Изобретение относится к способу получения замещенных производных пиразола.
В "The Chemistry of Heterocyclic Compounds", том 22, главы 3 и 5, описываются многочисленные возможности синтеза пиразола, как, например, конденсация α, β-дикарбонильных соединений с гидразинами, взаимодействие этилкарбонильного соединения с гидразинами и конденсация эфира гидразинуксусной кислоты с 1,2-дикетонами.
Известно далее, что 2-пиразолин можно дегидрировать с помощью хлора гипохлоритов щелочных либо щелочноземельных металлов (заявка DE-A 3035395), с помощью серы или селена (заявка DE-A 3029160) или же с помощью водного пероксида водорода (заявка DE-A 3415385), получая в результате пиразол. Известны далее такие способы получения пиразола, как термическое дегидрирование в газовой фазе 2-пиразолина в присутствии палладия либо платины (заявка DE-A 3209148) и термолиз N-сульфонил-2-пиразолина (заявка DE-A 3035394).
Кроме того, в ряде публикаций описано дегидрирование 2-пиразолинов в серной кислоте, осуществляемое в присутствии соединений йода. Согласно европейской заявке ЕР 0474037 пиразолин получают in situ из необязательно замещенного гидразина и бут-2-ендиола-(1.4), бут-1-ендиола-(3,4) или этинилалкилкарбинола. В международной заявке WO 95/06036 предлагается сначала из необязательно замещенного гидразина и ненасыщенного α, β-карбонильного соединения получать пиразолин и затем после смешения с серной кислотой и йодным катализатором осуществлять дегидрирование. Согласно европейской заявке ЕР 0402722 пиразолин получают предварительно либо in situ из необязательно замещенного пиразолина и глицерина, акролеина, соответственно винилалкилкетона или β-гидроксиэтилалкилкетона.
Все указанные способы, однако, являются неудовлетворительными в технологическом отношении, касается ли это необходимости использовать при их реализации крайне агрессивные окислители или дорогие катализаторы или образования ядовитых побочных продуктов, таких, как сероводород и селеноводород, или возможности получения применяемых в них соединений, связанной со значительными трудностями, или обязательного проведения при их осуществлении нескольких стадий.
С учетом вышеизложенного в основу изобретения была положена задача разработать более простой по технологии его осуществления и более экономичный способ получения производных пиразола.
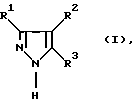
Неожиданным образом было установлено, что эта задача решается с помощью способа получения производных пиразола формулы I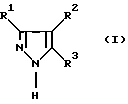
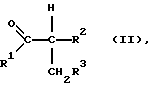
в которой R1, R2 и R3 независимо друг от друга обозначают атом водорода или необязательно замещенную алкильную, циклоалкильную, арильную либо аралкильную группу. Способ отличается тем, что карбонильное соединение формулы II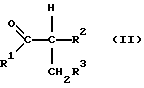
в которой R1, R2 и R3 имеют вышеуказанные значения, подвергают взаимодействию с гидразином, гидразингидратом либо с его кислотно-аддитивной солью в присутствии серной кислоты и йода или соединения, высвобождающего йод либо йодистый водород.
В способе по изобретению могут использоваться карбонильные соединения общей формулы II, в которой R1, R2 и R3 независимо друг от друга имеют значения, выбранные из группы, включающей атом водорода, прямоцепочечный либо разветвленный алкил, такой, как С1-С8алкил, прежде всего С1-С4алкил, такой, как метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метил-пропил и 1,1-диметилэтил; С3-С8циклоалкил, такой, как прежде всего циклопентил, циклогексил и циклогептил; С6-С14арил, такой прежде всего, как фенил; аралкил, такой прежде всего, как фенил-С1-С4алкил, причем алкильный остаток имеет значения согласно вышеприведенной расшифровке, например, бензил и 2-фенилэтил; и обозначают далее соответствующие органические радикалы, замещенные одним либо несколькими атомами галогена, такого, как фтор, хлор, бром или йод, нитро-, сульфо- или сульфокислотными группами, такими прежде всего, как хлорфенил, нитрофенил или толил.
В способе по изобретению предпочтительно использовать карбонильные соединения общей формулы II, в которой R2 не обозначает водород. Прежде всего R2 обозначает метил. R3 обозначает прежде всего атом водорода. Наиболее предпочтительными значениями R2 и R3 являются метил и водород соответственно. Наряду с указанными предпочтительны такие соединения общей формулы II, в которой R1 обозначает водород, метил, этил, н-пропил, трет-бутил, фенил, о-, м- либо п-толил, о-, м- либо п-хлорфенил, о-, м- либо п-нитрофенил, о-, м- либо п-сульфофенил или о-, м- либо п-сульфонилфенил.
Пригодными для использования в способе согласно изобретению являются в первую очередь следующие карбонильные соединения: изобутиральдегид, метилизопропилкетон (2-метилбутанон-3), этилизопропилкетон (2-метилпентанон-3), н-пропилизопропилкетон (2-метилгексанон-3), изопропил-трет-бутилкетон, фенилизопропилкетон, толилизопропилкетон, хлорфенилизопропилкетон, нитрофенилизопропилкетон, сульфофенилизопропилкетон и сульфонилфенилизопропилкетон.
В качестве второго компонента реакции используют гидразин. При этом можно применять как свободное основание гидразина, так и его гидраты или кислотно-аддитивные соли с минеральными кислотами, такие, например, как гидразиновые соли серной кислоты, соляной кислоты или фосфорной кислоты. Поскольку при применении солей, которые в реакционной среде не растворяются, может иметь место снижение выхода продукта, предпочтительно использовать гидрат либо свободное основание.
Серную кислоту используют в способе по изобретению в качестве разбавителя, агента конденсации и в качестве окислителя. Ее применяют в концентрации предпочтительно в пределах от 30 до 100 мас.%, прежде всего от 45 до 90 мас. %.
При необходимости можно использовать инертные, органические растворители, такие, как хлорированные углеводороды, например дихлорэтан, служащие дополнительными разбавителями.
В качестве катализатора наряду с элементарным йодом могут применяться также йодные соединения, такие, как йодистый водород, йодиды щелочных и щелочноземельных металлов, такие, как йодид лития, йодид натрия, йодид калия, йодид цезия, йодид магния и йодид кальция, равно как и другие йодиды металлов. Приемлемы и иные, неорганические йодные соединения, такие, как гипойодиты, йодиты, йодаты и перйодаты щелочных либо щелочноземельных металлов, или органические соединения йода, такие, как алкилйодиды, например метилйодид. Иод, соответственно йодное соединение применяют в этой реакции, как правило, в количествах от 0,01 до 10 мол.%, прежде всего от 0,05 до 5 мол.% на моль гидразина.
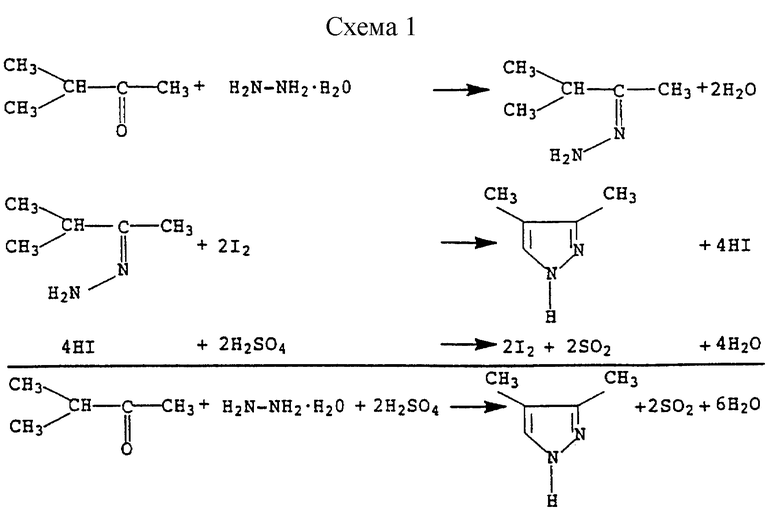
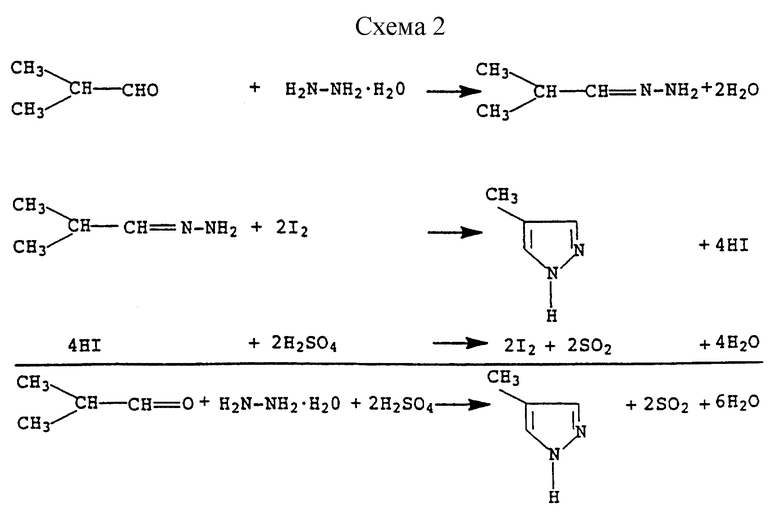
Указанную реакцию, осуществляемую с использованием гидразингидрата и метилизопропилкетона, а также йодистого водорода в качестве катализатора, можно представить схемой (см. в конце описания).
Реакцию целесообразно проводить таким образом, чтобы обеспечить успешное взаимодействие 1 моля гидразинового соединения с 0,5-2 молями, предпочтительно 0,8-1,5 моля карбонильного соединения формулы II в серной кислоте в присутствии каталитических количеств йодного соединения и чтобы при этом можно было частично удалять из реакционной смеси имеющуюся и дополнительно образующуюся воду. Предпочтительно такое удаление воды осуществлять путем перегонки, например, при нормальном давлении, т.е. при давлении порядка 1 атмосферы. Температура реакции находится в диапазоне от 50 до 250oС, предпочтительно от 80 до 200oС и прежде всего от 110 до 170oС. Обычно реакцию проводят при нормальном давлении. Возможны и варианты, в которых реакцию проводят при повышенном давлении или при соответственно повышенной температуре в серной кислоте с меньшей степенью концентрации, соответственно при пониженном давлении или соответственно при низкой температуре в серной кислоте с более высокой степенью концентрации.
Реакцию можно осуществлять следующим образом: либо все компоненты загружают одновременно в реакционный сосуд и нагревают до требуемой температуры, либо реагенты загружают в уже нагретый до соответствующей температуры реакционный сосуд в виде смеси или по отдельности, либо часть реагентов загружают в сосуд при определенной температуре, а остальное количество вводят в ходе реакции. Серную кислоту можно также загружать в реакционный сосуд как таковую или же совместно с гидразином.
Требуемую температуру реакции достигают предпочтительно при отгонке воды. Начало образования пиразола определяют по образованию диоксида серы. При абсорбции едким натром диоксид серы обеспечивает получение молярных количеств раствора гидросульфита натрия с высокой степенью чистоты. В отогнанной воде содержится большая часть используемого йодида в виде йодистого водорода, который можно повторно возвращать в процесс.
Последующую переработку реакционной смеси с целью выделения пиразола осуществляют с помощью обычных методов. Предпочтительно для проведения такой переработки темно-коричневую реакционную смесь нейтрализуют, например, едким натром, аммиаком или какими-либо другими неорганическими основаниями. Для выделения пиразола нейтрализованную реакционную смесь, например, несколько раз экстрагируют с помощью соответствующего растворителя. В качестве такого растворителя пригодны среди прочих изобутанол, хлорированные углеводороды или тетрагидрофуран. После сушки экстракционного раствора и упаривания досуха получают соответствующие пиразолы со степенью чистоты 80-90%. С целью повысить степень чистоты эти сырые продукты можно подвергать перегонке или перекристаллизации. Нейтрализованную реакционную смесь можно перерабатывать и путем перегонки, отгоняя при этом воду и чистый пиразол и получая в качестве остатка от перегонки загрязненный органическими побочными продуктами сульфат натрия (или сульфат аммония). При нейтрализации с использованием аммиака в кубовом погоне дистилляции получают содержащий загрязняющие примеси сульфат аммония, который оксидативным путем можно расщеплять на азот и диоксид серы. Последний можно через SO3 повторно превращать в серную кислоту.
Предлагаемый способ можно осуществлять в непрерывном либо перйодическом режиме, без давления, под давлением либо при слегка повышенном давлении.
Получаемые с помощью способа по изобретению пиразольные соединения формулы I представляют собой исходные вещества для органического синтеза, например, фармацевтических продуктов и средств защиты растений. Наиболее предпочтительно по способу согласно изобретению получают следующие соединения: 4-метилпиразол, 3,4-диметилпиразол, 3-этил-4-метилпиразол, 3-н-пропил-4-метилпиразол, 3-трет-бутил-4-метилпиразол, 3-фенил-4-метилпиразол, 3-толил-4-метилпиразол, 3-хлорфенил-4-метилпиразол и 3-нитрофенил - 4-метилпиразол.
Представленные ниже примеры служат для наглядного пояснения способа по изобретению.
Примеры
Пример 1. Получение 3,4-диметилпиразола (соединение формулы I, в которой R1 обозначает метил, R2 обозначает метил и R3 обозначает водород)
В суспензию из 560 г (4,0 моля) 70%-ной серной кислоты, 62,5 г (1,0 моль) 80%-ного гидразингидрата и 1 г (6,67 ммолей) йодида натрия при начальной температуре 120oС в течение 3 часов по каплям добавляют 111,8 г (1,3 моля) 3-метил-2-бутанона, в процессе введения которого температура реакции снижается до 110oС. По завершении добавления температура за счет отгонки 210 мл воды в течение 2 часов повышается до 130oС и в течение 30 мин при этой температуре смесь перемешивают. По завершении процесса добавления отогнанных 210 мл воды при температуре 100oС смесь охлаждают и с помощью 655 г (4,1 моля) 25%-ного едкого натра устанавливают на рН 9. После экстракции изобутанолом органическую фазу концентрируют и затем проводят перегонку под вакуумом. В результате получают 79,9 г 3,4-диметилпиразола с содержанием 99,2%. Этот показатель соответствует выходу, равному 82,6% от теории; tкип 90oС (при давлении 5 мбар). Идентификацию полученного продукта проводили на основании физико-химических данных.
Пример 2. Получение 3-фенил-4-метилпиразола (соединение формулы I, в которой R1 обозначает фенил, R2 обозначает метил и R3 обозначает водород)
В суспензию из 490 г (3,0 моля) 60%-ной серной кислоты, 31,25 г (0,5 моля) 80%-ного гидразингидрата и 0,5 г (3,33 моля) йодида натрия при 125oС в течение 2 часов по каплям добавляют 74,8 г (0,505 моля) изопропилфенилкетона. После перемешивания в течение одного часа при 125oС температуру за счет отгонки 155 мл воды доводят до 140oС. После охлаждения реакционной смеси значение рН последней с помощью 640 г (4,0 моля) едкого натра устанавливают равным 7,5. После фильтрации и сушки остаток на фильтре перекристаллизовывают из этанола. В результате получают 69,2 г светло-коричневых кристаллов с tпл 115oС и содержанием 97% (определение посредством ЖХВР), что соответствует выходу 85% от теории. Идентификацию полученного продукта осуществляли на основании физико-химических данных.
Пример 3. Получение 4-метилпиразола (соединение формулы I, в которой R1 обозначает водород, R2 обозначает метил и R3 обозначает водород)
К суспензии из 560 г (4,0 моля) 70%-ной серной кислоты и 62,5 г (1,0 моль) 80%-ного гидразингидрата добавляют 1,0 г (6,67 ммолей) йодида натрия и при 125oС в течение 2 часов с помощью дозирующего насоса под поверхность суспензии закачивают 86,4 г (1,2 моля) изобутиральдегида. В процессе добавления изобутиральдегида и в течение максимум 100 минут после его завершения отгоняют в общей сложности 175 мг воды и при этом температура реакционной смеси к концу повышается до 135oС. После охлаждения значение рН реакционного раствора с помощью 820 г (5,125 молей) 25%-ного едкого натра устанавливают равным 8,6 и проводят экстракцию изобутанолом. Объединенные экстракты концентрируют с помощью ротационного испарителя до количества в 82 г и затем осуществляют перегонку. 82% основного погона (tкип 82oС при давлении 7 мбар; 49 г) представляют собой 4-метилпиразол, который идентифицировали путем сравнения с аутентичным материалом. Выход составлял 49% от теории.
Пример 4. Получение 3-этил-4-метилпиразола (соединение формулы I, в которой R1 обозначает этил, R2 обозначает метил и R3 обозначает водород)
К суспензии из 280 г (2,0 моля) 70%-ной серной кислоты, 12,5 г (0,25 моля) 100%-ного гидразингидрата и 0,5 г (3,33 моля) йодида натрия при 125oС добавляют по каплям 27,5 г (0,275 моля) 2-метил-3-пентанона. По завершении добавления температуру в течение одного часа доводят до 110oС и в течение 6 часов при этой температуре смесь перемешивают. Затем реакционную смесь охлаждают и с помощью 480 г (3,0 моля) 25%-ного едкого натра устанавливают на рН 9. После экстракции изобутанолом органическую фазу концентрируют и затем проводят перегонку под вакуумом. В результате получают 18,5 г 3-этил-4-метилпиразола с tкип 90oС при давлении 5 мбар и содержанием 95% (определение посредством ЖХВР), что соответствует выходу 63,9% от теории. Идентификацию полученного продукта осуществляли сравнением физико-химических данных с аутентичным образцом.
Описывается способ получения производных пиразола формулы I, осуществляемый взаимодействием карбонильного соединения формулы II с гидразином, его гидратом либо с его солями в 30-100 мас.%-ной серной кислоте в присутствии каталитических количеств йода либо йодного соединения. Способ прост в технологическом отношении, так как проводится в одну стадию. 7 з.п.ф-лы.
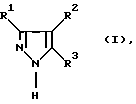
в которой R1 и R2 независимо друг от друга обозначают необязательно замещенную C1-С8алкильную, С3-С8циклоалкильную, С6-С14арильную или фенил-С1-С4алкильную группу;
R1 может дополнительно обозначать водород;
R3 обозначает водород,
отличающийся тем, что карбонильное соединение формулы II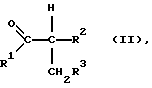
в которой R1, R2 и R3 имеют указанные выше значения,
подвергают взаимодействию с гидразином, гидразингидратом либо с его кислотно-аддитивной солью в присутствии 30-100 мас. %-ной серной кислоты и 0,05-5 мол. % йода, соответственно высвобождающего йод либо йодистый водород соединения, в пересчете на гидразиновое соединение, при температурах в интервале от 80 до 200oС, при этом гидразиновое соединение и карбонильное соединение применяют в молярном соотношении от 1: 0,8 до 1: 1,5.
| Способ получения 3,5-дифенилпиразола | 1978 |
|
SU707913A1 |
| ПРОИЗВОДНЫЕ 3-ФЕНИЛПИРАЗОЛА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ФЕНИЛПИРАЗОЛА И ФУНГИЦИДНАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2072991C1 |
| 0 |
|
SU402722A1 | |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Устройство для обучения крановщика навыкам по управлению краном | 1972 |
|
SU474037A1 |
