Настоящее изобретение относится к способу каталитического получения нитрила 2-гидрокси-4-(метилтио)масляной кислоты (ММП-CN) из 3-(метилтио)пропаналя (метилмеркаптопропионового альдегида, ММП) и цианистого водорода (HCN). В изобретении предлагается прежде всего способ получения стабильного при хранении ММП-CN с применением синильной кислоты в стехиометрических количествах, при этом продукт содержит синильную кислоту в количествах, сверхстехиометрических по отношению к непрореагировавшему ММП, соответственно к ММП, находящемуся в равновесии с ММП-CN.
Нитрил 2-гидрокси-4-(метилтио)масляной кислоты (ММП-циангидрин) представляет собой промежуточный продукт для получения D,L-метионина и 2-гидрокси-4-метилтиомасляной кислоты, являющейся гидроксианалогом метионина (ГАМ). Метионин представляет собой незаменимую аминокислоту, используемую помимо прочего в качестве добавки к кормам. ГАМ представляет собой жидкий заменитель метионина с меньшей биологической доступностью.
Уровень техники
Из ММП можно путем его реакции с цианистым водородом (синильной кислотой) с применением пригодных для этого катализаторов получать ММП-циангидрин (нитрил 2-гидрокси-4-(метилтио)масляной кислоты). К пригодным для применения в этих целях катализаторам относятся, например, пиридин или триэтиламин. Путем гидролиза ММП-циангидрина в присутствии, например, минеральных кислот получают ГАМ. Метионин образуется в результате реакции ММП-циангидрина с гидрокарбонатом аммония с образованием гидантоина, который можно омылять действием основания, например, карбоната калия или гидроксида натрия. Метионин выделяют действием диоксида углерода или серной кислоты.
Из, например, US 4960932 известно получение метионина четырехстадийным способом. На первой стадии путем присоединения HCN к ММП в присутствии триэтиламина получают ММП-циангидрин. HCN применяют в количестве, которое соответствует 1,05 моля по отношению к применяемому количеству ММП. Далее ММП-циангидрин на второй стадии подвергают взаимодействию с аммиаком, в результате чего образуется 2-амино-4-метилтиобутиронитрил, который затем на третьей стадии гидролизуют в присутствии кетона и гидроксида щелочного металла с образованием метилтиобутирамида, который в завершение омыляют до метионината щелочного металла.
При получении 2-гидрокси-4-метилтиомасляной кислоты (ГАМ) 2-гидрокси-4-метилтиобутиронитрил получают путем взаимодействия между собой ММП и HCN в среде, содержащей пиридин или амин (см. US 2745745, колонка 2, строки 52-55). Избыточный HCN при этом лишь отгоняют, например, в вакууме. Полученный 2-гидрокси-4-метилтиобутиронитрил затем гидролизуют в присутствии серной кислоты, в результате чего образуется непосредственно амид 2-гидрокси-4-метилтиомасляной кислоты, а в завершение образуется 2-гидрокси-4-метилтиомасляная кислота. Аналогичный способ описан также в ЕР 330527 А1, соответственно в US 4912257.
В WO 96/40631 А1 описано далее получение ММП-циангидрина взаимодействием ММП с цианистым водородом в присутствии пригодного для этого катализатора реакции присоединения. Согласно данной публикации было установлено, что в качестве такого катализатора реакции присоединения для получения ММП-циангидрина могут использоваться триизопропаноламин, никотинамид, имидазол, бензимидазол, 2-фторпиридин, поли-4-винилпиридин, 4-диметиламинопиридин, пиколин и пиразин. Помимо этого для катализа реакции между ММП и цианистым водородом с образованием ММП-циангидрина можно также использовать триалкиламины с 3-18 атомами углерода в каждом из связанных с атомом азота алкильных заместителей и третичные амины, в которых по меньшей мере один из связанных с атомом азота неводородных заместителей содержит согласно вышеуказанной публикации арильную группу.
Цианистый водород предпочтительно при этом использовать в молярном избытке около 2% в пересчете на ММП.
В WO 2006/015684 А2 описан, кроме того, способ прежде всего непрерывного получения ММП, соответственно ММП-циангидрина, предусматривающий в каждом случае использование гетерогенных аминных катализаторов при реакции присоединения.
Из US 5756803 известно далее проведение взаимодействия альдегида с цианистым водородом в присутствии буфера, с помощью которого величину рН раствора можно устанавливать на значение более 4, исключая при этом амины. В наиболее общем случае в качестве такого буфера могут использоваться соли кислот с щелочными металлами в смеси с кислотами или кислоты в смеси с гидроксидами щелочных металлов. Буфер применяют с той целью, чтобы, с одной стороны, воспрепятствовать разложению исходных веществ и целевого продукта, а с другой стороны, нейтрализовать кислоты, применяемые для стабилизации цианистого водорода. В данном случае HCN также дозируют в молярном избытке по отношению к ММП, составляющем преимущественно от 2 до 5%. При проведении взаимодействия ММП с HCN в присутствии обычно используемых оснований они хотя и ускоряют протекание реакции в указанных условиях, однако быстро приводят к разложению образовавшегося циангидрина и к разложению исходно применяемого альдегида с образованием интенсивно окрашенного раствора.
Для рекуперации непрореагировавших HCN и ММП, остаточные количества которых содержатся в отходящем газе из абсорбера-реактора, их вымывают в водяном газопромывателе, используемая в котором промывочная вода попадает при этом в продукт. Содержание воды в продукте составляет около 48 мас. %.
К существенным недостаткам ранее описанных в литературе способов следует отнести необходимость использования HCN в высоком молярном избытке для достижения высокого выхода ММП-CN. Избыточные количества HCN бесполезно теряются при осуществлении таких известных способов, что является их значительным экономическим недостатком. Помимо этого катализаторы, используемые при осуществлении известных способов, способствуют также образованию нежелательных побочных продуктов из исходно применяемых альдегидов, что приводит к загрязнению продукта примесями сверх допустимых пределов. Один из подходов к решению данной проблемы образования побочных продуктов описан в US 5756803, при этом, однако, в продукт в больших количествах попадает вода, которую, с одной стороны, для получения метионина необходимо по меньшей мере частично вновь удалять и которая, с другой стороны, опять же способствует разложению ММП-циангидрина, что в одном и другом случае является существенным недостатком. Поэтому описанный в US 5756803 продукт не стабилен при хранении и требует для его хранения и прежде всего для его транспортировки сложной и дорогостоящей переработки путем дистилляционного отделения от него воды, что является значительным экономическим недостатком данного способа.
Задача изобретения
В основу настоящего изобретения была положена задача разработать каталитический способ, который обеспечивал бы катализ реакции альдегида, прежде всего ММП, с цианистым водородом и одновременно получение стабильного при хранении циангидрина и который в то же время должен обеспечивать явное увеличение выхода продукта в пересчете на исходно используемый альдегид и цианистый водород.
Подробное описание изобретения
Указанная задача, а также другие задачи решаются с помощью способа получения нитрила 2-гидрокси-4-(метилтио)масляной кислоты, заключающегося в том, что 3-(метилмеркапто)пропионовый альдегид подвергают в зоне основной реакции взаимодействию с цианистым водородом в присутствии основания в качестве катализатора с получением нитрила и остаточный газообразный цианистый водород (HCN), выходящий из зоны основной реакции, абсорбируют в зоне абсорбции и последующей реакции, содержащей смесь из 3-(метилмеркапто)пропионового альдегида и катализатора, а также по выбору нитрила 2-гидрокси-4-(метилтио)масляной кислоты, и подвергают дальнейшему превращению.
Остаточный HCN реагирует вследствие абсорбции, соответственно конденсации с альдегидом с образованием циангидрина. Благодаря эффективному удалению HCN из газовой фазы цианистый водород можно в отличие от известных из литературы способов использовать в молярном соотношении между ним и альдегидом в пределах от 0,99 до 1,01, что является значительным экономическим преимуществом предлагаемого в изобретении способа.
В изобретении предлагается прежде всего способ проведения реакции присоединения цианистого водорода к ММП в присутствии основания, в первую очередь амина, при этом такой способ предусматривает абсорбцию остаточных количеств газообразного цианистого водорода вне зоны основной реакции при температуре примерно от 0 до 25°C в жидкой смеси, содержащей альдегид ММП и продукт реакции ММП с цианистым водородом, а также катализатор, и затем дальнейшее взаимодействие с ММП.
Предлагаемым в изобретении способом можно в целом эффективно подвергать взаимодействию с цианистым водородом альдегиды, содержащие от 1 до 6 атомов углерода и необязательно замещенные алкилом, алкоксигруппой или алкилтиогруппой.
При этом предпочтительно, чтобы присутствующая в зоне абсорбции и последующей реакции смесь по меньшей мере частично поступала из зоны основной реакции. Благодаря этому в отличие от US 5756803 предотвращается разбавление посторонними веществами или растворителями.
Зона основной реакции может содержать реактор с мешалкой или реактор с циркуляцией. В обоих вариантах обеспечивается быстрое и тщательное перемешивание и быстрое взаимодействие ММП и HCN между собой.
Зона основной реакции дополнительно может также содержать струйный насос. Такой насос обеспечивает дальнейшую интенсификацию смешения компонентов и может особо эффективно использоваться как дополнительное оборудование для всасывания HCN в зону основной реакции.
Последующая реакция может, как указано выше, происходить между HCN-содержащим газом и жидкостью. Такая реакция в этом случае происходит в зоне абсорбции и последующей реакции, содержащей в предпочтительном варианте устройство для введения газа в контакт с жидкостью, прежде всего колонну, такую, например, как тарельчатая колонна, насадочная колонна, барботажный реактор колонного типа, колонна с капельным течением, либо по выбору реактор с емкостью с механическим перемешиванием или реактор с погружным эжектором.
Зона абсорбции и последующей реакции может также являться частью реактора с циркуляцией, что способствует интенсивному перемешиванию и быстрому взаимодействию компонентов.
При осуществлении предлагаемого в изобретении способа в зону основной реакции подают в основном газообразный цианистый водород, а в предпочтительном варианте подают газ, содержащий цианистый водород в качестве продукта из процесса его получения.
Содержание цианистого водорода в применяемой газовой смеси составляет от 1 до 99 мас. %, предпочтительно от 5 до 75 мас. %, особенно предпочтительно от 6 до 22 мас. %. Цианистый водород получают прежде всего по методу Андрусова согласно DE 102007034715 А1 или по так называемому СМА-методу (процесс получения синильной кислоты из метана и аммиака) согласно DE 1041476 (реактор). Оба метода описаны также в Ullmann′s Encyclopedia of Industrial Chemistry, изд-во VCH-Verlagsgesellschaft mbH, 1987, глава ″Cyano Compounds Inorganic″, разделы 1.2.1 и 1.2.2. Присутствующий аммиак в каждом случае удаляют из газообразного продукта. Газообразный продукт из процесса Андрусова (газ из процесса Андрусова) после удаления аммиака обычно содержит примерно 10 мас. % цианистого водорода, тогда как газообразный продукт из СМА-процесса (газ из СМА-процесса) содержит примерно 70 мас. % цианистого водорода.
Так, в частности, газообразные продукты, получаемые в процессе Андрусова, обычно имеют следующий состав: 10,3 мас. % HCN, 3,7 мас. % Н2О, 1,3 мас. % Н2, 75,8 мас. % N2, 0,4 мас. % O2, 6,3 мас. % СО, 0,6 мас. % CO2, 0,4 мас. % СН4, 1,3 мас. % Ar, а газообразные продукты, получаемые в СМА-процессе, обычно имеют следующий состав: примерно 68,3 мас. % HCN, 6,7 мас. % H2O, 17,3 мас. % Н2, 3,6 мас. % N2, 4 мас. % СН4.
Непосредственное применение газообразного продукта обладает тем существенным преимуществом, что отсутствует необходимость в предшествующем и энергоемком сжижении цианистого водорода, а при соответствующем объединении с установкой для получения цианистого водорода достигается экономия существенных капиталовложений, неизбежных в противном случае на реализацию соответствующих технологических стадий абсорбции и отгонки HCN. Остальные компоненты газообразного продукта, содержащиеся в нем наряду с HCN, неожиданно не оказывают никакого отрицательного влияния на выход циангидрина.
Остаточные газы из процессов получения ММП-циангидрина и цианистого водорода можно затем совместно утилизировать или подавать на сжигание. В последнем случае полученную при этом энергию можно вновь использовать в обоих процессах, что предоставляет большую свободу выбора и является значительным экономическим преимуществом.
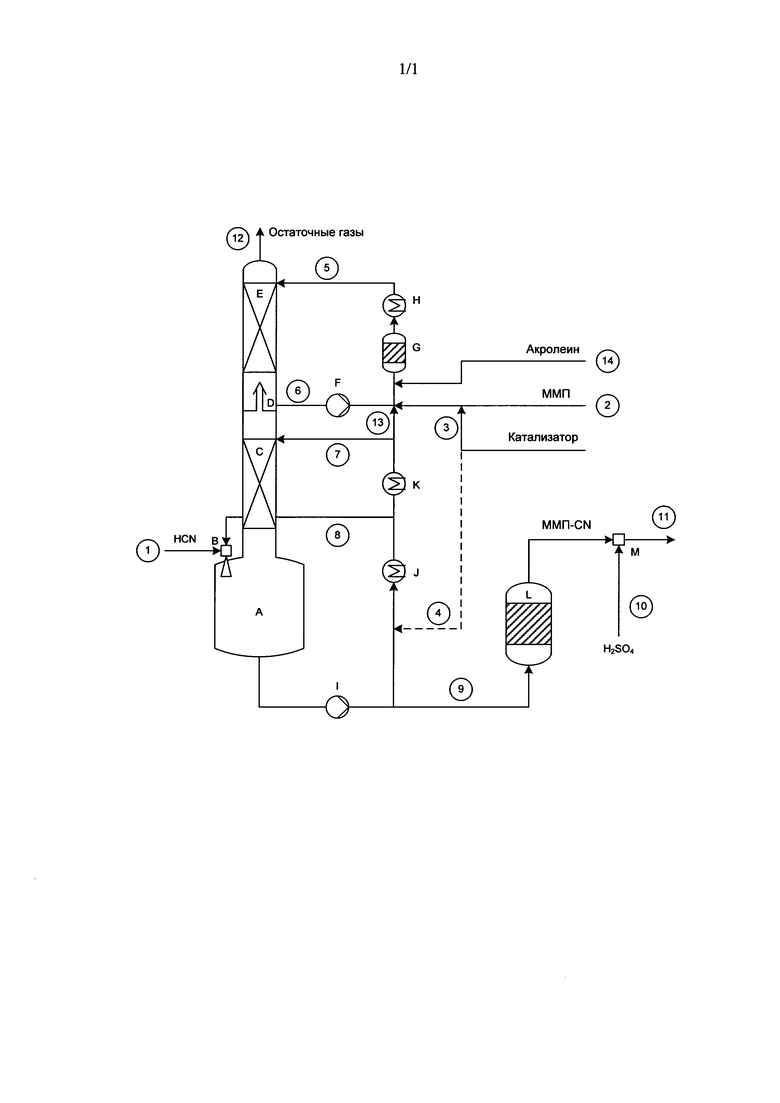
Ниже со ссылкой на прилагаемый к описанию единственный чертеж более подробно рассмотрен схематично проиллюстрированный на нем один из предпочтительных вариантов осуществления предлагаемого в изобретении способа, соответственно вариантов выполнения соответствующего устройства.
При применении тарельчатой колонны или насадочной колонны для абсорбции с химической реакцией (химической абсорбции) газовый поток, содержащий синильную кислоту, подается в низ (куб) (А) колонны (С) либо в предпочтительном варианте с помощью эксгаустера (или газодувки) (В) уже вводится в контакт с раствором альдегида, перекачиваемым насосом (I) по циркуляционному контуру (8). Температура в низу колонны регулируется с помощью теплообменника (J). Низ (А) и колонна (С) служат прежде всего в качестве зоны основной реакции, при этом колонну (С) можно отдельно термостатировать с помощью теплообменника (К). Температуру потоков (7) и (8) выбирают при этом такой, чтобы теплота реакции могла отдаваться с охлаждающей водой соответственно окружающей температуре, а реакция между альдегидом и HCN в части (С) колонны завершалась на 80-99,9%.
Альдегид можно подавать отдельно от катализатора или совместно с ним ((2), (3)). В предпочтительном варианте альдегид или смесь альдегида с катализатором (2)+(3) смешивают с частичным потоком (6) из абсорбционно-конденсационной части (Е) колонны, отбираемым из промежуточного сборника (D). Катализатор можно также подавать, например, по линии (4). В этом случае катализатор отчасти попадал бы также по линии (13) в верхний циркуляционный контур. Содержащийся в потоке (6) в остаточных количествах HCN подвергается в аппарате (G) с заданным временем пребывания в нем реагентов, т.е. в зоне (второй) последующей реакции, полному, соответственно почти полному превращению в циангидрин при своем взаимодействии с подаваемым альдегидом. Затем этот поток охлаждается в теплообменнике (Н) до температуры в пределах от 0 до 25°C с целью обеспечить конденсацию/абсорбцию HCN в максимально возможной степени. Промежуточный сборник (D), абсорбционно-конденсационная часть (Е) и аппарат (G) с заданным временем пребывания в нем реагентов прежде всего служат в качестве зоны абсорбции и последующей реакции. Поскольку поток (5) содержит в некоторых количествах циангидрин и был подвергнут охлаждению, выходящие из верха колонны остаточные газы содержат альдегид также лишь в очень малых остаточных количествах, и поэтому отсутствует необходимость в дополнительной промывке остаточного газа для рекуперации из него альдегида. Концентрацию циангидрина в потоке (5) можно путем соответствующего дозирования (13) из низа колонны устанавливать на значение, которое в предпочтительном варианте составляет от 10 до 70 мас. %. Очищенные газы в предпочтительном варианте направляют в сжигательную установку. В выходящем с потоком (9) продукте молярное соотношение между цианистым водородом и непрореагировавшим альдегидом превышает 1, что в существенной мере способствует стабилизации продукта. Помимо этого продукт прозрачен и лишь слабо окрашен, что свидетельствует об исключительно высокой избирательности данного варианта осуществления предлагаемого в изобретении способа.
После прохождения последующего реактора (L), в котором возможно присутствующий в остаточных количествах альдегид окончательно реагирует до достижения равновесия с цианистым водородом, полученный таким путем продуктсодержащий поток смешивают с кислотой. С этой целью используют пригодный для этого смеситель (М). Значение рН продукта (поток (11)) устанавливается при этом на величину в пределах от 1 до 4, преимущественно от 2 до 3.
В том случае, когда альдегид представляет собой ММП, как это имеет место в проиллюстрированном на чертеже варианте, поток исходного ММП при осуществлении описанного выше способа обычно содержит в небольшом количестве метилмеркаптан (ММ), который преобладающей частью попадал бы в поток отходящих газов (12). Такой избыточный ММ можно также по выбору подвергать взаимодействию с акролеином, который можно подавать в процесс, например, в виде потока (14), с получением ММП, который затем соответственно можно подвергать взаимодействию с HCN с получением ММП-CN, дополнительно повышая тем самым выход продукта.
При осуществлении предлагаемого в изобретении способа в качестве катализатора можно использовать низкомолекулярные или гетерогенные амины или растворы неорганических оснований либо смеси из кислот и низкомолекулярных аминов. Их используют также для установления величины рН на значение, которое лежит в необходимых для протекания реакции оптимальных пределах от примерно 4,5 до 6,0, преимущественно от 5,0 до 5,5, и которое измеряют рН-электродом (тип: ″Aquatrode Plus mit Pt 1000″, изготовитель: Metrohm Schweiz AG) непосредственно в циангидрине с типичным водосодержанием от 2 до 14 мас. %. Измерения проводят при температуре примерно 23°C в емкости с мешалкой, при этом измерение величины рН выполняется с температурной компенсацией. Для оперативного отслеживания условий реакции и для устранения погрешностей измерения с интервалом в один час проводят каждый раз по 4 измерения значения pH с вычислением среднего значения, при этом каждое измерение длится примерно 30 с. Однако измерения можно также проводить непосредственно в ходе реакции в режиме реального времени в реакционной системе при установленной в ней температуре и пересчитывать результаты измерений на значение рН при 23°C, что дополнительно упрощает контроль за процессом.
Низкомолекулярные амины, предпочтительно с 1-36 атомами углерода, обладают особым преимуществом, которое состоит в их практически неограниченной смешиваемости с реакционной средой, что в свою очередь способствует быстрому протеканию реакции.
К предпочтительным низкомолекулярным аминам при этом относятся три(C1-C12алкил)амины, преимущественно триэтиламин или триизопропаноламин, диалкиларалкиламины, преимущественно диметилбензиламин, диалкилариламины, преимущественно N,N-диметиланилин, гетероциклические амины, преимущественно никотинамид, имидазол, бензимидазол, 2-фторпиридин, 4-диметиламинопиридин, пиколин или пиразин.
В другом варианте можно также использовать гетерогенные амины общей формулы I
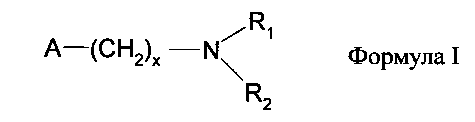
или поливинилпиридин, где
R1 и R2 обозначают водород, алкил с длиной цепи от C1 до C12, арил или
гетероарил, при этом R1 может иметь значения, отличные от R2,
x обозначает число от 0 до 6, а
А обозначает природную или синтетическую смолу, предпочтительно полистирол.
Эти и связанные с ними преимущества, такие, например, как более легкая отделяемость, малый унос на последующие стадии реакции, уже описаны в WO 2006/015684.
При этом в предпочтительном варианте катализатор формулы I представляет собой связанное с полимером основание, выбранное из группы гомологических диалкиламиноалкилполистиролов и несущих диалкиламиногруппы макросетчатых смол.
В особенно предпочтительном варианте катализатор формулы I представляет собой диэтиламиноэтилполистирол, диэтиламинометилполистирол, диметиламинометилполистирол, несущую диэтиламинометильные группы макросетчатую смолу или диметиламиноэтилполистирол.
В качестве неорганического основания можно эффективно использовать гидроксид щелочного металла, преимущественно NaOH или КОН, цианид щелочного металла, преимущественно NaCN или KCN, карбонат щелочного металла, преимущественно Na2CO3 или К2СО3, или гидрокарбонат щелочного металла, преимущественно NaHCO3 или КНСО3, индивидуально или в смешанном виде. Преимущество таких оснований состоит в наличии у них особо высокого каталитического действия, что в свою очередь способствует исключительно быстрому протеканию реакции, а также в том, что образующиеся из них в малых количествах солевые компоненты обладают низким потенциалом создавать помехи в последующем процессе. Однако в данном случае необходимо обеспечивать крайне тщательное перемешивание и контролировать температуру во избежание образования побочных продуктов в сколько-нибудь значительных количествах.
В качестве катализаторов можно с достижением соответствующих преимуществ использовать также смеси кислот и указанных выше низкомолекулярных аминов для возможности более эффективного регулирования значения рН с целью его установления на величину, лежащую в требуемых пределах, и его стабилизации благодаря буферному действию. Особенно предпочтительно при этом применение органических кислот, таких как жирные кислоты с короткой цепью, например, уксусная кислота, муравьиная кислота, лимонная кислота, и органические сульфокислоты, например, трифторметансульфоновая кислота, или применение минеральных кислот, таких, например, как серная кислота или фосфорная кислота, в сочетании с низкомолекулярными аминами.
В еще одном предпочтительном варианте осуществления изобретения температуру в зоне основной реакции выбирают таким образом, чтобы выделяющаяся теплота реакции могла передаваться охлаждающей воде соответственно окружающей температуре, в чем состоит еще одно существенное экономическое преимущество предлагаемого в изобретении способа.
В соответствии с этим температуру в зоне основной реакции устанавливают на величину в пределах от 20 до 80°C, предпочтительно от 30 до 70°C, особенно предпочтительно от 35 до 65°C. В таком интервале температур реакция также протекает сравнительно быстро.
При осуществлении предлагаемого в изобретении способа предпочтительно далее устанавливать температуру в зоне абсорбции и последующей реакции на величину в пределах от 0 до 30°C, предпочтительно от 4 до 15°C. Благодаря этому обеспечивается особо эффективная абсорбция цианистого водорода, а также все еще возможно достаточно полное превращение HCN при его взаимодействии с ММП в ММП-циангидрин.
Предпочтительно, кроме того, применение второй зоны последующей реакции непосредственно перед местом отбора продукта, которым является ММП-циангидрин. Температуру в этой зоне устанавливают на величину, которая аналогична температуре, создаваемой в зоне основной реакции, и которая в соответствии с этим лежит в пределах от 20 до 80°C, предпочтительно от 40 до 70°C, особенно предпочтительно от 45 до 65°C. Благодаря этому обеспечивается быстрое доведение полноты реакции между HCN и ММП с образованием ММП-циангидрина до практически количественной степени незадолго до отбора продукта.
Предлагаемый в изобретении способ в предпочтительном варианте осуществляют при абсолютном давлении в пределах от 0,9 до 5 бар, предпочтительно от 1,0 до 3 бар, особенно предпочтительно от 1 до 1,5 бара. Связанный с этим эффект состоит в предотвращении быстрого выделения абсорбированного HCN в газообразном виде из раствора и в предотвращении тем самым соответствующих потерь.
Предлагаемый в изобретении способ отличается далее тем, что молярное соотношение между синильной кислотой и 3-(метилтио)пропаналем можно устанавливать на величину в пределах от 0,98 до 1,03, предпочтительно от 0,99 до 1,01. Благодаря этому, во-первых, удается избежать потерь синильной кислоты, которыми именно в промышленном масштабе обусловлены существенные экономические убытки. Во-вторых, предотвращается образование нежелательных продуктов разложения синильной кислоты, таких, например, как полимерная синильная кислота или муравьиная кислота, которая является продуктом омыления синильной кислоты и обладает коррозионными свойствами по отношению к различным металлическим материалам, и тем самым предотвращаются соответствующие отрицательные последствия на последующих стадиях процесса получения метионина.
При осуществлении предлагаемого в изобретении способа катализатор в предпочтительном варианте используют в массовом соотношении между ним и 3-(метилтио)пропаналем в пределах от 0,00005 до 0,002, особенно предпочтительно от 0,0001 до 0,001. Связанный с этим эффект состоит в образовании побочных продуктов в особо низком количестве при одновременно высокой скорости реакции.
Предлагаемый в изобретении способ можно по выбору проводить в периодическом, полунепрерывном или же непрерывном режиме, при этом вариант с проведением предлагаемого в изобретении способа в непрерывном режиме является экономически особо эффективным при получении продукта в промышленном масштабе в количестве свыше 10000 тонн в год.
Получаемый предлагаемым в изобретении способом ММП-циангидрин обычно имеет следующий состав:
Молярный выход в пересчете на ММП составляет обычно от 99,50 до 99,99%.
Получаемый предлагаемым в изобретении способом продукт реакции, содержащий циангидрин метилтиопропионового альдегида (ММП-циангидрин), можно в особенно предпочтительном варианте непосредственно использовать для получения метионина и 2-гидрокси-4-метилтиомасляной кислоты. Для этого такой продукт реакции либо аминируют (аминонитрильный метод), либо подвергают взаимодействию со смесью аммиака и диоксида углерода (гидантоиновый метод) для образования метионина или непосредственно гидролизуют до 2-гидрокси-4-метилтиомасляной кислоты (гидроксианалога метионина, ГАМ). Помимо этого при создании изобретения неожиданно было установлено, что высококипящие ММП-олигомеры, уже присутствующие в ММП, при осуществлении предлагаемого в изобретении способа превращаются преобладающей частью в целевой ММП-циангидрин. Данный факт подтверждается, например, тем, что образующийся при дистилляции продуктов остаток после реакции выпадает в осадок в явно меньшем количестве, чем до реакции по превращению в ММП-циангидрин.
Ниже настоящее изобретение более подробно поясняется на примерах.
Применяемые аналитические методы
Содержание Н2О в ММП-CN определяли по методу титрования с биамперометрической индикацией конечного продукта (титрование методом Карла Фишера).
Для этого в сосуд для титрования помещали 20-30 мл титровальной среды (например, продукта Hydranal Solvent 5, фирма Fluka) и титровали в сухом состоянии титрантом (например, продуктом Hydranal Titrant 5, фирма Fluka). К оттитрованному первоначально помещенному в сосуд для титрования материалу добавляли (одноразовым пластмассовым шприцем) исследуемую пробу в количестве примерно 500 мг и титровали титрантом до конечной точки. Точную навеску пробы определяли путем разностного взвешивания.
Порядок проведения такого стандартного метода известен специалисту (см., например, P.A. Bruttel, R. Schlink, "Wasserbestimmung durch Karl-Fischer-Titration", Metrohm AG).
Содержание свободной синильной кислоты в продукте определяли в соответствии с принципом ионной хроматографии (ИХ) с амперометрическим определением цианида на рабочем электроде из серебра, при этом на стадии подготовки пробы от ее матрицы отделяли свободную синильную кислоту путем препаративной колоночной хроматографии.
Процесс препаративного отделения цианида проводили, например, при комнатной температуре на хроматографической колонке PRP-X 300 с длиной 250 мм и с внутренним диаметром 4,1 мм, выпускаемой фирмой Hamilton. В качестве подвижной фазы использовали 5-миллимолярную серную кислоту. При скорости потока 1,0 мл/мин вводили 100 мкл раствора пробы (0,1 г пробы в 10 мл подвижной фазы). Элюат из колонки на протяжении 4-8 мин собирали в мерную колбу емкостью 100 мл, объем содержимого колбы добавлением высокочистой воды доводили до метки и 100 мкл вводили в ИХ-колонку для определения цианида.
Аналогично раствору пробы препаративному разделению колоночной хроматографией подвергали калибровочный раствор NaCN известного состава и 100 мкл вводили в ИХ-колонку для определения цианида.
Процесс определения цианида путем ионной хроматографии проводили при комнатной температуре, например, на хроматографической колонке Carbo Рас РА1 с длиной 250 мм и с внутренним диаметром 4,0 мм, выпускаемой фирмой Dionex. В качестве подвижной фазы использовали раствор 1,5 г хлорида натрия и 1 мл этилендиамина в 1 л 50-миллимолярного раствора едкого натра. При скорости потока 1,0 мл/мин вводили 100 мкл раствора пробы, соответственно калибровочного раствора. Количественные значения определяли путем сравнения площадей пиков по методу внешнего стандарта.
Порядок проведения такого стандартного метода известен специалисту.
Содержание ММП-CN и ММП в продукте определяли ионоэксклюзионной хроматографией в изократическом режиме на катионообменнике с последующим УФ-обнаружением на длине волны 205 нм. Процесс определения указанных компонентов проводили, например, на хроматографической колонке PRP-X 300 с длиной 250 мм и с внутренним диаметром 4,1 мм, выпускаемой фирмой Hamilton, при температуре 25°C. В качестве подвижной фазы использовали 5-миллимолярную серную кислоту. При скорости потока 1,0 мл/мин вводили по 100 мкл соответствующего раствора пробы (0,5 г пробы для определения ММП, соответственно 0,06 г пробы для определения ММП-CN в 50 мл растворителя). Калибровку осуществляли путем ввода пригодных для этого калибровочных растворов (0,5 мг ММП в 50 мл растворителя, соответственно 50 мг ММП-CN в 50 мл растворителя).
В качестве растворителя использовали смесь из 500 мкл 0,5-молярной H2SO4 и 5 мл ацетонитрила, разбавленную высокочистой водой до объема 50 мл.
Количественные значения определяли путем сравнения площадей пиков по методу внешнего стандарта.
Порядок проведения такого стандартного метода известен специалисту.
Состав HCN-содержащего исходного газа для определения содержания в нем таких компонентов, как азот (N2), монооксид углерода (СО), диоксид углерода (СО2), метан (CH4), аммиак (NH3), синильная кислота (HCN), вода (Н2О), аргон (Ar)/кислород (О2) (либо/либо), водород (Н2) (лишь условно) и бензол в качестве внутреннего стандарта, анализировали газовой хроматографией. В этих целях использовали газовой хроматограф модели 6890 (Agilent, на базе HP 6890). Для проведения подобного анализа такой газовой хроматограф был оснащен тремя хроматографическими колонками: 1. колонкой HP-CPWAX 52СВ размером 25 м×0,32 мм при размере частиц набивки 0,2 мкм (на этой колонке отделяли NH3, HCN, воду и бензол), 2. колонкой с молекулярным ситом размером 30 м×0,32 мм при размере частиц набивки 12 мкм (на этой колонке отделяли Н2, N2, О2, СО и метан) и 3. колонкой Plot Q размером 30 м×0,32 мм при размере частиц набивки 20 мкм (на этой колонке отделяли СО2 и бензол), двумя детекторами теплопроводности, блоком измерения давления и массовым расходомером для гелия. Колонка 1 через заднее устройство для ввода пробы была соединена с задним детектором, колонки 2 и 3 передним устройством для ввода пробы были соединены с передним детектором.
Порядок проведения такого стандартного метода известен специалисту.
Состав выходящего из верха колонны остаточного газа для определения содержания в нем таких компонентов, как метилмеркаптан (ММ), метилмеркаптопропионовый альдегид (ММП) и акролеин (АК), анализировали газовой хроматографией. В этих целях использовали газовой хроматограф модели 7890А (Agilent). Для проведения подобного анализа такой газовой хроматограф был оснащен хроматографической колонкой (HP-INNOWAX размером 60 м×0,32 мм при размере частиц набивки 0,25 мкм) и задним детектором (пламенно-ионизационным детектором). Порядок проведения такого стандартного метода известен специалисту.
Пример 1
Для проведения эксперимента использовали показанное на прилагаемом к описанию чертеже оборудование с колонной диаметром 70 мм, оснащенной 2-мя насадками (С) и (Е) высотой соответственно 2500 и 1700 мм. Между насадками располагался промежуточный сборник (D), из которого для работы верхнего циркуляционного контура можно отбирать поток (6). Внизу колонны под ней находился ее куб объемом 4 л. Схема такого устройства прилагается (см. чертеж).
В низ (А) колонны эксгаустером (В) с расходом 8,98 кг/ч подавали поток (1), представлявший собой сырой газообразный продукт из процесса получения цианистого водорода по методу Андрусова, содержавший следующие компоненты в пересчете на массу: HCN: 8,87%, Н2О: 3,88%, Н2: 1,33%, N2: 76,01%, O2: 1,48%, СО: 5,67%, CO2: 1,13%, СН4: 0,39%. Входящий газ струйным. насосом (В) смешивали с потоком (8), циркулирующим с расходом 300 кг/ч. Температуру циркулирующего потока поддерживали при этом такой, чтобы в низу (А) колонны при его заполнении на 50% преобладала температура 50°C. Входной поток (7), подаваемый на насадку (С) с расходом 40 кг/ч, имел температуру 35°C.
Метилтиопропионовый альдегид подавали в реактор (G) по подводящей линии (2) с расходом 2,966 кг/ч. Этот альдегид содержал следующие компоненты в пересчете на массу: ММП: 96,46%, Н2О: 2,77%, ММ: 0,2%. Одновременно в реактор (G) по подводящей линии (3) с расходом 0,211 кг/ч подавали смесь из 99 мас. % ММП, имевшего вышеуказанный состав, и 1 мас. % триэтаноламина в качестве катализатора. Расход общего потока (5), состоявшего из исходных материалов (эдуктов) и циркуляционного потока (6), составлял в подводящей линии, ведущей к верхней насадке (Е), 40 кг/ч при температуре 6°C.
Молярное соотношение между исходными материалами HCN/ММП соответствовало 1. Продукт выходил из низа колонны с расходом 4,20 кг/ч и имел следующий состав в пересчете на массу: ММП-CN: 90,43%, Н2О: 7,82%, ММП: 0,14%, HCN: 0,16%, ММ: 0,01%. Отходящие газы выходили из верха колонны с расходом 8,07 кг/ч и имели следующий состав в пересчете на массу: HCN: 0,00%, ММП: 0,07%, ММ: 0,05%, H2O: 1,34%, Н2: 1,48%, N2: 86,02%, O2: 1,64%, СО: 6,31%, CO2: 1,26%, СН4: 0,44%. Эти отходящие газы подавали в сжигательную установку.
Пример 2
В данном примере использовали то же оборудование, что и в примере 1.
В низ (А) колонны эксгаустером (В) с расходом 8,94 кг/ч подавали поток (1), представлявший собой сырой газообразный продукт из процесса получения цианистого водорода по методу Андрусова, содержавший следующие компоненты в пересчете на массу: HCN: 8,9%, Н2О: 3,7%, Н2: 1,3%, N2: 76,3%, O2: 1,5%, СО: 5,6%», CO2: 1,1%, СН4: 0,4%. Входящий газ струйным насосом (В) смешивали с потоком (8), циркулирующим с расходом 280 кг/ч. Температуру циркулирующего потока поддерживали при этом такой, чтобы в низу (А) колонны при его заполнении на 50% преобладала температура 49,8°C. Входной поток (7), подаваемый на насадку (С) с расходом 40 кг/ч, имел температуру 35°C.
Метилтиопропионовый альдегид подавали в реактор (G) по подводящей линии (2) с расходом 2,976 кг/ч. Этот альдегид содержал следующие компоненты в пересчете на массу: ММП: 96,9%, Н2О: 2,8%, ММ: 0,2%. Одновременно в реактор (G) по подводящей линии (3) с расходом 0,2 кг/ч подавали смесь из 99 мас. % ММП, имевшего вышеуказанный состав, и 1 мас. % триэтаноламина в качестве катализатора. Помимо этого в реактор (G) по линии (13) подавали кубовый продукт с расходом 2 кг/ч. Расход общего потока (5), состоявшего из исходных материалов, циркуляционного потока (6) и потока (13) кубового продукта, составлял в подводящей линии, ведущей к верхней насадке (Е), 42 кг/ч при температуре 5,5°C.
Молярное соотношение между исходными материалами HCN/ММП соответствовало 1. Продукт выходил из низа колонны с расходом 4,25 кг/ч и имел следующий состав в пересчете на массу: ММП-CN: 90,06%, H2O: 8,81%, ММП: 0,75%, HCN: 0,21%, ММ: 0,01%. Отходящие газы выходили из верха колонны с расходом 7,88 кг/ч и имели следующий состав в пересчете на массу: HCN: 0,00%, ММП: 0,09%, ММ: 0,10%, H2O: 0,6%, Н2: 1,50%, N2: 86,60%, O2: 1,70%, СО: 6,40%, CO2: 1,20%, СН4: 0,50%. Эти отходящие газы подавали в сжигательную установку.
| название | год | авторы | номер документа |
|---|---|---|---|
| СТАБИЛЬНЫЙ ПРИ ХРАНЕНИИ НИТРИЛ 2-ГИДРОКСИ-4-(МЕТИЛТИО)МАСЛЯНОЙ КИСЛОТЫ | 2012 |
|
RU2597264C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛИ МЕТИОНИНА | 2012 |
|
RU2618042C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТИЛМЕРКАПТОПРОПИОНОВОГО АЛЬДЕГИДА, ПОЛУЧАЕМОГО ИЗ СЫРЫХ АКРОЛЕИНА И МЕТИЛМЕРКАПТАНА | 2012 |
|
RU2615734C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-(МЕТИЛТИО)ПРОПАНАЛЯ И 2-ГИДРОКСИ-4(МЕТИЛТИО)БУТАННИТРИЛА | 2005 |
|
RU2383531C2 |
| СПОСОБ ОЧИСТКИ СОДЕРЖАЩИХ ДИОКСИД УГЛЕРОДА ГАЗОВЫХ ПОТОКОВ | 2005 |
|
RU2388521C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-(МЕТИЛТИО)ПРОПАНАЛЯ И 2-ГИДРОКСИ-4-(МЕТИЛТИО)БУТАННИТРИЛА | 1996 |
|
RU2173681C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-(МЕТИЛТИО)ПРОПАНАЛЯ | 1996 |
|
RU2172734C2 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ 3-(МЕТИЛТИО)-ПРОПАНАЛЯ (ВАРИАНТЫ) | 1995 |
|
RU2149159C1 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ МЕТИОНИНА ИЛИ ЕГО СОЛИ | 1993 |
|
RU2116294C1 |
| СПОСОБ ОЧИСТКИ ЦИАНИСТОГО ВОДОРОДА | 2019 |
|
RU2800707C2 |
Изобретение относится к способу получения продукта, содержащего нитрил 2-гидрокси-4-(метилтио)масляной кислоты, заключающемуся в том, что 3-(метилмеркапто)-пропионовый альдегид подвергают взаимодействию с цианистым водородом в присутствии основания в качестве катализатора в зоне основной реакции с получением нитрила, и остаточный газообразный цианистый водород, выходящий из зоны основной реакции, абсорбируют в зоне абсорбции и последующей реакции, содержащей смесь из 3-(метилмеркапто)пропионового альдегида и катализатора, а также по выбору нитрила 2-гидрокси-4-(метилтио)масляной кислоты, и подвергают дальнейшему превращению с 3-(метилмеркапто)пропионовым альдегидом с последующим удалением продукта, содержащего нитрил 2-гидрокси-4-(метилтио)масляной кислоты, причем в выходящем продукте молярное соотношение между цианистым водородом и непрореагировавшим 3-(метилмеркапто)пропионовым альдегидом превышает 1. Способ позволяет получить стабильный при хранении циангидрин и повысить выход целевого продукта. 2 н. и 22 з.п. ф-лы, 1 ил., 2 пр.
1. Способ получения продукта, содержащего нитрил 2-гидрокси-4-(метилтио)масляной кислоты, заключающийся в том, что 3-(метилмеркапто)-пропионовый альдегид подвергают взаимодействию с цианистым водородом в присутствии основания в качестве катализатора в зоне основной реакции с получением нитрила, и остаточный газообразный цианистый водород, выходящий из зоны основной реакции, абсорбируют в зоне абсорбции и последующей реакции, содержащей смесь из 3-(метилмеркапто)пропионового альдегида и катализатора, а также по выбору нитрила 2-гидрокси-4-(метилтио)масляной кислоты, и подвергают дальнейшему превращению с 3-(метилмеркапто)пропионовым альдегидом с последующим удалением продукта, содержащего нитрил 2-гидрокси-4-(метилтио)масляной кислоты, причем в выходящем продукте молярное соотношение между цианистым водородом и непрореагировавшим 3-(метилмеркапто)пропионовым альдегидом превышает 1.
2. Способ по п. 1, отличающийся тем, что присутствующая в зоне абсорбции и последующей реакции смесь по меньшей мере частично поступает из зоны основной реакции.
3. Способ по п. 1, отличающийся тем, что зона основной реакции содержит реактор с мешалкой или реактор с циркуляцией.
4. Способ по п. 3, отличающийся тем, что зона основной реакции дополнительно содержит струйный насос.
5. Способ по одному из пп. 1-4, отличающийся тем, что зона абсорбции и последующей реакции содержит устройство для введения газа в контакт с жидкостью, предпочтительно колонну, прежде всего тарельчатую колонну, насадочную колонну, барботажный реактор колонного типа, колонну с капельным течением, либо по выбору реактор с емкостью с механическим перемешиванием, реактор с погружным эжектором или струйный насос.
6. Способ по одному из пп. 1-4, отличающийся тем, что зона абсорбции и последующей реакции является частью реактора с циркуляцией.
7. Способ по одному из пп. 1-4, отличающийся тем, что в зону основной реакции подают в основном газообразный цианистый водород, предпочтительно газ, содержащий цианистый водород в качестве продукта из процесса его получения.
8. Способ по п. 7, отличающийся тем, что содержание цианистого водорода в применяемом газе составляет от 1 до 99 мас. %, предпочтительно от 5 до 75 мас. %, особенно предпочтительно от 6 до 22 мас. %.
9. Способ по одному из пп. 1-4, 8, отличающийся тем, что в качестве катализатора используют низкомолекулярные или гетерогенные амины, растворы неорганических оснований или смеси из кислот и низкомолекулярных аминов.
10. Способ по п. 9, отличающийся тем, что в качестве низкомолекулярных аминов используют три(С1-С12алкил)амины, преимущественно триэтиламин или триизопропаноламин, диалкиларалкиламины, преимущественно диметилбензиламин, диалкилариламины, преимущественно N,N-диметиланилин, гетероциклические амины, преимущественно никотинамид, имидазол, бензимидазол, 2-фторпиридин, 4-диметиламинопиридин, пиколин или пиразин.
11. Способ по п. 9, отличающийся тем, что используют гетерогенные амины общей формулы
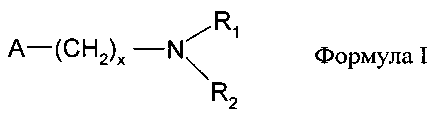
или поливинилпиридин, где
R1 и R2 обозначают водород, алкил с длиной цепи от С1 до С12, арил или гетероарил, при этом R1 может иметь значения, отличные от R2,
х обозначает число от 0 до 6, а
А обозначает природную или синтетическую смолу, предпочтительно полистирол.
12. Способ по п. 11, отличающийся тем, что катализатор формулы I представляет собой связанное с полимером основание, выбранное из группы гомологических диалкиламиноалкилполистиролов и несущих диалкиламиногруппы макросетчатых смол.
13. Способ по п. 12, отличающийся тем, что катализатор формулы I представляет собой диэтиламиноэтилполистирол, диэтиламинометилполистирол, диметиламинометилполистирол, несущую диэтиламинометильные группы макросетчатую смолу или диметиламиноэтилполистирол.
14. Способ по п. 9, отличающийся тем, что в качестве неорганического основания используют гидроксид щелочного металла, преимущественно NaOH или KOH, цианид щелочного металла, преимущественно NaCN или KCN, карбонат щелочного металла, преимущественно Na2CO3 или K2CO3, или гидрокарбонат щелочного металла, преимущественно NaHCO3 или KHCO3, индивидуально или в смешанном виде.
15. Способ по п. 9, отличающийся тем, что в качестве кислоты в смесях из кислот и низкомолекулярных аминов используют органические кислоты, преимущественно жирные кислоты с короткой цепью, прежде всего уксусную кислоту, муравьиную кислоту, лимонную кислоту, или органические сульфокислоты, преимущественно трифторметансульфоновую кислоту, либо минеральные кислоты, преимущественно серную кислоту или фосфорную кислоту.
16. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что в зоне основной реакции и в зоне абсорбции и последующей реакции величину рН, измеряемую рН-электродом при 23°С и при водосодержании от 2 до 14 мас. %, устанавливают на значение в пределах от 4,5 до 6,0, преимущественно от 5,0 до 5,5.
17. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что
температуру в зоне основной реакции устанавливают на величину в пределах от 20 до 80°С, предпочтительно от 30 до 70°С, особенно предпочтительно от 35 до 65°С.
18. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что температуру в зоне абсорбции и последующей реакции устанавливают на величину в пределах от 0 до 30°С, предпочтительно от 4 до 15°С.
19. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что перед местом отбора продукта, которым является ММП-циангидрин, для увеличения степени полноты превращения в него HCN и ММП предусматривают еще одну зону последующей реакции, температуру в которой устанавливают на величину в пределах от 20 до 80°С, предпочтительно от 40 до 70°С, особенно предпочтительно от 45 до 65°С.
20. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что его осуществляют при абсолютном давлении в пределах от 0,9 до 5 бар, предпочтительно от 1,0 до 3 бар, особенно предпочтительно от 1 до 1,5 бара.
21. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что молярное соотношение между синильной кислотой и 3-(метилтио)пропаналем составляет от 0,98 до 1,03, предпочтительно от 0,99 до 1,01.
22. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что катализатор используют в массовом соотношении между ним и 3-(метилтио)пропаналем в пределах 0,00005 до 0,002, предпочтительно от 0,0001 до 0,001.
23. Способ по одному из пп. 1-4, 8, 10-15, отличающийся тем, что его проводят в периодическом, полунепрерывном или непрерывном режиме.
24. Применение продукта реакции, содержащего ММП-циангидрин, полученного способом по одному из пп. 1-23, для получения D,L-метионина или 2-гидрокси-4-метилтиомасляной кислоты.
| US 4912257 A, 27.03.1990;US 5756803 A, 26.05.1998;US 4912257 A, 27.03.1990 | |||
| US 5756803 A, 26.05.1998;JP 2002105048 A, 10.04.2002;US 4960932 A, 10.02.1990;US 2745745 A, 15.05.1956;RU 2355678 С2, 20.05.2009. |

