ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому бром-фенил замещенному тиазолилдигидропиримидину, способу его получения и применения в качестве лекарственного средства, особенно для лечения и профилактики гепатита В. Настоящее изобретение также относится к композиции, содержащей дигидропиримидин, другое противовирусное средство и, когда это целесообразно, иммуномодулятор, и лекарственному средству, содержащему эту композицию, особенно для лечения и профилактики инфекций HBV, таких как гепатит В.
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
Вирус гепатита В принадлежит к семейству hepadna-вирусов. Он может вызывать острое и/или персистирующее, или прогрессирующее хроническое заболевание. Многие другие клинические проявления в патологическом состоянии также вызываются вирусом гепатита В, в частности хроническое воспаление печени, цирроз печени и печеночно-клеточная карцинома. Кроме того, совместная инфекция с вирусом гепатита дельта может оказывать неблагоприятное действие на прогрессирование заболевания.
Интерферон и ламивудин являются традиционными лекарственными средствами, одобренными для использования при лечении хронического гепатита. Однако интерферон имеет только умеренную активность, но имеет неблагоприятные побочные реакции. Хотя ламивудин обладает хорошей активностью, к нему быстро развивается резистентность во время лечения и зачастую появляются рецидивы после прекращения лечения. Значение IC50 ламивудина (3-TC) составляет 300 нM (Science, 299 (2003), 893-896).
В патенте США № 7074784 описывается 6-амидоалкилдигидропиримидин и его применение в качестве лекарственного средства в особенности для лечения и профилактики гепатита В.
В Примере 12 патента США № 7074784 описывается, что R1 представляет собой o-хлор, R2 представляет собой п-хлор, R6 представляет собой 3,5-дифтор-пиридин-2-илl, X представляет собой -CH2- и Z представляет собой морфолинил. Это соединение может ингибировать рост вируса гепатита В при культивировании клеток. Значение IC50 составляет 2 нM (протестировано самими авторами изобретения).
Основной заменой в Примере 12 является замена бис-хлора на R1 (o-бром) и R2 (п-фтор), что в результате приводит к IC50 Соединения 9, составляющей 7 нМ (описано в Примере 9 этого патента). А когда основные заместители меняются на R1 (o-хлор) и R2 (п-фтор), также получают приблизительное значение IC50 (IC50=2-4 нM в Примере 5).
Указывается, что значение IC50 не увеличивается и с изменением основных заместителей R1 и R2 (смотри Таблицу 1).
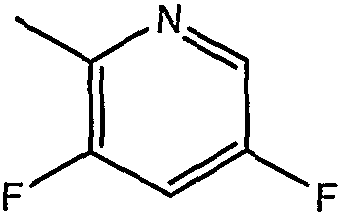
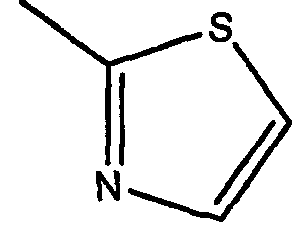
В патенте США № 7074784 B2 также описан пример, в котором дифтор-радикал замещен на тиазол-2-ил (описан в Примере 45 этого патента). Это производное имеет аналогичное значение IC50 (2 нM) (смотри Таблицу 1).



ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы изобретения неожиданно обнаружили, что производное с активностью в 10 раз выше и значением IC50 менее 1 нM может быть получено путем замещения тиазол-2-илом и изменения основных заместителей на R1=o-бром и R2=п-фтор. Это является неожиданным с учетом US 7074784 (смотри Таблицу 2).


Настоящее изобретение относится к соединению формулы (I) и его таутомеру (Iа),

где R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой C1-C4 алкил, R6 представляет собой тиазол-2-ил, Х представляет собой метилен и Z представляет собой морфолинил.
Предпочтительно, R1 соединения по настоящему изобретению формулы (I) и (Iа) представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой метил или этил, R6 представляет собой тиазол-2-ил, X представляет собой метилен и Z представляет собой морфолинил.
Настоящее изобретение также относится к энантиомеру соединения, описанного в настоящей заявке, и его смеси. Рацемат может быть отделен с помощью известного способа и, в основном, это гомогенное соединение в смеси стереоизомерных форм.
Соединения по настоящему изобретению включают изомеры формулы (I) и (Ia) и их смесь.
Соединение по настоящему изобретению также может быть в форме соли, предпочтительно физиологически приемлемой соли.
Физиологически приемлемая соль может быть солью неорганической кислоты или солью органической кислоты. Предпочтительно, это соль неорганической кислоты, такая как хлорид, бромид, фосфат или сульфат, и т.п., или карбоксилат, или сульфонат, например, ацетат, малеат, фумарат, малат, цитрат, тартрат, лактат, бензоат или метансульфонат, этансульфонат, бензолсульфонат, толуолсульфонат или нафталиндисульфонат, и т.п.
Физиологически приемлемая соль также может быть солью металла или аммонийной солью соединения по настоящему изобретению. В предпочтительном примере, это соль натрия, соль калия, соль магния или соль кальция, и аммонийная соль, образованная аммиаком или органическим амином, например, этиламином, диэтиламином или триэтиламином, диэтаноламином или триэтаноламином, дициклогексиламином, диметиламиноэтиловым спиртом, аргинином, лизином, этилендиамином или 2-фенилэтиламином, и т.п.
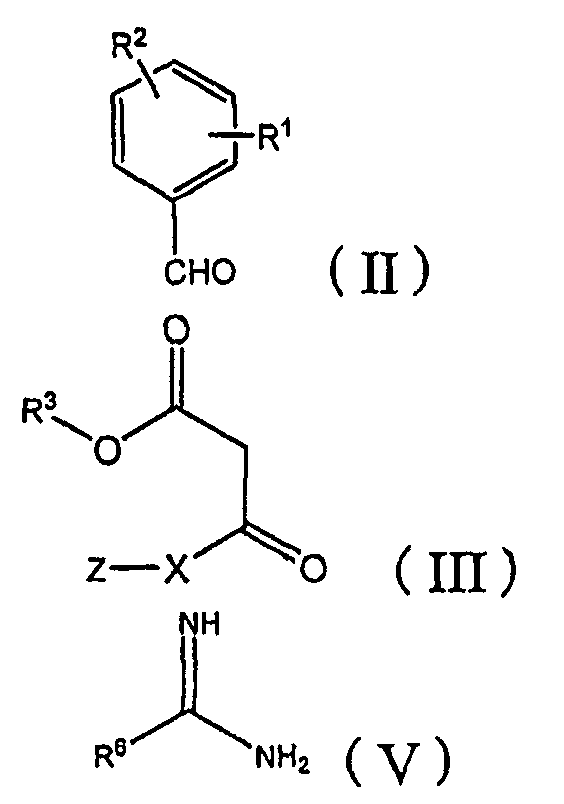
Соединение (I) по настоящему изобретению может быть получено следующими способами:
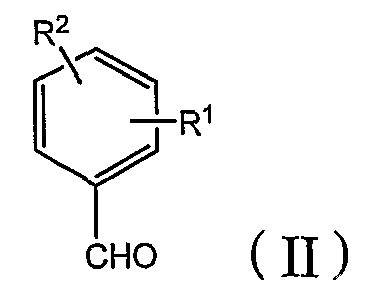
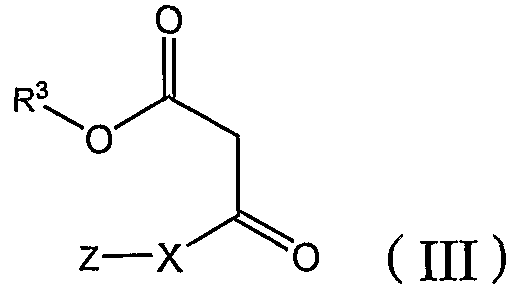
[A] сначала бензальдегид формулы (II) взаимодействует с β-кетоэфиром формулы (III) с добавлением или без добавления щелочи или кислоты, и, когда это целесообразно, в присутствии инертного органического растворителя с получением бензилиденового соединения формулы (IV):
 ,
,  ,
,  ,
,
где R1, R2, R3, X и Z определены в настоящей заявке, а затем бензилиденовое соединение взаимодействует с амидином формулы (V) или его солью (такой как гидрохлорид или ацетат) с добавлением или без добавления щелочи или кислоты и, когда это целесообразно, в присутствии инертного органического растворителя:
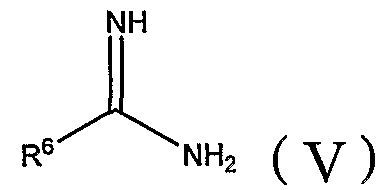
где R6 определен в настоящей заявке; или
[B] β-кетоэфир формулы (III) взаимодействует с бензальдегидом формулы (II) и амидином формулы (V) или его солью (такой как гидрохлорид или ацетат) с добавлением или без добавления щелочи или кислоты и, когда это целесообразно, в присутствии инертного органического растворителя на одной стадии; или
[C] в тех случаях, когда X в формуле (I) представляет собой метилен, соединение формулы (VI) взаимодействует с морфолином формулы (VII) с добавлением или без добавления щелочи и, когда это целесообразно, в присутствии инертного органического растворителя,
 ,
, 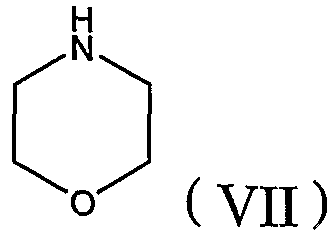
где R1, R2, R3 и R6 определены в настоящей заявке и Y представляет собой нуклеофильный заместитель, такой как хлор, бром, йод, метилсульфонил или толуолсульфонил; или
[D] бензальдегид формулы (II) взаимодействует с соединением формулы (X) и амидином формулы (V) с добавлением или без добавления щелочи и, когда это целесообразно, в инертном органическом растворителе,
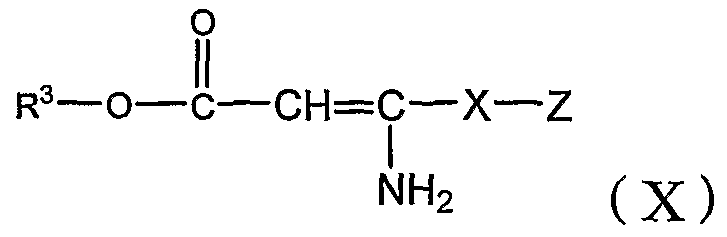
где R3, X и Z определены в настоящей заявке.
Соединение формулы (VI) может быть получено, например, путем взаимодействия соединения формулы (VIII)

где R1, R2, R3 и R6 определены в настоящей заявке, с бромирующим реагентом, таким как N-бромсукцинимид, предпочтительно в инертном органическом растворе, с получением соединения формулы (IX):

и взаимодействия соединения, имеющего нуклеофильный заместитель, непосредственно или после дополнительного превращения соединения в соответствии с общепринятым способом, описанным в литературе, с морфолином формулы (VII).
Для получения соединения формулы по настоящему изобретению (I), где X представляет собой метилен и Z представляет собой морфолинил, хлорацетат формулы (XI) взаимодействует с морфолином (VII) с получением β-кето карбоксилата формулы (III),

где R3 определен в настоящей заявке.
В качестве исходного вещества коммерчески доступным является 2-бром-4-фтор-бензальдегид (II).
В качестве исходного вещества, β-кетокарбоксилат (III) хорошо известен или может быть получен известными способами, опубликованными в литературе [например, D. Borrmann, "Umsetzung von Diketen mit Alkoholen, Phenolen und Mercaptanen", в "Methoden der organischen Chemie" (Houben-Weyl), vol. VII/4, 230 ff (1968); Y. Oikawa, K. Sugano und O. Yonemitsu, J. Org. Chem. 43, 2087 (1978)].
Соединение (V) хорошо известно и может быть получено в соответствии с описаниями WO-A-99/54326 и WO-A-99/54329.
Морфолин (VII) является коммерчески доступным.
Соединения (VIII) и (X) могут быть получены в соответствии со стадиями [A] или [B], описанными в WO-A-99/54326.
Все инертные органические растворители подходят для применения на стадиях A, B, C и D. Инертным органическим растворителем предпочтительно является спирт, такой как метанол, этанол и изопропиловый спирт, простой эфир, такой как диоксан, диэтиловый эфир, тетрагидрофуран, монометиловый эфир этиленгликоля, диметиловый эфир этиленгликоля, карбоновая кислота, такая как уксусная кислота, диметилформамид, диметилсульфоксид, ацетонитрил, пиридин или гексаметил-триамид фосфорной кислоты.
Температуру реакции можно изменять в пределах довольно широкого диапазона. Обычно температура составляет от 20°C до 150°C. Предпочтительно, температура равна температуре кипения выбранного растворителя.
Реакцию можно проводить при атмосферном давлении или под высоким давлением. Обычно реакцию проводят при атмосферном давлении.
Реакцию можно проводить с кислотой или щелочью, или без них. Предпочтительно проводить эту реакцию в присутствии слабой кислоты, такой как уксусная кислота, муравьиная кислота или подобное.
Один вариант осуществления настоящего изобретения относится к композиции, содержащей A) по меньшей мере один из указанных выше дигидропиримидинов и B) по меньшей мере одно из других противовирусных средств, отличное от A).
Конкретный вариант настоящего изобретения относится к композиции, содержащей A) указанный выше дигиропиримидин, B) ингибитор HBV полимеразы и, когда это целесообразно, C) иммуномодулятор.
Предпочтительно, иммуномодулятор C) выбран, например, из любых интерферонов, таких как α-интерферон, β-интерферон и γ-интерферон, главным образом, α-2a-интерферона и α-2b-интерферона, интерлейкина, например, интерлейкина-2, полипептида, такого как тимозин-α-1 и тимоктонан, производных имидазохинолина, таких как левамизол, иммуноглобулина и терапевтической вакцины.
Таким образом, настоящее изобретение также относится к композиции для лечения и профилактики HBV инфекций и ее применению для лечения заболеваний, индуцированных HBV.
Применение комбинаций настоящего изобретения обеспечивает важные преимущества для лечения заболеваний, индуцированных HBV, по сравнению с монотерапией отдельными соединениями, а именно, прежде всего, синергетическую противовирусную активность, а также хорошую переносимость комбинаций по настоящему изобретению, выраженную в Tox-50 (диапазон токсичности, в котором выживает 50% клеток).
Веществами, называемыми HBV ингибиторами полимеразы В для целей настоящего изобретения, являются те, которые в эндогенном полимеразном анализе, опубликованном Ph. A. Furman et al. in Antimicrobial Agents и Chemotherapy, Vol. 36 (No. 12), 2688 (1992) и которые описаны далее в этой заявке, приводят к ингибированию образования двойной спирали ДНК HBV так, чтобы приводить максимально к 50% активности от исходного значения.
HBV ингибиторы полимеразы B для применения в настоящем изобретении представляют собой вещества, описанные в эксперименте с эндогенной полимеразой, опубликованном в “Antimicrobial Agents и Chemotherapy” Vol.36 (No.12), 2688 (1992) автором Ph. A. Furman, и вещества, описанные ниже для ингибирования образования двухспиральной ДНК HBV, приводя в результате максимально к 50% значению активности, от исходного значения.
Вирионы HBV из культуральных супернатантов включают нуклеозид 5'-трифосфаты в плюс-цепи ДНК HBV in vitro. Применяя электрофорез в агарозном геле, включение [α-32P]-дезоксинуклеозид 5'-трифосфата в вирусный 3,2 тыс. осн. ДНК продукт наблюдают в присутствии и отсутствие вещества, потенциально обладающего ингибирующими свойствами в отношении полимеразы HBV. Вирионы HBV получают из супернатанта клеточной культуры клеток HepG2.2.15 путем осаждения полиэтиленгликолем и концентрируют. Одну часть по объему осветленного супернатанта клеточной культуры смешивают с 1/4 по объему водного раствора, содержащего 50% по массе полиэтиленгликоля 8000 и 0,6 M хлорида натрия. Вирионы осаждают центрифугированием при 2500×g/15 минут. Осадок ресуспендируют в 2 мл буфера, содержащего 0,05 M трис-HCl (pH 7,5) и диализируют против того же буфера, содержащего 100 мМ хлорида калия. Образцы могут быть заморожены при -80°C. Каждая реакционная смесь (100 мкл) содержит по меньшей мере 105 вирионов HBV; 50 мM трис-HCl (pH 7,5); 300 мM хлорида калия; 50 мM хлорида магния; 0,1% Nonident® P-40 (неионного детергента от Boehringer Mannheim); 10 мкM dATP, 10 мкM dGTP, 10 мкM dTTP; 10 мкКю [32P]dCTP (3000 Кю/ммоль; конечная концентрация 33 нM) и 1 мкM сильного ингибитора полимеразы в его трифосфорилированной форме. Образцы инкубируют при 37°C в течение одного часа, а затем реакцию останавливают добавлением 50 мМ EDTA. 10% масса/объем раствор SDS (содержащий 10 г SDS на 90 мл воды) добавляют до конечной концентрации 1% по объему (исходя из общего объема), и добавляют протеиназу K до конечной концентрации 1 мг/мл. После инкубирования при 37°C в течение одного часа, образцы экстрагируют таким же объемом смеси фенол/хлороформ/изоамиловый спирт (соотношение 25:24:1 по объему), и ДНК осаждают из водной фазы этанолом. Осадок ДНК ресуспендируют в 10 мкл буфера для геля (раствор 10,8 г триса, 5,5 г борной кислоты и 0,75 г EDTA в 1 литре воды (=TBE буфер)) и разделяют с помощью электрофореза в агарозном геле. Гель либо сушат, либо нуклеиновые кислоты, находящиеся в нем, переносят с помощью методики Саузерн-блоттинга на мембрану. Количество меченной образованной двухцепочечной ДНК затем определяют по отношению к отрицательному контролю (= реакция эндо-пол без вещества или с неактивным контрольным веществом). Ингибитор полимеразы HBV присутствует, если присутствует максимум 50% активности отрицательного контроля.
Предпочтительные HBV ингибиторы полимеразы B) включают, например, 3TC=ламивудин=4-амино-1-[(2R-цис)-2-(гидроксиметил)-1,3-оксатиолан-5-ил-]-пиримидин-2(1H)-он, ср. EP-B 382 526 (=Патент США № 5047407) и WO 91/11186 (=Патент США № 5204466); Адефовир дипивоксил = 9-{2-[[бис[(пивалоилокси)-метокси]-фосфинил]- метокси]-этил}-a-денин, ср. EP-B 481 214 (=Патенты США №№. 5663159 и 5792756), Патенты США №№. 4724233 и 4808716; BMS 200475=[1S-(1-α,3-α,4-β)]-2-амино-1,9-дигидро-9-[4-гидрокси-3-(гидроксиметил)-2-метилен-циклопентил]-6H-пурин-6-он, ср. EP-B 481 754 (=Патенты США №№. 5206244 и 5340816), WO 98/09964 и 99/41275; Абакавир=(-)-(1S-цис)-4-[2-амино-6-(циклопропиламино)-9H-пурин-9-ил]-2-циклопентен-1-метанол, ср. EP-B 349 242 (=Патент США № 5049671) и EP-B 434 450 (=Патент США № 5034394); FTC=(2R-цис)-4-амино-5-фтор-1-[2-(гидроксиметил)-1,3-оксатиолан-5-ил]-пиримидин-2(1H)-он, cf. WO 92/14743 (=Патенты США №№. 5204466, 5210085, 5539116, 5700937, 5728575, 5814639, 5827727, 5852027, 5892025, 5914331, 5914400) и WO 92/18517; β-L-FDDC =5-(6-амино-2-фтор-9H-пурин-9-ил)-тетрагидро-2-фуранметанол, ср. WO 94/27616 (=Патенты США №№. 5627160, 5561120, 5631239 и 5830881); L-FMAU=1-(2-дезокси-2-фтор-β-L-арабинофуранозил)-5-метил-пиримидин-2,4(1H,3H)-дион, ср. WO 99/05157, WO 99/05158 и Патент США № 5753789.
Другой предпочтительный вариант осуществления настоящего изобретения относится к композиции, содержащей A) указанные выше дигидропиримидины формулы (I) и (Ia); и B) ламивудин.
Другие предпочтительные средства B против вируса HBV включают, например, фенилпропенамиды следующей формулы:

где R1 и R2 каждый независимо представляют собой, C1-4 алкил или, вместе с атомом азота, на котором они расположены, образуют кольцо, имеющее от 5 до 6 кольцевых атомов, которые содержат углерод и/или кислород; R3-R12 каждый независимо представляют собой водород, галоген, C1-4 алкил, необязательно замещенный C1-4 алкокси, нитро, циано или трифторметил; и R13 представляет собой водород, C1-4 алкил, C1-7 ацил или аралкил и X представляет собой галоген или необязательно замещенный C1-4 алкил.
Фенил пропенамиды и способы их получения описаны в WO 98/33501 и упомянуты в настоящей заявке для публикации. AT-61 представляет собой соединение
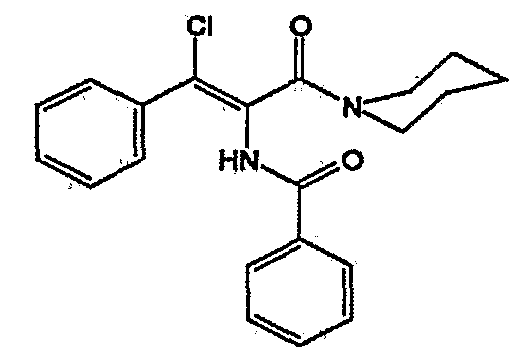
Предпочтительные иммуномодуляторы C) включают, например, любые интерфероны, такие как α-, β- и γ-интерфероны, в частности, также α-2a- и α-2b-интерфероны, интерлейкины, такие как интерлейкин-2, полипептиды, такие как астимозин-α-1 и тимоктонан, производные имидазохинолина, такие как Levamisole®, иммуноглобулины и терапевтические вакцины.
Еще один предпочтительный вариант осуществления настоящего изобретения относится к сочетаниям A) указанных выше дигидропиримидинов (I) и (Ia), B) ламивудина и, где это целесообразно, C) интерферона.
Описание тестов
Противовирусное действие соединений по настоящему изобретению на вирус гепатита В исследуют способами, основанными на способах, описанных M. A. Sells et al., Proc. Natl. Acad. Sci., 84, 1005-1009 (1987) и B. E. Korba et al., Antiviral Research 19, 55-70 (1992).
Противовирусные тесты проводят в 96-луночных микротитровальных планшетах. Первый вертикальный ряд планшета содержит только ростовую среду и клетки HepG2.2.15. Он служит в качестве вирусного контроля.
Маточные растворы тестируемых соединений (50 мМ) исходно растворяют в DMSO, и дальнейшие разведения готовят в ростовой среде HepG2.2.15. Соединения в соответствии с изобретением обычно раскапывают пипеткой в тестируемой концентрации 100 мкМ (1-я тестируемая концентрация) в каждом случае во второй вертикальный тестируемый ряд микротитровального планшета и впоследствии разбавляют в два приема 210 раз в ростовой среде плюс 2% масс. эмбриональной сыворотки теленка (объем 25 мкл).
Каждая лунка микротитровального планшета т.о. содержит 225 мкл суспензии клеток HepG2.2.15 (5×104 клеток/мл) в ростовой среде плюс 2% масс. эмбриональной сыворотки теленка. Тестируемую смесь инкубируют при 37°C и 5% CO2 (об./об.) в течение 4 дней.
Затем супернатант отсасывают и отбрасывают, и в лунки помещают 225 мкл свежеприготовленной ростовой среды. Соединения по настоящему изобретению каждое добавляют еще раз в виде 10-кратного концентрированного раствора в объеме 25 мкл. Смесь инкубируют в течение еще 4 дней.
Перед сбором супернатантов для определения противовирусного действия клетки HepG2.2.15 изучали под световым микроскопом или с помощью способов биохимического выявления (например, красителя Alamar Blue или красителя трипанового синего) цитотоксических изменений.
Супернатант и/или клетки затем собирают и втягивают с помощью вакуума в 96-луночные дот-блот камеры, покрытые нейлоновой мембраной (в соответствии с информацией производителя).
Определение цитотоксичности
Цитотоксические или цитостатические изменения, индуцированные веществом в клетках HepG2.2.15, выявляют, например, под световым микроскопом как изменения морфологии клетки. Такие изменения, индуцированные веществом в клетках HepG2.2.15 по сравнению с необработанными клетками, видны, например, как цитолиз, вакуолизация или изменение морфологии клетки. 50% цитотоксичность (Tox.-50) означает, что 50% клеток демонстрируют морфологию, сравнимую с соответствующим клеточным контролем.
Переносимость некоторых соединений в соответствии с настоящим изобретением дополнительно тестируют на других клетках-хозяевах, например, таких как клетки HeLa, первичных клетках периферической крови человека или трансформированных клеточных линиях, таких как клетки H-9.
При концентрациях соединений по настоящему изобретению >10 мкМ цитотоксические изменения не выявляются.
Определение противовирусного действия
После переноса супернатантов или лизированных клеток на нейлоновую мембрану блот-аппарата (смотри выше), внутри- или внеклеточные супернатанты клеток HepG2.2.15 денатурируют (1,5 M NaCl/0,5н NaOH), нейтрализуют (3 M NaCl/0,5M Трис HCl, pH 7,5) и промывают (2×SSC). Затем ДНК сушат на мембране путем инкубирования фильтров при 120°C в течение 2-4 часов.
Гибридизация ДНК
Выявление вирусной ДНК из обработанных клеток HepG2.2.15 на нейлоновых фильтрах обычно проводят с помощью нерадиоактивных, меченных дигоксигенином гепатит В-специфичных ДНК зондов, каждый из которых мечен дигоксигенином, очищенным и используемым для гибридизации в соответствии с информацией производителя.
Предварительную гибридизацию и гибридизацию проводят в 5×SSC, 1×блокирующем реагенте, 0,1% масс. N-лауроилсаркозина, 0,02% масс. SDS и 100 мкг ДНК молоки сельди. Предварительную гибридизацию проводят при 60°C в течение 30 минут и специфическую гибридизацию проводят с использованием от 20 до 40 нг/мл дигоксигенизированной, денатурированной HBV-специфичной ДНК (14 часов, 60°C). Затем фильтры промывают.
Выявление HBV-ДНК с помощью дигоксигенин-антител
Иммунологическое выявление ДНК, меченной дигоксигенином, проводят в соответствии с информацией производителя:
Фильтры промывали и предварительно гибридизировали в блокирующем реагенте (в соответствии с информацией производителя). Затем проводили гибридизацию с анти-DIG антителом, соединенным с щелочной фосфатазой в течение 30 минут. После стадии промывки добавляли субстрат щелочной фосфатазы, CSPD, инкубировали с фильтрами в течение 5 минут, затем упаковывали в пластиковую пленку и инкубировали при 37°C еще в течение 15 минут. Хемилюминесценцию сигналов гепатит В-специфичной ДНК визуализировали, экспонируя фильтры на рентгеновской пленке (выдержка зависит от силы сигнала: от 10 минут до 2 часов).
Половину максимальной ингибирующей концентрации (IC50, 50% ингибирующая концентрация) определяли как концентрацию, при которой внутри- и внеклеточная гепатит В-специфичная полоса уменьшалась под действием соединения по изобретению на 50% по сравнению с необработанным образцом.
Неожиданным является то, что соединение по настоящему изобретению демонстрирует эффективное противовирусное действие с IC50 менее 1 нM. Следовательно, соединение по настоящему изобретению подходит для применения при лечении заболеваний, вызванных вирусами, особенно острых и хронических персистентных HBV инфекций. Хронические вирусные заболевания, вызванные HBV, могут ухудшать клинические проявления и хроническая гепатит В вирусная инфекция может быть причиной цирроза и/или печеночно-клеточной карциномы во многих случаях.
Областями показаний, которые могут быть упомянуты для соединений по настоящему изобретению, являются, например: лечение острых и хронических вирусных инфекций, которые могут приводить к инфекционному гепатиту, например инфекции вирусами гепатита В. Соединения по настоящему изобретению особенно подходят для лечения хронических инфекций гепатита В и лечения острых и хронических гепатит В вирусных инфекций.
Настоящее изобретение включает фармацевтические препараты, которые, помимо нетоксичных инертных фармацевтически подходящих носителей, содержат одно или несколько соединений (I) или (Ia), или комбинацию по изобретению, или которые состоят из одного или нескольких активных ингредиентов (I) или (Ia) или комбинации по настоящему изобретению.
Предполагается, что активные ингредиенты (I) и (Ia) присутствуют в упомянутых выше фармацевтических препаратах в концентрации примерно от 0,1 до 99,5% масс., предпочтительно примерно от 0,5 до 95% масс. всей смеси.
Фармацевтические препараты, упомянутые выше, также могут содержать другие активные фармацевтические ингредиенты помимо соединений (I) и (Ia).
Соотношение количеств компонентов A, B и, где это целесообразно, C в композициях по настоящему изобретению может варьировать в широких пределах, предпочтительно это соотношение составляет от 5 до 500 мг A/10-1000 мг B, в частности от 10 до 200 мг A/20 до 400 мг B.
Компонент C, который также используется где это целесообразно, может быть использован в количествах, предпочтительно, от 1 до 10 миллионов, в частности, от 2 до 7 миллионов, МЕ (международных единиц), примерно три раза в неделю на протяжении периода времени вплоть до одного года.
Предполагается, что соединения или композиции по настоящему изобретению присутствуют в упомянутых выше фармацевтических препаратах в основном в концентрации примерно от 0,1 до 99,5, предпочтительно, примерно от 0,5 до 95% масс. всей смеси.
Фармацевтические препараты, упомянутые выше, могут быть получены обычным путем с помощью известных способов, например, путем смешивания активного ингредиента (ингредиентов) с носителем (носителями).
В основном, оказалось предпочтительным как у людей, так и в ветеринарной медицине, вводить активный ингредиент(ы) в суммарных количествах примерно от 0,5 примерно до 500, предпочтительно от 1 до 100 мг/кг массы тела каждые 24 часа, где это целесообразно, в виде ряда однократных доз, для достижения желаемых результатов. Однократная доза содержит активный ингредиент(ы), предпочтительно в количествах примерно от 1 примерно до 80, в частности, от 1 до 30 мг/кг массы тела. Однако может быть необходимым отклонение от упомянутых дозировок, в частности, в зависимости от видов и массы тела индивидуума, которому проводят лечение, природы и тяжести заболевания, типа препарата и пути введения лекарственного средства, и времени или интервала, в который происходит введение.
Следовательно, настоящее изобретение дополнительно относится к соединениям и композициям, охарактеризованным выше, для контроля за заболеваниями.
Настоящее изобретение дополнительно относится к лекарственным средствам, содержащим по меньшей мере одно из соединений или композиций, охарактеризованных выше, и, где это целесообразно, один или несколько других активных фармацевтических ингредиентов.
Настоящее изобретение дополнительно относится к применению соединений и композиций, охарактеризованных выше, для получения лекарственного средства для лечения и профилактики описанных выше заболеваний, предпочтительно вирусных заболеваний, в частности гепатита В.
Процентные величины в следующих примерах относятся в каждом случае к массе, если не указано иное. Соотношения растворителей в смесях растворителей в каждом случае основаны на объеме.
ПРИМЕРЫ
A. Получение промежуточных соединений
Промежуточное соединение 1
Этил 4-(2-бром-4-фторфенил)-2-(тиазол-2-ил)-6-метил-1,4-дигидропиримидин- 5-карбоновый эфир

Смесь 10,0 г (49,3 ммоль) 2-бром-4-фторбензальдегида, 6,4 г (49,3 ммоль) этилацетоацетата, 8,1 г (49,3 ммоль) 2-амидино-тиазолгидрохлорида и 4,8 г (58,5 ммоль) ацетата натрия растворяли или суспендировали в 400 мл этанола, а затем кипятили и дефлегмировали в течение 16 часов. Полученный раствор охлаждали до комнатной температуры и фильтровали. Остаток промывали водой для удаления неорганических солей. Получали продукт 10,8 г (51,6%). Температура плавления: 163-165°C.
Промежуточное соединение 2
Метил 4-(2-бром-4-фторфенил)-2-(тиазол-2-ил)-6-метил-1,4-дигидропиримидин- 5-карбоновый эфир
Промежуточное соединение 2 синтезировали из метилацетоацетата способом, аналогичным способу для промежуточного соединения 1. Выход: 53% (температура плавления: 155-157°C).
Промежуточное соединение 3
Этил 6-бромметил-4-(2-бром-4-фторфенил)-2-(тиазол-2-ил)-1,4-дигидропиримидин- 5-карбоновый эфир
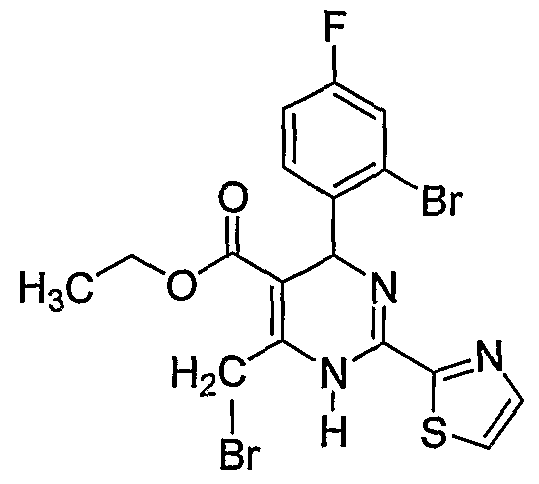
5,0 г (11,8 ммоль) промежуточного соединения 1 добавляли к 100 мл четырех хлористого углерода и нагревали до 50°C в атмосфере газообразного аргона с получением прозрачного раствора. При этой температуре 2,33 г (13,0 ммоль) N-бромсукцинимида добавляли в раствор и перемешивали при этой температуре в течение 10 минут. Полученный раствор затем сразу же охлаждали и фильтровали при комнатной температуре, и снижали давление для концентрирования. Чистота полученного продукта выше 90% согласно результатам тестирования с помощью ВЭЖХ, и этот продукт использовали в качестве исходного материала на следующей стадии. Rf=0,69 (соотношение петролейного эфира к этилацетату составляет 8:2).
Промежуточное соединение 4
Метил 6-бромметил-4-(2-бром-4-фторфенил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоновый эфир
Промежуточное соединение 4 синтезировали из промежуточного соединения 2 способом, аналогичным способу для получения промежуточного соединения 3. Rf=0,69 (соотношение петролейного эфира к этилацетату составляет 8:2).
В. Получения по Примерам
Пример 5
Этил 4-(2-бром-4-фторфенил)-2-(тиазол-2-ил)-6-(4-морфолинилметил)-1,4-дигидропиримидин-5-карбоновый эфир (b)

2,0 г промежуточного соединения 3 добавляли к 15 мл метанола с получением раствора. Раствор смешивали с 5-кратным объемом морфолина и перемешивали в течение 30 минут при комнатной температуре. Полученный раствор затем разбавляли водой и экстрагировали этилацетатом. Выход: 1,7 г. Температура плавления: 161-163°С. Rf=0,45 (соотношение петролейного эфира к этилацетату составляет 8:2)
Пример 6
Метил 4-(2-бром-4-фторфенил)-2-(тиазол-2-ил)-6-(4-морфолинилметил)-1,4-дигидропиримидин-5-карбоновый эфир (а)
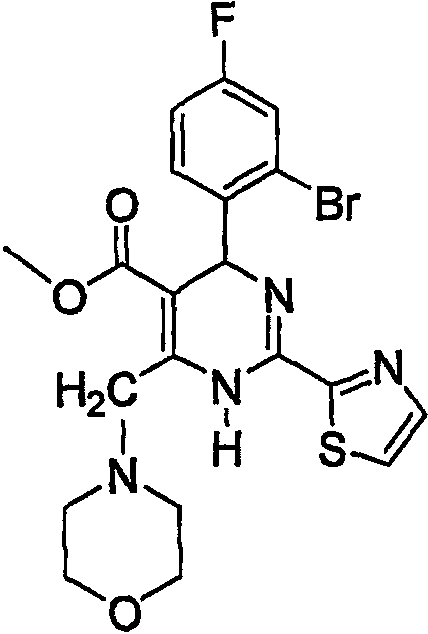
Соединение примера 6 синтезировали из промежуточного соединения 4 способом, аналогичным способу получения соединения примера 5. Температура плавления: 173-175°С. Rf=0,43 (соотношение петролейного эфира к этилацетату составляет 8:2).
Энантиомеры, полученные в Примере 5 и в Примере 6, разделяли на хиральной колонке (Daicel Chiralpak AS-H, подвижная фаза:н-гексан/этанол=99/1).
Активные соединения против HBV в этих двух примерах являются энантиомерами, имеющими относительно длительное время удерживания.
Данные, касающиеся активности соединений по настоящему изобретению, перечислены ниже:
Обработка клеток HepG2.2.15, продуцирующих вирус гепатита В соединениями по настоящему изобретению, может приводить к уменьшению внутри- и/или внеклеточной вирусной ДНК.
Пример 7

Стадия 1: Получение метил 4-(4-морфолинил)-3-оксобутаноата

К смеси метил-4-хлорацетоацетата (1,0 г, 6,64 ммоль) в дихлорметане (10 мл) добавляли морфолин (1,27 г, 14,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли воду, а затем смесь нейтрализовывали 2Н хлористоводородной кислотой до рН 7,0. Органическую фазу отделяли и сушили над безводным сульфатом натрия. Органическую фазу концентрировали в вакууме, затем осадок очищали колоночной хроматографией на силикагеле с использованием смеси 10:1 (об./об.) петролейный эфир/этилацетат в качестве элюента, с получением метил-4-(4-морфолинил)-3-оксобутаноата в виде бесцветной жидкости (0,48 г, 36%). Продукт был охарактеризован с помощью следующих спектроскопических данных: МС (ESI, положительный ион) m/z: 202,1 [M+l]; 1H-ЯMP (400 МГц, СDСl3): δ 2,52 (т, J=2,4 Гц, 4Н), 3,38 (с, 2Н), 3,50 (с, 2Н), 3,69 (т, J=2,4 Гц, 4Н), 3,76 (с, 3Н).
Стадия 2: Получение метил-2-(2-бром-4-фторбензилиден)-4-морфолино-3-оксобутаноата

К раствору 2-бром-4-фторбензальдегида (3,12 г, 15,4 ммоль) и метил-4-(4-морфолинил)-3-оксобутаноата (3,09 г, 15,4 ммоль) в изопропаноле (20 мл) добавляли пиперидин (0,1 мл) и ледяную уксусную кислоту (0,13 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем смесь концентрировали в вакууме и осадок очищали колоночной хроматографией на силикагеле с использованием смеси 10:1 (об./об.) дихлорметан/этилацетат в качестве элюента с получением желаемого соединения в виде желтого твердого вещества (3,32 г, 56%). Продукт использовали на следующей стадии в качестве смеси цис/транс-изомеров. Продукт был охарактеризован с помощью следующих спектроскопических данных: МС (ESI, положительный ион) m/z: 387,0 [М+2]; 1H-ЯМР (400 МГц, СDСl3): δ 2,52-2,60 (м, 4Н), 3,69-3,72 (м, 4Н), 3,77 (с, 3Н), 4,10 (с, 2Н), 7,17-7,36 (м, 3Н), 8,52 (с, 0,6Н), 8,90 (с, 0,4Н).
Стадия 3: Получение метил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоксилата (соед. а)
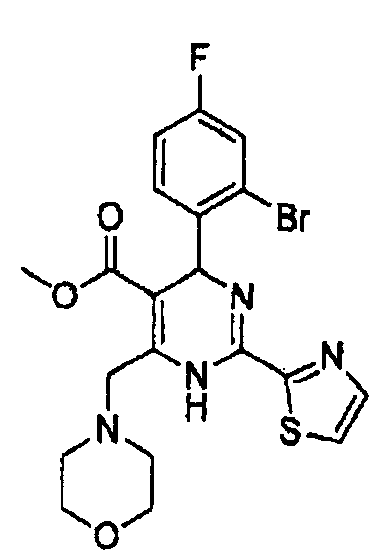
К раствору метил-2-(2-бром-4-фторбензилиден)-4-морфолино-3-оксобутаноата (0,77 г, 2,0 ммоль) в изопропаноле (10 мл) добавляли гидрохлорид тиазол-5-карбоксимидамида (0,32 г, 2 ммоль)) и ацетат натрия (0,20 г, 2,4 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 8 часов и концентрировали, затем осадок растворяли в дихлорметане (30 мл), органическую фазу промывали водным раствором хлорида натрия, а затем сушили над безводным сульфатом натрия и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с использованием смеси 4:1 (об./об.) петролейный эфир/этилацетат в качестве элюента с получением указанного в заголовке соединения в виде желтого твердого вещества (0,45 г, 45%). Продукт был охарактеризован с помощью следующих данных: температура плавления 173-175°С; МС (ESI, положительный ион) m/z: 496,0 [М+2]; 1H-ЯМР (400 МГц, DMSO): δ 3,30 (с, 3Н), 3,40 (с, 4Н), 3,90 (с, 4Н), 4,52-4,74 (м, J=16 Гц, 2Н), 6,02 (с, 1Н), 7,27-7,60 (м, 3Н), 8,08 (д, J=2,8 Гц, 1Н), 8,22 (с, 1Н), 9,98 (с, ушир. с, 1Н).
Пример 8
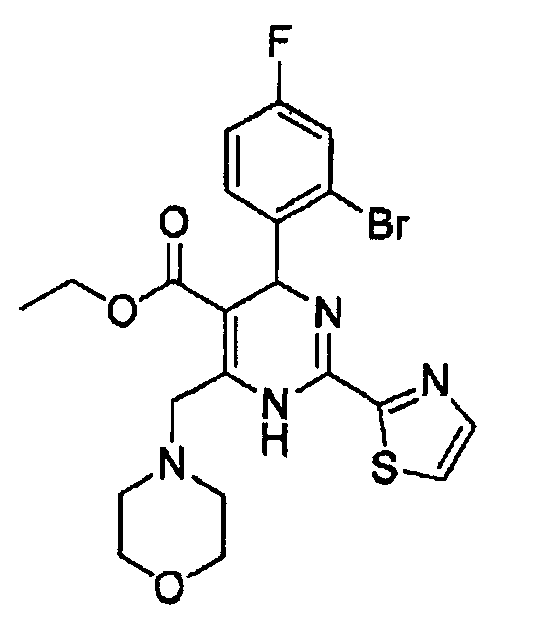
Стадия 1: Получение этил-4-морфолино-3-оксобутаноата
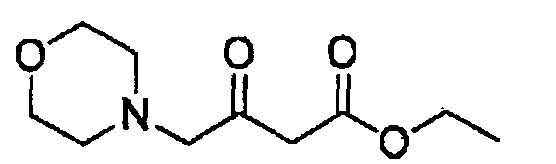
К раствору этил-4-хлорацетоацетата (1,09 г, 6,64 ммоль) в дихлорметане (10 мл) добавляли морфолин (1,27 г, 14,6 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли воду (20 мл) и смесь нейтрализовывали 2 Н хлористоводородной кислотой до РН 7,0. Органическую фазу отделяли и сушили над безводным сульфатом натрия, органическую фазу фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с использованием смеси 10:1 (об./об.) петролейный эфир/этилацетат в качестве элюента с получением указанного в заголовке соединения в виде бесцветной жидкости (0,4 г, 30%). Продукт был охарактеризован с помощью следующих спектроскопических данных: МС (ESI, положительный ион) m/z: 216,1 [М+1]; 1H-ЯМР (400 МГц, CDCl3): δ 1,29 (т, J=7,2 Гц, 3Н), 2,51(кв, J=2,4 Гц, 4Н), 3,28 (с, 2Н), 3,50 (с, 2Н), 3,73 (т, J=2,4 Гц, 4Н), 4,19 (т, J=7,2 Гц, 2Н).
Стадия 2: Получение этил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоксилата (соед. в)
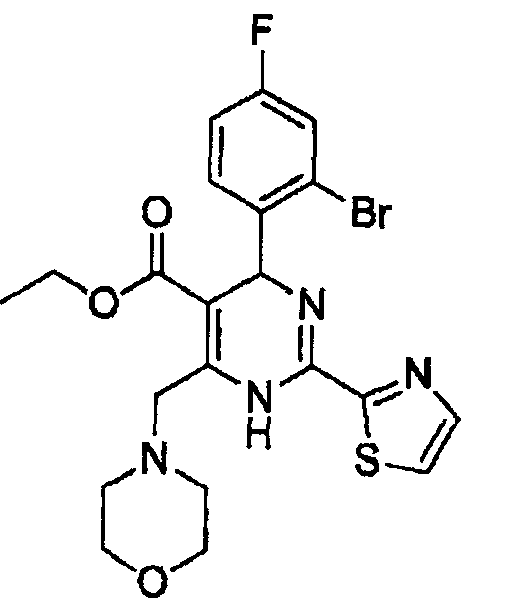
К раствору 2-бром-4-фторбензальдегида (0,40 г, 2,0 ммоль) в изопропаноле (10 мл) последовательно добавляли этил этил-4-морфолино-3-оксобутаноат (0,43 г, 2 ммоль), гидрохлорид тиазол-5-карбоксимидамида (0,32 г, 2 ммоль) и ацетат натрия (0,20 г, 2,4 ммоль). Затем реакционную смесь кипятили с обратным холодильником в течение 8 часов и концентрировали в вакууме, осадок растворяли в дихлорметане (30 мл). Органическую фазу промывали водным раствором хлорида натрия (20 мл×2), сушили над безводным сульфатом натрия, фильтровали, а затем концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с использованием смеси 4:1 (об./об.) петролейный эфир/этилацетат в качестве элюента с получением указанного в заголовке соединения в виде желтого твердого вещества (0,57 г, 56%). Продукт был охарактеризован с помощью следующих данных: температура плавления 161-163°С; МС (ESI, положительный ион) m/z: 510,0 [М+2]; 1H-ЯМР (400 МГц, DMSO): δ 1,08 (т, J=7,2 Гц, 3Н), 3,40 (с, 4Н), 3,90 (с, 4Н), 4,02 (kb, J=7,2 Гц, 2Н), 4,50-4,72 (m, J=16 Гц, 2H), 6,02 (с, 1Н), 7,27-7,60 (м, 3Н), 8,06 (д, J=2,8 Гц, 1Н), 8,12 (с, 1Н), 10,30 (с, ушир. с, 1Н).
Пример 9
Стадия 1: получение этил-3-амино-4-морфолинобут-2-еноата

К раствору этил-4-морфолино-3-оксобутаноата (8,56 г, 40 ммоль) в этаноле (30 мл) добавляли водный раствор NH3 (50 мл, 25%). Реакционную смесь перемешивали при 25°С в течение 1 часа после чего образовывался белый осадок. Белый осадок фильтровали, объединяли, промывали 10 мл воды, а затем сушили при комнатной температуре в течение 24 часов с получением указанного в заголовке соединения в виде белого твердого вещества (7,1 г, 83%). Продукт был охарактеризован с помощью следующих спектроскопических данных: МС (ESI, положительный ион) m/z: 215,1 [М+1]; 1H-ЯМР (400 МГц, СDСl3): δ 1,29 (т, J=7,2 Гц, 3Н), 2,50 (kb, J=2,4 Гц, 4Н), 3,10 (с, 2Н), 3,73 (т, J=2,4 Гц, 4Н), 4,19 (т, J=7,2 Гц, 2Н), 4,74 (с, 1Н), 8,62 (с, 2Н).
Стадия 2: Получение этил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоксилата (соед. в)
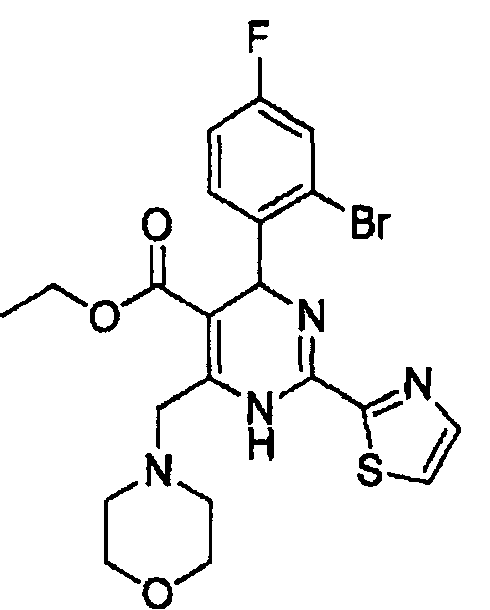
К раствору 2-бром-4-фторбензальдегида (0,40 г, 2,0 ммоль) в изопропаноле (10 мл) последовательно добавляли 3-амино-4-морфолинобут-2-еноат (0,43 г, 2 ммоль), гидрохлорид тиазол-5-карбоксимидамида (0,32 г, 2 ммоль) и ацетат натрия (0,20 г, 2,4 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 8 часов и концентрировали в вакууме, затем осадок растворяли в дихлорметане (30 мл). Органическую фазу промывали водным раствором хлорида натрия (20 мл×2), сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Осадок очищали колоночной хроматографией на силикагеле с использованием смеси 4:1 (об./об.) петролейный эфир/этилацетат в качестве элюента с получением указанного в заголовке соединения в виде желтого твердого вещества (0,57 г, 56%). Продукт был охарактеризован с помощью следующих данных: температура плавления 161-163°С; МС (ESI, положительный ион) m/z: 510,0 [М+2]; 1H-ЯМР (400 МГц, DMSO): δ 1,08 (т, J=7,2 Гц, 3Н), 3,40 (с, 4Н), 3,90 (с, 4Н), 4,02 (кв, J=7,2 Гц, 2Н), 4,50-4,72 (м, J=16 Гц, 2Н), 6,02 (с, 1Н), 7,27-7,60 (м, 3Н), 8,06 (д, J=2,8 Гц, 1Н), 8,12 (с, 1Н), 10,30 (с, ушир. с., 1Н).
m/z: 387,0 [M+2]; 1H-ЯМР (400 МГц, СDСl3): δ 2,52-2,60 (м, 4Н), 3,69-3,72 (м, 4Н), 3,77 (с, 3Н), 4,10 (с, 2Н), 7,17-7,36 (м, 3Н), 8,52 (с, 0,6Н), 8,90 (с, 0,4Н).
Примеры 10-12
Соединение: метил-4-(2-бром-4-фторфенил)-6-(морфолинометил) -2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоксилат (соед. а)
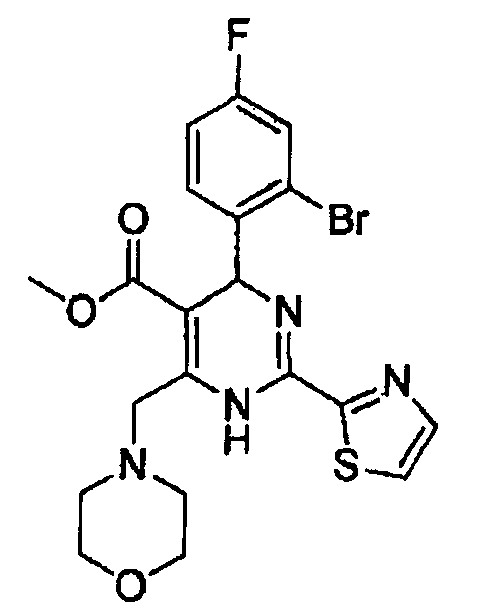
Соединение: этил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоксилат (соед. в)

Пример 10
Активные ингредиенты примера 10 представляют собой соединение (соед. а): (метил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-е-5-карбоксилат) и ингибитор полимеразы HBV (адефовир дипивоксил). Состав примера 10 является таким, как показано в таблице ниже.
Методика получения примера 10
1) Из polyvidone K30 приготавливали 10% водный раствор polyvidone K30.
2) Смесь микрокристаллической целлюлозы, прежелатинизированного крахмала, поливинилполипирролидона (первое добавление), соединения (соед. а) и адефовира дипивоксила смешивали в однородную смесь в высокоскоростном влажном грануляторе. В смесь добавляли связующее вещество и процесс грануляции продолжали в течение приблизительно 5 минут.Образовавшиеся гранулы доводили до требуемого размера с помощью 1-мм сита.
3) Гранулы сушили в сушильной печи или в псевдоожиженном слое.
4) Высушенные гранулы доводили до требуемого размера с помощью 1-мм сита.
5) К гранулам после доведения размера добавляли вторую часть поливинилполипирролидона. Смесь перемешивали в течение 5 минут.
6) К смеси, полученной на стадии 5, добавляли стеарат магния. Смесь перемешивали в течение 5 минут.
7) Из смеси, полученной на стадии 6, получали таблетки.
Пример 11
Активные ингредиенты примера 11 представляют собой соединение (соед. в):(этил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин е-5-карбоксилат) и ламивудин. Состав примера 11 является таким, как показано в таблице ниже.
Методика получения примера 11
1) Из гидроксипропилметилцеллюлозы получали 8% водный раствор гидроксипропилметилцеллюлозы.
2) Смесь микрокристаллической целлюлозы, лактозы, кроскармеллозы натрия (первое добавление), соединения (соед. в) и ламивудиневира перемешивали в однородную смесь на высокоскоростном влажном грануляторе. В смесь добавляли связующее вещество, и процесс грануляции продолжали в течение приблизительно 5 минут. Образовавшиеся гранулы доводили до требуемого размера с помощью 1-мм сита.
3) Гранулы сушили в сушильной печи или в псевдоожиженном слое.
4) Высушенные гранулы доводили до требуемого размера с помощью 1-мм сита.
5) К гранулам после доведения размера добавляли вторую часть кроскармеллозы натрия. Смесь перемешивали в течение 5 минут.
6) К смеси, полученной на стадии 5, добавляли стеарат магния. Смесь перемешивали в течение 5 минут.
7) Из смеси, полученной на стадии 6, получали таблетки.
8) Простые таблетки покрывали Opadry II.
Пример 12
Активные ингредиенты примера 12 представляют собой соединение (соед. в): (этил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин е-5-карбоксилат) и АТ-61. Состав примера 12 является таким, как показано в таблице ниже.
Методика получения примера 12
1) Из гидроксипропилметилцеллюлозы получали 8% водный раствор гидроксипропилметилцеллюлозы.
2) Смесь микрокристаллической целлюлозы, лактозы, кроскармеллозы натрия (первое добавление), соединения (соед. в) и АТ-61 перемешивали в однородную смесь на высокоскоростном влажном грануляторе. В смесь добавляли связующее вещество, и процесс грануляции продолжали в течение приблизительно 5 минут. Образовавшиеся гранулы доводили до требуемого размера с помощью 1-мм сита.
3) Гранулы сушили в сушильной печи или в псевдоожиженном слое.
4) Высушенные гранулы доводили до требуемого размера с помощью 1-мм сита.
5) К гранулам после доведения размера добавляли вторую часть кроскармеллозы натрия. Смесь перемешивали в течение 5 минут.
6) К смеси, полученной на стадии 5, добавляли стеарат магния. Смесь перемешивали в течение 5 минут.
7) Из смеси, полученной на стадии 6, получали таблетки.
8) Простые таблетки покрывали Opadry II.
Примеры 13-14
Пример 13
Активный ингредиент примера 13 представляет собой соединение (соед. а) (метил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоксилат), имеющее следующую структуру:

Состав примера 13 является таким, как представлено в таблице ниже.
Методика получения примера 13
Смесь соединения (соед. а), микрокристаллической целлюлозы, лактозы и карбоксиметилкрахмала натрия просеивали и перемешивали в течение 10 минут. К смеси добавляли стеарат магния и смесь перемешивали в течение 5 минут с получением перемешанного порошка. Оболочки капсулы заполняли перемешанным порошком, а затем запаивали с получением капсул.
Пример 14
Активным ингредиентом указанного примера является
соединение (соед. в) (этил-4-(2-бром-4-фторфенил)-6-(морфолинометил)-2-(тиазол-2-ил)-1,4-дигидропиримидин-5-карбоксилат), имеющее следующую структуру:
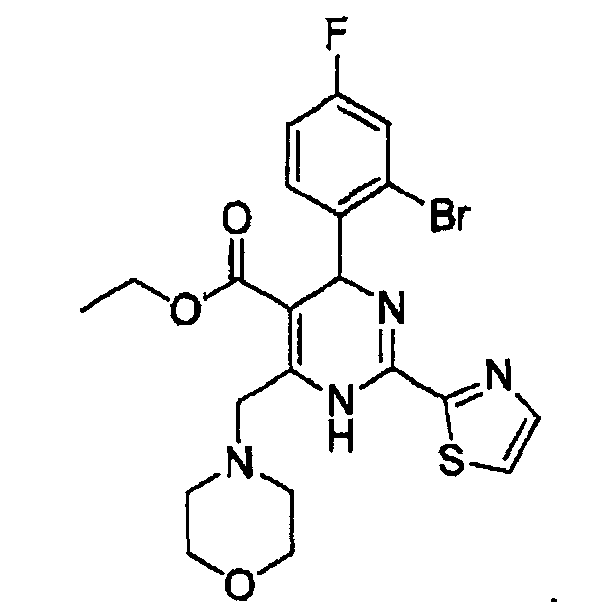
Состав примера 14 является таким, как показано в таблице ниже.
Методика получения примера 2
Смесь соединения (соед. в), микрокристаллической целлюлозы, прежелатинизированного крахмала и поливинилполипирролидона просеивали и перемешивали в течение 10 минут. К смеси добавляли стеарат магния, и смесь перемешивали в однородную смесь в течение 5 минут с получением перемешанного порошка. Из перемешанного порошка получали таблетки.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Примеры, описанные в настоящей заявке, показывают, что соединения, описанные в настоящем изобретении, демонстрируют эффективное противовирусное действие с IC50 менее 1 нМ. Следовательно, эти соединения могут быть использованы для лечения заболевания, индуцированного вирусами, главным образом, острых и хронических персистентных HBV инфекций в соответствии со способами по настоящему изобретению или любым способом, известным специалисту в данной области.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДИГИДРОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ | 2013 |
|
RU2655914C9 |
| ПРОЦЕССЫ ПРИГОТОВЛЕНИЯ ПРОИЗВОДНЫХ СОЕДИНЕНИЙ ДИГИДРОПИРИМИДИНА И ИХ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ | 2014 |
|
RU2697707C1 |
| ПРОЦЕССЫ ПРИГОТОВЛЕНИЯ ПРОИЗВОДНЫХ ДИГИДРОПИРИМИДИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2688193C1 |
| СОЕДИНЕНИЯ ДИГИДРОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ | 2015 |
|
RU2682672C2 |
| ДИГИДРОПИРИМИДИНЫ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2245881C9 |
| Ингибитор вируса гепатита В (ВГВ) | 2019 |
|
RU2736975C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЦЕТАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И ИНГИБИТОРЫ ПРОТЕАЗ НА ИХ ОСНОВЕ | 1997 |
|
RU2181360C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| ХРОМЕНОНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РI3-КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2598028C2 |
Изобретение относится к соединению формулы (I), или его таутомеру (Iа), или энантиомеру, или его физиологически приемлемой соли, где R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой C1-C4 алкил, R6 представляет собой тиазолил-2-ил, Х представляет собой метилен и Z представляет собой морфолинил. Также изобретение относится к способам получениям (вариантам) соединения формулы (I) или (Iа). Соединение формулы (I) или (Iа) применяют для получения фармацевтической композиции, для лечения и профилактики инфекций HBV и заболеваний, индуцированных HBV, таких как гепатит В. Технический результат - бром-фенил замещенные тиазолилдигидропиримидины для борьбы с HBV инфекциями. 11 н. и 9 з.п. ф-лы, 8 табл., 14 пр.
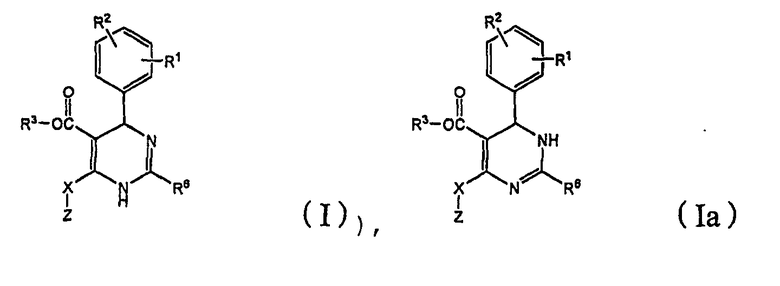
1. Соединение формулы (I) или его таутомер (Iа):

или энантиомер или его физиологически приемлемая соль, где
R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой C1-C4 алкил, R6 представляет собой тиазолил-2-ил, Х представляет собой метилен, и Z представляет собой морфолинил.
2. Соединение по п.1 или его энантиомер или физиологически приемлемая соль, где R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой метил или этил, R6 представляет собой тиазолил-2-ил, Х представляет собой метилен, и Z представляет собой морфолинил.
3. Соединение, имеющее одну из следующих структур или его энантиомер, таутомер или физиологически приемлемая соль:  , или
, или 
4. Соединение, имеющее одну из следующих структур или его левоизомер, таутомер или физиологически приемлемая соль:
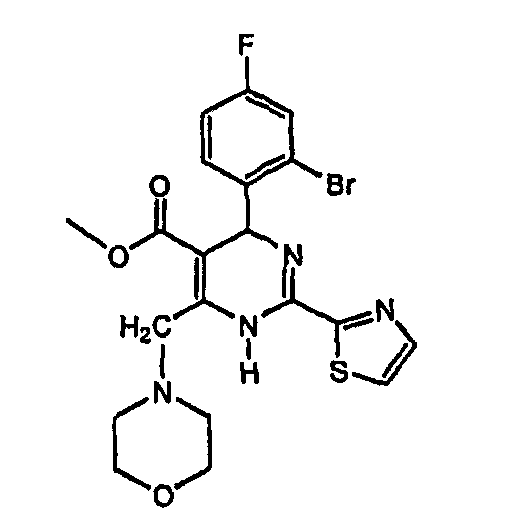 , или
, или 
5. Соединение по любому из пп.1-4 или его энантиомер или физиологически приемлемая соль, где соль представляет собой соль неорганической кислоты или соль органической кислоты.
6. Соединение по п.5 или его энантиомер или физиологически приемлемая соль, где соль неорганической кислоты представляет собой соль соляной кислоты, соль бромистоводородной кислоты, соль фосфорной кислоты или соль серной кислоты.
7. Соединение по любому из пп.1-4 или его энантиомер или физиологически приемлемая соль, где соль органической кислоты представляет собой карбоксилат или сульфонат.
8. Соединение по п.7 или его энантиомер или физиологически приемлемая соль, где карбоксилат представляет собой ацетат, малеат, фумарат, малат, цитрат, тартрат, лактат или бензоат.
9. Соединение по п.7 или его энантиомер или физиологически приемлемая соль, где сульфонат представляет собой метансульфонат, этансульфонат, бензолсульфонат, толуолсульфонат или нафталендисульфонат.
10. Способ получения соединения по п.1, где указанный способ характеризуется:
(а) взаимодействием бензальдегида формулы (II) с β-кетоэфиром формулы (III) с образованием бензилиденового соединения формулы (IV):
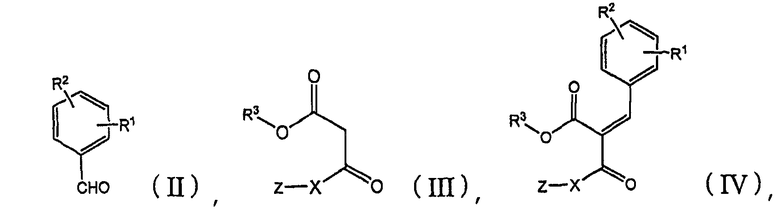 и
и
(b) взаимодействием бензилиденового соединения формулы (IV) с амидином формулы (V):
 или его солью
или его солью
где R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой С1-С4 алкил, R6 представляет собой тиазолил-2-ил, Х представляет собой метилен, и Z представляет собой морфолинил.
11. Способ получения соединения по п.1, где указанный способ характеризуется взаимодействием соединения формулы (III) с альдегидом (II) и амидином (V) или его солью на одной стадии;
где R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой C1-C4 алкил, R6 представляет собой тиазолил-2-ил, Х представляет собой метилен, и Z представляет собой морфолинил.
12. Способ получения соединения по п.1, где Х формулы (I) представляет собой метилен, и где указанный способ характеризуется взаимодействием соединения формулы (VI) с морфолином (VII) или его солью:

где Y представляет собой нуклеофильный заместитель, и R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой C1-C4 алкил, и R6 представляет собой тиазолил-2-ил.
13. Способ получения соединения по п.1, отличающийся стадией взаимодействия соединения формулы (II) с альдегидом формулы (X) и амидином формулы (V) или его солью:

где R1 представляет собой о-бром, R2 представляет собой п-фтор, R3 представляет собой C1-C4 алкил, R6 представляет собой тиазолил-2-ил, Х представляет собой метилен, и Z представляет собой морфолинил.
14. Фармацевтическая композиция, для лечения и профилактики инфекций HBV, и заболеваний, индуцированных HBV, содержащая следующие активные компоненты:
A) по меньшей мере одно соединение по любому из пп.1-9; и,
B) по меньшей мере одно средство против HBV, отличное от компонента А.
15. Фармацевтическая композиция по п.14, в которой компонент В представляет собой ингибитор полимеразы HBV, ламивудин или фенилпропенамидное соединение следующей формулы:

или его соль, где
каждый из R1 и R2 независимо представляет собой С1-4 алкил или вместе с атомом азота на котором они расположены, образуют кольцо, имеющее от 5 до 6 кольцевых атомов, которые содержат углерод и/или кислород; и
каждый из R3-R12 независимо представляет собой водород, галоген, С1-С4-алкил, необязательно замещенный С1-С4-алкокси, нитро, циано или трифторметил.
16. Фармацевтическая композиция по п.15, где компонент В представляет собой фенилпропенамидное соединение, которое имеет следующую структуру:

17. Применение соединения по любому из пп.1-9 для получения лекарственного средства для лечения и профилактики вирусного заболевания, гепатита В или заболевания, вызванного гепатитом В.
18. Применение композиции по любому из пп.14-16 для получения лекарственного средства для лечения и профилактики вирусного заболевания, гепатита В или заболевания, вызванного гепатитом В.
19. Применение по пп.17-18, где лекарственное средство представляет собой средство для лечения и профилактики заболевания, вызванного гепатитом В, выбранного из гепатита, цирроза или печеночно-клеточной карциномы.
20. Противовирусная фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-9 или по меньшей мере одну композицию по любому из пп.14-16 и фармацевтически приемлемый носитель.
| ДИГИДРОПИРИМИДИНЫ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2245881C9 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 4822798 A1, 18.04.1989. | |||

