Настоящее изобретение относится к производным глутаминовой кислоты, которые ингибируют фермент глицинамидрибонуклеотид-формилтрансферазу (GARFT), к способу ингибирования GARFT и к способу ингибирования роста и пролиферации клеток высших организмов или микроорганизмов таких, как бактерии, дрожжи и грибы. Такие действия включают противоопухолевую, противовоспалительную, противопсориазную и иммуноподавляющую активность.
Большой класс антипролиферативных средств включает антиметаболические соединения. Конкретный подкласс антиметаболитов, известных как антифолаты и антифолы, является антагонистом витаминной фолиевой кислоты. Обычно антифолаты имеют большое сходство по структуре с фолиевой кислотой и включают характерную P-бензоилглутаматную часть фолиевой кислоты. Глутаматная часть фолиевой кислоты принимает двойной отрицательный заряд при физиологическом значении pH. Поэтому это соединение и его аналоги обладают активной энергией, направляющей транспортную систему через клеточную мембрану, и оказывают метаболическое действие.
Глицинамидрибонуклеотид-формилтрансфераза (GARFT) является фолатзависимым ферментом в метаболическом пути биосинтеза пурина de novo. Этот путь является критическим для деления и пролиферации клеток. Известно, что остановка этого метаболического пути оказывает антипролиферативное действие, в частности, противоопухолевое действие. Поэтому был синтезирован ряд аналогов фолатов и изучена их способность ингибировать GARFT. Сообщалось, что прототипный, специфически, крепко связывающий ингибитор GARFT, 5,10-дидезазатетрагидрофолиевая кислота, обладает противоопухолевой активностью. Смотри F. M. Muggia, "Folat antimetabolites inhibitory to de novo purine synthesis" в New Drugs, Concepts and Results in Cancer Chemotherapy, p. 65 - 87, Kluwer Academic Peblishers, Boston (1991).
Настоящее изобретение относится к новому классу соединений, содержащих остаток глутаминовой кислоты или ее эфира. Эти соединения эффективны при ингибировании фермента глицинамидрибонуклеотид-формилтрансферазы (GARFT) и роста и пролиферации клеток высших организмов и микроорганизмов таких, как бактерии, дрожжей и грибов. Изобретение относится также к способам, использующим эти соединения в качестве ингибиторов фермента.
Как указано выше, соединение по изобретению обладают антипролиферативной активностью, свойством, которое может выражаться в виде противоопухолевой активности. Соединение по изобретению может быть активно само по себе или может быть предшественником, который превращается in vivo в активное содинение. Предпочтительные соединения изобретения активны при ингибировании фермента GARFT. В частности, предпочтительные соединения активны при ингибировании роста клеточной линии L1210, клеточной линии мышиной лейкемии, которую можно вырастить в тканевой культуре. Соединения по изобретению должны быть активны также при ингибировании роста бактерий таких, как грамотрицательных бактерий Escherichia coli, которые можно выращивать в культуре.
Другой аспект настоящего изобретения относится к терапевтическому способу ингибирования роста и пролиферации клеток высших организмов или микроорганизмов, который предусматривает введение хозяину эффективного количества соединения настоящего изобретения. Соединения изобретения, в частности, пригодны для лечения млекопитающих таких, как человек, и для лечения птиц. В частности, предпочтительный терапевтический способ предусматривает введение хозяину эффективного количества соединения по настоящему изобретению для ингибирования GARFT.
Многие из антипролиферативных лекарственных средств, описанных здесь, или их фармацевтически приемлемые соли можно использовать в способе терапии по изобретению. Соединения можно вводить в виде фармацевтически приемлемой композиции, содержащей разбавитель или носитель такой, как описанный выше.
Дозы соединений предпочтительно включают фармацевтические дозированные единицы, содержащие эффективное количество активного соединения. Термин "эффективное количество" обозначает количество, достаточное для ингибирования фолатных метаболических путей и достижения благоприятного действия при введении одной или нескольких фармацевтических дозированных единиц.
Примерная суточная дозированная единица для позвоночного хозяина содержит до одного грамма активного соединения на килограмм массы тела хозяина, предпочтительно полграмма, более предпочтительно 110 мг и наиболее предпочтительно около 50 мг или менее на килограмм массы тела хозяина. Выбранную дозу можно вводить теплокровному животному или млекопитающему, например человеку, нуждающемуся в лечении посредством ингибирования фолатных метаболических путей, любым известным методом введения дозы, включая местным образом, например, мазь или крем; перорально, ректально, например, в виде суппозиторий; парентерально инъекцией или длительно интравагинальным, внутриназальным, внутрибронхиальным, внутриушным или внутриглазным введением.
Соединения по настоящему изобретению можно охарактеризовать как проявляющие, одно или более, антипролиферативное действие, антибактериальное действие, антипаразитарное действие, антивирусное действие, антипсориазное действие, антипротозойное действие, антикокцидиальное действие, антивоспалительное действие, иммунодепрессивное действие или антигрибковое действие. Эти соединения особенно пригодны при проявлении протиоопухолевого действия у позвоночного хозяина, имеющего опухоль.
Настоящее изобретение относится к антипролиферативным соединениям, способным ингибировать GARFT и представленным [2-[4-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо[5,4-b] [1,4] тиазин-6-ил)этил] (бензоил или тиенилкарбонил)амино]пентадиовой кислотой формулы Ia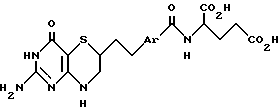
где Ar обозначает фенил, тиенил или тиенил, замещенный низшим алкилом;
или ее низшими алкиловыми эфирами.
Предпочтительно Ar выбран из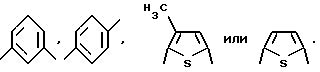
В частности, предпочтительно Ar представлен группой
и соединение является d-(2-[4-[2-(2-амино-4-оксо-4,6,7,8- тетрагидро-3H-пиримидо[5,4-b][1,4]тиазин-6-ил)этил)бензоиламино пентадиовой кислотой.
Предпочтительно также, что соединением является диэтиловый эфир d-(2-[4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо [5,4-b] [1,4]-тиазин-6-ил)этил]бензоиламино]пентадиовой кислоты.
Следует отметить, что применение указаний настоящего изобретения к конкретной проблеме или среде будет в пределах способности специалиста в данной области в свете содержащихся в изобретении указаний. Примеры продуктов настоящего изобретения и представительный способ их получения и выделения показаны в следующих примерах.
Пример 1. Стадия 1а. (1) 1,1-Диметоксибут-3-ин-2-ол
В перемешиваемый раствор 1,037 г (10,55 ммоль) триметилсилилацетилена в 50 мл сухого тетрагидрофурана (ТГФ) в атмосфере аргона при -78oC добавляют по каплям 6,6 мл 1,6 М н-бутиллития. Через 10 мин при -78oC добавляют по каплям раствор 1.21 г (10.46 ммоль) диметилацеталя глиоксаля в 5 мл тетрагидрофурана (ТГФ). После выдерживания 1 час при -78oC реакцию гасят 1 мл H2O и оставляют для нагревания до комнатной температуры, разбавляют этилацетатом и промывают насыщенным раствором NaCl. Водный слой снова экстрагируют этилацетатом и объединенный органический слой сушат над MgSO4 и концентрируют при пониженном давлении. К 785 мг полученного желтого масла, растворенного в тетрагидрофуране (ТГФ), добавляют 5.8 мл 1.0 М раствора фтористого тетрабутиламмония в тетрагидрофуране (ТГФ). После нагревания в течение 1 часа при 50oC летучую часть выпаривают и остаток очищают флаш-хроматографией на силикагеле при элюировании смесью метиленхлорида и этилацетата (9 : 1). Таким образом получают 412 мг (общий выход 60%) алкинового спирта, соединение (1), в виде бесцветного масла. ИК-спектр (соединение неразбавлено): 3441 (широкая), 3277, 2944, 2839, 1636, 1450, 1196, 1084 см-1.
1H ЯМР-спектр (CDCl3) δ 2.42 (широкий, 1H), 2.49 (с, 1H), 3.51 (с, 3H), 3.53 (с, 3H), 4.36 (широкий, 2H).
Элементный анализ:
Рассчитано для C6H10O3 • 0.35 H2O: C 52.81; H 7.90.
Найдено: C 52.87; H 7.86.
Стадия 1b
(2) Метиловый эфир 4-(3-гидрокси-4,4-диметоксибут-1-инил)-бензойной кислоты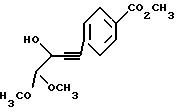
К перемешиваемому раствору 232 мг (1.78 ммоль) алкинового соединения (1) и 467 мг (1.78 ммоль) метил-4-иодбензоата в 5 мл диэтиламина добавляют 13 мг (0.18 ммоль) бис(трифенилфосфин)палладий (II) хлорида и 7 мг (0.036 ммоль) иодида меди (I). После выдерживания 15 час при комнатной температуре летучую часть удаляют при пониженном давлении и остаток очищают флаш-хроматографией на силикагеле при элюировании смесью метиленхлорида и этилацетата (12 : 1). Таким образом получают 347 мг (74%) соединения (2) в виде оранжевого масла.
ИК-спектр (соединение неразбавлено): 3451 (широкая), 2953, 2838, 1717, 1607, 1437, 1310, 1283, 1120, 1082 см-1.
1H ЯМР-спектр (CDCl3) δ 2.46 (широкий, 1H), 3.55 (с, 3H), 3.56 (с, 3H), 3.92 (с, 3H), 4.45 (д, 1H, J = 5.4 Гц), 4.60 (д, 1H, J = 5.3 Гц), 7.52 (д, 2H, J = 8.3 Гц), 7.98 (д, 2H, J = 8.3 Гц).
Элементный анализ:
Рассчитано для C14H16O5: C 63.62; H 6.10.
Найдено: C 63.14; H 6.14.
Стадия 1c
(3) Метиловый эфир 4-(3-гидрокси-4,4-диметоксибутил)бензойной кислоты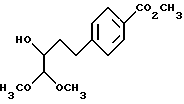
Раствор, содержащий 14.37 г (54.38 моль) соединения (2) и 1.40 г 5% Pd на угле в 175 мл этанола, гидрируют при давлении водорода 275790.28 Па в аппарате Парра. Через 2.5 часа реакционную смесь фильтруют и катализатор промывают этанолом и метанолом. После концентрирования при пониженном давлении остаток растворяют в хлористом метилене и фильтруют через небольшой слой силикагеля при элюировании метиленхлоридом, затем смесью метиленхлорида и этилацетата (1 : 1) для удаления остаточного угля. Таким образом получают 14.34 г (98%) насыщенного спирта, соединения (3), в виде желтого масла.
ИК-спектр (соединение неразбавлено): 3495 (широкая), 2953, 1721, 1611, 1437, 1283, 1109, 1080 см-1.
1H-ЯМР-спектр (CDCl3) δ 1.72 - 1.93 (м, 2), 2.75 - 2.93 (м, 2H), 3.39 (с, 3H), 3.44 (с, 3H), 3.58 (с, 1H), 3.90 (с, 3H), 4.13 (д, 1H, J = 6.1 Гц), 7.29 (д, 2H, J = 8.1 Гц), 7.95 (д, 2H, J = 8.2 Гц).
Элементный анализ:
Рассчитано для C14H20O5 • 0.20 H2O: C 61.84; H 7.56.
Найдено: C 61.83; H 7.57.
Стадия 1d
(4) Метиловый эфир 4-(3-метансульфонилокси-4,4-диметоксибутил)-бензойной кислоты
К перемешиваемому раствору 206 мг (0.77 ммоль) насыщенного спирта, соединения (3) и 0.16 мл (1.15 ммоль) триэтиламина в 5 мл хлористого метилена при 0oC добавляют 0.07 мл (0.085 ммоль) метансульфонилхлорида. После выдерживания 20 мин при 0oC добавляют еще 0.02 мл метансульфонилхлорида. Еще через 30 мин реакционную смесь выливают в насыщенный раствор NaHCO3 и экстрагируют дважды метиленхлоридом. Объединенные органические слои сушат над MgSO4 и растворитель удаляют при пониженном давлении. Это соединение было достаточно чистым для применения в следующей стадии. Аналитическую пробу получают очисткой флаш-хроматографией на силикагеле при элюировании смесью метиленхлорида и этилацетата (20 : 1). Таким образом получают мезилат, соединение (4), в виде бесцветного масла.
ИК-спектр (соединение неразбавлено): 2949, 2839, 1719, 1611, 1437, 1352, 1283, 1177, 1109, 1078 см-1.
1H-ЯМР-спектр (CDCl3) δ 2.04 (м, 2H), 2.77 - 2.91 (м, 2H), 3.09 (с. 3H), 3.41 (с, 3H), 3.45 (с. 3H), 3.90 (с, 3H), 4.38 (д, 1H, J = 5.5 Гц), 4.64 (м, 1H), 7.29 (д, 2H, J = 8.2 Гц), 7.96 (д, 2H, J = 8.2 Гц).
Элементный анализ:
Рассчитано для C15H22O7S: C 52.01; H 6.40; S 9.26.
Найдено: C 52.08; H 6.44; S 9.25.
Стадия 1e
(5) Метиловый эфир (4-(3-метансульфонилокси-4- оксибутил)-бензойной кислоты
В перемешиваемый раствор 600 мг (1.73 ммоль) диметилацеталя (мезилата) (соединение 4) в 5 мл хлористого метила при 0oC добавляют 1 мл H2O и 1 мл трифторуксусной кислоты. Реакционную смесь нагревают до комнатной температуры и затем кипятят 24 часа с обратным холодильником. Охлажденную реакционную смесь разбавляют этилацетатом и дважды последовательно промывают насыщенным раствором NaCl, насыщенным раствором NaHCO3, затем снова насыщенным раствором NaCl, сушат (MgSO4) и летучие продукты удаляют при пониженном давлении. Таким образом получают мезилат, соединение (5), которое применяют без очистки.
ЯМР-спектр (CDCl3) δ 2.20 (м, 2H), 2.85 (м, 2H), 3.17 (с, 3H), 3.91 (с, 3H), 4.95 (дд, 1H, J = 4.2 Гц, 8.4 Гц), 7.29 (д, 2H, J = 8.1 Гц), 7.99 (д, 2H, J = 8.2 Гц), 9.59 (с, 1H).
Стадия 1f
(6) Метиловый эфир 4-[3-(4-метоксибензилсульфанил)-4-оксо-бутил] бензойной кислоты
В перемешиваемый раствор 686 мг (2.28 моль) мезилата, соединения (5) и 0.40 мл (2.29 моль) N,N-диизопропилэтиламина в диметилформамиде (ДМФ) добавляют 0.48 мл (3.44 моль) 4-метокси -α- толуолтиола. После выдерживания 3 часа при комнатной температуре реакционную смесь выливают в 0.5 Н HCl и экстрагируют два раза этилацетатом. Объединенные органические слои промывают два раза насыщенным раствором NaCl, сушат (MgSO4) и концентрируют при пониженном давлении. Полученный альдегид, соединение (6), достаточно чистый для применения без дальнейшей очистки в следующей стадии.
ИК-спектр (KBr) 2930, 1715, 1703, 1611, 1512, 1282, 1244, 1107 см-1.
1H-ЯМР-спектр (CDCl3) δ 1.80 - 2.16 (м, 2H), 2.75 (м, 2H), 2.99 (м, 1H), 3.53 (AB, 2H, J = 13.4 Гц), 3.81 (с, 3H), 3.90 (с, 3H), 6.83 (д, 2H, J = 8.5 Гц), 7.12 (д, 2H, J = 8.2 Гц), 7.19 (д, 2H, J = 8.6 Гц), 7.90 (д, 2H, J = 8.2 Гц), 9.27 (д, 1H, J = 4.2 Гц).
Стадия 1g
(7) Метиловый эфир 4-[3-[1,3]диоксолан-2-ил-3-(4- метоксибензилсульфанил)пропил]бензойной кислоты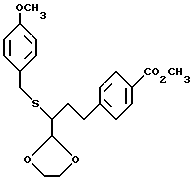
Сосуд, содержащий 301 мг (0.84 ммоль) альдегида, соединения (6), 94 мкл (1.68 ммоль) этиленгликоля и 42 мг (0.17 ммоль) п-толуолсульфоната пиридиния и 30 мл бензола, нагревают при кипячении с обратным холодильником, удаляя образуемую воду при помощи ловушки Дина-Старка. Через 3 часта реакционную смесь выливают в насыщенный раствор NaCl и экстрагируют два раза этилацетатом. Объединенные органические слои сушат (MgSO4) и концентрируют при пониженном давлении. Остаток очищают флаш-хроматографией на диоксиде кремния, элюируя смесью гексанов и этилацетата (5:1). Таким способом получают указанное в заголовке соединение (7) с общим выходом 84% в расчете на диметилацеталь, соединение (4).
ИК-спектр (соединение неразбавлено): 2949, 2886, 1721, 1611, 1510, 1435, 1279, 1248, 1177, 1111, 1034 см-1.
1H-ЯМР-спектр (CDCl3) δ 1.67 (м, 1H), 1.99 (м, 1H), 2.57 (м, 2H), 2.84 (м, 1H), 3.72 - 4.03 (м, 6H), 3.81 (с, 3H), 3.90 (с, 3H), 4.97 (д, 1H, J = 4.6 Гц), 6.83 (д, 2H, J = 8.6 Гц), 7.09 (д, 2H, J = 8.1 Гц), 7.23 (д, 2H, J = 8.5 Гц), 7.88 (д, 2H, J = 8.1 Гц).
Элементный анализ:
Рассчитано для C22H26O5S: C 65.65, H 6.51, S 7.97
Найдено: C 65.72, H 6.50, S 8.07
Стадия 1h
(8) Метиловый эфир 4-(3-[1,3]диоксолан-2-ил-3-меркаптопропил)бензойной кислоты
В перемешиваемый раствор 5.70 г (14.16 ммоль) соединения (7) и 5.42 г (17.00 ммоль) ацетата ртути (II) в метиленхлориде, охлажденном до 0oC, добавляют по каплям 5 мл трифторуксусной кислоты. После выдерживания 3 часа при 0oC добавляют насыщенный раствор сероводорода в метаноле и перемешивание продолжают при 0oC в течение 20 мин. Реакционную смесь выливают в насыщенный раствор NaCl и экстрагируют два раза хлористым метиленом. Объединенный органический слой сушат (MgSO4) и концентрируют при пониженном давлении. К сырому остатку, частично суспендированному в метаноле, добавляют по частям 1.07 г (28.28 ммоль) боргидрида натрия. Через около 30 минут реакционную смесь выливают в этилацетат и 0.5 Н HCl и слои разделяют. Металлическую ртуть, которая образовалась в реакции, промывают этилацетатом. Объединенный органический слой промывают насыщенным раствором NaCl, сушат (MgSO4) и концентрируют при пониженном давлении. Остаток очищают флаш-хроматографией на силикагеле при элюировании смесью гексанов и этилацетата (4:1). Таким образом получают 1.99 г (50%) тиола, соединения (8), в виде светло-желтого масла.
ИК-спектр (соединение неразбавлено): 2951, 2886, 1719, 1611, 1435, 1281, 1179, 1144, 1111 см-1.
1H-ЯМР-спектр (CDCl3) δ 1.64 (д, 1H, J = 7.8 Гц), 1.74 (м, 1H), 2.15 (м, 1H), 2.80 (м, 2H), 3.00 (м, 1H), 3.90 (с, 3H), 3.97 (м, 4H), 4.91 (д, 1H, J = 4.0 Гц), 7.28 (д, 2H, J = 8.1 Гц), 7.95 (д, 2H, J = 8.1 Гц).
Элементный анализ:
Рассчитано для C14H18O4S: C 59.55, H 6.14
Найдено: C 59.42, H 6.41
Стадия 1i
(9) Метиловый эфир 4-[3-(2,4-диамино-6-оксо-1,6-дигидропиримидин- 5-илсульфанил)-3-[1,3]диоксолан-2-ил]бензойной кислоты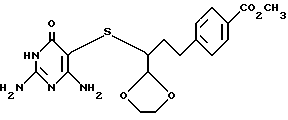
В перемешиваемый раствор 1.91 г (6.76 ммоль) тиола, соединения (8) и 1.39 г (6.78 ммоль) 5-бром-2,4-диамино-6-оксопиримидина в атмосфере аргона в дегазированном N, N-диметилформамиде добавляют 1.18 мл (6.77 ммоль) N,N-диизопропилэтиламина. Реакционную смесь нагревают при 90oC в течение 3 час. Охлажденную реакционную смесь выливают в насыщенный раствор NaCl и образованный осадок отделяют фильтрованием, промывают водой и сушат на воздухе. Фильтровальную лепешку диспергируют в метиленхлориде. Медленно добавляют гексаны и осадок снова отделяют фильтрованием, промывают гексанами и сушат. Таким образом получают 1.91 г (69%) целевого диоксолана, соединения (9), в виде несовсем белого твердого вещества с т.пл. 206 - 208oC (разложение).
ИК-спектр (KBr): 3439, 3341, 3154, 1701, 1636, 1591, 1470, 1447, 1287 см-1.
1H-ЯМР-спектр (DMSO) δ 1.64 (м, 1H), 1.84 (м, 1H), 2.58 (м, 1H), 2.79 (м, 1H), 3.19 (м, 1H), 3.82 (с, 3H), 3.85 (м, 4H), 4.83 (д, 1H, J = 4.3 Гц), 6.33 (ушир с, 4H), 7.33 (д, 2H, J = 8.1 Гц), 7.83 (д, 2H, J = 8.1 Гц), 10.03 (с, 1H).
Элементный анализ:
Рассчитано для C18H22N4O5S: C 53.19, H 5.46, N 13.78, S 7.89
Найдено: C 52.98, H 5.53, N 13.60, 7.76
Стадия 1j
(10) Метиловый эфир 4-[2-(2-амино-7-гидрокси-4-оксо-4,6,7,8- тетрагидро-3H-пиримидо[5,4-b][1,4]тиазин-6-ил)этил]бензойной кислоты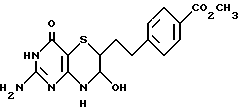
Перемешиваемую суспензию 1.20 г (2.85 ммоль) диоксолана, соединение (9), и 4 мл 2 Н HCl в 20 мл тетрагидрофурана (ТГФ) нагревают при кипячении с обратным холодильником 2.5 часа. Гомогенный раствор выливают медленно в насыщенный раствор NaHCO3 и образованный осадок отделяют. Фильтр экстрагируют этилацетатом. Осадок (58 мг), образованный между слоями, отделяют и объединяют с первым осадком. Этилацетатный слой сушат над MgSO4 и растворитель удаляют при пониженном давлении. Остаток (43 мг) также объединяют с первым осадком. Таким образом получают 984 (98%) аминоспирта, соединение (10), в виде оранжевого твердого вещества: т.пл. 213 - 216oC.
ИК-спектр (KBr): 3351, 3441, 1705, 1638, 1609, 1557, 1470, 1289, 1113, 1020 см-1.
1Н-ЯМH-спектр одной пары диастереомеров (DMSO) δ 1.39 и 1.96 (м, м, 1H), 1.70 (м, 1H), 2.56 - 2.89 (м, 3H), 3.82 (с, 3H), 4.71 и 4.84 (м, м, 1H), 5.37 и 5.40 (д, д, 1H, J = 6.6 Гц), 6.06 (с, 2H), 7.20 (д, 1H, J = 4.5 Гц), 7.31 и 7.36 (д, д, 2H, J = 8.1 Гц), 7.86 и 7.88 (д, д, 2H, J = 8.0 Гц), 10.16 и 10.19 (с, с, 1H).
Элементный анализ:
Рассчитано для C16H18N4O4S • 1.7 H2O; C 48.89, H 5.49, N 14.26, S 8.16
Найдено: C 48.78, H 5.18, N 14.00, S 8.03
Стадия 1k
(11) Метиловый эфир 4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо[5,4-b] [1,4]тиазин-6-ил]бензойной кислоты
При 0oC в суспензии 1.126 г (3.1 ммоль) аминоспирта, соединения (10), в тетрагидрофуране (ТГФ) добавляют 2.3 мл (18.64 ммоль) эфирата трехфтористого бора. После окончания этого добавления добавляют по частям 0,586 г (9.32 ммоль) цианоборогидрида натрия в течение 5 мин. Через 30 мин после добавления прибавляют 5 мл насыщенного раствора аммиака в метаноле, реакционную смесь разбавляют этилацетатом и промывают насыщенным раствором NaCl. Органический слой сушат (MgSO4) и растворитель удаляют при пониженном давлении. Остаток очищают флаш-хроматографией на диоксиде кремния при элюировании смесью хлористого метилена и метанола (9:1). Таким образом получают 542 мг (50%) дегидратированного эфира, соединения (11), в виде оранжевого твердого вещества с т.пл. 245 - 246oC (разложение).
ИК-спектр (KBr): 3358, 2936, 1721, 1644, 1595, 1537, 1447, 1346, 1281 см-1.
1H ЯМР-спектр (DMSO) δ 1.72 (м, 1H), 1.90 (м, 1H), 2.80 (м, 3H), 3.22 (м, 1H), 3.52 (м, 1H), 3.82 (с, 3H), 6.00 (с, 2H), 6.65 (с, 1H), 7.37 (д, 2H, J = 8.1 Гц), 7.87 (д, 2H, J = 8.1 Гц), 10.05 (с, 1H).
Элементный анализ:
Рассчитано для C16H18N4O3S: C 55.47, H 5.24, N 16.17, S 9.26
Найдено: C 55.31, H 5.29, N 16.09, S 9.17
Стадия 11
(12) 4-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо-[5,4-b] [1,4] тиазин-6-ил)этил)бензойная кислота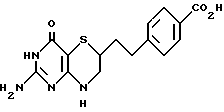
Раствор 530 мг (1.53 моль) эфира, соединения (11), и 10 мл и 1 H раствора NaOH перемешивают при комнатной температуре 30 мин. Гомогенный раствор осторожно подкисляют (pH 4) концентрированной HCl. После охлаждения на ледяной бане слабо-оранжевый осадок отделяют фильтрованием и сушат на воздухе, Осадок затем суспендируют в этаноле и этанол затем удаляют при пониженном давлении. Таким образом получают 468 мг (91%) кислоты, соединения (12), которая разлагается при температуре выше 310oC.
ИК-спектр (KBr): 3285, 3086, 2928, 1698, 1642, 1611, 1576, 1449, 1348 см-1.
1H ЯМР-спектр (DMSO) δ 1.72 (м, 1H), 1.89 (м, 1H), 2.78 (м, 3H), 3.20 (м, 1H), 3.48 (м, 1H), 6.07 (с, 2H), 6.68 (с, 1H), 7.33 (д, 2H, J = 8.1 Гц), 7.85 (д, 2H, J = 8.1 Гц), 10.11 (с, 1H), 12.77 (ушир. с, 1H).
Элементный анализ:
Рассчитано для C15H16N4O3S • 1.20 H2O: C 50.89, H 5.24, N 15.83, S 9.06
Найдено: C 50.70, H 4.92, N 15.58, S 8.87
Стадия 1m
(13) Диэтиловый эфир 2-[4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо[5,4-b] [1,4]тиазин-6-ил)этил]бензоиламино]-пентандиовой кислоты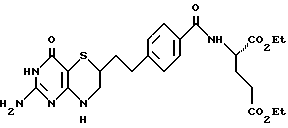
В перемешиваемый раствор 397 мг (1.19 ммоль) кислоты, соединения (12), 169 мг (1.25 ммоль) гидрата 1-гидроксибензотриазола (HOBT), 0.22 мл (1.25 ммоль) N, N-диизопропилэтиламина и 300 мг (1.25 ммоль) гидрохлорида диэтилового эфира L-глутаминовой кислоты в 15 мл N,N-диметилформамида добавляют 240 мг (1.25 ммоль) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC). После выдерживания 18 час при комнатной температуре реакционную смесь выливают в охлажденный льдом насыщенный раствор NaCl и образованный осадок отделяют, промывают водой и сушат на воздухе. Осадок очищают флаш-хроматографией на силикагеле при элюировании смесью метиленхлорида и метанола (9: 1). Таким образом получают 357 мг (58%) целевого продукта (13) в виде слабо-оранжевого твердого вещества с т.пл. 132 - 136oC.
ИК-спектр (KBr): 3333, 1732, 1645, 1572, 1535, 1449, 1343, 1203, 1020 см-1.
1H-ЯМР-спектр (DMSO) δ 1.15 (т, 3H, J = 7.3 Гц), 1.17 (т, 3H, J = 7.3 Гц), 1.72 (м, 1H), 1.88 - 2.10 (м, 3H), 2.42 (т, 2H, J = 7.4 Гц), 2.79 (м, 3H), 3.22 (м, 1H), 3.50 (м, 1H), 4.02 (кв, 2H, J = 7.3, 14.5 Гц), 4.09 (кв, 2H, J = 7.2, 14.3 Гц), 4.41 (м, 1H), 6.21 (с. 2H), 6.74 (с, 1H), 7.32 (д, 2H, J = 8.0 Гц), 7.80 (д, 2H, J = 8.0 Гц), 8.64 (д, 1H, J = 7.41 Гц), 10.24 (с, 1H).
Элементный анализ:
Рассчитано для: C24H31N5O6S:
C 55.69, H 6.04, N 13.53, S 6.19
Найдено: C 55.41, H 6.11, N 13.48, S 6.12
Стадия 1n
(14) 2-[4-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо-[5,4-b] [1,4]тиазин-6-ил)этил]бензоиламино]пентандиовая кислота
Смесь 320 мг (0,618 ммоль) глутамата, соединения (13), и 6 мл 1 H раствора NaOH перемешивают при комнатной температуре 3 часа, нейтрализуют концентрированной HCl, затем слегка подкисляют 2 H HCl. После охлаждения слабо-желтый осадок собирают и сушат на воздухе. Фильтровальную лепешку переносят в смесь этанола и ацетонитрила и остаточное количество воды удаляют азеотропной перегонкой. Таким образом получают 220 мг (77%) дикислоты, соединение (14), с. т.пл. 188 - 190oC.
ИК-спектр (KBr): 3348 (широкая), 2930, 1717, 1642, 1539, 1505, 1348 см-1.
1H ЯМР-спектр (DMSO) δ 1.71 (м, 1H), 1.92 (м, 2H), 2.08 (м, 1H), 2.34 (т, 2H, J = 7.4 Гц), 2.79 (м, 3H), 3.20 (м, 1H), 3.55 (м, 1H), 4.38 (м, 1H), 6.07 (с, 2H), 6.68 (с, 1H), 7.31 (д, 2H, J = 8.1 Гц), 7.80 (д, 2H, J = 8.2 Гц), 8.53 (д, 1H, J = 7.7 Гц), 10.11 (с, 1H), 12.40 (ушир. с, 2H).
Элементный анализ:
Рассчитано для C20H23N5O6S • 1.5 H2O
C 49.17, H 5.36, N 14.34, S 6.56
Найдено C 48.77, H 4.97, N 14.0,7, S 6.54
Пример 2
Получение тиофеновых производных осуществляли по методу, частично основанному на способе, описанном для 5-тиатериновых кольцевых систем [Henris, R. N. , Lazarus R.A., Benkovic, S.J. Preparation of 2-Amino-4(3H)-oxopyrimido[5,4-b] [1,4] thiazines (5-Thiapterins) and Their Evaluation as Cofactors for Phenylalanine Hydroxylase. J.Med. Chem., 1983, 26, 559 - 563].
Способ получения тиофеновых производных может быть представлен следующей реакционной схемой.
Схема. Синтез пиримидотиазинов

Условия проведения реакций
(a) этиловый эфир бромтиофена, (Ph3P)2PdCl2, CuJ, Et3N, CH3CN;
(b) H2, 5% Pd/C, EtOH;
(c) TsOH, EtOH;
(d) TsCl, Et3N, CH2Cl2;
(e) NaN3, ДМФ или CH3CN, Δ;
(f) (BOC)2O, H2, 5% Pd/C, ТГФ;
(g) TBSCl, Et3N, CH2Cl2;
(h) MsCl, Et3N, CH2Cl2;
(i) TBAF, ТГФ;
(j) NaH, ТГФ;
(k) MsCl, Et3N, CH2Cl2;
(l) KSAc, ацетон;
(m) (CH3OCO)2CHCl, K2CO3, MeOH;
(n) ТФУ, CH2Cl2;
(o) (CH3)3O • BF4, CH2Cl2;
(p) гидрохлорид гуанидина, NaOEt, EtOH;
(q) NaOH;
(r) диэтиловый эфир L-глутамата, ЕДС, НОВТ, ДИЭА, ДМФ;
(s) NaOH.
По вышеприведенной схеме получали оптически чистые тиофеновые производные, исходя из известного оптически чистого ацетилена [Nemec, M., Janda, M. , Srogl, J. , Stibor, I. The Synthesis of 4-Substituted 2-Thiophenecarboxylic Acids. Collect. Czech. Chem. Commun., 1974, 89, 3527-3581], полученного из чистого D(R)-глицеральдегидацетонида [Schmid, C.R., Bryant, J.D., D-(R)-Glyceraldehyde Acetonide. Org. Synth., 1993, 72, 6-13].
Процесс начинали со стадии катализируемого палладием присоединения к оптически чистой форме ацетилена 5 как этилового эфира 2-бромтиофен-5-карбоновой кислоты, так и 3-метильного производного с последующим восстановлением и гидролизом кислотой с получением диольных промежуточных соединений 4a или 4b. Для соединений C-6(R) атом азота в положении 8 вводили путем монотозилирования первичного спирта с последующим замещением с помощью азида натрия. Для соединений C-6(S) гидроксильную группу преобразовали путем защиты первичного спирта в виде силильного эфира, мезилирования вторичного спирта, удаления защитной группы и образования обращенного эпоксида с помощью гидрида натрия. Эпоксид раскрывали селективно по первому положению, где это менее затруднено, с помощью азида натрия. Азидоспирты восстанавливали в присутствии BOC-ангидрида с получением спиртов 3a-d. Атом серы в 5 положение вводили путем замещения соответствующего мезилата с помощью KSAc. Ацетильную защитную группу удаляли в щелочных условиях в присутствии диметилхлормалоната, и когда защитная группа БОК удалялась, свободный амин самопроизвольно циклизировался в лактамы 2a-d. Лактам алкилирования с помощью триметилоксонийтетрафторбората и полученный лактамный эфир подвергали взаимодействию с 3 эквивалентами свободного основания гуанидина в этаноле при кипячении с обратным холодильником с получением 2-амино-4-(3H)-оксопиримидо[5,4-b][1,4]-тиазинов. Гидролиз этиловых эфиров, пептидное связывание и гидролиз завершали синтез глютаминовых кислот 1a-d.
Спектры 1H ЯМР определяли, используя обычный электрически спектрометр QE-300, работающий в поле с 300 МГц. Химический сдвиг представляли в миллионных долях (δ) и устанавливали стандарты для сравнения так, что в CDCl3 CHCl3 соответствует 7,26 м.д., а в ДМСО-d6 ДМСО соответствует 2,49 м.д. Разрешение стандартов и пиков обозначают следующим образом: с - синглет, д - дублет, дд - дублет дублетов, т - триплет, шир.с. - широкий синглет, шир.д - широкий дублет, шир. - широкий сигнал, м - мультиплет. Масс-спектр определяли в центре масс-спектрометрии научно-исследовательского института Скриппа. Инфракрасные спектры поглощения были получены на FTIR корпорации MIDAC. Элементные микроанализы выполняли с помощью Atlantic Microlab Inc., Norcroas, GA или MHW Laboratories, Phoenix, AZ и представляли результаты по установленным элементам с точностью ± 0,4% от теоретических значений. N,N-диметилформамид (ДМФ) сушили через активированные (250oC)  молекулярные сита. Тетрагидрофуран подвергали дистилляции из натрийбензофенонкетила в атмосфере азота. Et2O относится к диэтиловому эфиру, ДИЭА относится к диизопропилэтиламину, TBSCl относится к трет-бутилдиметилсилилхлориду, НОВТ относится к 1-гидроксибензотриазолгидрату и EDC относится к 1-(3-(диметиламино)пропил)-3-этилкарбодиимида гидрохлориду. Хроматографию проводили с использованием силикагеля 60 (Merck Art 9385). Тонкослойную хроматографию (ТСХ) проводили на предварительно приготовленных листках двуокиси кремния 60 F264 (Merck Art 5719). Температура плавления определены на аппарате MelTcMp и они даны не в точке.
молекулярные сита. Тетрагидрофуран подвергали дистилляции из натрийбензофенонкетила в атмосфере азота. Et2O относится к диэтиловому эфиру, ДИЭА относится к диизопропилэтиламину, TBSCl относится к трет-бутилдиметилсилилхлориду, НОВТ относится к 1-гидроксибензотриазолгидрату и EDC относится к 1-(3-(диметиламино)пропил)-3-этилкарбодиимида гидрохлориду. Хроматографию проводили с использованием силикагеля 60 (Merck Art 9385). Тонкослойную хроматографию (ТСХ) проводили на предварительно приготовленных листках двуокиси кремния 60 F264 (Merck Art 5719). Температура плавления определены на аппарате MelTcMp и они даны не в точке.
5-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо- [5,4-b][1,4]тиазин-8(R)-ил)этил] тиофен-2-карбоновая кислота: не совсем белое твердое вещество: т.пл. 283 - 285oC разл.; [α]589 + 71,0o (с = 0,60, 1н NaOH); ИК (KBr) 3256 (шир), 2942, 1707, 1641, 1612, 1464, 1364 см-1; 1H ЯМР (ДМСО-d6) δ 1.72 (м, 1H), 1.89 (м, 1H), 2.81 - 3.04 (м, 2H), 3.16 - 6.2 (м, 3H, частичное наложение H2O), 6.08 (с, 2H), 6.68 (с, 1H), 6.92 (д, 1H, J = 4,0 Гц), 7.52 (д, 1H, J = 3,7 Гц), 10,12 (с, 1H), 12.80 (шир., 1H). Анал. (C13H14N4O3S2 • 0,60 H2O) C, H, N, S.
2(S) [[[5-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо- [5,4-b] [1,4]тиазин-6-(R)-ил]тиофен-2-ил]карбонил]амино]- пентандикарбоновая кислота (1a): не совсем белое твердое вещество; т.пл. 191 - 194oC, пенится; [α]589 + 61,9o (с = 0.65, 1Н NaOH); ИК (KBr) 3389, 3235, 2924, 1701, 1624, 1545, 1340 см-1; 1H ЯМР (ДМСО-d6) δ 1.70 - 2.04 (м, 4H), 2.29 (д, 2H, J = 7,5 Гц), 2.90 (м, 2H), 3.13 - 3.53 (м, 3H, частично затемняется H2O), 4.29 (м, 1H), 6.30 (с, 2H), 6.77 (с, 1H), 6.89 (д, 1H, J = 3,7 Гц), 7.56 (д, 1H, J = 3,7 Гц), 8,50 (д, 1H, J= 3,7 Гц), 8.50 (д, 1H, J=8,1 Гц), 10.30 (шир., 1H). Анал. (C18H21N3O8S2) • 1,8 H2O) C, H, N, S.
5-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо [5,4-b] [1,4] тиазин-6(S)-ил)этил]тиофен-2-карбоновая кислота: не совсем твердое вещество; т. пл. 258 - 261oC разл.; [ α ]589 - 81,3o (c=0,63, 1H NaOH); ИК (KBr) 3254 (шир. ), 2918, 1692, 1685, 1453, 1352 см-1; 1H ЯМР (ДМСО-d6) δ 1.72 (м, 1H), 1.89 (м, 1H), 2.80 - 3.03 (м, 2H), 3.18 - 3.52 (м, 3H, частичное наложение H2O), 6.09 (с, 2H), 6.80 (с, 1H), 6.92 (д, 1H, J=3,7 Гц), 7.52 (д, 1H, J=3,7 Гц), 10.20 (с, 1H), 12.80 (шир. , 1H); BMC, вычислено для C13H14N4O5S2 (M+Na+) 361,0405, найдено 361,0390.
2(S) [[[5-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H- пиримидо[5,4-b][1,4] тиазин-6(S)-ил)этил] тиофен-2-ил]-карбонил]амино] пентандикарбоновая кислота (1b): не совсем белое твердое вещество; т.пл. 220oC разл.; [ α ]589 -57,1o (с= 0,61, 1H NaOH); ИК (KBr) 3363, 3094, 2926, 1711, 1641, 1605, 1559, 1454, 1400, 1333, 1279 см-1; 1H ЯМР (ДМСО-d6) δ 1.70 - 2.05 (м, 4H), 2.29 (т, 2H, J= 7,4 Гц), 2.87 (м, 2H), 3.15 - 3.48 (м, 3H, частичное наложение H2O), 4.29 (м, 1H), 6.05 (с, 2H), 6.66 (с, 1H), 6.89 (д, 1H, J=3,7 Гц), 7.65 (д, 1H, J= 3,7 Гц), 8.50 (д, 1H, J=7,7 Гц), 10.05 (с, 1H), 12.50 (шир., 2H). Анал. (C18H27N5O8S2 • 1,4H2O) C, H, N, S.
5-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо [5,4-b] [1,4] тиазин-6(R)-ил)этил] -4-метилтиофен-2-карбоновая кислота: не совсем белое твердое вещество: т. пл. 253oC разл.; [ α ]589+80,7o (с=0,29, 1H NaOH); ИК (KBr) 3339 (шир), 2922, 1641, 1589, 1451, 1346, 1269 см-1; 1H ЯМР (ДМСО-d6) δ 1.64 (м, 1H), 1.82 (м, 1H), 2.09 (с, 3H), 2.78 - 2.94 (м, 2H), 3.10 - 3.55 (м, 3H, частичное наложение H2O), 6.10 (с, 2H), 6.69 (с, 1H), 7.42 (с, 1H), 10.15 (с, 1H), 12.75 (шир. с, 1H); BMC, вычислено для C14H16N4O2S2 (M+Na+) 370,0002, найдено 375,0570.
2(S) [[[5-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо [5,4-b][1,4] тиазин-6(R)-ил)этил]-4-метилтиофен-2-ил]-карбонил]-амино] пентандикарбоновая кислота (1c): не совсем белое твердое вещество; т.пл. 210oC разл.; [ α ] 589+64,4o (с= 0,45, 1H NaOH); ИК (KBr) 3341 (шир.), 2928, 1701, 1638, 1536, 1449, 1340 см-1; 1H ЯМР (ДМСО-d6) δ 1.78 - 2.05 (м, 4H), 2.10 (с, 3H), 2.28 (т, 2H, J=7,0 Гц), 2.82 (м, 2H), 3.45 (м, 3H, частичное наложение H2O), 4.25 (м, 1H), 5.98 (с, 2H), 6.6 (с, 1H), 7.54 (с, 1H), 8.38 (д, 1H, J=7,7 Гц), 10.05 (с, 1H), 12.5 (шир.с, 2H). Анал. (C19H23N6O6S2 • 0,7 H2O) C, H, N, S.
5-[2-(2-Амино-4-(3H)-оксо-5,6,7,8-тетрагидропиримидо [5,6-b] [1,4] тиазин-6(S)-ил)этил] -4-метилтиофен-2-карбоновая кислота: [ α ]589-77,9o (с= 0,58, 1H NaOH); 1H ЯМР (ДМСО-d6) δ 1.62 - 1.73 (м, 1H), 1.79 - 1.92 (м, 1H), 2.12 (с, 3H), 2.81 - 2.89 (м, 3H), 3.16 - 3.26 (м, 1H), 3.50 - 3.58 (м, 1H), 6.21 (шир. с, 2H), 6.76 (шир.с, 1H), 7.45 (с, 1H), 10.24 (шир., 1H), 12.76 (шир., 1H); Анал. (C14H18N4O3S2 • 1,4H2O) C, H, N, S.
2(S) [[[5-[2-(2-Амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо [5,4-b][1,4] тиазин-6(S)-ил)этил]-4-метилтиофен-2-ил]- карбонил]-амино]пентандикарбоновая кислота (1d): [ α ]589- 36,8o (с=0,57, 1H NaOH); 1H ЯМР (ДМСО-d6) δ 1.61 - 1.72 (м, 1H), 1.76 - 1.92 (м, 2H), 1.99 - 2.08 (м, 1H), 2.12 (с, 3H), 2.31 (т, 2H, J=7,0 Гц), 2.79 - 2.94 (м, 3H), 3.17 - 3.28 (м, 1H), 3.49 - 3.56 (м, 1H), 4.30 (ддд, 1H, J=5,7, 7,7 Гц), 6.08 (с, 2H), 6.70 (с, 1H), 7.58 (с, 1H), 8.44 (д, 1H, J= 7,7 Гц), 10.12 (шир.с, 1H), 12.45 (шир. 2H). Анал. (C19H23N3O8S2 • 0,75H2O) C, H, N, S.
Пример 3
Биологическая и биохимическая оценка
Испытание in vitro
Рост клеток в присутствии соединений по настоящему изобретению оценивают с использованием клеточной линии мышиной лейкемии L1210 (ATCC CCL219). Клеточную линию выдерживают в среде RPMI 1640, содержащей 5% инактивированной нагреванием сыворотки зародыша теленка без антибиотиков.
Величины IC50 определяют в 160 мкл микрокультурах, содержащих 1500 (L1210) клеток и установленных в планшете с 96 лунками в питательной среде, дополненной 50 ME/мл пенициллина и 50 мкг/мл стрептомицина. Рост измеряют в течение 3 дней при непрерывном экспонировании с различными концентрациями испытываемого соединения (14), добавленного через 4 часа после посева клеток, по методике Mosmann MITT-тетразолий-восставновления (Immunol. Meth. 65, 55 - 63 (1983)), модифицированной по Alley et al. (Cencer Res. 48, 589 - 601 (1988)). Нерастворимые в воде соединения растворяют в диметилсульфоксиде и разбавляют до конечной концентрации 0,5% растворителя в клеточных культурах.
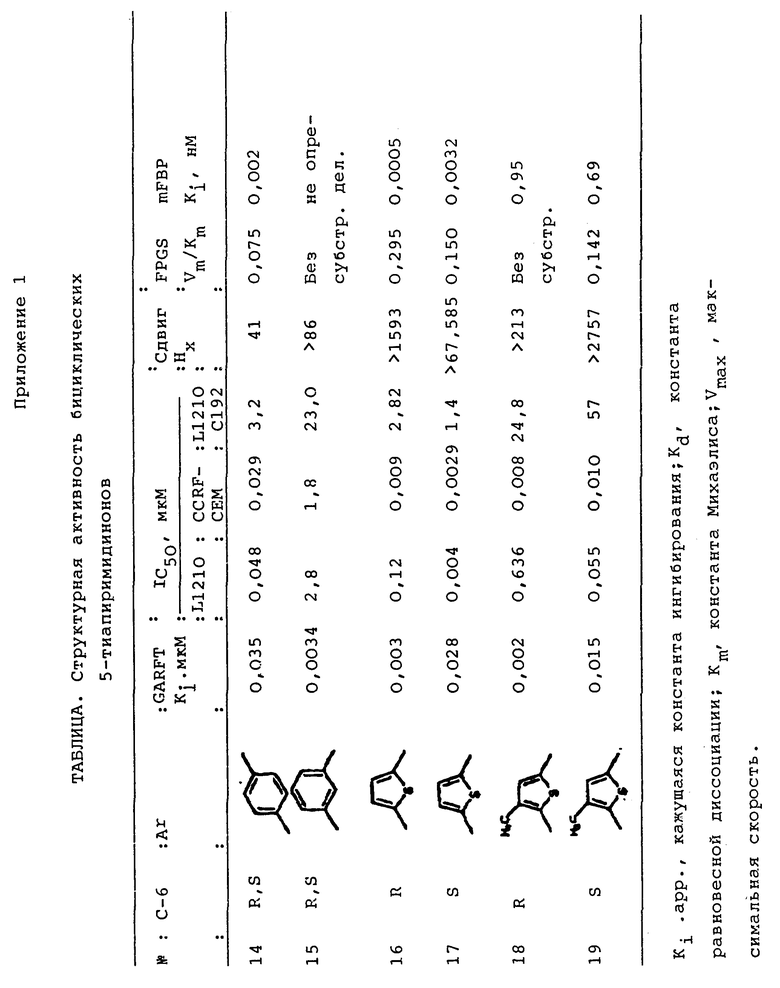
Определение констант ингибирования Gar-трансформилазы
Константу ингибирования GAR трансформилазы измеряют методом Cleland (Biochim. Biophys. Acta 67, 173 - 187 (1963)). Определения проводят при 22oC (начинают их добавлением фермента) путем спектрофотометрического анализа Young et al. (Biochemistry 23, 3979-3986 (1984)) и мониторинга реакции у 295 нм. Применяют GAR трансформилазный домен этого фермента человека. Изменяемым субстратом является 10-формил-5,8-дидезазафолат с концентрациями 0.83 мкМ, 1.25 мгМ, 2.5 мкМ и 5 мкМ тогда, как концентрацию другого субстрата, GAR (глицинамидрибонуклеотид), поддерживают постоянной на уровне 20 мкМ. Смесь для определения содержит 20 мкМ Hepes с pH 7.5, 20 мкМ GAR и изменяемые количества 10-формил-5,8-дидезазафолата и ингибитора. Ингибитор (14) применяют в виде пяти концентраций в пределах от 0 до приблизительно 3 Ki. Данные анализа наносят на график в виде зависимости скорости реакции от обратной концентрации 10-формил-5,6-дидезазафолата. Константу ингибирования определяют из снова построенного графика зависимости углов наклона этих линий, полученных для каждой концентрации ингибитора, от концентрации ингибитора.
Биохимические анализы. Активность GART измеряли с использованием модификации метода Young. Реакционные смеси содержали каталитический домен GART человека, 20 мкМ α, β- GAR, 10 или 20 мкМ FDDF, изменяемую концентрацию ингибитора GART 50 мМ ГЕПЕС-КОН, pH 7,5 и 50 мМ KCl. Реакцию инициировали добавлением фермента и затем контроль за реакцией проводили по поглощению y 294 нм при 20oC (ε294 = 18,9 мМ-1 см-1). Константу ингибирования GART(Ki) определяли из зависимости каталитической скорости в стационарной фазе от концентрации ингибитора и субстрата. На основании зависимости кажущейся Ki (Ki•app. ) от концентрации FDDF установили, что тип исследуемого ингибирования конкурентный относительно FDDF, и показали, что он описывается следующим образом: Ki•app.=Ki+(Ki/Km)[FDDF]. Константу Михаэлиса для FDDF,Km определяли независимо на основании зависимости каталитической скорости от концентрации FDDF, было показано, что она составляет 0,6 мкМ. Данные для определений как Km, так и Ki, были аппроксимированы нелинейными методами для управления Михаэлиса или уравнения Михаэлиса для конкурентного ингибирования, что подходит. Данные, являющиеся результатом ингибирования прочным связыванием, анализировали и Ki определяли аппроксимированием этих данных для уравнения прочного связывания Моррисона нелинейными методами.
Способность ингибиторов GART действовать в качестве субстратов для FPGS измеряли при помощи фермента, полученного из печени крыс. FPGS получали в частично очищенной форме из печени крыс по существу методом Moran и Colman. Крыс Sprague-Dawley умерщвляли ингаляцией CO2. Печень удаляли и через воротную вены ее перфузировали 20 мл охлажденного льдом 20 мМ ГЕПЕС. pH 7,4; 250 мМ сахарозы; 50 мМ 2-меркаптоэтанола. Ткань сохраняли при 4oC или на льду для оставшейся части процедуры. Печень промывали в свежем буфере для перфузии, взвешивали, затем смешивали в двукратном от его объемной массы (в сыром состоянии) объеме свежего буфера для перфузии, содержащего 0,5 мМ PMSF, 50 мкг/мл соевого ингибитора трипсина и 0.5 мМ ЭДТУ до образования однородной массы. Гомогенат фильтровали через марлю и фильтрат центрифугировали при 130000 x г в течение 60 мин. Получаемый супернатант фильтровали через тонкую стекловату для удаления суспендированных жиров и добавляли АТФ до 2,5 мМ. Белок осаждали 30% насыщенным сульфатом аммония и центрифугировали при 30000 x g в течение 15 мин. Получаемый осадок ресуспендировали в небольшом объеме смеси 20 мМ Трис-Cl с pH 7,4; 50 мМ 2-меркаптоэтанола; 5 мМ MgCl2; 5 мМ АТФ и 25 мкг/мл соевого ингибитора трипсина, затем центрифугировали, как указано выше. Из супернатанта удаляли суспендированные жиры и белки осаждали 50% насыщенным сульфатом аммония, затем центрифугировали, как указано выше, для получения осадка. Осажденный таким образом белок был стабильным при хранении при -70oC в течение более 4 месяцев. Поскольку FPGS совсем нестабилен в растворе, замороженный осадок FPGS ресуспендировали для анализа, как описано ниже, и сразу применяли. Осадок расплавляли и ресуспендировали в минимальном количестве буфера (20 мМ Трис-Cl, pH 7,4; 50 мМ 2-меркаптоэтанола; 5 мМ MgCl2, 5 мМ АТФ и 25 мкг/мл соевого ингибитора трипсина) и центрифугировали для удаления остаточных твердых компонентов. Супернатант обессоливали пропусканием через колонку с сефадексом G25, уравновешенным в 20 мМ Трис. pH 7,4; 50 мМ 2-меркаптоэтанола; 5 мМ MgCl2, 5 мМ АТФ и 25 мкг/мл соевого ингибитора трипсина и центрифугировали для удаления остаточных твердых компонентов. Супернатант обессоливали пропусканием через колонку с сефадексом G25, уравновешенным в 20 мМ Трис, pH 74; 50 мМ 2-меркатопэтанола и 5 мМ MgCl2. Фракции, содержащие белок, объединяли и концентрацию АТФ доводили до 5 мМ. Концентрацию белка определяли методом Bradford, применяя БСА (бычий сывороточный альбумин) в качестве белкового стандарта. Обычно концентрации белка были 3 мг/мл. Типичная смесь для анализа содержала 0,7 мг частично очищенного белка FPGS вместе с 200 мМ Трис-Cl с pH 8,5; изменяемой концентрацией ингибитора GART или фолиевой кислоты; 5 мМ АТФ; 10 мМ MgCl2; 30 мМ KCl; 50 мМ 2-меркаптоэтанола; 1 мМ глутаминовой кислоты и 2 мкКи 3H-глутаминовой кислоты в объеме 0,5 мл. Реакции инициировали добавлением фермента и инкубировали в течение 60 мин при 37oC. Реакцию затем гасили 0,5 мл раствора угля и помещали на лед для полной абсорбции продукта (получение раствора угля и значительную очистку 3H-глутаминовой кислоты проводили, как описывалось ранее). Уголь отделяли от реакционной смеси центрифугированием и получаемый осадок промывали 4 раза по 1 мл смеси 10 мМ глутамата с pH 6,8; 10 мМ 2-меркаптоэтанола, в каждом случае с ресуспендированием и центрифугированием. Продукты в конце элюировали из угля 1 мл раствора аммиака в этаноле (3 М NH4OH; 60% этанол) ресуспендировали и центрифугированием. Радиоактивность в 1 мл супернатанта элюента, содержащего меченый тритием продукт, измеряли сцинтилляционным счетом после добавления 1 мл воды. Кинетические параметры Km и Vmax определяли нелинейным аппроксимированием субстратной зависимости образования продукта для уравнения Михаэлиса.
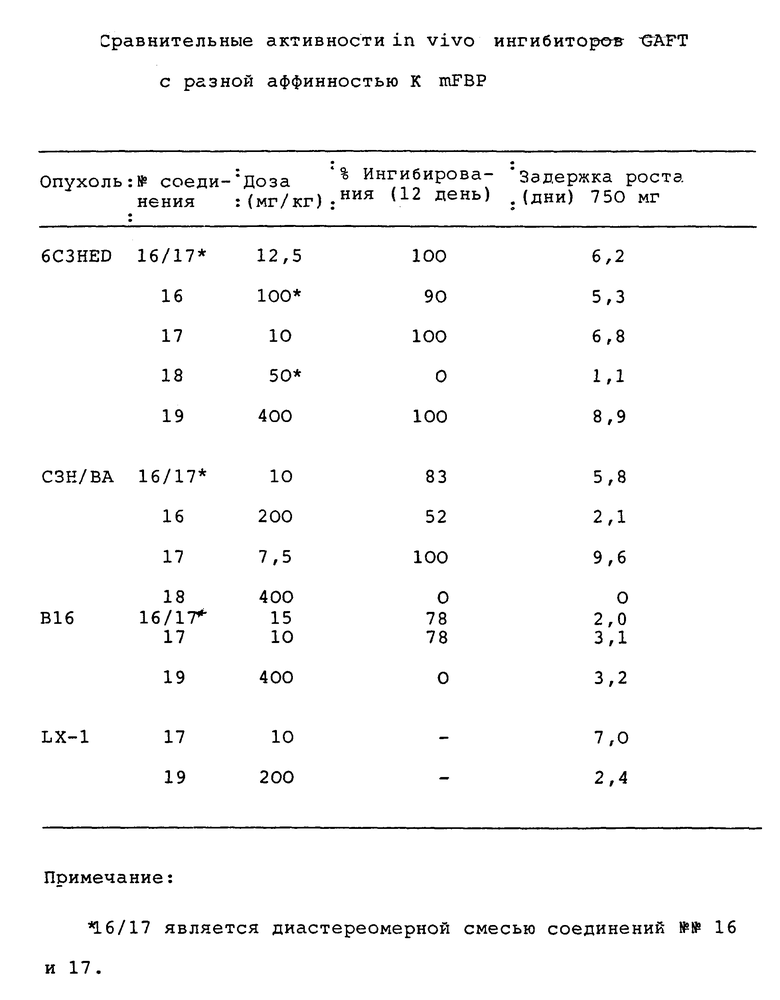
Константу диссоциации (Kd) mFBP определяли для ингибиторов GART методом конкурентного связывания с применением ассоциированных с мембраной клеток FBP, полученного из культивированных клеток KB. Прилипшие клетки соскабливали из колб, промывали один раз охлажденной льдом физиологическим раствором с фосфатным буфером (ФРФБ) и центрифугировали при 5000 x g в течение 5 мин при 4oC. Осадок клеток KB хранили при -70oC, активность mFBP была стабильной в течение по меньшей мере 6 месяцев. Клеточный осадок, содержащий приблизительно 2•108 клеток, ресуспендировали в 10 мл буфера для суспендирования (KH2PO4-KOH, pH 7,4; 10 мМ ЭДТУ; 10 мМ 2-меркаптоэтанола), быстро обрабатывали ультразвуком для завершения лизиса клеток и центрифугировали при 12000 x г в течение 10 мин при 4oC. Осадок освобождали от эндогенного связанного фолата ресуспендированием в 20 мл кислотного буфера (50 мМ KH2PO4-KOH, pH 3,5; 10 мМ ЭДТУ; 10 мМ 2-меркаптоэтанола) и центрифугировали, как указано выше. Осадок затем ресуспендировали в 20 мл буфера для суспендирования при pH 7,4 и центрифугировали, как указано выше. Осадок ресуспендировали в 5 мл буфере для суспендирования с pH 7,4, не содержащего ЭДТУ. Содержание белка количественно определяли методом Bradforda с применением БСА (бычьего сывороточного альбумина) в качестве белкового стандарта. Обычные выходы для этой методики были 4-5 мг общего мембранного белка на 2•108 клеток. Эту последнюю суспензию применяли в качестве источника ассоциированного с мембранами FBP человека. В этом анализе конкурентного связывания лиганду-конкуренту давали возможность конкурировать с 3H-фолиевой кислотой за связывание с mFBP. Реакционные смеси содержали 50-100 мкг клеточного мембранного белка, содержащего 3-6 пмоль (3-6 нМ) FBP, 17,25 пмоль 3H-фолиевой кислоты (17,25 нМ, 0,5 мкКи) и разные концентрации лиганда-конкурента в 1 мл смеси 50 мМ KH2PO4-KOH, pH 7,4; 10 мМ 2-меркаптоэтанола. Реакции связывания проводили при 25oC. Из-за быстрого связывания и очень медленного высвобождения связанной 3H-фолиевой кислоты лиганд-конкурент предварительно связывали в течение 30 мин в отсутствие 3H-фолиевой кислоты. Затем добавляли 3H-фолиевую кислоту и смесь оставляли для достижения равновесия в течение 2,5 часа. Полную реакционную смесь пропускали через нитроцеллюлозные фильтры в вакууме для улавливания клеточных мембран со связанной 3H-фолиевой кислотой. Уловленные мембраны затем промывали 4 раза по 1 мл буфера для реакции. Количество связанной 3H-фолиевой кислоты измеряли сцинтилляционным счетом нитроцеллюлозной мембраны. Полученные данные нелинейно аппроксимировали, как описано выше. mFBP Kd для 3H-фолиевой кислоты, применяемой для подсчета конкурента Kd, получали прямым титрованием mFBP 3H-фолатом и последующим нелинейным аппроксимированием данных для уравнения прочного связывания Kd, найдено, что он равен 60±14 мкМ.
Измерение IC50 в культуре ткани. Клеточные линии L1210 и CCRF-CEM были получены из Американской коллекции типовых культур. Клеточная линия L1210/C1920 была развита для устойчивости к форстриецину и не имела функции восстановленного фолантного носителя. Клетки L1210 выращивали в среде RPM1-1640, дополненной 5% диализованной фетальной бычьей сывороткой. Клеточные линии CEM и L1210/C1920 выращивали в среде RPMI, дополненной 10% диализованной фетальной бычьей сывороткой. Все культуры выдерживали при 37oC в воздухе с 5% CO2 в увлажненном термостате. Ингибирование роста клеток измеряли модифицированным методом Mosmann. Клетки в средней лог-фазе каждой клеточной линии разбавляли до 18500 клеток/мл в свежей питательной среде RPMI-1640, дополненной диализованной фетальной бычьей сывороткой, и затем аликвотные пробы вводили в колонки (ряды) от 2 до 12 титрационных микропланшетов с 96 лунками. Колонку 1 заполняли тем же объемом, 135 мкл, свежей среды без клеток для использования в качестве контрольного опыта. Микропланшеты затем помещали в термостат при 37oC в воздухе, содержащем 5% CO2. Через 4 часа микропланшеты убирали из термостата и затем раствор лекарственного средства, 15 мкл/лунку, при бинарных разведениях, добавляли в колонки от 12 до 4. Лунки, содержащие испытуемое соединение, получали в четырех повторностях на каждом планшете. В лунки в колонках 1 и 2 планшетов добавляли 15 мкл среды без испытуемого соединения. Колонка 3 получала 15 мкл разбавителя для лекарственного средства. Клетки затем возвращали в термостат на 72 часа (клеточные линии L1210 и C1920) или 120 часов (клеточная линия CEM). Для обратимых экспериментов среду дополняли 100 мкМ гипоксантина. После воздействия лекарственным средством в каждую лунку всех микропланшетов добавляли 50 мкл 0,8 мг/мл бромида (4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия (MTT), растворенного в среде для культуры тканей, после чего клетки были возвращены в термостат. Через 4 часа все планшеты убирали из инкубатора и центрифугировали при 1200 об./мин в течение 7 минут. Среду отсасывали и в каждую лунку всех планшетов добавляли 150 мкл ДМСО. Содержимое планшетов затем перемешивали при медленной скорости мешалкой типа вортекс в течение 1 часа в темноте при комнатной температуре. Содержание метаболизированного МТТ измеряли спектрофотометрическим способом при 540 нм на кинетическом микропланшетеридере Molecular Devices Vmax TM. Концентрацию лекарственного средства, требуемого для снижения роста клеток на 50%, измеряемую метаболизмом МТТ, определяли интерполяцией между O.D. (минус контрольный опыт) непосредственно выше и ниже O.D. 50% контроля (минус контрольный опыт).
Расчеты молекулярной механики на соединении 13. Эти расчеты проводили с применением batchmin rool с версией 3.5 программы Vacromodel. Были построены модели соединения 13 с псевдоэкваториальным и псевдоаксиальными ориентациями и загрузки генерировали при помощи МОРАК 6. Каждый из лигандов помещали в активный сайт фермента накладыванием пиримидиновой и глутаматной частей на верхнюю часть кристаллической структуры соединение 5 и субструктуру минимизировали при помощи силового поля (forcefield) AMBER с моделью условного растворителя.
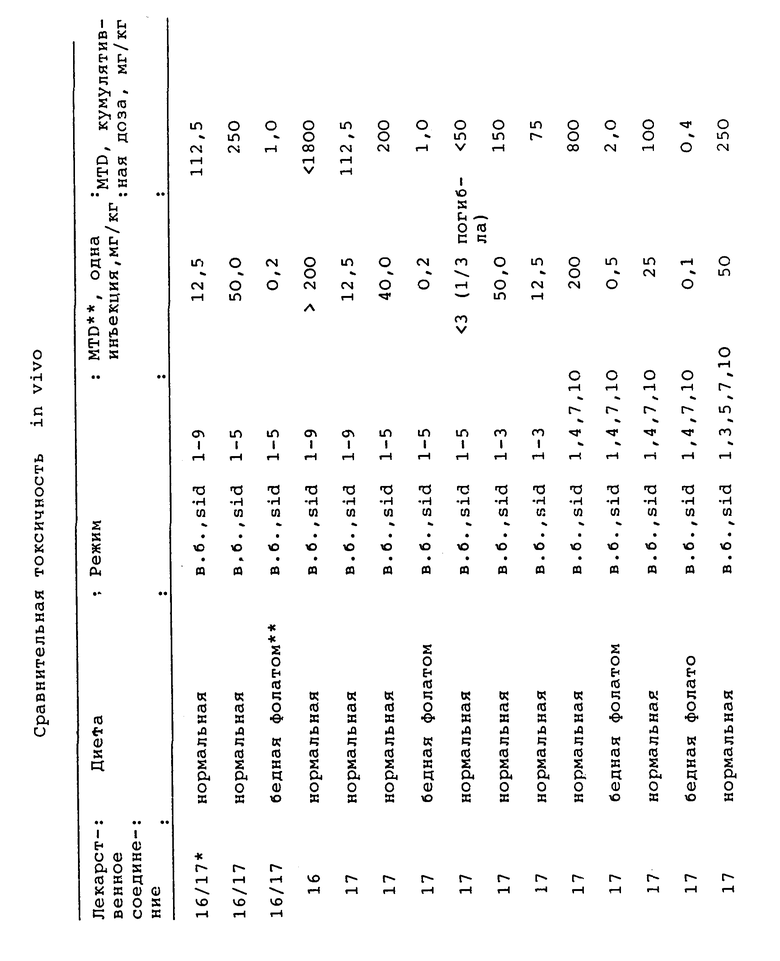
Результаты исследований приведены в таблицах.

Данное изобретение относится к новым соединениям обшей формулы I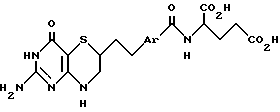
где Ar обозначает фенил, тиенил или тиенил, замещенный низшим алкилом, или ее низшему алкиловому эфиру, ингибирующим фермент глицинамидрибонуклеотидформилтрансферазу, а также к способу ингибирования роста и пролиферации клеток микроорганизмов или высших организмов путем введения хозяину эффективного количества соединения I или его эфира и к способу ингибирования in vitro глицинамидрибонуклеотидформилтарансферазы, воздействием на фермент эффективного количества соединения формулы I или его эфира. Соединения формулы I получают многостадийным способом, включающим взаимодействие соединения формулы III с соединением IV в присутствии основания с получением соединения V, которое при воздействии с кислотой в растворителе приводит к получению соединения VI, которое восстанавливают, и полученное соединение VII гидролизуют с получением соединения VIII и соединение VIII либо соединение VII, где R3 - водород, подвергают воздействию с гидрохлоридом диэфира глутаминовой кислоты и, если необходимо, полученное соединение гидролизуют. 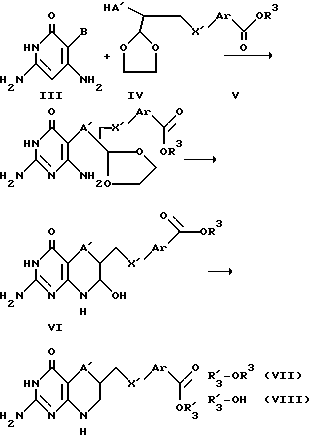 3 с. и 3 з.п.ф-лы, 3 табл.
3 с. и 3 з.п.ф-лы, 3 табл.

где Ar обозначает фенил, тиенил или тиенил, замещенный низшим алкилом,
или ее низший акриловый эфир.
3. Соединение по п.1, где Ar обозначает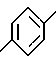
и являющееся d-2-[4-[2-(2-амино-4-оксо-4,6,7,8-тетрагидро-3H-пиримидо[5,4-b][1,4]тиазин-6-ил)этил]бензоиламино]пентандиовой кислотой.
| Способ получения производных 4(3Н)-оксо-5,6,7,8-тетрагидропиридо(2,3- @ )пиримидина или их таутомерных форм | 1987 |
|
SU1581222A3 |
| Способ получения производного пиридо[2,3- @ ]пиримидина, или его SS-, RS-изомеров, или смеси диастереомеров, или фармацевтически приемлемых солей с щелочными металлами | 1986 |
|
SU1676449A3 |
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| EP 431953 А2, 12.06.91 | |||
| Приспособление для нанизывания табачных листьев | 1934 |
|
SU43826A1 |
| Патогенез, лечение и эпидемиология лейкозов // Материалы Всесоюзного симпозиума по проблеме лейкозов | |||
| Рига, 23-25 марта 1971, с.49-51, 162-164, 181-183, 234-236 | |||
| Штрауб Ф.Б | |||
| Будапешт, изд-во "Биохимия" АН ВНР, 1965, с | |||
| Способ передачи радиотелеграфных сигналов | 1922 |
|
SU394A1 |
| F.M | |||
| Muggia "New Drugs, Concepts and Results in Cancer chemotherapy" Klumer Academic Puslishers, Boston, 1992, p.65-87. | |||

