Настоящее изобретение относится к способу получения 2-гидрокси-4-метилтиомасляной кислоты (гидроксианалог метионина или сокращенно ГАМ), в котором ГАМ выделяют из реакционной смеси, получаемой присоединением цианистоводородной кислоты (HCN) к метилмеркаптопропиональдегиду (ММП) и гидролизом получаемого при этом циангидрина метилмеркаптопропиональдегида (ММП-ЦГ) с помощью серной кислоты, причем реакционную смесь контактируют с практически несмешиваемым с водой органическим растворителем в экстракционной системе жидкость/жидкость с целью образования экстракционного раствора, содержащего растворитель и переведенный из реакционной смеси ГАМ, и ГАМ получают в виде экстракта из этого экстракционного раствора путем упаривания.
2-Гидрокси-4-метилтиомасляная кислота представляет собой гидроксианалог незаменимой аминокислоты метионин (ГАМ) в рацемической форме и является как и аминокислота важной добавкой к кормам для животных. В птицеводстве ГАМ проявляет ростстимулирующие свойства, сходные с таковыми у известной этой же способностью аминокислоты. Однако и в других областях, связанных с кормлением животных, этот аддитив вызывает повышенный интерес.
В большинстве случаев ГАМ применяют в виде водных концентратов, причем эти последние наряду с мономером содержат также некоторые количества олигомеров, главным образом ди- и тримерные линейные эфирокислоты. Содержание этих олигомеров зависит от условий получения и выбранной концентрации. Однако по причине их малой питательной ценности и неблагоприятного воздействия на текучесть, обусловленного повышением вязкости, желательно их процентную долю поддерживать на минимальном уровне. Коммерчески доступные композиции при общей концентрации 88-90 мас.% содержат в общей сложности до 24 мас.%, соответственно около 27 мол.% олигомеров, что соответствует соотношению мономеры/олигомеры 3:1.
При осуществлении общепринятого способа получения ГАМ исходят из 3-метилтиопропиональдегида, называемого также метилмеркаптопропиональдегидом или сокращенно ММП, который подвергают взаимодействию с цианистым водородом с получением 2-гидрокси-4-метилтиобутиронитрила, называемого также ММП- циангидрином или сокращенно ММП-ЦГ (уравнение I).
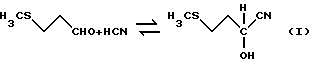
Затем образовавшийся ММП-циангидрин обычно с помощью сильных минеральных кислот, таких, как серная кислота или соляная кислота, через промежуточную стадию получения 2-гидрокси-4- метилтиобутирамида, называемого также ГАМ-амид (уравнение II),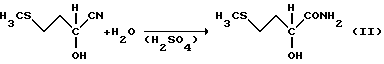
гидролизуют до гидроксианалога метионина (ГАМ) (уравнение III)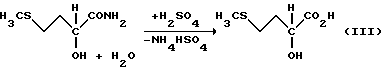
Этот гидролиз может проводиться как по одноступенчатому, так и по двухступенчатому механизму, причем под "ступенями" следует понимать то, что в реакцию гидролиза ММП-ЦГ либо один раз, либо дважды добавляют минеральную кислоту и/или воду, т.е. число ступеней соответствует числу введения добавок.
Касательно ближайшего уровня техники можно сослаться на следующие публикации:
EP-A-0142488 (далее документ D1),
EP-A-0143100 (далее документ D2),
EP-A-0330527 (далее документ D3) и
WO-A-94/28717, соответственно ЕР 93924374 (далее документ D4).
Один из стандартных способов получения ГАМ известен, например, из D1. В соответствии с техническим решением по получению ГАМ в жидкой форме, а именно в виде высококонцентрированного водного раствора, в этой публикации предлагается осуществлять двухступенчатый гидролиз с использованием серной кислоты.
Согласно D1 ГАМ получают реакцией гидролиза, проводимой при соответствующих условиях концентрирования и температурном режиме через амидную ступень с использованием избыточной минеральной кислоты, путем экстракции растворителем, причем в этих целях применяют определенные, частично смешиваемые с водой растворители.
Согласно приведенным в D1 данным отличительный признак описанного в этой публикации способа следует усматривать в получении ГАМ из экстракционного раствора, которое осуществляют таким образом, что процесс получения включает удаление органического растворителя в присутствии по крайней мере приблизительно 5 мас.% воды в пересчете на образующийся экстракт (ГАМ). ГАМ получают из экстракционного раствора путем перегонки (см. примеры), причем предпочтительна дистилляция с водяным паром. Полученный после удаления растворителя из экстракционного раствора путем перегонки с водяным паром погон представляет собой смесь ГАМ'а и воды. В соответствии с этим дистилляцию с водяным паром проводят таким образом, что содержание воды в погоне составляет по крайней мере 5 мас.%.
В указанной публикации говорится далее, что при проведении дистилляции условия в колонне регулируют таким образом, чтобы в ней повсюду, по крайней мере, однако, в кубовом остатке, содержание воды в жидкой фазе составляло 5 мас.%.
Из этого следует, что без присутствия достаточного количества воды во время получения ГАМ из экстракционного раствора следует опасаться увеличения образования нежелательных побочных продуктов (димеров и олигомеров).
При перегонке водяной пар, кроме того, служит в качестве активного агента, способствующего полному удалению экстрагирующего агента из ГАМ-раствора, например, за счет образования низкокипящей азеотропной смеси с соответствующим экстрагирующим агентом.
Другие варианты также в основном стандартного способа описаны в D2. В отличие от ограничительной части формулы изобретения D1 в этой заявке описывается гидролиз ММП-ЦГ с помощью минеральной кислоты, что можно рассматривать как альтернативную возможность применения HCl вместо H2SO4. В целом в данной публикации предлагаются три других варианта, представляющие собой в основном модификации выделения ГАМ из гидролизата минеральной кислоты или переработки экстракта при экстракции в системе жидкость/жидкость.
В одном из вариантов согласно D2 гидролизат без предварительного отделения каких-либо существенных содержащихся в нем фракций твердых веществ контактирует с органическим растворителем.
Кроме того, согласно D2 условия процесса экстракции регулируют таким образом, что экстракт и водный рафинат являются единственными жидкими фазами, которые образуются при разделении фаз после экстракции.
Недостаток этого варианта заключается в том, что экстракция осложняется наличием в гидролизате соли полностью в количестве, образующемся при гидролизе, что приводит к относительно высокому массопотоку гидролизата и соответственно также растворителя. Это в свою очередь обусловливает повышение энергозатрат на выпаривание растворителя и конденсацию, а также удорожание процесса, связанное с потерями растворителей и необходимостью применять соответствующие крупногабаритные аппараты для экстракции и упаривания. Тем самым, следовательно, снижение производственных и инвестиционных затрат в этой части осуществления способа было бы желательным (прежде всего имея в виду габариты такой установки и связанную с этим экономию средств).
Из рафината, полученного экстракцией в виде гомогенной жидкой фазы, согласно D1, соответственно D2, соответственно D4 необходимо путем упаривания или перегонки удалять остатки растворителя, что требует нежелательных дополнительных затрат.
Другой вариант согласно D2 касается удаления органического растворителя из экстракта. С этой целью предусматривается удалять растворитель за счет того, чтобы подвергать экстракт дистилляции с паром, при которой растворитель вытесняется и образуется кубовый остаток из водного ГАМ.
Недостатком при использовании пара является в первую очередь увеличение образующегося количества насыщенного растворителем водного конденсата вторичного пара, из которого затем нежелательными дополнительными операциями, такими, как перегонка или упаривание, необходимо удалять растворитель с тем, чтобы в последующем на соответствующем участке обеспечить его возврат в процесс, или который по соображениям экологии требуется утилизовать, например сжигать, что опять-таки связано со значительными материально-техническими затратами. Таким образом, было бы желательно избежать использования дополнительного стриппинг-пара.
И, наконец, в третьем варианте согласно D2 одним из главных факторов экстракции в системе жидкость/жидкость является тип применяемого растворителя. При выборе соответствующего растворителя заявители руководствуются при этом следующими критериями:
- температура кипения растворителя должна находиться в интервале от 60oC до 200oC;
- коэффициент распределения для ГАМ в равновесной системе гидролизат/растворитель должен составлять по меньшей мере приблизительно два;
- коэффициент распределения растворителя в равновесной системе экстракт/жидкая фаза должен составлять по меньшей мере приблизительно один;
- водорастворимость в растворителе при комнатной температуре не должна превышать приблизительно 12 мас.%.
Относительно высокий диапазон температур кипения применяемых растворителей 60-200oC требует при упаривании экстракта соответственно относительно высоких температур, которые могут повредить продукт, а также использования дополнительных вспомогательных средств, как, например, стриппинг-пар.
Один из существенных недостатков способов, описанных в D1 и D2, состоит, однако, в другом, а именно: при омылении образуется большое количество солевого "балласта", что осложняет в остальном в принципе относительно экономичном способе экстракции в системе жидкость/жидкость его осуществление в той части, которая касается выделения ГАМ. Переработка принудительно образующейся смеси аммониевых солей в большинстве случаев является нерентабельной, хранение по соображениям экологии весьма проблематично и даже в местах, где существуют менее строгие в этом отношении требования, в обозримом будущем, судя по всему, будет запрещено законом.
Неоднократные попытки снизить количество или даже полностью избежать образования солевых отходов в процессе омыления хотя и предпринимались и позволяли получать определенные преимущества, тем не менее их достигали, как правило, либо при наличии целого ряда других недостатков, присущих способам в D1 и D2, либо за счет отказа от предлагаемой технологии.
Так, в D3 описан одноступенчатый способ гидролиза с использованием серной кислоты в качестве агента омыления, в котором обходятся без растворителя и который приводит непосредственно к получению концентрированных водных ГАМ-растворов, причем в качестве побочного продукта получают кристаллический сульфат аммония в готовом для поставок на рынок виде. Указанную цель достигают за счет того, что омыляемую смесь нейтрализуют раствором гидроксида аммония до такой степени, которая обеспечивает перевод избыточной минеральной кислоты и образовавшегося бисульфата аммония в нейтральный сульфат, причем во время этого процесса образуются две жидкие фазы, которые в свою очередь разделяют и упаривают с целью получения, во-первых, жидкого ГАМ и, во-вторых, кристаллического сульфата аммония. При этом различные процессы фильтрации и возврата в цикл комбинируют таким образом, что практически не теряется ни один продукт и не образуются сточные воды, содержащие соль. Качество получаемого таким путем ГАМ сходно с таковым продукта, который получают согласно D1.
Однако и этот экологически относительно безвредный способ имеет ряд недостатков. Как было установлено заявителем, при работе по этому способу, для полной конверсии циангидрина требуются, что обусловлено необходимостью сравнительно более сильного разбавления серной кислоты (20-50%), гораздо большие избыточные количества кислоты, чем это указано. Кроме того, для предотвращения осаждения соли в процессе нейтрализации приходится работать с сильно разбавленной кислотой, что обусловлено необходимостью разделения обеих жидких фаз с требуемой степенью чистоты. Помимо этого, выделенный сульфат аммония имеет клейкую консистенцию и источает сильный запах, что обусловливает необходимость последующих операций по переработке, таких, например, как фильтрационная промывка или перекристаллизация, которые дополнительно удорожают способ. Более того, для проведения процесса упаривания - в отличие от указанного в заявке - требуются более высокие энергозатраты по сравнению со способом в D1, на который в заявке делаются ссылки. Значительное повышение материально-технических затрат связано помимо этого с работой с двумя раздельными потоками твердого материала, предусматривающей фильтрацию/центрифугирование, а также с не показанной на блок-схеме движения материала сушкой сульфата аммония.
Частичное решение указанной дилеммы предлагается в D4. В этой публикации описывается рекуперация серной кислоты из содержащего сульфат потока отходов, образующегося при получении 2-гидрокси-4- (метилтио)масляной кислоты, осуществляемая путем гидролиза 2- гидрокси-4-(метилтио)бутиронитрила с применением серной кислоты.
Рекуперация серной кислоты из содержащих сульфат аммония, бисульфат аммония и/или серную кислоту остатков давно уже относится к уровню техники касательно получения ГАМ и осуществляется, как это имеет место в случае остатков, образующихся при омылении, сжиганием образующихся в процессе омыления и экстракции потоков отходов в так называемой РК-установке (установка для расщепления и получения серной кислоты контактным способом).
При этом по известной специалисту в данной области технологии сначала получают SO2 в качестве продукта разложения, который контактным способом окисляют до SO3, который в завершении превращают в серную кислоту. Получаемую таким путем серную кислоту можно затем повторно возвращать в процесс омыления, тогда как другие первоначальные компоненты "солевых загрязнений" могут использоваться в основном в виде горючих газов.
Как ни прост и совершенен этот способ, он тем не менее не свободен от целого ряда недостатков. Так, в способах согласно D1 и D2 образуются потоки отходов, концентрация сульфатов в которых относительно невелика, однако ее в любом случае недостаточно, чтобы обеспечить непосредственное питание РК-установки. Для этого, как правило, требуется дополнительное концентрирование либо накопление, осуществляемое за счет смешения с более концентрированными потоками отходов из других процессов. Обычно используемые для работы РК-установок растворы содержат сульфат в количестве более 50%. Более высокие концентрации еще менее предпочтительны. Дополнительное концентрирование сточных вод, образующихся при выделении ГАМ, за счет упаривания является, однако, по причине высокой коррозионной агрессивности сточных вод относительно дорогостоящей и трудоемкой операцией, начиная от выбора специальных материалов для выпарных аппаратов и кончая особыми мерами безопасности.
В соответствии с указанным выше уровнем техники и с учетом недостатков известных способов в основу изобретения была положена задача разработать еще один способ получения 2-гидрокси-4-метилтиомасляной кислоты (ГАМ) описанного выше типа, который касательно переработки реакционных продуктов был бы максимально прост по своему осуществлению, не требовал высоких материально-технических затрат и обеспечивал бы получение максимально высококонцентрированного продукта с предельно низким содержанием димеров, олигомеров и побочных продуктов. При этом в новом способе предусматривалось сохранить по возможности преимущества простой по ее осуществлению операции по выделению ГАМ путем экстракции в системе жидкость/жидкость при одновременном обеспечении максимально простой и целенаправленной утилизации образующихся солевых загрязнений в содержащих сульфат сточных водах. Прежде всего необходимо было разработать процесс, который позволял бы при этом, в частности, непосредственно направлять потоки отходов в, например, установку для расщепления и получения серной кислоты контактным способом с целью возврата последней в производственный цикл, т. е. обеспечить возможность ее регенерации в процессе.
Эта равно как и другие не указанные более подробно задачи решаются с помощью способа, описанного выше типа, отличительный признак которого представлен в пункте 1 формулы изобретения.
Предпочтительные варианты способа представлены в пунктах формулы изобретения, зависимых от главного пункта 1.
Благодаря тому, что содержание соли в реакционной смеси до проведения экстракции в системе жидкость/жидкость доводят до уровня концентрации порядка более 50 мас. %, предпочтительно более 55 мас.% (масса/масса), соответственно в пересчете на общее количество неорганических компонентов в смеси, предлагается согласно изобретению такой способ, который обеспечивает получение жидкого ГАМ исключительно высокого качества и благодаря которому совершенно неожиданным образом решается проблема принудительно образующейся соли или по меньшей мере найдено неожиданно более эффективное решение проблемы.
Это обусловлено прежде всего тем, что достигнут целый ряд значительных преимуществ, обеспечивающих, в частности, возможность повышать соответствующим образом концентрацию соли после гидролиза, но до проведения экстракции в системе жидкость/жидкость и тем самым в любом случае не непосредственно перед возможным подключением РК-установки.
К указанным преимуществам согласно изобретению среди прочих относятся следующие:
Обычно осуществляемое перед подачей в РК-установку упаривание может быть полностью исключено.
Для дополнительного концентрирования перед экстракцией пригодны более простые в технологическом отношении и тем самым более дешевые аппараты, поскольку концентрируемый раствор в силу его специального состава во время осуществления этой части способа отличается существенно меньшей агрессивностью и тем самым прежде всего также более слабым коррозионным действием.
В случае по крайней мере частичного смещения процесса упаривания на более предпочтительный по времени момент получают в целом выигрыш касательно энергозатрат. Поскольку упариванию подвергают уже темперированный (горячий) гидролизат, а не рафинат, уже охлажденный в результате экстракции, снижается расход энергии.
При дополнительном концентрировании за счет упаривания более эффективно происходит отделение нежелательных легкокипящих компонентов гидролизата.
Гидролиз ГАМ-амида может проводиться с большей степенью разбавления, следствием чего является более полное химическое превращение. Поскольку при гидролизе ГАМ-амида с целью его максимально полной реализации предпочтительно работать с более разбавленной серной кислотой (<40 мас.%), можно использовать последнюю с меньшей степенью концентрации, не опасаясь благодаря этому, что последующая экстракция с помощью органического растворителя будет осложнена. Благодаря дополнительному концентрированию удается предотвратить ухудшение коэффициента распределения, прежде всего по той причине, что при экстракции в рафинате остается меньше ГАМ'а.
Таким образом, способ по изобретению отвечает прежде всего также требованию касательно предпочтительной концентрации серной кислоты при омылении, равно как и получения ГАМ из гидролизата в сочетании с получением рафината, который благодаря своему составу более пригоден для переработки в РК-установке, причем одновременно существенно улучшается энергетический баланс в целом.
Концентрация соли в гидролизате порядка >50 мас.% обеспечивает вполне приемлемую возможность последующей переработки с помощью РК-установки. Предпочтительно содержание соли в реакционной смеси должно составлять 55-60 мас. %, наиболее целесообразно выбирать этот показатель в пределах от приблизительно 60 до 80 мас.%, соответственно в пересчете на суммарное количество неорганических компонентов в смеси. При определении этого показателя, который можно обозначить как содержание по отношению к "не содержащему органических компонентов основному материалу", учитывается в основном содержание воды, содержание серной кислоты и содержание ионов сульфата, соответственно аммония. Все они и являются основными неорганическими компонентами гидролизата.
Под понятием "дополнительное концентрирование" в контексте настоящего изобретения имеется в виду в принципе повышение концентрации соли (по отношению к не содержащему органических компонентов основному материалу, выраженное в мас.%).
В одном из предпочтительных вариантов способа согласно изобретению в реакционную смесь (гидролизат), полученную присоединением HCN к ММП и гидролизом образовавшегося ММП-ЦГ кислотой H2SO4, с целью дополнительного концентрирования добавляют соответствующее количество сульфата аммония. Благодаря введенным добавкам концентрация соли повышается. Наряду с этим описанный вариант обладает рядом других существенных преимуществ.
Как уже упоминалось выше, следует учитывать два в принципе противоположных эффекта. С одной стороны, необходимо обеспечить максимально полное осуществление гидролизной реакции, для чего приемлема относительно более низкая концентрация амида в имеющейся воде при омылении ГАМ-амида. Отсюда вытекает принудительный вывод: требуется разбавленный раствор из ГАМ и гидросульфата аммония. С другой стороны, для экстракции более целесообразно снижать долю воды в гидролизате, т.е. другими словами, иметь максимально высокую концентрацию ГАМ в водной фазе.
Поэтому согласно D1 и D2 гидролизуют при концентрации серной кислоты порядка <40 мас.%, что, однако, принудительно приводит к использованию разбавленного раствора из ГАМ и гидросульфата аммония. Для улучшения последующей экстракции гидросульфат аммония согласно D1/D2 превращают за счет добавления безводного аммиака в нейтральный сульфат аммония. Таким путем хотя и снижают коррозионные свойства раствора, это в то же время может привести к осаждению твердых веществ, которые могут оказать негативное воздействие на процесс экстракции. Поэтому согласно D1/D2 для повторного растворения выпавших в осадок солей снова добавляют воду. Однако при этом недопустимо снижать концентрацию ГАМ, соответственно соли ниже определенного уровня, поскольку в противном случае это снова отрицательно скажется на эффективности экстракции.
В отличие от D1 и D2 благодаря добавлению сульфата аммония, как это предлагается согласно изобретению, обеспечивается возможность осуществлять гидролиз ГАМ-амида с более низкой степенью разбавления, что приводит к более полному химическому превращению на стадии гидролиза; одновременно не требуется более проводить нейтрализацию, т. е. отпадает необходимость в использовании безводного аммиака и в возможном повторном разбавлении.
В предпочтительном варианте выполнения способ по изобретению осуществляют таким образом, что перед выделением ГАМ'а добавляют определенное количество сульфата аммония, способствующее высаливанию с образованием двух фаз. Из D1/D2 хотя и известно, что наличие высокой концентрации соли (лучше всего гидросульфата аммония) "высаливает" ГАМ и тем самым оказывает положительный эффект на коэффициент распределения, однако согласно названным публикациям двухфазности следует избегать, поскольку этот фактор может отрицательно воздействовать на экстракцию. В противоположность этому согласно настоящему изобретению было установлено, что двухфазность, образующаяся в результате добавок сульфата аммония, особенно способствует успешному осуществлению экстракции и оказывает в целом положительное воздействие на этот процесс. Так, в частности, в одном из вариантов способа к полученной согласно изобретению двухфазной смеси можно добавлять такое количество растворителя, что в результате образуются две прозрачные фазы, разделение которых может проводиться только за одну операцию.
Кроме того, благодаря добавлению сульфата аммония на этой стадии способа получения ГАМ достигается одно общее преимущество, заключающееся в том, что целевой продукт получают без потерь и с высоким качеством. Добавки сульфата аммония предназначены только лишь для повышения концентрации аммониевой соли, тогда как концентрация ГАМ остается на прежнем уровне. Далее может отпасть необходимость в дополнительной термической нагрузке (обесцвечивание) на требуемый целевой продукт, более того, в результате растворения сульфата аммония происходит понижение температуры. Несмотря на это на коэффициент распределения оказывается положительное влияние.
Представленный вариант, в котором предусматривается добавление сульфата аммония, хотя и обладает неоспоримыми преимуществами касательно получения без потерь высококачественного продукта, в альтернативной модификации способа по изобретению может оказаться предпочтительным повышать концентрацию соли в реакционной смеси (гидролизате) путем упаривания.
В зависимости от степени концентрации соли в рафинате может иметь место увеличение кубового остатка растворителя, который согласно публикациям D1, D2 или D4 должен быть отделен на стадии упаривания рафината. В отличие от этого согласно изобретению было установлено, что возвращаемый таким путем в производственный цикл растворитель вследствие термической нагрузки, которой он был подвергнут в сильно кислой среде, в значительной степени загрязнен побочными продуктами и тем самым не пригоден для рециркуляции в экстракционную систему.
Неожиданным образом согласно изобретению было установлено, что даже незначительное охлаждение кубового продукта приводит к выходу двухфазной смеси, содержащей водный и органический рафинат, причем органический рафинат состоит на более чем 97% из растворителя, который непосредственно после простого отделения в разделительном сосуде без дальнейших дополнительных затрат направляют в экстракционную систему, минимизируя таким образом потери растворителя.
Далее один из особенно предпочтительных вариантов способа по изобретению, в котором концентрацию соли повышают за счет упаривания, отличается тем, что непосредственно из экстракционной системы поступают по крайней мере три жидкие фазы.
При этом хотя и имеется принципиальная возможность для образования гомогенного рафината и состоящего из двух жидких фаз экстракта, причем экстракт в качестве первой жидкой фазы включает в основном ГАМ, растворитель и незначительные количества воды, тогда как в качестве второй жидкой фазы в его состав входят в основном вода, ГАМ и незначительные количества соли, тем не менее гораздо предпочтительнее, чтобы при осуществлении одного из вариантов способа по изобретению образовывался гомогенный экстракт и состоящий из двух жидких фаз рафинат. В этом случае особенно целесообразно обеспечить возможность для образования рафината в качестве первой жидкой фазы, содержащего в основном аммониевую соль и воду, а также незначительные количества ГАМ и органического растворителя, тогда как в качестве второй жидкой фазы он должен состоять в основном из органического растворителя и предельно малых количеств воды и ГАМ'а.
Согласно предпочтительной модификации способа по изобретению последующие операции после процесса собственно упаривания осуществляют таким образом, что вторая жидкая фаза содержит ГАМ в количестве от 0,01 до 0,5 мас.%, растворитель в количестве от 90 до 99 мас.% и воду в количестве от 0,1 до 10 мас. %, тогда как первая жидкая фаза содержит воду в количестве от 20 до 50 мас.%, ГАМ в количестве от 0,01 до 0,5 мас.% и соль в количестве от 50 до 80 мас.%, причем компоненты каждой фазы должны составлять в сумме 100 мас.%.
Применяемый для упаривания с целью получения ГАМ экстракционный раствор получают из реакционной смеси путем экстракции. В принципе для этого могут естественно использоваться все известные из уровня техники органические растворители, обладающие указанными выше свойствами. Используемый для экстракции органический растворитель должен быть в основном несмешиваемым с водой. Допустима, правда, частичная смешиваемость этого органического растворителя с водой. Среди возможных растворителей, пригодных для разделения веществ способом экстракции в системе жидкость/жидкость, имеется множество таковых, которые отвечают условиям химической индифферентности и малой растворимости в воде. Предпочтительно, как правило, чтобы водорастворимость не превышала примерно 15 мас.%, преимущественно 10 мас.% при комнатной температуре. Среди пригодных для использования в указанных целях растворителей предпочтительны таковые, температура кипения которых находится в интервале от приблизительно 60oC до приблизительно 200oC, предпочтительно от приблизительно 70oC до приблизительно 150oC. Коэффициент распределения между растворителем, содержащим экстрагированный ГАМ, и водным рафинатом, образующимся после контактирования растворителя и ГАМ-гидролизата, должен составлять по меньшей мере примерно 2 для ГАМ в равновесной системе. Предпочтительно этот коэффициент распределения равен по меньшей мере 5. Также и коэффициент распределения ГАМ в равновесной системе экстракционный раствор/промывочная вода не должен быть ниже приблизительно 1,0. В дополнение к указанному раствор должен обладать также низкой токсичностью.
В качестве растворителей для проведения экстракции пригодны прежде всего многие кетоны, альдегиды и эфиры карбоновых кислот. К особенно предпочтительным растворителям относятся кетоны с относительно малой молекулярной массой, такие, например, как метил-н-пропилкетон, метилэтилкетон, метиламилкетон, метилизоамилкетон, метилизобутилкетон, этилбутилкетон и диизобутилкетон. Также пригодными для экстракции растворителями являются альдегиды, как, например, н-бутиральдегид, и сложные эфиры, например, этилацетат, н-бутилацетат, н- пропилацетат и изопропилацетат. Помимо названных, могут использоваться также спирты, хотя они по причине их взаиморастворимости с водой, медленного разделения фаз и тенденции реагировать с ГАМ менее предпочтительны.
В отличие от этих уже применяемых в уровне техники растворителей или сходных с ними, которые само собой разумеется также пусть и в ограниченной степени могут использоваться при реализации изобретения, неожиданным образом было установлено, что применение эфирных соединений в качестве растворителей при экстракции позволяет достигнуть существенных преимуществ.
К применяемым согласно изобретению простым эфирам относятся в первую очередь таковые общей формулы I
R1-O-R2, (I)
где R1 и R2 независимо друг от друга представляют собой одновременно либо порознь C1-C5-алкил, линейный либо разветвленный.
Некоторые из приемлемых эфирных соединений представлены в табл.А в конце описания.
Из эфирных соединений предпочтительны, во-первых, таковые, которые либо совсем не обладают склонностью к образованию пероксидов, либо обладают ею в незначительной степени; в качестве примера можно назвать МТБЭ (метил-трет-бутиловый эфир). Предпочтительны также асимметричные эфиры.
Во-вторых, наиболее целесообразно использовать такие соединения, температура кипения которых составляет менее 60oC, поскольку их полностью и простым путем можно удалять из целевого продукта.
Наибольшее предпочтение в рамках изобретения отдают применению метил-трет-бутилового эфира (МТБЭ), отвечающего всем вышеназванным критериям.
Собственно экстракцию можно осуществлять в непрерывном либо периодическом режиме. Для работы в периодическом режиме пригоден, например, смеситель-отстойник. Предпочтительно, однако, проводить экстракцию в установке непрерывного противоточного действия, в которой для ускорения массообмена между растворителем и водной фазой предусмотрена соответствующая экстракционная зона. Так, в частности, целесообразно осуществлять процесс экстракции в каскаде, состоящем из противоточных смесителей-отстойников, насадочной колонны, колонны с ситчатыми тарелками, выполненной предпочтительно в виде пульсационной колонны либо колонны с вибрирующим пакетом сит, роторно-дисковой колонны или центробежного экстрактора. В особенно предпочтительном варианте выполнения экстракцию проводят в колонне с ситчатыми тарелками для экстракции в системе жидкость/жидкость. Прерывистые или пульсирующие потоки, хотя и носят циклический характер, т.е. не являются непрерывными с точки зрения быстроты прохождения потока, в контексте настоящего описания рассматриваются как "непрерывные".
Процесс экстракции регулируют предпочтительно таким образом, чтобы фазу растворителя в экстракционной зоне образовывать в виде непрерывной фазы и соответственно сохранять ее в этом качестве.
Для снижения содержания соли в конечном продукте до минимума экстракт при необходимости промывают водой. Однако при уровне концентрации в определенных пределах от промывки можно отказаться, прежде всего при содержании сульфата менее 0,5 мас.% и с учетом концентрации соли в выходящем рафинате, которую по возможности не следует далее снижать за счет разбавления. В противоточной непрерывной экстракционной системе промывку экстракта можно проводить смешением с водой в месте, расположенном выше по ходу движения потока (имеется в виду направление движения органического потока), т.е. в месте, где гидролизат вводят в экстракционную систему жидкость/жидкость. Так, например, в вертикальной колонне с применением растворителя, удельный вес которого предпочтительно меньше 1, растворитель вводят в колонну в месте, расположенном ниже места подачи водного раствора гидролизата, а промывочную воду подают в колонну в месте, расположенном выше места подачи раствора гидролизата.
Производительность процесса экстракции повышают за счет работы при несколько повышенной температуре, что обусловлено необходимостью предусмотреть для фазы растворителя в экстракционной системе относительно низкую вязкость. Работа при температуре в диапазоне ниже температуры кипения используемого органического растворителя обеспечивает к тому же положительное воздействие на коэффициент распределения ГАМ'а между органической и водной фазами.
Согласно изобретению ГАМ можно получать из экстракционного раствора, как уже упоминалось выше, путем упаривания. Таким образом, одним из факторов реализации предлагаемого изобретения является, в частности, упаривание экстракционного раствора, получаемого жидкостно-жидкостной экстракцией реакционной смеси, которую в свою очередь получают, например, путем гидролиза ММП-ЦГ в присутствии серной кислоты. При этом процесс упаривания осуществляют предпочтительно таким образом, что содержание воды в образующемся экстракте составляет менее 4 мас.%, предпочтительно менее 2 мас.%. В результате совершенно неожиданно получают при этом высококонцентрированный жидкий ГАМ с особенно низким содержанием олигомеров и димеров. Принимая во внимание уровень техники, такой результат тем более неожидан, что может быть достигнут с меньшим содержанием воды по сравнению с тем, как это реализовано в известных публикациях (D1 и D2).
Согласно особенно предпочтительному варианту выполнения по изобретению органический растворитель удаляют при упаривании с помощью аппарата, позволяющего проводить стадию упаривания за короткое время нахождения в нем экстракционного раствора. Особенно предпочтительно поэтому удалять органический растворитель из экстракционного раствора при упаривании с помощью таких выпарных аппаратов, как испаритель с падающей пленкой, тонкопленочный испаритель и/или глубоковакуумный испаритель, либо с помощью аппарата подобного типа.
Под понятием "аппарат подобного типа" в контексте настоящего изобретения имеется в виду, что вышеназванные аппараты с коротким временем нахождения в них экстракционного раствора могут использоваться также в сочетании с другими, известными специалисту устройствами, для удаления растворителя из экстракционного раствора. При этом речь идет не обязательно о сочетании таких аппаратов и устройств, которые обеспечивают обработку находящегося в них материала за короткое время. В этой связи можно назвать, в частности, дистилляционные колонны, которые при необходимости могут оснащаться устройствами для подачи пара или какими-либо другими, пригодными для указанной цели средствами. Возможно также объединение нескольких аппаратов указанного выше типа в одну систему.
В предпочтительной модификации способа по изобретению предлагается упаривание экстракционного растворителя проводить преимущественно таким образом, чтобы свести до минимума остаточное содержание растворителя. Этого можно достичь, например, за счет объединения нескольких вышеназванных аппаратов со стадией упаривания, которая может дополнять выпарную систему или может быть интегрирована в указанные выпарные аппараты, как это имеет место, например, при непосредственной подаче стриппинг-среды в такой испаритель.
Специфические условия упаривания варьируют при необходимости в зависимости от особенностей применяемого для экстракции соответствующего растворителя. В принципе для упаривания, если при этом используют аппарат с кратким временем нахождения в нем экстракционвого раствора, предпочтительно, чтобы давление во время процесса упаривания не превышало 600 мбар, предпочтительно 400 мбар и особенно предпочтительно 200 мбар.
Температура при упаривании, как правило, также зависит от применяемого растворителя. Следует, однако, стремиться к тому (и это является поэтому также особенно предпочтительным согласно изобретению), чтобы температура в процессе упаривания не превышала 150oC. При существенном превышении этой температуры может иметь место термическое повреждение целевого продукта. При этом под температурой во время выпаривания подразумевается не температура, обусловленная контактом продукта с поверхностью выпарного аппарата, предусмотренного соответственно для такого кратковременного контакта. Под температурой во время упаривания имеется в виду скорее средняя температура в выпарном аппарате. В некоторых случаях температура на поверхности выпарного аппарата может находиться на уровне, значительно превышающем 150oC Решающим фактором является кратковременность контакта с применяемым выпарным аппаратом. Благодаря этому удается предотвратить термическое повреждение продукта даже в тех случаях, когда температура при контакте заметно превысит 150oC.
Касательно распределения температуры в рамках изобретения было установлено, что особенно качественный продукт получают, если температура образовавшегося экстракта непосредственно на выходе из выпарного аппарата находится в интервале от 30 до 100oC, предпочтительно от 50 до 95oC и особенно предпочтительно от 70 до 90oC.
Как уже указывалось, время нахождения образующегося экстракта в аппарате играет наряду с другими факторами решающую роль для качества и состава целевого ГАМ-продукта. В соответствии с дальнейшим предпочтительным развитием способа по изобретению время нахождения образующегося экстракта на стадии упаривания составляет не более 1,5 ч. Это относится ко времени нахождения во всей выпарной системе, включая по меньшей мере стадию упаривания с предельно краткой продолжительностью обработки. Эту кратковременность нахождения в аппарате в отличие от указанного общего показателя, равного 1,5 ч, следует скорее исчислять в минутном диапазоне или даже ниже. В любом случае согласно изобретению предпочтительно, чтобы при использовании для выпаривания только одного тонкопленочного испарителя и/или испарителя с падающей пленкой и/или глубоковакуумного испарителя время нахождения материала в этих аппаратах не превышало 1 ч, предпочтительно 40 мин.
Еще одно преимущество предлагаемого способа состоит в том, что не только более успешно решается проблема выделения ГАМ из реакционной смеси, получаемой гидролизом с помощью серной кислоты, но и одновременно совершенствуется также гидролиз самого ММП-ЦГ. Так, согласно предпочтительному варианту способа по изобретению гидролиз ММП-ЦГ осуществляют таким образом, что на первой ступени ММП-ЦГ гидролизуют 60-85 мас.%-ной, предпочтительно 65-80 мас.%- ной серной кислотой при молярном соотношении между ММП-ЦГ и H2SO4 от 1,0:0,5 до 1:1,0, предпочтительно от 1:0,6 до 1:0,95 при температурах в интервале от 30 до 90oC, предпочтительно от 50 до 70oC с получением в основном ГАМ-амида. При этом из ММП-циангидрина в основном образуется ГАМ-амид, а образующаяся смесь практически не содержит непрореагаровавший ММП-циангидрин. Другими словами, это означает, что имеет место практически количественный гидролиз.
C помощью описанных выше модификаций способа получения ГАМ можно выделять особенно высококачественный целевой продукт. Этот более совершенный ГАМ отличается согласно изобретению тем, что содержит в целом более 95 мас.% ГАМ, причем это количество представляет собой сумму мономерного ГАМ, димерного ГАМ и ГАМ- олигомеров (= общее количество ГАМ), а также более 0,1 и менее 5 мас.% воды. Особым преимуществом согласно изобретению оказалась прежде всего возможность получения ГАМ при сохранении практически всех качественных характеристик, который отличается тем, что содержит более 98 мас.% ГАМ как сумму мономерного ГАМ, димерного ГАМ и ГАМ-олигомеров, а также воду в количестве от 0,1 до менее 2 мас.% и имеет кинематическую вязкость >100 мм2/с при 25oC. При этом неожиданным образом было установлено, что кинематическая вязкость, определяемая по Кеннону-Фенске, высококонцентрированного продукта (т. е. ГАМ с содержанием по крайней мере 98 мас.% действующего вещества) после хранения и разбавления сравнима с кинематической вязкостью 88 мас.%-ного продукта. Несмотря на установившееся относительно высокое содержание димеров и олигомеров порядка 50 мас.%, которое имел высококонцентрированный продукт после хранения в течение приблизительно 300 дней при комнатной температуре, кинематическая вязкость помещенного на хранение коцентрата после разбавления водой до примерно 88 мас.% соответствует кинематической вязкости 88 мас.%-ного коммерческого продукта, имевшего в соответствующих экспериментальных условиях хранения равновесную концентрацию димеров и олигомеров, составлявшую лишь около 25 мас.%. В обоих случаях, т.е. как у разбавленного концентрата, так и у коммерческого продукта, было достигнуто состояние равновесия. Этот факт очень неожидан и является существенным преимуществом, обеспечивающим получение высококонцентрированного ГАМ в одном из вариантов выполнения изобретения. С учетом того, что количество димеров и олигомеров, как правило, оказывает отрицательное воздействие на вязкость ГАМ'а при практической переработке, тем более неожиданной оказалась возможность получения, несмотря на значительное исходное содержание димеров и олигомеров в так называемом высококонцентрированном продукте, легко перекачиваемой и тем самым транспортабельной смеси с вполне приемлемой вязкостью. Это дает целый ряд преимуществ: в частности, такая вязкость и прежде всего высокое содержание действующего вещества могут обеспечить более экономичную транспортировку высококонцентрированного продукта, поскольку при этом не требуется большого количества воды и, кроме того, в месте доставки, например, на мельнице по размолу кормов, его можно разбавлять водой до обычной для торговых поставок концентрации без повышения вязкости до нежелательных значений.
Далее согласно изобретению было установлено, что при соответствующем проведении реакции гидролиза в сочетании с применяемым согласно изобретению щадящим режимом упаривания при минимально кратковременной продолжительности пребывания материала в аппаратуре, можно получать особенно высокоценный в качественном отношении ГАМ. Получаемый по особенно предпочтительной технологии ГАМ отличается прежде всего суммарным количеством димеров и олигомеров, составляющим ≤10 мол.%, предпочтительно <7 мол.% по отношению к общему количеству ГАМ. Это означает, что вопреки распространенному в уровне техники ошибочному мнению можно получать высококонцентрированный ГАМ, представляющий собой благодаря крайне малому содержанию димеров и олигомеров очень удобный для непродолжительных перевозок продукт. В случае увеличения продолжительности транспортировки и связанной с этим необходимостью временного складирования, вследствие чего возрастает количество образующихся димеров и олигомеров, целесообразно эти последние добавлением воды и воздействием повышенной температуры снова переводить в мономерный ГАМ.
В соответствии с изобретением возможно далее высококонцентрированный ГАМ-продукт использовать при приготовлении добавок к кормам для животных. При этом оказывается, что смешением ГАМ-концентрата с водой, метионином и/или солями ГАМ'а (предпочтительно аммониевого ГАМ) (при необходимости NH3 для получения NH4-ГАМ) можно получать продукт с высокими питательными свойствами при сохранении качественных показателей, удовлетворяя тем самым в принципе потребности рынка.
Крайне неожиданным при осуществлении изобретения оказалось прежде всего то, что, начиная с момента выхода целевого ГАМ-продукта из стадии упаривания, возможно получение смесей не только за счет добавок соответствующих сокомпонентов, таких, как вода, метионин и/или аммониевый ГАМ, но также и обеспечивается то особое преимущество, что в случае смешения с аммониевым ГАМ аммиак можно непосредственно вводить в ГАМ-продукт после упаривания. При этом в зависимости от добавляемого количества аммиака требуемую долю ГАМ'а трансформируют в аммониевый ГАМ.
Ниже изобретение подробнее поясняется со ссылками на прилагаемые чертежи, на которых показано:
на фиг. 1 - блок-схема процесса выделения ГАМ после удаления соли и разделения фаз в системе жидкость/жидкость в ГАМ-гидролизате, причем независимо друг от друга возможны варианты, обозначенные линиями 1), 2) и 3),
на фиг. 2 - также блок-схема варианта осуществления способа по изобретению, в котором предусмотрено удаление соли без разделения фаз в системе жидкость/жидкость и где также линии 1) и 2) не зависят друг от друга,
на фиг. 3 - блок-схема другого варианта осуществления способа по изобретению, в котором ГАМ выделяют без удаления соли,
на фиг. 4 - блок-схема еще одного варианта изобретения, в котором ГАМ получают после накопления соли,
на фиг. 5 - технологическая схема оборудования, предназначенного для осуществления способа по изобретению, и
на фиг. 6 - схематическое изображение еще одного варианта оборудования, также предназначенного для осуществления способа по изобретению.
В представленном на фиг. 1 варианте способа циангидрин метилмеркаптопропиональдегида (ММП-ЦГ) по двухстадийной реакции гидролиза взаимодействием с водной серной кислотой превращают в кислоту, которая является гидроксианалогом метионина (ГАМ). Затем образующийся первичный ГАМ-гидролизат упаривают, повышая первоначальную концентрацию ГАМ от <40 мас.% до >40 мас.%, предпочтительно до >45 мас. %, и в результате образуются две жидкие фазы. Получаемую при упаривании воду конденсируют и возвращают на стадию гидролиза, причем температуру конденсации по энергосберегающим соображениям поддерживают на уровне, максимально близком температуре, при которой проводят гидролиз. Полученную при этом фракцию легкокипящих компонентов (ЛКК) с неприятным запахом практически полностью отделяют от водяного пара, удаляют через головную часть аппарата, при необходимости используя в этих целях стриппинг-газы, например воздух, и предпочтительно направляют непосредственно без предшествующей конденсации в печь для сжигания. Такая печь может быть составной частью установки для повторного извлечения серной кислоты (так называемой РК-установки).
Обе полученные из кубового остатка от упаривания жидкие фазы разделяют при температуре, превышающей комнатную температуру (КТ), однако максимально близкую температуре упаривания.
Нижнюю водную фазу, содержащую преимущественно образовавшуюся аммониевую соль, охлаждают до тех пор, пока основная часть растворенных солей не кристаллизуется [линия 1) или линия 2)]. Требуемая для этого температура составляет менее 30oC. Полученный солевой кристаллизат путем центрифугирования либо фильтрации отделяют от надосадочного раствора. Солевой кристаллизат с целью удаления все еще "сцепленного" с ним ценного вещества (ГАМ) можно промывать соответствующим органическим растворителем, но можно также и водой или же водным раствором соли.
Верхнюю органическую фазу, содержащую преимущественно ГАМ, а также водный фильтрат и необязательно органический фильтрат, разделяют либо совместно после частичного или полного предварительного смешения подают в экстракционную систему жидкость/жидкость [линия 1) или линия 2)] и с помощью органического растворителя разлагают по меньшей мере на две фазы, а именно на по меньшей мере в основном органический раствор экстракта, содержащий растворитель и ГАМ, а также незначительные количества воды и соли, соответственно на водный рафинат, который состоит преимущественно из соли и воды и который затем предпочтительно подают в установку для повторного извлечения серной кислоты [линия 1)] и необязательно, кроме того, на органический рафинат, который состоит преимущественно из растворителя и небольших количеств ГАМ и воды и который может быть повторно подан в экстракционную систему.
Органический раствор экстракта подают в систему для упаривания экстракта, причем выпаренный растворитель и при наличии таковых соответствующие количества воды рекуперируют путем конденсации и возвращают на стадию экстракции. Концентрацию ГАМ'а в получаемом при этом в виде кубового остатка от упаривания высококонцентрированном ГАМ-продукте кондиционированием за счет добавления требуемых количеств воды и/или введения соответствующих добавок, таких, как метионин или MHANH4-соль, доводят до требуемого уровня, предпочтительно до 78-98 мас.%.
Солевой кристаллизат после необязательной промывки может направляться на стадию очистки, соответственно кондиционирования [линия 1)], где за счет добавления соответствующих количеств NH3 и последующей кристаллизацией и сушкой получают готовый для поставок в торговую сеть сульфат аммония, или же указанный солевой кристаллизат в виде сырого продукта можно непосредственно направлять на сушку. Альтернативно этому солевой кристаллизат, в частности, после растворения в воде можно подавать в виде >60%-ного концентрированного раствора в установку для повторного извлечения серной кислоты [линия 2)]. Особенно предпочтительно при этом влажный после фильтрации солевой кристаллизат растворять в рафинате из стадии экстракции и полученный при этом высококонцентрированный солевой раствор с содержанием соли в количестве более 75 мас. % направлять на рекуперацию серной кислоты, поскольку минимальное содержание соли для этого должно составлять по меньшей мере 60 мас.% и, кроме того, любое превышение этого показателя концентрации способствует улучшению энергетического баланса рекуперационной установки. Такое дополнительное концентрирование возможно в данном случае прежде всего без проведения энергоемкого упаривания солевого раствора, получаемого по этому способу. Рекуперированную таким путем серную кислоту можно полностью или частично возвращать на стадию гидролиза ГАМ.
Целесообразным может оказаться также подавать водную фазу без отделения соли непосредственно в рекуперационную установку для серной кислоты совместно с рафинатом из экстракции [линия 3)]. И в этом случае содержание соли предпочтительно заметно превышает 60 мас.%. При этом теряется некоторое количество ГАМ (около 2,5% от теории), содержащегося в водной фазе в растворенном виде, однако это компенсируется тем существенным преимуществом, что достигается весьма значительная разгрузка стадии экстракции, соответственно стадии упаривания, поскольку обеспечена возможность в отличие от обычных способов (ср. D2) примерно наполовину уменьшить поступающий на экстракцию поток и тем самым расход применяемого растворителя, что позволяет резко снизить энергозатраты, связанные прежде всего с выпариванием растворителя, соответственно с его конденсацией.
В представленной на фиг. 2 двухстадийной реакции гидролиза циангидрин метилмеркаптопропиональдегида (ММП-ЦГ) взаимодействием с водной серной кислотой превращают в кислоту, которая является гидроксианалогом метионина (ГАМ). Затем образующийся первичный ГАМ-гидролизат упаривают, повышая первоначальную концентрацию ГАМ от <40 мас.% до >40 мас.%, предпочтительно до >45 мас.%, и в результате образуются две жидкие фазы. Получаемую при упаривании воду конденсируют и возвращают на стадию гидролиза, причем температуру конденсации по энергосберегающим соображениям поддерживают на уровне, максимально близком температуре, при которой проводят гидролиз. Полученную при этом фракцию легкокипящих компонентов (ЛКК) с неприятным запахом практически полностью отделяют от водяного пара, удаляют через головную часть аппарата, при необходимости используя в этих целях стриппинг-газы, например воздух, и предпочтительно направляют непосредственно без предшествующей конденсации в печь для сжигания. Такая печь может быть составной частью установки для повторного извлечения серной кислоты.
Обе полученные из кубового остатка от упаривания жидкие фазы совместно охлаждают по меньшей мере до тех пор, пока не образуется суспензия из солевого кристаллизата и гомогенной водно-органической жидкой фазы. Предпочтительно при этом охлаждать до комнатной температуры.
Солевой кристаллизат путем центрифугирования либо фильтрации отделяют от надосадочной жидкой фазы. Далее солевой кристаллизат с целью удаления все еще "сцепленного" с ним ценного вещества (ГАМ) промывают соответствующим органическим растворителем или же водой либо водным раствором соли.
Фильтрат и необязательно органический фильтрат раздельно либо совместно после частичного или полного предварительного смешения подают в экстракционную систему жидкость/жидкость и с помощью органического растворителя разлагают на по меньшей мере две фазы, а именно на по меньшей мере органический раствор экстракта, содержащий растворитель и ГАМ, а также незначительные количества воды и соли, и соответственно на водный рафинат, который состоит преимущественно из соли и воды и который затем предпочтительно подают в установку для повторного извлечения серной кислоты [линия 1)].
Органический раствор экстракта подают в систему для упаривания экстракта, причем выпаренный растворитель и при наличии таковых соответствующие количества воды рекуперируют путем конденсации и возвращают на стадию экстракции. Концентрацию ГАМ'а в получаемом при этом в виде кубового остатка от упаривания высококонцентрированном ГАМ-продукте кондиционированием за счет добавления требуемых количеств воды и/или введения соответствующих добавок, таких, как метионин или MHANH4-соль, доводят до требуемого уровня, предпочтительно до 78-98 мас.%.
Солевой кристаллизат после необязательной промывки может направляться на стадию очистки, соответственно кондиционирования [линия 1)], где за счет добавления соответствующих количеств NH3 и последующей кристаллизацией и сушкой получают готовый для поставок в торговую сеть сульфат аммония, или же указанный солевой кристаллизат в виде сырого продукта можно непосредственно направлять на сушку. Альтернативно этому солевой кристаллизат, в частности, после растворения в воде можно подавать в виде >60%-ного концентрированного раствора в установку для повторного извлечения серной кислоты [линия 2)]. Особенно предпочтительно при этом влажный после фильтрации солевой кристаллизат растворять в рафинате из стадии экстракции и полученный при этом высококонцентрированный солевой раствор с содержанием соли в количестве более 75 мас. % направлять на рекуперацию серной кислоты, поскольку минимальное содержание соли для этого должно составлять по меньшей мере 60 мас.% и, кроме того, любое превышение этого показателя концентрации способствует улучшению энергетического баланса рекуперационной установки. Такое дополнительное концентрирование возможно в данном случае прежде всего без проведения энергоемкого упаривания солевого раствора, получаемого по этому способу. Рекуперированную таким путем серную кислоту можно полностью или частично возвращать на стадию гидролиза ГАМ.
В представленном на фиг. 3 варианте способа циангидрин метилмеркаптопропиональдегида (ММП-ЦГ) по двухстадийной реакции гидролиза взаимодействием с водной серной кислотой превращают в кислоту, которая является гидроксианалогом метионина (ГАМ). Затем образующийся первичный ГАМ-гидролизат упаривают, повышая первоначальную концентрацию ГАМ от <40 мас.% до >40 мас.%, предпочтительно до >45 мас. %, и в результате образуются две жидкие фазы. Получаемую при упаривании воду конденсируют и возвращают на стадию гидролиза, причем температуру конденсации по энергосберегающим соображениям поддерживают на уровне, максимально близком температуре, при которой проводят гидролиз. Полученную при этом фракцию легкокипящих компонентов (ЛКК) с неприятным запахом практически полностью отделяют от водяного пара, удаляют через головную часть аппарата, при необходимости используя в этих целях стриппинг-газы, например воздух, и предпочтительно направляют непосредственно без предшествующей конденсации в печь для сжигания. Такая печь может быть составной частью установки для повторного извлечения серной кислоты (так называемой РК-установки).
Обе полученные из кубового остатка от упаривания жидкие фазы при необходимости совместно охлаждают, однако лишь до определенного предела, с тем, чтобы предотвратить образование солевого кристаллизата.
Продукт упаривания подают в экстракционную систему жидкость/жидкость и с помощью органического растворителя разлагают на по меньшей мере две фазы, а именно на по меньшей мере преимущественно органический раствор экстракта, содержащий растворитель и ГАМ, а также незначительные количества воды и соли, и соответственно на водный рафинат, который состоит преимущественно из соли и воды и который затем предпочтительно направляют в установку для повторного извлечения серной кислоты. Требуемая при этом концентрация соли минимум 60 мас. % зависит в основном от степени упаривания первичного гидролизата. При этом, правда, следует учитывать, что допустима только такая степень упаривания, при которой в экстракционной системе не должны образовываться солевые кристаллизаты как следствие слишком высокого концентрирования. Тем самым достигаемый таким путем уровень концентрации ниже по сравнению с этим показателем в вариантах способа, представленных на фиг. 1 и 2. Рекуперированную по такой методике серную кислоту можно полностью или частично возвращать на стадию гидролиза ГАМ.
Органический раствор экстракта подают в систему для упаривания экстракта, причем выпаренный растворитель и при наличии таковых соответствующие количества воды рекуперируют путем конденсации и возвращают на стадию экстракции. Концентрацию ГАМ'а в получаемом при этом в виде кубового остатка от упаривания высококонцентрированном ГАМ-продукте кондиционированием за счет добавления требуемых количеств воды и/или введения соответствующих добавок, таких, как метионин или MHANH4-соль, доводят до требуемого уровня, предпочтительно до 78-98 мас.%.
В представленном на фиг. 4 варианте циангидрин метилмеркаптопропиональдегида (ММП-ЦГ) по двухстадийной реакции гидролиза взаимодействием с водной серной кислотой превращают в кислоту - гидроксианалог метионина (ГАМ). Образовавшийся первичный ГАМ-гидролизат, концентрация ГАМ'а в котором менее 40 мас.%, подвергают затем испарительному охлаждению, снижая исходную температуру - реакционную температуру >100oC - до соответственно более низкой температуры, например до 60oC, и одновременно удаляя перегонкой, предпочтительно с использованием вакуума и необязательно стриппинг-газов, как, например воздух, фракцию легкокипящих компонентов (ЛКК) с неприятным запахом вместе с небольшим количеством водяного пара, и эту фракцию можно непосредственно, без предварительной конденсации направлять в печь для сжигания. Такая печь может быть также составной частью установки для повторного извлечения серной кислоты.
Последующим добавлением (NH4)2SO4 и/или NH4HSO4 к гомогенному раствору ГАМ-гидролизата, содержащуюся в нем концентрацию соли повышают до момента образования двух жидких фаз и при этом не должно оставаться никаких значительных количеств нерастворенных твердых веществ.
Обе жидкие фазы разделяют при температуре, превышающей КТ [линия 1)]. Верхнюю, содержащую преимущественно ГАМ органическую фазу, подают в экстракционную систему жидкость/жидкость [линия 1)] и с помощью органического растворителя разлагают по меньшей мере на две фазы, а именно на по меньшей мере преимущественно органический раствор экстракта, содержащий растворитель и ГАМ, а также незначительные количества воды и соли, и соответственно на водный рафинат, состоящий преимущественно из соли и воды.
Затем рафинат предпочтительно совместно с нижней, водной фазой, полученной в результате разделения фаз в системе жидкость/жидкость и содержащей в основном образовавшуюся аммониевую соль, подают в установку для повторного извлечения серной кислоты [линия 1)]. При этом теряется некоторое количество ГАМ'а (около 2,5% от теории), еще содержащегося в водной фазе в растворенном виде, однако это компенсируется тем существенным преимуществом, что достигается весьма значительная разгрузка стадии экстракции, соответственно стадии упаривания, поскольку в отличие от обычных способов (ср. D2) обеспечена возможность заметно уменьшить поступающий на экстракцию поток и тем самым также расход применяемого растворителя, что позволяет резко снизить энергозатраты, связанные прежде всего с последующим выпариванием растворителя, соответственно с его конденсацией.
Органический раствор экстракта подают в систему для упаривания экстракта, причем выпаренный растворитель и при наличии таковых соответствующие количества воды рекуперируют путем конденсации и возвращают на стадию экстракции. Концентрацию ГАМ'а в получаемом при этом в виде кубового остатка от упаривания высококонцентрированном ГАМ-продукте кондиционированием за счет добавления требуемых количеств воды и/или введения соответствующих добавок, таких, как метионин или MHANH4-соль, доводят до требуемого уровня, предпочтительно до 78- 98 мас.%.
Альтернативно этому можно также обе жидкие фазы совместно направлять в экстракционную систему жидкость/жидкость [линия 2)]. Образующийся при этом рафинат, >60%-ный концентрированный солевой раствор, может непосредственно подаваться в установку для повторного извлечения серной кислоты [линия 2)], поскольку минимальное содержание соли для этого должно составлять по меньшей мере 60 мас.% и, кроме того, любое превышение этого показателя концентрации способствует улучшению энергетического баланса рекуперационной установки. Такое дополнительное концентрирование возможно в данном случае прежде всего без проведения энергоемкого упаривания солевого раствора, получаемого по этому способу, что представляет собой существенное преимущество. Рекуперированную таким путем серную кислоту можно полностью или частично возвращать на стадию гидролиза ГАМ.
Ниже изобретение более подробно поясняется на примерах его выполнения.
Аналитические методы определения и пояснения
Количественное содержание ММП-циангидрина, ГАМ-амида, соответственно мономерного ГАМ определяли в обрабатывающих растворах посредством ЖХВР путем сравнения с внешним эталоном (чистая субстанция).
Общее количество ГАМ = ГАМ-амид (необязат.) + мономерный ГАМ (= ГАМ общ. ) + ГАМ-(димеры + олигомеры)
определяли путем титриметрии тиоэфирной функции с помощью стандартного раствора KBr/KBrO3 и выражали в виде суммы соответствующих ГАМ- мономерных эквивалентов в [мас. %] , соответственно в [г], соответственно в [молях], соответственно в [мол. %].
Содержание ГАМ-димеров + ГАМ-олигомеров (ДИМ + ОЛИ) определяли по разнице между общим количеством ГАМ и мономерного ГАМ (+ необязательно ГАМ-амид) и выражали в виде суммы соответствующих ГАМ-мономерных эквивалентов в [мас. %] , соответственно в [г], соответственно в [молях], соответственно в [мол. %].
С помощью стандартных методов определяли: содержание воды - титрованием по Карлу Фишеру, содержание растворителя - посредством ГХ или по разнице, содержание сульфата, соответственно аммония - посредством ионной хроматографии, а общее содержание соли определяли путем соответствующего пересчета по отношению к содержанию сульфата, соответственно аммония или по разнице.
Пример 1: Получение раствора ГАМ-гидролизата в непрерывном режиме
В двухступенчатом каскаде из смесителей-отстойников путем непрерывной подачи 4,2 кг/ч (31,3 моля/ч) 97,7% ММП-циангидрина и 4,5 кг/ч (29,7 молей/ч) 65% водной H2SO4 при температуре 50oC и общей продолжительности нахождения в аппаратуре в среднем 60 мин получали 8,7 кг/ч раствора ГАМ-амида. Реакцию по дальнейшему превращению этого раствора ГАМ-амида продолжали, непрерывно разбавляя 3,6 кг/ч воды, в двухступенчатом каскаде из смесителей-отстойников с подключенной за ними реакционной трубой при температуре 90-110oC и общей продолжительностью нахождения в аппаратуре в среднем 180 мин с получением в результате 12,3 кг/ч раствора ГАМ-гидролизата. Этот первично образующийся реакционный раствор непрерывно подавали в систему упаривания, где его концентрировали при давлении 100 мбар и охлаждали на выходе до температуры 50oC. Образовавшийся при этом предварительно упаренный ГАМ-гидролизат (10,8 кг/ч) имел согласно анализу следующий состав:
43,7 мас.% ГАМ общ.
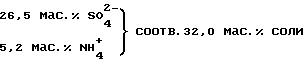
24,3 мас.% H2O
Пример 2: Получение экстракционного раствора ГАМ/МТБЭ
Опыт 1
В смесителе объемом 5 л со спускным клапаном в днище 2,5 кг ГАМ-гидролизата (43,7 мас.% ГАМ общ., полученного аналогично примеру 1) смешивали с 1,5 кг МТБЭ (технического) и в течение 10 мин интенсивно перемешивали при КТ. По завершении перемешивания обе образовавшиеся жидкие фазы разделяли. Эту операцию повторяли в общей сложности четыре раза, используя каждый раз свежий раствор.
Органические фазы, соответственно водные рафинатные фазы в каждом случае объединяли и анализировали. Состав фаз в [мас.%] представлен в таблице 1 (см. в конце описания).
Опыт 2
Опыт 1 повторяли с использованием 2,5 кг ГАМ-гидролизата и 1,5 кг МТБЭ, повторно извлеченного при упаривании экстракционного раствора ГАМ/МТБЭ (ср. пример 3). Состав фаз в [мас.%] представлен в таблице 2 (см. в конце описания).
Пример 3: Получение высококонцентрированного ГАМ-продукта
На фиг. 5 показана технологическая схема оборудования, применявшегося в примере 3 и состоявшего в основном из следующих аппаратов:
001 расходная емкость; испарители типа Sambay с соответственно площадью обменной реакции 0,06 м2 и обогреваемой двойной рубашкой;
испарители типа Sambay с соответственно площадью обменной реакции 0,06 м2 и обогреваемой двойной рубашкой;
004 сборник для ГАМ-продукта; конденсационная система для отогнанного растворителя, состоящая из соответственно охлаждаемого водой и охлаждаемого солевым раствором лабораторного холодильника, соответственно сборника и последовательно включенного водоструйного насоса с регулируемым вакуумом.
конденсационная система для отогнанного растворителя, состоящая из соответственно охлаждаемого водой и охлаждаемого солевым раствором лабораторного холодильника, соответственно сборника и последовательно включенного водоструйного насоса с регулируемым вакуумом.
Описание технологии со ссылкой на фиг. 5
Поступающий из стадии экстракции экстракционный раствор ГАМ/МТБЭ непрерывно подают из расходной емкости 001 в испаритель Sambay 002, обогреваемый извне. Погон из 002 с помощью игольчатого клапана подается также в обогреваемый испаритель Sambay 003, откуда образующийся ГАМ-продукт поступает в сборник 004 и там его анализируют. Содержащий в основном растворитель дистиллят собирают в сборниках обеих конденсационных систем 005 и 006 и оттуда его можно возвращать на экстракцию (ср. пример 2, опыт 2).
Опыт 3
Применение 0,95 л/ч (0,85 кг/ч) экстракционного раствора ГАМ/МТБЭ из примера 2, опыт 1
Испаритель 002:
- давление 250 мбар
- температуры:
обогреваемая рубашка 125oC
продукт на выходе 79oC
Состав высококонцентрированного ГАМ-продукта в кубовом остатке 002
ГАМ общ. - 98,0 мас.%
H2О - 0,5 мас.%
Испаритель 003:
- давление 50 мбар
- температуры:
обогреваемая рубашка 140oC
продукт на выходе 90oC
вторичный пар 30oC
Cостав высококонцентрированного ГАМ-продукта в кубовом остатке 003
ГАМ общ. - 99,0 мас.%
ГАМ - 83,9 мол.%
ДИМ+ОЛИ - 16,1 мол.%
H2О - 0,5 мас.%
МТБЭ - <10 част./млн
Из кубового остатка испарителя Sambay 003 получали 0,36 кг/ч высококонцентрированного ГАМ-продукта указанного выше состава.
Опыт 4
Применение 0,96 л/ч (0,86 кг/ч) экстракционного раствора ГАМ/МТБЭ из примера 2, опыт 2
Испаритель 002:
- давление 250 мбар
- температуры:
обогреваемая рубашка 125oC
продукт на выходе 96oC
Состав высококонцентрированного ГАМ-продукта в кубовом остатке 002
ГАМ общ. - 98,5 мас.%
H2О - 0,9 мас.%
Испаритель 003:
- давление 50 мбар
- температуры:
обогреваемая рубашка 120oC
продукт на выходе 100oC
вторичный пар 28oC
Состав высококонцентрированного ГАМ-продукта в кубовом остатке 003
ГАМ общ. - 100 мас.%
ГАМ - 85,7 мол.%
ДИМ + ОЛИ - 14,3 мол.%
H2О - 0,0 мас.%
МТБЭ - <1 част./млн.
Из кубового остатка испарителя Sambay 003 получали 0,36 кг/ч высококонцентрированного ГАМ-продукта указанного выше состава.
Пример 4: Повторное извлечение соли из ГАМ-гидролизата перед экстракцией путем разделения фаз в системе жидкость/жидкость и жидкость/твердое вещество (ср. фиг. 1)
Опыт 5: Выделение ГАМ путем разделения фаз в системе жидкость/жидкость и экстракцией в системе жидкость/жидкость с помощью МТБЭ
502 г ГАМ-гидролизата с содержанием 43,7 мас. % (219,4 г) ГАМ общ. (полученного аналогично примеру 1) упаривали при давлении 50 мбар до содержания ГАМ общ. 50 мас.%. Полученный концентрат (438,7 г) состоял из двух жидких фаз, которые разделяли при температуре 65oC. Состав обеих фаз представлен в таблице 3 (см. в конце описания).
Водную фазу охлаждали до температуры 26oC. Выпавший при этом в осадок солевой кристаллизат, состоявший из NH4HSO4 и (NH4)2SO4, отфильтровывали. В результате получили состав, представленный в таблице 4.
Солевой кристаллизат промывали на фильтре 10 г МТБЭ и полученный органический фильтрат (6,0 г) анализировали (потери МТБЭ при упаривании = 4,8 г):
ГАМ общ.= 7,4 мас.%, соотв. 0,44 г, соотв. 0,2% от теории
потери ГАМ'а через солевой кристаллизат: <0,2% от теории без промывки
и 0% от теории с промывкой.
Промытый солевой кристаллизат сушили (35,8 г) и анализировали:
SO4 2- 80,5 мас.%
NH4 + 18,5 мас.%
соль 22,3% от теории
Органическую фазу (292,4 г), водный фильтрат (103,6 г) и органический фильтрат (6,0 г) смешивали с 232 г МТБЭ и при КТ в течение короткого промежутка времени интенсивно перемешивали. По завершении перемешивания обе образовавшиеся жидкие фазы разделяли. Разделенные фазы имели состав, представленный в таблице 5 (см. в конце описания).
Остаточное содержание ГАМ общ. из рафината, полученного в результате описанной одноступенчатой экстракции, может быть снижено за счет однократного до многократного дополнительного экстрагирования с использованием свежего растворителя или путем непрерывного экстрагирования в системе с несколькими теоретическими разделительными ступенями до 0,1% от теории.
Солевой кристаллизат (35,8 г) полностью растворяли в рафинате (159 г) при 61oC. Полученный таким путем солевой раствор имел следующий состав:
ГАМ общ. 1,4 мас.%, соотв. 1,3% от теории
H2О 20,7 мас.%
соль 77,9 мас.%
Полученный описанным путем раствор можно направлять, что является особенно предпочтительным, в установку для повторного извлечения серной кислоты, поскольку содержание в нем соли заметно превышает 60 мас.%.
Опыт 6: Выделение ГАМ путем разделения фаз в системе жидкость/жидкость и экстракцией в системе жидкость/жидкость с помощью МИБК
505 г ГАМ-гидролизата с содержанием 43,7 мас. % (220,7 г) ГАМ общ. (полученного аналогично примеру 1) упаривали при давлении 50 мбар до содержания ГАМ общ. 49,9 мас.%. Полученный концентрат (440 г) состоял из двух жидких фаз, которые разделяли при температуре 60oC. Состав фаз представлен в таблице 6 (см. в конце описания).
Водную фазу охлаждали до температуры 20oC. Выпавший при этом в осадок солевой кристаллизат, состоящий из NH4HSO4 и (NH4)2SO4 отфильтровывали. Его состав представлен в таблице 7 (см. в конце описания).
Солевой кристаллизат промывали на фильтре 14 г МИБК и полученный органический фильтрат (13,8 г) анализировали.
ГАМ общ. = 9 мас.%, соотв. 1,2 г, соотв. 0,56% от теории, потери ГАМ через солевой кристаллизат <0,1% от теории.
Промытый солевой кристаллизат сушили (40 г) и анализировали:
SO4 2- 80,7 мас.%
NH4 + 18,8 мас.%
соль 24,7% от теории
Органическую фазу (299 г), водный фильтрат (83 г) и органический фильтрат (13,8 г) смешивали с 250 г МИБК и при КТ в течение короткого промежутка времени интенсивно перемешивали. По завершении перемешивания обе образовавшиеся жидкие фазы разделяли. Разделенные фазы имели состав, представленный в таблице 8 (см. в конце описания).
Остаточное содержание ГАМ общ. из рафината, полученного в результате описанной одноступенчатой экстракции, может быть снижено за счет однократного до многократного дополнительного экстрагирования с использованием свежего растворителя или путем непрерывного экстрагирования в системе с несколькими теоретическими разделительными ступенями до <0,1% от теории.
Пример 5: Повторное извлечение соли из ГАМ-гидролизата перед экстракцией путем разделения фаз в системе жидкость/твердое вещество (ср. фиг. 2)
Опыт 7: Выделение ГАМ без разделения фаз в системе жидкость/жидкость
505 г ГАМ-гидролизата с содержанием 43,7 мас. % (220,7 г) ГАМ общ. (полученного аналогично примеру 1) упаривали при давлении 50 мбар до содержания ГАМ общ. 49,9 мас.%. Полученный концентрат (440 г) охлаждали до КТ и при этом получали суспензию из солевого кристаллизата и гомогенную жидкую фазу, которую отделяли путем фильтрации. Установленный состав представлен в таблице 9 (см. в конце описания).
Солевой кристаллизат промывали на фильтре 20 г МИБК и органический фильтрат (41,6 г) анализировали:
ГАМ общ.: 52,5 мас.%, соотв. 21,8 г, соотв. 9,9% от теории.
Промытый солевой кристаллизат сушили (52,8 г) и анализировали:
ГАМ общ. 6,1 мас.% = 1,5% от теории
SO4 2- 75,0 мас.%
NH4 + 18,4 мас.%
соль 93,9 мас.% = 30,7% от теории.
Потери ГАМ через промытый солевой кристаллизат составляли 1,5% от теории.
Фильтрат (342,4 г) растворяли при КТ в 244 г МИБК и смешивали с органическим фильтратом (41,6 г) и при этом осаждалась водная жидкая фаза. Обе жидкие фазы разделяли и определяли состав, представленный в таблице 10 (см. в конце описания).
Остаточное содержание ГАМ общ. из рафината, полученного в результате описанной одноступенчатой экстракции, может быть снижено за счет однократного до многократного дополнительного экстрагирования с использованием свежего растворителя или путем непрерывного экстрагирования в системе с несколькими теоретическими разделительными ступенями до <0,1% от теории.
Остаточное содержание ГАМ общ. в солевом кристаллизате можно еще снизить за счет последующей дополнительной промывки растворителем либо водой. Такую дополнительную промывку предпочтительно проводить водным раствором NH4HSO4 и/или (NH4)2SO4, который в свою очередь предпочтительно можно повторно использовать несколько раз и затем по достижении его полного насыщения возвращать в экстракционную систему с целью повторной экстракции содержащегося в нем растворенного ГАМ общ.
Содержащий ГАМ органический фильтрат с целью выделения ГАМ'а из органического и/или водного фильтрата может непосредственно подаваться в экстракционную систему для растворителя. Преимущество этого состоит в том, что таким образом можно предотвратить потери ГАМ на примерно 0,5-12,5% от теории.
Солевые кристаллизаты из примеров 4 и 5 за счет добавок соответствующих количеств NH3 и последующей кристаллизации могут перерабатываться в готовый для коммерческих поставок продукт (NH4)2SO4. Указанные кристаллизаты могут также непосредственно или предпочтительно после растворения в воде либо в соответствующей NH4HSO4, соответственно (NH4)2SO4, равно как и раствор, содержащий обе соли, подаваться в установку для повторного извлечения H2SO4.
Пример 6: Экстракция в системе жидкость/жидкость ГАМ-гидролизата (ср. фиг. 3)
Опыт 8: Экстракция с помощью МТБЭ
100 г ГАМ-гидролизата с содержанием ГАМ общ. 43,7 мас.% (43,7 г) (полученного аналогично примеру 1) смешивали при КТ с 60 г МТБЭ и в течение короткого промежутка времени интенсивно перемешивали. По завершении перемешивания обе образовавшиеся жидкие фазы разделяли. Результаты представлены в таблице 11 (см. в конце описания).
Опыт 9: Экстракция с помощью МИБК
100 г ГАМ-гидролизата с содержанием ГАМ общ. 43,7 мас.% экстрагировали аналогично опыту 8 60 г МИБК (таблица 12):
Как показывает сравнение обеих одноступенчатых экстракций растворителем, органический раствор экстракта в случае МТБЭ (опыт 8) поглощает лишь примерно половину нежелательной неорганической аммониевой соли по сравнению с этим показателем в случае МИБК (опыт 9). При использовании МТБЭ, кроме того, еще больше снижаются потери ГАМ через рафинатную фазу.
Пример 7: Выделение ГАМ после накопления содержания соли (ср. фиг. 4)
Опыт 10: Выделение ГАМ путем разделения фаз в системе жидкость/жидкость и экстракцией в системе жидкость/жидкость
23 г (NH4)2SO4 растворяли в 598 г ГАМ- гидролизата с содержанием ГАМ общ. 43,7 мас.% (261,3 г) (полученного аналогично примеру 1) при температуре 60oC. Раствор (621 г) состоял из двух жидких фаз, которые разделяли при температуре 60oC. Анализ показал следующий состав, представленный в таблице 13 (см. в конце описания).
Водную фазу с содержанием соли примерно 60 мас.% можно непосредственно подавать в установку для повторного извлечения серной кислоты.
Органическую фазу (416 г) смешивали с 250 г МТБЭ и в течение короткого промежутка времени интенсивно перемешивали. По завершении перемешивания обе образовавшиеся жидкие фазы разделяли. Их состав представлен в таблице 14 (см. в конце описания).
Остаточное содержание ГАМ общ. из рафината, полученного в результате описанной одноступенчатой экстракции, может быть снижено за счет однократного до многократного дополнительного экстрагирования с использованием свежего растворителя или путем непрерывного экстрагирования в системе с несколькими теоретическими разделительными ступенями до <0,1% от теории.
Водную фазу (205 г) и рафинат (144 г) объединяли. Полученный таким путем солевой раствор (349 г) имел следующий состав:
ГАМ общ. 5,6 мас.%
H2О 35,3 мас.%
соль 59,1 мас.%
Примерно 60%-ный солевой раствор можно непосредственно подавать в установку для повторного извлечения серной кислоты. Дальнейшее концентрирование может быть достигнуто за счет повышения используемого количества соли в гидролизате и непрерывной экстракцией, а также полным выделением ГАМ из рафината.
Опыт 11: Выделение ГАМ экстракцией в системе жидкость/жидкость
Аналогично опыту 10 23 г (NH4)2SO4 растворяли в 598 г ГАМ-гидролизата. Образовавшуюся смесь из двух жидких фаз (620 г) смешивали с 372 г МТБЭ и при температуре 40oC интенсивно перемешивали. По завершении перемешивания обе образовавшиеся жидкие фазы разделяли. Результаты представлены в таблице 15 (см. в конце описания).
Остаточное содержание ГАМ общ. из рафината, полученного в результате описанной одноступенчатой экстракции, может быть снижено за счет однократного до многократного дополнительного экстрагирования с использованием свежего растворителя или путем непрерывного экстрагирования в системе с несколькими теоретическими разделительными ступенями до <0,1% от теории.
Рафинат с содержанием соли >60 мас.% может непосредственно подаваться в установку для повторного извлечения серной кислоты.
Как в опыте 10, так и в опыте 11 можно отказаться от промывки органического раствора экстракта водой, поскольку остаточное содержание сульфата уже исключительно низкое. Это дает большое преимущество, поскольку таким образом можно исключить необходимость в дополнительных затратах, обусловленных технологией процесса, равно как и нежелательного разбавления рафината.
Полученные в примерах 4, 5, 6 и 7 органические растворы экстракта можно подвергать аналогично примерам 2, соответственно 8 непрерывному упариванию до практически полного удаления растворителя и доводить содержание воды до менее 5 мас.%. Полученный таким путем высококонцентрированный ГАМ- продукт в результате соответствующего кондиционирования может быть переведен в различные смеси ГАМ-продукта.
Пример 8: Описание способа со ссылкой на фиг. 6
На фиг. 6 представлена технологическая схема используемого для примера 8 оборудования. Соответствующими позициями обозначены следующие аппараты, из которых в основном состоит это оборудование:
(001) экстракционная колонна, например пульсационная сетчатая колонна длиной 3 м, с внутренним диаметром 2,1 см, пакетом из 60 ситчатых тарелок и обогреваемой двойной рубашкой;
(002) тонкопленочный испаритель, например, испаритель типа Sambay с площадью обменной реакции 0,08 м2 и обогреваемой двойной рубашкой;
(003) конденсационная система, например, стеклянный холодильник с водяным охлаждением;
(004/005) сборники для возвращаемых в процесс воды и растворителя;
(006) разделитель фаз для органического и водного рафината;
(007) промывочная зона для сливного раствора экстракта.
Поступающий из стадии гидролиза ГАМ'а ГАМ-гидролизат, состоящий в основном из ГАМ (мономер + димеры + олигомеры + необязательно амид), (NH4)2SO4 и/или NH4HSO4, а также воды, после предварительного нагрева до температуры экстракции подают в соответствующей дозировке над 40-ной ситчатой тарелкой в экстракционную колонну 001. Растворитель (в данном случае метилизобутилкетон или МИБК) также после предварительного нагревания закачивают в нижнюю часть колонны (принцип противотока). Дополнительно сливной продукт, отбираемый из головной части колонны, обрабатывают в зоне промывки промывочной водой и водную фазу возвращают в подаваемый поток гидролизата. Содержащий в основном (NH4)2SO4 и/или NH4HSO4, а также воду водный рафинат и состоящий преимущественно из растворителя органический рафинат совместно отбирают при охлаждении из низа колонны. Обе фазы разделяют в разделителе фаз 006 и органический рафинат возвращают в экстракционную систему, а водный рафинат выводят из цикла. Экстракционный раствор, содержащий в основном ГАМ, растворитель, а также воду, отводят через верх колонны и затем после прохождения через зону промывки 007 подают в испаритель Sambay 002. В испарителе под вакуумом и при дополнительной продувке H2O-паром и N2-потоком перед выходом из испарителя МИБК и H2О совместно удаляют из экстракционного раствора. Упаривание осуществляли таким образом, что содержание воды в высококонцентрированном ГАМ-продукте на выходе из испарителя составляло менее 2 мас.% и он практически не содержал растворитель.
Поступающую из испарителя 002 смесь растворителя и воды сначала конденсировали в конденсационной системе 003 и для разделения подавали в сепарационную емкость. Воду и растворитель собирали соответственно в сборниках 004 и 005, откуда их возвращали в экстракционную систему. Продукт после выхода из испарителя охлаждали до комнатной температуры и направляли в предназначенный для него сборник.
Состав экстракционного раствора анализировали непосредственно за зоной промывки 007, а состав раствора водного и органического рафинатов определяли соответственно после выхода из разделителя фаз 006.
Состав высококонцентрированного ГАМ-продукта определяли непосредственно после выхода из испарителя.
Используемый для экстракции раствор ГАМ-гидролизата приготавливали в смесителе объемом 400 л, в котором поддерживали постоянное давление, из 114,7 кг (874 моля) ММП-циангидрина и 131,9 кг (874 моля, 1,00 мол.экв.) 65%-ной H2SO4 при температуре 50oC и времени нахождения в аппарате 60 мин, последующим разбавлением 96,7 кг H2О и продолжая реакцию при температуре 90oC и времени нахождения в аппарате 120 мин. По завершении реакции из раствора сырого гидролизата под вакуумом удаляли имеющиеся легколетучие побочные продукты, после чего проводили анализ. Полученный при этом состав использованного для экстракции ГАМ-гидролизата представлен в опыте 12, поясняющем пример 8.
Условия и результаты опыта 12 представлены ниже.
Опыт 12: Применение ГАМ-гидролизата из ММП-циангидрина и 1,0 мол.экв. H2SO4
Применение в экстракции:
Количественный расход:
МИБК - 6,7 кг/ч
ГАМ-гидролизат - 12,3 кг/ч
ГАМ общ. - 4,9 кг/ч
промыв. H2О - 1,3 кг/ч
МИБК/гидролизат - 0,55 [-]
Состав ГАМ-гидролизата:
ГАМ общ.: - 39,5 мас.%
ГАМ: - 94,7 мас.%
ДИМ+ОЛИ: - 5,3 мол.%
H2О - 28,7 мас.%
SO4 2-: - 27,5 мас.%
(NH4HSO4): - 33,2 мас.%
Экстракция (001):
температура: - 60oC (средняя)
состав экстракционного раствора:
МИБК - 44,7 мас.% (рассч.)
ГАМ общ. - 41,8 мас.%
H2О - 13,5 мас.%
Органический рафинат
Водный рафинат
МИБК - 97,5 мас.%
МИКБ - 77,5 част./млн.
ГАМ общ. - 0,1 мас.%
ГАМ общ. - 0,1 мас.%
H2O - 2,4 мас.%
NH4HSO4 - 59,1 мас.%
H2O - 40,7 мас.%
(количеств, расход 0,017 кг/ч)
Упаривание 002:
давление 600 мбар
испаритель - температура
в обогреваемой рубашке 180oC
в головной части 85oC
в нижней части не определ.
- стриппинг-пар 0,5 кг/ч
- стриппинг-газ N2 100 л/ч
Состав высококонцентрированного ГАМ-продукта в кубовом погоне:
ГАМ общ. - 98 мас.%
ГАМ - 86 мол.%
ДИМ+ОЛИ - 14 мол.%
H2О - 2 мас.%
МИБК - 40 част./млн.
Из кубового погона испарителя получали приблизительно 4,9 кг/ч высококонцентрированного ГАМ-продукта указанного выше состава. Органический рафинат возвращали в экстракционную колонну. Водный рафинат непосредственно и без дополнительной обработки выводили из цикла с целью утилизации. Таким образом из процесса можно было исключить стадию последующей перегонки, соответственно упаривания, предназначенную для удаления остаточного растворителя из кубового продукта колонны. Органический рафинат, который отбирали в мягких условиях в качестве третьей жидкой среды из экстракционной колонны, мог, кроме того, без дополнительной очистки непосредственно повторно подаваться в эту экстракционную колонну.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2-ГИДРОКСИ-4-МЕТИЛТИОМАСЛЯНОЙ КИСЛОТЫ (ГИДРОКСИАНАЛОГАМЕТИОНИНА, ГАМ), ГАМ | 1995 |
|
RU2130925C1 |
| СПОСОБ ПОЛУЧЕНИЯ АММОНИЕВОЙ СОЛИ 2-ГИДРОКСИ-4-МЕТИЛТИОМАСЛЯНОЙ КИСЛОТЫ | 2004 |
|
RU2355678C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНЫХ РАСТВОРОВ МЕТИОНИНАТА НАТРИЯ И ПРИМЕНЕНИЕ ЭТИХ РАСТВОРОВ ДЛЯ ПОЛУЧЕНИЯ ГРАНУЛЯТОВ | 1999 |
|
RU2222526C2 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛА 2-ГИДРОКСИ-4-(МЕТИЛТИО)МАСЛЯНОЙ КИСЛОТЫ ИЗ 3-(МЕТИЛТИО)ПРОПАНАЛЯ И ЦИАНИСТОГО ВОДОРОДА | 2012 |
|
RU2604534C2 |
| СТАБИЛЬНЫЙ ПРИ ХРАНЕНИИ НИТРИЛ 2-ГИДРОКСИ-4-(МЕТИЛТИО)МАСЛЯНОЙ КИСЛОТЫ | 2012 |
|
RU2597264C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАЗОЛА | 1998 |
|
RU2192418C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТИЛМЕРКАПТОПРОПИОНОВОГО АЛЬДЕГИДА, ПОЛУЧАЕМОГО ИЗ СЫРЫХ АКРОЛЕИНА И МЕТИЛМЕРКАПТАНА | 2012 |
|
RU2615734C2 |
| СПОСОБ ОЧИСТКИ СОДЕРЖАЩИХ ДИОКСИД УГЛЕРОДА ГАЗОВЫХ ПОТОКОВ | 2005 |
|
RU2388521C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ ОКСО-СПИРТОВ | 1997 |
|
RU2183210C2 |
| СПОСОБ ПОЛУЧЕНИЯ D,L-МЕТИОНИНА ИЛИ ЕГО СОЛИ (ВАРИАНТЫ) | 1996 |
|
RU2176240C2 |
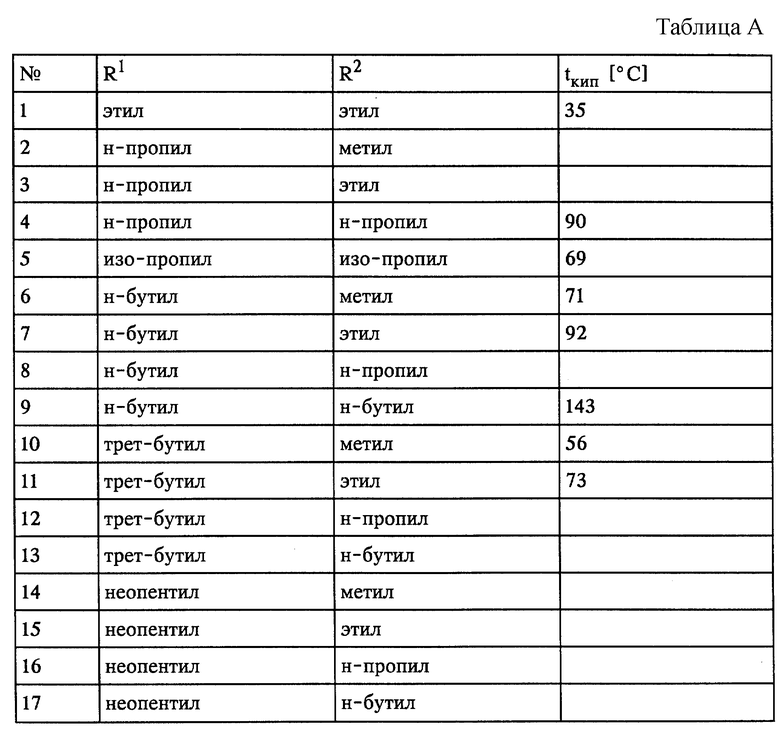
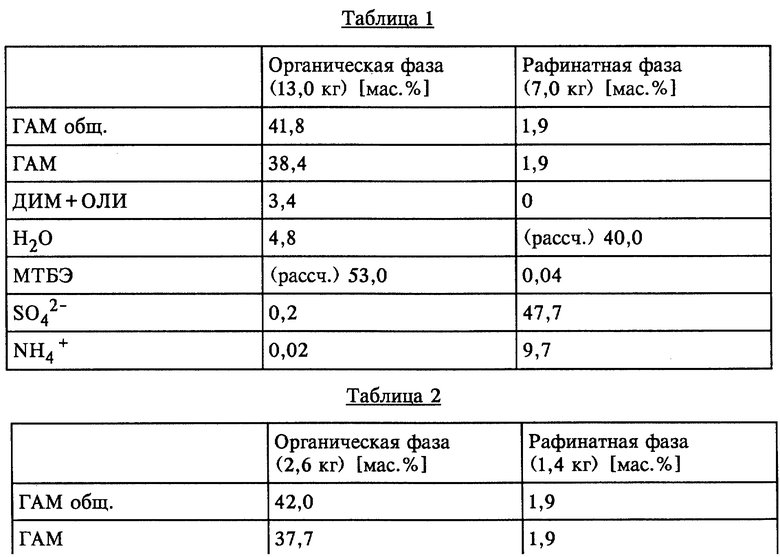
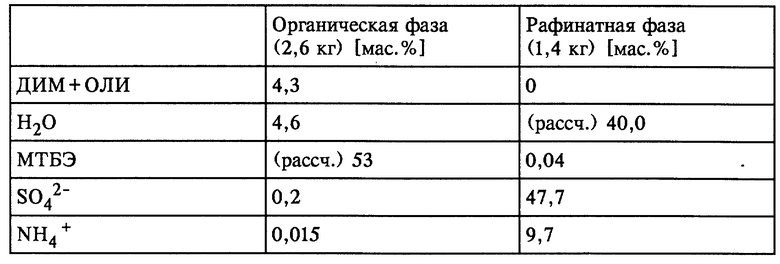


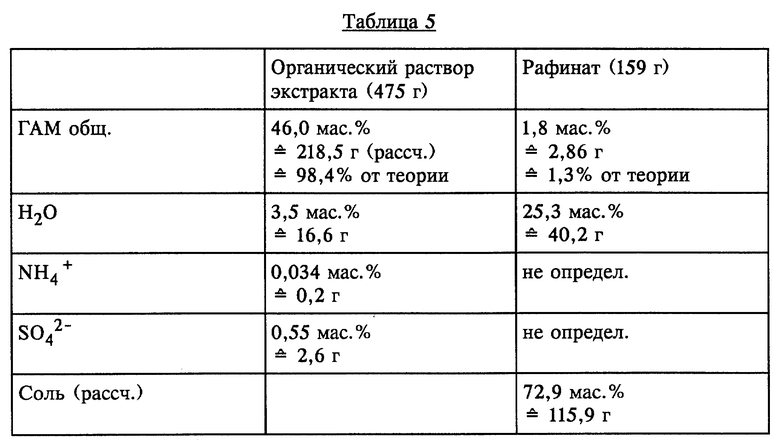
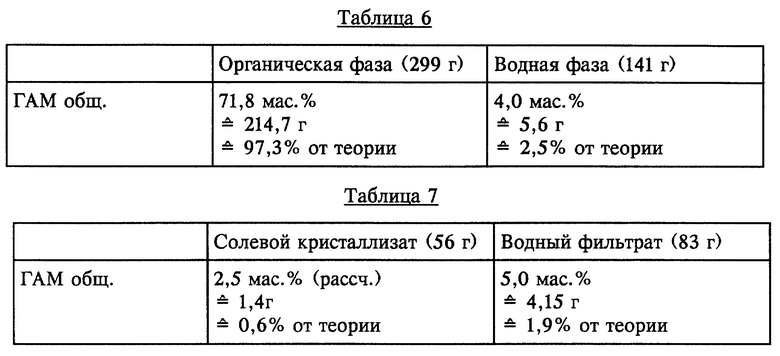
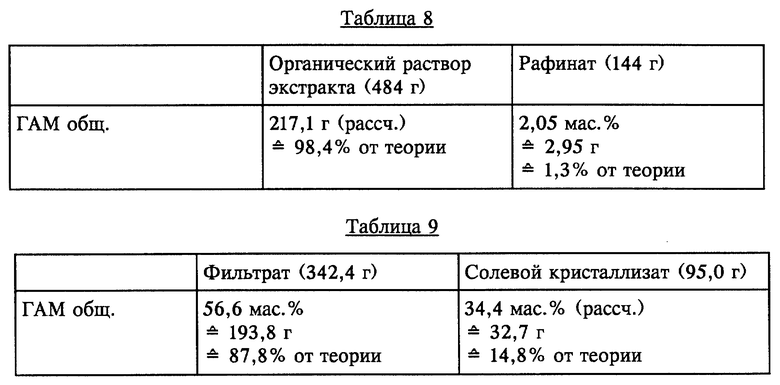

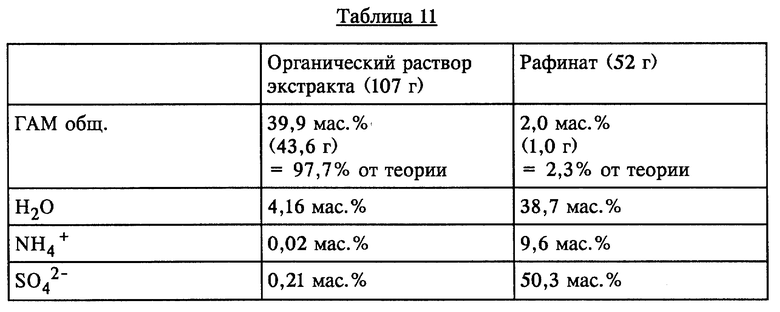
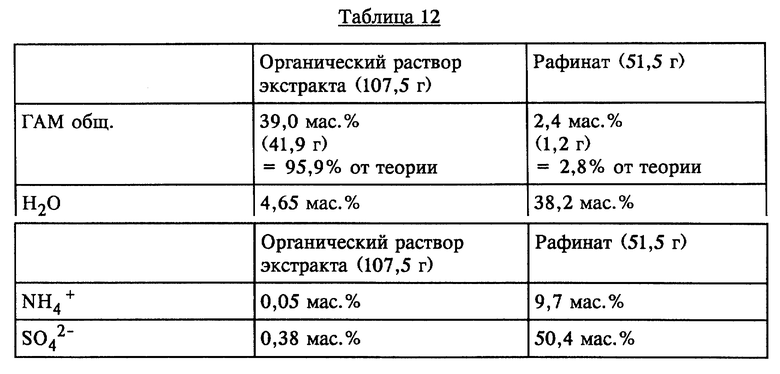
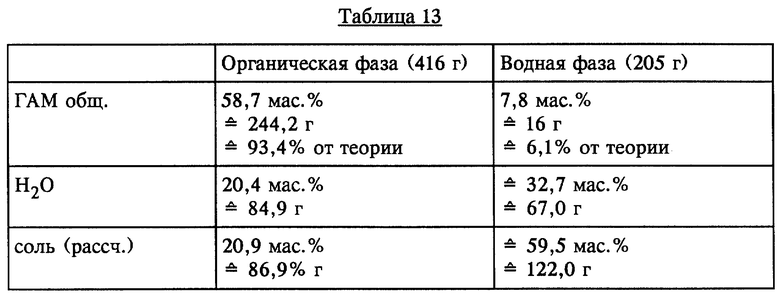

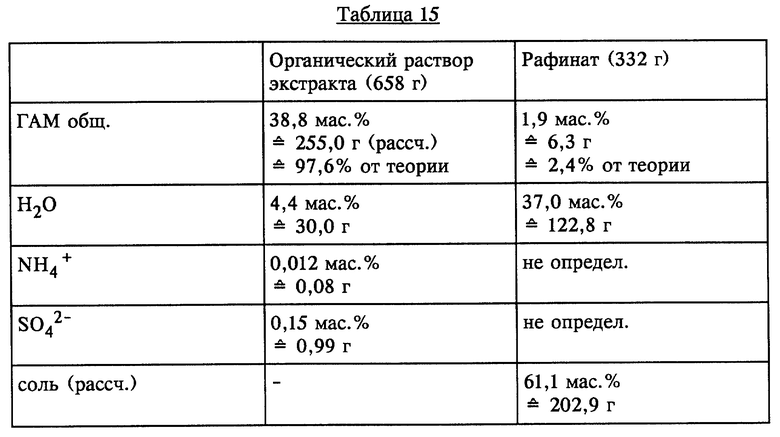
Описывается способ получения 2-гидрокси-4-метилтиомасляной кислоты (ГАМ) путем взаимодействия цианистоводородной кислоты (CHN) и метилмеркаптопропиональдегида (ММП) и гидролизом получаемого при этом циангидрина метилмеркаптопропиональдегида (ММП-ЦГ) с помощью серной кислоты, с последующим выделением ГАМ путем контактирования реакционной смеси с практически несмешиваемым с водой органическим растворителем в экстракционной системе жидкость/жидкость с целью образования экстракционного раствора, содержащего растворитель и выделенный из реакционной смеси ГАМ с получением экстракционного раствора, из которого путем упаривания получают ГАМ в виде экстракта. Способ отличается тем, что гидролиз ММП-ЦГ осуществляют таким образом, что на первой стадии ММП-ЦГ гидролизуют 60 - 85%-ной кислоты при молярном отношении ММП-ЦГ к Н2SO4 1:0,5 - 1:1,0, при температурах 30 - 90oC с получением в основном ГАМ-амида, который на второй стадии гидролизуют при добавлении воды без дальнейших добавок Н2SO4 при температурах до 140oC, содержание соли в реакционной смеси перед экстракцией в системе жидкость/жидкость доводят до > 50 мас.% (вес/вес), предпочтительно до > 55 мас.% (вес/вес), в пересчете на суммарное количество неорганических компонентов реакционной смеси. Технический результат - упрощение процесса, получение максимально высококонцентрированного продукта с предельно низким содержанием димеров, олигомеров и побочных продуктов. 16 з.п.ф-лы, 15 табл.
| Гидравлический распределитель | 1959 |
|
SU142488A1 |
| Бесконтактный электродвигатель постоянного тока | 1960 |
|
SU143100A1 |
| Способ получения 2-окси-4-(метилтио)-масляной кислоты | 1984 |
|
SU1428193A3 |

