Изобретение относится к ряду новых аналогов аминокислот, которые обнаруживают селективное ингибирование фермента, преобразующего фермент интерлейкин-1β, к композициям, содержащим новые аналоги аминокислот, и к способам использования их в лечебных целях. Конкретнее, ингибиторы фермента, преобразующего интерлейкин-1β, описанные в настоящем изобретении, включают новые аминокислотные метилкетоны, которые особенно полезны при лечении воспалительных, иммунных заболеваний и рака.
Интерлейкин-1β-протеаза (известная также как фермент, преобразующий интерлейкин-1β, или ICE) представляет собой фермент, ответственный за переработку биологически неактивного 31 к D-предшественника 1L-1β- в биологически активную форму 17 к D (Kostura, M.J; Tocci, M.J.; Limjuco, G; Chin, J.; Cameron P. ; Hillman, A.G.; Chartrain. N.A., Schmidt. J.A. Proc. Nat. Acad. Sci, 1989, 86, 5227-5231 и Black R.A.; Kronheim. S.R.; Sleath, P.R., FEBS Let 1989, 247, 386-391). В дополнение к действию как одного из ранних откликов организма на повреждение и заражение предполагается, что 1L-1β также действует как посредник при многих заболеваниях, включая ревматоидный артрит, остеоартрит, воспаление кишечника, сепсис и острый и хронический миелоидный лейкоз (Dinarello, C.A.; Wolff. S.M.; New Engl. J.Med., 1993, 328, 106). Чтобы продемонстрировать посредничество 1L-1β при некоторых заболеваниях человека и некоторых видов животных, используют встречающийся в природе антагонист 1L-1β рецептора (Hannum. C.H.; Wilcox. C.J.; Arend. W.P.; Joslin G.G.; Dripps. D.J.; Heimdal. P.L.; Armes. L.G.; Sommer. A.; Eisenberg. S. P.; Thompson. R.G, Nature, 1990, 343, 336-340; Eisenberg. S.P., Evans. R. J. ; Arend. W.P.; Verderber. E.; Brewer. M.T.; Hannum. C.H.; Thompson. R.G., Nature 1990, 343, 341-346; Ohlsson. K.; Bjork. P.; Bergenfeldt, M.; Hageman. R. ; Thompson. R. C., Nature 1990, 348, 550-552; и Wakabayashi. G., GASEB, 1991, 338-343). Специфическая роль 1L-1β при воспалении и иммуномодуляции подтверждается недавними наблюдениями, что вирус осповакцины использует ингибитор ICE для подавления воспалительной реакции его хозяина (Ray, C.A., и др., Cell, 1992, 69, 597-604).
Настоящее изобретение также относится к модуляции переработки 1L-1β для лечения ревматоидного артрита. Известно, что при данном заболевании содержание 1L-1β в синовиальной жидкости пациентов является повышенным. Кроме того, полагают, что 1L-1β стимулируют синтез ферментов, которые включаются в воспаление, таких как коллогеназа и PLA2, и продуцирует разрушение сустава, которое весьма похоже на ревматоидный артрит, следующий за интрартикулярной инъекцией у животных.
Ограниченное число аналогов пептидилметилкетонов составляет хорошо известный класс соединений, обладающих ингибирующей активностью по отношению к цистеинпротеазе (папаин, кетепсин B). Эти аналоги пептидилметилкетонов рассматриваются в D.Rich, в главе 4 "Proteinase Inhibitors", Barrett, A.J.; и Salvensen, G. , eds., Elsevier 1986. Позднее в качестве ингибиторов цистеинпротеазы также описаны α-арилокси- и α-арилацилоксиметилкетоны (Krantz A. и др., Biochemistry, 30, p. 4678-4687, 1991).
Однако эти аналоги пептидов по существу не обладают эффективностью и селективностью при ингибировании ICE.
Эффективные приемы для лечения воспалительных заболеваний, посредником в которых является 1L-1β, еще предстоит разработать. Следовательно, существует необходимость в лечебных средствах, эффективных для лечения и профилактики этих заболеваний.
В соответствии с настоящим изобретением предлагаются новые пептидные кетоны, имеющие формулу (I), и их фармацевтически приемлемые соли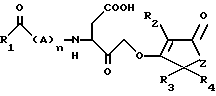
где R1 представляет собой (CR5R6)n, (CR5R6)n-арил, (CR5R6)n-гетероарил,
X-(CR5R6)n, X-(CR5R6)-арил или X-(CR5R6)n-гетероарил, в которых арил или гетероарил могут быть необязательно замещенными;
X представляет собой O или R5;
R5 и R6 независимо представляют собой H или низший алкил;
R2 представляет собой H, низший алкил или (CR5R6)-арил;
R3 и R4 независимо представляют собой H или алкил;
A представляет собой D- или L-изомер аминокислоты, выбираемой из группы, состоящей из аланина, валина, лейцина, изолейцина, пролина, фенилаланина, глицина, тирозина, метионина, аспарагина, глутамина, аспарагиновой кислоты, глутаминовой кислоты, лизина, аргинина, гистидина и β- тиенилаланина;
Z - представляет собой CH2 или O;
n равен 0 - 4.
Использованный здесь термин "аминокислота" охватывает как D-, так и L- их изомеры, а термин "фармацевтически приемлемые соли" означает соли присоединения кислот и оснований.
Термин "соли присоединения кислоты" или "кислотно-аддитивные соли" относится к таким солям, которые сохраняют биологическую эффективность и свойства свободного основания и которые не являются неподходящими биологически или по другим соображениям и образованы с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и с органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, п-толуолсульфоновая кислота, салициловая кислота и т.п.
Термин "соли присоединения основания" охватывает соли, образованные неорганическими основаниями, такие как соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.п. Особенно предпочтительными являются соли аммония, калия, натрия, кальция и магния, соли, образованные фармацевтически приемлемыми органическими нетоксичными основаниями, включая соли первичных, вторичных и третичных аминов, замещенных аминов, включая замещенные амины, встречающиеся в природе, циклических аминов и катионообменных смол, т.е. соли таких оснований, как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминовые смолы и т.п. Особенно предпочтительными органическими нетоксичными основаниями являются изопропиламин, диэтиламин, этаноламин, триметамин, дициклогексиламин, холин и кофеин.
"Алкил" означает радикал как насыщенного, так и ненасыщенного алифатического углеводорода, который может быть либо линейным, либо с разветвленной цепью. Предпочтительные группы содержат не более 12 атомов углерода и могут представлять собой метил, этил и структурные изомеры пропила, бутила, пентила, гексила, гептила, октила, нонила, децила, ундецила и додецила.
"Низший алкил" означает алкильную группу из числа упомянутых выше алкильных групп, содержащую от 1 до 7 атомов углерода. Подходящими низшими алкильными группами являются метил, этил, н-пропил, бутил, трет-бутил, н-пентил, неопентил, н-гексил и н-гептил.
"Арил" означает фенил, нафтил и замещенный фенил.
"Замещенный фенил" означает фенильную группу, в которой один или несколько атомов водорода заменены одинаковыми или разными заместителями, включая галоген, низший алкил, нитрогруппу, аминогруппу, ациламиногруппу, гидроксил, низшую алкоксигруппу, арил, гетероарил, низшую алкоксигруппу, алкилсульфонил, трифторметил, морфолинэтоксигруппу, морфолинсульфонил и карбобензоксиметилсульфамоил.
"Галоген" означает хлор, фтор, бром или йод.
"Гетероарил" означает пиридил, тиенил, фурил, тиазолил, имидазолил, пиразолил, триазинил, хинолил и изохинолил.
"Замещенный гетероарил" означает гетероарильную группу, в которой один или несколько атомов водорода замещены одинаковыми или разными заместителями, включая галоген, низший алкил, нитрогруппу, аминогруппу, ациламиногруппу, гидроксил, низшую алкоксигруппу, арил, гетероарил, алкилсульфонил, трифторметил, морфолинэтоксигруппу, морфолинсульфонил и карбобензоксиметилсульфамоил.
Настоящее изобретение касается способа ингибирования ICE у млекопитающего, нуждающегося в таком лечении, заключающегося во введении упомянутому млекопитающему эффективного ингибирующего количества соединения формулы (I) или фармацевтической композиции, содержащей соединение формулы (I) в фармацевтически приемлемом носителе. Способ ингибирования направлен на лечение болезненных состояний и расстройств, посредником в которых является 1L-1β, и которые включают инфекционные заболевания, такие как менингит и сальпингит; септический шок, респираторные заболевания; воспалительные состояния, такие как артрит, холангит, колит, энцефалит, эндоцеролит (endocerolitis), гепатит, панкреатит и реперфузионное повреждение, иммунные заболевания, такие как повышенная чувствительность; аутоиммунные заболевания, такие как рассеянный склероз; костные заболевания и некоторые опухоли.
Фармацевтическая композиция настоящего изобретения включает в себя активный ингредиент - соединение формулы (I) - в смеси с фармацевтически приемлемым нетоксичным носителем. Такие композиции могут быть приготовлены для применения посредством парентерального (подкожного, интраартикулярного, внутримышечного или внутривенного) введения, в частности в форме жидких растворов или суспензий; посредством орального или трансбуккального введения, в частности в форме таблеток или капсул; посредством трансдермального введения или интраназально, в частности в виде порошков, носовых капель или аэрозолей.
Для орального (или ректального) введения соединения обычно будут формулироваться в форме единичной дозы, такой как таблетка, капсула - желатиновая или крахмальная или суппозиторий. Такие препаративные формы (готовые формы препаратов), как правило, включают твердый, полутвердый или жидкий носитель или разбавитель. Примерами носителей и разбавителей являются лактоза, декстроза, сахароза, сорбит, маннит, крахмалы, аравийская камедь, фосфат кальция, минеральное масло, масло какао, алгинаты, трагакант, желатин, сироп, метилцеллюлоза, монолаурат полиоксиэтиленсобитана, метилгидроксибензоат, пропилгидроксибензоат, тальк и стеарат магния.
Композиции могут быть приготовлены любым из способов, хорошо известных в фармацевтической технике, например так, как описано в Remington's Pharmaceutical Sciences, 17th edition, Mack Publishing Company, Easton, PA, 1985. Готовые формы для парентерального введения могут содержать как обычные наполнители стерильную воду или солевой раствор, алкиленгликоли, такие как пропиленгликоль, полиалкиленгликоли, такие как полиэтиленгликоль, масла растительного происхождения, гидрированные нафталины и т.п. Примеры носителей для парентерального введения включают воду, водные носители, такие как физиологический раствор, раствор Рингера (Ringer), раствор декстрозы и раствор Хэнка (Hank), и неводные носители, такие как нелетучие масла (такие как кукурузное масло, хлопковое масло, арахисовое масло и сезамовое масло), этилолеат и изопропилмиристат. Стерильный физиологический раствор является предпочтительным носителем, а соединения являются достаточно водорастворимыми, чтобы составить растворы для всех поддающихся предвидению потребностей. Носитель может содержать небольшие количества добавок, таких как вещества, которые увеличивают растворимость, изотоничность и химическую устойчивость, например, антиоксидантов, буферных веществ и консерватов. Готовые формы для орального введения могут быть усилены добавлением солей желчи, а также добавлением ацилкарнитинов (Am.J.Physiol. 251:3332 (1986)). Формы для начального введения могут быть твердыми и содержать в качестве наполнителей, например, лактозу или декстран, или могут быть водными или масляными растворами для введения в нос в виде капель или распыления отмеренного количества. Типичные наполнители для трансбуккального введения включают сахара, стеарат кальция, стеарат магния, предварительно желатинированный крахмал и т.п.
При составлении готовых форм для назального введения для усиления поглощения через слизистую оболочку добавляют поверхностно-активные кислоты, такие как, например, гликохолевая кислота, холевая кислота, таурохолевая кислота, этохолевая кислота, дезоксихолевая кислота, хенодезоксихолевая кислота, дегидрохолевая кислота, гликодезоксихолевая кислота и т.п. (см. B.H. Vickery "LHPH and its Analogs-Contraception and Therapeutic Applications" Pt. 2. B.H.Vickery and J.S. Nester, Eds., MTP Press, Lancaster, UK 1987).
Вообще для такого применения, как описано в настоящем изобретении, подходящим является введение активного ингредиента в количестве от 0,1 до 30 мг на кг веса тела (мг/кг) при лечении человека, наиболее предпочтительно - от 0,1 до 30 мг/кг, причем предпочтительно вводить активный ингредиент в количестве от 0,1 до 20-50 мг/кг/день. Такое введение может быть осуществлено однократно, распределено на несколько раз или осуществлено путем постепенного высвобождения лекарственного средства, чтобы достичь наиболее эффективных результатов. При введении в виде единичной дозы наиболее предпочтительно вводить от 0,1 до 10 мг/кг.
Точная доза и режим введения этих соединений и композиций неизбежно будут зависеть от нужд отдельного пациента, которого лечат, вида лечения и степени развития болезни или необходимости. Вообще парентеральное введение требует меньших доз, чем другие способы введения, которые более зависимы от абсорбции.
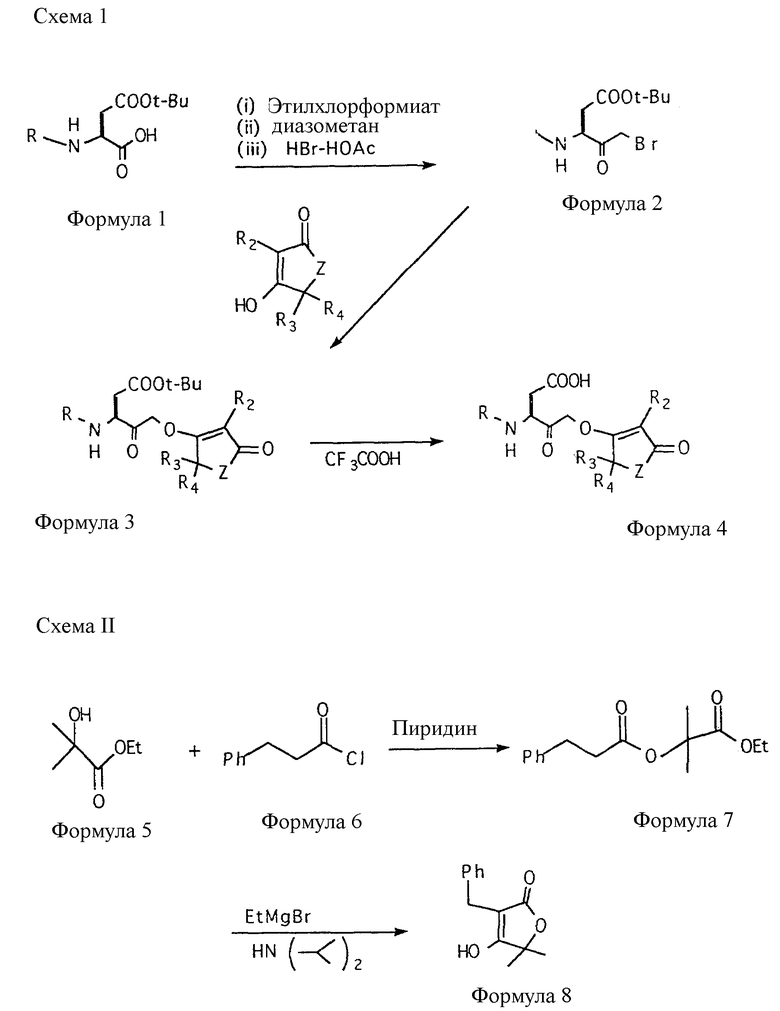
Соединения настоящего изобретения получают в соответствии со схемами I и II (см. в конце описания).
На этих схемах R2, R3, R4 и Z имеют значения, установленные выше для формулы (I), R представляет собой R1CO(A)n, где R1, A и n имеют значения, установленные выше для формулы (I).
Первая стадия этой процедуры заключается в синтезе бромметилкетона N-защищенной аминокислоты (формула 2). Способы получения различных аминокислот и пептидов (формула 1) хорошо разработаны в технике. N-защищенные аминокислоты, дипептиды и полипептиды, которые в некоторых случаях имеются в продаже или их получают по стандартным методикам, как описано в The Practice of Peptide Synthesis, M. Bodansky, Springer-Verlag, NU 1984, затем превращают в бромметилкетон аспарагиновой кислоты (формула 2) посредством катализированного кислотой разложения промежуточного диазометилкетона (Krantz, A. и др., Biochemistry 1991, 30, 4678-4687).
Бромметилкетон N-защищенной аминокислоты взаимодействует с различными тетроновыми кислотами или с циклопентадионами. Эту реакцию проводят, воздействуя бромметилкетоном на избыток тетроновой кислоты или циклопентадиона в ДМФА, содержащем гидрид натрия или калия или фторид калия. Реакцию удобно контролировать тонкослойной хроматографией (ТСХ) и как только ТСХ покажет, что замещение бромида тетроновой кислотой или циклопентадионом завершено, продукт выделяют, используя стандартные технические приемы. Нужный моно-трет-бутиловый эфир аспарагиновой кислоты тетроноилоксиметил- или циклопентадионилоксиметилкетона (формула 3) можно очистить обычными способами, включающими перекристаллизацию и колоночную хроматографию на силикагеле.
Тетрановые кислоты и циклопентадионы, используемые в реакции с бромметилкетонами, могут быть либо закуплены, либо синтезированы с помощью известных процедур (Haynes, L.J., J. Chem. Soc., Part 1, 1956, 4103-4106; White, J.D., и др., J. Amer. Chem. Soc. 1982, 104, 3923; Ramage. R. и др., J. Chem. Soc. Perkin Trans. 1, 1984, 1539-1545; Martinez. R.A. и др., Syn. Commun., 1989, 19, 373-377; Pandey B. и др., Syn. Commun, 1989, 19, 2741-2747). Эти синтезы легко могут быть разработаны специалистами в области органического синтеза. В качестве примера на схеме II представлено получение 3-бензил-5,5-диметилтетроновой кислоты (формула 8).
Следующие далее примеры иллюстрируют изобретение и не должны никоим образом истолковываться как ограничивающие описание и формулу изобретения.
Пример 1
2-Фенилтетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты (формула 1)
Готовят реакционную смесь, содержащую бромметилкетон β-третбутилового эфира N-бензилоксикарбонил-L-аспарагиновой кислоты (формула 2) (0,63 ммоль, 0,25 г), 1,2 экв. фенилтетроновой кислоты (0,75 ммоль, 0,13 г) и 2,5 экв. KF (1,57 ммоль, 0,09 г) в растворе безводного ДМФА (7 мл). Реакционную смесь перемешивают в течение ночи при 25oC. Реакционную смесь разбавляют этилацетатом и промывают водой, насыщенным водным NaHCO3, соляным раствором и сушат над Na2SO4. Экстракт фильтруют и растворитель удаляют при пониженном давлении, получают сырой продукт в виде масла. Масло растворяют в 2 мл этилацетата и добавляют гексан до тех пор, пока не получат слегка мутный раствор, который затем охлаждают при 4oC в течение 12 часов. Получают чистый для анализа β- трет-бутиловый эфир 2-фенилтетроноилоксиметилкетона N-бензилоксикарбонил-L-аспарагиновой кислоты (формула 3) в виде белого твердого вещества (0,2 г, 69%).
Т.пл. 85-87oC.
1H-ЯМР (300 МГц, CDCl3) δ: 7,82 (д., J = 7,57 Гц, 2H), 7,41-7,36 (м., 8H), 7,60 (д. , J = 8,0 Гц, 2H), 5,12-5,08 (м., 4H), 4,71-4,66 (м., 2H), 4,48-4,37 (д.д.д., J = 8,0, 5,1, 4,4 Гц, 1H), 3,08-3,00 (д.д., J = 17,7, 4,4 Гц, 1H), 2,73-2,67 (д.д., J = 17,7, 5,1 Гц, 1H), 1,43 (с., 9H).
Трет-бутиловый эфир (0,34 ммоль, 0,17 г) растворяют в 25% растворе трифторуксусной кислоты в метиленхлориде (объем. 15 мл) и в толуоле (2 мл). Реакционную смесь перемешивают при 25oC и оценивают полноту реакции (ТСХ) в пределах 1 часа. Растворители удаляют при пониженном давлении и остатки отгоняют азеотропно с метиленхлоридом несколько раз. Нужный конечный продукт получают в виде чисто белого твердого вещества (0,123 г, 82%).
Т.пл. 64-67oC.
1H-ЯМР (300 МГц, ДМСО) δ: 7,98 (д., J = 7,6 Гц, 2H), 7,87 (д., J = 7,15 Гц, 2H), 7,43-7,27 (м., 8H), 5,34 (с., 2H), 5,11 (с., 2H), 4,90 (м., 2H), 5,58-4,87 (д.д.д., J = 7,6, 7,1, 5,8 Гц, 1H), 2,84-2,77 (д.д., J = 16,9, 5,8 Гц, 1H), 2,67-2,58 (д.д., J = 17,0, 7,1 Гц, 1H).
Вычисляют для C23H21NO8 • 0,25 H2O:
Вычислено, %: C 62,23; H 4,88; N 3,16.
Найдено, %: C 62,20; H 4,89; N 3,07.
Используя соответствующие исходные материалы и реагенты и следуя методикам, описанным на схемах I и II и в примере 1, получают следующие далее соединения формулы 4.
Пример 2
2-(3,4-Дихлорфенил)тетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
Вычисляют для C23H19Cl2NO8:
Вычислено, %: С 54,35; Н 3,77; N 2,76.
Найдено, %: С 54,30; Н 3,80; N 2,67.
Пример 3
2-Бензил-5,5-диметилтетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
Вычисляют C, H, N для C26H27NO8 • 0,5 H2O:
Вычислено, %: C 63,67; H 5,75; N 2,86.
Найдено, %: C 63,93; H 5,70; N 2,88.
Пример 4
Тетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
Определяют содержание C, H, N для C17H17NO8
Вычислено, %: C 56,20; H 4,72; N 3,86.
Найдено, %: C 55,83; H 4,63; N 3,80.
Пример 5
2-(4-Метоксифенил)тетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
FAB масс-спектр: m/z = 470 [M+H]+.
1H-ЯМР (300 МГц, ДМСО) δ: 7,82 (д., J = 8,9 Гц, 2H), 7,38-7,34 (м., 5H), 6,97 (д., J = 8,9 Гц, 2H), 5,3 (с., 2H), 5,07 (с., 2H), 4,88-4,86 (м., 2H), 4,53-4,51 (м. , 1H), 3,75 (с., 3H), 2,84-2,77 (д.д., J = 17,0, 5,7 Гц, 1H), 2,66-2,58 (д.д., J = 17,0, 7,0 Гц, 1H).
Пример 6
2-Бензилтетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
1H ЯМР (300 МГц, ДМСО) δ: 7,96 (д., J = 7,4 Гц, 1H), 7,4-7,1 (м., 10H), 5,2 (с. , 2H), 5,06 (с., 2H), 4,77 (м., 2H), 4,50 (м., 1H), 3,44 (с., 1H), 2,80 (д.д., J = 17,0, 6,0 Гц, 1H), 2,62 (д.д., J = 17,0, 7,0 Гц, 1H).
Пример 7
2-Фенилтетроноилоксиметилкетон N-бензилоксикарбонил-L-валин-L- аспарагиновой кислоты
1H ЯМР (300 МГц, ДМСО) δ: 8,85 (д., J = 6,5 Гц, 1H), 7,86 (д., J = 7,6 Гц, 2H), 7,53 (д., J = 6,6 Гц, 1H), 7,43-7,33 (м., 8H), 5,24 (с., 2H), 5,02 (с. , 2H), 4,84-4,71 (м. , 2H), 4,58-4,51 (м., 1H), 3,85-3,80 (м., 1H), 2,88-2,81 (д. д., J = 17,0, 4,4 Гц, 1H), 2,62-2,54 (д.д., J = 17,3, 8,0 Гц, 1H), 1,97-1,90 (м., 1H), 0,86 (д., J = 6,9 Гц, 6H).
Пример 8
2-Фенил-5,5-диметилтетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
Масс-спектр низкого разрешения m/z 468 (M+H).
Пример 9
2-Бензилтетроноилоксиметилкетон N-бензилоксикарбонил-L-валин-L- аспарагиновой кислоты
Масс-спектр низкого разрешения m/z 553 (M+H), 509, 273.
Пример 10
5,5-Диметилтетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
Определяют содержание C, H, N для C19H21NO8 • 0,8 H2O:
Вычислено, %: C 56,23; H 5,61; N 3,45.
Найдено, %: C 56,22; H 5,37; N 3,42.
Пример 11
2-Хлортетроноилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
Определяют содержание C, H, N для C17H16ClNO8:
Вычислено, %:C 51,33; H 4,05; N 3,52.
Пример 12
2-Бензилтетроноилоксиметилкетон N-бензилоксикарбонил-L-валин-L- аланин-L-аспарагиновой кислоты
1H ЯМР (300 МГц, ДМСО), 0,82 (д., 3H), 90 (д., 3H), 1,20 (д., 3H), 2,55 (д. д. , 1H), 2,80 (д.д., 1H), 3,15 (д., 1H), 3,30 (д., 1H), 3,80 (м., 1H), 4,15 (м. , 1H), 4,40 (м., 1H), 4,60 (д., 1H), 4,70 (д., 1H), 5,0 (м., 2H), 5,15 (д. д., 1H), 5,25 (д.д., 1H), 7,25 (м., 10H), 8,20 (д., 1H), 8,85 (д., 1H).
Пример 13
2-Метилциклопентадионилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
Определяют содержание C, H, N для C19H21NO7:
Вычислено: %C %H %N
60,79 5,64 3,73
Найдено: %C %H %N
60,59 5,64 3,50
Пример 14
2-Фенилциклопентадионилоксиметилкетон N-бензилоксикарбонил-L-аспарагиновой кислоты
FAB масс-спектр: m/z = 438 [M+H]+.
1H ЯМР (300 МГц, ДМСО) δ: 7,99 (д., J = 7,6 Гц, 1H), 7,72 (д., J = 7,2 Гц, 2H), 7,37-7,34 (м., 8H), 5,35 (с., 2H), 5,07 (с., 2H), 4,52-4,50 (м., 1H), 2,83-2,77 (д.д., J = 17,0, 6,1 Гц, 1H), 2,63-2,58 (м., 4H), 2,49-2,43 (м., 2H).
В примере 15 представлена тетроновая кислота, которую используют в примере 3.
Пример 15
3-Бензил-5,5-диметилтетроновая кислота (формула 8, схема II)
Перемешивают вместе этил-2-гидроксиизобутират (3,69 г, 0,30 моль) (формула 5, схема II) и пиридин (80 мл) и охлаждают до 0oC. При охлаждении и механическом перемешивании добавляют по каплям гидроциннамоилхлорид (формула 6, схема II) (67,4 г, 0,40 моль). Образующуюся в результате неоднородную смесь перемешивают в течение 5 часов. Смесь выливают в воду. Добавление 10% H2SO4 помогает разрушить образующуюся эмульсию. Водный слой экстрагируют эфиром. Органический слой затем промывают 10% H2SO4 и насыщ. NaHCO3, сушат над Na2SO4 и концентрируют. Затем перегонкой получают сложный диэфир (формула 7, схема II) в виде бесцветного масла (26,8 г, 34%) посредством перегонки (112-115oC, 0,1 мм рт.ст).
К охлажденному льдом раствору этилмагнийбромида (- 2,0 М раствор в TMF, 150 мл, 0,30 моль) добавляют диизопропиламин (30,3 г, 0,30 моль) в 50 мл эфира. Реакционную смесь затем перемешивают при комнатной температуре в течение 20 мин. Раствор снова охлаждают до 0oC и в течение 20 мин добавляют раствор сложного диэфира, полученного выше (26,8 г, 0,1 моль). После нагревания до 40oC реакционная смесь становится однородной. После перемешивания в течение 20 мин реакционную смесь выливают в лед и концентрированную HCl. Кислый водный слой экстрагируют эфиром. Эфирную фазу затем промывают 5% HCl (2х) и экстрагируют 5% раствором K2CO3 (4х). Щелочную водную фазу затем промывают эфиром (2х) и подкисляют, добавляя разбавленную HCl. Масло, которое отделяют, растворяют в эфире. Испарение эфира дает желтое масло, которое после царапания постепенно затвердевает. Получают названное в заголовке соединение (формула 8, схема II).
Соединения настоящего изобретения испытывают на ингибирующую активность по отношению к 1L-1β-протеазе в соответствии со следующей далее методикой.
Частично очищенную 1L-1β-протеазу хранят при -80oC, оттаивают на льду и предварительно инкубируют в течение 10 минут при 37oC с 2,5 мМ дитиотрейтола в буферном растворе, содержащем 10 мМ в виде основных растворов в диметилсульфоксиде (ДМСО). Протеазу предварительно инкубируют с ингибитором в объеме 20 мкл в полипропиленовой пробирке для микроцентрифугирования объемом 1,5 мл в течение 15 минут при 37oC. Объем соединения, добавляемый к образцу, регулируют таким образом, чтобы при предварительной инкубации содержание ДМСО составило <15% (объемн.). Образец фермента затем стимулируют добавлением субстрата (TRITC-AYVHDAPVRS-NH2), чтобы получить конечную концентрацию 67 мкМ в конечном объеме 30 мкл. Реакции осуществляют в течение 60 минут при 37oC в темноте и обрывают их добавлением 10 мкл 10% трифторуксусной кислоты (ТФК). После добавления 115 мкл 0,1% ТФК образцы анализируют жидкостной хроматографией высокого давления, используя колонку с обращенной фазой (C18) и элюирование смесью ацетонитрила, воды и ТФК с градиентом. Субстрат и продукт отслеживают по их поглощению при 550 нм и элюировании за 4,2 и 5,2 минут соответственно.
Обнаружено, что испытуемые соединения имеют ингибирующую активность по отношению к 1L-1β- протеазе IC50 < 10 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СУЛЬФОНАМИДНЫЕ ИНГИБИТОРЫ HIY - АСПАРТИЛ-ПРОТЕАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ, СПОСОБ ИДЕНТИФИКАЦИИ ИНГИБИТОРА | 1993 |
|
RU2135496C1 |
| ПРОЛЕКАРСТВА ИНГИБИТОРА КАСПАЗЫ | 2006 |
|
RU2427582C2 |
| ПРОИЗВОДНЫЕ 1-(2-ОКСОАЦЕТИЛ)ПИПЕРИДИН- ИЛИ ПИРРОЛИДИН-2-КАРБОНОВЫХ КИСЛОТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ | 1993 |
|
RU2158258C2 |
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОДАВЛЕНИЯ МНОЖЕСТВЕННОЙ ЛЕКАРСТВЕННОЙ УСТОЙЧИВОСТИ | 1995 |
|
RU2165410C2 |
| ПРОИЗВОДНЫЕ 3-[2-(3-АЦИЛАМИНО-2-ОКСО-2Н-ПИРИДИН-1-ИЛ)-АЦЕТИЛАМИНО]-4-ОКСО-ПЕНТАНОВОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАСПАЗЫ | 2005 |
|
RU2412936C2 |
| ИНГИБИТОРЫ КАСПАЗ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2382780C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ СТИМУЛЯЦИИ РОСТА НЕВРИТОВ | 1996 |
|
RU2197240C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2005 |
|
RU2433127C2 |
| АМИНОПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2006 |
|
RU2427578C2 |
| ИНГИБИТОРЫ КАСПАЗ И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2372335C2 |
Пептидные кетоны формулы I или их фармацевтически приемлемые соли, где R2 - H, галоген, фенил или бензил; R3 и R4 - H или низший алкил; A - D или L-изомер аминокислоты (валин или аланин); Z - CH2 или O; n = 0-2, и фармокомпозиции на их основе обнаруживают селективное ингибирование фермента, преобразующего фермент интерлейкин-1β, и могут быть полезны при лечении воспалительных, иммуных заболеваний и рака. 3 с. и 12 з.п. ф-лы.
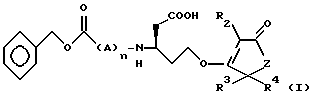
в которой R2 представляет Н, галоген, низший алкил, фенил или бензил;
R3 и R4 представляют независимо Н или низший алкил;
А представляет D или L изомер аминокислоты, выбранной из группы, состоящей из валина и аланина;
Z представляет СН2 или O;
n = 0-2.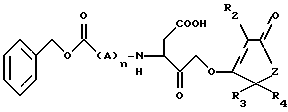
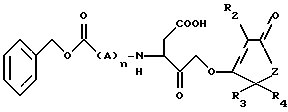
в которой R2 представляет Н, галоген, низший алкил, фенил или бензил;
R3 и R4 представляет независимо Н или низший алкил;
А представляет D или L изомер аминокислоты, выбранной из группы состоящей из валина и аланина,
Z представляет собой СН2 или O;
n = 0 - 2,
в фармацевтически приемлемом носителе.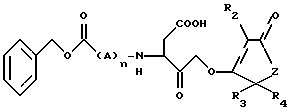
в которой R2 представляет Н, галоген, низший алкил, фенил или бензил;
R3 и R4 представляют независимо Н или низший алкил;
А представляет D или L изомер аминокислоты, выбранной из группы, состоящей из валина и аланина;
Z представляет СН2 или O;
n = 0 - 2,
в фармацевтически приемлемом носителе.
| Способ получения пептидов или их фармацевтически приемлемых солей | 1985 |
|
SU1676454A3 |
| Преобразователь "аналог-код | 1974 |
|
SU519748A1 |
| Огнетушитель | 0 |
|
SU91A1 |
| US 4582821 A, 1991 | |||
| SleaAh et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Biol | |||
| Chem | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| Рахмилевич А.П | |||
| Бюллетень экспериментальной биологии и медицины, 1988, 106, N12, c.698-701. | |||

