Область техники, к которой относится изобретение
Изобретение относится к получению производных бензотиофенкарбоксамида, полезных в качестве антагонистов простагландина D2 (называемого далее как "PGD2").
Предпосылки создания изобретения
Производные бензотиофенкарбоксамида общей формулы (I):
где R представляет водород или гидроксизащитную группу, Х представляет водород или алкил и двойная связь представляет Е- или Z-конфигурацию, являются антагонистами PGD2, специфическими для PGD2-рецепторов, и полезны в качестве лекарственного вещества для лечения заболеваний, связанных с дисфункцией мастоцита (тучных клеток), вызванной избыточной продукцией PGD2 (WO 97/00853, PCT/JP 97/04527 (WO 98/25919)). Следовательно, соединения описанной выше формулы могут быть использованы в качестве лекарственных средств для лечения системного мастоцитоза, нарушения системной активации тучных клеток, трахеального спазма, астмы, аллергического ринита, аллергического конъюнктивита, крапивницы, повреждения в результате ишемической реперфузии, воспаления и атопического дерматита. Среди таких соединений соединение, в котором OR представляет 5-гидрокси, Х представляет водород и двойная связь представляет Z-конфигурацию (далее называемое как "соединение А"), проявляет особенно высокий антагонистический эффект на PGD2, обладает превосходной активностью против носовой окклюзии и, как ожидается, будет многообещающим лекарственным средством для лечения носовой окклюзии.
Раскрытие сущности изобретения
Соединение (I) и способы его получения известны из литературы (WO 97/00853, PCT/JP 97/04527 (WO 98/25919)). Однако известные способы не всегда пригодны для промышленного производства с точки зрения производительности, безопасности для работников и окружающей среды и эффективного использования ресурсов по следующим, например, причинам:
1) использование хроматографии на силикагеле, не пригодной для массового производства;
2) низкий выход и большой расход времени;
3) наличие сложных процессов отделения и очистки продукта реакции;
4) попутное выделение вредного газа и запаха и/или образование вредных сточных жидкостей; и/или
5) потребность в качестве исходных соединений, реагентов и/или растворителей материалов, вредных или трудных в обращении.
В соответствии с настоящим изобретением предлагается способ получения соединения формулы (I)
где R представляет водород или гидроксизащитную группу, Х представляет водород или алкил и двойная связь представляет Е- или Z-конфигурацию, или его фармацевтически приемлемой соли или гидрата, включающий взаимодействие аминоспирта формулы (II)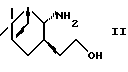
или его соли с соединением формулы (III)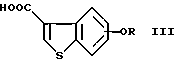
где R представляет водород или гидроксизащитную группу, или его реакционноспособным производным с получением соединения формулы (I-2)
где R - такой, как определено выше; окисление соединения (I-2) галогеноксокислотой в присутствии соединения из 2,2,6,6-тетраметилпиперидин-1-оксидов с получением соединения формулы (I-3)
где R - такой, как определено выше; осуществление взаимодействия соединения (I-3) с илидом в условиях реакции Виттига и, если требуется, снятие защиты у продукта реакции.
Наилучший вариант осуществления изобретения
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I-2)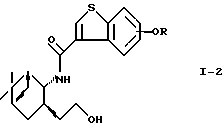
где R - такой, как определено выше, окисляют галогеноксокислотой в присутствии 2,2,6,6-тетраметилпиперидин-1-оксидов с получением соединения формулы (I-3)
где R - такой, как определенный выше.
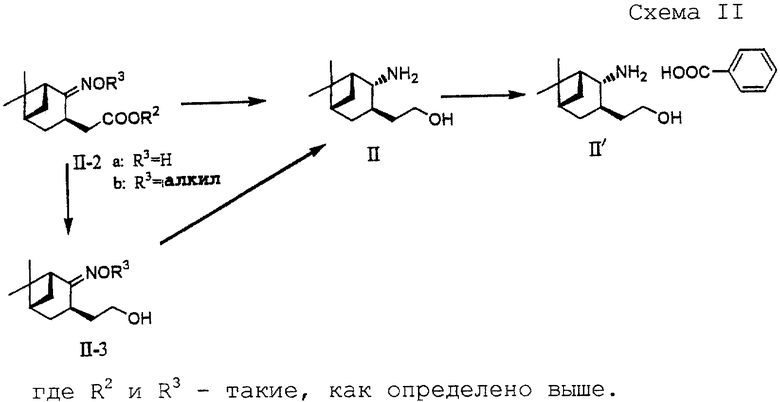
В другом предпочтительном варианте соединение формулы (II-2):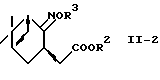
где R2 представляет алкил и R3 представляет водород или алкил, восстанавливают, используя систему восстановитель-кислота Льюиса, с получением аминоспирта формулы (II) или его соли.
В соответствии с предпочтительным вариантом используемый восстановитель выбирают из группы, состоящей из борогидридов, замещенных щелочным или щелочноземельным металлом, а кислоту Льюиса выбирают из группы, состоящей из галогенида олова, алюминия, титана, бора, циркония или никеля и их комплексов с простыми эфирами.
И в еще одном предпочтительном варианте соединение формулы (II-2)
где R2 и R3 - такие, как определены выше, преобразуют в спирт формулы (II-3)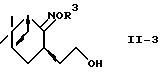
где R3 - такой, как определено выше, и спирт восстанавливают восстанавливающей системой металлический натрий-спирт или восстановитель-кислота Льюиса с получением аминоспирта формулы (II) или его соли.
Ниже дано определение терминов, использованных в данном описании.
Термин "гидроксизащитная группа" означает алкил, алкоксиалкил, ацил, аралкил, алкилсульфонил, арилсульфонил, алкилзамещенный силил, алкоксикарбонил, арилоксикарбонил, аралкилоксикарбонил или тетрагидропиранил.
Термин "алкил" означает C1-C20 неразветвленный или разветвленный алкил, в частности метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, трет-пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил, октадецил, нонадецил и икозил, причем предпочтительным является C1-С6 алкил. В качестве алкила для R2 предпочтительным является C1-С3 алкил.
Термин "алкокси" означает C1-C6 неразветвленный или разветвленный алкокси, в частности метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, изопентилокси, неопентилокси, втор-пентилокси, трет-пентилокси, н-гексилокси, неогексилокси, изогексилокси, втор-гексилокси, трет-гексилокси и тому подобное, причем предпочтительным является C1-С3 алкокси.
Термин "алкоксиалкил" означает алкильную группу, замещенную алкоксигруппой, и включает метоксиметил, этоксиметил, метоксиэтоксиметил, этоксиэтил, метоксипропил и тому подобное.
Термин "ацил" означает C1-С11 ацил, произведенный от алифатической или ароматической карбоновой кислоты. Примеры ацила алифатической кислоты включают ацетил, хлорацетил, трихлорацетил, пропионил, бутирил, валерил и тому подобное, а примеры ацила ароматической кислоты включают бензоил, п-нитробензоил, п-метоксибензоил, п-бромбензоил, толуоил, нафтоил и тому подобное.
Термин "арил" означает фенил, нафтил или полициклическую ароматическую углеводородную группу и тому подобное. Кроме того, арил может быть замещенным следующими заместителями.
Примеры заместителей включают алкил, такой как метил, этил, н-пропил, изопропил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил или трет-пентил, низший алкокси, такой как метокси или этокси, галоген, такой как фтор, хлор, бром или иод, нитро, гидрокси, карбокси, циано, сульфонил, амино, низший алкиламино, такой как метиламино, диметиламино, этилметиламино или диэтиламино, и тому подобное. Арильная группа может иметь один или несколько заместителей в любых возможных положениях. Конкретные примеры арила включают 2-метилфенил, 3-метилфенил, 4-метилфенил, 2-этилфенил, 3-этилфенил, 4-этилфенил, 4-пентилфенил, 4-карбоксифенил, 4-ацетилфенил, 4-(N, N-диметиламино)фенил, 4-нитрофенил, 4-гидроксифенил, 4-метоксифенил, 4-фторфенил, 4-хлорфенил, 4-иодфенил тому подобное.
Арильная группа в описанном ниже "аралкиле", "арилсульфониле", "арилоксикарбониле" или "аралкилоксикарбониле" может иметь заместители, аналогичные определенным выше.
Термин "аралкил" означает алкильную группу, замещенную арильной группой, и включает бензил, 4-метилбензил, 4-метоксибензил, 3,4-диметоксибензил, нафтилметил, фенетил и тому подобное.
Термин "алкилсульфонил" означает сульфонильную группу, замещенную алкильной группой, и включает метансульфонил, этансульфонил и тому подобное.
Термин "арилсульфонил" означает сульфонильную группу, замещенную арильной группой, и включает бензолсульфонил, п-толуолсульфонил и тому подобное.
Термин "алкилзамещенный силил" означает моно-, ди- или триалкилзамещенный силил, например метилсилил, диметилсилил, триметилсилил, трет-бутилдиметилсилил и тому подобное.
Термин "алкоксикарбонил" означает метоксикарбонил, изопропоксикарбонил, трет-бутоксикарбонил и тому подобное.
Термин "арилоксикарбонил" означает феноксикарбонил и тому подобное.
Термин "аралкилоксикарбонил" означает бензилоксикарбонил и тому подобное.
В качестве гидроксизащитной группы, представленной символом R, предпочтительным является алкил, алкоксиалкил, ацил, аралкил, алкилсульфонил, арилсульфонил, алкилзамещенный силил, алкоксикарбонил, арилоксикарбонил, аралкилоксикарбонил или тетрагидропиранил, а более предпочтительным является арилсульфонил.
Примеры солей соединения общей формулы (I) включают соли щелочных металлов, такие как литиевая соль, натриевая соль или калиевая соль и тому подобное, соли щелочноземельных металлов, такие как кальциевая соль и тому подобное, аммониевая соль, соли с органическими основаниями, такими как трометамин, триметиламин, триэтиламин, 2-аминобутан, трет-бутиламин, диизопропилэтиламин, н-бутилметиламин, н-бутилдиметиламин, три-н-бутиламин, циклогексиламин, дициклогексиламин, N-изопропилциклогексиламин, фурфуриламин, бензиламин, метилбензиламин, дибензиламин, N,N-диметилбензиламин, 2-хлорбензиламин, 4-метоксибензиламин, 1-нафталинметиламин, дифенилбензиламин, трифениламин, 1-нафтиламин, 1-аминоантрацен, 2-аминоантрацен, дегидроабиэтиламин, N-метилморфолин или пиридин, или аминокислотные соли, такие как лизиновая соль или аргининовая соль.
Соли аминоспиртов формулы (II) включают соли с органической кислотой, такой как бензойная кислота и т.д., и минеральной кислотой, такой как хлороводородная кислота, серная кислота и т.д.
Целевое соединение по настоящему изобретению представлено общей формулой (I), в которой двойная связь алкениленовой боковой цепи (т.е. 5-гептениленовой цепи) может находиться в Е- или Z-конфигурации.
Способ по настоящему изобретению описан ниже более подробно. При наличии заместителя(ей), возможно мешающего(их) реакции, его(их) можно подходящим образом защитить и на требуемой стадии освободить от защиты. Такие защиту или снятие защиты можно осуществить методом, известным в данной области техники.
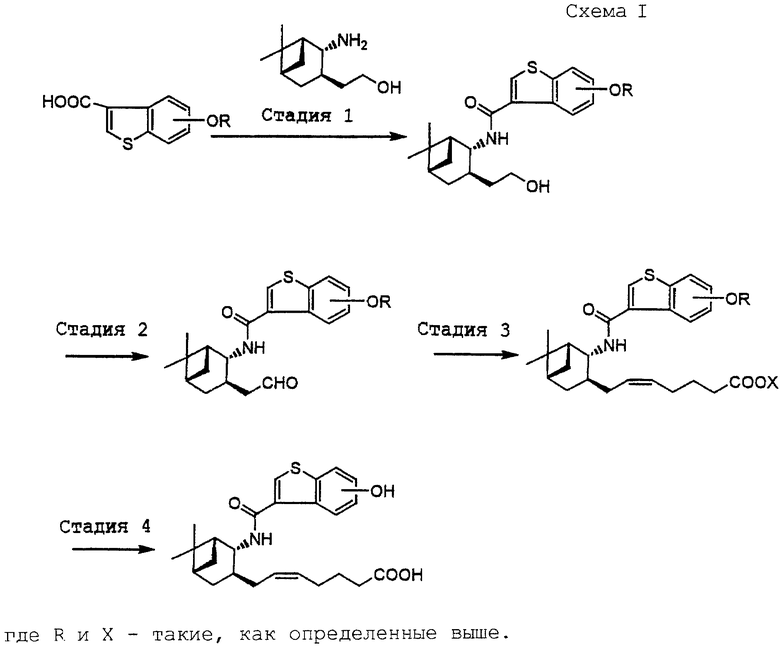
I. Получение соединения (I) (см. схему I в конце описания).
[Стадия 1]
Эта стадия связана с получением амида (I-2) путем ацилирования аминоспирта (II) или его соли карбоновой кислотой (III) или ее реакционноспособным производным.
Карбоновая кислота (соединение III), используемая при ацилировании, может быть синтезирована методом, известным в литературе [например, Nippon-Kagaku Zasshi, т. 88, 7, 758-763 (1967); Nippon-Kagaku Zasshi, т.86, 10, 1067-1072 (1965); J. Chem. Soc. (C), 1899-1905 (1967); J. Heterocycle. Chem. , т.10, 679-681 (1973)]. Термин "реакционноспособное производное" карбоновой кислоты (III) относится к соответствующим галогенангидридам кислоты (например, хлорангидриду, бромангидриду, иодангидриду), ангидридам кислоты (например, смешанным ангидридам кислоты с муравьиной или уксусной кислотой), активированным сложным эфирам (например, сукцинимидоэфиру) и тому подобному и включает ацилирующие агенты, обычно используемые для ацилирования аминогруппы. Например, чтобы получить галогенангидриды кислоты, карбоновую кислоту подвергают взаимодействию с тионилгалогенидом (например, тионилхлоридом), фосфористым галогенидом (например, фосфористым трихлоридом, фосфористым пентахлоридом), оксалилгалогенидом (например, оксалилхлоридом) или тому подобным в соответствии с известным методом (например, Shin-jikken Kagaku Koza, т. 14, стр. 1787 (1978); Synthesis, 852-854 (1986); Shin-jikken Kagaku Koza, т.22, стр.115 (1992)).
Ацилирование может быть осуществлено при обычных условиях, используемых для ацилирования аминогруппы. Например, при использовании галогенида карбоновой кислоты реакцию осуществляют согласно методу, общеизвестному как "реакция Шоттена-Баумана". Обычно галогенид карбоновой кислоты добавляют по каплям к водному щелочному раствору амина при перемешивании и охлаждении с удалением образующейся кислоты щелочью. В соответствии с другим вариантом, когда карбоновую кислоту используют в виде свободной кислоты, а не ее реакционноспособного производного, реакцию можно проводить традиционно в присутствии сочетающего агента, обычно используемого в реакции сочетания между амином и карбоновой кислотой, такого как дициклогексилкарбодиимид (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимид или N,N'-карбонилдиимидазол.
[Стадия 2]
Эта стадия относится к окислению спирта (I-2) с получением альдегида (I-3). До сих пор такую реакцию проводили с использованием окислителя из семейства хромовых кислот, такого как реактив Джонса (J. Org. Chem., 40, 1664-1665 (1975)), реактив Коллинза (J.C.S. Chem. Comm., 1972, 1126) или пиридинийхлорхромат (Tetrahedron Lett. , 2646-2650 (1975)). Известен также метод с использованием диоксида марганца (Helv. Chim. Acta., 39, 858-862 (1956)) или диметилсульфоксида (Swern oxidation, J. Org. Chem., 43, 2480-2482 (1978)). Однако эти существующие методы имеют недостатки. Например, хромовые кислоты ядовиты для организма человека и должны быть обезврежены после применения. Кроме того, окисление по Сверну (Swern) с использованием смеси диметилсульфоксид-оксалилхлорид не годится для крупносерийного производства, потому что оно сопровождается выделением моноксида углерода, опасного для работников, и запахом серы и к тому же его нужно осуществлять при низкой температуре, например между -50oС и -78oС.
В соответствии со способом по настоящему изобретению спирт (I-2) окисляют с помощью окислителя(ей), такого(их) как галогеноксокислота, в присутствии 2,2,6,6-тетраметилпиперидин-1-оксидов (называемых как "TEMPO") согласно описанию в литературе (например, J. Org. Chem., 52, 2559-2562 (1987)), что решает проблемы существующих методов. Примеры TEMPO включают 2,2,6,6-тетраметилпиперидин-1-оксид, 4-метокси-2,2,6,6-тетраметилпиперидин-1-оксид, 4-ацетиламино-2,2,6,6-тетраметилпиперидин-1-оксид, 4-бензоилокси-2,2,6,6-тетраметилпиперидин-1-оксид и 4-циано-2,2,2,2-тетраметилпиперидин-1-оксид. Примеры галогеноксокислот включают гипохлорит натрия, гипобромит натрия, бромит натрия и высшую хлорную известь. Раствор окислителя может быть доведен, например, до рН 8,5-9,5 добавлением минеральной кислоты, такой как гидрокарбонат натрия, хлористоводородная кислота или серная кислота. В соответствии с другим вариантом раствор окислителя может быть добавлен в присутствии гидрокарбоната натрия. Реакция может быть осуществлена за время от нескольких минут до нескольких десятков минут при температуре от температуры охлаждения льдом до комнатной температуры в растворителе, таком как этилацетат, ацетонитрил или дихлорметан.
Преимущество нового способа окисления по настоящему изобретению характеризуется следующим:
1) способ требует простых операций и мало времени, поскольку реакция дает продукт с высоким выходом при коротком времени реакции без поддержания очень низкой температуры;
2) способ безопасен, поскольку используемыми в реакции растворителями являются вода и этилацетат;
3) отделение и очистка продуктов реакции могут быть осуществлены с использованием лишь экстракции;
4) окисление осуществляют, используя дешевый реагент, гипохлорит натрия, и лишь весьма малое количество катализатора TEMPO при 1-0,2%-ном молярном эквиваленте спирту (I-2);
5) способ позволяет оператору работать в лучшей окружающей среде, потому что, в противоположность окислению по Сверну (Swern), реакция дает незначительное выделение моноксида углерода или запаха и, кроме того, хлорид натрия, образующийся из гипохлорита натрия, используемого в окислении, не нужно обезвреживать.
[Стадия 3]
Эта стадия связана с образованием двойной связи путем осуществления взаимодействия соединения формулы (I-3) с илидом (Рh3Р=СН(СН2)3СООН). Реакция образования двойной связи может быть осуществлена традиционным для реакции Виттига образом. Используемые в реакции илиды могут быть синтезированы в присутствии основания путем обработки фосфониевой соли, синтезированной из трифенилфосфина, и алкилгалогенида, имеющего требуемую для конденсации алкильную группу, например 5-бромпентановой кислоты. Примеры основания включают димсилнатрий, димсилкалий, гидрид натрия, н-бутиллитий, трет-бутоксид калия и диизопропиламид лития. Реакцию осуществляют в течение нескольких часов при комнатной температуре в растворителе, таком как диэтиловый эфир, тетрагидрофуран, н-гексан, 1,2-диметоксиэтан или диметилсульфоксид.
[Стадия 4]
На этой стадии производят снятие защиты у соединения (I), в котором R представляет гидроксизащитную группу, с получением соединения (I-1). Реакция может быть осуществлена традиционным образом с использованием в качестве катализатоpa хлористоводородной кислоты, серной кислоты, гидроксида натрия, гидроксида калия, или гидроксида бария, или тому подобное. Реакцию осуществляют в течение времени от нескольких десятков минут до нескольких часов при нагревании в растворителе, таком как метанол-вода, этанол-вода, ацетон-вода, ацетонитрил-вода или тому подобное, предпочтительно диметилсульфоксид-вода. Группировка OR может находиться в любом из положений 4, 5, 6 и 7, хотя предпочтительным является, чтобы она находилась в положении 5.
II. Получение соединения (II)
Исходный материал в этом процессе, аминоспирт (II), может быть получен известным способом из, например, (-)-миртенола. Предшествующий продукт, сложный метоксимэфир формулы (II-2), где R3 представляет метил, восстанавливают затем металлическим натрием в изопропаноле с получением соответствующего аминоспирта (II) (Hagishita, et al., Chem. Pharm, Bull., 37(6), 1524-1533 (1989)). Однако этот способ имеет недостатки, такие как низкий выход (39,6%) или плохая избирательность.
В качестве восстановителей, используемых при восстановлении сложных эфиров до спиртов, известны борогидрид натрия (J. Org. Chem., 28, 3261 (1982)), литийалюминийгидрид (Org. Syn., 63, 140), борогидрид лития (J. Org. Chem. , 47, 4702 (1982)) и тому подобное. Кроме того, в качестве способов восстановления оксимов до аминов используют уже известное каталитическое восстановление (Syn. Comm. , 27, 817 (1997); Org. Syn., сборник т. 5, 376 (1973)) или способы, в которых используют восстановитель(и), такой(ие) как диборан (J. Org. Chem., 30, 2877 (1965)), борогидрид натрия (J. Org. Chem., 48, 3412 (1983)), литийалюминийгидрид (Tetrahedron, 51, 8363 (1995)), система борогидрид натрия-хлорид титана (IV) (Synthesis. 1980, 695), система борогидрид натрия-хлорид никеля (II) (Chem. Ber., 117, 856 (1984)) или тому подобное. Ни в одном из указанных литературных источников, однако, не описан способ восстановления одновременно фрагментов как сложного эфира, так и оксима в одной молекуле, такой как соединение формулы (II-2), при высоком выходе и высокой стереоизбирательности.
Создатели настоящего изобретения добились успеха в восстановлении сложного оксимэфира формулы (II-2) до целевого аминоспирта (II) при высоком выходе и с высокой избирательностью путем использования системы восстановитель-кислота Льюиса (в частности, борогидрид натрия-кислота Льюиса), как показано на схеме II (см. в конце описания).
В соответствии с предлагаемым способом сложный оксимэфир (II-2) восстанавливают непосредственно или через спирт (II-3) с получением аминоспирта (II) или его соли. Восстановители, используемые в показанной выше реакции, включают борогидриды, замещенные щелочным или щелочноземельным металлом (борогидрид натрия, борогидрид лития, борогидрид кальция и т.д.).
Примеры кислоты Льюиса включают галогениды олова, алюминия, бора, титана, циркония или никеля (например, хлорид олова(II), хлорид олова(IV), хлорид алюминия, тетрахлорид титана, трифторид бора, тетрахлорид циркония, дихлорид никеля и т.д.) или их эфирные комплексы (например, бис(2-метоксиэтокси)алюминийгидрид натрия и т.д.).
Примеры растворителей включают простые эфиры (например, диэтиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан, диоксан, диметиловый эфир диэтиленгликоля и т.д.), углеводороды (например, толуол, ксилол и т.д.) и смешанные растворители, содержащие простые эфиры и углеводороды. Что касается восстановления спирта (II-3) до аминоспирта (II) или его соли, то существует также способ, в котором, кроме указанной выше системы восстановитель-кислота Льюиса, используют систему металлический натрий-спирт. Примеры спирта включают метанол, этанол, н-пропанол, изопропанол и тому подобное. Примеры растворителей включают углеводороды (например, толуол, ксилол и т.д.).
Ниже описан конкретный процесс реакции. Исходный материал, сложный оксимэфир (или алкилзамещенный оксим) (II-2а или II-2b), растворяют в 2 или более объемах растворителя. К раствору добавляют 2 или более молярных эквивалентов восстановителя и затем 0,1-0,4 молярного эквивалента (относительно восстановителя) кислоты Льюиса при 0oС-150oС. В соответствии с другим вариантом может быть добавлена смесь, заранее приготовленная путем смешивания кислоты Льюиса и растворителя. Кроме того, может быть изменен порядок добавления исходного материала (сложного оксимэфира), восстановителя и кислоты Льюиса. Затем реакционную смесь подвергают взаимодействию при 0oC-150oC в течение времени в пределах от нескольких минут до нескольких часов. Реакционный раствор может быть обработан путем добавления воды и разбавленной минеральной кислоты (например, разбавленной хлороводородной кислоты) с последующим перемешиванием, благодаря чему восстановитель разлагается. В соответствии с другим вариантом реакционный раствор может быть вылит в разбавленную минеральную кислоту.
Затем раствор нейтрализуют щелочью (например, гидроксидом натрия) и экстрагируют органическим растворителем (например, этилацетатом). После отгонки растворителя получают аминоспирт (II). При необходимости продукт может быть дополнительно очищен путем преобразования в кристаллическую соль (II') с помощью подходящей кислоты (например, бензойной кислоты) и затем нейтрализации щелочью с получением аминоспирта (II).
Описанным выше способом по настоящему изобретению целевой аминоспирт (II) может быть получен с высоким выходом (около 89%) при высокой стереоизбирательности (99% или выше).
Хотя способ получения соединения формулы (II), показанный на приведенной выше схеме II, является новым и полезным для получения соединения (II) как такового, он также способствует обеспечению безопасного и эффективного получения соединения (I), т.е. конечного продукта, при сочетании со способом получения соединения (I).
Для более подробной иллюстрации настоящего изобретения далее даны примеры, которые не следует считать сколь-нибудь ограничивающими объем изобретения. Используемые в примерах сокращения имеют следующие значения:
Рh: фенил
Ас: ацетил
TEMPO: 2,2,6,6-тетраметилпиперидин-1-оксил.
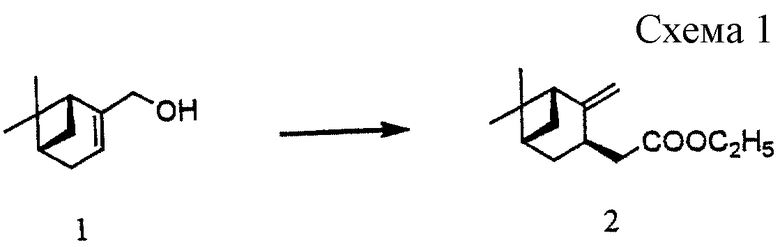
Ссылочный пример 1
Получение этил[(1R, 3R, 5S)-2-метилиден-10-норпинан-3-ил)]ацетата (2) (см. схему 1 в конце описания).
Смесь (-)-миртенола (1) (6,44 г, 42,3 ммоль), триэтилортоацетата (23 мл, 126 ммоль) и гидрохинона (27 мг) нагревали с перемешиванием при 165oС в течение 2 часов, при 185oС в течение 2 часов и при 195oС в течение 25 часов и отгоняли полученный этанол. Полученное масло очищали хроматографией на силикагеле (гексан: толуол = 10: 0-1:1) с получением 7,66 г указанного в заголовке соединения (2). Выход: 81,4%.
ИК (Пленка): 3070, 2980, 2921, 2869, 1737, 1638 см-1.
1Н ЯМР δ (CDCl3), 300 МГц: 0,76 и 1,24 (каждый 3Н, каждый с), 1,20 (1Н, д, J=9,9 Гц), 1,27 (3Н, т, J=7,2 Гц), 1,52 (1Н, м), 2,00 (1Н, м), 2,23-2,50 (3Н, м), 2,66 (1Н, дд, J=5,1 и 15,3 Гц), 3,03 (1Н, м), 4,16 (2Н, кв, J=7,2 Гц), 4,71 (2Н, д, 11,4 Гц).
Элементный анализ для C14H22О2.
Вычислено (%): С, 75,63; Н, 9,97.
Найдено (%): С, 75,61; Н, 9,99.
[α]D 24+29,1o (с=1,05, СН3ОН)
Ссылочный пример 2
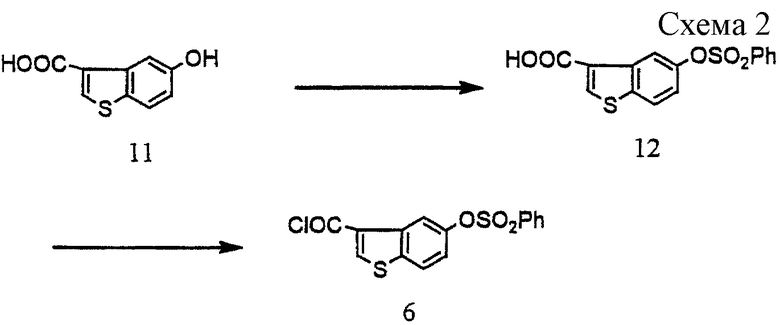
Получение 5-бензолсульфонилоксибензо[b]тиофен-3-карбонилхлорид (6) (см. схему 2 в конце описания).
5-Гидроксибензо[b] тиофен-3-карбоновую кислоту (11) (M. Martin-Smith et al. , J. Chem. Soc (С), 1899-1905 (1967), 8,63 г, 44,4 ммоль)) растворяли в водном тетрагидрофуране (содержание воды 20%; 160 мл) и 1 н. гидроксиде натрия (44 мл). К раствору добавляли по каплям 0,56 н. гидроксид натрия (87 мл) и бензолсульфонилхлорид (6,2 мл, 48,4 ммоль) при перемешивании и охлаждении на льду, поддерживая рН на уровне 11-12. По окончании реакции смесь разбавляли водой, подщелачивали и промывали толуолом. Водный слой слегка подкисляли добавлением концентрированной хлористоводородной кислоты при перемешивании и выпавшие в осадок кристаллы отфильтровывали, промывали водой и высушивали с получением 14,33 г 5-бензолсульфонилоксибензо[b]тиофен-3-карбоновой кислоты (12). Т.пл. 202-203oС.
ЯМР δ (CDCl3), 300 МГц: 7,16 (1H, дд, J=2,7 и 9,0 Гц), 7,55-7,61 (2Н, м), 7,73 (1H, м), 7,81 (1H, д, J=9,0 Гц), 7,90-7,94 (2Н, м), 8,16 (1Н, д, J= 2,7 Гц), 8,60 (1H, с).
ИК (Нуйол): 3102, 2925, 2854, 2744, 2640, 2577, 1672, 1599, 1558, 1500, 1460, 1451 см-1.
Элементный анализ для C15H10O5S2.
Вычислено (%): С, 53,88; Н, 3,01; S, 19,18.
Найдено (%): С, 53,83; Н, 3,03; S, 19,04.
5-Бензолсульфонилоксибензо[b] тиофен-3-карбоновую кислоту (12) (5,582 г, 16,7 ммоль), полученную так, как описано выше, нагревали с обратным холодильником в течение 1,5 часов с диметилформамидом (1 капля), тионилхлоридом (3,57 мл, 50 ммоль) и толуолом (22 мл). Удалив при пониженном давлении растворитель, получили 5,89 г целевого соединения (6).
ПРИМЕР 1
Получение аминоспирта
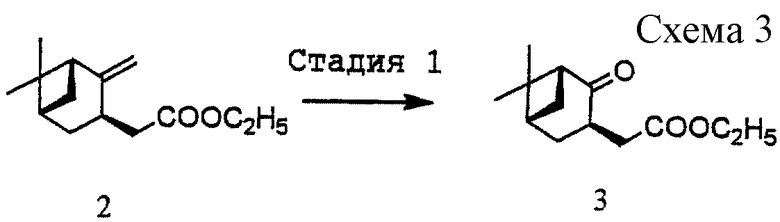
(1) Стадия 1: получение этил [(1R, 3R, 5S)-2-оксо-10-норпинан-3-ил]ацетата (3) (см. схему 3 в конце описания).
Соединение (2) (333,5 г, 1,5 моль), полученное в ссылочном примере 1, растворяли в дихлорметане (3,340 л) и метаноле (660 мл). Смесь охлаждали до температуры от -70oС до -73oС и вводили в нее газообразный азот в течение 4 часов. После введения в течение 1 часа газообразного азота добавляли триметилфосфит (265 мл, 2,26 моль) и давали реакционной смеси согреться до комнатной температуры. Затем реакционную смесь разделяли на два слоя добавлением ледяной воды (150 мл) и 10%-ной серной кислоты (300 г). Органический слой промывали водой (1,2 л), 2%-ным сульфитом натрия (1,2 кг) и водой (1,2 л). Водный слой экстрагировали этилацетатом (1,11 л). Органические слои объединяли и растворитель отгоняли при пониженном давлении с получением 456,51 г масла, которое затем растворяли в тетрагидрофуране (1,05 л). После добавления холодного 14%-ного водного гидроксида аммония (106,8 г) полученный бледно-желтый раствор перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавляли ледяной водой (750 мл). После добавления этилацетата (1,1 л) смесь перемешивали и разделяли на два слоя. Такую же процедуру повторяли еще раз и водный слой опять экстрагировали этилацетатом. Объединенный органический слой промывали 10%-ным рассолом (750 мл), сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель. Полученное масло растворяли в толуоле (500 мл) и отгоняли при пониженном давлении растворитель с получением 347,96 г масла. Выход неочищенного продукта: 103,4%.
1Н ЯМР δ (CDCl3), 300 МГц: 0,95 и 1,34 (каждый 3Н, каждый с), 1,27 (3Н, т, J=7,0 Гц), 1,40 (1Н, д, J=9,9 Гц), 1,67 (1Н, м), 2,25 (1Н, м), 2,33-2,42 (2Н, м), 2,56-2,65 (2Н, м), 2,86-3,02 (2Н, м), 4,14-4,21 (2Н, м).
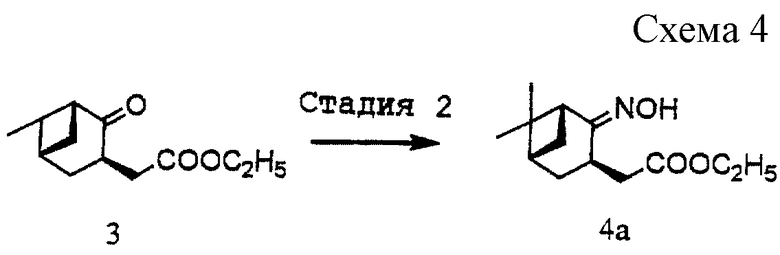
(2) Стадия 2 (см. схему 4 в конце описания).
(1) Получение этил [(1R, 3R, 5S)-2-гидроксиимино-10-норпинан-3-ил]ацетата (4а)
Соединение (3) (10,05 г, 44,9 ммоль) растворяли в этаноле (45 мл). К раствору добавляли гидроксиламингидрохлорид (4,99 г, 71,9 ммоль) и пиридин (4,7 мл, 58,1 ммоль) и смесь нагрели при 60oС в течение 2,5 часов при перемешивании. Реакционную смесь концентрировали при пониженном давлении, разбавляли водой и подкисляли хлористоводородной кислотой, после чего смесь экстрагировали этилацетатом. Органический слой промывали водой, водным раствором гидрокарбоната натрия и водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 10,72 г указанного в заголовке соединения (4а) в виде бесцветного масла. Выход неочищенного продукта: 100%.
[α]D 24+55,3o (c=l,01, СН3ОН)
(2) Получение этил [(1R, 3R, 5S)-2-метоксиимино-10-норпинан-3-ил]ацетата (4b) (см. схему 5 в конце описания).
Соединение (3) (107,0 г, 477 ммоль) растворяли в этаноле (500 мл). К раствору добавляли О-метилгидроксиламмонийхлорид (50,1 г, 600 ммоль) и пиридин (47,5 г, 600 ммоль) и смесь нагревали с обратным холодильником в течение 3 часов при перемешивании. Реакционную смесь концентрировали при пониженном давлении, разбавляли водой, подкисляли хлористоводородной кислотой и затем экстрагировали этилацетатом. Органический слой промывали водой, водным раствором гидрокарбоната натрия и водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель. Полученное бесцветное масло перегоняли при пониженном давлении с получением 106,1 г указанного в заголовке соединения (4b). Точка кипения: 118-123oС (пониженное давление 1,2 мм рт. столба). Выход: 87,8%.
ИК (пленка): 1738, 1630 см-1.
Элементный анализ для C14H23NO3.
Вычислено (%): С, 66,37; Н, 9,15; N, 5,53.
Найдено (%): С, 65,92; Н, 9,13; N, 5,60.
[α]D 24+69,5o (с=1,00%, СН3ОН)
(3) Стадия 3: получение бензойной кислой соли [(1R, 2R, 3R, 5S)-2-амино-10-норпинан-3-ил]этанола (II') (см. схему 6 в конце описания).
1) Получение из соединения (4b)
Борогидрид натрия (799 мг, 21,1 ммоль) суспендировали в 1,2-диметоксиэтане (5 мл). К суспензии добавляли при перемешивании и охлаждении льдом суспензию алюминийхлорида (507 мг, 3,8 ммоль) в 1,2-диметоксиэтане (5 мл), а затем раствор соединения (4b) (1,07 г, 4,2 ммоль) в 1,2-диметоксиэтане (3 мл) и смесь нагревали в ванне при 70oС в течение 3 часов. Затем к реакционной смеси добавляли воду (4 мл), 2 н. хлористоводородную кислоту (8 мл) и концентрированную хлористоводородную кислоту (1 мл) при перемешивании и охлаждении льдом, после чего смесь перемешивали при комнатной температуре в течение 40 минут. Затем реакционную смесь промывали эфиром, нейтрализовали гидроксидом натрия и экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и отгоняли растворитель при пониженном давлении с получением 789 мг указанного в заголовке соединения (II) в виде бесцветного масла. Продукт растворяли в диэтиловом эфире (5 мл). К раствору добавляли раствор бензойной кислоты (516 мг, 4,2 ммоль) в эфире (5 мл) и смесь перемешивали. Выпавшие в осадок кристаллы отфильтровывали, промывали эфиром и высушивали с получением 1,146 г бесцветной бензойной кислой соли аминоспирта (II'). Выход: 89% (чистота: 99,2%), т. пл. 183-185oС. Чистоту бензойной кислой соли аминоспирта (II') определяли путем преобразования соли в бензамид в присутствии конденсирующих агентов, таких как дициклогексилкарбодиимид (DCC, ДЦК) и 1-гидроксибензтриазола (НОВТ, ГОБТ) в тетрагидрофуране, и определения чистоты полученного амида с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ).
ИК (КВr): 3420, 2600 (ш), 1621, 1523, 1386 см-1.
1H ЯМР δ (CDCl3), 300 МГц: 0,72 (1Н, д, J=9,9 Гц), 1,06 и 1,13 (каждый 3Н, каждый с), 1,40 (1Н, м), 1,56-1,92 (3Н, м), 2,12-2,36 (4Н, м), 3,29 (1Н, м), 3,62 (1Н, м), 3,78 (1Н, м), 7,32-7,47 (3Н, м), 7,97-8,04 (2Н, м).
Элементный анализ для С18Н27NО3.
Вычислено (%): С, 70,79; Н, 8,91; N, 4,59.
Найдено (%): С, 70,54; Н, 8,93; N, 4,56.
[α]D 25+27,6o (с=1,00%, СН3ОН)
Эталонные значения: т. пл. 180-183oС, [α]D 26+27,lo (Chem. Pharm. Bull., 37, 1524 (1989)).
[Условия ВЭЖХ]: прибор: тип LC-6A (Shimazu); колонка: YMC-pack ODS-AMAM-302 (4,6 мм φ•150 мм); расход: 1,0 мл/мин; детектирование: УФ 254 нм; подвижная фаза: ацетонитрил-вода (1:1); время удерживания: 5,23 минуты.
2) Получение из соединения (4а) (часть 1)
Борогидрид натрия (1,55 г, 41,0 ммоль) суспендировали в диметиловом эфире диэтиленгликоля (13 мл). К суспензии добавляли при перемешивании и охлаждении льдом в течение 10 минут эфират трифторида бора (1,71 мл, 13,5 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. После добавления раствора соединения (4а) (1,015 г, 4,1 ммоль) в диметиловом эфире диэтиленгликоля (8 мл) смесь перемешивали при комнатной температуре в течение 20 минут. Затем смесь нагревали в ванне при 110oС при перемешивании в течение 2 часов. Раствор обрабатывали так, как описано выше в п.1, с получением 741 мг бензойной кислой соли аминоспирта (II'). Выход: 59% (чистота: 99,2%), т. пл. 178-180oС.
3) Получение из соединения (4а) (часть 2)
Борогидрид натрия (1,00 г, 26,4 ммоль) суспендировали в 1,2-диметоксиэтане (10 мл). К суспензии добавляли при перемешивании и охлаждении льдом соединение (4а) (1,00 г, 4,03 ммоль) и комплекс тетрахлорид титана-1,2-диметоксиэтан (1:1) (700 мг, 2,51 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут и затем нагревали в ванне при 70oС в течение 3 часов. Смесь обрабатывали так, как описано выше в п.1, с получением 750 мг бензойной кислой соли аминоспирта (II'). Выход: 61% (чистота: 94,2%), т. пл. 176-180oС.
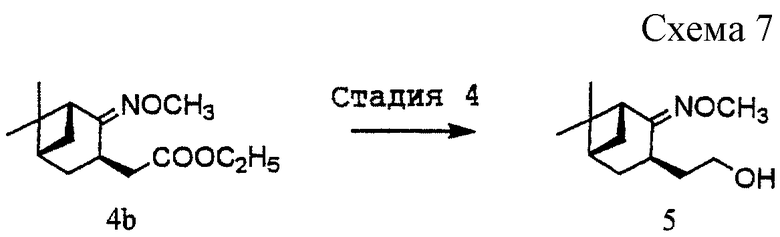
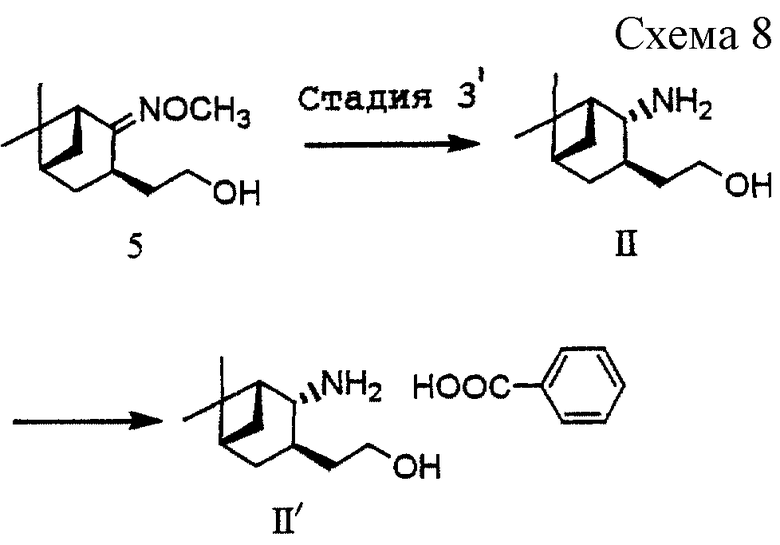
(4) Стадия 4: получение [(1R, 3R, 5S)-2-метоксиимино-10-норпинан-3-ил] этанола (5) (см. схему 7 в конце описания).
Соединение (4b) (23,8 г, 94 ммоль) растворяли в толуоле (111 мл). К раствору добавляли раствор 70%-ного бис(2-метоксиэтокси)алюминийгидрида натрия в толуоле (34,4 г, 119 ммоль) при температуре ниже 25oС в течение 20 минут и продолжали перемешивание в течение 30 минут при той же самой температуре. К реакционной смеси добавляли ацетон (7 г) для разложения реагента, после чего добавляли воду (30 мл) и затем 48%-ный раствор гидроксида натрия (43,8 г). Полученные два слоя разделяли и водный слой экстрагировали толуолом (111 мл). Объединенный органический слой промывали водой (3•30 мл). Органический слой сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 18,9 г указанного в заголовке соединения (5) в виде бесцветного масла. Выход: 95,1%. Продукт использовали в последующей реакции без очистки.
ИК (СНСl3): 3619, 3502, 3020, 2974, 2937, 2872, 2818, 1623, 1460 см-1.
[α]D 23,5+86,4o (с=1,00%, СН3ОН)
(5) Стадия 3': получение бензойной кислой соли [(1R, 2R, 3R, 5S)-2-амино-10-норпинан-3-ил]этанола (II') (см. схему 8 в конце описания).
Соединение (5) (9,63 г, 45,58 ммоль), полученное на стадии 4, растворяли в толуоле (33 мл) и н-пропаноле (72 мл). К раствору добавляли металлический натрий (7,47 г, 325 ммоль) порциями при нагревании с обратным холодильником в течение 25 минут. Через 1 час добавляли дополнительно металлический натрий (1,15 г, 50 ммоль) и продолжали перемешивание в течение 1 часа при нагревании с обратным холодильником. Реакционную смесь охлаждали и разделяли на два слоя путем добавления ледяной воды (39 мл) и толуола (95 мл). Водный слой экстрагировали толуолом (95 мл). Объединенный органический слой промывали рассолом (3•95 мл) и сушили над безводным сульфатом магния, после чего отгоняли при пониженном давлении растворитель с получением 8,4 г указанного в заголовке соединения (II) в виде бесцветного масла. Соединение (II) (8,4 г, 45,8 ммоль) растворяли в толуоле (33,3 мл) и ацетоне (111 мл) и раствор нагревали до 50oС. После добавления раствора бензойной кислоты (4,82 г, 39,47 ммоль) в ацетоне (22,2 мл) смесь перемешивали при той же температуре в течение 1 часа. Выпавшие в осадок кристаллы отфильтровывали, промывали холодным ацетоном (33,3 мл) и высушивали с получением 9,155 г бесцветной бензойной кислой соли (II') аминоспирта. Выход: 65,8%.
ИК (Нуйол): 3428, 2999, 2921, 2864, 2727, 2633, 2596, 2107, 1663, 1623, 1592, 1555, 1523, 1456, 1444 см-1.
[α]D 23,5+27,lo (с=1,01%, СН3ОН), т. пл. 181-183oС.
ПРИМЕР 2
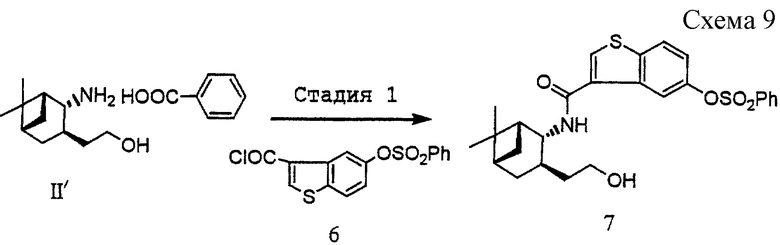
(1) Стадия 1: получение [3-[(1R, 2R, 3R, 5S)-3-(2-гидроксиэтил)-10-норпинан-2-ил]карбамоилбензо[b]тиофен-5-ил]бензолсульфоната (7) (см. схему 9 в конце описания).
Соль (+)-2-[(1R, 2R, 3R, 5S)-2-амино-10-норпинан-3-ил]этанола бензолсульфоновой кислоты (II', 5,1 г, 16,7 ммоль), полученную в примере 1, суспендировали в воде (10 мл). К суспензии добавляли 1 н. HCl (17 мл) и осажденную бензойную кислоту удаляли экстрагированием этилацетатом. Органический слой промывали водой (10 мл). К объединенному водному слою добавляли 4 н. раствор гидроксида натрия (9,2 мл, 36,8 ммоль)
при охлаждении льдом и затем добавляли по каплям в течение 15 минут при перемешивании раствор 5-бензолсульфонилоксибензо[b]тиофен-3-карбонилхлорида (6) (5,89 г, 16,7 ммоль), полученного в ссылочном примере 2, в тетрагидрофуране (36 мл). После перемешивания в течение еще 1 часа при той же самой температуре, добавляли 1 н. хлористоводородную кислоту (4 мл) и смесь экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 8,00 г (95,6%) указанного в заголовке соединения (7) в виде бесцветного аморфного вещества.
1H ЯМР δ (CDCl3), 300 МГц: 0,96 (1Н, д, J=9,9 Гц), 1,12 и 1,26 (каждый 3Н, каждый с), 1,50-2,42 (9Н, м), 3,69-3,82 (2Н, м), 4,40 (1Н, м), 6,21 (1Н, д, J= 8,1 Гц), 7,06 (1Н, дд, J=2,4 и 8,7 Гц), 7,51-7,56 (2Н, м), 7,67 (1Н, м), 7,73 (1Н, д, J=8,7 Гц), 7,85-7,88 (2Н, м), 7,88 (1Н, с), 8,06 (1Н, д, J= 2,4 Гц).
[α]D 25+35,7o (c=l,00%, СН3ОН)
(2) Стадия 2: получение [3-[(1R, 2R, 3R, 5S)-3-формилметил-10-норпинан-2-ил] карбамоилбензо[b]тиофен-5-ил]бензолсульфоната (8) (см. схему 10 в конце описания).
Соединение (7) (9,72 г, 18,3 ммоль) растворяли в этилацетате (70 мл). К раствору добавляли TEMPO (2,2,6,6-тетраметилпиперидин-1-оксил), 14,3 мг, 0,005 эквивалента) и бромид калия (218 мг, 0,1 эквивалента). Добавляли по каплям в течение 3 минут при перемешивании с поддержанием внутренней температуры в пределах от -1oС до -6oС 0,41 н. водный гипохлорит натрия (45 мл раствора, доведенного до рН 9,5 гидрокарбонатом натрия, 1 эквивалент). После 10 минут выдерживания при указанной температуре два слоя разделяли и водный слой экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 9,10 г (100%) указанного в заголовке соединения (8) в виде бесцветного аморфного вещества.
ИК (СНС13); 3443, 3093, 3066, 3030, 3016, 2925, 2871, 2828, 2729, 1720, 1655, 1599, 1558, 1513, 1377 см-1.
1H ЯМР δ (CDC13), 300 МГц: 0,97 (1Н, д, J=10,2 Гц), 1,17 и 1,28 (каждый 3Н, каждый с), 1,46 (1Н, м), 2,03 (1Н, м), 2,22 (1Н, м), 2,36-2,60 (3Н, м), 2,69 (1Н, ддд, J=1,2, 8,7 и 17,4 Гц), 3,14 (1Н, дд, J=4,5 и 17,4 Гц), 4,28 (1H, м), 6,18 (1Н, д, J=8,1 Гц), 7,09 (1H, дд, J=2,4 и 8,7 Гц), 7,50-7,55 (2Н, м), 7,67 (1H, м), 7,75 (1H, д, J=8,7 Гц), 7,85-7,89 (2Н, м), 7,89 (1H, с), 8,03 (1H, д, J=2,4 гц), 9,80 (1H, д, J-1,2 Гц).
[α]D 23+31,8o (с=1,00%, СН3ОН).
(3) Стадия 3: получение (5Z)-7-[(1R, 2R, 3S, 5S)-2-(5-бензолсульфонилоксибензо[b] тиофен-3-илкарбониламино)-10-норпинан-3-ил] -5-гептеновой кислоты (9) (см. схему 11 в конце описания).
4-Карбоксибутилтрифенилфосфонийбромид (12,17 г, 27,5 ммоль) и трет-бутоксид калия (7,19 г, 64,1 ммоль) суспендировали в тетрагидрофуране (64 мл) и перемешивали в течение 1 часа при охлаждении на льду. К реакционной смеси добавляли в течение 15 минут раствор соединения (8) (9,11 г, 18,3 ммоль), полученного на описанной выше стадии 2, в тетрагидрофуране (27 мл) и смесь непрерывно перемешивали 2 часа при той же самой температуре. Реакционную смесь разбавляли водой (80 мл) и промывали толуолом (2•105 мл). После доведения водного слоя до рН 8,1 добавлением 5 н. хлористоводородной кислоты (4,8 мл) добавляли безводный хлорид кальция (8,1 г, 73 ммоль), растворенный в воде (16 мл), и смесь экстрагировали этилацетатом (2•100 мл). В органический слой добавляли воду (100 мл) и водный слой доводили до рН ниже 2 добавлением 5 н. хлористоводородной кислоты и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и затем отгоняли при пониженном давлении растворитель с получением 11,06 г соединения (9), которое использовали без очистки.
(4) Стадия 4: получение (5Z)-7-[(1R, 2R, 3S, 5S)-2-(5-гидроксибензо[b] тиофен-3-илкарбониламино)-10-норпинан-3-ил] -5-гептеновой кислоты (10) (Соединение А) (см. схему 12 в конце описания).
Соединение (9) (11,06 г, 18,3 ммоль), полученное на описанной выше стадии (3), растворяли в диметилсульфоксиде (22 мл). К раствору добавляли 4 н. гидроксид натрия (27,5 мл) и смесь нагревали при 55oС в течение 2 часов при перемешивании. Реакционную смесь разбавляли водой (130 мл) и промывали толуолом (2•65 мл). Водный слой подкисляли 5 н. хлористоводородной кислотой и экстрагировали этилацетатом. Органический слой промывали водой, сушили над безводным сульфатом магния и отгоняли при пониженном давлении растворитель с получением 8,26 г неочищенного целевого соединения (10). Продукт растворяли в метаноле (40 мл) и воде (16 мл) и смесь затравляли и постепенно охлаждали при перемешивании. Выпавшие в осадок кристаллы отфильтровывали и промывали смесью вода-метанол (2: 5) с получением 6,35 г целевого соединения (10). Выход: 78,6%. Кристаллы растворяли в метаноле (40 мл) и к раствору добавляли воду (12 мл) в течение 7 минут при перемешивании. После добавления затравочных кристаллов раствор непрерывно перемешивали при 25oС в течение 1 часа. Добавляли дополнительную воду (7 мл) в течение 40 минут и продолжали перемешивание в течение 1,5 часов при 25oС. Выпавшие в осадок кристаллы отфильтровывали и промывали смесью вода:метанол (3:5) (8 мл) с получением 6,14 г почти бесцветного целевого соединения (10). Выход: 76,0%, т. п. 145-146oС.
ИК (Нуйол): 3313, 3096, 3059, 3001, 1717, 1627, 1603, 1548, 1469, 1440 см-1.
1H ЯМР δ (СОС13), 300 МГц: 1,02 (1Н, д, J=10,2 Гц), 1,12 и 1,24 (каждый 3Н, каждый с), 1,56-2,55 (14Н, м), 4,29 (1Н, м), 5,32-5,51 (2Н, м), 6,20 (1Н, д, J= 9,3 Гц), 7,01 (1Н, дд, J=2,4 и 9,0 Гц), 7,66 (1Н, д, J=9,0 Гц), 7,69 (1Н, с), 8,03 (1Н, д, J=2,4 Гц).
[α]D 24+50,70 (с=1,01%, СН3ОН).
Элементный анализ для C25H31NО4S.
Вычислено (%): С, 68,00; Н, 7,08; N, 3,17; S, 7,26.
Найдено (%): С, 67,84; Н, 7,08; N, 3,24; S, 7,31.





Изобретение относится к способу получения производных бензотиофенкарбоксамида формулы I, включающему взаимодействие аминоспирта формулы (II) или его соли с соединением формулы (III) или его реакционноспособным производным, окисление полученного продукта в присутствии 2,2,6,6-тетраметилпиперидин-1-оксидов и затем осуществление взаимодействия с илидом в условиях реакции Виттига с последующим необязательным снятием защиты. Описаны также способы получения промежуточных аминоспиртов формулы II. Соединения I могут быть использованы в качестве лекарственных средств при лечении системного мастоцитоза, астмы и других заболеваний. Заявленный способ требует простых операций и мало времени, безопасен, поскольку используемыми в реакции растворителями являются вода и этилацетат. 4 с. и 5 з.п. ф-лы.
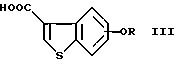

где R представляет водород или гидроксизащитную группу;
Х представляет водород или алкил и двойная связь представляет Е- или Z-конфигурацию,
или его фармацевтически приемлемой соли или гидрата, включающий взаимодействие аминоспирта формулы (II)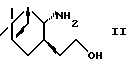
или его соли с соединением формулы (III)
где R представляет водород или гидроксизащитную группу,
или его реакционноспособным производным с получением соединения формулы (I-2)
где R - такой, как определено выше,
окисление соединения (I-2) галогеноксокислотой в присутствии соединения из 2,2,6,6-тетраметилпиперидин-1-оксидов с получением соединения формулы (I-3)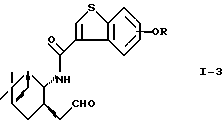
где R - такой, как определено выше,
последующее взаимодействие полученного соединения (I-3) с илидом в условиях реакции Виттига и, если требуется, снятие защиты у продукта реакции.
где R представляет водород или гидроксизащитную группу,
включающий окисление соединения формулы (I-2)
где R - такой, как определено выше,
галогеноксокислотой в присутствии 2,2,6,6-тетраметилпиперидин-1-оксидов.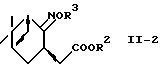
где R2 представляет алкил;
R3 представляет водород или алкил,
используя систему восстановитель-кислота Льюиса.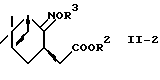
где R2 представляет алкил;
R3 представляет водород или алкил,
с использованием системы восстановитель-кислота Льюиса.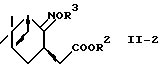
где R2 и R3 - такие, как определено выше,
в спирт формулы (II-3)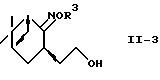
где R3 - такой, как определено выше,
и восстановления полученного спирта с помощью системы металлический натрий-спирт или восстановитель-кислота Льюиса.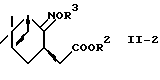
где R2 и R3 - такие, как определено выше,
в спирт формулы (II-3)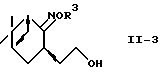
где R3 - такой, как определено выше,
и восстановление спирта с помощью системы металлический натрий-спирт или восстановитель-кислота Льюиса.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| SU 761471 A 07.09.1980. | |||

