Изобретение относится к бензотиофенкарбоксамидным производным, промежуточным соединениям для них, содержащим их фармацевтическим композициям, содержащим их антагонистам PGD2 (простагландина D2) и содержащим их лекарственным средствам для лечения носовой непроходимости.
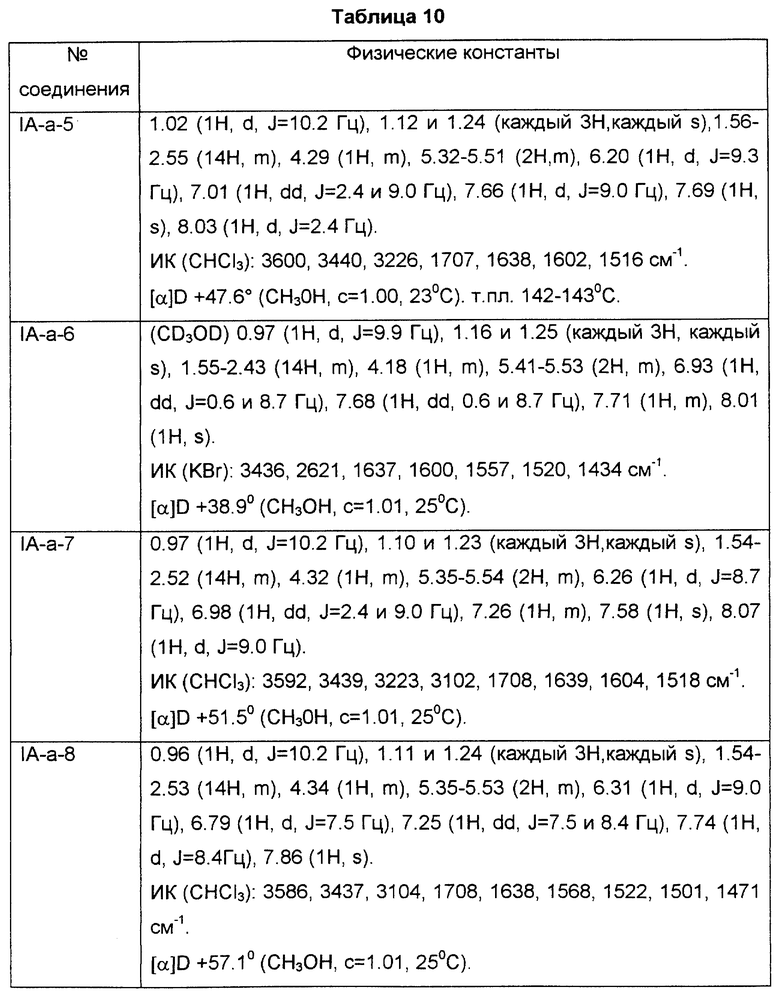
Известно [японская патентная публикация (Kokoku) N 53295/1991], что некоторые из бициклических амидных производных, аналогичных соединениям по настоящему изобретению, используются в качестве антагонистов тромбоксана A2 (ТХА2). Однако в японской патентной публикации (Kokoku) N 53295/1991 описано только то, что соединения используются в качестве антагонистов ТХА2, но не сделано предположения об их применимости в качестве антагонистов PGD2, как это обнаружено в настоящем изобретении. С другой стороны, известны [японская патентная публикация (Kokoku) N 79060/1993, Японская патентная публикация (Kokoku) N 23170/1994 и Chem. Pharm. Bull. Vol.37, N 6 1524-1533 (1989)] бициклические амидные производные, которые являются промежуточными соединениями для бициклических сульфонамидных производных. Однако раскрытые в этих документах соединения отличаются от соединений по настоящему изобретению типами заместителей в амидной части. Известно [WO 97/00853], что некоторые соединения, аналогичные соединениям по настоящему изобретению используются в качестве антагонистов PGD2. Однако не было сделано предположения, что соединения, раскрытые в WO 97/00853, обладают ингибиторной активностью против инфильтрации эозинофилов.
Известно, что ТХА2 обладает различными видами активности, такими как агрегация тромбоцитов, тромбогенез и т.д. Поэтому считается, что антагонисты ТХА2 пригодны в качестве антитромботических агентов, а также в качестве лекарственных средств при лечении инфаркта миокарда или астмы.
С другой стороны, антагонисты PGD2 по настоящему изобретению пригодны для улучшения состояния, обусловленного избыточным продуцированием PGD2, в частности в качестве лекарственных средств для лечения заболеваний, в которые вовлечена дисфункция тучных клеток, например системного мастоцитоза и расстройства системного активирования тучных клеток, а также трахеальной контракции, астмы, аллергического ринита, аллергического коньюктивита, крапивницы, ишемического реперфузионного повреждения, воспаления и атопического дерматита.
PGD2 является основным простаноидом, который продуцируется в и высвобождается из тучных клеток, в которых он продуцируется через PGG2 (простагландин G2) и PGH2 (простагландин H2) из арахидоновой кислоты под действием циклооксигеназы, активированной иммунологической или неиммунологической стимуляцией. PGD2 обладает разнообразной сильной физиологической и патологической активностью. Например, PGD2 может вызывать сильную трахеальную контракцию, приводящую к бронхиальной астме, а при системном аллергическом состоянии он расширяет периферические сосуды, вызывая анафилактический шок. Особенно большое внимание уделялось теории, согласно которой PGD2 является одним из причинных веществ, ответственных за возникновение носовой непроходимости при аллергическом рините. Поэтому было предложено разработать ингибитор биосинтеза PGD2 или антагониста рецептора PGD2 в качестве лекарственного средства для уменьшения носовой непроходимости. Однако возможно, что ингибитор биосинтеза PGD2 сильно влияет на синтез простагландинов в других частях организма, и поэтому желательно разработать антагонист (блокатор), специфичный к рецептору PGD2.
Описание изобретения
Для разработки антагонистов (блокаторов) рецептора PGD2, специфичных к рецептору PGD2, были проведены глубокие исследования и было обнаружено, что указанные ниже серии соединений формулы (I), их фармацевтически приемлемые соли или их гидраты обладают сильной активностью как антагонисты рецептора PGD2 и ингибиторной активностью против инфильтрации эозинофилов, и пригодны в качестве лекарственных средств для лечения носовой непроходимости. Соединения по настоящему изобретению, обладающие активностью антагонистов PGD2, отличаются от известных антагонистов ТХА2 активным сайтом и механизмом, применением и характером.
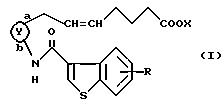
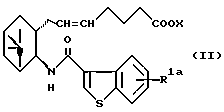
Соответственно, согласно настоящему изобретению предложено соединение формулы (I):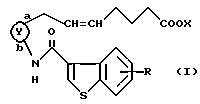
где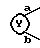
представляет собой
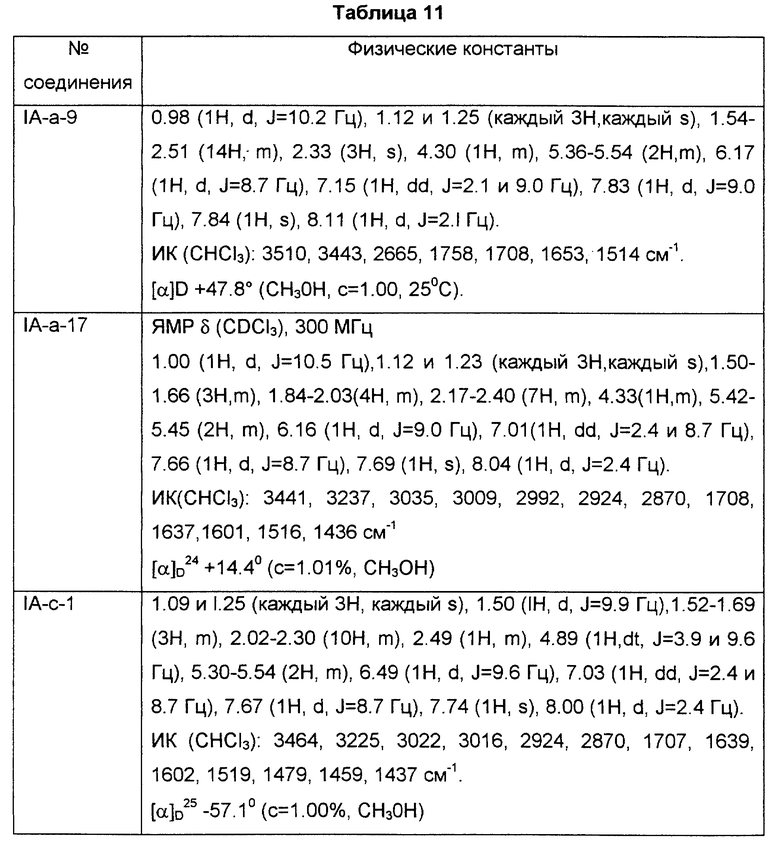
R представляет собой водород, алкил, алкокси, галоген, гидрокси, ацилокси или возможно замещенный арилсульфонилокси, X представляет собой водород или алкил, а двойная связь на α-цепи имеет E-конфигурацию или Z-конфигурацию, при условии, что соединение формулы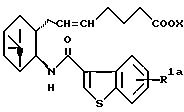
где R1a представляет собой водород, алкил или алкокси, X является таким, как определено выше, а двойная связь на α-цепи имеет E-конфигурацию или Z-конфигурацию,
исключено, его фармацевтически приемлемая соль или его гидрат.
В настоящем описании изобретения в формуле (I) на связь, представленную группой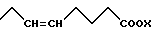
где X является таким, как определено выше, ссылаются как на α-цепь; на связь, представленную группой: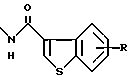
где R является таким, как определено выше,
ссылаются как на к ω-цепь.
Двойная связь на α- цепи имеет E-конфигурацию или Z-конфигурацию.
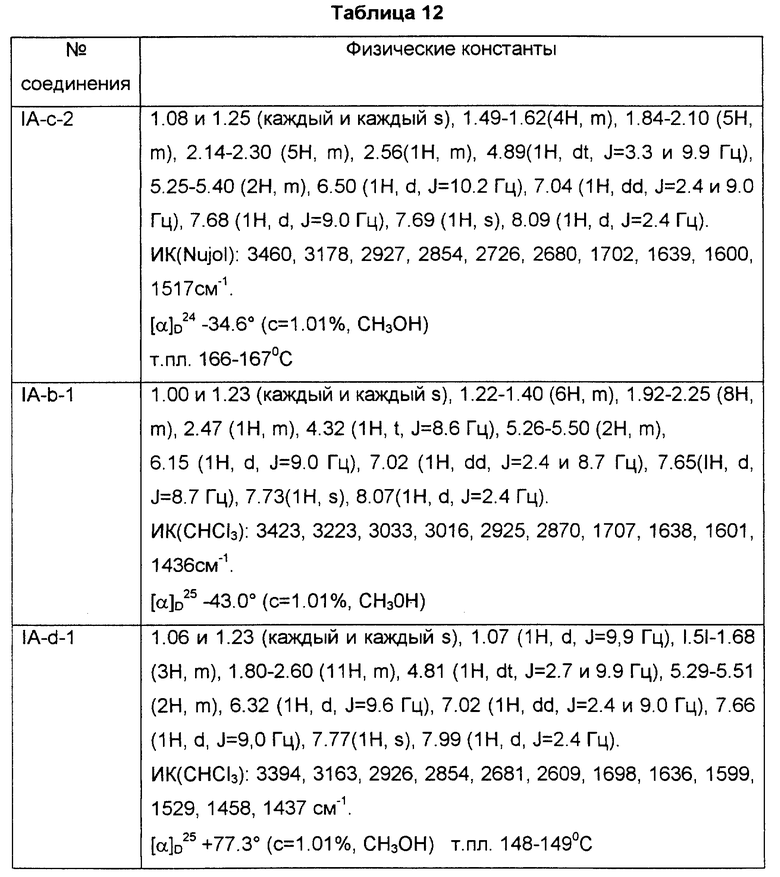
Краткое описание графических материалов
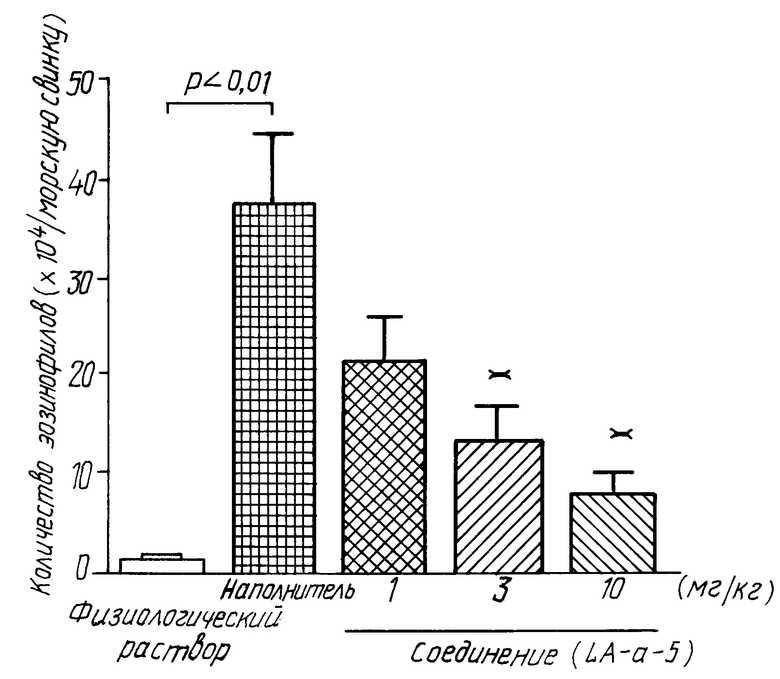
На чертеже показана активность соединения (IA-а-5) против индуцированной антигеном инфильтрации эозинофилов в носовой полости. На графике белый столбик указывает на группу, которой делали ингаляцию физиологическим раствором вместо овальбумина; черный столбик указывает на группу, которой делали ингаляцию антигеном для индуцирования воспалительной реакции, но не вводили соединение (IA-а-5); а серый столбик указывает на группы, которым делали ингаляцию антигеном для индуцирования воспалительной реакции и вводили соединение (IA-а-5). Звездочка ** указывает на значительное отличие от носителя при p < 0,01.
Наилучший способ осуществления изобретения
Более конкретно, примером соединений (I) является соединение формулы (IA):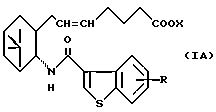
где R и X являются такими, как определено выше, а двойная связь на α--цепи имеет E-конфигурацию или Z-конфигурацию, при условии что соединение формулы;
где R1a представляет собой водород, алкил или алкокси, X является таким, как определено выше, а двойная связь на α-цепи имеет E-конфигурацию или Z-конфигурацию, исключено, его фармацевтически приемлемая соль или его гидрат.
Подобным образом, примером соединений (I) также является соединение формулы (IB):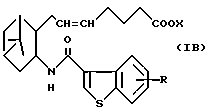
где R и X являются такими, как определено выше, а двойная связь на α--цепи имеет E-конфигурацию или Z-конфигурацию, его фармацевтически приемлемая соль или его гидрат.
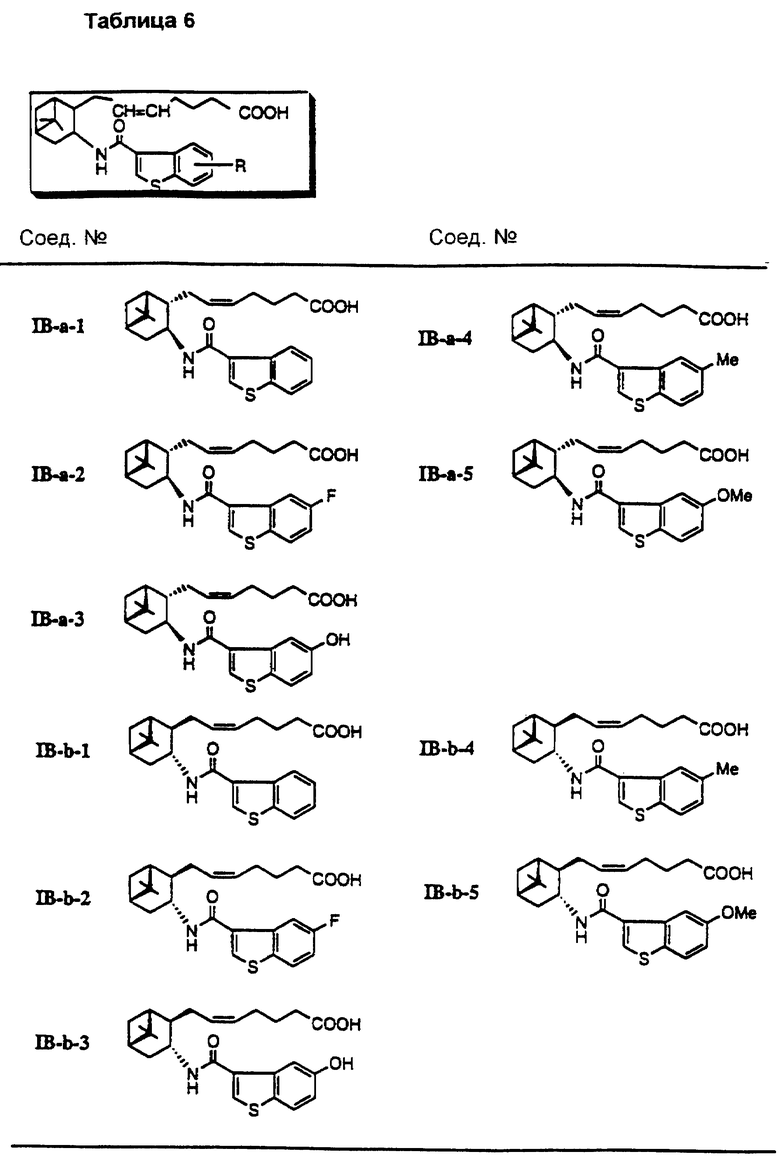
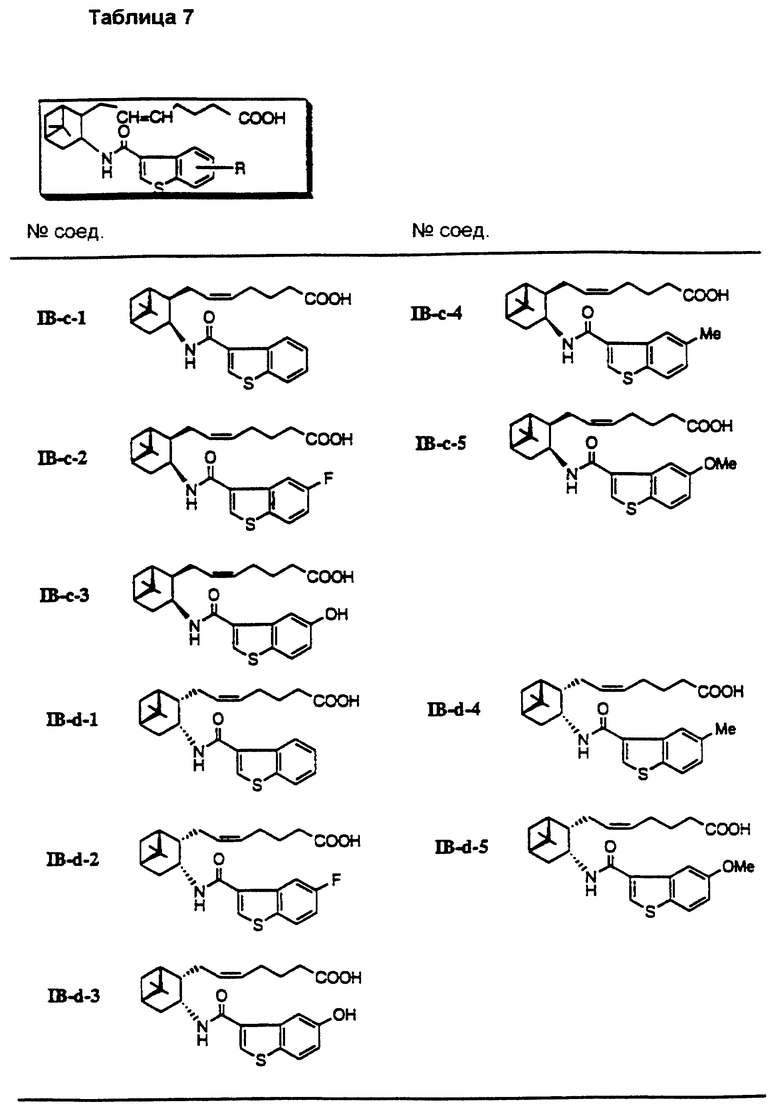
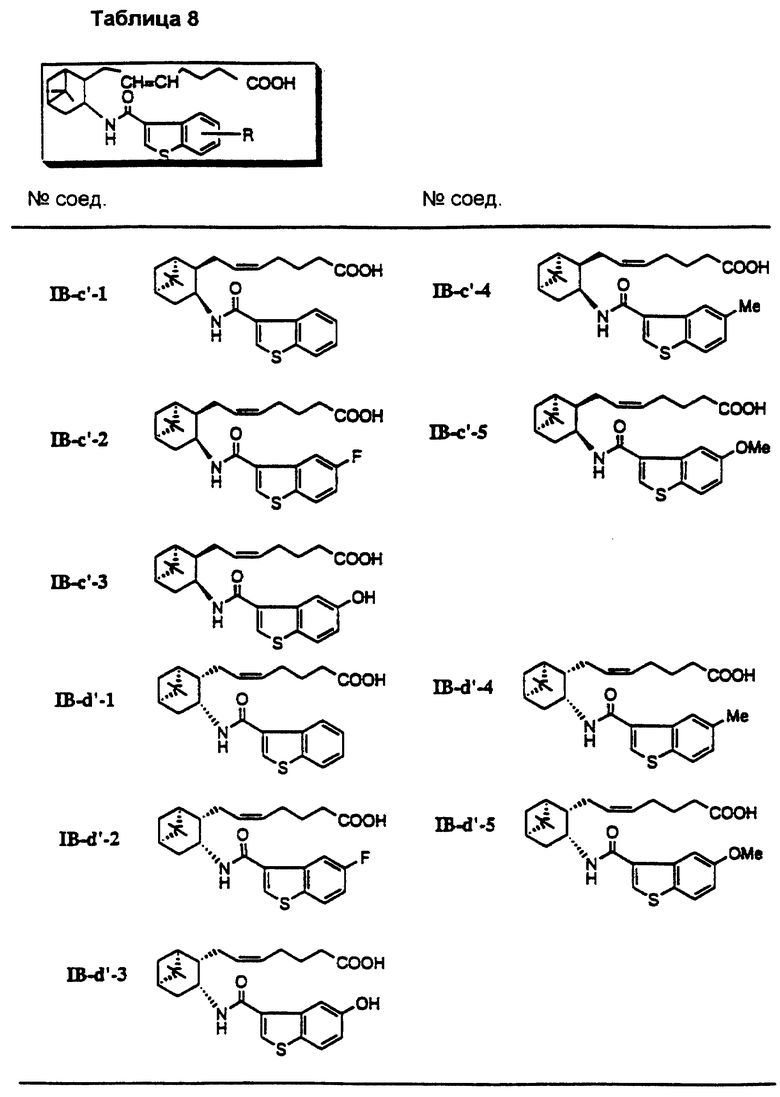
Более конкретно, примерами соединений формулы (IA) являются: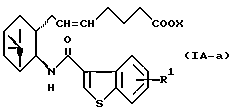
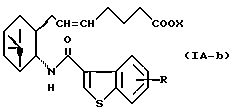
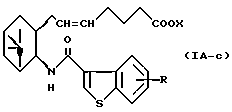
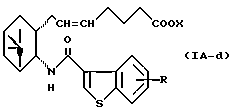
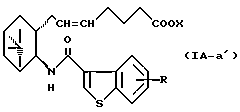
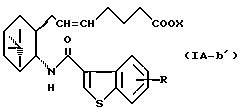
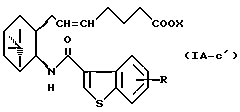
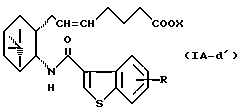
где R1 представляет собой галоген, гидрокси, ацилокси или возможно замещенный арилсульфонилокси, R и X являются такими, как определено выше, а двойная связь на α-цепи имеет E-конфигурацию или Z-конфигурацию.
Предпочтительные примеры соединений включают в себя соединения формулы (IA-a), (IA-b), (IA-c), (IA-d) и (IA-b'). В частности, предпочтительные примеры соединений включают в себя соединения формулы (IA-a).
Подобным образом, примером соединений формулы (IB) являются: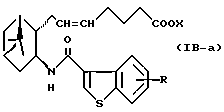
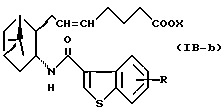
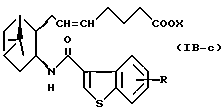
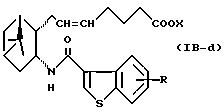
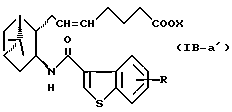

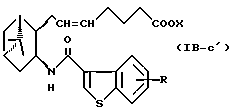
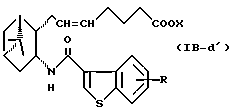
где R и X являются такими, как определено выше, и двойная связь на α-цепи имеет E-конфигурацию или Z-конфигурацию.
Предпочтительные примеры соединений включают в себя соединения формулы (IB-а') и (IB-b').
Другие примеры соединений включают в себя соединения, где двойная связь на α-цепи имеет E-конфигурацию, формулы (I), (IA), (IB), (IA-a), (IA-b), (IA-с), (IA-d), (IA-b'), (IB-а') и (IB-b').
Подобным образом, примеры соединений включают в себя соединения, где двойная связь на α-цепи имеет Z-конфигурацию, формулы (I), (IA), (IB), (IA-а), (IA-b), (IA-c), (IA-d), (IA-b'), (IB-а') и (IB-b').
Подобным образом, примеры соединений включают в себя соединения, где R представляет собой бромо, фторо, гидрокси, ацетокси или фенилсульфонил, а X является водородом, формулы (I), (IA), (IB), (IA-а), (IA-b), (IA-c), (IA-d), (IA-b'), (IB-a') и (IB-b').
Подобным образом, примеры соединений включают в себя соединения, где R является водородом, метилом или метокси, а X является водородом, формулы (I), (IA), (IB), (IA-b), (IA-c), (IA-d), (IA-b'), (IB-а') и (IB-b').
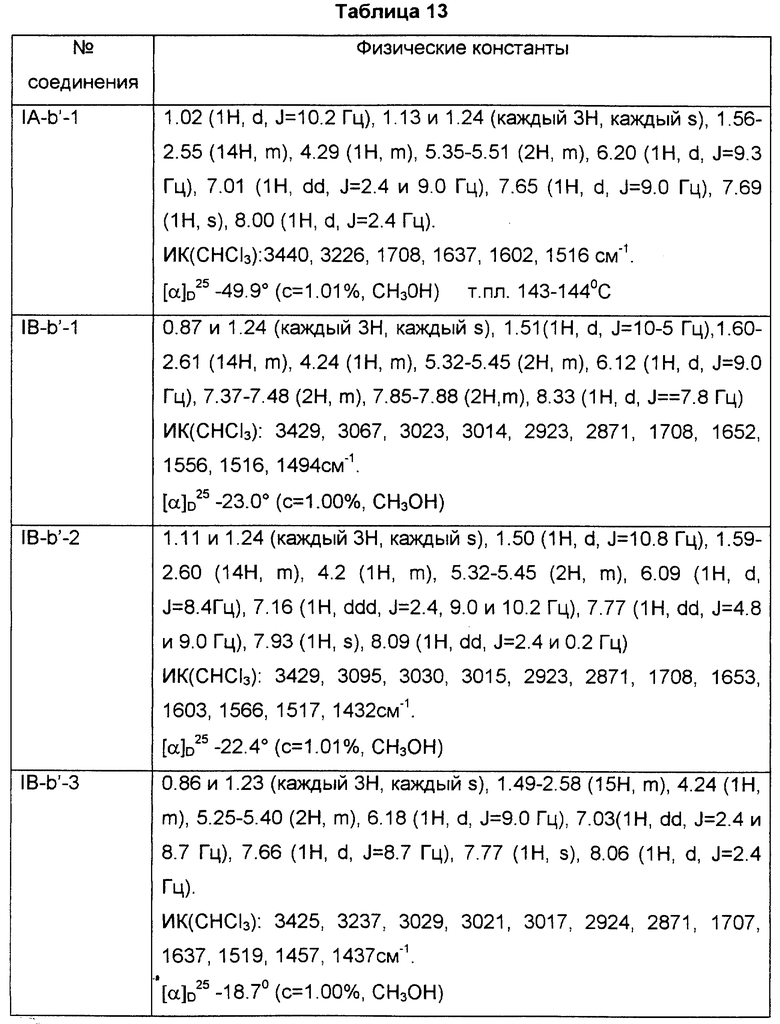
Примеры промежуточных соединений включают в себя соединения формулы (V):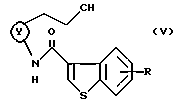
где кольцо Y и R являются такими, как определено выше.
Другие примеры промежуточных соединений включают в себя соединения формулы (VI):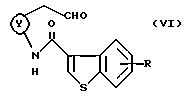
где кольцо Y и R являются такими, как определено выше.
Другие примеры промежуточных соединений включают в себя соединения формулы (III-а):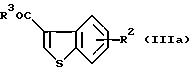
где R2 представляет собой ацилокси или возможно замещенный арилсульфонилокси, a R3 представляет собой гидрокси или галоген.
Предпочтительные примеры соединений включают в себя соединения (IIIb):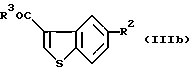
где R2 и R3 являются такими, как определено выше, или формулы (IIIc):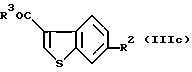
где R2 и R3 являются такими, как определено выше.
В частности, предпочтительные примеры соединений включают в себя соединения, где R3 представляет собой гидрокси, или R2 представляет собой фенилсульфонилокси или ацетокси, формулы (IIIa), (IIIb) и (IIIc).
Другим воплощением настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы (I) или содержащий его антагонист PGD2. В частности, соединения формулы (I) пригодны в качестве лекарственных средств для лечения носовой непроходимости. Антагонисты PGD2 по настоящему изобретению ингибируют инфильтрацию воспалительных клеток. Термин "воспалительные клетки" означает все лимфоциты, эозинофилы, нейтрофилы и макрофаги, и особенно эозинофилы.
Соединения (I) по настоящему изобретению демонстрируют PGD2-антагонистическую активность через связывание с рецептором PGD2, поэтому они пригодны в качестве лекарственных средств для лечения заболеваний, в которые вовлечена дисфункция тучных клеток, обусловленная избыточным продуцированием PGD2. Например, соединения (I) пригодны в качестве лекарственных средств для лечения заболеваний, таких как системный мастоцитоз и расстройство системной активации тучных клеток, а также трахеальной контракции, астмы, аллергического ринита, аллергического конъюктивита, крапивницы, ишемического реперфузионного повреждения, воспаления и атопического дерматита. Более того, соединения (I) по настоящему изобретению обладают активностью, ингибирующей инфильтрацию воспалительных клеток. Соединения (I) особенно пригодны в качестве лекарственных средств для лечения носовой непроходимости.
Термины, используемые в настоящем описании, являются такими, как определено ниже.
Термин "галоген" означает фторо, хлоро, бромо и иодо.
Термин "ацил" в "ацилокси" означает C1-C9-ацил, производный от алифатической карбоновой кислоты, например формил, ацетил, пропионил, бутирил, валерил и т. п. Термин "ацилокси" означает ацилокси, производный от вышеупомянутого ацила, например ацетокси, пропионилокси, бутирилокси, валерилокси и т.п.
Термин "арил" означает C6-C14-моноциклическое или конденсированное кольцо, например фенил, нафтил (например, 1-нафтил, 2-нафтил), антрил (например, 1-антрил, 2-антрил, 9-антрил) и т.п. Заместители на ариле включают в себя алкил, алкокси, галоген, гидрокси и т.п.
Термин "алкил" означает C1-C6-алкил с прямой или разветвленной цепью, например метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, н-пентил, изо-пентил, неопентил, трет-пентил, гексил и т.п.
Термин "алкокси" означает C1-C6-алкокси, например метокси, этокси, н-пропокси, изо-пропил, н-бутокси и т.п.
Примеры солей соединений (I) включают в себя соли, образованные щелочными металлами (например, литием, натрием или калием), щелочноземельными металлами (например, кальцием), органическим основанием (например, трометамином, триметиламином, триэтиламином, 2-аминобутаном, трет-бутиламином, диизопропилэтиламином, н-бутилметиламином, циклогексиламином, дициклогексиламином, N-изопропилциклогексиламином, фурфуриламином, бензиламином, метилбензиламином, дибензиламином, N, N-диметилбензиламином, 2-хлорбензиламином, 4-метоксибензиламином, 1-нафталинметиламином, дифенилбензиламином, трифениламином, 1-нафтиламином, 1-аминоантраценом, 2-аминоантраценом, дегидроабиэтиламином, N-метилморфолином или пиридином), аминокислотой (например, лизином или аргинином) и т.п.
Примеры гидратов соединений, представленных формулой (I), могут быть координированными с соединением (I) в возможном соотношении.
Соединения, представленные формулой (I), олицетворяют собой возможную стерическую конфигурацию, двойная связь на α--цепи имеет E-конфигурацию или Z-конфигурацию, связь с бициклическим кольцом представляет R-конфигурацию или S-конфигурацию, и включают в себя все изомеры (диастереомеры, эпимеры, энантиомеры и т.п.), рацематы и их смеси.
Общие способы получения соединений по настоящему изобретению могут быть проиллюстрированы следующим образом. В случае соединений, имеющих заместители, которые препятствуют взаимодействию, такие заместители предварительно могут быть защищены защитными группами, и они могут быть удалены на подходящей стадии.
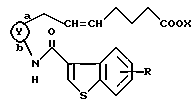
Способ 1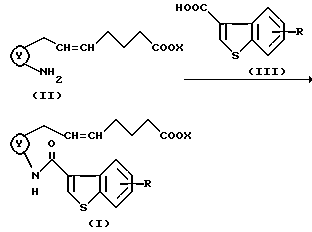
где кольцо Y, X и R являются такими, как определено выше, а двойная связь на α--цепи имеет E-конфигурацию или Z-конфигурацию.
Соединения формулы (I), как показано в вышеуказанном способе 1, могут быть получены взаимодействием карбоновой кислоты формулы (III) или их реакционноспособных производных с аминосоединениями формулы (II).
В этом способе исходные соединения (II), где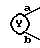
представляет собой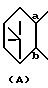
описаны в японской патентной публикации (Kokoku) N 23170/1994.
Соединения (II), где
представляет собой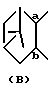
описаны в японской патентной публикации (Kokai) N 49/1986 и N 180862/1990.
Карбоновая кислота формулы (III) включает в себя
4-бромбензо[b]тиофен-3-карбоновую кислоту,
5-бромбензо[b]тиофен-3-карбоновую кислоту,
6-бромбензо[b]тиофен-3-карбоновую кислоту,
7-бромбензо[b]тиофен-3-карбоновую кислоту,
5-фторбензо[b]тиофен-3-карбоновую кислоту,
6-фторбензо[b]тиофен-3-карбоновую кислоту,
4-гидроксибензо[b]тиофен-3-карбоновую кислоту,
5-гидроксибензо[b]тиофен-3-карбоновую кислоту,
6-гидроксибензо[b]тиофен-3-карбоновую кислоту,
7-гидроксибензо[b]тиофен-3-карбоновую кислоту,
5-ацетоксибензо[b]тиофен-3-карбоновую кислоту,
бензо[b]тиофен-3-карбоновую кислоту и
5-бензосульфонилоксибензо[b]тиофен-3-карбоновую кислоту,
5-метилбензо[b]тиофен-3-карбоновую кислоту,
6-метоксибензо[b]тиофен-3-карбоновую кислоту,
5-метоксибензо[b]тиофен-3-карбоновую кислоту,
6-метоксибензо[b]тиофен-3-карбоновую кислоту.
Эти карбоновые кислоты могут иметь заместители, как определено выше. Эти карбоновые кислоты могут быть получены в соответствии со способами, описанными в Nippon Kagaku Zasshi Vol. 88, N 7, 758-763 (1967), Nippon Kagaku Zasshi Vol. 86, N 10, 1067-1072 (1965), J.Chem. Soc (с) 1899-1905 (1967), J. Heterocycle. Chem. Vol. 10, 679-681 (1973), J.Heterocyclic Chem. Vol. 19, 1131-1136 (1982) и J.Med. Chem. Vol. 29, 1637-1643(1986).
Реакционноспособное производное карбоновой кислоты формулы (III) означает соответствующий галогенангидрид кислоты (например, хлорангидрид, бромангидрид, иодангидрид), ангидрид кислоты (например, смешанный ангидрид с муравьиной кислотой или уксусной кислотой), активный эфир (например, сукцинимидный эфир) и т.п., и в общем может быть определено как ацилирующий агент, используемый для ацилирования аминогруппы. Например, когда используют галогенангидрид, соединение (III) подвергают взаимодействию с тионилгалогенидом (например, тионилхлоридом), галогенидом фосфора (например, трихлоридом фосфора, пентахлоридом фосфора), оксалилгалогенидом (например, оксалилхлоридом) и т.п., в соответствии с известными способами, описанными в литературе (например, Shin-Jikken-Kagaku-Koza, Vol. 14, 1787 (1978): Synthesis 852-854 (1986); Shin-Jikken-Kagaku-Koza, Vol. 22, 115 (1992)).
Взаимодействие можно проводить в условиях, обычно используемых для ацилирования аминогрупп. Например, в случае конденсации с галогенангидридом кислоты, взаимодействие осуществляют в растворителе, таком как эфирный растворитель (например, диэтиловый эфир, тетрагидрофуран, диоксан), бензольный растворитель (например, бензол, толуол, ксилол), галогенированный растворитель (например, дихлориметан, дихлорэтан, хлороформ), а также этилацетат, диметилформамид, диметилсульфоксид, ацетонитрил и водные растворители, или т. п. , если необходимо, в присутствии основания (например, органического основания, такого как триэтиламин, пиридин, N,N-диметиламинопиридин, N-метилморфолин; неорганического основания, такого как гидроксид натрия, гидроксид калия, карбонат калия или т.п.) при охлаждении, при комнатной температуре или при нагревании, предпочтительно при температуре в интервале от -20oC до температуры ледяной бани, или от комнатной температуры до температуры дефлегмации реакционной системы, в течение периода от нескольких минут до нескольких часов, предпочтительно от 0,5 часа до 24 часов, в частности от 1 часа до 12 часов. В случае использования карбоновой кислоты в свободной форме без превращения в реакционноспособные производные, взаимодействие проводят в присутствии конденсирующего агента (например, дициклогексилкарбодиимида (ДЦК), 1-этил-3-(3-метиламинопропил)карбодиимида, N,N'-карбонилдиимидазола), обычно используемого в реакции конденсации.
Соединение (I) по настоящему изобретению может быть также получено в соответствии со следующим способом.
Способ 2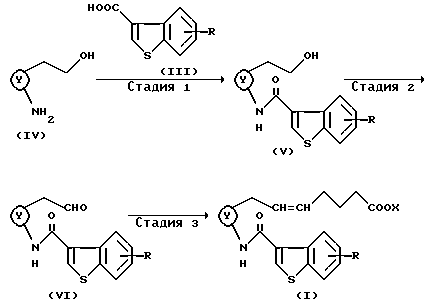
где кольцо Y, R и X являются такими, как определено выше, а двойная связь на α-цепи имеет E-конфигурацию или Z-конфигурацию.
(Стадия 1)
На этой стадии соединение формулы (V) может быть получено взаимодействием аминосоединения формулы (IV) с карбоновой кислотой формулы (III) или ее реакционноспособным производным в соответствии со способом, аналогичным способу 1. Некоторые аминосоединения формулы (IV) описаны в способе, раскрытом в Chem. Pharm. Bull. Vol. 37, N 6, 1524-1533 (1989).
(Стадия 2)
На этой стадии соединение формулы (V) окисляют до альдегидного соединения формулы (VI). Эта стадия может быть проведена с использованием хроматированных окисляющих агентов, таких как реагент Джонса, реагент Коллинза, хлорхромат пиридиния, дихромат пиридиния, в растворителе, таком как хлорированный углеводород (например, хлороформ, дихлорметан), эфир (например, этиловый эфир, тетрагидрофуран) или ацетон, бензол и т.п. при охлаждении или при комнатной температуре в течение нескольких часов. Эта стадия может быть также проведена с использованием окисляющих агентов в комбинации с подходящими активаторами (например, трифторуксусным ангидридом, оксалилхлоридом) и диметилсульфоксидом, при необходимости в присутствии основания (например, органического основания, такого как триэтиламин, диэтиламин).
(Стадия 3)
На этой стадии α-цепь альдегидного соединения формулы (VI) переводят в соединение формулы (I). На этой стадии соединение формулы (I) может быть получено взаимодействием альдегидного соединения формулы (VI) с илидным соединением, соответствующим остальной части α-цепи, в соответствии с условиями реакции Виттига. Далее, илидное соединение, соответствующее остальной части α-цепи, может быть синтезировано взаимодействием трифенилфосфина с соответствующей галогенированной алкановой кислотой или ее эфирным производным в присутствии основания согласно известному способу.
При взаимодействии других реакционноспособных производных или свободной кислоты с амином (II) или (IV), условия реакции определяют согласно свойствам отдельных реакционноспособных производных или свободной кислоты, в соответствии с известным способом. Продукт реакции может быть очищен общепринятым способом очистки, таким как экстракция растворителем, хроматография, перекристаллизация и т.п.
При желании, целевое соединение (I) по настоящему изобретению может быть превращено в соответствующее эфирное производное. Например, эфир может быть получен этерификацией карбоновой кислоты в соответствии с известным способом. При желании, могут быть получены E-изомер, Z-изомер или смеси в зависимости от реакционных условий.
Когда соединение (I) по настоящему изобретению используют при лечении, оно может быть приготовлено в форме обычных лекарственных препаратов для перорального и парентерального введения. Фармацевтическая композиция, содержащая соединение (I) по настоящему изобретению, может быть в форме для перорального или парентерального введения. Конкретно, она может быть приготовлена в форме препаратов для перорального введения, такие как таблетки, капсулы, гранулы, порошки, сироп и т.п.; препараты для парентерального введения, такие как растворы для инъекций или суспензии для внутривенной, внутримышечной или подкожной инъекции, ингалянт, глазные капли, капли в нос, суппозитории или препараты для чрескожного введения, такие как мазь.
При приготовлении препаратов могут быть использованы носители, эксципиенты, растворители и основания, обычно известные специалистам. В случае таблеток, их получают прессованием или формованием активного ингредиента вместе с вспомогательными компонентами. Примеры используемых вспомогательных компонентов включают в себя фармацевтически приемлемые эксципиенты, такие как связующие (например, кукурузный крахмал), наполнители (например, лактоза, микрокристаллическая целлюлоза), разрыхлители (например, крахмальный гликолят натрия) или смазки (например, стеарат магния). Таблетки могут быть покрыты подходящим образом. В случае жидких препаратов, таких как сиропы, растворы или суспензии, они могут содержать суспендирующие агенты (например, метилцеллюлозу), эмульгаторы (например, лецитин), консерванты и т.п. В случае инъекционных препаратов, они могут быть в форме раствора или суспензии, либо масляных или водных эмульсий, которые могут содержать стабилизирующий суспензию агент или диспергирующий агент и т.п. В случае ингалянта, его готовят в форме жидкого препарата, применяемого в ингаляторе. В случае глазных капель, их готовят в форме раствора или суспензии. В частности, в случае лекарственного средства для введения в нос для лечения носовой непроходимости, его можно использовать в форме раствора или суспензии, приготовленной общепринятым способом, или в форме порошка, приготовленного с использованием порошкового агента (например, гидроксипропилцеллюлозы, карбопола), которые вводят в носовую полость. Альтернативно, его можно использовать в форме аэрозоля после заполнения в специальный контейнер вместе с низкокипящим растворителем.
Хотя подходящая дозировка соединения (I) варьирует в зависимости от пути введения, возраста, веса тела, пола или состояния пациента и вида совместно используемого лекарственного средства (лекарственных средств), при его наличии, и окончательно должна определяться лечащим врачом. В случае перорального введения, суточная дозировка обычно может находиться между 0,01-100 мг, предпочтительно приблизительно 0,01-10 мг, более предпочтительно приблизительно 0,01-1 мг, на кг веса тела. В случае парентерального введения суточная дозировка может обычно находиться между приблизительно 0,001-100 мг, предпочтительно приблизительно 0,001-1 мг, более предпочтительно приблизительно 0,001-0,1 мг, на кг веса тела. Суточная дозировка может введена в 1-4 раздельных дозах.
Следующие примеры приведены для дальнейшего иллюстрирования настоящего изобретения и не должны быть истолкованы как ограничивающие объем изобретения.
Сокращения, используемые в примерах настоящего изобретения, приведены ниже
Me - метил
Ac - ацетил
Ph - фенил
Ссылка 1
Получение 5-бензолсульфонилоксибензо[b]тиофен-3-карбонилхлорида (3)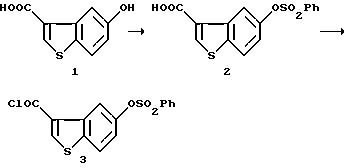
К раствору 8,63 г (44,4 ммоль) 5-гидроксибензо[b]тиофен-3-карбоновой кислоты (1) [J. Chem. Soc(C), 1899-1905 (1967), M.Martin-Smith et al] в 160 мл 80% водного тетрагидрофурана и 44 мл 1 н. гидроксида натрия добавляли 87 мл 0,56 н. гидроксида натрия и 6,2 мл (48,4 ммоль) бензолсульфонилхлорида, одновременно поддерживая pH 11-12, при перемешивании при охлаждении льдом. После завершения реакции, смесь разбавляли водой, подщелачивали и промывали толуолом. Водный слой слегка подкисляли концентрированной соляной кислотой при перемешивании. Выпавшие в осадок кристаллы отфильтровывали, промывали водой и сушили с получением 14,33 г 5-бензолсульфонилоксибензо[b]тиофен-3-карбоновой кислоты (2), т.пл. 202-203oC.
ЯМР δ (CDCl3), 300 МГц
7.16 (1Н, dd, J= 2.7 и 9.0 Гц), 7.55-7.61 (2Н, m), 7.73 (1Н, m), 7.81 (1H, d, J= 9.0 Гц), 7.90-7.94 (2Н, m), 8.16 (1H, d, J = 2.7 Гц), 8.60 (1Н, s).
ИК (Нуйол): 3102, 2925, 2854, 2744, 2640, 2577, 1672, 1599, 1558, 1500, 1460, 1451 см-1
Элементный анализ (для C15H10O5S2)
Вычислено, (%): C 53,88; H 3,01; S 19,18.
Обнаружено, (%): C 53,83; H 3,03; S 19,04.
Смесь 5,582 г (16,7 ммоль) полученной выше 5-бензолсульфонилоксибензо[b] -3-карбоновой кислоты (2), одной капли диметилформамида, 3,57 мл (50 ммоль) тионилхлорида и 22 мл толуола нагревали с обратным холодильником в течение 1,5 часов, а затем концентрировали при пониженном давлении с получением 5,89 г целевого соединения (3).
Ссылка 2
Получение 5-ацетоксибензо[b]тиофен-3-карбонилхлорида (5)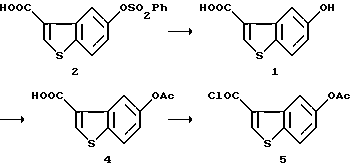
Раствор 100 мг (0,3 ммоль) полученной выше 5-бензолсульфонилоксибензо[b] тиофен-3-карбоновой кислоты (2) в 1,2 мл 1 н. гидроксида натрия оставляли стоять в течение 8 часов при 40oC. К этому добавляли соляную кислоту (1н., 1,2 мл), и выпавшие в осадок кристаллы отфильтровывали, промывали водой и сушили с получением 58 мг 5-гидроксибензо[b]тиофен-3-карбоновой кислоты (1). Выход 96,6%, т.пл. 262-263oC.
Раствор 1,140 мг полученной выше 5-гидроксибензо[b]тиофен-3-карбоновой кислоты (1) в 2 мл уксусного ангидрида и 4 мл пиридина оставляли стоять в течение 3 часов. После добавления воды смесь перемешивали в течение 1,5 часов при охлаждении льдом, и выпавшие в осадок кристаллы отфильтровывали, промывали водой и сушили с получением 1,349 мг 5-ацетоксибензо[b)тиофен-3-карбоновой кислоты (4). Выход 97,3%, т.пл. 239-240oC.
Смесь 1,349 мг полученной выше 5-ацетоксибензо[b]тиофен-3-карбоновой кислоты (4), одной капли диметилформамида, 1,22 мл (17,13 ммоль) тионилхлорида и 25 мл толуола нагревали с обратным холодильником в течение 1,5 часов, а затем концентрировали при пониженном давлении с получением 1,454 мг целевого соединения (5).
Ссылка 3
Получение (1R, 2S, 3S, 5S)-2-(2-амино-6,6-диметилбицикло[3.1.1]гепт-3-ил)этанола (IVA-b-1) и (1R,2R,3S,5S)-2-(2-амино-6,6-диметилбицикло[3.1.1] гепт-3-ил)этанола (IVA-c-1)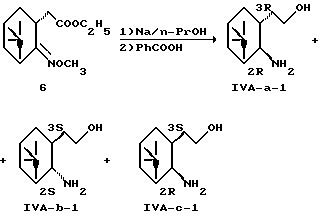
Соединение (6) [Chem. Pharm. Bull. Vol. 37, N 6, 1524-1533 (1989)] восстанавливали натрием в соответствии со способом, описанным в вышеуказанной литературе, и соединение (IV-a-1) удаляли фильтрацией в виде соли бензойной кислоты. Маточный раствор (79 г) суспендировали в 150 мл этилацетата, добавляли 260 мл 1 н. соляной кислоты и перемешивали. Водный слой, отделенный из двух слоев, подщелачивали 65 мл 4 н. гидроксида натрия и экстрагировали этилацетатом. Органический слой промывали водой и сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный маслянистый остаток (6.7 г из 30 г) растворяли в 40 мл 90% метанола, адсорбировали на 500 мл ионообменной смолы, Amberlite CG-50 (NH4+) тип I, и элюировали с использованием 2,2 л воды и 2,2 л 1 н. водного аммиака градиентным методом.
Одна фракция: 300 мл. Каждая фракция была исследована тонкослойной хроматографией (проявляющий растворитель; хлороформ:метанол:конц.водный аммиак = 90:10:1). Фракции 3-8 собирали и концентрировали при пониженном давлении. Остаток кристаллизовали из гексана; перекристаллизация давала 538 мг игл.
Т.пл. 117-118oC.
ЯМР δ (CDCl3), 300 МГц
1.01 и 1.21 (каждый 3Н, каждый s), 1.34 (1H, d, J=9.9 Гц), 1.52-1.66 (2Н, m), 1.90-2.07 (4H, m), 2.18 (1H, m), 2.48 (1H, m), 3.12 (3Н, bs), 3.49 (1H. dd, J=3.9 и 9.6 Гц), 3.61 (1H, dt, J=2.4 и 10.5 Гц), 3.84 (1H, ddd, J= 3.3, 4.8 и 10.5 Гц).
ИК (Нуйол): 3391, 3293, 3108, 2989, 2923, 2869, 2784, 2722, 2521, 1601, 1489, 1466 см-1
[α]
Элементный анализ для (C11H21NO)
Вычислено, (%): C 72,08; H 11,55; N 7,64.
Обнаружено, (%): C 72,04; H 11,58; N 7,58.
Посредством рентгеновского анализа кристаллов структурная формула была идентифицирована как формула (1R, 2R, 3S,5S)-2-(2-амино-6,6-диметилбицикло[3.1.1] гепт-3-ил)этанол (IVA-c-a-1). Маточный раствор (2,9 г) после перекристаллизации из гексана растворяли в 15 мл этилацетата, к которому добавляли раствор 3.0 мл этилацетата, содержащий 1,93 г бензойной кислоты. Выпавшие в осадок кристаллы отфильтровывали с получением 2,93 г соли бензойной кислоты соединения (IV-a-1).
Т.пл. 182-183oC.
Фракции 10-17 собирали и концентрировали при пониженном давлении. К раствору 2,66 г остатка в 15 мл этилацетата добавляли 11 мл этилацетата, содержащие 1,77 г бензойной кислоты. Выпавшие в осадок кристаллы отфильтровывали с получением 4,08 г игл.
Т.пл. 160-161oC.
ЯМР δ (CDCl3), 300 МГц
0.61 и 1.06 (каждый 3Н, каждый s), 1.36 (1H, m), 1.53-1.65 (2Н, m), 1.75-1.88 (2Н, m), 1.95-2.04 (4H, m), 3.18 (1H, d, J=6.3 Гц), 3.58 (1H, dt, J= 3.0 и 10.8 Гц), 3.81 (1H, m), 5.65 (4H, bs), 7.33-7.42 (3Н, m), 7.98-8.01 (2Н, m).
ИК (Нуйол): 3320, 2922, 2854, 2140, 1628, 1589, 1739, 1459, 1389 см-1
[α]
Элементный анализ (для C18H27NO3)
Вычислено, (%): C 70,79; H 8,91; N 4,59.
Обнаружено, (%): C 70,63; H 8,86; N 4,58.
Посредством рентгеновского анализа кристаллов структурная формула была идентифицирована как формула (1R, 2S, 3S,5S)-2-(2-амино-6,6-диметилбицикло[3.1.1]гепт-3-ил)этанол (IVA-b-1).
Пример
Получение (5Z)-7-{ (1R, 2R,3S,5S)-2-(5-гидроксибензо[b]тиофен-3-ил-карбониламино)-6,6-диметилбицикло[3.1.1]гепт-3-ил}-5-гептеноата натрия (IA-а-6)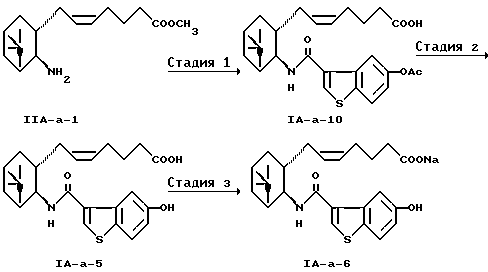
(Стадия 1)
К раствору 1,450 мг (5,2 ммоль) соединения (IIA-a-1) (японская патентная публикация (Kokoku) N 23170/1994) в 25 мл тетрагидрофурана добавляли 2,6 мл (18,7 ммоль) триэтиламина и 1,454 мг (1,1 ммоль) 5-ацетоксибензо[b]тиофен-3-карбонилхлорида (5), полученного по ссылке 2. После перемешивания в течение 1,5 часов смесь разбавляли водой и экстрагировали толуолом. Органический слой промывали разбавленной соляной кислотой и водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле (толуол:этилацетат=9:1) с получением 2,481 мг соединения (IA-а-10). Выход 96,1%.
[α]
Элементный анализ (для C28H35NO5S· 0,1H2O)
Вычислено, (%): C 67,34; H 7,10; N 2,80; S 6,42.
Обнаружено, (%): C 67,23; H 7,12; N 2,86; S 6,59.
(Стадия 2)
К раствору 2,357 мг (4,73 ммоль) полученного выше соединения (IA-a-10) в 25 мл метанола добавляли 4,1 мл (16,4 ммоль) 4 н. гидроксида натрия. После перемешивания в течение 6 часов смесь нейтрализовывали 17 мл 1 н. соляной кислоты, разбавляли водой и экстрагировали этилацетатом. Органический слой промывали водой, сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток перекристаллизовывали из этилацетата/н-гексана с получением 1,859 мг соединения (IA-а-5) в виде призм. Выход 86,5%.
Т.пл. 142-143oC.
[α]
Элементный анализ (для C25H31NO4S)
Вычислено, (%): C 68,900; H 7,08; N 3,17; S 7,26.
Обнаружено, (%): C 67,93; H 7,08; N 3,19; S 7,24.
(Стадия 3)
К раствору 203 мг (0,46 ммоль) полученного выше соединения (IA-а-5) в 3 мл метанола добавляли 0,42 мл (0,42 ммоль) 1 н. гидроксида натрия, и смесь концентрировали при пониженном давлении. Остаток растворяли в небольшом количестве этилацетата и разбавляли н-гексаном. Нерастворимое вещество растворяли в метаноле и концентрировали при пониженном давлении с получением 210 мг конечного соединения (IA-а-6). Выход 98,5%.
[α]
Элементный анализ (для C25H30NO4SNa·0,5H2O)
Вычислено, (%): C 63,54; H 6,61; N 2,96; S 6,78; Na 4,86.
Обнаружено, (%): C 63,40; H 6,69; N 3,13; S 6,73; Na 4,68.
Пример 2
Получение (5Z)-7-[(1R, 2S, 3R,5S)-2-(5-гидроксибензо[b]тиофен-3-ил-карбониламино)-6,6-диметилбицикло[3.1.1] гепт-3-ил]-5-гептеновой кислоты (IA-b-1)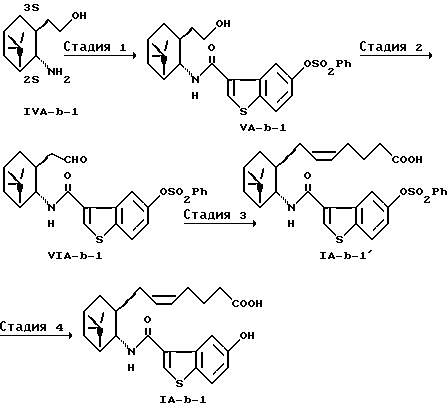
(Стадия 1)
К суспензии 916 мг (3 ммоль) соли бензойной кислоты (1R,2S,3S,5S)-2-(2-амино-6,6-диметилбицикло[3.1.1] гепт-3-ил)этанола в 3 мл воды добавляли 3,1 мл 1 н. соляной кислоты. Выпавшую в осадок бензойную кислоту экстрагировали этилацетатом. Водный слой доводили до pH 10,5 с использованием 700 мг безводного карбоната натрия, к которому по каплям добавляли раствор 1,06 г (3 ммоль) 5-бензолсульфонилоксибензо[b]тиофен-3-карбонилхлорида (3) в 6 мл тетрагидрофурана. Через 1,5 часов смесь разбавляли водой и экстрагировали толуолом. Органический слой промывали водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Таким образом полученный остаток (1.5 г) хроматографировали на силикагеле (гексан:этилацетат=1:1) с получением 1,497 г соединения (VA-b-1). Выход 99,8%.
[α]
Элементный анализ (для C26H29NO5S2 · 0,2H2O)
Вычислено, (%): C 62,05; H 5,89; N 2,78; S 12,74.
Обнаружено, (%): C 62,03; H 5,93; N 2,79; S 12,72.
(Стадия 2)
Раствор 0,61 мл (8,6 ммоль) диметилсульфоксида в 9,7 мл 1,2-диметоксиэтана охлаждали при -60oC и по каплям добавляли 0,37 мл (4,3 ммоль) оксалилхлорида. Через 15 минут к нему добавляли раствор 1,427 г (2,9 ммоль) полученного выше соединения (VA-b-1) в 11 мл 1,2-диметилоксиэтана. После перемешивания в течение 30 минут добавляли 1,2 мл триэтиламина и смесь перемешивали в течение 30 минут и нагревали постепенно до комнатной температуры. Смесь разбавляли водой и экстрагировали толуолом. Органический слой промывали водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный таким образом остаток хроматографировали на силикагеле (гексан:этилацетат=6:4) с получением 1,338 г соединения (VIA-b-1). Выход 94,1%.
[α]
Элементный анализ (для C26H27NO5S2·0,4H2O)
Вычислено, (%): C 61,85; H 5,55; N 2,77; S 12,70.
Обнаружено, (%): C 61,92; H 5,60; N 2,79; S 12,88.
(Стадия 3)
Суспензию 1,72 г (3,9 ммоль) 4-карбоксибутилтрифенилфосфония бромида и 1,016 г (9 ммоль) трет-бутоксида калия в 9 мл тетрагидрофурана перемешивали в течение 1 часа при охлаждении льдом. К смеси в течение 6 минут добавляли раствор 1,288 г (2,6 ммоль) полученного выше соединения (VIA-b-1) в 4 мл тетрагидрофурана и смесь перемешивали при той же температуре в течение 2 часов. Смесь разбавляли 15 мл воды, подкисляли до pH 10,5 1 н. соляной кислотой и дважды промывали 15 мл толуола. Водный слой подкисляли 1 н. соляной кислотой до pH 8,0, добавляли 1,15 г (10,4 ммоль) безводного хлорида кальция и дважды экстрагировали 15 мл этилацетата. Органический слой разбавляли 16 мл воды, подкисляли 1н. соляной кислоты до pH 2-3 и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 1,44 г соединения (IA-b-1'). Выход 95,5%.
Соединение использовали для следующей стадии без дальнейшей очистки.
(Стадия 4)
К раствору 1,44 г (2.6 ммоль) полученного выше соединения (IA-b-1') в 2,8 мл диметилсульфоксида добавляли 3,9 4 н. гидроксида натрия, и смесь перемешивали при 55oC в течение 3 часов. Смесь разбавляли водой и дважды промывали 15 мл толуола. Водный слой подкисляли 1 н. соляной кислотой и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 1,097 г соединения (IA-b-1). Выход 95,9%.
[α]
Элементный анализ (для C25H31NO4S·0,2H2O
Рассчитано, (%): C 67,45; H 7,11; N 3,15; S 7,20.
Обнаружено, (%): C 67,51; H 7,15; N 3,18; S 6,96.
Пример 3
Получение (5Е)-7-[(1R, 2R, 3S,5S)-2-(5-гидроксибензо[b]тиофен-3-ил-карбониламино)-6,6-диметилбицикло[3.1.1] гепт-3-ил] -5-гептеновой кислоты (IA-a-17)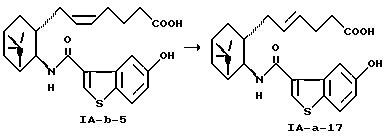
Раствор 11,04 г (25 ммоль) (5Z)-7-[(1R,2R,3S,5S)-2-(5-гидpoкcибензo[b] bтиoфен-3-ил-карбониламино)-6,6-диметилбицикло[3.1.1]гепт-3-ил]-5-гептеновой кислоты (IA-а-5), 4,32 г (18,8 ммоль) 1-метилтетразол-5-ил дисульфида (J. Org. Chem. , 50, 2794-2796 (1985), M. Narisada, Y. Terui, M. Yamakawa, F. Watanabe, M. Ohtani, and Н. Myazaki et al) и 2,84 г (17,3 ммоль) 2,2'-азобисизобутиронитрила в 1,1 л бензола нагревали с обратным холодильником при перемешивании в течение 8 часов. Смесь дважды экстрагировали 400 мл 0,4 н. гидроксида натрия. Водный слой подкисляли соляной кислотой и осадок собирали фильтрацией. Осадок (11,08 г) хроматографировали на силикагеле (хлороформ: метанол = 10:1) с получением 6,93 г соединения. Полученное соединение растворяли в 69 мл диметоксиэтана, к которому добавляли 2,15 г 4-метоксибензиламина и последовательно разбавляли 120 мл эфира при охлаждении льдом. Осадок отфильтровывали с получением 7,45 г кристаллического продукта, который перекристаллизовывали из изопропилового спирта/этилацетата/эфира (=2/10/5), чтобы очистить.
Т.пл. 108-111oC.
[α]
Чистоту изомера соли 4-метоксибензиламина анализировали высокоэффективной жидкостной хроматографией (ВЭЖХ). Результат: (Е-изомер): (Z-изомер) = 98,4:1,6
[Условия ВЭЖХ] Колонка: YMC-pack АМ-303-10 (10 мкм m.120A.ODS) (4,6 мм ⊘ × 250 мм); скорость потока: 1 мл/мин; детектирование: УФ 254 нм; подвижная фаза: уксусная кислота/вода/ацетонитрил = 0,1/52/48; время удерживания: (Е-изомер) 21 минута, (Z-изомер) 23 минуты.
Очищенную соль 4-метоксибензиламина (1,6 г) суспендировали в 25 мл воды, подкисляли 25 мл 1 н. соляной кислоты и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 1,21 г соединения (IA-a-17).
[α]
Элементный анализ (для C25H31NO4O·0,1H2O)
Вычислено, (%): C 67,72; H 7,09; N 3,16; S 7,23.
Обнаружено, (%): C 67,59; H 7,26; N 3,35; S 7,39.
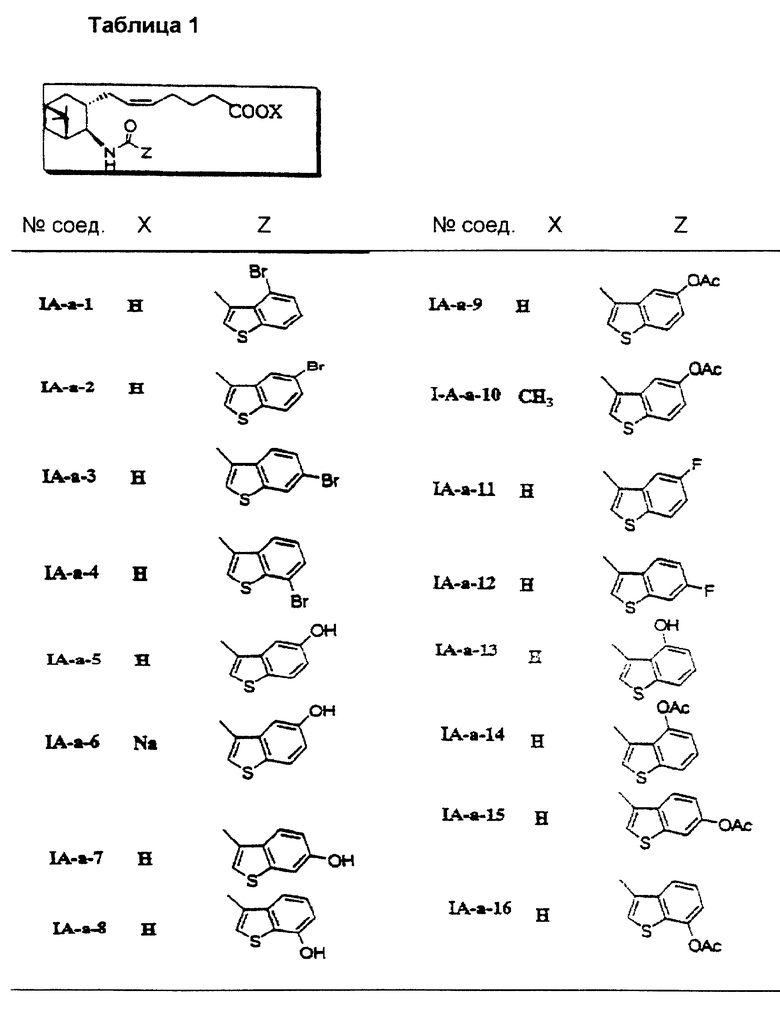
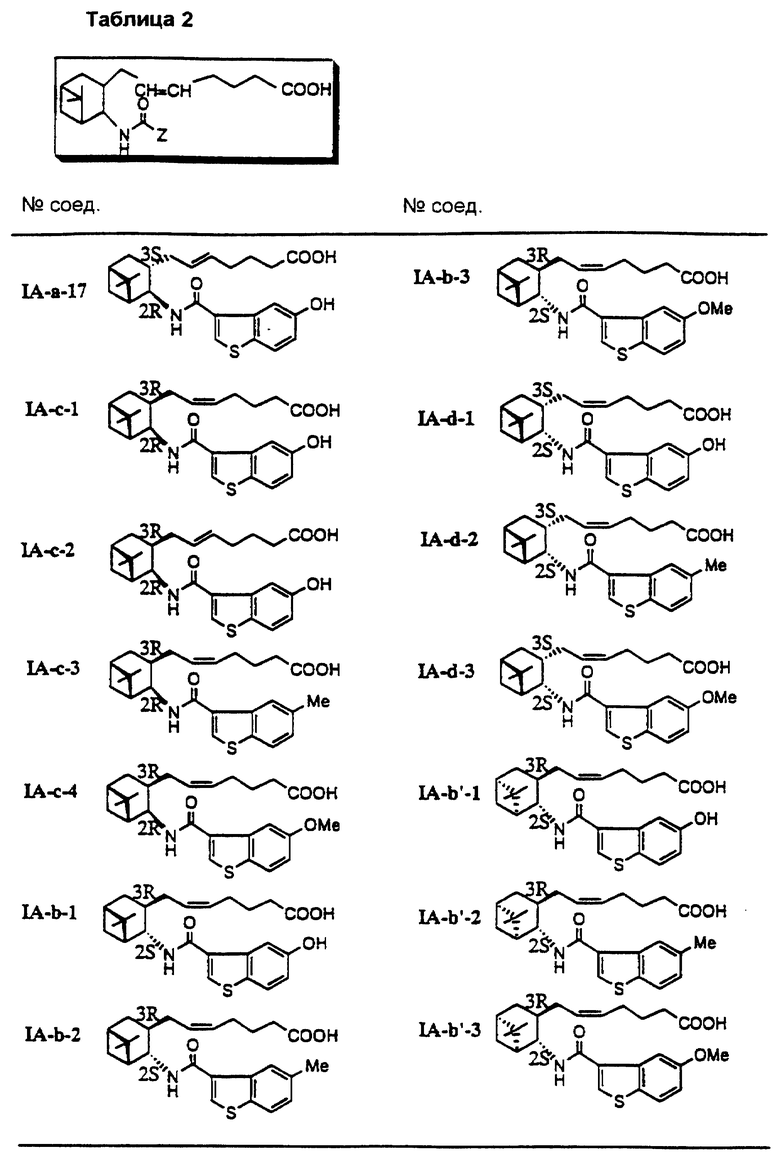
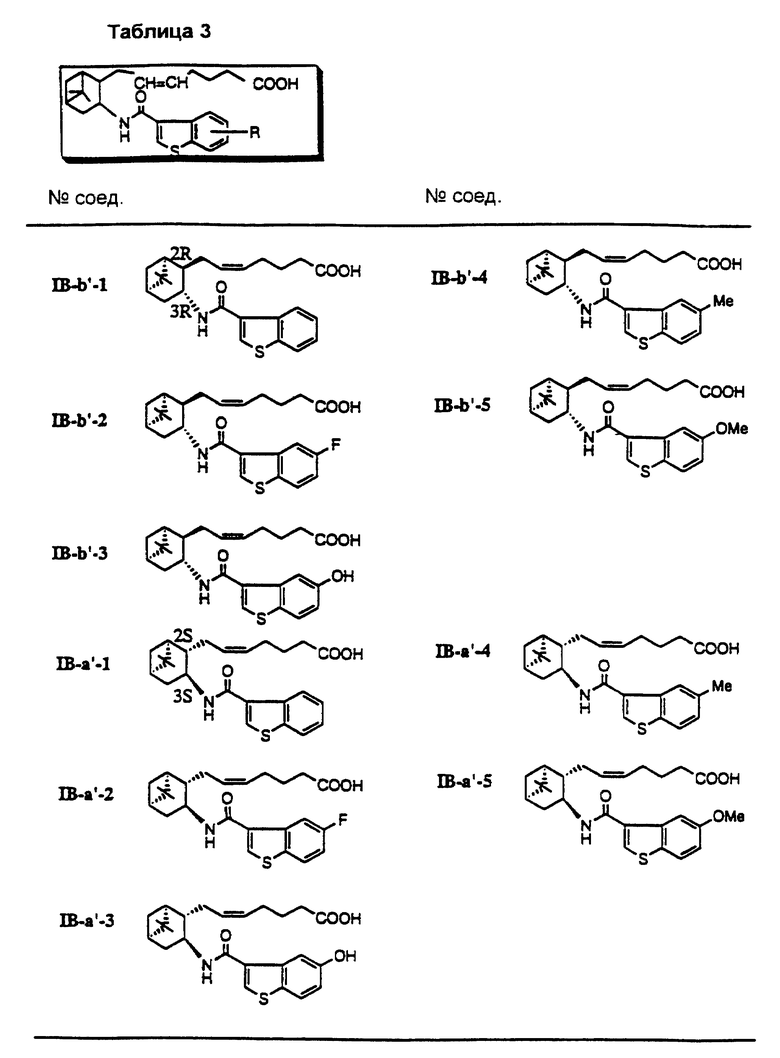
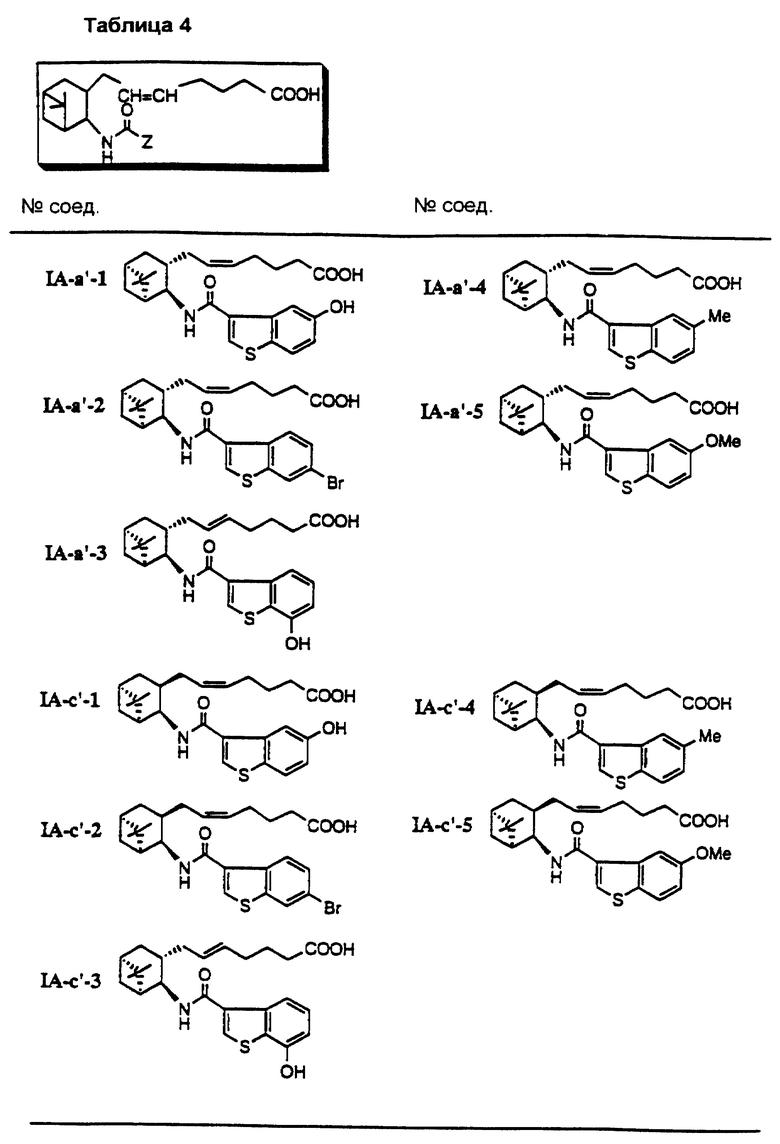
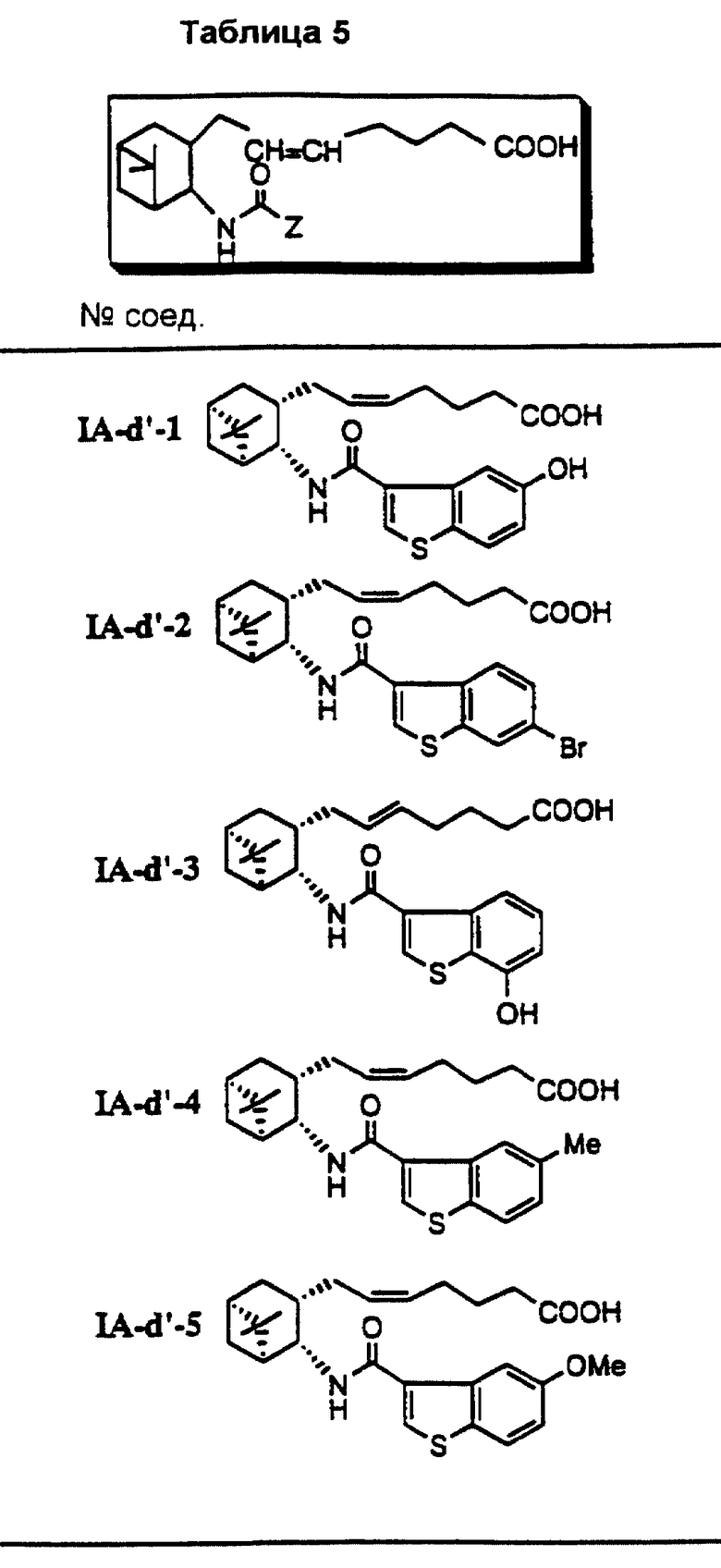
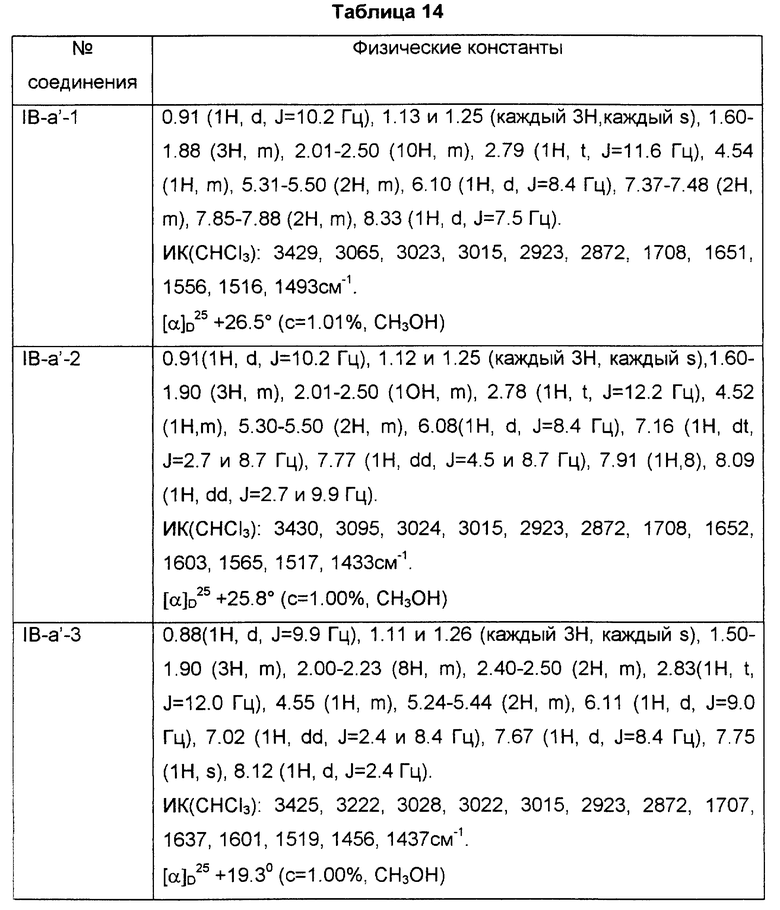
Соединения и физические константы, полученные таким же образом, как в приведенных выше примерах, показаны в следующих табл. 1-14.
Соединения, полученные в вышеуказанных примерах, были исследованы для определения активности in vivo и in vitro в соответствии со способом, как показано в примерах Экспериментов ниже.
Эксперимент 1. Связывание с рецептором PGD2
Материалы и Методы
(1) Получение мембранной фракции тромбоцитов человека
Образец крови получали из вены здоровых добровольцев (взрослые мужчина и женщина), используя пластиковый шприц, содержащий 3,8% цитрат натрия, помещали в пластиковую пробирку и осторожно перемешивали вращением. Образец затем центрифугировали при 1800 об./мин, 10 минут при комнатной температуре, и отбирали супернатант, содержащий ОТП (обогащенную тромбоцитами плазму). ОТП центрифугировали снова при 2300 об./мин, 22 минуты при комнатной температуре с получением тромбоцитов. Тромбоциты гомогенизировали, используя гомогенизатор (Ultra-Turrax), с последующим центрифугированием 3 раза при 20000 об. /мин, 10 минут при 4oC для получения мембранной фракции тромбоцитов. После определения белка мембранную фракцию доводили до 2 мг/мл и хранили в холодильнике при -80oC до использования.
(2) Связывание с рецептором PGD2
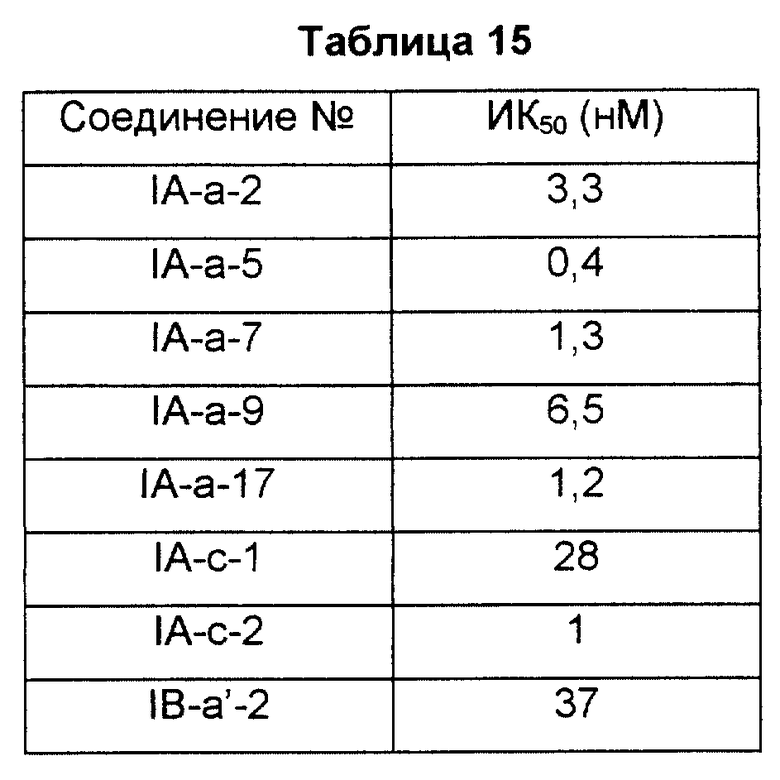
К раствору реакции связывания (50 мМ Трис/HCl, pH 7,4, 5 мМ MgCl2) (0,2 мл) добавляли мембранную фракцию тромбоцитов человека (0,1 мг) и 5 нМ [3H] PGD2 (115 Ки/ммоль) и давали возможность протекания реакции при 4oC в течение 90 минут. После завершения реакции реакционную смесь фильтровали через фильтровальную бумагу из стекловолокна, промывали несколько раз охлажденным солевым раствором и измеряли радиоактивность, оставшуюся на фильтровальной бумаге. Специфическое связывание вычисляли вычитанием неспецифического связывания (связывание в присутствии 10 мкМ PGD2) из общего связывания. Ингибиторную активность каждого соединения выражали как концентрацию, необходимую для 50% ингибирования (ИК50), которую определяли, рисуя кривую замещения нанесением на график степени связывания (%) в присутствии каждого соединения, где степень связывания в отсутствие исследуемого соединения равна 100%. Результаты показаны в табл. 15.
Эксперимент 2. Оценка антагонистической активности против рецептора PGD2 с использованием тромбоцитов человека
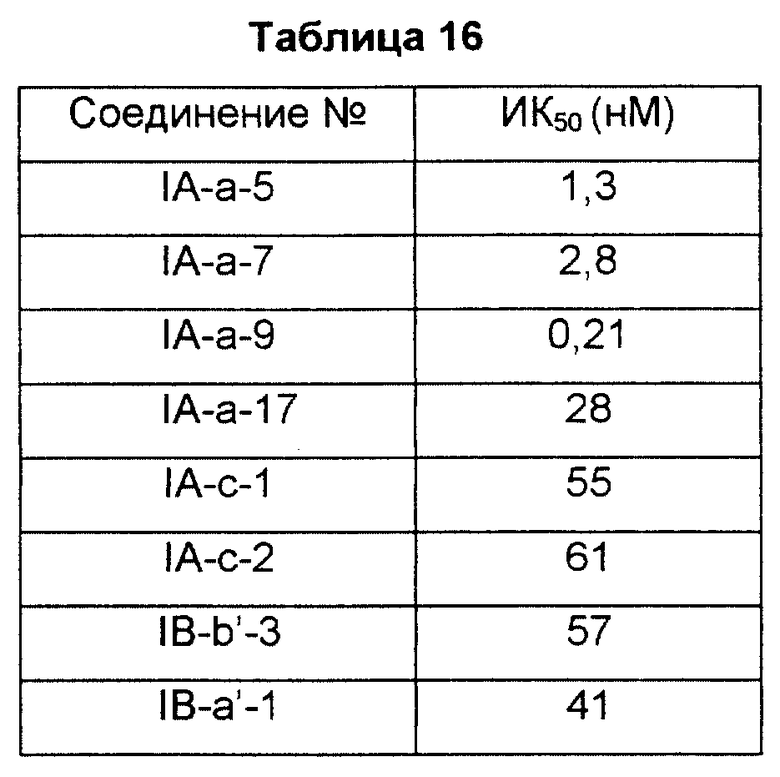
Периферическую кровь получали от здорового добровольца, используя шприц, в который предварительно добавляли раствор лимонной кислоты/декстрозы в объемном соотношении 1/9. Шприц подвергали центрифугированию при 180 g в течение 10 минут с получением супернатанта (ОТП: обогащенная тромбоцитами плазма). Полученную ОТП промывали три раза промывочным буфером, и количество тромбоцитов подсчитывали с помощью микросчетчика клеток. Суспензию, доведенную до конечной концентрации содержания тромбоцитов 5·108/мл, нагревали при 73oC, а затем подвергали предварительной обработке 3-изобутил-1-метилксантином (0,5 мМ) в течение 5 минут. К суспензии добавляли исследуемое соединение в различных концентрациях. Через десять минут реакцию индуцировали добавлением 0,1 мкМ PGD2 и через 2 минуты останавливали добавлением соляной кислоты. Тромбоциты разрушали на ультразвуковом гомогенизаторе. После центрифугирования цАМФ (циклический аденозинмонофосфат) в супернатанте определяли радиоиммуноанализом. Антагонизм рецептора PGD2 лекарственного средства оценивали следующим образом. Степень ингибирования по отношению к цАМФ, увеличенную с добавлением PGD2, определяли при отдельных концентрациях, а затем вычисляли концентрацию лекарственного средства, требуемую для 50% ингибирования (ИК50).
Результаты показаны в табл. 16.
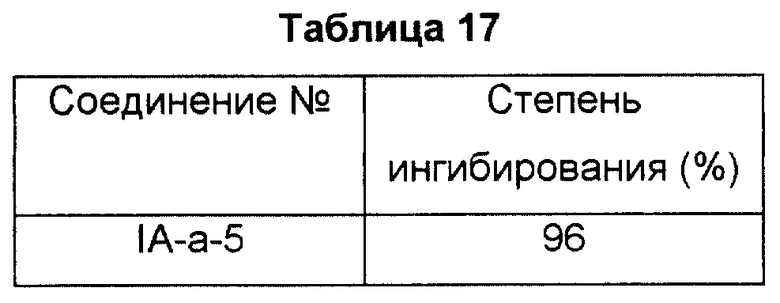
Эксперимент 3. Эксперимент с использованием модели носовой непроходимости
Ниже описан способ, используемый для измерения внутриносового давления для оценки противодействия носовой непроходимости с использованием морских свинок.
Раствор 1% овальбумина (ОВА) обрабатывали ультразвуковым распылителем для получения аэрозоля. Самца морской свинки Хартли (Hartley) сенсибилизировали ингаляцией аэрозолем дважды в течение 10 минут с интервалом в одну неделю. Через семь дней после сенсибилизации морской свинке экспонировали антиген для инициирования реакции. Коротко, трахею рассекали под анестезией пентобарбиталом (30 мг/кг, внутрибрюшинно) и канюли вводили в трахею со стороны легких и со стороны носовой полости. Канюлю, введенную со стороны легких, соединяли с аппаратом искусственного дыхания, который обеспечивает 4 мл воздуха 60 раз/мин. После остановки самостоятельного дыхания у морской свинки с использованием Галламина (Gallamin) (2 мг/кг, внутривенно), воздух подавали со стороны морды с помощью аппарата искусственного дыхания с частотой 70 раз/мин и скоростью потока 4 мл воздуха за раз, и атмосферное давление, требуемое для аэрации, измеряли с помощью датчика, укрепленного в ветви бронха. Результаты измерения использовали как параметр сопротивления носовой полости. Экспозицию антигена выполняли, генерируя аэрозоль 3% раствора ОВА в течение 3 минут между респиратором и канюлей носовой полости. Исследуемое соединение вводили перорально за 60 минут до экспонирования антигена. Внутриносовое давление измеряли постоянно в интервале между 0 и 30 минутами, и эффект выражали как степень ингибирования к степени, полученной для носителя, используя AUC (площадь под кривой) в течение 30 минут (на вертикальной оси внутриносовое давление (см H2O), а на горизонтальной оси время (0-30 мин)), как показание прибора. Результаты показаны в табл. 17.
Эксперимент 4. Активность по отношению к инфильтрации эозинофилов в носовую полость путем введения антигена.
Самцу морской свинки Хартли (Hartley) внутрибрюшинно инъецировали циклофосфамид (30 мг/кг), через 2 дня внутрибрюшинно инъецировали 1 мл суспензии, содержащей 1 мг овальбумина (ОВА) и 100 мг гидроксида алюминия. Через 3 недели внутрибрюшинно инъецировали 1 мл смеси ОВА (10 мкг) и гидроксида алюминия (100 мг) как дополнительную иммунизацию для системной сенсибилизации. По истечении 3 недель местной сенсибилизации в обе ноздри закапывали по 10 мкл 1% раствора ОВА четыре раза с 2-4-дневным интервалом. Через 5-7 дней после конечной сенсибилизации осуществляли введение антигена в нос путем закапывания 10 мкл 1% раствора ОВА морским свинкам в обе ноздри. Через пять часов после введения антигена в нос морские свинки были обескровлены под анестезией. Носовые воздушные пути промывали, вливая 10 мл солевого раствора, и промывные воды собирали. Промывные воды центрифугировали, осадки клеток ресуспендировали в 100 мкл солевого раствора, и общее количество клеток подсчитывали окрашиванием по Турк (Turk). Затем готовили образцы мазков, и клетки классифицировали после окрашивания по Мей-Грюнвальд-Гимза (Мау-Grunwald-Giemsa). Количество эозинофилов определяли умножением коэффициента эозинофилов на общее количество клеток. Исследуемое соединение (IA-а-5) суспендировали в 0,5% метилцеллюлозе и вводили перорально в дозе 1 мг/кг, 3 мг/кг и 10 мг/кг соответственно, за 1 час до введения антигена. Результаты показаны на чертеже.
Мы подтвердили то, что из вышеуказанных экспериментов 1 и 2 следует, что соединение по настоящему изобретению имеет PGD2-антагонистическую активность; эксперимент 4 подтверждает, что соединение по настоящему изобретению в значительной степени подавляет инфильтрацию эозинофилов, и эксперимент 3 подтверждает, что соединение по настоящему изобретению пригодно в качестве лекарственного средства для лечения носовой непроходимости.
Промышленная применимость
Согласно настоящему изобретению антагонисты PGD2 и ингибиторы для инфильтрации эозинофилов пригодны в качестве лекарственных средств для лечения заболеваний, таких как системный мастоцитоз и расстройство системной активации тучных клеток, а также трахеальной контракции, астмы, аллергического ринита, аллергического конъюктивита, крапивницы, ишемического реперфузионного повреждения, воспаления и атопического дерматита.
Описываются новые бензотиофенкарбоксамидные производные общей формулы I. R представляет собой водород, алкил, алкокси, галоген, гидрокси, ацилокси или фенилсульфонилокси, Х представляет собой водород или алкил, а двойная связь на α-цепи имеет Е-конфигурацию или Z-конфигурацию, при условии, что соединение формулы II, где R10 представляет собой водород, алкил или алкокси, Х является таким, как определено выше, а двойная связь на α-цепи имеет Е-конфигурацию или Z-конфигурацию, исключено, их фармацевтически приемлемые соли или гидраты. Соединения благодаря своей биологической активности могут быть использованы для лечения носовой непроходимости, а также использоваться в качестве антагонистов PGD2. Описываются также фармацевтическая композиция и лекарственное средство на основе соединений формулы I. 8 с. и 34 з.п. ф-лы, 1 ил., 17 табл.
где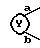
представляет собой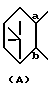
или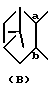
где
представляет собой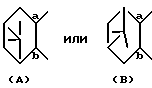
R представляет собой водород, алкил, алкокси, галоген, гидрокси, ацилокси или фенилсульфонилокси;
X представляет собой водород или алкил;
двойная связь на α-цепи имеет E конфигурацию или Z конфигурацию,
при условии, что соединение формулы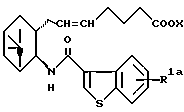
где R1а представляет собой водород, алкил или алкокси;
X является таким, как определено выше;
двойная связь на α-цепи имеет E конфигурацию или Z конфигурацию,
исключено, их фармацевтически приемлемые соли или гидраты.
представляет собой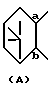
3. Соединение, его фармацевтически приемлемая соль или его гидрат по п. 1, где
представляет собой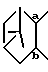
4. Соединение, его фармацевтически приемлемая соль или его гидрат по п. 1, где
представляет собой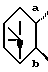
и R1 является галогеном, гидрокси, ацилокси или возможно замещенным арилсульфонилокси.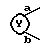
представляет собой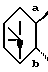
6. Соединение, его фармацевтически приемлемая соль или его гидрат по п. 1, где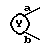
представляет собой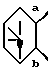
7. Соединение, его фармацевтически приемлемая соль или его гидрат по п. 1, где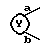
представляет собой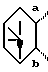
8. Соединение, его фармацевтически приемлемая соль или его гидрат по п. 1, где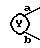
представляет собой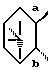
9. Соединение, его фармацевтически приемлемая соль или его гидрат по п. 1, где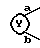
представляет собой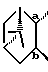
10. Соединение, его фармацевтически приемлемая соль или его гидрат по п. 1, где
представляет собой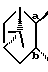
11. Соединение, его фармацевтически приемлемая соль или его гидрат по любому из пп.1 - 10, где двойная связь на α-цепи имеет E конфигурацию.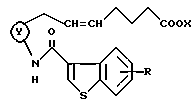
где кольцевой остаток Y представляет собой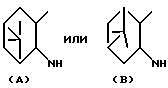
R представляет собой водород, галоген, гидрокси, ацилокси или фенилсульфонилокси;
X представляет собой водород или алкил;
двойная связь имеет E конфигурацию или Z конфигурацию,
при условии, что соединение формулы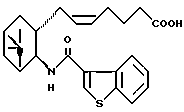
исключено, его фармацевтически приемлемая соль или его гидрат.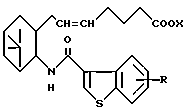
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
при условии, что соединение формулы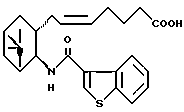
исключено, его фармацевтически приемлемая соль или его гидрат.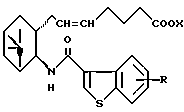
где R представляет собой галоген, гидрокси или ацилокси;
X представляет собой водород или алкил,
его фармацевтически приемлемая соль или его гидрат.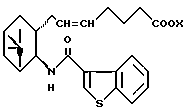
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
при условии, что соединение формулы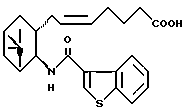
исключено,
его фармацевтически приемлемая соль или его гидрат.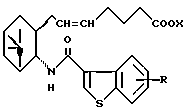
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
его фармацевтически приемлемая соль или его гидрат.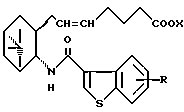
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
его фармацевтически приемлемая соль или его гидрат.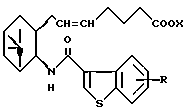
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
его фармацевтически приемлемая соль или его гидрат.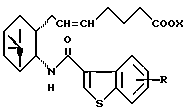
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
его фармацевтически приемлемая соль или его гидрат.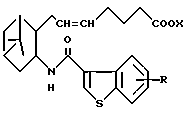
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
его фармацевтически приемлемая соль или его гидрат.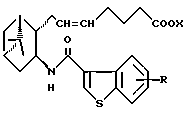
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
его фармацевтически приемлемая соль или его гидрат.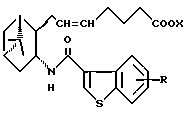
где R и X являются такими, как определено выше;
двойная связь имеет E конфигурацию или Z конфигурацию,
его фармацевтически приемлемая соль или его гидрат.
где кольцо Y и R являются такими, как определено выше.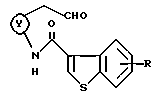
где кольцо Y и R являются такими, как определено выше.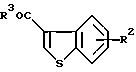
где R2 представляет собой ацилокси или фенилсульфонилокси;
R3 представляет собой гидрокси или галоген.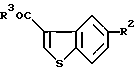
по п.33, где R2 и R3 являются такими, как определено в п.33.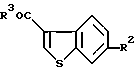
по п.33, где R2 и R3 являются такими, как определено в п.33.
Приоритет по пунктам и признакам:
13.12.96 по пп.17 и 18, 38 в части, касающейся фармацевтической композиции, содержащей соединение, его фармацевтически приемлемую соль или его гидрат по п. 17 или 18; п.39 в части, касающейся антагониста, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по п.17 или 18; п. 40 в части, касающейся антагониста PGD2, ингибирующего инфильтрацию воспалительных клеток, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по п.17 или 18; п.41 в части, касающейся антагониста PGD2, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по п.17 или 18; п.42 в части, касающейся лекарственного средства для лечения носовой непроходимости, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по п.17 или 18;
19.09.97 по п.15, 16, 19 - 32, 38 в части, касающейся фармацевтической композиции, содержащей соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.15, 16 или 19 - 30; п.39 в части, касающейся антагониста PGD2, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.15, 16 или 19 - 30; п.40 в части, касающейся антагониста PGD2, ингибирующего инфильтрацию воспалительных клеток, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.15, 16 и 19 - 30; п.41 в части, касающейся антагониста PGD2, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп. 15, 16 или 19 - 30; п.42 в части, касающейся лекарственного средства для лечения носовой непроходимости, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.15, 16 или 19 - 30;
10.12.97 по пп. 1 - 14, 33 - 37; п.38 в части, касающейся фармацевтической композиции, содержащей соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.1 - 14; п.39 в части, касающейся антагониста PGD2, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.1 - 14, п.40 в части, касающейся антагониста PGD2, ингибирующего инфильтрацию воспалительных клеток, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп. 1 - 14; п.41 в части, касающейся антагониста PGD2, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.1 - 14; п.42 в части, касающейся лекарственного средства для лечения носовой непроходимости, содержащего соединение, его фармацевтически приемлемую соль или его гидрат по любому из пп.1 - 14.
| SU, 341231 А, 1972 | |||
| EP, 0158380 А1, 1985 | |||
| EP, 01604086 А1, 1985 |

