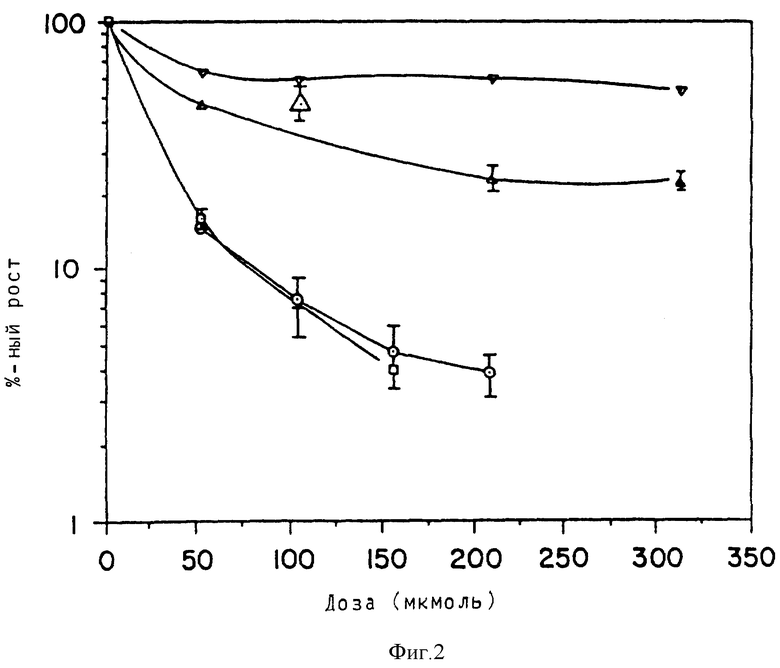
Чувствительность контрольных клеток XP (клетки xeroderma pigmentosa, трансфецированные pZipneoSV(X)1) (Fan и др., Nucleic Acids Res., 18, стр. 5723, 1990 г.), экспримирующих почти не определяемое содержание АТазы, к темозоломиду в 4 - 5 раз превышает чувствительность трансфецированных кДНК АТазы клеток человека к темозоломиду (см. табл. 1). В опыте, включающем образование колоний, по цитотоксичности темозоломида (см. фиг. 2, показывающую кривую по цитотоксичности темозоломида в клеточных линиях, трансфецированных pZipneoSV(X)1. (○,□) или phAT (Δ,▿) и производных от XP, в отсутствии (○,▿) БГ соответственно присутствии (□,Δ) 10 мкмоль БГ, знаки ошибки указывают на стандартное отклонение +/-1) предварительная обработка БГ привела к подобной степени потенциации клеток XP человека, трансфецированных АТазой, как в опухолевых клетках, но не имела измеримого эффекта на контрольные клетки XP, не экспримирующие АТазу. Хотя БГ снизил активность АТазы в трансфецированных клетках (см. ниже), они остались более устойчивыми к темозоломиду, чем контрольные фибробласты, трансфецированные pZip.
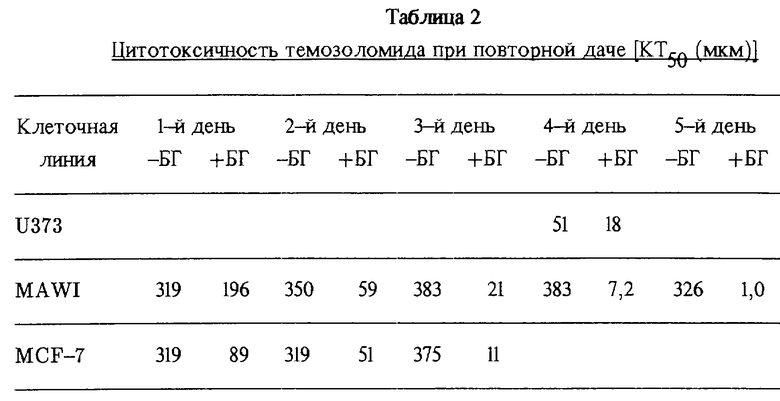
Данные, полученные при режиме повторной дачи, указаны ниже в табл. 2.
Перед дачей повторных ежедневных доз темозоломида клетки в тканевых культурах подвергали воздействию О6-бензилгуанина (БГ).
Схема повторной дачи привела к сильной потенциации токсичности темозоломида в клетках MAWI и MCF-7 (см. также фиг. 3, показывающую кривую соотношения цитотоксичности ежедневно повторяющихся доз темозоломида в опухолевых клеточных линиях человека MAWI (х), MCF-7 (▿) и U373 (#) при даче лишь самого лекарства (KT50', +БТ) по сравнению с предварительной инкубацией вместе с БГ (KT50', +БГ).
После дачи пяти доз, каждой на 24 часа, чувствительность клеточной линии MAWI к воздействию темозоломида имела более, чем 300-кратную величину в присутствии БГ. При повторной даче лишь темозоломида токсичность не больше, чем при одной единственной дозе в течение 24 часов. В подобном опыте на клетках U373, имеющих нижнее содержание АТазы, наличие БГ привело лишь к трехкратному увеличению после четырех доз по 24 часа.
Содержание алкилтрансферазы
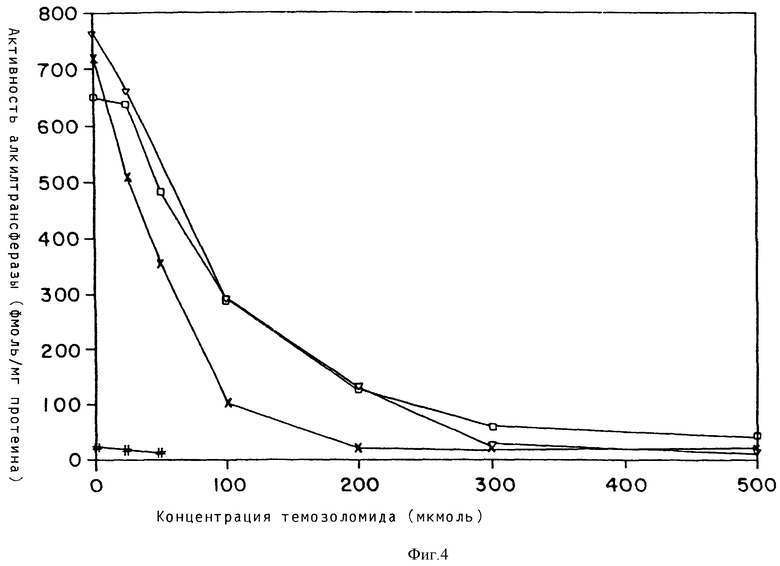
Используемые количества БГ быстро снизили первоначально большое содержание АТазы в клетках MAWI и трансфецированных кДНК АТазы фибробластах XP человека до необнаружимого уровня. В результате жидкостной хроматографии высокого разрешения оказалось, что БГ явился устойчивым в культурной среде ткани по меньшей мере в течение 24 часов при температуре 37oC.
Оказалось также, что после инкубации в течение трех часов темозоломид приводит к уменьшению содержания АТазы в клеточных линиях U373, MCF-7, LOVO и MAWI. В каждой клеточной линии при концентрации 50 - 100 мкмоль наблюдалось 50%-ное снижение (см. фиг. 4, на которой показана кривая, указывающая влияние растущей концентрации темозоломида на содержание АТазы в опухолевых клеточных линиях человека: LOVO (□) , MAWI (х), MCF-7 (▿) , U373 (#), несмотря на 3 - 4-кратную разницу цитотоксичности темозоломида при даче отдельной дозы между MCF-7 и клеточными линиями толстой и прямой кишок (LOVO и MAWI)). Подобное снижение наблюдалось в более чувствительной линии U373, хотя содержание АТазы находилось на грани обнаружимости опыта.
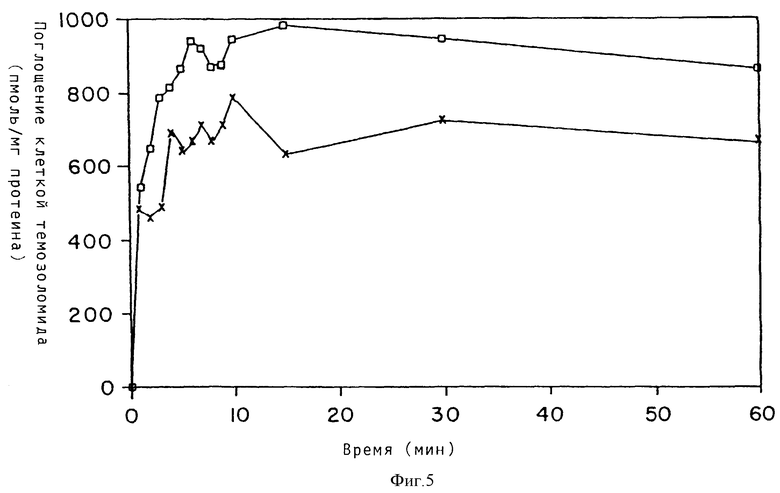
Для исключения возможности разницы в транспорте темозоломида исследовали поглощение [14C]-маркированного соединения наиболее чувствительными и наиболее устойчивыми клеточными линиями [ZR-75-1 (х) соответственно MAWI (□) ]. Результаты, приведенные на фиг. 5, показывают, что поглощение быстро осуществлялось при температуре 4oC, и в обеих клеточных линиях оно завершалось в течение пяти минут. В обеих клеточных линиях обнаружили подобное количество лекарства при исследовании концентрации протеина. Быстрое поглощение при температуре 4oC соответствовало пассивной диффузии темозоломида, ранее доказанному в двух лимфоидных клеточных линиях (Sull и др., Biochem. Pharmacol., 36, стр. 3215, 1987 г.).


Фармацевтическая композиция с антиопухолевой активностью содержит в качестве активного ингредиента темозоломид-8-карбамоил-3-метилимидазо[5,1-d] -1,2,3,5-тетразин-4-(3H)-он и ингибитор O6-алкилгуанин-ДНК-алкилтрансферазы в соотношении от 1 : 1 до 1 : 20000. Фармацевтическая композиция также содержит, по меньшей мере, один фармацевтически приемлемый носитель. Ингибитор выбран из группы, включающей O6-алкилгуанины, O6-арилгуанины и O6-бензилированные гуанины, гуанозины и 2'-деоксигуанозины. Способ получения фармацевтической композиции включает смешение темозоломида с ингибитором O6-алкилгуанин-ДНК-алкилтрансферазы и фармацевтически приемлемым носителем. Композиция обеспечивает повышенную антиопухолевую активность. 2 с. и 2 з.п. ф-лы, 5 ил., 2 табл.
| NEWLANDS ET AL., Br.J.CANCER, 65, 1992, p.287 | |||
| Огнетушитель | 0 |
|
SU91A1 |
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, ч.2, 1986, с.453-456. | |||

